User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Using lipid guidelines to manage metabolic syndrome for patients taking an antipsychotic
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?

Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
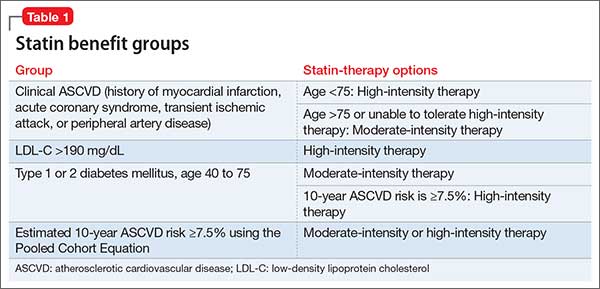
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
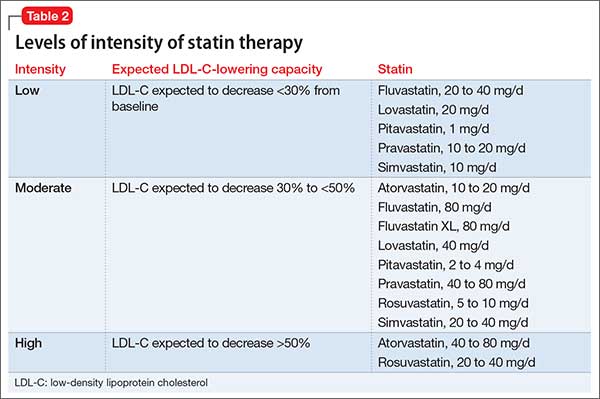
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
How to talk to patients and families about brain stimulation
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).

Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
Rediscovering clozapine: After a turbulent history, current guidance on initiating and monitoring
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
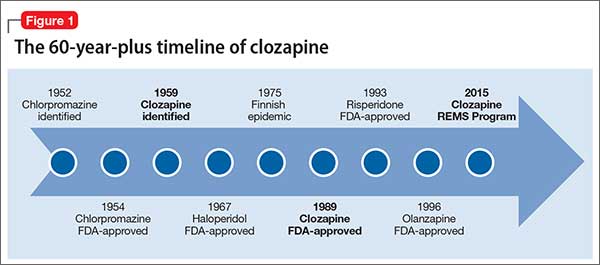
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11

New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.

REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
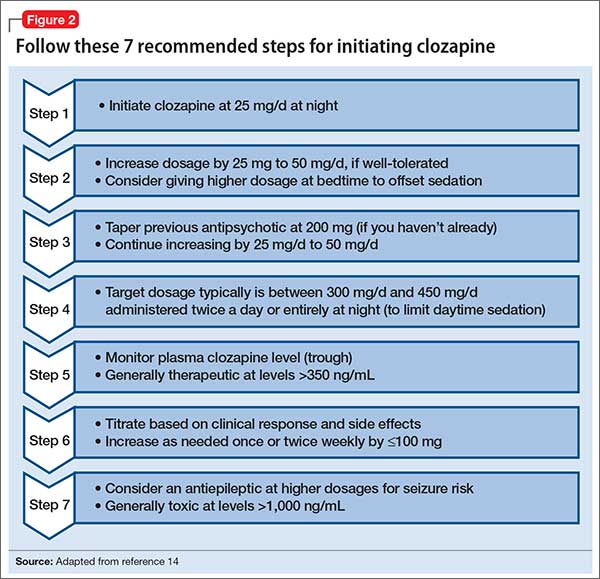
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).

Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.

Long-acting injectable aripiprazole lauroxil for schizophrenia
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
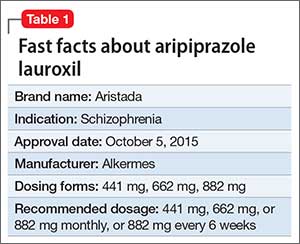
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5

Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2

After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
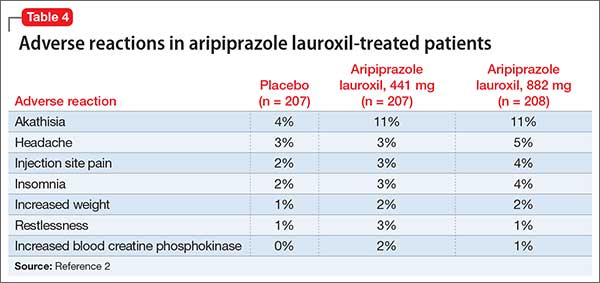
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Counseling geriatric patients about opportunity and risk when ‘digital dating’
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.

Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.
Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.
Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
Scopolamine-induced mania: ‘Theoretically possible, but statistically improbable'
Dr. Emjay Tan’s case study of a 36-year-old man who became “Manic after taking a vacation” (Cases That Test Your Skills, Current Psychiatry. April 2016, p. 45-50) is off the mark by attributing the manic episode to scopolamine—theoretically possible, but statistically improbable.
Dr. Tan may be unaware of a more frequent event: vacation hypomania. About one-third of my bipolar disorder patients had their first manic episode while on an overseas vacation or upon their return. It isn’t the fun, excitement, or novelty of a vacation that triggers the episode, but sleep deprivation, which is part and parcel of such events, particularly when they involve a holiday in a substantially different time zone.
Few people get to sleep more than a few hours the night before departing on a vacation; there’s so much to do: packing, getting to the airport hours before the flight, etc. Not many people sleep soundly on the plane, and many experience the effects of jet lag both during the first few days of vacation and when the vacationer returns home. Many vacations come with substantial and protracted sleep deprivation, and sleep deprivation is an excellent way to trigger a hypomanic episode. I suspect that is why Dr. Tan’s patient, who did not have a history of psychiatric symptoms, but who might have been genetically predisposed, became manifestly symptomatic shortly following his return from an overseas holiday.
Of course, it isn’t just first episodes of hypomania that are triggered by sleep deprivation in patients with undiagnosed bipolar disorder; the event is common in the lives of people who already receive treatment. Accordingly, my patients know that I might increase their lithium dosage for at least a few days to give them added protection as they head overseas, coupled with advice to do their best to get proper sleep.
Despite such prophylaxis, many of my bipolar disorder patients have taken a long flight overseas and, then, after half a day in the air, continued “flying.” To the best of my knowledge, none ever took scopolamine.
Martin Blinder, MD
Past Assistant Clinical Professor of Psychiatry
University of California, San Francisco
Past Adjunct Professor of Law
University of California
Hastings College of Law
San Francisco, California
Dr. Emjay Tan’s case study of a 36-year-old man who became “Manic after taking a vacation” (Cases That Test Your Skills, Current Psychiatry. April 2016, p. 45-50) is off the mark by attributing the manic episode to scopolamine—theoretically possible, but statistically improbable.
Dr. Tan may be unaware of a more frequent event: vacation hypomania. About one-third of my bipolar disorder patients had their first manic episode while on an overseas vacation or upon their return. It isn’t the fun, excitement, or novelty of a vacation that triggers the episode, but sleep deprivation, which is part and parcel of such events, particularly when they involve a holiday in a substantially different time zone.
Few people get to sleep more than a few hours the night before departing on a vacation; there’s so much to do: packing, getting to the airport hours before the flight, etc. Not many people sleep soundly on the plane, and many experience the effects of jet lag both during the first few days of vacation and when the vacationer returns home. Many vacations come with substantial and protracted sleep deprivation, and sleep deprivation is an excellent way to trigger a hypomanic episode. I suspect that is why Dr. Tan’s patient, who did not have a history of psychiatric symptoms, but who might have been genetically predisposed, became manifestly symptomatic shortly following his return from an overseas holiday.
Of course, it isn’t just first episodes of hypomania that are triggered by sleep deprivation in patients with undiagnosed bipolar disorder; the event is common in the lives of people who already receive treatment. Accordingly, my patients know that I might increase their lithium dosage for at least a few days to give them added protection as they head overseas, coupled with advice to do their best to get proper sleep.
Despite such prophylaxis, many of my bipolar disorder patients have taken a long flight overseas and, then, after half a day in the air, continued “flying.” To the best of my knowledge, none ever took scopolamine.
Martin Blinder, MD
Past Assistant Clinical Professor of Psychiatry
University of California, San Francisco
Past Adjunct Professor of Law
University of California
Hastings College of Law
San Francisco, California
Dr. Emjay Tan’s case study of a 36-year-old man who became “Manic after taking a vacation” (Cases That Test Your Skills, Current Psychiatry. April 2016, p. 45-50) is off the mark by attributing the manic episode to scopolamine—theoretically possible, but statistically improbable.
Dr. Tan may be unaware of a more frequent event: vacation hypomania. About one-third of my bipolar disorder patients had their first manic episode while on an overseas vacation or upon their return. It isn’t the fun, excitement, or novelty of a vacation that triggers the episode, but sleep deprivation, which is part and parcel of such events, particularly when they involve a holiday in a substantially different time zone.
Few people get to sleep more than a few hours the night before departing on a vacation; there’s so much to do: packing, getting to the airport hours before the flight, etc. Not many people sleep soundly on the plane, and many experience the effects of jet lag both during the first few days of vacation and when the vacationer returns home. Many vacations come with substantial and protracted sleep deprivation, and sleep deprivation is an excellent way to trigger a hypomanic episode. I suspect that is why Dr. Tan’s patient, who did not have a history of psychiatric symptoms, but who might have been genetically predisposed, became manifestly symptomatic shortly following his return from an overseas holiday.
Of course, it isn’t just first episodes of hypomania that are triggered by sleep deprivation in patients with undiagnosed bipolar disorder; the event is common in the lives of people who already receive treatment. Accordingly, my patients know that I might increase their lithium dosage for at least a few days to give them added protection as they head overseas, coupled with advice to do their best to get proper sleep.
Despite such prophylaxis, many of my bipolar disorder patients have taken a long flight overseas and, then, after half a day in the air, continued “flying.” To the best of my knowledge, none ever took scopolamine.
Martin Blinder, MD
Past Assistant Clinical Professor of Psychiatry
University of California, San Francisco
Past Adjunct Professor of Law
University of California
Hastings College of Law
San Francisco, California

5 Myths of tobacco cessation
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
No evidence of pregnancy, but she is suicidal and depressed after ‘my baby died’
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.

Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
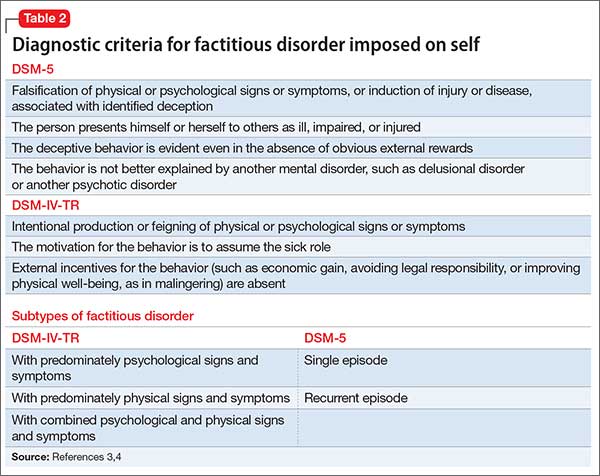
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.
Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.
Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.
Pregnant and nursing patients benefit from ‘ambitious’ changes to drug labeling for safety
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).

Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.

Lessons learned working in the clinical trial industry
As a resident in psychiatry, I am being trained in the art of diagnosis, treatment, and prevention of mental illness and emotional problems. As part of my training, research and scholarly activities are encouraged—reminding us that clinical medicine is always evolving and that it is every physician’s duty to be at the forefront of advancements in medical science.
Last year, I worked in the clinical trial industry under a seasoned principal investigator. I learned several lessons from my time with him and in the industry. Here, I present these lessons as a starting point for residents who are looking to gain experience or contemplating a career as an expert trialist or principal investigator.
Lesson 1: Know the lingo
To make the transition from physician to principal investigator go more smoothly, I recommend taking the time to learn the language of the industry. The good news? Clinical trials involve patients who have a medical history and take medications, which you are well acquainted with. In addition to medical jargon, the industry has developed its own distinctive terminology and abbreviations: adverse drug reaction (ADR), good clinical practice (GCP), contract research organization (CRO), and more.
Don’t stop there, however. I recommend that you read FDA research guidelines and guidelines of the International Conference on Harmonisation of Good Clinical Practice (ICH-GCP) to be familiar with the ethics and standard regulations of the industry.
Lesson 2: When in doubt, refer to the Protocol
Every clinical trial has a manual, so to speak, known as the Study Protocol, which outlines approved methods of performing diagnostic tests and procedures; provides information on the study timeline; and specifies patient inclusion and exclusion criteria. This document ensures conformity across all study sites, helps prevent errors, limits bias, and answers questions that might come up during the study. It’s worth noting that, in my experience, many of the questions about exclusionary medications arise when psychiatric drugs are involved.
Lesson 3: Document. Document. And document.
The golden rule in clinical practice and research is: “If it isn’t documented, it didn’t happen.” (Recall what I said about reading FDA and ICH-GCP guidelines to learn about regulations.) Documentation of all study-related activities must be meticulous. At any time, your documents might be subject to external or internal audit, conducted to preserve conformity to the protocol and maintain patient safety. Improper documentation can delay, even invalidate, your research.
Lesson 4: Remember that advertising is an art
The real work begins when your site is ready to accept patients. To fill the study, patients need to be aware that you are recruiting participants. A good starting point is to inform likely candidates from your existing patient population about any studies from which they might benefit.
Most times, however, recruiting among your patients is not enough to meet necessary enrollment numbers. You will have to advertise the study to the general public. Advertisements must target the specific patient population, informing them of the study but, at the same time, not be coercive or make false promises. The advertisements must be approved by the study’s institutional review board, which is responsible for protecting the rights and welfare of study participants.
Advertising can be tricky. If an advertisement is too vague, you will get a huge response, causing time and resources to be spent screening patients—most of whom might not be suitable for the study. If an advertisement is too specific, on the other hand, the response might be poor or none at all.
Advertising is its own industry. It might be best to hire an advertising expert who can help you decide on the selection of media (radio, television, print, digital) and can design a campaign that best suits your needs. If you decide to hire a professional, I recommend close collaboration with him (her), to help him understand the medical nature of the study.
Related Resources
• ClinicalTrials.gov. About clinical studies. https://clinicaltrials.gov/ct2/about-studies.
• U.S. Food and Drug Administration. Clinical trials and human subject protection. http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/default.htm.
• ICH GCP. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. http://ichgcp.net/.
As a resident in psychiatry, I am being trained in the art of diagnosis, treatment, and prevention of mental illness and emotional problems. As part of my training, research and scholarly activities are encouraged—reminding us that clinical medicine is always evolving and that it is every physician’s duty to be at the forefront of advancements in medical science.
Last year, I worked in the clinical trial industry under a seasoned principal investigator. I learned several lessons from my time with him and in the industry. Here, I present these lessons as a starting point for residents who are looking to gain experience or contemplating a career as an expert trialist or principal investigator.
Lesson 1: Know the lingo
To make the transition from physician to principal investigator go more smoothly, I recommend taking the time to learn the language of the industry. The good news? Clinical trials involve patients who have a medical history and take medications, which you are well acquainted with. In addition to medical jargon, the industry has developed its own distinctive terminology and abbreviations: adverse drug reaction (ADR), good clinical practice (GCP), contract research organization (CRO), and more.
Don’t stop there, however. I recommend that you read FDA research guidelines and guidelines of the International Conference on Harmonisation of Good Clinical Practice (ICH-GCP) to be familiar with the ethics and standard regulations of the industry.
Lesson 2: When in doubt, refer to the Protocol
Every clinical trial has a manual, so to speak, known as the Study Protocol, which outlines approved methods of performing diagnostic tests and procedures; provides information on the study timeline; and specifies patient inclusion and exclusion criteria. This document ensures conformity across all study sites, helps prevent errors, limits bias, and answers questions that might come up during the study. It’s worth noting that, in my experience, many of the questions about exclusionary medications arise when psychiatric drugs are involved.
Lesson 3: Document. Document. And document.
The golden rule in clinical practice and research is: “If it isn’t documented, it didn’t happen.” (Recall what I said about reading FDA and ICH-GCP guidelines to learn about regulations.) Documentation of all study-related activities must be meticulous. At any time, your documents might be subject to external or internal audit, conducted to preserve conformity to the protocol and maintain patient safety. Improper documentation can delay, even invalidate, your research.
Lesson 4: Remember that advertising is an art
The real work begins when your site is ready to accept patients. To fill the study, patients need to be aware that you are recruiting participants. A good starting point is to inform likely candidates from your existing patient population about any studies from which they might benefit.
Most times, however, recruiting among your patients is not enough to meet necessary enrollment numbers. You will have to advertise the study to the general public. Advertisements must target the specific patient population, informing them of the study but, at the same time, not be coercive or make false promises. The advertisements must be approved by the study’s institutional review board, which is responsible for protecting the rights and welfare of study participants.
Advertising can be tricky. If an advertisement is too vague, you will get a huge response, causing time and resources to be spent screening patients—most of whom might not be suitable for the study. If an advertisement is too specific, on the other hand, the response might be poor or none at all.
Advertising is its own industry. It might be best to hire an advertising expert who can help you decide on the selection of media (radio, television, print, digital) and can design a campaign that best suits your needs. If you decide to hire a professional, I recommend close collaboration with him (her), to help him understand the medical nature of the study.
Related Resources
• ClinicalTrials.gov. About clinical studies. https://clinicaltrials.gov/ct2/about-studies.
• U.S. Food and Drug Administration. Clinical trials and human subject protection. http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/default.htm.
• ICH GCP. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. http://ichgcp.net/.
As a resident in psychiatry, I am being trained in the art of diagnosis, treatment, and prevention of mental illness and emotional problems. As part of my training, research and scholarly activities are encouraged—reminding us that clinical medicine is always evolving and that it is every physician’s duty to be at the forefront of advancements in medical science.
Last year, I worked in the clinical trial industry under a seasoned principal investigator. I learned several lessons from my time with him and in the industry. Here, I present these lessons as a starting point for residents who are looking to gain experience or contemplating a career as an expert trialist or principal investigator.
Lesson 1: Know the lingo
To make the transition from physician to principal investigator go more smoothly, I recommend taking the time to learn the language of the industry. The good news? Clinical trials involve patients who have a medical history and take medications, which you are well acquainted with. In addition to medical jargon, the industry has developed its own distinctive terminology and abbreviations: adverse drug reaction (ADR), good clinical practice (GCP), contract research organization (CRO), and more.
Don’t stop there, however. I recommend that you read FDA research guidelines and guidelines of the International Conference on Harmonisation of Good Clinical Practice (ICH-GCP) to be familiar with the ethics and standard regulations of the industry.
Lesson 2: When in doubt, refer to the Protocol
Every clinical trial has a manual, so to speak, known as the Study Protocol, which outlines approved methods of performing diagnostic tests and procedures; provides information on the study timeline; and specifies patient inclusion and exclusion criteria. This document ensures conformity across all study sites, helps prevent errors, limits bias, and answers questions that might come up during the study. It’s worth noting that, in my experience, many of the questions about exclusionary medications arise when psychiatric drugs are involved.
Lesson 3: Document. Document. And document.
The golden rule in clinical practice and research is: “If it isn’t documented, it didn’t happen.” (Recall what I said about reading FDA and ICH-GCP guidelines to learn about regulations.) Documentation of all study-related activities must be meticulous. At any time, your documents might be subject to external or internal audit, conducted to preserve conformity to the protocol and maintain patient safety. Improper documentation can delay, even invalidate, your research.
Lesson 4: Remember that advertising is an art
The real work begins when your site is ready to accept patients. To fill the study, patients need to be aware that you are recruiting participants. A good starting point is to inform likely candidates from your existing patient population about any studies from which they might benefit.
Most times, however, recruiting among your patients is not enough to meet necessary enrollment numbers. You will have to advertise the study to the general public. Advertisements must target the specific patient population, informing them of the study but, at the same time, not be coercive or make false promises. The advertisements must be approved by the study’s institutional review board, which is responsible for protecting the rights and welfare of study participants.
Advertising can be tricky. If an advertisement is too vague, you will get a huge response, causing time and resources to be spent screening patients—most of whom might not be suitable for the study. If an advertisement is too specific, on the other hand, the response might be poor or none at all.
Advertising is its own industry. It might be best to hire an advertising expert who can help you decide on the selection of media (radio, television, print, digital) and can design a campaign that best suits your needs. If you decide to hire a professional, I recommend close collaboration with him (her), to help him understand the medical nature of the study.
Related Resources
• ClinicalTrials.gov. About clinical studies. https://clinicaltrials.gov/ct2/about-studies.
• U.S. Food and Drug Administration. Clinical trials and human subject protection. http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/default.htm.
• ICH GCP. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. http://ichgcp.net/.
