User login
Cardiovascular complications of systemic sclerosis: What to look for
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS

Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
KEY POINTS
- Pulmonary hypertension is common in systemic sclerosis and carries a poor prognosis. Patients with systemic sclerosis should be screened regularly with echocardiography, followed, when necessary, by right heart catheterization to detect it early.
- Myocardial infarction and stroke are more common in patients with systemic sclerosis, and preventive measures are the same as for the general population.
- Right ventricular dysfunction secondary to pulmonary hypertension is common in systemic sclerosis; left ventricular dysfunction is less so. Routine echocardiography should include assessment of right and left ventricular function.
- Electrocardiography should be performed periodically, and urgently when indicated, to look for potentially dangerous arrhythmias.
Bony bumps in the mouth
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
Antisynthetase syndrome: Not just an inflammatory myopathy
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
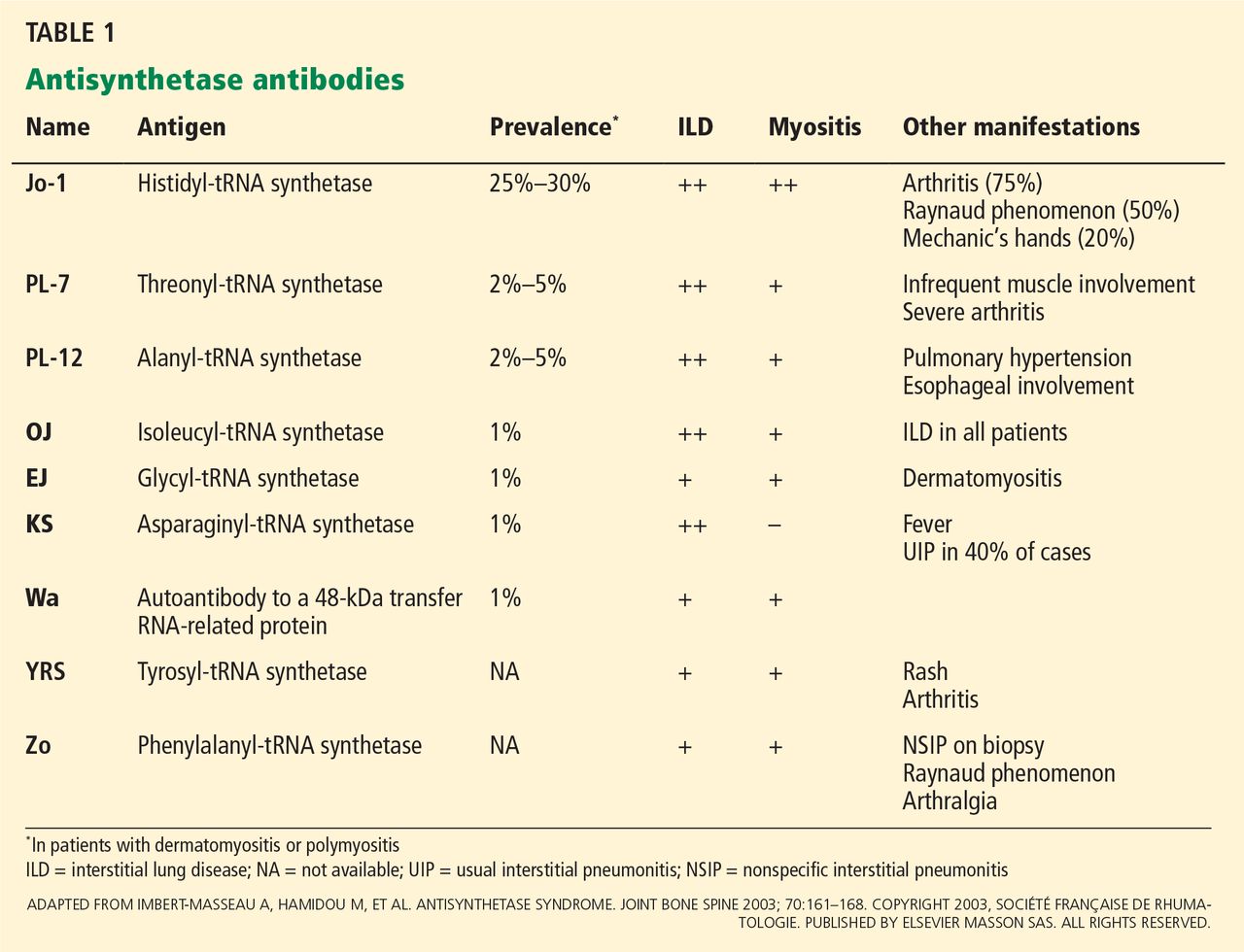
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
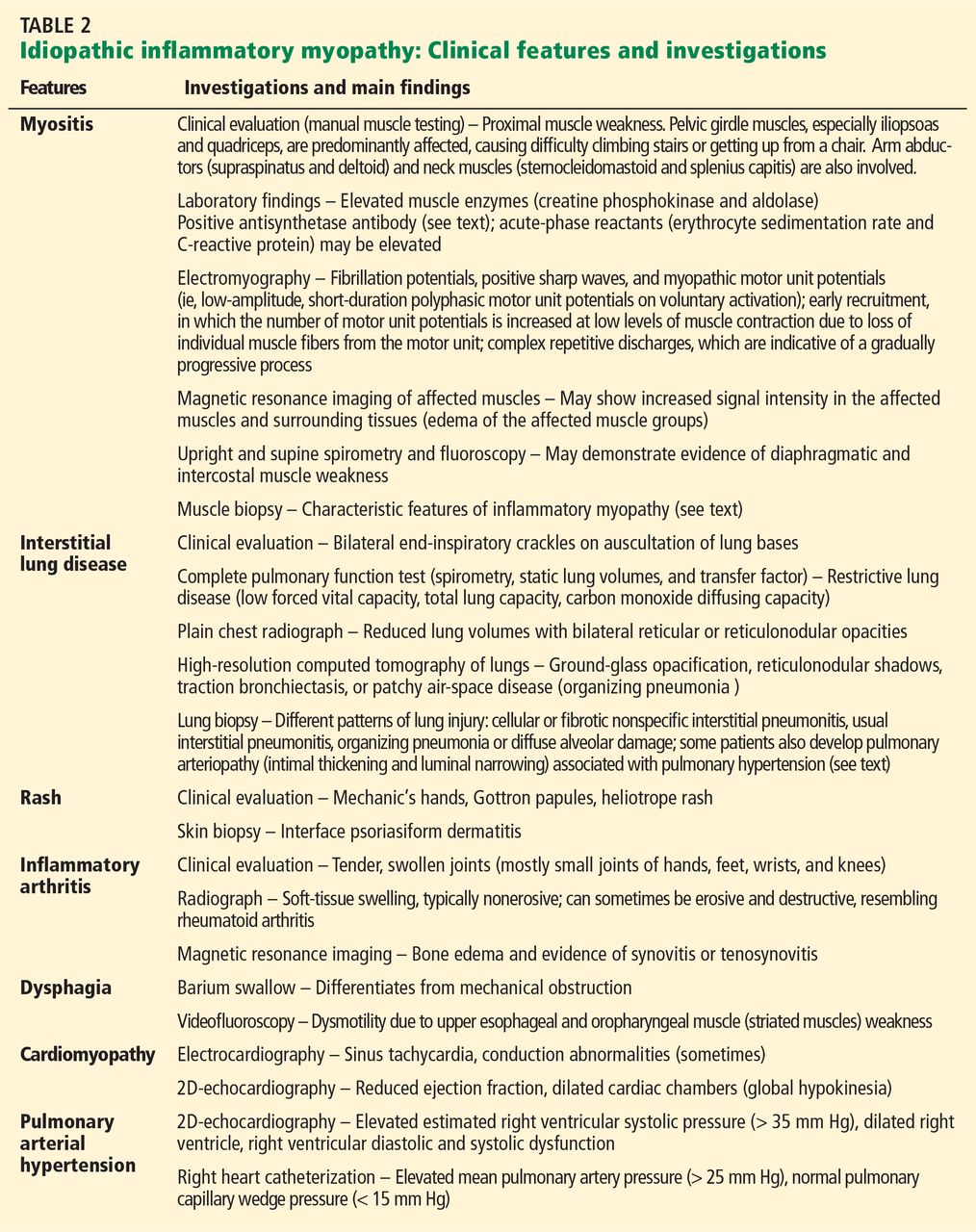
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
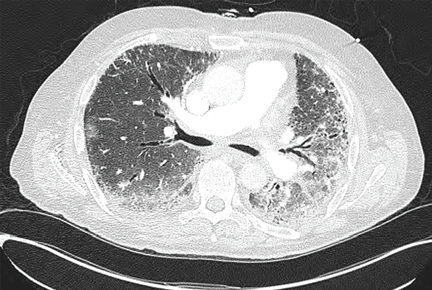
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
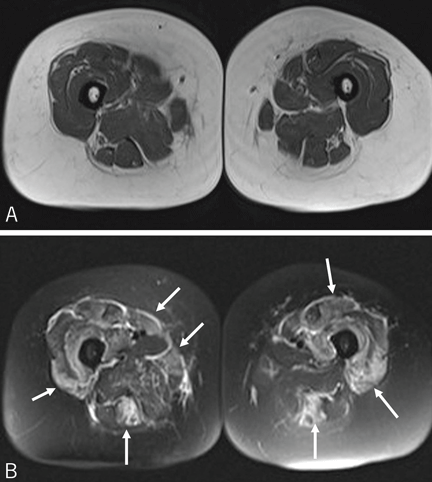
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
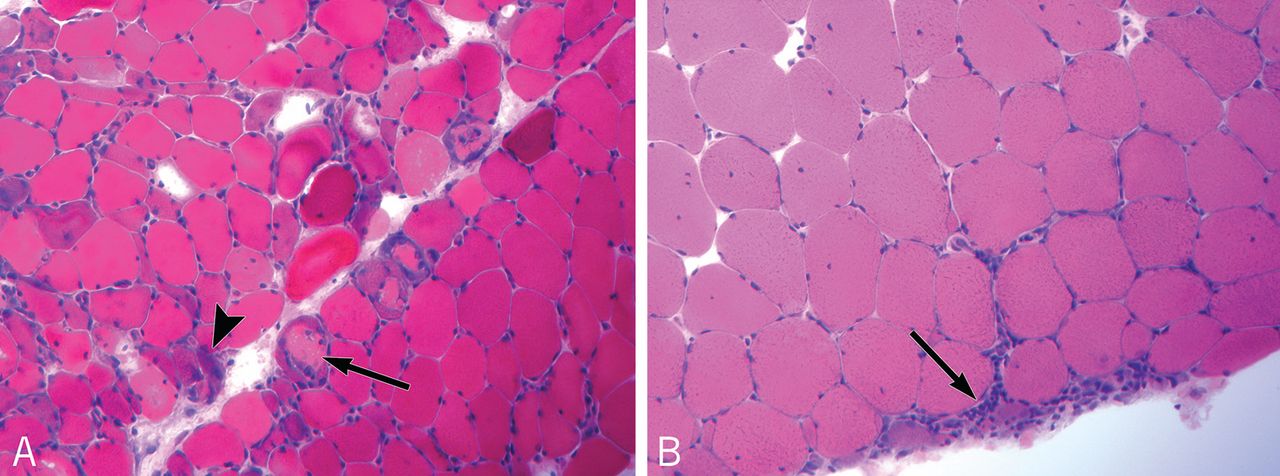
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
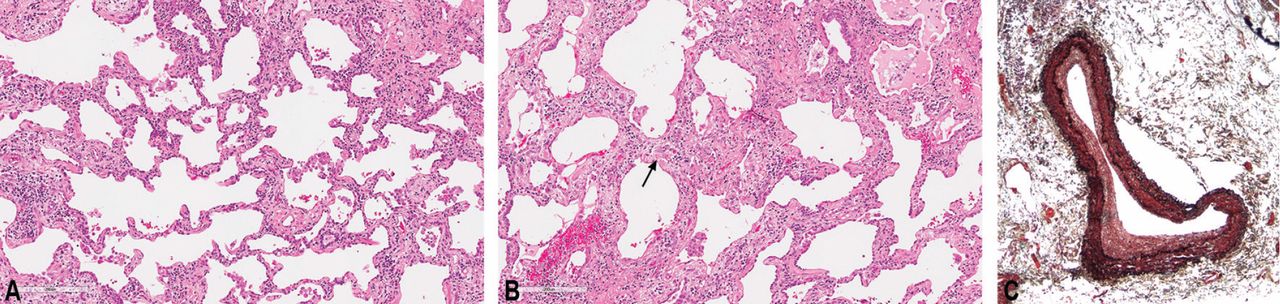
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
KEY POINTS
- Antisynthetase syndrome can present with a wide variety of clinical manifestations, including myositis and interstitial lung disease.
- The type and severity of interstitial lung disease usually determine the long-term outcome.
- In the appropriate clinical setting, the diagnosis is usually confirmed by the detection of antibodies to various aminoacyl-transfer RNA synthetases, anti-Jo-1 antibody being the most common.
- Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine or methotrexate are often required as steroid-sparing agents and also to achieve disease control.
- In the case of severe pulmonary involvement, cyclophosphamide is recommended.
When do Raynaud symptoms merit a workup for autoimmune rheumatic disease?
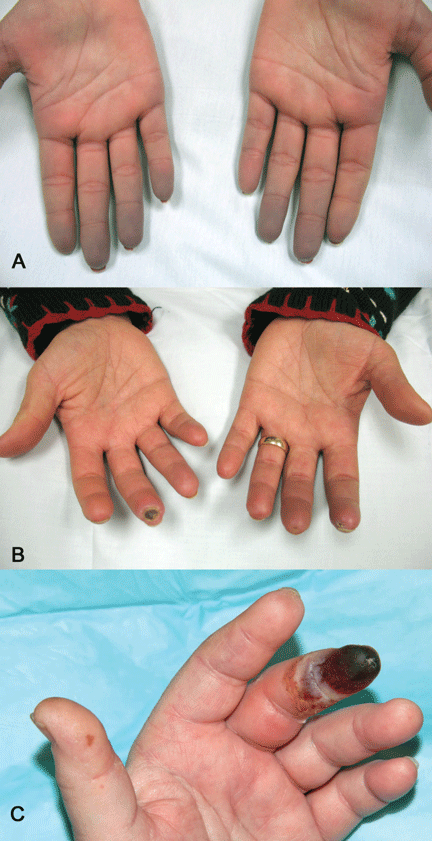
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
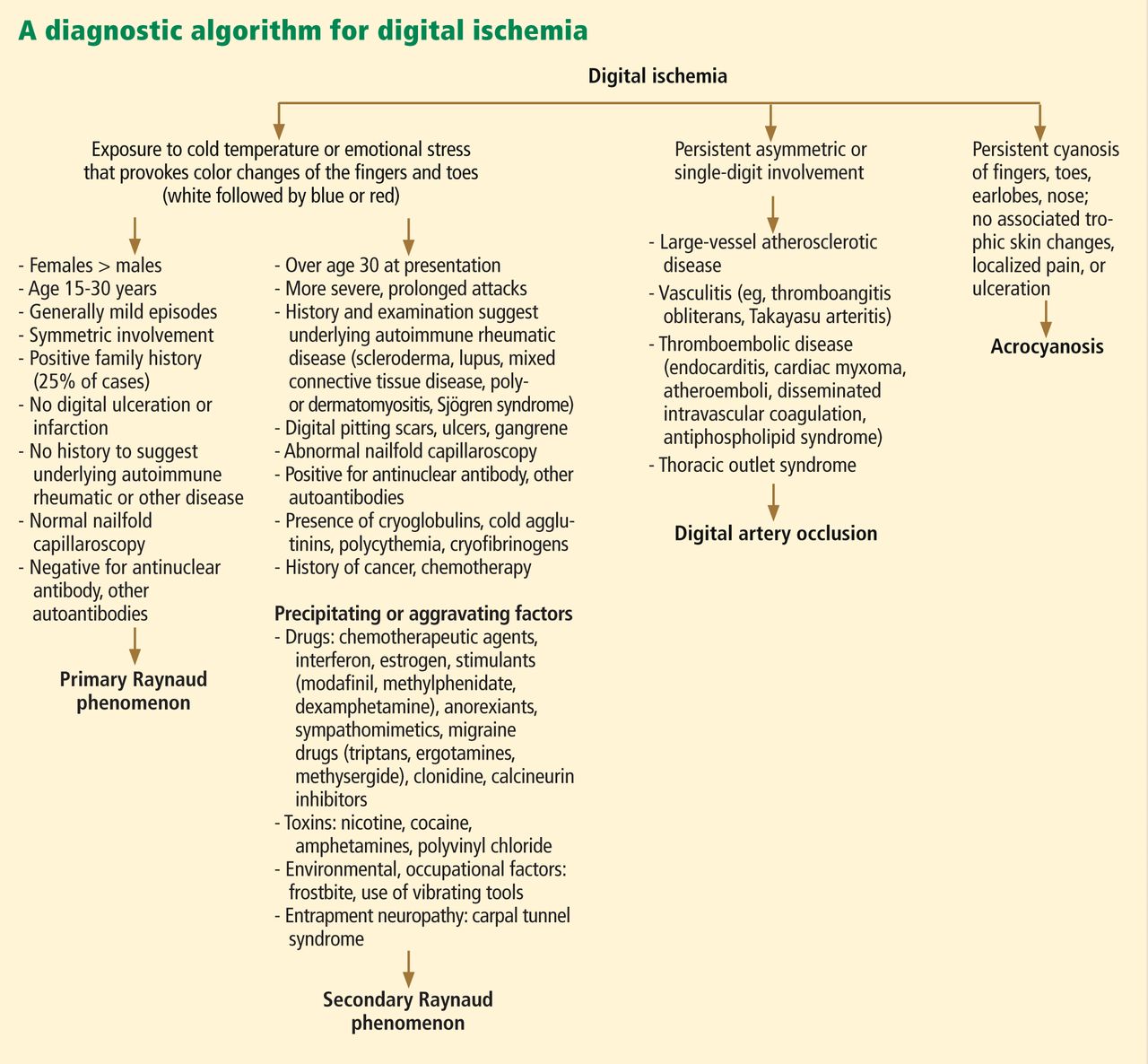
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
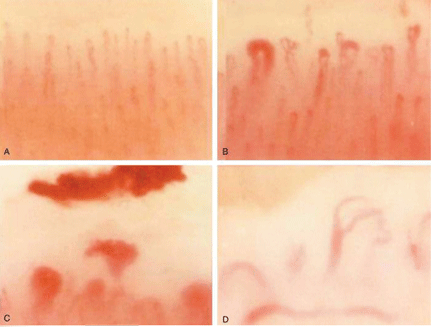
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
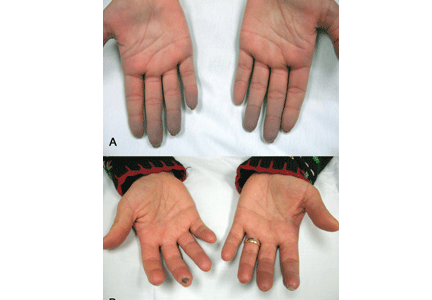
A 48-year-old woman with an ecchymotic rash
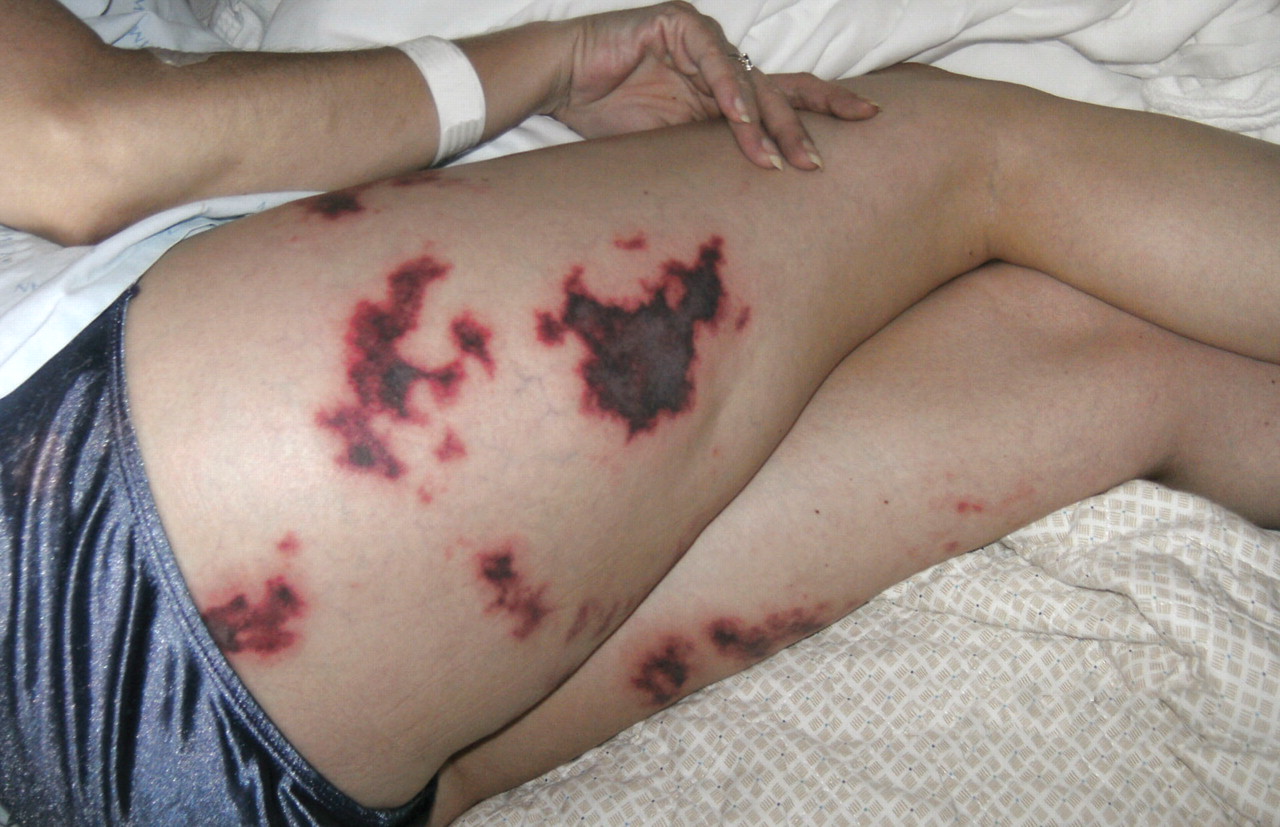
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
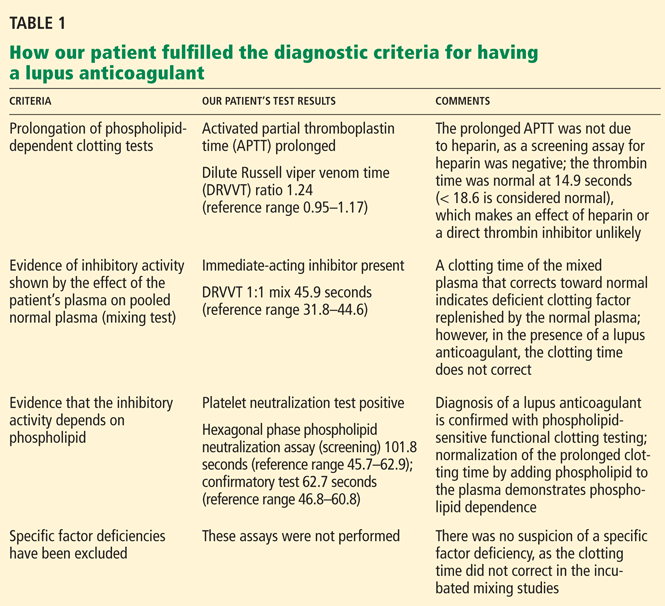
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
A 51-year-old man with nodular lesions
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.