User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Assessing tremor to rule out psychogenic origin: It’s tricky
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.

Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3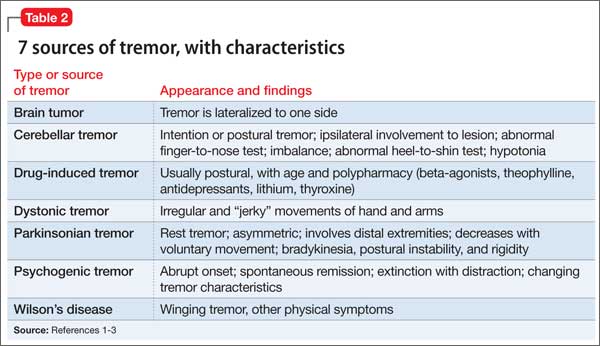
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.

Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.
Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.
Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.
Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.
Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
Suvorexant for sleep-onset insomnia or sleep-maintenance insomnia, or both
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2

Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Akathisia: Is restlessness a primary condition or an adverse drug effect?
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112). 
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
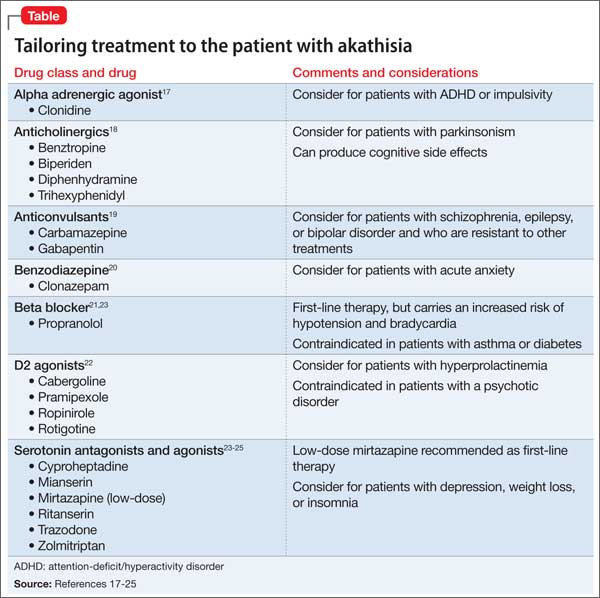
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.

From bedlam to biomarkers: The transformation of psychiatry’s terminology reflects its 4 conceptual earthquakes
Consider here my journey in psychiatry since my adolescence. Growing up in the 1960s and 1970s, I did not watch much television; my father was convinced TV would be “too distracting” for us children. At first, I was angry about his rule, and would occasionally watch programs such as Bonanza at sleep-overs.
The lure of psychoanalysis
Gradually, I became grateful to my father because—in contrast to my classmates, who sat passively for hours watching TV after school—I voraciously read the piles of fiction and nonfiction books that I checked out from the school library every week, expanding my general knowledge and perspectives. One of my favorite genres became psychology and psychiatry, including many of Sigmund Freud’s works.
I was enchanted by psychoanalysis and its explanation of mental illness because, growing up, I had been told that madness is caused by demonic spirits and bad behavior and it is completely untreatable. By the time I was in high school, I had decided to become a psychiatrist, and was practicing what I read by “counseling” my classmates about family conflicts, raging drives, and frustrating relationships with girlfriends.
Rising tide of psychopharmacology
My love for psychiatry never wavered during my undergraduate years. I focused not only on required pre-med courses but enthusiastically took many psychology, sociology, and anthropology electives to expand my understanding of human behavior. In medical school, I enjoyed all rotations, but psychiatry was simply sublime. Often, I offered (to my classmates’ delight) to take their weekend call at the psychiatric hospital so I could see more patients.
After my internship, I married my wife (a behavioral psychologist) and embarked on psychiatry residency training with gusto. I was far better prepared, I realized, than my fellow residents; my faculty supervisors noticed that I answered questions more often than many others during rounds and lectures. (Thanks, Dad, for banning television!) I relished every psychotherapy session and spent hours listening to audiotapes of my patients’ sessions to improve my skills and to discover the psychodynamic nuances of their psychopathology. Being supervised by expert psychoanalysts was the highlight of my week as I honed my psychodynamic psychotherapy skills.
But something interesting happened during my residency: Psychopharmacology and electroconvulsive therapy were helping my severely ill psychotic, manic, and depressed patients much faster than psychotherapy could. Length of stay in the wards typically was 30 days (there was no managed care back then to limit stay to an absurd 5 days), and I saw substantial improvement in many of my patients before discharge.
I was so enthralled by the rising tide of psychopharmacology that I decided in PGY-2 to conduct psychopharmacology research—which, I came to realize, was easier than research on psychotherapy. I secured a mentor from the department of pharmacology. In PGY-3, I presented my data at the Annual Meeting of the American Psychiatric Association; in PGY-4, the paper was published in the American Journal of Psychiatry.
By the end of residency, I had applied to the National Institute of Mental Health (NIMH) to pursue a research fellowship in the neuropharmacology of schizophrenia to prepare me for an academic career. I participated in numerous studies on the NIMH research ward, brimming with patients who had refractory schizophrenia (before the advent of clozapine in 1989), and I published many articles with mentors and fellow researchers.
Investigating brain biology
Then another funny thing happened: During my fellowship, one of my mentors shared with me some early studies about postmortem structural changes in the brain of schizophrenia patients. That prompted me to spend hours in the basement of the pathology department examining the brains of dozens of patients with schizophrenia, noting atrophic changes and performing measurements and histopathologic studies.
Consequently, I embarked on neuroimaging research to study the morphological abnormalities of cortical and subcortical regions in living patients. I found myself going beyond neuropsychopharmacology and diving into neuroanatomy books and neuroscience journals. I realized that I was continuously learning and using a new scientific language in my daily work.
After I left NIMH to begin a career of teaching, research, and patient care in a medical school setting, I was engulfed by meteoric advances in neuroscience producing unprecedented insights about the molecular biology of schizophrenia and other severe neuropsychiatric disorders, leading me to pursue new opportunities in neurobiology while continuing my psychopharmacology research.
The rate of transformation is mind-boggling
Looking back at the span of time from childhood through the exciting journey of my psychiatry career, I realize how massive a transformation I have witnessed and experienced. The specialty has shifted its clinical and scientific paradigms through several conceptual models—from demonic possession to psychoanalysis to psychopharmacology and, last, to molecular neurobiology. Four times in my life, the lexicon of psychiatry has undergone a complete makeover. This is a light-speed pace of scientific progress over a few decades—truly breathtaking! It’s like rewriting a dictionary over and over, with no 2 successive editions resembling each other whatsoever.
The Table shows 4 sets of examples of psychiatric terminology, each representing 1 of the 4 paradigmatic models that my generation of psychiatrists has had to adopt and use in clinical care and research. I cannot think of any other medical specialty that has come close to evolving and transforming its language and conceptual models of etiology and treatment at such a rapid pace.
When I embraced psychiatry in adolescence as my future career, I never imagined, in my wildest dreams, that I would experience such successive scientific earthquakes in my beloved medical specialty. Perhaps that’s what kept me stimulated and eager to come to work every day; I use all the models and treatment tools I have learned in understanding and helping my patients with evolving psychotherapeutic and biopharmaceutical tools; I also teach my students and residents about the multifaceted wonders of the human mind and the magnificent complexities of the brain in health and disease.
Psychiatry has been, and will continue to be, a Pandora’s box of medicine, full of stunning scientific twists and surprises and a transformative lexicon to match.
Consider here my journey in psychiatry since my adolescence. Growing up in the 1960s and 1970s, I did not watch much television; my father was convinced TV would be “too distracting” for us children. At first, I was angry about his rule, and would occasionally watch programs such as Bonanza at sleep-overs.
The lure of psychoanalysis
Gradually, I became grateful to my father because—in contrast to my classmates, who sat passively for hours watching TV after school—I voraciously read the piles of fiction and nonfiction books that I checked out from the school library every week, expanding my general knowledge and perspectives. One of my favorite genres became psychology and psychiatry, including many of Sigmund Freud’s works.
I was enchanted by psychoanalysis and its explanation of mental illness because, growing up, I had been told that madness is caused by demonic spirits and bad behavior and it is completely untreatable. By the time I was in high school, I had decided to become a psychiatrist, and was practicing what I read by “counseling” my classmates about family conflicts, raging drives, and frustrating relationships with girlfriends.
Rising tide of psychopharmacology
My love for psychiatry never wavered during my undergraduate years. I focused not only on required pre-med courses but enthusiastically took many psychology, sociology, and anthropology electives to expand my understanding of human behavior. In medical school, I enjoyed all rotations, but psychiatry was simply sublime. Often, I offered (to my classmates’ delight) to take their weekend call at the psychiatric hospital so I could see more patients.
After my internship, I married my wife (a behavioral psychologist) and embarked on psychiatry residency training with gusto. I was far better prepared, I realized, than my fellow residents; my faculty supervisors noticed that I answered questions more often than many others during rounds and lectures. (Thanks, Dad, for banning television!) I relished every psychotherapy session and spent hours listening to audiotapes of my patients’ sessions to improve my skills and to discover the psychodynamic nuances of their psychopathology. Being supervised by expert psychoanalysts was the highlight of my week as I honed my psychodynamic psychotherapy skills.
But something interesting happened during my residency: Psychopharmacology and electroconvulsive therapy were helping my severely ill psychotic, manic, and depressed patients much faster than psychotherapy could. Length of stay in the wards typically was 30 days (there was no managed care back then to limit stay to an absurd 5 days), and I saw substantial improvement in many of my patients before discharge.
I was so enthralled by the rising tide of psychopharmacology that I decided in PGY-2 to conduct psychopharmacology research—which, I came to realize, was easier than research on psychotherapy. I secured a mentor from the department of pharmacology. In PGY-3, I presented my data at the Annual Meeting of the American Psychiatric Association; in PGY-4, the paper was published in the American Journal of Psychiatry.
By the end of residency, I had applied to the National Institute of Mental Health (NIMH) to pursue a research fellowship in the neuropharmacology of schizophrenia to prepare me for an academic career. I participated in numerous studies on the NIMH research ward, brimming with patients who had refractory schizophrenia (before the advent of clozapine in 1989), and I published many articles with mentors and fellow researchers.
Investigating brain biology
Then another funny thing happened: During my fellowship, one of my mentors shared with me some early studies about postmortem structural changes in the brain of schizophrenia patients. That prompted me to spend hours in the basement of the pathology department examining the brains of dozens of patients with schizophrenia, noting atrophic changes and performing measurements and histopathologic studies.
Consequently, I embarked on neuroimaging research to study the morphological abnormalities of cortical and subcortical regions in living patients. I found myself going beyond neuropsychopharmacology and diving into neuroanatomy books and neuroscience journals. I realized that I was continuously learning and using a new scientific language in my daily work.
After I left NIMH to begin a career of teaching, research, and patient care in a medical school setting, I was engulfed by meteoric advances in neuroscience producing unprecedented insights about the molecular biology of schizophrenia and other severe neuropsychiatric disorders, leading me to pursue new opportunities in neurobiology while continuing my psychopharmacology research.
The rate of transformation is mind-boggling
Looking back at the span of time from childhood through the exciting journey of my psychiatry career, I realize how massive a transformation I have witnessed and experienced. The specialty has shifted its clinical and scientific paradigms through several conceptual models—from demonic possession to psychoanalysis to psychopharmacology and, last, to molecular neurobiology. Four times in my life, the lexicon of psychiatry has undergone a complete makeover. This is a light-speed pace of scientific progress over a few decades—truly breathtaking! It’s like rewriting a dictionary over and over, with no 2 successive editions resembling each other whatsoever.
The Table shows 4 sets of examples of psychiatric terminology, each representing 1 of the 4 paradigmatic models that my generation of psychiatrists has had to adopt and use in clinical care and research. I cannot think of any other medical specialty that has come close to evolving and transforming its language and conceptual models of etiology and treatment at such a rapid pace.
When I embraced psychiatry in adolescence as my future career, I never imagined, in my wildest dreams, that I would experience such successive scientific earthquakes in my beloved medical specialty. Perhaps that’s what kept me stimulated and eager to come to work every day; I use all the models and treatment tools I have learned in understanding and helping my patients with evolving psychotherapeutic and biopharmaceutical tools; I also teach my students and residents about the multifaceted wonders of the human mind and the magnificent complexities of the brain in health and disease.
Psychiatry has been, and will continue to be, a Pandora’s box of medicine, full of stunning scientific twists and surprises and a transformative lexicon to match.
Consider here my journey in psychiatry since my adolescence. Growing up in the 1960s and 1970s, I did not watch much television; my father was convinced TV would be “too distracting” for us children. At first, I was angry about his rule, and would occasionally watch programs such as Bonanza at sleep-overs.
The lure of psychoanalysis
Gradually, I became grateful to my father because—in contrast to my classmates, who sat passively for hours watching TV after school—I voraciously read the piles of fiction and nonfiction books that I checked out from the school library every week, expanding my general knowledge and perspectives. One of my favorite genres became psychology and psychiatry, including many of Sigmund Freud’s works.
I was enchanted by psychoanalysis and its explanation of mental illness because, growing up, I had been told that madness is caused by demonic spirits and bad behavior and it is completely untreatable. By the time I was in high school, I had decided to become a psychiatrist, and was practicing what I read by “counseling” my classmates about family conflicts, raging drives, and frustrating relationships with girlfriends.
Rising tide of psychopharmacology
My love for psychiatry never wavered during my undergraduate years. I focused not only on required pre-med courses but enthusiastically took many psychology, sociology, and anthropology electives to expand my understanding of human behavior. In medical school, I enjoyed all rotations, but psychiatry was simply sublime. Often, I offered (to my classmates’ delight) to take their weekend call at the psychiatric hospital so I could see more patients.
After my internship, I married my wife (a behavioral psychologist) and embarked on psychiatry residency training with gusto. I was far better prepared, I realized, than my fellow residents; my faculty supervisors noticed that I answered questions more often than many others during rounds and lectures. (Thanks, Dad, for banning television!) I relished every psychotherapy session and spent hours listening to audiotapes of my patients’ sessions to improve my skills and to discover the psychodynamic nuances of their psychopathology. Being supervised by expert psychoanalysts was the highlight of my week as I honed my psychodynamic psychotherapy skills.
But something interesting happened during my residency: Psychopharmacology and electroconvulsive therapy were helping my severely ill psychotic, manic, and depressed patients much faster than psychotherapy could. Length of stay in the wards typically was 30 days (there was no managed care back then to limit stay to an absurd 5 days), and I saw substantial improvement in many of my patients before discharge.
I was so enthralled by the rising tide of psychopharmacology that I decided in PGY-2 to conduct psychopharmacology research—which, I came to realize, was easier than research on psychotherapy. I secured a mentor from the department of pharmacology. In PGY-3, I presented my data at the Annual Meeting of the American Psychiatric Association; in PGY-4, the paper was published in the American Journal of Psychiatry.
By the end of residency, I had applied to the National Institute of Mental Health (NIMH) to pursue a research fellowship in the neuropharmacology of schizophrenia to prepare me for an academic career. I participated in numerous studies on the NIMH research ward, brimming with patients who had refractory schizophrenia (before the advent of clozapine in 1989), and I published many articles with mentors and fellow researchers.
Investigating brain biology
Then another funny thing happened: During my fellowship, one of my mentors shared with me some early studies about postmortem structural changes in the brain of schizophrenia patients. That prompted me to spend hours in the basement of the pathology department examining the brains of dozens of patients with schizophrenia, noting atrophic changes and performing measurements and histopathologic studies.
Consequently, I embarked on neuroimaging research to study the morphological abnormalities of cortical and subcortical regions in living patients. I found myself going beyond neuropsychopharmacology and diving into neuroanatomy books and neuroscience journals. I realized that I was continuously learning and using a new scientific language in my daily work.
After I left NIMH to begin a career of teaching, research, and patient care in a medical school setting, I was engulfed by meteoric advances in neuroscience producing unprecedented insights about the molecular biology of schizophrenia and other severe neuropsychiatric disorders, leading me to pursue new opportunities in neurobiology while continuing my psychopharmacology research.
The rate of transformation is mind-boggling
Looking back at the span of time from childhood through the exciting journey of my psychiatry career, I realize how massive a transformation I have witnessed and experienced. The specialty has shifted its clinical and scientific paradigms through several conceptual models—from demonic possession to psychoanalysis to psychopharmacology and, last, to molecular neurobiology. Four times in my life, the lexicon of psychiatry has undergone a complete makeover. This is a light-speed pace of scientific progress over a few decades—truly breathtaking! It’s like rewriting a dictionary over and over, with no 2 successive editions resembling each other whatsoever.
The Table shows 4 sets of examples of psychiatric terminology, each representing 1 of the 4 paradigmatic models that my generation of psychiatrists has had to adopt and use in clinical care and research. I cannot think of any other medical specialty that has come close to evolving and transforming its language and conceptual models of etiology and treatment at such a rapid pace.
When I embraced psychiatry in adolescence as my future career, I never imagined, in my wildest dreams, that I would experience such successive scientific earthquakes in my beloved medical specialty. Perhaps that’s what kept me stimulated and eager to come to work every day; I use all the models and treatment tools I have learned in understanding and helping my patients with evolving psychotherapeutic and biopharmaceutical tools; I also teach my students and residents about the multifaceted wonders of the human mind and the magnificent complexities of the brain in health and disease.
Psychiatry has been, and will continue to be, a Pandora’s box of medicine, full of stunning scientific twists and surprises and a transformative lexicon to match.
Here’s what we can do to minimize the daily hassle of prior authorizations
Insistence on prior authorization (PA) when prescribing certain pharmaceuticals has grown considerably over the past 5 years. Most requests for PA are issued by pharmacy benefit management (PBM) companies that have been contracted by an insurer. A PA can be triggered when a physician orders:
• a brand-name medication
• a medication not on the formulary of the PBM
• a quantity above an arbitrary ceiling
• a medication that has multiple indications (that is, the PBM won’t pay for indication X but will pay for indication Y).
What are the problems caused by PAs? I outline a number of them, and their potential consequences, in this “Commentary.” What can we do, in our practices, to lessen the disruption they cause and the time and money they cost us (Box)? 
’Prior authorization’—a misleading, disingenuous term
The physician’s prescription is legal authorization for the patient to receive the medicine. It would be more accurate if PBMs labeled what they do “prior approval for reimbursement.”
PBMs exist to manipulate and coordinate the demand for medication generated, on one hand, by patients and their physician and, on the other, by the cost of supplying portion of that demand. The cost of a medication to the PBM is controlled by:
• negotiating rebates with drug manufacturers
• advantageous contracting with pharmacies
• denying payment, when feasible, using the PA system.
The goal of the PA is to boost the profits of the PBM—not to pay for the best fit between the needs of the patient and the medications available, as determined by the treating physician.
The games begin!
The PA process usually begins when the patient goes to the pharmacy, prescription in hand, and gives it to the pharmacist, who enters it into the computer. At that point, if the PBM has put a block on paying for the medication, 3 things happen in sequence:
1. The computer alerts the pharmacist about the block (or that a higher copay is required).
2. The pharmacist tells the patient something about the block—although not necessarily the whole story.
3. The pharmacist tells the physician’s office (by fax, e-mail, or telephone) that the PBM wants authorization and that the physician must call a toll-free telephone number to obtain that authorization.
The physician’s office then makes the initial call to the PBM. That call can take 10 to 20 minutes, answering preliminary questions. The call generates a questionnaire from the PBM that is faxed to the office, filled with questions that one could characterize as loaded. The questionnaire is intended to provide grounds for disapproval or approval—not to obtain in-depth understanding of the individual patient’s needs.
Playing pieces on a chessboard
Note that the physician and pharmacist, thrust unwillingly into the middle of this gambit, spend considerable uncompensated time on the PA process. (Primary care physicians and their nursing and clerical staff, spent, on average, 19.8 hours a week obtaining PAs in 2006.1)
PBMs have shifted responsibility for communication to physicians and pharmacists by requiring that the physician always contact the PBM. A PBM will not contact a physician directly, either to begin the PA or ask questions during the process.
If the request for authorization is denied, what’s the outcome? The physician’s office and the pharmacist have spent uncompensated time taking action that resulted in the PBM and the insurer improving their bottom line without benefit to anyone else.
Communication breakdown. The cumbersome, multistep PA process opens the door to miscommunication. This happens often, I’ve found: The physician wastes time because the pharmacist passed along an incomplete message, or a patient gives vague or confusing information in trying to transmit what the pharmacist said. Sometimes, when physicians get through to a live person at the PBM, they are told that the pharmacist misinformed the office: No, the medication didn’t require PA after all.
Why can’t PBMs streamline the process, sparing busy physicians’ offices the time spent on initial telephone calls, by installing software that would allow the pharmacist who first encounters a payment block to, with a few keystrokes, instantly send the relevant questionnaire to the physician’s fax machine or computer?
Obstacles to satisfaction
From the perspective of the patient, the word that probably best characterizes his emotional response to the PA process is “helpless.” He wasn’t expecting a denial; it’s likely that he hadn’t been fully or clearly informed at the time he selected the insurer that he might someday face such an obstacle. Even though he had a legal prescription, written by a physician, any attempt to go back to the insurer or the PBM to complain is rarely successful. If he tried, he would likely get no satisfaction: The clerk at the other end of the telephone would swiftly inform him that there were a number of complicated rules, policies, or “step programs” that must be adhered to before the PBM pays for a prescription.
Even if the medication is covered, the patient might be told that there are “quantity limits” that prevent reimbursement for the prescription as written—limits that were not made explicit when he signed up for the insurance plan. All these obstacles can generate confusion, anxiety, frustration, and anger—understandably so.
The ‘safety’ catch. Obstacles do not necessarily end when the medication is approved; such approval is merely a “coverage eligibility review.” In addition, PBMs make it clear that every prescription also undergoes a so-called safety review by a pharmacist before it is dispensed. If the PBM’s pharmacist identifies a safety concern, the medication “might not be dispensed,” Express Scripts says, “or your patient could receive less than what you prescribed.”
That is an ominous statement: The PBM is openly and arrogantly taking for itself the right to unilaterally determine what is safe and to override the physician’s judgment as it sees fit. We all know that there are relative risks in taking most medications that we prescribe; the degree of that risk needs to be carefully calculated against the likely benefits for a given patient, whose detailed history is known to the treating physician. History and risk-benefit calculation are not available to the reviewing pharmacist. The existence of “safety concerns” by itself, outside of the full context of care, is insufficient justification for a PBM to stop payment for a medication.
“Approved”—but… Equally ominous is that, after a medication has been approved through the PA process, some PBMs add these words in their notification to the physician:
This medication is approved for coverage until [insert date],
or until coverage for the medication is no longer available
under the benefit plan or the medication becomes subject
to a pharmacy benefit coverage requirement, such as supply
limits or notification, whichever occurs first.
In other words, the approval is provisional, and shouldn’t be counted on to remain in place for the entire period for which dispensing has been approved. Imagine the uncertainty and anxiety of a patient who reads that statement and realizes that the medication that, at last, has relieved her symptoms might be withdrawn from coverage at any time for reasons unrelated to effectiveness.
The patient can appeal the decision of a PBM or insurer that refuses to pay for a medication, but that patient, and his physician, might ask themselves whether the considerable time required to appeal is justified, given that the criteria used for denials are arbitrary and one-sided.
Serious consequences can ensue after a PBM denies coverage for a medication. Some patients cannot afford hundreds of dollars out of pocket for 1 month of 1 medicine. When their supply runs out, they become vulnerable to symptoms of withdrawal or exacerbation of underlying illness.
Armchair care. A PBM, after it has denied approval of payment, might “ask” the physician to choose another medication that the PBM does cover. For a non-physician administrator who has never seen the patient to propose such a switch is micromanagement—to say the least. Such an action is also disrespectful of the physician’s judgment.
Loss of possible placebo effect. If the physician goes along and makes the switch proposed by the PBM, the patient will know that the new medication is the physician’s second (or third) choice. Any potential positive placebo effect is thus lost. Does that matter? It might—a lot.
Most physicians would be glad to have a positive placebo effect assist or augment the physiologic effects of a medication, especially at the start of treatment when the patient might feel helpless or hopeless. Such negative feelings are likely to be magnified if the patient knows that he has been coerced into taking a second-line therapy. A positive placebo effect, on the other hand, might well have lowered levels of his stress hormones for a few weeks—and that effect could have made a positive difference.
Casualties for the physician are time, money, and morale. PAs consume large chunks of time. Some of the PA forms require that 20 or more questions be answered; a few of those questions can take significant time to answer, having to look through a thick chart to research prior medications.
PAs also cost money: directly to pay the salary of staff that share the PA work, indirectly by crowding out the doctor’s potential billing time and replacing it with uncompensated PA work.
Worse, in my opinion, is the cost to morale. Physicians express their annoyance, aggravation, frustration, and anger at meetings and in postings at the end of journal articles on the subject. Some speak of becoming numb from the daily hassle of dealing with PAs.2 The disrespect for the physician’s decisions inherent in the PA process, the implicit humiliation of appealing to someone who doesn’t know the patient to approve payment for a medication that’s been legally prescribed, and the cost in time and money all provoke emotions that are damaging to morale.
Much to do in limited time
Time isn’t elastic; setting priorities is vital. Most physicians would, I think, agree that their priorities are:
• giving patients adequate time at office visits
• returning calls from patients with urgent messages
• communicating with professional colleagues about shared patients
• returning calls from pharmacists who have questions about prescriptions
• researching solutions to clinical problems
• keeping up with the literature.
Physicians must decide where completing PAs—intrusive, time-consuming, and a threat to morale—fits in that list. Should PAs be allowed to supplant, or delay, the completion of other vital, positive clinical priorities?
Until we are able to introduce improvements that speed up the PA process, patients will have the supply of their medications disrupted and physicians will pay in time, money, and morale.
Disclosure
Dr. Mode reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Casalino L, Nicholson S, Gans DN, et al. What does it cost physician practices to interact with health insurance plans? Health Aff (Millwood). 2009;28(4):533-542.
2. Bendix J. Curing the prior authorization headache. Med Econ. October 10, 2013. http://medicaleconomics.modernmedicine.com/medical-economics/content/ tags/americas-health-insurance-plans/curing-priorauthorization-headache?page=full. Accessed December 2, 2014.
Insistence on prior authorization (PA) when prescribing certain pharmaceuticals has grown considerably over the past 5 years. Most requests for PA are issued by pharmacy benefit management (PBM) companies that have been contracted by an insurer. A PA can be triggered when a physician orders:
• a brand-name medication
• a medication not on the formulary of the PBM
• a quantity above an arbitrary ceiling
• a medication that has multiple indications (that is, the PBM won’t pay for indication X but will pay for indication Y).
What are the problems caused by PAs? I outline a number of them, and their potential consequences, in this “Commentary.” What can we do, in our practices, to lessen the disruption they cause and the time and money they cost us (Box)?
’Prior authorization’—a misleading, disingenuous term
The physician’s prescription is legal authorization for the patient to receive the medicine. It would be more accurate if PBMs labeled what they do “prior approval for reimbursement.”
PBMs exist to manipulate and coordinate the demand for medication generated, on one hand, by patients and their physician and, on the other, by the cost of supplying portion of that demand. The cost of a medication to the PBM is controlled by:
• negotiating rebates with drug manufacturers
• advantageous contracting with pharmacies
• denying payment, when feasible, using the PA system.
The goal of the PA is to boost the profits of the PBM—not to pay for the best fit between the needs of the patient and the medications available, as determined by the treating physician.
The games begin!
The PA process usually begins when the patient goes to the pharmacy, prescription in hand, and gives it to the pharmacist, who enters it into the computer. At that point, if the PBM has put a block on paying for the medication, 3 things happen in sequence:
1. The computer alerts the pharmacist about the block (or that a higher copay is required).
2. The pharmacist tells the patient something about the block—although not necessarily the whole story.
3. The pharmacist tells the physician’s office (by fax, e-mail, or telephone) that the PBM wants authorization and that the physician must call a toll-free telephone number to obtain that authorization.
The physician’s office then makes the initial call to the PBM. That call can take 10 to 20 minutes, answering preliminary questions. The call generates a questionnaire from the PBM that is faxed to the office, filled with questions that one could characterize as loaded. The questionnaire is intended to provide grounds for disapproval or approval—not to obtain in-depth understanding of the individual patient’s needs.
Playing pieces on a chessboard
Note that the physician and pharmacist, thrust unwillingly into the middle of this gambit, spend considerable uncompensated time on the PA process. (Primary care physicians and their nursing and clerical staff, spent, on average, 19.8 hours a week obtaining PAs in 2006.1)
PBMs have shifted responsibility for communication to physicians and pharmacists by requiring that the physician always contact the PBM. A PBM will not contact a physician directly, either to begin the PA or ask questions during the process.
If the request for authorization is denied, what’s the outcome? The physician’s office and the pharmacist have spent uncompensated time taking action that resulted in the PBM and the insurer improving their bottom line without benefit to anyone else.
Communication breakdown. The cumbersome, multistep PA process opens the door to miscommunication. This happens often, I’ve found: The physician wastes time because the pharmacist passed along an incomplete message, or a patient gives vague or confusing information in trying to transmit what the pharmacist said. Sometimes, when physicians get through to a live person at the PBM, they are told that the pharmacist misinformed the office: No, the medication didn’t require PA after all.
Why can’t PBMs streamline the process, sparing busy physicians’ offices the time spent on initial telephone calls, by installing software that would allow the pharmacist who first encounters a payment block to, with a few keystrokes, instantly send the relevant questionnaire to the physician’s fax machine or computer?
Obstacles to satisfaction
From the perspective of the patient, the word that probably best characterizes his emotional response to the PA process is “helpless.” He wasn’t expecting a denial; it’s likely that he hadn’t been fully or clearly informed at the time he selected the insurer that he might someday face such an obstacle. Even though he had a legal prescription, written by a physician, any attempt to go back to the insurer or the PBM to complain is rarely successful. If he tried, he would likely get no satisfaction: The clerk at the other end of the telephone would swiftly inform him that there were a number of complicated rules, policies, or “step programs” that must be adhered to before the PBM pays for a prescription.
Even if the medication is covered, the patient might be told that there are “quantity limits” that prevent reimbursement for the prescription as written—limits that were not made explicit when he signed up for the insurance plan. All these obstacles can generate confusion, anxiety, frustration, and anger—understandably so.
The ‘safety’ catch. Obstacles do not necessarily end when the medication is approved; such approval is merely a “coverage eligibility review.” In addition, PBMs make it clear that every prescription also undergoes a so-called safety review by a pharmacist before it is dispensed. If the PBM’s pharmacist identifies a safety concern, the medication “might not be dispensed,” Express Scripts says, “or your patient could receive less than what you prescribed.”
That is an ominous statement: The PBM is openly and arrogantly taking for itself the right to unilaterally determine what is safe and to override the physician’s judgment as it sees fit. We all know that there are relative risks in taking most medications that we prescribe; the degree of that risk needs to be carefully calculated against the likely benefits for a given patient, whose detailed history is known to the treating physician. History and risk-benefit calculation are not available to the reviewing pharmacist. The existence of “safety concerns” by itself, outside of the full context of care, is insufficient justification for a PBM to stop payment for a medication.
“Approved”—but… Equally ominous is that, after a medication has been approved through the PA process, some PBMs add these words in their notification to the physician:
This medication is approved for coverage until [insert date],
or until coverage for the medication is no longer available
under the benefit plan or the medication becomes subject
to a pharmacy benefit coverage requirement, such as supply
limits or notification, whichever occurs first.
In other words, the approval is provisional, and shouldn’t be counted on to remain in place for the entire period for which dispensing has been approved. Imagine the uncertainty and anxiety of a patient who reads that statement and realizes that the medication that, at last, has relieved her symptoms might be withdrawn from coverage at any time for reasons unrelated to effectiveness.
The patient can appeal the decision of a PBM or insurer that refuses to pay for a medication, but that patient, and his physician, might ask themselves whether the considerable time required to appeal is justified, given that the criteria used for denials are arbitrary and one-sided.
Serious consequences can ensue after a PBM denies coverage for a medication. Some patients cannot afford hundreds of dollars out of pocket for 1 month of 1 medicine. When their supply runs out, they become vulnerable to symptoms of withdrawal or exacerbation of underlying illness.
Armchair care. A PBM, after it has denied approval of payment, might “ask” the physician to choose another medication that the PBM does cover. For a non-physician administrator who has never seen the patient to propose such a switch is micromanagement—to say the least. Such an action is also disrespectful of the physician’s judgment.
Loss of possible placebo effect. If the physician goes along and makes the switch proposed by the PBM, the patient will know that the new medication is the physician’s second (or third) choice. Any potential positive placebo effect is thus lost. Does that matter? It might—a lot.
Most physicians would be glad to have a positive placebo effect assist or augment the physiologic effects of a medication, especially at the start of treatment when the patient might feel helpless or hopeless. Such negative feelings are likely to be magnified if the patient knows that he has been coerced into taking a second-line therapy. A positive placebo effect, on the other hand, might well have lowered levels of his stress hormones for a few weeks—and that effect could have made a positive difference.
Casualties for the physician are time, money, and morale. PAs consume large chunks of time. Some of the PA forms require that 20 or more questions be answered; a few of those questions can take significant time to answer, having to look through a thick chart to research prior medications.
PAs also cost money: directly to pay the salary of staff that share the PA work, indirectly by crowding out the doctor’s potential billing time and replacing it with uncompensated PA work.
Worse, in my opinion, is the cost to morale. Physicians express their annoyance, aggravation, frustration, and anger at meetings and in postings at the end of journal articles on the subject. Some speak of becoming numb from the daily hassle of dealing with PAs.2 The disrespect for the physician’s decisions inherent in the PA process, the implicit humiliation of appealing to someone who doesn’t know the patient to approve payment for a medication that’s been legally prescribed, and the cost in time and money all provoke emotions that are damaging to morale.
Much to do in limited time
Time isn’t elastic; setting priorities is vital. Most physicians would, I think, agree that their priorities are:
• giving patients adequate time at office visits
• returning calls from patients with urgent messages
• communicating with professional colleagues about shared patients
• returning calls from pharmacists who have questions about prescriptions
• researching solutions to clinical problems
• keeping up with the literature.
Physicians must decide where completing PAs—intrusive, time-consuming, and a threat to morale—fits in that list. Should PAs be allowed to supplant, or delay, the completion of other vital, positive clinical priorities?
Until we are able to introduce improvements that speed up the PA process, patients will have the supply of their medications disrupted and physicians will pay in time, money, and morale.
Disclosure
Dr. Mode reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Insistence on prior authorization (PA) when prescribing certain pharmaceuticals has grown considerably over the past 5 years. Most requests for PA are issued by pharmacy benefit management (PBM) companies that have been contracted by an insurer. A PA can be triggered when a physician orders:
• a brand-name medication
• a medication not on the formulary of the PBM
• a quantity above an arbitrary ceiling
• a medication that has multiple indications (that is, the PBM won’t pay for indication X but will pay for indication Y).
What are the problems caused by PAs? I outline a number of them, and their potential consequences, in this “Commentary.” What can we do, in our practices, to lessen the disruption they cause and the time and money they cost us (Box)?
’Prior authorization’—a misleading, disingenuous term
The physician’s prescription is legal authorization for the patient to receive the medicine. It would be more accurate if PBMs labeled what they do “prior approval for reimbursement.”
PBMs exist to manipulate and coordinate the demand for medication generated, on one hand, by patients and their physician and, on the other, by the cost of supplying portion of that demand. The cost of a medication to the PBM is controlled by:
• negotiating rebates with drug manufacturers
• advantageous contracting with pharmacies
• denying payment, when feasible, using the PA system.
The goal of the PA is to boost the profits of the PBM—not to pay for the best fit between the needs of the patient and the medications available, as determined by the treating physician.
The games begin!
The PA process usually begins when the patient goes to the pharmacy, prescription in hand, and gives it to the pharmacist, who enters it into the computer. At that point, if the PBM has put a block on paying for the medication, 3 things happen in sequence:
1. The computer alerts the pharmacist about the block (or that a higher copay is required).
2. The pharmacist tells the patient something about the block—although not necessarily the whole story.
3. The pharmacist tells the physician’s office (by fax, e-mail, or telephone) that the PBM wants authorization and that the physician must call a toll-free telephone number to obtain that authorization.
The physician’s office then makes the initial call to the PBM. That call can take 10 to 20 minutes, answering preliminary questions. The call generates a questionnaire from the PBM that is faxed to the office, filled with questions that one could characterize as loaded. The questionnaire is intended to provide grounds for disapproval or approval—not to obtain in-depth understanding of the individual patient’s needs.
Playing pieces on a chessboard
Note that the physician and pharmacist, thrust unwillingly into the middle of this gambit, spend considerable uncompensated time on the PA process. (Primary care physicians and their nursing and clerical staff, spent, on average, 19.8 hours a week obtaining PAs in 2006.1)
PBMs have shifted responsibility for communication to physicians and pharmacists by requiring that the physician always contact the PBM. A PBM will not contact a physician directly, either to begin the PA or ask questions during the process.
If the request for authorization is denied, what’s the outcome? The physician’s office and the pharmacist have spent uncompensated time taking action that resulted in the PBM and the insurer improving their bottom line without benefit to anyone else.
Communication breakdown. The cumbersome, multistep PA process opens the door to miscommunication. This happens often, I’ve found: The physician wastes time because the pharmacist passed along an incomplete message, or a patient gives vague or confusing information in trying to transmit what the pharmacist said. Sometimes, when physicians get through to a live person at the PBM, they are told that the pharmacist misinformed the office: No, the medication didn’t require PA after all.
Why can’t PBMs streamline the process, sparing busy physicians’ offices the time spent on initial telephone calls, by installing software that would allow the pharmacist who first encounters a payment block to, with a few keystrokes, instantly send the relevant questionnaire to the physician’s fax machine or computer?
Obstacles to satisfaction
From the perspective of the patient, the word that probably best characterizes his emotional response to the PA process is “helpless.” He wasn’t expecting a denial; it’s likely that he hadn’t been fully or clearly informed at the time he selected the insurer that he might someday face such an obstacle. Even though he had a legal prescription, written by a physician, any attempt to go back to the insurer or the PBM to complain is rarely successful. If he tried, he would likely get no satisfaction: The clerk at the other end of the telephone would swiftly inform him that there were a number of complicated rules, policies, or “step programs” that must be adhered to before the PBM pays for a prescription.
Even if the medication is covered, the patient might be told that there are “quantity limits” that prevent reimbursement for the prescription as written—limits that were not made explicit when he signed up for the insurance plan. All these obstacles can generate confusion, anxiety, frustration, and anger—understandably so.
The ‘safety’ catch. Obstacles do not necessarily end when the medication is approved; such approval is merely a “coverage eligibility review.” In addition, PBMs make it clear that every prescription also undergoes a so-called safety review by a pharmacist before it is dispensed. If the PBM’s pharmacist identifies a safety concern, the medication “might not be dispensed,” Express Scripts says, “or your patient could receive less than what you prescribed.”
That is an ominous statement: The PBM is openly and arrogantly taking for itself the right to unilaterally determine what is safe and to override the physician’s judgment as it sees fit. We all know that there are relative risks in taking most medications that we prescribe; the degree of that risk needs to be carefully calculated against the likely benefits for a given patient, whose detailed history is known to the treating physician. History and risk-benefit calculation are not available to the reviewing pharmacist. The existence of “safety concerns” by itself, outside of the full context of care, is insufficient justification for a PBM to stop payment for a medication.
“Approved”—but… Equally ominous is that, after a medication has been approved through the PA process, some PBMs add these words in their notification to the physician:
This medication is approved for coverage until [insert date],
or until coverage for the medication is no longer available
under the benefit plan or the medication becomes subject
to a pharmacy benefit coverage requirement, such as supply
limits or notification, whichever occurs first.
In other words, the approval is provisional, and shouldn’t be counted on to remain in place for the entire period for which dispensing has been approved. Imagine the uncertainty and anxiety of a patient who reads that statement and realizes that the medication that, at last, has relieved her symptoms might be withdrawn from coverage at any time for reasons unrelated to effectiveness.
The patient can appeal the decision of a PBM or insurer that refuses to pay for a medication, but that patient, and his physician, might ask themselves whether the considerable time required to appeal is justified, given that the criteria used for denials are arbitrary and one-sided.
Serious consequences can ensue after a PBM denies coverage for a medication. Some patients cannot afford hundreds of dollars out of pocket for 1 month of 1 medicine. When their supply runs out, they become vulnerable to symptoms of withdrawal or exacerbation of underlying illness.
Armchair care. A PBM, after it has denied approval of payment, might “ask” the physician to choose another medication that the PBM does cover. For a non-physician administrator who has never seen the patient to propose such a switch is micromanagement—to say the least. Such an action is also disrespectful of the physician’s judgment.
Loss of possible placebo effect. If the physician goes along and makes the switch proposed by the PBM, the patient will know that the new medication is the physician’s second (or third) choice. Any potential positive placebo effect is thus lost. Does that matter? It might—a lot.
Most physicians would be glad to have a positive placebo effect assist or augment the physiologic effects of a medication, especially at the start of treatment when the patient might feel helpless or hopeless. Such negative feelings are likely to be magnified if the patient knows that he has been coerced into taking a second-line therapy. A positive placebo effect, on the other hand, might well have lowered levels of his stress hormones for a few weeks—and that effect could have made a positive difference.
Casualties for the physician are time, money, and morale. PAs consume large chunks of time. Some of the PA forms require that 20 or more questions be answered; a few of those questions can take significant time to answer, having to look through a thick chart to research prior medications.
PAs also cost money: directly to pay the salary of staff that share the PA work, indirectly by crowding out the doctor’s potential billing time and replacing it with uncompensated PA work.
Worse, in my opinion, is the cost to morale. Physicians express their annoyance, aggravation, frustration, and anger at meetings and in postings at the end of journal articles on the subject. Some speak of becoming numb from the daily hassle of dealing with PAs.2 The disrespect for the physician’s decisions inherent in the PA process, the implicit humiliation of appealing to someone who doesn’t know the patient to approve payment for a medication that’s been legally prescribed, and the cost in time and money all provoke emotions that are damaging to morale.
Much to do in limited time
Time isn’t elastic; setting priorities is vital. Most physicians would, I think, agree that their priorities are:
• giving patients adequate time at office visits
• returning calls from patients with urgent messages
• communicating with professional colleagues about shared patients
• returning calls from pharmacists who have questions about prescriptions
• researching solutions to clinical problems
• keeping up with the literature.
Physicians must decide where completing PAs—intrusive, time-consuming, and a threat to morale—fits in that list. Should PAs be allowed to supplant, or delay, the completion of other vital, positive clinical priorities?
Until we are able to introduce improvements that speed up the PA process, patients will have the supply of their medications disrupted and physicians will pay in time, money, and morale.
Disclosure
Dr. Mode reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Casalino L, Nicholson S, Gans DN, et al. What does it cost physician practices to interact with health insurance plans? Health Aff (Millwood). 2009;28(4):533-542.
2. Bendix J. Curing the prior authorization headache. Med Econ. October 10, 2013. http://medicaleconomics.modernmedicine.com/medical-economics/content/ tags/americas-health-insurance-plans/curing-priorauthorization-headache?page=full. Accessed December 2, 2014.
1. Casalino L, Nicholson S, Gans DN, et al. What does it cost physician practices to interact with health insurance plans? Health Aff (Millwood). 2009;28(4):533-542.
2. Bendix J. Curing the prior authorization headache. Med Econ. October 10, 2013. http://medicaleconomics.modernmedicine.com/medical-economics/content/ tags/americas-health-insurance-plans/curing-priorauthorization-headache?page=full. Accessed December 2, 2014.

Treatment-resistant schizophrenia
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Identify withdrawal-emergent syndromes

He’s been making new ‘friends’
CASE Seeing friends
Mr. B, age 91, presents to the emergency room (ER) for hip pain. As he is being evaluated, he asks a nurse to tell the “other people” around her to leave so that he can have privacy. As clarification, Mr. B reports visual hallucinations, which prompts the ER physician to request a psychiatry consult.
Mr. B is alert and oriented to time, place, and person when he is evaluated by the on-call psychiatry resident. He reports that he has been seeing several unusual things for the last 4 to 5 months. Asked to elaborate, Mr. B admits seeing colorful and vivid images of people around him. These people come and go as they like; rarely, they talk to him. He describes the conversations as “a constant chatter” in the background and adds that it is difficult to understand what they are talking about.
Mr. B states that he has been “seeing” a couple of people on a regular basis, and they are “sort of like my friends.” He endorses that these people often sing songs or dance for him. He states that, sometimes, these “friends” bring 3 or 4 friends and, although he could not make out their faces clearly, “they all are around me.” He describes the people he sees as “nice people” and does not report being scared or frightened by them.
Mr. B does not report paranoia, and denies command-type hallucinations. He and his family report no unusual changes in behavior in recent months. The medical history is remarkable for atrial fibrillation, coronary artery disease, chronic obstructive pulmonary disease, age-related macular degeneration, and glaucoma.
Mr. B denies having any ongoing mood or anxiety symptoms. He states that he knows these people are “probably not real,” and they do not bother him and just keep him company.
What could be causing Mr. B’s hallucinations?
a) a stroke
b) late-onset schizophrenia
c) dementia
d) Charles Bonnet syndrome
The authors’ observations
Visual hallucinations among geriatric pa-tients are a common and confusing presentation. In addition to several medical causes for this presentation (Table 1), consider Charles Bonnet syndrome in patients with visual loss, presenting as visual hallucinations with intact insight and absence of a mental illness. Other conditions to consider in the differential diagnosis include Parkinson’s disease, dementia with Lewy bodies, schizophrenia, seizures, migraine, and stroke, including lesions of the thalamus or brain stem.

Charles Bonnet syndrome was first described by Swiss philosopher Charles Bonnet in the 18th century. He reported vivid visual hallucinations in his visually impaired grandfather (bilateral cataracts).1
It is important to recognize this syndrome because patients can present across different specialties, including psychiatry, ophthalmology, neurology, geriatric medicine, and family medicine.2 As life expectancy increases, this condition might be seen more often. It is prudent to identify, intervene, and refer as appropriate, in addition to educating patients and caregivers about the nature and course of the condition.
EVALUATION Not psychotic
Mr. B reports good sleep and appetite. He denies using alcohol or illicit drugs. He states he slipped in the bathroom the day before coming to the ER, but denies other recent falls or injuries. Other than hip pain, he has no other physical complaints. His medication regimen includes aspirin, lisinopril, lovastatin, and metoprolol.
The ER team diagnoses a hip fracture. Mr. B is transferred to the orthopedic service; the psychiatry consult team continues to follow him. Mental status examination is unremarkable other than the visual hallucinations. His speech is clear, non-pressured, with goal-directed thought processing. Mini-Mental State Examination score is 23/30 with Mr. B having difficulty with object drawing and 3-object recall. Brief cognitive examination in the ER is unremarkable.
The orthopedic team decides on conservative management of the hip fracture. There is no evidence of infection. Mr. B is afebrile with clear sensorium; complete blood cell count and normal liver function tests are normal; urinalysis and urine drug screen are negative; and chest radiography is unremarkable. CT and MRI of the head are unremarkable.
After 1 week in the hospital, Mr. B continues to experience vivid visual imagery. No signs of active infection are found. An ophthalmologist is consulted, who confirms Mr. B’s earlier diagnosis of glaucoma and age-related macular degeneration but does not recommend further treatment. Visual field test by confrontation is normal, with normal visual reflexes.
The authors’ observations
The reported prevalence of Charles Bonnet syndrome among visually impaired people varies from study to study—from as low as 0.4% to as high as 63%.3-6 The reason for such variation can be attributed to several variables:
• underdiagnosis
• misdiagnosis
• underreporting by patients because of the benign nature of the hallucinations
• patients’ reluctance to report visual hallucinations because of fear of being labeled “mentally ill.”7,8
Symptoms
There are no specific diagnostic criteria for Charles Bonnet syndrome (Table 2). However, the following are generally accepted for diagnosis9:
• grossly intact cognition, although mild cognitive impairment may be present in some cases10
• underlying visual disorder, usually acquired, such as glaucoma, age-related macular degeneration, diabetic retinopathy, central retinal artery occlusion, and optic neuritis3,4,11
• no hallucinations or perceptive difficulties in other sensory modalities
• generally intact insight
• absence of delusions
• absence of other neurologic, psychiatric, toxic, or metabolic conditions; medical causes of delirium must be ruled out.

Hallucinations might not be disturbing to the patient. Hallucinations could be simple (light flashes, lines, or geometric shapes) or complex (faces, figures, or scenes),12 and perceived as in color or in black and white. Hallucinations mostly are pleasant and rarely have any emotional impact or meaning. Although hallucinations are almost exclusively visual, they can be accompanied by noise or auditory hallucinations.13,14
Other characteristics of Charles Bonnet syndrome include:
• typical age of onset is approximately 72 years (range, 70 to 92 years)
• no sex distinction has been identified
• episodes can last from a few seconds to few hours; the syndrome may last a few days or a few years5
• it is not uncommon for episodes to occur in clusters, followed by symptom-free intervals and recurrences
• symptoms tend to fade away as patients progress to complete loss of sight.15
The course of Charles Bonnet syndrome is uncertain and unpredictable and the episodic nature can be frustrating for both patient and clinician. The syndrome could be misdiagnosed as a psychiatric condition.
Pathophysiology
The precise mechanism behind simple or complex vivid hallucinations in persons with Charles Bonnet syndrome is unclear. Several theories have been proposed.
Release theory proposes a loss of input to the primary visual areas, which decreases cortical inhibition and further causes disinhibition of visual association areas, thereby “releasing” visual hallucinations.16 Research suggests that this might be an attempt by surviving neurons to recover vision. Loss of input somehow causes surviving neurons to adapt by increased sensitivity to residual visual stimuli.
Deafferentation theory. This relatively new theory proposes deafferentation of the visual sensory pathway, which, in turn, causes disinhibition of neurons in the visual cortical regions, thereby causing them to fire spontaneously. This could cause a sensation analogous to phantom limb pain, which would be called “phantom vision presence of brain activity in the absence of an actual visual input.” Further, biochemical and molecular changes have been proposed to explain the deafferentation theory.17
Neurobiological evidence. Limited data are available for a neurobiological basis to visual hallucinations in Charles Bonnet syndrome. A few studies have used functional MRI and single-photon emission CT and reported possible association of visual hallucinations to specific visual areas.18,19
Risk factors
Social or physical isolation, loneliness, low extraversion, and shyness are risk factors for Charles Bonnet syndrome in visually impaired people.20 Sensory deprivation and low level of arousal favor the occurrence of hallucinations.5 Rate of vision loss—not the nature of pathology or severity of visual impairment—has been suggested to increase the risk of developing Charles Bonnet syndrome.21
What are the treatment options for Charles Bonnet syndrome?
a) begin an antipsychotic
b) do nothing; there is no cure
c) educate the patient about the nature of the hallucinations
d) refer the patient to an ophthalmologist for evaluation of vision loss
Treatment
There are several modalities to manage visual hallucinations in a patient with Charles Bonnet syndrome (Table 3). After ruling out medical and other psychiatric causes of visual hallucinations, treatment might not be indicated if the patient is not disturbed by the hallucinations. In most cases, reassurance and educating the patient and family about the benign nature of the visual hallucinations is all that is needed.

For patients who are disturbed by these visions or for whom there is a treatable cause, treatment could include cataract removal, medical therapy to reduce intraocular pressure in glaucoma, treatment of diabetic retinopathy, or laser photocoagulation. These treatments are associated with a reduction in hallucinations.22
In some cases, hallucinations disappear as visual acuity deteriorates. Psychotropics have been used to treat Charles Bonnet syndrome, including:
• antipsychotics, including haloperidol, risperidone, and olanzapine
• anticonvulsants, including valproic acid, gabapentin, and carbamazepine
• antidepressants, including mirtazapine and venlafaxine.23-30
Some experts recommend a conservative approach, which might be justified because some cases of Charles Bonnet syndrome are episodic and remit spontaneously.31 Again, however, consider pharmacotherapy if a patient is disturbed by hallucinations or if hallucinations impair overall functioning.
TREATMENT Education
After discussion with Mr. B and his family, he is started on risperidone, 1 mg at bedtime, and the psychiatric team provides information about the nature of Charles Bonnet syndrome. Mr. B reportedly takes this medication for a few days and then stops because he does not want the visual hallucinations to go away.
The psychiatry team sees Mr. B before discharge. He and his family are educated about the benign nature of the syndrome, the need for continued family support, and the fact that hallucinations will have minimal or no implications for his life.
The authors’ observations
It is important to remember that a visual description of hallucinations in Charles Bonnet syndrome can be quite vivid, and that the patient might not identify his hallucinations as such or consider them as a problem. Be careful not to dismiss the patient’s complaints as a primary psychiatric condition. It also is important to be mindful of the patient’s concerns with a psychiatric diagnosis; detailed discussion with the patient is helpful in most cases. A more comprehensive and empathetic approach to care could go a long way to sustain quality of life for these patients.
Bottom Line
Charles Bonnet syndrome is characterized by visual hallucinations in patients with visual impairment who have intact insight and an absence of mental illness. Taking a thorough history can help rule out medical and psychiatric causes of visual hallucinations. Educate patients and family about the nature of the hallucinations. In some cases, a psychotropic may be indicated.
Related Resources
• Nguyen ND, Osterweil D, Hoffman J. Charles Bonnet syndrome: treating nonpsychiatric hallucinations. Consult Pharm. 2013;28(3):184-188.
• Lapid MI, Burton MC, Chang MT, et al. Clinical phenomenology and mortality in Charles Bonnet syndrome. J Geriatr Psychiatry Neurol. 2013;26(1):3-9.
Drug Brand Names
Carbamazepine • Tegretol Mirtazapine • Remeron
Gabapentin • Neurontin Olanzapine • Zyprexa
Haloperidol • Haldol Risperidone • Risperdal
Lisinopril • Prinivil, Zestril Valproic acid • Depakene
Lovastatin • Mevacor Venlafaxine • Effexor
Metoprolol • Lopressor
Acknowledgement
The authors acknowledge Barry Liskow, MD, Vice Chair of Psychiatry, Kansas University Medical Center, Kansas City, Kansas, for providing both insight into the topic and useful feedback on the manuscript.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bonnet C. Essai analytique sur les facultes de l’ame. Copenhagen, Denmark: Chez le Ferres CI. & Ant. Philibert; 1760:426-429.
2. Plummer C, Kleinitz A, Vroomen P, et al. Of Roman chariots and goats in overcoats: the syndrome of Charles Bonnet. J Clin Neurosci. 2007;14(8):709-714.
3. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients with macular degeneration. Am J Psychiatry. 1992;149(12):1701-1706.
4. Tan CS, Lim VS, Ho DY, et al. Charles Bonnet syndrome in Asian patients in a tertiary ophthalmic centre. Br J Ophthalmol. 2004;88(10):1325-1329.
5. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Visual hallucinations in psychologically normal people: Charles Bonnet’s syndrome. Lancet. 1996;347(9004):794-797.
6. Menon GJ. Complex visual hallucinations in the visually impaired: a structured history-taking approach. Arch Ophthalmol. 2005;123(3):349-355.
7. Hart CT. Formed visual hallucinations: a symptom of cranial arteritis. Br Med J. 1967;3(5566):643-644.
8. Norton-Wilson L, Munir M. Visual perceptual disorders resembling the Charles Bonnet syndrome. A study of 434 consecutive patients referred to a psychogeriatric unit. Fam Pract. 1987;4(1):27-35.
9. Eperjesi F, Akbarali N. Rehabilitation in Charles Bonnet syndrome: a review of treatment options. Clin Exp Optom. 2004;87(3):149-152.
10. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients from an ophthalmology clinic and medical clinic population. J Nerv Ment Dis. 1994;182(5):273-276.
11. Manford M, Andermann F. Complex visual hallucinations. Clinical and neurobiological insights. Brain. 1998;121(pt 10):1819-1840.
12. Kester EM. Charles Bonnet syndrome: case presentation and literature review. Optometry. 2009;80(7):360-366.
13. Hori H, Terao T, Nakamura JL. Charles Bonnet syndrome with auditory hallucinations: a diagnostic dilemma. Psychopathology. 2001;34(3):164-166.
14. Menon GJ, Rahman I, Menon SJ, et al. Complex visual hallucinations in the visually impaired: the Charles Bonnet Syndrome. Surv Ophthalmol. 2003;48(1):58-72.
15. Fernandez A, Lichtshein G, Vieweg WV. The Charles Bonnet syndrome: a review. J Nerv Ment Dis. 1997;185(3):195-200.
16. Cogan DG. Visual hallucinations as release phenomena. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1973;188(2):139-150.
17. Burke W. The neural basis of Charles Bonnet hallucinations: a hypothesis. J Neurol Neurosurg Psychiatry. 2002;73(5):535-541.
18. Ffytche DH, Howard RJ, Brammer MJ, et al. The anatomy of conscious vision: an fMRI study of visual hallucinations. Nat Neurosci. 1998;1(8):738-742.
19. Adachi N, Watanabe T, Matsuda H, et al. Hyperperfusion in the lateral temporal cortex, the striatum and the thalamus during complex visual hallucinations: single photon emission computed tomography findings in patients with Charles Bonnet syndrome. Psychiatry Clin Neurosci. 2000;54(2):157-162.
20. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Social and psychological characteristics of elderly visually handicapped patients with the Charles Bonnet Syndrome. Compr Psychiatry. 1999;40(4):315-319.
21. Shiraishi Y, Terao T, Ibi K, et al. Charles Bonnet syndrome and visual acuity—the involvement of dynamic or acute sensory deprivation. Eur Arch Psychiatry Clin Neurosci. 2004;254(6):362-364.
22. Tueth MJ, Cheong JA, Samander J. The Charles Bonnet syndrome: a type of organic visual hallucinosis. J Geriatr Psychiatry Neurol. 1995;8(1):1-3.
23. Nguyen H, Le C, Nguyen H. Charles Bonnet syndrome in an elderly patient concurrent with acute cerebellar infarction treated successfully with haloperidol. J Am Geriatr Soc. 2011;59(4):761-762.
24. Campbell JJ, Ngo G. Risperidone treatment of complex hallucinations in a patient with posterior cortical atrophy. J Neuropsychiatry Clin Neurosci. 2008;20(3):378-379.
25. Colletti Moja M, Milano E, Gasverde S, et al. Olanzapine therapy in hallucinatory visions related to Bonnet syndrome. Neurol Sci. 2005;26(3):168-170.
26. Jang JW, Youn YC, Seok JW, et al. Hypermetabolism in the left thalamus and right inferior temporal area on positron emission tomography-statistical parametric mapping (PET-SPM) in a patient with Charles Bonnet syndrome resolving after treatment with valproic acid. J Clin Neurosci. 2011;18(8):1130-1132.
27. Paulig M, Mentrup H. Charles Bonnet’s syndrome; Complete remission of complex visual hallucinations treated by gabapentin. J Neurol Neurosurg Psychiatry. 2001;70(6):813-814.
28. Terao T. Effect of carbamazepine and clonazepam combination on Charles Bonnet syndrome: a case report. Hum Psychopharmacol. 1998;13(6):451-453.
29. Siddiqui Z, Ramaswmay S, Petty F. Mirtazapine for Charles Bonnet syndrome. Can J Psychiatry. 2004;49(11):787-788.
30. Lang UE, Stogowski D, Schulze D, et al. Charles Bonnet Syndrome: successful treatment of visual hallucinations due to vision loss with selective serotonin reuptake inhibitors. J Psychopharmacol. 2007;21(5):553-555.
31. Hartney KE, Catalano G, Catalano MC. Charles Bonnet syndrome: are medications necessary? J Psychiatr Pract. 2011;17(2):137-141.
CASE Seeing friends
Mr. B, age 91, presents to the emergency room (ER) for hip pain. As he is being evaluated, he asks a nurse to tell the “other people” around her to leave so that he can have privacy. As clarification, Mr. B reports visual hallucinations, which prompts the ER physician to request a psychiatry consult.
Mr. B is alert and oriented to time, place, and person when he is evaluated by the on-call psychiatry resident. He reports that he has been seeing several unusual things for the last 4 to 5 months. Asked to elaborate, Mr. B admits seeing colorful and vivid images of people around him. These people come and go as they like; rarely, they talk to him. He describes the conversations as “a constant chatter” in the background and adds that it is difficult to understand what they are talking about.
Mr. B states that he has been “seeing” a couple of people on a regular basis, and they are “sort of like my friends.” He endorses that these people often sing songs or dance for him. He states that, sometimes, these “friends” bring 3 or 4 friends and, although he could not make out their faces clearly, “they all are around me.” He describes the people he sees as “nice people” and does not report being scared or frightened by them.
Mr. B does not report paranoia, and denies command-type hallucinations. He and his family report no unusual changes in behavior in recent months. The medical history is remarkable for atrial fibrillation, coronary artery disease, chronic obstructive pulmonary disease, age-related macular degeneration, and glaucoma.
Mr. B denies having any ongoing mood or anxiety symptoms. He states that he knows these people are “probably not real,” and they do not bother him and just keep him company.
What could be causing Mr. B’s hallucinations?
a) a stroke
b) late-onset schizophrenia
c) dementia
d) Charles Bonnet syndrome
The authors’ observations
Visual hallucinations among geriatric pa-tients are a common and confusing presentation. In addition to several medical causes for this presentation (Table 1), consider Charles Bonnet syndrome in patients with visual loss, presenting as visual hallucinations with intact insight and absence of a mental illness. Other conditions to consider in the differential diagnosis include Parkinson’s disease, dementia with Lewy bodies, schizophrenia, seizures, migraine, and stroke, including lesions of the thalamus or brain stem.
Charles Bonnet syndrome was first described by Swiss philosopher Charles Bonnet in the 18th century. He reported vivid visual hallucinations in his visually impaired grandfather (bilateral cataracts).1
It is important to recognize this syndrome because patients can present across different specialties, including psychiatry, ophthalmology, neurology, geriatric medicine, and family medicine.2 As life expectancy increases, this condition might be seen more often. It is prudent to identify, intervene, and refer as appropriate, in addition to educating patients and caregivers about the nature and course of the condition.
EVALUATION Not psychotic
Mr. B reports good sleep and appetite. He denies using alcohol or illicit drugs. He states he slipped in the bathroom the day before coming to the ER, but denies other recent falls or injuries. Other than hip pain, he has no other physical complaints. His medication regimen includes aspirin, lisinopril, lovastatin, and metoprolol.
The ER team diagnoses a hip fracture. Mr. B is transferred to the orthopedic service; the psychiatry consult team continues to follow him. Mental status examination is unremarkable other than the visual hallucinations. His speech is clear, non-pressured, with goal-directed thought processing. Mini-Mental State Examination score is 23/30 with Mr. B having difficulty with object drawing and 3-object recall. Brief cognitive examination in the ER is unremarkable.
The orthopedic team decides on conservative management of the hip fracture. There is no evidence of infection. Mr. B is afebrile with clear sensorium; complete blood cell count and normal liver function tests are normal; urinalysis and urine drug screen are negative; and chest radiography is unremarkable. CT and MRI of the head are unremarkable.
After 1 week in the hospital, Mr. B continues to experience vivid visual imagery. No signs of active infection are found. An ophthalmologist is consulted, who confirms Mr. B’s earlier diagnosis of glaucoma and age-related macular degeneration but does not recommend further treatment. Visual field test by confrontation is normal, with normal visual reflexes.
The authors’ observations
The reported prevalence of Charles Bonnet syndrome among visually impaired people varies from study to study—from as low as 0.4% to as high as 63%.3-6 The reason for such variation can be attributed to several variables:
• underdiagnosis
• misdiagnosis
• underreporting by patients because of the benign nature of the hallucinations
• patients’ reluctance to report visual hallucinations because of fear of being labeled “mentally ill.”7,8
Symptoms
There are no specific diagnostic criteria for Charles Bonnet syndrome (Table 2). However, the following are generally accepted for diagnosis9:
• grossly intact cognition, although mild cognitive impairment may be present in some cases10
• underlying visual disorder, usually acquired, such as glaucoma, age-related macular degeneration, diabetic retinopathy, central retinal artery occlusion, and optic neuritis3,4,11
• no hallucinations or perceptive difficulties in other sensory modalities
• generally intact insight
• absence of delusions
• absence of other neurologic, psychiatric, toxic, or metabolic conditions; medical causes of delirium must be ruled out.
Hallucinations might not be disturbing to the patient. Hallucinations could be simple (light flashes, lines, or geometric shapes) or complex (faces, figures, or scenes),12 and perceived as in color or in black and white. Hallucinations mostly are pleasant and rarely have any emotional impact or meaning. Although hallucinations are almost exclusively visual, they can be accompanied by noise or auditory hallucinations.13,14
Other characteristics of Charles Bonnet syndrome include:
• typical age of onset is approximately 72 years (range, 70 to 92 years)
• no sex distinction has been identified
• episodes can last from a few seconds to few hours; the syndrome may last a few days or a few years5
• it is not uncommon for episodes to occur in clusters, followed by symptom-free intervals and recurrences
• symptoms tend to fade away as patients progress to complete loss of sight.15
The course of Charles Bonnet syndrome is uncertain and unpredictable and the episodic nature can be frustrating for both patient and clinician. The syndrome could be misdiagnosed as a psychiatric condition.
Pathophysiology
The precise mechanism behind simple or complex vivid hallucinations in persons with Charles Bonnet syndrome is unclear. Several theories have been proposed.
Release theory proposes a loss of input to the primary visual areas, which decreases cortical inhibition and further causes disinhibition of visual association areas, thereby “releasing” visual hallucinations.16 Research suggests that this might be an attempt by surviving neurons to recover vision. Loss of input somehow causes surviving neurons to adapt by increased sensitivity to residual visual stimuli.
Deafferentation theory. This relatively new theory proposes deafferentation of the visual sensory pathway, which, in turn, causes disinhibition of neurons in the visual cortical regions, thereby causing them to fire spontaneously. This could cause a sensation analogous to phantom limb pain, which would be called “phantom vision presence of brain activity in the absence of an actual visual input.” Further, biochemical and molecular changes have been proposed to explain the deafferentation theory.17
Neurobiological evidence. Limited data are available for a neurobiological basis to visual hallucinations in Charles Bonnet syndrome. A few studies have used functional MRI and single-photon emission CT and reported possible association of visual hallucinations to specific visual areas.18,19
Risk factors
Social or physical isolation, loneliness, low extraversion, and shyness are risk factors for Charles Bonnet syndrome in visually impaired people.20 Sensory deprivation and low level of arousal favor the occurrence of hallucinations.5 Rate of vision loss—not the nature of pathology or severity of visual impairment—has been suggested to increase the risk of developing Charles Bonnet syndrome.21
What are the treatment options for Charles Bonnet syndrome?
a) begin an antipsychotic
b) do nothing; there is no cure
c) educate the patient about the nature of the hallucinations
d) refer the patient to an ophthalmologist for evaluation of vision loss
Treatment
There are several modalities to manage visual hallucinations in a patient with Charles Bonnet syndrome (Table 3). After ruling out medical and other psychiatric causes of visual hallucinations, treatment might not be indicated if the patient is not disturbed by the hallucinations. In most cases, reassurance and educating the patient and family about the benign nature of the visual hallucinations is all that is needed.
For patients who are disturbed by these visions or for whom there is a treatable cause, treatment could include cataract removal, medical therapy to reduce intraocular pressure in glaucoma, treatment of diabetic retinopathy, or laser photocoagulation. These treatments are associated with a reduction in hallucinations.22
In some cases, hallucinations disappear as visual acuity deteriorates. Psychotropics have been used to treat Charles Bonnet syndrome, including:
• antipsychotics, including haloperidol, risperidone, and olanzapine
• anticonvulsants, including valproic acid, gabapentin, and carbamazepine
• antidepressants, including mirtazapine and venlafaxine.23-30
Some experts recommend a conservative approach, which might be justified because some cases of Charles Bonnet syndrome are episodic and remit spontaneously.31 Again, however, consider pharmacotherapy if a patient is disturbed by hallucinations or if hallucinations impair overall functioning.
TREATMENT Education
After discussion with Mr. B and his family, he is started on risperidone, 1 mg at bedtime, and the psychiatric team provides information about the nature of Charles Bonnet syndrome. Mr. B reportedly takes this medication for a few days and then stops because he does not want the visual hallucinations to go away.
The psychiatry team sees Mr. B before discharge. He and his family are educated about the benign nature of the syndrome, the need for continued family support, and the fact that hallucinations will have minimal or no implications for his life.
The authors’ observations
It is important to remember that a visual description of hallucinations in Charles Bonnet syndrome can be quite vivid, and that the patient might not identify his hallucinations as such or consider them as a problem. Be careful not to dismiss the patient’s complaints as a primary psychiatric condition. It also is important to be mindful of the patient’s concerns with a psychiatric diagnosis; detailed discussion with the patient is helpful in most cases. A more comprehensive and empathetic approach to care could go a long way to sustain quality of life for these patients.
Bottom Line
Charles Bonnet syndrome is characterized by visual hallucinations in patients with visual impairment who have intact insight and an absence of mental illness. Taking a thorough history can help rule out medical and psychiatric causes of visual hallucinations. Educate patients and family about the nature of the hallucinations. In some cases, a psychotropic may be indicated.
Related Resources
• Nguyen ND, Osterweil D, Hoffman J. Charles Bonnet syndrome: treating nonpsychiatric hallucinations. Consult Pharm. 2013;28(3):184-188.
• Lapid MI, Burton MC, Chang MT, et al. Clinical phenomenology and mortality in Charles Bonnet syndrome. J Geriatr Psychiatry Neurol. 2013;26(1):3-9.
Drug Brand Names
Carbamazepine • Tegretol Mirtazapine • Remeron
Gabapentin • Neurontin Olanzapine • Zyprexa
Haloperidol • Haldol Risperidone • Risperdal
Lisinopril • Prinivil, Zestril Valproic acid • Depakene
Lovastatin • Mevacor Venlafaxine • Effexor
Metoprolol • Lopressor
Acknowledgement
The authors acknowledge Barry Liskow, MD, Vice Chair of Psychiatry, Kansas University Medical Center, Kansas City, Kansas, for providing both insight into the topic and useful feedback on the manuscript.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Seeing friends
Mr. B, age 91, presents to the emergency room (ER) for hip pain. As he is being evaluated, he asks a nurse to tell the “other people” around her to leave so that he can have privacy. As clarification, Mr. B reports visual hallucinations, which prompts the ER physician to request a psychiatry consult.
Mr. B is alert and oriented to time, place, and person when he is evaluated by the on-call psychiatry resident. He reports that he has been seeing several unusual things for the last 4 to 5 months. Asked to elaborate, Mr. B admits seeing colorful and vivid images of people around him. These people come and go as they like; rarely, they talk to him. He describes the conversations as “a constant chatter” in the background and adds that it is difficult to understand what they are talking about.
Mr. B states that he has been “seeing” a couple of people on a regular basis, and they are “sort of like my friends.” He endorses that these people often sing songs or dance for him. He states that, sometimes, these “friends” bring 3 or 4 friends and, although he could not make out their faces clearly, “they all are around me.” He describes the people he sees as “nice people” and does not report being scared or frightened by them.
Mr. B does not report paranoia, and denies command-type hallucinations. He and his family report no unusual changes in behavior in recent months. The medical history is remarkable for atrial fibrillation, coronary artery disease, chronic obstructive pulmonary disease, age-related macular degeneration, and glaucoma.
Mr. B denies having any ongoing mood or anxiety symptoms. He states that he knows these people are “probably not real,” and they do not bother him and just keep him company.
What could be causing Mr. B’s hallucinations?
a) a stroke
b) late-onset schizophrenia
c) dementia
d) Charles Bonnet syndrome
The authors’ observations
Visual hallucinations among geriatric pa-tients are a common and confusing presentation. In addition to several medical causes for this presentation (Table 1), consider Charles Bonnet syndrome in patients with visual loss, presenting as visual hallucinations with intact insight and absence of a mental illness. Other conditions to consider in the differential diagnosis include Parkinson’s disease, dementia with Lewy bodies, schizophrenia, seizures, migraine, and stroke, including lesions of the thalamus or brain stem.
Charles Bonnet syndrome was first described by Swiss philosopher Charles Bonnet in the 18th century. He reported vivid visual hallucinations in his visually impaired grandfather (bilateral cataracts).1
It is important to recognize this syndrome because patients can present across different specialties, including psychiatry, ophthalmology, neurology, geriatric medicine, and family medicine.2 As life expectancy increases, this condition might be seen more often. It is prudent to identify, intervene, and refer as appropriate, in addition to educating patients and caregivers about the nature and course of the condition.
EVALUATION Not psychotic
Mr. B reports good sleep and appetite. He denies using alcohol or illicit drugs. He states he slipped in the bathroom the day before coming to the ER, but denies other recent falls or injuries. Other than hip pain, he has no other physical complaints. His medication regimen includes aspirin, lisinopril, lovastatin, and metoprolol.
The ER team diagnoses a hip fracture. Mr. B is transferred to the orthopedic service; the psychiatry consult team continues to follow him. Mental status examination is unremarkable other than the visual hallucinations. His speech is clear, non-pressured, with goal-directed thought processing. Mini-Mental State Examination score is 23/30 with Mr. B having difficulty with object drawing and 3-object recall. Brief cognitive examination in the ER is unremarkable.
The orthopedic team decides on conservative management of the hip fracture. There is no evidence of infection. Mr. B is afebrile with clear sensorium; complete blood cell count and normal liver function tests are normal; urinalysis and urine drug screen are negative; and chest radiography is unremarkable. CT and MRI of the head are unremarkable.
After 1 week in the hospital, Mr. B continues to experience vivid visual imagery. No signs of active infection are found. An ophthalmologist is consulted, who confirms Mr. B’s earlier diagnosis of glaucoma and age-related macular degeneration but does not recommend further treatment. Visual field test by confrontation is normal, with normal visual reflexes.
The authors’ observations
The reported prevalence of Charles Bonnet syndrome among visually impaired people varies from study to study—from as low as 0.4% to as high as 63%.3-6 The reason for such variation can be attributed to several variables:
• underdiagnosis
• misdiagnosis
• underreporting by patients because of the benign nature of the hallucinations
• patients’ reluctance to report visual hallucinations because of fear of being labeled “mentally ill.”7,8
Symptoms
There are no specific diagnostic criteria for Charles Bonnet syndrome (Table 2). However, the following are generally accepted for diagnosis9:
• grossly intact cognition, although mild cognitive impairment may be present in some cases10
• underlying visual disorder, usually acquired, such as glaucoma, age-related macular degeneration, diabetic retinopathy, central retinal artery occlusion, and optic neuritis3,4,11
• no hallucinations or perceptive difficulties in other sensory modalities
• generally intact insight
• absence of delusions
• absence of other neurologic, psychiatric, toxic, or metabolic conditions; medical causes of delirium must be ruled out.
Hallucinations might not be disturbing to the patient. Hallucinations could be simple (light flashes, lines, or geometric shapes) or complex (faces, figures, or scenes),12 and perceived as in color or in black and white. Hallucinations mostly are pleasant and rarely have any emotional impact or meaning. Although hallucinations are almost exclusively visual, they can be accompanied by noise or auditory hallucinations.13,14
Other characteristics of Charles Bonnet syndrome include:
• typical age of onset is approximately 72 years (range, 70 to 92 years)
• no sex distinction has been identified
• episodes can last from a few seconds to few hours; the syndrome may last a few days or a few years5
• it is not uncommon for episodes to occur in clusters, followed by symptom-free intervals and recurrences
• symptoms tend to fade away as patients progress to complete loss of sight.15
The course of Charles Bonnet syndrome is uncertain and unpredictable and the episodic nature can be frustrating for both patient and clinician. The syndrome could be misdiagnosed as a psychiatric condition.
Pathophysiology
The precise mechanism behind simple or complex vivid hallucinations in persons with Charles Bonnet syndrome is unclear. Several theories have been proposed.
Release theory proposes a loss of input to the primary visual areas, which decreases cortical inhibition and further causes disinhibition of visual association areas, thereby “releasing” visual hallucinations.16 Research suggests that this might be an attempt by surviving neurons to recover vision. Loss of input somehow causes surviving neurons to adapt by increased sensitivity to residual visual stimuli.
Deafferentation theory. This relatively new theory proposes deafferentation of the visual sensory pathway, which, in turn, causes disinhibition of neurons in the visual cortical regions, thereby causing them to fire spontaneously. This could cause a sensation analogous to phantom limb pain, which would be called “phantom vision presence of brain activity in the absence of an actual visual input.” Further, biochemical and molecular changes have been proposed to explain the deafferentation theory.17
Neurobiological evidence. Limited data are available for a neurobiological basis to visual hallucinations in Charles Bonnet syndrome. A few studies have used functional MRI and single-photon emission CT and reported possible association of visual hallucinations to specific visual areas.18,19
Risk factors
Social or physical isolation, loneliness, low extraversion, and shyness are risk factors for Charles Bonnet syndrome in visually impaired people.20 Sensory deprivation and low level of arousal favor the occurrence of hallucinations.5 Rate of vision loss—not the nature of pathology or severity of visual impairment—has been suggested to increase the risk of developing Charles Bonnet syndrome.21
What are the treatment options for Charles Bonnet syndrome?
a) begin an antipsychotic
b) do nothing; there is no cure
c) educate the patient about the nature of the hallucinations
d) refer the patient to an ophthalmologist for evaluation of vision loss
Treatment
There are several modalities to manage visual hallucinations in a patient with Charles Bonnet syndrome (Table 3). After ruling out medical and other psychiatric causes of visual hallucinations, treatment might not be indicated if the patient is not disturbed by the hallucinations. In most cases, reassurance and educating the patient and family about the benign nature of the visual hallucinations is all that is needed.
For patients who are disturbed by these visions or for whom there is a treatable cause, treatment could include cataract removal, medical therapy to reduce intraocular pressure in glaucoma, treatment of diabetic retinopathy, or laser photocoagulation. These treatments are associated with a reduction in hallucinations.22
In some cases, hallucinations disappear as visual acuity deteriorates. Psychotropics have been used to treat Charles Bonnet syndrome, including:
• antipsychotics, including haloperidol, risperidone, and olanzapine
• anticonvulsants, including valproic acid, gabapentin, and carbamazepine
• antidepressants, including mirtazapine and venlafaxine.23-30
Some experts recommend a conservative approach, which might be justified because some cases of Charles Bonnet syndrome are episodic and remit spontaneously.31 Again, however, consider pharmacotherapy if a patient is disturbed by hallucinations or if hallucinations impair overall functioning.
TREATMENT Education
After discussion with Mr. B and his family, he is started on risperidone, 1 mg at bedtime, and the psychiatric team provides information about the nature of Charles Bonnet syndrome. Mr. B reportedly takes this medication for a few days and then stops because he does not want the visual hallucinations to go away.
The psychiatry team sees Mr. B before discharge. He and his family are educated about the benign nature of the syndrome, the need for continued family support, and the fact that hallucinations will have minimal or no implications for his life.
The authors’ observations
It is important to remember that a visual description of hallucinations in Charles Bonnet syndrome can be quite vivid, and that the patient might not identify his hallucinations as such or consider them as a problem. Be careful not to dismiss the patient’s complaints as a primary psychiatric condition. It also is important to be mindful of the patient’s concerns with a psychiatric diagnosis; detailed discussion with the patient is helpful in most cases. A more comprehensive and empathetic approach to care could go a long way to sustain quality of life for these patients.
Bottom Line
Charles Bonnet syndrome is characterized by visual hallucinations in patients with visual impairment who have intact insight and an absence of mental illness. Taking a thorough history can help rule out medical and psychiatric causes of visual hallucinations. Educate patients and family about the nature of the hallucinations. In some cases, a psychotropic may be indicated.
Related Resources
• Nguyen ND, Osterweil D, Hoffman J. Charles Bonnet syndrome: treating nonpsychiatric hallucinations. Consult Pharm. 2013;28(3):184-188.
• Lapid MI, Burton MC, Chang MT, et al. Clinical phenomenology and mortality in Charles Bonnet syndrome. J Geriatr Psychiatry Neurol. 2013;26(1):3-9.
Drug Brand Names
Carbamazepine • Tegretol Mirtazapine • Remeron
Gabapentin • Neurontin Olanzapine • Zyprexa
Haloperidol • Haldol Risperidone • Risperdal
Lisinopril • Prinivil, Zestril Valproic acid • Depakene
Lovastatin • Mevacor Venlafaxine • Effexor
Metoprolol • Lopressor
Acknowledgement
The authors acknowledge Barry Liskow, MD, Vice Chair of Psychiatry, Kansas University Medical Center, Kansas City, Kansas, for providing both insight into the topic and useful feedback on the manuscript.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bonnet C. Essai analytique sur les facultes de l’ame. Copenhagen, Denmark: Chez le Ferres CI. & Ant. Philibert; 1760:426-429.
2. Plummer C, Kleinitz A, Vroomen P, et al. Of Roman chariots and goats in overcoats: the syndrome of Charles Bonnet. J Clin Neurosci. 2007;14(8):709-714.
3. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients with macular degeneration. Am J Psychiatry. 1992;149(12):1701-1706.
4. Tan CS, Lim VS, Ho DY, et al. Charles Bonnet syndrome in Asian patients in a tertiary ophthalmic centre. Br J Ophthalmol. 2004;88(10):1325-1329.
5. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Visual hallucinations in psychologically normal people: Charles Bonnet’s syndrome. Lancet. 1996;347(9004):794-797.
6. Menon GJ. Complex visual hallucinations in the visually impaired: a structured history-taking approach. Arch Ophthalmol. 2005;123(3):349-355.
7. Hart CT. Formed visual hallucinations: a symptom of cranial arteritis. Br Med J. 1967;3(5566):643-644.
8. Norton-Wilson L, Munir M. Visual perceptual disorders resembling the Charles Bonnet syndrome. A study of 434 consecutive patients referred to a psychogeriatric unit. Fam Pract. 1987;4(1):27-35.
9. Eperjesi F, Akbarali N. Rehabilitation in Charles Bonnet syndrome: a review of treatment options. Clin Exp Optom. 2004;87(3):149-152.
10. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients from an ophthalmology clinic and medical clinic population. J Nerv Ment Dis. 1994;182(5):273-276.
11. Manford M, Andermann F. Complex visual hallucinations. Clinical and neurobiological insights. Brain. 1998;121(pt 10):1819-1840.
12. Kester EM. Charles Bonnet syndrome: case presentation and literature review. Optometry. 2009;80(7):360-366.
13. Hori H, Terao T, Nakamura JL. Charles Bonnet syndrome with auditory hallucinations: a diagnostic dilemma. Psychopathology. 2001;34(3):164-166.
14. Menon GJ, Rahman I, Menon SJ, et al. Complex visual hallucinations in the visually impaired: the Charles Bonnet Syndrome. Surv Ophthalmol. 2003;48(1):58-72.
15. Fernandez A, Lichtshein G, Vieweg WV. The Charles Bonnet syndrome: a review. J Nerv Ment Dis. 1997;185(3):195-200.
16. Cogan DG. Visual hallucinations as release phenomena. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1973;188(2):139-150.
17. Burke W. The neural basis of Charles Bonnet hallucinations: a hypothesis. J Neurol Neurosurg Psychiatry. 2002;73(5):535-541.
18. Ffytche DH, Howard RJ, Brammer MJ, et al. The anatomy of conscious vision: an fMRI study of visual hallucinations. Nat Neurosci. 1998;1(8):738-742.
19. Adachi N, Watanabe T, Matsuda H, et al. Hyperperfusion in the lateral temporal cortex, the striatum and the thalamus during complex visual hallucinations: single photon emission computed tomography findings in patients with Charles Bonnet syndrome. Psychiatry Clin Neurosci. 2000;54(2):157-162.
20. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Social and psychological characteristics of elderly visually handicapped patients with the Charles Bonnet Syndrome. Compr Psychiatry. 1999;40(4):315-319.
21. Shiraishi Y, Terao T, Ibi K, et al. Charles Bonnet syndrome and visual acuity—the involvement of dynamic or acute sensory deprivation. Eur Arch Psychiatry Clin Neurosci. 2004;254(6):362-364.
22. Tueth MJ, Cheong JA, Samander J. The Charles Bonnet syndrome: a type of organic visual hallucinosis. J Geriatr Psychiatry Neurol. 1995;8(1):1-3.
23. Nguyen H, Le C, Nguyen H. Charles Bonnet syndrome in an elderly patient concurrent with acute cerebellar infarction treated successfully with haloperidol. J Am Geriatr Soc. 2011;59(4):761-762.
24. Campbell JJ, Ngo G. Risperidone treatment of complex hallucinations in a patient with posterior cortical atrophy. J Neuropsychiatry Clin Neurosci. 2008;20(3):378-379.
25. Colletti Moja M, Milano E, Gasverde S, et al. Olanzapine therapy in hallucinatory visions related to Bonnet syndrome. Neurol Sci. 2005;26(3):168-170.
26. Jang JW, Youn YC, Seok JW, et al. Hypermetabolism in the left thalamus and right inferior temporal area on positron emission tomography-statistical parametric mapping (PET-SPM) in a patient with Charles Bonnet syndrome resolving after treatment with valproic acid. J Clin Neurosci. 2011;18(8):1130-1132.
27. Paulig M, Mentrup H. Charles Bonnet’s syndrome; Complete remission of complex visual hallucinations treated by gabapentin. J Neurol Neurosurg Psychiatry. 2001;70(6):813-814.
28. Terao T. Effect of carbamazepine and clonazepam combination on Charles Bonnet syndrome: a case report. Hum Psychopharmacol. 1998;13(6):451-453.
29. Siddiqui Z, Ramaswmay S, Petty F. Mirtazapine for Charles Bonnet syndrome. Can J Psychiatry. 2004;49(11):787-788.
30. Lang UE, Stogowski D, Schulze D, et al. Charles Bonnet Syndrome: successful treatment of visual hallucinations due to vision loss with selective serotonin reuptake inhibitors. J Psychopharmacol. 2007;21(5):553-555.
31. Hartney KE, Catalano G, Catalano MC. Charles Bonnet syndrome: are medications necessary? J Psychiatr Pract. 2011;17(2):137-141.
1. Bonnet C. Essai analytique sur les facultes de l’ame. Copenhagen, Denmark: Chez le Ferres CI. & Ant. Philibert; 1760:426-429.
2. Plummer C, Kleinitz A, Vroomen P, et al. Of Roman chariots and goats in overcoats: the syndrome of Charles Bonnet. J Clin Neurosci. 2007;14(8):709-714.
3. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients with macular degeneration. Am J Psychiatry. 1992;149(12):1701-1706.
4. Tan CS, Lim VS, Ho DY, et al. Charles Bonnet syndrome in Asian patients in a tertiary ophthalmic centre. Br J Ophthalmol. 2004;88(10):1325-1329.
5. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Visual hallucinations in psychologically normal people: Charles Bonnet’s syndrome. Lancet. 1996;347(9004):794-797.
6. Menon GJ. Complex visual hallucinations in the visually impaired: a structured history-taking approach. Arch Ophthalmol. 2005;123(3):349-355.
7. Hart CT. Formed visual hallucinations: a symptom of cranial arteritis. Br Med J. 1967;3(5566):643-644.
8. Norton-Wilson L, Munir M. Visual perceptual disorders resembling the Charles Bonnet syndrome. A study of 434 consecutive patients referred to a psychogeriatric unit. Fam Pract. 1987;4(1):27-35.
9. Eperjesi F, Akbarali N. Rehabilitation in Charles Bonnet syndrome: a review of treatment options. Clin Exp Optom. 2004;87(3):149-152.
10. Holroyd S, Rabins PV, Finkelstein D, et al. Visual hallucinations in patients from an ophthalmology clinic and medical clinic population. J Nerv Ment Dis. 1994;182(5):273-276.
11. Manford M, Andermann F. Complex visual hallucinations. Clinical and neurobiological insights. Brain. 1998;121(pt 10):1819-1840.
12. Kester EM. Charles Bonnet syndrome: case presentation and literature review. Optometry. 2009;80(7):360-366.
13. Hori H, Terao T, Nakamura JL. Charles Bonnet syndrome with auditory hallucinations: a diagnostic dilemma. Psychopathology. 2001;34(3):164-166.
14. Menon GJ, Rahman I, Menon SJ, et al. Complex visual hallucinations in the visually impaired: the Charles Bonnet Syndrome. Surv Ophthalmol. 2003;48(1):58-72.
15. Fernandez A, Lichtshein G, Vieweg WV. The Charles Bonnet syndrome: a review. J Nerv Ment Dis. 1997;185(3):195-200.
16. Cogan DG. Visual hallucinations as release phenomena. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1973;188(2):139-150.
17. Burke W. The neural basis of Charles Bonnet hallucinations: a hypothesis. J Neurol Neurosurg Psychiatry. 2002;73(5):535-541.
18. Ffytche DH, Howard RJ, Brammer MJ, et al. The anatomy of conscious vision: an fMRI study of visual hallucinations. Nat Neurosci. 1998;1(8):738-742.
19. Adachi N, Watanabe T, Matsuda H, et al. Hyperperfusion in the lateral temporal cortex, the striatum and the thalamus during complex visual hallucinations: single photon emission computed tomography findings in patients with Charles Bonnet syndrome. Psychiatry Clin Neurosci. 2000;54(2):157-162.
20. Teunisse RJ, Cruysberg JR, Hoefnagels WH, et al. Social and psychological characteristics of elderly visually handicapped patients with the Charles Bonnet Syndrome. Compr Psychiatry. 1999;40(4):315-319.
21. Shiraishi Y, Terao T, Ibi K, et al. Charles Bonnet syndrome and visual acuity—the involvement of dynamic or acute sensory deprivation. Eur Arch Psychiatry Clin Neurosci. 2004;254(6):362-364.
22. Tueth MJ, Cheong JA, Samander J. The Charles Bonnet syndrome: a type of organic visual hallucinosis. J Geriatr Psychiatry Neurol. 1995;8(1):1-3.
23. Nguyen H, Le C, Nguyen H. Charles Bonnet syndrome in an elderly patient concurrent with acute cerebellar infarction treated successfully with haloperidol. J Am Geriatr Soc. 2011;59(4):761-762.
24. Campbell JJ, Ngo G. Risperidone treatment of complex hallucinations in a patient with posterior cortical atrophy. J Neuropsychiatry Clin Neurosci. 2008;20(3):378-379.
25. Colletti Moja M, Milano E, Gasverde S, et al. Olanzapine therapy in hallucinatory visions related to Bonnet syndrome. Neurol Sci. 2005;26(3):168-170.
26. Jang JW, Youn YC, Seok JW, et al. Hypermetabolism in the left thalamus and right inferior temporal area on positron emission tomography-statistical parametric mapping (PET-SPM) in a patient with Charles Bonnet syndrome resolving after treatment with valproic acid. J Clin Neurosci. 2011;18(8):1130-1132.
27. Paulig M, Mentrup H. Charles Bonnet’s syndrome; Complete remission of complex visual hallucinations treated by gabapentin. J Neurol Neurosurg Psychiatry. 2001;70(6):813-814.
28. Terao T. Effect of carbamazepine and clonazepam combination on Charles Bonnet syndrome: a case report. Hum Psychopharmacol. 1998;13(6):451-453.
29. Siddiqui Z, Ramaswmay S, Petty F. Mirtazapine for Charles Bonnet syndrome. Can J Psychiatry. 2004;49(11):787-788.
30. Lang UE, Stogowski D, Schulze D, et al. Charles Bonnet Syndrome: successful treatment of visual hallucinations due to vision loss with selective serotonin reuptake inhibitors. J Psychopharmacol. 2007;21(5):553-555.
31. Hartney KE, Catalano G, Catalano MC. Charles Bonnet syndrome: are medications necessary? J Psychiatr Pract. 2011;17(2):137-141.
How to modify psychotropic therapy for patients who have liver dysfunction
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3

Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
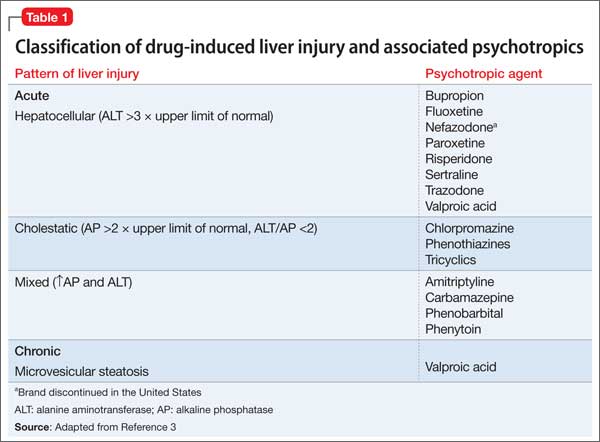
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5

When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
Good, bad, and ugly: Prior authorization and medicolegal risk
Dear Dr. Mossman,
Where I practice, most health care plans won’t pay for certain medications without giving prior authorization (PA). Completing PA forms and making telephone calls take up time that could be better spent treating patients. I’m tempted to set a new policy of not doing PAs. If I do, might I face legal trouble?
Submitted by “Dr. A”
If you provide clinical care, you’ve probably dealt with third-party payers who require prior authorization (PA) before they will pay for certain treatments. Dr. A is not alone in feeling exasperated about the time it takes to complete a PA.1 After spending several hours each month waiting on hold and wading through stacks of paperwork, you may feel like Dr. A and consider refusing to do any more PAs.
But is Dr. A’s proposed solution a good idea? To address this question and the frustration that lies behind it, we’ll take a cue from Italian film director Sergio Leone and discuss:
• how PAs affect psychiatric care: the good, the bad, and the ugly
• potential exposure to professional liability and ethics complaints that might result from refusing or failing to seek PA
• strategies to reduce the burden of PAs while providing efficient, effective care.
The good
Recent decades have witnessed huge increases in spending on prescription medication. Psychotropics are no exception; state Medicaid spending for anti-psychotic medication grew from <$1 billion in 1995 to >$5.5 billion in 2005.2
Requiring a PA for expensive drugs is one way that third-party payers try to rein in costs and hold down insurance premiums. Imposing financial constraints often is just one aim of a pharmacy benefit management (PBM) program. Insurers also justify PBMs by pointing out that feedback to practitioners whose prescribing falls well outside the norm—in the form of mailed warnings, physician second opinions, or pharmacist consultation—can improve patient safety and encourage appropriate treatment options for enrolled patients.3,4 Examples of such benefits include reducing overuse of prescription opioids5 and antipsychotics among children,3 misuse of buprenorphine,6 and adverse effects from potentially inappropriate prescriptions.7
The bad
The bad news for doctors: Cost savings for payers come at the expense of providers and their practices, in the form of time spent doing paperwork and talking on the phone to complete PAs or contest PA decisions.8 Addressing PA requests costs an estimated $83,000 per physician per year. The total administrative burden for all 835,000 physicians who practice in the United States therefore is 868,000,000 hours, or $69 billion annually.9
To make matters worse, PA requirements may increase the overall cost of care. After Georgia Medicaid instituted PA requirements for second-generation antipsychotics (SGAs), average monthly per member drug costs fell $19.62, but average monthly outpatient treatment costs rose $31.59 per member.10 Pharmacy savings that result from requiring PAs for SGAs can be offset quickly by small increases in the hospitalization rate or emergency department visits.9,11
The ugly
Many physicians believe that the PA process undermines patient care by decreasing time devoted to direct patient contact, incentivizing suboptimal treatment, and limiting medication access.1,12,13 But do any data support this belief? Do PAs impede treatment for vulnerable persons with severe mental illnesses?
The answer, some studies suggest, is “Yes.” A Maine Medicaid PA policy slowed initiation of treatment for bipolar disorder by reducing the rate of starting non-preferred medications, although the same policy had no impact on patients already receiving treatment.14 Another study examined the effect of PA processes for inpatient psychiatry treatment and found that patients were less likely to be admitted on weekends, probably because PA review was not available on those days.15 A third study showed that PA requirements and resulting impediments to getting refills were correlated with medication discontinuation by patients with schizophrenia or bipolar disorder, which can increase the risk of decompensation, work-related problems, and hospitalization.16
Problems with PAs
Whether they are helpful or counterproductive, PAs are a practice reality. Dr. A’s proposed solution sounds appealing, but it might create ethical and legal problems.
Among the fundamental elements of ethical medical practice is physicians’ obligation to give patients “guidance … as to the optimal course of action” and to “advocate for patients in dealing with third parties when appropriate.”17 It’s fine for psychiatrists to consider prescribing treatments that patients’ health care coverage favors, but we also have to help patients weigh and evaluate costs, particularly when patients’ circumstances and medical interests militate strongly for options that third-party payers balk at paying for. Patients’ interests—not what’s expedient—are always physicians’ foremost concern.18
Beyond purely ethical considerations, you might face legal consequences if you refuse or fail to seek PAs for what you think is the proper medication. As Table 1 shows, one key factor is whether you are under contract with the patient’s insurance carrier; if you are, failure to seek a PA when appropriate may constitute a breach of the contract (Donna Vanderpool, written communication, October 5, 2014).
If the prescribed medication does not meet the standard of care and your patient suffers some harm, a licensing board complaint and investigation are possible. You also face exposure to a medical malpractice action. Although we do not know of any instances in which such an action has succeeded, 2 recent court decisions suggest that harm to a patient stemmed from failing to seek PA for a medication could constitute grounds for a lawsuit.19,20 Efforts to contain medical costs have been around for decades, and courts have held that physicians, third-party payers, and utilization review intermediaries are bound by “the standard of reasonable community practice”21 and should not let cost limitations “corrupt medical judgment.”22 Physicians who do not appeal limitations at odds with their medical judgment might bear responsibility for any injuries that occur.18,22
Managing PA requests
Given the inevitability of encountering PA requests and your ethical and professional obligations to help patients, what can you do (Table 29,23,27)?
Some practitioners charge patients for time spent completing PAs.23 Although physicians should “complete without charge the appropriate ‘simplified’ insurance claim form as a part of service to the patient;” they also may consider “a charge for more complex or multiple forms … in conformity with local custom.”24 Legally, physicians’ contracts with insurance panels may preclude charging such fees, but if a patient is being seen out of network, the physician does not have a contractual obligation and may charge.9 If your practice setting lets you choose which insurances you accept, the impact and burden of seeking PAs is a factor to consider when deciding whether to participate in a particular panel.23
In an interesting twist, an Ohio physician successfully sued a medical insurance administrator for the cost of his time responding to PA inquiries.25 Reasoning that the insurance administrator “should expect to pay for the reasonable value of” the doctor’s time because the PAs “were solely intended for the benefit of the insurance administrator” or parties whom the administrator served, the judge awarded the doctor $187.50 plus 8% interest.
Considerations that are more practical relate to how to triage and address the volume of PA requests. Some large medical practices centralize PAs and try to set up pre-approved plans of care or blanket approvals for frequently encountered conditions. Centralization also allows one key administrative assistant to develop skills in processing PA requests and to build relationships with payers.26
The administrative assistant also can compile lists of preferred alternative medications, PA forms, and payer Web sites. Using and submitting requests through payer Web sites can speed up PA processing, which saves time and money.27 As electronic health records improve, they may incorporate patients’ formularies and provide automatic alerts for required PAs.23
Patients should be involved, too. They can help to obtain relevant formulary information and to weigh alternative therapies. You can help them understand your role in the PA process, the reasoning behind your treatment recommendations, and the delays in picking up prescribed medications that waiting for PA approval can create.
It’s easy to get angry about PAs
Your best response, however, is to practice prudent and—within reason— cost-effective medicine. When generic or insurer-preferred medications are clinically appropriate and meet treatment guidelines, trying them first is sensible and defensible. If your patient fails the initial low-cost treatment, or if a low-cost choice isn’t appropriate, document this clearly and seek approval for a costlier treatment.9
BOTTOM LINE
Physicians have ethical and legal obligations to advocate for their patients’ needs and best interests. This sometimes includes completing prior authorization requests. Find strategies that minimize hassle and make sense in your practice, and seek efficient ways to document the medical necessity of requested tests, procedures, or therapies.
Acknowledgment
Drs. Marett and Mossman thanks Donna Vanderpool, MBA, JD, and Annette Reynolds, MD, for their helpful input in preparing this article.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brown CM, Richards K, Rascati KL, et al. Effects of a psychotherapeutic drug prior authorization (PA) requirement on patients and providers: a providers’ perspective. Adm Policy Ment Health. 2008;35(3):181-188.
2. Law MR, Ross-Degnan D, Soumerai SB. Effect of prior authorization of second-generation antipsychotic agents on pharmacy utilization and reimbursements. Psychiatr Serv. 2008;59(5):540-546.
3. Stein BD, Leckman-Westin E, Okeke E, et al. The effects of prior authorization policies on Medicaid-enrolled children’s use of antipsychotic medications: evidence from two Mid-Atlantic states. J Child Adolesc Psychopharmacol. 2014;24(7):374-381.
4. Adams KT. Prior authorization–still used, still an issue. Biotechnol Healthc. 2010;7(4):28.
5. Garcia MM, Angelini MC, Thomas T, et al. Implementation of an opioid management initiative by a state Medicaid program. J Manag Care Pharm. 2014;20(5):447-454.
6. Clark RE, Baxter JD, Barton BA, et al. The impact of prior authorization on buprenorphine dose, relapse rates, and cost for Massachusetts Medicaid beneficiaries with opioid dependence [published online July 9, 2014]. Health Serv Res. doi: 10.1111/1475-6773.12201.
7. Dunn RL, Harrison D, Ripley TL. The beers criteria as an outpatient screening tool for potentially inappropriate medications. Consult Pharm. 2011;26(10):754-763.
8. Lennertz MD, Wertheimer AI. Is prior authorization for prescribed drugs cost-effective? Drug Benefit Trends. 2008;20:136-139.
9. Bendix J. The prior authorization predicament. Med Econ. 2014;91(13)29-30,32,34-35.
10. Farley JF, Cline RR, Schommer JC, et al. Retrospective assessment of Medicaid step-therapy prior authorization policy for atypical antipsychotic medications. Clin Ther. 2008;30(8):1524-1539; discussion 1506-1507.
11. Abouzaid S, Jutkowitz E, Foley KA, et al. Economic impact of prior authorization policies for atypical antipsychotics in the treatment of schizophrenia. Popul Health Manag. 2010;13(5):247-254.
12. Brown CM, Nwokeji E, Rascati KL, et al. Development of the burden of prior authorization of psychotherapeutics (BoPAP) scale to assess the effects of prior authorization among Texas Medicaid providers. Adm Policy Ment Health. 2009;36(4):278-287.
13. Rascati KL, Brown CM. Prior authorization for antipsychotic medications—It’s not just about the money. Clin Ther. 2008;30(8):1506-1507.
14. Lu CY, Soumerai SB, Ross-Degnan D, et al. Unintended impacts of a Medicaid prior authorization policy on access to medications for bipolar disorder. Med Care. 2010;48(1):4-9.
15. Stephens RJ, White SE, Cudnik M, et al. Factors associated with longer lengths of stay for mental health emergency department patients. J Emerg Med. 2014; 47(4):412-419.
16. Brown JD, Barrett A, Caffery E, et al. Medication continuity among Medicaid beneficiaries with schizophrenia and bipolar disorder. Psychiatr Serv. 2013;64(9):878-885.
17. American Medical Association. Opinion 10.01– Fundamental elements of the patient-physician relationship. http://www.ama-assn.org/ama/pub/ physician-resources/medical-ethics/code-medical-ethics/opinion1001.page?. Accessed October 11, 2014.
18. Hall RC. Ethical and legal implications of managed care. Gen Hosp Psychiatry. 1997;19(3):200-208.
19. Porter v Thadani, 2010 U.S. Dist. LEXIS 35145 (NH 2010).
20. NB ex rel Peacock v District of Columbia, 682 F3d 77 (DC Cir 2012).
21. Wilson v Blue Cross of Southern California, 222 Cal App 3d 660, 271 Cal Rptr 876 (1990).
22. Wickline v State of California, 192 Cal App 3d 1630, 239 Cal Rptr 810 (1986).
23. Terry K. Prior authorization made easier. Med Econ. 2007;84(20):34,38,40.
24. American Medical Association. Ethics Opinion 6.07– Insurance forms completion charges. http://www. ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion607.page? Updated June 1994. Accessed October 11, 2014.
25. Gibson v Medco Health Solutions, 06-CVF-106 (OH 2008).
26. Bendix J. Curing the prior authorization headache. Med Econ. 2013;90(19):24,26-27,29-31.
27. American Medical Association. Electronic prior authorization toolkit. Available at http://www.ama-assn.org/ama/pub/advocacy/topics/administrative-simplification-initiatives/electronic-transactions-toolkit/ prior-authorization.page. Accessed October 11, 2014.
Dear Dr. Mossman,
Where I practice, most health care plans won’t pay for certain medications without giving prior authorization (PA). Completing PA forms and making telephone calls take up time that could be better spent treating patients. I’m tempted to set a new policy of not doing PAs. If I do, might I face legal trouble?
Submitted by “Dr. A”
If you provide clinical care, you’ve probably dealt with third-party payers who require prior authorization (PA) before they will pay for certain treatments. Dr. A is not alone in feeling exasperated about the time it takes to complete a PA.1 After spending several hours each month waiting on hold and wading through stacks of paperwork, you may feel like Dr. A and consider refusing to do any more PAs.
But is Dr. A’s proposed solution a good idea? To address this question and the frustration that lies behind it, we’ll take a cue from Italian film director Sergio Leone and discuss:
• how PAs affect psychiatric care: the good, the bad, and the ugly
• potential exposure to professional liability and ethics complaints that might result from refusing or failing to seek PA
• strategies to reduce the burden of PAs while providing efficient, effective care.
The good
Recent decades have witnessed huge increases in spending on prescription medication. Psychotropics are no exception; state Medicaid spending for anti-psychotic medication grew from <$1 billion in 1995 to >$5.5 billion in 2005.2
Requiring a PA for expensive drugs is one way that third-party payers try to rein in costs and hold down insurance premiums. Imposing financial constraints often is just one aim of a pharmacy benefit management (PBM) program. Insurers also justify PBMs by pointing out that feedback to practitioners whose prescribing falls well outside the norm—in the form of mailed warnings, physician second opinions, or pharmacist consultation—can improve patient safety and encourage appropriate treatment options for enrolled patients.3,4 Examples of such benefits include reducing overuse of prescription opioids5 and antipsychotics among children,3 misuse of buprenorphine,6 and adverse effects from potentially inappropriate prescriptions.7
The bad
The bad news for doctors: Cost savings for payers come at the expense of providers and their practices, in the form of time spent doing paperwork and talking on the phone to complete PAs or contest PA decisions.8 Addressing PA requests costs an estimated $83,000 per physician per year. The total administrative burden for all 835,000 physicians who practice in the United States therefore is 868,000,000 hours, or $69 billion annually.9
To make matters worse, PA requirements may increase the overall cost of care. After Georgia Medicaid instituted PA requirements for second-generation antipsychotics (SGAs), average monthly per member drug costs fell $19.62, but average monthly outpatient treatment costs rose $31.59 per member.10 Pharmacy savings that result from requiring PAs for SGAs can be offset quickly by small increases in the hospitalization rate or emergency department visits.9,11
The ugly
Many physicians believe that the PA process undermines patient care by decreasing time devoted to direct patient contact, incentivizing suboptimal treatment, and limiting medication access.1,12,13 But do any data support this belief? Do PAs impede treatment for vulnerable persons with severe mental illnesses?
The answer, some studies suggest, is “Yes.” A Maine Medicaid PA policy slowed initiation of treatment for bipolar disorder by reducing the rate of starting non-preferred medications, although the same policy had no impact on patients already receiving treatment.14 Another study examined the effect of PA processes for inpatient psychiatry treatment and found that patients were less likely to be admitted on weekends, probably because PA review was not available on those days.15 A third study showed that PA requirements and resulting impediments to getting refills were correlated with medication discontinuation by patients with schizophrenia or bipolar disorder, which can increase the risk of decompensation, work-related problems, and hospitalization.16
Problems with PAs
Whether they are helpful or counterproductive, PAs are a practice reality. Dr. A’s proposed solution sounds appealing, but it might create ethical and legal problems.
Among the fundamental elements of ethical medical practice is physicians’ obligation to give patients “guidance … as to the optimal course of action” and to “advocate for patients in dealing with third parties when appropriate.”17 It’s fine for psychiatrists to consider prescribing treatments that patients’ health care coverage favors, but we also have to help patients weigh and evaluate costs, particularly when patients’ circumstances and medical interests militate strongly for options that third-party payers balk at paying for. Patients’ interests—not what’s expedient—are always physicians’ foremost concern.18
Beyond purely ethical considerations, you might face legal consequences if you refuse or fail to seek PAs for what you think is the proper medication. As Table 1 shows, one key factor is whether you are under contract with the patient’s insurance carrier; if you are, failure to seek a PA when appropriate may constitute a breach of the contract (Donna Vanderpool, written communication, October 5, 2014).
If the prescribed medication does not meet the standard of care and your patient suffers some harm, a licensing board complaint and investigation are possible. You also face exposure to a medical malpractice action. Although we do not know of any instances in which such an action has succeeded, 2 recent court decisions suggest that harm to a patient stemmed from failing to seek PA for a medication could constitute grounds for a lawsuit.19,20 Efforts to contain medical costs have been around for decades, and courts have held that physicians, third-party payers, and utilization review intermediaries are bound by “the standard of reasonable community practice”21 and should not let cost limitations “corrupt medical judgment.”22 Physicians who do not appeal limitations at odds with their medical judgment might bear responsibility for any injuries that occur.18,22
Managing PA requests
Given the inevitability of encountering PA requests and your ethical and professional obligations to help patients, what can you do (Table 29,23,27)?
Some practitioners charge patients for time spent completing PAs.23 Although physicians should “complete without charge the appropriate ‘simplified’ insurance claim form as a part of service to the patient;” they also may consider “a charge for more complex or multiple forms … in conformity with local custom.”24 Legally, physicians’ contracts with insurance panels may preclude charging such fees, but if a patient is being seen out of network, the physician does not have a contractual obligation and may charge.9 If your practice setting lets you choose which insurances you accept, the impact and burden of seeking PAs is a factor to consider when deciding whether to participate in a particular panel.23
In an interesting twist, an Ohio physician successfully sued a medical insurance administrator for the cost of his time responding to PA inquiries.25 Reasoning that the insurance administrator “should expect to pay for the reasonable value of” the doctor’s time because the PAs “were solely intended for the benefit of the insurance administrator” or parties whom the administrator served, the judge awarded the doctor $187.50 plus 8% interest.
Considerations that are more practical relate to how to triage and address the volume of PA requests. Some large medical practices centralize PAs and try to set up pre-approved plans of care or blanket approvals for frequently encountered conditions. Centralization also allows one key administrative assistant to develop skills in processing PA requests and to build relationships with payers.26
The administrative assistant also can compile lists of preferred alternative medications, PA forms, and payer Web sites. Using and submitting requests through payer Web sites can speed up PA processing, which saves time and money.27 As electronic health records improve, they may incorporate patients’ formularies and provide automatic alerts for required PAs.23
Patients should be involved, too. They can help to obtain relevant formulary information and to weigh alternative therapies. You can help them understand your role in the PA process, the reasoning behind your treatment recommendations, and the delays in picking up prescribed medications that waiting for PA approval can create.
It’s easy to get angry about PAs
Your best response, however, is to practice prudent and—within reason— cost-effective medicine. When generic or insurer-preferred medications are clinically appropriate and meet treatment guidelines, trying them first is sensible and defensible. If your patient fails the initial low-cost treatment, or if a low-cost choice isn’t appropriate, document this clearly and seek approval for a costlier treatment.9
BOTTOM LINE
Physicians have ethical and legal obligations to advocate for their patients’ needs and best interests. This sometimes includes completing prior authorization requests. Find strategies that minimize hassle and make sense in your practice, and seek efficient ways to document the medical necessity of requested tests, procedures, or therapies.
Acknowledgment
Drs. Marett and Mossman thanks Donna Vanderpool, MBA, JD, and Annette Reynolds, MD, for their helpful input in preparing this article.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dear Dr. Mossman,
Where I practice, most health care plans won’t pay for certain medications without giving prior authorization (PA). Completing PA forms and making telephone calls take up time that could be better spent treating patients. I’m tempted to set a new policy of not doing PAs. If I do, might I face legal trouble?
Submitted by “Dr. A”
If you provide clinical care, you’ve probably dealt with third-party payers who require prior authorization (PA) before they will pay for certain treatments. Dr. A is not alone in feeling exasperated about the time it takes to complete a PA.1 After spending several hours each month waiting on hold and wading through stacks of paperwork, you may feel like Dr. A and consider refusing to do any more PAs.
But is Dr. A’s proposed solution a good idea? To address this question and the frustration that lies behind it, we’ll take a cue from Italian film director Sergio Leone and discuss:
• how PAs affect psychiatric care: the good, the bad, and the ugly
• potential exposure to professional liability and ethics complaints that might result from refusing or failing to seek PA
• strategies to reduce the burden of PAs while providing efficient, effective care.
The good
Recent decades have witnessed huge increases in spending on prescription medication. Psychotropics are no exception; state Medicaid spending for anti-psychotic medication grew from <$1 billion in 1995 to >$5.5 billion in 2005.2
Requiring a PA for expensive drugs is one way that third-party payers try to rein in costs and hold down insurance premiums. Imposing financial constraints often is just one aim of a pharmacy benefit management (PBM) program. Insurers also justify PBMs by pointing out that feedback to practitioners whose prescribing falls well outside the norm—in the form of mailed warnings, physician second opinions, or pharmacist consultation—can improve patient safety and encourage appropriate treatment options for enrolled patients.3,4 Examples of such benefits include reducing overuse of prescription opioids5 and antipsychotics among children,3 misuse of buprenorphine,6 and adverse effects from potentially inappropriate prescriptions.7
The bad
The bad news for doctors: Cost savings for payers come at the expense of providers and their practices, in the form of time spent doing paperwork and talking on the phone to complete PAs or contest PA decisions.8 Addressing PA requests costs an estimated $83,000 per physician per year. The total administrative burden for all 835,000 physicians who practice in the United States therefore is 868,000,000 hours, or $69 billion annually.9
To make matters worse, PA requirements may increase the overall cost of care. After Georgia Medicaid instituted PA requirements for second-generation antipsychotics (SGAs), average monthly per member drug costs fell $19.62, but average monthly outpatient treatment costs rose $31.59 per member.10 Pharmacy savings that result from requiring PAs for SGAs can be offset quickly by small increases in the hospitalization rate or emergency department visits.9,11
The ugly
Many physicians believe that the PA process undermines patient care by decreasing time devoted to direct patient contact, incentivizing suboptimal treatment, and limiting medication access.1,12,13 But do any data support this belief? Do PAs impede treatment for vulnerable persons with severe mental illnesses?
The answer, some studies suggest, is “Yes.” A Maine Medicaid PA policy slowed initiation of treatment for bipolar disorder by reducing the rate of starting non-preferred medications, although the same policy had no impact on patients already receiving treatment.14 Another study examined the effect of PA processes for inpatient psychiatry treatment and found that patients were less likely to be admitted on weekends, probably because PA review was not available on those days.15 A third study showed that PA requirements and resulting impediments to getting refills were correlated with medication discontinuation by patients with schizophrenia or bipolar disorder, which can increase the risk of decompensation, work-related problems, and hospitalization.16
Problems with PAs
Whether they are helpful or counterproductive, PAs are a practice reality. Dr. A’s proposed solution sounds appealing, but it might create ethical and legal problems.
Among the fundamental elements of ethical medical practice is physicians’ obligation to give patients “guidance … as to the optimal course of action” and to “advocate for patients in dealing with third parties when appropriate.”17 It’s fine for psychiatrists to consider prescribing treatments that patients’ health care coverage favors, but we also have to help patients weigh and evaluate costs, particularly when patients’ circumstances and medical interests militate strongly for options that third-party payers balk at paying for. Patients’ interests—not what’s expedient—are always physicians’ foremost concern.18
Beyond purely ethical considerations, you might face legal consequences if you refuse or fail to seek PAs for what you think is the proper medication. As Table 1 shows, one key factor is whether you are under contract with the patient’s insurance carrier; if you are, failure to seek a PA when appropriate may constitute a breach of the contract (Donna Vanderpool, written communication, October 5, 2014).
If the prescribed medication does not meet the standard of care and your patient suffers some harm, a licensing board complaint and investigation are possible. You also face exposure to a medical malpractice action. Although we do not know of any instances in which such an action has succeeded, 2 recent court decisions suggest that harm to a patient stemmed from failing to seek PA for a medication could constitute grounds for a lawsuit.19,20 Efforts to contain medical costs have been around for decades, and courts have held that physicians, third-party payers, and utilization review intermediaries are bound by “the standard of reasonable community practice”21 and should not let cost limitations “corrupt medical judgment.”22 Physicians who do not appeal limitations at odds with their medical judgment might bear responsibility for any injuries that occur.18,22
Managing PA requests
Given the inevitability of encountering PA requests and your ethical and professional obligations to help patients, what can you do (Table 29,23,27)?
Some practitioners charge patients for time spent completing PAs.23 Although physicians should “complete without charge the appropriate ‘simplified’ insurance claim form as a part of service to the patient;” they also may consider “a charge for more complex or multiple forms … in conformity with local custom.”24 Legally, physicians’ contracts with insurance panels may preclude charging such fees, but if a patient is being seen out of network, the physician does not have a contractual obligation and may charge.9 If your practice setting lets you choose which insurances you accept, the impact and burden of seeking PAs is a factor to consider when deciding whether to participate in a particular panel.23
In an interesting twist, an Ohio physician successfully sued a medical insurance administrator for the cost of his time responding to PA inquiries.25 Reasoning that the insurance administrator “should expect to pay for the reasonable value of” the doctor’s time because the PAs “were solely intended for the benefit of the insurance administrator” or parties whom the administrator served, the judge awarded the doctor $187.50 plus 8% interest.
Considerations that are more practical relate to how to triage and address the volume of PA requests. Some large medical practices centralize PAs and try to set up pre-approved plans of care or blanket approvals for frequently encountered conditions. Centralization also allows one key administrative assistant to develop skills in processing PA requests and to build relationships with payers.26
The administrative assistant also can compile lists of preferred alternative medications, PA forms, and payer Web sites. Using and submitting requests through payer Web sites can speed up PA processing, which saves time and money.27 As electronic health records improve, they may incorporate patients’ formularies and provide automatic alerts for required PAs.23
Patients should be involved, too. They can help to obtain relevant formulary information and to weigh alternative therapies. You can help them understand your role in the PA process, the reasoning behind your treatment recommendations, and the delays in picking up prescribed medications that waiting for PA approval can create.
It’s easy to get angry about PAs
Your best response, however, is to practice prudent and—within reason— cost-effective medicine. When generic or insurer-preferred medications are clinically appropriate and meet treatment guidelines, trying them first is sensible and defensible. If your patient fails the initial low-cost treatment, or if a low-cost choice isn’t appropriate, document this clearly and seek approval for a costlier treatment.9
BOTTOM LINE
Physicians have ethical and legal obligations to advocate for their patients’ needs and best interests. This sometimes includes completing prior authorization requests. Find strategies that minimize hassle and make sense in your practice, and seek efficient ways to document the medical necessity of requested tests, procedures, or therapies.
Acknowledgment
Drs. Marett and Mossman thanks Donna Vanderpool, MBA, JD, and Annette Reynolds, MD, for their helpful input in preparing this article.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brown CM, Richards K, Rascati KL, et al. Effects of a psychotherapeutic drug prior authorization (PA) requirement on patients and providers: a providers’ perspective. Adm Policy Ment Health. 2008;35(3):181-188.
2. Law MR, Ross-Degnan D, Soumerai SB. Effect of prior authorization of second-generation antipsychotic agents on pharmacy utilization and reimbursements. Psychiatr Serv. 2008;59(5):540-546.
3. Stein BD, Leckman-Westin E, Okeke E, et al. The effects of prior authorization policies on Medicaid-enrolled children’s use of antipsychotic medications: evidence from two Mid-Atlantic states. J Child Adolesc Psychopharmacol. 2014;24(7):374-381.
4. Adams KT. Prior authorization–still used, still an issue. Biotechnol Healthc. 2010;7(4):28.
5. Garcia MM, Angelini MC, Thomas T, et al. Implementation of an opioid management initiative by a state Medicaid program. J Manag Care Pharm. 2014;20(5):447-454.
6. Clark RE, Baxter JD, Barton BA, et al. The impact of prior authorization on buprenorphine dose, relapse rates, and cost for Massachusetts Medicaid beneficiaries with opioid dependence [published online July 9, 2014]. Health Serv Res. doi: 10.1111/1475-6773.12201.
7. Dunn RL, Harrison D, Ripley TL. The beers criteria as an outpatient screening tool for potentially inappropriate medications. Consult Pharm. 2011;26(10):754-763.
8. Lennertz MD, Wertheimer AI. Is prior authorization for prescribed drugs cost-effective? Drug Benefit Trends. 2008;20:136-139.
9. Bendix J. The prior authorization predicament. Med Econ. 2014;91(13)29-30,32,34-35.
10. Farley JF, Cline RR, Schommer JC, et al. Retrospective assessment of Medicaid step-therapy prior authorization policy for atypical antipsychotic medications. Clin Ther. 2008;30(8):1524-1539; discussion 1506-1507.
11. Abouzaid S, Jutkowitz E, Foley KA, et al. Economic impact of prior authorization policies for atypical antipsychotics in the treatment of schizophrenia. Popul Health Manag. 2010;13(5):247-254.
12. Brown CM, Nwokeji E, Rascati KL, et al. Development of the burden of prior authorization of psychotherapeutics (BoPAP) scale to assess the effects of prior authorization among Texas Medicaid providers. Adm Policy Ment Health. 2009;36(4):278-287.
13. Rascati KL, Brown CM. Prior authorization for antipsychotic medications—It’s not just about the money. Clin Ther. 2008;30(8):1506-1507.
14. Lu CY, Soumerai SB, Ross-Degnan D, et al. Unintended impacts of a Medicaid prior authorization policy on access to medications for bipolar disorder. Med Care. 2010;48(1):4-9.
15. Stephens RJ, White SE, Cudnik M, et al. Factors associated with longer lengths of stay for mental health emergency department patients. J Emerg Med. 2014; 47(4):412-419.
16. Brown JD, Barrett A, Caffery E, et al. Medication continuity among Medicaid beneficiaries with schizophrenia and bipolar disorder. Psychiatr Serv. 2013;64(9):878-885.
17. American Medical Association. Opinion 10.01– Fundamental elements of the patient-physician relationship. http://www.ama-assn.org/ama/pub/ physician-resources/medical-ethics/code-medical-ethics/opinion1001.page?. Accessed October 11, 2014.
18. Hall RC. Ethical and legal implications of managed care. Gen Hosp Psychiatry. 1997;19(3):200-208.
19. Porter v Thadani, 2010 U.S. Dist. LEXIS 35145 (NH 2010).
20. NB ex rel Peacock v District of Columbia, 682 F3d 77 (DC Cir 2012).
21. Wilson v Blue Cross of Southern California, 222 Cal App 3d 660, 271 Cal Rptr 876 (1990).
22. Wickline v State of California, 192 Cal App 3d 1630, 239 Cal Rptr 810 (1986).
23. Terry K. Prior authorization made easier. Med Econ. 2007;84(20):34,38,40.
24. American Medical Association. Ethics Opinion 6.07– Insurance forms completion charges. http://www. ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion607.page? Updated June 1994. Accessed October 11, 2014.
25. Gibson v Medco Health Solutions, 06-CVF-106 (OH 2008).
26. Bendix J. Curing the prior authorization headache. Med Econ. 2013;90(19):24,26-27,29-31.
27. American Medical Association. Electronic prior authorization toolkit. Available at http://www.ama-assn.org/ama/pub/advocacy/topics/administrative-simplification-initiatives/electronic-transactions-toolkit/ prior-authorization.page. Accessed October 11, 2014.
1. Brown CM, Richards K, Rascati KL, et al. Effects of a psychotherapeutic drug prior authorization (PA) requirement on patients and providers: a providers’ perspective. Adm Policy Ment Health. 2008;35(3):181-188.
2. Law MR, Ross-Degnan D, Soumerai SB. Effect of prior authorization of second-generation antipsychotic agents on pharmacy utilization and reimbursements. Psychiatr Serv. 2008;59(5):540-546.
3. Stein BD, Leckman-Westin E, Okeke E, et al. The effects of prior authorization policies on Medicaid-enrolled children’s use of antipsychotic medications: evidence from two Mid-Atlantic states. J Child Adolesc Psychopharmacol. 2014;24(7):374-381.
4. Adams KT. Prior authorization–still used, still an issue. Biotechnol Healthc. 2010;7(4):28.
5. Garcia MM, Angelini MC, Thomas T, et al. Implementation of an opioid management initiative by a state Medicaid program. J Manag Care Pharm. 2014;20(5):447-454.
6. Clark RE, Baxter JD, Barton BA, et al. The impact of prior authorization on buprenorphine dose, relapse rates, and cost for Massachusetts Medicaid beneficiaries with opioid dependence [published online July 9, 2014]. Health Serv Res. doi: 10.1111/1475-6773.12201.
7. Dunn RL, Harrison D, Ripley TL. The beers criteria as an outpatient screening tool for potentially inappropriate medications. Consult Pharm. 2011;26(10):754-763.
8. Lennertz MD, Wertheimer AI. Is prior authorization for prescribed drugs cost-effective? Drug Benefit Trends. 2008;20:136-139.
9. Bendix J. The prior authorization predicament. Med Econ. 2014;91(13)29-30,32,34-35.
10. Farley JF, Cline RR, Schommer JC, et al. Retrospective assessment of Medicaid step-therapy prior authorization policy for atypical antipsychotic medications. Clin Ther. 2008;30(8):1524-1539; discussion 1506-1507.
11. Abouzaid S, Jutkowitz E, Foley KA, et al. Economic impact of prior authorization policies for atypical antipsychotics in the treatment of schizophrenia. Popul Health Manag. 2010;13(5):247-254.
12. Brown CM, Nwokeji E, Rascati KL, et al. Development of the burden of prior authorization of psychotherapeutics (BoPAP) scale to assess the effects of prior authorization among Texas Medicaid providers. Adm Policy Ment Health. 2009;36(4):278-287.
13. Rascati KL, Brown CM. Prior authorization for antipsychotic medications—It’s not just about the money. Clin Ther. 2008;30(8):1506-1507.
14. Lu CY, Soumerai SB, Ross-Degnan D, et al. Unintended impacts of a Medicaid prior authorization policy on access to medications for bipolar disorder. Med Care. 2010;48(1):4-9.
15. Stephens RJ, White SE, Cudnik M, et al. Factors associated with longer lengths of stay for mental health emergency department patients. J Emerg Med. 2014; 47(4):412-419.
16. Brown JD, Barrett A, Caffery E, et al. Medication continuity among Medicaid beneficiaries with schizophrenia and bipolar disorder. Psychiatr Serv. 2013;64(9):878-885.
17. American Medical Association. Opinion 10.01– Fundamental elements of the patient-physician relationship. http://www.ama-assn.org/ama/pub/ physician-resources/medical-ethics/code-medical-ethics/opinion1001.page?. Accessed October 11, 2014.
18. Hall RC. Ethical and legal implications of managed care. Gen Hosp Psychiatry. 1997;19(3):200-208.
19. Porter v Thadani, 2010 U.S. Dist. LEXIS 35145 (NH 2010).
20. NB ex rel Peacock v District of Columbia, 682 F3d 77 (DC Cir 2012).
21. Wilson v Blue Cross of Southern California, 222 Cal App 3d 660, 271 Cal Rptr 876 (1990).
22. Wickline v State of California, 192 Cal App 3d 1630, 239 Cal Rptr 810 (1986).
23. Terry K. Prior authorization made easier. Med Econ. 2007;84(20):34,38,40.
24. American Medical Association. Ethics Opinion 6.07– Insurance forms completion charges. http://www. ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion607.page? Updated June 1994. Accessed October 11, 2014.
25. Gibson v Medco Health Solutions, 06-CVF-106 (OH 2008).
26. Bendix J. Curing the prior authorization headache. Med Econ. 2013;90(19):24,26-27,29-31.
27. American Medical Association. Electronic prior authorization toolkit. Available at http://www.ama-assn.org/ama/pub/advocacy/topics/administrative-simplification-initiatives/electronic-transactions-toolkit/ prior-authorization.page. Accessed October 11, 2014.