User login
How do clinical prediction rules compare with joint fluid analysis in diagnosing gout?
EVIDENCE-BASED ANSWER:
Clinical prediction rules effectively diagnose gout without joint fluid analysis. The American College of Rheumatology clinical prediction rules, the most accurate rules developed for research purposes, have a sensitivity of 92%, specificity of 89%, positive likelihood ratio of 8.36, and negative likelihood ratio of 0.09 (strength of recommendation [SOR]: A, prospective cohort studies).
The Netherlands criteria, developed for use in primary care, have a positive predictive value of more than 80%, a positive likelihood ratio of 3.48, and a negative likelihood ratio of 0.17 (SOR: A, prospective cohort study).
EVIDENCE SUMMARY
In 2015, the American College of Rheumatology (ACR) redefined the clinical criteria for diagnosis of gout based on a 3-step system1 that can be found at: http://goutclassificationcalculator.auckland.ac.nz. The ACR rule was derived from a cross-sectional study of 983 patients in 25 rheumatology centers in 16 countries who presented with a swollen joint.2 Of the 983 patients, 509 had gout; the prevalence was 52%. Data from 653 of these patients were used to develop the rule and then validated in the remaining 330 patients.
Compared with the gold standard of monosodium urate crystals in synovial fluid, the ACR rule has a sensitivity of 92% and a specificity of 89%. The rule, designed for the research setting, involves using synovial fluid analysis, ultrasound imaging, and radiography, which makes it less useful in a primary care setting.
The Netherlands rule for primary care
A prospective diagnostic study in 328 family medicine patients (74% male; mean age 57) with monoarthritis tested the ability of multiple clinical variables to diagnose gout using monosodium urate crystals in synovial fluid as the gold standard.3 The prevalence of gout in this population was 57%.
The best diagnostic rule (Netherlands rule) comprised the following predefined variables: male sex, previous patient-reported arthritis attack, onset within one day, joint redness, first metatarsophalangeal joint (MTP1) involvement, hypertension or cardiovascular disease (angina pectoris, myocardial infarction, heart failure, cerebrovascular accident, transient ischemic attack, or peripheral vascular disease), and serum uric acid level above 5.88 mg/dL. The rule gives one point for each item. A score >8 had a positive likelihood ratio for diagnosing gout of 3.48 (TABLE1) and a higher positive predictive value (PPV) than family physicians’ clinical impressions (83% vs 64%).
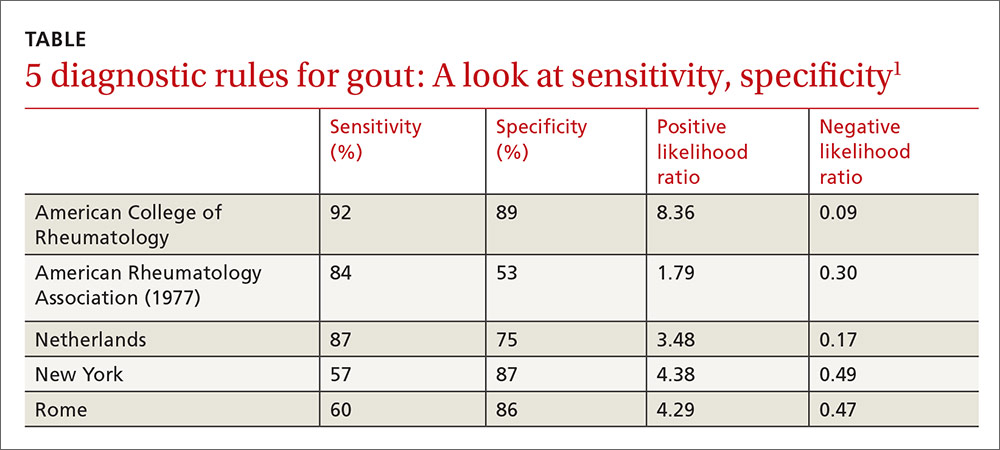
The prevalence of gout in patients with scores of <4, 4 to 8, and >8 were 2.8%, 27%, and 80%, respectively. For scores of 4 to 8, the probability of gout is indeterminate, and synovial fluid analysis is recommended.
The Netherlands rule, validated in a secondary care practice of 390 patients with monoarthritis, found that a score >8 had a PPV of 87% and a score <4 had a negative predictive value of 95%.4 The probability of gout based on this rule can be calculated at http://www.umcn.nl/goutcalc.
In the study used to develop the Netherlands rule, no patients with a high probability of gout had septic arthritis. The ability of the rule to differentiate between gout and septic arthritis was tested retrospectively in 33 patients with acute gout (podagra excluded) diagnosed by the presence of monosodium urate joint crystals and 27 patients with septic arthritis diagnosed by positive bacterial culture.5 Patients with gout had significantly higher scores than patients with septic arthritis (7.8 ± 1.59 vs 3.4 ± 2.3; P<.001).
American Rheumatology Association, New York, and Rome prediction rules
A study of 82 Veterans Administration patients compared the American Rheumatology Association (ARA), New York, and Rome prediction rules with regard to their ability to diagnose gout with synovial urate crystals.6 The ARA criteria for gout diagnosis require either tophi or monosodium urate crystals in synovial fluid, or 6 out of a list of 12 other criteria.7
The New York prediction rule requires that patients meet 2 or more of the following criteria: at least 2 attacks of painful joint swelling with complete resolution within 2 weeks, podagra, tophi, and rapid response to colchicine treatment, defined as a major reduction in the objective signs of inflammation within 48 hours.
The Rome prediction rule requires meeting 2 of 3 criteria: serum uric acid >7 mg/dL in men and >6 mg/dL in women, presence of tophi, and history of attacks of painful joint swelling with abrupt onset and resolution within 2 weeks.
The New York prediction rule had the highest positive likelihood ratio of 4.4 compared with the ARA (1.8) and Rome (4.3) rules.6 The utility of the New York and Rome rules, although they have fewer criteria than ARA, is limited by the fact that they include a previous episode of joint swelling and tophi. These criteria increase their specificity but make them less useful in diagnosing a first episode of gout, when tophi are unlikely to have developed.
Prediction rules are more sensitive in established gout
The new ACR prediction rule was compared with the ARA, Rome, and New York clinical prediction rules using urate crystals as the gold standard in early (less than 2 years) and established disease (longer than 2 years).8 All clinical prediction rules were more sensitive in established disease than early disease (95.3% vs 84.1%; P<.001) and more specific in early disease than established disease (79.9% vs 52.5%; P<.001).
1. Neogi T, Jansen TL, Dalbeth N, et al. 2015 Gout Classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2015;74:1789-1798.
2. Taylor WJ, Fransen J, Jansen TL, et al. Study for Updated Gout Classification Criteria (SUGAR): identification of features to classify gout. Arthritis Care Res (Hoboken). 2015;67:1304-1315.
3. Janssens HJ, Fransen J, van de Lisdonk EH, et al. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170:1120-1126.
4. Kienhorst LB, Janssens HJ, Fransen J, et al. The validation of a diagnostic rule for gout without joint fluid analysis: a prospective study. Rheumatology (Oxford). 2015;54:609-614.
5. Lee K, Choi ST, Kang EJ, et al. SAT0377 The performance of a novel scoring system in the differential diagnosis between acute gout and septic arthritis. Ann Rheum Dis. 2013;72:A711.
6. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Taylor WJ, Fransen J, Dalbeth N, et al. Performance of classification criteria for gout in early and established disease. Ann Rheum Dis. 2016;75:178-182.
EVIDENCE-BASED ANSWER:
Clinical prediction rules effectively diagnose gout without joint fluid analysis. The American College of Rheumatology clinical prediction rules, the most accurate rules developed for research purposes, have a sensitivity of 92%, specificity of 89%, positive likelihood ratio of 8.36, and negative likelihood ratio of 0.09 (strength of recommendation [SOR]: A, prospective cohort studies).
The Netherlands criteria, developed for use in primary care, have a positive predictive value of more than 80%, a positive likelihood ratio of 3.48, and a negative likelihood ratio of 0.17 (SOR: A, prospective cohort study).
EVIDENCE SUMMARY
In 2015, the American College of Rheumatology (ACR) redefined the clinical criteria for diagnosis of gout based on a 3-step system1 that can be found at: http://goutclassificationcalculator.auckland.ac.nz. The ACR rule was derived from a cross-sectional study of 983 patients in 25 rheumatology centers in 16 countries who presented with a swollen joint.2 Of the 983 patients, 509 had gout; the prevalence was 52%. Data from 653 of these patients were used to develop the rule and then validated in the remaining 330 patients.
Compared with the gold standard of monosodium urate crystals in synovial fluid, the ACR rule has a sensitivity of 92% and a specificity of 89%. The rule, designed for the research setting, involves using synovial fluid analysis, ultrasound imaging, and radiography, which makes it less useful in a primary care setting.
The Netherlands rule for primary care
A prospective diagnostic study in 328 family medicine patients (74% male; mean age 57) with monoarthritis tested the ability of multiple clinical variables to diagnose gout using monosodium urate crystals in synovial fluid as the gold standard.3 The prevalence of gout in this population was 57%.
The best diagnostic rule (Netherlands rule) comprised the following predefined variables: male sex, previous patient-reported arthritis attack, onset within one day, joint redness, first metatarsophalangeal joint (MTP1) involvement, hypertension or cardiovascular disease (angina pectoris, myocardial infarction, heart failure, cerebrovascular accident, transient ischemic attack, or peripheral vascular disease), and serum uric acid level above 5.88 mg/dL. The rule gives one point for each item. A score >8 had a positive likelihood ratio for diagnosing gout of 3.48 (TABLE1) and a higher positive predictive value (PPV) than family physicians’ clinical impressions (83% vs 64%).
The prevalence of gout in patients with scores of <4, 4 to 8, and >8 were 2.8%, 27%, and 80%, respectively. For scores of 4 to 8, the probability of gout is indeterminate, and synovial fluid analysis is recommended.
The Netherlands rule, validated in a secondary care practice of 390 patients with monoarthritis, found that a score >8 had a PPV of 87% and a score <4 had a negative predictive value of 95%.4 The probability of gout based on this rule can be calculated at http://www.umcn.nl/goutcalc.
In the study used to develop the Netherlands rule, no patients with a high probability of gout had septic arthritis. The ability of the rule to differentiate between gout and septic arthritis was tested retrospectively in 33 patients with acute gout (podagra excluded) diagnosed by the presence of monosodium urate joint crystals and 27 patients with septic arthritis diagnosed by positive bacterial culture.5 Patients with gout had significantly higher scores than patients with septic arthritis (7.8 ± 1.59 vs 3.4 ± 2.3; P<.001).
American Rheumatology Association, New York, and Rome prediction rules
A study of 82 Veterans Administration patients compared the American Rheumatology Association (ARA), New York, and Rome prediction rules with regard to their ability to diagnose gout with synovial urate crystals.6 The ARA criteria for gout diagnosis require either tophi or monosodium urate crystals in synovial fluid, or 6 out of a list of 12 other criteria.7
The New York prediction rule requires that patients meet 2 or more of the following criteria: at least 2 attacks of painful joint swelling with complete resolution within 2 weeks, podagra, tophi, and rapid response to colchicine treatment, defined as a major reduction in the objective signs of inflammation within 48 hours.
The Rome prediction rule requires meeting 2 of 3 criteria: serum uric acid >7 mg/dL in men and >6 mg/dL in women, presence of tophi, and history of attacks of painful joint swelling with abrupt onset and resolution within 2 weeks.
The New York prediction rule had the highest positive likelihood ratio of 4.4 compared with the ARA (1.8) and Rome (4.3) rules.6 The utility of the New York and Rome rules, although they have fewer criteria than ARA, is limited by the fact that they include a previous episode of joint swelling and tophi. These criteria increase their specificity but make them less useful in diagnosing a first episode of gout, when tophi are unlikely to have developed.
Prediction rules are more sensitive in established gout
The new ACR prediction rule was compared with the ARA, Rome, and New York clinical prediction rules using urate crystals as the gold standard in early (less than 2 years) and established disease (longer than 2 years).8 All clinical prediction rules were more sensitive in established disease than early disease (95.3% vs 84.1%; P<.001) and more specific in early disease than established disease (79.9% vs 52.5%; P<.001).
EVIDENCE-BASED ANSWER:
Clinical prediction rules effectively diagnose gout without joint fluid analysis. The American College of Rheumatology clinical prediction rules, the most accurate rules developed for research purposes, have a sensitivity of 92%, specificity of 89%, positive likelihood ratio of 8.36, and negative likelihood ratio of 0.09 (strength of recommendation [SOR]: A, prospective cohort studies).
The Netherlands criteria, developed for use in primary care, have a positive predictive value of more than 80%, a positive likelihood ratio of 3.48, and a negative likelihood ratio of 0.17 (SOR: A, prospective cohort study).
EVIDENCE SUMMARY
In 2015, the American College of Rheumatology (ACR) redefined the clinical criteria for diagnosis of gout based on a 3-step system1 that can be found at: http://goutclassificationcalculator.auckland.ac.nz. The ACR rule was derived from a cross-sectional study of 983 patients in 25 rheumatology centers in 16 countries who presented with a swollen joint.2 Of the 983 patients, 509 had gout; the prevalence was 52%. Data from 653 of these patients were used to develop the rule and then validated in the remaining 330 patients.
Compared with the gold standard of monosodium urate crystals in synovial fluid, the ACR rule has a sensitivity of 92% and a specificity of 89%. The rule, designed for the research setting, involves using synovial fluid analysis, ultrasound imaging, and radiography, which makes it less useful in a primary care setting.
The Netherlands rule for primary care
A prospective diagnostic study in 328 family medicine patients (74% male; mean age 57) with monoarthritis tested the ability of multiple clinical variables to diagnose gout using monosodium urate crystals in synovial fluid as the gold standard.3 The prevalence of gout in this population was 57%.
The best diagnostic rule (Netherlands rule) comprised the following predefined variables: male sex, previous patient-reported arthritis attack, onset within one day, joint redness, first metatarsophalangeal joint (MTP1) involvement, hypertension or cardiovascular disease (angina pectoris, myocardial infarction, heart failure, cerebrovascular accident, transient ischemic attack, or peripheral vascular disease), and serum uric acid level above 5.88 mg/dL. The rule gives one point for each item. A score >8 had a positive likelihood ratio for diagnosing gout of 3.48 (TABLE1) and a higher positive predictive value (PPV) than family physicians’ clinical impressions (83% vs 64%).
The prevalence of gout in patients with scores of <4, 4 to 8, and >8 were 2.8%, 27%, and 80%, respectively. For scores of 4 to 8, the probability of gout is indeterminate, and synovial fluid analysis is recommended.
The Netherlands rule, validated in a secondary care practice of 390 patients with monoarthritis, found that a score >8 had a PPV of 87% and a score <4 had a negative predictive value of 95%.4 The probability of gout based on this rule can be calculated at http://www.umcn.nl/goutcalc.
In the study used to develop the Netherlands rule, no patients with a high probability of gout had septic arthritis. The ability of the rule to differentiate between gout and septic arthritis was tested retrospectively in 33 patients with acute gout (podagra excluded) diagnosed by the presence of monosodium urate joint crystals and 27 patients with septic arthritis diagnosed by positive bacterial culture.5 Patients with gout had significantly higher scores than patients with septic arthritis (7.8 ± 1.59 vs 3.4 ± 2.3; P<.001).
American Rheumatology Association, New York, and Rome prediction rules
A study of 82 Veterans Administration patients compared the American Rheumatology Association (ARA), New York, and Rome prediction rules with regard to their ability to diagnose gout with synovial urate crystals.6 The ARA criteria for gout diagnosis require either tophi or monosodium urate crystals in synovial fluid, or 6 out of a list of 12 other criteria.7
The New York prediction rule requires that patients meet 2 or more of the following criteria: at least 2 attacks of painful joint swelling with complete resolution within 2 weeks, podagra, tophi, and rapid response to colchicine treatment, defined as a major reduction in the objective signs of inflammation within 48 hours.
The Rome prediction rule requires meeting 2 of 3 criteria: serum uric acid >7 mg/dL in men and >6 mg/dL in women, presence of tophi, and history of attacks of painful joint swelling with abrupt onset and resolution within 2 weeks.
The New York prediction rule had the highest positive likelihood ratio of 4.4 compared with the ARA (1.8) and Rome (4.3) rules.6 The utility of the New York and Rome rules, although they have fewer criteria than ARA, is limited by the fact that they include a previous episode of joint swelling and tophi. These criteria increase their specificity but make them less useful in diagnosing a first episode of gout, when tophi are unlikely to have developed.
Prediction rules are more sensitive in established gout
The new ACR prediction rule was compared with the ARA, Rome, and New York clinical prediction rules using urate crystals as the gold standard in early (less than 2 years) and established disease (longer than 2 years).8 All clinical prediction rules were more sensitive in established disease than early disease (95.3% vs 84.1%; P<.001) and more specific in early disease than established disease (79.9% vs 52.5%; P<.001).
1. Neogi T, Jansen TL, Dalbeth N, et al. 2015 Gout Classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2015;74:1789-1798.
2. Taylor WJ, Fransen J, Jansen TL, et al. Study for Updated Gout Classification Criteria (SUGAR): identification of features to classify gout. Arthritis Care Res (Hoboken). 2015;67:1304-1315.
3. Janssens HJ, Fransen J, van de Lisdonk EH, et al. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170:1120-1126.
4. Kienhorst LB, Janssens HJ, Fransen J, et al. The validation of a diagnostic rule for gout without joint fluid analysis: a prospective study. Rheumatology (Oxford). 2015;54:609-614.
5. Lee K, Choi ST, Kang EJ, et al. SAT0377 The performance of a novel scoring system in the differential diagnosis between acute gout and septic arthritis. Ann Rheum Dis. 2013;72:A711.
6. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Taylor WJ, Fransen J, Dalbeth N, et al. Performance of classification criteria for gout in early and established disease. Ann Rheum Dis. 2016;75:178-182.
1. Neogi T, Jansen TL, Dalbeth N, et al. 2015 Gout Classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2015;74:1789-1798.
2. Taylor WJ, Fransen J, Jansen TL, et al. Study for Updated Gout Classification Criteria (SUGAR): identification of features to classify gout. Arthritis Care Res (Hoboken). 2015;67:1304-1315.
3. Janssens HJ, Fransen J, van de Lisdonk EH, et al. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170:1120-1126.
4. Kienhorst LB, Janssens HJ, Fransen J, et al. The validation of a diagnostic rule for gout without joint fluid analysis: a prospective study. Rheumatology (Oxford). 2015;54:609-614.
5. Lee K, Choi ST, Kang EJ, et al. SAT0377 The performance of a novel scoring system in the differential diagnosis between acute gout and septic arthritis. Ann Rheum Dis. 2013;72:A711.
6. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Taylor WJ, Fransen J, Dalbeth N, et al. Performance of classification criteria for gout in early and established disease. Ann Rheum Dis. 2016;75:178-182.
Evidence-based answers from the Family Physicians Inquiries Network
Yeast Infection in Pregnancy? Think Twice About Fluconazole

A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although the latter are recommended as firstline therapy, the ease of oral therapy makes it an attractive option.3,4
However, the safety of oral fluconazole during pregnancy has recently come under scrutiny. Case reports have linked high-dose use with congenital malformation.5,6 These case reports led to epidemiologic studies in which no such association was found.7,8
A large cohort study involving 1,079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk for congenital malformation or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriage.10 However, the validity of both studies’ findings was limited by small numbers of participants.
The current study is the largest to date to evaluate whether use of fluconazole in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth, compared to topical azoles.
STUDY SUMMARY
Increased risk for miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole–exposed pregnancy was matched with up to four unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented by filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. Of the total cohort, 3,315 pregnancies were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortion occurred in 147 of these pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR], 1.48).
Rates of stillbirth. Of 5,382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR, 1.32). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were four times more likely than lower doses (150 mg) to be associated with stillbirth (HRs, 4.10 and 0.99, respectively).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk for spontaneous abortion, compared to topical azole use (130 of 2,823 pregnancies vs 118 of 2,823 pregnancies; HR, 1.62)—but not an increased risk for stillbirth (20 of 4,301 pregnancies vs 22 of 4,301 pregnancies; HR, 1.18).
WHAT'S NEW
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk for spontaneous abortion. By comparing treatments in a sensitivity analysis, the researchers were able to eliminate Candida infections causing spontaneous abortion as a confounding factor. In addition, this study challenges the balance between ease of use and safety.
CAVEATS
A skewed population?
This cohort study using a Danish hospital registry may not be generalizable to a larger, non-Scandinavian population. Those not seeking care through a hospital were likely missed; if those seeking care through the hospital had a higher risk for abortion, the results could be biased. However, this would not have affected the results of the comparison between the two active treatments.
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically.
In all, given the large sample size and the care taken to match each exposed pregnancy with up to four unexposed pregnancies, these limitations likely had little influence on the overall findings.
CHALLENGES TO IMPLEMENTATION
Balancing ease of use with safety
Given the ease of using oral fluconazole, compared with daily topical azole therapy, many clinicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(9):624-626.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al; Vaginal Infections and Prematurity Study Group. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidiasis. Practitioner. 1985;229:655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although the latter are recommended as firstline therapy, the ease of oral therapy makes it an attractive option.3,4
However, the safety of oral fluconazole during pregnancy has recently come under scrutiny. Case reports have linked high-dose use with congenital malformation.5,6 These case reports led to epidemiologic studies in which no such association was found.7,8
A large cohort study involving 1,079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk for congenital malformation or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriage.10 However, the validity of both studies’ findings was limited by small numbers of participants.
The current study is the largest to date to evaluate whether use of fluconazole in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth, compared to topical azoles.
STUDY SUMMARY
Increased risk for miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole–exposed pregnancy was matched with up to four unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented by filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. Of the total cohort, 3,315 pregnancies were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortion occurred in 147 of these pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR], 1.48).
Rates of stillbirth. Of 5,382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR, 1.32). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were four times more likely than lower doses (150 mg) to be associated with stillbirth (HRs, 4.10 and 0.99, respectively).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk for spontaneous abortion, compared to topical azole use (130 of 2,823 pregnancies vs 118 of 2,823 pregnancies; HR, 1.62)—but not an increased risk for stillbirth (20 of 4,301 pregnancies vs 22 of 4,301 pregnancies; HR, 1.18).
WHAT'S NEW
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk for spontaneous abortion. By comparing treatments in a sensitivity analysis, the researchers were able to eliminate Candida infections causing spontaneous abortion as a confounding factor. In addition, this study challenges the balance between ease of use and safety.
CAVEATS
A skewed population?
This cohort study using a Danish hospital registry may not be generalizable to a larger, non-Scandinavian population. Those not seeking care through a hospital were likely missed; if those seeking care through the hospital had a higher risk for abortion, the results could be biased. However, this would not have affected the results of the comparison between the two active treatments.
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically.
In all, given the large sample size and the care taken to match each exposed pregnancy with up to four unexposed pregnancies, these limitations likely had little influence on the overall findings.
CHALLENGES TO IMPLEMENTATION
Balancing ease of use with safety
Given the ease of using oral fluconazole, compared with daily topical azole therapy, many clinicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(9):624-626.
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although the latter are recommended as firstline therapy, the ease of oral therapy makes it an attractive option.3,4
However, the safety of oral fluconazole during pregnancy has recently come under scrutiny. Case reports have linked high-dose use with congenital malformation.5,6 These case reports led to epidemiologic studies in which no such association was found.7,8
A large cohort study involving 1,079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk for congenital malformation or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriage.10 However, the validity of both studies’ findings was limited by small numbers of participants.
The current study is the largest to date to evaluate whether use of fluconazole in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth, compared to topical azoles.
STUDY SUMMARY
Increased risk for miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole–exposed pregnancy was matched with up to four unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented by filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. Of the total cohort, 3,315 pregnancies were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortion occurred in 147 of these pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR], 1.48).
Rates of stillbirth. Of 5,382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR, 1.32). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were four times more likely than lower doses (150 mg) to be associated with stillbirth (HRs, 4.10 and 0.99, respectively).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk for spontaneous abortion, compared to topical azole use (130 of 2,823 pregnancies vs 118 of 2,823 pregnancies; HR, 1.62)—but not an increased risk for stillbirth (20 of 4,301 pregnancies vs 22 of 4,301 pregnancies; HR, 1.18).
WHAT'S NEW
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk for spontaneous abortion. By comparing treatments in a sensitivity analysis, the researchers were able to eliminate Candida infections causing spontaneous abortion as a confounding factor. In addition, this study challenges the balance between ease of use and safety.
CAVEATS
A skewed population?
This cohort study using a Danish hospital registry may not be generalizable to a larger, non-Scandinavian population. Those not seeking care through a hospital were likely missed; if those seeking care through the hospital had a higher risk for abortion, the results could be biased. However, this would not have affected the results of the comparison between the two active treatments.
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically.
In all, given the large sample size and the care taken to match each exposed pregnancy with up to four unexposed pregnancies, these limitations likely had little influence on the overall findings.
CHALLENGES TO IMPLEMENTATION
Balancing ease of use with safety
Given the ease of using oral fluconazole, compared with daily topical azole therapy, many clinicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(9):624-626.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al; Vaginal Infections and Prematurity Study Group. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidiasis. Practitioner. 1985;229:655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al; Vaginal Infections and Prematurity Study Group. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidiasis. Practitioner. 1985;229:655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
Yeast infection in pregnancy? Think twice about fluconazole
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

Light Therapy For Nonseasonal Major Depressive Disorder?
PRACTICE CHANGER
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder.1
Strength of Recommendation
B: Based on a single moderate-quality randomized controlled trial.1
A 38-year-old woman recently diagnosed with major depressive disorder (MDD) without a seasonal pattern presents to discuss treatment options. Her Hamilton Depression Rating Scale (HAM-D) score is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in five adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective firstline pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with firstline or secondline drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N = 620) demonstrated the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD but found no benefit to light used as monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted less than four weeks.
In a five-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline (50 mg/d) or sham light treatment (using a dim red light) plus sertraline (50 mg/d). The investigators found a statistically significant reduction in depression score in the active treatment group compared to the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the seven atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Continue for the study summary >>
STUDY SUMMARY
Light therapy improves nonseasonal depression
This latest study was an eight-week, randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19 to 60) from outpatient psychiatry clinics who had a diagnosis of MDD (diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least two weeks prior to the first visit; they were subsequently monitored for one week to identify spontaneous responders and give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10 PM and 8 AM daily).
The investigators randomly assigned patients to one of four treatment groups:
• Active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill
• Fluoxetine (20 mg/d) plus sham light therapy
• Placebo pills with sham light therapy; or
• Combined active light therapy with fluoxetine (20 mg/d).
Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat. Adherence was assessed through review of patients’ daily logs of device treatment times and through pill counts.
The primary outcome at eight weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥ 50% reduction in MADRS score) and remission (MADRS score ≤ 10) at the final eighth-week visit. MADRS scoring was used because of its sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of eight weeks, the mean changes in MADRS scores from baseline were: light monotherapy, 13.4; fluoxetine monotherapy, 8.8; combination therapy, 16.9; and placebo, 6.5. The improvement was significant in the light monotherapy treatment group and in the combination therapy group, compared with the placebo group, and in the combination group, compared with the fluoxetine treatment group; improvement was not significant for the fluoxetine treatment group compared with the placebo group, however.
The treatment response (≥ 50% MADRS improvement) rate was highest in the combination treatment group (75.9%), followed by light monotherapy (50%), placebo (33.3%), and fluoxetine monotherapy (29%). There was a significant response effect for the combination versus placebo treatment group.
Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively). Combination therapy was superior to placebo in treatment response (≥ 50% reduction in the MADRS score) and remission (MADRS ≤ 10), with numbers needed to treat of 2.4 and 3.5, respectively.
By the end of the eight-week study period, 16 of 122 patients had dropped out. Two reported lack of efficacy, five reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
WHAT’S NEW
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious for increasing the remission rate of nonseasonal MDD.
Continue for caveats >>
CAVEATS
Variables may have affected results
Among the study’s limitations: use of a single SSRI (other, more potent SSRIs might work better); location (southern Canada; benefits may differ in regions farther south); and exclusion of pregnant and breastfeeding women from the study population.
Furthermore, the trial duration was relatively short, and the investigators did not attain their preplanned sample size for the study. This limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
CHALLENGES TO IMPLEMENTATION
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.10 Since few studies have tested light therapy for nonseasonal MDD, uncertainty remains about patient selection, as well as optimal dose, timing, and duration in the management of nonseasonal MDD.11 Although the associated risks are minimal, bright light therapy can lead to mania or hypomania; clinicians need to monitor for such effects when initiating therapy.3
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which are available in medical supply stores and online, ranges from $118 to $237.4,11 However, such devices are reusable, making the amortized cost almost negligible and perhaps negating this concern.12
REFERENCES
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. American Psychiatric Association practice guideline for the treatment of patients with major depressive disorder. 3rd ed. 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed July 5, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed July 5, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. Prim Care Companion CNS Disord. 2014;16(3). Epub 2014 Jun 26.
11. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
12. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(7):486-488.
PRACTICE CHANGER
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder.1
Strength of Recommendation
B: Based on a single moderate-quality randomized controlled trial.1
A 38-year-old woman recently diagnosed with major depressive disorder (MDD) without a seasonal pattern presents to discuss treatment options. Her Hamilton Depression Rating Scale (HAM-D) score is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in five adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective firstline pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with firstline or secondline drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N = 620) demonstrated the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD but found no benefit to light used as monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted less than four weeks.
In a five-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline (50 mg/d) or sham light treatment (using a dim red light) plus sertraline (50 mg/d). The investigators found a statistically significant reduction in depression score in the active treatment group compared to the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the seven atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Continue for the study summary >>
STUDY SUMMARY
Light therapy improves nonseasonal depression
This latest study was an eight-week, randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19 to 60) from outpatient psychiatry clinics who had a diagnosis of MDD (diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least two weeks prior to the first visit; they were subsequently monitored for one week to identify spontaneous responders and give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10 PM and 8 AM daily).
The investigators randomly assigned patients to one of four treatment groups:
• Active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill
• Fluoxetine (20 mg/d) plus sham light therapy
• Placebo pills with sham light therapy; or
• Combined active light therapy with fluoxetine (20 mg/d).
Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat. Adherence was assessed through review of patients’ daily logs of device treatment times and through pill counts.
The primary outcome at eight weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥ 50% reduction in MADRS score) and remission (MADRS score ≤ 10) at the final eighth-week visit. MADRS scoring was used because of its sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of eight weeks, the mean changes in MADRS scores from baseline were: light monotherapy, 13.4; fluoxetine monotherapy, 8.8; combination therapy, 16.9; and placebo, 6.5. The improvement was significant in the light monotherapy treatment group and in the combination therapy group, compared with the placebo group, and in the combination group, compared with the fluoxetine treatment group; improvement was not significant for the fluoxetine treatment group compared with the placebo group, however.
The treatment response (≥ 50% MADRS improvement) rate was highest in the combination treatment group (75.9%), followed by light monotherapy (50%), placebo (33.3%), and fluoxetine monotherapy (29%). There was a significant response effect for the combination versus placebo treatment group.
Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively). Combination therapy was superior to placebo in treatment response (≥ 50% reduction in the MADRS score) and remission (MADRS ≤ 10), with numbers needed to treat of 2.4 and 3.5, respectively.
By the end of the eight-week study period, 16 of 122 patients had dropped out. Two reported lack of efficacy, five reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
WHAT’S NEW
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious for increasing the remission rate of nonseasonal MDD.
Continue for caveats >>
CAVEATS
Variables may have affected results
Among the study’s limitations: use of a single SSRI (other, more potent SSRIs might work better); location (southern Canada; benefits may differ in regions farther south); and exclusion of pregnant and breastfeeding women from the study population.
Furthermore, the trial duration was relatively short, and the investigators did not attain their preplanned sample size for the study. This limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
CHALLENGES TO IMPLEMENTATION
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.10 Since few studies have tested light therapy for nonseasonal MDD, uncertainty remains about patient selection, as well as optimal dose, timing, and duration in the management of nonseasonal MDD.11 Although the associated risks are minimal, bright light therapy can lead to mania or hypomania; clinicians need to monitor for such effects when initiating therapy.3
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which are available in medical supply stores and online, ranges from $118 to $237.4,11 However, such devices are reusable, making the amortized cost almost negligible and perhaps negating this concern.12
REFERENCES
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. American Psychiatric Association practice guideline for the treatment of patients with major depressive disorder. 3rd ed. 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed July 5, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed July 5, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. Prim Care Companion CNS Disord. 2014;16(3). Epub 2014 Jun 26.
11. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
12. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(7):486-488.
PRACTICE CHANGER
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder.1
Strength of Recommendation
B: Based on a single moderate-quality randomized controlled trial.1
A 38-year-old woman recently diagnosed with major depressive disorder (MDD) without a seasonal pattern presents to discuss treatment options. Her Hamilton Depression Rating Scale (HAM-D) score is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in five adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective firstline pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with firstline or secondline drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N = 620) demonstrated the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD but found no benefit to light used as monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted less than four weeks.
In a five-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline (50 mg/d) or sham light treatment (using a dim red light) plus sertraline (50 mg/d). The investigators found a statistically significant reduction in depression score in the active treatment group compared to the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the seven atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Continue for the study summary >>
STUDY SUMMARY
Light therapy improves nonseasonal depression
This latest study was an eight-week, randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19 to 60) from outpatient psychiatry clinics who had a diagnosis of MDD (diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least two weeks prior to the first visit; they were subsequently monitored for one week to identify spontaneous responders and give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10 PM and 8 AM daily).
The investigators randomly assigned patients to one of four treatment groups:
• Active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill
• Fluoxetine (20 mg/d) plus sham light therapy
• Placebo pills with sham light therapy; or
• Combined active light therapy with fluoxetine (20 mg/d).
Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat. Adherence was assessed through review of patients’ daily logs of device treatment times and through pill counts.
The primary outcome at eight weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥ 50% reduction in MADRS score) and remission (MADRS score ≤ 10) at the final eighth-week visit. MADRS scoring was used because of its sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of eight weeks, the mean changes in MADRS scores from baseline were: light monotherapy, 13.4; fluoxetine monotherapy, 8.8; combination therapy, 16.9; and placebo, 6.5. The improvement was significant in the light monotherapy treatment group and in the combination therapy group, compared with the placebo group, and in the combination group, compared with the fluoxetine treatment group; improvement was not significant for the fluoxetine treatment group compared with the placebo group, however.
The treatment response (≥ 50% MADRS improvement) rate was highest in the combination treatment group (75.9%), followed by light monotherapy (50%), placebo (33.3%), and fluoxetine monotherapy (29%). There was a significant response effect for the combination versus placebo treatment group.
Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively). Combination therapy was superior to placebo in treatment response (≥ 50% reduction in the MADRS score) and remission (MADRS ≤ 10), with numbers needed to treat of 2.4 and 3.5, respectively.
By the end of the eight-week study period, 16 of 122 patients had dropped out. Two reported lack of efficacy, five reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
WHAT’S NEW
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious for increasing the remission rate of nonseasonal MDD.
Continue for caveats >>
CAVEATS
Variables may have affected results
Among the study’s limitations: use of a single SSRI (other, more potent SSRIs might work better); location (southern Canada; benefits may differ in regions farther south); and exclusion of pregnant and breastfeeding women from the study population.
Furthermore, the trial duration was relatively short, and the investigators did not attain their preplanned sample size for the study. This limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
CHALLENGES TO IMPLEMENTATION
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.10 Since few studies have tested light therapy for nonseasonal MDD, uncertainty remains about patient selection, as well as optimal dose, timing, and duration in the management of nonseasonal MDD.11 Although the associated risks are minimal, bright light therapy can lead to mania or hypomania; clinicians need to monitor for such effects when initiating therapy.3
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which are available in medical supply stores and online, ranges from $118 to $237.4,11 However, such devices are reusable, making the amortized cost almost negligible and perhaps negating this concern.12
REFERENCES
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. American Psychiatric Association practice guideline for the treatment of patients with major depressive disorder. 3rd ed. 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed July 5, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed July 5, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. Prim Care Companion CNS Disord. 2014;16(3). Epub 2014 Jun 26.
11. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
12. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(7):486-488.
Does oseltamivir shorten flu symptom duration?
Yes. Treatment of influenza virus infection with oral oseltamivir reduces time to alleviation of symptoms in adults and children by approximately one day compared with placebo. It reduces symptom duration even when initiated more than 2 days after symptom onset (strength of recommendation: A, systematic review of randomized controlled trials [RCTs], meta-analysis of observation trials, RCT).
Evidence summary
A 2014 systematic review included 8 RCTs in adults (3954 patients) and one RCT in children (669 patients) with influenza and compared time to alleviation of symptoms with oseltamivir and placebo.1 Symptoms were defined as local (nasal discharge, dry cough, sore throat) and systemic (fever, myalgia, headache, fatigue). Methodology for diagnosis varied by trial.
Oral oseltamivir (75, 150, or 300 mg for 5 to 10 days) reduced time to first alleviation of symptoms by 17 hours (95% confidence interval [CI], 8.4-25 hours) compared with placebo for adults and 29 hours (95% CI, 12-47 hours) for the otherwise healthy children.
The systematic review also included 2 RCTs involving 660 children with chronic asthma who received treatment with oseltamivir. Researchers found no reduction in time to symptom alleviation with the oseltamivir.
Treatment with oseltamivir increased the risk of nausea (number needed to harm [NNH]=28) and vomiting (NNH=22) in adults and the risk of vomiting (NNH=19) in children. Sources of bias included industry sponsorship of all trials, differing placebo components, inadequate recruitment, and use of other medication.
Shorter fever duration?
A 2012 meta-analysis of 6 observational studies (5842 patients) compared the effect of oral oseltamivir with no treatment on duration of signs and symptoms (definition not given) in patients with influenza (method of diagnosis not stated).2 Oseltamivir reduced fever duration by 33 hours (95% CI, 21-45 hours) compared with no treatment.
The authors describe the evidence as being of very low quality because of study heterogeneity, lack of control for confounding variables, selection bias, and study sources (many unpublished industry studies).
There’s benefit even with late Tx
A 2014 double-blind RCT, not included in the previously described reviews, of 130 adults and 1070 children with a positive rapid influenza test examined the effect of oseltamivir and placebo on symptom duration.3 Research assistants visited participants at home each day until patients were asymptomatic for 7 consecutive days.
Treatment with oseltamivir reduced symptom duration by a median of one day compared with no treatment (hazard ratio=0.87; 95% CI, 0.79-0.95). This benefit was observed regardless of whether treatment was initiated fewer or more than 48 hours after symptom onset. One notable limitation was failure to control for paracetamol (acetaminophen) usage, a possible confounder for duration of symptoms, such as fever.
1. Jefferson T, Jones MA, Doshi P, et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev. 2014;(4):CD008965.
2. Hsu J, Santesso N, Mustafa R, et al. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann Intern Med. 2012;156:512-524.
3. Fry AM, Goswami D, Nahar K, et al. Efficacy of oseltamivir treatment started within 5 days of symptom onset to reduce influenza illness duration and virus shedding in an urban setting in Bangladesh: a randomised placebo-controlled trial. Lancet Infect Dis. 2014;14:109-118.
Yes. Treatment of influenza virus infection with oral oseltamivir reduces time to alleviation of symptoms in adults and children by approximately one day compared with placebo. It reduces symptom duration even when initiated more than 2 days after symptom onset (strength of recommendation: A, systematic review of randomized controlled trials [RCTs], meta-analysis of observation trials, RCT).
Evidence summary
A 2014 systematic review included 8 RCTs in adults (3954 patients) and one RCT in children (669 patients) with influenza and compared time to alleviation of symptoms with oseltamivir and placebo.1 Symptoms were defined as local (nasal discharge, dry cough, sore throat) and systemic (fever, myalgia, headache, fatigue). Methodology for diagnosis varied by trial.
Oral oseltamivir (75, 150, or 300 mg for 5 to 10 days) reduced time to first alleviation of symptoms by 17 hours (95% confidence interval [CI], 8.4-25 hours) compared with placebo for adults and 29 hours (95% CI, 12-47 hours) for the otherwise healthy children.
The systematic review also included 2 RCTs involving 660 children with chronic asthma who received treatment with oseltamivir. Researchers found no reduction in time to symptom alleviation with the oseltamivir.
Treatment with oseltamivir increased the risk of nausea (number needed to harm [NNH]=28) and vomiting (NNH=22) in adults and the risk of vomiting (NNH=19) in children. Sources of bias included industry sponsorship of all trials, differing placebo components, inadequate recruitment, and use of other medication.
Shorter fever duration?
A 2012 meta-analysis of 6 observational studies (5842 patients) compared the effect of oral oseltamivir with no treatment on duration of signs and symptoms (definition not given) in patients with influenza (method of diagnosis not stated).2 Oseltamivir reduced fever duration by 33 hours (95% CI, 21-45 hours) compared with no treatment.
The authors describe the evidence as being of very low quality because of study heterogeneity, lack of control for confounding variables, selection bias, and study sources (many unpublished industry studies).
There’s benefit even with late Tx
A 2014 double-blind RCT, not included in the previously described reviews, of 130 adults and 1070 children with a positive rapid influenza test examined the effect of oseltamivir and placebo on symptom duration.3 Research assistants visited participants at home each day until patients were asymptomatic for 7 consecutive days.
Treatment with oseltamivir reduced symptom duration by a median of one day compared with no treatment (hazard ratio=0.87; 95% CI, 0.79-0.95). This benefit was observed regardless of whether treatment was initiated fewer or more than 48 hours after symptom onset. One notable limitation was failure to control for paracetamol (acetaminophen) usage, a possible confounder for duration of symptoms, such as fever.
Yes. Treatment of influenza virus infection with oral oseltamivir reduces time to alleviation of symptoms in adults and children by approximately one day compared with placebo. It reduces symptom duration even when initiated more than 2 days after symptom onset (strength of recommendation: A, systematic review of randomized controlled trials [RCTs], meta-analysis of observation trials, RCT).
Evidence summary
A 2014 systematic review included 8 RCTs in adults (3954 patients) and one RCT in children (669 patients) with influenza and compared time to alleviation of symptoms with oseltamivir and placebo.1 Symptoms were defined as local (nasal discharge, dry cough, sore throat) and systemic (fever, myalgia, headache, fatigue). Methodology for diagnosis varied by trial.
Oral oseltamivir (75, 150, or 300 mg for 5 to 10 days) reduced time to first alleviation of symptoms by 17 hours (95% confidence interval [CI], 8.4-25 hours) compared with placebo for adults and 29 hours (95% CI, 12-47 hours) for the otherwise healthy children.
The systematic review also included 2 RCTs involving 660 children with chronic asthma who received treatment with oseltamivir. Researchers found no reduction in time to symptom alleviation with the oseltamivir.
Treatment with oseltamivir increased the risk of nausea (number needed to harm [NNH]=28) and vomiting (NNH=22) in adults and the risk of vomiting (NNH=19) in children. Sources of bias included industry sponsorship of all trials, differing placebo components, inadequate recruitment, and use of other medication.
Shorter fever duration?
A 2012 meta-analysis of 6 observational studies (5842 patients) compared the effect of oral oseltamivir with no treatment on duration of signs and symptoms (definition not given) in patients with influenza (method of diagnosis not stated).2 Oseltamivir reduced fever duration by 33 hours (95% CI, 21-45 hours) compared with no treatment.
The authors describe the evidence as being of very low quality because of study heterogeneity, lack of control for confounding variables, selection bias, and study sources (many unpublished industry studies).
There’s benefit even with late Tx
A 2014 double-blind RCT, not included in the previously described reviews, of 130 adults and 1070 children with a positive rapid influenza test examined the effect of oseltamivir and placebo on symptom duration.3 Research assistants visited participants at home each day until patients were asymptomatic for 7 consecutive days.
Treatment with oseltamivir reduced symptom duration by a median of one day compared with no treatment (hazard ratio=0.87; 95% CI, 0.79-0.95). This benefit was observed regardless of whether treatment was initiated fewer or more than 48 hours after symptom onset. One notable limitation was failure to control for paracetamol (acetaminophen) usage, a possible confounder for duration of symptoms, such as fever.
1. Jefferson T, Jones MA, Doshi P, et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev. 2014;(4):CD008965.
2. Hsu J, Santesso N, Mustafa R, et al. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann Intern Med. 2012;156:512-524.
3. Fry AM, Goswami D, Nahar K, et al. Efficacy of oseltamivir treatment started within 5 days of symptom onset to reduce influenza illness duration and virus shedding in an urban setting in Bangladesh: a randomised placebo-controlled trial. Lancet Infect Dis. 2014;14:109-118.
1. Jefferson T, Jones MA, Doshi P, et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev. 2014;(4):CD008965.
2. Hsu J, Santesso N, Mustafa R, et al. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann Intern Med. 2012;156:512-524.
3. Fry AM, Goswami D, Nahar K, et al. Efficacy of oseltamivir treatment started within 5 days of symptom onset to reduce influenza illness duration and virus shedding in an urban setting in Bangladesh: a randomised placebo-controlled trial. Lancet Infect Dis. 2014;14:109-118.
Evidence-based answers from the Family Physicians Inquiries Network

Light therapy for nonseasonal major depressive disorder?
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

Resistant Hypertension? Time to Consider This Fourth-line Drug
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
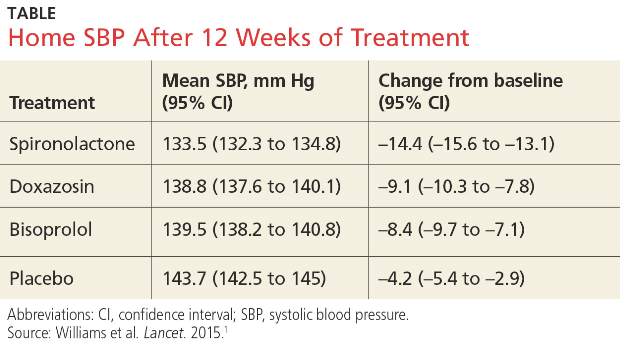
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
Resistant hypertension? Time to consider this fourth-line drug
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
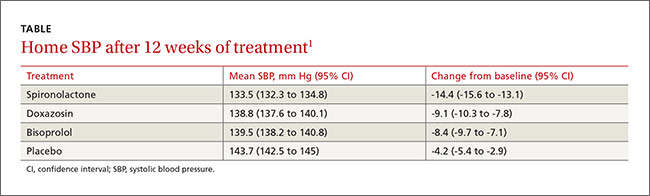
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

What Next When Metformin Isn't Enough For Type 2 Diabetes?
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5
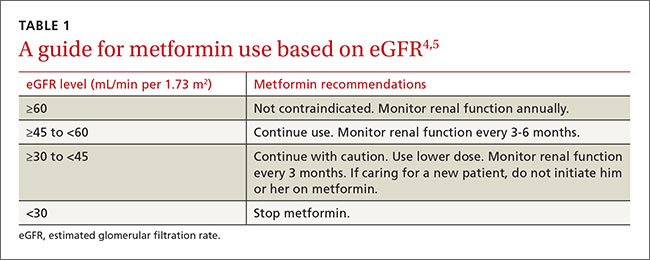
What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1
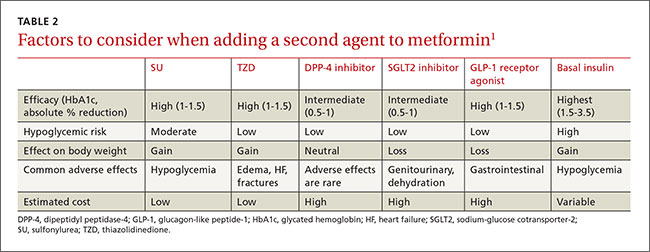
Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1
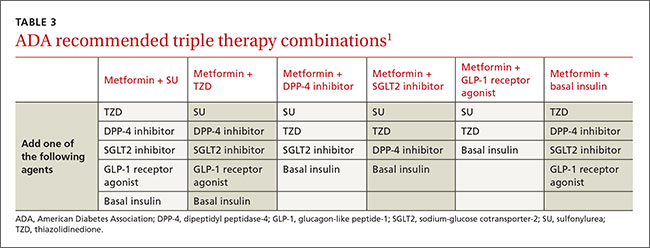
In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral meds
For type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12
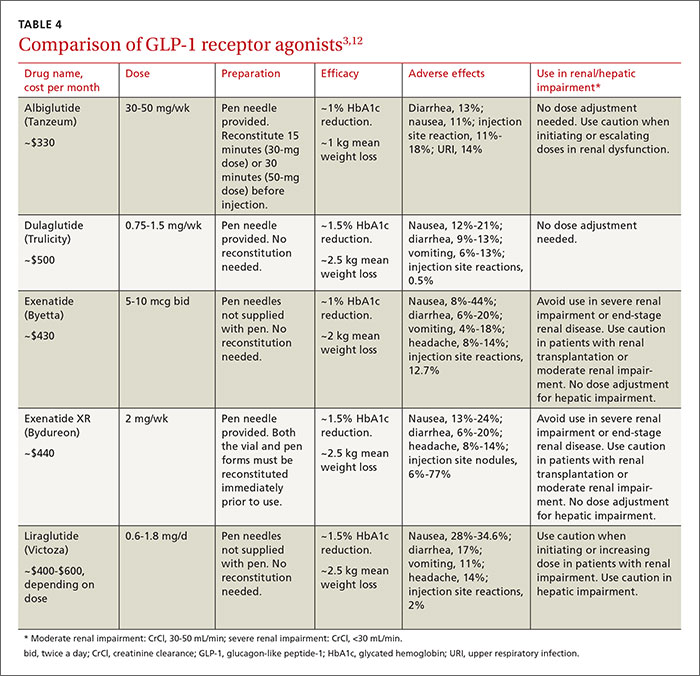
All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merck
manuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5
What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1
Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1
In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral meds
For type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12
All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5
What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1
Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1
In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral meds
For type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12
All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merck
manuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merck
manuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.

What next when metformin isn't enough for type 2 diabetes?
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5

What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1

Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1

In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral medsFor type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12

All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merckmanuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5
What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1
Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1
In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral medsFor type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12
All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
› Turn first to metformin for pharmacologic treatment of type 2 diabetes. A
› Add a second oral agent (such as a sulfonylurea, thiazolidinedione, sodium-glucose cotransporter-2 inhibitor, or dipeptidyl peptidase 4 inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or basal insulin if metformin at a maximum tolerated dose does not achieve the HbA1c target over 3 months. A
› Progress to bolus mealtime insulin or a GLP-1 agonist to cover postprandial glycemic excursions if HbA1c remains above goal despite an adequate trial of basal insulin. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The "Standards of Medical Care in Diabetes" guidelines published in 2015 by the American Diabetes Association (ADA) state that metformin is the preferred initial pharmacotherapy for managing type 2 diabetes.1 Metformin, a biguanide, enhances insulin sensitivity in muscle and fat tissue and inhibits hepatic glucose production. Advantages of metformin include the longstanding research supporting its efficacy and safety, an expected decrease in the glycated hemoglobin (HbA1c) level of 1% to 1.5%, low cost, minimal hypoglycemic risk, and potential reductions in cardiovascular (CV) events due to decreased low-density lipoprotein (LDL) cholesterol.1,2
To minimize adverse gastrointestinal effects, start metformin at 500 mg once or twice a day and titrate upward every one to 2 weeks to the target dose.3 To help guide dosing decisions, use the estimated glomerular filtration rate (eGFR) instead of the serum creatinine (SCr) level, because the SCr can translate into a variable range of eGFRs (TABLE 1).4,5
What if metformin alone isn't enough?
CASE › Richard C, age 50, has type 2 diabetes, hypertension, hyperlipidemia, and obesity. He takes metformin 1 g twice a day for his diabetes. After 3 months on this regimen, his HbA1c is 8.8%. How would you manage Mr. C's diabetes going forward?
If metformin at a maximum tolerated dose does not achieve the HbA1c target after 3 months, add a second oral agent (a sulfonylurea [SU], thiazolidinedione [TZD], dipeptidyl peptidase 4 [DPP-4] inhibitor, or sodium-glucose cotransporter-2 [SGLT2] inhibitor), a glucagon-like peptide-1 (GLP-1) receptor agonist, or a basal insulin (TABLE 2).1
Factors that will affect the choice of the second agent include patient preference, cost, potential adverse effects, impact on weight, efficacy, and risk of hypoglycemia.
Based on cost, familiarity, and longstanding safety data, you decide to give Mr. C an SU, while cautioning him about hypoglycemia.
CASE › Mr. C has now been taking metformin and an SU at maximum doses for 2 years and continues with lifestyle modifications. Though his HbA1c level dropped after adding the SU, over 2 years it has crept up to 8.6% and his mean blood glucose is 186 mg/dL. What are your treatment options now?
If the target HbA1c level is not achieved on dual therapy, consider triple therapy combinations (TABLE 3).1
In Mr. C's case, a third oral agent could be added, but DPP-4 and SGLT2 are unlikely to get his HbA1c below 7%. TZD may get his HbA1c into the desired range but is associated with adverse effects such as heart failure, edema, and weight gain. Mr. C agrees instead to start a basal insulin in conjunction with metformin. You could continue the SU, but you decide to stop it because the additive effect of these medications increases the risk of hypoglycemia.
CASE › Six months later Mr. C is taking metformin and insulin glargine, a basal insulin, adjusted to a fasting blood glucose of 80 to 130 mg/dL. His HbA1c is still above target at 8.4%, and the mean postprandial blood glucose is 232 mg/dL.
Mr. C is still above target for HbA1c and for postprandial blood glucose (goal: <180 mg/dL), so he needs pharmacotherapy that targets postprandial glucose elevations.1 His fasting blood glucose readings are at goal, so increasing his insulin glargine is not recommended because it could cause hypoglycemia. An oral agent other than SU could be added, but none is potent enough to lower the HbA1c to goal (TABLE 2).1 There are 3 other options:
- add a mealtime bolus of insulin
- add a GLP-1 receptor agonist
- switch to premixed (biphasic) insulin.
What to do when basal insulin isn’t enough—with or without oral medsFor type 2 diabetes poorly controlled on basal insulin with or without oral agents, the 2015 ADA treatment guidelines recommend adding a GLP-1 receptor agonist or mealtime insulin.1 A less desirable alternative is to switch from basal insulin to a twice-daily premixed (biphasic) insulin analog (70/30 aspart mix or 75/25 or 50/50 lispro mix). The human NPH-Regular premixed formulations (70/30) are less costly alternatives. The disadvantage with all premixed insulins is they only cover 2 postprandial glucose elevations a day.1,6,7
Insulin requires multiple daily injections, can lead to weight gain, and carries the risk of hypoglycemia, which causes significant morbidity.8,9 Daily or weekly administration of a GLP-1 receptor agonist combined with basal insulin can offer a more convenient alternative to mealtime boluses of insulin.
What are GLP-1 receptor agonists?
GLP-1 receptor agonists exert their maximum influence on blood glucose levels during the postprandial period by mimicking the body’s natural incretin hormonal response to oral glucose ingestion.10 They delay gastric emptying, promote satiety, decrease glucagon secretion, and increase insulin secretion.10,11 This mechanism blunts the spiking of postprandial blood glucose after a meal and improves blood glucose control and weight reduction.1,6,7
A systematic review and meta-analysis by Eng and colleagues compared the safety and efficacy of combined GLP-1 agonist and basal insulin with other treatment regimens.7 Fifteen randomized controlled trials were included involving 4348 participants with a mean trial duration of 25 weeks.
Compared with all other treatment regimens, the GLP-1 receptor agonist and basal insulin combination not only significantly reduced HbA1c by 0.44% (95% confidence interval [CI], -0.60 to -0.29) and increased the likelihood of attaining an HbA1c of <7.0% (relative risk [RR]=1.92; 95% CI, 1.43 to 2.56) but also reduced weight by 3.22 kg (-4.90 to -1.54) with no increased risk of hypoglycemia (RR=0.99; 0.76 to 1.29).7
GLP-1 agonist vs bolus insulin
Compared with basal-bolus insulin regimens, the combination of a GLP-1 receptor agonist with basal insulin has led to a significantly lowered risk of hypoglycemia (RR=0.67; 95% CI, 0.56 to 0.80), greater weight loss (-5.66 kg; 95% CI, -9.8 to -1.51) and an average reduction in HbA1c of 0.1% (95% CI, -0.17 to -0.02).7
There are 5 GLP-1 receptor agonists that have US Food and Drug Administration approval for the treatment of type 2 diabetes: albiglutide, dulaglutide, exenatide, exenatide XR, and liraglutide (TABLE 4).3,12
All 5 agents are administered subcutaneously and packaged in pen-injector form. Adverse effects include nausea, which is transient and diminishes within the first few weeks of therapy, and less commonly, pancreatitis.3,12
All of the GLP-1 receptor agonists, except short-acting exenatide, carry a warning about the risk of worsening renal function and a possible association with medullary thyroid carcinomas, which were identified in rats, but have not been observed in humans.3,12 Medications in this drug class have a low risk for precipitating hypoglycemia.11 Cost is their chief disadvantage, although copay reduction cards are available online for most of the products. Evaluate efficacy, ease of use, tolerability, and cost when selecting a GLP-1 receptor agonist.3,12
CASE › Mr. C prefers a more convenient option than adding another daily injection. Given his obesity, a GLP-1 receptor agonist can help with weight loss and lower his risk for hypoglycemia. To further increase the convenience in dosing, you lean toward either weekly exenatide XR or dulaglutide over basal-bolus combination insulin. Weekly albiglutide is less potent than exenatide XR and dulaglutide in decreasing HbA1c.12 Mr. C’s insurance plan provides preferred coverage for exenatide XR and he is eligible for a copay savings card, meaning he will pay no more than $25 per month for this new prescription. You prescribe exenatide XR and ask him to record his postprandial blood glucose levels. You follow up in one month to assess his response.
CORRESPONDENCE
Anne Mounsey, MD, University of North Carolina School of Medicine, Department of Family Medicine, 590 Manning Drive, Campus Box 7595, Chapel Hill, NC 27599; [email protected].
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merckmanuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.
1. American Diabetes Association. Standards of medical care in diabetes - 2015. Diabetes Care. 2015;38 (Suppl):S1-S94.
2. Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602-613.
3. Merck Manual. Metformin. Available at: http://www.merckmanuals.com/professional/appendixes/brand-names-of-some-commonly-used-drugs. Accessed April 18, 2015.
4. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011;34:1431-1437.
5. Philbrick AM, Ernst ME, McDanel DL, et al. Metformin use in renal dysfunction: is a serum creatinine threshold appropriate? Am J Health Syst Pharm. 2009;66:2017-2023.
6. Pharmacist’s Letter. Drugs for Type 2 Diabetes [detail document]. September 2015. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/ArticleDD.aspx?nidchk=1&cs=&s=PL&pt=2&segment=4407&dd=280601. Accessed December 28, 2015.
7. Eng C, Kramer CK, Zinman B, et al. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014;384:2228-2234.
8. Inzucchi SE, Burgenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364-1379.
9. Bonds DE, Miller ME, Bergenstal RM, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the ACCORD study. BMJ. 2010:340:b4909.
10. Garber AJ. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34 (Suppl 2):S279-S284.
11. Young LA, Buse JB. GLP-1 receptor agonists and basal insulin in type 2 diabetes. Lancet. 2014;384:2180-2181.
12. Pharmacist’s Letter. Comparison of GLP-1 Agonists [detail document]. December 2014. Available at: http://pharmacistsletter.therapeuticresearch.com/pl/Browse.aspx?cs=&s=PL&pt=6&fpt=31&dd=300804&pb=PL&cat=5718#dd. Accessed December 28, 2015.
