User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Weight gain and antidepressants

Treating opioid addiction in emerging adults
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Crossing your ‘t’s: Practice policies for the private practitioner
Developing your practice policies and sharing them with your patients is essential to building long-term, trusting relationships. Having a clear starting point helps avert disagreement down the road and allows patients to feel comfortable knowing what they are getting in to, which will provide a foundation on which you and the patient can focus on clinical matters.
What’s in a policy?
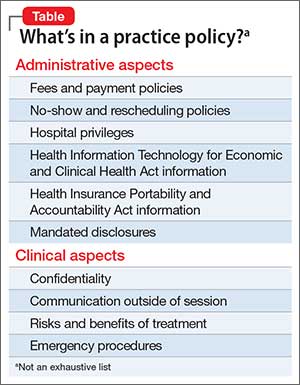
Policies should cover administrative aspects of care, such as mandated disclosures; relevant Health Insurance Portability and Accountability Act and Health Information Technology for Economic and Clinical Health Act information; hospital privilege status; and fees and payment policies. Your policies also will touch on areas where business overlaps with patient care, such as confidentiality and its limits, communication methods outside of session, and the risks and benefits of treatment (Table).
Address communication and billing policies for complex scenarios. Although these scenarios might not come up often, if you wait until you are confronted with the situation, the patient might (rightly) feel that she (he) wasn’t properly informed before giving consent. For example:
- For college students. Do you try to build college students’ autonomy by sending them all billing statements directly? If not, how will you handle the diagnosis code that appears on the statement, which their parents could see? What if the student doesn’t act on the statements—will you start mailing them to the parents? Should you mandate that you be able to talk with their parents?
- For adolescents. Consider whether you will allow them to communicate with you directly. Will they be able to e-mail you? How will you communicate with her (his) parents if your relationship is primarily with the teenager? How will you handle medication changes when the teenager prefers you keep everything private, but the parents have the right to informed consent?
- Will you charge for the time it takes you to talk with other providers (CPT 90887); review reports (CPT 90885); for e-mails or phone calls that are only a minute, or 10 minutes (e-mail, CPT 99444; brief phone calls, CPT 99441); or out-of-session refills? What if an insurance company does, or doesn’t, cover these codes? Is it different for patients you see occasionally for medication checks and for those whom you see weekly for therapy?
Psychodynamics of policies
Nowhere does being both a business and a service intersect more than when discussing how much you charge, and for what services. Patients may have little understanding of all the time you spend on their care, and why you choose to bill or not to bill for certain services. They could naturally develop transference reactions based on your policies, or might not even read them and just sign off, which also can give you useful clinical data.
Patients should review and accept your policies before the first appointment is booked. However, it is still meaningful to extend the opportunity to discuss them with a patient at the first session—but if they do not want to ask questions or discuss administrative matters, then follow their lead. By at least offering, this conveys to the patient that you wish to develop a trusting relationship, and that you are open to addressing conflicts or confusion at the beginning.
A valuable investment in time
Spending a bit of time now to create or review your current policies will save a lot of time—and perhaps money or legal action—later. If you can’t think of every scenario or issue today, don’t fret. Your experience in practice will inevitably lead you to recalibrate and update your policies. What’s most important is that your patients know where you stand and that they can trust you over the long-term.
Developing your practice policies and sharing them with your patients is essential to building long-term, trusting relationships. Having a clear starting point helps avert disagreement down the road and allows patients to feel comfortable knowing what they are getting in to, which will provide a foundation on which you and the patient can focus on clinical matters.
What’s in a policy?
Policies should cover administrative aspects of care, such as mandated disclosures; relevant Health Insurance Portability and Accountability Act and Health Information Technology for Economic and Clinical Health Act information; hospital privilege status; and fees and payment policies. Your policies also will touch on areas where business overlaps with patient care, such as confidentiality and its limits, communication methods outside of session, and the risks and benefits of treatment (Table).
Address communication and billing policies for complex scenarios. Although these scenarios might not come up often, if you wait until you are confronted with the situation, the patient might (rightly) feel that she (he) wasn’t properly informed before giving consent. For example:
- For college students. Do you try to build college students’ autonomy by sending them all billing statements directly? If not, how will you handle the diagnosis code that appears on the statement, which their parents could see? What if the student doesn’t act on the statements—will you start mailing them to the parents? Should you mandate that you be able to talk with their parents?
- For adolescents. Consider whether you will allow them to communicate with you directly. Will they be able to e-mail you? How will you communicate with her (his) parents if your relationship is primarily with the teenager? How will you handle medication changes when the teenager prefers you keep everything private, but the parents have the right to informed consent?
- Will you charge for the time it takes you to talk with other providers (CPT 90887); review reports (CPT 90885); for e-mails or phone calls that are only a minute, or 10 minutes (e-mail, CPT 99444; brief phone calls, CPT 99441); or out-of-session refills? What if an insurance company does, or doesn’t, cover these codes? Is it different for patients you see occasionally for medication checks and for those whom you see weekly for therapy?
Psychodynamics of policies
Nowhere does being both a business and a service intersect more than when discussing how much you charge, and for what services. Patients may have little understanding of all the time you spend on their care, and why you choose to bill or not to bill for certain services. They could naturally develop transference reactions based on your policies, or might not even read them and just sign off, which also can give you useful clinical data.
Patients should review and accept your policies before the first appointment is booked. However, it is still meaningful to extend the opportunity to discuss them with a patient at the first session—but if they do not want to ask questions or discuss administrative matters, then follow their lead. By at least offering, this conveys to the patient that you wish to develop a trusting relationship, and that you are open to addressing conflicts or confusion at the beginning.
A valuable investment in time
Spending a bit of time now to create or review your current policies will save a lot of time—and perhaps money or legal action—later. If you can’t think of every scenario or issue today, don’t fret. Your experience in practice will inevitably lead you to recalibrate and update your policies. What’s most important is that your patients know where you stand and that they can trust you over the long-term.
Developing your practice policies and sharing them with your patients is essential to building long-term, trusting relationships. Having a clear starting point helps avert disagreement down the road and allows patients to feel comfortable knowing what they are getting in to, which will provide a foundation on which you and the patient can focus on clinical matters.
What’s in a policy?
Policies should cover administrative aspects of care, such as mandated disclosures; relevant Health Insurance Portability and Accountability Act and Health Information Technology for Economic and Clinical Health Act information; hospital privilege status; and fees and payment policies. Your policies also will touch on areas where business overlaps with patient care, such as confidentiality and its limits, communication methods outside of session, and the risks and benefits of treatment (Table).
Address communication and billing policies for complex scenarios. Although these scenarios might not come up often, if you wait until you are confronted with the situation, the patient might (rightly) feel that she (he) wasn’t properly informed before giving consent. For example:
- For college students. Do you try to build college students’ autonomy by sending them all billing statements directly? If not, how will you handle the diagnosis code that appears on the statement, which their parents could see? What if the student doesn’t act on the statements—will you start mailing them to the parents? Should you mandate that you be able to talk with their parents?
- For adolescents. Consider whether you will allow them to communicate with you directly. Will they be able to e-mail you? How will you communicate with her (his) parents if your relationship is primarily with the teenager? How will you handle medication changes when the teenager prefers you keep everything private, but the parents have the right to informed consent?
- Will you charge for the time it takes you to talk with other providers (CPT 90887); review reports (CPT 90885); for e-mails or phone calls that are only a minute, or 10 minutes (e-mail, CPT 99444; brief phone calls, CPT 99441); or out-of-session refills? What if an insurance company does, or doesn’t, cover these codes? Is it different for patients you see occasionally for medication checks and for those whom you see weekly for therapy?
Psychodynamics of policies
Nowhere does being both a business and a service intersect more than when discussing how much you charge, and for what services. Patients may have little understanding of all the time you spend on their care, and why you choose to bill or not to bill for certain services. They could naturally develop transference reactions based on your policies, or might not even read them and just sign off, which also can give you useful clinical data.
Patients should review and accept your policies before the first appointment is booked. However, it is still meaningful to extend the opportunity to discuss them with a patient at the first session—but if they do not want to ask questions or discuss administrative matters, then follow their lead. By at least offering, this conveys to the patient that you wish to develop a trusting relationship, and that you are open to addressing conflicts or confusion at the beginning.
A valuable investment in time
Spending a bit of time now to create or review your current policies will save a lot of time—and perhaps money or legal action—later. If you can’t think of every scenario or issue today, don’t fret. Your experience in practice will inevitably lead you to recalibrate and update your policies. What’s most important is that your patients know where you stand and that they can trust you over the long-term.
Why I keep fortune cookies on my desk
Many of my patients ask, “Why do you have fortune cookies on your desk?” Then, I offer them one. I considered having other treats, but decided on fortune cookies because of:
Comfort. The cookie is a small treat for those who want one.
Diet. You don’t have to eat the cookie to enjoy it; you can still read the fortune. For patients who have an eating disorder, the cookie allows us to naturally transition the conversation to issues they are experiencing.
Cultural competency. I treat patients of many backgrounds. Some have never seen a fortune cookie (remember to warn them there is a fortune inside!). Others know the fortune cookie is not a Chinese invention,1 as it is popularly thought to be.
Impulsivity. Do patients grab a cookie immediately, wait for one to be offered, or ask for one?
At this point, I ask patients to tell me their fortune. This allows me to assess:
Fine motor skills. Do they have a hand tremor or weakness, or a problem with involuntary movement? How well do they open the individually wrapped cookie?
Problem solving. On the slip of paper in the cookie, fortunes are printed on one side; on the other side are lucky numbers and a Chinese phrase. Some patients fail to turn the slip of paper over; they look it and say, “There are only numbers on this piece of paper.”
Eyesight. Can they see without glasses? Did they bring their glasses? (By extension, I can gauge whether they need, and use, glasses when reaching for a pill bottle in the medicine cabinet.)
Literacy. Can they read their fortune aloud?
Last, I ask what the fortune means and how it might apply to them. This helps me understand their:
Thought process. I am looking for how they think: Abstractly? Concretely? How well do they articulate and explain the meaning of the fortune?
Insight. Having them explain how the fortune applies to them can be helpful to understanding their thinking.
1. Lee J8. Solving a riddle wrapped in a mystery inside a cookie. The New York Times. http://www.nytimes.com/2008/01/16/dining/16fort.html?_r=2&pagewanted=1. Published January 16, 2008. Accessed April 22, 2016.
Many of my patients ask, “Why do you have fortune cookies on your desk?” Then, I offer them one. I considered having other treats, but decided on fortune cookies because of:
Comfort. The cookie is a small treat for those who want one.
Diet. You don’t have to eat the cookie to enjoy it; you can still read the fortune. For patients who have an eating disorder, the cookie allows us to naturally transition the conversation to issues they are experiencing.
Cultural competency. I treat patients of many backgrounds. Some have never seen a fortune cookie (remember to warn them there is a fortune inside!). Others know the fortune cookie is not a Chinese invention,1 as it is popularly thought to be.
Impulsivity. Do patients grab a cookie immediately, wait for one to be offered, or ask for one?
At this point, I ask patients to tell me their fortune. This allows me to assess:
Fine motor skills. Do they have a hand tremor or weakness, or a problem with involuntary movement? How well do they open the individually wrapped cookie?
Problem solving. On the slip of paper in the cookie, fortunes are printed on one side; on the other side are lucky numbers and a Chinese phrase. Some patients fail to turn the slip of paper over; they look it and say, “There are only numbers on this piece of paper.”
Eyesight. Can they see without glasses? Did they bring their glasses? (By extension, I can gauge whether they need, and use, glasses when reaching for a pill bottle in the medicine cabinet.)
Literacy. Can they read their fortune aloud?
Last, I ask what the fortune means and how it might apply to them. This helps me understand their:
Thought process. I am looking for how they think: Abstractly? Concretely? How well do they articulate and explain the meaning of the fortune?
Insight. Having them explain how the fortune applies to them can be helpful to understanding their thinking.
Many of my patients ask, “Why do you have fortune cookies on your desk?” Then, I offer them one. I considered having other treats, but decided on fortune cookies because of:
Comfort. The cookie is a small treat for those who want one.
Diet. You don’t have to eat the cookie to enjoy it; you can still read the fortune. For patients who have an eating disorder, the cookie allows us to naturally transition the conversation to issues they are experiencing.
Cultural competency. I treat patients of many backgrounds. Some have never seen a fortune cookie (remember to warn them there is a fortune inside!). Others know the fortune cookie is not a Chinese invention,1 as it is popularly thought to be.
Impulsivity. Do patients grab a cookie immediately, wait for one to be offered, or ask for one?
At this point, I ask patients to tell me their fortune. This allows me to assess:
Fine motor skills. Do they have a hand tremor or weakness, or a problem with involuntary movement? How well do they open the individually wrapped cookie?
Problem solving. On the slip of paper in the cookie, fortunes are printed on one side; on the other side are lucky numbers and a Chinese phrase. Some patients fail to turn the slip of paper over; they look it and say, “There are only numbers on this piece of paper.”
Eyesight. Can they see without glasses? Did they bring their glasses? (By extension, I can gauge whether they need, and use, glasses when reaching for a pill bottle in the medicine cabinet.)
Literacy. Can they read their fortune aloud?
Last, I ask what the fortune means and how it might apply to them. This helps me understand their:
Thought process. I am looking for how they think: Abstractly? Concretely? How well do they articulate and explain the meaning of the fortune?
Insight. Having them explain how the fortune applies to them can be helpful to understanding their thinking.
1. Lee J8. Solving a riddle wrapped in a mystery inside a cookie. The New York Times. http://www.nytimes.com/2008/01/16/dining/16fort.html?_r=2&pagewanted=1. Published January 16, 2008. Accessed April 22, 2016.
1. Lee J8. Solving a riddle wrapped in a mystery inside a cookie. The New York Times. http://www.nytimes.com/2008/01/16/dining/16fort.html?_r=2&pagewanted=1. Published January 16, 2008. Accessed April 22, 2016.
Treated with a mood stabilizer, he becomes incontinent and walks oddly
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
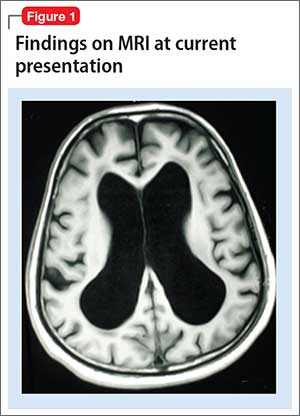
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
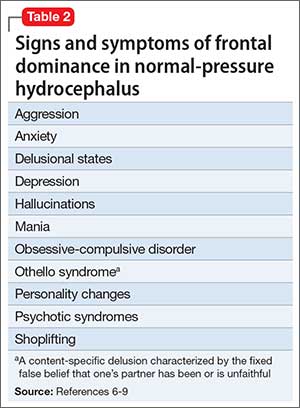
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.

Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
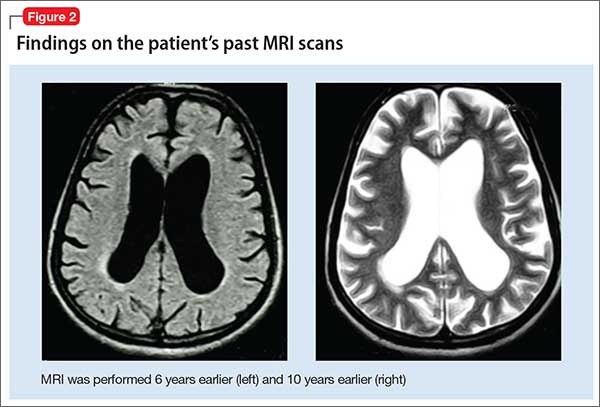
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
Would better policing of metabolic status help you avoid medicolegal worries?
Dear Dr. Mossman,
All the psychiatrists at our clinic agree: It is hard to remember when our patients who take an antipsychotic are due for metabolic monitoring, and it’s even harder to get many of them to follow through with timely blood tests. For many, stopping their medication would be a bad idea. If we keep a patient on an antipsychotic and a metabolic problem results, how serious is our malpractice liability risk?
Submitted by “Dr. V”
Antipsychotics, the mainstay of treatment for schizophrenia,1 put patients at risk of gaining weight and developing metabolic syndrome, including type 2 diabetes mellitus, hypertension, and dyslipidemia.2 Second-generation antipsychotics are the biggest offenders, but taking a first-generation antipsychotic also can lead to these adverse effects.3
Most psychiatrists are aware of these risks and prefer that their patients do not experience them. However, many psychiatrists neglect proper monitoring or, like Dr. V, find it hard to ensure it happens and thus worry about clinical deterioration if patients stop taking an antipsychotic.4 If you are in the same situation as Dr. V, what medicolegal risks are you facing?
To answer this question, we will:
- review the clinical guidelines and standards for monitoring metabolic effects of antipsychotics
- examine how well (or poorly) physicians adhere to these standards
- discuss what “standard of care” means and how a practice guideline affects the standard effects
- propose how psychiatrists can do better at policing the metabolic effects of antipsychotics.
I’ll be watching you: Following guidelines
Several medical specialty societies have published guidelines for monitoring the metabolic effects of antipsychotics.5-8 These guidelines instruct physicians to obtain a thorough personal and family history; consider metabolic risks when starting a medication; and monitor weight, waist circumference, blood pressure, glucose, hemoglobin A1c, and lipids at various intervals. They also advise referral for management of detected metabolic problems.
Although the recommendations seem clear, many physicians don’t follow them. A 2012 meta-analysis of 48 studies, covering >200,000 antipsychotic-treated patients, showed that baseline measurements of cholesterol, glucose, and weight occurred in <50% of cases.9 A more recent review found that, among adults with a serious mental illness, the rate of lipid testing varied from 6% to 85% and for glucose monitoring, between 18% and 75%.10 In the first years after antipsychotic monitoring guidelines were established, they had only a modest impact on practice,9,11 and some studies showed the guidelines made no difference at all.12-14
Monitoring compliance varies with the type of insurance coverage patients have but remains suboptimal among the commercially insured,11 Medicaid patients,14-16 and veterans.17,18 Studies on antipsychotic treatment in children, adolescents, patients with dementia, and patients with an intellectual disability show insufficient monitoring as well.9,14,17,19-21 The reasons for these gaps are manifold, but one commonly cited factor is uncertainty about whether the psychiatrist or primary care physician should handle monitoring.22
Every claim you stake: The ‘standard of care’
In a medical malpractice case, the party claiming injury must show that the accused physician failed to follow “the generally recognized practices and procedures which would be exercised by ordinary competent practitioners in a defendant doctor’s field of medicine under the same or similar circumstances.”23 In the studies mentioned above,9-14 a large fraction of psychiatrists—many of whom, we can presume, are “competent practitioners”—don’t follow the antipsychotic monitoring guidelines in actual practice. Could failing to follow those guidelines still be the basis for a successful lawsuit?
The answer seems to be ‘yes.’ Published legal decisions describe malpractice lawsuits alleging physicians’ failure to follow antipsychotic guidelines,24,25 and online advertisements show that attorneys believe such cases can generate a payout.26,27 This may seem odd, given what studies say about psychiatrists’ monitoring practices. But determining the “standard of care” in a malpractice case is not an empirical question; it is a legal matter that is decided based on the testimony of expert witnesses.28 Here, customary practice matters, but it’s not the whole story.
Although the standard of care against which courts measure a physician’s actions “is that of a reasonably prudent practitioner …, The degree of care actually practiced by members of the profession is only some evidence of what is reasonably prudent—it is not dispositive.”29 To support their opinion concerning the standard of care, testifying medical witnesses sometimes use practice guidelines. In this case, an explanation of why a particular guideline was chosen is crucial.30
Using guidelines to establish the standard is controversial. On one hand, using guidelines in malpractice litigation allows for some consistency about expectations of practitioners.31,32 Although guidelines are not identical to evidenced-based medicine, they generally reflect an evidence-based expert consensus about sound medical practice. If a hospital uses a guideline to train its employees, the guideline provides the courts with clear information on what should have happened.33,34 Laws in some states allow clinicians to invoke their adherence to a guideline in defense against malpractice claims.35
On the other hand, critics contend that guidelines may not set an accurate standard for the quality of care, nor do they necessarily reflect a proper balance of the conflicting interests of patients and the health care system.36 The American Psychiatric Association states that its practice guidelines “are not intended to serve or be construed as a ‘standard of medical care.’”37
Conformity is not the only measure of prudent practice, and following guidelines does not immunize a clinician from lawsuit if a particular clinical situation demands a different course of action.32 Guidelines can be costly to implement,36 compliance with guidelines generally is low,35 and national guidelines do not necessarily improve the quality of care.38 Last, relying on guidelines to determine the standard of care might stifle innovation or development of alternate approaches by silencing viewpoints.39,40 Table 133-35,39,41 (page 60)summarizes variables that make a guideline more indicative of the standard of care.

Every step you take: Better monitoring
Medical professionals often are slow to update their practice to reflect new knowledge about optimal treatment. But practice guidelines influence the court’s views about the standard of care, and Dr. V’s question shows that he and his colleagues agree that metabolic status needs to be better monitored when patients take antipsychotic drugs. The following discussion and Table 242-45 offer suggestions for how psychiatrists and their practice settings could better accomplish this.
Electronic health records (EHRs). Monitoring health indices often is the largest hurdle that health care professionals face.46 However, large health care systems with EHRs are in a good position to develop and implement automated computer routines that track which patients need monitoring and note due dates, abnormal results, and management interventions.42 Some studies suggest that monitoring rates in both inpatient47 and outpatient48 settings improve with built-in EHR reminders. However, if a system uses too many reminders, the resulting “alert fatigue” will limit their value.22 Providing individual feedback about monitoring practices may enhance physicians’ buy-in to reminder systems.48

Integrated care systems can improve patient outcomes, particularly antipsychotic monitoring. Advantages include shared funding streams, a unified medical record, coordinated scheduling of psychiatric and primary care appointments, and addressing blood-draw refusals.43 More frequent primary care visits make antipsychotic monitoring more likely.11 Ultimately, integrated care could resolve problems related to determining which clinicians are responsible for monitoring and managing adverse metabolic effects.
Third-party payers. Managed care interventions also could improve monitoring rates.44 Prior authorization often requires physicians to obtain appropriate lab work. Insurers might contact physicians with educational interventions, including free webinars, provider alerts, and letters about monitoring rates in their region. Some insurers also provide disease management programs for patients and their caregivers.
Individual and small group practices. Psychiatrists who practice outside a large health care system might designate 2 months each year as “physical health months.” In the “Let’s Get Physical” program,45 physicians were given longer appointment times during these months to address metabolic monitoring, provide education about managing side effects of medication, and encourage better diets and exercise.
Overall, the best techniques might be those implicit to good doctoring: clear and open communication with patients, effective patient education, respect of informed consent, and thorough follow-up.49
1. Mossman D, Steinberg JL. Promoting, prescribing, and pushing pills: understanding the lessons of antipsychotic drug litigation. Michigan St U J Med & Law. 2009;13:263-334.
2. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation: evaluating the risk/benefit equation and improving the standard of care. J Clin Psychopharmacol. 2004;24(5 suppl 1):S7-S14.
3. De Hert M, Schreurs V, Sweers K, et al. Typical and atypical antipsychotics differentially affect long-term incidence rates of the metabolic syndrome in first-episode patients with schizophrenia: a retrospective chart review. Schizophr Res. 2008;101(1-3):295-303.
4. Appelbaum PS, Gutheil TG. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007.
5. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry. 2004;65(2):267-272.
6. Pappadopulos E, Macintyre JC II, Crismon ML, et al. Treatment recommendations for the use of antipsychotics for aggressive youth (TRAAY). Part II. J Am Acad Child Adolesc Psychiatry. 2003;42(2):145-161.
7. Pringsheim T, Panagiotopoulos C, Davidson J, et al; CAMESA guideline group. Evidence-based recommendations for monitoring safety of second generation antipsychotics in children and youth [Erratum in: J Can Acad Adolesc Psychiatry. 2011;20(3):1-2]. J Can Acad Child Adolesc Psychiatry. 2011;20(3):218-233.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Mitchell AJ, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med. 2012;42(1):125-147.
10. Baller JB, McGinty EE, Azrin ST, et al. Screening for cardiovascular risk factors in adults with serious mental illness: a review of the evidence. BMC Psychiatry. 2015;15:55.
11. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry. 2009;166(3):345-353.
12. Dhamane AD, Martin BC, Brixner DI, et al. Metabolic monitoring of patients prescribed second-generation antipsychotics. J Psychiatr Pract. 2013;19(5):360-374.
13. Morrato EH, Newcomer JW, Kamat S, et al. Metabolic screening after the American Diabetes Association’s consensus statement on antipsychotic drugs and diabetes. Diabetes Care. 2009;32(6):1037-1042.
14. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry. 2010;67(1):17-24.
15. Moeller KE, Rigler SK, Mayorga A, et al. Quality of monitoring for metabolic effects associated with second generation antipsychotics in patients with schizophrenia on public insurance. Schizophr Res. 2011;126(1-3):117-123.
16. Barnett M, VonMuenster S, Wehring H, et al. Assessment of monitoring for glucose and lipid dysregulation in adult Medi-Cal patients newly started on antipsychotics. Ann Clin Psychiatry. 2010;22(1):9-18.
17. Mittal D, Li C, Viverito K, et al. Monitoring for metabolic side effects among outpatients with dementia receiving antipsychotics. Psychiatr Serv. 2014;65(9):1147-1153.
18. Hsu C, Ried LD, Bengtson MA, et al. Metabolic monitoring in veterans with schizophrenia-related disorders and treated with second-generation antipsychotics: findings from a Veterans Affairs-based population. J Am Pharm Assoc. 2008;48(3):393-400.
19. Raebel MA, Penfold R, McMahon AW, et al. Adherence to guidelines for glucose assessment in starting second-generation antipsychotics. Pediatrics. 2014;134(5):e1308-e1314.
20. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003-2011. Psychiatr Serv. 2015;66(6):604-609.
21. Teeluckdharry S, Sharma S, O’Rourke E, et al. Monitoring metabolic side effects of atypical antipsychotics in people with an intellectual disability. J Intellect Disabil. 2013;17(3):223-235.
22. Lee J, Dalack GW, Casher MI, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second-generation antipsychotics utilizing a computer-based intervention. J Clin Pharm Ther. 2016;41(2):209-213.
23. McCourt v Abernathy, 457 SE2d 603 (SC 1995).
24. Schultz v AstraZeneca Pharma LP, LEXIS 94534, 2006 WL 3797932, (ND Cal 2006).
25. Redmond v AstraZeneca Pharma LP, 492 F Supp 2d 575 (SD Miss 2007).
26. Goguen D. Risperdal, Seroquel, Symbyax, Zyprexa, and other antipsychotic drugs. http://www.nolo.com/legal-encyclopedia/risperdal-seroquel-symbyax-zyprexa-antipsychotics-29866.html. Accessed April 4, 2016.
27. FreeAdvice staff. Risperdal medical malpractice lawsuits: Risperdal injury lawyer explains what you need to know. http://injury-law.freeadvice.com/injury-law/drug-toxic_chemicals/risperdal.htm. Accessed April 4, 2016.
28. Lewis MK, Gohagan JK, Merenstein DJ. The locality rule and the physician’s dilemma: local medical practices vs the national standard of care. JAMA. 2007;297(23):2633-2637.
29. Harris v Groth, 99 Wn2d 438, 663 P2d 113 (1983).
30. Moffett P, Moore G. The standard of care: legal history and definitions: the bad and good news. West J Emerg Med. 2011;12(1):109-112.
31. Taylor C. The use of clinical practice guidelines in determining standard of care. J Legal Med. 2014;35(2):273-290.
32. Bal BS, Brenner LH. Medicolegal sidebar: the law and social values: conformity to norms. Clin Orthop Relat Res. 2015;473(5):1555-1559.
33. Recupero PR. Clinical practice guidelines as learned treatises: understanding their use as evidence in the courtroom. J Am Acad Psychiatry Law. 2008;36(3):290-301.
34. Price v Cleveland Clinic Found, 515 NE2d 931 (Ohio Ct App 1986).
35. Zonana H. Commentary: when is a practice guideline only a guideline? J Am Acad Psychiatry Law. 2008;36(3):302-305.
36. Guillod O. Clinical guidelines and professional liability: a short comment from the legal side. ORL J Otorhinolaryngol Relat Spec. 2010;72(3):133-136; discussion 136-137.
37. American Psychiatric Association. Practice guidelines for the psychiatric evaluation of adults. 3rd ed. Arlington, VA: American Psychiatric Association; 2016.
38. Brouwers MC, Kho ME, Browman GP, et al; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842.
39. Vermaas AM. Liability in relation to the use of professional medical guidelines. Med Law. 2003;22(2):233-238.
40. Strauss DC, Thomas JM. What does the medical profession mean by “standard of care?”. J Clin Oncol. 2009;27(32):e192-e193.
41. Kozlick D. Clinical practice guidelines and the legal standard of care: warnings, predictions, and interdisciplinary encounters. Health Law J. 2011;19:125-151.
42. Owen RR, Drummond KL, Viverito KM, et al. Monitoring and managing metabolic effects of antipsychotics: a cluster randomized trial of an intervention combining evidence-based quality improvement and external facilitation. Implement Sci. 2013;8:120.
43. Ruiz LM, Damron M, Jones KB, et al. Antipsychotic use and metabolic monitoring in individuals with developmental disabilities served in a Medicaid medical home [published online January 27, 2016]. J Autism Dev Disord. doi: 10.1007/s10803-016-2712-x.
44. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manag Care Spec Pharm. 2015;21(9):769-77,777a-777cc.
45. Wilson E, Randall C, Patterson S, et al. Monitoring and management of metabolic abnormalities: mixed-method evaluation of a successful intervention. Australas Psychiatry. 2014;22(3):248-253.
46. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
47. DelMonte MT, Bostwick JR, Bess JD, et al. Evaluation of a computer-based intervention to enhance metabolic monitoring in psychiatry inpatients treated with second-generation antipsychotics. J Clin Pharm Ther. 2012;37(6):668-673.
48. Lai CL, Chan HY, Pan YJ, et al. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry. 2015;48(1):25-29.
49. Bailey RK, Adams JB, Unger DM. Atypical antipsychotics: a case study in new era risk management. J Psychiatr Pract. 2006;12(4):253-258.
Dear Dr. Mossman,
All the psychiatrists at our clinic agree: It is hard to remember when our patients who take an antipsychotic are due for metabolic monitoring, and it’s even harder to get many of them to follow through with timely blood tests. For many, stopping their medication would be a bad idea. If we keep a patient on an antipsychotic and a metabolic problem results, how serious is our malpractice liability risk?
Submitted by “Dr. V”
Antipsychotics, the mainstay of treatment for schizophrenia,1 put patients at risk of gaining weight and developing metabolic syndrome, including type 2 diabetes mellitus, hypertension, and dyslipidemia.2 Second-generation antipsychotics are the biggest offenders, but taking a first-generation antipsychotic also can lead to these adverse effects.3
Most psychiatrists are aware of these risks and prefer that their patients do not experience them. However, many psychiatrists neglect proper monitoring or, like Dr. V, find it hard to ensure it happens and thus worry about clinical deterioration if patients stop taking an antipsychotic.4 If you are in the same situation as Dr. V, what medicolegal risks are you facing?
To answer this question, we will:
- review the clinical guidelines and standards for monitoring metabolic effects of antipsychotics
- examine how well (or poorly) physicians adhere to these standards
- discuss what “standard of care” means and how a practice guideline affects the standard effects
- propose how psychiatrists can do better at policing the metabolic effects of antipsychotics.
I’ll be watching you: Following guidelines
Several medical specialty societies have published guidelines for monitoring the metabolic effects of antipsychotics.5-8 These guidelines instruct physicians to obtain a thorough personal and family history; consider metabolic risks when starting a medication; and monitor weight, waist circumference, blood pressure, glucose, hemoglobin A1c, and lipids at various intervals. They also advise referral for management of detected metabolic problems.
Although the recommendations seem clear, many physicians don’t follow them. A 2012 meta-analysis of 48 studies, covering >200,000 antipsychotic-treated patients, showed that baseline measurements of cholesterol, glucose, and weight occurred in <50% of cases.9 A more recent review found that, among adults with a serious mental illness, the rate of lipid testing varied from 6% to 85% and for glucose monitoring, between 18% and 75%.10 In the first years after antipsychotic monitoring guidelines were established, they had only a modest impact on practice,9,11 and some studies showed the guidelines made no difference at all.12-14
Monitoring compliance varies with the type of insurance coverage patients have but remains suboptimal among the commercially insured,11 Medicaid patients,14-16 and veterans.17,18 Studies on antipsychotic treatment in children, adolescents, patients with dementia, and patients with an intellectual disability show insufficient monitoring as well.9,14,17,19-21 The reasons for these gaps are manifold, but one commonly cited factor is uncertainty about whether the psychiatrist or primary care physician should handle monitoring.22
Every claim you stake: The ‘standard of care’
In a medical malpractice case, the party claiming injury must show that the accused physician failed to follow “the generally recognized practices and procedures which would be exercised by ordinary competent practitioners in a defendant doctor’s field of medicine under the same or similar circumstances.”23 In the studies mentioned above,9-14 a large fraction of psychiatrists—many of whom, we can presume, are “competent practitioners”—don’t follow the antipsychotic monitoring guidelines in actual practice. Could failing to follow those guidelines still be the basis for a successful lawsuit?
The answer seems to be ‘yes.’ Published legal decisions describe malpractice lawsuits alleging physicians’ failure to follow antipsychotic guidelines,24,25 and online advertisements show that attorneys believe such cases can generate a payout.26,27 This may seem odd, given what studies say about psychiatrists’ monitoring practices. But determining the “standard of care” in a malpractice case is not an empirical question; it is a legal matter that is decided based on the testimony of expert witnesses.28 Here, customary practice matters, but it’s not the whole story.
Although the standard of care against which courts measure a physician’s actions “is that of a reasonably prudent practitioner …, The degree of care actually practiced by members of the profession is only some evidence of what is reasonably prudent—it is not dispositive.”29 To support their opinion concerning the standard of care, testifying medical witnesses sometimes use practice guidelines. In this case, an explanation of why a particular guideline was chosen is crucial.30
Using guidelines to establish the standard is controversial. On one hand, using guidelines in malpractice litigation allows for some consistency about expectations of practitioners.31,32 Although guidelines are not identical to evidenced-based medicine, they generally reflect an evidence-based expert consensus about sound medical practice. If a hospital uses a guideline to train its employees, the guideline provides the courts with clear information on what should have happened.33,34 Laws in some states allow clinicians to invoke their adherence to a guideline in defense against malpractice claims.35
On the other hand, critics contend that guidelines may not set an accurate standard for the quality of care, nor do they necessarily reflect a proper balance of the conflicting interests of patients and the health care system.36 The American Psychiatric Association states that its practice guidelines “are not intended to serve or be construed as a ‘standard of medical care.’”37
Conformity is not the only measure of prudent practice, and following guidelines does not immunize a clinician from lawsuit if a particular clinical situation demands a different course of action.32 Guidelines can be costly to implement,36 compliance with guidelines generally is low,35 and national guidelines do not necessarily improve the quality of care.38 Last, relying on guidelines to determine the standard of care might stifle innovation or development of alternate approaches by silencing viewpoints.39,40 Table 133-35,39,41 (page 60)summarizes variables that make a guideline more indicative of the standard of care.
Every step you take: Better monitoring
Medical professionals often are slow to update their practice to reflect new knowledge about optimal treatment. But practice guidelines influence the court’s views about the standard of care, and Dr. V’s question shows that he and his colleagues agree that metabolic status needs to be better monitored when patients take antipsychotic drugs. The following discussion and Table 242-45 offer suggestions for how psychiatrists and their practice settings could better accomplish this.
Electronic health records (EHRs). Monitoring health indices often is the largest hurdle that health care professionals face.46 However, large health care systems with EHRs are in a good position to develop and implement automated computer routines that track which patients need monitoring and note due dates, abnormal results, and management interventions.42 Some studies suggest that monitoring rates in both inpatient47 and outpatient48 settings improve with built-in EHR reminders. However, if a system uses too many reminders, the resulting “alert fatigue” will limit their value.22 Providing individual feedback about monitoring practices may enhance physicians’ buy-in to reminder systems.48
Integrated care systems can improve patient outcomes, particularly antipsychotic monitoring. Advantages include shared funding streams, a unified medical record, coordinated scheduling of psychiatric and primary care appointments, and addressing blood-draw refusals.43 More frequent primary care visits make antipsychotic monitoring more likely.11 Ultimately, integrated care could resolve problems related to determining which clinicians are responsible for monitoring and managing adverse metabolic effects.
Third-party payers. Managed care interventions also could improve monitoring rates.44 Prior authorization often requires physicians to obtain appropriate lab work. Insurers might contact physicians with educational interventions, including free webinars, provider alerts, and letters about monitoring rates in their region. Some insurers also provide disease management programs for patients and their caregivers.
Individual and small group practices. Psychiatrists who practice outside a large health care system might designate 2 months each year as “physical health months.” In the “Let’s Get Physical” program,45 physicians were given longer appointment times during these months to address metabolic monitoring, provide education about managing side effects of medication, and encourage better diets and exercise.
Overall, the best techniques might be those implicit to good doctoring: clear and open communication with patients, effective patient education, respect of informed consent, and thorough follow-up.49
Dear Dr. Mossman,
All the psychiatrists at our clinic agree: It is hard to remember when our patients who take an antipsychotic are due for metabolic monitoring, and it’s even harder to get many of them to follow through with timely blood tests. For many, stopping their medication would be a bad idea. If we keep a patient on an antipsychotic and a metabolic problem results, how serious is our malpractice liability risk?
Submitted by “Dr. V”
Antipsychotics, the mainstay of treatment for schizophrenia,1 put patients at risk of gaining weight and developing metabolic syndrome, including type 2 diabetes mellitus, hypertension, and dyslipidemia.2 Second-generation antipsychotics are the biggest offenders, but taking a first-generation antipsychotic also can lead to these adverse effects.3
Most psychiatrists are aware of these risks and prefer that their patients do not experience them. However, many psychiatrists neglect proper monitoring or, like Dr. V, find it hard to ensure it happens and thus worry about clinical deterioration if patients stop taking an antipsychotic.4 If you are in the same situation as Dr. V, what medicolegal risks are you facing?
To answer this question, we will:
- review the clinical guidelines and standards for monitoring metabolic effects of antipsychotics
- examine how well (or poorly) physicians adhere to these standards
- discuss what “standard of care” means and how a practice guideline affects the standard effects
- propose how psychiatrists can do better at policing the metabolic effects of antipsychotics.
I’ll be watching you: Following guidelines
Several medical specialty societies have published guidelines for monitoring the metabolic effects of antipsychotics.5-8 These guidelines instruct physicians to obtain a thorough personal and family history; consider metabolic risks when starting a medication; and monitor weight, waist circumference, blood pressure, glucose, hemoglobin A1c, and lipids at various intervals. They also advise referral for management of detected metabolic problems.
Although the recommendations seem clear, many physicians don’t follow them. A 2012 meta-analysis of 48 studies, covering >200,000 antipsychotic-treated patients, showed that baseline measurements of cholesterol, glucose, and weight occurred in <50% of cases.9 A more recent review found that, among adults with a serious mental illness, the rate of lipid testing varied from 6% to 85% and for glucose monitoring, between 18% and 75%.10 In the first years after antipsychotic monitoring guidelines were established, they had only a modest impact on practice,9,11 and some studies showed the guidelines made no difference at all.12-14
Monitoring compliance varies with the type of insurance coverage patients have but remains suboptimal among the commercially insured,11 Medicaid patients,14-16 and veterans.17,18 Studies on antipsychotic treatment in children, adolescents, patients with dementia, and patients with an intellectual disability show insufficient monitoring as well.9,14,17,19-21 The reasons for these gaps are manifold, but one commonly cited factor is uncertainty about whether the psychiatrist or primary care physician should handle monitoring.22
Every claim you stake: The ‘standard of care’
In a medical malpractice case, the party claiming injury must show that the accused physician failed to follow “the generally recognized practices and procedures which would be exercised by ordinary competent practitioners in a defendant doctor’s field of medicine under the same or similar circumstances.”23 In the studies mentioned above,9-14 a large fraction of psychiatrists—many of whom, we can presume, are “competent practitioners”—don’t follow the antipsychotic monitoring guidelines in actual practice. Could failing to follow those guidelines still be the basis for a successful lawsuit?
The answer seems to be ‘yes.’ Published legal decisions describe malpractice lawsuits alleging physicians’ failure to follow antipsychotic guidelines,24,25 and online advertisements show that attorneys believe such cases can generate a payout.26,27 This may seem odd, given what studies say about psychiatrists’ monitoring practices. But determining the “standard of care” in a malpractice case is not an empirical question; it is a legal matter that is decided based on the testimony of expert witnesses.28 Here, customary practice matters, but it’s not the whole story.
Although the standard of care against which courts measure a physician’s actions “is that of a reasonably prudent practitioner …, The degree of care actually practiced by members of the profession is only some evidence of what is reasonably prudent—it is not dispositive.”29 To support their opinion concerning the standard of care, testifying medical witnesses sometimes use practice guidelines. In this case, an explanation of why a particular guideline was chosen is crucial.30
Using guidelines to establish the standard is controversial. On one hand, using guidelines in malpractice litigation allows for some consistency about expectations of practitioners.31,32 Although guidelines are not identical to evidenced-based medicine, they generally reflect an evidence-based expert consensus about sound medical practice. If a hospital uses a guideline to train its employees, the guideline provides the courts with clear information on what should have happened.33,34 Laws in some states allow clinicians to invoke their adherence to a guideline in defense against malpractice claims.35
On the other hand, critics contend that guidelines may not set an accurate standard for the quality of care, nor do they necessarily reflect a proper balance of the conflicting interests of patients and the health care system.36 The American Psychiatric Association states that its practice guidelines “are not intended to serve or be construed as a ‘standard of medical care.’”37
Conformity is not the only measure of prudent practice, and following guidelines does not immunize a clinician from lawsuit if a particular clinical situation demands a different course of action.32 Guidelines can be costly to implement,36 compliance with guidelines generally is low,35 and national guidelines do not necessarily improve the quality of care.38 Last, relying on guidelines to determine the standard of care might stifle innovation or development of alternate approaches by silencing viewpoints.39,40 Table 133-35,39,41 (page 60)summarizes variables that make a guideline more indicative of the standard of care.
Every step you take: Better monitoring
Medical professionals often are slow to update their practice to reflect new knowledge about optimal treatment. But practice guidelines influence the court’s views about the standard of care, and Dr. V’s question shows that he and his colleagues agree that metabolic status needs to be better monitored when patients take antipsychotic drugs. The following discussion and Table 242-45 offer suggestions for how psychiatrists and their practice settings could better accomplish this.
Electronic health records (EHRs). Monitoring health indices often is the largest hurdle that health care professionals face.46 However, large health care systems with EHRs are in a good position to develop and implement automated computer routines that track which patients need monitoring and note due dates, abnormal results, and management interventions.42 Some studies suggest that monitoring rates in both inpatient47 and outpatient48 settings improve with built-in EHR reminders. However, if a system uses too many reminders, the resulting “alert fatigue” will limit their value.22 Providing individual feedback about monitoring practices may enhance physicians’ buy-in to reminder systems.48
Integrated care systems can improve patient outcomes, particularly antipsychotic monitoring. Advantages include shared funding streams, a unified medical record, coordinated scheduling of psychiatric and primary care appointments, and addressing blood-draw refusals.43 More frequent primary care visits make antipsychotic monitoring more likely.11 Ultimately, integrated care could resolve problems related to determining which clinicians are responsible for monitoring and managing adverse metabolic effects.
Third-party payers. Managed care interventions also could improve monitoring rates.44 Prior authorization often requires physicians to obtain appropriate lab work. Insurers might contact physicians with educational interventions, including free webinars, provider alerts, and letters about monitoring rates in their region. Some insurers also provide disease management programs for patients and their caregivers.
Individual and small group practices. Psychiatrists who practice outside a large health care system might designate 2 months each year as “physical health months.” In the “Let’s Get Physical” program,45 physicians were given longer appointment times during these months to address metabolic monitoring, provide education about managing side effects of medication, and encourage better diets and exercise.
Overall, the best techniques might be those implicit to good doctoring: clear and open communication with patients, effective patient education, respect of informed consent, and thorough follow-up.49
1. Mossman D, Steinberg JL. Promoting, prescribing, and pushing pills: understanding the lessons of antipsychotic drug litigation. Michigan St U J Med & Law. 2009;13:263-334.
2. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation: evaluating the risk/benefit equation and improving the standard of care. J Clin Psychopharmacol. 2004;24(5 suppl 1):S7-S14.
3. De Hert M, Schreurs V, Sweers K, et al. Typical and atypical antipsychotics differentially affect long-term incidence rates of the metabolic syndrome in first-episode patients with schizophrenia: a retrospective chart review. Schizophr Res. 2008;101(1-3):295-303.
4. Appelbaum PS, Gutheil TG. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007.
5. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry. 2004;65(2):267-272.
6. Pappadopulos E, Macintyre JC II, Crismon ML, et al. Treatment recommendations for the use of antipsychotics for aggressive youth (TRAAY). Part II. J Am Acad Child Adolesc Psychiatry. 2003;42(2):145-161.
7. Pringsheim T, Panagiotopoulos C, Davidson J, et al; CAMESA guideline group. Evidence-based recommendations for monitoring safety of second generation antipsychotics in children and youth [Erratum in: J Can Acad Adolesc Psychiatry. 2011;20(3):1-2]. J Can Acad Child Adolesc Psychiatry. 2011;20(3):218-233.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Mitchell AJ, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med. 2012;42(1):125-147.
10. Baller JB, McGinty EE, Azrin ST, et al. Screening for cardiovascular risk factors in adults with serious mental illness: a review of the evidence. BMC Psychiatry. 2015;15:55.
11. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry. 2009;166(3):345-353.
12. Dhamane AD, Martin BC, Brixner DI, et al. Metabolic monitoring of patients prescribed second-generation antipsychotics. J Psychiatr Pract. 2013;19(5):360-374.
13. Morrato EH, Newcomer JW, Kamat S, et al. Metabolic screening after the American Diabetes Association’s consensus statement on antipsychotic drugs and diabetes. Diabetes Care. 2009;32(6):1037-1042.
14. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry. 2010;67(1):17-24.
15. Moeller KE, Rigler SK, Mayorga A, et al. Quality of monitoring for metabolic effects associated with second generation antipsychotics in patients with schizophrenia on public insurance. Schizophr Res. 2011;126(1-3):117-123.
16. Barnett M, VonMuenster S, Wehring H, et al. Assessment of monitoring for glucose and lipid dysregulation in adult Medi-Cal patients newly started on antipsychotics. Ann Clin Psychiatry. 2010;22(1):9-18.
17. Mittal D, Li C, Viverito K, et al. Monitoring for metabolic side effects among outpatients with dementia receiving antipsychotics. Psychiatr Serv. 2014;65(9):1147-1153.
18. Hsu C, Ried LD, Bengtson MA, et al. Metabolic monitoring in veterans with schizophrenia-related disorders and treated with second-generation antipsychotics: findings from a Veterans Affairs-based population. J Am Pharm Assoc. 2008;48(3):393-400.
19. Raebel MA, Penfold R, McMahon AW, et al. Adherence to guidelines for glucose assessment in starting second-generation antipsychotics. Pediatrics. 2014;134(5):e1308-e1314.
20. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003-2011. Psychiatr Serv. 2015;66(6):604-609.
21. Teeluckdharry S, Sharma S, O’Rourke E, et al. Monitoring metabolic side effects of atypical antipsychotics in people with an intellectual disability. J Intellect Disabil. 2013;17(3):223-235.
22. Lee J, Dalack GW, Casher MI, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second-generation antipsychotics utilizing a computer-based intervention. J Clin Pharm Ther. 2016;41(2):209-213.
23. McCourt v Abernathy, 457 SE2d 603 (SC 1995).
24. Schultz v AstraZeneca Pharma LP, LEXIS 94534, 2006 WL 3797932, (ND Cal 2006).
25. Redmond v AstraZeneca Pharma LP, 492 F Supp 2d 575 (SD Miss 2007).
26. Goguen D. Risperdal, Seroquel, Symbyax, Zyprexa, and other antipsychotic drugs. http://www.nolo.com/legal-encyclopedia/risperdal-seroquel-symbyax-zyprexa-antipsychotics-29866.html. Accessed April 4, 2016.
27. FreeAdvice staff. Risperdal medical malpractice lawsuits: Risperdal injury lawyer explains what you need to know. http://injury-law.freeadvice.com/injury-law/drug-toxic_chemicals/risperdal.htm. Accessed April 4, 2016.
28. Lewis MK, Gohagan JK, Merenstein DJ. The locality rule and the physician’s dilemma: local medical practices vs the national standard of care. JAMA. 2007;297(23):2633-2637.
29. Harris v Groth, 99 Wn2d 438, 663 P2d 113 (1983).
30. Moffett P, Moore G. The standard of care: legal history and definitions: the bad and good news. West J Emerg Med. 2011;12(1):109-112.
31. Taylor C. The use of clinical practice guidelines in determining standard of care. J Legal Med. 2014;35(2):273-290.
32. Bal BS, Brenner LH. Medicolegal sidebar: the law and social values: conformity to norms. Clin Orthop Relat Res. 2015;473(5):1555-1559.
33. Recupero PR. Clinical practice guidelines as learned treatises: understanding their use as evidence in the courtroom. J Am Acad Psychiatry Law. 2008;36(3):290-301.
34. Price v Cleveland Clinic Found, 515 NE2d 931 (Ohio Ct App 1986).
35. Zonana H. Commentary: when is a practice guideline only a guideline? J Am Acad Psychiatry Law. 2008;36(3):302-305.
36. Guillod O. Clinical guidelines and professional liability: a short comment from the legal side. ORL J Otorhinolaryngol Relat Spec. 2010;72(3):133-136; discussion 136-137.
37. American Psychiatric Association. Practice guidelines for the psychiatric evaluation of adults. 3rd ed. Arlington, VA: American Psychiatric Association; 2016.
38. Brouwers MC, Kho ME, Browman GP, et al; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842.
39. Vermaas AM. Liability in relation to the use of professional medical guidelines. Med Law. 2003;22(2):233-238.
40. Strauss DC, Thomas JM. What does the medical profession mean by “standard of care?”. J Clin Oncol. 2009;27(32):e192-e193.
41. Kozlick D. Clinical practice guidelines and the legal standard of care: warnings, predictions, and interdisciplinary encounters. Health Law J. 2011;19:125-151.
42. Owen RR, Drummond KL, Viverito KM, et al. Monitoring and managing metabolic effects of antipsychotics: a cluster randomized trial of an intervention combining evidence-based quality improvement and external facilitation. Implement Sci. 2013;8:120.
43. Ruiz LM, Damron M, Jones KB, et al. Antipsychotic use and metabolic monitoring in individuals with developmental disabilities served in a Medicaid medical home [published online January 27, 2016]. J Autism Dev Disord. doi: 10.1007/s10803-016-2712-x.
44. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manag Care Spec Pharm. 2015;21(9):769-77,777a-777cc.
45. Wilson E, Randall C, Patterson S, et al. Monitoring and management of metabolic abnormalities: mixed-method evaluation of a successful intervention. Australas Psychiatry. 2014;22(3):248-253.
46. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
47. DelMonte MT, Bostwick JR, Bess JD, et al. Evaluation of a computer-based intervention to enhance metabolic monitoring in psychiatry inpatients treated with second-generation antipsychotics. J Clin Pharm Ther. 2012;37(6):668-673.
48. Lai CL, Chan HY, Pan YJ, et al. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry. 2015;48(1):25-29.
49. Bailey RK, Adams JB, Unger DM. Atypical antipsychotics: a case study in new era risk management. J Psychiatr Pract. 2006;12(4):253-258.
1. Mossman D, Steinberg JL. Promoting, prescribing, and pushing pills: understanding the lessons of antipsychotic drug litigation. Michigan St U J Med & Law. 2009;13:263-334.
2. Nasrallah HA, Newcomer JW. Atypical antipsychotics and metabolic dysregulation: evaluating the risk/benefit equation and improving the standard of care. J Clin Psychopharmacol. 2004;24(5 suppl 1):S7-S14.
3. De Hert M, Schreurs V, Sweers K, et al. Typical and atypical antipsychotics differentially affect long-term incidence rates of the metabolic syndrome in first-episode patients with schizophrenia: a retrospective chart review. Schizophr Res. 2008;101(1-3):295-303.
4. Appelbaum PS, Gutheil TG. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007.
5. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry. 2004;65(2):267-272.
6. Pappadopulos E, Macintyre JC II, Crismon ML, et al. Treatment recommendations for the use of antipsychotics for aggressive youth (TRAAY). Part II. J Am Acad Child Adolesc Psychiatry. 2003;42(2):145-161.
7. Pringsheim T, Panagiotopoulos C, Davidson J, et al; CAMESA guideline group. Evidence-based recommendations for monitoring safety of second generation antipsychotics in children and youth [Erratum in: J Can Acad Adolesc Psychiatry. 2011;20(3):1-2]. J Can Acad Child Adolesc Psychiatry. 2011;20(3):218-233.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Mitchell AJ, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med. 2012;42(1):125-147.
10. Baller JB, McGinty EE, Azrin ST, et al. Screening for cardiovascular risk factors in adults with serious mental illness: a review of the evidence. BMC Psychiatry. 2015;15:55.
11. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry. 2009;166(3):345-353.
12. Dhamane AD, Martin BC, Brixner DI, et al. Metabolic monitoring of patients prescribed second-generation antipsychotics. J Psychiatr Pract. 2013;19(5):360-374.
13. Morrato EH, Newcomer JW, Kamat S, et al. Metabolic screening after the American Diabetes Association’s consensus statement on antipsychotic drugs and diabetes. Diabetes Care. 2009;32(6):1037-1042.
14. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry. 2010;67(1):17-24.
15. Moeller KE, Rigler SK, Mayorga A, et al. Quality of monitoring for metabolic effects associated with second generation antipsychotics in patients with schizophrenia on public insurance. Schizophr Res. 2011;126(1-3):117-123.
16. Barnett M, VonMuenster S, Wehring H, et al. Assessment of monitoring for glucose and lipid dysregulation in adult Medi-Cal patients newly started on antipsychotics. Ann Clin Psychiatry. 2010;22(1):9-18.
17. Mittal D, Li C, Viverito K, et al. Monitoring for metabolic side effects among outpatients with dementia receiving antipsychotics. Psychiatr Serv. 2014;65(9):1147-1153.
18. Hsu C, Ried LD, Bengtson MA, et al. Metabolic monitoring in veterans with schizophrenia-related disorders and treated with second-generation antipsychotics: findings from a Veterans Affairs-based population. J Am Pharm Assoc. 2008;48(3):393-400.
19. Raebel MA, Penfold R, McMahon AW, et al. Adherence to guidelines for glucose assessment in starting second-generation antipsychotics. Pediatrics. 2014;134(5):e1308-e1314.
20. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003-2011. Psychiatr Serv. 2015;66(6):604-609.
21. Teeluckdharry S, Sharma S, O’Rourke E, et al. Monitoring metabolic side effects of atypical antipsychotics in people with an intellectual disability. J Intellect Disabil. 2013;17(3):223-235.
22. Lee J, Dalack GW, Casher MI, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second-generation antipsychotics utilizing a computer-based intervention. J Clin Pharm Ther. 2016;41(2):209-213.
23. McCourt v Abernathy, 457 SE2d 603 (SC 1995).
24. Schultz v AstraZeneca Pharma LP, LEXIS 94534, 2006 WL 3797932, (ND Cal 2006).
25. Redmond v AstraZeneca Pharma LP, 492 F Supp 2d 575 (SD Miss 2007).
26. Goguen D. Risperdal, Seroquel, Symbyax, Zyprexa, and other antipsychotic drugs. http://www.nolo.com/legal-encyclopedia/risperdal-seroquel-symbyax-zyprexa-antipsychotics-29866.html. Accessed April 4, 2016.
27. FreeAdvice staff. Risperdal medical malpractice lawsuits: Risperdal injury lawyer explains what you need to know. http://injury-law.freeadvice.com/injury-law/drug-toxic_chemicals/risperdal.htm. Accessed April 4, 2016.
28. Lewis MK, Gohagan JK, Merenstein DJ. The locality rule and the physician’s dilemma: local medical practices vs the national standard of care. JAMA. 2007;297(23):2633-2637.
29. Harris v Groth, 99 Wn2d 438, 663 P2d 113 (1983).
30. Moffett P, Moore G. The standard of care: legal history and definitions: the bad and good news. West J Emerg Med. 2011;12(1):109-112.
31. Taylor C. The use of clinical practice guidelines in determining standard of care. J Legal Med. 2014;35(2):273-290.
32. Bal BS, Brenner LH. Medicolegal sidebar: the law and social values: conformity to norms. Clin Orthop Relat Res. 2015;473(5):1555-1559.
33. Recupero PR. Clinical practice guidelines as learned treatises: understanding their use as evidence in the courtroom. J Am Acad Psychiatry Law. 2008;36(3):290-301.
34. Price v Cleveland Clinic Found, 515 NE2d 931 (Ohio Ct App 1986).
35. Zonana H. Commentary: when is a practice guideline only a guideline? J Am Acad Psychiatry Law. 2008;36(3):302-305.
36. Guillod O. Clinical guidelines and professional liability: a short comment from the legal side. ORL J Otorhinolaryngol Relat Spec. 2010;72(3):133-136; discussion 136-137.
37. American Psychiatric Association. Practice guidelines for the psychiatric evaluation of adults. 3rd ed. Arlington, VA: American Psychiatric Association; 2016.
38. Brouwers MC, Kho ME, Browman GP, et al; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842.
39. Vermaas AM. Liability in relation to the use of professional medical guidelines. Med Law. 2003;22(2):233-238.
40. Strauss DC, Thomas JM. What does the medical profession mean by “standard of care?”. J Clin Oncol. 2009;27(32):e192-e193.
41. Kozlick D. Clinical practice guidelines and the legal standard of care: warnings, predictions, and interdisciplinary encounters. Health Law J. 2011;19:125-151.
42. Owen RR, Drummond KL, Viverito KM, et al. Monitoring and managing metabolic effects of antipsychotics: a cluster randomized trial of an intervention combining evidence-based quality improvement and external facilitation. Implement Sci. 2013;8:120.
43. Ruiz LM, Damron M, Jones KB, et al. Antipsychotic use and metabolic monitoring in individuals with developmental disabilities served in a Medicaid medical home [published online January 27, 2016]. J Autism Dev Disord. doi: 10.1007/s10803-016-2712-x.
44. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manag Care Spec Pharm. 2015;21(9):769-77,777a-777cc.
45. Wilson E, Randall C, Patterson S, et al. Monitoring and management of metabolic abnormalities: mixed-method evaluation of a successful intervention. Australas Psychiatry. 2014;22(3):248-253.
46. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
47. DelMonte MT, Bostwick JR, Bess JD, et al. Evaluation of a computer-based intervention to enhance metabolic monitoring in psychiatry inpatients treated with second-generation antipsychotics. J Clin Pharm Ther. 2012;37(6):668-673.
48. Lai CL, Chan HY, Pan YJ, et al. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry. 2015;48(1):25-29.
49. Bailey RK, Adams JB, Unger DM. Atypical antipsychotics: a case study in new era risk management. J Psychiatr Pract. 2006;12(4):253-258.

Advances in transcranial magnetic stimulation for managing major depressive disorders
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).

Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
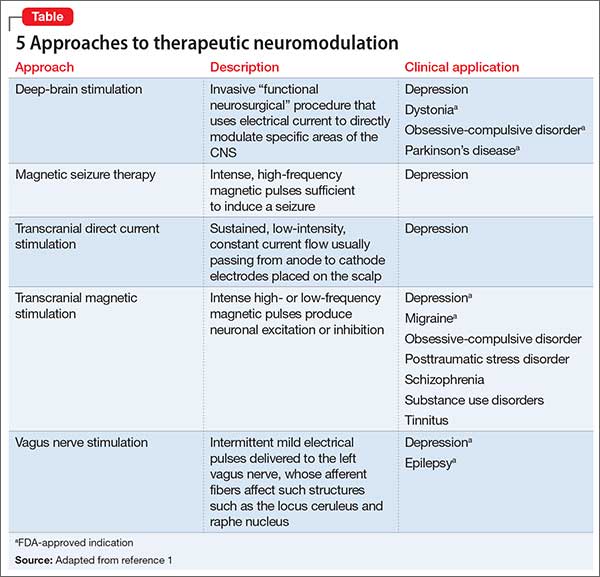
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).
Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).
Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.

How to control weight gain when prescribing antidepressants
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
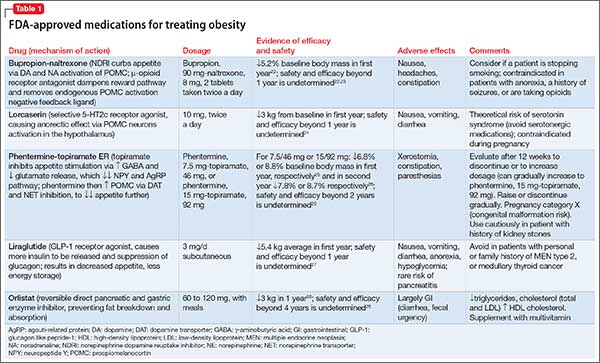
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
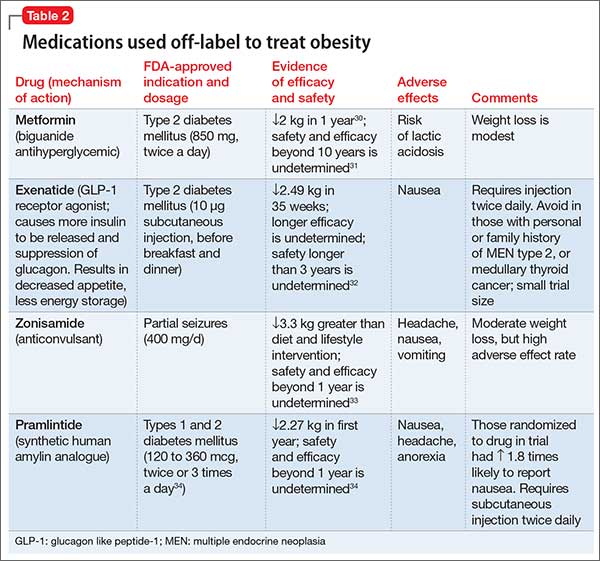
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.

Benzodiazepines might succeed as monotherapy for cannabinoid-induced catatonia
We found the case report “Unresponsive and mute after he smoked ‘Spice’” (Cases That Test Your Skills, Current Psychiatry. March 2016 p. 65-70) intriguing because we recently published an article that discusses 3 similar cases of DSM-5 unspecified catatonia.1 The diagnosis of unspecified catatonia applies to catatonia that does not fully meet criteria for either catatonic disorder associated with another mental disorder or catatonic disorder associated with another medical disorder.
In Case 3 of our article, we described a patient who presented with unspecified catatonia after smoking a synthetic cannabinoid. The patient had been diagnosed with schizophrenia 4 years prior, but had not been adherent to treatment with an antipsychotic regimen since his diagnosis. His companion reported he had been smoking “K2” before he presented to the hospital. He was admitted to acute psychiatry, treated with oral lorazepam, 1 mg, 3 times a day, and improved within 3 days. He did not require electroconvulsive therapy (ECT). We have followed this patient through electronic medical records for 2.5 years after this hospitalization. He has not presented with re-emergent signs or symptoms of catatonia.
We also have conducted a literature review of synthetic cannabinoids and catatonia. As synthetic cannabinoids are a relatively recent phenomena, we recognize that reviews on catatonia might not include such substances.2 Our literature review shows there are no other published cases of patients who returned to baseline functioning after treat<hl name="3"/>ment with benzodiazepine (ie, lorazepam) monotherapy.
It appears that, in similar cases of Cannabis-induced or synthetic cannabinoid-induced catatonia, return to baseline level of functioning before hospitalization required ECT in addition to benzodiazepines. Therefore, we feel it is important to note that in some cases, such as with our patient, Cannabis-induced catatonia might resolve with benzodiazepine monotherapy.
Shannon M. O’Connell, DO
PGY-3 Psychiatry Resident
Brendan T. Carroll, MD
Clinical Assistant Professor
Ohio University Heritage College of Osteopathic Medicine
Athens, Ohio
Chillicothe Veterans Affairs Medical Center
Chillicothe, Ohio
1. Bottoms J, Carroll BT. Unspecified catatonia: 3 cases. In Carroll BT, Spiegel DR, eds. Catatonia on the consultation liaison service and other clinical settings. New York, NY: Nova Science Publishers; 2015.
2. The Guidelines and Evidence-Based Medicine Subcommittee of the Academy of Psycho-somatic Medicine (APM); The European Association of Psychosomatic Medicine (EAPM). Catatonia in medically ill patients: evidence-based medicine (EBM) monograph for psychosomatic medicine practice. http://www.apm.org/library/monographs/catatonia/Catatonia_APM-EAPM_2015-04-17.pdf. Published April 17, 2015. Accessed May 3, 2016.
We found the case report “Unresponsive and mute after he smoked ‘Spice’” (Cases That Test Your Skills, Current Psychiatry. March 2016 p. 65-70) intriguing because we recently published an article that discusses 3 similar cases of DSM-5 unspecified catatonia.1 The diagnosis of unspecified catatonia applies to catatonia that does not fully meet criteria for either catatonic disorder associated with another mental disorder or catatonic disorder associated with another medical disorder.
In Case 3 of our article, we described a patient who presented with unspecified catatonia after smoking a synthetic cannabinoid. The patient had been diagnosed with schizophrenia 4 years prior, but had not been adherent to treatment with an antipsychotic regimen since his diagnosis. His companion reported he had been smoking “K2” before he presented to the hospital. He was admitted to acute psychiatry, treated with oral lorazepam, 1 mg, 3 times a day, and improved within 3 days. He did not require electroconvulsive therapy (ECT). We have followed this patient through electronic medical records for 2.5 years after this hospitalization. He has not presented with re-emergent signs or symptoms of catatonia.
We also have conducted a literature review of synthetic cannabinoids and catatonia. As synthetic cannabinoids are a relatively recent phenomena, we recognize that reviews on catatonia might not include such substances.2 Our literature review shows there are no other published cases of patients who returned to baseline functioning after treat<hl name="3"/>ment with benzodiazepine (ie, lorazepam) monotherapy.
It appears that, in similar cases of Cannabis-induced or synthetic cannabinoid-induced catatonia, return to baseline level of functioning before hospitalization required ECT in addition to benzodiazepines. Therefore, we feel it is important to note that in some cases, such as with our patient, Cannabis-induced catatonia might resolve with benzodiazepine monotherapy.
Shannon M. O’Connell, DO
PGY-3 Psychiatry Resident
Brendan T. Carroll, MD
Clinical Assistant Professor
Ohio University Heritage College of Osteopathic Medicine
Athens, Ohio
Chillicothe Veterans Affairs Medical Center
Chillicothe, Ohio
We found the case report “Unresponsive and mute after he smoked ‘Spice’” (Cases That Test Your Skills, Current Psychiatry. March 2016 p. 65-70) intriguing because we recently published an article that discusses 3 similar cases of DSM-5 unspecified catatonia.1 The diagnosis of unspecified catatonia applies to catatonia that does not fully meet criteria for either catatonic disorder associated with another mental disorder or catatonic disorder associated with another medical disorder.
In Case 3 of our article, we described a patient who presented with unspecified catatonia after smoking a synthetic cannabinoid. The patient had been diagnosed with schizophrenia 4 years prior, but had not been adherent to treatment with an antipsychotic regimen since his diagnosis. His companion reported he had been smoking “K2” before he presented to the hospital. He was admitted to acute psychiatry, treated with oral lorazepam, 1 mg, 3 times a day, and improved within 3 days. He did not require electroconvulsive therapy (ECT). We have followed this patient through electronic medical records for 2.5 years after this hospitalization. He has not presented with re-emergent signs or symptoms of catatonia.
We also have conducted a literature review of synthetic cannabinoids and catatonia. As synthetic cannabinoids are a relatively recent phenomena, we recognize that reviews on catatonia might not include such substances.2 Our literature review shows there are no other published cases of patients who returned to baseline functioning after treat<hl name="3"/>ment with benzodiazepine (ie, lorazepam) monotherapy.
It appears that, in similar cases of Cannabis-induced or synthetic cannabinoid-induced catatonia, return to baseline level of functioning before hospitalization required ECT in addition to benzodiazepines. Therefore, we feel it is important to note that in some cases, such as with our patient, Cannabis-induced catatonia might resolve with benzodiazepine monotherapy.
Shannon M. O’Connell, DO
PGY-3 Psychiatry Resident
Brendan T. Carroll, MD
Clinical Assistant Professor
Ohio University Heritage College of Osteopathic Medicine
Athens, Ohio
Chillicothe Veterans Affairs Medical Center
Chillicothe, Ohio
1. Bottoms J, Carroll BT. Unspecified catatonia: 3 cases. In Carroll BT, Spiegel DR, eds. Catatonia on the consultation liaison service and other clinical settings. New York, NY: Nova Science Publishers; 2015.
2. The Guidelines and Evidence-Based Medicine Subcommittee of the Academy of Psycho-somatic Medicine (APM); The European Association of Psychosomatic Medicine (EAPM). Catatonia in medically ill patients: evidence-based medicine (EBM) monograph for psychosomatic medicine practice. http://www.apm.org/library/monographs/catatonia/Catatonia_APM-EAPM_2015-04-17.pdf. Published April 17, 2015. Accessed May 3, 2016.
1. Bottoms J, Carroll BT. Unspecified catatonia: 3 cases. In Carroll BT, Spiegel DR, eds. Catatonia on the consultation liaison service and other clinical settings. New York, NY: Nova Science Publishers; 2015.
2. The Guidelines and Evidence-Based Medicine Subcommittee of the Academy of Psycho-somatic Medicine (APM); The European Association of Psychosomatic Medicine (EAPM). Catatonia in medically ill patients: evidence-based medicine (EBM) monograph for psychosomatic medicine practice. http://www.apm.org/library/monographs/catatonia/Catatonia_APM-EAPM_2015-04-17.pdf. Published April 17, 2015. Accessed May 3, 2016.

The scourge of societal anosognosia about the mentally ill
What if this increase had occurred in cardiovascular disease or cancer (both on the decline, in fact, thanks to the intense attention they receive)? I think there would have been a public outcry, followed by demands by Congress that the National Institutes of Health and the CDC address this catastrophic rise immediately. And billions of dollars would then be earmarked to prevent these 2 diseases.
How sad that society has “forgotten” that mental illness has deadly consequences, often leading to suicide (42,773 deaths in 2014 alone2—the second most common cause among people age 15 to 253)! Hundreds of thousands of people attempt suicide every year, and those who do not lose their life often end up injured or maimed. Millions who suffer depression, bipolar disorder, schizophrenia, anxiety, posttraumatic stress disorder, or a substance use disorder are at high risk of suicide, and many never receive the timely intervention that might save their life.
Our national blind spot
It is poignant that the CDC report was released in spring: The rate of suicide is highest in April and May, when the light-dark cycle is reversed. This springtime peak runs contrary to the common belief that the rate of suicide is highest during winter months. The Annual Meeting of the American Psychiatric Association convenes in May, such that, ironically, thousands of psychiatrists are away from their office exactly when their patients might need them most
Lack of attention to the high risk of suicide among all ages and both sexes is emblematic of society’s inexplicable neglect of the needs of the mentally ill. That neglect is fueled, and exacerbated, by the destructive stigma attached to brain disorders that display psychiatric symptoms. As a neuropsychiatrist, I label this neglect societal anosognosia—the same as the lack of insight seen in patients with acute schizophrenia, who are unaware of how impaired they are and insist that they are not sick. (Anosognosia also occurs in stroke patients who deny that their limb is paralyzed and insist that all is well.)
Loss of insight can have serious consequences for patients who lose the ability to monitor and evaluate their physical and mental health. Just as patients with anosognosia think they do not need help, a society that fails to attend to the mental illness of its citizens endangers their overall health and welfare.
From neglect of mental illness many hazards arise
Tens of millions of Americans suffer from mental illness, according to the National Institute of Mental Health-sponsored Epidemiologic Catchment Area Study.4 The last thing these people can afford is societal anosognosia, which deprives them of necessary and timely access to psychiatric care.
Societal anosognosia is associated with numerous hazards for persons with mental illness, including:
- Lack of compassion, which is readily available for people with a medical ailment (broken bones, cardiovascular disease, cancer).
- Lack of adequate, affordable health insurance and financial support, compared with what is available for non-psychiatric disorders.
- Shortage of publicly funded programs and mental health practitioners to provide prevention and intervention for those who consider ending their life during an episode of depression, psychosis, stress, or a panic attack.
- Allowing the stigma to continue unabated. Why are there strict laws about hate crimes, but not about stigma? Why does society continue to portray depression and anxiety as a personal weakness or failure, while patients with Parkinson’s disease or multiple sclerosis who have motor weakness are not stigmatized for their physical deficits?
- Transforming the seriously mentally ill into felons by arresting and jailing them because of erratic behavior—instead of hospitalizing them for the medical care they need. The transinstitutionalization of the mentally ill—from state hospitals to prisons—is one of the most shameful consequences of societal anosognosia, burdening our patients with the dual stigma of being a criminal and mentally ill.
- Turning a blind eye to abuses by insurance companies. More appalling is the perpetuation of restricted health coverage despite the passage of parity laws! Why are sensory and motor disorders of brain lesions covered fully, while the thought, emotional, and behavior disorders of the brain covered only partially?
- Consent laws that restrict psychiatrists from medicating acutely psychotic or depressed patients unless they consent—but no laws that restrict a cardiologist from immediately treating an unconscious heart attack patient who cannot consent, or an obtunded stroke patient who cannot communicate? The duration of untreated psychosis or depression has been shown repeatedly to have deleterious effects on brain tissue and functional outcomes, yet treatment of an acutely ill psychiatric patient is often delayed until a court order is obtained. When was the last time a court order was needed to treat an acute myocardial infarction?
- Failure to recognize that premature mortality (by approximately 25 years) is a devastating consequence of mental illness, whether from suicide or cardiometabolic risk factors due to smoking, substance use (often used to self-medicate because proper treatment is lacking), poor diet, and sedentary living.
- Failure to provide basic primary care to people with severe mental illness, and the much lower use of life-saving diagnostic and treatment procedures offered to these patients, compared with non-psychiatric patients.
- Inadequate funding for research on psychiatric disorders, compared with other medical disorders—even though direct and indirect costs of mental illness to society (hundreds of billions of dollars a year) far exceed costs of most medical disorders.
- Severe shortage of rehabilitation programs for the mentally ill, compared with many other medical disorders. Why does paralysis of the mind receive far less support than paralysis of the legs or arms?
The rising suicide rate reflects poorly on us
Societal anosognosia is a global scourge, affecting many underdeveloped countries. Why do developed nations, like ours, have the same blind spot for mental illness? Might ignorance and discrimination be universal?
The tragic rise in the rate of death by suicide in men and women, among all age groups, year after year, is stunningly incongruent when juxtaposed against the elimination of smallpox and other communicable diseases through a concerted societal effort to support scientific advances in vaccine development. Societal anosognosia appears to be selective: We have comprehensive insight about diseases of the body but not diseases of the mind.
The essence and soul of a society are the collective minds of its citizens, not their bodies. Societal anosognosia is a serious dysfunction of its mind, and a rising suicide rate is a symptom of that pathological dysfunction.
1. Curtin SC, Warner M, Hedegaard H, et al. Increase in suicide in the United States, 1999-2014. National Center for Health Statistics Data Brief No. 241. Atlanta, GA: National Center for Health Statistics, U.S. Department of Health and Human Services; 2016.
2. Ten leading causes of death by age group, United States – 2014. Centers for Disease Control and Prevention. http://www.cdc.gov/injury/images/lc-charts/leading_causes_of_death_age_group_2014_1050w760h.gif. Accessed May 20, 2016.
3. Morris M. Stemming the rising tide of suicide. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/specialty-focus/depression/single-article-page/stemming-the-rising-tide-of-suicide/01cd45cabfc693bedb0e30bb6cb0b89e.html. Published April 26, 2016. Accessed May 13, 2016.
4. Robins LN, Regier DA, eds. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: Free Press; 1990.
What if this increase had occurred in cardiovascular disease or cancer (both on the decline, in fact, thanks to the intense attention they receive)? I think there would have been a public outcry, followed by demands by Congress that the National Institutes of Health and the CDC address this catastrophic rise immediately. And billions of dollars would then be earmarked to prevent these 2 diseases.
How sad that society has “forgotten” that mental illness has deadly consequences, often leading to suicide (42,773 deaths in 2014 alone2—the second most common cause among people age 15 to 253)! Hundreds of thousands of people attempt suicide every year, and those who do not lose their life often end up injured or maimed. Millions who suffer depression, bipolar disorder, schizophrenia, anxiety, posttraumatic stress disorder, or a substance use disorder are at high risk of suicide, and many never receive the timely intervention that might save their life.
Our national blind spot
It is poignant that the CDC report was released in spring: The rate of suicide is highest in April and May, when the light-dark cycle is reversed. This springtime peak runs contrary to the common belief that the rate of suicide is highest during winter months. The Annual Meeting of the American Psychiatric Association convenes in May, such that, ironically, thousands of psychiatrists are away from their office exactly when their patients might need them most
Lack of attention to the high risk of suicide among all ages and both sexes is emblematic of society’s inexplicable neglect of the needs of the mentally ill. That neglect is fueled, and exacerbated, by the destructive stigma attached to brain disorders that display psychiatric symptoms. As a neuropsychiatrist, I label this neglect societal anosognosia—the same as the lack of insight seen in patients with acute schizophrenia, who are unaware of how impaired they are and insist that they are not sick. (Anosognosia also occurs in stroke patients who deny that their limb is paralyzed and insist that all is well.)
Loss of insight can have serious consequences for patients who lose the ability to monitor and evaluate their physical and mental health. Just as patients with anosognosia think they do not need help, a society that fails to attend to the mental illness of its citizens endangers their overall health and welfare.
From neglect of mental illness many hazards arise
Tens of millions of Americans suffer from mental illness, according to the National Institute of Mental Health-sponsored Epidemiologic Catchment Area Study.4 The last thing these people can afford is societal anosognosia, which deprives them of necessary and timely access to psychiatric care.
Societal anosognosia is associated with numerous hazards for persons with mental illness, including:
- Lack of compassion, which is readily available for people with a medical ailment (broken bones, cardiovascular disease, cancer).
- Lack of adequate, affordable health insurance and financial support, compared with what is available for non-psychiatric disorders.
- Shortage of publicly funded programs and mental health practitioners to provide prevention and intervention for those who consider ending their life during an episode of depression, psychosis, stress, or a panic attack.
- Allowing the stigma to continue unabated. Why are there strict laws about hate crimes, but not about stigma? Why does society continue to portray depression and anxiety as a personal weakness or failure, while patients with Parkinson’s disease or multiple sclerosis who have motor weakness are not stigmatized for their physical deficits?
- Transforming the seriously mentally ill into felons by arresting and jailing them because of erratic behavior—instead of hospitalizing them for the medical care they need. The transinstitutionalization of the mentally ill—from state hospitals to prisons—is one of the most shameful consequences of societal anosognosia, burdening our patients with the dual stigma of being a criminal and mentally ill.
- Turning a blind eye to abuses by insurance companies. More appalling is the perpetuation of restricted health coverage despite the passage of parity laws! Why are sensory and motor disorders of brain lesions covered fully, while the thought, emotional, and behavior disorders of the brain covered only partially?
- Consent laws that restrict psychiatrists from medicating acutely psychotic or depressed patients unless they consent—but no laws that restrict a cardiologist from immediately treating an unconscious heart attack patient who cannot consent, or an obtunded stroke patient who cannot communicate? The duration of untreated psychosis or depression has been shown repeatedly to have deleterious effects on brain tissue and functional outcomes, yet treatment of an acutely ill psychiatric patient is often delayed until a court order is obtained. When was the last time a court order was needed to treat an acute myocardial infarction?
- Failure to recognize that premature mortality (by approximately 25 years) is a devastating consequence of mental illness, whether from suicide or cardiometabolic risk factors due to smoking, substance use (often used to self-medicate because proper treatment is lacking), poor diet, and sedentary living.
- Failure to provide basic primary care to people with severe mental illness, and the much lower use of life-saving diagnostic and treatment procedures offered to these patients, compared with non-psychiatric patients.
- Inadequate funding for research on psychiatric disorders, compared with other medical disorders—even though direct and indirect costs of mental illness to society (hundreds of billions of dollars a year) far exceed costs of most medical disorders.
- Severe shortage of rehabilitation programs for the mentally ill, compared with many other medical disorders. Why does paralysis of the mind receive far less support than paralysis of the legs or arms?
The rising suicide rate reflects poorly on us
Societal anosognosia is a global scourge, affecting many underdeveloped countries. Why do developed nations, like ours, have the same blind spot for mental illness? Might ignorance and discrimination be universal?
The tragic rise in the rate of death by suicide in men and women, among all age groups, year after year, is stunningly incongruent when juxtaposed against the elimination of smallpox and other communicable diseases through a concerted societal effort to support scientific advances in vaccine development. Societal anosognosia appears to be selective: We have comprehensive insight about diseases of the body but not diseases of the mind.
The essence and soul of a society are the collective minds of its citizens, not their bodies. Societal anosognosia is a serious dysfunction of its mind, and a rising suicide rate is a symptom of that pathological dysfunction.
What if this increase had occurred in cardiovascular disease or cancer (both on the decline, in fact, thanks to the intense attention they receive)? I think there would have been a public outcry, followed by demands by Congress that the National Institutes of Health and the CDC address this catastrophic rise immediately. And billions of dollars would then be earmarked to prevent these 2 diseases.
How sad that society has “forgotten” that mental illness has deadly consequences, often leading to suicide (42,773 deaths in 2014 alone2—the second most common cause among people age 15 to 253)! Hundreds of thousands of people attempt suicide every year, and those who do not lose their life often end up injured or maimed. Millions who suffer depression, bipolar disorder, schizophrenia, anxiety, posttraumatic stress disorder, or a substance use disorder are at high risk of suicide, and many never receive the timely intervention that might save their life.
Our national blind spot
It is poignant that the CDC report was released in spring: The rate of suicide is highest in April and May, when the light-dark cycle is reversed. This springtime peak runs contrary to the common belief that the rate of suicide is highest during winter months. The Annual Meeting of the American Psychiatric Association convenes in May, such that, ironically, thousands of psychiatrists are away from their office exactly when their patients might need them most
Lack of attention to the high risk of suicide among all ages and both sexes is emblematic of society’s inexplicable neglect of the needs of the mentally ill. That neglect is fueled, and exacerbated, by the destructive stigma attached to brain disorders that display psychiatric symptoms. As a neuropsychiatrist, I label this neglect societal anosognosia—the same as the lack of insight seen in patients with acute schizophrenia, who are unaware of how impaired they are and insist that they are not sick. (Anosognosia also occurs in stroke patients who deny that their limb is paralyzed and insist that all is well.)
Loss of insight can have serious consequences for patients who lose the ability to monitor and evaluate their physical and mental health. Just as patients with anosognosia think they do not need help, a society that fails to attend to the mental illness of its citizens endangers their overall health and welfare.
From neglect of mental illness many hazards arise
Tens of millions of Americans suffer from mental illness, according to the National Institute of Mental Health-sponsored Epidemiologic Catchment Area Study.4 The last thing these people can afford is societal anosognosia, which deprives them of necessary and timely access to psychiatric care.
Societal anosognosia is associated with numerous hazards for persons with mental illness, including:
- Lack of compassion, which is readily available for people with a medical ailment (broken bones, cardiovascular disease, cancer).
- Lack of adequate, affordable health insurance and financial support, compared with what is available for non-psychiatric disorders.
- Shortage of publicly funded programs and mental health practitioners to provide prevention and intervention for those who consider ending their life during an episode of depression, psychosis, stress, or a panic attack.
- Allowing the stigma to continue unabated. Why are there strict laws about hate crimes, but not about stigma? Why does society continue to portray depression and anxiety as a personal weakness or failure, while patients with Parkinson’s disease or multiple sclerosis who have motor weakness are not stigmatized for their physical deficits?
- Transforming the seriously mentally ill into felons by arresting and jailing them because of erratic behavior—instead of hospitalizing them for the medical care they need. The transinstitutionalization of the mentally ill—from state hospitals to prisons—is one of the most shameful consequences of societal anosognosia, burdening our patients with the dual stigma of being a criminal and mentally ill.
- Turning a blind eye to abuses by insurance companies. More appalling is the perpetuation of restricted health coverage despite the passage of parity laws! Why are sensory and motor disorders of brain lesions covered fully, while the thought, emotional, and behavior disorders of the brain covered only partially?
- Consent laws that restrict psychiatrists from medicating acutely psychotic or depressed patients unless they consent—but no laws that restrict a cardiologist from immediately treating an unconscious heart attack patient who cannot consent, or an obtunded stroke patient who cannot communicate? The duration of untreated psychosis or depression has been shown repeatedly to have deleterious effects on brain tissue and functional outcomes, yet treatment of an acutely ill psychiatric patient is often delayed until a court order is obtained. When was the last time a court order was needed to treat an acute myocardial infarction?
- Failure to recognize that premature mortality (by approximately 25 years) is a devastating consequence of mental illness, whether from suicide or cardiometabolic risk factors due to smoking, substance use (often used to self-medicate because proper treatment is lacking), poor diet, and sedentary living.
- Failure to provide basic primary care to people with severe mental illness, and the much lower use of life-saving diagnostic and treatment procedures offered to these patients, compared with non-psychiatric patients.
- Inadequate funding for research on psychiatric disorders, compared with other medical disorders—even though direct and indirect costs of mental illness to society (hundreds of billions of dollars a year) far exceed costs of most medical disorders.
- Severe shortage of rehabilitation programs for the mentally ill, compared with many other medical disorders. Why does paralysis of the mind receive far less support than paralysis of the legs or arms?
The rising suicide rate reflects poorly on us
Societal anosognosia is a global scourge, affecting many underdeveloped countries. Why do developed nations, like ours, have the same blind spot for mental illness? Might ignorance and discrimination be universal?
The tragic rise in the rate of death by suicide in men and women, among all age groups, year after year, is stunningly incongruent when juxtaposed against the elimination of smallpox and other communicable diseases through a concerted societal effort to support scientific advances in vaccine development. Societal anosognosia appears to be selective: We have comprehensive insight about diseases of the body but not diseases of the mind.
The essence and soul of a society are the collective minds of its citizens, not their bodies. Societal anosognosia is a serious dysfunction of its mind, and a rising suicide rate is a symptom of that pathological dysfunction.
1. Curtin SC, Warner M, Hedegaard H, et al. Increase in suicide in the United States, 1999-2014. National Center for Health Statistics Data Brief No. 241. Atlanta, GA: National Center for Health Statistics, U.S. Department of Health and Human Services; 2016.
2. Ten leading causes of death by age group, United States – 2014. Centers for Disease Control and Prevention. http://www.cdc.gov/injury/images/lc-charts/leading_causes_of_death_age_group_2014_1050w760h.gif. Accessed May 20, 2016.
3. Morris M. Stemming the rising tide of suicide. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/specialty-focus/depression/single-article-page/stemming-the-rising-tide-of-suicide/01cd45cabfc693bedb0e30bb6cb0b89e.html. Published April 26, 2016. Accessed May 13, 2016.
4. Robins LN, Regier DA, eds. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: Free Press; 1990.
1. Curtin SC, Warner M, Hedegaard H, et al. Increase in suicide in the United States, 1999-2014. National Center for Health Statistics Data Brief No. 241. Atlanta, GA: National Center for Health Statistics, U.S. Department of Health and Human Services; 2016.
2. Ten leading causes of death by age group, United States – 2014. Centers for Disease Control and Prevention. http://www.cdc.gov/injury/images/lc-charts/leading_causes_of_death_age_group_2014_1050w760h.gif. Accessed May 20, 2016.
3. Morris M. Stemming the rising tide of suicide. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/specialty-focus/depression/single-article-page/stemming-the-rising-tide-of-suicide/01cd45cabfc693bedb0e30bb6cb0b89e.html. Published April 26, 2016. Accessed May 13, 2016.
4. Robins LN, Regier DA, eds. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: Free Press; 1990.