User login
Pigmenting Purpuric Dermatoses: Striking But Not a Manifestation of COVID-19 Infection
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6

Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1

Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24

Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.

Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6
Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1
Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24
Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.
Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6
Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1
Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24
Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.
Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
Practice Points
- Dermatologists should be aware of the clinical presentations of pigmenting purpuric dermatoses (PPDs).
- Certain PPDs may resemble the thromboembolic events seen in COVID-19. Clinicians should especially be aware of how to differentiate these benign pigmentary disorders from other serious conditions.
- Teledermatology is widely utilized, but caution may be prudent when evaluating erythematous or purpuric dermatoses, especially those of the lower extremities.
- Pigmenting purpuric dermatoses generally are benign and do not require immediate treatment.

Treatment Delay in Melanoma: A Risk Factor Analysis of an Impending Crisis
Melanoma is the most lethal skin cancer and is the second most common cancer in adolescents and young adults.1 It is the fifth most common cancer in the United States based on incidence, which has steadily risen for the last 2 decades.2,3 For melanoma management, delayed initial diagnosis has been associated with more advanced lesions at presentation and poorer outcomes.4 However, the prognostic implications of delaying melanoma management after diagnosis merits further scrutiny.
This study investigates the associations between melanoma treatment delay (MTD) and patient and tumor characteristics. Although most cases undergo surgical treatment first, more advanced stages may require initiating chemotherapy, radiation therapy, or immunotherapy. In addition, patients who are poor surgical candidates may opt for topical field therapy, such as imiquimod for superficial lesions, prior to more definitive treatment.5 In the Medicaid population, patients who are older than 85 years, married, and previously diagnosed with another melanoma and who also have an increased comorbidity burden have a higher likelihood of MTD.6 For nonmelanoma skin cancers, patient denial is the most common patient-specific factor accounting for treatment delay.7 For this study, our aim was to further evaluate the independent risk factors associated with MTD.
Methods
Case Selection
The National Cancer Database (NCDB) was queried for all cutaneous melanoma cases from 2004 to 2015 (N=525,271). The NCDB is an oncology database sourced from more than 1500 accredited cancer facilities in the United States and Puerto Rico. It receives cases from academic hospitals, Veterans Health Administration hospitals, and community centers.8 Annually, the database collects approximately 70% of cancer diagnoses and 48% of melanoma diagnoses in the United States.9,10 Per institutional guidelines, this analysis was determined to be exempt from institutional review board approval due to the deidentified nature of the dataset.
The selection scheme is illustrated in Table 1. International Statistical Classification of Diseases and Related Health Problems histology codes 8720/3 through 8780/3 combined with the site and morphology primary codes C44.0 through C44.9 identified all patients with a diagnosis of cutaneous melanoma. Primary site was established with the histology codes in the following manner: C44.0 through C44.4 for head/neck primary, C44.5 for trunk primary, C44.6 through C44.7 for extremity primary, and C44.8 through C44.9 for not otherwise specified. Because the NCDB does not specify cause of death, any cases in which the melanoma diagnosis was not the patient’s primary (or first) cancer diagnosis were excluded because of potential ambiguity. Cases lacking histologic confirmation of the diagnosis after primary site biopsy or cases diagnosed from autopsy reports also were excluded. Reports missing staging data or undergoing palliative management were removed. In total, 104,118 cases met the inclusion criteria.

Variables of Interest
The NCDB database codes for a variable “Treatment Started, Days from Dx” are defined as the number of days between the date of diagnosis and the date on which treatment—surgery, radiation, systemic, or other therapy—of the patient began at any facility.11 Treatment delays were classified as more than 45 days or more than 90 days. These thresholds were chosen based on previous studies citing a 45-day recommendation as the timeframe in which primary site excision of melanoma should occur for improved outcomes.1,6,12 Additionally, the postponement cutoffs were aligned with prior studies on surgical delay in melanoma for the Medicaid population.6 Delays of 45 days were labeled as moderate MTD (mMTD), whereas postponements more than 90 days were designated as severe MTD (sMTD).
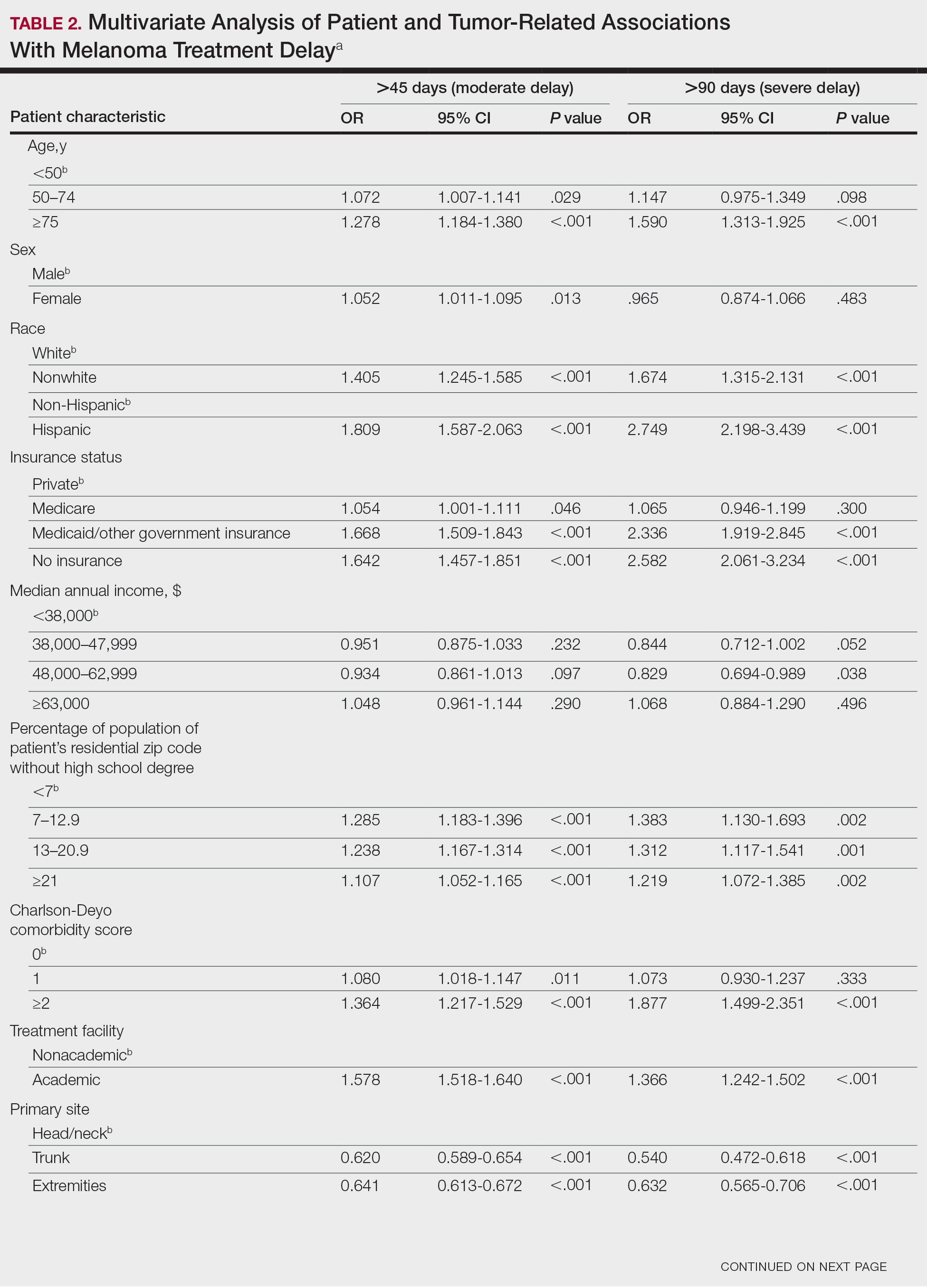
Patient and tumor characteristics were analyzed for associations with MTD (Table 2). Covariates included age, sex, race (white vs nonwhite), Hispanic ethnicity, insurance status (private; Medicare, Medicaid or other government insurance; and no insurance), median annual income of the patient’s residential zip code (based on 2008-2012 census data), percentage of the population of the patient’s residential zip code without a high school degree (based on 2008-2012 census data), Charlson-Deyo (CD) comorbidity score (a weighted score derived from the sum scores for comorbid conditions), geographic location (rural, urban, and metropolitan), and treatment facility (academic vs nonacademic). Tumor characteristics included primary site (head/neck, trunk, and extremities), stage, and Breslow depth of invasion. Tumor stage was determined using the American Joint Committee on Cancer 6th and 7th editions, depending on the patient’s year of diagnosis.

Statistical Methods
χ2 and Fisher exact tests were used to analyze categorical variables involving patient demographics and tumor characteristics by bivariate analysis (Tables 3 and 4). Multivariate analysis determined the relative impact on MTD by including variables that significantly differed on bivariate χ2 analysis (Table 2). Multivariate modeling determined odds ratio (OR) and corresponding 95% CI for the risk-adjusted associations of the variables with MTD. All statistical analyses were performed using SPSS Statistics version 23 (IBM). P<.05 was considered statistically significant, and all statistical tests were 2-tailed. Line graph figures by year of diagnosis were modeled by SPSS using the mean days of delay per year. Independent sample t tests assessed for differences in mean values.
Results
The final study population included 104,118 patients, most of whom were male (56.4%), white (96.6%), and aged 50 to 74 years (54.4%). Most patients were privately insured (52.6%), had no CD comorbidities (87.5%), and lived in metropolitan cities (80.4%)(Table 3). A large majority (95,473 [91.7%]) of patients received surgery as the first means of treatment, with a smaller portion (863 [0.8%]) having unspecified systemic therapy first. The remaining cases were first treated with chemotherapy (1738 [1.7%]), immunotherapy (382 [0.4%]), or radiation (490 [0.5%]), and the rest did not specify treatment sequence. The tumors were most commonly located on the extremities (40.7%), were stage I (41.2%), and had a Breslow depth of less than 1 mm (41.6%).
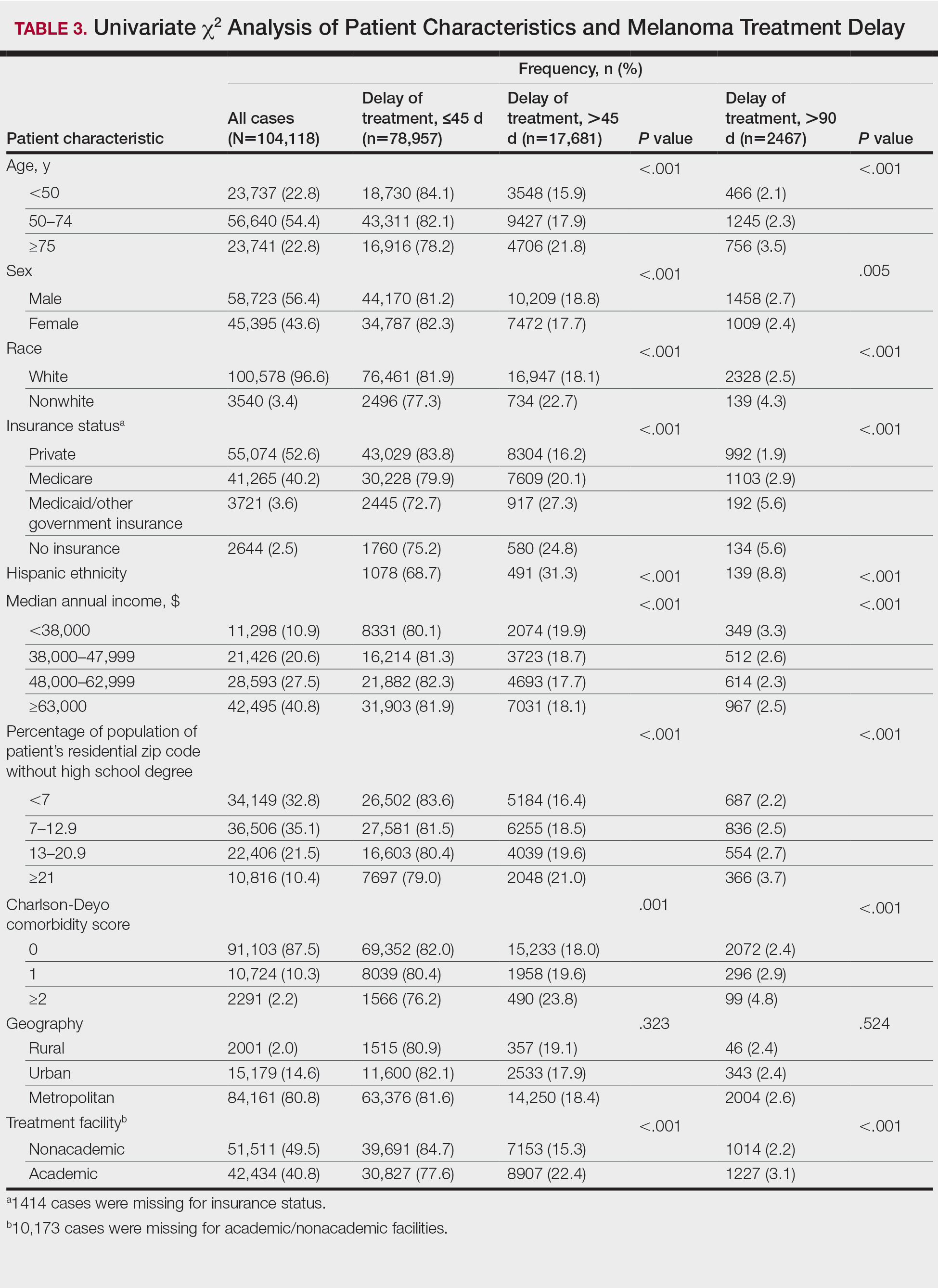
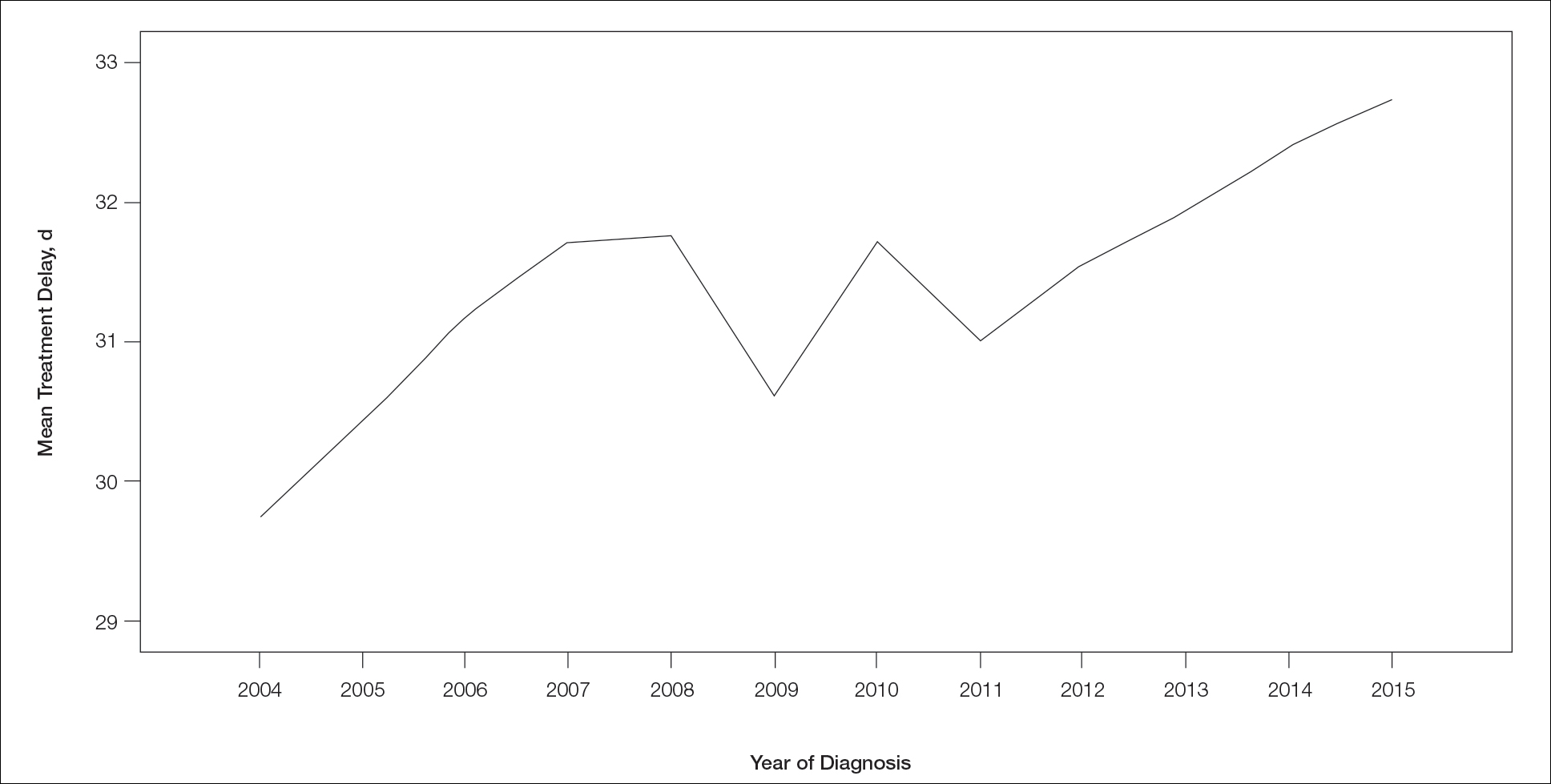
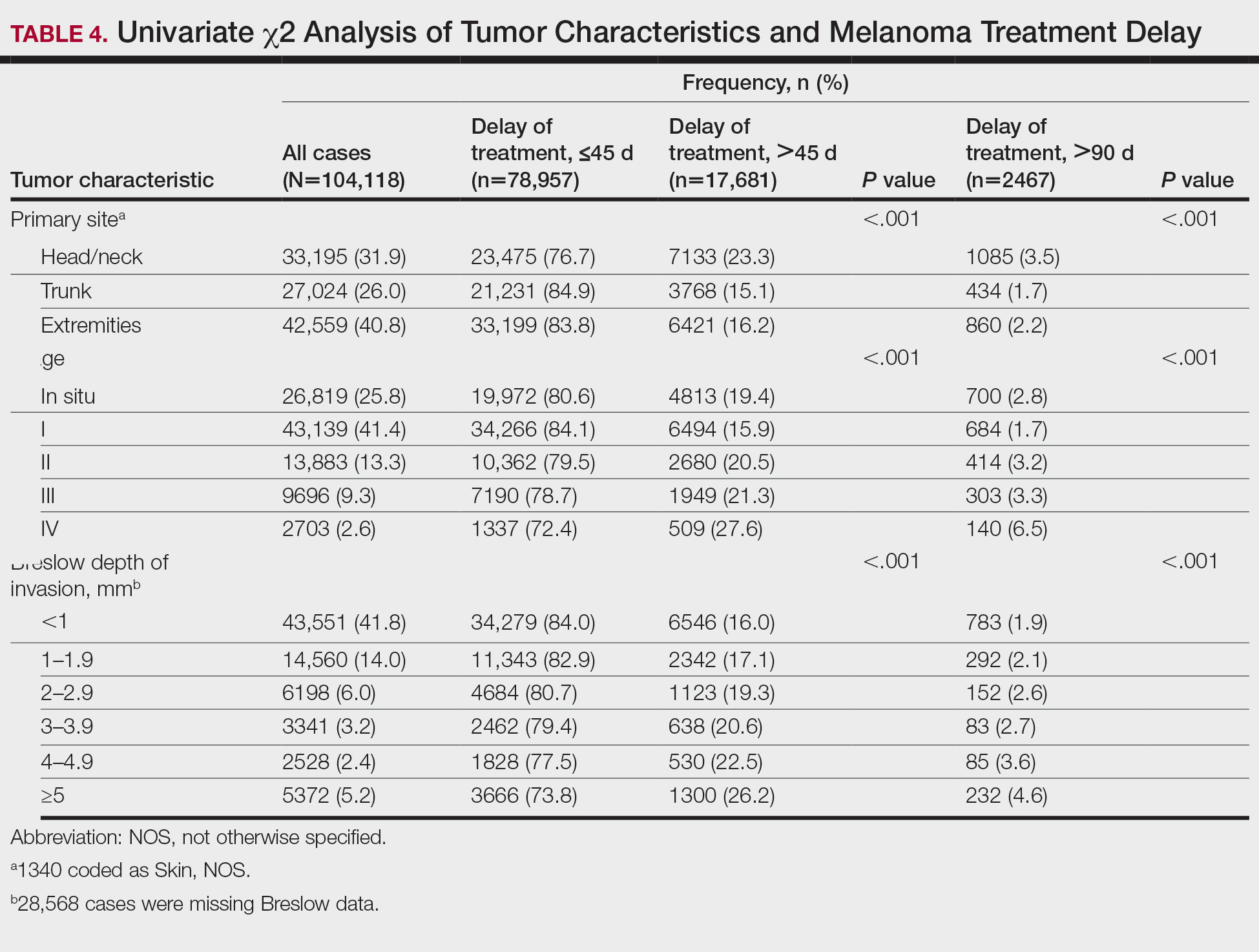
Treatment delay averaged 31.55 days, with a median of 27 days. Overall mean MTD increased significantly from 29.74 days in 2004 to 32.55 days in 2015 (2-tailed t test; P<.001)(Figure). A total of 78,957 cases (75.8%) received treatment within 45 days, whereas 2467 cases (2.5%) were postponed past 90 days. On bivariate analysis, age, sex, race, insurance status, Hispanic ethnicity, median annual income of residential zip code, percentage of the population of the patient’s residential zip code with high school degrees, CD score, and academic treatment facility held significant associations with mMTD and sMTD (P<.05)(Table 3). Analyzing bivariate associations with pertinent tumor characteristics—primary site, stage, and Breslow depth—also held significant associations with mMTD and sMTD (P<.001)(Table 4).
On multivariate analysis, controlling for the variables significant on bivariate analysis, multiple factors showed independent associations with MTD (Table 2). Patients aged 50 to 74 years were more likely to have mMTD (reference: <50 years; P=.029; OR=1.072). Patients 75 years and older showed greater rates of mMTD (reference: <50 years; P<.001; OR=1.278) and sMTD (P<.001; OR=1.590). Women had more mMTD (P=.013; OR=1.052). Nonwhite patients had greater rates of both mMTD (reference: white; P<.001; OR=1.405) and sMTD (P<.001; OR=1.674). Hispanic patients also had greater mMTD (reference: non-Hispanic: P<.001; OR=1.809) and sMTD (P<.001; OR=2.749). Compared to patients with private insurance, those with Medicare were more likely to have mMTD (P=.046; OR=1.054). Patients with no insurance or Medicaid/other government insurance showed more mMTD (no insurance: P<.001, OR=1.642; Medicaid/other: P<.001, OR=1.668) and sMTD (no insurance: P<.001, OR=2.582; Medicaid/other: P<.001, OR=2.336).
With respect to the median annual income of the patient’s residential zip code, patients residing in areas with a median income of $48,000 to $62,999 were less likely to have an sMTD (reference: <$38,000; P=.038; OR=0.829). Compared with patients residing in zip codes where a high percentage of the population had high school degrees, areas with higher nongraduate rates had greater overall rates of MTD (P<.001). Patients with more CD comorbidities also held an association with mMTD (CD1 with reference: CD0; P=.011; OR=1.080)(CD2 with reference: CD0; P<.001; OR=1.364) and sMTD (CD2 with reference: CD0; P<.001; OR=1.877). Academic facilities had greater rates of mMTD (reference: nonacademic facilities; P<.001; OR=1.578) and sMTD (P<.001; OR=1.366). In reference to head/neck primaries, primary sites on the trunk and extremities showed fewer mMTD (trunk: P<.001, OR=0.620; extremities: P<.001, OR=0.641) and sMTD (trunk: P<.001, OR=0.540; extremities: P<.001, OR=0.632). Compared with in situ disease, stage I melanomas were less likely to have treatment delay (mMTD: P<.001, OR=0.902; sMTD: P<.001, OR=0.690), whereas stages II (mMTD: P<.001, OR=1.130), III (mMTD: P<.001, OR=1.196; sMTD: P=.023, OR=1.204), and IV (mMTD: P<.001, OR=1.690; sMTD: P<.001, OR=2.240) were more highly associated with treatments delays.
Comment
The path to successful melanoma management involves 2 timeframes. One is time to diagnosis and the other is time to treatment. With 24.2% of patients receiving treatment later than 45 days after diagnosis, MTD is common and, according to our results, has increased on average from 2004 to 2015. This delay may be partially explained by a shortage of dermatologists, leading to longer wait times and follow-up.13,14 Melanoma treatment delay also varied based on insurance status. Unsurprisingly, those with private insurance showed the lowest rates of MTD. Those with no insurance, Medicare, or Medicaid/other government insurance likely faced greater socioeconomic barriers to health care, such as coverage issues.15 Transportation, low health literacy, and limited work schedule flexibility have been described as additional hurdles to health care that could contribute to this finding.16,17 Similarly, nonwhite patients, Hispanic patients, and those from zip codes with low high school graduation rates had more MTD. Although these findings may be explained by socioeconomic barriers and heightened distrust of the health care system, it also is important to consider physician accessibility.18,19
Considering the 2011 Affordable Care Act along with the 2014 Medicaid expansion, our study holds implications on the impact of these legislations on melanoma treatment. Studies have supported expected rises in Medicaid coverage.20,21 The overall uninsured rate in the United States declined from 16% in 2010 to 9.1% in 2015.22 In our study, the uninsured population showed the highest average MTD rates, though those with Medicaid also had significant MTD. Another treacherous hurdle for patients is the coordination of care among dermatologists, oncologists, general surgeons, plastic surgeons, and Mohs surgeons as a multidisciplinary team. Lott et al6 found that patients who received both biopsy and excision from a dermatologist had the shortest treatment delays, whereas those who had a dermatologist biopsy the site and a different surgeon—including Mohs surgeons—excise it experienced significantly greater MTDs (probablility of MTD >45 days was 31% [95% CI, 24%-37%]. This discordant care and referrals could explain the surprising finding that treatment at an academic facility was independently associated with more MTD, possibly due to the care transitions and referrals that disproportionately affect academic centers and multidisciplinary teams, as mentioned above, regarding the transition of care to other physicians (eg, plastic surgeon). A total of 70.1% of our cases treated at academic facilities reported a prior diagnosis at another facility. These results should not dissuade the pursuit of multidisciplinary treatment teams but should raise caution to untimely referrals.
Age, sex, and race were all associated with more MTD. Patients older than 50 years likely face more complex decisions regarding treatment burden, quality of life, and functional outcomes of more aggressive treatments. High rates of surgical refusal for a number of malignancies have been documented in the elderly population,23-25 which is of particular concern for the high surgery burden of head and neck melanomas,26 as further supported by the findings of more MTD for head and neck primaries. As with elderly patients, patients with higher comorbidity scores and more advanced tumors face similar family–patient care discussions to guide treatment. Additionally, women were more likely to experience MTD, which may be connected to a greater concern for cosmesis27 and necessitate more complex management options, such as Mohs micrographic surgery (a procedure that has gained some support for melanoma excision with the help of immunostaining).28
There are several limitations to this study. Accurate data rely on precise record keeping, reporting, and coding by the contributing institutions. The NCDB case diagnosis is derived from data entry without a centralized review process by experienced dermatopathologists. We could not assess the effects of tumor diameter, as these data were inadequately recorded within the dataset. The NCDB also does not provide details on specific immunotherapy or chemotherapy agents. The NCDB also is a facility-based data source, potentially biasing the melanoma data toward thicker advanced tumors more readily managed at such institutions. Lastly, it is impossible to distinguish between patient-related (ie, difficult decision-making) and health care–related (ie, health care accessibility) delays. Nonetheless, we maintain that minimizing MTD is important for survival outcomes and for limiting the progression of melanomas, regardless of the underlying rationale. We believe that our study expands on conclusions previously limited to a Medicare population.
Conclusion
According to the NCDB, mean MTD has increased significantly from 2004 to 2015. Our results suggest that MTD is relatively common in the United States, thereby increasing the risk for metastases. Higher MTD rates are independently associated with being older than 50 years, female, nonwhite, not privately insured, Hispanic, and treated at an academic facility; having a positive comorbidity history and stage II to IV tumors; and residing in a zip code with a low high school graduation rate. Stage I tumors, primaries not located on the head or neck, and residing in a zip code with a higher median income are associated with lower MTD rates. Policymakers, patients, and dermatologists should better recognize these risk factors to facilitate patient guidance and health equity.
- Huff LS, Chang CA, Thomas JF, et al. Defining an acceptable period of time from melanoma biopsy to excision. Dermatol Reports. 2012;4:E2.
- Matthews NH, Li WQ, Qureshi AA, et al. Epidemiology of Melanoma. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
- Nelson BR, Hamlet KR, Gillard M, et al. Sebaceous carcinoma. J Am Acad Dermatol. 1995;33:1-15.
- Fan Q, Cohen S, John B, et al. Melanoma in situ treated with topical imiquimod for management of persistently positive margins: a review of treatment methods. Ochsner J. 2015;15:443-447.
- Lott JP, Narayan D, Soulos PR, et al. Delay of surgery for melanoma among Medicare beneficiaries. JAMA Dermatol. 2015;151:731-741.
- Renzi C, Mastroeni S, Mannooranparampil TJ, et al. Delay in diagnosis and treatment of squamous cell carcinoma of the skin. Acta Derm Venereol. 2010;90:595-601.
- Winchester DP, Stewart AK, Phillips JL, et al. The National Cancer Database: past, present, and future. Ann Surg Oncol. 2010;17:4-7.
- Raval MV, Bilimoria KY, Stewart AK, et al. Using the NCDB for cancer care improvement: an introduction to available quality assessment tools. J Surg Oncol. 2009;99:488-490.
- Turkeltaub AE, Pezzi TA, Pezzi CM, et al. Characteristics, treatment, and survival of invasive malignant melanoma (MM) in giant pigmented nevi (GPN) in adults: 976 cases from the National Cancer Data Base (NCDB). J Am Acad Dermatol. 2016;74:1128-1134.
- Boffa DJ, Rosen JE, Mallin K, et al. Using the National Cancer Database for outcomes research: a review. JAMA Oncol. 2017;3:1722-1728.
- Riker AI, Glass F, Perez I, et al. Cutaneous melanoma: methods of biopsy and definitive surgical excision. Dermatol Ther. 2005;18:387-393.
- Kimball AB, Resneck JS Jr. The US dermatology workforce: a specialty remains in shortage. J Am Acad Dermatol. 2008;59:741-745.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.
- Okoro CA, Zhao G, Dhingra SS, et al. Peer reviewed: lack of health insurance among adults aged 18 to 64 years: findings from the 2013 Behavioral Risk Factor Surveillance System. Prev Chronic Dis. 2015;12:E231.
- Syed ST, Gerber BS, Sharp LK. Traveling towards disease: transportation barriers to health care access. J Community Health. 2013;38:976-993.
- Valerio M, Cabana MD, White DF, et al. Understanding of asthma management: Medicaid parents’ perspectives. Chest. 2006;129:594-601.
- Kaplan CP, Nápoles A, Davis S, et al. Latinos and cancer information: perspectives of patients, health professionals and telephone cancer information specialists. J Health Dispar Res Pract. 2016;9:154-167.
- Armstrong K, Ravenell KL, McMurphy S, et al. Racial/ethnic differences in physician distrust in the United States. Am J Public Health. 2007;97:1283-1289.
- Moss HA, Havrilesky LJ, Chino J. Insurance coverage among women diagnosed with a gynecologic malignancy before and after implementation of the Affordable Care Act. Gynecol Oncol. 2017;146:457-464.
- Moss HA, Havrilesky LJ, Zafar SY, et al. Trends in insurance status among patients diagnosed with cancer before and after implementation of the Affordable Care Act. J Oncol Pract. 2018;14:E92-E102.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316:525-532.
- Crippen MM, Brady JS, Mozeika AM, et al. Impact of body mass index on operative outcomes in head and neck free flap surgery. Otolaryngol Head Neck Surg. 2018;159:817-823.
- Verkooijen HM, Fioretta GM, Rapiti E, et al. Patients’ refusal of surgery strongly impairs breast cancer survival. Ann Surg. 2005;242:276-280.
- Wang J, Wang FW. Refusal of cancer-directed surgery strongly impairs survival of patients with localized hepatocellular carcinoma. Int J Surg Oncol. 2010;2010:381795.
- Zito PM, Scharf R. Cancer, melanoma, head and neck. StatPearls. StatPearls Publishing; 2018.
- Al-Dujaili Z, Henry M, Dorizas A, et al. Skin cancer concerns particular to women. Int J Womens Dermatol. 2017;3:S49-S51.
- Etzkorn JR, Jew OS, Shin TM, et al. Mohs micrographic surgery with melanoma antigen recognized by T cells 1 (MART-1) immunostaining for atypical intraepidermal melanocytic proliferation. J Am Acad Dermatol. 2018;79:1109-1116.e1
Melanoma is the most lethal skin cancer and is the second most common cancer in adolescents and young adults.1 It is the fifth most common cancer in the United States based on incidence, which has steadily risen for the last 2 decades.2,3 For melanoma management, delayed initial diagnosis has been associated with more advanced lesions at presentation and poorer outcomes.4 However, the prognostic implications of delaying melanoma management after diagnosis merits further scrutiny.
This study investigates the associations between melanoma treatment delay (MTD) and patient and tumor characteristics. Although most cases undergo surgical treatment first, more advanced stages may require initiating chemotherapy, radiation therapy, or immunotherapy. In addition, patients who are poor surgical candidates may opt for topical field therapy, such as imiquimod for superficial lesions, prior to more definitive treatment.5 In the Medicaid population, patients who are older than 85 years, married, and previously diagnosed with another melanoma and who also have an increased comorbidity burden have a higher likelihood of MTD.6 For nonmelanoma skin cancers, patient denial is the most common patient-specific factor accounting for treatment delay.7 For this study, our aim was to further evaluate the independent risk factors associated with MTD.
Methods
Case Selection
The National Cancer Database (NCDB) was queried for all cutaneous melanoma cases from 2004 to 2015 (N=525,271). The NCDB is an oncology database sourced from more than 1500 accredited cancer facilities in the United States and Puerto Rico. It receives cases from academic hospitals, Veterans Health Administration hospitals, and community centers.8 Annually, the database collects approximately 70% of cancer diagnoses and 48% of melanoma diagnoses in the United States.9,10 Per institutional guidelines, this analysis was determined to be exempt from institutional review board approval due to the deidentified nature of the dataset.
The selection scheme is illustrated in Table 1. International Statistical Classification of Diseases and Related Health Problems histology codes 8720/3 through 8780/3 combined with the site and morphology primary codes C44.0 through C44.9 identified all patients with a diagnosis of cutaneous melanoma. Primary site was established with the histology codes in the following manner: C44.0 through C44.4 for head/neck primary, C44.5 for trunk primary, C44.6 through C44.7 for extremity primary, and C44.8 through C44.9 for not otherwise specified. Because the NCDB does not specify cause of death, any cases in which the melanoma diagnosis was not the patient’s primary (or first) cancer diagnosis were excluded because of potential ambiguity. Cases lacking histologic confirmation of the diagnosis after primary site biopsy or cases diagnosed from autopsy reports also were excluded. Reports missing staging data or undergoing palliative management were removed. In total, 104,118 cases met the inclusion criteria.
Variables of Interest
The NCDB database codes for a variable “Treatment Started, Days from Dx” are defined as the number of days between the date of diagnosis and the date on which treatment—surgery, radiation, systemic, or other therapy—of the patient began at any facility.11 Treatment delays were classified as more than 45 days or more than 90 days. These thresholds were chosen based on previous studies citing a 45-day recommendation as the timeframe in which primary site excision of melanoma should occur for improved outcomes.1,6,12 Additionally, the postponement cutoffs were aligned with prior studies on surgical delay in melanoma for the Medicaid population.6 Delays of 45 days were labeled as moderate MTD (mMTD), whereas postponements more than 90 days were designated as severe MTD (sMTD).
Patient and tumor characteristics were analyzed for associations with MTD (Table 2). Covariates included age, sex, race (white vs nonwhite), Hispanic ethnicity, insurance status (private; Medicare, Medicaid or other government insurance; and no insurance), median annual income of the patient’s residential zip code (based on 2008-2012 census data), percentage of the population of the patient’s residential zip code without a high school degree (based on 2008-2012 census data), Charlson-Deyo (CD) comorbidity score (a weighted score derived from the sum scores for comorbid conditions), geographic location (rural, urban, and metropolitan), and treatment facility (academic vs nonacademic). Tumor characteristics included primary site (head/neck, trunk, and extremities), stage, and Breslow depth of invasion. Tumor stage was determined using the American Joint Committee on Cancer 6th and 7th editions, depending on the patient’s year of diagnosis.
Statistical Methods
χ2 and Fisher exact tests were used to analyze categorical variables involving patient demographics and tumor characteristics by bivariate analysis (Tables 3 and 4). Multivariate analysis determined the relative impact on MTD by including variables that significantly differed on bivariate χ2 analysis (Table 2). Multivariate modeling determined odds ratio (OR) and corresponding 95% CI for the risk-adjusted associations of the variables with MTD. All statistical analyses were performed using SPSS Statistics version 23 (IBM). P<.05 was considered statistically significant, and all statistical tests were 2-tailed. Line graph figures by year of diagnosis were modeled by SPSS using the mean days of delay per year. Independent sample t tests assessed for differences in mean values.
Results
The final study population included 104,118 patients, most of whom were male (56.4%), white (96.6%), and aged 50 to 74 years (54.4%). Most patients were privately insured (52.6%), had no CD comorbidities (87.5%), and lived in metropolitan cities (80.4%)(Table 3). A large majority (95,473 [91.7%]) of patients received surgery as the first means of treatment, with a smaller portion (863 [0.8%]) having unspecified systemic therapy first. The remaining cases were first treated with chemotherapy (1738 [1.7%]), immunotherapy (382 [0.4%]), or radiation (490 [0.5%]), and the rest did not specify treatment sequence. The tumors were most commonly located on the extremities (40.7%), were stage I (41.2%), and had a Breslow depth of less than 1 mm (41.6%).
Treatment delay averaged 31.55 days, with a median of 27 days. Overall mean MTD increased significantly from 29.74 days in 2004 to 32.55 days in 2015 (2-tailed t test; P<.001)(Figure). A total of 78,957 cases (75.8%) received treatment within 45 days, whereas 2467 cases (2.5%) were postponed past 90 days. On bivariate analysis, age, sex, race, insurance status, Hispanic ethnicity, median annual income of residential zip code, percentage of the population of the patient’s residential zip code with high school degrees, CD score, and academic treatment facility held significant associations with mMTD and sMTD (P<.05)(Table 3). Analyzing bivariate associations with pertinent tumor characteristics—primary site, stage, and Breslow depth—also held significant associations with mMTD and sMTD (P<.001)(Table 4).
On multivariate analysis, controlling for the variables significant on bivariate analysis, multiple factors showed independent associations with MTD (Table 2). Patients aged 50 to 74 years were more likely to have mMTD (reference: <50 years; P=.029; OR=1.072). Patients 75 years and older showed greater rates of mMTD (reference: <50 years; P<.001; OR=1.278) and sMTD (P<.001; OR=1.590). Women had more mMTD (P=.013; OR=1.052). Nonwhite patients had greater rates of both mMTD (reference: white; P<.001; OR=1.405) and sMTD (P<.001; OR=1.674). Hispanic patients also had greater mMTD (reference: non-Hispanic: P<.001; OR=1.809) and sMTD (P<.001; OR=2.749). Compared to patients with private insurance, those with Medicare were more likely to have mMTD (P=.046; OR=1.054). Patients with no insurance or Medicaid/other government insurance showed more mMTD (no insurance: P<.001, OR=1.642; Medicaid/other: P<.001, OR=1.668) and sMTD (no insurance: P<.001, OR=2.582; Medicaid/other: P<.001, OR=2.336).
With respect to the median annual income of the patient’s residential zip code, patients residing in areas with a median income of $48,000 to $62,999 were less likely to have an sMTD (reference: <$38,000; P=.038; OR=0.829). Compared with patients residing in zip codes where a high percentage of the population had high school degrees, areas with higher nongraduate rates had greater overall rates of MTD (P<.001). Patients with more CD comorbidities also held an association with mMTD (CD1 with reference: CD0; P=.011; OR=1.080)(CD2 with reference: CD0; P<.001; OR=1.364) and sMTD (CD2 with reference: CD0; P<.001; OR=1.877). Academic facilities had greater rates of mMTD (reference: nonacademic facilities; P<.001; OR=1.578) and sMTD (P<.001; OR=1.366). In reference to head/neck primaries, primary sites on the trunk and extremities showed fewer mMTD (trunk: P<.001, OR=0.620; extremities: P<.001, OR=0.641) and sMTD (trunk: P<.001, OR=0.540; extremities: P<.001, OR=0.632). Compared with in situ disease, stage I melanomas were less likely to have treatment delay (mMTD: P<.001, OR=0.902; sMTD: P<.001, OR=0.690), whereas stages II (mMTD: P<.001, OR=1.130), III (mMTD: P<.001, OR=1.196; sMTD: P=.023, OR=1.204), and IV (mMTD: P<.001, OR=1.690; sMTD: P<.001, OR=2.240) were more highly associated with treatments delays.
Comment
The path to successful melanoma management involves 2 timeframes. One is time to diagnosis and the other is time to treatment. With 24.2% of patients receiving treatment later than 45 days after diagnosis, MTD is common and, according to our results, has increased on average from 2004 to 2015. This delay may be partially explained by a shortage of dermatologists, leading to longer wait times and follow-up.13,14 Melanoma treatment delay also varied based on insurance status. Unsurprisingly, those with private insurance showed the lowest rates of MTD. Those with no insurance, Medicare, or Medicaid/other government insurance likely faced greater socioeconomic barriers to health care, such as coverage issues.15 Transportation, low health literacy, and limited work schedule flexibility have been described as additional hurdles to health care that could contribute to this finding.16,17 Similarly, nonwhite patients, Hispanic patients, and those from zip codes with low high school graduation rates had more MTD. Although these findings may be explained by socioeconomic barriers and heightened distrust of the health care system, it also is important to consider physician accessibility.18,19
Considering the 2011 Affordable Care Act along with the 2014 Medicaid expansion, our study holds implications on the impact of these legislations on melanoma treatment. Studies have supported expected rises in Medicaid coverage.20,21 The overall uninsured rate in the United States declined from 16% in 2010 to 9.1% in 2015.22 In our study, the uninsured population showed the highest average MTD rates, though those with Medicaid also had significant MTD. Another treacherous hurdle for patients is the coordination of care among dermatologists, oncologists, general surgeons, plastic surgeons, and Mohs surgeons as a multidisciplinary team. Lott et al6 found that patients who received both biopsy and excision from a dermatologist had the shortest treatment delays, whereas those who had a dermatologist biopsy the site and a different surgeon—including Mohs surgeons—excise it experienced significantly greater MTDs (probablility of MTD >45 days was 31% [95% CI, 24%-37%]. This discordant care and referrals could explain the surprising finding that treatment at an academic facility was independently associated with more MTD, possibly due to the care transitions and referrals that disproportionately affect academic centers and multidisciplinary teams, as mentioned above, regarding the transition of care to other physicians (eg, plastic surgeon). A total of 70.1% of our cases treated at academic facilities reported a prior diagnosis at another facility. These results should not dissuade the pursuit of multidisciplinary treatment teams but should raise caution to untimely referrals.
Age, sex, and race were all associated with more MTD. Patients older than 50 years likely face more complex decisions regarding treatment burden, quality of life, and functional outcomes of more aggressive treatments. High rates of surgical refusal for a number of malignancies have been documented in the elderly population,23-25 which is of particular concern for the high surgery burden of head and neck melanomas,26 as further supported by the findings of more MTD for head and neck primaries. As with elderly patients, patients with higher comorbidity scores and more advanced tumors face similar family–patient care discussions to guide treatment. Additionally, women were more likely to experience MTD, which may be connected to a greater concern for cosmesis27 and necessitate more complex management options, such as Mohs micrographic surgery (a procedure that has gained some support for melanoma excision with the help of immunostaining).28
There are several limitations to this study. Accurate data rely on precise record keeping, reporting, and coding by the contributing institutions. The NCDB case diagnosis is derived from data entry without a centralized review process by experienced dermatopathologists. We could not assess the effects of tumor diameter, as these data were inadequately recorded within the dataset. The NCDB also does not provide details on specific immunotherapy or chemotherapy agents. The NCDB also is a facility-based data source, potentially biasing the melanoma data toward thicker advanced tumors more readily managed at such institutions. Lastly, it is impossible to distinguish between patient-related (ie, difficult decision-making) and health care–related (ie, health care accessibility) delays. Nonetheless, we maintain that minimizing MTD is important for survival outcomes and for limiting the progression of melanomas, regardless of the underlying rationale. We believe that our study expands on conclusions previously limited to a Medicare population.
Conclusion
According to the NCDB, mean MTD has increased significantly from 2004 to 2015. Our results suggest that MTD is relatively common in the United States, thereby increasing the risk for metastases. Higher MTD rates are independently associated with being older than 50 years, female, nonwhite, not privately insured, Hispanic, and treated at an academic facility; having a positive comorbidity history and stage II to IV tumors; and residing in a zip code with a low high school graduation rate. Stage I tumors, primaries not located on the head or neck, and residing in a zip code with a higher median income are associated with lower MTD rates. Policymakers, patients, and dermatologists should better recognize these risk factors to facilitate patient guidance and health equity.
Melanoma is the most lethal skin cancer and is the second most common cancer in adolescents and young adults.1 It is the fifth most common cancer in the United States based on incidence, which has steadily risen for the last 2 decades.2,3 For melanoma management, delayed initial diagnosis has been associated with more advanced lesions at presentation and poorer outcomes.4 However, the prognostic implications of delaying melanoma management after diagnosis merits further scrutiny.
This study investigates the associations between melanoma treatment delay (MTD) and patient and tumor characteristics. Although most cases undergo surgical treatment first, more advanced stages may require initiating chemotherapy, radiation therapy, or immunotherapy. In addition, patients who are poor surgical candidates may opt for topical field therapy, such as imiquimod for superficial lesions, prior to more definitive treatment.5 In the Medicaid population, patients who are older than 85 years, married, and previously diagnosed with another melanoma and who also have an increased comorbidity burden have a higher likelihood of MTD.6 For nonmelanoma skin cancers, patient denial is the most common patient-specific factor accounting for treatment delay.7 For this study, our aim was to further evaluate the independent risk factors associated with MTD.
Methods
Case Selection
The National Cancer Database (NCDB) was queried for all cutaneous melanoma cases from 2004 to 2015 (N=525,271). The NCDB is an oncology database sourced from more than 1500 accredited cancer facilities in the United States and Puerto Rico. It receives cases from academic hospitals, Veterans Health Administration hospitals, and community centers.8 Annually, the database collects approximately 70% of cancer diagnoses and 48% of melanoma diagnoses in the United States.9,10 Per institutional guidelines, this analysis was determined to be exempt from institutional review board approval due to the deidentified nature of the dataset.
The selection scheme is illustrated in Table 1. International Statistical Classification of Diseases and Related Health Problems histology codes 8720/3 through 8780/3 combined with the site and morphology primary codes C44.0 through C44.9 identified all patients with a diagnosis of cutaneous melanoma. Primary site was established with the histology codes in the following manner: C44.0 through C44.4 for head/neck primary, C44.5 for trunk primary, C44.6 through C44.7 for extremity primary, and C44.8 through C44.9 for not otherwise specified. Because the NCDB does not specify cause of death, any cases in which the melanoma diagnosis was not the patient’s primary (or first) cancer diagnosis were excluded because of potential ambiguity. Cases lacking histologic confirmation of the diagnosis after primary site biopsy or cases diagnosed from autopsy reports also were excluded. Reports missing staging data or undergoing palliative management were removed. In total, 104,118 cases met the inclusion criteria.
Variables of Interest
The NCDB database codes for a variable “Treatment Started, Days from Dx” are defined as the number of days between the date of diagnosis and the date on which treatment—surgery, radiation, systemic, or other therapy—of the patient began at any facility.11 Treatment delays were classified as more than 45 days or more than 90 days. These thresholds were chosen based on previous studies citing a 45-day recommendation as the timeframe in which primary site excision of melanoma should occur for improved outcomes.1,6,12 Additionally, the postponement cutoffs were aligned with prior studies on surgical delay in melanoma for the Medicaid population.6 Delays of 45 days were labeled as moderate MTD (mMTD), whereas postponements more than 90 days were designated as severe MTD (sMTD).
Patient and tumor characteristics were analyzed for associations with MTD (Table 2). Covariates included age, sex, race (white vs nonwhite), Hispanic ethnicity, insurance status (private; Medicare, Medicaid or other government insurance; and no insurance), median annual income of the patient’s residential zip code (based on 2008-2012 census data), percentage of the population of the patient’s residential zip code without a high school degree (based on 2008-2012 census data), Charlson-Deyo (CD) comorbidity score (a weighted score derived from the sum scores for comorbid conditions), geographic location (rural, urban, and metropolitan), and treatment facility (academic vs nonacademic). Tumor characteristics included primary site (head/neck, trunk, and extremities), stage, and Breslow depth of invasion. Tumor stage was determined using the American Joint Committee on Cancer 6th and 7th editions, depending on the patient’s year of diagnosis.
Statistical Methods
χ2 and Fisher exact tests were used to analyze categorical variables involving patient demographics and tumor characteristics by bivariate analysis (Tables 3 and 4). Multivariate analysis determined the relative impact on MTD by including variables that significantly differed on bivariate χ2 analysis (Table 2). Multivariate modeling determined odds ratio (OR) and corresponding 95% CI for the risk-adjusted associations of the variables with MTD. All statistical analyses were performed using SPSS Statistics version 23 (IBM). P<.05 was considered statistically significant, and all statistical tests were 2-tailed. Line graph figures by year of diagnosis were modeled by SPSS using the mean days of delay per year. Independent sample t tests assessed for differences in mean values.
Results
The final study population included 104,118 patients, most of whom were male (56.4%), white (96.6%), and aged 50 to 74 years (54.4%). Most patients were privately insured (52.6%), had no CD comorbidities (87.5%), and lived in metropolitan cities (80.4%)(Table 3). A large majority (95,473 [91.7%]) of patients received surgery as the first means of treatment, with a smaller portion (863 [0.8%]) having unspecified systemic therapy first. The remaining cases were first treated with chemotherapy (1738 [1.7%]), immunotherapy (382 [0.4%]), or radiation (490 [0.5%]), and the rest did not specify treatment sequence. The tumors were most commonly located on the extremities (40.7%), were stage I (41.2%), and had a Breslow depth of less than 1 mm (41.6%).
Treatment delay averaged 31.55 days, with a median of 27 days. Overall mean MTD increased significantly from 29.74 days in 2004 to 32.55 days in 2015 (2-tailed t test; P<.001)(Figure). A total of 78,957 cases (75.8%) received treatment within 45 days, whereas 2467 cases (2.5%) were postponed past 90 days. On bivariate analysis, age, sex, race, insurance status, Hispanic ethnicity, median annual income of residential zip code, percentage of the population of the patient’s residential zip code with high school degrees, CD score, and academic treatment facility held significant associations with mMTD and sMTD (P<.05)(Table 3). Analyzing bivariate associations with pertinent tumor characteristics—primary site, stage, and Breslow depth—also held significant associations with mMTD and sMTD (P<.001)(Table 4).
On multivariate analysis, controlling for the variables significant on bivariate analysis, multiple factors showed independent associations with MTD (Table 2). Patients aged 50 to 74 years were more likely to have mMTD (reference: <50 years; P=.029; OR=1.072). Patients 75 years and older showed greater rates of mMTD (reference: <50 years; P<.001; OR=1.278) and sMTD (P<.001; OR=1.590). Women had more mMTD (P=.013; OR=1.052). Nonwhite patients had greater rates of both mMTD (reference: white; P<.001; OR=1.405) and sMTD (P<.001; OR=1.674). Hispanic patients also had greater mMTD (reference: non-Hispanic: P<.001; OR=1.809) and sMTD (P<.001; OR=2.749). Compared to patients with private insurance, those with Medicare were more likely to have mMTD (P=.046; OR=1.054). Patients with no insurance or Medicaid/other government insurance showed more mMTD (no insurance: P<.001, OR=1.642; Medicaid/other: P<.001, OR=1.668) and sMTD (no insurance: P<.001, OR=2.582; Medicaid/other: P<.001, OR=2.336).
With respect to the median annual income of the patient’s residential zip code, patients residing in areas with a median income of $48,000 to $62,999 were less likely to have an sMTD (reference: <$38,000; P=.038; OR=0.829). Compared with patients residing in zip codes where a high percentage of the population had high school degrees, areas with higher nongraduate rates had greater overall rates of MTD (P<.001). Patients with more CD comorbidities also held an association with mMTD (CD1 with reference: CD0; P=.011; OR=1.080)(CD2 with reference: CD0; P<.001; OR=1.364) and sMTD (CD2 with reference: CD0; P<.001; OR=1.877). Academic facilities had greater rates of mMTD (reference: nonacademic facilities; P<.001; OR=1.578) and sMTD (P<.001; OR=1.366). In reference to head/neck primaries, primary sites on the trunk and extremities showed fewer mMTD (trunk: P<.001, OR=0.620; extremities: P<.001, OR=0.641) and sMTD (trunk: P<.001, OR=0.540; extremities: P<.001, OR=0.632). Compared with in situ disease, stage I melanomas were less likely to have treatment delay (mMTD: P<.001, OR=0.902; sMTD: P<.001, OR=0.690), whereas stages II (mMTD: P<.001, OR=1.130), III (mMTD: P<.001, OR=1.196; sMTD: P=.023, OR=1.204), and IV (mMTD: P<.001, OR=1.690; sMTD: P<.001, OR=2.240) were more highly associated with treatments delays.
Comment
The path to successful melanoma management involves 2 timeframes. One is time to diagnosis and the other is time to treatment. With 24.2% of patients receiving treatment later than 45 days after diagnosis, MTD is common and, according to our results, has increased on average from 2004 to 2015. This delay may be partially explained by a shortage of dermatologists, leading to longer wait times and follow-up.13,14 Melanoma treatment delay also varied based on insurance status. Unsurprisingly, those with private insurance showed the lowest rates of MTD. Those with no insurance, Medicare, or Medicaid/other government insurance likely faced greater socioeconomic barriers to health care, such as coverage issues.15 Transportation, low health literacy, and limited work schedule flexibility have been described as additional hurdles to health care that could contribute to this finding.16,17 Similarly, nonwhite patients, Hispanic patients, and those from zip codes with low high school graduation rates had more MTD. Although these findings may be explained by socioeconomic barriers and heightened distrust of the health care system, it also is important to consider physician accessibility.18,19
Considering the 2011 Affordable Care Act along with the 2014 Medicaid expansion, our study holds implications on the impact of these legislations on melanoma treatment. Studies have supported expected rises in Medicaid coverage.20,21 The overall uninsured rate in the United States declined from 16% in 2010 to 9.1% in 2015.22 In our study, the uninsured population showed the highest average MTD rates, though those with Medicaid also had significant MTD. Another treacherous hurdle for patients is the coordination of care among dermatologists, oncologists, general surgeons, plastic surgeons, and Mohs surgeons as a multidisciplinary team. Lott et al6 found that patients who received both biopsy and excision from a dermatologist had the shortest treatment delays, whereas those who had a dermatologist biopsy the site and a different surgeon—including Mohs surgeons—excise it experienced significantly greater MTDs (probablility of MTD >45 days was 31% [95% CI, 24%-37%]. This discordant care and referrals could explain the surprising finding that treatment at an academic facility was independently associated with more MTD, possibly due to the care transitions and referrals that disproportionately affect academic centers and multidisciplinary teams, as mentioned above, regarding the transition of care to other physicians (eg, plastic surgeon). A total of 70.1% of our cases treated at academic facilities reported a prior diagnosis at another facility. These results should not dissuade the pursuit of multidisciplinary treatment teams but should raise caution to untimely referrals.
Age, sex, and race were all associated with more MTD. Patients older than 50 years likely face more complex decisions regarding treatment burden, quality of life, and functional outcomes of more aggressive treatments. High rates of surgical refusal for a number of malignancies have been documented in the elderly population,23-25 which is of particular concern for the high surgery burden of head and neck melanomas,26 as further supported by the findings of more MTD for head and neck primaries. As with elderly patients, patients with higher comorbidity scores and more advanced tumors face similar family–patient care discussions to guide treatment. Additionally, women were more likely to experience MTD, which may be connected to a greater concern for cosmesis27 and necessitate more complex management options, such as Mohs micrographic surgery (a procedure that has gained some support for melanoma excision with the help of immunostaining).28
There are several limitations to this study. Accurate data rely on precise record keeping, reporting, and coding by the contributing institutions. The NCDB case diagnosis is derived from data entry without a centralized review process by experienced dermatopathologists. We could not assess the effects of tumor diameter, as these data were inadequately recorded within the dataset. The NCDB also does not provide details on specific immunotherapy or chemotherapy agents. The NCDB also is a facility-based data source, potentially biasing the melanoma data toward thicker advanced tumors more readily managed at such institutions. Lastly, it is impossible to distinguish between patient-related (ie, difficult decision-making) and health care–related (ie, health care accessibility) delays. Nonetheless, we maintain that minimizing MTD is important for survival outcomes and for limiting the progression of melanomas, regardless of the underlying rationale. We believe that our study expands on conclusions previously limited to a Medicare population.
Conclusion
According to the NCDB, mean MTD has increased significantly from 2004 to 2015. Our results suggest that MTD is relatively common in the United States, thereby increasing the risk for metastases. Higher MTD rates are independently associated with being older than 50 years, female, nonwhite, not privately insured, Hispanic, and treated at an academic facility; having a positive comorbidity history and stage II to IV tumors; and residing in a zip code with a low high school graduation rate. Stage I tumors, primaries not located on the head or neck, and residing in a zip code with a higher median income are associated with lower MTD rates. Policymakers, patients, and dermatologists should better recognize these risk factors to facilitate patient guidance and health equity.
- Huff LS, Chang CA, Thomas JF, et al. Defining an acceptable period of time from melanoma biopsy to excision. Dermatol Reports. 2012;4:E2.
- Matthews NH, Li WQ, Qureshi AA, et al. Epidemiology of Melanoma. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
- Nelson BR, Hamlet KR, Gillard M, et al. Sebaceous carcinoma. J Am Acad Dermatol. 1995;33:1-15.
- Fan Q, Cohen S, John B, et al. Melanoma in situ treated with topical imiquimod for management of persistently positive margins: a review of treatment methods. Ochsner J. 2015;15:443-447.
- Lott JP, Narayan D, Soulos PR, et al. Delay of surgery for melanoma among Medicare beneficiaries. JAMA Dermatol. 2015;151:731-741.
- Renzi C, Mastroeni S, Mannooranparampil TJ, et al. Delay in diagnosis and treatment of squamous cell carcinoma of the skin. Acta Derm Venereol. 2010;90:595-601.
- Winchester DP, Stewart AK, Phillips JL, et al. The National Cancer Database: past, present, and future. Ann Surg Oncol. 2010;17:4-7.
- Raval MV, Bilimoria KY, Stewart AK, et al. Using the NCDB for cancer care improvement: an introduction to available quality assessment tools. J Surg Oncol. 2009;99:488-490.
- Turkeltaub AE, Pezzi TA, Pezzi CM, et al. Characteristics, treatment, and survival of invasive malignant melanoma (MM) in giant pigmented nevi (GPN) in adults: 976 cases from the National Cancer Data Base (NCDB). J Am Acad Dermatol. 2016;74:1128-1134.
- Boffa DJ, Rosen JE, Mallin K, et al. Using the National Cancer Database for outcomes research: a review. JAMA Oncol. 2017;3:1722-1728.
- Riker AI, Glass F, Perez I, et al. Cutaneous melanoma: methods of biopsy and definitive surgical excision. Dermatol Ther. 2005;18:387-393.
- Kimball AB, Resneck JS Jr. The US dermatology workforce: a specialty remains in shortage. J Am Acad Dermatol. 2008;59:741-745.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.
- Okoro CA, Zhao G, Dhingra SS, et al. Peer reviewed: lack of health insurance among adults aged 18 to 64 years: findings from the 2013 Behavioral Risk Factor Surveillance System. Prev Chronic Dis. 2015;12:E231.
- Syed ST, Gerber BS, Sharp LK. Traveling towards disease: transportation barriers to health care access. J Community Health. 2013;38:976-993.
- Valerio M, Cabana MD, White DF, et al. Understanding of asthma management: Medicaid parents’ perspectives. Chest. 2006;129:594-601.
- Kaplan CP, Nápoles A, Davis S, et al. Latinos and cancer information: perspectives of patients, health professionals and telephone cancer information specialists. J Health Dispar Res Pract. 2016;9:154-167.
- Armstrong K, Ravenell KL, McMurphy S, et al. Racial/ethnic differences in physician distrust in the United States. Am J Public Health. 2007;97:1283-1289.
- Moss HA, Havrilesky LJ, Chino J. Insurance coverage among women diagnosed with a gynecologic malignancy before and after implementation of the Affordable Care Act. Gynecol Oncol. 2017;146:457-464.
- Moss HA, Havrilesky LJ, Zafar SY, et al. Trends in insurance status among patients diagnosed with cancer before and after implementation of the Affordable Care Act. J Oncol Pract. 2018;14:E92-E102.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316:525-532.
- Crippen MM, Brady JS, Mozeika AM, et al. Impact of body mass index on operative outcomes in head and neck free flap surgery. Otolaryngol Head Neck Surg. 2018;159:817-823.
- Verkooijen HM, Fioretta GM, Rapiti E, et al. Patients’ refusal of surgery strongly impairs breast cancer survival. Ann Surg. 2005;242:276-280.
- Wang J, Wang FW. Refusal of cancer-directed surgery strongly impairs survival of patients with localized hepatocellular carcinoma. Int J Surg Oncol. 2010;2010:381795.
- Zito PM, Scharf R. Cancer, melanoma, head and neck. StatPearls. StatPearls Publishing; 2018.
- Al-Dujaili Z, Henry M, Dorizas A, et al. Skin cancer concerns particular to women. Int J Womens Dermatol. 2017;3:S49-S51.
- Etzkorn JR, Jew OS, Shin TM, et al. Mohs micrographic surgery with melanoma antigen recognized by T cells 1 (MART-1) immunostaining for atypical intraepidermal melanocytic proliferation. J Am Acad Dermatol. 2018;79:1109-1116.e1
- Huff LS, Chang CA, Thomas JF, et al. Defining an acceptable period of time from melanoma biopsy to excision. Dermatol Reports. 2012;4:E2.
- Matthews NH, Li WQ, Qureshi AA, et al. Epidemiology of Melanoma. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
- Nelson BR, Hamlet KR, Gillard M, et al. Sebaceous carcinoma. J Am Acad Dermatol. 1995;33:1-15.
- Fan Q, Cohen S, John B, et al. Melanoma in situ treated with topical imiquimod for management of persistently positive margins: a review of treatment methods. Ochsner J. 2015;15:443-447.
- Lott JP, Narayan D, Soulos PR, et al. Delay of surgery for melanoma among Medicare beneficiaries. JAMA Dermatol. 2015;151:731-741.
- Renzi C, Mastroeni S, Mannooranparampil TJ, et al. Delay in diagnosis and treatment of squamous cell carcinoma of the skin. Acta Derm Venereol. 2010;90:595-601.
- Winchester DP, Stewart AK, Phillips JL, et al. The National Cancer Database: past, present, and future. Ann Surg Oncol. 2010;17:4-7.
- Raval MV, Bilimoria KY, Stewart AK, et al. Using the NCDB for cancer care improvement: an introduction to available quality assessment tools. J Surg Oncol. 2009;99:488-490.
- Turkeltaub AE, Pezzi TA, Pezzi CM, et al. Characteristics, treatment, and survival of invasive malignant melanoma (MM) in giant pigmented nevi (GPN) in adults: 976 cases from the National Cancer Data Base (NCDB). J Am Acad Dermatol. 2016;74:1128-1134.
- Boffa DJ, Rosen JE, Mallin K, et al. Using the National Cancer Database for outcomes research: a review. JAMA Oncol. 2017;3:1722-1728.
- Riker AI, Glass F, Perez I, et al. Cutaneous melanoma: methods of biopsy and definitive surgical excision. Dermatol Ther. 2005;18:387-393.
- Kimball AB, Resneck JS Jr. The US dermatology workforce: a specialty remains in shortage. J Am Acad Dermatol. 2008;59:741-745.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.
- Okoro CA, Zhao G, Dhingra SS, et al. Peer reviewed: lack of health insurance among adults aged 18 to 64 years: findings from the 2013 Behavioral Risk Factor Surveillance System. Prev Chronic Dis. 2015;12:E231.
- Syed ST, Gerber BS, Sharp LK. Traveling towards disease: transportation barriers to health care access. J Community Health. 2013;38:976-993.
- Valerio M, Cabana MD, White DF, et al. Understanding of asthma management: Medicaid parents’ perspectives. Chest. 2006;129:594-601.
- Kaplan CP, Nápoles A, Davis S, et al. Latinos and cancer information: perspectives of patients, health professionals and telephone cancer information specialists. J Health Dispar Res Pract. 2016;9:154-167.
- Armstrong K, Ravenell KL, McMurphy S, et al. Racial/ethnic differences in physician distrust in the United States. Am J Public Health. 2007;97:1283-1289.
- Moss HA, Havrilesky LJ, Chino J. Insurance coverage among women diagnosed with a gynecologic malignancy before and after implementation of the Affordable Care Act. Gynecol Oncol. 2017;146:457-464.
- Moss HA, Havrilesky LJ, Zafar SY, et al. Trends in insurance status among patients diagnosed with cancer before and after implementation of the Affordable Care Act. J Oncol Pract. 2018;14:E92-E102.
- Obama B. United States health care reform: progress to date and next steps. JAMA. 2016;316:525-532.
- Crippen MM, Brady JS, Mozeika AM, et al. Impact of body mass index on operative outcomes in head and neck free flap surgery. Otolaryngol Head Neck Surg. 2018;159:817-823.
- Verkooijen HM, Fioretta GM, Rapiti E, et al. Patients’ refusal of surgery strongly impairs breast cancer survival. Ann Surg. 2005;242:276-280.
- Wang J, Wang FW. Refusal of cancer-directed surgery strongly impairs survival of patients with localized hepatocellular carcinoma. Int J Surg Oncol. 2010;2010:381795.
- Zito PM, Scharf R. Cancer, melanoma, head and neck. StatPearls. StatPearls Publishing; 2018.
- Al-Dujaili Z, Henry M, Dorizas A, et al. Skin cancer concerns particular to women. Int J Womens Dermatol. 2017;3:S49-S51.
- Etzkorn JR, Jew OS, Shin TM, et al. Mohs micrographic surgery with melanoma antigen recognized by T cells 1 (MART-1) immunostaining for atypical intraepidermal melanocytic proliferation. J Am Acad Dermatol. 2018;79:1109-1116.e1
Practice Points
- Melanoma treatment delays (MTDs) have been linked to poor outcomes.
- Based on the National Cancer Database, the mean MTD has increased significantly from 2004 to 2015 (P11<.001).
- More delays are seen in patients who are older than 50 years, female, nonwhite, not privately insured, and treated at an academic facility and who have more advanced tumor stage and head/neck primaries.
Botanical Briefs: Phytophotodermatitis Is an Occupational and Recreational Dermatosis in the Limelight
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
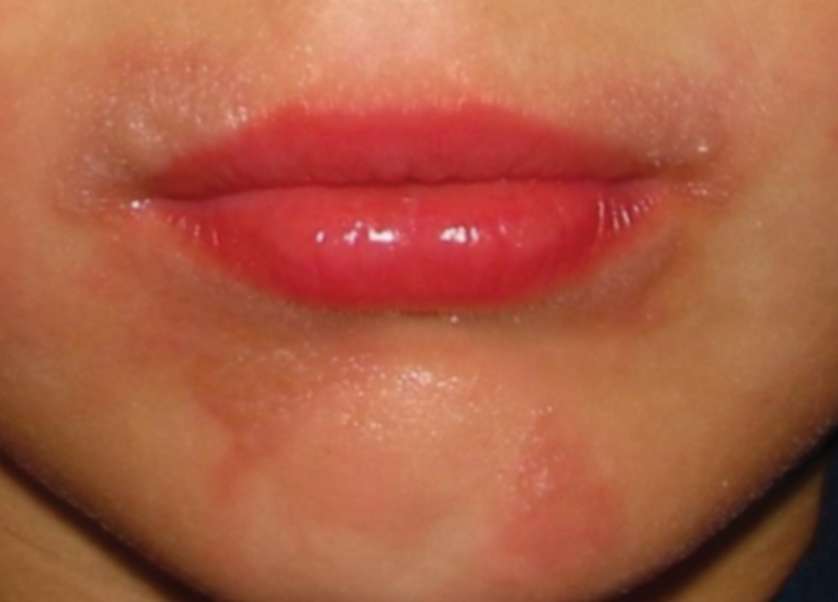
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.

- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.
- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
Practice Points
- Phytophotodermatitis (PPD) can be both an occupational and recreational dermatosis.
- Phytophotodermatitis is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.
- Individuals who work with plants should be aware of PPD and methods of prevention.
- Phytophotodermatitis may be evident only as asymptomatic hyperpigmentation.
Shedding Light on Onychomadesis
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23

Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Practice Points
- Onychomadesis in a child may be a cutaneous sign of systemic disease.
- In childhood, onychomadesis is sometimes linked with hand-foot-and-mouth disease.
- Spontaneous nail regrowth usually occurs within 12 weeks but may occur faster in children.
Neonatal and Infantile Acne Vulgaris: An Update
Acne vulgaris typically is associated with adolescence and young adulthood; however, it also can affect neonates, infants, and small children.1 Acne neonatorum occurs in up to 20% of newborns. The clinical importance of neonatal acne lies in its differentiation from infectious diseases, the exclusion of virilization as its underlying cause, and the possible implication of severe acne in adolescence.2 Neonatal acne also must be distinguished from acne that is induced by application of topical oils and ointments (acne venenata) and from acneform eruptions induced by acnegenic maternal medications such as hydantoin (fetal hydantoin syndrome) and lithium.3
Neonatal Acne (Acne Neonatorum)
Clinical Presentation
Neonatal acne (acne neonatorum) typically presents as small closed comedones on the forehead, nose, and cheeks (Figure 1).4 Accompanying sebaceous hyperplasia often is noted.5 Less frequently, open comedones, inflammatory papules, and pustules may develop.6 Neonatal acne may be evident at birth or appear during the first 4 weeks of life7 and is more commonly seen in boys.8
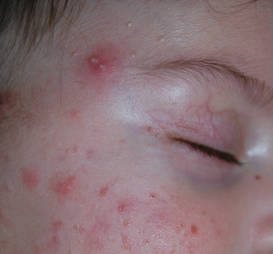
Etiology
Several factors may be pivotal in the etiology of neonatal acne, including increased sebum excretion, stimulation of the sebaceous glands by maternal or neonatal androgens,4 and colonization of sebaceous glands by Malassezia species.2 Increased sebum excretion occurs during the neonatal period due to enlarged sebaceous glands,2 which may result from the substantial production of β-hydroxysteroids from the relatively large adrenal glands.9,10 After 6 months of age, the size of the sebaceous glands and the sebum excretion rate decrease.9,10
Both maternal and neonatal androgens have been implicated in the stimulation of sebaceous glands in neonatal acne.2 The neonatal adrenal gland produces high levels of dehydroepiandrosterone,2 which stimulate sebaceous glands until around 1 year of age when dehydroepiandrosterone levels drop off as a consequence of involution of the neonatal adrenal gland.11 Testicular androgens provide additional stimulation to the sebaceous glands, which may explain why neonatal acne is more common in boys.1 Neonatal acne may be an inflammatory response to Malassezia species; however, Malassezia was not isolated in a series of patients,12 suggesting that neonatal acne is an early presentation of comedonal acne and not a response to Malassezia.2,12
Differential Diagnosis
There are a number of acneform eruptions that should be considered in the differential diagnosis,3 including bacterial folliculitis, secondary syphilis,13 herpes simplex virus and varicella zoster virus,14 and skin colonization by fungi of Malassezia species.15 Other neonatal eruptions such as erythema toxicum neonatorum,16 transient neonatal pustular melanosis, and milia and pustular miliaria, as well as a drug eruption associated with hydantoin, lithium, or halogens should be considered.17 The relationship between neonatal acne and neonatal cephalic pustulosis, which is characterized by papules and pustules without comedones, is controversial; some consider them to be 2 different entities,14 while others do not.18
Treatment
Guardians should be reassured that neonatal acne is mild, self-limited, and generally resolves spontaneously without scarring in approximately 1 to 3 months.1,2 In most cases, no treatment is needed.19 If necessary, comedones may be treated with azelaic acid cream 20% or tretinoin cream 0.025% to 0.05%.1,2 For inflammatory lesions, erythromycin solution 2% and benzoyl peroxide gel 2.5% may be used.1,20 Severe or recalcitrant disease warrants a workup for congenital adrenal hyperplasia, a virilizing tumor, or underlying endocrinopathy.19
Infantile Acne Vulgaris
Clinical Presentation
Infantile acne vulgaris shares similarities with neonatal acne21,22 in that they both affect the face, predominantly the cheeks, and have a male predominance (Figure 2).1,10 However, by definition, onset of infantile acne typically occurs later than acne neonatorum, usually at 3 to 6 months of age.1,4 Lesions are more pleomorphic and inflammatory than in neonatal acne. In addition to closed and open comedones, infantile acne may be first evident with papules, pustules, severe nodules, and cysts with scarring potential (Figure 3).1,2,5 Accordingly, treatment may be required. Most cases of infantile acne resolve by 4 or 5 years of age, but some remain active into puberty.1 Patients with a history of infantile acne have an increased incidence of acne vulgaris during adolescence compared to their peers, with greater severity and enhanced risk for scarring.4,23

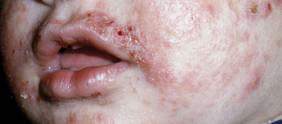
Etiology
The etiology of infantile acne remains unclear.2 Similar to neonatal acne, infantile acne may be a result of elevated androgens produced by the fetal adrenal glands as well as by the testes in males.11 For example, a child with infantile acne had elevated luteinizing hormone, follicle-stimulating hormone, and testosterone levels.24 Therefore, hyperandrogenism should be considered as an etiology. Other causes also have been suggested. Rarely, an adrenocortical tumor may be associated with persistent infantile acne with signs of virilization and rapid development.25Malassezia was implicated in infantile acne in a 6-month-old infant who was successfully treated with ketoconazole cream 2%.26
Differential Diagnosis
Infantile acne often is misdiagnosed because it is rarely considered in the differential diagnosis. When closed comedones predominate, acne venenata induced by topical creams, lotions, or oils may be etiologic. Chloracne also should be considered.14
Treatment
Guardians should be educated about the likely chronicity of infantile acne, which may require long-term treatment, as well as the possibility that acne may recur in severe form during puberty.1 The treatment strategy for infantile acne is similar to treatment of acne at any age, with topical agents including retinoids (eg, tretinoin, benzoyl peroxide) and topical antibacterials (eg, erythromycin). Twice-daily erythromycin 125 to 250 mg is the treatment of choice when oral antibiotics are indicated. Tetracyclines are contraindicated in treatment of neonatal and infantile acne. Intralesional injections with low-concentration triamcinolone acetonide, cryotherapy, or topical corticosteroids for a short period of time can be used to treat deep nodules and cysts.2 Acne that is refractory to treatment with oral antibiotics alone or combined with topical treatments poses a dilemma, given the potential cosmetic sequelae of scarring and quality-of-life concerns. Because reducing or eliminating dairy intake appears beneficial for adolescents with moderate to severe acne,27 this approach may represent a good option for infantile acne.
Conclusion
Neonatal and infantile acne vulgaris may be overlooked or misdiagnosed. It is important to consider and treat. Early childhood acne may represent a virilization syndrome.
- Jansen T, Burgdorf WH, Plewig G. Pathogenesis and treatment of acne in childhood. Pediatr Dermatol. 1997;14:17-21.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Kuflik JH, Schwartz RA. Acneiform eruptions. Cutis. 2000;66:97-100.
- Barbareschi M, Benardon S, Guanziroli E, et al. Classification and grading. In: Schwartz RA, Micali G, eds. Acne. Gurgaon, India: Nature Publishing Group; 2013:67-75.
- Mengesha YM, Bennett ML. Pustular skin disorders: diagnosis and treatment. Am J Clin Dermatol. 2002;3:389-400.
- O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. common rashes. Am Fam Physician. 2008;77:47-52.
- Nanda S, Reddy BS, Ramji S, et al. Analytical study of pustular eruptions in neonates. Pediatr Dermatol. 2002;19:210-215.
- Yonkosky DM, Pochi PE. Acne vulgaris in childhood. pathogenesis and management. Dermatol Clin. 1986;4:127-136.
- Agache P, Blanc D, Barrand C, et al. Sebum levels during the first year of life. Br J Dermatol. 1980;103:643-649.
- Herane MI, Ando I. Acne in infancy and acne genetics. Dermatology. 2003;206:24-28.
- Lucky AW. A review of infantile and pediatric acne. Dermatology (Basel, Switzerland). 1998;103:643-649.
- Bernier V, Weill FX, Hirigoyen V, et al. Skin colonization by Malassezia species in neonates: a prospective study and relationship with neonatal cephalic pustulosis. Arch Dermatol. 2002;138:215-218.
- Lambert WC, Bagley MP, Khan Y, et al. Pustular acneiform secondary syphilis. Cutis. 1986;37:69-70.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Borton LK, Schwartz RA. Pityrosporum folliculitis: a common acneiform condition of middle age. Ariz Med. 1981;38:598-601.
- Morgan AJ, Steen CJ, Schwartz RA, et al. Erythema toxicum neonatorum revisited. Cutis. 2009;83:13-16.
- Brodkin RH, Schwartz RA. Cutaneous signs of dioxin exposure. Am Fam Physician. 1984;30:189-194.
- Mancini AJ, Baldwin HE, Eichenfield LF, et al. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg. 2011;30(suppl 3):S2-S5.
- Katsambas AD, Katoulis AC, Stavropoulos P. Acne neonatorum: a study of 22 cases. Int J Dermatol. 1999;38:128-130.
- Van Praag MC, Van Rooij RW, Folkers E, et al. Diagnosis and treatment of pustular disorders in the neonate. Pediatr Dermatol. 1997;14:131-143.
- Barnes CJ, Eichenfield LF, Lee J, et al. A practical approach for the use of oral isotretinoin for infantile acne. Pediatr Dermatol. 2005;22:166-169.
- Janniger CK. Neonatal and infantile acne vulgaris. Cutis. 1993;52:16.
- Chew EW, Bingham A, Burrows D. Incidence of acne vulgaris in patients with infantile acne. Clin Exp Dermatol. 1990;15:376-377.
- Duke EM. Infantile acne associated with transient increases in plasma concentrations of luteinising hormone, follicle-stimulating hormone, and testosterone. Br Med J (Clinical Res Ed). 1981;282:1275-1276.
- Mann MW, Ellis SS, Mallory SB. Infantile acne as the initial sign of an adrenocortical tumor [published online ahead of print September 14, 2006]. J Am Acad Dermatol. 2007;56(suppl 2):S15-S18.
- Kang SK, Jee MS, Choi JH, et al. A case of infantile acne due to Pityrosporum. Pediatr Dermatol. 2003;20:68-70.
- Di Landro A, Cazzaniga S, Parazzini F, et al. Family history, body mass index, selected dietary factors, menstrual history, and risk of moderate to severe acne in adolescents and young adults [published online ahead of print March 3, 2012]. J Am Acad Dermatol. 2012;67:1129-1135.
Acne vulgaris typically is associated with adolescence and young adulthood; however, it also can affect neonates, infants, and small children.1 Acne neonatorum occurs in up to 20% of newborns. The clinical importance of neonatal acne lies in its differentiation from infectious diseases, the exclusion of virilization as its underlying cause, and the possible implication of severe acne in adolescence.2 Neonatal acne also must be distinguished from acne that is induced by application of topical oils and ointments (acne venenata) and from acneform eruptions induced by acnegenic maternal medications such as hydantoin (fetal hydantoin syndrome) and lithium.3
Neonatal Acne (Acne Neonatorum)
Clinical Presentation
Neonatal acne (acne neonatorum) typically presents as small closed comedones on the forehead, nose, and cheeks (Figure 1).4 Accompanying sebaceous hyperplasia often is noted.5 Less frequently, open comedones, inflammatory papules, and pustules may develop.6 Neonatal acne may be evident at birth or appear during the first 4 weeks of life7 and is more commonly seen in boys.8
Etiology
Several factors may be pivotal in the etiology of neonatal acne, including increased sebum excretion, stimulation of the sebaceous glands by maternal or neonatal androgens,4 and colonization of sebaceous glands by Malassezia species.2 Increased sebum excretion occurs during the neonatal period due to enlarged sebaceous glands,2 which may result from the substantial production of β-hydroxysteroids from the relatively large adrenal glands.9,10 After 6 months of age, the size of the sebaceous glands and the sebum excretion rate decrease.9,10
Both maternal and neonatal androgens have been implicated in the stimulation of sebaceous glands in neonatal acne.2 The neonatal adrenal gland produces high levels of dehydroepiandrosterone,2 which stimulate sebaceous glands until around 1 year of age when dehydroepiandrosterone levels drop off as a consequence of involution of the neonatal adrenal gland.11 Testicular androgens provide additional stimulation to the sebaceous glands, which may explain why neonatal acne is more common in boys.1 Neonatal acne may be an inflammatory response to Malassezia species; however, Malassezia was not isolated in a series of patients,12 suggesting that neonatal acne is an early presentation of comedonal acne and not a response to Malassezia.2,12
Differential Diagnosis
There are a number of acneform eruptions that should be considered in the differential diagnosis,3 including bacterial folliculitis, secondary syphilis,13 herpes simplex virus and varicella zoster virus,14 and skin colonization by fungi of Malassezia species.15 Other neonatal eruptions such as erythema toxicum neonatorum,16 transient neonatal pustular melanosis, and milia and pustular miliaria, as well as a drug eruption associated with hydantoin, lithium, or halogens should be considered.17 The relationship between neonatal acne and neonatal cephalic pustulosis, which is characterized by papules and pustules without comedones, is controversial; some consider them to be 2 different entities,14 while others do not.18
Treatment
Guardians should be reassured that neonatal acne is mild, self-limited, and generally resolves spontaneously without scarring in approximately 1 to 3 months.1,2 In most cases, no treatment is needed.19 If necessary, comedones may be treated with azelaic acid cream 20% or tretinoin cream 0.025% to 0.05%.1,2 For inflammatory lesions, erythromycin solution 2% and benzoyl peroxide gel 2.5% may be used.1,20 Severe or recalcitrant disease warrants a workup for congenital adrenal hyperplasia, a virilizing tumor, or underlying endocrinopathy.19
Infantile Acne Vulgaris
Clinical Presentation
Infantile acne vulgaris shares similarities with neonatal acne21,22 in that they both affect the face, predominantly the cheeks, and have a male predominance (Figure 2).1,10 However, by definition, onset of infantile acne typically occurs later than acne neonatorum, usually at 3 to 6 months of age.1,4 Lesions are more pleomorphic and inflammatory than in neonatal acne. In addition to closed and open comedones, infantile acne may be first evident with papules, pustules, severe nodules, and cysts with scarring potential (Figure 3).1,2,5 Accordingly, treatment may be required. Most cases of infantile acne resolve by 4 or 5 years of age, but some remain active into puberty.1 Patients with a history of infantile acne have an increased incidence of acne vulgaris during adolescence compared to their peers, with greater severity and enhanced risk for scarring.4,23
Etiology
The etiology of infantile acne remains unclear.2 Similar to neonatal acne, infantile acne may be a result of elevated androgens produced by the fetal adrenal glands as well as by the testes in males.11 For example, a child with infantile acne had elevated luteinizing hormone, follicle-stimulating hormone, and testosterone levels.24 Therefore, hyperandrogenism should be considered as an etiology. Other causes also have been suggested. Rarely, an adrenocortical tumor may be associated with persistent infantile acne with signs of virilization and rapid development.25Malassezia was implicated in infantile acne in a 6-month-old infant who was successfully treated with ketoconazole cream 2%.26
Differential Diagnosis
Infantile acne often is misdiagnosed because it is rarely considered in the differential diagnosis. When closed comedones predominate, acne venenata induced by topical creams, lotions, or oils may be etiologic. Chloracne also should be considered.14
Treatment
Guardians should be educated about the likely chronicity of infantile acne, which may require long-term treatment, as well as the possibility that acne may recur in severe form during puberty.1 The treatment strategy for infantile acne is similar to treatment of acne at any age, with topical agents including retinoids (eg, tretinoin, benzoyl peroxide) and topical antibacterials (eg, erythromycin). Twice-daily erythromycin 125 to 250 mg is the treatment of choice when oral antibiotics are indicated. Tetracyclines are contraindicated in treatment of neonatal and infantile acne. Intralesional injections with low-concentration triamcinolone acetonide, cryotherapy, or topical corticosteroids for a short period of time can be used to treat deep nodules and cysts.2 Acne that is refractory to treatment with oral antibiotics alone or combined with topical treatments poses a dilemma, given the potential cosmetic sequelae of scarring and quality-of-life concerns. Because reducing or eliminating dairy intake appears beneficial for adolescents with moderate to severe acne,27 this approach may represent a good option for infantile acne.
Conclusion
Neonatal and infantile acne vulgaris may be overlooked or misdiagnosed. It is important to consider and treat. Early childhood acne may represent a virilization syndrome.
Acne vulgaris typically is associated with adolescence and young adulthood; however, it also can affect neonates, infants, and small children.1 Acne neonatorum occurs in up to 20% of newborns. The clinical importance of neonatal acne lies in its differentiation from infectious diseases, the exclusion of virilization as its underlying cause, and the possible implication of severe acne in adolescence.2 Neonatal acne also must be distinguished from acne that is induced by application of topical oils and ointments (acne venenata) and from acneform eruptions induced by acnegenic maternal medications such as hydantoin (fetal hydantoin syndrome) and lithium.3
Neonatal Acne (Acne Neonatorum)
Clinical Presentation
Neonatal acne (acne neonatorum) typically presents as small closed comedones on the forehead, nose, and cheeks (Figure 1).4 Accompanying sebaceous hyperplasia often is noted.5 Less frequently, open comedones, inflammatory papules, and pustules may develop.6 Neonatal acne may be evident at birth or appear during the first 4 weeks of life7 and is more commonly seen in boys.8
Etiology
Several factors may be pivotal in the etiology of neonatal acne, including increased sebum excretion, stimulation of the sebaceous glands by maternal or neonatal androgens,4 and colonization of sebaceous glands by Malassezia species.2 Increased sebum excretion occurs during the neonatal period due to enlarged sebaceous glands,2 which may result from the substantial production of β-hydroxysteroids from the relatively large adrenal glands.9,10 After 6 months of age, the size of the sebaceous glands and the sebum excretion rate decrease.9,10
Both maternal and neonatal androgens have been implicated in the stimulation of sebaceous glands in neonatal acne.2 The neonatal adrenal gland produces high levels of dehydroepiandrosterone,2 which stimulate sebaceous glands until around 1 year of age when dehydroepiandrosterone levels drop off as a consequence of involution of the neonatal adrenal gland.11 Testicular androgens provide additional stimulation to the sebaceous glands, which may explain why neonatal acne is more common in boys.1 Neonatal acne may be an inflammatory response to Malassezia species; however, Malassezia was not isolated in a series of patients,12 suggesting that neonatal acne is an early presentation of comedonal acne and not a response to Malassezia.2,12
Differential Diagnosis
There are a number of acneform eruptions that should be considered in the differential diagnosis,3 including bacterial folliculitis, secondary syphilis,13 herpes simplex virus and varicella zoster virus,14 and skin colonization by fungi of Malassezia species.15 Other neonatal eruptions such as erythema toxicum neonatorum,16 transient neonatal pustular melanosis, and milia and pustular miliaria, as well as a drug eruption associated with hydantoin, lithium, or halogens should be considered.17 The relationship between neonatal acne and neonatal cephalic pustulosis, which is characterized by papules and pustules without comedones, is controversial; some consider them to be 2 different entities,14 while others do not.18
Treatment
Guardians should be reassured that neonatal acne is mild, self-limited, and generally resolves spontaneously without scarring in approximately 1 to 3 months.1,2 In most cases, no treatment is needed.19 If necessary, comedones may be treated with azelaic acid cream 20% or tretinoin cream 0.025% to 0.05%.1,2 For inflammatory lesions, erythromycin solution 2% and benzoyl peroxide gel 2.5% may be used.1,20 Severe or recalcitrant disease warrants a workup for congenital adrenal hyperplasia, a virilizing tumor, or underlying endocrinopathy.19
Infantile Acne Vulgaris
Clinical Presentation
Infantile acne vulgaris shares similarities with neonatal acne21,22 in that they both affect the face, predominantly the cheeks, and have a male predominance (Figure 2).1,10 However, by definition, onset of infantile acne typically occurs later than acne neonatorum, usually at 3 to 6 months of age.1,4 Lesions are more pleomorphic and inflammatory than in neonatal acne. In addition to closed and open comedones, infantile acne may be first evident with papules, pustules, severe nodules, and cysts with scarring potential (Figure 3).1,2,5 Accordingly, treatment may be required. Most cases of infantile acne resolve by 4 or 5 years of age, but some remain active into puberty.1 Patients with a history of infantile acne have an increased incidence of acne vulgaris during adolescence compared to their peers, with greater severity and enhanced risk for scarring.4,23
Etiology
The etiology of infantile acne remains unclear.2 Similar to neonatal acne, infantile acne may be a result of elevated androgens produced by the fetal adrenal glands as well as by the testes in males.11 For example, a child with infantile acne had elevated luteinizing hormone, follicle-stimulating hormone, and testosterone levels.24 Therefore, hyperandrogenism should be considered as an etiology. Other causes also have been suggested. Rarely, an adrenocortical tumor may be associated with persistent infantile acne with signs of virilization and rapid development.25Malassezia was implicated in infantile acne in a 6-month-old infant who was successfully treated with ketoconazole cream 2%.26
Differential Diagnosis
Infantile acne often is misdiagnosed because it is rarely considered in the differential diagnosis. When closed comedones predominate, acne venenata induced by topical creams, lotions, or oils may be etiologic. Chloracne also should be considered.14
Treatment
Guardians should be educated about the likely chronicity of infantile acne, which may require long-term treatment, as well as the possibility that acne may recur in severe form during puberty.1 The treatment strategy for infantile acne is similar to treatment of acne at any age, with topical agents including retinoids (eg, tretinoin, benzoyl peroxide) and topical antibacterials (eg, erythromycin). Twice-daily erythromycin 125 to 250 mg is the treatment of choice when oral antibiotics are indicated. Tetracyclines are contraindicated in treatment of neonatal and infantile acne. Intralesional injections with low-concentration triamcinolone acetonide, cryotherapy, or topical corticosteroids for a short period of time can be used to treat deep nodules and cysts.2 Acne that is refractory to treatment with oral antibiotics alone or combined with topical treatments poses a dilemma, given the potential cosmetic sequelae of scarring and quality-of-life concerns. Because reducing or eliminating dairy intake appears beneficial for adolescents with moderate to severe acne,27 this approach may represent a good option for infantile acne.
Conclusion
Neonatal and infantile acne vulgaris may be overlooked or misdiagnosed. It is important to consider and treat. Early childhood acne may represent a virilization syndrome.
- Jansen T, Burgdorf WH, Plewig G. Pathogenesis and treatment of acne in childhood. Pediatr Dermatol. 1997;14:17-21.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Kuflik JH, Schwartz RA. Acneiform eruptions. Cutis. 2000;66:97-100.
- Barbareschi M, Benardon S, Guanziroli E, et al. Classification and grading. In: Schwartz RA, Micali G, eds. Acne. Gurgaon, India: Nature Publishing Group; 2013:67-75.
- Mengesha YM, Bennett ML. Pustular skin disorders: diagnosis and treatment. Am J Clin Dermatol. 2002;3:389-400.
- O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. common rashes. Am Fam Physician. 2008;77:47-52.
- Nanda S, Reddy BS, Ramji S, et al. Analytical study of pustular eruptions in neonates. Pediatr Dermatol. 2002;19:210-215.
- Yonkosky DM, Pochi PE. Acne vulgaris in childhood. pathogenesis and management. Dermatol Clin. 1986;4:127-136.
- Agache P, Blanc D, Barrand C, et al. Sebum levels during the first year of life. Br J Dermatol. 1980;103:643-649.
- Herane MI, Ando I. Acne in infancy and acne genetics. Dermatology. 2003;206:24-28.
- Lucky AW. A review of infantile and pediatric acne. Dermatology (Basel, Switzerland). 1998;103:643-649.
- Bernier V, Weill FX, Hirigoyen V, et al. Skin colonization by Malassezia species in neonates: a prospective study and relationship with neonatal cephalic pustulosis. Arch Dermatol. 2002;138:215-218.
- Lambert WC, Bagley MP, Khan Y, et al. Pustular acneiform secondary syphilis. Cutis. 1986;37:69-70.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Borton LK, Schwartz RA. Pityrosporum folliculitis: a common acneiform condition of middle age. Ariz Med. 1981;38:598-601.
- Morgan AJ, Steen CJ, Schwartz RA, et al. Erythema toxicum neonatorum revisited. Cutis. 2009;83:13-16.
- Brodkin RH, Schwartz RA. Cutaneous signs of dioxin exposure. Am Fam Physician. 1984;30:189-194.
- Mancini AJ, Baldwin HE, Eichenfield LF, et al. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg. 2011;30(suppl 3):S2-S5.
- Katsambas AD, Katoulis AC, Stavropoulos P. Acne neonatorum: a study of 22 cases. Int J Dermatol. 1999;38:128-130.
- Van Praag MC, Van Rooij RW, Folkers E, et al. Diagnosis and treatment of pustular disorders in the neonate. Pediatr Dermatol. 1997;14:131-143.
- Barnes CJ, Eichenfield LF, Lee J, et al. A practical approach for the use of oral isotretinoin for infantile acne. Pediatr Dermatol. 2005;22:166-169.
- Janniger CK. Neonatal and infantile acne vulgaris. Cutis. 1993;52:16.
- Chew EW, Bingham A, Burrows D. Incidence of acne vulgaris in patients with infantile acne. Clin Exp Dermatol. 1990;15:376-377.
- Duke EM. Infantile acne associated with transient increases in plasma concentrations of luteinising hormone, follicle-stimulating hormone, and testosterone. Br Med J (Clinical Res Ed). 1981;282:1275-1276.
- Mann MW, Ellis SS, Mallory SB. Infantile acne as the initial sign of an adrenocortical tumor [published online ahead of print September 14, 2006]. J Am Acad Dermatol. 2007;56(suppl 2):S15-S18.
- Kang SK, Jee MS, Choi JH, et al. A case of infantile acne due to Pityrosporum. Pediatr Dermatol. 2003;20:68-70.
- Di Landro A, Cazzaniga S, Parazzini F, et al. Family history, body mass index, selected dietary factors, menstrual history, and risk of moderate to severe acne in adolescents and young adults [published online ahead of print March 3, 2012]. J Am Acad Dermatol. 2012;67:1129-1135.
- Jansen T, Burgdorf WH, Plewig G. Pathogenesis and treatment of acne in childhood. Pediatr Dermatol. 1997;14:17-21.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Kuflik JH, Schwartz RA. Acneiform eruptions. Cutis. 2000;66:97-100.
- Barbareschi M, Benardon S, Guanziroli E, et al. Classification and grading. In: Schwartz RA, Micali G, eds. Acne. Gurgaon, India: Nature Publishing Group; 2013:67-75.
- Mengesha YM, Bennett ML. Pustular skin disorders: diagnosis and treatment. Am J Clin Dermatol. 2002;3:389-400.
- O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. common rashes. Am Fam Physician. 2008;77:47-52.
- Nanda S, Reddy BS, Ramji S, et al. Analytical study of pustular eruptions in neonates. Pediatr Dermatol. 2002;19:210-215.
- Yonkosky DM, Pochi PE. Acne vulgaris in childhood. pathogenesis and management. Dermatol Clin. 1986;4:127-136.
- Agache P, Blanc D, Barrand C, et al. Sebum levels during the first year of life. Br J Dermatol. 1980;103:643-649.
- Herane MI, Ando I. Acne in infancy and acne genetics. Dermatology. 2003;206:24-28.
- Lucky AW. A review of infantile and pediatric acne. Dermatology (Basel, Switzerland). 1998;103:643-649.
- Bernier V, Weill FX, Hirigoyen V, et al. Skin colonization by Malassezia species in neonates: a prospective study and relationship with neonatal cephalic pustulosis. Arch Dermatol. 2002;138:215-218.
- Lambert WC, Bagley MP, Khan Y, et al. Pustular acneiform secondary syphilis. Cutis. 1986;37:69-70.
- Antoniou C, Dessinioti C, Stratigos AJ, et al. Clinical and therapeutic approach to childhood acne: an update. Pediatr Dermatol. 2009;26:373-380.
- Borton LK, Schwartz RA. Pityrosporum folliculitis: a common acneiform condition of middle age. Ariz Med. 1981;38:598-601.
- Morgan AJ, Steen CJ, Schwartz RA, et al. Erythema toxicum neonatorum revisited. Cutis. 2009;83:13-16.
- Brodkin RH, Schwartz RA. Cutaneous signs of dioxin exposure. Am Fam Physician. 1984;30:189-194.
- Mancini AJ, Baldwin HE, Eichenfield LF, et al. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg. 2011;30(suppl 3):S2-S5.
- Katsambas AD, Katoulis AC, Stavropoulos P. Acne neonatorum: a study of 22 cases. Int J Dermatol. 1999;38:128-130.
- Van Praag MC, Van Rooij RW, Folkers E, et al. Diagnosis and treatment of pustular disorders in the neonate. Pediatr Dermatol. 1997;14:131-143.
- Barnes CJ, Eichenfield LF, Lee J, et al. A practical approach for the use of oral isotretinoin for infantile acne. Pediatr Dermatol. 2005;22:166-169.
- Janniger CK. Neonatal and infantile acne vulgaris. Cutis. 1993;52:16.
- Chew EW, Bingham A, Burrows D. Incidence of acne vulgaris in patients with infantile acne. Clin Exp Dermatol. 1990;15:376-377.
- Duke EM. Infantile acne associated with transient increases in plasma concentrations of luteinising hormone, follicle-stimulating hormone, and testosterone. Br Med J (Clinical Res Ed). 1981;282:1275-1276.
- Mann MW, Ellis SS, Mallory SB. Infantile acne as the initial sign of an adrenocortical tumor [published online ahead of print September 14, 2006]. J Am Acad Dermatol. 2007;56(suppl 2):S15-S18.
- Kang SK, Jee MS, Choi JH, et al. A case of infantile acne due to Pityrosporum. Pediatr Dermatol. 2003;20:68-70.
- Di Landro A, Cazzaniga S, Parazzini F, et al. Family history, body mass index, selected dietary factors, menstrual history, and risk of moderate to severe acne in adolescents and young adults [published online ahead of print March 3, 2012]. J Am Acad Dermatol. 2012;67:1129-1135.
Practice Points
- Infantile acne needs to be recognized and treated.
- Acne in early childhood may represent virilization.
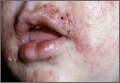
Rosacea in the Pediatric Population
Epidemiology
Rosacea in childhood is most likely underreported because of its clinical similarity to other erythematous facial disorders.1 Rosacea is generally thought of as a disease of fair-skinned, young to middle-aged adults, though it has been noted to affect people of other complexions and ages.2 Most full-blown cases in the pediatric population have been in light-skinned children ranging from infants to adolescents.
Etiology
The etiology of rosacea is unknown, though certain exacerbating factors undoubtedly have a role in predisposed individuals.3,4 Emotions such as anger, anxiety, and embarrassment can lead to flushing. Environmental conditions such as wind, cold, humidity, or heat from any source (eg, sun, sauna, whirlpool, vigorous exercise) can do the same. Vasodilators such as alcohol or vasodilatory medications can lead to flushing, though these are not likely causes in children. Spicy foods such as chili, curry, and peppers, as well as hot foods and beverages including coffee, tea, and hot chocolate, may contribute to symptoms. Irritants such as alcohol-based cleansers, astringents, perfume, shaving lotion, certain soaps, sunscreen, and facecloths may aggravate rosacea.3,4 Saprophytic mites (Demodex folliculorum and Demodex brevis) may cause an inflammatory or allergic reaction by either blocking hair follicles or acting as vectors for microorganisms that some believe may be responsible for or may trigger rosacea.5 Immunodeficiency, as in patients with human immunodeficiency virus, also may contribute to the development of rosacea.6
Genetics plays an uncertain role in the development of blushing and ultimately rosacea. If vasodilator substances or mediators are implicated in the development of rosacea as postulated, the disease may have a genetic basis because such mediators are often under the control of single genes.7 In one study, 20% of children with rosacea were found to have a history of rosacea in their immediate families, though this number may be an underestimate because only one parent of each patient was examined and half of the parents clinically diagnosed with rosacea reported no familial involvement.8 A family history of perioral dermatitis also may be important, as this condition may be related to rosacea.9
Pathophysiology
Chronic transient vasodilation, as occurs with blushing, is the earliest representation of rosacea. The warmth and redness associated with flushing is caused by vasodilation, allowing excess blood flow, and by engorgement of the subpapillary venous plexus.2 Flushing without sweating is typically seen in children and is likely due to circulating vasodilator substances or mediators such as bradykinin, catecholamines, cytokines, endorphins, gastrin, histamine, neuropeptides, serotonin, substance P, and vascular endothelial growth factor.10 A flaw in the autonomic innervation of the cutaneous vasculature also is a likely mechanism.10
Clinical Description
The first stage of rosacea consists of blushing, in which the face becomes bright red in response to certain stimuli (Figure). Episodes of erythema are recurrent and last longer than normal physiologic flushing, which typically subsides within minutes.11 Telangiectasias can become apparent over time. Children in this early stage of the condition may complain of burning or irritation.
Please refer to the PDF to view the figure
In the second, or intermediate, stage of rosacea, the rash consists of papules and pustules on a background of erythema with telangiectasias confined to the child's face. Although peripheral involvement of the back, upper chest, and scalp may be seen in adults, these areas seem to be spared in the pediatric population.12
The third, or late, stage of rosacea involves coarse skin, inflammatory nodules, or gross enlargement of facial features.11 Such chronic changes do not occur in children as they do in adults, presumably because the disease process takes more time to evolve.13
Eye involvement can occur in children.14 It may include manifestations such as blepharoconjunctivitis, episcleritis, keratitis, meibomianitis, chalazia, hordeola, and hyperemic conjunctivae.15,16 Although any of these eye conditions can potentially occur in children, meibomian gland inflammation and keratitis are the common findings noted.17 Peripheral vascularization followed by subepithelial infiltrates can lead to scarring or perforation in the lower two thirds of the cornea. The disease may be unilateral, but most commonly it affects both eyes.17
Steroid-induced rosacea also has been termed iatrosacea because of its mode of acquisition.18 Topical fluorinated and low-dose corticosteroids can cause a rosacealike dermatitis of the face consisting of persistent erythema with papules, pustules, telangiectasias, and sometimes atrophy.19-22 Corticosteroids may be an exacerbating factor leading to classic rosacea rather than the cause of an independent disease.2 The distribution of steroid rosacea to the eyelids and lateral face may help distinguish it from the centrally located typical rosacea.23 A case of pediatric rosacea associated with the use of topical fluorinated glucocorticosteroids was identified in a 9-month-old boy and 16-year-old girl, both with erythematous patches and papules on the cheeks, paranasal areas, and chin.18,23 Forty-six boys and 60 girls, ranging in age of onset from 6 months to 13 years (average, 7 years), were diagnosed with steroid rosacea. Nearly all of the children had perinasal and perioral involvement of erythematous skin interspersed with papules and/or pustules. The lower eyelids were affected in roughly half of the patients.8
Diagnosis
Consistent flushing in children may be a sign of vasomotor instability and early rosacea. These children may blush more frequently and with greater intensity for longer periods than their peers exposed to the same stimuli.24 Thus, blushing in the early stage of rosacea may be an accentuation of the body's normal physiologic response system. A diagnosis of pediatric rosacea beyond the initial stage should be considered when a healthy child has acuminate papules and small pustules of the face, especially if there also exists flushing, telangiectasias, or a family history of rosacea.14
Differential Diagnosis
The earliest form of rosacea, facial blushing, may be difficult to distinguish from flushing due to other causes. Blushing due to emotions such as embarrassment or anger and to exercise-induced flushing are both appropriate reactions to such stimuli, whereas blushing in the first stage of rosacea may be an exaggeration of this phenomenon.2 The main pathway for thermoregulatory and emotional flushing is the cervical sympathetic outflow tract.25 Gustatory blushing, as occurs with consumption of spicy foods, is mediated by autonomic neurons via a branch of the trigeminal nerve.2 Sweating often occurs in conjunction with the aforementioned causes of flushing, but it is rarely associated with rosacea flushing. Thus, sweating may be helpful in reaching a diagnosis; however, exceptions exist.2 Frey syndrome (auriculotemporal syndrome), which is characterized by warmth and sweating in the malar region caused by aberrant autonomic fiber connections after damage in the parotid region, may mimic the early stage of rosacea.
The intermediate stage of pediatric rosacea may be confused with other papulopustular disorders such as acne vulgaris, perioral dermatitis, and lupus erythematosus (Table). Careful attention to symptoms, distribution of facial lesions, and potential biopsy results are warranted to distinguish between the conditions. Steroid rosacea and perioral dermatitis may be variants of rosacea or completely separate conditions.9,26
Please refer to the PDF to view the table
Perioral dermatitis is a rosacealike dermatitis characterized by erythematous papules and pustules usually confined to the perioral region, though the perinasal and periocular areas may be involved.27 A granulomatous perioral dermatitis with tiny, closely spaced, flesh-colored papules in the perioral, perinasal, and periorbital areas was described in children aged 3 to 11 years.28 All cases had spontaneous resolution of symptoms regardless of treatment. The patients did not exhibit flushing or telangiectasias.28
The classic butterfly rash, consisting of erythema and telangiectasia of the malar region and associated with systemic lupus erythematosus, also can be confused with rosacea. Histopathologic examination and direct immunofluorescence of the lesion may help to differentiate lupus from rosacea.14
Laboratory Diagnosis
There is no specific histologic change unique to rosacea.12 The most common findings are telangiectasia, edema, elastosis, a variable amount of superficial and deep perivascular lymphohistiocytic inflammatory infiltrate loosely arranged around the hair follicles, and especially architectural disruption of the upper dermis.12,28 Depending on the variant of rosacea, there may be an exaggeration of one or more histopathologic signs.12 For example, granulomatous rosacea may contain collections of granulomas with multinucleated giant cells.28
Treatment
Treatment is gradual and largely determined by the clinical type of rosacea. Children with early or intermediate stages of rosacea are encouraged to avoid their individual local triggers to prevent flares. Topical corticosteroids, especially fluorinated medications, should be discouraged, because even low-potency steroids, including over-the-counter preparations and hydrocortisone 1%, have been shown to cause worsening of the condition.8
Traditional therapy for rosacea includes topical and systemic antibiotics, topical metronidazole, and topical retinoids. Oral tetracycline can be used for adolescents in doses similar to those prescribed for adults. It should not be used in children younger than approximately 9 years17 because it is known to cause dental staining and to be deposited in the skeletal system where it can cause temporary depression in bone growth.29,30 Azithromycin or a low dose of doxycycline can be used with good results.31,32 For younger children, oral erythromycin is safe and effective to eliminate the erythema, papules, and pustules of rosacea.32 Topical erythromycin and clindamycin have been used with varying results. Azelaic acid and isotretinoin also may be effective.33 Topical metronidazole 0.75% gel has proven effective in clinical trials.34,35 A combination of systemic antibiotics and topical treatment may lead to a substantial reduction in inflammatory lesions, erythema, and the size and diameter of telangiectatic vessels.32
Eyelid hygiene and erythromycin or bacitracin ointment, to improve meibomian gland function, are appropriate initial treatments for the ocular manifestations of childhood rosacea.17,36 A low dose of steroid drops can manage significant irritation when needed. Systemic therapy with tetracycline or other oral pharmaceuticals used to treat the face also may work for ocular symptoms.17
The treatment of steroid rosacea is a slow process often involving antiacne agents such as benzoyl peroxide and oral or topical antibiotics.23 Abrupt discontinuation of topical steroids followed by administration of antibiotics is a suitable treatment option. Prior recommendation has been to taper all topical steroids to prevent rebound flare; however, one study found clearing of symptoms by week 3 in 22% of patients, by week 4 in 86% of patients, and by week 8 in 100% of patients following abrupt cessation of topical steroids and a regimen of oral erythromycin stearate or topical clindamycin phosphate in children with erythromycin allergy or intolerance.8 Thus, a gradual withdrawal of topical nonfluorinated steroids may not be necessary. However, it is more common than not for children to experience an initial flare of their condition upon withdrawal from topical fluorinated steroids. This is followed by a slow and steady fading.23
- Buxton PK. Acne and rosacea. BMJ. 1988;296:43-45.
- Greaves MW. Flushing and flushing syndromes, rosacea and perioral dermatitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. Vol 3. Oxford, England: Blackwell Science; 1998:2099-2111.
- McDonnell JK, Tomecki KJ. Rosacea: an update. Cleve Clin J Med. 2000;67:587-590.
- Zuber TJ. Rosacea. Prim Care. 2000;27:309-318.
- Roihu T, Kariniemi A-L. Demodex mites in acne rosacea. J Cutan Pathol. 1998;25:550-552.
- Vin-Christian K, Maurer TA, Berger TG. Acne rosacea as a cutaneous manifestation of HIV infection. J Am Acad Dermatol. 1994;30:139-140.
- Bamford JT. Rosacea: current thoughts on origin. Semin Cutan Med Surg. 2001;20:199-206.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Weston WL, Morelli JG. Identical twins with perioral dermatitis. Pediatr Dermatol. 1998;15:144.
- Landow K. Unraveling the mystery of rosacea. Postgrad Med. 2002;112:51-58.
- Jansen T, Plewig G. Rosacea. In: DJ Demis, ed. Clinical Dermatology. Philadelphia, Pa: Lippincott Williams and Wilkins; 1997:1-11.
- Marks R, Harcourt-Webster JN. Histopathology of rosacea. Arch Dermatol. 1969;100:683-691.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Derm. 1972;87:425-429.
- Drolet B, Paller AS. Childhood rosacea. Pediatr Dermatol. 1992;9:22-26.
- Browning DJ, Proia AD. Ocular rosacea. Surv Ophthalmol. 1986;31:145-158.
- Jenkins MS, Brown SI, Lempert SL, et al. Ocular rosacea. Am J Ophthalmol. 1979;88:618-622.
- Erzurum SA, Feder RS, Greenwald MJ. Acne rosacea with keratitis in childhood. Arch Ophthalmol. 1993;111:228-230.
- Litt JZ. Steroid-induced rosacea. Am Fam Physician. 1993;48:67-71.
- Leyden JJ, Thew M, Kligman AM. Steroid rosacea. Arch Dermatol. 1974;110:619-622.
- Hogan DJ, Epstein JD, Lane PR. Perioral dermatitis: an uncommon condition? Can Med Assoc J. 1986;134:1025-1028.
- Hogan DJ, Rooney ME. Facial telangiectasia associated with long-term application of a topical corticosteroid to the scalp. J Am Acad Dermatol. 1989;20:1129-1130.
- Coskey RJ. Perioral dermatitis. Cutis. 1984;34:55-56, 58.
- Franco HL, Weson WL. Steroid rosacea in children. Pediatrics. 1979;64:36-38.
- Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
- Drummond PD, Lance JW. Facial flushing mediated by the sympathetic nervous system. Brain. 1987;110:793-803.
- Manders SM, Lucky AW. Perioral dermatitis in childhood.J Am Acad Dermatol. 1992;27:688-692.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Howard R, Tsuchiya A. Adult skin disease in the pediatric patient. Dermatol Clin. 1998;16:593-608.
- Gruber GG, Callen JP. Systemic complications of commonly used dermatologic drugs. Cutis. 1978;21:825-829.
- Caputo R, Barbareschi M, Veraldi S. Azithromycin: a new drug for systemic treatment of inflammatory acneic lesions. G Ital Dermatol Venereol.2003;138:327-331.
- Bikowski JB. Subantimicrobial dose for acne and rosacea. SKINmed. 2003;2:234-245.
- Szepietowski J. Azelaic acid gel: use in the treatment of rosacea. Dermatol Klin (Wroclaw). 2003;3:150.
- Anonymous. Topical metronidazole for rosacea. Med Lett Drugs Ther. 1989;31:75-76.
- Micali G, Licastro R, Lembo D. Treatment of rosacea with metronidazole gel 0.75%. G Ital Dermatol Venereol. 1992;127:247-250.
- McCully JP, Dougherty JM, Deneau DG. Classification of chronic blepharitis.
Ophthalmology. 1982;89:1173-1180.
Epidemiology
Rosacea in childhood is most likely underreported because of its clinical similarity to other erythematous facial disorders.1 Rosacea is generally thought of as a disease of fair-skinned, young to middle-aged adults, though it has been noted to affect people of other complexions and ages.2 Most full-blown cases in the pediatric population have been in light-skinned children ranging from infants to adolescents.
Etiology
The etiology of rosacea is unknown, though certain exacerbating factors undoubtedly have a role in predisposed individuals.3,4 Emotions such as anger, anxiety, and embarrassment can lead to flushing. Environmental conditions such as wind, cold, humidity, or heat from any source (eg, sun, sauna, whirlpool, vigorous exercise) can do the same. Vasodilators such as alcohol or vasodilatory medications can lead to flushing, though these are not likely causes in children. Spicy foods such as chili, curry, and peppers, as well as hot foods and beverages including coffee, tea, and hot chocolate, may contribute to symptoms. Irritants such as alcohol-based cleansers, astringents, perfume, shaving lotion, certain soaps, sunscreen, and facecloths may aggravate rosacea.3,4 Saprophytic mites (Demodex folliculorum and Demodex brevis) may cause an inflammatory or allergic reaction by either blocking hair follicles or acting as vectors for microorganisms that some believe may be responsible for or may trigger rosacea.5 Immunodeficiency, as in patients with human immunodeficiency virus, also may contribute to the development of rosacea.6
Genetics plays an uncertain role in the development of blushing and ultimately rosacea. If vasodilator substances or mediators are implicated in the development of rosacea as postulated, the disease may have a genetic basis because such mediators are often under the control of single genes.7 In one study, 20% of children with rosacea were found to have a history of rosacea in their immediate families, though this number may be an underestimate because only one parent of each patient was examined and half of the parents clinically diagnosed with rosacea reported no familial involvement.8 A family history of perioral dermatitis also may be important, as this condition may be related to rosacea.9
Pathophysiology
Chronic transient vasodilation, as occurs with blushing, is the earliest representation of rosacea. The warmth and redness associated with flushing is caused by vasodilation, allowing excess blood flow, and by engorgement of the subpapillary venous plexus.2 Flushing without sweating is typically seen in children and is likely due to circulating vasodilator substances or mediators such as bradykinin, catecholamines, cytokines, endorphins, gastrin, histamine, neuropeptides, serotonin, substance P, and vascular endothelial growth factor.10 A flaw in the autonomic innervation of the cutaneous vasculature also is a likely mechanism.10
Clinical Description
The first stage of rosacea consists of blushing, in which the face becomes bright red in response to certain stimuli (Figure). Episodes of erythema are recurrent and last longer than normal physiologic flushing, which typically subsides within minutes.11 Telangiectasias can become apparent over time. Children in this early stage of the condition may complain of burning or irritation.
Please refer to the PDF to view the figure
In the second, or intermediate, stage of rosacea, the rash consists of papules and pustules on a background of erythema with telangiectasias confined to the child's face. Although peripheral involvement of the back, upper chest, and scalp may be seen in adults, these areas seem to be spared in the pediatric population.12
The third, or late, stage of rosacea involves coarse skin, inflammatory nodules, or gross enlargement of facial features.11 Such chronic changes do not occur in children as they do in adults, presumably because the disease process takes more time to evolve.13
Eye involvement can occur in children.14 It may include manifestations such as blepharoconjunctivitis, episcleritis, keratitis, meibomianitis, chalazia, hordeola, and hyperemic conjunctivae.15,16 Although any of these eye conditions can potentially occur in children, meibomian gland inflammation and keratitis are the common findings noted.17 Peripheral vascularization followed by subepithelial infiltrates can lead to scarring or perforation in the lower two thirds of the cornea. The disease may be unilateral, but most commonly it affects both eyes.17
Steroid-induced rosacea also has been termed iatrosacea because of its mode of acquisition.18 Topical fluorinated and low-dose corticosteroids can cause a rosacealike dermatitis of the face consisting of persistent erythema with papules, pustules, telangiectasias, and sometimes atrophy.19-22 Corticosteroids may be an exacerbating factor leading to classic rosacea rather than the cause of an independent disease.2 The distribution of steroid rosacea to the eyelids and lateral face may help distinguish it from the centrally located typical rosacea.23 A case of pediatric rosacea associated with the use of topical fluorinated glucocorticosteroids was identified in a 9-month-old boy and 16-year-old girl, both with erythematous patches and papules on the cheeks, paranasal areas, and chin.18,23 Forty-six boys and 60 girls, ranging in age of onset from 6 months to 13 years (average, 7 years), were diagnosed with steroid rosacea. Nearly all of the children had perinasal and perioral involvement of erythematous skin interspersed with papules and/or pustules. The lower eyelids were affected in roughly half of the patients.8
Diagnosis
Consistent flushing in children may be a sign of vasomotor instability and early rosacea. These children may blush more frequently and with greater intensity for longer periods than their peers exposed to the same stimuli.24 Thus, blushing in the early stage of rosacea may be an accentuation of the body's normal physiologic response system. A diagnosis of pediatric rosacea beyond the initial stage should be considered when a healthy child has acuminate papules and small pustules of the face, especially if there also exists flushing, telangiectasias, or a family history of rosacea.14
Differential Diagnosis
The earliest form of rosacea, facial blushing, may be difficult to distinguish from flushing due to other causes. Blushing due to emotions such as embarrassment or anger and to exercise-induced flushing are both appropriate reactions to such stimuli, whereas blushing in the first stage of rosacea may be an exaggeration of this phenomenon.2 The main pathway for thermoregulatory and emotional flushing is the cervical sympathetic outflow tract.25 Gustatory blushing, as occurs with consumption of spicy foods, is mediated by autonomic neurons via a branch of the trigeminal nerve.2 Sweating often occurs in conjunction with the aforementioned causes of flushing, but it is rarely associated with rosacea flushing. Thus, sweating may be helpful in reaching a diagnosis; however, exceptions exist.2 Frey syndrome (auriculotemporal syndrome), which is characterized by warmth and sweating in the malar region caused by aberrant autonomic fiber connections after damage in the parotid region, may mimic the early stage of rosacea.
The intermediate stage of pediatric rosacea may be confused with other papulopustular disorders such as acne vulgaris, perioral dermatitis, and lupus erythematosus (Table). Careful attention to symptoms, distribution of facial lesions, and potential biopsy results are warranted to distinguish between the conditions. Steroid rosacea and perioral dermatitis may be variants of rosacea or completely separate conditions.9,26
Please refer to the PDF to view the table
Perioral dermatitis is a rosacealike dermatitis characterized by erythematous papules and pustules usually confined to the perioral region, though the perinasal and periocular areas may be involved.27 A granulomatous perioral dermatitis with tiny, closely spaced, flesh-colored papules in the perioral, perinasal, and periorbital areas was described in children aged 3 to 11 years.28 All cases had spontaneous resolution of symptoms regardless of treatment. The patients did not exhibit flushing or telangiectasias.28
The classic butterfly rash, consisting of erythema and telangiectasia of the malar region and associated with systemic lupus erythematosus, also can be confused with rosacea. Histopathologic examination and direct immunofluorescence of the lesion may help to differentiate lupus from rosacea.14
Laboratory Diagnosis
There is no specific histologic change unique to rosacea.12 The most common findings are telangiectasia, edema, elastosis, a variable amount of superficial and deep perivascular lymphohistiocytic inflammatory infiltrate loosely arranged around the hair follicles, and especially architectural disruption of the upper dermis.12,28 Depending on the variant of rosacea, there may be an exaggeration of one or more histopathologic signs.12 For example, granulomatous rosacea may contain collections of granulomas with multinucleated giant cells.28
Treatment
Treatment is gradual and largely determined by the clinical type of rosacea. Children with early or intermediate stages of rosacea are encouraged to avoid their individual local triggers to prevent flares. Topical corticosteroids, especially fluorinated medications, should be discouraged, because even low-potency steroids, including over-the-counter preparations and hydrocortisone 1%, have been shown to cause worsening of the condition.8
Traditional therapy for rosacea includes topical and systemic antibiotics, topical metronidazole, and topical retinoids. Oral tetracycline can be used for adolescents in doses similar to those prescribed for adults. It should not be used in children younger than approximately 9 years17 because it is known to cause dental staining and to be deposited in the skeletal system where it can cause temporary depression in bone growth.29,30 Azithromycin or a low dose of doxycycline can be used with good results.31,32 For younger children, oral erythromycin is safe and effective to eliminate the erythema, papules, and pustules of rosacea.32 Topical erythromycin and clindamycin have been used with varying results. Azelaic acid and isotretinoin also may be effective.33 Topical metronidazole 0.75% gel has proven effective in clinical trials.34,35 A combination of systemic antibiotics and topical treatment may lead to a substantial reduction in inflammatory lesions, erythema, and the size and diameter of telangiectatic vessels.32
Eyelid hygiene and erythromycin or bacitracin ointment, to improve meibomian gland function, are appropriate initial treatments for the ocular manifestations of childhood rosacea.17,36 A low dose of steroid drops can manage significant irritation when needed. Systemic therapy with tetracycline or other oral pharmaceuticals used to treat the face also may work for ocular symptoms.17
The treatment of steroid rosacea is a slow process often involving antiacne agents such as benzoyl peroxide and oral or topical antibiotics.23 Abrupt discontinuation of topical steroids followed by administration of antibiotics is a suitable treatment option. Prior recommendation has been to taper all topical steroids to prevent rebound flare; however, one study found clearing of symptoms by week 3 in 22% of patients, by week 4 in 86% of patients, and by week 8 in 100% of patients following abrupt cessation of topical steroids and a regimen of oral erythromycin stearate or topical clindamycin phosphate in children with erythromycin allergy or intolerance.8 Thus, a gradual withdrawal of topical nonfluorinated steroids may not be necessary. However, it is more common than not for children to experience an initial flare of their condition upon withdrawal from topical fluorinated steroids. This is followed by a slow and steady fading.23
Epidemiology
Rosacea in childhood is most likely underreported because of its clinical similarity to other erythematous facial disorders.1 Rosacea is generally thought of as a disease of fair-skinned, young to middle-aged adults, though it has been noted to affect people of other complexions and ages.2 Most full-blown cases in the pediatric population have been in light-skinned children ranging from infants to adolescents.
Etiology
The etiology of rosacea is unknown, though certain exacerbating factors undoubtedly have a role in predisposed individuals.3,4 Emotions such as anger, anxiety, and embarrassment can lead to flushing. Environmental conditions such as wind, cold, humidity, or heat from any source (eg, sun, sauna, whirlpool, vigorous exercise) can do the same. Vasodilators such as alcohol or vasodilatory medications can lead to flushing, though these are not likely causes in children. Spicy foods such as chili, curry, and peppers, as well as hot foods and beverages including coffee, tea, and hot chocolate, may contribute to symptoms. Irritants such as alcohol-based cleansers, astringents, perfume, shaving lotion, certain soaps, sunscreen, and facecloths may aggravate rosacea.3,4 Saprophytic mites (Demodex folliculorum and Demodex brevis) may cause an inflammatory or allergic reaction by either blocking hair follicles or acting as vectors for microorganisms that some believe may be responsible for or may trigger rosacea.5 Immunodeficiency, as in patients with human immunodeficiency virus, also may contribute to the development of rosacea.6
Genetics plays an uncertain role in the development of blushing and ultimately rosacea. If vasodilator substances or mediators are implicated in the development of rosacea as postulated, the disease may have a genetic basis because such mediators are often under the control of single genes.7 In one study, 20% of children with rosacea were found to have a history of rosacea in their immediate families, though this number may be an underestimate because only one parent of each patient was examined and half of the parents clinically diagnosed with rosacea reported no familial involvement.8 A family history of perioral dermatitis also may be important, as this condition may be related to rosacea.9
Pathophysiology
Chronic transient vasodilation, as occurs with blushing, is the earliest representation of rosacea. The warmth and redness associated with flushing is caused by vasodilation, allowing excess blood flow, and by engorgement of the subpapillary venous plexus.2 Flushing without sweating is typically seen in children and is likely due to circulating vasodilator substances or mediators such as bradykinin, catecholamines, cytokines, endorphins, gastrin, histamine, neuropeptides, serotonin, substance P, and vascular endothelial growth factor.10 A flaw in the autonomic innervation of the cutaneous vasculature also is a likely mechanism.10
Clinical Description
The first stage of rosacea consists of blushing, in which the face becomes bright red in response to certain stimuli (Figure). Episodes of erythema are recurrent and last longer than normal physiologic flushing, which typically subsides within minutes.11 Telangiectasias can become apparent over time. Children in this early stage of the condition may complain of burning or irritation.
Please refer to the PDF to view the figure
In the second, or intermediate, stage of rosacea, the rash consists of papules and pustules on a background of erythema with telangiectasias confined to the child's face. Although peripheral involvement of the back, upper chest, and scalp may be seen in adults, these areas seem to be spared in the pediatric population.12
The third, or late, stage of rosacea involves coarse skin, inflammatory nodules, or gross enlargement of facial features.11 Such chronic changes do not occur in children as they do in adults, presumably because the disease process takes more time to evolve.13
Eye involvement can occur in children.14 It may include manifestations such as blepharoconjunctivitis, episcleritis, keratitis, meibomianitis, chalazia, hordeola, and hyperemic conjunctivae.15,16 Although any of these eye conditions can potentially occur in children, meibomian gland inflammation and keratitis are the common findings noted.17 Peripheral vascularization followed by subepithelial infiltrates can lead to scarring or perforation in the lower two thirds of the cornea. The disease may be unilateral, but most commonly it affects both eyes.17
Steroid-induced rosacea also has been termed iatrosacea because of its mode of acquisition.18 Topical fluorinated and low-dose corticosteroids can cause a rosacealike dermatitis of the face consisting of persistent erythema with papules, pustules, telangiectasias, and sometimes atrophy.19-22 Corticosteroids may be an exacerbating factor leading to classic rosacea rather than the cause of an independent disease.2 The distribution of steroid rosacea to the eyelids and lateral face may help distinguish it from the centrally located typical rosacea.23 A case of pediatric rosacea associated with the use of topical fluorinated glucocorticosteroids was identified in a 9-month-old boy and 16-year-old girl, both with erythematous patches and papules on the cheeks, paranasal areas, and chin.18,23 Forty-six boys and 60 girls, ranging in age of onset from 6 months to 13 years (average, 7 years), were diagnosed with steroid rosacea. Nearly all of the children had perinasal and perioral involvement of erythematous skin interspersed with papules and/or pustules. The lower eyelids were affected in roughly half of the patients.8
Diagnosis
Consistent flushing in children may be a sign of vasomotor instability and early rosacea. These children may blush more frequently and with greater intensity for longer periods than their peers exposed to the same stimuli.24 Thus, blushing in the early stage of rosacea may be an accentuation of the body's normal physiologic response system. A diagnosis of pediatric rosacea beyond the initial stage should be considered when a healthy child has acuminate papules and small pustules of the face, especially if there also exists flushing, telangiectasias, or a family history of rosacea.14
Differential Diagnosis
The earliest form of rosacea, facial blushing, may be difficult to distinguish from flushing due to other causes. Blushing due to emotions such as embarrassment or anger and to exercise-induced flushing are both appropriate reactions to such stimuli, whereas blushing in the first stage of rosacea may be an exaggeration of this phenomenon.2 The main pathway for thermoregulatory and emotional flushing is the cervical sympathetic outflow tract.25 Gustatory blushing, as occurs with consumption of spicy foods, is mediated by autonomic neurons via a branch of the trigeminal nerve.2 Sweating often occurs in conjunction with the aforementioned causes of flushing, but it is rarely associated with rosacea flushing. Thus, sweating may be helpful in reaching a diagnosis; however, exceptions exist.2 Frey syndrome (auriculotemporal syndrome), which is characterized by warmth and sweating in the malar region caused by aberrant autonomic fiber connections after damage in the parotid region, may mimic the early stage of rosacea.
The intermediate stage of pediatric rosacea may be confused with other papulopustular disorders such as acne vulgaris, perioral dermatitis, and lupus erythematosus (Table). Careful attention to symptoms, distribution of facial lesions, and potential biopsy results are warranted to distinguish between the conditions. Steroid rosacea and perioral dermatitis may be variants of rosacea or completely separate conditions.9,26
Please refer to the PDF to view the table
Perioral dermatitis is a rosacealike dermatitis characterized by erythematous papules and pustules usually confined to the perioral region, though the perinasal and periocular areas may be involved.27 A granulomatous perioral dermatitis with tiny, closely spaced, flesh-colored papules in the perioral, perinasal, and periorbital areas was described in children aged 3 to 11 years.28 All cases had spontaneous resolution of symptoms regardless of treatment. The patients did not exhibit flushing or telangiectasias.28
The classic butterfly rash, consisting of erythema and telangiectasia of the malar region and associated with systemic lupus erythematosus, also can be confused with rosacea. Histopathologic examination and direct immunofluorescence of the lesion may help to differentiate lupus from rosacea.14
Laboratory Diagnosis
There is no specific histologic change unique to rosacea.12 The most common findings are telangiectasia, edema, elastosis, a variable amount of superficial and deep perivascular lymphohistiocytic inflammatory infiltrate loosely arranged around the hair follicles, and especially architectural disruption of the upper dermis.12,28 Depending on the variant of rosacea, there may be an exaggeration of one or more histopathologic signs.12 For example, granulomatous rosacea may contain collections of granulomas with multinucleated giant cells.28
Treatment
Treatment is gradual and largely determined by the clinical type of rosacea. Children with early or intermediate stages of rosacea are encouraged to avoid their individual local triggers to prevent flares. Topical corticosteroids, especially fluorinated medications, should be discouraged, because even low-potency steroids, including over-the-counter preparations and hydrocortisone 1%, have been shown to cause worsening of the condition.8
Traditional therapy for rosacea includes topical and systemic antibiotics, topical metronidazole, and topical retinoids. Oral tetracycline can be used for adolescents in doses similar to those prescribed for adults. It should not be used in children younger than approximately 9 years17 because it is known to cause dental staining and to be deposited in the skeletal system where it can cause temporary depression in bone growth.29,30 Azithromycin or a low dose of doxycycline can be used with good results.31,32 For younger children, oral erythromycin is safe and effective to eliminate the erythema, papules, and pustules of rosacea.32 Topical erythromycin and clindamycin have been used with varying results. Azelaic acid and isotretinoin also may be effective.33 Topical metronidazole 0.75% gel has proven effective in clinical trials.34,35 A combination of systemic antibiotics and topical treatment may lead to a substantial reduction in inflammatory lesions, erythema, and the size and diameter of telangiectatic vessels.32
Eyelid hygiene and erythromycin or bacitracin ointment, to improve meibomian gland function, are appropriate initial treatments for the ocular manifestations of childhood rosacea.17,36 A low dose of steroid drops can manage significant irritation when needed. Systemic therapy with tetracycline or other oral pharmaceuticals used to treat the face also may work for ocular symptoms.17
The treatment of steroid rosacea is a slow process often involving antiacne agents such as benzoyl peroxide and oral or topical antibiotics.23 Abrupt discontinuation of topical steroids followed by administration of antibiotics is a suitable treatment option. Prior recommendation has been to taper all topical steroids to prevent rebound flare; however, one study found clearing of symptoms by week 3 in 22% of patients, by week 4 in 86% of patients, and by week 8 in 100% of patients following abrupt cessation of topical steroids and a regimen of oral erythromycin stearate or topical clindamycin phosphate in children with erythromycin allergy or intolerance.8 Thus, a gradual withdrawal of topical nonfluorinated steroids may not be necessary. However, it is more common than not for children to experience an initial flare of their condition upon withdrawal from topical fluorinated steroids. This is followed by a slow and steady fading.23
- Buxton PK. Acne and rosacea. BMJ. 1988;296:43-45.
- Greaves MW. Flushing and flushing syndromes, rosacea and perioral dermatitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. Vol 3. Oxford, England: Blackwell Science; 1998:2099-2111.
- McDonnell JK, Tomecki KJ. Rosacea: an update. Cleve Clin J Med. 2000;67:587-590.
- Zuber TJ. Rosacea. Prim Care. 2000;27:309-318.
- Roihu T, Kariniemi A-L. Demodex mites in acne rosacea. J Cutan Pathol. 1998;25:550-552.
- Vin-Christian K, Maurer TA, Berger TG. Acne rosacea as a cutaneous manifestation of HIV infection. J Am Acad Dermatol. 1994;30:139-140.
- Bamford JT. Rosacea: current thoughts on origin. Semin Cutan Med Surg. 2001;20:199-206.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Weston WL, Morelli JG. Identical twins with perioral dermatitis. Pediatr Dermatol. 1998;15:144.
- Landow K. Unraveling the mystery of rosacea. Postgrad Med. 2002;112:51-58.
- Jansen T, Plewig G. Rosacea. In: DJ Demis, ed. Clinical Dermatology. Philadelphia, Pa: Lippincott Williams and Wilkins; 1997:1-11.
- Marks R, Harcourt-Webster JN. Histopathology of rosacea. Arch Dermatol. 1969;100:683-691.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Derm. 1972;87:425-429.
- Drolet B, Paller AS. Childhood rosacea. Pediatr Dermatol. 1992;9:22-26.
- Browning DJ, Proia AD. Ocular rosacea. Surv Ophthalmol. 1986;31:145-158.
- Jenkins MS, Brown SI, Lempert SL, et al. Ocular rosacea. Am J Ophthalmol. 1979;88:618-622.
- Erzurum SA, Feder RS, Greenwald MJ. Acne rosacea with keratitis in childhood. Arch Ophthalmol. 1993;111:228-230.
- Litt JZ. Steroid-induced rosacea. Am Fam Physician. 1993;48:67-71.
- Leyden JJ, Thew M, Kligman AM. Steroid rosacea. Arch Dermatol. 1974;110:619-622.
- Hogan DJ, Epstein JD, Lane PR. Perioral dermatitis: an uncommon condition? Can Med Assoc J. 1986;134:1025-1028.
- Hogan DJ, Rooney ME. Facial telangiectasia associated with long-term application of a topical corticosteroid to the scalp. J Am Acad Dermatol. 1989;20:1129-1130.
- Coskey RJ. Perioral dermatitis. Cutis. 1984;34:55-56, 58.
- Franco HL, Weson WL. Steroid rosacea in children. Pediatrics. 1979;64:36-38.
- Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
- Drummond PD, Lance JW. Facial flushing mediated by the sympathetic nervous system. Brain. 1987;110:793-803.
- Manders SM, Lucky AW. Perioral dermatitis in childhood.J Am Acad Dermatol. 1992;27:688-692.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Howard R, Tsuchiya A. Adult skin disease in the pediatric patient. Dermatol Clin. 1998;16:593-608.
- Gruber GG, Callen JP. Systemic complications of commonly used dermatologic drugs. Cutis. 1978;21:825-829.
- Caputo R, Barbareschi M, Veraldi S. Azithromycin: a new drug for systemic treatment of inflammatory acneic lesions. G Ital Dermatol Venereol.2003;138:327-331.
- Bikowski JB. Subantimicrobial dose for acne and rosacea. SKINmed. 2003;2:234-245.
- Szepietowski J. Azelaic acid gel: use in the treatment of rosacea. Dermatol Klin (Wroclaw). 2003;3:150.
- Anonymous. Topical metronidazole for rosacea. Med Lett Drugs Ther. 1989;31:75-76.
- Micali G, Licastro R, Lembo D. Treatment of rosacea with metronidazole gel 0.75%. G Ital Dermatol Venereol. 1992;127:247-250.
- McCully JP, Dougherty JM, Deneau DG. Classification of chronic blepharitis.
Ophthalmology. 1982;89:1173-1180.
- Buxton PK. Acne and rosacea. BMJ. 1988;296:43-45.
- Greaves MW. Flushing and flushing syndromes, rosacea and perioral dermatitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Textbook of Dermatology. Vol 3. Oxford, England: Blackwell Science; 1998:2099-2111.
- McDonnell JK, Tomecki KJ. Rosacea: an update. Cleve Clin J Med. 2000;67:587-590.
- Zuber TJ. Rosacea. Prim Care. 2000;27:309-318.
- Roihu T, Kariniemi A-L. Demodex mites in acne rosacea. J Cutan Pathol. 1998;25:550-552.
- Vin-Christian K, Maurer TA, Berger TG. Acne rosacea as a cutaneous manifestation of HIV infection. J Am Acad Dermatol. 1994;30:139-140.
- Bamford JT. Rosacea: current thoughts on origin. Semin Cutan Med Surg. 2001;20:199-206.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Weston WL, Morelli JG. Identical twins with perioral dermatitis. Pediatr Dermatol. 1998;15:144.
- Landow K. Unraveling the mystery of rosacea. Postgrad Med. 2002;112:51-58.
- Jansen T, Plewig G. Rosacea. In: DJ Demis, ed. Clinical Dermatology. Philadelphia, Pa: Lippincott Williams and Wilkins; 1997:1-11.
- Marks R, Harcourt-Webster JN. Histopathology of rosacea. Arch Dermatol. 1969;100:683-691.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Derm. 1972;87:425-429.
- Drolet B, Paller AS. Childhood rosacea. Pediatr Dermatol. 1992;9:22-26.
- Browning DJ, Proia AD. Ocular rosacea. Surv Ophthalmol. 1986;31:145-158.
- Jenkins MS, Brown SI, Lempert SL, et al. Ocular rosacea. Am J Ophthalmol. 1979;88:618-622.
- Erzurum SA, Feder RS, Greenwald MJ. Acne rosacea with keratitis in childhood. Arch Ophthalmol. 1993;111:228-230.
- Litt JZ. Steroid-induced rosacea. Am Fam Physician. 1993;48:67-71.
- Leyden JJ, Thew M, Kligman AM. Steroid rosacea. Arch Dermatol. 1974;110:619-622.
- Hogan DJ, Epstein JD, Lane PR. Perioral dermatitis: an uncommon condition? Can Med Assoc J. 1986;134:1025-1028.
- Hogan DJ, Rooney ME. Facial telangiectasia associated with long-term application of a topical corticosteroid to the scalp. J Am Acad Dermatol. 1989;20:1129-1130.
- Coskey RJ. Perioral dermatitis. Cutis. 1984;34:55-56, 58.
- Franco HL, Weson WL. Steroid rosacea in children. Pediatrics. 1979;64:36-38.
- Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
- Drummond PD, Lance JW. Facial flushing mediated by the sympathetic nervous system. Brain. 1987;110:793-803.
- Manders SM, Lucky AW. Perioral dermatitis in childhood.J Am Acad Dermatol. 1992;27:688-692.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Howard R, Tsuchiya A. Adult skin disease in the pediatric patient. Dermatol Clin. 1998;16:593-608.
- Gruber GG, Callen JP. Systemic complications of commonly used dermatologic drugs. Cutis. 1978;21:825-829.
- Caputo R, Barbareschi M, Veraldi S. Azithromycin: a new drug for systemic treatment of inflammatory acneic lesions. G Ital Dermatol Venereol.2003;138:327-331.
- Bikowski JB. Subantimicrobial dose for acne and rosacea. SKINmed. 2003;2:234-245.
- Szepietowski J. Azelaic acid gel: use in the treatment of rosacea. Dermatol Klin (Wroclaw). 2003;3:150.
- Anonymous. Topical metronidazole for rosacea. Med Lett Drugs Ther. 1989;31:75-76.
- Micali G, Licastro R, Lembo D. Treatment of rosacea with metronidazole gel 0.75%. G Ital Dermatol Venereol. 1992;127:247-250.
- McCully JP, Dougherty JM, Deneau DG. Classification of chronic blepharitis.
Ophthalmology. 1982;89:1173-1180.