User login
Managing clozapine-induced neutropenia and agranulocytosis
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to 
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
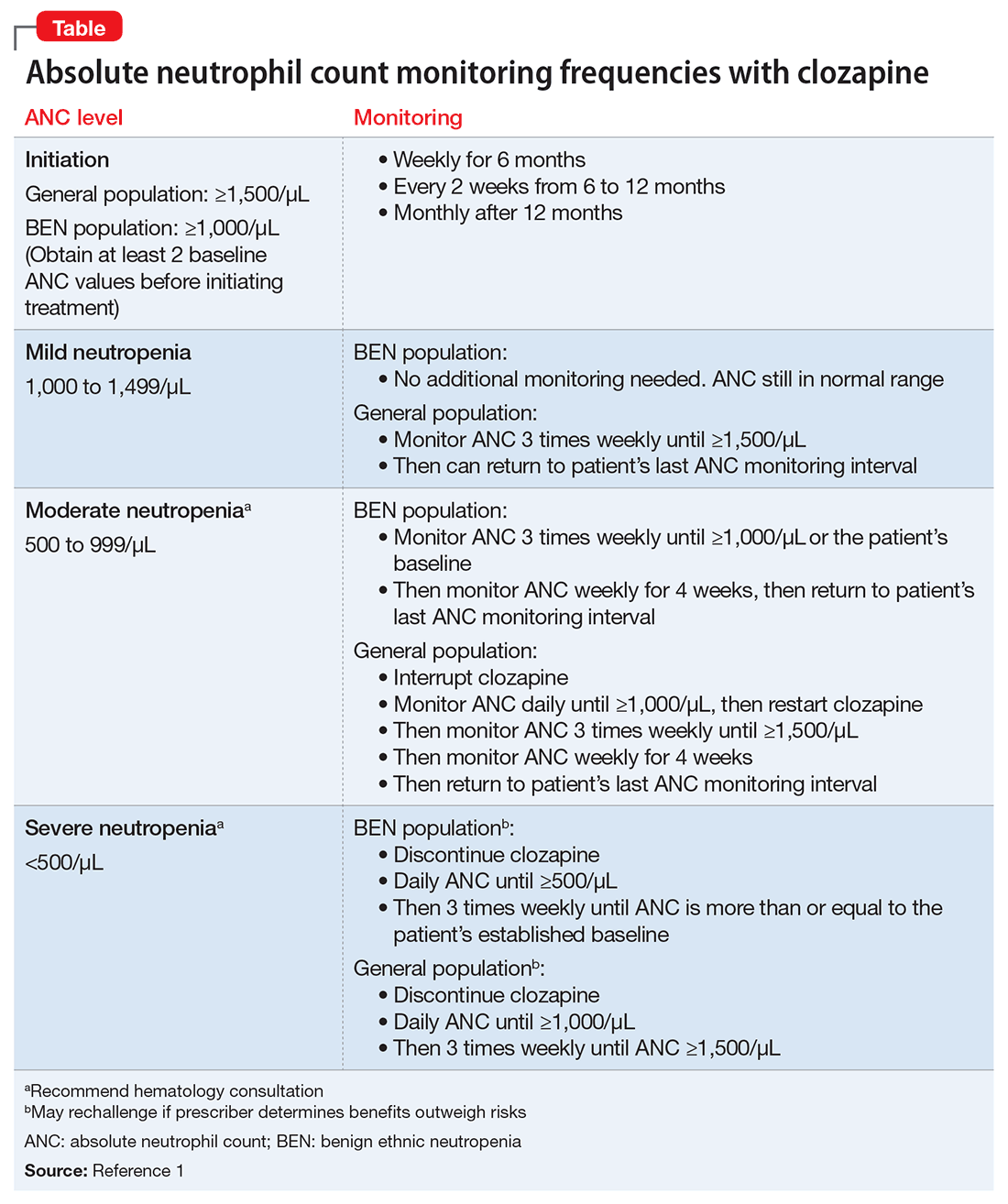
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
How to assess and manage high cholesterol in patients with mental illness
High serum cholesterol is a leading cause of heart attack and stroke,1,2 yet remains one of the most under-screened and undertreated modifiable risk factors in persons with mental illness. Well tolerated and effective treatments can considerably lower the risk of cardiovascular events, and should be offered to psychiatric patients who are at high risk, while considering possible adverse effects and potential interactions between psychotropics and medications used to lower cholesterol.
Systematic lowering of total cholesterol and, particularly, atherogenic low-density lipoprotein (LDL) and non-high density lipoprotein (HDL) cholesterol, results in consistent and significant reduction in risk of cardiovascular events in persons at risk for developing cardiovascular disease (CVD) and in preventing reoccurrence of these events.1,3,4 Even individuals who have relatively lower levels of total cholesterol but are at high risk (such as if a cardiovascular event has occurred) could reduce their CVD risk (known as secondary prevention) through lipid lowering therapies.5,6
Adults with psychiatric illness shoulder a disproportionate burden of CVD morbidity and mortality, especially those with severe mental illness (SMI, schizophrenia, schizoaffective disorder, bipolar disorder, treatment-resistant depression).7-9 Among modifiable CVD risk factors, dyslipidemia has the highest rates of missed screenings and treatment within psychiatric populations. In one analysis, up to 90% of adults with SMI and identified lipid disorders did not receive treatment.10 Persons with SMI generally do not receive guideline-concordant, systematic quality preventive care, which contributes to a widening mortality gap for this population.11,12
This review aims to provide clinicians with practical guidance on the assessment and management of high cholesterol to improve recognition and treatment, lower CVD risk, and reduce this observed mortality gap.
Screening and diagnosis
In 2013, the American College of Cardiology (ACC) and the American Heart Association (AHA) released updated guidelines on diagnosing and managing high cholesterol to reduce CVD risk.1 These guidelines focus on updated 10-year CVD risk assessment models with treatment goals reliant on adherence to statin therapy rather than pre-specified cholesterol targets listed in previous guidelines.13
Updates to assessment and treatment guidelines have removed some barriers to screening and diagnosing high cholesterol—namely, fasting lipid panels are no longer required to determine 10-year CVD risk and initiate treatment.14 For adults taking a second-generation antipsychotic that is associated with weight gain and metabolic syndrome, experts generally recommend yearly non-fasting lipid panels.6,14
The United States Preventive Services Task Force recommends screening:
- men age ≥35 at average risk for CVD every 5 years
- women age ≥45 every 5 years15
- adults as young as age 20 who have accelerated risk factors, such as cigarette smoking and hypertension
- adults with a family history of heart attack or stroke in male first-degree relative age ≥50 and female first-degree relatives age ≥60.
Many adults receiving care in behavioral health settings, regardless of their medication regimen, qualify for screening at least every 5 years, if not more frequently. Although statin treatment before age 40 is less beneficial and likely not necessary for primary prevention, monitoring could help identify alternative therapies and prioritize more intensive diet and lifestyle modifications.
At a routine office visit, clinicians can collect vital signs, record smoking status, and reconcile all medications, which provides the data needed to calculate a patient’s 10-year CVD risk (Table 1). Coupled with laboratory testing, which includes a non-fasting total cholesterol, HDL, and hemoglobin A1c (representative of a 3-month blood sugar average, ≥6.5% is diagnostic of type 2 diabetes mellitus [T2DM]), all data points can be entered into online risk calculators (search “ASCVD risk calculator” or visit http://tools.acc.org/ASCVD-Risk-Estimator to access the ACC/AHA risk calculator). Persons scoring >20% 10-year risk are considered at extremely high risk, and are in the same risk category as adults with existing CVD or who have had a cardiovascular event. Persons at <5% 10-year risk generally are considered low risk, and primary prevention with a statin medication is not indicated.
Treatment and management
Dietary modification and lifestyle changes (exercise, quitting smoking), lowering high cholesterol with medications, and switching from highly metabolically active drugs to less metabolically active ones can help lower total cholesterol in patients at risk of CVD.
Statins
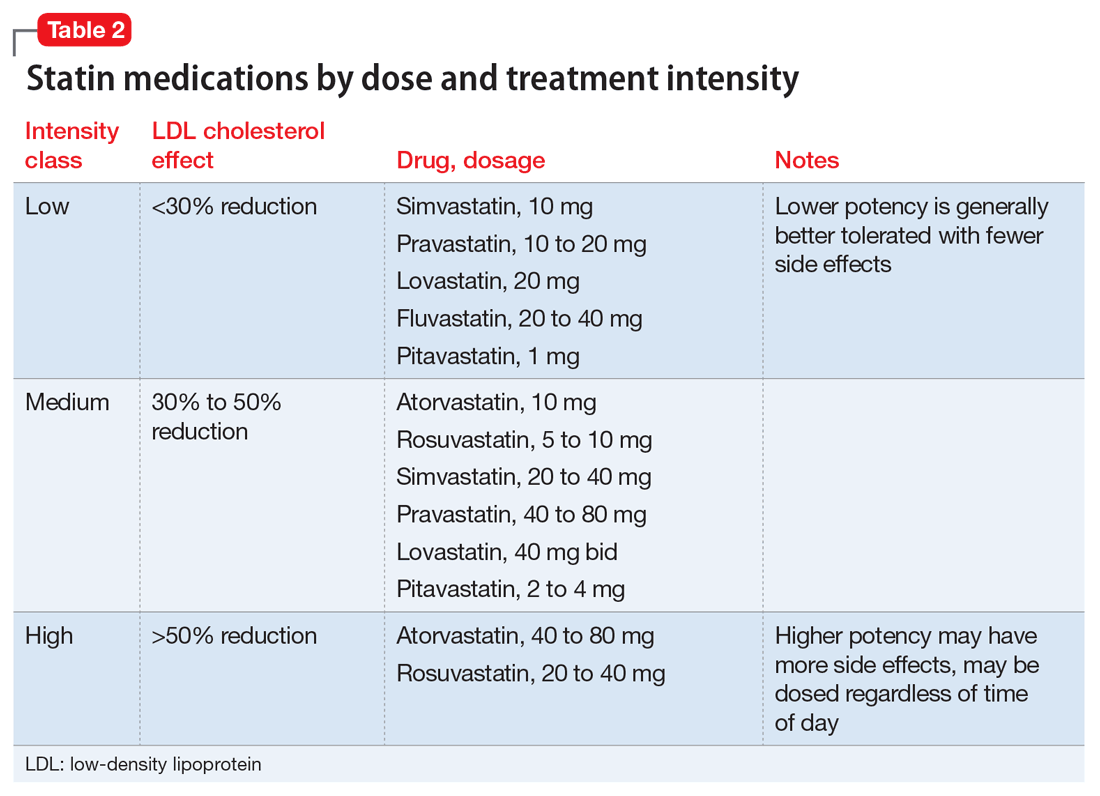
HMG-CoA reductase inhibitors (statins) consistently reduce total cholesterol and non-HDL cholesterol by 30% to 50%, depending on drug and dosage (potency, listed as low, medium, and high). Not all statins are equally effective at lowering cholesterol; some are more potent than others (Table 2).16
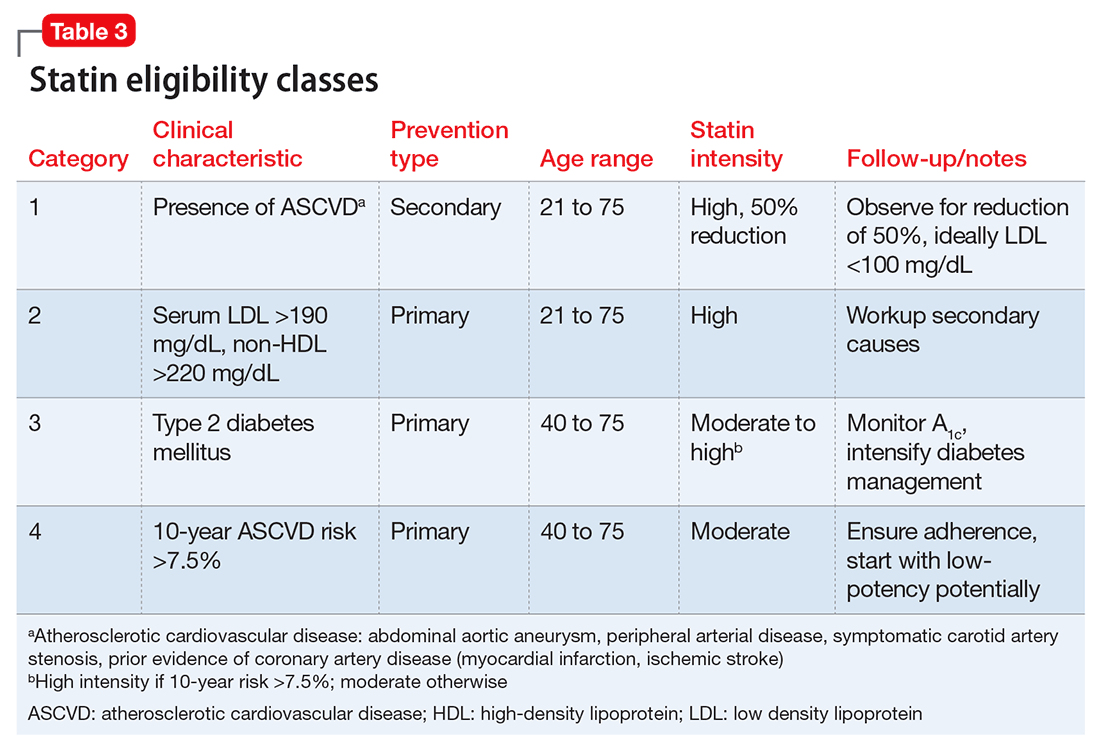
Individuals are eligible for statin therapy based on their level of CVD risk. Persons at higher risk generally benefit from greater intensity statin treatment and cholesterol reduction; highest intensity statin regimens can lower total cholesterol by approximately 50%.
There are 4 statin eligibility classes (Table 3). Most adults fall into category 4: 10-year risk of >7.5% and needing primary prevention. In addition to removing specific LDL targets as therapy goals, calculation of this risk percentage and the specific cut-off values have been the most controversial aspects of the new cholesterol guidelines. Most experts agree that, in adults age 40 to 75, 10-year risk >10% indicates daily statin use as tolerated for primary prevention, and 10-year risk <5% does not warrant statin use. Recent large studies have validated these new techniques for calculating risk, and found them to be beneficial in potential for cost savings and risk classification.17,18
Considerations in psychiatric patients. Statins have been associated with depression in case series, but larger analyses have not confirmed this association.19 Emerging evidence has identified a potential correlation between statin use and accelerated onset of T2DM, but the absolute risk is relatively low and most experts continue to recommend statin therapy despite this potential risk.13 Many statins, including atorvastatin, are available as a generic and can be taken once daily. Some, such as simvastatin, have notable interactions with commonly prescribed psychotropics including risperidone and quetiapine. Pravastatin is dually excreted by the liver and kidneys and may have fewer drug-drug interactions in patients with psychiatric illness taking common psychotropic therapies, but is not considered a high-potency statin and might not confer adequate benefits in CVD risk reduction.
Contraindications. Statins are pregnancy category X, and generally should not be prescribed for women of childbearing age without intensive counseling. The most notable adverse effects for statins include muscle aches and cramps (myalgia), but generally are not severe. If encountered, consider checking a serum creatinine kinase (CK) level, and if significantly elevated above 10 times the upper limit, stopping statin therapy would be advised. If the CK is only mildly elevated, consider lowering the dosage or switching to a lower potency agent. Lovastatin and pravastatin generally are better tolerated than atorvastatin and are considered lower potency (Table 2).
Statins can be safely used in the presence of liver conditions, such as hepatitis C and alcohol use, although periodic monitoring of transaminase levels is recommended. For adults in the general population without liver disease, regular monitoring of transaminase levels is not necessary.
Alternate lipid-lowering pharmacotherapies unfortunately have fallen out of favor. Fibrates, niacin, ezetimibe, and omega-3 fatty acids once were recommended to lower triglycerides or raise HDL cholesterol levels, but since have been shown to have little effect on cardiovascular morbidity or mortality. Adding further medications, other than statins, to lower cholesterol values to pre-defined targets is not the current standard of care.
High triglyceride concentrations traditionally have been addressed directly, but failure to improve CVD mortality or morbidity by treating triglycerides alone has resulted in refocusing clinical efforts in dyslipidemia management on atherogenic cholesterol, including LDL and non-HDL fractions.20 Non-fasting triglycerides >500 mg/dL should be retested when fasting, and levels that remain >500 mg/dL could place the patient at risk for pancreatitis and might warrant intervention with fibrates at that time. This scenario is not common, and referral to a primary care physician or endocrinologist may be warranted.
Lifestyle changes
With or without statin therapies, diet and lifestyle changes are the cornerstone of healthy living and should be encouraged in all patients. Most overweight or obese patients will benefit from exercise and dietary modifications. Such interventions have shown potential for reducing total cholesterol and non-HDL and HDL cholesterol, but rarely are these interventions sustained long enough to produce meaningful reduction in CVD risk through lipid lowering. Diets rich in isocaloric tree nuts and red-yeast rice extract—a form of a statin—have shown promise in reducing cholesterol, but typically take excessive personal resources and are not sustained to the degree necessary to reduce CVD risk over time.21 Similarly, regular exercise routines can help lower overall cholesterol numbers, but rarely reduce total cholesterol by >10%.
Because individuals with SMI smoke at a higher rate than the general population, it should be noted that smoking cessation is associated with a reduction in total cholesterol and a trial of smoking cessation therapy is warranted before initiating a statin medication for primary prevention of CVD. Many patients would discover that their 10-year ASCVD risk would fall under the level needed for statin therapy if they could successfully stop smoking.
Switching pharmacotherapies
Switching antipsychotic agents from highly metabolically risky compounds, such as risperidone and olanzapine, to less metabolically active compounds, such as aripiprazole, ziprasidone, or haloperidol, have been associated with improvements in lipid profiles.22-24 Clinicians must weigh the potential benefits of switching therapies against the risk of psychiatric destabilization and long-term adherence, keeping in mind that changes in lipids seen with switching could be mild (approximately 10% reduction in total cholesterol).
Summing up
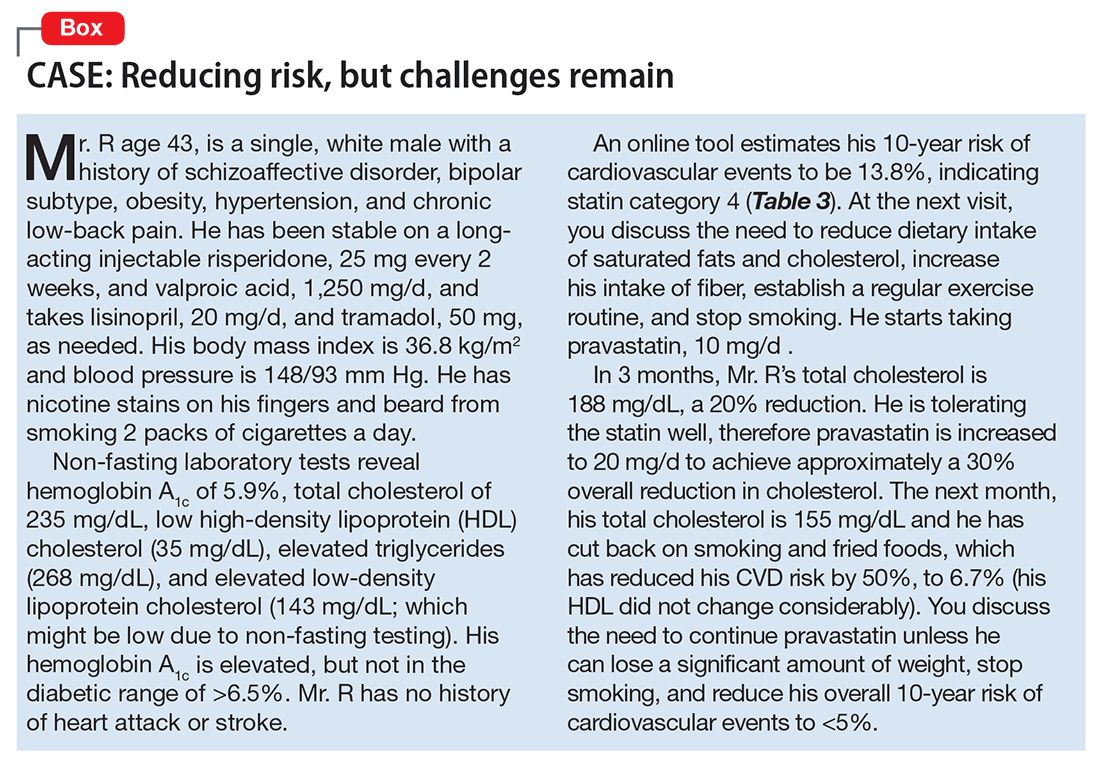
Cholesterol management is considered part of a program to systematically lower CVD risk. Statin therapy usually is indicated for life, or until the age of 75, at which point treatment risks and benefits change because of life expectancy. Other components of CVD risk reduction include a focus on blood pressure control, smoking cessation, T2DM management, and weight loss. Tracking lipid profiles over time to ensure broad targets of 30% to 50% reduction in total cholesterol, approximately 3 months after initiation and yearly thereafter, can help ensure adherence to therapy. With systematic lowering of modifiable CVD risk factors, we can hope to gradually improve the quality of life for our patients with mental illnesses (see the Box for a case example illustrating successful use of these strategies).
1. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 suppl B):S1-S45.
2. LaRosa JC, Hunninghake D, Bush D, et al. The cholesterol facts. A summary of the evidence relating dietary fats, serum cholesterol, and coronary heart disease. A joint statement by the American Heart Association and the National Heart, Lung, and Blood Institute. The Task Force on Cholesterol Issues, American Heart Association. Circulation. 1990;81(5):1721-1733.
3. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.e2.
4. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816. doi: 10.1002/14651858.CD004816.pub5.
5. Gaziano JM, Gaziano TA. What’s new with measuring cholesterol? JAMA. 2013;310(19):2043-2044.
6. Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
7. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
8. Crump C, Winkleby MA, Sundquist K, et al. Comorbidities and mortality in persons with schizophrenia: a Swedish national cohort study. Am J Psychiatry. 2013;170(3):324-333.
9. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states [published online March 15, 2006]. Prev Chronic Dis. 2006;3(2):A42.
10. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
11. Osby U, Correia N, Brandt L, et al. Time trends in schizophrenia mortality in Stockholm county, Sweden: cohort study. BMJ. 2000;321(7259):483-484.
12. Mitchell AJ, Lord O. Do deficits in cardiac care influence high mortality rates in schizophrenia? A systematic review and pooled analysis. J Psychopharmacol. 2010;24(suppl 4):69-80.
13. Ganda OP. Deciphering cholesterol treatment guidelines: a clinician’s perspective. JAMA. 2015;313(10):1009-1010.
14. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv. 2014;65(5):573-576.
15. U.S. Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia). http://www.uspreventiveservicestaskforce.org/uspstf/uspschol.htm. Published June 2008. Accessed October 12, 2016.
16. Cupp M. Characteristics of the various statins. Pharmacist’s Letter. 2012;28(6):280606.
17. Pursnani A, Massaro JM, D’Agostino RB Sr, et al. Guideline-based statin eligibility, coronary artery calcification, and cardiovascular events. JAMA. 2015;314(2):134-141.
18. Pandya A, Sy S, Cho S, et al. Cost-effectiveness of 10-year risk thresholds for initiation of statin therapy for primary prevention of cardiovascular disease. JAMA. 2015;314(2):142-150.
19. You H, Lu W, Zhao S, et al. The relationship between statins and depression: a review of the literature. Expert Opin Pharmacother. 2013;14(11):1467-1476.
20. Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-1861.
21. Kelly RB. Diet and exercise in the management of hyperlipidemia. Am Fam Physician. 2010;81(9):1097-1102.
22. Erhardt L. Cigarette smoking: an undertreated risk factor for cardiovascular disease. Atherosclerosis. 2009;205(1):23-32.
23. Weiden PJ. Switching antipsychotics as a treatment strategy for antipsychotic-induced weight gain and dyslipidemia. J Clin Psychiatry. 2007;68(suppl 4):34-39.
24. Stroup TS, McEvoy JP, Ring KD, et al; Schizophrenia Trials Network. A randomized trial examining the effectiveness of switching from olanzapine, quetiapine, or risperidone to aripiprazole to reduce metabolic risk: comparison of antipsychotics for metabolic problems (CAMP). Am J Psychiatry. 2011;168(9):947-956.
High serum cholesterol is a leading cause of heart attack and stroke,1,2 yet remains one of the most under-screened and undertreated modifiable risk factors in persons with mental illness. Well tolerated and effective treatments can considerably lower the risk of cardiovascular events, and should be offered to psychiatric patients who are at high risk, while considering possible adverse effects and potential interactions between psychotropics and medications used to lower cholesterol.
Systematic lowering of total cholesterol and, particularly, atherogenic low-density lipoprotein (LDL) and non-high density lipoprotein (HDL) cholesterol, results in consistent and significant reduction in risk of cardiovascular events in persons at risk for developing cardiovascular disease (CVD) and in preventing reoccurrence of these events.1,3,4 Even individuals who have relatively lower levels of total cholesterol but are at high risk (such as if a cardiovascular event has occurred) could reduce their CVD risk (known as secondary prevention) through lipid lowering therapies.5,6
Adults with psychiatric illness shoulder a disproportionate burden of CVD morbidity and mortality, especially those with severe mental illness (SMI, schizophrenia, schizoaffective disorder, bipolar disorder, treatment-resistant depression).7-9 Among modifiable CVD risk factors, dyslipidemia has the highest rates of missed screenings and treatment within psychiatric populations. In one analysis, up to 90% of adults with SMI and identified lipid disorders did not receive treatment.10 Persons with SMI generally do not receive guideline-concordant, systematic quality preventive care, which contributes to a widening mortality gap for this population.11,12
This review aims to provide clinicians with practical guidance on the assessment and management of high cholesterol to improve recognition and treatment, lower CVD risk, and reduce this observed mortality gap.
Screening and diagnosis
In 2013, the American College of Cardiology (ACC) and the American Heart Association (AHA) released updated guidelines on diagnosing and managing high cholesterol to reduce CVD risk.1 These guidelines focus on updated 10-year CVD risk assessment models with treatment goals reliant on adherence to statin therapy rather than pre-specified cholesterol targets listed in previous guidelines.13
Updates to assessment and treatment guidelines have removed some barriers to screening and diagnosing high cholesterol—namely, fasting lipid panels are no longer required to determine 10-year CVD risk and initiate treatment.14 For adults taking a second-generation antipsychotic that is associated with weight gain and metabolic syndrome, experts generally recommend yearly non-fasting lipid panels.6,14
The United States Preventive Services Task Force recommends screening:
- men age ≥35 at average risk for CVD every 5 years
- women age ≥45 every 5 years15
- adults as young as age 20 who have accelerated risk factors, such as cigarette smoking and hypertension
- adults with a family history of heart attack or stroke in male first-degree relative age ≥50 and female first-degree relatives age ≥60.
Many adults receiving care in behavioral health settings, regardless of their medication regimen, qualify for screening at least every 5 years, if not more frequently. Although statin treatment before age 40 is less beneficial and likely not necessary for primary prevention, monitoring could help identify alternative therapies and prioritize more intensive diet and lifestyle modifications.
At a routine office visit, clinicians can collect vital signs, record smoking status, and reconcile all medications, which provides the data needed to calculate a patient’s 10-year CVD risk (Table 1). Coupled with laboratory testing, which includes a non-fasting total cholesterol, HDL, and hemoglobin A1c (representative of a 3-month blood sugar average, ≥6.5% is diagnostic of type 2 diabetes mellitus [T2DM]), all data points can be entered into online risk calculators (search “ASCVD risk calculator” or visit http://tools.acc.org/ASCVD-Risk-Estimator to access the ACC/AHA risk calculator). Persons scoring >20% 10-year risk are considered at extremely high risk, and are in the same risk category as adults with existing CVD or who have had a cardiovascular event. Persons at <5% 10-year risk generally are considered low risk, and primary prevention with a statin medication is not indicated.
Treatment and management
Dietary modification and lifestyle changes (exercise, quitting smoking), lowering high cholesterol with medications, and switching from highly metabolically active drugs to less metabolically active ones can help lower total cholesterol in patients at risk of CVD.
Statins
HMG-CoA reductase inhibitors (statins) consistently reduce total cholesterol and non-HDL cholesterol by 30% to 50%, depending on drug and dosage (potency, listed as low, medium, and high). Not all statins are equally effective at lowering cholesterol; some are more potent than others (Table 2).16
Individuals are eligible for statin therapy based on their level of CVD risk. Persons at higher risk generally benefit from greater intensity statin treatment and cholesterol reduction; highest intensity statin regimens can lower total cholesterol by approximately 50%.
There are 4 statin eligibility classes (Table 3). Most adults fall into category 4: 10-year risk of >7.5% and needing primary prevention. In addition to removing specific LDL targets as therapy goals, calculation of this risk percentage and the specific cut-off values have been the most controversial aspects of the new cholesterol guidelines. Most experts agree that, in adults age 40 to 75, 10-year risk >10% indicates daily statin use as tolerated for primary prevention, and 10-year risk <5% does not warrant statin use. Recent large studies have validated these new techniques for calculating risk, and found them to be beneficial in potential for cost savings and risk classification.17,18
Considerations in psychiatric patients. Statins have been associated with depression in case series, but larger analyses have not confirmed this association.19 Emerging evidence has identified a potential correlation between statin use and accelerated onset of T2DM, but the absolute risk is relatively low and most experts continue to recommend statin therapy despite this potential risk.13 Many statins, including atorvastatin, are available as a generic and can be taken once daily. Some, such as simvastatin, have notable interactions with commonly prescribed psychotropics including risperidone and quetiapine. Pravastatin is dually excreted by the liver and kidneys and may have fewer drug-drug interactions in patients with psychiatric illness taking common psychotropic therapies, but is not considered a high-potency statin and might not confer adequate benefits in CVD risk reduction.
Contraindications. Statins are pregnancy category X, and generally should not be prescribed for women of childbearing age without intensive counseling. The most notable adverse effects for statins include muscle aches and cramps (myalgia), but generally are not severe. If encountered, consider checking a serum creatinine kinase (CK) level, and if significantly elevated above 10 times the upper limit, stopping statin therapy would be advised. If the CK is only mildly elevated, consider lowering the dosage or switching to a lower potency agent. Lovastatin and pravastatin generally are better tolerated than atorvastatin and are considered lower potency (Table 2).
Statins can be safely used in the presence of liver conditions, such as hepatitis C and alcohol use, although periodic monitoring of transaminase levels is recommended. For adults in the general population without liver disease, regular monitoring of transaminase levels is not necessary.
Alternate lipid-lowering pharmacotherapies unfortunately have fallen out of favor. Fibrates, niacin, ezetimibe, and omega-3 fatty acids once were recommended to lower triglycerides or raise HDL cholesterol levels, but since have been shown to have little effect on cardiovascular morbidity or mortality. Adding further medications, other than statins, to lower cholesterol values to pre-defined targets is not the current standard of care.
High triglyceride concentrations traditionally have been addressed directly, but failure to improve CVD mortality or morbidity by treating triglycerides alone has resulted in refocusing clinical efforts in dyslipidemia management on atherogenic cholesterol, including LDL and non-HDL fractions.20 Non-fasting triglycerides >500 mg/dL should be retested when fasting, and levels that remain >500 mg/dL could place the patient at risk for pancreatitis and might warrant intervention with fibrates at that time. This scenario is not common, and referral to a primary care physician or endocrinologist may be warranted.
Lifestyle changes
With or without statin therapies, diet and lifestyle changes are the cornerstone of healthy living and should be encouraged in all patients. Most overweight or obese patients will benefit from exercise and dietary modifications. Such interventions have shown potential for reducing total cholesterol and non-HDL and HDL cholesterol, but rarely are these interventions sustained long enough to produce meaningful reduction in CVD risk through lipid lowering. Diets rich in isocaloric tree nuts and red-yeast rice extract—a form of a statin—have shown promise in reducing cholesterol, but typically take excessive personal resources and are not sustained to the degree necessary to reduce CVD risk over time.21 Similarly, regular exercise routines can help lower overall cholesterol numbers, but rarely reduce total cholesterol by >10%.
Because individuals with SMI smoke at a higher rate than the general population, it should be noted that smoking cessation is associated with a reduction in total cholesterol and a trial of smoking cessation therapy is warranted before initiating a statin medication for primary prevention of CVD. Many patients would discover that their 10-year ASCVD risk would fall under the level needed for statin therapy if they could successfully stop smoking.
Switching pharmacotherapies
Switching antipsychotic agents from highly metabolically risky compounds, such as risperidone and olanzapine, to less metabolically active compounds, such as aripiprazole, ziprasidone, or haloperidol, have been associated with improvements in lipid profiles.22-24 Clinicians must weigh the potential benefits of switching therapies against the risk of psychiatric destabilization and long-term adherence, keeping in mind that changes in lipids seen with switching could be mild (approximately 10% reduction in total cholesterol).
Summing up
Cholesterol management is considered part of a program to systematically lower CVD risk. Statin therapy usually is indicated for life, or until the age of 75, at which point treatment risks and benefits change because of life expectancy. Other components of CVD risk reduction include a focus on blood pressure control, smoking cessation, T2DM management, and weight loss. Tracking lipid profiles over time to ensure broad targets of 30% to 50% reduction in total cholesterol, approximately 3 months after initiation and yearly thereafter, can help ensure adherence to therapy. With systematic lowering of modifiable CVD risk factors, we can hope to gradually improve the quality of life for our patients with mental illnesses (see the Box for a case example illustrating successful use of these strategies).
High serum cholesterol is a leading cause of heart attack and stroke,1,2 yet remains one of the most under-screened and undertreated modifiable risk factors in persons with mental illness. Well tolerated and effective treatments can considerably lower the risk of cardiovascular events, and should be offered to psychiatric patients who are at high risk, while considering possible adverse effects and potential interactions between psychotropics and medications used to lower cholesterol.
Systematic lowering of total cholesterol and, particularly, atherogenic low-density lipoprotein (LDL) and non-high density lipoprotein (HDL) cholesterol, results in consistent and significant reduction in risk of cardiovascular events in persons at risk for developing cardiovascular disease (CVD) and in preventing reoccurrence of these events.1,3,4 Even individuals who have relatively lower levels of total cholesterol but are at high risk (such as if a cardiovascular event has occurred) could reduce their CVD risk (known as secondary prevention) through lipid lowering therapies.5,6
Adults with psychiatric illness shoulder a disproportionate burden of CVD morbidity and mortality, especially those with severe mental illness (SMI, schizophrenia, schizoaffective disorder, bipolar disorder, treatment-resistant depression).7-9 Among modifiable CVD risk factors, dyslipidemia has the highest rates of missed screenings and treatment within psychiatric populations. In one analysis, up to 90% of adults with SMI and identified lipid disorders did not receive treatment.10 Persons with SMI generally do not receive guideline-concordant, systematic quality preventive care, which contributes to a widening mortality gap for this population.11,12
This review aims to provide clinicians with practical guidance on the assessment and management of high cholesterol to improve recognition and treatment, lower CVD risk, and reduce this observed mortality gap.
Screening and diagnosis
In 2013, the American College of Cardiology (ACC) and the American Heart Association (AHA) released updated guidelines on diagnosing and managing high cholesterol to reduce CVD risk.1 These guidelines focus on updated 10-year CVD risk assessment models with treatment goals reliant on adherence to statin therapy rather than pre-specified cholesterol targets listed in previous guidelines.13
Updates to assessment and treatment guidelines have removed some barriers to screening and diagnosing high cholesterol—namely, fasting lipid panels are no longer required to determine 10-year CVD risk and initiate treatment.14 For adults taking a second-generation antipsychotic that is associated with weight gain and metabolic syndrome, experts generally recommend yearly non-fasting lipid panels.6,14
The United States Preventive Services Task Force recommends screening:
- men age ≥35 at average risk for CVD every 5 years
- women age ≥45 every 5 years15
- adults as young as age 20 who have accelerated risk factors, such as cigarette smoking and hypertension
- adults with a family history of heart attack or stroke in male first-degree relative age ≥50 and female first-degree relatives age ≥60.
Many adults receiving care in behavioral health settings, regardless of their medication regimen, qualify for screening at least every 5 years, if not more frequently. Although statin treatment before age 40 is less beneficial and likely not necessary for primary prevention, monitoring could help identify alternative therapies and prioritize more intensive diet and lifestyle modifications.
At a routine office visit, clinicians can collect vital signs, record smoking status, and reconcile all medications, which provides the data needed to calculate a patient’s 10-year CVD risk (Table 1). Coupled with laboratory testing, which includes a non-fasting total cholesterol, HDL, and hemoglobin A1c (representative of a 3-month blood sugar average, ≥6.5% is diagnostic of type 2 diabetes mellitus [T2DM]), all data points can be entered into online risk calculators (search “ASCVD risk calculator” or visit http://tools.acc.org/ASCVD-Risk-Estimator to access the ACC/AHA risk calculator). Persons scoring >20% 10-year risk are considered at extremely high risk, and are in the same risk category as adults with existing CVD or who have had a cardiovascular event. Persons at <5% 10-year risk generally are considered low risk, and primary prevention with a statin medication is not indicated.
Treatment and management
Dietary modification and lifestyle changes (exercise, quitting smoking), lowering high cholesterol with medications, and switching from highly metabolically active drugs to less metabolically active ones can help lower total cholesterol in patients at risk of CVD.
Statins
HMG-CoA reductase inhibitors (statins) consistently reduce total cholesterol and non-HDL cholesterol by 30% to 50%, depending on drug and dosage (potency, listed as low, medium, and high). Not all statins are equally effective at lowering cholesterol; some are more potent than others (Table 2).16
Individuals are eligible for statin therapy based on their level of CVD risk. Persons at higher risk generally benefit from greater intensity statin treatment and cholesterol reduction; highest intensity statin regimens can lower total cholesterol by approximately 50%.
There are 4 statin eligibility classes (Table 3). Most adults fall into category 4: 10-year risk of >7.5% and needing primary prevention. In addition to removing specific LDL targets as therapy goals, calculation of this risk percentage and the specific cut-off values have been the most controversial aspects of the new cholesterol guidelines. Most experts agree that, in adults age 40 to 75, 10-year risk >10% indicates daily statin use as tolerated for primary prevention, and 10-year risk <5% does not warrant statin use. Recent large studies have validated these new techniques for calculating risk, and found them to be beneficial in potential for cost savings and risk classification.17,18
Considerations in psychiatric patients. Statins have been associated with depression in case series, but larger analyses have not confirmed this association.19 Emerging evidence has identified a potential correlation between statin use and accelerated onset of T2DM, but the absolute risk is relatively low and most experts continue to recommend statin therapy despite this potential risk.13 Many statins, including atorvastatin, are available as a generic and can be taken once daily. Some, such as simvastatin, have notable interactions with commonly prescribed psychotropics including risperidone and quetiapine. Pravastatin is dually excreted by the liver and kidneys and may have fewer drug-drug interactions in patients with psychiatric illness taking common psychotropic therapies, but is not considered a high-potency statin and might not confer adequate benefits in CVD risk reduction.
Contraindications. Statins are pregnancy category X, and generally should not be prescribed for women of childbearing age without intensive counseling. The most notable adverse effects for statins include muscle aches and cramps (myalgia), but generally are not severe. If encountered, consider checking a serum creatinine kinase (CK) level, and if significantly elevated above 10 times the upper limit, stopping statin therapy would be advised. If the CK is only mildly elevated, consider lowering the dosage or switching to a lower potency agent. Lovastatin and pravastatin generally are better tolerated than atorvastatin and are considered lower potency (Table 2).
Statins can be safely used in the presence of liver conditions, such as hepatitis C and alcohol use, although periodic monitoring of transaminase levels is recommended. For adults in the general population without liver disease, regular monitoring of transaminase levels is not necessary.
Alternate lipid-lowering pharmacotherapies unfortunately have fallen out of favor. Fibrates, niacin, ezetimibe, and omega-3 fatty acids once were recommended to lower triglycerides or raise HDL cholesterol levels, but since have been shown to have little effect on cardiovascular morbidity or mortality. Adding further medications, other than statins, to lower cholesterol values to pre-defined targets is not the current standard of care.
High triglyceride concentrations traditionally have been addressed directly, but failure to improve CVD mortality or morbidity by treating triglycerides alone has resulted in refocusing clinical efforts in dyslipidemia management on atherogenic cholesterol, including LDL and non-HDL fractions.20 Non-fasting triglycerides >500 mg/dL should be retested when fasting, and levels that remain >500 mg/dL could place the patient at risk for pancreatitis and might warrant intervention with fibrates at that time. This scenario is not common, and referral to a primary care physician or endocrinologist may be warranted.
Lifestyle changes
With or without statin therapies, diet and lifestyle changes are the cornerstone of healthy living and should be encouraged in all patients. Most overweight or obese patients will benefit from exercise and dietary modifications. Such interventions have shown potential for reducing total cholesterol and non-HDL and HDL cholesterol, but rarely are these interventions sustained long enough to produce meaningful reduction in CVD risk through lipid lowering. Diets rich in isocaloric tree nuts and red-yeast rice extract—a form of a statin—have shown promise in reducing cholesterol, but typically take excessive personal resources and are not sustained to the degree necessary to reduce CVD risk over time.21 Similarly, regular exercise routines can help lower overall cholesterol numbers, but rarely reduce total cholesterol by >10%.
Because individuals with SMI smoke at a higher rate than the general population, it should be noted that smoking cessation is associated with a reduction in total cholesterol and a trial of smoking cessation therapy is warranted before initiating a statin medication for primary prevention of CVD. Many patients would discover that their 10-year ASCVD risk would fall under the level needed for statin therapy if they could successfully stop smoking.
Switching pharmacotherapies
Switching antipsychotic agents from highly metabolically risky compounds, such as risperidone and olanzapine, to less metabolically active compounds, such as aripiprazole, ziprasidone, or haloperidol, have been associated with improvements in lipid profiles.22-24 Clinicians must weigh the potential benefits of switching therapies against the risk of psychiatric destabilization and long-term adherence, keeping in mind that changes in lipids seen with switching could be mild (approximately 10% reduction in total cholesterol).
Summing up
Cholesterol management is considered part of a program to systematically lower CVD risk. Statin therapy usually is indicated for life, or until the age of 75, at which point treatment risks and benefits change because of life expectancy. Other components of CVD risk reduction include a focus on blood pressure control, smoking cessation, T2DM management, and weight loss. Tracking lipid profiles over time to ensure broad targets of 30% to 50% reduction in total cholesterol, approximately 3 months after initiation and yearly thereafter, can help ensure adherence to therapy. With systematic lowering of modifiable CVD risk factors, we can hope to gradually improve the quality of life for our patients with mental illnesses (see the Box for a case example illustrating successful use of these strategies).
1. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 suppl B):S1-S45.
2. LaRosa JC, Hunninghake D, Bush D, et al. The cholesterol facts. A summary of the evidence relating dietary fats, serum cholesterol, and coronary heart disease. A joint statement by the American Heart Association and the National Heart, Lung, and Blood Institute. The Task Force on Cholesterol Issues, American Heart Association. Circulation. 1990;81(5):1721-1733.
3. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.e2.
4. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816. doi: 10.1002/14651858.CD004816.pub5.
5. Gaziano JM, Gaziano TA. What’s new with measuring cholesterol? JAMA. 2013;310(19):2043-2044.
6. Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
7. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
8. Crump C, Winkleby MA, Sundquist K, et al. Comorbidities and mortality in persons with schizophrenia: a Swedish national cohort study. Am J Psychiatry. 2013;170(3):324-333.
9. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states [published online March 15, 2006]. Prev Chronic Dis. 2006;3(2):A42.
10. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
11. Osby U, Correia N, Brandt L, et al. Time trends in schizophrenia mortality in Stockholm county, Sweden: cohort study. BMJ. 2000;321(7259):483-484.
12. Mitchell AJ, Lord O. Do deficits in cardiac care influence high mortality rates in schizophrenia? A systematic review and pooled analysis. J Psychopharmacol. 2010;24(suppl 4):69-80.
13. Ganda OP. Deciphering cholesterol treatment guidelines: a clinician’s perspective. JAMA. 2015;313(10):1009-1010.
14. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv. 2014;65(5):573-576.
15. U.S. Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia). http://www.uspreventiveservicestaskforce.org/uspstf/uspschol.htm. Published June 2008. Accessed October 12, 2016.
16. Cupp M. Characteristics of the various statins. Pharmacist’s Letter. 2012;28(6):280606.
17. Pursnani A, Massaro JM, D’Agostino RB Sr, et al. Guideline-based statin eligibility, coronary artery calcification, and cardiovascular events. JAMA. 2015;314(2):134-141.
18. Pandya A, Sy S, Cho S, et al. Cost-effectiveness of 10-year risk thresholds for initiation of statin therapy for primary prevention of cardiovascular disease. JAMA. 2015;314(2):142-150.
19. You H, Lu W, Zhao S, et al. The relationship between statins and depression: a review of the literature. Expert Opin Pharmacother. 2013;14(11):1467-1476.
20. Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-1861.
21. Kelly RB. Diet and exercise in the management of hyperlipidemia. Am Fam Physician. 2010;81(9):1097-1102.
22. Erhardt L. Cigarette smoking: an undertreated risk factor for cardiovascular disease. Atherosclerosis. 2009;205(1):23-32.
23. Weiden PJ. Switching antipsychotics as a treatment strategy for antipsychotic-induced weight gain and dyslipidemia. J Clin Psychiatry. 2007;68(suppl 4):34-39.
24. Stroup TS, McEvoy JP, Ring KD, et al; Schizophrenia Trials Network. A randomized trial examining the effectiveness of switching from olanzapine, quetiapine, or risperidone to aripiprazole to reduce metabolic risk: comparison of antipsychotics for metabolic problems (CAMP). Am J Psychiatry. 2011;168(9):947-956.
1. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 suppl B):S1-S45.
2. LaRosa JC, Hunninghake D, Bush D, et al. The cholesterol facts. A summary of the evidence relating dietary fats, serum cholesterol, and coronary heart disease. A joint statement by the American Heart Association and the National Heart, Lung, and Blood Institute. The Task Force on Cholesterol Issues, American Heart Association. Circulation. 1990;81(5):1721-1733.
3. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.e2.
4. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816. doi: 10.1002/14651858.CD004816.pub5.
5. Gaziano JM, Gaziano TA. What’s new with measuring cholesterol? JAMA. 2013;310(19):2043-2044.
6. Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
7. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
8. Crump C, Winkleby MA, Sundquist K, et al. Comorbidities and mortality in persons with schizophrenia: a Swedish national cohort study. Am J Psychiatry. 2013;170(3):324-333.
9. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states [published online March 15, 2006]. Prev Chronic Dis. 2006;3(2):A42.
10. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
11. Osby U, Correia N, Brandt L, et al. Time trends in schizophrenia mortality in Stockholm county, Sweden: cohort study. BMJ. 2000;321(7259):483-484.
12. Mitchell AJ, Lord O. Do deficits in cardiac care influence high mortality rates in schizophrenia? A systematic review and pooled analysis. J Psychopharmacol. 2010;24(suppl 4):69-80.
13. Ganda OP. Deciphering cholesterol treatment guidelines: a clinician’s perspective. JAMA. 2015;313(10):1009-1010.
14. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv. 2014;65(5):573-576.
15. U.S. Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia). http://www.uspreventiveservicestaskforce.org/uspstf/uspschol.htm. Published June 2008. Accessed October 12, 2016.
16. Cupp M. Characteristics of the various statins. Pharmacist’s Letter. 2012;28(6):280606.
17. Pursnani A, Massaro JM, D’Agostino RB Sr, et al. Guideline-based statin eligibility, coronary artery calcification, and cardiovascular events. JAMA. 2015;314(2):134-141.
18. Pandya A, Sy S, Cho S, et al. Cost-effectiveness of 10-year risk thresholds for initiation of statin therapy for primary prevention of cardiovascular disease. JAMA. 2015;314(2):142-150.
19. You H, Lu W, Zhao S, et al. The relationship between statins and depression: a review of the literature. Expert Opin Pharmacother. 2013;14(11):1467-1476.
20. Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-1861.
21. Kelly RB. Diet and exercise in the management of hyperlipidemia. Am Fam Physician. 2010;81(9):1097-1102.
22. Erhardt L. Cigarette smoking: an undertreated risk factor for cardiovascular disease. Atherosclerosis. 2009;205(1):23-32.
23. Weiden PJ. Switching antipsychotics as a treatment strategy for antipsychotic-induced weight gain and dyslipidemia. J Clin Psychiatry. 2007;68(suppl 4):34-39.
24. Stroup TS, McEvoy JP, Ring KD, et al; Schizophrenia Trials Network. A randomized trial examining the effectiveness of switching from olanzapine, quetiapine, or risperidone to aripiprazole to reduce metabolic risk: comparison of antipsychotics for metabolic problems (CAMP). Am J Psychiatry. 2011;168(9):947-956.
Assess and treat catatonia using this systematic approach
Catatonia is a neuropsychiatric condition with varying presentations that involve behavioral, motoric, cognitive, affective, and, occasionally, autonomic disturbances. Underlying causes of the syndrome include:
- mood disorders
- psychotic disorders
- neurologic disease
- general medical conditions
- metabolic abnormalities
- drug intoxication or withdrawal.

- deep vein thrombosis and pulmonary embolism
- pressure sores or ulcers
- muscle contractures
- nutritional deficiencies and dehydration from decreased oral intake.1
Prompt recognition, assessment, and treatment are vital.
We recommend the following systematic approach to evaluate and treat catatonia (Table).
Assess
Appropriate assessment of catatonia requires recognition of the array of potential underlying causes of the syndrome.
Obtain a complete history, including:
- recent changes in behavior
- past psychiatric illness and hospitalization
- past or current neurologic or medical disease
- prescription and illicit drug use.
Collateral informants, such as family members and caregivers, could provide valuable information. This history could reveal causative factors and identify appropriate targets for treatment.
Physical and mental status examinations can help characterize the type and severity of motoric and behavioral symptoms, such as rigidity, waxy flexibility, negativism, automatic obedience, ambitendency, and perseveration. Monitoring vital signs is crucial because of the risk of medical complications and malignant catatonia, which can be lethal if not treated.
Laboratory testing and imaging might be indicated to rule out medical causes, such as infection, metabolic disturbances, drug intoxication and withdrawal, and acute neurologic etiologies.
Rate
Identify and rate symptom severity. After determining that a patient has catatonia, consider using a standardized instrument, such as the Bush Francis Catatonia Rating Scale (BFCRS),2 to assess the patient’s type of symptoms and degree of impairment. Scores obtained on such instruments can be tracked as the patient receives treatment. Although the BFCRS is imperfect because of ambiguous symptom descriptions and because symptoms can remain after effective treatment, it is the most widely researched catatonia scale.
Treat and monitor
Although there are no published data from large-scale, randomized, controlled trials, clinical experience shows that the mainstays of treatment still are benzodiazepines and electroconvulsive therapy (ECT). A benzodiazepine challenge of IV lorazepam, 2 mg, can lead to rapid, substantial symptomatic relief with relatively low risk of harm. An estimated 50% to 70% of patients with catatonia respond within 5 days to IV lorazepam, 2 mg, every 3 to 8 hours.3
When patients do not respond to benzodiazepines, consider ECT. For patients with medical, neurologic, and toxic metabolic causes of catatonia, treat the underlying disturbance first.
1. Clinebell K, Azzam PN, Gopalan P, et al. Guidelines for preventing common medical complications of catatonia: case report and literature review. J Clin Psychiatry. 2014;75(6):644-651.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Fink M. Catatonia: syndrome or schizophrenia subtype? Recognition and treatment. J Neural Transmission (Vienna). 2001;108(6):637-644.
Catatonia is a neuropsychiatric condition with varying presentations that involve behavioral, motoric, cognitive, affective, and, occasionally, autonomic disturbances. Underlying causes of the syndrome include:
- mood disorders
- psychotic disorders
- neurologic disease
- general medical conditions
- metabolic abnormalities
- drug intoxication or withdrawal.
- deep vein thrombosis and pulmonary embolism
- pressure sores or ulcers
- muscle contractures
- nutritional deficiencies and dehydration from decreased oral intake.1
Prompt recognition, assessment, and treatment are vital.
We recommend the following systematic approach to evaluate and treat catatonia (Table).
Assess
Appropriate assessment of catatonia requires recognition of the array of potential underlying causes of the syndrome.
Obtain a complete history, including:
- recent changes in behavior
- past psychiatric illness and hospitalization
- past or current neurologic or medical disease
- prescription and illicit drug use.
Collateral informants, such as family members and caregivers, could provide valuable information. This history could reveal causative factors and identify appropriate targets for treatment.
Physical and mental status examinations can help characterize the type and severity of motoric and behavioral symptoms, such as rigidity, waxy flexibility, negativism, automatic obedience, ambitendency, and perseveration. Monitoring vital signs is crucial because of the risk of medical complications and malignant catatonia, which can be lethal if not treated.
Laboratory testing and imaging might be indicated to rule out medical causes, such as infection, metabolic disturbances, drug intoxication and withdrawal, and acute neurologic etiologies.
Rate
Identify and rate symptom severity. After determining that a patient has catatonia, consider using a standardized instrument, such as the Bush Francis Catatonia Rating Scale (BFCRS),2 to assess the patient’s type of symptoms and degree of impairment. Scores obtained on such instruments can be tracked as the patient receives treatment. Although the BFCRS is imperfect because of ambiguous symptom descriptions and because symptoms can remain after effective treatment, it is the most widely researched catatonia scale.
Treat and monitor
Although there are no published data from large-scale, randomized, controlled trials, clinical experience shows that the mainstays of treatment still are benzodiazepines and electroconvulsive therapy (ECT). A benzodiazepine challenge of IV lorazepam, 2 mg, can lead to rapid, substantial symptomatic relief with relatively low risk of harm. An estimated 50% to 70% of patients with catatonia respond within 5 days to IV lorazepam, 2 mg, every 3 to 8 hours.3
When patients do not respond to benzodiazepines, consider ECT. For patients with medical, neurologic, and toxic metabolic causes of catatonia, treat the underlying disturbance first.
Catatonia is a neuropsychiatric condition with varying presentations that involve behavioral, motoric, cognitive, affective, and, occasionally, autonomic disturbances. Underlying causes of the syndrome include:
- mood disorders
- psychotic disorders
- neurologic disease
- general medical conditions
- metabolic abnormalities
- drug intoxication or withdrawal.
- deep vein thrombosis and pulmonary embolism
- pressure sores or ulcers
- muscle contractures
- nutritional deficiencies and dehydration from decreased oral intake.1
Prompt recognition, assessment, and treatment are vital.
We recommend the following systematic approach to evaluate and treat catatonia (Table).
Assess
Appropriate assessment of catatonia requires recognition of the array of potential underlying causes of the syndrome.
Obtain a complete history, including:
- recent changes in behavior
- past psychiatric illness and hospitalization
- past or current neurologic or medical disease
- prescription and illicit drug use.
Collateral informants, such as family members and caregivers, could provide valuable information. This history could reveal causative factors and identify appropriate targets for treatment.
Physical and mental status examinations can help characterize the type and severity of motoric and behavioral symptoms, such as rigidity, waxy flexibility, negativism, automatic obedience, ambitendency, and perseveration. Monitoring vital signs is crucial because of the risk of medical complications and malignant catatonia, which can be lethal if not treated.
Laboratory testing and imaging might be indicated to rule out medical causes, such as infection, metabolic disturbances, drug intoxication and withdrawal, and acute neurologic etiologies.
Rate
Identify and rate symptom severity. After determining that a patient has catatonia, consider using a standardized instrument, such as the Bush Francis Catatonia Rating Scale (BFCRS),2 to assess the patient’s type of symptoms and degree of impairment. Scores obtained on such instruments can be tracked as the patient receives treatment. Although the BFCRS is imperfect because of ambiguous symptom descriptions and because symptoms can remain after effective treatment, it is the most widely researched catatonia scale.
Treat and monitor
Although there are no published data from large-scale, randomized, controlled trials, clinical experience shows that the mainstays of treatment still are benzodiazepines and electroconvulsive therapy (ECT). A benzodiazepine challenge of IV lorazepam, 2 mg, can lead to rapid, substantial symptomatic relief with relatively low risk of harm. An estimated 50% to 70% of patients with catatonia respond within 5 days to IV lorazepam, 2 mg, every 3 to 8 hours.3
When patients do not respond to benzodiazepines, consider ECT. For patients with medical, neurologic, and toxic metabolic causes of catatonia, treat the underlying disturbance first.
1. Clinebell K, Azzam PN, Gopalan P, et al. Guidelines for preventing common medical complications of catatonia: case report and literature review. J Clin Psychiatry. 2014;75(6):644-651.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Fink M. Catatonia: syndrome or schizophrenia subtype? Recognition and treatment. J Neural Transmission (Vienna). 2001;108(6):637-644.
1. Clinebell K, Azzam PN, Gopalan P, et al. Guidelines for preventing common medical complications of catatonia: case report and literature review. J Clin Psychiatry. 2014;75(6):644-651.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Fink M. Catatonia: syndrome or schizophrenia subtype? Recognition and treatment. J Neural Transmission (Vienna). 2001;108(6):637-644.
Consider Rx metformin to prevent metabolic syndrome
Many atypical antipsychotics, particularly clozapine and olanzapine, are associated with weight gain, insulin resistance, and metabolic syndrome. Metabolic syndrome is associated with type 2 diabetes mellitus (T2DM) and cardiovascular disease, which are among the leading causes of morbidity and mortality in persons with severe mental illness.1
Clinicians should take measures to prevent T2DM and weight gain in individuals taking antipsychotics before these conditions develop. Metformin re-sensitizes the body to insulin and is a first-line treatment for T2DM. Adding metformin when patients start metabolically high-risk antipsychotics or shortly after they begin gaining weight is an evidence-based strategy to prevent metabolic syndrome.
Evaluate the evidence
In randomized controlled trials, metformin was associated with modest weight loss and improvement in metabolic parameters (eg, fasting blood glucose, serum triglycerides, and total cholesterol) in patients with schizophrenia receiving antipsychotics.1,2 Metformin is effective for preventing metabolic syndrome and as a treatment intervention; therefore, it may prove most beneficial early in treatment before weight gain or insulin resistance develop.
Importantly, weight gain and metabolic syndrome are risk factors for cardiovascular disease, but the number needed to treat for metformin to prevent cardiovascular outcomes, such as myocardial infarction, is not known. Also, metformin is not FDA-approved for this indication. Clinicians should discuss with the patient the risks and benefits of prophylactic metformin, and consider his (her) treatment preferences.
Tolerability and adverse effects
Metformin generally is well-tolerated. Gastrointestinal (GI) symptoms, including nausea and vomiting (14%) and diarrhea (7%), are the most common adverse effects.2 Lactic acidosis is rare and is associated with alcohol use disorders and impaired renal, hepatic, or cardiopulmonary function.3 Because metformin is excreted renally, toxicity could occur in patients with impaired renal function.
Before initiating prophylactic metformin, confirm that the patient does not have T2DM (eg, hemoglobin A1c <6.5%). A thorough medical history, including alcohol use and kidney and liver function tests, are needed to reduce the risk of lactic acidosis.3
Dosing
Although metformin has been studied at many dosages,2 we recommend gradual titration to 1,000 mg, twice daily, taken with meals to reduce the risk of GI effects.
Additional interventions
Metformin alone is not sufficient to mitigate metabolic risk. Providers should address dietary interventions, exercise, and smoking cessation at each visit, and communicate actively with other providers to create a comprehensive treatment plan.
1. Jarskog LF, Hamer RF, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight patients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
2. Zheng W, Li X-B, Tang Y-L, et al. Metformin for weight gain and metabolic abnormalities associated with antipsychotic treatment: meta-analysis of randomized placebo-controlled trials. J Clin Psychopharmacol. 2015;35(5):499-509.
3. Wang M, Tong J-H, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
Many atypical antipsychotics, particularly clozapine and olanzapine, are associated with weight gain, insulin resistance, and metabolic syndrome. Metabolic syndrome is associated with type 2 diabetes mellitus (T2DM) and cardiovascular disease, which are among the leading causes of morbidity and mortality in persons with severe mental illness.1
Clinicians should take measures to prevent T2DM and weight gain in individuals taking antipsychotics before these conditions develop. Metformin re-sensitizes the body to insulin and is a first-line treatment for T2DM. Adding metformin when patients start metabolically high-risk antipsychotics or shortly after they begin gaining weight is an evidence-based strategy to prevent metabolic syndrome.
Evaluate the evidence
In randomized controlled trials, metformin was associated with modest weight loss and improvement in metabolic parameters (eg, fasting blood glucose, serum triglycerides, and total cholesterol) in patients with schizophrenia receiving antipsychotics.1,2 Metformin is effective for preventing metabolic syndrome and as a treatment intervention; therefore, it may prove most beneficial early in treatment before weight gain or insulin resistance develop.
Importantly, weight gain and metabolic syndrome are risk factors for cardiovascular disease, but the number needed to treat for metformin to prevent cardiovascular outcomes, such as myocardial infarction, is not known. Also, metformin is not FDA-approved for this indication. Clinicians should discuss with the patient the risks and benefits of prophylactic metformin, and consider his (her) treatment preferences.
Tolerability and adverse effects
Metformin generally is well-tolerated. Gastrointestinal (GI) symptoms, including nausea and vomiting (14%) and diarrhea (7%), are the most common adverse effects.2 Lactic acidosis is rare and is associated with alcohol use disorders and impaired renal, hepatic, or cardiopulmonary function.3 Because metformin is excreted renally, toxicity could occur in patients with impaired renal function.
Before initiating prophylactic metformin, confirm that the patient does not have T2DM (eg, hemoglobin A1c <6.5%). A thorough medical history, including alcohol use and kidney and liver function tests, are needed to reduce the risk of lactic acidosis.3
Dosing
Although metformin has been studied at many dosages,2 we recommend gradual titration to 1,000 mg, twice daily, taken with meals to reduce the risk of GI effects.
Additional interventions
Metformin alone is not sufficient to mitigate metabolic risk. Providers should address dietary interventions, exercise, and smoking cessation at each visit, and communicate actively with other providers to create a comprehensive treatment plan.
Many atypical antipsychotics, particularly clozapine and olanzapine, are associated with weight gain, insulin resistance, and metabolic syndrome. Metabolic syndrome is associated with type 2 diabetes mellitus (T2DM) and cardiovascular disease, which are among the leading causes of morbidity and mortality in persons with severe mental illness.1
Clinicians should take measures to prevent T2DM and weight gain in individuals taking antipsychotics before these conditions develop. Metformin re-sensitizes the body to insulin and is a first-line treatment for T2DM. Adding metformin when patients start metabolically high-risk antipsychotics or shortly after they begin gaining weight is an evidence-based strategy to prevent metabolic syndrome.
Evaluate the evidence
In randomized controlled trials, metformin was associated with modest weight loss and improvement in metabolic parameters (eg, fasting blood glucose, serum triglycerides, and total cholesterol) in patients with schizophrenia receiving antipsychotics.1,2 Metformin is effective for preventing metabolic syndrome and as a treatment intervention; therefore, it may prove most beneficial early in treatment before weight gain or insulin resistance develop.
Importantly, weight gain and metabolic syndrome are risk factors for cardiovascular disease, but the number needed to treat for metformin to prevent cardiovascular outcomes, such as myocardial infarction, is not known. Also, metformin is not FDA-approved for this indication. Clinicians should discuss with the patient the risks and benefits of prophylactic metformin, and consider his (her) treatment preferences.
Tolerability and adverse effects
Metformin generally is well-tolerated. Gastrointestinal (GI) symptoms, including nausea and vomiting (14%) and diarrhea (7%), are the most common adverse effects.2 Lactic acidosis is rare and is associated with alcohol use disorders and impaired renal, hepatic, or cardiopulmonary function.3 Because metformin is excreted renally, toxicity could occur in patients with impaired renal function.
Before initiating prophylactic metformin, confirm that the patient does not have T2DM (eg, hemoglobin A1c <6.5%). A thorough medical history, including alcohol use and kidney and liver function tests, are needed to reduce the risk of lactic acidosis.3
Dosing
Although metformin has been studied at many dosages,2 we recommend gradual titration to 1,000 mg, twice daily, taken with meals to reduce the risk of GI effects.
Additional interventions
Metformin alone is not sufficient to mitigate metabolic risk. Providers should address dietary interventions, exercise, and smoking cessation at each visit, and communicate actively with other providers to create a comprehensive treatment plan.
1. Jarskog LF, Hamer RF, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight patients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
2. Zheng W, Li X-B, Tang Y-L, et al. Metformin for weight gain and metabolic abnormalities associated with antipsychotic treatment: meta-analysis of randomized placebo-controlled trials. J Clin Psychopharmacol. 2015;35(5):499-509.
3. Wang M, Tong J-H, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
1. Jarskog LF, Hamer RF, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight patients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
2. Zheng W, Li X-B, Tang Y-L, et al. Metformin for weight gain and metabolic abnormalities associated with antipsychotic treatment: meta-analysis of randomized placebo-controlled trials. J Clin Psychopharmacol. 2015;35(5):499-509.
3. Wang M, Tong J-H, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
High-value intervention: Providing colorectal cancer screening
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
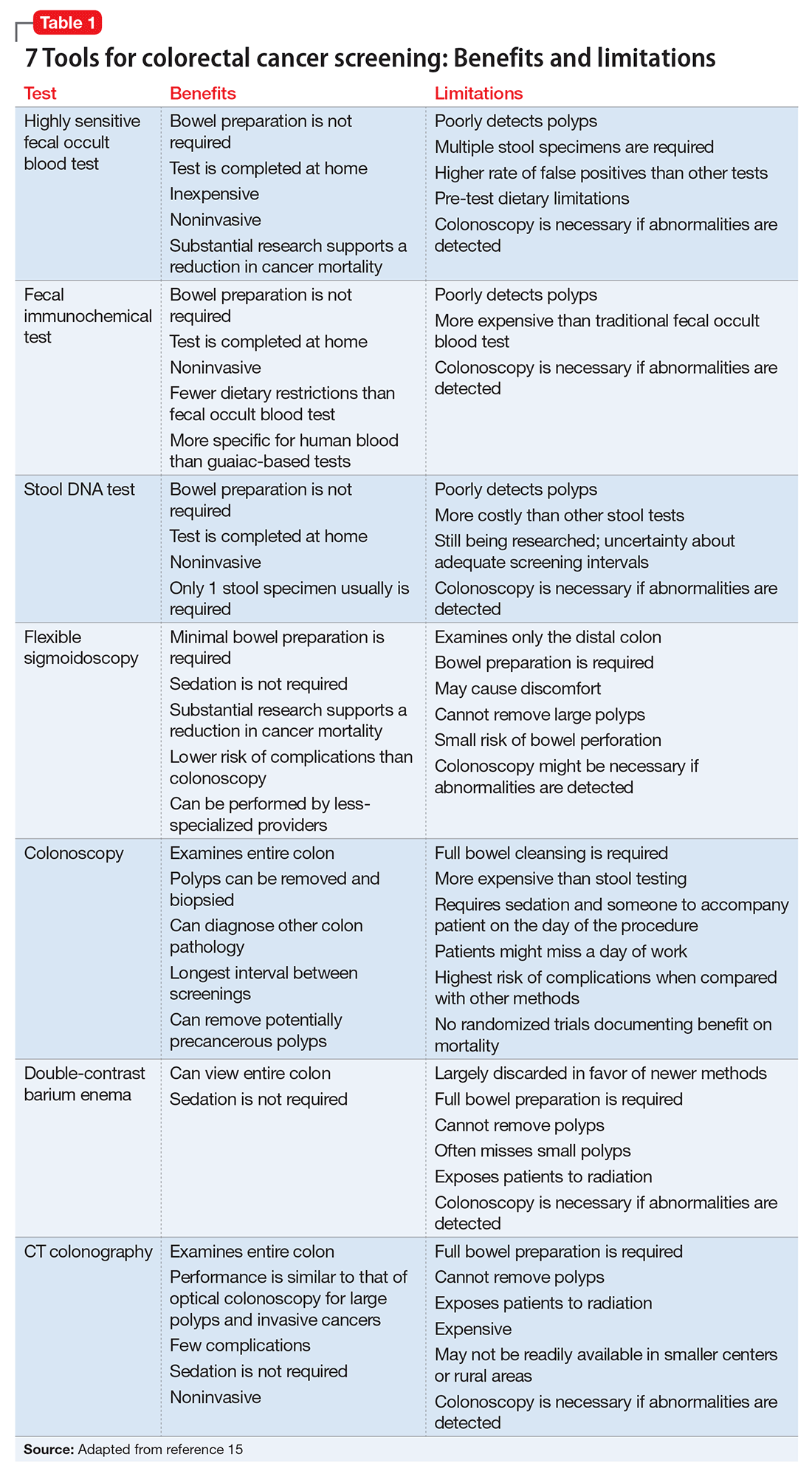
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major 
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.
Take steps to relieve ataxia in patients with alcohol use disorder
Ataxia is a well-known complication of chronic alcohol abuse, which is attributed to degeneration of the cerebellar vermis. However, effective treatment approaches, as well as the timing and level of recovery, remain unclear. One cross-sectional study found that long-term abstainers from alcohol had less severe ataxia than short-term abstainers,1 suggesting that improvement is possible with continued sobriety. However, a recent longitudinal study contradicts this finding, reporting no improvement in ataxia in patients abstinent for 10 weeks to 1 year.2
CASE REPORT
Unable to walk, heavy alcohol use
Mr. G, a 59-year-old white male with a history of daily, heavy alcohol use, presents to the emergency room reporting that he has “not been able to walk right” for 3 weeks. He is in a wheelchair because of ataxia and difficulty balancing. He denies headaches, visual changes, weakness, numbness, and difficulty speaking or swallowing.
Mr. G reports drinking one 40-oz bottle of malt liquor and 2 pints of vodka per day for more than 40 years. His alcohol abuse led to homelessness, unemployment, and divorce. Despite heavy drinking, he denies signs of withdrawal, including shaking, sweating, seizures, and delirium.
Mr. G has no other medical conditions. He denies a family history of neurologic disorders or substance abuse.
His pulse is 100 beats per minute, respirations of 16 breaths per minute, temperature of 37°C, and blood pressure of 143/89 mm Hg. Physical examination reveals a wide-based gait.
Mr. G is admitted to the inpatient psychiatric unit to monitor and treat his alcohol withdrawal and to undergo further workup of the gait disturbance.
A head CT scan shows non-specific changes; an EEG also is within normal limits. Complete blood count, basic metabolic panel, liver function test, HIV test, acute hepatitis panel, thyroid function test, erythrocyte sedimentation rate, and vitamin B12 tests are within normal ranges.
A full neurologic exam reveals a wide-based gait, impaired heel-shin test, and dysmetria on finger-nose-finger test. Mr. G is given a diagnosis of ataxia due to alcoholic cerebellar degeneration. Thiamine repletion is suggested.
Treatment and outcome
Mr. G continues on thiamine, 100 mg, twice daily, and oxazepam, 15 mg, as needed, to manage withdrawal symptoms. He receives gait training 3 times per week.
Approximately 10 days after admission, Mr. G is able to ambulate with a walker. Three weeks after admission, his gait has improved and he walks with a cane. (See the video at CurrentPsychiatry.com for an illustration of this progressive recovery.)
After discharge, Mr. G is referred to an addiction psychiatrist and addiction psychotherapist for ongoing treatment of alcohol use disorder.
Making the diagnosis
In a patient complaining of balance difficulties, consider ataxia secondary to cerebellar degeneration.
- Take a complete history. Ask about the onset and progression of ataxia.
- Obtain a family history. Some types of ataxia are genetic.
- Perform a neurologic examination, which may reveal signs of cerebellar deficits, particularly characteristic wide-based gait. These patients will have difficulty when walking in tandem. Other impairments on the neurologic exam that may raise suspicion for a cerebellar disorder include: impaired heel-shin test, impaired finger-nose-finger test (dysmetria), impaired rapid alternating movements (dysdiadochokinesia), nystagmus, impaired smooth pursuits, intention tremor, or speech abnormalities.
- Perform head imaging, such as a CT scan or MRI. In patients with ataxia secondary to alcohol abuse, imaging might reveal degeneration of the cerebellar vermis.
- Perform laboratory tests, such as inflammatory markers, vitamin levels, and thyroid function testing to detect possible toxic-metabolic or inflammatory causes.
Alcohol-induced ataxia can be diagnosed in patients with a history of heavy drinking if the workup does not reveal another possible cause for the gait disturbance. Other less common deficits associated with alcohol-induced cerebellar injury include:
- dysarthria
- abnormal rate and force of movement
- limb ataxia.3
Recommendations
- Be able to recognize the characteristic gait of patients with alcohol-induced ataxia.
- Provide thiamine supplementation.
- Refer patients to physical therapy.
- Educate your patients that their gait will not improve and may worsen if they continue to drink.
- Refer patients for ongoing treatment for alcohol use disorder, including medication management and psychotherapy.
Our experience suggests that patients with alcohol use disorder with cerebellar ataxia could have a good prognosis for ambulation. Improvement could occur over several weeks; it is unclear whether further gains can be expected with months or years of abstinence.
1. Smith S, Fein G. Persistent but less severe ataxia in long-term versus short-term abstinent alcoholic men and women: a cross-sectional analysis. Alcohol Clin Exp Res. 2011;35(12):2184-2192.
2. Fein G, Greenstein D. Gait and balance deficits in chronic alcoholics: no improvement from 10 weeks through 1 year abstinence. Alcohol Clin Exp Res. 2013;37(1):86-95.
3. Fitzpatrick LE, Jackson M, Crowe SF. Characterization of cerebellar ataxia in chronic alcoholics using the International Cooperative Ataxia Rating Scale (ICARS). Alcohol Clin Exp Res. 2012;36(11):1942-1951.
Ataxia is a well-known complication of chronic alcohol abuse, which is attributed to degeneration of the cerebellar vermis. However, effective treatment approaches, as well as the timing and level of recovery, remain unclear. One cross-sectional study found that long-term abstainers from alcohol had less severe ataxia than short-term abstainers,1 suggesting that improvement is possible with continued sobriety. However, a recent longitudinal study contradicts this finding, reporting no improvement in ataxia in patients abstinent for 10 weeks to 1 year.2
CASE REPORT
Unable to walk, heavy alcohol use
Mr. G, a 59-year-old white male with a history of daily, heavy alcohol use, presents to the emergency room reporting that he has “not been able to walk right” for 3 weeks. He is in a wheelchair because of ataxia and difficulty balancing. He denies headaches, visual changes, weakness, numbness, and difficulty speaking or swallowing.
Mr. G reports drinking one 40-oz bottle of malt liquor and 2 pints of vodka per day for more than 40 years. His alcohol abuse led to homelessness, unemployment, and divorce. Despite heavy drinking, he denies signs of withdrawal, including shaking, sweating, seizures, and delirium.
Mr. G has no other medical conditions. He denies a family history of neurologic disorders or substance abuse.
His pulse is 100 beats per minute, respirations of 16 breaths per minute, temperature of 37°C, and blood pressure of 143/89 mm Hg. Physical examination reveals a wide-based gait.
Mr. G is admitted to the inpatient psychiatric unit to monitor and treat his alcohol withdrawal and to undergo further workup of the gait disturbance.
A head CT scan shows non-specific changes; an EEG also is within normal limits. Complete blood count, basic metabolic panel, liver function test, HIV test, acute hepatitis panel, thyroid function test, erythrocyte sedimentation rate, and vitamin B12 tests are within normal ranges.
A full neurologic exam reveals a wide-based gait, impaired heel-shin test, and dysmetria on finger-nose-finger test. Mr. G is given a diagnosis of ataxia due to alcoholic cerebellar degeneration. Thiamine repletion is suggested.
Treatment and outcome
Mr. G continues on thiamine, 100 mg, twice daily, and oxazepam, 15 mg, as needed, to manage withdrawal symptoms. He receives gait training 3 times per week.
Approximately 10 days after admission, Mr. G is able to ambulate with a walker. Three weeks after admission, his gait has improved and he walks with a cane. (See the video at CurrentPsychiatry.com for an illustration of this progressive recovery.)
After discharge, Mr. G is referred to an addiction psychiatrist and addiction psychotherapist for ongoing treatment of alcohol use disorder.
Making the diagnosis
In a patient complaining of balance difficulties, consider ataxia secondary to cerebellar degeneration.
- Take a complete history. Ask about the onset and progression of ataxia.
- Obtain a family history. Some types of ataxia are genetic.
- Perform a neurologic examination, which may reveal signs of cerebellar deficits, particularly characteristic wide-based gait. These patients will have difficulty when walking in tandem. Other impairments on the neurologic exam that may raise suspicion for a cerebellar disorder include: impaired heel-shin test, impaired finger-nose-finger test (dysmetria), impaired rapid alternating movements (dysdiadochokinesia), nystagmus, impaired smooth pursuits, intention tremor, or speech abnormalities.
- Perform head imaging, such as a CT scan or MRI. In patients with ataxia secondary to alcohol abuse, imaging might reveal degeneration of the cerebellar vermis.
- Perform laboratory tests, such as inflammatory markers, vitamin levels, and thyroid function testing to detect possible toxic-metabolic or inflammatory causes.
Alcohol-induced ataxia can be diagnosed in patients with a history of heavy drinking if the workup does not reveal another possible cause for the gait disturbance. Other less common deficits associated with alcohol-induced cerebellar injury include:
- dysarthria
- abnormal rate and force of movement
- limb ataxia.3
Recommendations
- Be able to recognize the characteristic gait of patients with alcohol-induced ataxia.
- Provide thiamine supplementation.
- Refer patients to physical therapy.
- Educate your patients that their gait will not improve and may worsen if they continue to drink.
- Refer patients for ongoing treatment for alcohol use disorder, including medication management and psychotherapy.
Our experience suggests that patients with alcohol use disorder with cerebellar ataxia could have a good prognosis for ambulation. Improvement could occur over several weeks; it is unclear whether further gains can be expected with months or years of abstinence.
Ataxia is a well-known complication of chronic alcohol abuse, which is attributed to degeneration of the cerebellar vermis. However, effective treatment approaches, as well as the timing and level of recovery, remain unclear. One cross-sectional study found that long-term abstainers from alcohol had less severe ataxia than short-term abstainers,1 suggesting that improvement is possible with continued sobriety. However, a recent longitudinal study contradicts this finding, reporting no improvement in ataxia in patients abstinent for 10 weeks to 1 year.2
CASE REPORT
Unable to walk, heavy alcohol use
Mr. G, a 59-year-old white male with a history of daily, heavy alcohol use, presents to the emergency room reporting that he has “not been able to walk right” for 3 weeks. He is in a wheelchair because of ataxia and difficulty balancing. He denies headaches, visual changes, weakness, numbness, and difficulty speaking or swallowing.
Mr. G reports drinking one 40-oz bottle of malt liquor and 2 pints of vodka per day for more than 40 years. His alcohol abuse led to homelessness, unemployment, and divorce. Despite heavy drinking, he denies signs of withdrawal, including shaking, sweating, seizures, and delirium.
Mr. G has no other medical conditions. He denies a family history of neurologic disorders or substance abuse.
His pulse is 100 beats per minute, respirations of 16 breaths per minute, temperature of 37°C, and blood pressure of 143/89 mm Hg. Physical examination reveals a wide-based gait.
Mr. G is admitted to the inpatient psychiatric unit to monitor and treat his alcohol withdrawal and to undergo further workup of the gait disturbance.
A head CT scan shows non-specific changes; an EEG also is within normal limits. Complete blood count, basic metabolic panel, liver function test, HIV test, acute hepatitis panel, thyroid function test, erythrocyte sedimentation rate, and vitamin B12 tests are within normal ranges.
A full neurologic exam reveals a wide-based gait, impaired heel-shin test, and dysmetria on finger-nose-finger test. Mr. G is given a diagnosis of ataxia due to alcoholic cerebellar degeneration. Thiamine repletion is suggested.
Treatment and outcome
Mr. G continues on thiamine, 100 mg, twice daily, and oxazepam, 15 mg, as needed, to manage withdrawal symptoms. He receives gait training 3 times per week.
Approximately 10 days after admission, Mr. G is able to ambulate with a walker. Three weeks after admission, his gait has improved and he walks with a cane. (See the video at CurrentPsychiatry.com for an illustration of this progressive recovery.)
After discharge, Mr. G is referred to an addiction psychiatrist and addiction psychotherapist for ongoing treatment of alcohol use disorder.
Making the diagnosis
In a patient complaining of balance difficulties, consider ataxia secondary to cerebellar degeneration.
- Take a complete history. Ask about the onset and progression of ataxia.
- Obtain a family history. Some types of ataxia are genetic.
- Perform a neurologic examination, which may reveal signs of cerebellar deficits, particularly characteristic wide-based gait. These patients will have difficulty when walking in tandem. Other impairments on the neurologic exam that may raise suspicion for a cerebellar disorder include: impaired heel-shin test, impaired finger-nose-finger test (dysmetria), impaired rapid alternating movements (dysdiadochokinesia), nystagmus, impaired smooth pursuits, intention tremor, or speech abnormalities.
- Perform head imaging, such as a CT scan or MRI. In patients with ataxia secondary to alcohol abuse, imaging might reveal degeneration of the cerebellar vermis.
- Perform laboratory tests, such as inflammatory markers, vitamin levels, and thyroid function testing to detect possible toxic-metabolic or inflammatory causes.
Alcohol-induced ataxia can be diagnosed in patients with a history of heavy drinking if the workup does not reveal another possible cause for the gait disturbance. Other less common deficits associated with alcohol-induced cerebellar injury include:
- dysarthria
- abnormal rate and force of movement
- limb ataxia.3
Recommendations
- Be able to recognize the characteristic gait of patients with alcohol-induced ataxia.
- Provide thiamine supplementation.
- Refer patients to physical therapy.
- Educate your patients that their gait will not improve and may worsen if they continue to drink.
- Refer patients for ongoing treatment for alcohol use disorder, including medication management and psychotherapy.
Our experience suggests that patients with alcohol use disorder with cerebellar ataxia could have a good prognosis for ambulation. Improvement could occur over several weeks; it is unclear whether further gains can be expected with months or years of abstinence.
1. Smith S, Fein G. Persistent but less severe ataxia in long-term versus short-term abstinent alcoholic men and women: a cross-sectional analysis. Alcohol Clin Exp Res. 2011;35(12):2184-2192.
2. Fein G, Greenstein D. Gait and balance deficits in chronic alcoholics: no improvement from 10 weeks through 1 year abstinence. Alcohol Clin Exp Res. 2013;37(1):86-95.
3. Fitzpatrick LE, Jackson M, Crowe SF. Characterization of cerebellar ataxia in chronic alcoholics using the International Cooperative Ataxia Rating Scale (ICARS). Alcohol Clin Exp Res. 2012;36(11):1942-1951.
1. Smith S, Fein G. Persistent but less severe ataxia in long-term versus short-term abstinent alcoholic men and women: a cross-sectional analysis. Alcohol Clin Exp Res. 2011;35(12):2184-2192.
2. Fein G, Greenstein D. Gait and balance deficits in chronic alcoholics: no improvement from 10 weeks through 1 year abstinence. Alcohol Clin Exp Res. 2013;37(1):86-95.
3. Fitzpatrick LE, Jackson M, Crowe SF. Characterization of cerebellar ataxia in chronic alcoholics using the International Cooperative Ataxia Rating Scale (ICARS). Alcohol Clin Exp Res. 2012;36(11):1942-1951.
Ataxia due to alcohol abuse
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Pseudobulbar affect: When patients laugh or cry, but don’t know why
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3

Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).

Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14


2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.

Pregnant nearly a year? The patient has symptoms but evidence is lacking
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).

Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).
Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).
Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
Pimavanserin for psychosis in patients with Parkinson’s disease
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.

As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.