User login
Reticular Hyperpigmentation With Keratotic Papules in the Axillae and Groin
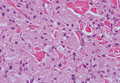
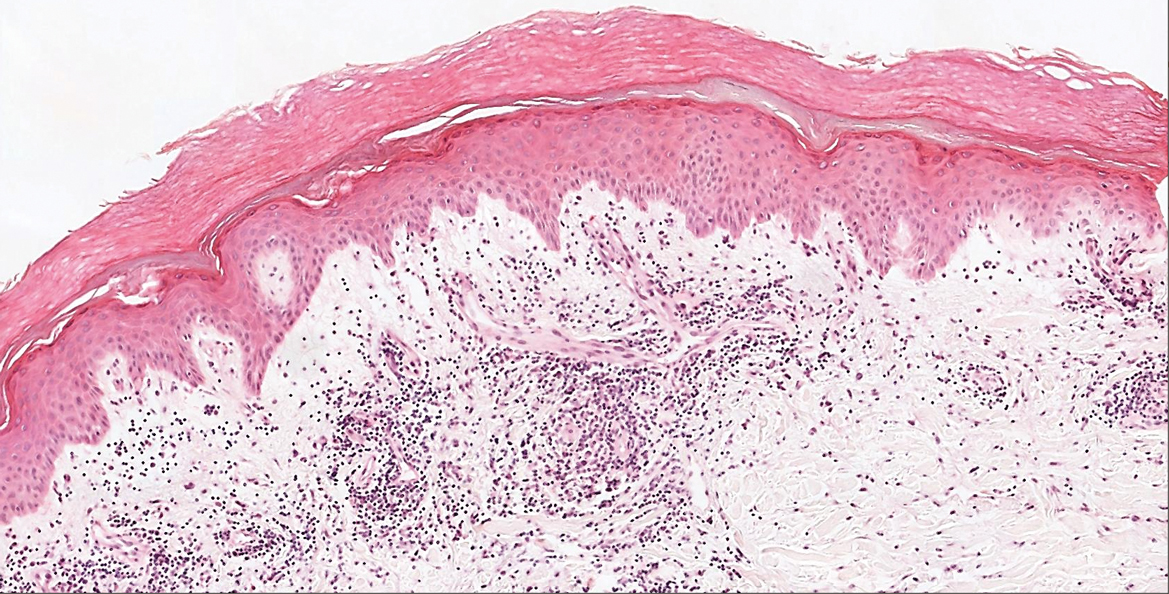
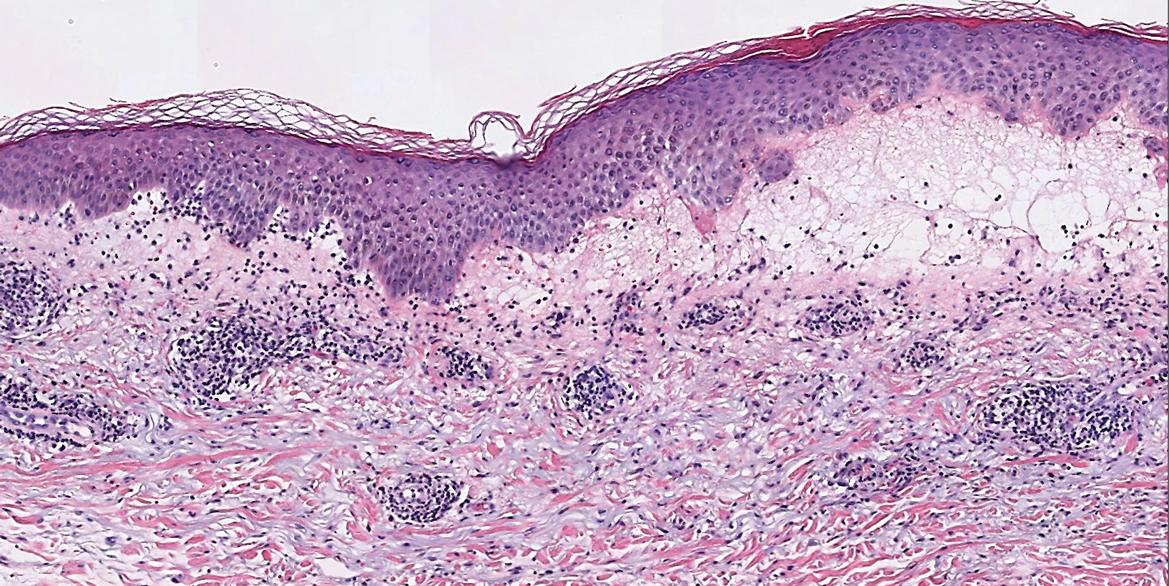
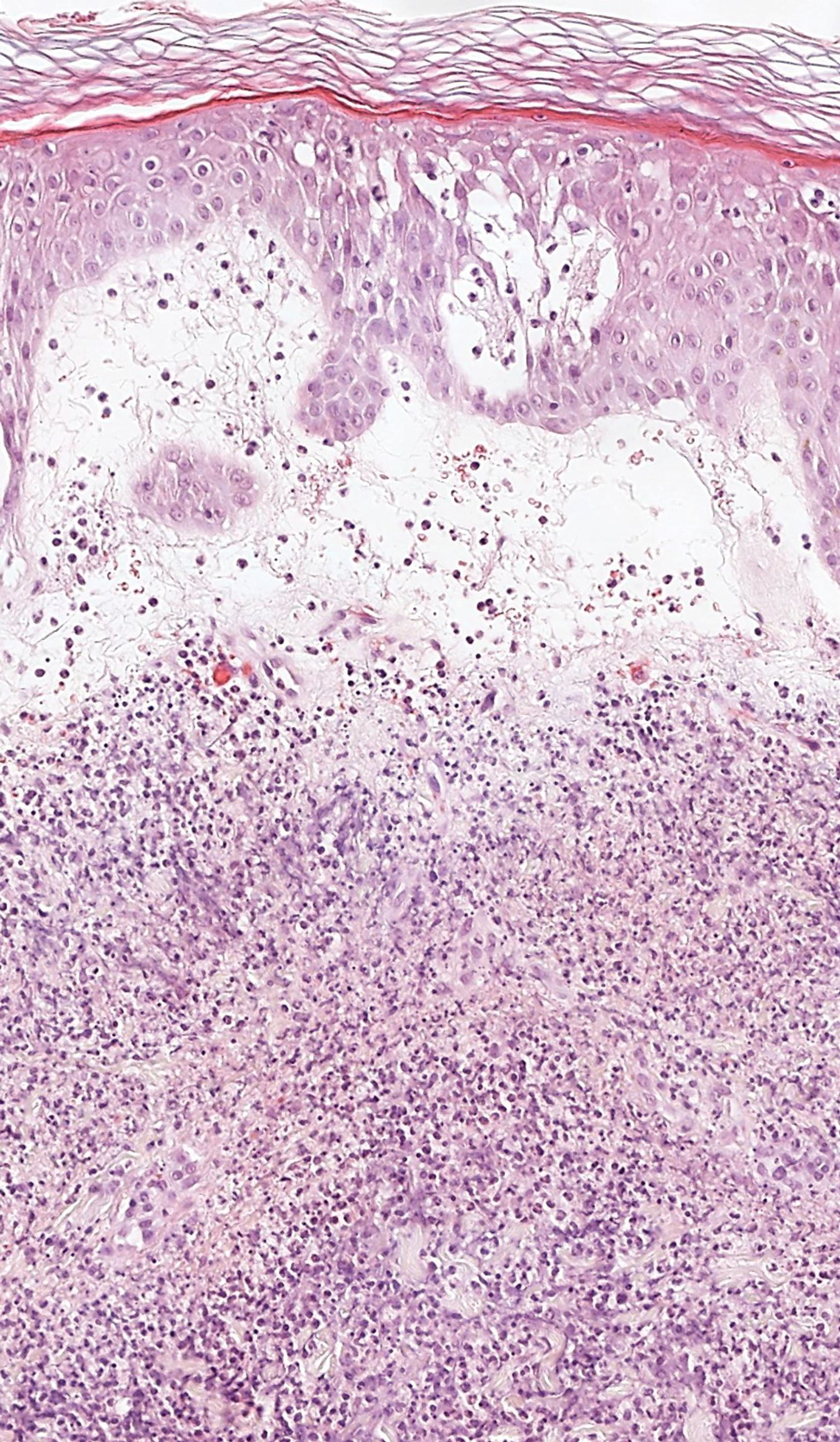
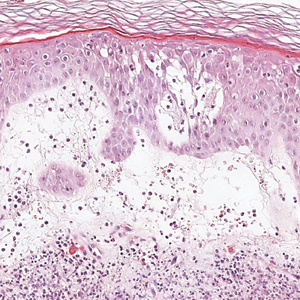
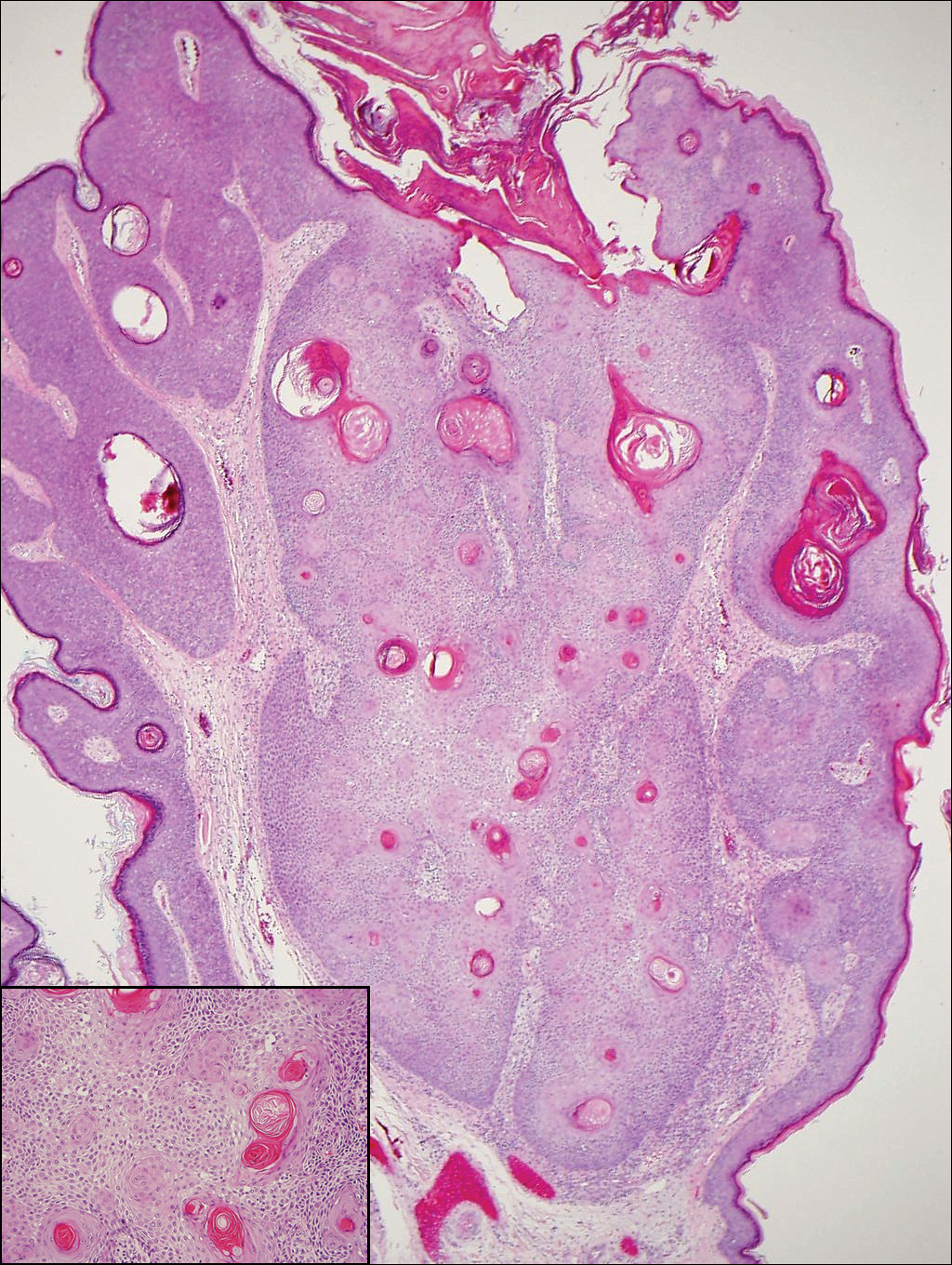
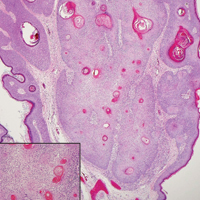
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
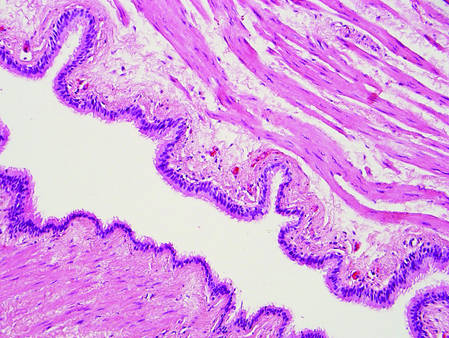
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.

Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8

Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.

Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10

- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.
Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8
Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.
Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10
The Diagnosis: Galli-Galli Disease
Several cutaneous conditions can present as reticulated hyperpigmentation or keratotic papules. Although genetic testing can help identify some of these dermatoses, biopsy typically is sufficient for diagnosis, and genetic testing can be considered for more clinically challenging cases. In our case, the clinical evidence and histopathologic findings were diagnostic of Galli-Galli disease (GGD), an autosomal-dominant genodermatosis with incomplete penetrance. Our patient was unaware of any family members with a diagnosis of GGD; however, she reported a great uncle with similar clinical findings.
Galli-Galli disease is a rare allelic variant of Dowling- Degos disease (DDD), both caused by a loss-of-function mutation in the keratin 5 gene, KRT5. Both conditions present as reticulated papules distributed symmetrically in the flexural regions, most commonly the axillae and groin, but also as comedolike papules, typically in patients aged 30 to 50 years.1 Cutaneous lesions primarily are of cosmetic concern but can be extremely pruritic, especially for patients with GGD. Gene mutations in protein O-fucosyltransferase 1, POFUT1; protein O-glucosyltransferase 1, POGLUT1; and presenilin enhancer 2, PSENEN, also have been discovered in cases of DDD and GGD.2,3
Galli-Galli disease and DDD are distinguishable by their histologic appearance. Both diseases show elongated fingerlike rete ridges and a thin suprapapillary epidermis. The basal projections often are described as bulbous or resembling antler horns.4 Galli-Galli disease can be differentiated from DDD by focal suprabasal acantholysis with minimal dyskeratosis (quiz images).5 Due to the genetic and clinical similarities, many consider GGD an acantholytic variant of DDD rather than its own entity. Indeed, some patients have shown acantholysis in one area of biopsy but not others.6
Hailey-Hailey disease (HHD)(also known as benign familial or benign chronic pemphigus) is an autosomaldominant disorder caused by mutation of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1. Clinically, patients tend to present at a wide age range with fragile flaccid vesicles that commonly develop on the neck, axillae, and groin. Histologically, the epidermis is acanthotic with a dilapidated brick wall– like appearance from a few persistent intercellular connections amid widespread acantholysis (Figure 1).7 Unlike in autoimmune pemphigus, direct immunofluorescence is negative, and acantholysis spares the adnexal structures. Hailey-Hailey disease does not involve reticulated hyperpigmentation or the elongated bulbous rete seen in GGD. Confluent and reticulated papillomatosis is a rare, typically asymptomatic, hyperpigmented dermatosis. It presents as a conglomeration of scaly hyperpigmented macules or papillomatous papules that coalesce centrally and are reticulated toward the periphery.
Confluent and reticulated papillomatosis most commonly is seen on the trunk, initially presenting in adolescents and young adults. Confluent and reticulated papillomatosis is histologically similar to acanthosis nigricans. Histopathology will show hyperkeratosis, papillomatosis, and minimal to no inflammatory infiltrate, with no elongated rete ridges or acantholysis (Figure 2).8
Pemphigus vulgaris is a blistering disease resulting from the development of autoantibodies against desmogleins 1 and 3. Similar to GGD, there is suprabasal acantholysis, which often results in a tombstonelike appearance consisting of separation between the basal layer cells of the epidermis but with maintained attachment to the underlying basement membrane zone. Unlike HHD, the acantholysis tends to involve the follicular epithelium in pemphigus vulgaris (Figure 3). Clinically, the blisters are positive for Nikolsky sign and can be both cutaneous or mucosal, commonly arising initially in the mouth during the fourth or fifth decades of life. Ruptured blisters can result in painful and hemorrhagic erosions.9 Direct immunofluorescence exhibits a classic chicken wire–like deposition of IgG and C3 between keratinocytes of the epidermis. Although sometimes difficult to appreciate, the deposition can be more prominent in the lower epidermis, in contrast to pemphigus foliaceus, which can have more prominent deposition in the upper epidermis.
Darier disease (or dyskeratosis follicularis) is an autosomal-dominant genodermatosis caused by mutation of the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2. Clinically, this disorder arises in adolescents as red-brown, greasy, crusted papules in seborrheic areas that may coalesce into papillomatous clusters. Palmar punctate keratoses and pits also are common. Histologically, Darier disease can appear similar to GGD, as both can show acantholysis and dyskeratosis. Darier disease will tend to show more prominent dyskeratosis with corps ronds and grains, as well as thicker villilike projections of keratinocytes into the papillary dermis, in contrast to the thinner, fingerlike or bulbous projections that hang down from the epidermis in GGD (Figure 4).10
- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
- Hanneken S, Rütten A, Eigelshoven S, et al. Morbus Galli-Galli. Hautarzt. 2013;64:282.
- Wilson NJ, Cole C, Kroboth K, et al. Mutations in POGLUT1 in Galli- Galli/Dowling-Degos disease. Br J Dermatol. 2017;176:270-274.
- Ralser DJ, Basmanav FB, Tafazzoli A, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-1490.
- Desai CA, Virmani N, Sakhiya J, et al. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol. 2016; 82:720-723.
- Joshi TP, Shaver S, Tschen J. Exacerbation of Galli-Galli disease following dialysis treatment: a case report and review of aggravating factors. Cureus. 2021;13:E15401.
- Muller CS, Pfohler C, Tilgen W. Changing a concept—controversy on the confusion spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol. 2008;36:44-48.
- Dai Y, Yu L, Wang Y, et al. Case report: a case of Hailey-Hailey disease mimicking condyloma acuminatum and a novel splice-site mutation of ATP2C1 gene. Front Genet. 2021;12:777630.
- Banjar TA, Abdulwahab RA, Al Hawsawi KA. Confluent and reticulated papillomatosis of Gougerot and Carteaud: a case report and review of the literature. Cureus. 2022;14:E24557.
- Porro AM, Seque CA, Ferreira MCC, et al. Pemphigus vulgaris. An Bras Dermatol. 2019;94:264-278.
- Bachar-Wikström E, Wikström JD. Darier disease—a multi-organ condition? Acta Derm Venereol. 2021;101:adv00430.
A 37-year-old woman presented with multiple hyperkeratotic small papules in the axillae and groin of 1 year’s duration. She reported pruritus and occasional sleep disruption. Subtle background reticulated hyperpigmentation was present. The patient reported that she had a great uncle with similar findings.



Tender, Diffuse, Edematous, and Erythematous Papules on the Face, Neck, Chest, and Extremities
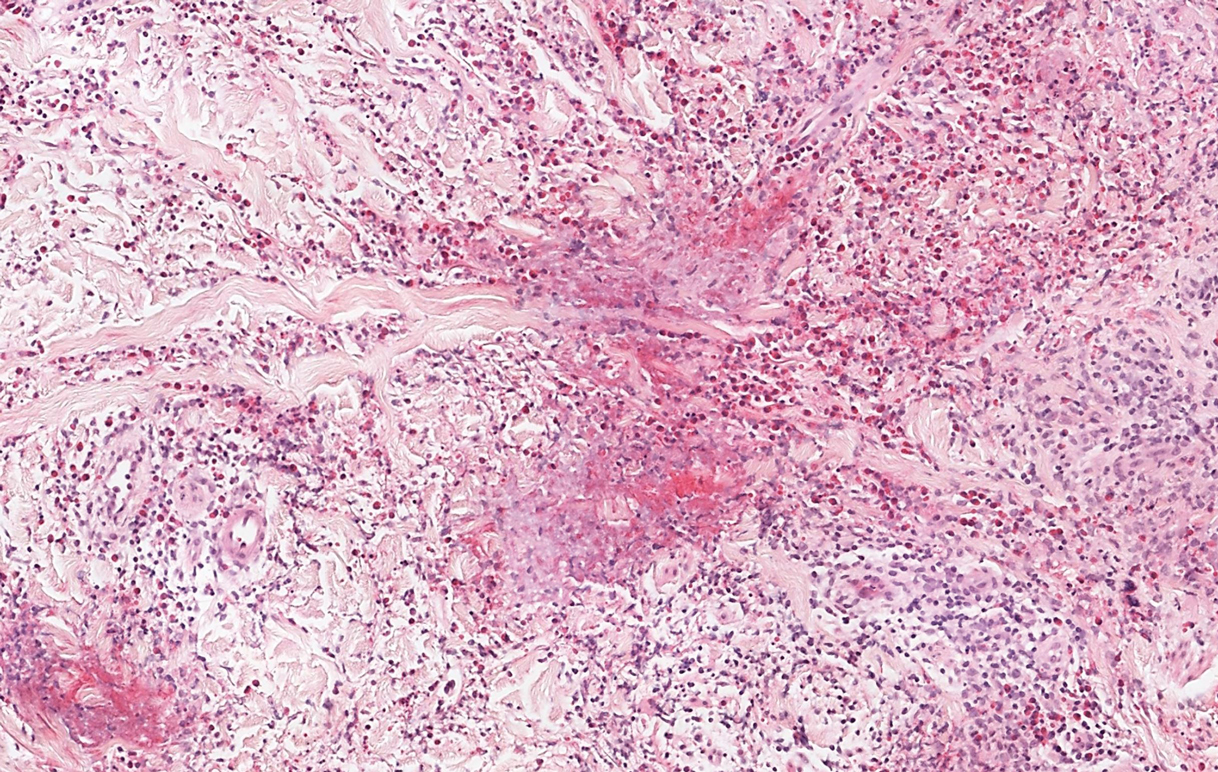
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.

Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
A 62-year-old woman presented with a tender diffuse eruption of erythematous and edematous papules and plaques on the face, neck, chest, and extremities, some appearing vesiculopustular.
Verrucoid Lesion on the Eyelid
The Diagnosis: Inverted Follicular Keratosis
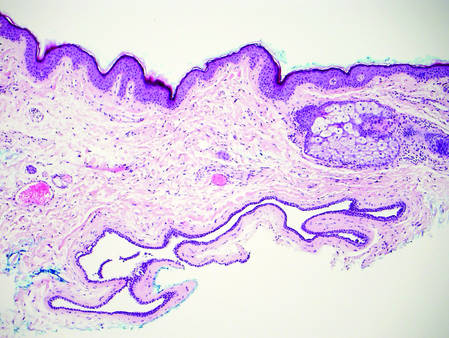
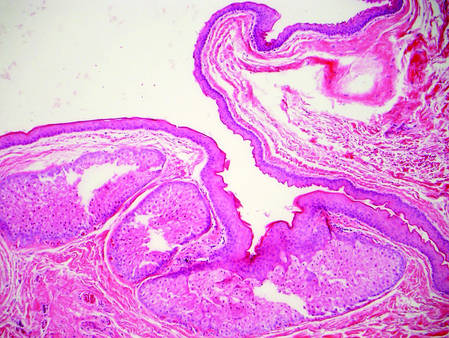
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.

Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11

Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).

Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
A 60-year-old man presented with a 3-mm verrucous papule on the right upper eyelid of 2 years' duration.
Cyst on the Eyebrow
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
| H&E, original magnification ×40. |
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
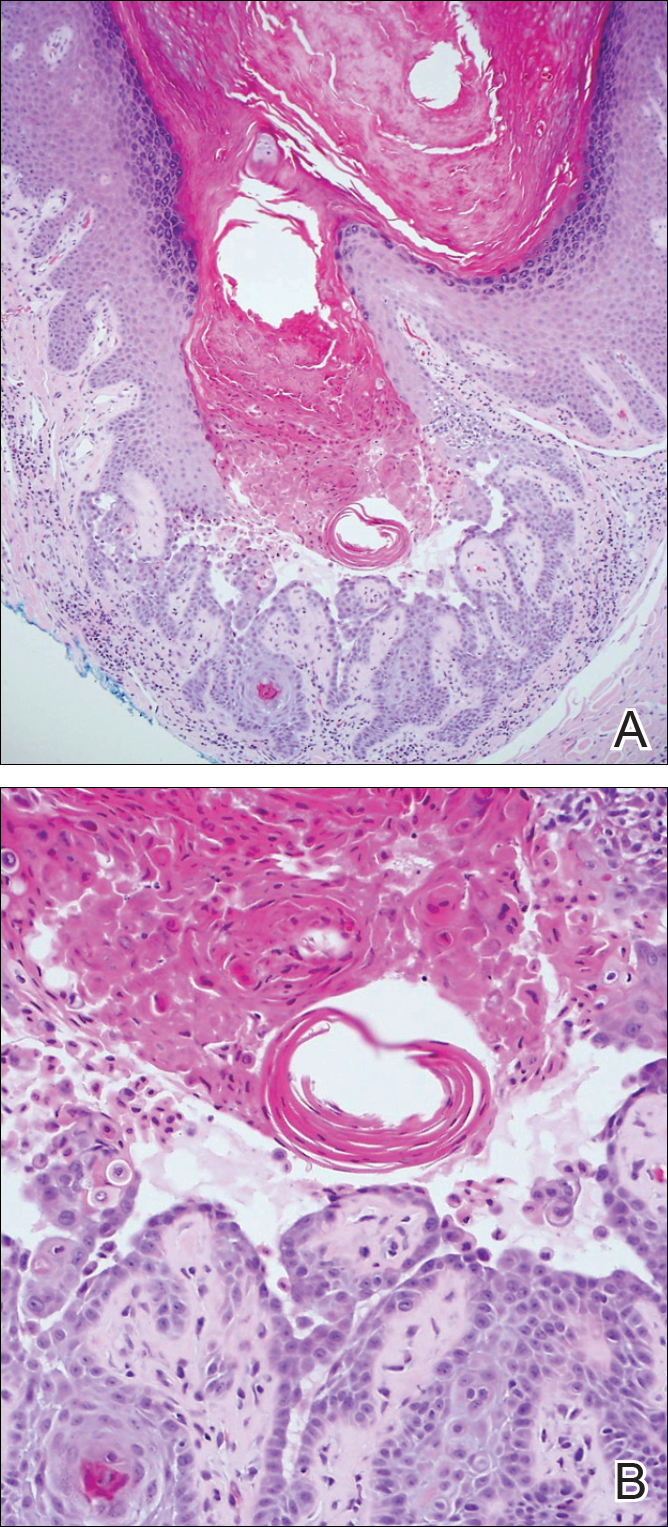
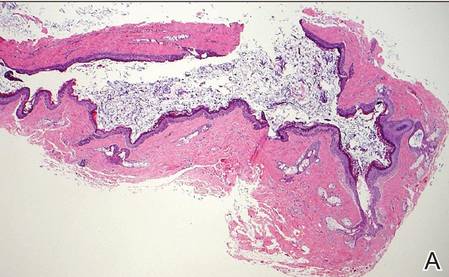
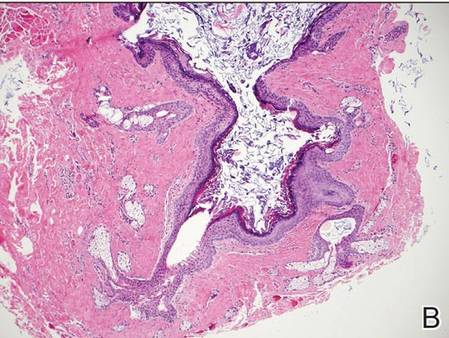
Dermoid Cyst
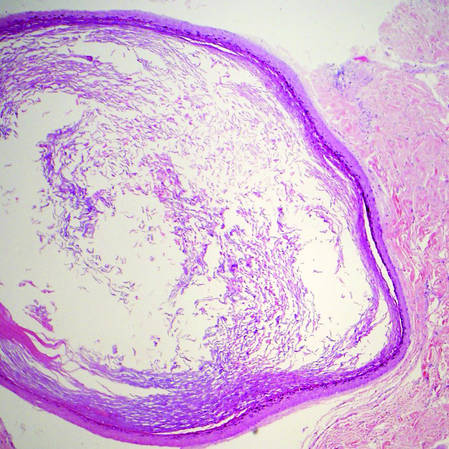
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
| 
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
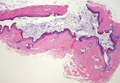
Desmoplastic Melanoma
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
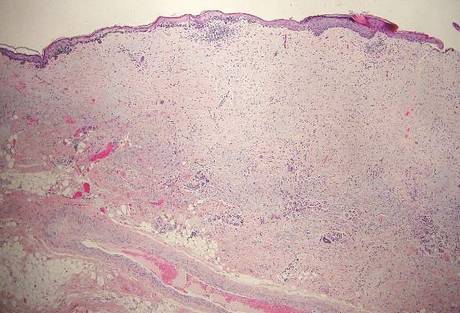
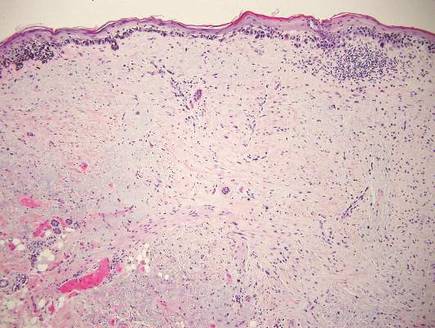
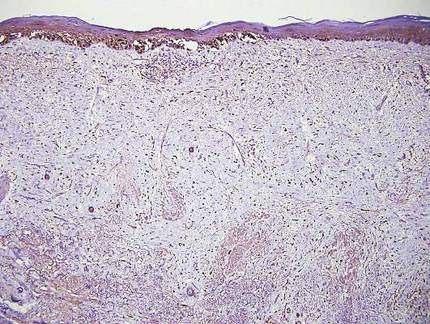
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
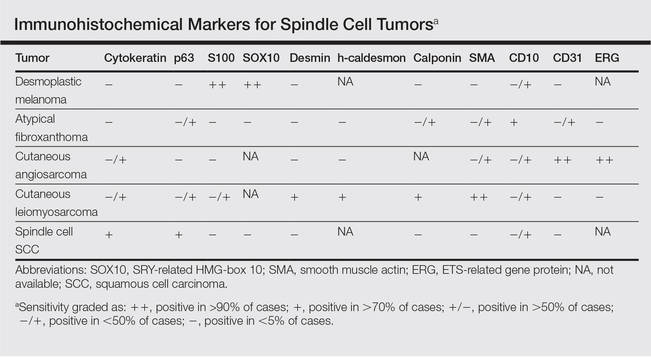
| 
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
| 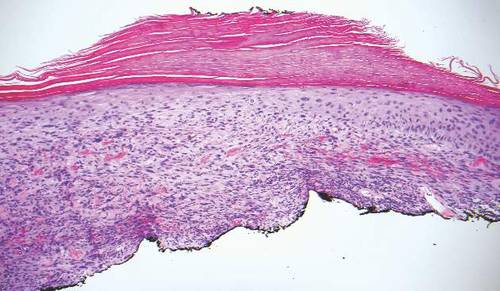
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
|
|
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
|
|
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
|
|
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
|
|
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
Chromoblastomycosis
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7

Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
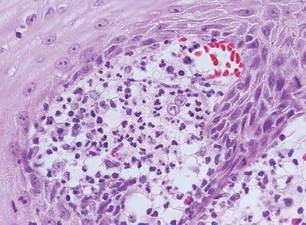
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
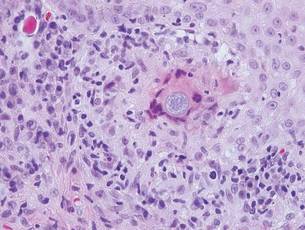
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
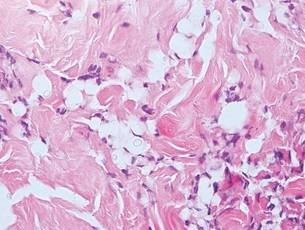
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
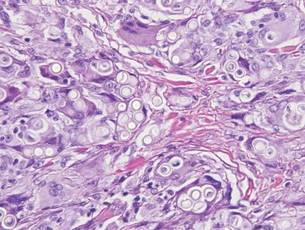
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Leading Innovation
Life is not the way it was 20 years ago, or even 10 years ago, and it will not be the same 20 years from now. Health care is changing all around us and it behooves us to be informed and proactive to keep up with the inevitable evolution. Awareness of the many factors that currently impinge or will impinge on our daily practice and patient care is a perfect opportunity for innovation.
“Mindless habitual behavior is the enemy of innovation.”
—Rosabeth Moss Kanter
Take a minute to think about a roadblock in your practice or something that occurs every day and ask yourself why it is done that way and if it could be done better. Keep it in mind for later or jot it down on scrap paper. Maybe prior authorizations are monopolizing your nurses’ time, or you have always sutured punch biopsies.
Innovation is not a conscious part of most people’s routine daily activities and is not a focus during the course of medical training. As a matter of fact, “As medicine has become more standardized and increasingly regulated, it turns out there is much less room for innovation.”1 It often falls into the category of “when I have some time,” but time never seems to come. It may not seem urgent, but it certainly is important and requires attention. Time, however small, should be allotted for innovation.
“Creativity is thinking up new things. Innovation is doing new things.”
—Theodore Levitt
Often people think of new drug and device development or technological advances when they hear “innovation,” but it could come in the form of new job descriptions, new models of care, or better processes and approaches to what we have always done. It can be thought of as fixing what is broken or creating something new.2
Innovation is not just the generation of creative ideas but their distillation and implementation. It is an active process that starts with inspiration; identifying what is broken, or better yet, what we can do better. The challenge that follows from this inspiration is the gauntlet that is thrown down to the team. The group should be given free reign to generate ideas, practical or outlandish, that can then be combined, amalgamated, and considered before implementation.3
“If you look at history, innovation doesn’t come just from giving people incentives; it comes from creating environments where their ideas can
connect.”
—Steven Johnson
We may not be able to teach creativity or dictate innovation, but we can foster it or at least stop hindering it. Innovation typically occurs from brainstorming and interacting when ideas are assimilated and put into action. How do you foster it? Hold meetings, or parts of meetings, on opportunities instead of problems. Improve mingling of participants at all levels to stimulate the collaboration of ideas and acknowledge that everyone’s perspective and creativity is valuable.
It is essential to empower the team and encourage an open, receptive, and questioning culture. Encourage the team to challenge assumptions and inferred rules that really are only habit. Once the ideas start flowing, do not stop with the first “good” idea. Allow the brainstorming to continue and refine it into the “best” idea. Leaders should work to remove as many roadblocks of implementation as possible and strive to tolerate the ambiguity that will remain. Insistence on hard data can result in analysis paralysis and lack of follow-through. Consider any failure to be a discovery of what does not work without looking for blame. Recognize and reward successful and attempted innovation to create a supportive atmosphere.
Creating such a culture is often more about conscious avoidance of actions that stifle innovation. Leaders may naturally avoid conflict by surrounding themselves with yes men, but without the lateral thinkers the team will be stuck in groupthink. If necessary, assign someone to play devil’s advocate. As adults, we tend to compare ideas with our internal database for what is wrong rather than asking what is right about the idea and playing with the possibilities. Avoid playing whack-a-mole with ideas and using phrases such as “it will never work,” “we tried that before,” “but we have always done it this way,” or “if it ain’t broke, don’t fix it.”
When change is imminent we can argue, complain, and wait for others to find ways to adjust, or we can make innovation a deliberate focus by establishing a culture that fosters it and educates the team about the innovative process. Go back to the roadblocks and/or habits in your practice that you considered earlier, present the challenge to your group, and get innovating!
- Shaywitz DA, Ausiello DA. Preserving creativity in medicine. PLoS Med. 2004;1:e34
- Prather C. Manager’s Guide to Fostering Innovation and Creativity in Teams. New York, NY: McGraw-Hill Companies, Inc; 2010.
- Baumgartner J. The innovation process. Jeffrey Baumgartner Web site. http://www.creativejeffrey.com /creative/innovationprocess.php?topic=creative. Accessed September 21, 2015.
Life is not the way it was 20 years ago, or even 10 years ago, and it will not be the same 20 years from now. Health care is changing all around us and it behooves us to be informed and proactive to keep up with the inevitable evolution. Awareness of the many factors that currently impinge or will impinge on our daily practice and patient care is a perfect opportunity for innovation.
“Mindless habitual behavior is the enemy of innovation.”
—Rosabeth Moss Kanter
Take a minute to think about a roadblock in your practice or something that occurs every day and ask yourself why it is done that way and if it could be done better. Keep it in mind for later or jot it down on scrap paper. Maybe prior authorizations are monopolizing your nurses’ time, or you have always sutured punch biopsies.
Innovation is not a conscious part of most people’s routine daily activities and is not a focus during the course of medical training. As a matter of fact, “As medicine has become more standardized and increasingly regulated, it turns out there is much less room for innovation.”1 It often falls into the category of “when I have some time,” but time never seems to come. It may not seem urgent, but it certainly is important and requires attention. Time, however small, should be allotted for innovation.
“Creativity is thinking up new things. Innovation is doing new things.”
—Theodore Levitt
Often people think of new drug and device development or technological advances when they hear “innovation,” but it could come in the form of new job descriptions, new models of care, or better processes and approaches to what we have always done. It can be thought of as fixing what is broken or creating something new.2
Innovation is not just the generation of creative ideas but their distillation and implementation. It is an active process that starts with inspiration; identifying what is broken, or better yet, what we can do better. The challenge that follows from this inspiration is the gauntlet that is thrown down to the team. The group should be given free reign to generate ideas, practical or outlandish, that can then be combined, amalgamated, and considered before implementation.3
“If you look at history, innovation doesn’t come just from giving people incentives; it comes from creating environments where their ideas can
connect.”
—Steven Johnson
We may not be able to teach creativity or dictate innovation, but we can foster it or at least stop hindering it. Innovation typically occurs from brainstorming and interacting when ideas are assimilated and put into action. How do you foster it? Hold meetings, or parts of meetings, on opportunities instead of problems. Improve mingling of participants at all levels to stimulate the collaboration of ideas and acknowledge that everyone’s perspective and creativity is valuable.
It is essential to empower the team and encourage an open, receptive, and questioning culture. Encourage the team to challenge assumptions and inferred rules that really are only habit. Once the ideas start flowing, do not stop with the first “good” idea. Allow the brainstorming to continue and refine it into the “best” idea. Leaders should work to remove as many roadblocks of implementation as possible and strive to tolerate the ambiguity that will remain. Insistence on hard data can result in analysis paralysis and lack of follow-through. Consider any failure to be a discovery of what does not work without looking for blame. Recognize and reward successful and attempted innovation to create a supportive atmosphere.
Creating such a culture is often more about conscious avoidance of actions that stifle innovation. Leaders may naturally avoid conflict by surrounding themselves with yes men, but without the lateral thinkers the team will be stuck in groupthink. If necessary, assign someone to play devil’s advocate. As adults, we tend to compare ideas with our internal database for what is wrong rather than asking what is right about the idea and playing with the possibilities. Avoid playing whack-a-mole with ideas and using phrases such as “it will never work,” “we tried that before,” “but we have always done it this way,” or “if it ain’t broke, don’t fix it.”
When change is imminent we can argue, complain, and wait for others to find ways to adjust, or we can make innovation a deliberate focus by establishing a culture that fosters it and educates the team about the innovative process. Go back to the roadblocks and/or habits in your practice that you considered earlier, present the challenge to your group, and get innovating!
Life is not the way it was 20 years ago, or even 10 years ago, and it will not be the same 20 years from now. Health care is changing all around us and it behooves us to be informed and proactive to keep up with the inevitable evolution. Awareness of the many factors that currently impinge or will impinge on our daily practice and patient care is a perfect opportunity for innovation.
“Mindless habitual behavior is the enemy of innovation.”
—Rosabeth Moss Kanter
Take a minute to think about a roadblock in your practice or something that occurs every day and ask yourself why it is done that way and if it could be done better. Keep it in mind for later or jot it down on scrap paper. Maybe prior authorizations are monopolizing your nurses’ time, or you have always sutured punch biopsies.
Innovation is not a conscious part of most people’s routine daily activities and is not a focus during the course of medical training. As a matter of fact, “As medicine has become more standardized and increasingly regulated, it turns out there is much less room for innovation.”1 It often falls into the category of “when I have some time,” but time never seems to come. It may not seem urgent, but it certainly is important and requires attention. Time, however small, should be allotted for innovation.
“Creativity is thinking up new things. Innovation is doing new things.”
—Theodore Levitt
Often people think of new drug and device development or technological advances when they hear “innovation,” but it could come in the form of new job descriptions, new models of care, or better processes and approaches to what we have always done. It can be thought of as fixing what is broken or creating something new.2
Innovation is not just the generation of creative ideas but their distillation and implementation. It is an active process that starts with inspiration; identifying what is broken, or better yet, what we can do better. The challenge that follows from this inspiration is the gauntlet that is thrown down to the team. The group should be given free reign to generate ideas, practical or outlandish, that can then be combined, amalgamated, and considered before implementation.3
“If you look at history, innovation doesn’t come just from giving people incentives; it comes from creating environments where their ideas can
connect.”
—Steven Johnson
We may not be able to teach creativity or dictate innovation, but we can foster it or at least stop hindering it. Innovation typically occurs from brainstorming and interacting when ideas are assimilated and put into action. How do you foster it? Hold meetings, or parts of meetings, on opportunities instead of problems. Improve mingling of participants at all levels to stimulate the collaboration of ideas and acknowledge that everyone’s perspective and creativity is valuable.
It is essential to empower the team and encourage an open, receptive, and questioning culture. Encourage the team to challenge assumptions and inferred rules that really are only habit. Once the ideas start flowing, do not stop with the first “good” idea. Allow the brainstorming to continue and refine it into the “best” idea. Leaders should work to remove as many roadblocks of implementation as possible and strive to tolerate the ambiguity that will remain. Insistence on hard data can result in analysis paralysis and lack of follow-through. Consider any failure to be a discovery of what does not work without looking for blame. Recognize and reward successful and attempted innovation to create a supportive atmosphere.
Creating such a culture is often more about conscious avoidance of actions that stifle innovation. Leaders may naturally avoid conflict by surrounding themselves with yes men, but without the lateral thinkers the team will be stuck in groupthink. If necessary, assign someone to play devil’s advocate. As adults, we tend to compare ideas with our internal database for what is wrong rather than asking what is right about the idea and playing with the possibilities. Avoid playing whack-a-mole with ideas and using phrases such as “it will never work,” “we tried that before,” “but we have always done it this way,” or “if it ain’t broke, don’t fix it.”
When change is imminent we can argue, complain, and wait for others to find ways to adjust, or we can make innovation a deliberate focus by establishing a culture that fosters it and educates the team about the innovative process. Go back to the roadblocks and/or habits in your practice that you considered earlier, present the challenge to your group, and get innovating!
- Shaywitz DA, Ausiello DA. Preserving creativity in medicine. PLoS Med. 2004;1:e34
- Prather C. Manager’s Guide to Fostering Innovation and Creativity in Teams. New York, NY: McGraw-Hill Companies, Inc; 2010.
- Baumgartner J. The innovation process. Jeffrey Baumgartner Web site. http://www.creativejeffrey.com /creative/innovationprocess.php?topic=creative. Accessed September 21, 2015.
- Shaywitz DA, Ausiello DA. Preserving creativity in medicine. PLoS Med. 2004;1:e34
- Prather C. Manager’s Guide to Fostering Innovation and Creativity in Teams. New York, NY: McGraw-Hill Companies, Inc; 2010.
- Baumgartner J. The innovation process. Jeffrey Baumgartner Web site. http://www.creativejeffrey.com /creative/innovationprocess.php?topic=creative. Accessed September 21, 2015.

Extramammary Paget Disease
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
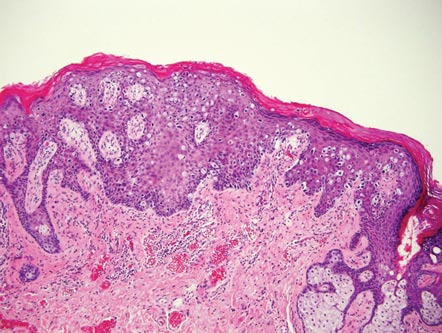
| 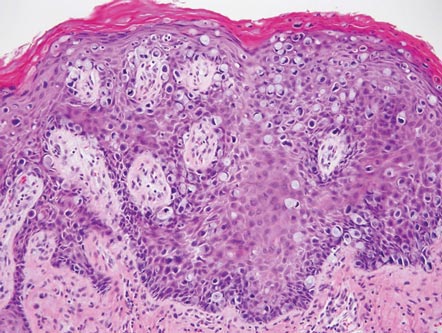
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
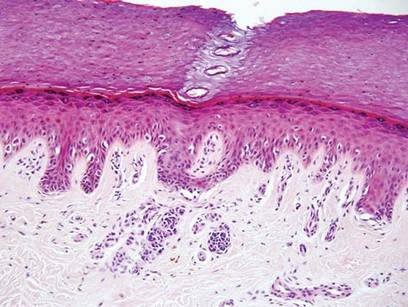
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
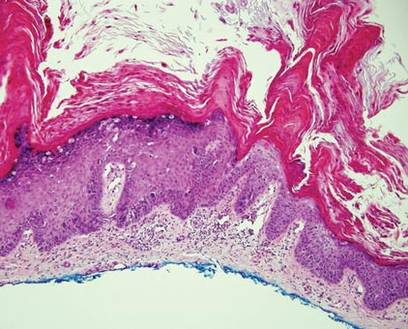
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
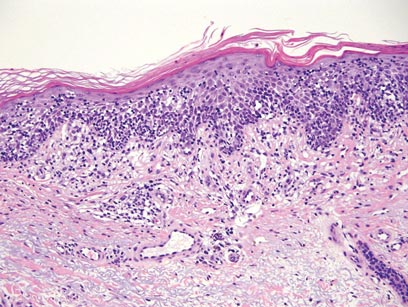
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
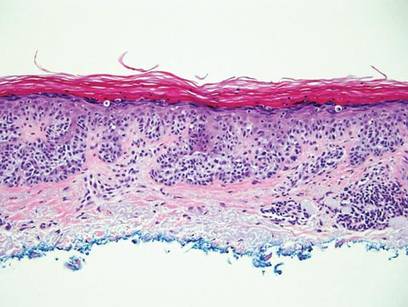
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
Granular Cell Tumor
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
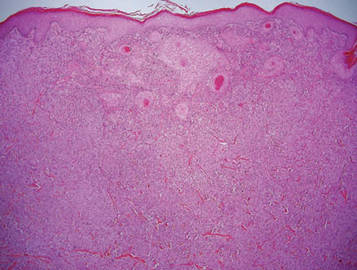
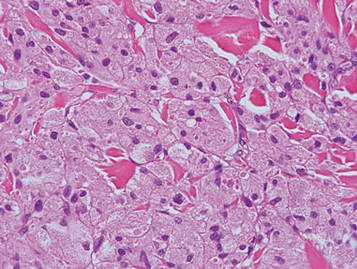
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
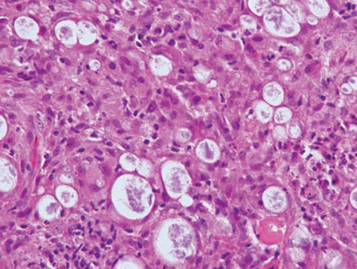

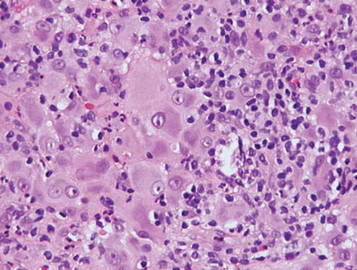

1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.