User login
Exuberant Lymphomatoid Papulosis of the Head and Upper Trunk
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
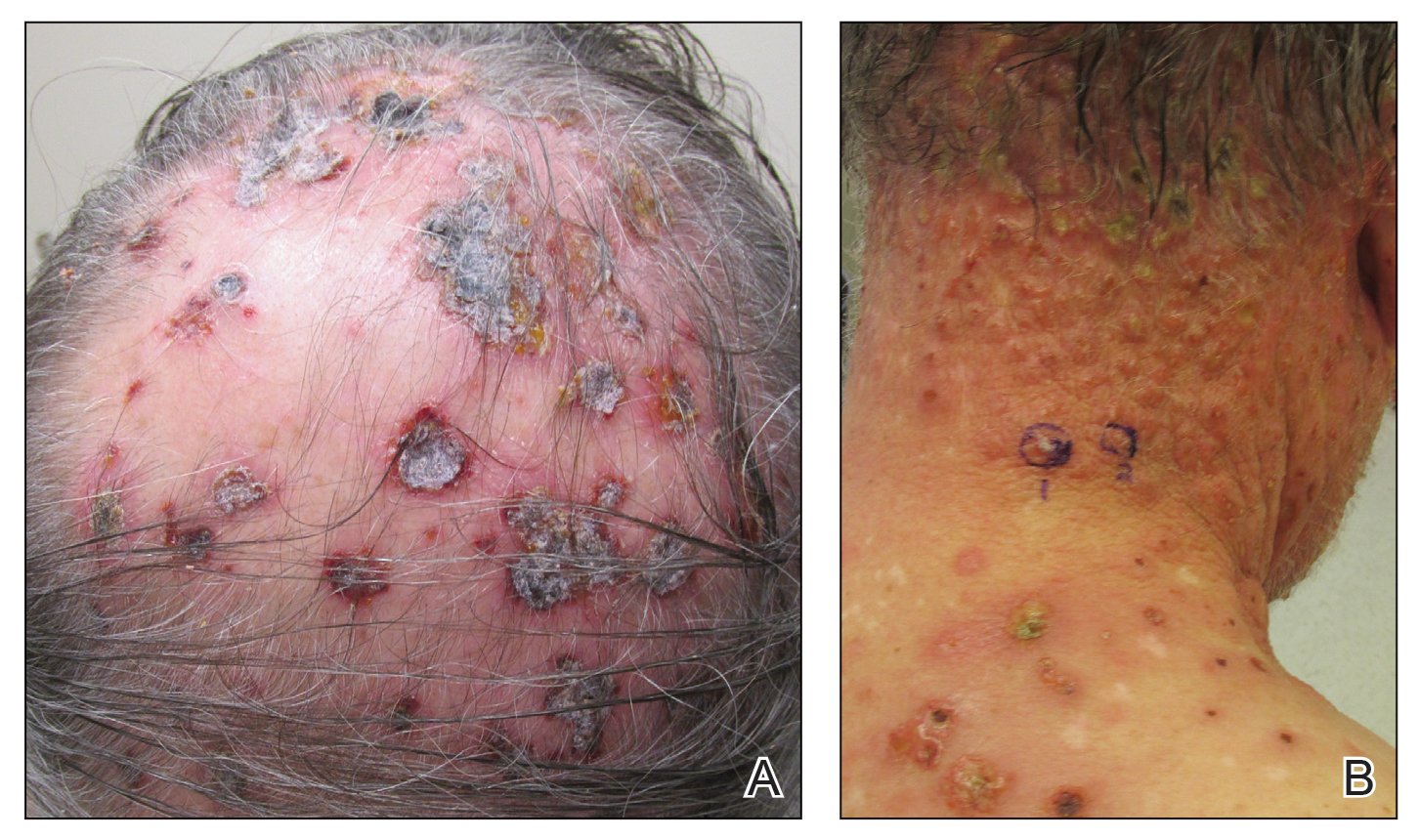
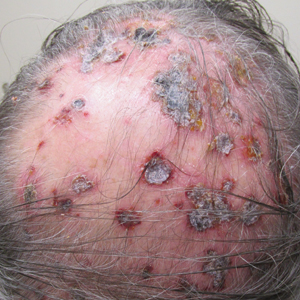
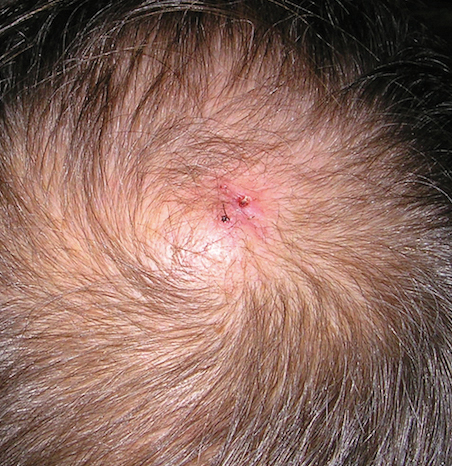
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
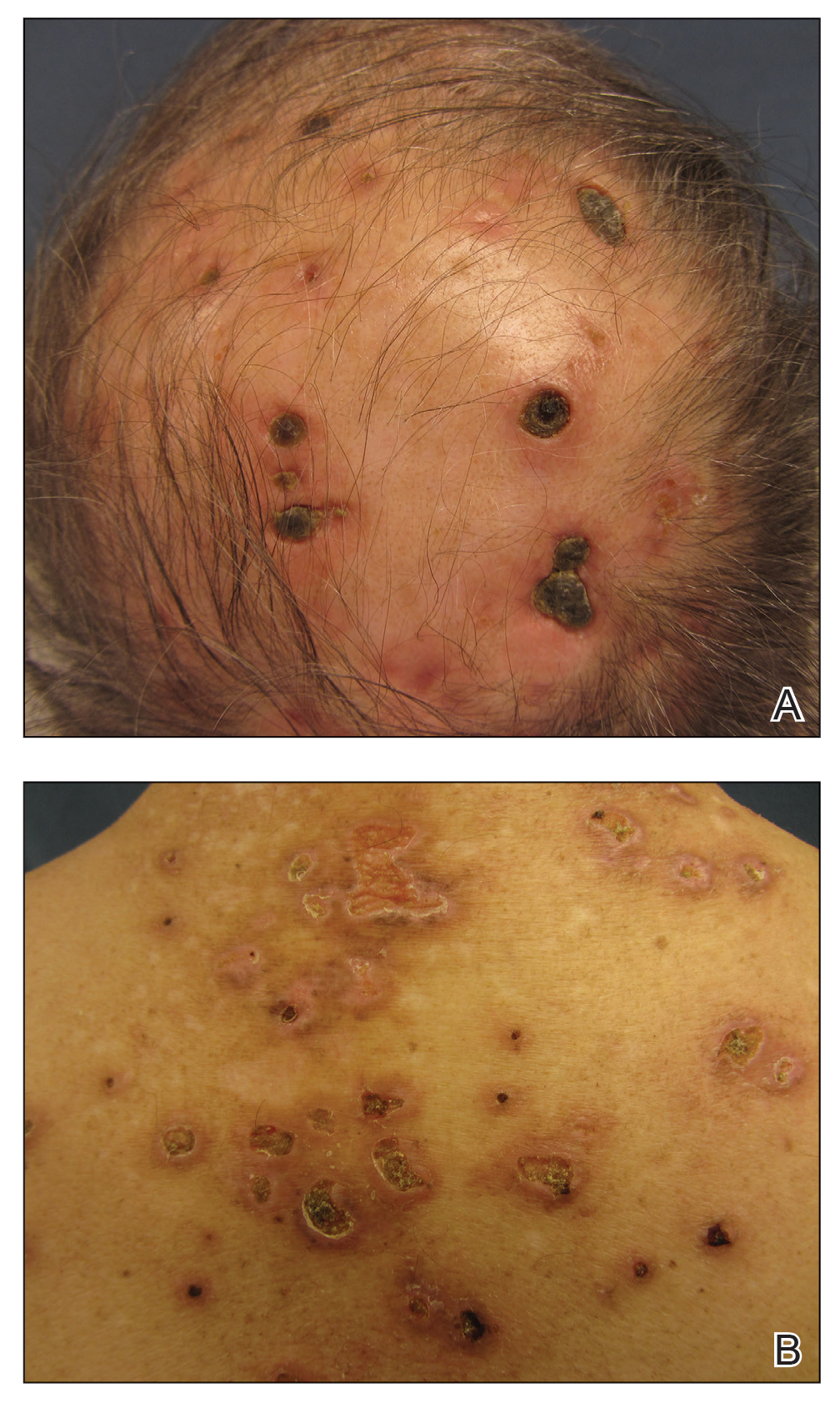
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
Practice Points
- Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder characterized by red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution.
- Histopathologically, LyP consists of a frequently CD30Mathematical Pi LT Std+ lymphocytic proliferation in multiple described patterns.
- Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy.
Scalp Arteriovenous Fistula With Intracranial Communication
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
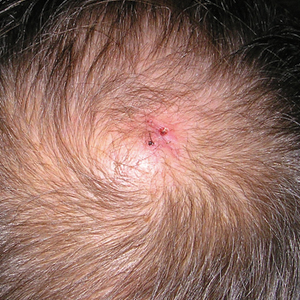
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
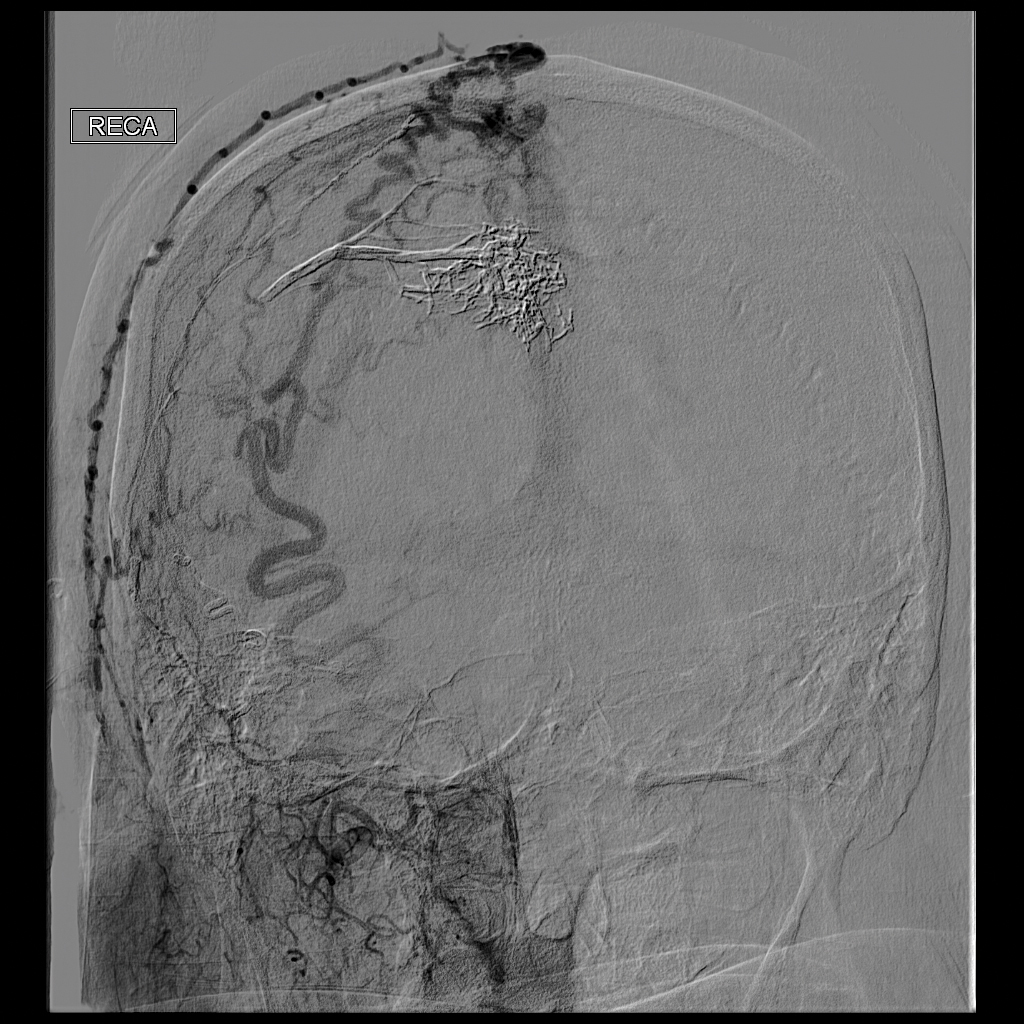
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
Practice Points
- Scalp arteriovenous fistulas may be traumatic or spontaneous and present as either an innocuous-looking scalp nodule or as a progressively enlarging pulsatile mass on the scalp.
- Clinical detection followed by appropriate imaging and referral to neurosurgery or interventional neuroradiology is vital to patient safety.
Field Cancerization With Multiple Keratoacanthomas Successfully Treated With Topical and Intralesional 5-Fluorouracil
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
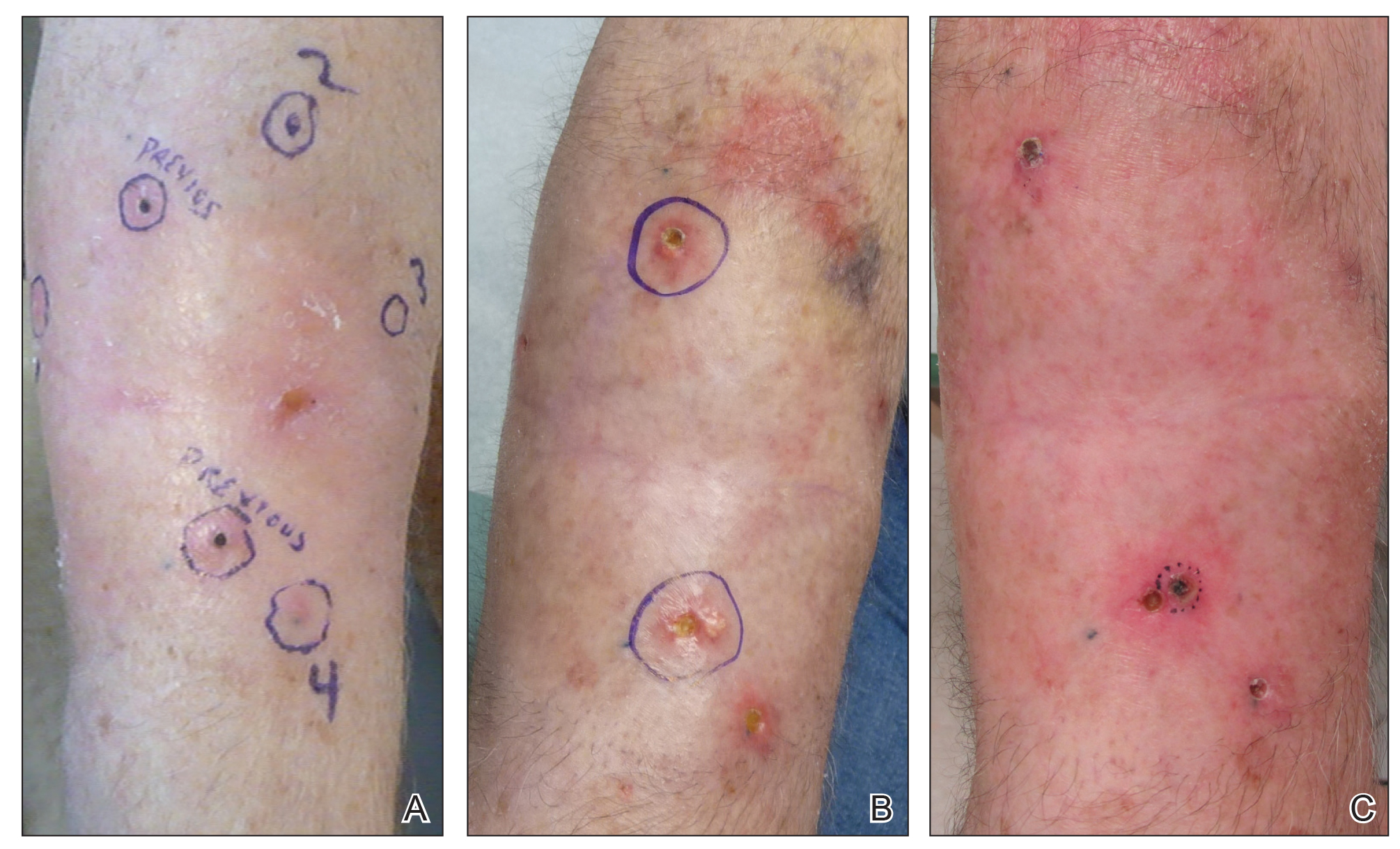
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
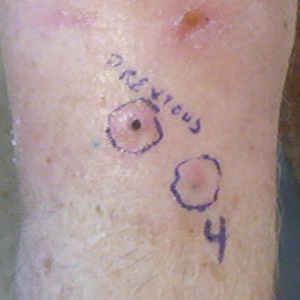
Darkening and Eruptive Nevi During Treatment With Erlotinib
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
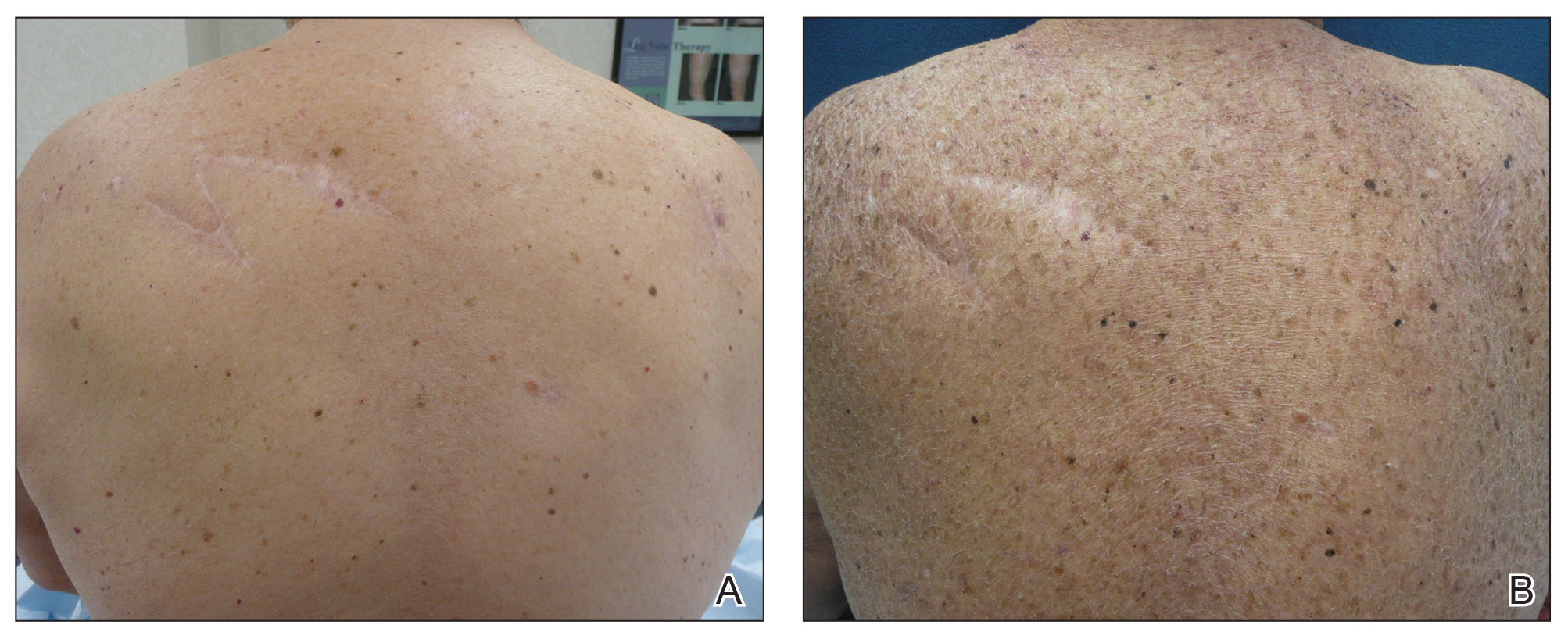
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
Practice Points
- Cutaneous side effects of erlotinib include acneform eruption, xerosis, paronychia, and pruritus.
- Clinicians should monitor patients for darkening and/or eruptive nevi as well as melanoma during treatment with erlotinib.