User login
Data-based Recommendations for Dialysis
Q) I work in a cardiology practice. Recently, a patient on dialysis mentioned that her nephrology practitioner recommended either home therapy or nocturnal dialysis. Why would someone recommend these, and what are the differences between home, nocturnal, and regular daytime dialysis?
Patients usually require dialysis when 90% or more of their renal function is lost.5 This can happen acutely or result from a chronic process. Dialysis performs many of the functions of a kidney, such as removing waste and fluid buildup that damaged kidneys cannot. It also helps maintain electrolyte balance.
There are several forms of hemodialysis including home, incenter, and nocturnal; the most frequently used is in-center hemodialysis.5 Patients on in-center hemodialysis visit the center three times a week, and their treatments last from three to five hours; the nationwide average is four hours. These patients have very restricted schedules and must maintain their appointments with limited flexibility. Food, drinks, and nonmedical personnel may not be allowed in the treatment area. Between treatments, patients must follow a diet that restricts fluid, sodium, and potassium intake.
Home dialysis has become a popular alternative, since it may be done in a location and at a time that is convenient for the patient. With more flexibility, many patients are able to continue working and feel like they have a more “normal” life. Types of home dialysis include home hemodialysis (HHD) or peritoneal dialysis (PD). A relative or friend may need to assist the patient during HHD, which is undergone more frequently (between five and seven days per week) and for a shorter duration of time than in-center dialysis. PD is done every day, either at night or multiple times throughout the day. Although no partner is needed for PD, a medical provider is available by phone to address any concerns that may arise during treatment.
Nocturnal hemodialysis is similar to daytime in-center hemodialysis, but it occurs while the patient is asleep. The treatment duration is longer (an average of eight hours per treatment). The slower blood flow allows for gentler dialysis. Patients who undergo nocturnal hemodialysis have higher survival and lower hospitalization rates, with better phosphorus control and blood pressure.6 This is attributed to the slower removal of excess fluid and more effective clearance of toxins.
So, why is your patient being encouraged to consider home or nocturnal dialysis? Studies have shown that for the cardiac patient, slower, gentler dialysis is preferable.7 The clinician who recommended it has the patient’s best interest in mind. —TAH
Tricia A. Howard, MHS, PA-C, DFAAPA
PA Program, South University, Savannah, Georgia
5. Gilbert S, Weiner DE. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier; 2014.
6. Lacson E, Wang W, Lester K, et al. Outcomes associated with in-center nocturnal hemodialysis from a large multicenter program. Clin J Am Soc Nephrol. 2010;5(2):220-226.
7. Lin J, Berns JS. Is hemodialysis bad for the heart? Semin Dial. 2012;25(1):86-87.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Q) I work in a cardiology practice. Recently, a patient on dialysis mentioned that her nephrology practitioner recommended either home therapy or nocturnal dialysis. Why would someone recommend these, and what are the differences between home, nocturnal, and regular daytime dialysis?
Patients usually require dialysis when 90% or more of their renal function is lost.5 This can happen acutely or result from a chronic process. Dialysis performs many of the functions of a kidney, such as removing waste and fluid buildup that damaged kidneys cannot. It also helps maintain electrolyte balance.
There are several forms of hemodialysis including home, incenter, and nocturnal; the most frequently used is in-center hemodialysis.5 Patients on in-center hemodialysis visit the center three times a week, and their treatments last from three to five hours; the nationwide average is four hours. These patients have very restricted schedules and must maintain their appointments with limited flexibility. Food, drinks, and nonmedical personnel may not be allowed in the treatment area. Between treatments, patients must follow a diet that restricts fluid, sodium, and potassium intake.
Home dialysis has become a popular alternative, since it may be done in a location and at a time that is convenient for the patient. With more flexibility, many patients are able to continue working and feel like they have a more “normal” life. Types of home dialysis include home hemodialysis (HHD) or peritoneal dialysis (PD). A relative or friend may need to assist the patient during HHD, which is undergone more frequently (between five and seven days per week) and for a shorter duration of time than in-center dialysis. PD is done every day, either at night or multiple times throughout the day. Although no partner is needed for PD, a medical provider is available by phone to address any concerns that may arise during treatment.
Nocturnal hemodialysis is similar to daytime in-center hemodialysis, but it occurs while the patient is asleep. The treatment duration is longer (an average of eight hours per treatment). The slower blood flow allows for gentler dialysis. Patients who undergo nocturnal hemodialysis have higher survival and lower hospitalization rates, with better phosphorus control and blood pressure.6 This is attributed to the slower removal of excess fluid and more effective clearance of toxins.
So, why is your patient being encouraged to consider home or nocturnal dialysis? Studies have shown that for the cardiac patient, slower, gentler dialysis is preferable.7 The clinician who recommended it has the patient’s best interest in mind. —TAH
Tricia A. Howard, MHS, PA-C, DFAAPA
PA Program, South University, Savannah, Georgia
Q) I work in a cardiology practice. Recently, a patient on dialysis mentioned that her nephrology practitioner recommended either home therapy or nocturnal dialysis. Why would someone recommend these, and what are the differences between home, nocturnal, and regular daytime dialysis?
Patients usually require dialysis when 90% or more of their renal function is lost.5 This can happen acutely or result from a chronic process. Dialysis performs many of the functions of a kidney, such as removing waste and fluid buildup that damaged kidneys cannot. It also helps maintain electrolyte balance.
There are several forms of hemodialysis including home, incenter, and nocturnal; the most frequently used is in-center hemodialysis.5 Patients on in-center hemodialysis visit the center three times a week, and their treatments last from three to five hours; the nationwide average is four hours. These patients have very restricted schedules and must maintain their appointments with limited flexibility. Food, drinks, and nonmedical personnel may not be allowed in the treatment area. Between treatments, patients must follow a diet that restricts fluid, sodium, and potassium intake.
Home dialysis has become a popular alternative, since it may be done in a location and at a time that is convenient for the patient. With more flexibility, many patients are able to continue working and feel like they have a more “normal” life. Types of home dialysis include home hemodialysis (HHD) or peritoneal dialysis (PD). A relative or friend may need to assist the patient during HHD, which is undergone more frequently (between five and seven days per week) and for a shorter duration of time than in-center dialysis. PD is done every day, either at night or multiple times throughout the day. Although no partner is needed for PD, a medical provider is available by phone to address any concerns that may arise during treatment.
Nocturnal hemodialysis is similar to daytime in-center hemodialysis, but it occurs while the patient is asleep. The treatment duration is longer (an average of eight hours per treatment). The slower blood flow allows for gentler dialysis. Patients who undergo nocturnal hemodialysis have higher survival and lower hospitalization rates, with better phosphorus control and blood pressure.6 This is attributed to the slower removal of excess fluid and more effective clearance of toxins.
So, why is your patient being encouraged to consider home or nocturnal dialysis? Studies have shown that for the cardiac patient, slower, gentler dialysis is preferable.7 The clinician who recommended it has the patient’s best interest in mind. —TAH
Tricia A. Howard, MHS, PA-C, DFAAPA
PA Program, South University, Savannah, Georgia
5. Gilbert S, Weiner DE. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier; 2014.
6. Lacson E, Wang W, Lester K, et al. Outcomes associated with in-center nocturnal hemodialysis from a large multicenter program. Clin J Am Soc Nephrol. 2010;5(2):220-226.
7. Lin J, Berns JS. Is hemodialysis bad for the heart? Semin Dial. 2012;25(1):86-87.
5. Gilbert S, Weiner DE. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier; 2014.
6. Lacson E, Wang W, Lester K, et al. Outcomes associated with in-center nocturnal hemodialysis from a large multicenter program. Clin J Am Soc Nephrol. 2010;5(2):220-226.
7. Lin J, Berns JS. Is hemodialysis bad for the heart? Semin Dial. 2012;25(1):86-87.
Data-based Recommendations for CKD Screening
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
An abnormal peripheral blood smear and altered mental status
A 72-year-old woman with type 2 diabetes mellitus, hypertension, and atrial fibrillation on anticoagulation was brought to the emergency department by her husband after 1 day of altered mental status with acute onset. Her husband reported that she had been minimally arousable, and the physical examination revealed that she was stuporous and withdrew extremities only from noxious stimuli.
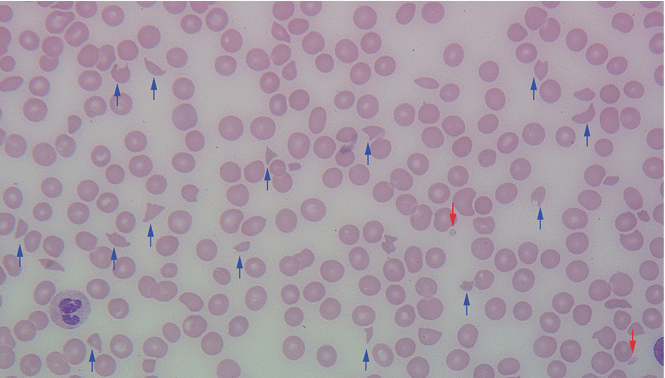
Results of initial laboratory tests revealed a creatinine level of 2.4 mg/dL (reference range 0.7–1.4), hemoglobin 12.1 g/dL (12–16), platelet count 16 × 109/L (150–400), white blood cell count of 7.7 × 109/L (3.7–11), and international normalized ratio of 2.1. A peripheral blood smear is shown in Figure 1.
Computed tomography showed evidence of chronic small vascular ischemia. Magnetic resonance imaging of the brain showed numerous foci of restricted diffusion within the supratentorial and infratentorial areas, suggesting microembolic phenomena.
The peripheral blood smear was compatible with microangiopathic hemolytic anemia, which can occur in thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome, malignant hypertension, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, eclampsia, renal allograft rejection, hematopoietic stem cell transplant, and severe sepsis.1,2
In addition to hemolytic anemia, the patient also had neurologic abnormalities, renal involvement, and thrombocytopenia. The hemolytic anemia and thrombocytopenia were sufficient to raise our suspicion of TTP and to consider initiation of plasma exchange. Only 5% of patients with TTP demonstrate the classic pentad of clinical features,1 ie, thrombocytopenia, microangiopathic hemolytic anemia, fluctuating neurologic signs, renal impairment, and fever.
In 1991, when plasma exchange was introduced for TTP, the survival rate of patients increased from 10% to 78%.1,3 Thus, the diagnosis of TTP is an urgent indication for plasma exchange. We normally do plasma exchange daily until the platelet levels improve.
Our patient received methylprednisone 125 mg intravenously every 12 hours and plasma exchange daily. After three cycles of plasma exchange, she regained normal consciousness, and her platelet count had increased to 20.5 × 109/L on the day of discharge from our hospital.
TTP is a life-threatening hematologic disorder. Evidence of microangiopathic hemolytic anemia on a peripheral blood smear is vital to the suspicion of TTP. The diagnosis should be confirmed by ADAMTS13 testing, which should show decreased activity (< 10%) or increased inhibition, or both. Rapid management with plasma exchange and steroids can lead to a satisfactory outcome.
Acknowledgment: We are particularly grateful to Dr. Vivian Arguello (Director of Flow Cytometry, Department of Pathology, Einstein Medical Center, Philadelphia) for her kind support with the blood smear image.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116:4060–4069.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991; 325:393–397.
A 72-year-old woman with type 2 diabetes mellitus, hypertension, and atrial fibrillation on anticoagulation was brought to the emergency department by her husband after 1 day of altered mental status with acute onset. Her husband reported that she had been minimally arousable, and the physical examination revealed that she was stuporous and withdrew extremities only from noxious stimuli.
Results of initial laboratory tests revealed a creatinine level of 2.4 mg/dL (reference range 0.7–1.4), hemoglobin 12.1 g/dL (12–16), platelet count 16 × 109/L (150–400), white blood cell count of 7.7 × 109/L (3.7–11), and international normalized ratio of 2.1. A peripheral blood smear is shown in Figure 1.
Computed tomography showed evidence of chronic small vascular ischemia. Magnetic resonance imaging of the brain showed numerous foci of restricted diffusion within the supratentorial and infratentorial areas, suggesting microembolic phenomena.
The peripheral blood smear was compatible with microangiopathic hemolytic anemia, which can occur in thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome, malignant hypertension, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, eclampsia, renal allograft rejection, hematopoietic stem cell transplant, and severe sepsis.1,2
In addition to hemolytic anemia, the patient also had neurologic abnormalities, renal involvement, and thrombocytopenia. The hemolytic anemia and thrombocytopenia were sufficient to raise our suspicion of TTP and to consider initiation of plasma exchange. Only 5% of patients with TTP demonstrate the classic pentad of clinical features,1 ie, thrombocytopenia, microangiopathic hemolytic anemia, fluctuating neurologic signs, renal impairment, and fever.
In 1991, when plasma exchange was introduced for TTP, the survival rate of patients increased from 10% to 78%.1,3 Thus, the diagnosis of TTP is an urgent indication for plasma exchange. We normally do plasma exchange daily until the platelet levels improve.
Our patient received methylprednisone 125 mg intravenously every 12 hours and plasma exchange daily. After three cycles of plasma exchange, she regained normal consciousness, and her platelet count had increased to 20.5 × 109/L on the day of discharge from our hospital.
TTP is a life-threatening hematologic disorder. Evidence of microangiopathic hemolytic anemia on a peripheral blood smear is vital to the suspicion of TTP. The diagnosis should be confirmed by ADAMTS13 testing, which should show decreased activity (< 10%) or increased inhibition, or both. Rapid management with plasma exchange and steroids can lead to a satisfactory outcome.
Acknowledgment: We are particularly grateful to Dr. Vivian Arguello (Director of Flow Cytometry, Department of Pathology, Einstein Medical Center, Philadelphia) for her kind support with the blood smear image.
A 72-year-old woman with type 2 diabetes mellitus, hypertension, and atrial fibrillation on anticoagulation was brought to the emergency department by her husband after 1 day of altered mental status with acute onset. Her husband reported that she had been minimally arousable, and the physical examination revealed that she was stuporous and withdrew extremities only from noxious stimuli.
Results of initial laboratory tests revealed a creatinine level of 2.4 mg/dL (reference range 0.7–1.4), hemoglobin 12.1 g/dL (12–16), platelet count 16 × 109/L (150–400), white blood cell count of 7.7 × 109/L (3.7–11), and international normalized ratio of 2.1. A peripheral blood smear is shown in Figure 1.
Computed tomography showed evidence of chronic small vascular ischemia. Magnetic resonance imaging of the brain showed numerous foci of restricted diffusion within the supratentorial and infratentorial areas, suggesting microembolic phenomena.
The peripheral blood smear was compatible with microangiopathic hemolytic anemia, which can occur in thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome, malignant hypertension, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, eclampsia, renal allograft rejection, hematopoietic stem cell transplant, and severe sepsis.1,2
In addition to hemolytic anemia, the patient also had neurologic abnormalities, renal involvement, and thrombocytopenia. The hemolytic anemia and thrombocytopenia were sufficient to raise our suspicion of TTP and to consider initiation of plasma exchange. Only 5% of patients with TTP demonstrate the classic pentad of clinical features,1 ie, thrombocytopenia, microangiopathic hemolytic anemia, fluctuating neurologic signs, renal impairment, and fever.
In 1991, when plasma exchange was introduced for TTP, the survival rate of patients increased from 10% to 78%.1,3 Thus, the diagnosis of TTP is an urgent indication for plasma exchange. We normally do plasma exchange daily until the platelet levels improve.
Our patient received methylprednisone 125 mg intravenously every 12 hours and plasma exchange daily. After three cycles of plasma exchange, she regained normal consciousness, and her platelet count had increased to 20.5 × 109/L on the day of discharge from our hospital.
TTP is a life-threatening hematologic disorder. Evidence of microangiopathic hemolytic anemia on a peripheral blood smear is vital to the suspicion of TTP. The diagnosis should be confirmed by ADAMTS13 testing, which should show decreased activity (< 10%) or increased inhibition, or both. Rapid management with plasma exchange and steroids can lead to a satisfactory outcome.
Acknowledgment: We are particularly grateful to Dr. Vivian Arguello (Director of Flow Cytometry, Department of Pathology, Einstein Medical Center, Philadelphia) for her kind support with the blood smear image.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116:4060–4069.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991; 325:393–397.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116:4060–4069.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991; 325:393–397.
Calcineurin inhibitor proves effective against lupus nephritis
Patients with highly active lupus nephritis who took the investigational oral calcineurin inhibitor voclosporin plus mycophenolate mofetil and tapered corticosteroids were twice as likely to achieve complete remission by 24 weeks, compared against placebo-treated patients who also received standard of care treatment in a phase IIb study trial reported by Aurinia Pharmaceuticals.
The 24-week complete remission primary endpoint of the AURA-LV(Aurinia Urinary Protein Reduction Active–Lupus With Voclosporin) study – defined as a urine protein/creatinine ratio of 0.5 mg/mg or less as well as normal stable renal function (estimated glomerular filtration rate of 60 mL/min per 1.73 m2 or greater or no confirmed decrease from baseline in eGFR of 20% or greater) – occurred in 32.6% of patients who were randomized to take 23.7 mg of voclosporin twice daily, which was significantly higher than the 19.3% rate observed in the placebo-treated group. The rate was 27.3% in a higher-dose group that received 39.5 mg of voclosporin twice daily.
Serious adverse events occurred at higher rates in both voclosporin arms of the trial than in the placebo arm, but Aurinia said in its statement announcing the results that the nature of the events was consistent with highly active lupus nephritis. A total of 13 deaths occurred, including 2 in the high-dose arm, 10 in the low-dose arm, and 1 in the placebo arm, but the company said that the investigator deemed the deaths as unrelated to voclosporin. Eleven of the deaths occurred in Asia.
Both low- and high-dose voclosporin arms attained a partial response by 24 weeks (50% drop in urine protein per creatinine ratio) in a significantly higher percentage of patients than did the placebo arm (69.7% and 65.9%, respectively, vs. 49.4%).
The Lupus Research Alliance welcomed the results of the study but noted that more needs to be known about the risk-benefit profile of the drug, specifically in reference to the 12 deaths reported in those who took voclosporin. “The magnitude of benefit is quite striking and unprecedented in lupus nephritis, but the number of deaths is a concern that must be taken seriously. We are very hopeful that further analysis of the safety data will confirm that voclosporin can provide a safe and effective treatment,” Margaret G. Dowd, co–chief executive officer of the Lupus Research Alliance, said in a statement.
The trial enrolled and randomized 265 patients diagnosed with highly active lupus nephritis (according to clinical signs and renal biopsy features) across centers in more than 20 countries. Besides being randomized to either active treatment arm or placebo, all patients received mycophenolate mofetil (CellCept) and oral corticosteroids that started at 20-25 mg/daily and then tapered down to 5 mg daily by week 8 and 2.5 mg daily by week 16. All patients also had an initial 500-1,000 mg intravenous dose of steroids.
Aurinia said that the study will continue to 48 weeks, and these data will be available in early 2017.
Patients with highly active lupus nephritis who took the investigational oral calcineurin inhibitor voclosporin plus mycophenolate mofetil and tapered corticosteroids were twice as likely to achieve complete remission by 24 weeks, compared against placebo-treated patients who also received standard of care treatment in a phase IIb study trial reported by Aurinia Pharmaceuticals.
The 24-week complete remission primary endpoint of the AURA-LV(Aurinia Urinary Protein Reduction Active–Lupus With Voclosporin) study – defined as a urine protein/creatinine ratio of 0.5 mg/mg or less as well as normal stable renal function (estimated glomerular filtration rate of 60 mL/min per 1.73 m2 or greater or no confirmed decrease from baseline in eGFR of 20% or greater) – occurred in 32.6% of patients who were randomized to take 23.7 mg of voclosporin twice daily, which was significantly higher than the 19.3% rate observed in the placebo-treated group. The rate was 27.3% in a higher-dose group that received 39.5 mg of voclosporin twice daily.
Serious adverse events occurred at higher rates in both voclosporin arms of the trial than in the placebo arm, but Aurinia said in its statement announcing the results that the nature of the events was consistent with highly active lupus nephritis. A total of 13 deaths occurred, including 2 in the high-dose arm, 10 in the low-dose arm, and 1 in the placebo arm, but the company said that the investigator deemed the deaths as unrelated to voclosporin. Eleven of the deaths occurred in Asia.
Both low- and high-dose voclosporin arms attained a partial response by 24 weeks (50% drop in urine protein per creatinine ratio) in a significantly higher percentage of patients than did the placebo arm (69.7% and 65.9%, respectively, vs. 49.4%).
The Lupus Research Alliance welcomed the results of the study but noted that more needs to be known about the risk-benefit profile of the drug, specifically in reference to the 12 deaths reported in those who took voclosporin. “The magnitude of benefit is quite striking and unprecedented in lupus nephritis, but the number of deaths is a concern that must be taken seriously. We are very hopeful that further analysis of the safety data will confirm that voclosporin can provide a safe and effective treatment,” Margaret G. Dowd, co–chief executive officer of the Lupus Research Alliance, said in a statement.
The trial enrolled and randomized 265 patients diagnosed with highly active lupus nephritis (according to clinical signs and renal biopsy features) across centers in more than 20 countries. Besides being randomized to either active treatment arm or placebo, all patients received mycophenolate mofetil (CellCept) and oral corticosteroids that started at 20-25 mg/daily and then tapered down to 5 mg daily by week 8 and 2.5 mg daily by week 16. All patients also had an initial 500-1,000 mg intravenous dose of steroids.
Aurinia said that the study will continue to 48 weeks, and these data will be available in early 2017.
Patients with highly active lupus nephritis who took the investigational oral calcineurin inhibitor voclosporin plus mycophenolate mofetil and tapered corticosteroids were twice as likely to achieve complete remission by 24 weeks, compared against placebo-treated patients who also received standard of care treatment in a phase IIb study trial reported by Aurinia Pharmaceuticals.
The 24-week complete remission primary endpoint of the AURA-LV(Aurinia Urinary Protein Reduction Active–Lupus With Voclosporin) study – defined as a urine protein/creatinine ratio of 0.5 mg/mg or less as well as normal stable renal function (estimated glomerular filtration rate of 60 mL/min per 1.73 m2 or greater or no confirmed decrease from baseline in eGFR of 20% or greater) – occurred in 32.6% of patients who were randomized to take 23.7 mg of voclosporin twice daily, which was significantly higher than the 19.3% rate observed in the placebo-treated group. The rate was 27.3% in a higher-dose group that received 39.5 mg of voclosporin twice daily.
Serious adverse events occurred at higher rates in both voclosporin arms of the trial than in the placebo arm, but Aurinia said in its statement announcing the results that the nature of the events was consistent with highly active lupus nephritis. A total of 13 deaths occurred, including 2 in the high-dose arm, 10 in the low-dose arm, and 1 in the placebo arm, but the company said that the investigator deemed the deaths as unrelated to voclosporin. Eleven of the deaths occurred in Asia.
Both low- and high-dose voclosporin arms attained a partial response by 24 weeks (50% drop in urine protein per creatinine ratio) in a significantly higher percentage of patients than did the placebo arm (69.7% and 65.9%, respectively, vs. 49.4%).
The Lupus Research Alliance welcomed the results of the study but noted that more needs to be known about the risk-benefit profile of the drug, specifically in reference to the 12 deaths reported in those who took voclosporin. “The magnitude of benefit is quite striking and unprecedented in lupus nephritis, but the number of deaths is a concern that must be taken seriously. We are very hopeful that further analysis of the safety data will confirm that voclosporin can provide a safe and effective treatment,” Margaret G. Dowd, co–chief executive officer of the Lupus Research Alliance, said in a statement.
The trial enrolled and randomized 265 patients diagnosed with highly active lupus nephritis (according to clinical signs and renal biopsy features) across centers in more than 20 countries. Besides being randomized to either active treatment arm or placebo, all patients received mycophenolate mofetil (CellCept) and oral corticosteroids that started at 20-25 mg/daily and then tapered down to 5 mg daily by week 8 and 2.5 mg daily by week 16. All patients also had an initial 500-1,000 mg intravenous dose of steroids.
Aurinia said that the study will continue to 48 weeks, and these data will be available in early 2017.
Monitoring renal function during daily oral HIV PrEP
DURBAN, SOUTH AFRICA – The optimal frequency of kidney safety monitoring in patients using oral daily tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection is every 6 months, but less frequent monitoring may be reasonable in most low-risk patients, Renee Heffron, PhD, said at the 21st International AIDS Conference.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The risk of a clinically relevant decline in creatinine clearance during the first 12 months of therapy appears to be largely confined to the subgroup of patients on tenofovir/emtricitabine (Truvada) for pre-exposure prophylaxis (PrEP) who weigh 55 kg or less, have a baseline creatinine clearance rate of 60-90 mL/min, or are at least 45 years old, according to Dr. Heffron of the University of Washington, Seattle.

The question of how frequently to monitor renal function is a key issue as PrEP with tenofovir/emtricitabine is ramped up to scale in sub-Saharan Africa and other parts of the developing world where the majority of new HIV infections occur – and where laboratory resources are often limited. The randomized clinical trials that led to marketing approval of tenofovir/emtricitabine for PrEP in the United States and elsewhere monitored creatinine clearance every 3 months. But the confirmatory demonstration projects used a range of kidney monitoring schedules, she explained.
She presented an analysis of clinically relevant kidney toxicity in 4,404 initially HIV-negative subjects on tenofovir/emtricitabine in the Partners PrEP Study, in which creatinine clearance was measured every 3 months, and in 955 participants in the Partners Demonstration Study, in which monitoring was performed every 6 months. All participants were at high risk for HIV acquisition because they were members of serodiscordant couples.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The cumulative rate in the randomized trial was 0.4%, 0.5%, and 0.7% at 3, 6, and 12 months, and it was 0.2% at both 6 and 12 months in the demonstration project, Dr. Heffron reported.
These renal events were not only rare, they were reassuringly nonprogressive and resolved within a few weeks of PrEP discontinuation, she added.
Her analysis of the combined 5,359 subjects in the two Partners studies identified three independent predictors of a fall in creatinine clearance to below 60 mL/min during the first 12 months of therapy. A baseline age of 45 years or more was associated with an adjusted 2.5-fold increase, compared with younger patients. Subjects with a creatinine clearance of 60-90 mL/min at enrollment were 74 times more likely to experience a significant drop in creatinine clearance than those who started on PrEP with a creatinine clearance rate in excess of 90 mL/min. And patients weighing 55 kg or less had a 2.7-fold greater risk than those weighing more. But fewer than 5% of patients with any of these three predictors actually experienced a drop in creatinine clearance to below 60 mL/min.
The data from the two Partners studies support guidelines from the Centers for Disease Control and Prevention recommending creatinine monitoring every 6 months for people on oral daily PrEP. Still, patients with one of the defined risk factors might logically be candidates for targeted monitoring, Dr. Heffron observed.
The Partners studies were funded by the National Institutes of Health, the Bill and Melinda Gates Foundation, and the U.S. Agency for International Development. Dr. Heffron reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – The optimal frequency of kidney safety monitoring in patients using oral daily tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection is every 6 months, but less frequent monitoring may be reasonable in most low-risk patients, Renee Heffron, PhD, said at the 21st International AIDS Conference.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The risk of a clinically relevant decline in creatinine clearance during the first 12 months of therapy appears to be largely confined to the subgroup of patients on tenofovir/emtricitabine (Truvada) for pre-exposure prophylaxis (PrEP) who weigh 55 kg or less, have a baseline creatinine clearance rate of 60-90 mL/min, or are at least 45 years old, according to Dr. Heffron of the University of Washington, Seattle.
The question of how frequently to monitor renal function is a key issue as PrEP with tenofovir/emtricitabine is ramped up to scale in sub-Saharan Africa and other parts of the developing world where the majority of new HIV infections occur – and where laboratory resources are often limited. The randomized clinical trials that led to marketing approval of tenofovir/emtricitabine for PrEP in the United States and elsewhere monitored creatinine clearance every 3 months. But the confirmatory demonstration projects used a range of kidney monitoring schedules, she explained.
She presented an analysis of clinically relevant kidney toxicity in 4,404 initially HIV-negative subjects on tenofovir/emtricitabine in the Partners PrEP Study, in which creatinine clearance was measured every 3 months, and in 955 participants in the Partners Demonstration Study, in which monitoring was performed every 6 months. All participants were at high risk for HIV acquisition because they were members of serodiscordant couples.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The cumulative rate in the randomized trial was 0.4%, 0.5%, and 0.7% at 3, 6, and 12 months, and it was 0.2% at both 6 and 12 months in the demonstration project, Dr. Heffron reported.
These renal events were not only rare, they were reassuringly nonprogressive and resolved within a few weeks of PrEP discontinuation, she added.
Her analysis of the combined 5,359 subjects in the two Partners studies identified three independent predictors of a fall in creatinine clearance to below 60 mL/min during the first 12 months of therapy. A baseline age of 45 years or more was associated with an adjusted 2.5-fold increase, compared with younger patients. Subjects with a creatinine clearance of 60-90 mL/min at enrollment were 74 times more likely to experience a significant drop in creatinine clearance than those who started on PrEP with a creatinine clearance rate in excess of 90 mL/min. And patients weighing 55 kg or less had a 2.7-fold greater risk than those weighing more. But fewer than 5% of patients with any of these three predictors actually experienced a drop in creatinine clearance to below 60 mL/min.
The data from the two Partners studies support guidelines from the Centers for Disease Control and Prevention recommending creatinine monitoring every 6 months for people on oral daily PrEP. Still, patients with one of the defined risk factors might logically be candidates for targeted monitoring, Dr. Heffron observed.
The Partners studies were funded by the National Institutes of Health, the Bill and Melinda Gates Foundation, and the U.S. Agency for International Development. Dr. Heffron reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – The optimal frequency of kidney safety monitoring in patients using oral daily tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection is every 6 months, but less frequent monitoring may be reasonable in most low-risk patients, Renee Heffron, PhD, said at the 21st International AIDS Conference.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The risk of a clinically relevant decline in creatinine clearance during the first 12 months of therapy appears to be largely confined to the subgroup of patients on tenofovir/emtricitabine (Truvada) for pre-exposure prophylaxis (PrEP) who weigh 55 kg or less, have a baseline creatinine clearance rate of 60-90 mL/min, or are at least 45 years old, according to Dr. Heffron of the University of Washington, Seattle.
The question of how frequently to monitor renal function is a key issue as PrEP with tenofovir/emtricitabine is ramped up to scale in sub-Saharan Africa and other parts of the developing world where the majority of new HIV infections occur – and where laboratory resources are often limited. The randomized clinical trials that led to marketing approval of tenofovir/emtricitabine for PrEP in the United States and elsewhere monitored creatinine clearance every 3 months. But the confirmatory demonstration projects used a range of kidney monitoring schedules, she explained.
She presented an analysis of clinically relevant kidney toxicity in 4,404 initially HIV-negative subjects on tenofovir/emtricitabine in the Partners PrEP Study, in which creatinine clearance was measured every 3 months, and in 955 participants in the Partners Demonstration Study, in which monitoring was performed every 6 months. All participants were at high risk for HIV acquisition because they were members of serodiscordant couples.
The occurrence and pattern of detection of a drop in creatinine clearance to less than 60 mL/min during the first 12 months of therapy didn’t differ significantly regardless of whether monitoring was done at 3- or 6-month intervals. The cumulative rate in the randomized trial was 0.4%, 0.5%, and 0.7% at 3, 6, and 12 months, and it was 0.2% at both 6 and 12 months in the demonstration project, Dr. Heffron reported.
These renal events were not only rare, they were reassuringly nonprogressive and resolved within a few weeks of PrEP discontinuation, she added.
Her analysis of the combined 5,359 subjects in the two Partners studies identified three independent predictors of a fall in creatinine clearance to below 60 mL/min during the first 12 months of therapy. A baseline age of 45 years or more was associated with an adjusted 2.5-fold increase, compared with younger patients. Subjects with a creatinine clearance of 60-90 mL/min at enrollment were 74 times more likely to experience a significant drop in creatinine clearance than those who started on PrEP with a creatinine clearance rate in excess of 90 mL/min. And patients weighing 55 kg or less had a 2.7-fold greater risk than those weighing more. But fewer than 5% of patients with any of these three predictors actually experienced a drop in creatinine clearance to below 60 mL/min.
The data from the two Partners studies support guidelines from the Centers for Disease Control and Prevention recommending creatinine monitoring every 6 months for people on oral daily PrEP. Still, patients with one of the defined risk factors might logically be candidates for targeted monitoring, Dr. Heffron observed.
The Partners studies were funded by the National Institutes of Health, the Bill and Melinda Gates Foundation, and the U.S. Agency for International Development. Dr. Heffron reported having no financial conflicts of interest.
AT AIDS 2016
Key clinical point: Monitoring creatinine clearance every 6 months is optimal in patients taking tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection.
Major finding: Fewer than 1% of patients experienced a decline in creatinine clearance to below 60 mL/min during their first 12 months on oral daily tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection.
Data source: This was a secondary analysis of 5,359 adults whose creatinine clearance was measured every 3 or 6 months while on oral daily tenofovir/emtricitabine for pre-exposure prophylaxis against HIV infection in a randomized trial or open-label demonstration project.
Disclosures: The studies were funded by NIH, the Bill and Melinda Gates Foundation, and the U.S. Agency for International Development. The presenter reported having no financial conflicts of interest.

UTIs not caused by E. coli more likely in certain children
Certain children are more highly predisposed to contracting a urinary tract infection caused by a pathogen other than Escherichia coli, which is typically the most common cause of UTIs, a study showed.
“It may be clinically important to predict which children have UTIs caused by organisms other than E. coli because these organisms differ in their patterns of antimicrobial susceptibility,” wrote the study authors led by Nader Shaikh, MD, of the University of Pittsburgh. “Furthermore, some guidelines have suggested that screening for vesicoureteral reflux (VUR) with a voiding cystourethrogram (VCUG) should, at least in part, be based on whether an organism other than E. coli is recovered,” they wrote.

Dr. Shaikh and his coinvestigators examined the medical records of children in the Randomized Intervention for Children With Vesicoureteral Reflux (RIVUR) trial and the Careful Urinary Tract Infection Evaluation (CUTIE), both of which were prospective multicenter studies. Children included in both studies were 2-71 months of age; RIVUR subjects had VUR grades 1-4 and presented with either a first or second febrile or symptomatic UTI, while CUTIE subjects presented with either their first or second UTI but not VUR (Ped Inf Dis J. 2016. doi:10.1097/INF.0000000000001301).
In total, 769 children from 19 centers were included from both studies, of which 703 (91%) were female and 596 (78%) were white. Nine percent of all the children had UTIs that were not caused by E. coli. Circumcised males had the highest odds ratio associated with non–E. coli UTIs, with an OR of 5.5 (95% CI, 1.18-17.1; P = .003). significantly higher than the 1.6 odds ratio for uncircumcised males (95% CI, 0.6-4.6; P = .35).
Hispanic children also had a higher risk (OR = 2.3; 95% CI, 1.1-4.6; P = .02) than either non-Hispanic children or females, which were reference cohorts. Other groups found to be at higher-than-normal risk for non–E. coli UTIs were children without fever (OR = 2.8; 95% CI, 1.2-6.6; P = .02) and children with VUR grade 3 or 4 (OR = 2.2; 95% CI, 1.2-4.1; P = .01).
While more than 90% of children’s UTIs were caused by E. coli, the most common pathogens causing UTIs, causative organisms in the other 70 children were Proteus species (21 children, 30%), Klebsiella species (16, 23%), Enterococcus species (14, 20%), Enterobacter species (8, 11%), and “other species” (11, 16%).
“Children with UTIs caused by organisms other than E. coli were twice as likely to have high-grade VUR (grade 3 and 4), which is consistent with prior studies,” Dr. Shaikh and his coauthors noted, adding, “the association between Hispanic ethnicity and non-E. coli pathogens is novel and may be due to differences in genes involved with susceptibility to UTIs.”
There were no disclosures or sources of funding provided.
In this study of almost 800 children in the Pittsburgh area, the investigators sought to identify children at risk for urinary tract infections that more likely would have a bacterial organism not susceptible to standard first-line empiric antibiotic treatment.
They found that circumcised males, children with grade 3-4 vesicoureteral reflux, Hispanic children, and children without fever were more likely to have a UTI caused by organisms other than Escherichia coli and, therefore, less likely to respond to standard first-line antibiotic therapy. These investigators are the preeminent authorities in UTI management for children, so their findings should be viewed in that light.

|
Dr. Michael E. Pichichero |
The advance from the study is not a major one because all children with a suspected UTI should have a suitable culture specimen obtained before starting antibiotics, and the treatment choice continued or changed based on culture results. So really, the findings apply only to a decision about initial empiric treatment while awaiting culture results.
As a guide, if a clinician were to consider the diagnosis of UTI based on history, examination, and urinalysis, and the child was a circumcised male, a child with known grade 3 or 4 vesicoureteral reflux, Hispanic, or without fever, then the empiric antibiotic selected should be broader spectrum while awaiting urine culture results.
Michael E. Pichichero, MD, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
In this study of almost 800 children in the Pittsburgh area, the investigators sought to identify children at risk for urinary tract infections that more likely would have a bacterial organism not susceptible to standard first-line empiric antibiotic treatment.
They found that circumcised males, children with grade 3-4 vesicoureteral reflux, Hispanic children, and children without fever were more likely to have a UTI caused by organisms other than Escherichia coli and, therefore, less likely to respond to standard first-line antibiotic therapy. These investigators are the preeminent authorities in UTI management for children, so their findings should be viewed in that light.
|
|
Dr. Michael E. Pichichero |
The advance from the study is not a major one because all children with a suspected UTI should have a suitable culture specimen obtained before starting antibiotics, and the treatment choice continued or changed based on culture results. So really, the findings apply only to a decision about initial empiric treatment while awaiting culture results.
As a guide, if a clinician were to consider the diagnosis of UTI based on history, examination, and urinalysis, and the child was a circumcised male, a child with known grade 3 or 4 vesicoureteral reflux, Hispanic, or without fever, then the empiric antibiotic selected should be broader spectrum while awaiting urine culture results.
Michael E. Pichichero, MD, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
In this study of almost 800 children in the Pittsburgh area, the investigators sought to identify children at risk for urinary tract infections that more likely would have a bacterial organism not susceptible to standard first-line empiric antibiotic treatment.
They found that circumcised males, children with grade 3-4 vesicoureteral reflux, Hispanic children, and children without fever were more likely to have a UTI caused by organisms other than Escherichia coli and, therefore, less likely to respond to standard first-line antibiotic therapy. These investigators are the preeminent authorities in UTI management for children, so their findings should be viewed in that light.
|
|
Dr. Michael E. Pichichero |
The advance from the study is not a major one because all children with a suspected UTI should have a suitable culture specimen obtained before starting antibiotics, and the treatment choice continued or changed based on culture results. So really, the findings apply only to a decision about initial empiric treatment while awaiting culture results.
As a guide, if a clinician were to consider the diagnosis of UTI based on history, examination, and urinalysis, and the child was a circumcised male, a child with known grade 3 or 4 vesicoureteral reflux, Hispanic, or without fever, then the empiric antibiotic selected should be broader spectrum while awaiting urine culture results.
Michael E. Pichichero, MD, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
Certain children are more highly predisposed to contracting a urinary tract infection caused by a pathogen other than Escherichia coli, which is typically the most common cause of UTIs, a study showed.
“It may be clinically important to predict which children have UTIs caused by organisms other than E. coli because these organisms differ in their patterns of antimicrobial susceptibility,” wrote the study authors led by Nader Shaikh, MD, of the University of Pittsburgh. “Furthermore, some guidelines have suggested that screening for vesicoureteral reflux (VUR) with a voiding cystourethrogram (VCUG) should, at least in part, be based on whether an organism other than E. coli is recovered,” they wrote.
Dr. Shaikh and his coinvestigators examined the medical records of children in the Randomized Intervention for Children With Vesicoureteral Reflux (RIVUR) trial and the Careful Urinary Tract Infection Evaluation (CUTIE), both of which were prospective multicenter studies. Children included in both studies were 2-71 months of age; RIVUR subjects had VUR grades 1-4 and presented with either a first or second febrile or symptomatic UTI, while CUTIE subjects presented with either their first or second UTI but not VUR (Ped Inf Dis J. 2016. doi:10.1097/INF.0000000000001301).
In total, 769 children from 19 centers were included from both studies, of which 703 (91%) were female and 596 (78%) were white. Nine percent of all the children had UTIs that were not caused by E. coli. Circumcised males had the highest odds ratio associated with non–E. coli UTIs, with an OR of 5.5 (95% CI, 1.18-17.1; P = .003). significantly higher than the 1.6 odds ratio for uncircumcised males (95% CI, 0.6-4.6; P = .35).
Hispanic children also had a higher risk (OR = 2.3; 95% CI, 1.1-4.6; P = .02) than either non-Hispanic children or females, which were reference cohorts. Other groups found to be at higher-than-normal risk for non–E. coli UTIs were children without fever (OR = 2.8; 95% CI, 1.2-6.6; P = .02) and children with VUR grade 3 or 4 (OR = 2.2; 95% CI, 1.2-4.1; P = .01).
While more than 90% of children’s UTIs were caused by E. coli, the most common pathogens causing UTIs, causative organisms in the other 70 children were Proteus species (21 children, 30%), Klebsiella species (16, 23%), Enterococcus species (14, 20%), Enterobacter species (8, 11%), and “other species” (11, 16%).
“Children with UTIs caused by organisms other than E. coli were twice as likely to have high-grade VUR (grade 3 and 4), which is consistent with prior studies,” Dr. Shaikh and his coauthors noted, adding, “the association between Hispanic ethnicity and non-E. coli pathogens is novel and may be due to differences in genes involved with susceptibility to UTIs.”
There were no disclosures or sources of funding provided.
Certain children are more highly predisposed to contracting a urinary tract infection caused by a pathogen other than Escherichia coli, which is typically the most common cause of UTIs, a study showed.
“It may be clinically important to predict which children have UTIs caused by organisms other than E. coli because these organisms differ in their patterns of antimicrobial susceptibility,” wrote the study authors led by Nader Shaikh, MD, of the University of Pittsburgh. “Furthermore, some guidelines have suggested that screening for vesicoureteral reflux (VUR) with a voiding cystourethrogram (VCUG) should, at least in part, be based on whether an organism other than E. coli is recovered,” they wrote.
Dr. Shaikh and his coinvestigators examined the medical records of children in the Randomized Intervention for Children With Vesicoureteral Reflux (RIVUR) trial and the Careful Urinary Tract Infection Evaluation (CUTIE), both of which were prospective multicenter studies. Children included in both studies were 2-71 months of age; RIVUR subjects had VUR grades 1-4 and presented with either a first or second febrile or symptomatic UTI, while CUTIE subjects presented with either their first or second UTI but not VUR (Ped Inf Dis J. 2016. doi:10.1097/INF.0000000000001301).
In total, 769 children from 19 centers were included from both studies, of which 703 (91%) were female and 596 (78%) were white. Nine percent of all the children had UTIs that were not caused by E. coli. Circumcised males had the highest odds ratio associated with non–E. coli UTIs, with an OR of 5.5 (95% CI, 1.18-17.1; P = .003). significantly higher than the 1.6 odds ratio for uncircumcised males (95% CI, 0.6-4.6; P = .35).
Hispanic children also had a higher risk (OR = 2.3; 95% CI, 1.1-4.6; P = .02) than either non-Hispanic children or females, which were reference cohorts. Other groups found to be at higher-than-normal risk for non–E. coli UTIs were children without fever (OR = 2.8; 95% CI, 1.2-6.6; P = .02) and children with VUR grade 3 or 4 (OR = 2.2; 95% CI, 1.2-4.1; P = .01).
While more than 90% of children’s UTIs were caused by E. coli, the most common pathogens causing UTIs, causative organisms in the other 70 children were Proteus species (21 children, 30%), Klebsiella species (16, 23%), Enterococcus species (14, 20%), Enterobacter species (8, 11%), and “other species” (11, 16%).
“Children with UTIs caused by organisms other than E. coli were twice as likely to have high-grade VUR (grade 3 and 4), which is consistent with prior studies,” Dr. Shaikh and his coauthors noted, adding, “the association between Hispanic ethnicity and non-E. coli pathogens is novel and may be due to differences in genes involved with susceptibility to UTIs.”
There were no disclosures or sources of funding provided.
FROM THE PEDIATRIC INFECTIOUS DISEASE JOURNAL
Key clinical point: Non–Escherichia coli urinary tract infections are more likely to occur in children who are uncircumcised, are Hispanic, have no fever, or have grade 3-4 vesicoureteral reflux.
Major finding: Circumcised males had an odds ratio of 5.5 (95% CI, 1.8-17.1; P = .003) of infection by pathogens other than E. coli; the odds ratio for Hispanic children (OR = 2.3; 95% CI, 1.1-4.6; P = .02), children without fever (OR = 2.8; 95% CI, 1.2-6.6; P = .02), and children with grade 3-4 VUR (OR = 2.2; 95% CI, 1.2-4.1; P = .01) also were relatively high.
Data source: A review of data from two prospective multicenter studies involving 769 children with a UTI aged 2-71 months .
Disclosures: Funding sources and individual disclosures were not provided.

In septic shock, vasopressin not better than norepinephrine
Vasopressin was no better than norepinephrine in preventing kidney failure when used as a first-line treatment for septic shock, according to a report published online Aug. 2 in JAMA.
In a multicenter, double-blind, randomized trial comparing the two approaches in 408 ICU patients with septic shock, the early use of vasopressin didn’t reduce the number of days free of kidney failure, compared with standard norepinephrine.

However, “the 95% confidence intervals of the difference between [study] groups has an upper limit of 5 days in favor of vasopressin, which could be clinically important,” said Anthony C. Gordon, MD, of Charing Cross Hospital and Imperial College London, and his associates. “Therefore, these results are still consistent with a potentially clinically important benefit for vasopressin; but a larger trial would be needed to confirm or refute this.”
Norepinephrine is the recommended first-line vasopressor for septic shock, but “there has been a growing interest in the use of vasopressin” ever since researchers described a relative deficiency of vasopressin in the disorder, Dr. Gordon and his associates noted.
“Preclinical and small clinical studies have suggested that vasopressin may be better able to maintain glomerular filtration rate and improve creatinine clearance, compared with norepinephrine,” the investigators said, and other studies have suggested that combining vasopressin with corticosteroids may prevent deterioration in organ function and reduce the duration of shock, thereby improving survival.
To examine those possibilities, they performed the VANISH (Vasopressin vs. Norepinephrine as Initial Therapy in Septic Shock) trial, assessing patients age 16 years and older at 18 general adult ICUs in the United Kingdom during a 2-year period. The study participants were randomly assigned to receive vasopressin plus hydrocortisone (100 patients), vasopressin plus matching placebo (104 patients), norepinephrine plus hydrocortisone (101 patients), or norepinephrine plus matching placebo (103 patients).
The primary outcome measure was the number of days alive and free of kidney failure during the 28 days following randomization. There was no significant difference among the four study groups in the number or the distribution of kidney-failure–free days, the investigators said (JAMA. 2016 Aug 2. doi: 10.1001/jama.2016.10485).
In addition, the percentage of survivors who never developed kidney failure was not significantly different between the two groups who received vasopressin (57.0%) and the two who received norepinephrine (59.2%). And the median number of days free of kidney failure in the subgroup of patients who died or developed kidney failure was not significantly different between those receiving vasopressin (9 days) and those receiving norepinephrine (13 days).
The quantities of IV fluids administered, the total fluid balance, serum lactate levels, and heart rate were all similar across the four study groups. There also was no significant difference in 28-day mortality between patients who received vasopressin (30.9%) and those who received norepinephrine (27.5%). Adverse event profiles also were comparable.
However, the rate of renal replacement therapy was 25.4% with vasopressin, significantly lower than the 35.3% rate in the norepinephrine group. The use of such therapy was not controlled in the trial and was initiated according to the treating physicians’ preference. “It is therefore not possible to know why renal replacement therapy was or was not started,” Dr. Gordon and his associates noted.
The use of renal replacement therapy wasn’t a primary outcome of the trial. Nevertheless, it is an important patient-centered outcome and may be a factor to consider when treating adults who have septic shock, the researchers added.
The study was supported by the U.K. National Institute for Health Research and the U.K. Intensive Care Foundation. Dr. Gordon reported ties to Ferring, HCA International, Orion, and Tenax Therapeutics; his associates reported having no relevant financial disclosures.
Vasopressin was no better than norepinephrine in preventing kidney failure when used as a first-line treatment for septic shock, according to a report published online Aug. 2 in JAMA.
In a multicenter, double-blind, randomized trial comparing the two approaches in 408 ICU patients with septic shock, the early use of vasopressin didn’t reduce the number of days free of kidney failure, compared with standard norepinephrine.
However, “the 95% confidence intervals of the difference between [study] groups has an upper limit of 5 days in favor of vasopressin, which could be clinically important,” said Anthony C. Gordon, MD, of Charing Cross Hospital and Imperial College London, and his associates. “Therefore, these results are still consistent with a potentially clinically important benefit for vasopressin; but a larger trial would be needed to confirm or refute this.”
Norepinephrine is the recommended first-line vasopressor for septic shock, but “there has been a growing interest in the use of vasopressin” ever since researchers described a relative deficiency of vasopressin in the disorder, Dr. Gordon and his associates noted.
“Preclinical and small clinical studies have suggested that vasopressin may be better able to maintain glomerular filtration rate and improve creatinine clearance, compared with norepinephrine,” the investigators said, and other studies have suggested that combining vasopressin with corticosteroids may prevent deterioration in organ function and reduce the duration of shock, thereby improving survival.
To examine those possibilities, they performed the VANISH (Vasopressin vs. Norepinephrine as Initial Therapy in Septic Shock) trial, assessing patients age 16 years and older at 18 general adult ICUs in the United Kingdom during a 2-year period. The study participants were randomly assigned to receive vasopressin plus hydrocortisone (100 patients), vasopressin plus matching placebo (104 patients), norepinephrine plus hydrocortisone (101 patients), or norepinephrine plus matching placebo (103 patients).
The primary outcome measure was the number of days alive and free of kidney failure during the 28 days following randomization. There was no significant difference among the four study groups in the number or the distribution of kidney-failure–free days, the investigators said (JAMA. 2016 Aug 2. doi: 10.1001/jama.2016.10485).
In addition, the percentage of survivors who never developed kidney failure was not significantly different between the two groups who received vasopressin (57.0%) and the two who received norepinephrine (59.2%). And the median number of days free of kidney failure in the subgroup of patients who died or developed kidney failure was not significantly different between those receiving vasopressin (9 days) and those receiving norepinephrine (13 days).
The quantities of IV fluids administered, the total fluid balance, serum lactate levels, and heart rate were all similar across the four study groups. There also was no significant difference in 28-day mortality between patients who received vasopressin (30.9%) and those who received norepinephrine (27.5%). Adverse event profiles also were comparable.
However, the rate of renal replacement therapy was 25.4% with vasopressin, significantly lower than the 35.3% rate in the norepinephrine group. The use of such therapy was not controlled in the trial and was initiated according to the treating physicians’ preference. “It is therefore not possible to know why renal replacement therapy was or was not started,” Dr. Gordon and his associates noted.
The use of renal replacement therapy wasn’t a primary outcome of the trial. Nevertheless, it is an important patient-centered outcome and may be a factor to consider when treating adults who have septic shock, the researchers added.
The study was supported by the U.K. National Institute for Health Research and the U.K. Intensive Care Foundation. Dr. Gordon reported ties to Ferring, HCA International, Orion, and Tenax Therapeutics; his associates reported having no relevant financial disclosures.
Vasopressin was no better than norepinephrine in preventing kidney failure when used as a first-line treatment for septic shock, according to a report published online Aug. 2 in JAMA.
In a multicenter, double-blind, randomized trial comparing the two approaches in 408 ICU patients with septic shock, the early use of vasopressin didn’t reduce the number of days free of kidney failure, compared with standard norepinephrine.
However, “the 95% confidence intervals of the difference between [study] groups has an upper limit of 5 days in favor of vasopressin, which could be clinically important,” said Anthony C. Gordon, MD, of Charing Cross Hospital and Imperial College London, and his associates. “Therefore, these results are still consistent with a potentially clinically important benefit for vasopressin; but a larger trial would be needed to confirm or refute this.”
Norepinephrine is the recommended first-line vasopressor for septic shock, but “there has been a growing interest in the use of vasopressin” ever since researchers described a relative deficiency of vasopressin in the disorder, Dr. Gordon and his associates noted.
“Preclinical and small clinical studies have suggested that vasopressin may be better able to maintain glomerular filtration rate and improve creatinine clearance, compared with norepinephrine,” the investigators said, and other studies have suggested that combining vasopressin with corticosteroids may prevent deterioration in organ function and reduce the duration of shock, thereby improving survival.
To examine those possibilities, they performed the VANISH (Vasopressin vs. Norepinephrine as Initial Therapy in Septic Shock) trial, assessing patients age 16 years and older at 18 general adult ICUs in the United Kingdom during a 2-year period. The study participants were randomly assigned to receive vasopressin plus hydrocortisone (100 patients), vasopressin plus matching placebo (104 patients), norepinephrine plus hydrocortisone (101 patients), or norepinephrine plus matching placebo (103 patients).
The primary outcome measure was the number of days alive and free of kidney failure during the 28 days following randomization. There was no significant difference among the four study groups in the number or the distribution of kidney-failure–free days, the investigators said (JAMA. 2016 Aug 2. doi: 10.1001/jama.2016.10485).
In addition, the percentage of survivors who never developed kidney failure was not significantly different between the two groups who received vasopressin (57.0%) and the two who received norepinephrine (59.2%). And the median number of days free of kidney failure in the subgroup of patients who died or developed kidney failure was not significantly different between those receiving vasopressin (9 days) and those receiving norepinephrine (13 days).
The quantities of IV fluids administered, the total fluid balance, serum lactate levels, and heart rate were all similar across the four study groups. There also was no significant difference in 28-day mortality between patients who received vasopressin (30.9%) and those who received norepinephrine (27.5%). Adverse event profiles also were comparable.
However, the rate of renal replacement therapy was 25.4% with vasopressin, significantly lower than the 35.3% rate in the norepinephrine group. The use of such therapy was not controlled in the trial and was initiated according to the treating physicians’ preference. “It is therefore not possible to know why renal replacement therapy was or was not started,” Dr. Gordon and his associates noted.
The use of renal replacement therapy wasn’t a primary outcome of the trial. Nevertheless, it is an important patient-centered outcome and may be a factor to consider when treating adults who have septic shock, the researchers added.
The study was supported by the U.K. National Institute for Health Research and the U.K. Intensive Care Foundation. Dr. Gordon reported ties to Ferring, HCA International, Orion, and Tenax Therapeutics; his associates reported having no relevant financial disclosures.
FROM JAMA
Key clinical point: Vasopressin didn’t perform better than norepinephrine in preventing kidney failure when used as a first-line treatment for septic shock.
Major finding: The primary outcome measure – the number of days alive and free of kidney failure during the first month of treatment – was not significantly different among the four study groups.
Data source: A multicenter, double-blind, randomized clinical trial involving 408 ICU patients treated in the United Kingdom during a 2-year period.
Disclosures: The study was supported by the U.K. National Institute for Health Research and the U.K. Intensive Care Foundation. Dr. Gordon reported ties to Ferring, HCA International, Orion, and Tenax Therapeutics; his associates reported having no relevant financial disclosures.
Anemia of chronic kidney disease: Treat it, but not too aggressively
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
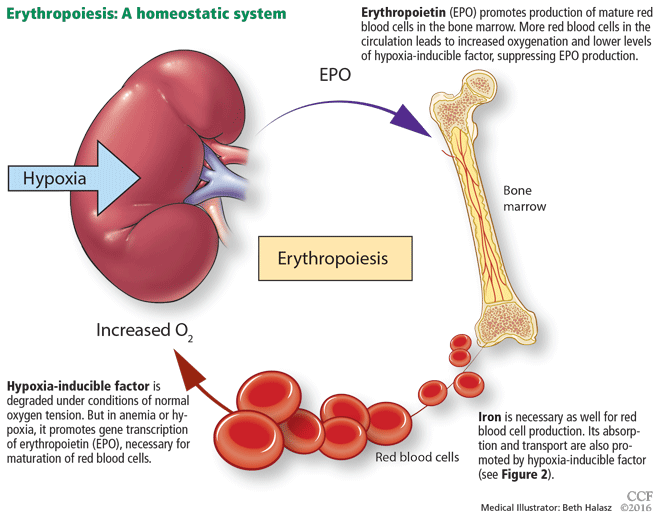
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
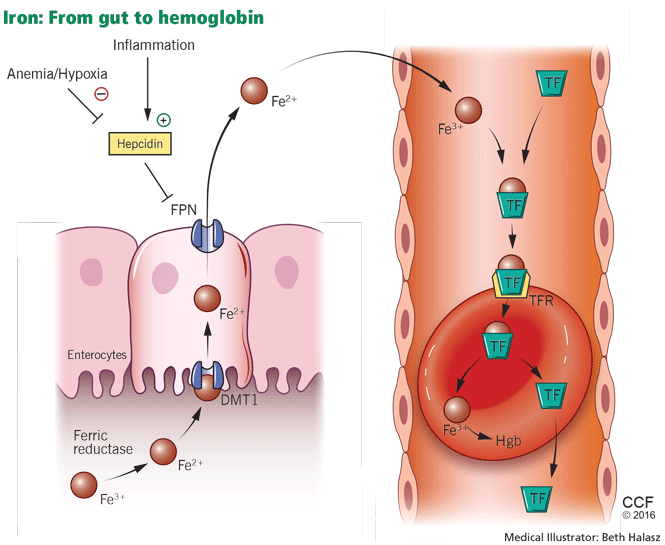
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
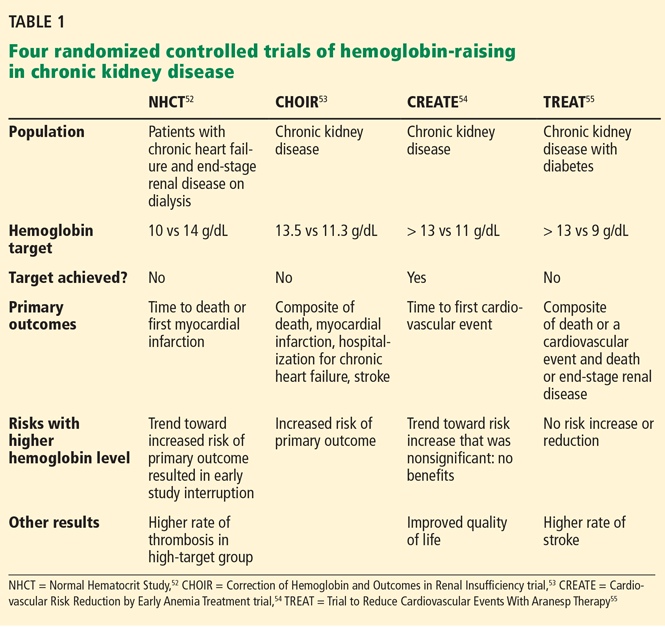
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
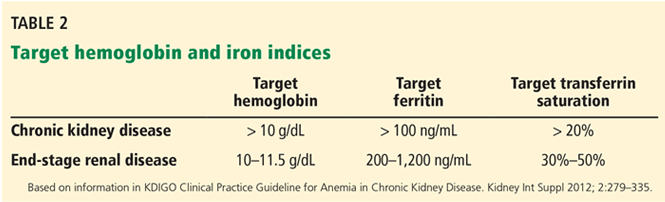
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
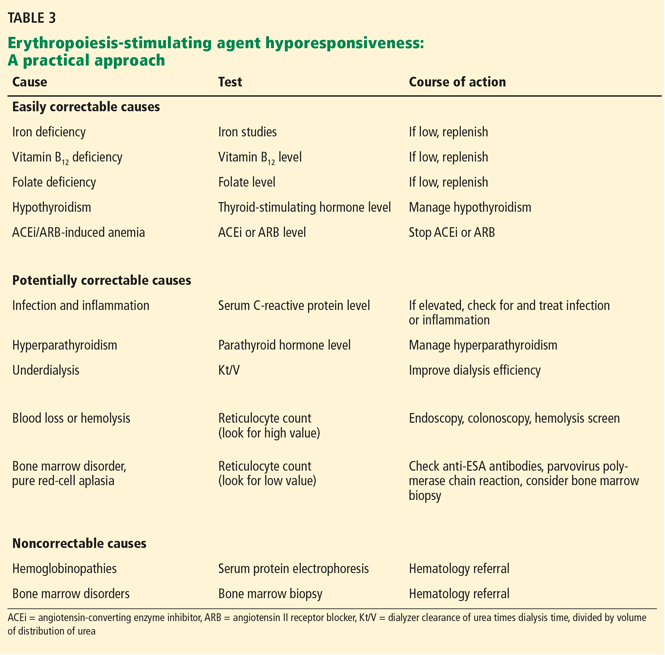
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
KEY POINTS
- Before treating with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies.
- Recognizing anemia in chronic kidney disease is important and often involves participation by the primary care physician, especially in early disease when chronic kidney disease may be mild.
- The only proven benefit of ESA therapy is avoidance of blood transfusions.
- ESAs should not be used to increase the hemoglobin concentration above 13 g/dL. In end-stage renal disease, the goal of therapy is to maintain levels at a target no higher than 11.5 g/dL. In nondialysis-dependent chronic kidney disease, the decision to prescribe ESA therapy should be individualized.
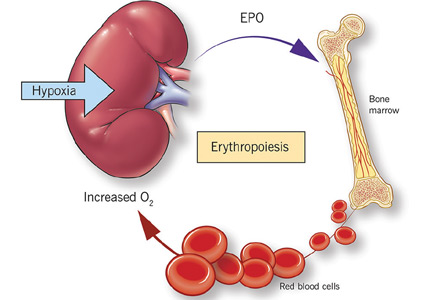
Thrombotic thrombocytopenic purpura: The role of ADAMTS13
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
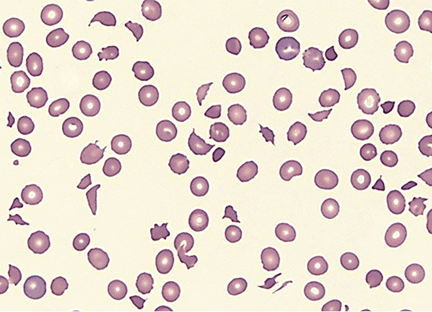
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4


ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
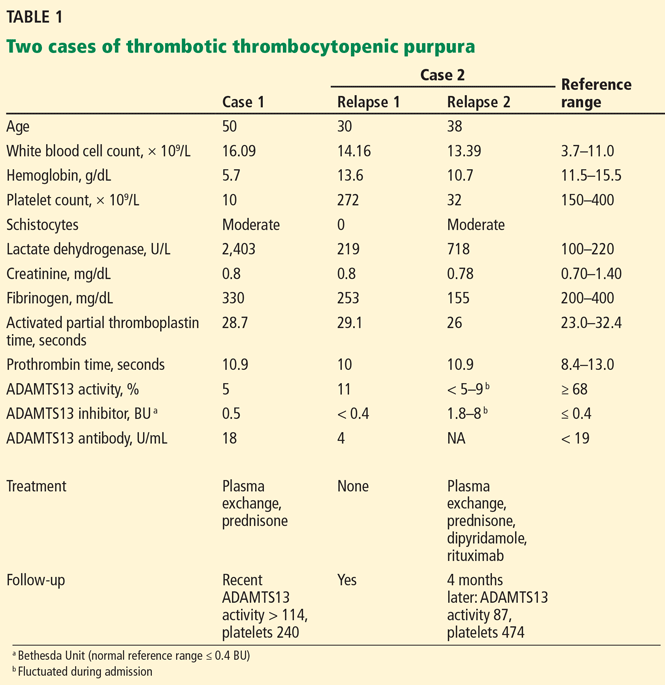
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9

Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS

Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
KEY POINTS
- Symptoms of TTP are usually neurologic but can also be cardiac or abdominal. Thrombocytopenia and unexplained microangiopathic hemolytic anemia are sufficient to highly suspect the disease.
- In the appropriate clinical setting, an ADAMTS13 activity level lower than 10% is highly indicative of TTP.
- ADAMTS13 inhibitor and ADAMTS13 antibody assays provide more diagnostic clues. ADAMTS13 antibody is generally absent in the congenital form.
- The ADAMTS13 assay can help distinguish TTP from hemolytic-uremic syndrome, which presents similarly but typically involves normal or only mildly reduced ADAMTS13 activity.
- A strong clinical suspicion of TTP warrants immediate initiation of therapeutic plasma exchange without waiting for ADAMTS13 test results.
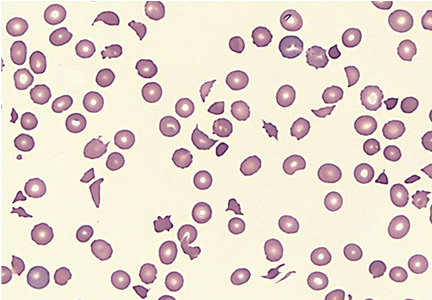
Advanced-stage calciphylaxis: Think before you punch
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
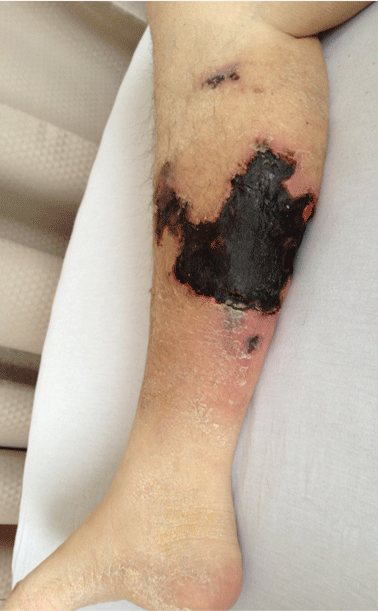
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
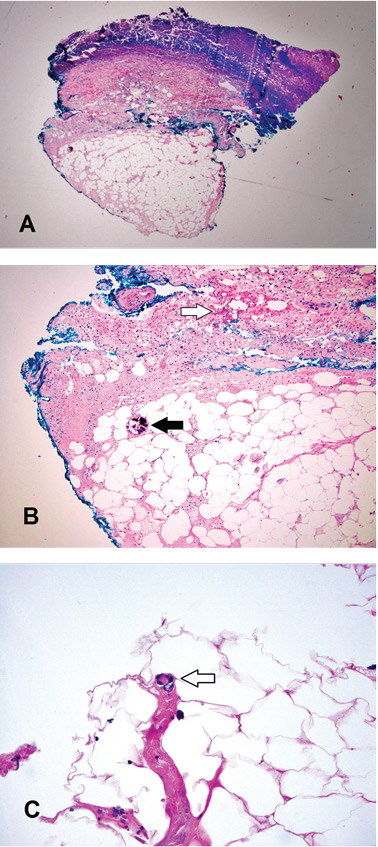
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.