User login
Dental Health: What It Means in Kidney Disease
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.

Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.
Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.
Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.

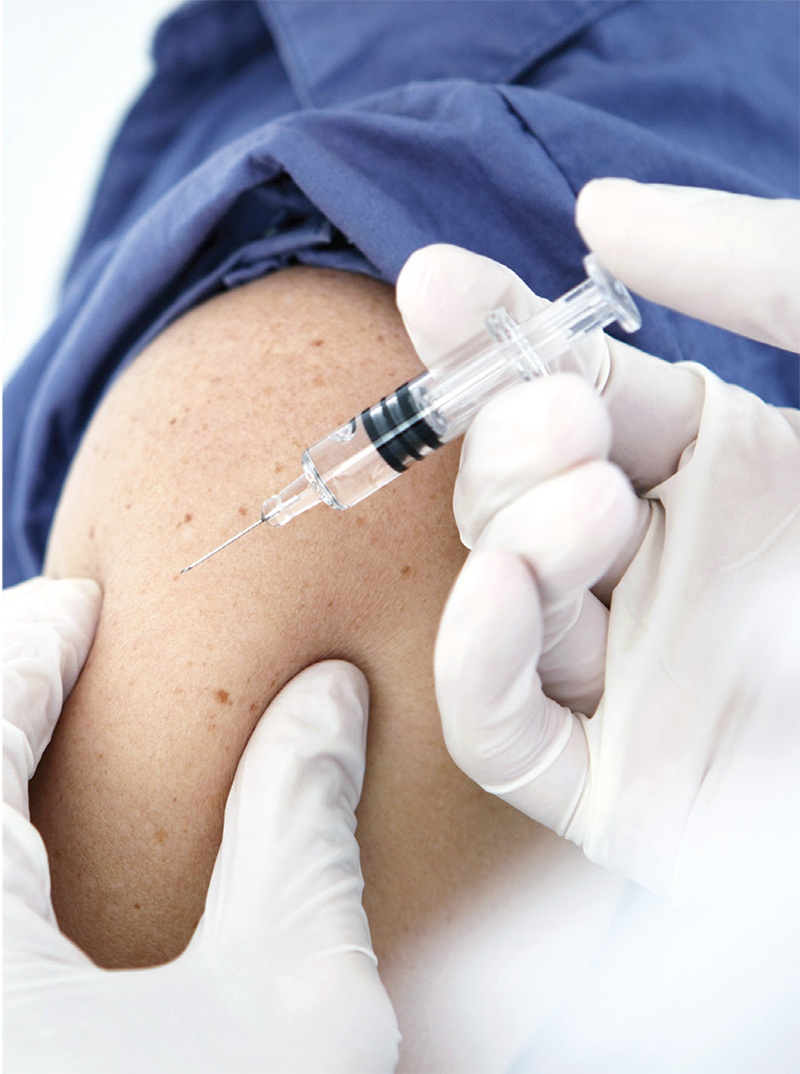
For Patients With CKD, Don’t Wait—Vaccinate!
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
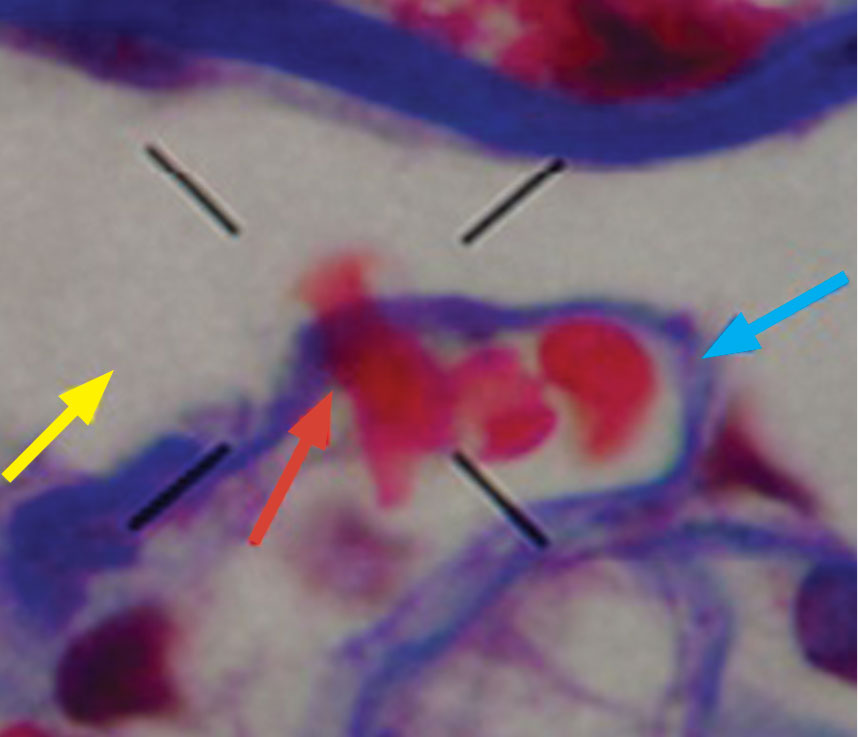
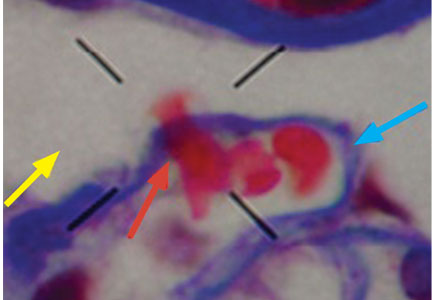
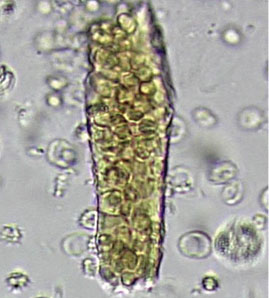
Dysmorphic red blood cell formation
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
.")
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
Quality in urine microscopy: The eyes of the beholder
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE


URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE
URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE
URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
Idiopathic hypercalciuria: Can we prevent stones and protect bones?
A 65-year-old woman was recently diagnosed with osteoporosis after a screening bone mineral density test. She has hypertension (treated with lisinopril), and she had an episode of passing a kidney stone 10 years ago. A 24-hour urine study reveals an elevated urinary calcium level.
What should the physician keep in mind in managing this patient?
IDIOPATHIC HYPERCALCIURIA
Many potential causes of secondary hypercalciuria must be ruled out before deciding that a patient has idiopathic hypercalciuria, which was first noted as a distinct entity by Albright et al in 1953.1 Causes of secondary hypercalciuria include primary hyperparathyroidism, hyperthyroidism, Paget disease, myeloma, malignancy, immobility, accelerated osteoporosis, sarcoidosis, renal tubular acidosis, and drug-induced urinary calcium loss such as that seen with loop diuretics.
Idiopathic hypercalciuria is identified by the following:
- Persistent hypercalciuria despite normal or restricted calcium intake2,3
- Normal levels of parathyroid hormone (PTH), phosphorus, and 1,25-dihydroxy-vitamin D (the active form of vitamin D, also called calcitriol) in the presence of hypercalciuria; serum calcium levels are also normal.
An alias for idiopathic hypercalciuria is “fasting hypercalciuria,” as increased urinary calcium persists and sometimes worsens while fasting or on a low-calcium diet, with increased bone turnover, reduced bone density, and normal serum PTH levels.4,5
Mineral loss from bone predominates in idiopathic hypercalciuria, but there is also a minor component of intestinal hyperabsorption of calcium and reduced renal calcium reabsorption.6 Distinguishing among intestinal hyperabsorptive hypercalciuria, renal leak hypercalciuria, and idiopathic or fasting hypercalciuria can be difficult and subtle. It has been argued that differentiating among hypercalciuric subtypes (hyperabsorptive, renal leak, idiopathic) is not useful; in general clinical practice, it is impractical to collect multiple 24-hour urine samples in the setting of controlled high- vs low-calcium diets.
COMPLICATIONS OF IDIOPATHIC HYPERCALCIURIA
Calcium is an important component in many physiologic processes, including coagulation, cell membrane transfer, hormone release, neuromuscular activation, and myocardial contraction. A sophisticated system of hormonally mediated interactions normally maintains stable extracellular calcium levels. Calcium is vital for bone strength, but the bones are the body’s calcium “bank,” and withdrawals from this bank are made at the expense of bone strength and integrity.
Renal stones
Patients with idiopathic hypercalciuria have a high incidence of renal stones. Conversely, 40% to 50% of patients with recurrent kidney stones have evidence of idiopathic hypercalciuria, the most common metabolic abnormality in “stone-formers.”7,8 Further, 35% to 40% of first- and second-degree relatives of stone-formers who have idiopathic hypercalciuria also have the condition.9 In the general population without kidney stones and without first-degree relatives with stones, the prevalence is approximately 5% to 10%.10,11
Bone loss
People with idiopathic hypercalciuria have lower bone density and a higher incidence of fracture than their normocalciuric peers. This relationship has been observed in both sexes and all ages. Idiopathic hypercalciuria has been noted in 10% to 19% of otherwise healthy men with low bone mass, in postmenopausal women with osteoporosis,10–12 and in up to 40% of postmenopausal women with osteoporotic fractures and no history of kidney stones.13
LABORATORY DEFINITION
Urinary calcium excretion
Heaney et al14 measured 24-hour urinary calcium excretion in a group of early postmenopausal women, whom he divided into 3 groups by dietary calcium intake:
- Low intake (< 500 mg/day)
- Moderate intake (500–1,000 mg/day)
- High intake (> 1,000 mg/day).
In the women who were estrogen-deprived (ie, postmenopausal and not on estrogen replacement therapy), the 95% probability ranges for urinary calcium excretion were:
- 32–252 mg/day (0.51–4.06 mg/kg/day) with low calcium intake
- 36–286 mg/day (0.57–4.52 mg/kg/day) with moderate calcium intake
- 45–357 mg/day (0.69–5.47 mg/kg/day) with high calcium intake.
For estrogen-replete women (perimenopausal or postmenopausal on estrogen replacement), using the same categories of dietary calcium intake, calcium excretion was:
- 39–194 mg/day (0.65–3.23 mg/kg/day) with low calcium intake
- 54–269 mg/day (0.77–3.84 mg/kg/day) with moderate calcium intake
- 66–237 mg/day (0.98–4.89 mg/kg/day) with high calcium intake.
In the estrogen-deprived group, urinary calcium excretion increased by only 55 mg/day per 1,000-mg increase in dietary intake, though there was individual variability. These data suggest that hypercalciuria should be defined as:
- Greater than 250 mg/day (> 4.1 mg/kg/day) in estrogen-replete women
- Greater than 300 mg/day (> 5.0 mg/kg/day) in estrogen-deprived women.
Urinary calcium-to-creatinine ratio
Use of a spot urinary calcium-to-creatinine ratio has been advocated as an alternative to the more labor-intensive 24-hour urine collection.15 However, the spot urine calcium-creatinine ratio correlates poorly with 24-hour urine criteria for hypercalciuria whether by absolute, weight-based, or menopausal and calcium-adjusted definitions.
Importantly, spot urine measurements show poor sensitivity and specificity for hypercalciuria. Spot urine samples underestimate the 24-hour urinary calcium (Bland-Altman bias –71 mg/24 hours), and postprandial sampling overestimates it (Bland-Altman bias +61 mg/24 hours).15
WHAT IS THE MECHANISM OF IDIOPATHIC HYPERCALCIURIA?
The pathophysiology of idiopathic hypercalciuria has been difficult to establish.
Increased sensitivity to vitamin D? In the hyperabsorbing population, activated vitamin D levels are often robust, but a few studies of rats with hyperabsorbing, hyperexcreting physiology have shown normal calcitriol levels, suggesting an increased sensitivity to the actions of 1,25-dihydroxyvitamin D.16
Another study found that hypercalciuric stone-forming rats have more 1,25-dihydroxyvitamin D receptors than do controls.17
These changes have not been demonstrated in patients with idiopathic hypercalciuria.
High sodium intake has been proposed as the cause of idiopathic hypercalciuria. High sodium intake leads to increased urinary sodium excretion, and the increased tubular sodium load can decrease tubular calcium reabsorption, possibly favoring a reduction in bone mineral density over time.18–20
In healthy people, urine calcium excretion increases by about 0.6 mmol/day (20–40 mg/day) for each 100-mmol (2,300 mg) increment in daily sodium ingestion.21,22 But high sodium intake is seldom the principal cause of idiopathic hypercalciuria.
High protein intake, often observed in patients with nephrolithiasis, increases dietary acid load, stimulating release of calcium from bone and inhibiting renal reabsorption of calcium.23,24 Increasing dietary protein from 0.5 to 2.0 mg/kg/day can double the urinary calcium output.25
In mice, induction of metabolic acidosis, thought to mimic a high-protein diet, inhibits osteoblastic alkaline phosphatase activity while stimulating prostaglandin E2 production.26 This in turn increases osteoblastic expression of receptor activator for nuclear factor kappa b (RANK) ligand, thereby potentially contributing to osteoclastogenesis and osteoclast activity.26
Decreasing dietary protein decreases the recurrence of nephrolithiasis in established stone-formers.27 Still, urine calcium levels are higher in those with idiopathic hypercalciuria than in normal controls at comparable levels of acid excretion, so while protein ingestion could potentially exacerbate the hypercalciuria, it is unlikely to be the sole cause.
Renal calcium leak? The frequent finding of low to low-normal PTH levels in patients with idiopathic hypercalciuria contradicts the potential etiologic mechanism of renal calcium “leak.” In idiopathic hypercalciuria, the PTH response to an oral calcium load is abnormal. If given an oral calcium load, the PTH level should decline if this were due to renal leak, but in the setting of idiopathic hypercalciuria, no clinically meaningful change in PTH occurs. This lack of response of PTH to oral calcium load has been seen in both rat and human studies. Patients also excrete normal to high amounts of urine calcium after prolonged fasting or a low-calcium diet. Low-calcium diets do not induce hyperparathyroidism in these patients, and so the source of the elevated calcium in the urine must be primarily from bone. Increased levels of 1,25-dihydroxyvitamin D in patients with idiopathic hypercalciuria have been noted.28,29
Whether the cytokine milieu also contributes to the calcitriol levels is unclear, but the high or high-normal plasma level of 1,25-dihydroxyvitamin D may be the reason that the PTH is unperturbed.
IMPACT ON BONE HEALTH
Nephrolithiasis is strongly linked to fracture risk.
The bone mineral density of trabecular bone is more affected by calcium excretion than that of cortical bone.18,20,30 However, lumbar spine bone mineral density has not been consistently found to be lower in patients with hyperabsorptive hypercalciuria. Rather, bone mineral density is correlated inversely with urine calcium excretion in men and women who form stones, but not in patients without nephrolithiasis.
In children
In children, idiopathic hypercalciuria is well known to be linked to osteopenia. This is an important group to study, as adult idiopathic hypercalciuria often begins in childhood. However, the trajectory of bone loss vs gain in children is fraught with variables such as growth, puberty, and body mass index, making this a difficult group from which to extrapolate conclusions to adults.
In men
There is more information on the relationship between hypercalciuria and osteoporosis in men than in women.
In 1998, Melton et al31 published the findings of a 25-year population-based cohort study of 624 patients, 442 (71%) of whom were men, referred for new-onset urolithiasis. The incidence of vertebral fracture was 4 times higher in this group than in patients without stone disease, but there was no difference in the rate of hip, forearm, or nonvertebral fractures. This is consistent with earlier data that report a loss of predominantly cancellous bone associated with urolithiasis.
National Health and Nutrition Examination Survey III data in 2001 focused on a potential relationship between kidney stones and bone mineral density or prevalent spine or wrist fracture.32 More than 14,000 people had hip bone mineral density measurements, of whom 793 (477 men, 316 women) had kidney stones. Men with previous nephrolithiasis had lower femoral neck bone mineral density than those without. Men with kidney stones were also more likely to report prevalent wrist and spine fractures. In women, no difference was noted between those with or without stone disease with respect to femoral neck bone mineral density or fracture incidence.
Cauley et al33 also evaluated a relationship between kidney stones and bone mineral density in the Osteoporotic Fractures in Men (MrOS) study. Of approximately 6,000 men, 13.2% reported a history of kidney stones. These men had lower spine and total hip bone mineral density than controls who had not had kidney stones, and the difference persisted after adjusting for age, race, weight, and other variables. However, further data from this cohort revealed that so few men with osteoporosis had hypercalciuria that its routine measurement was not recommended.34
In women
The relationship between idiopathic hypercalciuria and fractures has been more difficult to establish in women.
Sowers et al35 performed an observational study of 1,309 women ages 20 to 92 with a history of nephrolithiasis. No association was noted between stone disease and reduced bone mineral density in the femoral neck, lumbar spine, or radius.
These epidemiologic studies did not include the cause of the kidney stones (eg, whether or not there was associated hypercalciuria or primary hyperparathyroidism), and typically a diagnosis of idiopathic hypercalciuria was not established.
The difference in association between low bone mineral density or fracture with nephrolithiasis between men and women is not well understood, but the most consistent hypothesis is that the influence of hypoestrogenemia in women is much stronger than that of the hypercalciuria.20
Does the degree of hypercalciuria influence the amount of bone loss?
A few trials have tried to determine whether the amount of calcium in the urine influences the magnitude of bone loss.
In 2003, Asplin et al36 reported that bone mineral density Z-scores differed significantly by urinary calcium excretion, but only in stone-formers. In patients without stone disease, there was no difference in Z-scores according to the absolute value of hypercalciuria. This may be due to a self-selection bias in which stone-formers avoid calcium in the diet and those without stone disease do not.
Three studies looking solely at men with idiopathic hypercalciuria also did not detect a significant difference in bone mineral loss according to degree of hypercalciuria.20,30,37
A POLYGENIC DISORDER?
The potential contribution of genetic changes to the development of idiopathic hypercalciuria has been studied. While there is an increased risk of idiopathic hypercalciuria in first-degree relatives of patients with nephrolithiasis, most experts believe that idiopathic hypercalciuria is likely a polygenic disorder.9,38
EVALUATION AND TREATMENT
The 2014 revised version of the National Osteoporosis Foundation’s “Clinician’s guide to prevention and treatment of osteoporosis”39 noted that hypercalciuria is a risk factor that contributes to the development of osteoporosis and possibly osteoporotic fractures, and that consideration should be given to evaluating for hypercalciuria, but only in selected cases. In patients with kidney stones, the link between hypercalciuria and bone loss and fracture is recognized and should be explored in both women and men at risk of osteoporosis, as 45% to 50% of patients who form calcium stones have hypercalciuria.
Patients with kidney stones who have low bone mass and idiopathic hypercalciuria should increase their daily fluid intake, follow a low-salt and low-animal-protein diet, and take thiazide diuretics to reduce the incidence of further calcium stones. Whether this approach also improves bone mass and strength and reduces the risk of fractures within this cohort requires further study.
Dietary interventions
Don’t restrict calcium intake. Despite the connection between hypercalciuria and nephrolithiasis, restriction of dietary calcium to prevent relapse of nephrolithiasis is a risk factor for negative calcium balance and bone demineralization. Observational studies and prospective clinical trials have demonstrated an increased risk of stone formation with low calcium intake.27,30 Nevertheless, this practice seems logical to many patients with kidney stones, and this process may independently contribute to lower bone mineral density.
A low-sodium, low-animal-protein diet is beneficial. Though increased intake of sodium or protein is not the main cause of idiopathic hypercalciuria, pharmacologic therapy, especially with thiazide diuretics, is more likely to be successful in the setting of a low-sodium, low-protein diet.
Borghi et al27 studied 2 diets in men with nephrolithiasis and idiopathic hypercalciuria: a low-calcium diet and a low-salt, low-animal-protein, normal-calcium diet. Men on the latter diet experienced a greater reduction in urinary calcium excretion than those on the low-calcium diet.
Breslau et al40 found that urinary calcium excretion fell by 50% in 15 people when they switched from an animal-based to a plant-based protein diet.
Thiazide diuretics
Several epidemiologic and randomized studies41–45 found that thiazide therapy decreased the likelihood of hip fracture in postmenopausal women, men, and premenopausal women. Doses ranged from 12.5 to 50 mg of hydrochlorothiazide. Bone density increased in the radius, total body, total hip, and lumbar spine. One prospective trial noted that fracture risk declined with longer duration of thiazide use, with the largest reduction in those who used thiazides for 8 or more years.46
Thiazides have anticalciuric actions.47 In addition, they have positive effects on osteoblastic cell proliferation and activity, inhibiting osteocalcin expression by osteoblasts, thereby possibly improving bone formation and mineralization.48 The effects of thiazides on bone was reviewed by Sakhaee et al.49
However, fewer studies have looked at thiazides in patients with idiopathic hypercalciuria.
García-Nieto et al50 looked retrospectively at 22 children (average age 11.7) with idiopathic hypercalciuria and osteopenia who had received thiazides (19 received chlorthalidone 25 mg daily, and 3 received hydrochlorothiazide 25 mg daily) for an average of 2.4 years, and at 32 similar patients who had not received thiazides. Twelve (55%) of the patients receiving thiazides had an improvement in bone mineral density Z-scores, compared with 23 (72%) of the controls. This finding is confounded by growth that occurred during the study, and both groups demonstrated a significantly increased body mass index and bone mineral apparent density at the end of the trial.
Bushinsky and Favus51 evaluated whether chlorthalidone improved bone quality or structure in rats that were genetically prone to hypercalciuric stones. These rats are uniformly stone-formers, and while they have components of calcium hyperabsorption, they also demonstrate renal hyperexcretion (leak) and enhanced bone mineral resorption.51 When fed a high-calcium diet, they maintain a reduction in bone mineral density and bone strength. Study rats were given chlorthalidone 4 to 5 mg/kg/day. After 18 weeks of therapy, significant improvements were observed in trabecular thickness and connectivity as well as increased vertebral compressive strength.52 No difference in cortical bone was noted.
No randomized, blinded, placebo-controlled trial has yet been done to study the impact of thiazides on bone mineral density or fracture risk in patients with idiopathic hypercalciuria.
In practice, many physicians choose chlorthalidone over hydrochlorothiazide because of chlorthalidone’s longer half-life. Combinations of a thiazide diuretic and potassium-sparing medications are also employed, such as hydrochlorothiazide plus either triamterene or spironolactone to reduce the number of pills the patient has to take.
Potassium citrate
When prescribing thiazide diuretics, one should also consider prescribing potassium citrate, as this agent not only prevents hypokalemia but also increases urinary citrate excretion, which can help to inhibit crystallization of calcium salts.6
In a longitudinal study of 28 patients with hypercalciuria,53 combined therapy with a thiazide or indapamide and potassium citrate over a mean of 7 years increased bone density of the lumbar spine by 7.1% and of the femoral neck by 4.1%, compared with treatment in age- and sex-matched normocalcemic peers. In the same study, daily urinary calcium excretion decreased and urinary pH and citrate levels increased; urinary saturation of calcium oxalate decreased by 46%, and stone formation was decreased.
Another trial evaluated 120 patients with idiopathic calcium nephrolithiasis, half of whom were given potassium citrate. Those given potassium citrate experienced an increase in distal radius bone mineral density over 2 years.54 It is theorized that alkalinization may decrease bone turnover in these patients.
Bisphosphonates
As one of the proposed main mechanisms of bone loss in idiopathic hypercalciuria is direct bone resorption, a potential target for therapy is the osteoclast, which bisphosphonates inhibit.
Ruml et al55 studied the impact of alendronate vs placebo in 16 normal men undergoing 3 weeks of strict bedrest. Compared with the placebo group, those who received alendronate had significantly lower 24-hour urine calcium excretion and higher levels of PTH and 1,25-dihydroxyvitamin D.
Weisinger et al56 evaluated the effects of alendronate 10 mg daily in 10 patients who had stone disease with documented idiopathic hypercalciuria and also in 8 normocalciuric patients without stone disease. Alendronate resulted in a sustained reduction of calcium in the urine in the patients with idiopathic hypercalciuria but not in the normocalciuric patients.
Data are somewhat scant as to the effect of bisphosphonates on bone health in the setting of idiopathic hypercalciuria,57,58 and therapy with bisphosphonates is not recommended in patients with idiopathic hypercalciuria outside the realm of postmenopausal osteoporosis or other indications for bisphosphonates approved by the US Food and Drug Administration (FDA).
Calcimimetics
Calcium-sensing receptors are found not only in parathyroid tissue but also in the intestines and kidneys. Locally, elevated plasma calcium in the kidney causes activation of the calcium-sensing receptor, diminishing further calcium reabsorption.59 Agents that increase the sensitivity of the calcium-sensing receptors are classified as calcimimetics.
Cinacalcet is a calcimimetic approved by the FDA for treatment of secondary hyperparathyroidism in patients with chronic kidney disease on dialysis, for the treatment of hypercalcemia in patients with parathyroid carcinoma, and for patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. In an uncontrolled 5-year study of cinacalcet in patients with primary hyperparathyroidism, there was no significant change in bone density.60
Anti-inflammatory drugs
The role of cytokines in stimulating bone resorption in idiopathic hypercalciuria has led to the investigation of several anti-inflammatory drugs (eg, diclofenac, indomethacin) as potential treatments, but studies have been limited in number and scope.61,62
Omega-3 fatty acids
Omega-3 fatty acids are thought to alter prostaglandin metabolism and to potentially reduce stone formation.63
A retrospective study of 29 patients with stone disease found that, combined with dietary counseling, omega-3 fatty acids could potentially reduce urinary calcium and oxalate excretion and increase urinary citrate in hypercalciuric stone-formers.64
A review of published randomized controlled trials of omega-3 fatty acids in skeletal health discovered that 4 studies found positive effects on bone mineral density or bone turnover markers, whereas 5 studies reported no differences. All trials were small, and none evaluated fracture outcome.65
- Albright F, Henneman P, Benedict PH, Forbes AP. Idiopathic hypercalciuria: a preliminary report. Proc R Soc Med 1953; 46:1077–1081.
- Pak CY. Pathophysiology of calcium nephrolithiasis. In: Seldin DW, Giebiscg G, eds. The Kidney: Physiology and Pathophysiology. New York, NY: Raven Press; 1992:2461–2480.
- Frick KK, Bushinsky DA. Molecular mechanisms of primary hypercalciuria. J Am Soc Nephrol 2003; 14:1082–1095.
- Pacifici R, Rothstein M, Rifas L, et al. Increased monocyte interleukin-1 activity and decreased vertebral bone density in patients with fasting idiopathic hypercalciuria. J Clin Endocrinol Metab 1990; 71:138–145.
- Messa P, Mioni G, Montanaro D, et al. About a primitive osseous origin of the so-called ‘renal hypercalciuria.’ Contrib Nephrol 1987; 58:106–110.
- Zerwekh JE. Bone disease and idiopathic hypercalciuria. Semin Nephrol 2008; 28:133–142.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Lemann J Jr. Pathogenesis of idiopathic hypercalciuria and nephrolithiasis. In: Coe FL, Favus MJ, eds. Disorders of Bone and Mineral Metabolism. New York, NY: Raven Press; 1992:685-706.
- Coe FL, Parks JH, Moore ES. Familial idiopathic hypercalciuria. N Engl J Med 1979; 300:337–340.
- Giannini S, Nobile M, Dalle Carbonare L, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. Eur J Endocrinol 2003; 149:209–213.
- Tannenbaum C, Clark J, Schwartzman K, et al. Yield of laboratory testing to identify secondary contributors to osteoporosis in otherwise healthy women. J Clin Endocrinol Metab 2002; 87:4431–4437.
- Cerda Gabaroi D, Peris P, Monegal A, et al. Search for hidden secondary causes in postmenopausal women with osteoporosis. Menopause 2010; 17:135–139.
- Rull MA, Cano-García Mdel C, Arrabal-Martín M, Arrabal-Polo MA. The importance of urinary calcium in postmenopausal women with osteoporotic fracture. Can Urol Assoc J 2015; 9:E183–E186.
- Heaney RP, Recker RR, Ryan RA. Urinary calcium in perimenopausal women: normative values. Osteoporos Int 1999; 9:13–18.
- Bleich HL, Moore MJ, Lemann J Jr, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Li XQ, Tembe V, Horwitz GM, Bushinsky DA, Favus MJ. Increased intestinal vitamin D receptor in genetic hypercalciuric rats. A cause of intestinal calcium hyperabsorption. J Clin Invest 1993; 91:661–667.
- Yao J, Kathpalia P, Bushinsky DA, Favus MJ. Hyperresponsiveness of vitamin D receptor gene expression to 1,25-dihydroxyvitamin D3. A new characteristic of genetic hypercalciuric stone-forming rats. J Clin Invest 1998; 101:2223–2232.
- Pietschmann F, Breslau NA, Pak CY. Reduced vertebral bone density in hypercalciuric nephrolithiasis. J Bone Miner Res 1992; 7:1383–1388.
- Jaeger P, Lippuner K, Casez JP, Hess B, Ackermann D, Hug C. Low bone mass in idiopathic renal stone formers: magnitude and significance. J Bone Miner Res 1994; 9:1525–1532.
- Vezzoli G, Soldati L, Arcidiacono T, et al. Urinary calcium is a determinant of bone mineral density in elderly men participating in the InCHIANTI study. Kidney Int 2005; 67:2006–2014.
- Lemann J Jr, Worcester EM, Gray RW. Hypercalciuria and stones. Am J Kidney Dis 1991; 17:386–391.
- Gokce C, Gokce O, Baydinc C, et al. Use of random urine samples to estimate total urinary calcium and phosphate excretion. Arch Intern Med 1991; 151:1587–1588.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Siener R, Schade N, Nicolay C, von Unruh GE, Hesse A. The efficacy of dietary intervention on urinary risk factors for stone formation in recurrent calcium oxalate stone patients. J Urol 2005; 173:1601–1605.
- Jones AN, Shafer MM, Keuler NS, Crone EM, Hansen KE. Fasting and postprandial spot urine calcium-to-creatinine ratios do not detect hypercalciuria. Osteoporos Int 2012; 23:553–562.
- Frick KK, Bushinsky DA. Metabolic acidosis stimulates RANKL RNA expression in bone through a cyclo-oxygenase-dependent mechanism. J Bone Miner Res 2003; 18:1317–1325.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Ghazali A, Fuentes V, Desaint C, et al. Low bone mineral density and peripheral blood monocyte activation profile in calcium stone formers with idiopathic hypercalciuria. J Clin Endocrinol Metab 1997; 82:32–38.
- Broadus AE, Insogna KL, Lang R, Ellison AF, Dreyer BE. Evidence for disordered control of 1,25-dihydroxyvitamin D production in absorptive hypercalciuria. N Engl J Med 1984; 311:73–80.
- Tasca A, Cacciola A, Ferrarese P, et al. Bone alterations in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Urology 2002; 59:865–869.
- Melton LJ 3rd, Crowson CS, Khosla S, Wilson DM, O’Fallon WM. Fracture risk among patients with urolithiasis: a population-based cohort study. Kidney Int 1998; 53:459–464.
- Lauderdale DS, Thisted RA, Wen M, Favus MJ. Bone mineral density and fracture among prevalent kidney stone cases in the Third National Health and Nutrition Examination Survey. J Bone Miner Res 2001; 16:1893–1898.
- Cauley JA, Fullman RL, Stone KL, et al; MrOS Research Group. Factors associated with the lumbar spine and proximal femur bone mineral density in older men. Osteoporos Int 2005; 16:1525–1537.
- Fink HA, Litwack-Harrison S, Taylor BC, et al; Osteoporotic Fractures in Men (MrOS) Study Group. Clinical utility of routine laboratory testing to identify possible secondary causes in older men with osteoporosis: the Osteoporotic Fractures in Men (MrOS) Study. Osteoporos Int 2016: 27:331–338.
- Sowers MR, Jannausch M, Wood C, Pope SK, Lachance LL, Peterson B. Prevalence of renal stones in a population-based study with dietary calcium, oxalate and medication exposures. Am J Epidemiol 1998; 147:914–920.
- Asplin JR, Bauer KA, Kinder J, et al. Bone mineral density and urine calcium excretion among subjects with and without nephrolithiasis. Kidney Int 2003; 63:662–669.
- Letavernier E, Traxer O, Daudon M, et al. Determinants of osteopenia in male renal-stone-disease patients with idiopathic hypercalciuria. Clin J Am Soc Nephrol 2011; 6:1149–1154.
- Vezzoli G, Soldati L, Gambaro G. Update on primary hypercalciuria from a genetic perspective. J Urol 2008; 179:1676–1682.
- Cosman F, de Beur SJ, LeBoff MS, et al; National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int 2014: 25:2359–2381.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Reid IR, Ames RW, Orr-Walker BJ, et al. Hydrochlorothiazide reduces loss of cortical bone in normal postmenopausal women: a randomized controlled trial. Am J Med 2000; 109:362–370.
- Bolland MJ, Ames RW, Horne AM, Orr-Walker BJ, Gamble GD, Reid IR. The effect of treatment with a thiazide diuretic for 4 years on bone density in normal postmenopausal women. Osteoporos Int 2007; 18:479–486.
- LaCroix AZ, Ott SM, Ichikawa L, Scholes D, Barlow WE. Low-dose hydrochlorothiazide and preservation of bone mineral density in older adults. Ann Intern Med 2000; 133:516–526.
- Wasnich RD, Davis JW, He YF, Petrovich H, Ross PD. A randomized, double-masked, placebo-controlled trial of chlorthalidone and bone loss in elderly women. Osteoporos Int 1995; 5:247–251.
- Adams JS, Song CF, Kantorovich V. Rapid recovery of bone mass in hypercalciuric, osteoporotic men treated with hydrochlorothiazide. Ann Intern Med 1999; 130:658–660.
- Feskanich D, Willett WC, Stampfer MJ, Colditz GA. A prospective study of thiazide use and fractures in women. Osteoporos Int 1997; 7:79–84.
- Lamberg BA, Kuhlback B. Effect of chlorothiazide and hydrochlorothiazide on the excretion of calcium in the urine. Scand J Clin Lab Invest 1959; 11:351–357.
- Lajeunesse D, Delalandre A, Guggino SE. Thiazide diuretics affect osteocalcin production in human osteoblasts at the transcription level without affecting vitamin D3 receptors. J Bone Miner Res 2000; 15:894–901.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2001; 79:393–403.
- García-Nieto V, Monge-Zamorano M, González-García M, Luis-Yanes MI. Effect of thiazides on bone mineral density in children with idiopathic hypercalciuria. Pediatr Nephrol 2012; 27:261–268.
- Bushinsky DA, Favus MJ. Mechanism of hypercalciuria in genetic hypercalciuric rats. Inherited defect in intestinal calcium transport. J Clin Invest 1988; 82:1585–1591.
- Bushinsky DA, Willett T, Asplin JR, Culbertson C, Che SP, Grynpas M. Chlorthalidone improves vertebral bone quality in genetic hypercalciuric stone-forming rats. J Bone Miner Res 2011; 26:1904–1912.
- Pak CY, Heller HJ, Pearle MS, Odvina CV, Poindexter JR, Peterson RD. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol 2003; 169:465–469.
- Vescini F, Buffa A, LaManna G, et al. Long-term potassium citrate therapy and bone mineral density in idiopathic calcium stone formers. J Endocrinol Invest 2005; 28:218–222.
- Ruml LA, Dubois SK, Roberts ML, Pak CY. Prevention of hypercalciuria and stone-forming propensity during prolonged bedrest by alendronate. J Bone Miner Res 1995; 10:655–662.
- Weisinger JR, Alonzo E, Machado C, et al. Role of bones in the physiopathology of idiopathic hypercalciuria: effect of amino-bisphosphonate alendronate. Medicina (B Aires) 1997; 57(suppl 1):45–48. Spanish.
- Heilberg IP, Martini LA, Teixeira SH, et al. Effect of etidronate treatment on bone mass of male nephrolithiasis patients with idiopathic hypercalciuria and osteopenia. Nephron 1998; 79:430–437.
- Bushinsky DA, Neumann KJ, Asplin J, Krieger NS. Alendronate decreases urine calcium and supersaturation in genetic hypercalciuric rats. Kidney Int 1999; 55:234–243.
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 1995; 92:131–145.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009; 94:4860–4867.
- Filipponi P, Mannarelli C, Pacifici R, et al. Evidence for a prostaglandin-mediated bone resorptive mechanism in subjects with fasting hypercalciuria. Calcif Tissue Int 1988; 43:61–66.
- Gomaa AA, Hassan HA, Ghaneimah SA. Effect of aspirin and indomethacin on the serum and urinary calcium, magnesium and phosphate. Pharmacol Res 1990; 22:59–70.
- Buck AC, Davies RL, Harrison T. The protective role of eicosapentaenoic acid (EPA) in the pathogenesis of nephrolithiasis. J Urol 1991; 146:188–194.
- Ortiz-Alvarado O, Miyaoka R, Kriedberg C, et al. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid in the management of hypercalciuric stone formers. Urology 2012; 79:282–286.
- Orchard TS, Pan X, Cheek F, Ing SW, Jackson RD. A systematic review of omega-3 fatty acids and osteoporosis. Br J Nutr 2012; 107(suppl 2):S253–S260.
A 65-year-old woman was recently diagnosed with osteoporosis after a screening bone mineral density test. She has hypertension (treated with lisinopril), and she had an episode of passing a kidney stone 10 years ago. A 24-hour urine study reveals an elevated urinary calcium level.
What should the physician keep in mind in managing this patient?
IDIOPATHIC HYPERCALCIURIA
Many potential causes of secondary hypercalciuria must be ruled out before deciding that a patient has idiopathic hypercalciuria, which was first noted as a distinct entity by Albright et al in 1953.1 Causes of secondary hypercalciuria include primary hyperparathyroidism, hyperthyroidism, Paget disease, myeloma, malignancy, immobility, accelerated osteoporosis, sarcoidosis, renal tubular acidosis, and drug-induced urinary calcium loss such as that seen with loop diuretics.
Idiopathic hypercalciuria is identified by the following:
- Persistent hypercalciuria despite normal or restricted calcium intake2,3
- Normal levels of parathyroid hormone (PTH), phosphorus, and 1,25-dihydroxy-vitamin D (the active form of vitamin D, also called calcitriol) in the presence of hypercalciuria; serum calcium levels are also normal.
An alias for idiopathic hypercalciuria is “fasting hypercalciuria,” as increased urinary calcium persists and sometimes worsens while fasting or on a low-calcium diet, with increased bone turnover, reduced bone density, and normal serum PTH levels.4,5
Mineral loss from bone predominates in idiopathic hypercalciuria, but there is also a minor component of intestinal hyperabsorption of calcium and reduced renal calcium reabsorption.6 Distinguishing among intestinal hyperabsorptive hypercalciuria, renal leak hypercalciuria, and idiopathic or fasting hypercalciuria can be difficult and subtle. It has been argued that differentiating among hypercalciuric subtypes (hyperabsorptive, renal leak, idiopathic) is not useful; in general clinical practice, it is impractical to collect multiple 24-hour urine samples in the setting of controlled high- vs low-calcium diets.
COMPLICATIONS OF IDIOPATHIC HYPERCALCIURIA
Calcium is an important component in many physiologic processes, including coagulation, cell membrane transfer, hormone release, neuromuscular activation, and myocardial contraction. A sophisticated system of hormonally mediated interactions normally maintains stable extracellular calcium levels. Calcium is vital for bone strength, but the bones are the body’s calcium “bank,” and withdrawals from this bank are made at the expense of bone strength and integrity.
Renal stones
Patients with idiopathic hypercalciuria have a high incidence of renal stones. Conversely, 40% to 50% of patients with recurrent kidney stones have evidence of idiopathic hypercalciuria, the most common metabolic abnormality in “stone-formers.”7,8 Further, 35% to 40% of first- and second-degree relatives of stone-formers who have idiopathic hypercalciuria also have the condition.9 In the general population without kidney stones and without first-degree relatives with stones, the prevalence is approximately 5% to 10%.10,11
Bone loss
People with idiopathic hypercalciuria have lower bone density and a higher incidence of fracture than their normocalciuric peers. This relationship has been observed in both sexes and all ages. Idiopathic hypercalciuria has been noted in 10% to 19% of otherwise healthy men with low bone mass, in postmenopausal women with osteoporosis,10–12 and in up to 40% of postmenopausal women with osteoporotic fractures and no history of kidney stones.13
LABORATORY DEFINITION
Urinary calcium excretion
Heaney et al14 measured 24-hour urinary calcium excretion in a group of early postmenopausal women, whom he divided into 3 groups by dietary calcium intake:
- Low intake (< 500 mg/day)
- Moderate intake (500–1,000 mg/day)
- High intake (> 1,000 mg/day).
In the women who were estrogen-deprived (ie, postmenopausal and not on estrogen replacement therapy), the 95% probability ranges for urinary calcium excretion were:
- 32–252 mg/day (0.51–4.06 mg/kg/day) with low calcium intake
- 36–286 mg/day (0.57–4.52 mg/kg/day) with moderate calcium intake
- 45–357 mg/day (0.69–5.47 mg/kg/day) with high calcium intake.
For estrogen-replete women (perimenopausal or postmenopausal on estrogen replacement), using the same categories of dietary calcium intake, calcium excretion was:
- 39–194 mg/day (0.65–3.23 mg/kg/day) with low calcium intake
- 54–269 mg/day (0.77–3.84 mg/kg/day) with moderate calcium intake
- 66–237 mg/day (0.98–4.89 mg/kg/day) with high calcium intake.
In the estrogen-deprived group, urinary calcium excretion increased by only 55 mg/day per 1,000-mg increase in dietary intake, though there was individual variability. These data suggest that hypercalciuria should be defined as:
- Greater than 250 mg/day (> 4.1 mg/kg/day) in estrogen-replete women
- Greater than 300 mg/day (> 5.0 mg/kg/day) in estrogen-deprived women.
Urinary calcium-to-creatinine ratio
Use of a spot urinary calcium-to-creatinine ratio has been advocated as an alternative to the more labor-intensive 24-hour urine collection.15 However, the spot urine calcium-creatinine ratio correlates poorly with 24-hour urine criteria for hypercalciuria whether by absolute, weight-based, or menopausal and calcium-adjusted definitions.
Importantly, spot urine measurements show poor sensitivity and specificity for hypercalciuria. Spot urine samples underestimate the 24-hour urinary calcium (Bland-Altman bias –71 mg/24 hours), and postprandial sampling overestimates it (Bland-Altman bias +61 mg/24 hours).15
WHAT IS THE MECHANISM OF IDIOPATHIC HYPERCALCIURIA?
The pathophysiology of idiopathic hypercalciuria has been difficult to establish.
Increased sensitivity to vitamin D? In the hyperabsorbing population, activated vitamin D levels are often robust, but a few studies of rats with hyperabsorbing, hyperexcreting physiology have shown normal calcitriol levels, suggesting an increased sensitivity to the actions of 1,25-dihydroxyvitamin D.16
Another study found that hypercalciuric stone-forming rats have more 1,25-dihydroxyvitamin D receptors than do controls.17
These changes have not been demonstrated in patients with idiopathic hypercalciuria.
High sodium intake has been proposed as the cause of idiopathic hypercalciuria. High sodium intake leads to increased urinary sodium excretion, and the increased tubular sodium load can decrease tubular calcium reabsorption, possibly favoring a reduction in bone mineral density over time.18–20
In healthy people, urine calcium excretion increases by about 0.6 mmol/day (20–40 mg/day) for each 100-mmol (2,300 mg) increment in daily sodium ingestion.21,22 But high sodium intake is seldom the principal cause of idiopathic hypercalciuria.
High protein intake, often observed in patients with nephrolithiasis, increases dietary acid load, stimulating release of calcium from bone and inhibiting renal reabsorption of calcium.23,24 Increasing dietary protein from 0.5 to 2.0 mg/kg/day can double the urinary calcium output.25
In mice, induction of metabolic acidosis, thought to mimic a high-protein diet, inhibits osteoblastic alkaline phosphatase activity while stimulating prostaglandin E2 production.26 This in turn increases osteoblastic expression of receptor activator for nuclear factor kappa b (RANK) ligand, thereby potentially contributing to osteoclastogenesis and osteoclast activity.26
Decreasing dietary protein decreases the recurrence of nephrolithiasis in established stone-formers.27 Still, urine calcium levels are higher in those with idiopathic hypercalciuria than in normal controls at comparable levels of acid excretion, so while protein ingestion could potentially exacerbate the hypercalciuria, it is unlikely to be the sole cause.
Renal calcium leak? The frequent finding of low to low-normal PTH levels in patients with idiopathic hypercalciuria contradicts the potential etiologic mechanism of renal calcium “leak.” In idiopathic hypercalciuria, the PTH response to an oral calcium load is abnormal. If given an oral calcium load, the PTH level should decline if this were due to renal leak, but in the setting of idiopathic hypercalciuria, no clinically meaningful change in PTH occurs. This lack of response of PTH to oral calcium load has been seen in both rat and human studies. Patients also excrete normal to high amounts of urine calcium after prolonged fasting or a low-calcium diet. Low-calcium diets do not induce hyperparathyroidism in these patients, and so the source of the elevated calcium in the urine must be primarily from bone. Increased levels of 1,25-dihydroxyvitamin D in patients with idiopathic hypercalciuria have been noted.28,29
Whether the cytokine milieu also contributes to the calcitriol levels is unclear, but the high or high-normal plasma level of 1,25-dihydroxyvitamin D may be the reason that the PTH is unperturbed.
IMPACT ON BONE HEALTH
Nephrolithiasis is strongly linked to fracture risk.
The bone mineral density of trabecular bone is more affected by calcium excretion than that of cortical bone.18,20,30 However, lumbar spine bone mineral density has not been consistently found to be lower in patients with hyperabsorptive hypercalciuria. Rather, bone mineral density is correlated inversely with urine calcium excretion in men and women who form stones, but not in patients without nephrolithiasis.
In children
In children, idiopathic hypercalciuria is well known to be linked to osteopenia. This is an important group to study, as adult idiopathic hypercalciuria often begins in childhood. However, the trajectory of bone loss vs gain in children is fraught with variables such as growth, puberty, and body mass index, making this a difficult group from which to extrapolate conclusions to adults.
In men
There is more information on the relationship between hypercalciuria and osteoporosis in men than in women.
In 1998, Melton et al31 published the findings of a 25-year population-based cohort study of 624 patients, 442 (71%) of whom were men, referred for new-onset urolithiasis. The incidence of vertebral fracture was 4 times higher in this group than in patients without stone disease, but there was no difference in the rate of hip, forearm, or nonvertebral fractures. This is consistent with earlier data that report a loss of predominantly cancellous bone associated with urolithiasis.
National Health and Nutrition Examination Survey III data in 2001 focused on a potential relationship between kidney stones and bone mineral density or prevalent spine or wrist fracture.32 More than 14,000 people had hip bone mineral density measurements, of whom 793 (477 men, 316 women) had kidney stones. Men with previous nephrolithiasis had lower femoral neck bone mineral density than those without. Men with kidney stones were also more likely to report prevalent wrist and spine fractures. In women, no difference was noted between those with or without stone disease with respect to femoral neck bone mineral density or fracture incidence.
Cauley et al33 also evaluated a relationship between kidney stones and bone mineral density in the Osteoporotic Fractures in Men (MrOS) study. Of approximately 6,000 men, 13.2% reported a history of kidney stones. These men had lower spine and total hip bone mineral density than controls who had not had kidney stones, and the difference persisted after adjusting for age, race, weight, and other variables. However, further data from this cohort revealed that so few men with osteoporosis had hypercalciuria that its routine measurement was not recommended.34
In women
The relationship between idiopathic hypercalciuria and fractures has been more difficult to establish in women.
Sowers et al35 performed an observational study of 1,309 women ages 20 to 92 with a history of nephrolithiasis. No association was noted between stone disease and reduced bone mineral density in the femoral neck, lumbar spine, or radius.
These epidemiologic studies did not include the cause of the kidney stones (eg, whether or not there was associated hypercalciuria or primary hyperparathyroidism), and typically a diagnosis of idiopathic hypercalciuria was not established.
The difference in association between low bone mineral density or fracture with nephrolithiasis between men and women is not well understood, but the most consistent hypothesis is that the influence of hypoestrogenemia in women is much stronger than that of the hypercalciuria.20
Does the degree of hypercalciuria influence the amount of bone loss?
A few trials have tried to determine whether the amount of calcium in the urine influences the magnitude of bone loss.
In 2003, Asplin et al36 reported that bone mineral density Z-scores differed significantly by urinary calcium excretion, but only in stone-formers. In patients without stone disease, there was no difference in Z-scores according to the absolute value of hypercalciuria. This may be due to a self-selection bias in which stone-formers avoid calcium in the diet and those without stone disease do not.
Three studies looking solely at men with idiopathic hypercalciuria also did not detect a significant difference in bone mineral loss according to degree of hypercalciuria.20,30,37
A POLYGENIC DISORDER?
The potential contribution of genetic changes to the development of idiopathic hypercalciuria has been studied. While there is an increased risk of idiopathic hypercalciuria in first-degree relatives of patients with nephrolithiasis, most experts believe that idiopathic hypercalciuria is likely a polygenic disorder.9,38
EVALUATION AND TREATMENT
The 2014 revised version of the National Osteoporosis Foundation’s “Clinician’s guide to prevention and treatment of osteoporosis”39 noted that hypercalciuria is a risk factor that contributes to the development of osteoporosis and possibly osteoporotic fractures, and that consideration should be given to evaluating for hypercalciuria, but only in selected cases. In patients with kidney stones, the link between hypercalciuria and bone loss and fracture is recognized and should be explored in both women and men at risk of osteoporosis, as 45% to 50% of patients who form calcium stones have hypercalciuria.
Patients with kidney stones who have low bone mass and idiopathic hypercalciuria should increase their daily fluid intake, follow a low-salt and low-animal-protein diet, and take thiazide diuretics to reduce the incidence of further calcium stones. Whether this approach also improves bone mass and strength and reduces the risk of fractures within this cohort requires further study.
Dietary interventions
Don’t restrict calcium intake. Despite the connection between hypercalciuria and nephrolithiasis, restriction of dietary calcium to prevent relapse of nephrolithiasis is a risk factor for negative calcium balance and bone demineralization. Observational studies and prospective clinical trials have demonstrated an increased risk of stone formation with low calcium intake.27,30 Nevertheless, this practice seems logical to many patients with kidney stones, and this process may independently contribute to lower bone mineral density.
A low-sodium, low-animal-protein diet is beneficial. Though increased intake of sodium or protein is not the main cause of idiopathic hypercalciuria, pharmacologic therapy, especially with thiazide diuretics, is more likely to be successful in the setting of a low-sodium, low-protein diet.
Borghi et al27 studied 2 diets in men with nephrolithiasis and idiopathic hypercalciuria: a low-calcium diet and a low-salt, low-animal-protein, normal-calcium diet. Men on the latter diet experienced a greater reduction in urinary calcium excretion than those on the low-calcium diet.
Breslau et al40 found that urinary calcium excretion fell by 50% in 15 people when they switched from an animal-based to a plant-based protein diet.
Thiazide diuretics
Several epidemiologic and randomized studies41–45 found that thiazide therapy decreased the likelihood of hip fracture in postmenopausal women, men, and premenopausal women. Doses ranged from 12.5 to 50 mg of hydrochlorothiazide. Bone density increased in the radius, total body, total hip, and lumbar spine. One prospective trial noted that fracture risk declined with longer duration of thiazide use, with the largest reduction in those who used thiazides for 8 or more years.46
Thiazides have anticalciuric actions.47 In addition, they have positive effects on osteoblastic cell proliferation and activity, inhibiting osteocalcin expression by osteoblasts, thereby possibly improving bone formation and mineralization.48 The effects of thiazides on bone was reviewed by Sakhaee et al.49
However, fewer studies have looked at thiazides in patients with idiopathic hypercalciuria.
García-Nieto et al50 looked retrospectively at 22 children (average age 11.7) with idiopathic hypercalciuria and osteopenia who had received thiazides (19 received chlorthalidone 25 mg daily, and 3 received hydrochlorothiazide 25 mg daily) for an average of 2.4 years, and at 32 similar patients who had not received thiazides. Twelve (55%) of the patients receiving thiazides had an improvement in bone mineral density Z-scores, compared with 23 (72%) of the controls. This finding is confounded by growth that occurred during the study, and both groups demonstrated a significantly increased body mass index and bone mineral apparent density at the end of the trial.
Bushinsky and Favus51 evaluated whether chlorthalidone improved bone quality or structure in rats that were genetically prone to hypercalciuric stones. These rats are uniformly stone-formers, and while they have components of calcium hyperabsorption, they also demonstrate renal hyperexcretion (leak) and enhanced bone mineral resorption.51 When fed a high-calcium diet, they maintain a reduction in bone mineral density and bone strength. Study rats were given chlorthalidone 4 to 5 mg/kg/day. After 18 weeks of therapy, significant improvements were observed in trabecular thickness and connectivity as well as increased vertebral compressive strength.52 No difference in cortical bone was noted.
No randomized, blinded, placebo-controlled trial has yet been done to study the impact of thiazides on bone mineral density or fracture risk in patients with idiopathic hypercalciuria.
In practice, many physicians choose chlorthalidone over hydrochlorothiazide because of chlorthalidone’s longer half-life. Combinations of a thiazide diuretic and potassium-sparing medications are also employed, such as hydrochlorothiazide plus either triamterene or spironolactone to reduce the number of pills the patient has to take.
Potassium citrate
When prescribing thiazide diuretics, one should also consider prescribing potassium citrate, as this agent not only prevents hypokalemia but also increases urinary citrate excretion, which can help to inhibit crystallization of calcium salts.6
In a longitudinal study of 28 patients with hypercalciuria,53 combined therapy with a thiazide or indapamide and potassium citrate over a mean of 7 years increased bone density of the lumbar spine by 7.1% and of the femoral neck by 4.1%, compared with treatment in age- and sex-matched normocalcemic peers. In the same study, daily urinary calcium excretion decreased and urinary pH and citrate levels increased; urinary saturation of calcium oxalate decreased by 46%, and stone formation was decreased.
Another trial evaluated 120 patients with idiopathic calcium nephrolithiasis, half of whom were given potassium citrate. Those given potassium citrate experienced an increase in distal radius bone mineral density over 2 years.54 It is theorized that alkalinization may decrease bone turnover in these patients.
Bisphosphonates
As one of the proposed main mechanisms of bone loss in idiopathic hypercalciuria is direct bone resorption, a potential target for therapy is the osteoclast, which bisphosphonates inhibit.
Ruml et al55 studied the impact of alendronate vs placebo in 16 normal men undergoing 3 weeks of strict bedrest. Compared with the placebo group, those who received alendronate had significantly lower 24-hour urine calcium excretion and higher levels of PTH and 1,25-dihydroxyvitamin D.
Weisinger et al56 evaluated the effects of alendronate 10 mg daily in 10 patients who had stone disease with documented idiopathic hypercalciuria and also in 8 normocalciuric patients without stone disease. Alendronate resulted in a sustained reduction of calcium in the urine in the patients with idiopathic hypercalciuria but not in the normocalciuric patients.
Data are somewhat scant as to the effect of bisphosphonates on bone health in the setting of idiopathic hypercalciuria,57,58 and therapy with bisphosphonates is not recommended in patients with idiopathic hypercalciuria outside the realm of postmenopausal osteoporosis or other indications for bisphosphonates approved by the US Food and Drug Administration (FDA).
Calcimimetics
Calcium-sensing receptors are found not only in parathyroid tissue but also in the intestines and kidneys. Locally, elevated plasma calcium in the kidney causes activation of the calcium-sensing receptor, diminishing further calcium reabsorption.59 Agents that increase the sensitivity of the calcium-sensing receptors are classified as calcimimetics.
Cinacalcet is a calcimimetic approved by the FDA for treatment of secondary hyperparathyroidism in patients with chronic kidney disease on dialysis, for the treatment of hypercalcemia in patients with parathyroid carcinoma, and for patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. In an uncontrolled 5-year study of cinacalcet in patients with primary hyperparathyroidism, there was no significant change in bone density.60
Anti-inflammatory drugs
The role of cytokines in stimulating bone resorption in idiopathic hypercalciuria has led to the investigation of several anti-inflammatory drugs (eg, diclofenac, indomethacin) as potential treatments, but studies have been limited in number and scope.61,62
Omega-3 fatty acids
Omega-3 fatty acids are thought to alter prostaglandin metabolism and to potentially reduce stone formation.63
A retrospective study of 29 patients with stone disease found that, combined with dietary counseling, omega-3 fatty acids could potentially reduce urinary calcium and oxalate excretion and increase urinary citrate in hypercalciuric stone-formers.64
A review of published randomized controlled trials of omega-3 fatty acids in skeletal health discovered that 4 studies found positive effects on bone mineral density or bone turnover markers, whereas 5 studies reported no differences. All trials were small, and none evaluated fracture outcome.65
A 65-year-old woman was recently diagnosed with osteoporosis after a screening bone mineral density test. She has hypertension (treated with lisinopril), and she had an episode of passing a kidney stone 10 years ago. A 24-hour urine study reveals an elevated urinary calcium level.
What should the physician keep in mind in managing this patient?
IDIOPATHIC HYPERCALCIURIA
Many potential causes of secondary hypercalciuria must be ruled out before deciding that a patient has idiopathic hypercalciuria, which was first noted as a distinct entity by Albright et al in 1953.1 Causes of secondary hypercalciuria include primary hyperparathyroidism, hyperthyroidism, Paget disease, myeloma, malignancy, immobility, accelerated osteoporosis, sarcoidosis, renal tubular acidosis, and drug-induced urinary calcium loss such as that seen with loop diuretics.
Idiopathic hypercalciuria is identified by the following:
- Persistent hypercalciuria despite normal or restricted calcium intake2,3
- Normal levels of parathyroid hormone (PTH), phosphorus, and 1,25-dihydroxy-vitamin D (the active form of vitamin D, also called calcitriol) in the presence of hypercalciuria; serum calcium levels are also normal.
An alias for idiopathic hypercalciuria is “fasting hypercalciuria,” as increased urinary calcium persists and sometimes worsens while fasting or on a low-calcium diet, with increased bone turnover, reduced bone density, and normal serum PTH levels.4,5
Mineral loss from bone predominates in idiopathic hypercalciuria, but there is also a minor component of intestinal hyperabsorption of calcium and reduced renal calcium reabsorption.6 Distinguishing among intestinal hyperabsorptive hypercalciuria, renal leak hypercalciuria, and idiopathic or fasting hypercalciuria can be difficult and subtle. It has been argued that differentiating among hypercalciuric subtypes (hyperabsorptive, renal leak, idiopathic) is not useful; in general clinical practice, it is impractical to collect multiple 24-hour urine samples in the setting of controlled high- vs low-calcium diets.
COMPLICATIONS OF IDIOPATHIC HYPERCALCIURIA
Calcium is an important component in many physiologic processes, including coagulation, cell membrane transfer, hormone release, neuromuscular activation, and myocardial contraction. A sophisticated system of hormonally mediated interactions normally maintains stable extracellular calcium levels. Calcium is vital for bone strength, but the bones are the body’s calcium “bank,” and withdrawals from this bank are made at the expense of bone strength and integrity.
Renal stones
Patients with idiopathic hypercalciuria have a high incidence of renal stones. Conversely, 40% to 50% of patients with recurrent kidney stones have evidence of idiopathic hypercalciuria, the most common metabolic abnormality in “stone-formers.”7,8 Further, 35% to 40% of first- and second-degree relatives of stone-formers who have idiopathic hypercalciuria also have the condition.9 In the general population without kidney stones and without first-degree relatives with stones, the prevalence is approximately 5% to 10%.10,11
Bone loss
People with idiopathic hypercalciuria have lower bone density and a higher incidence of fracture than their normocalciuric peers. This relationship has been observed in both sexes and all ages. Idiopathic hypercalciuria has been noted in 10% to 19% of otherwise healthy men with low bone mass, in postmenopausal women with osteoporosis,10–12 and in up to 40% of postmenopausal women with osteoporotic fractures and no history of kidney stones.13
LABORATORY DEFINITION
Urinary calcium excretion
Heaney et al14 measured 24-hour urinary calcium excretion in a group of early postmenopausal women, whom he divided into 3 groups by dietary calcium intake:
- Low intake (< 500 mg/day)
- Moderate intake (500–1,000 mg/day)
- High intake (> 1,000 mg/day).
In the women who were estrogen-deprived (ie, postmenopausal and not on estrogen replacement therapy), the 95% probability ranges for urinary calcium excretion were:
- 32–252 mg/day (0.51–4.06 mg/kg/day) with low calcium intake
- 36–286 mg/day (0.57–4.52 mg/kg/day) with moderate calcium intake
- 45–357 mg/day (0.69–5.47 mg/kg/day) with high calcium intake.
For estrogen-replete women (perimenopausal or postmenopausal on estrogen replacement), using the same categories of dietary calcium intake, calcium excretion was:
- 39–194 mg/day (0.65–3.23 mg/kg/day) with low calcium intake
- 54–269 mg/day (0.77–3.84 mg/kg/day) with moderate calcium intake
- 66–237 mg/day (0.98–4.89 mg/kg/day) with high calcium intake.
In the estrogen-deprived group, urinary calcium excretion increased by only 55 mg/day per 1,000-mg increase in dietary intake, though there was individual variability. These data suggest that hypercalciuria should be defined as:
- Greater than 250 mg/day (> 4.1 mg/kg/day) in estrogen-replete women
- Greater than 300 mg/day (> 5.0 mg/kg/day) in estrogen-deprived women.
Urinary calcium-to-creatinine ratio
Use of a spot urinary calcium-to-creatinine ratio has been advocated as an alternative to the more labor-intensive 24-hour urine collection.15 However, the spot urine calcium-creatinine ratio correlates poorly with 24-hour urine criteria for hypercalciuria whether by absolute, weight-based, or menopausal and calcium-adjusted definitions.
Importantly, spot urine measurements show poor sensitivity and specificity for hypercalciuria. Spot urine samples underestimate the 24-hour urinary calcium (Bland-Altman bias –71 mg/24 hours), and postprandial sampling overestimates it (Bland-Altman bias +61 mg/24 hours).15
WHAT IS THE MECHANISM OF IDIOPATHIC HYPERCALCIURIA?
The pathophysiology of idiopathic hypercalciuria has been difficult to establish.
Increased sensitivity to vitamin D? In the hyperabsorbing population, activated vitamin D levels are often robust, but a few studies of rats with hyperabsorbing, hyperexcreting physiology have shown normal calcitriol levels, suggesting an increased sensitivity to the actions of 1,25-dihydroxyvitamin D.16
Another study found that hypercalciuric stone-forming rats have more 1,25-dihydroxyvitamin D receptors than do controls.17
These changes have not been demonstrated in patients with idiopathic hypercalciuria.
High sodium intake has been proposed as the cause of idiopathic hypercalciuria. High sodium intake leads to increased urinary sodium excretion, and the increased tubular sodium load can decrease tubular calcium reabsorption, possibly favoring a reduction in bone mineral density over time.18–20
In healthy people, urine calcium excretion increases by about 0.6 mmol/day (20–40 mg/day) for each 100-mmol (2,300 mg) increment in daily sodium ingestion.21,22 But high sodium intake is seldom the principal cause of idiopathic hypercalciuria.
High protein intake, often observed in patients with nephrolithiasis, increases dietary acid load, stimulating release of calcium from bone and inhibiting renal reabsorption of calcium.23,24 Increasing dietary protein from 0.5 to 2.0 mg/kg/day can double the urinary calcium output.25
In mice, induction of metabolic acidosis, thought to mimic a high-protein diet, inhibits osteoblastic alkaline phosphatase activity while stimulating prostaglandin E2 production.26 This in turn increases osteoblastic expression of receptor activator for nuclear factor kappa b (RANK) ligand, thereby potentially contributing to osteoclastogenesis and osteoclast activity.26
Decreasing dietary protein decreases the recurrence of nephrolithiasis in established stone-formers.27 Still, urine calcium levels are higher in those with idiopathic hypercalciuria than in normal controls at comparable levels of acid excretion, so while protein ingestion could potentially exacerbate the hypercalciuria, it is unlikely to be the sole cause.
Renal calcium leak? The frequent finding of low to low-normal PTH levels in patients with idiopathic hypercalciuria contradicts the potential etiologic mechanism of renal calcium “leak.” In idiopathic hypercalciuria, the PTH response to an oral calcium load is abnormal. If given an oral calcium load, the PTH level should decline if this were due to renal leak, but in the setting of idiopathic hypercalciuria, no clinically meaningful change in PTH occurs. This lack of response of PTH to oral calcium load has been seen in both rat and human studies. Patients also excrete normal to high amounts of urine calcium after prolonged fasting or a low-calcium diet. Low-calcium diets do not induce hyperparathyroidism in these patients, and so the source of the elevated calcium in the urine must be primarily from bone. Increased levels of 1,25-dihydroxyvitamin D in patients with idiopathic hypercalciuria have been noted.28,29
Whether the cytokine milieu also contributes to the calcitriol levels is unclear, but the high or high-normal plasma level of 1,25-dihydroxyvitamin D may be the reason that the PTH is unperturbed.
IMPACT ON BONE HEALTH
Nephrolithiasis is strongly linked to fracture risk.
The bone mineral density of trabecular bone is more affected by calcium excretion than that of cortical bone.18,20,30 However, lumbar spine bone mineral density has not been consistently found to be lower in patients with hyperabsorptive hypercalciuria. Rather, bone mineral density is correlated inversely with urine calcium excretion in men and women who form stones, but not in patients without nephrolithiasis.
In children
In children, idiopathic hypercalciuria is well known to be linked to osteopenia. This is an important group to study, as adult idiopathic hypercalciuria often begins in childhood. However, the trajectory of bone loss vs gain in children is fraught with variables such as growth, puberty, and body mass index, making this a difficult group from which to extrapolate conclusions to adults.
In men
There is more information on the relationship between hypercalciuria and osteoporosis in men than in women.
In 1998, Melton et al31 published the findings of a 25-year population-based cohort study of 624 patients, 442 (71%) of whom were men, referred for new-onset urolithiasis. The incidence of vertebral fracture was 4 times higher in this group than in patients without stone disease, but there was no difference in the rate of hip, forearm, or nonvertebral fractures. This is consistent with earlier data that report a loss of predominantly cancellous bone associated with urolithiasis.
National Health and Nutrition Examination Survey III data in 2001 focused on a potential relationship between kidney stones and bone mineral density or prevalent spine or wrist fracture.32 More than 14,000 people had hip bone mineral density measurements, of whom 793 (477 men, 316 women) had kidney stones. Men with previous nephrolithiasis had lower femoral neck bone mineral density than those without. Men with kidney stones were also more likely to report prevalent wrist and spine fractures. In women, no difference was noted between those with or without stone disease with respect to femoral neck bone mineral density or fracture incidence.
Cauley et al33 also evaluated a relationship between kidney stones and bone mineral density in the Osteoporotic Fractures in Men (MrOS) study. Of approximately 6,000 men, 13.2% reported a history of kidney stones. These men had lower spine and total hip bone mineral density than controls who had not had kidney stones, and the difference persisted after adjusting for age, race, weight, and other variables. However, further data from this cohort revealed that so few men with osteoporosis had hypercalciuria that its routine measurement was not recommended.34
In women
The relationship between idiopathic hypercalciuria and fractures has been more difficult to establish in women.
Sowers et al35 performed an observational study of 1,309 women ages 20 to 92 with a history of nephrolithiasis. No association was noted between stone disease and reduced bone mineral density in the femoral neck, lumbar spine, or radius.
These epidemiologic studies did not include the cause of the kidney stones (eg, whether or not there was associated hypercalciuria or primary hyperparathyroidism), and typically a diagnosis of idiopathic hypercalciuria was not established.
The difference in association between low bone mineral density or fracture with nephrolithiasis between men and women is not well understood, but the most consistent hypothesis is that the influence of hypoestrogenemia in women is much stronger than that of the hypercalciuria.20
Does the degree of hypercalciuria influence the amount of bone loss?
A few trials have tried to determine whether the amount of calcium in the urine influences the magnitude of bone loss.
In 2003, Asplin et al36 reported that bone mineral density Z-scores differed significantly by urinary calcium excretion, but only in stone-formers. In patients without stone disease, there was no difference in Z-scores according to the absolute value of hypercalciuria. This may be due to a self-selection bias in which stone-formers avoid calcium in the diet and those without stone disease do not.
Three studies looking solely at men with idiopathic hypercalciuria also did not detect a significant difference in bone mineral loss according to degree of hypercalciuria.20,30,37
A POLYGENIC DISORDER?
The potential contribution of genetic changes to the development of idiopathic hypercalciuria has been studied. While there is an increased risk of idiopathic hypercalciuria in first-degree relatives of patients with nephrolithiasis, most experts believe that idiopathic hypercalciuria is likely a polygenic disorder.9,38
EVALUATION AND TREATMENT
The 2014 revised version of the National Osteoporosis Foundation’s “Clinician’s guide to prevention and treatment of osteoporosis”39 noted that hypercalciuria is a risk factor that contributes to the development of osteoporosis and possibly osteoporotic fractures, and that consideration should be given to evaluating for hypercalciuria, but only in selected cases. In patients with kidney stones, the link between hypercalciuria and bone loss and fracture is recognized and should be explored in both women and men at risk of osteoporosis, as 45% to 50% of patients who form calcium stones have hypercalciuria.
Patients with kidney stones who have low bone mass and idiopathic hypercalciuria should increase their daily fluid intake, follow a low-salt and low-animal-protein diet, and take thiazide diuretics to reduce the incidence of further calcium stones. Whether this approach also improves bone mass and strength and reduces the risk of fractures within this cohort requires further study.
Dietary interventions
Don’t restrict calcium intake. Despite the connection between hypercalciuria and nephrolithiasis, restriction of dietary calcium to prevent relapse of nephrolithiasis is a risk factor for negative calcium balance and bone demineralization. Observational studies and prospective clinical trials have demonstrated an increased risk of stone formation with low calcium intake.27,30 Nevertheless, this practice seems logical to many patients with kidney stones, and this process may independently contribute to lower bone mineral density.
A low-sodium, low-animal-protein diet is beneficial. Though increased intake of sodium or protein is not the main cause of idiopathic hypercalciuria, pharmacologic therapy, especially with thiazide diuretics, is more likely to be successful in the setting of a low-sodium, low-protein diet.
Borghi et al27 studied 2 diets in men with nephrolithiasis and idiopathic hypercalciuria: a low-calcium diet and a low-salt, low-animal-protein, normal-calcium diet. Men on the latter diet experienced a greater reduction in urinary calcium excretion than those on the low-calcium diet.
Breslau et al40 found that urinary calcium excretion fell by 50% in 15 people when they switched from an animal-based to a plant-based protein diet.
Thiazide diuretics
Several epidemiologic and randomized studies41–45 found that thiazide therapy decreased the likelihood of hip fracture in postmenopausal women, men, and premenopausal women. Doses ranged from 12.5 to 50 mg of hydrochlorothiazide. Bone density increased in the radius, total body, total hip, and lumbar spine. One prospective trial noted that fracture risk declined with longer duration of thiazide use, with the largest reduction in those who used thiazides for 8 or more years.46
Thiazides have anticalciuric actions.47 In addition, they have positive effects on osteoblastic cell proliferation and activity, inhibiting osteocalcin expression by osteoblasts, thereby possibly improving bone formation and mineralization.48 The effects of thiazides on bone was reviewed by Sakhaee et al.49
However, fewer studies have looked at thiazides in patients with idiopathic hypercalciuria.
García-Nieto et al50 looked retrospectively at 22 children (average age 11.7) with idiopathic hypercalciuria and osteopenia who had received thiazides (19 received chlorthalidone 25 mg daily, and 3 received hydrochlorothiazide 25 mg daily) for an average of 2.4 years, and at 32 similar patients who had not received thiazides. Twelve (55%) of the patients receiving thiazides had an improvement in bone mineral density Z-scores, compared with 23 (72%) of the controls. This finding is confounded by growth that occurred during the study, and both groups demonstrated a significantly increased body mass index and bone mineral apparent density at the end of the trial.
Bushinsky and Favus51 evaluated whether chlorthalidone improved bone quality or structure in rats that were genetically prone to hypercalciuric stones. These rats are uniformly stone-formers, and while they have components of calcium hyperabsorption, they also demonstrate renal hyperexcretion (leak) and enhanced bone mineral resorption.51 When fed a high-calcium diet, they maintain a reduction in bone mineral density and bone strength. Study rats were given chlorthalidone 4 to 5 mg/kg/day. After 18 weeks of therapy, significant improvements were observed in trabecular thickness and connectivity as well as increased vertebral compressive strength.52 No difference in cortical bone was noted.
No randomized, blinded, placebo-controlled trial has yet been done to study the impact of thiazides on bone mineral density or fracture risk in patients with idiopathic hypercalciuria.
In practice, many physicians choose chlorthalidone over hydrochlorothiazide because of chlorthalidone’s longer half-life. Combinations of a thiazide diuretic and potassium-sparing medications are also employed, such as hydrochlorothiazide plus either triamterene or spironolactone to reduce the number of pills the patient has to take.
Potassium citrate
When prescribing thiazide diuretics, one should also consider prescribing potassium citrate, as this agent not only prevents hypokalemia but also increases urinary citrate excretion, which can help to inhibit crystallization of calcium salts.6
In a longitudinal study of 28 patients with hypercalciuria,53 combined therapy with a thiazide or indapamide and potassium citrate over a mean of 7 years increased bone density of the lumbar spine by 7.1% and of the femoral neck by 4.1%, compared with treatment in age- and sex-matched normocalcemic peers. In the same study, daily urinary calcium excretion decreased and urinary pH and citrate levels increased; urinary saturation of calcium oxalate decreased by 46%, and stone formation was decreased.
Another trial evaluated 120 patients with idiopathic calcium nephrolithiasis, half of whom were given potassium citrate. Those given potassium citrate experienced an increase in distal radius bone mineral density over 2 years.54 It is theorized that alkalinization may decrease bone turnover in these patients.
Bisphosphonates
As one of the proposed main mechanisms of bone loss in idiopathic hypercalciuria is direct bone resorption, a potential target for therapy is the osteoclast, which bisphosphonates inhibit.
Ruml et al55 studied the impact of alendronate vs placebo in 16 normal men undergoing 3 weeks of strict bedrest. Compared with the placebo group, those who received alendronate had significantly lower 24-hour urine calcium excretion and higher levels of PTH and 1,25-dihydroxyvitamin D.
Weisinger et al56 evaluated the effects of alendronate 10 mg daily in 10 patients who had stone disease with documented idiopathic hypercalciuria and also in 8 normocalciuric patients without stone disease. Alendronate resulted in a sustained reduction of calcium in the urine in the patients with idiopathic hypercalciuria but not in the normocalciuric patients.
Data are somewhat scant as to the effect of bisphosphonates on bone health in the setting of idiopathic hypercalciuria,57,58 and therapy with bisphosphonates is not recommended in patients with idiopathic hypercalciuria outside the realm of postmenopausal osteoporosis or other indications for bisphosphonates approved by the US Food and Drug Administration (FDA).
Calcimimetics
Calcium-sensing receptors are found not only in parathyroid tissue but also in the intestines and kidneys. Locally, elevated plasma calcium in the kidney causes activation of the calcium-sensing receptor, diminishing further calcium reabsorption.59 Agents that increase the sensitivity of the calcium-sensing receptors are classified as calcimimetics.
Cinacalcet is a calcimimetic approved by the FDA for treatment of secondary hyperparathyroidism in patients with chronic kidney disease on dialysis, for the treatment of hypercalcemia in patients with parathyroid carcinoma, and for patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. In an uncontrolled 5-year study of cinacalcet in patients with primary hyperparathyroidism, there was no significant change in bone density.60
Anti-inflammatory drugs
The role of cytokines in stimulating bone resorption in idiopathic hypercalciuria has led to the investigation of several anti-inflammatory drugs (eg, diclofenac, indomethacin) as potential treatments, but studies have been limited in number and scope.61,62
Omega-3 fatty acids
Omega-3 fatty acids are thought to alter prostaglandin metabolism and to potentially reduce stone formation.63
A retrospective study of 29 patients with stone disease found that, combined with dietary counseling, omega-3 fatty acids could potentially reduce urinary calcium and oxalate excretion and increase urinary citrate in hypercalciuric stone-formers.64
A review of published randomized controlled trials of omega-3 fatty acids in skeletal health discovered that 4 studies found positive effects on bone mineral density or bone turnover markers, whereas 5 studies reported no differences. All trials were small, and none evaluated fracture outcome.65
- Albright F, Henneman P, Benedict PH, Forbes AP. Idiopathic hypercalciuria: a preliminary report. Proc R Soc Med 1953; 46:1077–1081.
- Pak CY. Pathophysiology of calcium nephrolithiasis. In: Seldin DW, Giebiscg G, eds. The Kidney: Physiology and Pathophysiology. New York, NY: Raven Press; 1992:2461–2480.
- Frick KK, Bushinsky DA. Molecular mechanisms of primary hypercalciuria. J Am Soc Nephrol 2003; 14:1082–1095.
- Pacifici R, Rothstein M, Rifas L, et al. Increased monocyte interleukin-1 activity and decreased vertebral bone density in patients with fasting idiopathic hypercalciuria. J Clin Endocrinol Metab 1990; 71:138–145.
- Messa P, Mioni G, Montanaro D, et al. About a primitive osseous origin of the so-called ‘renal hypercalciuria.’ Contrib Nephrol 1987; 58:106–110.
- Zerwekh JE. Bone disease and idiopathic hypercalciuria. Semin Nephrol 2008; 28:133–142.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Lemann J Jr. Pathogenesis of idiopathic hypercalciuria and nephrolithiasis. In: Coe FL, Favus MJ, eds. Disorders of Bone and Mineral Metabolism. New York, NY: Raven Press; 1992:685-706.
- Coe FL, Parks JH, Moore ES. Familial idiopathic hypercalciuria. N Engl J Med 1979; 300:337–340.
- Giannini S, Nobile M, Dalle Carbonare L, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. Eur J Endocrinol 2003; 149:209–213.
- Tannenbaum C, Clark J, Schwartzman K, et al. Yield of laboratory testing to identify secondary contributors to osteoporosis in otherwise healthy women. J Clin Endocrinol Metab 2002; 87:4431–4437.
- Cerda Gabaroi D, Peris P, Monegal A, et al. Search for hidden secondary causes in postmenopausal women with osteoporosis. Menopause 2010; 17:135–139.
- Rull MA, Cano-García Mdel C, Arrabal-Martín M, Arrabal-Polo MA. The importance of urinary calcium in postmenopausal women with osteoporotic fracture. Can Urol Assoc J 2015; 9:E183–E186.
- Heaney RP, Recker RR, Ryan RA. Urinary calcium in perimenopausal women: normative values. Osteoporos Int 1999; 9:13–18.
- Bleich HL, Moore MJ, Lemann J Jr, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Li XQ, Tembe V, Horwitz GM, Bushinsky DA, Favus MJ. Increased intestinal vitamin D receptor in genetic hypercalciuric rats. A cause of intestinal calcium hyperabsorption. J Clin Invest 1993; 91:661–667.
- Yao J, Kathpalia P, Bushinsky DA, Favus MJ. Hyperresponsiveness of vitamin D receptor gene expression to 1,25-dihydroxyvitamin D3. A new characteristic of genetic hypercalciuric stone-forming rats. J Clin Invest 1998; 101:2223–2232.
- Pietschmann F, Breslau NA, Pak CY. Reduced vertebral bone density in hypercalciuric nephrolithiasis. J Bone Miner Res 1992; 7:1383–1388.
- Jaeger P, Lippuner K, Casez JP, Hess B, Ackermann D, Hug C. Low bone mass in idiopathic renal stone formers: magnitude and significance. J Bone Miner Res 1994; 9:1525–1532.
- Vezzoli G, Soldati L, Arcidiacono T, et al. Urinary calcium is a determinant of bone mineral density in elderly men participating in the InCHIANTI study. Kidney Int 2005; 67:2006–2014.
- Lemann J Jr, Worcester EM, Gray RW. Hypercalciuria and stones. Am J Kidney Dis 1991; 17:386–391.
- Gokce C, Gokce O, Baydinc C, et al. Use of random urine samples to estimate total urinary calcium and phosphate excretion. Arch Intern Med 1991; 151:1587–1588.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Siener R, Schade N, Nicolay C, von Unruh GE, Hesse A. The efficacy of dietary intervention on urinary risk factors for stone formation in recurrent calcium oxalate stone patients. J Urol 2005; 173:1601–1605.
- Jones AN, Shafer MM, Keuler NS, Crone EM, Hansen KE. Fasting and postprandial spot urine calcium-to-creatinine ratios do not detect hypercalciuria. Osteoporos Int 2012; 23:553–562.
- Frick KK, Bushinsky DA. Metabolic acidosis stimulates RANKL RNA expression in bone through a cyclo-oxygenase-dependent mechanism. J Bone Miner Res 2003; 18:1317–1325.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Ghazali A, Fuentes V, Desaint C, et al. Low bone mineral density and peripheral blood monocyte activation profile in calcium stone formers with idiopathic hypercalciuria. J Clin Endocrinol Metab 1997; 82:32–38.
- Broadus AE, Insogna KL, Lang R, Ellison AF, Dreyer BE. Evidence for disordered control of 1,25-dihydroxyvitamin D production in absorptive hypercalciuria. N Engl J Med 1984; 311:73–80.
- Tasca A, Cacciola A, Ferrarese P, et al. Bone alterations in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Urology 2002; 59:865–869.
- Melton LJ 3rd, Crowson CS, Khosla S, Wilson DM, O’Fallon WM. Fracture risk among patients with urolithiasis: a population-based cohort study. Kidney Int 1998; 53:459–464.
- Lauderdale DS, Thisted RA, Wen M, Favus MJ. Bone mineral density and fracture among prevalent kidney stone cases in the Third National Health and Nutrition Examination Survey. J Bone Miner Res 2001; 16:1893–1898.
- Cauley JA, Fullman RL, Stone KL, et al; MrOS Research Group. Factors associated with the lumbar spine and proximal femur bone mineral density in older men. Osteoporos Int 2005; 16:1525–1537.
- Fink HA, Litwack-Harrison S, Taylor BC, et al; Osteoporotic Fractures in Men (MrOS) Study Group. Clinical utility of routine laboratory testing to identify possible secondary causes in older men with osteoporosis: the Osteoporotic Fractures in Men (MrOS) Study. Osteoporos Int 2016: 27:331–338.
- Sowers MR, Jannausch M, Wood C, Pope SK, Lachance LL, Peterson B. Prevalence of renal stones in a population-based study with dietary calcium, oxalate and medication exposures. Am J Epidemiol 1998; 147:914–920.
- Asplin JR, Bauer KA, Kinder J, et al. Bone mineral density and urine calcium excretion among subjects with and without nephrolithiasis. Kidney Int 2003; 63:662–669.
- Letavernier E, Traxer O, Daudon M, et al. Determinants of osteopenia in male renal-stone-disease patients with idiopathic hypercalciuria. Clin J Am Soc Nephrol 2011; 6:1149–1154.
- Vezzoli G, Soldati L, Gambaro G. Update on primary hypercalciuria from a genetic perspective. J Urol 2008; 179:1676–1682.
- Cosman F, de Beur SJ, LeBoff MS, et al; National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int 2014: 25:2359–2381.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Reid IR, Ames RW, Orr-Walker BJ, et al. Hydrochlorothiazide reduces loss of cortical bone in normal postmenopausal women: a randomized controlled trial. Am J Med 2000; 109:362–370.
- Bolland MJ, Ames RW, Horne AM, Orr-Walker BJ, Gamble GD, Reid IR. The effect of treatment with a thiazide diuretic for 4 years on bone density in normal postmenopausal women. Osteoporos Int 2007; 18:479–486.
- LaCroix AZ, Ott SM, Ichikawa L, Scholes D, Barlow WE. Low-dose hydrochlorothiazide and preservation of bone mineral density in older adults. Ann Intern Med 2000; 133:516–526.
- Wasnich RD, Davis JW, He YF, Petrovich H, Ross PD. A randomized, double-masked, placebo-controlled trial of chlorthalidone and bone loss in elderly women. Osteoporos Int 1995; 5:247–251.
- Adams JS, Song CF, Kantorovich V. Rapid recovery of bone mass in hypercalciuric, osteoporotic men treated with hydrochlorothiazide. Ann Intern Med 1999; 130:658–660.
- Feskanich D, Willett WC, Stampfer MJ, Colditz GA. A prospective study of thiazide use and fractures in women. Osteoporos Int 1997; 7:79–84.
- Lamberg BA, Kuhlback B. Effect of chlorothiazide and hydrochlorothiazide on the excretion of calcium in the urine. Scand J Clin Lab Invest 1959; 11:351–357.
- Lajeunesse D, Delalandre A, Guggino SE. Thiazide diuretics affect osteocalcin production in human osteoblasts at the transcription level without affecting vitamin D3 receptors. J Bone Miner Res 2000; 15:894–901.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2001; 79:393–403.
- García-Nieto V, Monge-Zamorano M, González-García M, Luis-Yanes MI. Effect of thiazides on bone mineral density in children with idiopathic hypercalciuria. Pediatr Nephrol 2012; 27:261–268.
- Bushinsky DA, Favus MJ. Mechanism of hypercalciuria in genetic hypercalciuric rats. Inherited defect in intestinal calcium transport. J Clin Invest 1988; 82:1585–1591.
- Bushinsky DA, Willett T, Asplin JR, Culbertson C, Che SP, Grynpas M. Chlorthalidone improves vertebral bone quality in genetic hypercalciuric stone-forming rats. J Bone Miner Res 2011; 26:1904–1912.
- Pak CY, Heller HJ, Pearle MS, Odvina CV, Poindexter JR, Peterson RD. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol 2003; 169:465–469.
- Vescini F, Buffa A, LaManna G, et al. Long-term potassium citrate therapy and bone mineral density in idiopathic calcium stone formers. J Endocrinol Invest 2005; 28:218–222.
- Ruml LA, Dubois SK, Roberts ML, Pak CY. Prevention of hypercalciuria and stone-forming propensity during prolonged bedrest by alendronate. J Bone Miner Res 1995; 10:655–662.
- Weisinger JR, Alonzo E, Machado C, et al. Role of bones in the physiopathology of idiopathic hypercalciuria: effect of amino-bisphosphonate alendronate. Medicina (B Aires) 1997; 57(suppl 1):45–48. Spanish.
- Heilberg IP, Martini LA, Teixeira SH, et al. Effect of etidronate treatment on bone mass of male nephrolithiasis patients with idiopathic hypercalciuria and osteopenia. Nephron 1998; 79:430–437.
- Bushinsky DA, Neumann KJ, Asplin J, Krieger NS. Alendronate decreases urine calcium and supersaturation in genetic hypercalciuric rats. Kidney Int 1999; 55:234–243.
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 1995; 92:131–145.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009; 94:4860–4867.
- Filipponi P, Mannarelli C, Pacifici R, et al. Evidence for a prostaglandin-mediated bone resorptive mechanism in subjects with fasting hypercalciuria. Calcif Tissue Int 1988; 43:61–66.
- Gomaa AA, Hassan HA, Ghaneimah SA. Effect of aspirin and indomethacin on the serum and urinary calcium, magnesium and phosphate. Pharmacol Res 1990; 22:59–70.
- Buck AC, Davies RL, Harrison T. The protective role of eicosapentaenoic acid (EPA) in the pathogenesis of nephrolithiasis. J Urol 1991; 146:188–194.
- Ortiz-Alvarado O, Miyaoka R, Kriedberg C, et al. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid in the management of hypercalciuric stone formers. Urology 2012; 79:282–286.
- Orchard TS, Pan X, Cheek F, Ing SW, Jackson RD. A systematic review of omega-3 fatty acids and osteoporosis. Br J Nutr 2012; 107(suppl 2):S253–S260.
- Albright F, Henneman P, Benedict PH, Forbes AP. Idiopathic hypercalciuria: a preliminary report. Proc R Soc Med 1953; 46:1077–1081.
- Pak CY. Pathophysiology of calcium nephrolithiasis. In: Seldin DW, Giebiscg G, eds. The Kidney: Physiology and Pathophysiology. New York, NY: Raven Press; 1992:2461–2480.
- Frick KK, Bushinsky DA. Molecular mechanisms of primary hypercalciuria. J Am Soc Nephrol 2003; 14:1082–1095.
- Pacifici R, Rothstein M, Rifas L, et al. Increased monocyte interleukin-1 activity and decreased vertebral bone density in patients with fasting idiopathic hypercalciuria. J Clin Endocrinol Metab 1990; 71:138–145.
- Messa P, Mioni G, Montanaro D, et al. About a primitive osseous origin of the so-called ‘renal hypercalciuria.’ Contrib Nephrol 1987; 58:106–110.
- Zerwekh JE. Bone disease and idiopathic hypercalciuria. Semin Nephrol 2008; 28:133–142.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Lemann J Jr. Pathogenesis of idiopathic hypercalciuria and nephrolithiasis. In: Coe FL, Favus MJ, eds. Disorders of Bone and Mineral Metabolism. New York, NY: Raven Press; 1992:685-706.
- Coe FL, Parks JH, Moore ES. Familial idiopathic hypercalciuria. N Engl J Med 1979; 300:337–340.
- Giannini S, Nobile M, Dalle Carbonare L, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. Eur J Endocrinol 2003; 149:209–213.
- Tannenbaum C, Clark J, Schwartzman K, et al. Yield of laboratory testing to identify secondary contributors to osteoporosis in otherwise healthy women. J Clin Endocrinol Metab 2002; 87:4431–4437.
- Cerda Gabaroi D, Peris P, Monegal A, et al. Search for hidden secondary causes in postmenopausal women with osteoporosis. Menopause 2010; 17:135–139.
- Rull MA, Cano-García Mdel C, Arrabal-Martín M, Arrabal-Polo MA. The importance of urinary calcium in postmenopausal women with osteoporotic fracture. Can Urol Assoc J 2015; 9:E183–E186.
- Heaney RP, Recker RR, Ryan RA. Urinary calcium in perimenopausal women: normative values. Osteoporos Int 1999; 9:13–18.
- Bleich HL, Moore MJ, Lemann J Jr, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Li XQ, Tembe V, Horwitz GM, Bushinsky DA, Favus MJ. Increased intestinal vitamin D receptor in genetic hypercalciuric rats. A cause of intestinal calcium hyperabsorption. J Clin Invest 1993; 91:661–667.
- Yao J, Kathpalia P, Bushinsky DA, Favus MJ. Hyperresponsiveness of vitamin D receptor gene expression to 1,25-dihydroxyvitamin D3. A new characteristic of genetic hypercalciuric stone-forming rats. J Clin Invest 1998; 101:2223–2232.
- Pietschmann F, Breslau NA, Pak CY. Reduced vertebral bone density in hypercalciuric nephrolithiasis. J Bone Miner Res 1992; 7:1383–1388.
- Jaeger P, Lippuner K, Casez JP, Hess B, Ackermann D, Hug C. Low bone mass in idiopathic renal stone formers: magnitude and significance. J Bone Miner Res 1994; 9:1525–1532.
- Vezzoli G, Soldati L, Arcidiacono T, et al. Urinary calcium is a determinant of bone mineral density in elderly men participating in the InCHIANTI study. Kidney Int 2005; 67:2006–2014.
- Lemann J Jr, Worcester EM, Gray RW. Hypercalciuria and stones. Am J Kidney Dis 1991; 17:386–391.
- Gokce C, Gokce O, Baydinc C, et al. Use of random urine samples to estimate total urinary calcium and phosphate excretion. Arch Intern Med 1991; 151:1587–1588.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Siener R, Schade N, Nicolay C, von Unruh GE, Hesse A. The efficacy of dietary intervention on urinary risk factors for stone formation in recurrent calcium oxalate stone patients. J Urol 2005; 173:1601–1605.
- Jones AN, Shafer MM, Keuler NS, Crone EM, Hansen KE. Fasting and postprandial spot urine calcium-to-creatinine ratios do not detect hypercalciuria. Osteoporos Int 2012; 23:553–562.
- Frick KK, Bushinsky DA. Metabolic acidosis stimulates RANKL RNA expression in bone through a cyclo-oxygenase-dependent mechanism. J Bone Miner Res 2003; 18:1317–1325.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Ghazali A, Fuentes V, Desaint C, et al. Low bone mineral density and peripheral blood monocyte activation profile in calcium stone formers with idiopathic hypercalciuria. J Clin Endocrinol Metab 1997; 82:32–38.
- Broadus AE, Insogna KL, Lang R, Ellison AF, Dreyer BE. Evidence for disordered control of 1,25-dihydroxyvitamin D production in absorptive hypercalciuria. N Engl J Med 1984; 311:73–80.
- Tasca A, Cacciola A, Ferrarese P, et al. Bone alterations in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Urology 2002; 59:865–869.
- Melton LJ 3rd, Crowson CS, Khosla S, Wilson DM, O’Fallon WM. Fracture risk among patients with urolithiasis: a population-based cohort study. Kidney Int 1998; 53:459–464.
- Lauderdale DS, Thisted RA, Wen M, Favus MJ. Bone mineral density and fracture among prevalent kidney stone cases in the Third National Health and Nutrition Examination Survey. J Bone Miner Res 2001; 16:1893–1898.
- Cauley JA, Fullman RL, Stone KL, et al; MrOS Research Group. Factors associated with the lumbar spine and proximal femur bone mineral density in older men. Osteoporos Int 2005; 16:1525–1537.
- Fink HA, Litwack-Harrison S, Taylor BC, et al; Osteoporotic Fractures in Men (MrOS) Study Group. Clinical utility of routine laboratory testing to identify possible secondary causes in older men with osteoporosis: the Osteoporotic Fractures in Men (MrOS) Study. Osteoporos Int 2016: 27:331–338.
- Sowers MR, Jannausch M, Wood C, Pope SK, Lachance LL, Peterson B. Prevalence of renal stones in a population-based study with dietary calcium, oxalate and medication exposures. Am J Epidemiol 1998; 147:914–920.
- Asplin JR, Bauer KA, Kinder J, et al. Bone mineral density and urine calcium excretion among subjects with and without nephrolithiasis. Kidney Int 2003; 63:662–669.
- Letavernier E, Traxer O, Daudon M, et al. Determinants of osteopenia in male renal-stone-disease patients with idiopathic hypercalciuria. Clin J Am Soc Nephrol 2011; 6:1149–1154.
- Vezzoli G, Soldati L, Gambaro G. Update on primary hypercalciuria from a genetic perspective. J Urol 2008; 179:1676–1682.
- Cosman F, de Beur SJ, LeBoff MS, et al; National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int 2014: 25:2359–2381.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Reid IR, Ames RW, Orr-Walker BJ, et al. Hydrochlorothiazide reduces loss of cortical bone in normal postmenopausal women: a randomized controlled trial. Am J Med 2000; 109:362–370.
- Bolland MJ, Ames RW, Horne AM, Orr-Walker BJ, Gamble GD, Reid IR. The effect of treatment with a thiazide diuretic for 4 years on bone density in normal postmenopausal women. Osteoporos Int 2007; 18:479–486.
- LaCroix AZ, Ott SM, Ichikawa L, Scholes D, Barlow WE. Low-dose hydrochlorothiazide and preservation of bone mineral density in older adults. Ann Intern Med 2000; 133:516–526.
- Wasnich RD, Davis JW, He YF, Petrovich H, Ross PD. A randomized, double-masked, placebo-controlled trial of chlorthalidone and bone loss in elderly women. Osteoporos Int 1995; 5:247–251.
- Adams JS, Song CF, Kantorovich V. Rapid recovery of bone mass in hypercalciuric, osteoporotic men treated with hydrochlorothiazide. Ann Intern Med 1999; 130:658–660.
- Feskanich D, Willett WC, Stampfer MJ, Colditz GA. A prospective study of thiazide use and fractures in women. Osteoporos Int 1997; 7:79–84.
- Lamberg BA, Kuhlback B. Effect of chlorothiazide and hydrochlorothiazide on the excretion of calcium in the urine. Scand J Clin Lab Invest 1959; 11:351–357.
- Lajeunesse D, Delalandre A, Guggino SE. Thiazide diuretics affect osteocalcin production in human osteoblasts at the transcription level without affecting vitamin D3 receptors. J Bone Miner Res 2000; 15:894–901.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2001; 79:393–403.
- García-Nieto V, Monge-Zamorano M, González-García M, Luis-Yanes MI. Effect of thiazides on bone mineral density in children with idiopathic hypercalciuria. Pediatr Nephrol 2012; 27:261–268.
- Bushinsky DA, Favus MJ. Mechanism of hypercalciuria in genetic hypercalciuric rats. Inherited defect in intestinal calcium transport. J Clin Invest 1988; 82:1585–1591.
- Bushinsky DA, Willett T, Asplin JR, Culbertson C, Che SP, Grynpas M. Chlorthalidone improves vertebral bone quality in genetic hypercalciuric stone-forming rats. J Bone Miner Res 2011; 26:1904–1912.
- Pak CY, Heller HJ, Pearle MS, Odvina CV, Poindexter JR, Peterson RD. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol 2003; 169:465–469.
- Vescini F, Buffa A, LaManna G, et al. Long-term potassium citrate therapy and bone mineral density in idiopathic calcium stone formers. J Endocrinol Invest 2005; 28:218–222.
- Ruml LA, Dubois SK, Roberts ML, Pak CY. Prevention of hypercalciuria and stone-forming propensity during prolonged bedrest by alendronate. J Bone Miner Res 1995; 10:655–662.
- Weisinger JR, Alonzo E, Machado C, et al. Role of bones in the physiopathology of idiopathic hypercalciuria: effect of amino-bisphosphonate alendronate. Medicina (B Aires) 1997; 57(suppl 1):45–48. Spanish.
- Heilberg IP, Martini LA, Teixeira SH, et al. Effect of etidronate treatment on bone mass of male nephrolithiasis patients with idiopathic hypercalciuria and osteopenia. Nephron 1998; 79:430–437.
- Bushinsky DA, Neumann KJ, Asplin J, Krieger NS. Alendronate decreases urine calcium and supersaturation in genetic hypercalciuric rats. Kidney Int 1999; 55:234–243.
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 1995; 92:131–145.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009; 94:4860–4867.
- Filipponi P, Mannarelli C, Pacifici R, et al. Evidence for a prostaglandin-mediated bone resorptive mechanism in subjects with fasting hypercalciuria. Calcif Tissue Int 1988; 43:61–66.
- Gomaa AA, Hassan HA, Ghaneimah SA. Effect of aspirin and indomethacin on the serum and urinary calcium, magnesium and phosphate. Pharmacol Res 1990; 22:59–70.
- Buck AC, Davies RL, Harrison T. The protective role of eicosapentaenoic acid (EPA) in the pathogenesis of nephrolithiasis. J Urol 1991; 146:188–194.
- Ortiz-Alvarado O, Miyaoka R, Kriedberg C, et al. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid in the management of hypercalciuric stone formers. Urology 2012; 79:282–286.
- Orchard TS, Pan X, Cheek F, Ing SW, Jackson RD. A systematic review of omega-3 fatty acids and osteoporosis. Br J Nutr 2012; 107(suppl 2):S253–S260.
KEY POINTS
- Idiopathic hypercalciuria is common in patients with kidney stones and is also present in up to 20% of postmenopausal women with osteoporosis but no history of kidney stones.
- Idiopathic hypercalciuria has been directly implicated as a cause of loss of trabecular bone, especially in men. But reversing the hypercalciuria in this condition has not been definitively shown to diminish fracture incidence.
- Patients with kidney stones who have low bone mass and idiopathic hypercalciuria should increase their daily fluid intake, follow a diet low in salt and animal protein, and take thiazide diuretics to reduce the risk of further calcium stone formation. Whether this approach also improves bone mass and strength and reduces fracture risk in this patient group requires further study.
What is the hepatitis B vaccination regimen in chronic kidney disease?
For patients age 16 and older with advanced chronic kidney disease (CKD), including those undergoing hemodialysis, we recommend a higher dose of hepatitis B virus (HBV) vaccine, more doses, or both. Vaccination with a higher dose may improve the immune response. The hepatitis B surface antibody (anti-HBs) titer should be monitored 1 to 2 months after completion of the vaccination schedule and annually thereafter, with a target titer of 10 IU/mL or greater. For patients who do not develop a protective antibody titer after completing the initial vaccination schedule, the vaccination schedule should be repeated.
RECOMMENDED DOSES AND SCHEDULES
Recommendation 1
Give higher vaccine doses, increase the number of doses, or both.

Recommendation 2
A 4-dose regimen may provide a better antibody response than a 3-dose regimen. (Note: This recommendation applies only to Engerix-B; 4 doses of Recombivax-HB would be an off-label use.)
Rationale. The US Centers for Disease Control and Prevention reported that after completion of a 3-dose vaccination schedule, the median proportion of patients developing a protective antibody response was 64% (range 34%–88%) vs a median of 86% (range 40%–98%) after a 4-dose schedule.3
Lacson et al5 compared antibody response rates after 3 doses of Recombivax-HB and after 4 doses of Engerix-B and found a better response rate with the 4-dose schedule. The rate of persistent protective anti-HBs titer after 1 year was 77% for Engerix-B vs 53% for Recombivax-HB.
Agarwal et al6 evaluated response rates in patients who had mild CKD (serum creatinine levels 1.5–3.0 mg/dL), moderate CKD (creatinine 3.0–6.0 mg/dL), and severe CKD (creatinine > 6.0 mg/dL). The seroconversion rates after 3 doses of 40-μg HBV vaccine were 87.5% in those with mild CKD, 66.6% in those with moderate CKD, and 35.7% in those with severe disease. After a fourth dose, rates improved significantly to 100%, 77%, and 36.4%, respectively.
Recommendation 3
In patients with CKD, vaccination should be done early, before they become dependent on hemodialysis.
Rationale. Patients with advanced CKD may have a lower seroconversion rate. Fraser et al7 found that after a 4-dose series, the seroprotection rate in adult prehemodialysis patients with serum creatinine levels of 4 mg/dL or less was 86%, compared with 37% in patients with serum creatinine levels above 4 mg/dL, of whom 88% were on hemodialysis.7
In a 2003 prospective cohort study by DaRoza et al,8 patients with higher levels of kidney function were more likely to respond to HBV vaccination, and the level of kidney function was found to be an independent predictor of seroconversion.8
A 2012 prospective study by Ghadiani et al9 compared seroconversion rates in patients with stage 3 or 4 CKD vs patients on hemodialysis, with medical staff as controls. The authors reported seroprotection rates of 26.1% in patients on hemodialysis, 55.2% in patients with stage 3 or 4 CKD, and 96.2% in controls. They concluded that vaccination is more likely to induce seroconversion in earlier stages of kidney disease.9
MONITORING THE RESPONSE TO VACCINATION AND REVACCINATION
Testing after vaccination is recommended to determine response. Testing should be done 1 to 2 months after the last dose of the vaccination schedule.1–3 Anti-HBs levels 10 IU/mL and greater are considered protective.10
Revaccination with a full vaccination series is recommended for patients who do not develop adequate levels of protective antibodies after completion of the vaccination schedule.2 Reported response rates to revaccination have varied from 40% to 50% after 2 or 3 additional intramuscular doses of 40 µg, to 64% after 4 additional intramuscular doses of 10 µg.3 Serologic testing should be repeated after the last dose of the vaccination series, as serologic testing after only 1 or 2 additional doses appears to be no more cost-effective.2,3
To the best of our knowledge, no data exist to indicate that in nonresponders, further doses given after completion of 2 full vaccination schedules would induce an antibody response.
ANTIBODY PERSISTENCE AND BOOSTER DOSES
Antibody levels fall with time in patients on hemodialysis. Limited data suggest that in patients who respond to the primary vaccination series, antibodies remain detectable for 6 months in 80% to 100% (median 100%) of patients and for 12 months in 58% to 100% (median 70%) of patients.3 The need for booster doses should be assessed by annual monitoring.2,11 Booster doses should be given when the anti-HBs titer declines to below 10 IU/mL. Limited data indicate that nearly all such patients would respond to a booster dose.3
OTHER WAYS TO IMPROVE VACCINE RESPONSE
Other strategies to improve vaccine response, such as the addition of adjuvants or immunostimulants, have shown variable success.12 Intradermal HBV vaccination in patients on chronic hemodialysis has also been proposed. The efficacy of intradermal vaccination may be related to the dense network of immunologic dendritic cells within the dermis. After intradermal administration, the antigen is taken up by dendritic cells residing in the dermis, which mature and travel to the regional lymph node where further immunostimulation takes place.13
In a systematic review of four prospective trials with a total of 204 hemodialysis patients,13 a significantly higher proportion of patients achieved seroconversion with intradermal HBV vaccine administration than with intramuscular administration. The authors concluded that the intradermal route in primary nonresponders undergoing hemodialysis provides an effective alternative to the intramuscular route to protect against HBV infection in this highly susceptible population.
Additional well-designed, double-blinded, randomized trials are needed to establish clear guidelines on intradermal HBV vaccine dosing and vaccination schedules.
- Grzegorzewska AE. Hepatitis B vaccination in chronic kidney disease: review of evidence in non-dialyzed patients. Hepat Mon 2012; 12:e7359.
- Chi C, Patel P, Pilishvili T, Moore M, Murphy T, Strikas R. Guidelines for vaccinating kidney dialysis patients and patients with chronic kidney disease. www.cdc.gov/dialysis/PDFs/Vaccinating_Dialysis_Patients_and_Patients_dec2012.pdf. Accessed September 6, 2017.
- Recommendations for preventing transmission of infections among chronic hemodialysis patients. MMWR Recomm Rep 2001; 50:1–43.
- Kim DK, Riley LE, Harriman KH, Hunter P, Bridges CB; Advisory Committee on Immunization Practices. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. Ann Intern Med 2017; 166:209–219.
- Lacson E, Teng M, Ong J, Vienneau L, Ofsthun N, Lazarus JM. Antibody response to Engerix-B and Recombivax-HB hepatitis B vaccination in end-stage renal disease. Hemodialysis international. Hemodial Int 2005; 9:367–375.
- Agarwal SK, Irshad M, Dash SC. Comparison of two schedules of hepatitis B vaccination in patients with mild, moderate and severe renal failure. J Assoc Physicians India 1999; 47:183–185.
- Fraser GM, Ochana N, Fenyves D, et al. Increasing serum creatinine and age reduce the response to hepatitis B vaccine in renal failure patients. J Hepatol 1994; 21:450–454.
- DaRoza G, Loewen A, Djurdjev O, et al. Stage of chronic kidney disease predicts seroconversion after hepatitis B immunization: earlier is better. Am J Kidney Dis 2003; 42:1184–1192.
- Ghadiani MH, Besharati S, Mousavinasab N, Jalalzadeh M. Response rates to HB vaccine in CKD stages 3-4 and hemodialysis patients. J Res Med Sci 2012; 17:527–533.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Guidelines for vaccination in patients with chronic kidney disease. Indian J Nephrol 2016; 26(suppl 1):S15–S18.
- Somi MH, Hajipour B. Improving hepatitis B vaccine efficacy in end-stage renal diseases patients and role of adjuvants. ISRN Gastroenterol 2012; 2012:960413.
- Yousaf F, Gandham S, Galler M, Spinowitz B, Charytan C. Systematic review of the efficacy and safety of intradermal versus intramuscular hepatitis B vaccination in end-stage renal disease population unresponsive to primary vaccination series. Ren Fail 2015; 37:1080–1088.
For patients age 16 and older with advanced chronic kidney disease (CKD), including those undergoing hemodialysis, we recommend a higher dose of hepatitis B virus (HBV) vaccine, more doses, or both. Vaccination with a higher dose may improve the immune response. The hepatitis B surface antibody (anti-HBs) titer should be monitored 1 to 2 months after completion of the vaccination schedule and annually thereafter, with a target titer of 10 IU/mL or greater. For patients who do not develop a protective antibody titer after completing the initial vaccination schedule, the vaccination schedule should be repeated.
RECOMMENDED DOSES AND SCHEDULES
Recommendation 1
Give higher vaccine doses, increase the number of doses, or both.
Recommendation 2
A 4-dose regimen may provide a better antibody response than a 3-dose regimen. (Note: This recommendation applies only to Engerix-B; 4 doses of Recombivax-HB would be an off-label use.)
Rationale. The US Centers for Disease Control and Prevention reported that after completion of a 3-dose vaccination schedule, the median proportion of patients developing a protective antibody response was 64% (range 34%–88%) vs a median of 86% (range 40%–98%) after a 4-dose schedule.3
Lacson et al5 compared antibody response rates after 3 doses of Recombivax-HB and after 4 doses of Engerix-B and found a better response rate with the 4-dose schedule. The rate of persistent protective anti-HBs titer after 1 year was 77% for Engerix-B vs 53% for Recombivax-HB.
Agarwal et al6 evaluated response rates in patients who had mild CKD (serum creatinine levels 1.5–3.0 mg/dL), moderate CKD (creatinine 3.0–6.0 mg/dL), and severe CKD (creatinine > 6.0 mg/dL). The seroconversion rates after 3 doses of 40-μg HBV vaccine were 87.5% in those with mild CKD, 66.6% in those with moderate CKD, and 35.7% in those with severe disease. After a fourth dose, rates improved significantly to 100%, 77%, and 36.4%, respectively.
Recommendation 3
In patients with CKD, vaccination should be done early, before they become dependent on hemodialysis.
Rationale. Patients with advanced CKD may have a lower seroconversion rate. Fraser et al7 found that after a 4-dose series, the seroprotection rate in adult prehemodialysis patients with serum creatinine levels of 4 mg/dL or less was 86%, compared with 37% in patients with serum creatinine levels above 4 mg/dL, of whom 88% were on hemodialysis.7
In a 2003 prospective cohort study by DaRoza et al,8 patients with higher levels of kidney function were more likely to respond to HBV vaccination, and the level of kidney function was found to be an independent predictor of seroconversion.8
A 2012 prospective study by Ghadiani et al9 compared seroconversion rates in patients with stage 3 or 4 CKD vs patients on hemodialysis, with medical staff as controls. The authors reported seroprotection rates of 26.1% in patients on hemodialysis, 55.2% in patients with stage 3 or 4 CKD, and 96.2% in controls. They concluded that vaccination is more likely to induce seroconversion in earlier stages of kidney disease.9
MONITORING THE RESPONSE TO VACCINATION AND REVACCINATION
Testing after vaccination is recommended to determine response. Testing should be done 1 to 2 months after the last dose of the vaccination schedule.1–3 Anti-HBs levels 10 IU/mL and greater are considered protective.10
Revaccination with a full vaccination series is recommended for patients who do not develop adequate levels of protective antibodies after completion of the vaccination schedule.2 Reported response rates to revaccination have varied from 40% to 50% after 2 or 3 additional intramuscular doses of 40 µg, to 64% after 4 additional intramuscular doses of 10 µg.3 Serologic testing should be repeated after the last dose of the vaccination series, as serologic testing after only 1 or 2 additional doses appears to be no more cost-effective.2,3
To the best of our knowledge, no data exist to indicate that in nonresponders, further doses given after completion of 2 full vaccination schedules would induce an antibody response.
ANTIBODY PERSISTENCE AND BOOSTER DOSES
Antibody levels fall with time in patients on hemodialysis. Limited data suggest that in patients who respond to the primary vaccination series, antibodies remain detectable for 6 months in 80% to 100% (median 100%) of patients and for 12 months in 58% to 100% (median 70%) of patients.3 The need for booster doses should be assessed by annual monitoring.2,11 Booster doses should be given when the anti-HBs titer declines to below 10 IU/mL. Limited data indicate that nearly all such patients would respond to a booster dose.3
OTHER WAYS TO IMPROVE VACCINE RESPONSE
Other strategies to improve vaccine response, such as the addition of adjuvants or immunostimulants, have shown variable success.12 Intradermal HBV vaccination in patients on chronic hemodialysis has also been proposed. The efficacy of intradermal vaccination may be related to the dense network of immunologic dendritic cells within the dermis. After intradermal administration, the antigen is taken up by dendritic cells residing in the dermis, which mature and travel to the regional lymph node where further immunostimulation takes place.13
In a systematic review of four prospective trials with a total of 204 hemodialysis patients,13 a significantly higher proportion of patients achieved seroconversion with intradermal HBV vaccine administration than with intramuscular administration. The authors concluded that the intradermal route in primary nonresponders undergoing hemodialysis provides an effective alternative to the intramuscular route to protect against HBV infection in this highly susceptible population.
Additional well-designed, double-blinded, randomized trials are needed to establish clear guidelines on intradermal HBV vaccine dosing and vaccination schedules.
For patients age 16 and older with advanced chronic kidney disease (CKD), including those undergoing hemodialysis, we recommend a higher dose of hepatitis B virus (HBV) vaccine, more doses, or both. Vaccination with a higher dose may improve the immune response. The hepatitis B surface antibody (anti-HBs) titer should be monitored 1 to 2 months after completion of the vaccination schedule and annually thereafter, with a target titer of 10 IU/mL or greater. For patients who do not develop a protective antibody titer after completing the initial vaccination schedule, the vaccination schedule should be repeated.
RECOMMENDED DOSES AND SCHEDULES
Recommendation 1
Give higher vaccine doses, increase the number of doses, or both.
Recommendation 2
A 4-dose regimen may provide a better antibody response than a 3-dose regimen. (Note: This recommendation applies only to Engerix-B; 4 doses of Recombivax-HB would be an off-label use.)
Rationale. The US Centers for Disease Control and Prevention reported that after completion of a 3-dose vaccination schedule, the median proportion of patients developing a protective antibody response was 64% (range 34%–88%) vs a median of 86% (range 40%–98%) after a 4-dose schedule.3
Lacson et al5 compared antibody response rates after 3 doses of Recombivax-HB and after 4 doses of Engerix-B and found a better response rate with the 4-dose schedule. The rate of persistent protective anti-HBs titer after 1 year was 77% for Engerix-B vs 53% for Recombivax-HB.
Agarwal et al6 evaluated response rates in patients who had mild CKD (serum creatinine levels 1.5–3.0 mg/dL), moderate CKD (creatinine 3.0–6.0 mg/dL), and severe CKD (creatinine > 6.0 mg/dL). The seroconversion rates after 3 doses of 40-μg HBV vaccine were 87.5% in those with mild CKD, 66.6% in those with moderate CKD, and 35.7% in those with severe disease. After a fourth dose, rates improved significantly to 100%, 77%, and 36.4%, respectively.
Recommendation 3
In patients with CKD, vaccination should be done early, before they become dependent on hemodialysis.
Rationale. Patients with advanced CKD may have a lower seroconversion rate. Fraser et al7 found that after a 4-dose series, the seroprotection rate in adult prehemodialysis patients with serum creatinine levels of 4 mg/dL or less was 86%, compared with 37% in patients with serum creatinine levels above 4 mg/dL, of whom 88% were on hemodialysis.7
In a 2003 prospective cohort study by DaRoza et al,8 patients with higher levels of kidney function were more likely to respond to HBV vaccination, and the level of kidney function was found to be an independent predictor of seroconversion.8
A 2012 prospective study by Ghadiani et al9 compared seroconversion rates in patients with stage 3 or 4 CKD vs patients on hemodialysis, with medical staff as controls. The authors reported seroprotection rates of 26.1% in patients on hemodialysis, 55.2% in patients with stage 3 or 4 CKD, and 96.2% in controls. They concluded that vaccination is more likely to induce seroconversion in earlier stages of kidney disease.9
MONITORING THE RESPONSE TO VACCINATION AND REVACCINATION
Testing after vaccination is recommended to determine response. Testing should be done 1 to 2 months after the last dose of the vaccination schedule.1–3 Anti-HBs levels 10 IU/mL and greater are considered protective.10
Revaccination with a full vaccination series is recommended for patients who do not develop adequate levels of protective antibodies after completion of the vaccination schedule.2 Reported response rates to revaccination have varied from 40% to 50% after 2 or 3 additional intramuscular doses of 40 µg, to 64% after 4 additional intramuscular doses of 10 µg.3 Serologic testing should be repeated after the last dose of the vaccination series, as serologic testing after only 1 or 2 additional doses appears to be no more cost-effective.2,3
To the best of our knowledge, no data exist to indicate that in nonresponders, further doses given after completion of 2 full vaccination schedules would induce an antibody response.
ANTIBODY PERSISTENCE AND BOOSTER DOSES
Antibody levels fall with time in patients on hemodialysis. Limited data suggest that in patients who respond to the primary vaccination series, antibodies remain detectable for 6 months in 80% to 100% (median 100%) of patients and for 12 months in 58% to 100% (median 70%) of patients.3 The need for booster doses should be assessed by annual monitoring.2,11 Booster doses should be given when the anti-HBs titer declines to below 10 IU/mL. Limited data indicate that nearly all such patients would respond to a booster dose.3
OTHER WAYS TO IMPROVE VACCINE RESPONSE
Other strategies to improve vaccine response, such as the addition of adjuvants or immunostimulants, have shown variable success.12 Intradermal HBV vaccination in patients on chronic hemodialysis has also been proposed. The efficacy of intradermal vaccination may be related to the dense network of immunologic dendritic cells within the dermis. After intradermal administration, the antigen is taken up by dendritic cells residing in the dermis, which mature and travel to the regional lymph node where further immunostimulation takes place.13
In a systematic review of four prospective trials with a total of 204 hemodialysis patients,13 a significantly higher proportion of patients achieved seroconversion with intradermal HBV vaccine administration than with intramuscular administration. The authors concluded that the intradermal route in primary nonresponders undergoing hemodialysis provides an effective alternative to the intramuscular route to protect against HBV infection in this highly susceptible population.
Additional well-designed, double-blinded, randomized trials are needed to establish clear guidelines on intradermal HBV vaccine dosing and vaccination schedules.
- Grzegorzewska AE. Hepatitis B vaccination in chronic kidney disease: review of evidence in non-dialyzed patients. Hepat Mon 2012; 12:e7359.
- Chi C, Patel P, Pilishvili T, Moore M, Murphy T, Strikas R. Guidelines for vaccinating kidney dialysis patients and patients with chronic kidney disease. www.cdc.gov/dialysis/PDFs/Vaccinating_Dialysis_Patients_and_Patients_dec2012.pdf. Accessed September 6, 2017.
- Recommendations for preventing transmission of infections among chronic hemodialysis patients. MMWR Recomm Rep 2001; 50:1–43.
- Kim DK, Riley LE, Harriman KH, Hunter P, Bridges CB; Advisory Committee on Immunization Practices. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. Ann Intern Med 2017; 166:209–219.
- Lacson E, Teng M, Ong J, Vienneau L, Ofsthun N, Lazarus JM. Antibody response to Engerix-B and Recombivax-HB hepatitis B vaccination in end-stage renal disease. Hemodialysis international. Hemodial Int 2005; 9:367–375.
- Agarwal SK, Irshad M, Dash SC. Comparison of two schedules of hepatitis B vaccination in patients with mild, moderate and severe renal failure. J Assoc Physicians India 1999; 47:183–185.
- Fraser GM, Ochana N, Fenyves D, et al. Increasing serum creatinine and age reduce the response to hepatitis B vaccine in renal failure patients. J Hepatol 1994; 21:450–454.
- DaRoza G, Loewen A, Djurdjev O, et al. Stage of chronic kidney disease predicts seroconversion after hepatitis B immunization: earlier is better. Am J Kidney Dis 2003; 42:1184–1192.
- Ghadiani MH, Besharati S, Mousavinasab N, Jalalzadeh M. Response rates to HB vaccine in CKD stages 3-4 and hemodialysis patients. J Res Med Sci 2012; 17:527–533.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Guidelines for vaccination in patients with chronic kidney disease. Indian J Nephrol 2016; 26(suppl 1):S15–S18.
- Somi MH, Hajipour B. Improving hepatitis B vaccine efficacy in end-stage renal diseases patients and role of adjuvants. ISRN Gastroenterol 2012; 2012:960413.
- Yousaf F, Gandham S, Galler M, Spinowitz B, Charytan C. Systematic review of the efficacy and safety of intradermal versus intramuscular hepatitis B vaccination in end-stage renal disease population unresponsive to primary vaccination series. Ren Fail 2015; 37:1080–1088.
- Grzegorzewska AE. Hepatitis B vaccination in chronic kidney disease: review of evidence in non-dialyzed patients. Hepat Mon 2012; 12:e7359.
- Chi C, Patel P, Pilishvili T, Moore M, Murphy T, Strikas R. Guidelines for vaccinating kidney dialysis patients and patients with chronic kidney disease. www.cdc.gov/dialysis/PDFs/Vaccinating_Dialysis_Patients_and_Patients_dec2012.pdf. Accessed September 6, 2017.
- Recommendations for preventing transmission of infections among chronic hemodialysis patients. MMWR Recomm Rep 2001; 50:1–43.
- Kim DK, Riley LE, Harriman KH, Hunter P, Bridges CB; Advisory Committee on Immunization Practices. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. Ann Intern Med 2017; 166:209–219.
- Lacson E, Teng M, Ong J, Vienneau L, Ofsthun N, Lazarus JM. Antibody response to Engerix-B and Recombivax-HB hepatitis B vaccination in end-stage renal disease. Hemodialysis international. Hemodial Int 2005; 9:367–375.
- Agarwal SK, Irshad M, Dash SC. Comparison of two schedules of hepatitis B vaccination in patients with mild, moderate and severe renal failure. J Assoc Physicians India 1999; 47:183–185.
- Fraser GM, Ochana N, Fenyves D, et al. Increasing serum creatinine and age reduce the response to hepatitis B vaccine in renal failure patients. J Hepatol 1994; 21:450–454.
- DaRoza G, Loewen A, Djurdjev O, et al. Stage of chronic kidney disease predicts seroconversion after hepatitis B immunization: earlier is better. Am J Kidney Dis 2003; 42:1184–1192.
- Ghadiani MH, Besharati S, Mousavinasab N, Jalalzadeh M. Response rates to HB vaccine in CKD stages 3-4 and hemodialysis patients. J Res Med Sci 2012; 17:527–533.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Guidelines for vaccination in patients with chronic kidney disease. Indian J Nephrol 2016; 26(suppl 1):S15–S18.
- Somi MH, Hajipour B. Improving hepatitis B vaccine efficacy in end-stage renal diseases patients and role of adjuvants. ISRN Gastroenterol 2012; 2012:960413.
- Yousaf F, Gandham S, Galler M, Spinowitz B, Charytan C. Systematic review of the efficacy and safety of intradermal versus intramuscular hepatitis B vaccination in end-stage renal disease population unresponsive to primary vaccination series. Ren Fail 2015; 37:1080–1088.

Tamsulosin for patients with ureteral stones?
ILLUSTRATIVE CASE
A 54-year-old man presents to the emergency department (ED) with acute onset left flank pain that radiates to the groin. A computed tomography (CT) scan of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed appropriate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8% with a self-reported prevalence in men of 10.6% and a self-reported prevalence in women of 7.1%.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with nonsteroidal anti-inflammatory medications as first-line treatment and opioids as a second-line option.3 In addition, alpha-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the alpha-blocker tamsulosin. Two systematic reviews, limited by heterogeneity because some of the studies lacked a placebo control and blinding, concluded that alpha-blockers increased stone passage within one to 6 weeks when compared with placebo or no additional therapy.4,5 However, a recent large multicenter, randomized controlled trial (RCT) revealed no difference between tamsulosin and nifedipine or either one compared with placebo at decreasing the need for further treatment to achieve stone passage within 4 weeks.6
[polldaddy:9906038]
STUDY SUMMARY
New meta-analysis breaks down results by stone size
This meta-analysis by Wang et al, consisting of 8 randomized, double-blind, placebo-controlled trials of adult patients (N=1384), examined the effect of oral tamsulosin 0.4 mg/d (average of a 28-day course) on distal ureteral stone passage.1 A subgroup analysis comparing stone size (<5 mm and 5-10 mm) was also conducted to determine if stone size modified the effect of tamsulosin.
Although the initial search included studies published between 1966 and 2015, the 8 that were eventually analyzed were published between 2009 and 2015, were conducted in multiple countries (and included regardless of language), and were conducted in ED and outpatient urology settings. The main outcome measure was the risk difference in stone passage between the tamsulosin group and placebo group after follow-up imaging at 3 weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk of stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; risk difference [RD]=17%; 95% confidence interval [CI], 6%-27%), but significant heterogeneity existed across the trials (I2=80.2%). After subgroup analysis by stone size, the researchers found that tamsulosin was beneficial for larger stones, 5 to 10 mm in size (6 trials, N=514; RD=22%; 95% CI, 12%-33%; number needed to treat=5), compared with placebo, but not for smaller stones, <5 mm in size (4 trials, N=533; RD=-0.3%; 95% CI, -4% to 3%). The measure of heterogeneity in the 5- to 10-mm subgroup demonstrated a less heterogeneous population of studies (I2=33%) than that for the <5-mm subgroup (I2=0%).
In terms of adverse events, tamsulosin did not increase the risk of dizziness (RD=.2%; 95% CI, -2.1% to 2.5%) or postural hypotension (RD=.1%; 95% CI, -0.4% to 0.5%) compared with placebo.
WHAT’S NEW
Passage of larger stones increases with tamsulosin
This meta-analysis included only randomized, double-blind, placebo-controlled trials. Prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.) that recommended against the alpha-blockers tamsulosin and nifedipine for ureteral stones measuring <10 mm.6,7
But the subgroup analysis in this more recent review went one step further in the investigation of tamsulosin’s effect by examining passage rates by stone size (<5 mm vs 5-10 mm) and revealing that passage of larger stones (5-10 mm) increased with tamsulosin. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
Study doesn’t address proximal, or extra-large stones
Only distal stones were included in 7 of the 8 trials. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones >10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69:353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69:468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015;386:341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.
ILLUSTRATIVE CASE
A 54-year-old man presents to the emergency department (ED) with acute onset left flank pain that radiates to the groin. A computed tomography (CT) scan of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed appropriate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8% with a self-reported prevalence in men of 10.6% and a self-reported prevalence in women of 7.1%.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with nonsteroidal anti-inflammatory medications as first-line treatment and opioids as a second-line option.3 In addition, alpha-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the alpha-blocker tamsulosin. Two systematic reviews, limited by heterogeneity because some of the studies lacked a placebo control and blinding, concluded that alpha-blockers increased stone passage within one to 6 weeks when compared with placebo or no additional therapy.4,5 However, a recent large multicenter, randomized controlled trial (RCT) revealed no difference between tamsulosin and nifedipine or either one compared with placebo at decreasing the need for further treatment to achieve stone passage within 4 weeks.6
[polldaddy:9906038]
STUDY SUMMARY
New meta-analysis breaks down results by stone size
This meta-analysis by Wang et al, consisting of 8 randomized, double-blind, placebo-controlled trials of adult patients (N=1384), examined the effect of oral tamsulosin 0.4 mg/d (average of a 28-day course) on distal ureteral stone passage.1 A subgroup analysis comparing stone size (<5 mm and 5-10 mm) was also conducted to determine if stone size modified the effect of tamsulosin.
Although the initial search included studies published between 1966 and 2015, the 8 that were eventually analyzed were published between 2009 and 2015, were conducted in multiple countries (and included regardless of language), and were conducted in ED and outpatient urology settings. The main outcome measure was the risk difference in stone passage between the tamsulosin group and placebo group after follow-up imaging at 3 weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk of stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; risk difference [RD]=17%; 95% confidence interval [CI], 6%-27%), but significant heterogeneity existed across the trials (I2=80.2%). After subgroup analysis by stone size, the researchers found that tamsulosin was beneficial for larger stones, 5 to 10 mm in size (6 trials, N=514; RD=22%; 95% CI, 12%-33%; number needed to treat=5), compared with placebo, but not for smaller stones, <5 mm in size (4 trials, N=533; RD=-0.3%; 95% CI, -4% to 3%). The measure of heterogeneity in the 5- to 10-mm subgroup demonstrated a less heterogeneous population of studies (I2=33%) than that for the <5-mm subgroup (I2=0%).
In terms of adverse events, tamsulosin did not increase the risk of dizziness (RD=.2%; 95% CI, -2.1% to 2.5%) or postural hypotension (RD=.1%; 95% CI, -0.4% to 0.5%) compared with placebo.
WHAT’S NEW
Passage of larger stones increases with tamsulosin
This meta-analysis included only randomized, double-blind, placebo-controlled trials. Prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.) that recommended against the alpha-blockers tamsulosin and nifedipine for ureteral stones measuring <10 mm.6,7
But the subgroup analysis in this more recent review went one step further in the investigation of tamsulosin’s effect by examining passage rates by stone size (<5 mm vs 5-10 mm) and revealing that passage of larger stones (5-10 mm) increased with tamsulosin. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
Study doesn’t address proximal, or extra-large stones
Only distal stones were included in 7 of the 8 trials. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones >10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 54-year-old man presents to the emergency department (ED) with acute onset left flank pain that radiates to the groin. A computed tomography (CT) scan of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed appropriate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8% with a self-reported prevalence in men of 10.6% and a self-reported prevalence in women of 7.1%.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with nonsteroidal anti-inflammatory medications as first-line treatment and opioids as a second-line option.3 In addition, alpha-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the alpha-blocker tamsulosin. Two systematic reviews, limited by heterogeneity because some of the studies lacked a placebo control and blinding, concluded that alpha-blockers increased stone passage within one to 6 weeks when compared with placebo or no additional therapy.4,5 However, a recent large multicenter, randomized controlled trial (RCT) revealed no difference between tamsulosin and nifedipine or either one compared with placebo at decreasing the need for further treatment to achieve stone passage within 4 weeks.6
[polldaddy:9906038]
STUDY SUMMARY
New meta-analysis breaks down results by stone size
This meta-analysis by Wang et al, consisting of 8 randomized, double-blind, placebo-controlled trials of adult patients (N=1384), examined the effect of oral tamsulosin 0.4 mg/d (average of a 28-day course) on distal ureteral stone passage.1 A subgroup analysis comparing stone size (<5 mm and 5-10 mm) was also conducted to determine if stone size modified the effect of tamsulosin.
Although the initial search included studies published between 1966 and 2015, the 8 that were eventually analyzed were published between 2009 and 2015, were conducted in multiple countries (and included regardless of language), and were conducted in ED and outpatient urology settings. The main outcome measure was the risk difference in stone passage between the tamsulosin group and placebo group after follow-up imaging at 3 weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk of stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; risk difference [RD]=17%; 95% confidence interval [CI], 6%-27%), but significant heterogeneity existed across the trials (I2=80.2%). After subgroup analysis by stone size, the researchers found that tamsulosin was beneficial for larger stones, 5 to 10 mm in size (6 trials, N=514; RD=22%; 95% CI, 12%-33%; number needed to treat=5), compared with placebo, but not for smaller stones, <5 mm in size (4 trials, N=533; RD=-0.3%; 95% CI, -4% to 3%). The measure of heterogeneity in the 5- to 10-mm subgroup demonstrated a less heterogeneous population of studies (I2=33%) than that for the <5-mm subgroup (I2=0%).
In terms of adverse events, tamsulosin did not increase the risk of dizziness (RD=.2%; 95% CI, -2.1% to 2.5%) or postural hypotension (RD=.1%; 95% CI, -0.4% to 0.5%) compared with placebo.
WHAT’S NEW
Passage of larger stones increases with tamsulosin
This meta-analysis included only randomized, double-blind, placebo-controlled trials. Prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.) that recommended against the alpha-blockers tamsulosin and nifedipine for ureteral stones measuring <10 mm.6,7
But the subgroup analysis in this more recent review went one step further in the investigation of tamsulosin’s effect by examining passage rates by stone size (<5 mm vs 5-10 mm) and revealing that passage of larger stones (5-10 mm) increased with tamsulosin. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
Study doesn’t address proximal, or extra-large stones
Only distal stones were included in 7 of the 8 trials. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones >10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69:353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69:468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015;386:341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69:353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69:468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015;386:341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65:118-120.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
PRACTICE CHANGER
Prescribe tamsulosin for stone expulsion in patients with distal ureteral stones 5 to 10 mm in size.1
STRENGTH OF RECOMMENDATION
A: Based on a meta-analysis of randomized controlled trials.
Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69:353-361.
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.

Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
The National Kidney Foundation Names Clinician Reviews® Recipient of the 2018 Nostradamus Award
Frontline Medical Communications today announced that its journal, Clinician Reviews (CR), dedicated to nurse practitioners and physician assistants, has been named recipient of the 2018 Nostradamus Award.
Annually, National Kidney Foundation’s Council of Advanced Practitioners (CAP) presents this award to an individual, group, or organization that, through forethought and vision, recognizes, supports, and promotes the contributions of Advance Practitioners in nephrology. Clinician Reviews is being recognized for its Q&A feature Renal Consult, which provides expert advice to help clinicians address the complexities of renal diseases.
“Clinician Reviews joins a list of outstanding winners, including nephrologists, a United States senator, and others who have recognized CAP’s worth and supported its advancement,” said Karen Clemments, Editorial Director of clinical publications and Editor of Clinician Reviews. She continued, “We are excited to be among an esteemed group of past recipients for our ongoing endeavors to educate advanced practitioners in support of their clinical, professional needs in preventing, diagnosing, and treating kidney diseases.”
In announcing the award, Ms. Clemments noted that Renal Consult aligns with CAP’s goal to improve patient outcomes by enhancing advanced practitioners’ knowledge base and skills that will have a direct impact on clinical practice in a variety of settings. Renal Consult appears quarterly in print and online in CR’s robust, interactive website, digital edition, and mobile app.
The National Kidney Foundation is the leading organization in the United States dedicated to the awareness, prevention, and treatment of kidney disease for hundreds of thousands of healthcare professionals, millions of patients and their families, and tens of millions of Americans at risk. Clinician Reviews will be recognized during an awards luncheon at the NKF 2018 Spring Clinical Meeting in April.
Frontline Medical Communications today announced that its journal, Clinician Reviews (CR), dedicated to nurse practitioners and physician assistants, has been named recipient of the 2018 Nostradamus Award.
Annually, National Kidney Foundation’s Council of Advanced Practitioners (CAP) presents this award to an individual, group, or organization that, through forethought and vision, recognizes, supports, and promotes the contributions of Advance Practitioners in nephrology. Clinician Reviews is being recognized for its Q&A feature Renal Consult, which provides expert advice to help clinicians address the complexities of renal diseases.
“Clinician Reviews joins a list of outstanding winners, including nephrologists, a United States senator, and others who have recognized CAP’s worth and supported its advancement,” said Karen Clemments, Editorial Director of clinical publications and Editor of Clinician Reviews. She continued, “We are excited to be among an esteemed group of past recipients for our ongoing endeavors to educate advanced practitioners in support of their clinical, professional needs in preventing, diagnosing, and treating kidney diseases.”
In announcing the award, Ms. Clemments noted that Renal Consult aligns with CAP’s goal to improve patient outcomes by enhancing advanced practitioners’ knowledge base and skills that will have a direct impact on clinical practice in a variety of settings. Renal Consult appears quarterly in print and online in CR’s robust, interactive website, digital edition, and mobile app.
The National Kidney Foundation is the leading organization in the United States dedicated to the awareness, prevention, and treatment of kidney disease for hundreds of thousands of healthcare professionals, millions of patients and their families, and tens of millions of Americans at risk. Clinician Reviews will be recognized during an awards luncheon at the NKF 2018 Spring Clinical Meeting in April.
Frontline Medical Communications today announced that its journal, Clinician Reviews (CR), dedicated to nurse practitioners and physician assistants, has been named recipient of the 2018 Nostradamus Award.
Annually, National Kidney Foundation’s Council of Advanced Practitioners (CAP) presents this award to an individual, group, or organization that, through forethought and vision, recognizes, supports, and promotes the contributions of Advance Practitioners in nephrology. Clinician Reviews is being recognized for its Q&A feature Renal Consult, which provides expert advice to help clinicians address the complexities of renal diseases.
“Clinician Reviews joins a list of outstanding winners, including nephrologists, a United States senator, and others who have recognized CAP’s worth and supported its advancement,” said Karen Clemments, Editorial Director of clinical publications and Editor of Clinician Reviews. She continued, “We are excited to be among an esteemed group of past recipients for our ongoing endeavors to educate advanced practitioners in support of their clinical, professional needs in preventing, diagnosing, and treating kidney diseases.”
In announcing the award, Ms. Clemments noted that Renal Consult aligns with CAP’s goal to improve patient outcomes by enhancing advanced practitioners’ knowledge base and skills that will have a direct impact on clinical practice in a variety of settings. Renal Consult appears quarterly in print and online in CR’s robust, interactive website, digital edition, and mobile app.
The National Kidney Foundation is the leading organization in the United States dedicated to the awareness, prevention, and treatment of kidney disease for hundreds of thousands of healthcare professionals, millions of patients and their families, and tens of millions of Americans at risk. Clinician Reviews will be recognized during an awards luncheon at the NKF 2018 Spring Clinical Meeting in April.
