User login
FDA Approves Bosutinib for Previously Treated CML
Bosutinib, a tyrosine kinase inhibitor, has been approved by the Food and Drug Administration as a treatment for chronic myeloid leukemia, based on the results of a study of 546 patients, the agency announced on Sept. 4.
The approved indication is for the treatment of adults with chronic, accelerated, or blast-phase Philadelphia chromosome–positive (Ph+) chronic myeloid leukemia (CML) who are resistant to or cannot tolerate other treatments, including imatinib, according to the FDA and a statement issued by the manufacturer, Pfizer Inc.

Pfizer will be marketing bosutinib as Bosulif. The new agent inhibits the Abl and Src signaling pathways, and is taken once a day at a dose of 500 mg by mouth, according to the company.
The FDA has approved other tyrosine kinase inhibitors to treat CML, including imatinib (Gleevec) in 2001, dasatinib (Sprycel) in 2006, and nilotinib (Tasigna) in 2007. With these approvals, "we are seeing improvements in the treatment of CML based on a better understanding of the molecular basis of the disease," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement. He noted that these improvements have been seen in people in chronic and accelerated phases of CML.
In the international, phase I/II study, all 546 adults enrolled were treated with bosutinib; they were diagnosed with chronic, accelerated or blast phase CML, and had progressed after treatment with imatinib or with imatinib followed by dasatinib and/or nilotinib, or they could not tolerate previous treatments.
Among the patients with chronic phase CML who had been treated previously with imatinib (266 patients), almost 34% achieved a major cytogenic response (MCyR) by 24 weeks of treatment with bosutinib. Almost 53% of the patients who achieved a MCyR had a response that lasted at least 18 months, and a median duration of MCyR was not reached, according to the FDA and company statements.
Of the 108 patients with chronic phase CML who had been treated with imatinib and at least one other tyrosine kinase inhibitor, almost 27% achieved a MCyR by 24 weeks of treatment with bosutinib. About 51% of those who achieved a MCyR had a response that lasted at least 9 months, and in this group of patients, the median duration of MCyR also was not reached, according to the FDA and company statements.
The FDA also noted that among the patients with accelerated CML, who had been treated previously with a regimen that included at least imatinib, 33% had a complete hematologic response, and 55% achieved an overall hematologic response within the first 48 weeks of treatment.
Among those with blast phase CML, 15% achieved a complete hematologic response, and 28% achieved an overall hematologic response, according to the FDA.
Of the total 374 evaluable patients with chronic CML, 16 patients (4%) had confirmed disease transformation to advanced or blast phase while undergoing treatment with bosutinib.
Diarrhea, nausea, thrombocytopenia, vomiting, abdominal pain, rash, anemia, fever, and fatigue were among the most common adverse events associated with bosutinib treatment, according to the FDA.
Bosutinib was approved as an orphan drug, a drug that is intended for a disease that affects fewer than 200,000 people in the United States. In 2012, about 5,430 people will be diagnosed with CML, according to the FDA. With improvements in therapy, as many as 26,000 people are living with the disease, and that number is expected to increase 10-fold by 2040, Pfizer added.
Bosutinib, a tyrosine kinase inhibitor, has been approved by the Food and Drug Administration as a treatment for chronic myeloid leukemia, based on the results of a study of 546 patients, the agency announced on Sept. 4.
The approved indication is for the treatment of adults with chronic, accelerated, or blast-phase Philadelphia chromosome–positive (Ph+) chronic myeloid leukemia (CML) who are resistant to or cannot tolerate other treatments, including imatinib, according to the FDA and a statement issued by the manufacturer, Pfizer Inc.
Pfizer will be marketing bosutinib as Bosulif. The new agent inhibits the Abl and Src signaling pathways, and is taken once a day at a dose of 500 mg by mouth, according to the company.
The FDA has approved other tyrosine kinase inhibitors to treat CML, including imatinib (Gleevec) in 2001, dasatinib (Sprycel) in 2006, and nilotinib (Tasigna) in 2007. With these approvals, "we are seeing improvements in the treatment of CML based on a better understanding of the molecular basis of the disease," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement. He noted that these improvements have been seen in people in chronic and accelerated phases of CML.
In the international, phase I/II study, all 546 adults enrolled were treated with bosutinib; they were diagnosed with chronic, accelerated or blast phase CML, and had progressed after treatment with imatinib or with imatinib followed by dasatinib and/or nilotinib, or they could not tolerate previous treatments.
Among the patients with chronic phase CML who had been treated previously with imatinib (266 patients), almost 34% achieved a major cytogenic response (MCyR) by 24 weeks of treatment with bosutinib. Almost 53% of the patients who achieved a MCyR had a response that lasted at least 18 months, and a median duration of MCyR was not reached, according to the FDA and company statements.
Of the 108 patients with chronic phase CML who had been treated with imatinib and at least one other tyrosine kinase inhibitor, almost 27% achieved a MCyR by 24 weeks of treatment with bosutinib. About 51% of those who achieved a MCyR had a response that lasted at least 9 months, and in this group of patients, the median duration of MCyR also was not reached, according to the FDA and company statements.
The FDA also noted that among the patients with accelerated CML, who had been treated previously with a regimen that included at least imatinib, 33% had a complete hematologic response, and 55% achieved an overall hematologic response within the first 48 weeks of treatment.
Among those with blast phase CML, 15% achieved a complete hematologic response, and 28% achieved an overall hematologic response, according to the FDA.
Of the total 374 evaluable patients with chronic CML, 16 patients (4%) had confirmed disease transformation to advanced or blast phase while undergoing treatment with bosutinib.
Diarrhea, nausea, thrombocytopenia, vomiting, abdominal pain, rash, anemia, fever, and fatigue were among the most common adverse events associated with bosutinib treatment, according to the FDA.
Bosutinib was approved as an orphan drug, a drug that is intended for a disease that affects fewer than 200,000 people in the United States. In 2012, about 5,430 people will be diagnosed with CML, according to the FDA. With improvements in therapy, as many as 26,000 people are living with the disease, and that number is expected to increase 10-fold by 2040, Pfizer added.
Bosutinib, a tyrosine kinase inhibitor, has been approved by the Food and Drug Administration as a treatment for chronic myeloid leukemia, based on the results of a study of 546 patients, the agency announced on Sept. 4.
The approved indication is for the treatment of adults with chronic, accelerated, or blast-phase Philadelphia chromosome–positive (Ph+) chronic myeloid leukemia (CML) who are resistant to or cannot tolerate other treatments, including imatinib, according to the FDA and a statement issued by the manufacturer, Pfizer Inc.
Pfizer will be marketing bosutinib as Bosulif. The new agent inhibits the Abl and Src signaling pathways, and is taken once a day at a dose of 500 mg by mouth, according to the company.
The FDA has approved other tyrosine kinase inhibitors to treat CML, including imatinib (Gleevec) in 2001, dasatinib (Sprycel) in 2006, and nilotinib (Tasigna) in 2007. With these approvals, "we are seeing improvements in the treatment of CML based on a better understanding of the molecular basis of the disease," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement. He noted that these improvements have been seen in people in chronic and accelerated phases of CML.
In the international, phase I/II study, all 546 adults enrolled were treated with bosutinib; they were diagnosed with chronic, accelerated or blast phase CML, and had progressed after treatment with imatinib or with imatinib followed by dasatinib and/or nilotinib, or they could not tolerate previous treatments.
Among the patients with chronic phase CML who had been treated previously with imatinib (266 patients), almost 34% achieved a major cytogenic response (MCyR) by 24 weeks of treatment with bosutinib. Almost 53% of the patients who achieved a MCyR had a response that lasted at least 18 months, and a median duration of MCyR was not reached, according to the FDA and company statements.
Of the 108 patients with chronic phase CML who had been treated with imatinib and at least one other tyrosine kinase inhibitor, almost 27% achieved a MCyR by 24 weeks of treatment with bosutinib. About 51% of those who achieved a MCyR had a response that lasted at least 9 months, and in this group of patients, the median duration of MCyR also was not reached, according to the FDA and company statements.
The FDA also noted that among the patients with accelerated CML, who had been treated previously with a regimen that included at least imatinib, 33% had a complete hematologic response, and 55% achieved an overall hematologic response within the first 48 weeks of treatment.
Among those with blast phase CML, 15% achieved a complete hematologic response, and 28% achieved an overall hematologic response, according to the FDA.
Of the total 374 evaluable patients with chronic CML, 16 patients (4%) had confirmed disease transformation to advanced or blast phase while undergoing treatment with bosutinib.
Diarrhea, nausea, thrombocytopenia, vomiting, abdominal pain, rash, anemia, fever, and fatigue were among the most common adverse events associated with bosutinib treatment, according to the FDA.
Bosutinib was approved as an orphan drug, a drug that is intended for a disease that affects fewer than 200,000 people in the United States. In 2012, about 5,430 people will be diagnosed with CML, according to the FDA. With improvements in therapy, as many as 26,000 people are living with the disease, and that number is expected to increase 10-fold by 2040, Pfizer added.

FDA Panel Recommends TNF Blocker for Ulcerative Colitis
SILVER SPRING, MD. – The majority of a Food and Drug Administration panel on Aug. 28 agreed that data on adalimumab from two studies of patients with moderately to severely active ulcerative colitis who have not had an adequate response to conventional treatments, support its approval for this indication.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 15-2 that, based on these studies, the expected benefits of adalimumab, a subcutaneously administered tumor necrosis factor blocker, outweighed its potential risks as a treatment for such patients. While most of the panel agreed that the recommended dose had been shown to be clinically effective, they agreed that the optimal dose for treating ulcerative colitis (UC) had not yet been determined and that postapproval studies addressing optimal dosing, subpopulations of patients who may benefit most from treatment, and long-term safety are needed.
Those voting in favor of approval cited the need for more treatments and the availability of a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects. And although the differences in remission rates between placebo and treatment at 8 weeks and at 1 year were less than 10% – one of the main issues raised by FDA reviewers – most of the panel members agreed that these differences still represented a clinically meaningful benefit. Infliximab (Remicade), an intravenous TNF blocker, is approved for treating UC.
One of the panelists voting in favor of approval, Dr. Andelka LoSavio of the department of gastroenterology and nutrition, Loyola University, Maywood, Ill., said that once adalimumab became available for patients with UC, its use would provide more insight into which subpopulations of patients with UC can benefit most, which was the case with Crohn’s disease and adalimumab. She and other panel members supported the company’s recommendation that if a patient does not respond after 8 weeks of treatment, that treatment be stopped.
The two statisticians on the panel voted no on the risk-benefit question for reasons that included inadequate long-term data, missing data, and uncertainties about the dose.
Since adalimumab – marketed as Humira by Abbott Laboratories – was first approved in 2002 for treating adults with moderately to severely active rheumatoid arthritis, it has been approved for treating moderately to severely active Crohn’s disease, as well as psoriasis, ankylosing spondylitis, and juvenile idiopathic arthritis indications. Based on the same data submitted to the FDA, adalimumab was approved in April 2012 in the European Union for treating adults with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy, according to Abbott.
The recommended dosing schedule for UC is a single 160 mg starting dose, followed by 80 mg on day 15, and then 40 mg every other week starting at day 29, continuing treatment only in people who have responded within the first 8 weeks of treatment.
Abbott filed for approval in January 2011, but the FDA raised questions about whether the appropriate dose had been studied and the strength of the results in the two pivotal trials, including whether the differences in clinical remission rates between placebo and treatment were clinically meaningful.
The two double-blind placebo-controlled phase III studies compared adalimumab to placebo in 1,094 treatment-refractory patients with moderately to severely active UC. In one 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% among those on placebo, a 9.3% difference that was statistically significant.
In the second study, which followed patients for 1 year and included patients who had been treated with infliximab (40%), the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference that was statistically significant. At 52 weeks, the remission rate was 17.3% among those on adalimumab, vs. 8.5% among those on placebo, an 8.8% difference that also was statistically significant. No new safety signals for adalimumab were identified in the trials. If approved, the company plans to start a postmarketing registry of patients with UC treated with adalimumab.
If approved for UC, adalimumab will be the only self-administered biologic therapy available for patients with UC, the company pointed out.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The majority of a Food and Drug Administration panel on Aug. 28 agreed that data on adalimumab from two studies of patients with moderately to severely active ulcerative colitis who have not had an adequate response to conventional treatments, support its approval for this indication.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 15-2 that, based on these studies, the expected benefits of adalimumab, a subcutaneously administered tumor necrosis factor blocker, outweighed its potential risks as a treatment for such patients. While most of the panel agreed that the recommended dose had been shown to be clinically effective, they agreed that the optimal dose for treating ulcerative colitis (UC) had not yet been determined and that postapproval studies addressing optimal dosing, subpopulations of patients who may benefit most from treatment, and long-term safety are needed.
Those voting in favor of approval cited the need for more treatments and the availability of a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects. And although the differences in remission rates between placebo and treatment at 8 weeks and at 1 year were less than 10% – one of the main issues raised by FDA reviewers – most of the panel members agreed that these differences still represented a clinically meaningful benefit. Infliximab (Remicade), an intravenous TNF blocker, is approved for treating UC.
One of the panelists voting in favor of approval, Dr. Andelka LoSavio of the department of gastroenterology and nutrition, Loyola University, Maywood, Ill., said that once adalimumab became available for patients with UC, its use would provide more insight into which subpopulations of patients with UC can benefit most, which was the case with Crohn’s disease and adalimumab. She and other panel members supported the company’s recommendation that if a patient does not respond after 8 weeks of treatment, that treatment be stopped.
The two statisticians on the panel voted no on the risk-benefit question for reasons that included inadequate long-term data, missing data, and uncertainties about the dose.
Since adalimumab – marketed as Humira by Abbott Laboratories – was first approved in 2002 for treating adults with moderately to severely active rheumatoid arthritis, it has been approved for treating moderately to severely active Crohn’s disease, as well as psoriasis, ankylosing spondylitis, and juvenile idiopathic arthritis indications. Based on the same data submitted to the FDA, adalimumab was approved in April 2012 in the European Union for treating adults with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy, according to Abbott.
The recommended dosing schedule for UC is a single 160 mg starting dose, followed by 80 mg on day 15, and then 40 mg every other week starting at day 29, continuing treatment only in people who have responded within the first 8 weeks of treatment.
Abbott filed for approval in January 2011, but the FDA raised questions about whether the appropriate dose had been studied and the strength of the results in the two pivotal trials, including whether the differences in clinical remission rates between placebo and treatment were clinically meaningful.
The two double-blind placebo-controlled phase III studies compared adalimumab to placebo in 1,094 treatment-refractory patients with moderately to severely active UC. In one 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% among those on placebo, a 9.3% difference that was statistically significant.
In the second study, which followed patients for 1 year and included patients who had been treated with infliximab (40%), the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference that was statistically significant. At 52 weeks, the remission rate was 17.3% among those on adalimumab, vs. 8.5% among those on placebo, an 8.8% difference that also was statistically significant. No new safety signals for adalimumab were identified in the trials. If approved, the company plans to start a postmarketing registry of patients with UC treated with adalimumab.
If approved for UC, adalimumab will be the only self-administered biologic therapy available for patients with UC, the company pointed out.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The majority of a Food and Drug Administration panel on Aug. 28 agreed that data on adalimumab from two studies of patients with moderately to severely active ulcerative colitis who have not had an adequate response to conventional treatments, support its approval for this indication.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 15-2 that, based on these studies, the expected benefits of adalimumab, a subcutaneously administered tumor necrosis factor blocker, outweighed its potential risks as a treatment for such patients. While most of the panel agreed that the recommended dose had been shown to be clinically effective, they agreed that the optimal dose for treating ulcerative colitis (UC) had not yet been determined and that postapproval studies addressing optimal dosing, subpopulations of patients who may benefit most from treatment, and long-term safety are needed.
Those voting in favor of approval cited the need for more treatments and the availability of a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects. And although the differences in remission rates between placebo and treatment at 8 weeks and at 1 year were less than 10% – one of the main issues raised by FDA reviewers – most of the panel members agreed that these differences still represented a clinically meaningful benefit. Infliximab (Remicade), an intravenous TNF blocker, is approved for treating UC.
One of the panelists voting in favor of approval, Dr. Andelka LoSavio of the department of gastroenterology and nutrition, Loyola University, Maywood, Ill., said that once adalimumab became available for patients with UC, its use would provide more insight into which subpopulations of patients with UC can benefit most, which was the case with Crohn’s disease and adalimumab. She and other panel members supported the company’s recommendation that if a patient does not respond after 8 weeks of treatment, that treatment be stopped.
The two statisticians on the panel voted no on the risk-benefit question for reasons that included inadequate long-term data, missing data, and uncertainties about the dose.
Since adalimumab – marketed as Humira by Abbott Laboratories – was first approved in 2002 for treating adults with moderately to severely active rheumatoid arthritis, it has been approved for treating moderately to severely active Crohn’s disease, as well as psoriasis, ankylosing spondylitis, and juvenile idiopathic arthritis indications. Based on the same data submitted to the FDA, adalimumab was approved in April 2012 in the European Union for treating adults with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy, according to Abbott.
The recommended dosing schedule for UC is a single 160 mg starting dose, followed by 80 mg on day 15, and then 40 mg every other week starting at day 29, continuing treatment only in people who have responded within the first 8 weeks of treatment.
Abbott filed for approval in January 2011, but the FDA raised questions about whether the appropriate dose had been studied and the strength of the results in the two pivotal trials, including whether the differences in clinical remission rates between placebo and treatment were clinically meaningful.
The two double-blind placebo-controlled phase III studies compared adalimumab to placebo in 1,094 treatment-refractory patients with moderately to severely active UC. In one 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% among those on placebo, a 9.3% difference that was statistically significant.
In the second study, which followed patients for 1 year and included patients who had been treated with infliximab (40%), the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference that was statistically significant. At 52 weeks, the remission rate was 17.3% among those on adalimumab, vs. 8.5% among those on placebo, an 8.8% difference that also was statistically significant. No new safety signals for adalimumab were identified in the trials. If approved, the company plans to start a postmarketing registry of patients with UC treated with adalimumab.
If approved for UC, adalimumab will be the only self-administered biologic therapy available for patients with UC, the company pointed out.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
AT A MEETING OF THE FDA'S GASTROINTESTINAL DRUGS ADVISORY COMMITTEE
First Generic Pioglitazone Approved, With HF Warning
The first generic formulation of the orally administered type 2 diabetes drug pioglitazone has been approved by the Food and Drug Administration, the agency announced on Aug. 17.
Pioglitazone, a thiazolidinedione (TZD), was approved in 1999, as an adjunct to diet and exercise, to improve glycemic control in adults with type 2 diabetes mellitus, and has been marketed as Actos by Takeda Pharmaceuticals America. It is taken once daily without regard to meals.
The generic formulation is manufactured by Mylan Pharmaceuticals. Approved are 15-mg, 30-mg, and 45-mg tablets, the three Actos doses available. As with the brand-name version, patients will receive a Medication Guide with all filled generic pioglitazone prescriptions, which provides information about the drug’s use and safety, according to the FDA statement.
The pioglitazone label has a boxed warning stating that treatment with TZDs, including pioglitazone, can cause or exacerbate heart failure, and that patients on treatment with pioglitazone should be monitored closely for heart failure; it is contraindicated in patients with New York Heart Association (NYHA) III or IV heart failure. The label also includes the statement that the risk of bladder cancer may be increased after 1 year of treatment with pioglitazone, which was added to the drug’s label in 2011. This boxed warning was added to the TZD labels in 2007.
Pioglitazone is also being studied as a treatment for Alzheimer’s disease.
The other TZD on the U.S. market is rosiglitazone (Avandia), but its use is highly restricted because of its associated cardiovascular risks. The Avandia label also includes a statement on the increased risk of myocardial infarction associated with treatment.
Serious adverse events associated with pioglitazone should be reported to FDA’s MedWatch adverse event reporting program at 800-332-1088 or here.
The first generic formulation of the orally administered type 2 diabetes drug pioglitazone has been approved by the Food and Drug Administration, the agency announced on Aug. 17.
Pioglitazone, a thiazolidinedione (TZD), was approved in 1999, as an adjunct to diet and exercise, to improve glycemic control in adults with type 2 diabetes mellitus, and has been marketed as Actos by Takeda Pharmaceuticals America. It is taken once daily without regard to meals.
The generic formulation is manufactured by Mylan Pharmaceuticals. Approved are 15-mg, 30-mg, and 45-mg tablets, the three Actos doses available. As with the brand-name version, patients will receive a Medication Guide with all filled generic pioglitazone prescriptions, which provides information about the drug’s use and safety, according to the FDA statement.
The pioglitazone label has a boxed warning stating that treatment with TZDs, including pioglitazone, can cause or exacerbate heart failure, and that patients on treatment with pioglitazone should be monitored closely for heart failure; it is contraindicated in patients with New York Heart Association (NYHA) III or IV heart failure. The label also includes the statement that the risk of bladder cancer may be increased after 1 year of treatment with pioglitazone, which was added to the drug’s label in 2011. This boxed warning was added to the TZD labels in 2007.
Pioglitazone is also being studied as a treatment for Alzheimer’s disease.
The other TZD on the U.S. market is rosiglitazone (Avandia), but its use is highly restricted because of its associated cardiovascular risks. The Avandia label also includes a statement on the increased risk of myocardial infarction associated with treatment.
Serious adverse events associated with pioglitazone should be reported to FDA’s MedWatch adverse event reporting program at 800-332-1088 or here.
The first generic formulation of the orally administered type 2 diabetes drug pioglitazone has been approved by the Food and Drug Administration, the agency announced on Aug. 17.
Pioglitazone, a thiazolidinedione (TZD), was approved in 1999, as an adjunct to diet and exercise, to improve glycemic control in adults with type 2 diabetes mellitus, and has been marketed as Actos by Takeda Pharmaceuticals America. It is taken once daily without regard to meals.
The generic formulation is manufactured by Mylan Pharmaceuticals. Approved are 15-mg, 30-mg, and 45-mg tablets, the three Actos doses available. As with the brand-name version, patients will receive a Medication Guide with all filled generic pioglitazone prescriptions, which provides information about the drug’s use and safety, according to the FDA statement.
The pioglitazone label has a boxed warning stating that treatment with TZDs, including pioglitazone, can cause or exacerbate heart failure, and that patients on treatment with pioglitazone should be monitored closely for heart failure; it is contraindicated in patients with New York Heart Association (NYHA) III or IV heart failure. The label also includes the statement that the risk of bladder cancer may be increased after 1 year of treatment with pioglitazone, which was added to the drug’s label in 2011. This boxed warning was added to the TZD labels in 2007.
Pioglitazone is also being studied as a treatment for Alzheimer’s disease.
The other TZD on the U.S. market is rosiglitazone (Avandia), but its use is highly restricted because of its associated cardiovascular risks. The Avandia label also includes a statement on the increased risk of myocardial infarction associated with treatment.
Serious adverse events associated with pioglitazone should be reported to FDA’s MedWatch adverse event reporting program at 800-332-1088 or here.
FDA Warns About Pediatric Codeine-Related Deaths
The Food and Drug Administration issued a warning on Aug. 15 about the risk of death in children who receive codeine for postoperative pain, particularly after tonsillectomy and/or adenoidectomy, based on three deaths and a nonfatal case of life-threatening respiratory depression.
The three children who died had evidence of being ultrarapid metabolizers of codeine, and the fourth case was in a child with evidence of being an "extensive" metabolizer, the FDA said in a statement announcing the warning. All four children, aged 2-5 years, underwent a tonsillectomy and/or adenoidectomy for treating obstructive sleep apnea syndrome, and received codeine within the typical dose range.

"The FDA is currently conducting a review of adverse event reports and other information to determine if there are additional cases of inadvertent overdose or death in children taking codeine, and if these adverse events occur during treatment of other kinds of pain, such as postoperative pain following other types of surgery or procedures," Dr. Bob Rappaport, director of the Division of Anesthesia, Analgesia, and Addiction Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
For now, the FDA is advising health care professionals and parents to be aware of the risks of codeine in children, "particularly in those who have undergone tonsillectomy and/or adenoidectomy for obstructive sleep apnea syndrome," and – when they prescribe medications that contain codeine to children – to use "the lowest effective dose for the shortest time on an as-needed basis." Parents and caregivers should stop codeine and get immediate medical attention if a child who has been receiving codeine after surgery has any symptoms of an overdose.
CYP2D6 (cytochrome P450 2D6) metabolizes codeine to morphine. Ultrametabolizers of codeine have a genetic variation in this enzyme and are more likely to have abnormally high levels of morphine after taking codeine, and therefore are at greater risk of related adverse events and death.
The four children started to show signs of morphine toxicity within 1-2 days of starting the codeine, and in the three children who died, postmortem morphine levels were "substantially higher" than the normal therapeutic range, according to the FDA. The four cases were also described in Pediatrics (2012;129:e1343-7).
An estimated 1%-7% of the general population are ultrarapid metabolizers, but the rate is much higher in certain ethnic groups, with the highest prevalence (29%) reported for people of Ethiopian descent. The reported prevalence is 6% in the Greek population, 3.4%-6.5% among the African American population, 3.6% in whites, and 1%-2% in northern Europeans.
When the FDA review is completed, the agency plans to provide an update.
The risk of morphine overdose in nursing infants whose mothers are taking codeine and who are ultrarapid metabolizers was recognized several years ago. In 2007, a year after the first case report was described in the Lancet (2006;368:704), the FDA issued a warning about this risk.
The FDA has also posted a consumer update on its website.
The full alert is available, and possible cases should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
The Food and Drug Administration issued a warning on Aug. 15 about the risk of death in children who receive codeine for postoperative pain, particularly after tonsillectomy and/or adenoidectomy, based on three deaths and a nonfatal case of life-threatening respiratory depression.
The three children who died had evidence of being ultrarapid metabolizers of codeine, and the fourth case was in a child with evidence of being an "extensive" metabolizer, the FDA said in a statement announcing the warning. All four children, aged 2-5 years, underwent a tonsillectomy and/or adenoidectomy for treating obstructive sleep apnea syndrome, and received codeine within the typical dose range.
"The FDA is currently conducting a review of adverse event reports and other information to determine if there are additional cases of inadvertent overdose or death in children taking codeine, and if these adverse events occur during treatment of other kinds of pain, such as postoperative pain following other types of surgery or procedures," Dr. Bob Rappaport, director of the Division of Anesthesia, Analgesia, and Addiction Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
For now, the FDA is advising health care professionals and parents to be aware of the risks of codeine in children, "particularly in those who have undergone tonsillectomy and/or adenoidectomy for obstructive sleep apnea syndrome," and – when they prescribe medications that contain codeine to children – to use "the lowest effective dose for the shortest time on an as-needed basis." Parents and caregivers should stop codeine and get immediate medical attention if a child who has been receiving codeine after surgery has any symptoms of an overdose.
CYP2D6 (cytochrome P450 2D6) metabolizes codeine to morphine. Ultrametabolizers of codeine have a genetic variation in this enzyme and are more likely to have abnormally high levels of morphine after taking codeine, and therefore are at greater risk of related adverse events and death.
The four children started to show signs of morphine toxicity within 1-2 days of starting the codeine, and in the three children who died, postmortem morphine levels were "substantially higher" than the normal therapeutic range, according to the FDA. The four cases were also described in Pediatrics (2012;129:e1343-7).
An estimated 1%-7% of the general population are ultrarapid metabolizers, but the rate is much higher in certain ethnic groups, with the highest prevalence (29%) reported for people of Ethiopian descent. The reported prevalence is 6% in the Greek population, 3.4%-6.5% among the African American population, 3.6% in whites, and 1%-2% in northern Europeans.
When the FDA review is completed, the agency plans to provide an update.
The risk of morphine overdose in nursing infants whose mothers are taking codeine and who are ultrarapid metabolizers was recognized several years ago. In 2007, a year after the first case report was described in the Lancet (2006;368:704), the FDA issued a warning about this risk.
The FDA has also posted a consumer update on its website.
The full alert is available, and possible cases should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
The Food and Drug Administration issued a warning on Aug. 15 about the risk of death in children who receive codeine for postoperative pain, particularly after tonsillectomy and/or adenoidectomy, based on three deaths and a nonfatal case of life-threatening respiratory depression.
The three children who died had evidence of being ultrarapid metabolizers of codeine, and the fourth case was in a child with evidence of being an "extensive" metabolizer, the FDA said in a statement announcing the warning. All four children, aged 2-5 years, underwent a tonsillectomy and/or adenoidectomy for treating obstructive sleep apnea syndrome, and received codeine within the typical dose range.
"The FDA is currently conducting a review of adverse event reports and other information to determine if there are additional cases of inadvertent overdose or death in children taking codeine, and if these adverse events occur during treatment of other kinds of pain, such as postoperative pain following other types of surgery or procedures," Dr. Bob Rappaport, director of the Division of Anesthesia, Analgesia, and Addiction Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
For now, the FDA is advising health care professionals and parents to be aware of the risks of codeine in children, "particularly in those who have undergone tonsillectomy and/or adenoidectomy for obstructive sleep apnea syndrome," and – when they prescribe medications that contain codeine to children – to use "the lowest effective dose for the shortest time on an as-needed basis." Parents and caregivers should stop codeine and get immediate medical attention if a child who has been receiving codeine after surgery has any symptoms of an overdose.
CYP2D6 (cytochrome P450 2D6) metabolizes codeine to morphine. Ultrametabolizers of codeine have a genetic variation in this enzyme and are more likely to have abnormally high levels of morphine after taking codeine, and therefore are at greater risk of related adverse events and death.
The four children started to show signs of morphine toxicity within 1-2 days of starting the codeine, and in the three children who died, postmortem morphine levels were "substantially higher" than the normal therapeutic range, according to the FDA. The four cases were also described in Pediatrics (2012;129:e1343-7).
An estimated 1%-7% of the general population are ultrarapid metabolizers, but the rate is much higher in certain ethnic groups, with the highest prevalence (29%) reported for people of Ethiopian descent. The reported prevalence is 6% in the Greek population, 3.4%-6.5% among the African American population, 3.6% in whites, and 1%-2% in northern Europeans.
When the FDA review is completed, the agency plans to provide an update.
The risk of morphine overdose in nursing infants whose mothers are taking codeine and who are ultrarapid metabolizers was recognized several years ago. In 2007, a year after the first case report was described in the Lancet (2006;368:704), the FDA issued a warning about this risk.
The FDA has also posted a consumer update on its website.
The full alert is available, and possible cases should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.

Report Cites Ways to Improve Obesity Drug Studies
Measures that take into account the effects of weight loss on specific areas of "feeling and function" should be included in the evaluation of drugs being developed to treat obesity, according to a report on obesity drug outcome measures, released by the George Washington University School of Public Health and Health Services.
"The FDA should consider patient improvements in feeling and function associated with weight loss as part of the risk-benefit calculus in its evaluation of drugs for the treatment of obesity where data are provided demonstrating benefit in a drug-specific clinical trial," concludes the report, called "Obesity Drug Outcome Measures."
The specific areas that can be measured using validated tools include osteoarthritis, mobility, and urinary incontinence, and could be considered secondary end points for obesity drug trials, the report pointed out.
The report is a result of the discussions and recommendations made by stakeholders convened by GWU – which included clinicians who treat obesity, Food and Drug Administration officials who review these drugs, patient advocates, and pharmaceutical companies – to evaluate the issues and challenges related to FDA approval of drugs for obesity and the appropriate use of obesity drugs, including the need for more treatments to help fill the void between lifestyle interventions and bariatric surgery.

The risks associated with a drug treatment may be more acceptable to an obese patient with "feeling and function abnormalities," Dr. Donna Ryan said during a webinar to discuss the report. Dr. Ryan, who is involved in clinical trials and research of obesity at the Pennington Biomedical Research Center, Baton Rouge, La., noted that one of the main focuses of the panel was on the various conditions that can be improved with modest weight loss, and cited a study that found a modest amount of weight loss at 1 year was associated with improvements in urinary incontinence. The effect of weight loss on improvements in sleep apnea was also mentioned during the webinar.
Among the other recommendations in the report were considering the drugs for chronic weight management as obesity treatments, not weight loss drugs, and limiting the availability of these drugs to people "for whom they are medically appropriate," and studying obesity drugs in pediatric patients.
Currently, the FDA’s draft guidance for industry for developing products for weight management are based on changes in body weight, as a primary end point, and changes in cardiometabolic parameters, as a secondary end point. The guidance, used since 2007, states that a drug can be considered effective if after 1 year of treatment, the difference in mean weight loss between patients treated with the product and those treated with placebo is at least 5% and the difference is statistically significant; or the proportion of subjects who lose at least 5% of their baseline body weight on the active product is at least 35%, is about double the proportion in the placebo-treated group – and the difference between the groups is statistically significant.
The FDA does not accept any quality of life or patient-reported outcome measurements when considering obesity drugs for approval, because the tools that measure these outcomes do not meet the agency’s standards.
The research project was supported by unrestricted gifts from Eisai Co.; Novo Nordisk Worldwide; Obesity Action Coalition; Orexigen Therapeutics; Takeda Pharmaceuticals, USA; and Vivus.
Measures that take into account the effects of weight loss on specific areas of "feeling and function" should be included in the evaluation of drugs being developed to treat obesity, according to a report on obesity drug outcome measures, released by the George Washington University School of Public Health and Health Services.
"The FDA should consider patient improvements in feeling and function associated with weight loss as part of the risk-benefit calculus in its evaluation of drugs for the treatment of obesity where data are provided demonstrating benefit in a drug-specific clinical trial," concludes the report, called "Obesity Drug Outcome Measures."
The specific areas that can be measured using validated tools include osteoarthritis, mobility, and urinary incontinence, and could be considered secondary end points for obesity drug trials, the report pointed out.
The report is a result of the discussions and recommendations made by stakeholders convened by GWU – which included clinicians who treat obesity, Food and Drug Administration officials who review these drugs, patient advocates, and pharmaceutical companies – to evaluate the issues and challenges related to FDA approval of drugs for obesity and the appropriate use of obesity drugs, including the need for more treatments to help fill the void between lifestyle interventions and bariatric surgery.
The risks associated with a drug treatment may be more acceptable to an obese patient with "feeling and function abnormalities," Dr. Donna Ryan said during a webinar to discuss the report. Dr. Ryan, who is involved in clinical trials and research of obesity at the Pennington Biomedical Research Center, Baton Rouge, La., noted that one of the main focuses of the panel was on the various conditions that can be improved with modest weight loss, and cited a study that found a modest amount of weight loss at 1 year was associated with improvements in urinary incontinence. The effect of weight loss on improvements in sleep apnea was also mentioned during the webinar.
Among the other recommendations in the report were considering the drugs for chronic weight management as obesity treatments, not weight loss drugs, and limiting the availability of these drugs to people "for whom they are medically appropriate," and studying obesity drugs in pediatric patients.
Currently, the FDA’s draft guidance for industry for developing products for weight management are based on changes in body weight, as a primary end point, and changes in cardiometabolic parameters, as a secondary end point. The guidance, used since 2007, states that a drug can be considered effective if after 1 year of treatment, the difference in mean weight loss between patients treated with the product and those treated with placebo is at least 5% and the difference is statistically significant; or the proportion of subjects who lose at least 5% of their baseline body weight on the active product is at least 35%, is about double the proportion in the placebo-treated group – and the difference between the groups is statistically significant.
The FDA does not accept any quality of life or patient-reported outcome measurements when considering obesity drugs for approval, because the tools that measure these outcomes do not meet the agency’s standards.
The research project was supported by unrestricted gifts from Eisai Co.; Novo Nordisk Worldwide; Obesity Action Coalition; Orexigen Therapeutics; Takeda Pharmaceuticals, USA; and Vivus.
Measures that take into account the effects of weight loss on specific areas of "feeling and function" should be included in the evaluation of drugs being developed to treat obesity, according to a report on obesity drug outcome measures, released by the George Washington University School of Public Health and Health Services.
"The FDA should consider patient improvements in feeling and function associated with weight loss as part of the risk-benefit calculus in its evaluation of drugs for the treatment of obesity where data are provided demonstrating benefit in a drug-specific clinical trial," concludes the report, called "Obesity Drug Outcome Measures."
The specific areas that can be measured using validated tools include osteoarthritis, mobility, and urinary incontinence, and could be considered secondary end points for obesity drug trials, the report pointed out.
The report is a result of the discussions and recommendations made by stakeholders convened by GWU – which included clinicians who treat obesity, Food and Drug Administration officials who review these drugs, patient advocates, and pharmaceutical companies – to evaluate the issues and challenges related to FDA approval of drugs for obesity and the appropriate use of obesity drugs, including the need for more treatments to help fill the void between lifestyle interventions and bariatric surgery.
The risks associated with a drug treatment may be more acceptable to an obese patient with "feeling and function abnormalities," Dr. Donna Ryan said during a webinar to discuss the report. Dr. Ryan, who is involved in clinical trials and research of obesity at the Pennington Biomedical Research Center, Baton Rouge, La., noted that one of the main focuses of the panel was on the various conditions that can be improved with modest weight loss, and cited a study that found a modest amount of weight loss at 1 year was associated with improvements in urinary incontinence. The effect of weight loss on improvements in sleep apnea was also mentioned during the webinar.
Among the other recommendations in the report were considering the drugs for chronic weight management as obesity treatments, not weight loss drugs, and limiting the availability of these drugs to people "for whom they are medically appropriate," and studying obesity drugs in pediatric patients.
Currently, the FDA’s draft guidance for industry for developing products for weight management are based on changes in body weight, as a primary end point, and changes in cardiometabolic parameters, as a secondary end point. The guidance, used since 2007, states that a drug can be considered effective if after 1 year of treatment, the difference in mean weight loss between patients treated with the product and those treated with placebo is at least 5% and the difference is statistically significant; or the proportion of subjects who lose at least 5% of their baseline body weight on the active product is at least 35%, is about double the proportion in the placebo-treated group – and the difference between the groups is statistically significant.
The FDA does not accept any quality of life or patient-reported outcome measurements when considering obesity drugs for approval, because the tools that measure these outcomes do not meet the agency’s standards.
The research project was supported by unrestricted gifts from Eisai Co.; Novo Nordisk Worldwide; Obesity Action Coalition; Orexigen Therapeutics; Takeda Pharmaceuticals, USA; and Vivus.
FROM A WEBINAR SPONSORED BY THE GEORGE WASHINGTON UNIVERSITY SCHOOL OF PUBLIC HEALTH AND HEALTH SERVICES

Flu Vaccine Approved for Upcoming Season
The influenza vaccine for the 2012-13 season has been approved by the Food and Drug Administration, the agency has announced.
The three strains that are included are an A/California/7/2009 (H1N1)-like virus, which was included in the 2011-2012 influenza vaccine, and two new strains: an A/Victoria/361/2011 (H3N2)-like virus, and a B/Wisconsin/1/2010-like virus.

"It is especially important to get vaccinated this year because two of the three virus strains used in this season’s influenza vaccines differ from the strains included in last year’s vaccines," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in an Aug. 13 statement.
Six manufacturers are licensed to produce and distribute influenza vaccine in the United States. The approved products are Afluria (CSL Limited); Fluarix (GlaxoSmithKline Biologicals); FluLaval (ID Biomedical Corp.); FluMist (MedImmune Vaccines Inc.); Fluvirin (Novartis Vaccines and Diagnostics Limited); and Fluzone, Fluzone High-Dose, and Fluzone Intradermal (Sanofi Pasteur).
Each year, the selection of strains to be included in the influenza vaccine is based on information about influenza virus circulating worldwide during the previous season and on recommendations from the FDA’s Vaccines and Related Biological Products Advisory Committee.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommends an annual influenza vaccine for everyone aged 6 months and older.
The influenza vaccine for the 2012-13 season has been approved by the Food and Drug Administration, the agency has announced.
The three strains that are included are an A/California/7/2009 (H1N1)-like virus, which was included in the 2011-2012 influenza vaccine, and two new strains: an A/Victoria/361/2011 (H3N2)-like virus, and a B/Wisconsin/1/2010-like virus.
"It is especially important to get vaccinated this year because two of the three virus strains used in this season’s influenza vaccines differ from the strains included in last year’s vaccines," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in an Aug. 13 statement.
Six manufacturers are licensed to produce and distribute influenza vaccine in the United States. The approved products are Afluria (CSL Limited); Fluarix (GlaxoSmithKline Biologicals); FluLaval (ID Biomedical Corp.); FluMist (MedImmune Vaccines Inc.); Fluvirin (Novartis Vaccines and Diagnostics Limited); and Fluzone, Fluzone High-Dose, and Fluzone Intradermal (Sanofi Pasteur).
Each year, the selection of strains to be included in the influenza vaccine is based on information about influenza virus circulating worldwide during the previous season and on recommendations from the FDA’s Vaccines and Related Biological Products Advisory Committee.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommends an annual influenza vaccine for everyone aged 6 months and older.
The influenza vaccine for the 2012-13 season has been approved by the Food and Drug Administration, the agency has announced.
The three strains that are included are an A/California/7/2009 (H1N1)-like virus, which was included in the 2011-2012 influenza vaccine, and two new strains: an A/Victoria/361/2011 (H3N2)-like virus, and a B/Wisconsin/1/2010-like virus.
"It is especially important to get vaccinated this year because two of the three virus strains used in this season’s influenza vaccines differ from the strains included in last year’s vaccines," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in an Aug. 13 statement.
Six manufacturers are licensed to produce and distribute influenza vaccine in the United States. The approved products are Afluria (CSL Limited); Fluarix (GlaxoSmithKline Biologicals); FluLaval (ID Biomedical Corp.); FluMist (MedImmune Vaccines Inc.); Fluvirin (Novartis Vaccines and Diagnostics Limited); and Fluzone, Fluzone High-Dose, and Fluzone Intradermal (Sanofi Pasteur).
Each year, the selection of strains to be included in the influenza vaccine is based on information about influenza virus circulating worldwide during the previous season and on recommendations from the FDA’s Vaccines and Related Biological Products Advisory Committee.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommends an annual influenza vaccine for everyone aged 6 months and older.

Mechanical Embolectomy Device Cleared for Acute Stroke Treatment
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed ; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.

Courtesy Stryker Neurovascular
An illustration of the Trevo Pro Retriever engaging a thrombus.
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed ; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.
Courtesy Stryker Neurovascular
An illustration of the Trevo Pro Retriever engaging a thrombus.
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed ; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.
Courtesy Stryker Neurovascular
An illustration of the Trevo Pro Retriever engaging a thrombus.
Mechanical Embolectomy Device Cleared for Acute Stroke Treatment
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
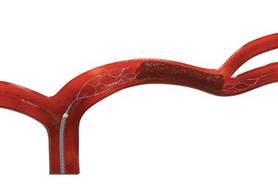
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.
A thrombus retrieval device for use in patients experiencing an acute ischemic stroke has been cleared for marketing in the United States by the Food and Drug Administration, the manufacturer announced on Aug. 13.
The device will be marketed as the "Trevo Pro Retriever," according to the statement issued by Stryker Neurovascular.
In May, the company announced the results of the TREVO 2 study, a randomized, clinical trial that compared the Trevo device with another Stryker device, the Merci Retriever, in patients experiencing an acute ischemic stroke. The inclusion criteria were patients aged 18-85 years presenting with clinical signs and symptoms consistent with an acute ischemic stroke, who had either failed; tissue plasminogen activator (TPA) therapy or in whom TPA was contraindicated, according to the description of the study.
The postprocedure revascularization rate was 92% among patients randomized to treatment with the Trevo device, compared with 77% among those treated with the Merci Retriever device, a significant difference, according to the statement announcing the device’s premarket notification clearance. Performance measures that included shorter hospital stays and improvement in the National Institutes of Health Stroke Scale also favored the Trevo Pro Retriever device, the statement said.
The device was developed by Concentric Medical, which was acquired by Stryker in October 2011.

Giant Cell Arteritis: Temporal Artery Biopsy Trumps ACR Criteria
The current American College of Rheumatology criteria for diagnosing giant cell arteritis have low sensitivity and specificity and should be abandoned, according to the authors of a study that analyzed the clinical utility of the guidelines in a retrospective chart review.
The current ACR criteria, published in 1990, "should not be used to determine the presence or absence of GCA," concluded Dr. Ann P. Murchison of the Wills Eye Institute, Philadelphia, and her coauthors. Instead, they recommended that a temporal artery biopsy at least 2 cm long be done for all patients suspected of having GCA, and all patients with a positive results "must be treated with systemic corticosteroids, even in the absence of other" ACR criteria (Am. J. Ophthalmol. 2012 July 20 [doi:10.1016/j.ajo.2012.03.045]).
They also recommended that all physicians who treat GCA should be educated about the importance of the temporal artery biopsy in diagnosing GCA, and that biopsy results – not the ACR criteria – "should be used as the only indicator of the presence or absence of disease in research regarding GCA."
The current ACR criteria, which the authors pointed out are frequently used to diagnose GCA, do not require a positive biopsy for diagnosis: The criteria state that the diagnosis of GCA can be made when patients meet three of five criteria: age older than 50 years; new onset of a localized headache; temporal artery tenderness or a decreased temporal artery pulse; an ESR (erythrocyte sedimentation rate) equal to or greater than 50 mm/hr; or a positive temporal biopsy.
The investigators reviewed the charts of 112 patients who had a temporal artery biopsy between October 2001 and 2006 at the Wills Eye Institute as part of an evaluation for possible GCA, and looked for the presence or absence of the ACR GCA criteria, the results of biopsies, and the progression of vision loss after the cases were diagnosed.
Of the 112 patients, 74 met their criteria for inclusion in the analysis. (Exclusion criteria included having been treated with corticosteroids before presentation and having a biopsy specimen that was shorter than 2 cm in length.) Of those 74 patients, 35 had a positive biopsy, and 39 had a negative biopsy.
Of the 35 patients with a positive biopsy, 9 (25.7%) met only two of the ACR criteria (age older than 50 years and a positive biopsy), and so would not have been diagnosed if the ACR criteria alone had been used to make the diagnosis, despite the positive biopsy. Based on the criteria, these patients would not have been biopsied or treated "and would have been at higher risk for further catastrophic visual loss," the authors wrote.
In the biopsy-positive group, 16 (4.7%) met two criteria before having a biopsy and then required a biopsy for the diagnosis. Another 10 (28.5%) met three or four criteria before having a biopsy.
Of the 39 patients with a negative biopsy, 11 (28.2%) met three or four of the ACR criteria. In these patients, if the criteria alone (without the biopsy) had been used to make the diagnosis, they would have been treated with corticosteroids, which would have exposed them "to potential adverse events from chronic corticosteroid administration," the investigators said. Another 17 (43.6%) met two criteria and had a biopsy to rule out GCA. And 11 (28.2%) met only one of the criteria, so the biopsy would not have changed the diagnosis, based on the ACR criteria, they noted.
Based on their calculations, the sensitivity of the ACR criteria was 74.3% and specificity was 71.8%. Sensitivity dropped to 28.6% when the biopsy results were not included. These calculations, they pointed out, are significantly lower than the sensitivity (93.5%) and specificity (91.2%) cited by the ACR in 1990.
The authors noted that questions regarding the ability of the ACR criteria to reliably diagnose GCA are not new, and cited studies finding that "none of the criteria alone or in combination are as sensitive or specific as a temporal artery biopsy." However, they added, "temporal artery biopsy still is not considered essential" in diagnosing GCA by some physicians.
The authors of the study had no potential conflicts of interest to disclose.
The current American College of Rheumatology criteria for diagnosing giant cell arteritis have low sensitivity and specificity and should be abandoned, according to the authors of a study that analyzed the clinical utility of the guidelines in a retrospective chart review.
The current ACR criteria, published in 1990, "should not be used to determine the presence or absence of GCA," concluded Dr. Ann P. Murchison of the Wills Eye Institute, Philadelphia, and her coauthors. Instead, they recommended that a temporal artery biopsy at least 2 cm long be done for all patients suspected of having GCA, and all patients with a positive results "must be treated with systemic corticosteroids, even in the absence of other" ACR criteria (Am. J. Ophthalmol. 2012 July 20 [doi:10.1016/j.ajo.2012.03.045]).
They also recommended that all physicians who treat GCA should be educated about the importance of the temporal artery biopsy in diagnosing GCA, and that biopsy results – not the ACR criteria – "should be used as the only indicator of the presence or absence of disease in research regarding GCA."
The current ACR criteria, which the authors pointed out are frequently used to diagnose GCA, do not require a positive biopsy for diagnosis: The criteria state that the diagnosis of GCA can be made when patients meet three of five criteria: age older than 50 years; new onset of a localized headache; temporal artery tenderness or a decreased temporal artery pulse; an ESR (erythrocyte sedimentation rate) equal to or greater than 50 mm/hr; or a positive temporal biopsy.
The investigators reviewed the charts of 112 patients who had a temporal artery biopsy between October 2001 and 2006 at the Wills Eye Institute as part of an evaluation for possible GCA, and looked for the presence or absence of the ACR GCA criteria, the results of biopsies, and the progression of vision loss after the cases were diagnosed.
Of the 112 patients, 74 met their criteria for inclusion in the analysis. (Exclusion criteria included having been treated with corticosteroids before presentation and having a biopsy specimen that was shorter than 2 cm in length.) Of those 74 patients, 35 had a positive biopsy, and 39 had a negative biopsy.
Of the 35 patients with a positive biopsy, 9 (25.7%) met only two of the ACR criteria (age older than 50 years and a positive biopsy), and so would not have been diagnosed if the ACR criteria alone had been used to make the diagnosis, despite the positive biopsy. Based on the criteria, these patients would not have been biopsied or treated "and would have been at higher risk for further catastrophic visual loss," the authors wrote.
In the biopsy-positive group, 16 (4.7%) met two criteria before having a biopsy and then required a biopsy for the diagnosis. Another 10 (28.5%) met three or four criteria before having a biopsy.
Of the 39 patients with a negative biopsy, 11 (28.2%) met three or four of the ACR criteria. In these patients, if the criteria alone (without the biopsy) had been used to make the diagnosis, they would have been treated with corticosteroids, which would have exposed them "to potential adverse events from chronic corticosteroid administration," the investigators said. Another 17 (43.6%) met two criteria and had a biopsy to rule out GCA. And 11 (28.2%) met only one of the criteria, so the biopsy would not have changed the diagnosis, based on the ACR criteria, they noted.
Based on their calculations, the sensitivity of the ACR criteria was 74.3% and specificity was 71.8%. Sensitivity dropped to 28.6% when the biopsy results were not included. These calculations, they pointed out, are significantly lower than the sensitivity (93.5%) and specificity (91.2%) cited by the ACR in 1990.
The authors noted that questions regarding the ability of the ACR criteria to reliably diagnose GCA are not new, and cited studies finding that "none of the criteria alone or in combination are as sensitive or specific as a temporal artery biopsy." However, they added, "temporal artery biopsy still is not considered essential" in diagnosing GCA by some physicians.
The authors of the study had no potential conflicts of interest to disclose.
The current American College of Rheumatology criteria for diagnosing giant cell arteritis have low sensitivity and specificity and should be abandoned, according to the authors of a study that analyzed the clinical utility of the guidelines in a retrospective chart review.
The current ACR criteria, published in 1990, "should not be used to determine the presence or absence of GCA," concluded Dr. Ann P. Murchison of the Wills Eye Institute, Philadelphia, and her coauthors. Instead, they recommended that a temporal artery biopsy at least 2 cm long be done for all patients suspected of having GCA, and all patients with a positive results "must be treated with systemic corticosteroids, even in the absence of other" ACR criteria (Am. J. Ophthalmol. 2012 July 20 [doi:10.1016/j.ajo.2012.03.045]).
They also recommended that all physicians who treat GCA should be educated about the importance of the temporal artery biopsy in diagnosing GCA, and that biopsy results – not the ACR criteria – "should be used as the only indicator of the presence or absence of disease in research regarding GCA."
The current ACR criteria, which the authors pointed out are frequently used to diagnose GCA, do not require a positive biopsy for diagnosis: The criteria state that the diagnosis of GCA can be made when patients meet three of five criteria: age older than 50 years; new onset of a localized headache; temporal artery tenderness or a decreased temporal artery pulse; an ESR (erythrocyte sedimentation rate) equal to or greater than 50 mm/hr; or a positive temporal biopsy.
The investigators reviewed the charts of 112 patients who had a temporal artery biopsy between October 2001 and 2006 at the Wills Eye Institute as part of an evaluation for possible GCA, and looked for the presence or absence of the ACR GCA criteria, the results of biopsies, and the progression of vision loss after the cases were diagnosed.
Of the 112 patients, 74 met their criteria for inclusion in the analysis. (Exclusion criteria included having been treated with corticosteroids before presentation and having a biopsy specimen that was shorter than 2 cm in length.) Of those 74 patients, 35 had a positive biopsy, and 39 had a negative biopsy.
Of the 35 patients with a positive biopsy, 9 (25.7%) met only two of the ACR criteria (age older than 50 years and a positive biopsy), and so would not have been diagnosed if the ACR criteria alone had been used to make the diagnosis, despite the positive biopsy. Based on the criteria, these patients would not have been biopsied or treated "and would have been at higher risk for further catastrophic visual loss," the authors wrote.
In the biopsy-positive group, 16 (4.7%) met two criteria before having a biopsy and then required a biopsy for the diagnosis. Another 10 (28.5%) met three or four criteria before having a biopsy.
Of the 39 patients with a negative biopsy, 11 (28.2%) met three or four of the ACR criteria. In these patients, if the criteria alone (without the biopsy) had been used to make the diagnosis, they would have been treated with corticosteroids, which would have exposed them "to potential adverse events from chronic corticosteroid administration," the investigators said. Another 17 (43.6%) met two criteria and had a biopsy to rule out GCA. And 11 (28.2%) met only one of the criteria, so the biopsy would not have changed the diagnosis, based on the ACR criteria, they noted.
Based on their calculations, the sensitivity of the ACR criteria was 74.3% and specificity was 71.8%. Sensitivity dropped to 28.6% when the biopsy results were not included. These calculations, they pointed out, are significantly lower than the sensitivity (93.5%) and specificity (91.2%) cited by the ACR in 1990.
The authors noted that questions regarding the ability of the ACR criteria to reliably diagnose GCA are not new, and cited studies finding that "none of the criteria alone or in combination are as sensitive or specific as a temporal artery biopsy." However, they added, "temporal artery biopsy still is not considered essential" in diagnosing GCA by some physicians.
The authors of the study had no potential conflicts of interest to disclose.
FROM THE ANNALS OF OPHTHALMOLOGY
FDA Approves Aflibercept for Advanced Colorectal Cancer
Aflibercept, an angiogenesis inhibitor, has been approved as a second-line treatment for metastatic colorectal cancer in combination with the FOLFIRI chemotherapy regimen, the Food and Drug Administration announced on Aug. 3.
The indication is for patients whose tumors are resistant to, or have progressed after, an oxaliplatin-containing chemotherapy regimen, according to the FDA statement. The FOLFIRI regimen comprises folinic acid (leucovorin), 5-fluorouracil, and irinotecan.
Approval was based on the phase III VELOUR study of 1,226 patients with metastatic colorectal cancer whose disease progressed during treatment with oxaliplatin-based combination chemotherapy or whose tumor was surgically removed but recurred within 6 months after treatment with oxaliplatin-based combination chemotherapy. Patients continued treatment until cancer progression or unacceptable adverse effects.
The addition of aflibercept was associated with improvements in median overall survival and response rate and with a delay in tumor progression and growth: Overall survival reached a median of 13.5 months among those treated with aflibercept and FOLFIRI, vs. 12 months among those treated with placebo and FOLFIRI. Tumor size was reduced in 20% of those on the combination regimen and 11% of those receiving FOLFIRI plus placebo. In addition, median progression-free survival among patients given aflibercept and FOLIRI was 6.9 months, compared with 4.7 months in the placebo group.
Leucopenia, diarrhea, mouth ulcers, fatigue, hypertension, proteinuria, weight loss, decreased appetite, abdominal pain, and headache were among the most common side effects associated with treatment. A boxed warning covers the increased risk of severe and sometimes fatal bleeding, including gastrointestinal bleeding, GI perforations, and impaired wound healing.
Aflibercept targets the vascular endothelial growth factor (VEGF) and is also referred to as VEGF Trap. It will be marketed as Zaltrap by Sanofi-Aventis in collaboration with Regeneron Pharmaceuticals.
Another VEGF-specific angiogenesis inhibitor, bevacizumab (Avastin), was previously approved as a first- or second- line treatment of patients with metastatic colorectal cancer, in combination with intravenous 5-fluorouracil–based chemotherapy.
"This approval demonstrates the benefits of adding a biological agent, Zaltrap, to a commonly used chemotherapy drug regimen, FOLFIRI," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in the FDA announcement.
Click here to see a discussion of the VELOUR trial results in the journal Community Oncology.
Click here for prescribing information from the FDA.
Aflibercept, an angiogenesis inhibitor, has been approved as a second-line treatment for metastatic colorectal cancer in combination with the FOLFIRI chemotherapy regimen, the Food and Drug Administration announced on Aug. 3.
The indication is for patients whose tumors are resistant to, or have progressed after, an oxaliplatin-containing chemotherapy regimen, according to the FDA statement. The FOLFIRI regimen comprises folinic acid (leucovorin), 5-fluorouracil, and irinotecan.
Approval was based on the phase III VELOUR study of 1,226 patients with metastatic colorectal cancer whose disease progressed during treatment with oxaliplatin-based combination chemotherapy or whose tumor was surgically removed but recurred within 6 months after treatment with oxaliplatin-based combination chemotherapy. Patients continued treatment until cancer progression or unacceptable adverse effects.
The addition of aflibercept was associated with improvements in median overall survival and response rate and with a delay in tumor progression and growth: Overall survival reached a median of 13.5 months among those treated with aflibercept and FOLFIRI, vs. 12 months among those treated with placebo and FOLFIRI. Tumor size was reduced in 20% of those on the combination regimen and 11% of those receiving FOLFIRI plus placebo. In addition, median progression-free survival among patients given aflibercept and FOLIRI was 6.9 months, compared with 4.7 months in the placebo group.
Leucopenia, diarrhea, mouth ulcers, fatigue, hypertension, proteinuria, weight loss, decreased appetite, abdominal pain, and headache were among the most common side effects associated with treatment. A boxed warning covers the increased risk of severe and sometimes fatal bleeding, including gastrointestinal bleeding, GI perforations, and impaired wound healing.
Aflibercept targets the vascular endothelial growth factor (VEGF) and is also referred to as VEGF Trap. It will be marketed as Zaltrap by Sanofi-Aventis in collaboration with Regeneron Pharmaceuticals.
Another VEGF-specific angiogenesis inhibitor, bevacizumab (Avastin), was previously approved as a first- or second- line treatment of patients with metastatic colorectal cancer, in combination with intravenous 5-fluorouracil–based chemotherapy.
"This approval demonstrates the benefits of adding a biological agent, Zaltrap, to a commonly used chemotherapy drug regimen, FOLFIRI," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in the FDA announcement.
Click here to see a discussion of the VELOUR trial results in the journal Community Oncology.
Click here for prescribing information from the FDA.
Aflibercept, an angiogenesis inhibitor, has been approved as a second-line treatment for metastatic colorectal cancer in combination with the FOLFIRI chemotherapy regimen, the Food and Drug Administration announced on Aug. 3.
The indication is for patients whose tumors are resistant to, or have progressed after, an oxaliplatin-containing chemotherapy regimen, according to the FDA statement. The FOLFIRI regimen comprises folinic acid (leucovorin), 5-fluorouracil, and irinotecan.
Approval was based on the phase III VELOUR study of 1,226 patients with metastatic colorectal cancer whose disease progressed during treatment with oxaliplatin-based combination chemotherapy or whose tumor was surgically removed but recurred within 6 months after treatment with oxaliplatin-based combination chemotherapy. Patients continued treatment until cancer progression or unacceptable adverse effects.
The addition of aflibercept was associated with improvements in median overall survival and response rate and with a delay in tumor progression and growth: Overall survival reached a median of 13.5 months among those treated with aflibercept and FOLFIRI, vs. 12 months among those treated with placebo and FOLFIRI. Tumor size was reduced in 20% of those on the combination regimen and 11% of those receiving FOLFIRI plus placebo. In addition, median progression-free survival among patients given aflibercept and FOLIRI was 6.9 months, compared with 4.7 months in the placebo group.
Leucopenia, diarrhea, mouth ulcers, fatigue, hypertension, proteinuria, weight loss, decreased appetite, abdominal pain, and headache were among the most common side effects associated with treatment. A boxed warning covers the increased risk of severe and sometimes fatal bleeding, including gastrointestinal bleeding, GI perforations, and impaired wound healing.
Aflibercept targets the vascular endothelial growth factor (VEGF) and is also referred to as VEGF Trap. It will be marketed as Zaltrap by Sanofi-Aventis in collaboration with Regeneron Pharmaceuticals.
Another VEGF-specific angiogenesis inhibitor, bevacizumab (Avastin), was previously approved as a first- or second- line treatment of patients with metastatic colorectal cancer, in combination with intravenous 5-fluorouracil–based chemotherapy.
"This approval demonstrates the benefits of adding a biological agent, Zaltrap, to a commonly used chemotherapy drug regimen, FOLFIRI," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in the FDA announcement.
Click here to see a discussion of the VELOUR trial results in the journal Community Oncology.
Click here for prescribing information from the FDA.