TORONTO – Hospitalized patients with acute kidney injury had more than double the inpatient rate of venous thromboembolism as had patients without acute kidney injury in a prospective, observational study of more than 6,000 hospitalized U.S. soldiers.
Mitchel L. Zoler/Frontline Medical News
Dr. Michael McMahon
“I think this should lower our threshold for investigating [possible cases of] venous thromboembolism in patients with acute kidney injury,” Michael McMahon, MD, said at the CHEST annual meeting. Acute kidney injury (AKI) “may require new prophylactic or diagnostic strategies” to prevent in-hospital venous thromboembolism (VTE) or to detect it early, said Dr. McMahon, a pulmonologist and critical care medicine physician at Walter Reed National Military Medical Center in Bethesda, Md.
He offered four possible mechanisms to explain a link between AKI and VTE:
Patients with AKI are in a hypercoagulable state.
AKI alters the pharmacodynamics or pharmacokinetics of VTE prophylactic treatments.
AKI is a marker of an illness that causes VTE.
VTE leads to an increased rate of AKI rather than the other way around.
Dr. McMahon’s analysis also revealed that two other clinical conditions that are generally believed to raise VTE risk – obesity and impaired overall renal function identified with stagnant measures – did not correspond with a significantly elevated VTE rate in this study.
The data came from 6,552 adults hospitalized for at least 2 days at Walter Reed between September 2009 and March 2011. The study excluded patients with VTE at the time of admission and also those who had been treated with an anticoagulant at the time of admission. The patients averaged 55 years of age and were hospitalized for a median of 4 days. About 22% of patients received VTE prophylaxis with unfractionated heparin, about 41% received prophylaxis with low-molecular-weight heparin, and about 39% received no VTE prophylaxis (percentages total 102% because of rounding).
About 16% of the patients had been diagnosed with AKI at the time of admission, and an additional 8% developed AKI while hospitalized, defined as an increase in serum creatinine during hospitalization of at least 50% above baseline levels or an increase of more than 0.3 mg/dL above the level at time of admission. During hospitalization, 160 patients (2%) developed a new onset VTE.
In an analysis that adjusted for baseline differences in type of surgery, body mass index, sex, age, and prior hospitalizations during the prior 90 days, the results showed that patients with preexisting or new onset AKI had a 2.2-fold higher rate of VTE, compared with patients without AKI, and this difference was statistically significant, Dr. McMahon reported.
The analysis also showed a significant 62% relatively higher rate of VTE among soldiers hospitalized for a deployment-related event, as well as a significant 63% relatively lower VTE rate among patients not receiving medical prophylaxis, compared with patients receiving an anticoagulant. Dr. McMahon suggested that this lower rate of VTEs among patients not on prophylaxis reflected success in identifying which patients had an increased risk for VTE and hence received prophylaxis.
TORONTO – Hospitalized patients with acute kidney injury had more than double the inpatient rate of venous thromboembolism as had patients without acute kidney injury in a prospective, observational study of more than 6,000 hospitalized U.S. soldiers.
Mitchel L. Zoler/Frontline Medical News
Dr. Michael McMahon
“I think this should lower our threshold for investigating [possible cases of] venous thromboembolism in patients with acute kidney injury,” Michael McMahon, MD, said at the CHEST annual meeting. Acute kidney injury (AKI) “may require new prophylactic or diagnostic strategies” to prevent in-hospital venous thromboembolism (VTE) or to detect it early, said Dr. McMahon, a pulmonologist and critical care medicine physician at Walter Reed National Military Medical Center in Bethesda, Md.
He offered four possible mechanisms to explain a link between AKI and VTE:
Patients with AKI are in a hypercoagulable state.
AKI alters the pharmacodynamics or pharmacokinetics of VTE prophylactic treatments.
AKI is a marker of an illness that causes VTE.
VTE leads to an increased rate of AKI rather than the other way around.
Dr. McMahon’s analysis also revealed that two other clinical conditions that are generally believed to raise VTE risk – obesity and impaired overall renal function identified with stagnant measures – did not correspond with a significantly elevated VTE rate in this study.
The data came from 6,552 adults hospitalized for at least 2 days at Walter Reed between September 2009 and March 2011. The study excluded patients with VTE at the time of admission and also those who had been treated with an anticoagulant at the time of admission. The patients averaged 55 years of age and were hospitalized for a median of 4 days. About 22% of patients received VTE prophylaxis with unfractionated heparin, about 41% received prophylaxis with low-molecular-weight heparin, and about 39% received no VTE prophylaxis (percentages total 102% because of rounding).
About 16% of the patients had been diagnosed with AKI at the time of admission, and an additional 8% developed AKI while hospitalized, defined as an increase in serum creatinine during hospitalization of at least 50% above baseline levels or an increase of more than 0.3 mg/dL above the level at time of admission. During hospitalization, 160 patients (2%) developed a new onset VTE.
In an analysis that adjusted for baseline differences in type of surgery, body mass index, sex, age, and prior hospitalizations during the prior 90 days, the results showed that patients with preexisting or new onset AKI had a 2.2-fold higher rate of VTE, compared with patients without AKI, and this difference was statistically significant, Dr. McMahon reported.
The analysis also showed a significant 62% relatively higher rate of VTE among soldiers hospitalized for a deployment-related event, as well as a significant 63% relatively lower VTE rate among patients not receiving medical prophylaxis, compared with patients receiving an anticoagulant. Dr. McMahon suggested that this lower rate of VTEs among patients not on prophylaxis reflected success in identifying which patients had an increased risk for VTE and hence received prophylaxis.
TORONTO – Hospitalized patients with acute kidney injury had more than double the inpatient rate of venous thromboembolism as had patients without acute kidney injury in a prospective, observational study of more than 6,000 hospitalized U.S. soldiers.
Mitchel L. Zoler/Frontline Medical News
Dr. Michael McMahon
“I think this should lower our threshold for investigating [possible cases of] venous thromboembolism in patients with acute kidney injury,” Michael McMahon, MD, said at the CHEST annual meeting. Acute kidney injury (AKI) “may require new prophylactic or diagnostic strategies” to prevent in-hospital venous thromboembolism (VTE) or to detect it early, said Dr. McMahon, a pulmonologist and critical care medicine physician at Walter Reed National Military Medical Center in Bethesda, Md.
He offered four possible mechanisms to explain a link between AKI and VTE:
Patients with AKI are in a hypercoagulable state.
AKI alters the pharmacodynamics or pharmacokinetics of VTE prophylactic treatments.
AKI is a marker of an illness that causes VTE.
VTE leads to an increased rate of AKI rather than the other way around.
Dr. McMahon’s analysis also revealed that two other clinical conditions that are generally believed to raise VTE risk – obesity and impaired overall renal function identified with stagnant measures – did not correspond with a significantly elevated VTE rate in this study.
The data came from 6,552 adults hospitalized for at least 2 days at Walter Reed between September 2009 and March 2011. The study excluded patients with VTE at the time of admission and also those who had been treated with an anticoagulant at the time of admission. The patients averaged 55 years of age and were hospitalized for a median of 4 days. About 22% of patients received VTE prophylaxis with unfractionated heparin, about 41% received prophylaxis with low-molecular-weight heparin, and about 39% received no VTE prophylaxis (percentages total 102% because of rounding).
About 16% of the patients had been diagnosed with AKI at the time of admission, and an additional 8% developed AKI while hospitalized, defined as an increase in serum creatinine during hospitalization of at least 50% above baseline levels or an increase of more than 0.3 mg/dL above the level at time of admission. During hospitalization, 160 patients (2%) developed a new onset VTE.
In an analysis that adjusted for baseline differences in type of surgery, body mass index, sex, age, and prior hospitalizations during the prior 90 days, the results showed that patients with preexisting or new onset AKI had a 2.2-fold higher rate of VTE, compared with patients without AKI, and this difference was statistically significant, Dr. McMahon reported.
The analysis also showed a significant 62% relatively higher rate of VTE among soldiers hospitalized for a deployment-related event, as well as a significant 63% relatively lower VTE rate among patients not receiving medical prophylaxis, compared with patients receiving an anticoagulant. Dr. McMahon suggested that this lower rate of VTEs among patients not on prophylaxis reflected success in identifying which patients had an increased risk for VTE and hence received prophylaxis.
Key clinical point: Hospitalized patients with acute kidney injury had a significantly higher rate of venous thromboembolism than did inpatients without AKI.
Major finding: Inpatients with AKI had an adjusted 2.2-fold higher rate of VTE, compared with other inpatients.
Data source: Prospective, observational data from 6,552 inpatients at a single U.S. military hospital.
Intense monitoring of urine output could be a useful tool in detecting acute kidney injury (AKI), according to a study conducted at the University of Pittsburgh.
Kui Jin, MD, of the University of Pittsburgh and his associates found that, after adjustment for baseline characteristics, intensive monitoring of urine output (UO) was associated with higher rates of AKI, with an odds ratio of 1.22. Intensive UO monitoring also was strongly associated with improved 30-day survival among patients developing AKI.
shironosov/Thinkstock
“Treatment for AKI is focused on supportive care and identification of the underlying etiology. Both of these priorities might be improved by earlier detection of AKI and closer monitoring of kidney function,” wrote Dr. Jin and his associates.
This retrospective cohort study included 15,724 adult patients admitted to the center’s ICUs during 2000-2008. All patients had either their UO or serum creatinine (SC) monitored. These patients were then divided into subcohorts that were monitored at one of two different intensities. UO intensive monitoring was defined by hourly recordings, with gaps no greater than 3 hours for the first 48 hours after ICU admission. The group receiving less intensive UO monitoring comprised patients who did not meet intensive monitoring criteria, regardless of their UO in the 7 days following ICU admission. The patients who had their SC intensively monitored had 3 calendar days of samples taken after their ICU admissions. Those who did not meet SC intensive monitoring criteria were placed into the less intensive SC monitoring group.
To understand the effect of the monitoring strategies on detecting the development of AKI, the researchers determined each patient’s baseline, admission, and reference serum creatinine levels. Baseline creatinine was defined as the lowest value in the year prior to hospital admission. Reference creatinine was the baseline creatinine, if available, or the lowest creatinine level recorded within 24 hours after ICU admission. A third method for determining reference creatinine levels was used for some patients, which involved making an estimation based on the Modification of Diet in Renal Disease equation for serum creatinine.
The crude rates of stage 2-3 AKI 7 days after admission to the ICU were similar between patients from both groups that had their UO monitored; 62.5% of intensive and 63.9% of less intensive patients displayed symptoms. After the researchers adjusted for baseline characteristics, however, intensive monitoring of UO was associated with greater rates of stage 2-3 AKI (OR, 1.22; P less than .001). Crude rates were higher in the patients who received intensive monitoring for SC, compared with patients who received less intensive monitoring for SC. Ultimately, Dr. Jin and his associates found that, when caring for patients with or without AKI, fluid management is one of the most important factors. Patients who underwent intensive UO monitoring received less fluid in their first 24 hours (3.6 L) in the ICU, compared with patients who received less intense UO monitoring (4.2 L). Patients who received intensive monitoring of their UO also were less likely to use vasopressors (29.9% vs. 43.3%; P less than .001), suggesting these patients were more hemodynamically stable. Further, the percentage of patients at or above 10% of fluid overload was lower in the group who received intensive monitoring of their UO (2.49% vs. 5.68%; P less than .001), during the first 72 hours in the ICU.
“Our results should help inform clinical decisions and ICU policy around frequency of monitoring of UO, especially for patients at high risk of AKI,” Dr. Jin and his colleagues wrote.
None of the authors had financial disclosures to report. Partial funding was provided by a research grant from C.R. Bard.
Intense monitoring of urine output could be a useful tool in detecting acute kidney injury (AKI), according to a study conducted at the University of Pittsburgh.
Kui Jin, MD, of the University of Pittsburgh and his associates found that, after adjustment for baseline characteristics, intensive monitoring of urine output (UO) was associated with higher rates of AKI, with an odds ratio of 1.22. Intensive UO monitoring also was strongly associated with improved 30-day survival among patients developing AKI.
shironosov/Thinkstock
“Treatment for AKI is focused on supportive care and identification of the underlying etiology. Both of these priorities might be improved by earlier detection of AKI and closer monitoring of kidney function,” wrote Dr. Jin and his associates.
This retrospective cohort study included 15,724 adult patients admitted to the center’s ICUs during 2000-2008. All patients had either their UO or serum creatinine (SC) monitored. These patients were then divided into subcohorts that were monitored at one of two different intensities. UO intensive monitoring was defined by hourly recordings, with gaps no greater than 3 hours for the first 48 hours after ICU admission. The group receiving less intensive UO monitoring comprised patients who did not meet intensive monitoring criteria, regardless of their UO in the 7 days following ICU admission. The patients who had their SC intensively monitored had 3 calendar days of samples taken after their ICU admissions. Those who did not meet SC intensive monitoring criteria were placed into the less intensive SC monitoring group.
To understand the effect of the monitoring strategies on detecting the development of AKI, the researchers determined each patient’s baseline, admission, and reference serum creatinine levels. Baseline creatinine was defined as the lowest value in the year prior to hospital admission. Reference creatinine was the baseline creatinine, if available, or the lowest creatinine level recorded within 24 hours after ICU admission. A third method for determining reference creatinine levels was used for some patients, which involved making an estimation based on the Modification of Diet in Renal Disease equation for serum creatinine.
The crude rates of stage 2-3 AKI 7 days after admission to the ICU were similar between patients from both groups that had their UO monitored; 62.5% of intensive and 63.9% of less intensive patients displayed symptoms. After the researchers adjusted for baseline characteristics, however, intensive monitoring of UO was associated with greater rates of stage 2-3 AKI (OR, 1.22; P less than .001). Crude rates were higher in the patients who received intensive monitoring for SC, compared with patients who received less intensive monitoring for SC. Ultimately, Dr. Jin and his associates found that, when caring for patients with or without AKI, fluid management is one of the most important factors. Patients who underwent intensive UO monitoring received less fluid in their first 24 hours (3.6 L) in the ICU, compared with patients who received less intense UO monitoring (4.2 L). Patients who received intensive monitoring of their UO also were less likely to use vasopressors (29.9% vs. 43.3%; P less than .001), suggesting these patients were more hemodynamically stable. Further, the percentage of patients at or above 10% of fluid overload was lower in the group who received intensive monitoring of their UO (2.49% vs. 5.68%; P less than .001), during the first 72 hours in the ICU.
“Our results should help inform clinical decisions and ICU policy around frequency of monitoring of UO, especially for patients at high risk of AKI,” Dr. Jin and his colleagues wrote.
None of the authors had financial disclosures to report. Partial funding was provided by a research grant from C.R. Bard.
Intense monitoring of urine output could be a useful tool in detecting acute kidney injury (AKI), according to a study conducted at the University of Pittsburgh.
Kui Jin, MD, of the University of Pittsburgh and his associates found that, after adjustment for baseline characteristics, intensive monitoring of urine output (UO) was associated with higher rates of AKI, with an odds ratio of 1.22. Intensive UO monitoring also was strongly associated with improved 30-day survival among patients developing AKI.
shironosov/Thinkstock
“Treatment for AKI is focused on supportive care and identification of the underlying etiology. Both of these priorities might be improved by earlier detection of AKI and closer monitoring of kidney function,” wrote Dr. Jin and his associates.
This retrospective cohort study included 15,724 adult patients admitted to the center’s ICUs during 2000-2008. All patients had either their UO or serum creatinine (SC) monitored. These patients were then divided into subcohorts that were monitored at one of two different intensities. UO intensive monitoring was defined by hourly recordings, with gaps no greater than 3 hours for the first 48 hours after ICU admission. The group receiving less intensive UO monitoring comprised patients who did not meet intensive monitoring criteria, regardless of their UO in the 7 days following ICU admission. The patients who had their SC intensively monitored had 3 calendar days of samples taken after their ICU admissions. Those who did not meet SC intensive monitoring criteria were placed into the less intensive SC monitoring group.
To understand the effect of the monitoring strategies on detecting the development of AKI, the researchers determined each patient’s baseline, admission, and reference serum creatinine levels. Baseline creatinine was defined as the lowest value in the year prior to hospital admission. Reference creatinine was the baseline creatinine, if available, or the lowest creatinine level recorded within 24 hours after ICU admission. A third method for determining reference creatinine levels was used for some patients, which involved making an estimation based on the Modification of Diet in Renal Disease equation for serum creatinine.
The crude rates of stage 2-3 AKI 7 days after admission to the ICU were similar between patients from both groups that had their UO monitored; 62.5% of intensive and 63.9% of less intensive patients displayed symptoms. After the researchers adjusted for baseline characteristics, however, intensive monitoring of UO was associated with greater rates of stage 2-3 AKI (OR, 1.22; P less than .001). Crude rates were higher in the patients who received intensive monitoring for SC, compared with patients who received less intensive monitoring for SC. Ultimately, Dr. Jin and his associates found that, when caring for patients with or without AKI, fluid management is one of the most important factors. Patients who underwent intensive UO monitoring received less fluid in their first 24 hours (3.6 L) in the ICU, compared with patients who received less intense UO monitoring (4.2 L). Patients who received intensive monitoring of their UO also were less likely to use vasopressors (29.9% vs. 43.3%; P less than .001), suggesting these patients were more hemodynamically stable. Further, the percentage of patients at or above 10% of fluid overload was lower in the group who received intensive monitoring of their UO (2.49% vs. 5.68%; P less than .001), during the first 72 hours in the ICU.
“Our results should help inform clinical decisions and ICU policy around frequency of monitoring of UO, especially for patients at high risk of AKI,” Dr. Jin and his colleagues wrote.
None of the authors had financial disclosures to report. Partial funding was provided by a research grant from C.R. Bard.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
Decreased distal delivery of sodium
Mineralocorticoid deficiency
Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Figure 1. A number of pharmacologic agents and conditions can interfere with the renin-angiotensin-aldosterone system, altering renal potassium excretion. Reabsorption of sodium in the collecting duct increases the luminal electronegativity, providing a more favorable gradient for potassium secretion. Aldosterone is critical for this reabsorptive process. A number of drugs and conditions interfere with the production of aldosterone and, as a result, reduce renal potassium secretion. In some patients, more than 1 disturbance may be present. NSAIDs = nonsteroidal anti-inflammatory drugs.
Decreased mineralocorticoid levels or activity due to disturbances in the renin-angiotensin-aldosterone system will impair renal potassium secretion. Such disturbances can be the result of diseases or drugs (Figure 1).13,16,17
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Figure 2. Electrocardiographic signs of hyperkalemia
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
ST-segment depression
Widening of the PR interval
Widening of the QRS interval
Loss of the P wave
A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
References
Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
Biff F. Palmer, MD Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX
Deborah J. Clegg, PhD Professor of Internal Medicine, Biomedical Research Department, Diabetes and Obesity Research Division, Cedars-Sinai Medical Center, Los Angeles, CA
Address: Biff F. Palmer, MD, Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390; [email protected]
Biff F. Palmer, MD Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX
Deborah J. Clegg, PhD Professor of Internal Medicine, Biomedical Research Department, Diabetes and Obesity Research Division, Cedars-Sinai Medical Center, Los Angeles, CA
Address: Biff F. Palmer, MD, Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390; [email protected]
Author and Disclosure Information
Biff F. Palmer, MD Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX
Deborah J. Clegg, PhD Professor of Internal Medicine, Biomedical Research Department, Diabetes and Obesity Research Division, Cedars-Sinai Medical Center, Los Angeles, CA
Address: Biff F. Palmer, MD, Professor of Internal Medicine, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390; [email protected]
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
Decreased distal delivery of sodium
Mineralocorticoid deficiency
Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Figure 1. A number of pharmacologic agents and conditions can interfere with the renin-angiotensin-aldosterone system, altering renal potassium excretion. Reabsorption of sodium in the collecting duct increases the luminal electronegativity, providing a more favorable gradient for potassium secretion. Aldosterone is critical for this reabsorptive process. A number of drugs and conditions interfere with the production of aldosterone and, as a result, reduce renal potassium secretion. In some patients, more than 1 disturbance may be present. NSAIDs = nonsteroidal anti-inflammatory drugs.
Decreased mineralocorticoid levels or activity due to disturbances in the renin-angiotensin-aldosterone system will impair renal potassium secretion. Such disturbances can be the result of diseases or drugs (Figure 1).13,16,17
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Figure 2. Electrocardiographic signs of hyperkalemia
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
ST-segment depression
Widening of the PR interval
Widening of the QRS interval
Loss of the P wave
A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
Decreased distal delivery of sodium
Mineralocorticoid deficiency
Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Figure 1. A number of pharmacologic agents and conditions can interfere with the renin-angiotensin-aldosterone system, altering renal potassium excretion. Reabsorption of sodium in the collecting duct increases the luminal electronegativity, providing a more favorable gradient for potassium secretion. Aldosterone is critical for this reabsorptive process. A number of drugs and conditions interfere with the production of aldosterone and, as a result, reduce renal potassium secretion. In some patients, more than 1 disturbance may be present. NSAIDs = nonsteroidal anti-inflammatory drugs.
Decreased mineralocorticoid levels or activity due to disturbances in the renin-angiotensin-aldosterone system will impair renal potassium secretion. Such disturbances can be the result of diseases or drugs (Figure 1).13,16,17
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Figure 2. Electrocardiographic signs of hyperkalemia
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
ST-segment depression
Widening of the PR interval
Widening of the QRS interval
Loss of the P wave
A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
References
Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
References
Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
Exclude pseudohyperkalemia in patients who have a normal electrocardiogram and no risk factors for the development of hyperkalemia.
Decreased distal delivery of sodium, reduced mineralocorticoid levels or activity, and a distal tubular defect are causes of impaired renal potassium secretion.
Medical conditions and medications that alter the renin-angiotensin-aldosterone system can give rise to hyperkalemia.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Author and Disclosure Information
Denise Link, MPAS, PA-C Nephrology PA University of Texas Southwestern Medical Center Dallas Outreach Chair for Council of Advanced Practitioners Executive Committee National Kidney Foundation AAPA Liaison to RPA (Renal Physicians Association) American Academy of PAs Dallas, Texas
Denise Link, MPAS, PA-C Nephrology PA University of Texas Southwestern Medical Center Dallas Outreach Chair for Council of Advanced Practitioners Executive Committee National Kidney Foundation AAPA Liaison to RPA (Renal Physicians Association) American Academy of PAs Dallas, Texas
Author and Disclosure Information
Denise Link, MPAS, PA-C Nephrology PA University of Texas Southwestern Medical Center Dallas Outreach Chair for Council of Advanced Practitioners Executive Committee National Kidney Foundation AAPA Liaison to RPA (Renal Physicians Association) American Academy of PAs Dallas, Texas
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Periprocedural administration of intravenous sodium bicarbonate did not improve outcomes compared with standard sodium chloride in patients with impaired kidney function undergoing angiography, according to results of a randomized study of 5,177 patients.
In addition, there was no benefit for oral acetylcysteine administration over placebo for mitigating those same postangiography risks, Steven D. Weisbord, MD, said at the American Heart Association scientific sessions in Anaheim, Calif.
Hypothetically, both sodium bicarbonate and acetylcysteine could help prevent acute kidney injury associated with contrast material used during angiography, said Dr. Weisbord of the University of Pittsburgh.
However, multiple studies of the two agents have yielded “inconsistent results … consequently, equipoise exists regarding these interventions, despite their widespread use in clinical practice,” Dr. Weisbord said.
To provide more definitive evidence, Dr. Weisbord and his colleagues conducted PRESERVE, a multicenter, randomized, controlled trial comprising 5,177 patients scheduled for angiography who were at high risk of renal complications. Using a 2-by-2 factorial design, patients were randomized to receive intravenous 1.26% sodium bicarbonate or intravenous 0.9% sodium chloride, and to 5 days of oral acetylcysteine or oral placebo.
They found no significant differences between arms in the study’s composite primary endpoint of death, need for dialysis, or persistent increase in serum creatinine by 50% or more.
That composite endpoint occurred in 4.4% of patients receiving sodium bicarbonate, and similarly in 4.7% of patients receiving sodium chloride.
Likewise, the endpoint occurred in 4.6% of patients in the acetylcysteine group and 4.5% of the placebo group, Dr. Weisbord reported.
The investigators had planned to enroll 7,680 patients, but the sponsor of the trial stopped the study after enrollment of 5,177 based on the results showing no significant benefit of either treatment, he noted.
There are a few reasons why results of PRESERVE might show a lack of benefit for these agents, in contrast to some previous studies suggesting both the treatments might reduce risk of contrast-associated renal complications in high-risk patients.
Notably, “most of these interventions have been underpowered,” Dr. Weisbord noted.
Also, most previous trials used a primary endpoint of increase in blood creatinine level within days of the angiography. By contrast, the primary endpoint of the current study was a composite of serious adverse events “that are recognized sequelae of acute kidney injury,” he added.
Although subsequent investigations could shed new light on the controversy, the findings of PRESERVE support the “strong likelihood that these interventions are not clinically effective” in preventing acute kidney injury or longer-term adverse outcomes after angiography, he concluded.
The PRESERVE results were published simultaneously with Dr. Weisbord’s presentation (N Engl J Med. 2017 Nov 12. doi: 10.1056/NEJMoa1710933).
The study was supported by the U.S. Department of Veterans Affairs Office of Research and Development and the National Health and Medical Research Council of Australia. Dr. Weisbord reported receiving personal fees from Durect outside the submitted work.
Periprocedural administration of intravenous sodium bicarbonate did not improve outcomes compared with standard sodium chloride in patients with impaired kidney function undergoing angiography, according to results of a randomized study of 5,177 patients.
In addition, there was no benefit for oral acetylcysteine administration over placebo for mitigating those same postangiography risks, Steven D. Weisbord, MD, said at the American Heart Association scientific sessions in Anaheim, Calif.
Hypothetically, both sodium bicarbonate and acetylcysteine could help prevent acute kidney injury associated with contrast material used during angiography, said Dr. Weisbord of the University of Pittsburgh.
However, multiple studies of the two agents have yielded “inconsistent results … consequently, equipoise exists regarding these interventions, despite their widespread use in clinical practice,” Dr. Weisbord said.
To provide more definitive evidence, Dr. Weisbord and his colleagues conducted PRESERVE, a multicenter, randomized, controlled trial comprising 5,177 patients scheduled for angiography who were at high risk of renal complications. Using a 2-by-2 factorial design, patients were randomized to receive intravenous 1.26% sodium bicarbonate or intravenous 0.9% sodium chloride, and to 5 days of oral acetylcysteine or oral placebo.
They found no significant differences between arms in the study’s composite primary endpoint of death, need for dialysis, or persistent increase in serum creatinine by 50% or more.
That composite endpoint occurred in 4.4% of patients receiving sodium bicarbonate, and similarly in 4.7% of patients receiving sodium chloride.
Likewise, the endpoint occurred in 4.6% of patients in the acetylcysteine group and 4.5% of the placebo group, Dr. Weisbord reported.
The investigators had planned to enroll 7,680 patients, but the sponsor of the trial stopped the study after enrollment of 5,177 based on the results showing no significant benefit of either treatment, he noted.
There are a few reasons why results of PRESERVE might show a lack of benefit for these agents, in contrast to some previous studies suggesting both the treatments might reduce risk of contrast-associated renal complications in high-risk patients.
Notably, “most of these interventions have been underpowered,” Dr. Weisbord noted.
Also, most previous trials used a primary endpoint of increase in blood creatinine level within days of the angiography. By contrast, the primary endpoint of the current study was a composite of serious adverse events “that are recognized sequelae of acute kidney injury,” he added.
Although subsequent investigations could shed new light on the controversy, the findings of PRESERVE support the “strong likelihood that these interventions are not clinically effective” in preventing acute kidney injury or longer-term adverse outcomes after angiography, he concluded.
The PRESERVE results were published simultaneously with Dr. Weisbord’s presentation (N Engl J Med. 2017 Nov 12. doi: 10.1056/NEJMoa1710933).
The study was supported by the U.S. Department of Veterans Affairs Office of Research and Development and the National Health and Medical Research Council of Australia. Dr. Weisbord reported receiving personal fees from Durect outside the submitted work.
Periprocedural administration of intravenous sodium bicarbonate did not improve outcomes compared with standard sodium chloride in patients with impaired kidney function undergoing angiography, according to results of a randomized study of 5,177 patients.
In addition, there was no benefit for oral acetylcysteine administration over placebo for mitigating those same postangiography risks, Steven D. Weisbord, MD, said at the American Heart Association scientific sessions in Anaheim, Calif.
Hypothetically, both sodium bicarbonate and acetylcysteine could help prevent acute kidney injury associated with contrast material used during angiography, said Dr. Weisbord of the University of Pittsburgh.
However, multiple studies of the two agents have yielded “inconsistent results … consequently, equipoise exists regarding these interventions, despite their widespread use in clinical practice,” Dr. Weisbord said.
To provide more definitive evidence, Dr. Weisbord and his colleagues conducted PRESERVE, a multicenter, randomized, controlled trial comprising 5,177 patients scheduled for angiography who were at high risk of renal complications. Using a 2-by-2 factorial design, patients were randomized to receive intravenous 1.26% sodium bicarbonate or intravenous 0.9% sodium chloride, and to 5 days of oral acetylcysteine or oral placebo.
They found no significant differences between arms in the study’s composite primary endpoint of death, need for dialysis, or persistent increase in serum creatinine by 50% or more.
That composite endpoint occurred in 4.4% of patients receiving sodium bicarbonate, and similarly in 4.7% of patients receiving sodium chloride.
Likewise, the endpoint occurred in 4.6% of patients in the acetylcysteine group and 4.5% of the placebo group, Dr. Weisbord reported.
The investigators had planned to enroll 7,680 patients, but the sponsor of the trial stopped the study after enrollment of 5,177 based on the results showing no significant benefit of either treatment, he noted.
There are a few reasons why results of PRESERVE might show a lack of benefit for these agents, in contrast to some previous studies suggesting both the treatments might reduce risk of contrast-associated renal complications in high-risk patients.
Notably, “most of these interventions have been underpowered,” Dr. Weisbord noted.
Also, most previous trials used a primary endpoint of increase in blood creatinine level within days of the angiography. By contrast, the primary endpoint of the current study was a composite of serious adverse events “that are recognized sequelae of acute kidney injury,” he added.
Although subsequent investigations could shed new light on the controversy, the findings of PRESERVE support the “strong likelihood that these interventions are not clinically effective” in preventing acute kidney injury or longer-term adverse outcomes after angiography, he concluded.
The PRESERVE results were published simultaneously with Dr. Weisbord’s presentation (N Engl J Med. 2017 Nov 12. doi: 10.1056/NEJMoa1710933).
The study was supported by the U.S. Department of Veterans Affairs Office of Research and Development and the National Health and Medical Research Council of Australia. Dr. Weisbord reported receiving personal fees from Durect outside the submitted work.
Key clinical point: In patients with impaired kidney function, periprocedural sodium bicarbonate did not improve postangiography clinical outcomes compared with standard sodium chloride, and neither did oral acetylcysteine when compared to placebo.
Major finding: The composite primary endpoint of death, need for dialysis, or persistent increase in serum creatinine was similar regardless of which treatments the patients received.
Data source: PRESERVE, a randomized study using a 2-by-2 factorial design to evaluate intravenous sodium bicarbonate versus sodium chloride and acetylcysteine versus placebo in 5,177 patients at high risk of renal complications.
Disclosures: PRESERVE was supported by the U.S. Department of Veterans Affairs Office of Research and Development and the National Health and Medical Research Council of Australia. Dr. Weisbord reported receiving personal fees from Durect outside the submitted work.
SAN DIEGO – Among patients with systemic lupus erythematosus, a low level of vitamin D is associated with an increased risk of end-stage renal disease and total organ damage, results from a single-center cohort study showed.
“We had previously proved that vitamin D supplementation helped lupus activity,” lead study author Michelle Petri, MD, MPH, said in an interview in advance of the annual meeting of the American College of Rheumatology. “Now, we prove that it specifically helps renal activity as measured by the urine protein. By helping to reduce urine protein, it helps to prevent permanent renal damage and end-stage renal disease.”
Dr. Michelle Petri
In an effort to determine whether low vitamin D predicted later renal damage, Dr. Petri of the department of rheumatology at Johns Hopkins University, Baltimore, and her associates evaluated 1,392 patients in the Hopkins Lupus Cohort, a longitudinal study of over 2,000 systemic lupus erythematosus (SLE) patients who are seen quarterly.
The first measure of vitamin D typically occurred in late 2009 or 2010 for existing patients and at the first visit of new patients after that. The researchers categorized patients based on their first measure of vitamin D as less than 20 ng/mL or 20 ng/mL or higher. At the first visit when vitamin D was measured, 27.3% had levels of 25-hydroxyvitamin D less than 20 ng/mL. The mean age of patients was 47.3 years, 92% were female, 50% were white, and 41% were African American.
In the study, Dr. Petri and her associates used the Systemic Lupus International Collaborative Clinics/American College of Rheumatology Damage Index to calculate the risk of lifetime organ damage. After adjusting for age, gender, and ethnicity, low levels of vitamin D were significantly associated with increased risk of renal damage (RR, 1.66; P = .0206) and total organ damage (RR, 1.17; P = .0245), they found.
Skin damage was another concern, with an adjusted relative risk of 1.22, though it was not statistically significant (P = .3561). The investigators observed no association between low vitamin D and musculoskeletal damage, including osteoporotic fractures.
“There is a lot of interest in lupus right now, due to [singer Selena] Gomez’s kidney transplant for lupus nephritis,” said Dr. Petri, who also directs the Johns Hopkins Lupus Center. “So, I think there is interest in how to prevent the need for kidney transplant. Vitamin D helps kidney lupus – and we only need to achieve a level of 40 ng/mL, [which is] safe and easy to do.” She acknowledged the study’s single-center design as a limitation but underscored its large sample size as a strength.
The Hopkins Lupus Cohort is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Petri disclosed having received research support from Anthera, GlaxoSmithKline, EMD Serono, Eli Lilly, Bristol-Myers Squibb, Amgen, United Rheumatology, Global Academy, and Exagen.
SAN DIEGO – Among patients with systemic lupus erythematosus, a low level of vitamin D is associated with an increased risk of end-stage renal disease and total organ damage, results from a single-center cohort study showed.
“We had previously proved that vitamin D supplementation helped lupus activity,” lead study author Michelle Petri, MD, MPH, said in an interview in advance of the annual meeting of the American College of Rheumatology. “Now, we prove that it specifically helps renal activity as measured by the urine protein. By helping to reduce urine protein, it helps to prevent permanent renal damage and end-stage renal disease.”
Dr. Michelle Petri
In an effort to determine whether low vitamin D predicted later renal damage, Dr. Petri of the department of rheumatology at Johns Hopkins University, Baltimore, and her associates evaluated 1,392 patients in the Hopkins Lupus Cohort, a longitudinal study of over 2,000 systemic lupus erythematosus (SLE) patients who are seen quarterly.
The first measure of vitamin D typically occurred in late 2009 or 2010 for existing patients and at the first visit of new patients after that. The researchers categorized patients based on their first measure of vitamin D as less than 20 ng/mL or 20 ng/mL or higher. At the first visit when vitamin D was measured, 27.3% had levels of 25-hydroxyvitamin D less than 20 ng/mL. The mean age of patients was 47.3 years, 92% were female, 50% were white, and 41% were African American.
In the study, Dr. Petri and her associates used the Systemic Lupus International Collaborative Clinics/American College of Rheumatology Damage Index to calculate the risk of lifetime organ damage. After adjusting for age, gender, and ethnicity, low levels of vitamin D were significantly associated with increased risk of renal damage (RR, 1.66; P = .0206) and total organ damage (RR, 1.17; P = .0245), they found.
Skin damage was another concern, with an adjusted relative risk of 1.22, though it was not statistically significant (P = .3561). The investigators observed no association between low vitamin D and musculoskeletal damage, including osteoporotic fractures.
“There is a lot of interest in lupus right now, due to [singer Selena] Gomez’s kidney transplant for lupus nephritis,” said Dr. Petri, who also directs the Johns Hopkins Lupus Center. “So, I think there is interest in how to prevent the need for kidney transplant. Vitamin D helps kidney lupus – and we only need to achieve a level of 40 ng/mL, [which is] safe and easy to do.” She acknowledged the study’s single-center design as a limitation but underscored its large sample size as a strength.
The Hopkins Lupus Cohort is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Petri disclosed having received research support from Anthera, GlaxoSmithKline, EMD Serono, Eli Lilly, Bristol-Myers Squibb, Amgen, United Rheumatology, Global Academy, and Exagen.
SAN DIEGO – Among patients with systemic lupus erythematosus, a low level of vitamin D is associated with an increased risk of end-stage renal disease and total organ damage, results from a single-center cohort study showed.
“We had previously proved that vitamin D supplementation helped lupus activity,” lead study author Michelle Petri, MD, MPH, said in an interview in advance of the annual meeting of the American College of Rheumatology. “Now, we prove that it specifically helps renal activity as measured by the urine protein. By helping to reduce urine protein, it helps to prevent permanent renal damage and end-stage renal disease.”
Dr. Michelle Petri
In an effort to determine whether low vitamin D predicted later renal damage, Dr. Petri of the department of rheumatology at Johns Hopkins University, Baltimore, and her associates evaluated 1,392 patients in the Hopkins Lupus Cohort, a longitudinal study of over 2,000 systemic lupus erythematosus (SLE) patients who are seen quarterly.
The first measure of vitamin D typically occurred in late 2009 or 2010 for existing patients and at the first visit of new patients after that. The researchers categorized patients based on their first measure of vitamin D as less than 20 ng/mL or 20 ng/mL or higher. At the first visit when vitamin D was measured, 27.3% had levels of 25-hydroxyvitamin D less than 20 ng/mL. The mean age of patients was 47.3 years, 92% were female, 50% were white, and 41% were African American.
In the study, Dr. Petri and her associates used the Systemic Lupus International Collaborative Clinics/American College of Rheumatology Damage Index to calculate the risk of lifetime organ damage. After adjusting for age, gender, and ethnicity, low levels of vitamin D were significantly associated with increased risk of renal damage (RR, 1.66; P = .0206) and total organ damage (RR, 1.17; P = .0245), they found.
Skin damage was another concern, with an adjusted relative risk of 1.22, though it was not statistically significant (P = .3561). The investigators observed no association between low vitamin D and musculoskeletal damage, including osteoporotic fractures.
“There is a lot of interest in lupus right now, due to [singer Selena] Gomez’s kidney transplant for lupus nephritis,” said Dr. Petri, who also directs the Johns Hopkins Lupus Center. “So, I think there is interest in how to prevent the need for kidney transplant. Vitamin D helps kidney lupus – and we only need to achieve a level of 40 ng/mL, [which is] safe and easy to do.” She acknowledged the study’s single-center design as a limitation but underscored its large sample size as a strength.
The Hopkins Lupus Cohort is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Petri disclosed having received research support from Anthera, GlaxoSmithKline, EMD Serono, Eli Lilly, Bristol-Myers Squibb, Amgen, United Rheumatology, Global Academy, and Exagen.
Key clinical point: Supplemental vitamin D should be part of the treatment plan for patients with systemic lupus erythematosus (SLE).
Major finding: SLE patients with low vitamin D levels face a significantly increased risk of renal damage (relatve risk, 1.66; P = .0206) and total organ damage (RR, 1.17; P = .0245).
Study details: A single-center cohort study of 1,392 patients with SLE.
Disclosures: The Hopkins Lupus Cohort is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Petri disclosed having received research support from Anthera, GlaxoSmithKline, EMD Serono, Eli Lilly, Bristol-Myers Squibb, Amgen, United Rheumatology, Global Academy, and Exagen.
Approximately one in three US adults, or about 75 million people, have high blood pressure (BP), which has been defined as a BP of 140/90 mm Hg or higher.1 Unfortunately, only about half (54%) of those affected have their condition under optimal control.1 From an epidemiologic standpoint, hypertension has the distinction of being the most common chronic condition in the US, affecting about 54% of persons ages 55 to 64 and about 73% of those 75 and older.2,3 It is the number one reason patients schedule office visits with physicians; it accounts for the most prescriptions; and it is a major risk factor for heart disease and stroke, as well as a significant contributor to mortality throughout the world.4
HYPERTENSIVE URGENCY VS EMERGENCY
Hypertensive urgencies and emergencies account for approximately 27% of all medical emergencies and 2% to 3% of all annual visits to the emergency department (ED).5 Hypertensive urgency, or severe asymptomatic hypertension, is a common complaint in urgent care clinics and primary care offices as well. It is often defined as a systolic BP (SBP) of ≥ 160 mm Hg and/or a diastolic BP (DBP) ≥ 100 mm Hg with no associated end-organ damage.5-7 Patients may experience hypertensive urgency if they have been noncompliant with their antihypertensive drug regimen; present with pain; have white-coat hypertension or anxiety; or use recreational drugs (eg, sympathomimetics).5,8-10
Alternatively, hypertensive emergency, also known as hypertensive crisis, is generally defined as elevated BP > 180/120 mm Hg. Equally important, it is associated with signs, symptoms, or laboratory values indicative of target end-organ damage, such as cerebrovascular accident, myocardial infarction (MI), aortic dissection, acute left ventricular failure, acute pulmonary edema, acute renal failure, acute mental status changes (hypertensive encephalopathy), and eclampsia.5,7,8,11,12
Determining appropriate management for patients with hypertensive urgency is controversial among clinicians. Practice patterns range from full screening and “rule-outs”—with prompt initiation of antihypertensive agents, regardless of whether the patient is symptomatic—to sending the patient home with minimal screening, laboratory testing, or treatment.
This article offers a guided approach to managing patients with hypertensive urgency in a logical fashion, based on risk stratification, thereby avoiding both extremes (extensive unnecessary workup or discharge without workup resulting in adverse outcomes). It is vital to differentiate between patients with hypertensive emergency, in which BP should be lowered in minutes, and patients with hypertensive urgency, in which BP can be lowered more slowly.12
PATHOPHYSIOLOGY
Normally, when BP increases, blood vessel diameter changes in response; this autoregulation serves to limit damage. However, when BP increases abruptly, the body’s ability to hemodynamically calibrate to such a rapid change is impeded, thus allowing for potential end-organ damage.5,12 The increased vascular resistance observed in many patients with hypertension appears to be an autoregulatory process that helps to maintain a normal or viable level of tissue blood flow and organ perfusion despite the increased BP, rather than a primary cause of the hypertension.13
The exact physiology of hypertensive urgencies is not clearly understood, because of the multifactorial nature of the process. One leading theory is that circulating humoral vasoconstrictors cause an abrupt increase in systemic vascular resistance, which in turn causes mechanical shear stress to the endothelial wall. This endothelial damage promotes more vasoconstriction, platelet aggregation, and activation of the renin-angiotensin-aldosterone system, which thereby increases release of angiotensin II and various cytokines.14
HISTORY AND PHYSICAL
A detailed medical history is of utmost importance in distinguishing patients who present with asymptomatic hypertensive urgency from those experiencing a hypertensive emergency. In addition, obtain a full medication list, including any nutritional supplements or illicit drugs the patient may be taking. Question the patient regarding medication adherence; some may not be taking antihypertensive agents as prescribed or may have altered the dosing frequency in an effort to extend the duration of their prescription.5,8 Table 1 lists pertinent questions to ask at presentation; the answers will dictate who needs further workup and possible admission as well as who will require screening for end-organ damage.7
The physical exam should focus primarily on a thorough cardiopulmonary and neurologic examination, as well as funduscopic examination, if needed. A complete set of vital signs should be recorded upon the patient’s arrival to the ED or clinic and should be repeated on the opposite arm for verification. Beginning with the eyes, conduct a thorough funduscopic examination to evaluate for papilledema or hemorrhages.5 During the cardiopulmonary exam, attention should be focused on signs of congestive heart failure and/or pulmonary edema, such as increased jugular vein distension, an S3 gallop, peripheral edema, and pulmonary rales. The neurologic exam is essential in evaluating for cerebrovascular accident, transient ischemic attack, or intracranial hemorrhage. A full cranial nerve examination is necessary, in addition to motor and sensory testing, at minimum.5,9
RISK STRATIFICATION
According to the 2013 Task Force of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC), several risk factors contribute to overall cardiovascular risk in asymptomatic patients presenting with severe hypertension (see Table 2).8 This report has been monumental in linking grades of hypertension directly to cardiovascular risk factors, but it differs from that recently published by the Eighth Joint National Committee (JNC 8), which offers evidence-based guidelines for the management of high BP in the general population of adults (with some modifications for individuals with diabetes or chronic kidney disease or of black ethnicity).15
According to the ESH/ESC study, patients with one or two risk factors who have grade 1 hypertension (SBP 140-159 mm Hg) are at moderate risk for cardiovascular disease (CVD) and patients with grade 2 (SBP 160-179 mm Hg) or grade 3 (SBP ≥ 180 mm Hg) hypertension are at moderate-to-high risk and high risk, respectively.8 Patients with three or more risk factors, or who already have end-organ damage, diabetes, or chronic kidney disease, enter the high-risk category for CVD even at grade 1 hypertension.8
These cardiovascular risk factors can and should be used as guidelines for deciding who needs further screening and who may have benign causes of severe hypertension (eg, white-coat hypertension, anxiety) that can be managed safely in an outpatient setting. In the author’s opinion, patients with known cardiovascular risk factors, those with signs or symptoms of end-organ damage, and those with test results suggestive of end-organ damage should have a more immediate treatment strategy initiated.
Numerous observational studies have shown a direct relationship between systemic hypertension and CVD risk in men and women of various ages, races, and ethnicities, regardless of other risk factors for CVD.12 In patients with diabetes, uncontrolled hypertension is a strong predictor of cardiovascular morbidity and mortality and of progressive nephropathy leading to chronic kidney disease.8
SCREENING
Results from the following tests may provide useful clues in the workup of a patient with hypertensive urgency.
Basic metabolic panel.Many EDs and primary care offices offer point-of-care testing that can typically give a rapid (< 10 min) result of a basic metabolic panel. This useful, quick screening tool can identify renal failure due to chronic untreated hypertension, acute renal failure, or other disease states that cause electrolyte abnormalities such as hyperaldosteronism (hypertension with hypokalemia) or Cushing syndrome (hypertension with hypernatremia and hyperkalemia).7
Cardiac enzymes. Measurement of cardiac troponins (T or I) may provide confirmatory evidence of myocardial necrosis within two to three hours of suspected acute MI.16,17 These tests are now available in most EDs and some clinics with point-of-care testing. A variety of current guidelines advocate repeat cardiac enzyme measurements at various time points, depending on results of initial testing and concomitant risk factors. These protocols vary by facility.
ECG. Obtaining an ECG is another quick, easy, and useful way to screen patients presenting with severe hypertensive urgency. Evidence of left ventricular hypertrophy suggests an increased risk for MI, stroke, heart failure, and sudden death.7,18-20 The Cornell criteria of summing the R wave in aVL and the S wave in V3, with a cutoff of 2.8 mV in men and 2.0 mV in women, has been shown to be the best predictor of future cardiovascular mortality.7 While an isolated finding of left ventricular hypertrophy on an ECG—in and of itself—may have limited value for an individual patient, this finding coupled with other risk factors may alter the provider’s assessment.
Chest radiograph. A chest radiograph can be helpful when used in conjunction with physical exam findings that suggest pulmonary edema and cardiomegaly.7 Widened mediastinum and tortuous aorta may also be evident on chest x-ray, necessitating further workup and imaging.
Urinalysis. In a patient presenting with asymptomatic hypertensive urgency, a urine dipstick result that shows new-onset proteinuria, while not definitive for diagnosis of nephrotic syndrome, may certainly prove helpful in the patient’s workup.5,13
Urine drug screen. In patients without a history of hypertension who present with asymptomatic hypertensive urgency, the urine drug screen may ascertain exposure to cocaine, amphetamine, or phencyclidine.
Pregnancy test. A pregnancy test is essential for any female patient of childbearing age presenting to the ED, and a positive result may be concerning for preeclampsia in a hypertensive patient with no prior history of the condition.7
TREATMENT
Knowing who to treat and when is a vast area of debate among emergency and primary care providers. Patients with hypertension who have established risk factors are known to have worse outcomes than those who may be otherwise healthy. Some clinicians believe that patients presenting with hypertensive urgency should be discharged home without screening and/or treatment. However, because uncontrolled severe hypertension can lead to acute complications (eg, MI, cerebrovascular accident), in practice, many providers are unwilling to send the patient home without workup.12 The patient’s condition must be viewed in the context of the entire disease spectrum, including risk factors.
The Figure offers a disposition pathway of recommendations based on risk stratification as well as screening tools for some of the less common causes of hypertensive urgency. Regardless of the results of screening tests or the decision to treat, affected patients require close primary care follow-up. Many of these patients may need further testing and careful management of their BP medication regimen.
How to treat
For patients with severe asymptomatic hypertension, if the history, physical, and screening tests do not show evidence of end-organ damage, BP can be controlled within 24 to 48 hours.5,10,11,21 In adults with hypertensive urgency, the most reasonable goal is to reduce the BP to ≤ 160/100 mm Hg5-7; however, the mean arterial pressure should not be lowered by more than 25% within the first two to three hours.13
Patients at high risk for imminent neurovascular, cardiovascular, renovascular, or pulmonary events should have their BP lowered over a period of hours, not minutes. In fact, there is evidence that rapid lowering of BP in asymptomatic patients may cause adverse outcomes.6 For example, in patients with acute ischemic stroke, increases in cerebral perfusion pressure promote an increase in vascular resistance—but decreasing the cerebral perfusion pressure abruptly will thereby decrease the cerebral blood flow, potentially causing cerebral ischemia or a worsening of the stroke.9,14
Treatment options
A broad spectrum of therapeutic options has proven helpful in lowering BP over a short period of time, including oral captopril, clonidine, hydralazine, labetalol, and hydrochlorothiazide (see Table 3).7,9,12,15 Nifedipine is contraindicated because of the abrupt and often unpredictable reduction in BP and associated myocardial ischemia, especially in patients with MI or left ventricular hypertrophy.14,22,23 In cases of hypertensive urgency secondary to cocaine abuse, benzodiazepines would be the drug of choice and ß-blockers should be avoided due to the risk for coronary vasoconstriction.7
For patients with previously treated hypertension, the following options are reasonable: Increase the dose of the current antihypertensive medication; add another agent; reinstitute prior antihypertensive medications in nonadherent patients; or add a diuretic.
In patients with previously untreated hypertension, no clear evidence supports using one particular agent over another. However, initial treatment options that are generally considered safe include an ACE inhibitor, an angiotensin receptor blocker, a calcium channel blocker, or a thiazide diuretic.15 A few examples of medications within these categories include lisinopril (10 mg PO qd), losartan (50 mg PO qd), amlodipine (2.5 mg PO qd), or hydrochlorothiazide (25 mg PO qd).
Close follow-up is essential when an antihypertensive medication is started or reinstituted. Encourage the patient to reestablish care with their primary care provider (if you do not fill that role). You may need to refer the patient to a new provider or, in some cases, have the patient return to the ED for a repeat BP check.
CONCLUSION
The challenges of managing patients with hypertensive urgency are complicated by low follow-up rates with primary physicians, difficulty in obtaining referrals and follow-up for the patient, and hesitancy of providers to start patients on new BP medications. This article clarifies a well-defined algorithm for how to screen and risk-stratify patients who present to the ED or primary care office with hypertensive urgency.
References
1. CDC. High blood pressure fact sheet. www.cdc.gov/dhdsp/data_statistics/fact_sheets/fs_bloodpressure.htm. Accessed September 26, 2017. 2. Decker WW, Godwin SA, Hess EP, et al; American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Asymptomatic Hypertension in the ED. Clinical policy: critical issues in the evaluation and management of adult patients with asymptomatic hypertension in the emergency department. Ann Emerg Med. 2006;47(3):237-249. 3. CDC. High blood pressure facts. www.cdc.gov/bloodpressure/facts.htm. Accessed October 19, 2017. 4. World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks. Geneva, Switzerland: WHO; 2009. www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf. Accessed October 19, 2017. 5. Stewart DL, Feinstein SE, Colgan R. Hypertensive urgencies and emergencies. Prim Care. 2006;33(3):613-623. 6. Wolf SJ, Lo B, Shih RD, et al; American College of Emergency Physicians Clinical Policies Committee. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med. 2013;62(1):59-68. 7. McKinnon M, O’Neill JM. Hypertension in the emergency department: treat now, later, or not at all. Emerg Med Pract. 2010;12(6):1-22. 8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7): 1281-1357. 9. Shayne PH, Pitts SR. Severely increased blood pressure in the emergency department. Ann Emerg Med. 2003;41(4): 513-529. 10. Aggarwal M, Khan IA. Hypertensive crisis: hypertensive emergencies and urgencies. Cardiol Clin. 2006;24(1):135-146. 11. Houston MC. The comparative effects of clonidine hydrochloride and nifedipine in the treatment of hypertensive crises. Am Heart J. 1998;115(1 pt 1):152-159. 12. Kitiyakara C, Guaman NJ. Malignant hypertension and hypertensive emergencies. J Am Soc Nephrol. 1998;9(1):133-142. 13. Elliott WJ. Hypertensive emergencies. Crit Care Clin. 2001;17(2):435-451. 14. Papadopoulos DP, Mourouzis I, Thomopoulos C, et al. Hypertension crisis. Blood Press. 2010;19(6):328-336. 15. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. 16. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361(9):868-877. 17. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361(9):858-867. 18. Ghali JK, Kadakia S, Cooper RS, Liao YL. Impact of left ventricular hypertrophy on ventricular arrhythmias in the absence of coronary artery disease. J Am Coll Cardiol. 1991;17(6):1277-1282. 19. Bang CN, Soliman EZ, Simpson LM, et al. Electrocardiographic left ventricular hypertrophy predicts cardiovascular morbidity and mortality in hypertensive patients: the ALLHAT study. Am J Hypertens. 2017;30(9):914-922. 20. Hsieh BP, Pham MX, Froelicher VF. Prognostic value of electrocardiographic criteria for left ventricular hypertrophy. Am Heart J. 2005;150(1):161-167. 21. Kinsella K, Baraff LJ. Initiation of therapy for asymptomatic hypertension in the emergency department. Ann Emerg Med. 2009;54(6):791-792. 22. O’Mailia JJ, Sander GE, Giles TD. Nifedipine-associated myocardial ischemia or infarction in the treatment of hypertensive urgencies. Ann Intern Med. 1987;107(2):185-186. 23. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331.
David Indarawis is Assistant Professor and Director of Clinical Education in the School of Physician Assistant Studies at the University of Florida College of Medicine, Gainesville.
The author has no financial relationships to disclose.
David Indarawis is Assistant Professor and Director of Clinical Education in the School of Physician Assistant Studies at the University of Florida College of Medicine, Gainesville.
The author has no financial relationships to disclose.
Author and Disclosure Information
David Indarawis is Assistant Professor and Director of Clinical Education in the School of Physician Assistant Studies at the University of Florida College of Medicine, Gainesville.
The author has no financial relationships to disclose.
Approximately one in three US adults, or about 75 million people, have high blood pressure (BP), which has been defined as a BP of 140/90 mm Hg or higher.1 Unfortunately, only about half (54%) of those affected have their condition under optimal control.1 From an epidemiologic standpoint, hypertension has the distinction of being the most common chronic condition in the US, affecting about 54% of persons ages 55 to 64 and about 73% of those 75 and older.2,3 It is the number one reason patients schedule office visits with physicians; it accounts for the most prescriptions; and it is a major risk factor for heart disease and stroke, as well as a significant contributor to mortality throughout the world.4
HYPERTENSIVE URGENCY VS EMERGENCY
Hypertensive urgencies and emergencies account for approximately 27% of all medical emergencies and 2% to 3% of all annual visits to the emergency department (ED).5 Hypertensive urgency, or severe asymptomatic hypertension, is a common complaint in urgent care clinics and primary care offices as well. It is often defined as a systolic BP (SBP) of ≥ 160 mm Hg and/or a diastolic BP (DBP) ≥ 100 mm Hg with no associated end-organ damage.5-7 Patients may experience hypertensive urgency if they have been noncompliant with their antihypertensive drug regimen; present with pain; have white-coat hypertension or anxiety; or use recreational drugs (eg, sympathomimetics).5,8-10
Alternatively, hypertensive emergency, also known as hypertensive crisis, is generally defined as elevated BP > 180/120 mm Hg. Equally important, it is associated with signs, symptoms, or laboratory values indicative of target end-organ damage, such as cerebrovascular accident, myocardial infarction (MI), aortic dissection, acute left ventricular failure, acute pulmonary edema, acute renal failure, acute mental status changes (hypertensive encephalopathy), and eclampsia.5,7,8,11,12
Determining appropriate management for patients with hypertensive urgency is controversial among clinicians. Practice patterns range from full screening and “rule-outs”—with prompt initiation of antihypertensive agents, regardless of whether the patient is symptomatic—to sending the patient home with minimal screening, laboratory testing, or treatment.
This article offers a guided approach to managing patients with hypertensive urgency in a logical fashion, based on risk stratification, thereby avoiding both extremes (extensive unnecessary workup or discharge without workup resulting in adverse outcomes). It is vital to differentiate between patients with hypertensive emergency, in which BP should be lowered in minutes, and patients with hypertensive urgency, in which BP can be lowered more slowly.12
PATHOPHYSIOLOGY
Normally, when BP increases, blood vessel diameter changes in response; this autoregulation serves to limit damage. However, when BP increases abruptly, the body’s ability to hemodynamically calibrate to such a rapid change is impeded, thus allowing for potential end-organ damage.5,12 The increased vascular resistance observed in many patients with hypertension appears to be an autoregulatory process that helps to maintain a normal or viable level of tissue blood flow and organ perfusion despite the increased BP, rather than a primary cause of the hypertension.13
The exact physiology of hypertensive urgencies is not clearly understood, because of the multifactorial nature of the process. One leading theory is that circulating humoral vasoconstrictors cause an abrupt increase in systemic vascular resistance, which in turn causes mechanical shear stress to the endothelial wall. This endothelial damage promotes more vasoconstriction, platelet aggregation, and activation of the renin-angiotensin-aldosterone system, which thereby increases release of angiotensin II and various cytokines.14
HISTORY AND PHYSICAL
A detailed medical history is of utmost importance in distinguishing patients who present with asymptomatic hypertensive urgency from those experiencing a hypertensive emergency. In addition, obtain a full medication list, including any nutritional supplements or illicit drugs the patient may be taking. Question the patient regarding medication adherence; some may not be taking antihypertensive agents as prescribed or may have altered the dosing frequency in an effort to extend the duration of their prescription.5,8 Table 1 lists pertinent questions to ask at presentation; the answers will dictate who needs further workup and possible admission as well as who will require screening for end-organ damage.7
The physical exam should focus primarily on a thorough cardiopulmonary and neurologic examination, as well as funduscopic examination, if needed. A complete set of vital signs should be recorded upon the patient’s arrival to the ED or clinic and should be repeated on the opposite arm for verification. Beginning with the eyes, conduct a thorough funduscopic examination to evaluate for papilledema or hemorrhages.5 During the cardiopulmonary exam, attention should be focused on signs of congestive heart failure and/or pulmonary edema, such as increased jugular vein distension, an S3 gallop, peripheral edema, and pulmonary rales. The neurologic exam is essential in evaluating for cerebrovascular accident, transient ischemic attack, or intracranial hemorrhage. A full cranial nerve examination is necessary, in addition to motor and sensory testing, at minimum.5,9
RISK STRATIFICATION
According to the 2013 Task Force of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC), several risk factors contribute to overall cardiovascular risk in asymptomatic patients presenting with severe hypertension (see Table 2).8 This report has been monumental in linking grades of hypertension directly to cardiovascular risk factors, but it differs from that recently published by the Eighth Joint National Committee (JNC 8), which offers evidence-based guidelines for the management of high BP in the general population of adults (with some modifications for individuals with diabetes or chronic kidney disease or of black ethnicity).15
According to the ESH/ESC study, patients with one or two risk factors who have grade 1 hypertension (SBP 140-159 mm Hg) are at moderate risk for cardiovascular disease (CVD) and patients with grade 2 (SBP 160-179 mm Hg) or grade 3 (SBP ≥ 180 mm Hg) hypertension are at moderate-to-high risk and high risk, respectively.8 Patients with three or more risk factors, or who already have end-organ damage, diabetes, or chronic kidney disease, enter the high-risk category for CVD even at grade 1 hypertension.8
These cardiovascular risk factors can and should be used as guidelines for deciding who needs further screening and who may have benign causes of severe hypertension (eg, white-coat hypertension, anxiety) that can be managed safely in an outpatient setting. In the author’s opinion, patients with known cardiovascular risk factors, those with signs or symptoms of end-organ damage, and those with test results suggestive of end-organ damage should have a more immediate treatment strategy initiated.
Numerous observational studies have shown a direct relationship between systemic hypertension and CVD risk in men and women of various ages, races, and ethnicities, regardless of other risk factors for CVD.12 In patients with diabetes, uncontrolled hypertension is a strong predictor of cardiovascular morbidity and mortality and of progressive nephropathy leading to chronic kidney disease.8
SCREENING
Results from the following tests may provide useful clues in the workup of a patient with hypertensive urgency.
Basic metabolic panel.Many EDs and primary care offices offer point-of-care testing that can typically give a rapid (< 10 min) result of a basic metabolic panel. This useful, quick screening tool can identify renal failure due to chronic untreated hypertension, acute renal failure, or other disease states that cause electrolyte abnormalities such as hyperaldosteronism (hypertension with hypokalemia) or Cushing syndrome (hypertension with hypernatremia and hyperkalemia).7
Cardiac enzymes. Measurement of cardiac troponins (T or I) may provide confirmatory evidence of myocardial necrosis within two to three hours of suspected acute MI.16,17 These tests are now available in most EDs and some clinics with point-of-care testing. A variety of current guidelines advocate repeat cardiac enzyme measurements at various time points, depending on results of initial testing and concomitant risk factors. These protocols vary by facility.
ECG. Obtaining an ECG is another quick, easy, and useful way to screen patients presenting with severe hypertensive urgency. Evidence of left ventricular hypertrophy suggests an increased risk for MI, stroke, heart failure, and sudden death.7,18-20 The Cornell criteria of summing the R wave in aVL and the S wave in V3, with a cutoff of 2.8 mV in men and 2.0 mV in women, has been shown to be the best predictor of future cardiovascular mortality.7 While an isolated finding of left ventricular hypertrophy on an ECG—in and of itself—may have limited value for an individual patient, this finding coupled with other risk factors may alter the provider’s assessment.
Chest radiograph. A chest radiograph can be helpful when used in conjunction with physical exam findings that suggest pulmonary edema and cardiomegaly.7 Widened mediastinum and tortuous aorta may also be evident on chest x-ray, necessitating further workup and imaging.
Urinalysis. In a patient presenting with asymptomatic hypertensive urgency, a urine dipstick result that shows new-onset proteinuria, while not definitive for diagnosis of nephrotic syndrome, may certainly prove helpful in the patient’s workup.5,13
Urine drug screen. In patients without a history of hypertension who present with asymptomatic hypertensive urgency, the urine drug screen may ascertain exposure to cocaine, amphetamine, or phencyclidine.
Pregnancy test. A pregnancy test is essential for any female patient of childbearing age presenting to the ED, and a positive result may be concerning for preeclampsia in a hypertensive patient with no prior history of the condition.7
TREATMENT
Knowing who to treat and when is a vast area of debate among emergency and primary care providers. Patients with hypertension who have established risk factors are known to have worse outcomes than those who may be otherwise healthy. Some clinicians believe that patients presenting with hypertensive urgency should be discharged home without screening and/or treatment. However, because uncontrolled severe hypertension can lead to acute complications (eg, MI, cerebrovascular accident), in practice, many providers are unwilling to send the patient home without workup.12 The patient’s condition must be viewed in the context of the entire disease spectrum, including risk factors.
The Figure offers a disposition pathway of recommendations based on risk stratification as well as screening tools for some of the less common causes of hypertensive urgency. Regardless of the results of screening tests or the decision to treat, affected patients require close primary care follow-up. Many of these patients may need further testing and careful management of their BP medication regimen.
How to treat
For patients with severe asymptomatic hypertension, if the history, physical, and screening tests do not show evidence of end-organ damage, BP can be controlled within 24 to 48 hours.5,10,11,21 In adults with hypertensive urgency, the most reasonable goal is to reduce the BP to ≤ 160/100 mm Hg5-7; however, the mean arterial pressure should not be lowered by more than 25% within the first two to three hours.13
Patients at high risk for imminent neurovascular, cardiovascular, renovascular, or pulmonary events should have their BP lowered over a period of hours, not minutes. In fact, there is evidence that rapid lowering of BP in asymptomatic patients may cause adverse outcomes.6 For example, in patients with acute ischemic stroke, increases in cerebral perfusion pressure promote an increase in vascular resistance—but decreasing the cerebral perfusion pressure abruptly will thereby decrease the cerebral blood flow, potentially causing cerebral ischemia or a worsening of the stroke.9,14
Treatment options
A broad spectrum of therapeutic options has proven helpful in lowering BP over a short period of time, including oral captopril, clonidine, hydralazine, labetalol, and hydrochlorothiazide (see Table 3).7,9,12,15 Nifedipine is contraindicated because of the abrupt and often unpredictable reduction in BP and associated myocardial ischemia, especially in patients with MI or left ventricular hypertrophy.14,22,23 In cases of hypertensive urgency secondary to cocaine abuse, benzodiazepines would be the drug of choice and ß-blockers should be avoided due to the risk for coronary vasoconstriction.7
For patients with previously treated hypertension, the following options are reasonable: Increase the dose of the current antihypertensive medication; add another agent; reinstitute prior antihypertensive medications in nonadherent patients; or add a diuretic.
In patients with previously untreated hypertension, no clear evidence supports using one particular agent over another. However, initial treatment options that are generally considered safe include an ACE inhibitor, an angiotensin receptor blocker, a calcium channel blocker, or a thiazide diuretic.15 A few examples of medications within these categories include lisinopril (10 mg PO qd), losartan (50 mg PO qd), amlodipine (2.5 mg PO qd), or hydrochlorothiazide (25 mg PO qd).
Close follow-up is essential when an antihypertensive medication is started or reinstituted. Encourage the patient to reestablish care with their primary care provider (if you do not fill that role). You may need to refer the patient to a new provider or, in some cases, have the patient return to the ED for a repeat BP check.
CONCLUSION
The challenges of managing patients with hypertensive urgency are complicated by low follow-up rates with primary physicians, difficulty in obtaining referrals and follow-up for the patient, and hesitancy of providers to start patients on new BP medications. This article clarifies a well-defined algorithm for how to screen and risk-stratify patients who present to the ED or primary care office with hypertensive urgency.
IN THIS ARTICLE
Patient history; what to ask
Cardiovascular risk factors
Disposition pathway
Oral medications
Approximately one in three US adults, or about 75 million people, have high blood pressure (BP), which has been defined as a BP of 140/90 mm Hg or higher.1 Unfortunately, only about half (54%) of those affected have their condition under optimal control.1 From an epidemiologic standpoint, hypertension has the distinction of being the most common chronic condition in the US, affecting about 54% of persons ages 55 to 64 and about 73% of those 75 and older.2,3 It is the number one reason patients schedule office visits with physicians; it accounts for the most prescriptions; and it is a major risk factor for heart disease and stroke, as well as a significant contributor to mortality throughout the world.4
HYPERTENSIVE URGENCY VS EMERGENCY
Hypertensive urgencies and emergencies account for approximately 27% of all medical emergencies and 2% to 3% of all annual visits to the emergency department (ED).5 Hypertensive urgency, or severe asymptomatic hypertension, is a common complaint in urgent care clinics and primary care offices as well. It is often defined as a systolic BP (SBP) of ≥ 160 mm Hg and/or a diastolic BP (DBP) ≥ 100 mm Hg with no associated end-organ damage.5-7 Patients may experience hypertensive urgency if they have been noncompliant with their antihypertensive drug regimen; present with pain; have white-coat hypertension or anxiety; or use recreational drugs (eg, sympathomimetics).5,8-10
Alternatively, hypertensive emergency, also known as hypertensive crisis, is generally defined as elevated BP > 180/120 mm Hg. Equally important, it is associated with signs, symptoms, or laboratory values indicative of target end-organ damage, such as cerebrovascular accident, myocardial infarction (MI), aortic dissection, acute left ventricular failure, acute pulmonary edema, acute renal failure, acute mental status changes (hypertensive encephalopathy), and eclampsia.5,7,8,11,12
Determining appropriate management for patients with hypertensive urgency is controversial among clinicians. Practice patterns range from full screening and “rule-outs”—with prompt initiation of antihypertensive agents, regardless of whether the patient is symptomatic—to sending the patient home with minimal screening, laboratory testing, or treatment.
This article offers a guided approach to managing patients with hypertensive urgency in a logical fashion, based on risk stratification, thereby avoiding both extremes (extensive unnecessary workup or discharge without workup resulting in adverse outcomes). It is vital to differentiate between patients with hypertensive emergency, in which BP should be lowered in minutes, and patients with hypertensive urgency, in which BP can be lowered more slowly.12
PATHOPHYSIOLOGY
Normally, when BP increases, blood vessel diameter changes in response; this autoregulation serves to limit damage. However, when BP increases abruptly, the body’s ability to hemodynamically calibrate to such a rapid change is impeded, thus allowing for potential end-organ damage.5,12 The increased vascular resistance observed in many patients with hypertension appears to be an autoregulatory process that helps to maintain a normal or viable level of tissue blood flow and organ perfusion despite the increased BP, rather than a primary cause of the hypertension.13
The exact physiology of hypertensive urgencies is not clearly understood, because of the multifactorial nature of the process. One leading theory is that circulating humoral vasoconstrictors cause an abrupt increase in systemic vascular resistance, which in turn causes mechanical shear stress to the endothelial wall. This endothelial damage promotes more vasoconstriction, platelet aggregation, and activation of the renin-angiotensin-aldosterone system, which thereby increases release of angiotensin II and various cytokines.14
HISTORY AND PHYSICAL
A detailed medical history is of utmost importance in distinguishing patients who present with asymptomatic hypertensive urgency from those experiencing a hypertensive emergency. In addition, obtain a full medication list, including any nutritional supplements or illicit drugs the patient may be taking. Question the patient regarding medication adherence; some may not be taking antihypertensive agents as prescribed or may have altered the dosing frequency in an effort to extend the duration of their prescription.5,8 Table 1 lists pertinent questions to ask at presentation; the answers will dictate who needs further workup and possible admission as well as who will require screening for end-organ damage.7
The physical exam should focus primarily on a thorough cardiopulmonary and neurologic examination, as well as funduscopic examination, if needed. A complete set of vital signs should be recorded upon the patient’s arrival to the ED or clinic and should be repeated on the opposite arm for verification. Beginning with the eyes, conduct a thorough funduscopic examination to evaluate for papilledema or hemorrhages.5 During the cardiopulmonary exam, attention should be focused on signs of congestive heart failure and/or pulmonary edema, such as increased jugular vein distension, an S3 gallop, peripheral edema, and pulmonary rales. The neurologic exam is essential in evaluating for cerebrovascular accident, transient ischemic attack, or intracranial hemorrhage. A full cranial nerve examination is necessary, in addition to motor and sensory testing, at minimum.5,9
RISK STRATIFICATION
According to the 2013 Task Force of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC), several risk factors contribute to overall cardiovascular risk in asymptomatic patients presenting with severe hypertension (see Table 2).8 This report has been monumental in linking grades of hypertension directly to cardiovascular risk factors, but it differs from that recently published by the Eighth Joint National Committee (JNC 8), which offers evidence-based guidelines for the management of high BP in the general population of adults (with some modifications for individuals with diabetes or chronic kidney disease or of black ethnicity).15
According to the ESH/ESC study, patients with one or two risk factors who have grade 1 hypertension (SBP 140-159 mm Hg) are at moderate risk for cardiovascular disease (CVD) and patients with grade 2 (SBP 160-179 mm Hg) or grade 3 (SBP ≥ 180 mm Hg) hypertension are at moderate-to-high risk and high risk, respectively.8 Patients with three or more risk factors, or who already have end-organ damage, diabetes, or chronic kidney disease, enter the high-risk category for CVD even at grade 1 hypertension.8
These cardiovascular risk factors can and should be used as guidelines for deciding who needs further screening and who may have benign causes of severe hypertension (eg, white-coat hypertension, anxiety) that can be managed safely in an outpatient setting. In the author’s opinion, patients with known cardiovascular risk factors, those with signs or symptoms of end-organ damage, and those with test results suggestive of end-organ damage should have a more immediate treatment strategy initiated.
Numerous observational studies have shown a direct relationship between systemic hypertension and CVD risk in men and women of various ages, races, and ethnicities, regardless of other risk factors for CVD.12 In patients with diabetes, uncontrolled hypertension is a strong predictor of cardiovascular morbidity and mortality and of progressive nephropathy leading to chronic kidney disease.8
SCREENING
Results from the following tests may provide useful clues in the workup of a patient with hypertensive urgency.
Basic metabolic panel.Many EDs and primary care offices offer point-of-care testing that can typically give a rapid (< 10 min) result of a basic metabolic panel. This useful, quick screening tool can identify renal failure due to chronic untreated hypertension, acute renal failure, or other disease states that cause electrolyte abnormalities such as hyperaldosteronism (hypertension with hypokalemia) or Cushing syndrome (hypertension with hypernatremia and hyperkalemia).7
Cardiac enzymes. Measurement of cardiac troponins (T or I) may provide confirmatory evidence of myocardial necrosis within two to three hours of suspected acute MI.16,17 These tests are now available in most EDs and some clinics with point-of-care testing. A variety of current guidelines advocate repeat cardiac enzyme measurements at various time points, depending on results of initial testing and concomitant risk factors. These protocols vary by facility.
ECG. Obtaining an ECG is another quick, easy, and useful way to screen patients presenting with severe hypertensive urgency. Evidence of left ventricular hypertrophy suggests an increased risk for MI, stroke, heart failure, and sudden death.7,18-20 The Cornell criteria of summing the R wave in aVL and the S wave in V3, with a cutoff of 2.8 mV in men and 2.0 mV in women, has been shown to be the best predictor of future cardiovascular mortality.7 While an isolated finding of left ventricular hypertrophy on an ECG—in and of itself—may have limited value for an individual patient, this finding coupled with other risk factors may alter the provider’s assessment.
Chest radiograph. A chest radiograph can be helpful when used in conjunction with physical exam findings that suggest pulmonary edema and cardiomegaly.7 Widened mediastinum and tortuous aorta may also be evident on chest x-ray, necessitating further workup and imaging.
Urinalysis. In a patient presenting with asymptomatic hypertensive urgency, a urine dipstick result that shows new-onset proteinuria, while not definitive for diagnosis of nephrotic syndrome, may certainly prove helpful in the patient’s workup.5,13
Urine drug screen. In patients without a history of hypertension who present with asymptomatic hypertensive urgency, the urine drug screen may ascertain exposure to cocaine, amphetamine, or phencyclidine.
Pregnancy test. A pregnancy test is essential for any female patient of childbearing age presenting to the ED, and a positive result may be concerning for preeclampsia in a hypertensive patient with no prior history of the condition.7
TREATMENT
Knowing who to treat and when is a vast area of debate among emergency and primary care providers. Patients with hypertension who have established risk factors are known to have worse outcomes than those who may be otherwise healthy. Some clinicians believe that patients presenting with hypertensive urgency should be discharged home without screening and/or treatment. However, because uncontrolled severe hypertension can lead to acute complications (eg, MI, cerebrovascular accident), in practice, many providers are unwilling to send the patient home without workup.12 The patient’s condition must be viewed in the context of the entire disease spectrum, including risk factors.
The Figure offers a disposition pathway of recommendations based on risk stratification as well as screening tools for some of the less common causes of hypertensive urgency. Regardless of the results of screening tests or the decision to treat, affected patients require close primary care follow-up. Many of these patients may need further testing and careful management of their BP medication regimen.
How to treat
For patients with severe asymptomatic hypertension, if the history, physical, and screening tests do not show evidence of end-organ damage, BP can be controlled within 24 to 48 hours.5,10,11,21 In adults with hypertensive urgency, the most reasonable goal is to reduce the BP to ≤ 160/100 mm Hg5-7; however, the mean arterial pressure should not be lowered by more than 25% within the first two to three hours.13
Patients at high risk for imminent neurovascular, cardiovascular, renovascular, or pulmonary events should have their BP lowered over a period of hours, not minutes. In fact, there is evidence that rapid lowering of BP in asymptomatic patients may cause adverse outcomes.6 For example, in patients with acute ischemic stroke, increases in cerebral perfusion pressure promote an increase in vascular resistance—but decreasing the cerebral perfusion pressure abruptly will thereby decrease the cerebral blood flow, potentially causing cerebral ischemia or a worsening of the stroke.9,14
Treatment options
A broad spectrum of therapeutic options has proven helpful in lowering BP over a short period of time, including oral captopril, clonidine, hydralazine, labetalol, and hydrochlorothiazide (see Table 3).7,9,12,15 Nifedipine is contraindicated because of the abrupt and often unpredictable reduction in BP and associated myocardial ischemia, especially in patients with MI or left ventricular hypertrophy.14,22,23 In cases of hypertensive urgency secondary to cocaine abuse, benzodiazepines would be the drug of choice and ß-blockers should be avoided due to the risk for coronary vasoconstriction.7
For patients with previously treated hypertension, the following options are reasonable: Increase the dose of the current antihypertensive medication; add another agent; reinstitute prior antihypertensive medications in nonadherent patients; or add a diuretic.
In patients with previously untreated hypertension, no clear evidence supports using one particular agent over another. However, initial treatment options that are generally considered safe include an ACE inhibitor, an angiotensin receptor blocker, a calcium channel blocker, or a thiazide diuretic.15 A few examples of medications within these categories include lisinopril (10 mg PO qd), losartan (50 mg PO qd), amlodipine (2.5 mg PO qd), or hydrochlorothiazide (25 mg PO qd).
Close follow-up is essential when an antihypertensive medication is started or reinstituted. Encourage the patient to reestablish care with their primary care provider (if you do not fill that role). You may need to refer the patient to a new provider or, in some cases, have the patient return to the ED for a repeat BP check.
CONCLUSION
The challenges of managing patients with hypertensive urgency are complicated by low follow-up rates with primary physicians, difficulty in obtaining referrals and follow-up for the patient, and hesitancy of providers to start patients on new BP medications. This article clarifies a well-defined algorithm for how to screen and risk-stratify patients who present to the ED or primary care office with hypertensive urgency.
References
1. CDC. High blood pressure fact sheet. www.cdc.gov/dhdsp/data_statistics/fact_sheets/fs_bloodpressure.htm. Accessed September 26, 2017. 2. Decker WW, Godwin SA, Hess EP, et al; American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Asymptomatic Hypertension in the ED. Clinical policy: critical issues in the evaluation and management of adult patients with asymptomatic hypertension in the emergency department. Ann Emerg Med. 2006;47(3):237-249. 3. CDC. High blood pressure facts. www.cdc.gov/bloodpressure/facts.htm. Accessed October 19, 2017. 4. World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks. Geneva, Switzerland: WHO; 2009. www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf. Accessed October 19, 2017. 5. Stewart DL, Feinstein SE, Colgan R. Hypertensive urgencies and emergencies. Prim Care. 2006;33(3):613-623. 6. Wolf SJ, Lo B, Shih RD, et al; American College of Emergency Physicians Clinical Policies Committee. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med. 2013;62(1):59-68. 7. McKinnon M, O’Neill JM. Hypertension in the emergency department: treat now, later, or not at all. Emerg Med Pract. 2010;12(6):1-22. 8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7): 1281-1357. 9. Shayne PH, Pitts SR. Severely increased blood pressure in the emergency department. Ann Emerg Med. 2003;41(4): 513-529. 10. Aggarwal M, Khan IA. Hypertensive crisis: hypertensive emergencies and urgencies. Cardiol Clin. 2006;24(1):135-146. 11. Houston MC. The comparative effects of clonidine hydrochloride and nifedipine in the treatment of hypertensive crises. Am Heart J. 1998;115(1 pt 1):152-159. 12. Kitiyakara C, Guaman NJ. Malignant hypertension and hypertensive emergencies. J Am Soc Nephrol. 1998;9(1):133-142. 13. Elliott WJ. Hypertensive emergencies. Crit Care Clin. 2001;17(2):435-451. 14. Papadopoulos DP, Mourouzis I, Thomopoulos C, et al. Hypertension crisis. Blood Press. 2010;19(6):328-336. 15. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. 16. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361(9):868-877. 17. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361(9):858-867. 18. Ghali JK, Kadakia S, Cooper RS, Liao YL. Impact of left ventricular hypertrophy on ventricular arrhythmias in the absence of coronary artery disease. J Am Coll Cardiol. 1991;17(6):1277-1282. 19. Bang CN, Soliman EZ, Simpson LM, et al. Electrocardiographic left ventricular hypertrophy predicts cardiovascular morbidity and mortality in hypertensive patients: the ALLHAT study. Am J Hypertens. 2017;30(9):914-922. 20. Hsieh BP, Pham MX, Froelicher VF. Prognostic value of electrocardiographic criteria for left ventricular hypertrophy. Am Heart J. 2005;150(1):161-167. 21. Kinsella K, Baraff LJ. Initiation of therapy for asymptomatic hypertension in the emergency department. Ann Emerg Med. 2009;54(6):791-792. 22. O’Mailia JJ, Sander GE, Giles TD. Nifedipine-associated myocardial ischemia or infarction in the treatment of hypertensive urgencies. Ann Intern Med. 1987;107(2):185-186. 23. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331.
References
1. CDC. High blood pressure fact sheet. www.cdc.gov/dhdsp/data_statistics/fact_sheets/fs_bloodpressure.htm. Accessed September 26, 2017. 2. Decker WW, Godwin SA, Hess EP, et al; American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Asymptomatic Hypertension in the ED. Clinical policy: critical issues in the evaluation and management of adult patients with asymptomatic hypertension in the emergency department. Ann Emerg Med. 2006;47(3):237-249. 3. CDC. High blood pressure facts. www.cdc.gov/bloodpressure/facts.htm. Accessed October 19, 2017. 4. World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks. Geneva, Switzerland: WHO; 2009. www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf. Accessed October 19, 2017. 5. Stewart DL, Feinstein SE, Colgan R. Hypertensive urgencies and emergencies. Prim Care. 2006;33(3):613-623. 6. Wolf SJ, Lo B, Shih RD, et al; American College of Emergency Physicians Clinical Policies Committee. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med. 2013;62(1):59-68. 7. McKinnon M, O’Neill JM. Hypertension in the emergency department: treat now, later, or not at all. Emerg Med Pract. 2010;12(6):1-22. 8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7): 1281-1357. 9. Shayne PH, Pitts SR. Severely increased blood pressure in the emergency department. Ann Emerg Med. 2003;41(4): 513-529. 10. Aggarwal M, Khan IA. Hypertensive crisis: hypertensive emergencies and urgencies. Cardiol Clin. 2006;24(1):135-146. 11. Houston MC. The comparative effects of clonidine hydrochloride and nifedipine in the treatment of hypertensive crises. Am Heart J. 1998;115(1 pt 1):152-159. 12. Kitiyakara C, Guaman NJ. Malignant hypertension and hypertensive emergencies. J Am Soc Nephrol. 1998;9(1):133-142. 13. Elliott WJ. Hypertensive emergencies. Crit Care Clin. 2001;17(2):435-451. 14. Papadopoulos DP, Mourouzis I, Thomopoulos C, et al. Hypertension crisis. Blood Press. 2010;19(6):328-336. 15. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. 16. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361(9):868-877. 17. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361(9):858-867. 18. Ghali JK, Kadakia S, Cooper RS, Liao YL. Impact of left ventricular hypertrophy on ventricular arrhythmias in the absence of coronary artery disease. J Am Coll Cardiol. 1991;17(6):1277-1282. 19. Bang CN, Soliman EZ, Simpson LM, et al. Electrocardiographic left ventricular hypertrophy predicts cardiovascular morbidity and mortality in hypertensive patients: the ALLHAT study. Am J Hypertens. 2017;30(9):914-922. 20. Hsieh BP, Pham MX, Froelicher VF. Prognostic value of electrocardiographic criteria for left ventricular hypertrophy. Am Heart J. 2005;150(1):161-167. 21. Kinsella K, Baraff LJ. Initiation of therapy for asymptomatic hypertension in the emergency department. Ann Emerg Med. 2009;54(6):791-792. 22. O’Mailia JJ, Sander GE, Giles TD. Nifedipine-associated myocardial ischemia or infarction in the treatment of hypertensive urgencies. Ann Intern Med. 1987;107(2):185-186. 23. Grossman E, Messerli FH, Grodzicki T, Kowey P. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-1331.
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
Dr. Bennett P. deBoisblanc
The SMART and SALT-ED trials were awesome and beautifully planned. The researchers used a pragmatic design that is the wave of the future. The incremental benefit from balanced crystalloid fluids was small, about 1%, but it’s a cheap solution. If you administer 7 L of fluid to a patient the incremental cost compared with 0.9% saline is about $45. Based on the number needed to treat that the studies found, this means it would cost less than $5,000 extra to prevent one major adverse kidney event. Nothing else in the ICU or ED compares with that. It’s a phenomenal impact from a low-tech intervention.
Bennett P. deBoisblanc, MD, is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
Dr. Bennett P. deBoisblanc
The SMART and SALT-ED trials were awesome and beautifully planned. The researchers used a pragmatic design that is the wave of the future. The incremental benefit from balanced crystalloid fluids was small, about 1%, but it’s a cheap solution. If you administer 7 L of fluid to a patient the incremental cost compared with 0.9% saline is about $45. Based on the number needed to treat that the studies found, this means it would cost less than $5,000 extra to prevent one major adverse kidney event. Nothing else in the ICU or ED compares with that. It’s a phenomenal impact from a low-tech intervention.
Bennett P. deBoisblanc, MD, is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
Body
Mitchel L. Zoler/Frontline Medical News
Dr. Bennett P. deBoisblanc
The SMART and SALT-ED trials were awesome and beautifully planned. The researchers used a pragmatic design that is the wave of the future. The incremental benefit from balanced crystalloid fluids was small, about 1%, but it’s a cheap solution. If you administer 7 L of fluid to a patient the incremental cost compared with 0.9% saline is about $45. Based on the number needed to treat that the studies found, this means it would cost less than $5,000 extra to prevent one major adverse kidney event. Nothing else in the ICU or ED compares with that. It’s a phenomenal impact from a low-tech intervention.
Bennett P. deBoisblanc, MD, is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
Title
Fluid switch has big impact for small cost
Fluid switch has big impact for small cost
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
Key clinical point: IV fluids with balanced crystalloids outperformed 0.9% saline for preventing death, need for renal replacement therapy, and persistent renal dysfunction in a pair of randomized trials with more than 29,000 patients.
Major finding: Balanced crystalloids reduced combined adverse renal events by 1.1% in ICU patients and 0.9% in ED patients.
Data source: The SMART and SALT-ED trials, both single-center studies with a total of 29,149 patients.
Disclosures: The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
Randomized trials sit at the pinnacle of the clinical research pyramid. Yet for decades we have recognized that a specific therapy given to an individual patient in the real world may not have the result observed in a clinical trial. Trial medicine differs from real-world medicine in many ways, including rigorous attention to monitoring for compliance and safety. In addition, historically, volunteers have differed from real-world patients in several obvious ways, including demographics. For years, many cardiovascular trials in the United States were performed in populations of limited diversity, lacking appropriate numbers of women, Asians, and African Americans.
Clinical experience and observational studies made us aware that African American patients responded differently to some treatments than the white male patients in the clinical trials. This awareness led to some interesting biologic hypotheses and, over the past 13 years, has led to trials focused on the treatment of heart failure and hypertension in African Americans. But a full biologic understanding of the apparent racial differences in clinical response to specific therapies has for the most part remained elusive.
Contributing to this understanding gap was that we historically did not fully appreciate the differences according to race (and likely sex) in the clinical progression of diseases such as hypertension, heart failure, and, as discussed in this issue of the Journal by Dr. Joseph V. Nally, Jr., chronic kidney disease. African Americans with congestive heart failure seem to fare worse than their white counterparts with the same disease. Given the strong link between heart failure and chronic kidney disease and the crosstalk between the heart and kidneys, it is no surprise that African Americans with chronic kidney disease progress to end-stage renal disease at a higher rate than whites. Yet, as Dr. Nally points out, once on dialysis, African Americans live longer—an intriguing observation that came from analysis of large databases devoted to the study of patients with chronic kidney disease.
As a patient’s self-defined racial identity may not be biologically accurate, using molecular genetic techniques to delve more deeply into the characteristics of patients in these chronic kidney disease registries is starting to yield fascinating results—and even more questions. Links between APOL1 gene polymorphisms and the occurrence of renal disease and the survival of transplanted kidneys is assuredly just the start of a journey of genomic discovery and understanding.
Readers will note the short editor’s note at the start of Dr. Nally’s article, indicating that it was based on a Medicine Grand Rounds lecture at Cleveland Clinic, the 14th annual Lawrence “Chris” Crain Memorial Lecture. In 1997, Chris became the first African American chief resident in internal medicine at Cleveland Clinic, and I had the pleasure of interacting with him while he was in that role. Chris was a natural leader. He was soft-spoken, curious, and passionate about delivering and understanding the basics of high-quality clinical care.
After his residency, with Byron Hoogwerf as the internal medicine program director, Chris trained with Joe Nally as his program director in nephrology, and further developed his interest in renal and cardiovascular disease in African Americans. He moved to Atlanta, where he died far too prematurely in July 2003. That year, in conjunction with Chris’s mother, wife, extended family, and other faculty, Drs. Hoogwerf and Nally established the Lawrence “Chris” Crain Memorial Lectureship, devoted to Chris’s passion of furthering our understanding and our ability to deliver optimal care to African American patients with cardiovascular and renal disease.
chronic kidney disease, CKD, congestive heart failure, CHF, African American, black, disparities, racial, Lawrence Chris Crain, Joseph Nally, Brian Mandell,
Randomized trials sit at the pinnacle of the clinical research pyramid. Yet for decades we have recognized that a specific therapy given to an individual patient in the real world may not have the result observed in a clinical trial. Trial medicine differs from real-world medicine in many ways, including rigorous attention to monitoring for compliance and safety. In addition, historically, volunteers have differed from real-world patients in several obvious ways, including demographics. For years, many cardiovascular trials in the United States were performed in populations of limited diversity, lacking appropriate numbers of women, Asians, and African Americans.
Clinical experience and observational studies made us aware that African American patients responded differently to some treatments than the white male patients in the clinical trials. This awareness led to some interesting biologic hypotheses and, over the past 13 years, has led to trials focused on the treatment of heart failure and hypertension in African Americans. But a full biologic understanding of the apparent racial differences in clinical response to specific therapies has for the most part remained elusive.
Contributing to this understanding gap was that we historically did not fully appreciate the differences according to race (and likely sex) in the clinical progression of diseases such as hypertension, heart failure, and, as discussed in this issue of the Journal by Dr. Joseph V. Nally, Jr., chronic kidney disease. African Americans with congestive heart failure seem to fare worse than their white counterparts with the same disease. Given the strong link between heart failure and chronic kidney disease and the crosstalk between the heart and kidneys, it is no surprise that African Americans with chronic kidney disease progress to end-stage renal disease at a higher rate than whites. Yet, as Dr. Nally points out, once on dialysis, African Americans live longer—an intriguing observation that came from analysis of large databases devoted to the study of patients with chronic kidney disease.
As a patient’s self-defined racial identity may not be biologically accurate, using molecular genetic techniques to delve more deeply into the characteristics of patients in these chronic kidney disease registries is starting to yield fascinating results—and even more questions. Links between APOL1 gene polymorphisms and the occurrence of renal disease and the survival of transplanted kidneys is assuredly just the start of a journey of genomic discovery and understanding.
Readers will note the short editor’s note at the start of Dr. Nally’s article, indicating that it was based on a Medicine Grand Rounds lecture at Cleveland Clinic, the 14th annual Lawrence “Chris” Crain Memorial Lecture. In 1997, Chris became the first African American chief resident in internal medicine at Cleveland Clinic, and I had the pleasure of interacting with him while he was in that role. Chris was a natural leader. He was soft-spoken, curious, and passionate about delivering and understanding the basics of high-quality clinical care.
After his residency, with Byron Hoogwerf as the internal medicine program director, Chris trained with Joe Nally as his program director in nephrology, and further developed his interest in renal and cardiovascular disease in African Americans. He moved to Atlanta, where he died far too prematurely in July 2003. That year, in conjunction with Chris’s mother, wife, extended family, and other faculty, Drs. Hoogwerf and Nally established the Lawrence “Chris” Crain Memorial Lectureship, devoted to Chris’s passion of furthering our understanding and our ability to deliver optimal care to African American patients with cardiovascular and renal disease.
I am pleased to share this lecture with you.
Randomized trials sit at the pinnacle of the clinical research pyramid. Yet for decades we have recognized that a specific therapy given to an individual patient in the real world may not have the result observed in a clinical trial. Trial medicine differs from real-world medicine in many ways, including rigorous attention to monitoring for compliance and safety. In addition, historically, volunteers have differed from real-world patients in several obvious ways, including demographics. For years, many cardiovascular trials in the United States were performed in populations of limited diversity, lacking appropriate numbers of women, Asians, and African Americans.
Clinical experience and observational studies made us aware that African American patients responded differently to some treatments than the white male patients in the clinical trials. This awareness led to some interesting biologic hypotheses and, over the past 13 years, has led to trials focused on the treatment of heart failure and hypertension in African Americans. But a full biologic understanding of the apparent racial differences in clinical response to specific therapies has for the most part remained elusive.
Contributing to this understanding gap was that we historically did not fully appreciate the differences according to race (and likely sex) in the clinical progression of diseases such as hypertension, heart failure, and, as discussed in this issue of the Journal by Dr. Joseph V. Nally, Jr., chronic kidney disease. African Americans with congestive heart failure seem to fare worse than their white counterparts with the same disease. Given the strong link between heart failure and chronic kidney disease and the crosstalk between the heart and kidneys, it is no surprise that African Americans with chronic kidney disease progress to end-stage renal disease at a higher rate than whites. Yet, as Dr. Nally points out, once on dialysis, African Americans live longer—an intriguing observation that came from analysis of large databases devoted to the study of patients with chronic kidney disease.
As a patient’s self-defined racial identity may not be biologically accurate, using molecular genetic techniques to delve more deeply into the characteristics of patients in these chronic kidney disease registries is starting to yield fascinating results—and even more questions. Links between APOL1 gene polymorphisms and the occurrence of renal disease and the survival of transplanted kidneys is assuredly just the start of a journey of genomic discovery and understanding.
Readers will note the short editor’s note at the start of Dr. Nally’s article, indicating that it was based on a Medicine Grand Rounds lecture at Cleveland Clinic, the 14th annual Lawrence “Chris” Crain Memorial Lecture. In 1997, Chris became the first African American chief resident in internal medicine at Cleveland Clinic, and I had the pleasure of interacting with him while he was in that role. Chris was a natural leader. He was soft-spoken, curious, and passionate about delivering and understanding the basics of high-quality clinical care.
After his residency, with Byron Hoogwerf as the internal medicine program director, Chris trained with Joe Nally as his program director in nephrology, and further developed his interest in renal and cardiovascular disease in African Americans. He moved to Atlanta, where he died far too prematurely in July 2003. That year, in conjunction with Chris’s mother, wife, extended family, and other faculty, Drs. Hoogwerf and Nally established the Lawrence “Chris” Crain Memorial Lectureship, devoted to Chris’s passion of furthering our understanding and our ability to deliver optimal care to African American patients with cardiovascular and renal disease.
Toward understanding chronic kidney disease in African Americans
Display Headline
Toward understanding chronic kidney disease in African Americans
Legacy Keywords
chronic kidney disease, CKD, congestive heart failure, CHF, African American, black, disparities, racial, Lawrence Chris Crain, Joseph Nally, Brian Mandell,
Legacy Keywords
chronic kidney disease, CKD, congestive heart failure, CHF, African American, black, disparities, racial, Lawrence Chris Crain, Joseph Nally, Brian Mandell,
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.
GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.
More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.
The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.
The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Author and Disclosure Information
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.
GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.
More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.
The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.
The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.
GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.
More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.
The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.
The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
Patients with chronic kidney disease are more likely to die than to progress to end-stage disease, and cardiovascular disease and cancer are the leading causes of death.
As kidney function declines, the chance of dying from cardiovascular disease increases.
African Americans tend to develop kidney disease at a younger age than whites and are much more likely to progress to dialysis.
About 15% of African Americans are homozygous for a variant of the APOL1 gene. They are more likely to develop kidney disease and to have worse outcomes.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo






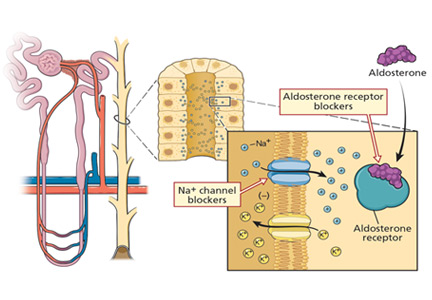


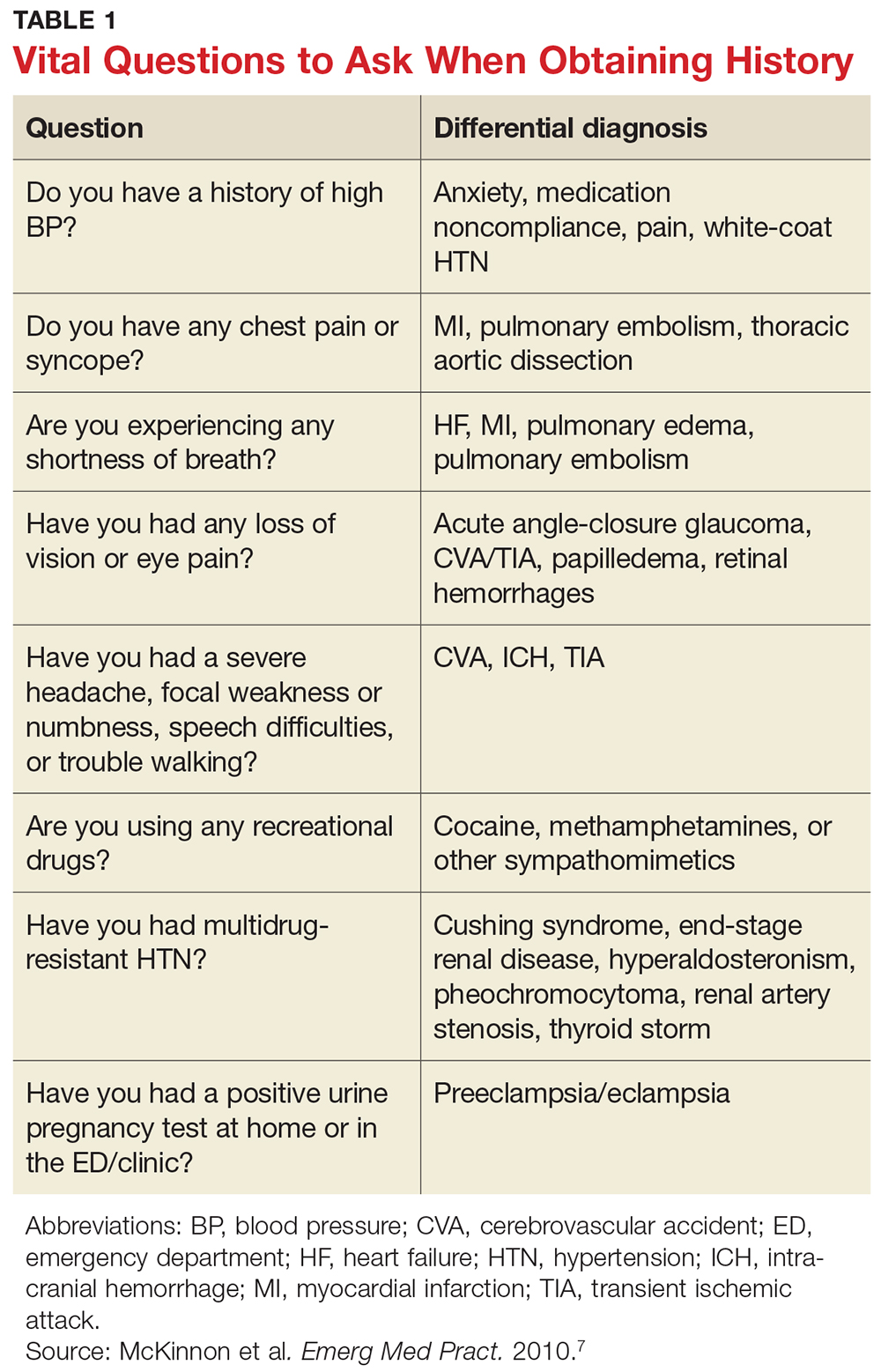
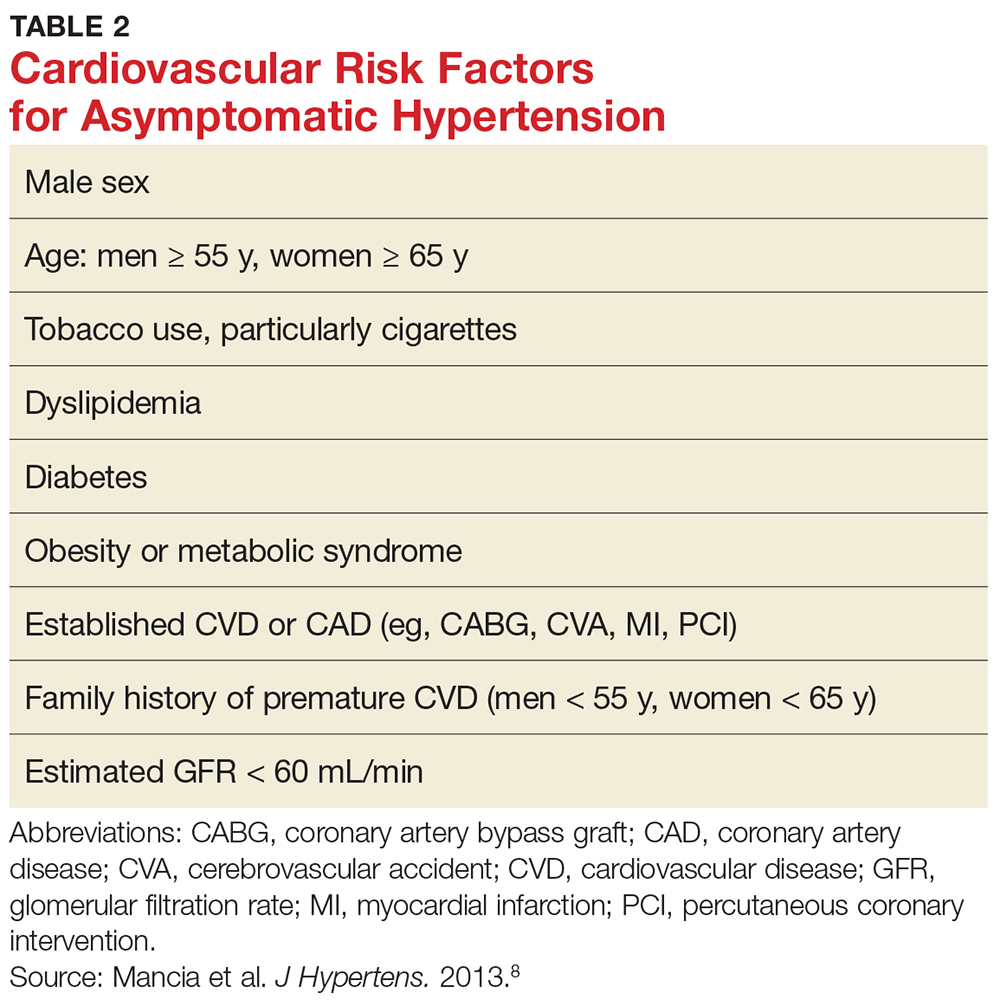
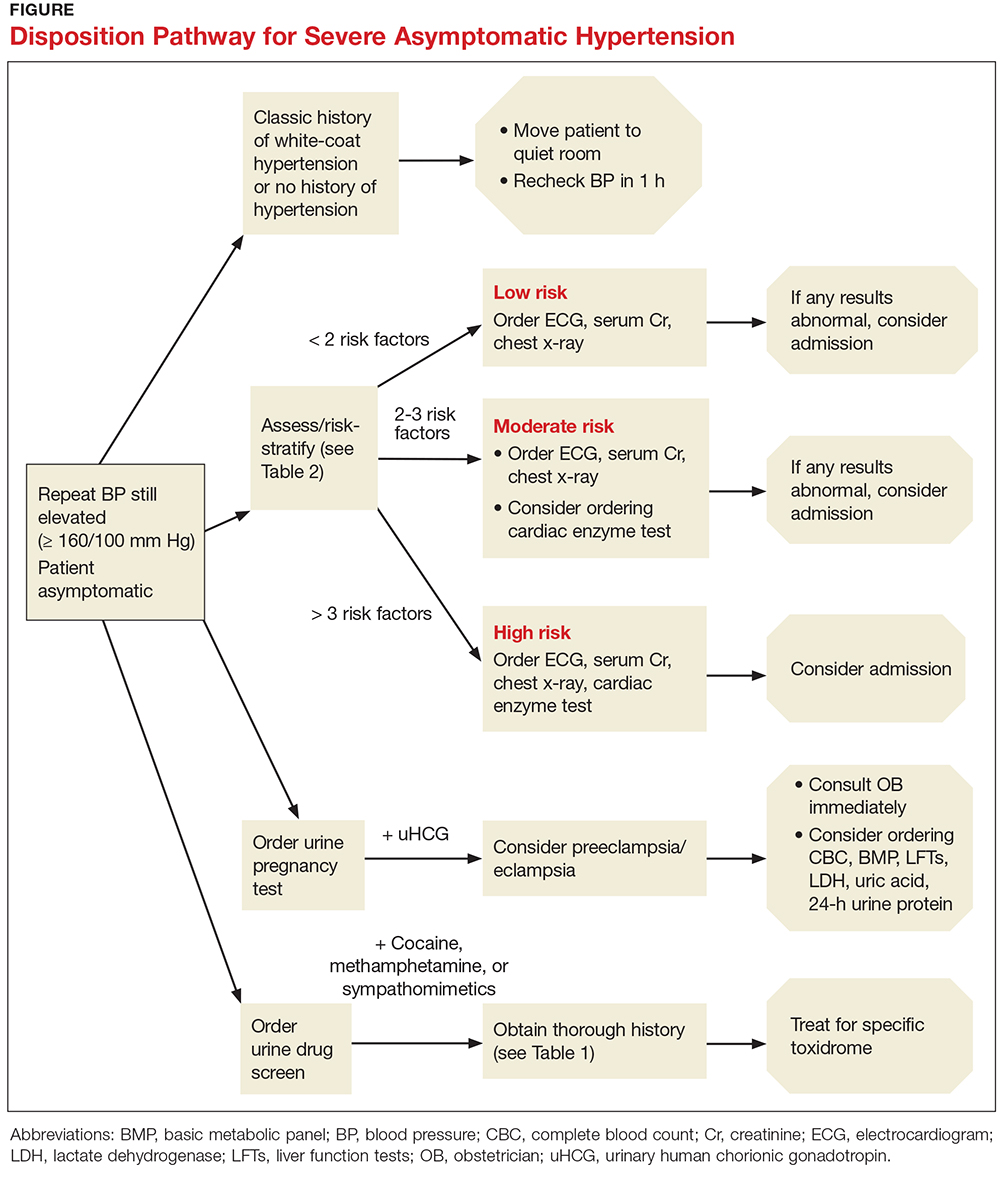
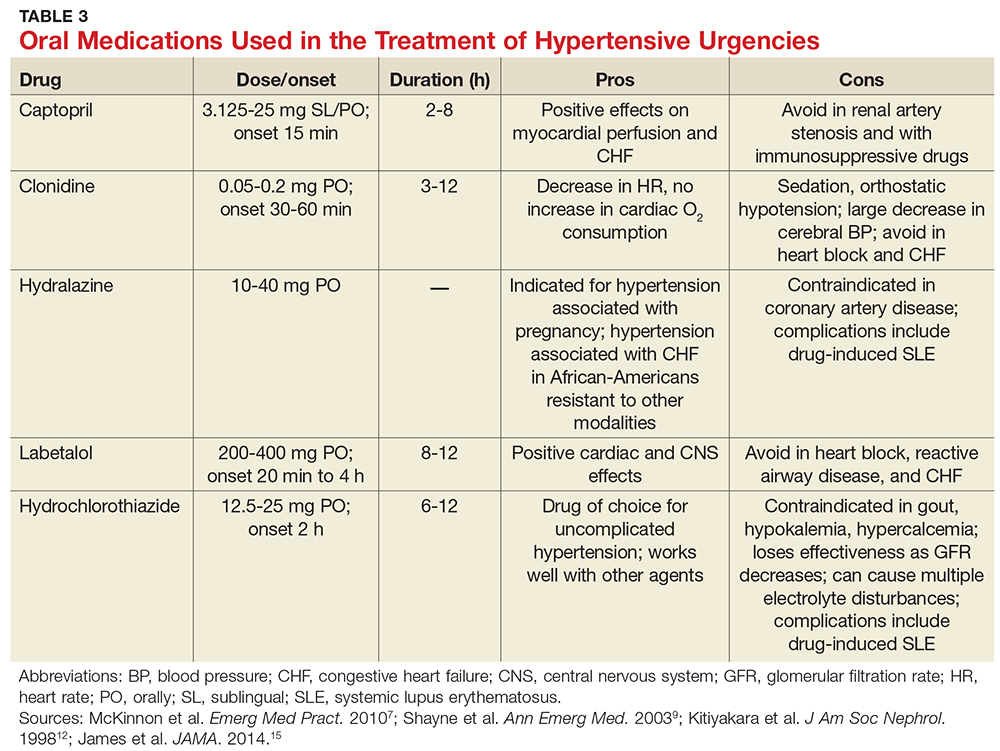








 Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.