User login
Official news magazine of the Society of Hospital Medicine
Copyright by Society of Hospital Medicine or related companies. All rights reserved. ISSN 1553-085X
nav[contains(@class, 'nav-ce-stack nav-ce-stack__large-screen')]
header[@id='header']
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
div[contains(@class, 'pane-pub-article-hospitalist')]
Caution with IVC filters in elderly
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.

Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.
Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.
Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Labeling of medication warnings
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.

The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at [email protected].
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at [email protected].
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at [email protected].
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
FDA adds diabetic kidney disease, heart failure indications to canagliflozin
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.

FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.
FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.
FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.
Ticagrelor monotherapy tops DAPT for high-risk PCI patients
SAN FRANCISCO – After 3 months of ticagrelor (Brilinta) plus aspirin following cardiac stenting, stopping the aspirin but continuing the ticagrelor resulted in less bleeding with no increase in ischemic events in a randomized trial with more than 7,000 drug-eluting stent patients at high risk for both.

“This was a superior therapy” to staying on both drugs, the more usual approach, said lead investigator Roxana Mehran, MD, director of interventional cardiovascular research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York.
“We can’t say this is for all comers, but for patients whose physician felt comfortable putting them on aspirin and ticagrelor,” who tolerated it well for the first 3 months, and who had clinical and angiographic indications of risk, “I think these patients can be peeled away” from aspirin, she said in a presentation at the Transcatheter Cardiovascular Therapeutics annual meeting that coincided with publication of the trial, dubbed TWILIGHT (Ticagrelor with Aspirin or Alone in High-Risk Patients after Coronary Intervention).
Interventional cardiologists have long sought the sweet spot for dual-antiplatelet therapy (DAPT) after stenting; the idea is to maximize thrombosis prevention while minimizing bleeding risk. The trial supports the trend in recent years towards shorter DAPT. Often, however, it’s the P2Y12 inhibitor – ticagrelor, clopidogrel (Plavix), or prasugrel (Effient) – that goes first, not the aspirin.
Responding to an audience question about why the trial didn’t include an aspirin monotherapy arm, Dr. Mehran said that aspirin alone wouldn’t have been sufficient in high-risk patients “in whom you have almost 70% acute coronary syndrome.” She added that her team has data showing that aspirin itself doesn’t have much effect on blood thrombogenicity.
The 7,119 patients in TWILIGHT were on ticagrelor 90 mg twice daily and aspirin 81-100 mg daily for 3 months, then evenly randomized to continued treatment or ticagrelor plus an aspirin placebo for a year.
Subjects had to have at least one clinical and angiographic finding that put them at high risk for bleeding or an ischemic event, such as chronic kidney disease, acute coronary syndrome, diabetes, or a bifurcated target lesion treated with two stents.
One year after randomization, 4% in the ticagrelor monotherapy group versus 7.1% in the ticagrelor plus aspirin arm reached the primary end point, actionable (type 2), severe (type 3), or fatal (type 5) bleeding on the Bleeding Academic Research Consortium scale (hazard ratio, 0.56; 95% confidence interval, 0.45 - 0.68, P less than .001).
The incidence of death from any cause, nonfatal myocardial infarction, or nonfatal stroke was 3.9% in both groups (HR, 0.99; 95% CI, 0.78-1.25; P less than .001 for noninferiority).
There were more ischemic strokes in the ticagrelor monotherapy arm (0.5% versus 0.2%). All-cause mortality (1.3% versus 1%) and stent thrombosis (0.6% versus 0.4%) were more frequent in the ticagrelor/aspirin group, but the differences were not statistically significant.
The two groups were well balanced. The mean age was 65 years, 23.8% of the patients were female, 37% had diabetes, and 65% had percutaneous coronary intervention for an acute coronary syndrome. Almost two-thirds had multivessel disease. Mean stent length was about 40 mm. The trial excluded patients with prior strokes.
Almost 2,000 patients originally enrolled in the trial never made it to randomization because they had a major bleeding or ischemic event in the 3-month run up, or dyspnea or some other reaction to ticagrelor.
The recent STOPDAPT-2 trial had a similar outcome – less bleeding with no increase in ischemic events – with clopidogrel monotherapy after a month-long run in of dual therapy with aspirin, versus continued treatment with both, in patients at low risk for ischemic events after stenting (JAMA. 2019 Jun 25;321[24]:2414-27).
Another recent study, GLOBAL LEADERS, concluded that 1 month of DAPT followed by ticagrelor monotherapy for 23 months was not superior to 12 months of DAPT followed by a year of aspirin. There was a numerical advantage for solo ticagrelor on death, myocardial infarction, and bleeding, but it did not reach statistical significance (Lancet. 2018 Sep 15;392[10151]:940-9).
The work was funded by ticagrelor’s maker, AstraZeneca. Dr. Mehran reported consulting and other relationships with Abbott, Janssen, and other companies.
SOURCE: Mehran A et al. N Engl J Med. 2019 Sep 26. doi: 10.1056/NEJMoa1908419.
SAN FRANCISCO – After 3 months of ticagrelor (Brilinta) plus aspirin following cardiac stenting, stopping the aspirin but continuing the ticagrelor resulted in less bleeding with no increase in ischemic events in a randomized trial with more than 7,000 drug-eluting stent patients at high risk for both.
“This was a superior therapy” to staying on both drugs, the more usual approach, said lead investigator Roxana Mehran, MD, director of interventional cardiovascular research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York.
“We can’t say this is for all comers, but for patients whose physician felt comfortable putting them on aspirin and ticagrelor,” who tolerated it well for the first 3 months, and who had clinical and angiographic indications of risk, “I think these patients can be peeled away” from aspirin, she said in a presentation at the Transcatheter Cardiovascular Therapeutics annual meeting that coincided with publication of the trial, dubbed TWILIGHT (Ticagrelor with Aspirin or Alone in High-Risk Patients after Coronary Intervention).
Interventional cardiologists have long sought the sweet spot for dual-antiplatelet therapy (DAPT) after stenting; the idea is to maximize thrombosis prevention while minimizing bleeding risk. The trial supports the trend in recent years towards shorter DAPT. Often, however, it’s the P2Y12 inhibitor – ticagrelor, clopidogrel (Plavix), or prasugrel (Effient) – that goes first, not the aspirin.
Responding to an audience question about why the trial didn’t include an aspirin monotherapy arm, Dr. Mehran said that aspirin alone wouldn’t have been sufficient in high-risk patients “in whom you have almost 70% acute coronary syndrome.” She added that her team has data showing that aspirin itself doesn’t have much effect on blood thrombogenicity.
The 7,119 patients in TWILIGHT were on ticagrelor 90 mg twice daily and aspirin 81-100 mg daily for 3 months, then evenly randomized to continued treatment or ticagrelor plus an aspirin placebo for a year.
Subjects had to have at least one clinical and angiographic finding that put them at high risk for bleeding or an ischemic event, such as chronic kidney disease, acute coronary syndrome, diabetes, or a bifurcated target lesion treated with two stents.
One year after randomization, 4% in the ticagrelor monotherapy group versus 7.1% in the ticagrelor plus aspirin arm reached the primary end point, actionable (type 2), severe (type 3), or fatal (type 5) bleeding on the Bleeding Academic Research Consortium scale (hazard ratio, 0.56; 95% confidence interval, 0.45 - 0.68, P less than .001).
The incidence of death from any cause, nonfatal myocardial infarction, or nonfatal stroke was 3.9% in both groups (HR, 0.99; 95% CI, 0.78-1.25; P less than .001 for noninferiority).
There were more ischemic strokes in the ticagrelor monotherapy arm (0.5% versus 0.2%). All-cause mortality (1.3% versus 1%) and stent thrombosis (0.6% versus 0.4%) were more frequent in the ticagrelor/aspirin group, but the differences were not statistically significant.
The two groups were well balanced. The mean age was 65 years, 23.8% of the patients were female, 37% had diabetes, and 65% had percutaneous coronary intervention for an acute coronary syndrome. Almost two-thirds had multivessel disease. Mean stent length was about 40 mm. The trial excluded patients with prior strokes.
Almost 2,000 patients originally enrolled in the trial never made it to randomization because they had a major bleeding or ischemic event in the 3-month run up, or dyspnea or some other reaction to ticagrelor.
The recent STOPDAPT-2 trial had a similar outcome – less bleeding with no increase in ischemic events – with clopidogrel monotherapy after a month-long run in of dual therapy with aspirin, versus continued treatment with both, in patients at low risk for ischemic events after stenting (JAMA. 2019 Jun 25;321[24]:2414-27).
Another recent study, GLOBAL LEADERS, concluded that 1 month of DAPT followed by ticagrelor monotherapy for 23 months was not superior to 12 months of DAPT followed by a year of aspirin. There was a numerical advantage for solo ticagrelor on death, myocardial infarction, and bleeding, but it did not reach statistical significance (Lancet. 2018 Sep 15;392[10151]:940-9).
The work was funded by ticagrelor’s maker, AstraZeneca. Dr. Mehran reported consulting and other relationships with Abbott, Janssen, and other companies.
SOURCE: Mehran A et al. N Engl J Med. 2019 Sep 26. doi: 10.1056/NEJMoa1908419.
SAN FRANCISCO – After 3 months of ticagrelor (Brilinta) plus aspirin following cardiac stenting, stopping the aspirin but continuing the ticagrelor resulted in less bleeding with no increase in ischemic events in a randomized trial with more than 7,000 drug-eluting stent patients at high risk for both.
“This was a superior therapy” to staying on both drugs, the more usual approach, said lead investigator Roxana Mehran, MD, director of interventional cardiovascular research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York.
“We can’t say this is for all comers, but for patients whose physician felt comfortable putting them on aspirin and ticagrelor,” who tolerated it well for the first 3 months, and who had clinical and angiographic indications of risk, “I think these patients can be peeled away” from aspirin, she said in a presentation at the Transcatheter Cardiovascular Therapeutics annual meeting that coincided with publication of the trial, dubbed TWILIGHT (Ticagrelor with Aspirin or Alone in High-Risk Patients after Coronary Intervention).
Interventional cardiologists have long sought the sweet spot for dual-antiplatelet therapy (DAPT) after stenting; the idea is to maximize thrombosis prevention while minimizing bleeding risk. The trial supports the trend in recent years towards shorter DAPT. Often, however, it’s the P2Y12 inhibitor – ticagrelor, clopidogrel (Plavix), or prasugrel (Effient) – that goes first, not the aspirin.
Responding to an audience question about why the trial didn’t include an aspirin monotherapy arm, Dr. Mehran said that aspirin alone wouldn’t have been sufficient in high-risk patients “in whom you have almost 70% acute coronary syndrome.” She added that her team has data showing that aspirin itself doesn’t have much effect on blood thrombogenicity.
The 7,119 patients in TWILIGHT were on ticagrelor 90 mg twice daily and aspirin 81-100 mg daily for 3 months, then evenly randomized to continued treatment or ticagrelor plus an aspirin placebo for a year.
Subjects had to have at least one clinical and angiographic finding that put them at high risk for bleeding or an ischemic event, such as chronic kidney disease, acute coronary syndrome, diabetes, or a bifurcated target lesion treated with two stents.
One year after randomization, 4% in the ticagrelor monotherapy group versus 7.1% in the ticagrelor plus aspirin arm reached the primary end point, actionable (type 2), severe (type 3), or fatal (type 5) bleeding on the Bleeding Academic Research Consortium scale (hazard ratio, 0.56; 95% confidence interval, 0.45 - 0.68, P less than .001).
The incidence of death from any cause, nonfatal myocardial infarction, or nonfatal stroke was 3.9% in both groups (HR, 0.99; 95% CI, 0.78-1.25; P less than .001 for noninferiority).
There were more ischemic strokes in the ticagrelor monotherapy arm (0.5% versus 0.2%). All-cause mortality (1.3% versus 1%) and stent thrombosis (0.6% versus 0.4%) were more frequent in the ticagrelor/aspirin group, but the differences were not statistically significant.
The two groups were well balanced. The mean age was 65 years, 23.8% of the patients were female, 37% had diabetes, and 65% had percutaneous coronary intervention for an acute coronary syndrome. Almost two-thirds had multivessel disease. Mean stent length was about 40 mm. The trial excluded patients with prior strokes.
Almost 2,000 patients originally enrolled in the trial never made it to randomization because they had a major bleeding or ischemic event in the 3-month run up, or dyspnea or some other reaction to ticagrelor.
The recent STOPDAPT-2 trial had a similar outcome – less bleeding with no increase in ischemic events – with clopidogrel monotherapy after a month-long run in of dual therapy with aspirin, versus continued treatment with both, in patients at low risk for ischemic events after stenting (JAMA. 2019 Jun 25;321[24]:2414-27).
Another recent study, GLOBAL LEADERS, concluded that 1 month of DAPT followed by ticagrelor monotherapy for 23 months was not superior to 12 months of DAPT followed by a year of aspirin. There was a numerical advantage for solo ticagrelor on death, myocardial infarction, and bleeding, but it did not reach statistical significance (Lancet. 2018 Sep 15;392[10151]:940-9).
The work was funded by ticagrelor’s maker, AstraZeneca. Dr. Mehran reported consulting and other relationships with Abbott, Janssen, and other companies.
SOURCE: Mehran A et al. N Engl J Med. 2019 Sep 26. doi: 10.1056/NEJMoa1908419.
REPORTING FROM TCT 2019

CDC reports most vaping lung disease linked to THC-containing cartridges
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.

The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.
The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.
The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
Judge dismisses doctors’ lawsuit against ABIM
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”

In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”
In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”
In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
Palliative care programs continue growth in U.S. hospitals
Growth continues among palliative care programs in the United States, although access often depends “more upon accidents of geography than it does upon the needs of patients,” according to the Center to Advance Palliative Care and the National Palliative Care Research Center.
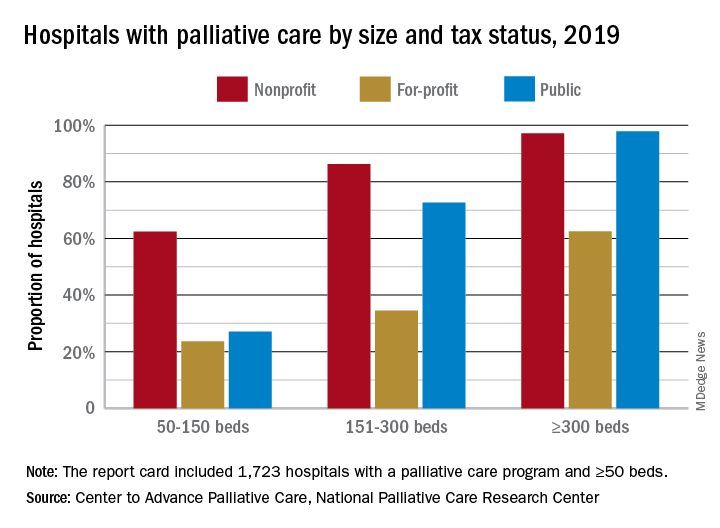
“As is true for many aspects of health care, geography is destiny. Where you live determines your access to the best quality of life and highest quality of care during a serious illness,” said Diane E. Meier, MD, director of the Center to Advance Palliative Care, in a written statement.
the two organizations said in their 2019 report card on palliative care access. What hasn’t changed since 2015, however, is the country’s overall grade, which remains a B.
Delaware, New Hampshire, Rhode Island, and Vermont have a palliative care program in all of their hospitals with 50 or more beds and each earned a grade of A (palliative care rate of greater than 80%), along with 17 other states. The lowest-performing states – Alabama, Mississippi, New Mexico, Oklahoma, and Wyoming – all received Ds for having a rate below 40%, the CAPC said.
The urban/rural divide also is prominent in palliative care: “90% of hospitals with palliative care are in urban areas. Only 17% of rural hospitals with fifty or more beds report palliative care programs,” the report said.
Hospital type is another source of disparity. Small, nonprofit hospitals are much more likely to offer access to palliative care than either for-profit or public facilities of the same size, but the gap closes as size increases, at least between nonprofit and public hospitals. For the largest institutions, the public hospitals pull into the lead, 98% versus 97%, over the nonprofits, with the for-profit facilities well behind at 63%.
“High quality palliative care has been shown to improve patient and family quality of life, improve patients’ and families’ health care experiences, and in certain diseases, prolong life. Palliative care has also been shown to improve hospital efficiency and reduce unnecessary spending,” said R. Sean Morrison, MD, director of the National Palliative Care Research Center.
The report card is based on data from the American Hospital Association’s Annual Survey Database, with additional data from the National Palliative Care Registry and Center to Advance Palliative Care’s Mapping Community Palliative Care initiative. The final sample included 2,409 hospitals with 50 or more beds.
Growth continues among palliative care programs in the United States, although access often depends “more upon accidents of geography than it does upon the needs of patients,” according to the Center to Advance Palliative Care and the National Palliative Care Research Center.
“As is true for many aspects of health care, geography is destiny. Where you live determines your access to the best quality of life and highest quality of care during a serious illness,” said Diane E. Meier, MD, director of the Center to Advance Palliative Care, in a written statement.
the two organizations said in their 2019 report card on palliative care access. What hasn’t changed since 2015, however, is the country’s overall grade, which remains a B.
Delaware, New Hampshire, Rhode Island, and Vermont have a palliative care program in all of their hospitals with 50 or more beds and each earned a grade of A (palliative care rate of greater than 80%), along with 17 other states. The lowest-performing states – Alabama, Mississippi, New Mexico, Oklahoma, and Wyoming – all received Ds for having a rate below 40%, the CAPC said.
The urban/rural divide also is prominent in palliative care: “90% of hospitals with palliative care are in urban areas. Only 17% of rural hospitals with fifty or more beds report palliative care programs,” the report said.
Hospital type is another source of disparity. Small, nonprofit hospitals are much more likely to offer access to palliative care than either for-profit or public facilities of the same size, but the gap closes as size increases, at least between nonprofit and public hospitals. For the largest institutions, the public hospitals pull into the lead, 98% versus 97%, over the nonprofits, with the for-profit facilities well behind at 63%.
“High quality palliative care has been shown to improve patient and family quality of life, improve patients’ and families’ health care experiences, and in certain diseases, prolong life. Palliative care has also been shown to improve hospital efficiency and reduce unnecessary spending,” said R. Sean Morrison, MD, director of the National Palliative Care Research Center.
The report card is based on data from the American Hospital Association’s Annual Survey Database, with additional data from the National Palliative Care Registry and Center to Advance Palliative Care’s Mapping Community Palliative Care initiative. The final sample included 2,409 hospitals with 50 or more beds.
Growth continues among palliative care programs in the United States, although access often depends “more upon accidents of geography than it does upon the needs of patients,” according to the Center to Advance Palliative Care and the National Palliative Care Research Center.
“As is true for many aspects of health care, geography is destiny. Where you live determines your access to the best quality of life and highest quality of care during a serious illness,” said Diane E. Meier, MD, director of the Center to Advance Palliative Care, in a written statement.
the two organizations said in their 2019 report card on palliative care access. What hasn’t changed since 2015, however, is the country’s overall grade, which remains a B.
Delaware, New Hampshire, Rhode Island, and Vermont have a palliative care program in all of their hospitals with 50 or more beds and each earned a grade of A (palliative care rate of greater than 80%), along with 17 other states. The lowest-performing states – Alabama, Mississippi, New Mexico, Oklahoma, and Wyoming – all received Ds for having a rate below 40%, the CAPC said.
The urban/rural divide also is prominent in palliative care: “90% of hospitals with palliative care are in urban areas. Only 17% of rural hospitals with fifty or more beds report palliative care programs,” the report said.
Hospital type is another source of disparity. Small, nonprofit hospitals are much more likely to offer access to palliative care than either for-profit or public facilities of the same size, but the gap closes as size increases, at least between nonprofit and public hospitals. For the largest institutions, the public hospitals pull into the lead, 98% versus 97%, over the nonprofits, with the for-profit facilities well behind at 63%.
“High quality palliative care has been shown to improve patient and family quality of life, improve patients’ and families’ health care experiences, and in certain diseases, prolong life. Palliative care has also been shown to improve hospital efficiency and reduce unnecessary spending,” said R. Sean Morrison, MD, director of the National Palliative Care Research Center.
The report card is based on data from the American Hospital Association’s Annual Survey Database, with additional data from the National Palliative Care Registry and Center to Advance Palliative Care’s Mapping Community Palliative Care initiative. The final sample included 2,409 hospitals with 50 or more beds.
Apixaban prevents clots with cancer
Background: Active cancer places patients at increased risk for VTE. Ambulatory patients can be risk stratified using the validated Khorana score to assess risk for VTE, a complication resulting in significant morbidity, mortality, and health care costs.

Study design: Randomized, placebo-controlled, double-blind clinical trial.
Setting: Ambulatory; Canada.
Synopsis: A total of 1,809 patients were assessed for eligibility, 1,235 were excluded, and 574 with a Khorana score of 2 or higher were randomized to apixaban 2.5 mg twice daily or placebo. Treatment or placebo was given within 24 hours after the initiation of chemotherapy and continued for 180 days. The primary efficacy outcome – first episode of major VTE within 180 days of randomization – occurred in 4.2% of the apixaban group and in 10.2% of the placebo group (hazard ratio, 0.41; 95% confidence interval, 0.26-0.65; P less than .001). Major bleeding in the modified intention-to-treat analysis occurred in 3.5% in the apixaban group and 1.8% in the placebo group (HR, 2.00; 95% CI, 1.01-3.95; P = .046).
There was no significant difference in overall survival, with 87% of deaths were related to cancer or cancer progression.
Bottom line: VTE is significantly lower with the use of apixaban, compared with placebo, in intermediate- to high-risk ambulatory patients with active cancer who are initiating chemotherapy.
Citation: Carrier M et al. Apixaban to prevent venous thromboembolism in patients with cancer. N Engl J Med. 2018 Dec 4. doi: 10.1056/NEJMoa1814468.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Active cancer places patients at increased risk for VTE. Ambulatory patients can be risk stratified using the validated Khorana score to assess risk for VTE, a complication resulting in significant morbidity, mortality, and health care costs.
Study design: Randomized, placebo-controlled, double-blind clinical trial.
Setting: Ambulatory; Canada.
Synopsis: A total of 1,809 patients were assessed for eligibility, 1,235 were excluded, and 574 with a Khorana score of 2 or higher were randomized to apixaban 2.5 mg twice daily or placebo. Treatment or placebo was given within 24 hours after the initiation of chemotherapy and continued for 180 days. The primary efficacy outcome – first episode of major VTE within 180 days of randomization – occurred in 4.2% of the apixaban group and in 10.2% of the placebo group (hazard ratio, 0.41; 95% confidence interval, 0.26-0.65; P less than .001). Major bleeding in the modified intention-to-treat analysis occurred in 3.5% in the apixaban group and 1.8% in the placebo group (HR, 2.00; 95% CI, 1.01-3.95; P = .046).
There was no significant difference in overall survival, with 87% of deaths were related to cancer or cancer progression.
Bottom line: VTE is significantly lower with the use of apixaban, compared with placebo, in intermediate- to high-risk ambulatory patients with active cancer who are initiating chemotherapy.
Citation: Carrier M et al. Apixaban to prevent venous thromboembolism in patients with cancer. N Engl J Med. 2018 Dec 4. doi: 10.1056/NEJMoa1814468.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Active cancer places patients at increased risk for VTE. Ambulatory patients can be risk stratified using the validated Khorana score to assess risk for VTE, a complication resulting in significant morbidity, mortality, and health care costs.
Study design: Randomized, placebo-controlled, double-blind clinical trial.
Setting: Ambulatory; Canada.
Synopsis: A total of 1,809 patients were assessed for eligibility, 1,235 were excluded, and 574 with a Khorana score of 2 or higher were randomized to apixaban 2.5 mg twice daily or placebo. Treatment or placebo was given within 24 hours after the initiation of chemotherapy and continued for 180 days. The primary efficacy outcome – first episode of major VTE within 180 days of randomization – occurred in 4.2% of the apixaban group and in 10.2% of the placebo group (hazard ratio, 0.41; 95% confidence interval, 0.26-0.65; P less than .001). Major bleeding in the modified intention-to-treat analysis occurred in 3.5% in the apixaban group and 1.8% in the placebo group (HR, 2.00; 95% CI, 1.01-3.95; P = .046).
There was no significant difference in overall survival, with 87% of deaths were related to cancer or cancer progression.
Bottom line: VTE is significantly lower with the use of apixaban, compared with placebo, in intermediate- to high-risk ambulatory patients with active cancer who are initiating chemotherapy.
Citation: Carrier M et al. Apixaban to prevent venous thromboembolism in patients with cancer. N Engl J Med. 2018 Dec 4. doi: 10.1056/NEJMoa1814468.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
SHM and Jefferson College of Population Health partner to provide vital education for hospitalists
Both the Society of Hospital Medicine and Jefferson College of Population Health, of Thomas Jefferson University in Philadelphia, share a goal to educate physicians to be effective leaders and managers in the pursuit of health care quality, safety, and population health, and they have entered into a partnership with this in mind.

Alexis Skoufalos, EdD, associate dean, strategic development, for Jefferson College of Population Health, recently spoke with The Hospitalist to discuss the importance of population health to hospital medicine professionals, the health care landscape as a whole, and the benefits of this new partnership with SHM.
Can you explain the importance of population health in the current health care landscape?
Many people confuse population health with public health. While they are related, they are different disciplines. Public health focuses on prevention and health promotion (clean water, vaccines, exercise, using seat belts, and so on), but it stops there.
Population health builds on the foundation of public health and goes a step further, working to connect health and health care delivery. It takes a more holistic approach, looking at what we need to do inside and outside the delivery system to help people to get and stay healthy, as well as take better care of them when they do get sick.
We work to identify and understand the health impact of social and environmental factors, while also looking for ways to make health care delivery safer, better, and more affordable and accessible.
This can get complicated. It involves sorting through lots of information to uncover the best way to meet the needs of a specific group, whether that is a community, a neighborhood, or a patient with a particular condition.
It’s about taking the time to really look at things from different vantage points. You won’t see the same view if you are looking at something through a telescope as you would looking through a microscope. That information can help you to adjust your perspective to identify the best course of action.
In order to be successful in improving population health, providers need to understand how to work with the other stakeholders in the health care ecosystem. Collaboration and coordination are the best ways to optimize the resources available.
It is important for delivery systems to establish good working relationships with community nonprofit and service organizations, faith-based organizations, social service providers, school systems, and federal, state, and local government.
At Jefferson, we thought it was important to create a college and programs that would prepare professionals across the workforce for this new challenge.
How did this partnership between SHM and Jefferson College of Population Health come to fruition?
Hospitalists are an important link with a person’s primary care team. The work they do to prepare a person and their family for successful discharge to the community after a hospital stay can make all the difference in a person’s recovery, condition management, and preventing readmission to the hospital.
Because both of our organizations are based in Philadelphia, we have had longstanding connections with SHM leadership. It was only natural for us to talk with SHM about how we can build upon the society’s excellent continuing education offerings and work together to provide members with additional content that can equip them to advance their careers.
How did SHM and Jefferson College of Population Health identify the mutually beneficial educational offerings in each institution that are included in this partnership?
Members of our respective leadership teams got together to complete a detailed review of the offerings from each organization. SHM’s Leadership Academy and JCPH’s Population Health Academy are rigorous continuing education programs that can provide physicians with excellent just-in-time information they can put to use right away.
After a careful examination of the curriculum, JCPH determined that SHM members can apply the credits they earn from completing two qualified sessions from the Leadership Academy to satisfy the elective course requirement for a Master’s degree. (Note: This does not apply to the Population Health Intelligence Program, which does not include an elective course.)
How will this partnership benefit Jefferson College of Population Health?
Our mission is to prepare health care leaders with the skills and tools they need to be effective in improving population health. Clinicians who work in a hospital setting have a key role to play.
We are also dedicated to making a difference right here in Philadelphia. The more students we have in our programs, the more of an impact we (and they) will have in improving outcomes in our own community.
We need to move the needle and get Philadelphia County out of the basement in terms of health rankings. We have a responsibility to do what we can to make a difference, and we appreciate the partnership with SHM to make it happen.
What other components of the partnership are especially noteworthy to highlight?
In addition to what I’ve already discussed, the following are some of the significant benefits that SHM members are entitled to as a result of the partnership with JCPH:
- 15% discount on tuition for any JCPH online graduate degree program.
- Registration discount for JCPH’s Population Health Academy in Philadelphia.
- Special registration rate for Annual Population Health Colloquium.
For more information about this partnership, visit hospitalmedicine.org/jefferson.
Both the Society of Hospital Medicine and Jefferson College of Population Health, of Thomas Jefferson University in Philadelphia, share a goal to educate physicians to be effective leaders and managers in the pursuit of health care quality, safety, and population health, and they have entered into a partnership with this in mind.
Alexis Skoufalos, EdD, associate dean, strategic development, for Jefferson College of Population Health, recently spoke with The Hospitalist to discuss the importance of population health to hospital medicine professionals, the health care landscape as a whole, and the benefits of this new partnership with SHM.
Can you explain the importance of population health in the current health care landscape?
Many people confuse population health with public health. While they are related, they are different disciplines. Public health focuses on prevention and health promotion (clean water, vaccines, exercise, using seat belts, and so on), but it stops there.
Population health builds on the foundation of public health and goes a step further, working to connect health and health care delivery. It takes a more holistic approach, looking at what we need to do inside and outside the delivery system to help people to get and stay healthy, as well as take better care of them when they do get sick.
We work to identify and understand the health impact of social and environmental factors, while also looking for ways to make health care delivery safer, better, and more affordable and accessible.
This can get complicated. It involves sorting through lots of information to uncover the best way to meet the needs of a specific group, whether that is a community, a neighborhood, or a patient with a particular condition.
It’s about taking the time to really look at things from different vantage points. You won’t see the same view if you are looking at something through a telescope as you would looking through a microscope. That information can help you to adjust your perspective to identify the best course of action.
In order to be successful in improving population health, providers need to understand how to work with the other stakeholders in the health care ecosystem. Collaboration and coordination are the best ways to optimize the resources available.
It is important for delivery systems to establish good working relationships with community nonprofit and service organizations, faith-based organizations, social service providers, school systems, and federal, state, and local government.
At Jefferson, we thought it was important to create a college and programs that would prepare professionals across the workforce for this new challenge.
How did this partnership between SHM and Jefferson College of Population Health come to fruition?
Hospitalists are an important link with a person’s primary care team. The work they do to prepare a person and their family for successful discharge to the community after a hospital stay can make all the difference in a person’s recovery, condition management, and preventing readmission to the hospital.
Because both of our organizations are based in Philadelphia, we have had longstanding connections with SHM leadership. It was only natural for us to talk with SHM about how we can build upon the society’s excellent continuing education offerings and work together to provide members with additional content that can equip them to advance their careers.
How did SHM and Jefferson College of Population Health identify the mutually beneficial educational offerings in each institution that are included in this partnership?
Members of our respective leadership teams got together to complete a detailed review of the offerings from each organization. SHM’s Leadership Academy and JCPH’s Population Health Academy are rigorous continuing education programs that can provide physicians with excellent just-in-time information they can put to use right away.
After a careful examination of the curriculum, JCPH determined that SHM members can apply the credits they earn from completing two qualified sessions from the Leadership Academy to satisfy the elective course requirement for a Master’s degree. (Note: This does not apply to the Population Health Intelligence Program, which does not include an elective course.)
How will this partnership benefit Jefferson College of Population Health?
Our mission is to prepare health care leaders with the skills and tools they need to be effective in improving population health. Clinicians who work in a hospital setting have a key role to play.
We are also dedicated to making a difference right here in Philadelphia. The more students we have in our programs, the more of an impact we (and they) will have in improving outcomes in our own community.
We need to move the needle and get Philadelphia County out of the basement in terms of health rankings. We have a responsibility to do what we can to make a difference, and we appreciate the partnership with SHM to make it happen.
What other components of the partnership are especially noteworthy to highlight?
In addition to what I’ve already discussed, the following are some of the significant benefits that SHM members are entitled to as a result of the partnership with JCPH:
- 15% discount on tuition for any JCPH online graduate degree program.
- Registration discount for JCPH’s Population Health Academy in Philadelphia.
- Special registration rate for Annual Population Health Colloquium.
For more information about this partnership, visit hospitalmedicine.org/jefferson.
Both the Society of Hospital Medicine and Jefferson College of Population Health, of Thomas Jefferson University in Philadelphia, share a goal to educate physicians to be effective leaders and managers in the pursuit of health care quality, safety, and population health, and they have entered into a partnership with this in mind.
Alexis Skoufalos, EdD, associate dean, strategic development, for Jefferson College of Population Health, recently spoke with The Hospitalist to discuss the importance of population health to hospital medicine professionals, the health care landscape as a whole, and the benefits of this new partnership with SHM.
Can you explain the importance of population health in the current health care landscape?
Many people confuse population health with public health. While they are related, they are different disciplines. Public health focuses on prevention and health promotion (clean water, vaccines, exercise, using seat belts, and so on), but it stops there.
Population health builds on the foundation of public health and goes a step further, working to connect health and health care delivery. It takes a more holistic approach, looking at what we need to do inside and outside the delivery system to help people to get and stay healthy, as well as take better care of them when they do get sick.
We work to identify and understand the health impact of social and environmental factors, while also looking for ways to make health care delivery safer, better, and more affordable and accessible.
This can get complicated. It involves sorting through lots of information to uncover the best way to meet the needs of a specific group, whether that is a community, a neighborhood, or a patient with a particular condition.
It’s about taking the time to really look at things from different vantage points. You won’t see the same view if you are looking at something through a telescope as you would looking through a microscope. That information can help you to adjust your perspective to identify the best course of action.
In order to be successful in improving population health, providers need to understand how to work with the other stakeholders in the health care ecosystem. Collaboration and coordination are the best ways to optimize the resources available.
It is important for delivery systems to establish good working relationships with community nonprofit and service organizations, faith-based organizations, social service providers, school systems, and federal, state, and local government.
At Jefferson, we thought it was important to create a college and programs that would prepare professionals across the workforce for this new challenge.
How did this partnership between SHM and Jefferson College of Population Health come to fruition?
Hospitalists are an important link with a person’s primary care team. The work they do to prepare a person and their family for successful discharge to the community after a hospital stay can make all the difference in a person’s recovery, condition management, and preventing readmission to the hospital.
Because both of our organizations are based in Philadelphia, we have had longstanding connections with SHM leadership. It was only natural for us to talk with SHM about how we can build upon the society’s excellent continuing education offerings and work together to provide members with additional content that can equip them to advance their careers.
How did SHM and Jefferson College of Population Health identify the mutually beneficial educational offerings in each institution that are included in this partnership?
Members of our respective leadership teams got together to complete a detailed review of the offerings from each organization. SHM’s Leadership Academy and JCPH’s Population Health Academy are rigorous continuing education programs that can provide physicians with excellent just-in-time information they can put to use right away.
After a careful examination of the curriculum, JCPH determined that SHM members can apply the credits they earn from completing two qualified sessions from the Leadership Academy to satisfy the elective course requirement for a Master’s degree. (Note: This does not apply to the Population Health Intelligence Program, which does not include an elective course.)
How will this partnership benefit Jefferson College of Population Health?
Our mission is to prepare health care leaders with the skills and tools they need to be effective in improving population health. Clinicians who work in a hospital setting have a key role to play.
We are also dedicated to making a difference right here in Philadelphia. The more students we have in our programs, the more of an impact we (and they) will have in improving outcomes in our own community.
We need to move the needle and get Philadelphia County out of the basement in terms of health rankings. We have a responsibility to do what we can to make a difference, and we appreciate the partnership with SHM to make it happen.
What other components of the partnership are especially noteworthy to highlight?
In addition to what I’ve already discussed, the following are some of the significant benefits that SHM members are entitled to as a result of the partnership with JCPH:
- 15% discount on tuition for any JCPH online graduate degree program.
- Registration discount for JCPH’s Population Health Academy in Philadelphia.
- Special registration rate for Annual Population Health Colloquium.
For more information about this partnership, visit hospitalmedicine.org/jefferson.
Dual therapy best for AFib with ACS no matter the treatment strategy
SAN FRANCISCO – Anticoagulation with apixaban and a P2Y12 inhibitor without aspirin provides superior safety and similar efficacy in patients with atrial fibrillation who have an acute coronary syndrome, compared with regimens that include vitamin K antagonists, aspirin, or both.

The findings come from a prespecified analysis of data from the AUGUSTUS trial presented by Stephan Windecker, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting.
“This study adds very important information [to the notion] that triple therapy in the setting of atrial fibrillation and PCI [percutaneous coronary intervention] is really not the way to go,” Ori Ben-Yehuda, MD, FACC, executive director of the Cardiovascular Research Foundation’s Clinical Trials Center, said during a media briefing.
In the recent multicenter AUGUSTUS trial, Dr. Windecker, of the department of cardiology at Bern University Hospital, Switzerland, and colleagues found that among 4,614 patients with atrial fibrillation and a recent acute coronary syndrome or PCI treated with a P2Y12 inhibitor, apixaban without aspirin resulted in less bleeding, fewer hospitalizations, and no significant differences in ischemic events compared with regimens that included a vitamin K antagonist (VKA), aspirin, or both (N Engl J Med. 2019;380:1509-24). For this prespecified analysis, the researchers used a 2×2 factorial design to compare apixaban with VKA and aspirin with placebo in the AUGUSTUS trial participants with ACS treated medically (group 1; 1,097 patients, or 24%), those with ACS treated with PCI (group 2; 1,714 patients, or 37%), and those undergoing elective PCI (group 3; 1,784 patients, or 39%). The outcomes of interest were bleeding, death, and hospitalization as well as death and ischemic events by antithrombotic strategy in the study participants. This marks the only trial in the field that included patients with ACS managed medically, Dr. Windecker said.
At baseline, the median age of patients was 71 years, 30% were female, 36% had diabetes, and 45% had heart failure. Patients managed medically were younger (a median age of 70) and more frequently female; 57% presented with heart failure. The groups had identical CHA2DS2VASc scores (4), and very similar HAS-BLED scores (2 in groups 1 and 2, and 3 in group 3).
Apixaban compared with VKA showed lower International Society on Thrombosis and Haemostasis–defined major or clinically relevant nonmajor bleeding among patients in group 1 (HR, 0.44), group 2 (HR, 0.68), and group 3 (HR, 0.82) (P for interaction = .052). Apixaban compared with VKA reduced death or hospitalization among patients in group 1 (HR, 0.71), group 2 (HR 0.88), and group 3 (HR, 0.87) (P for interaction = .345). Compared with VKA, apixaban resulted in a similar effect on death and ischemic events among patients in all three treatment groups (P for interaction = .356).
Compared with placebo, aspirin had a higher rate of bleeding among patients in group 1 (HR, 1.49), group 2 (HR, 2.02) and group 3 (HR, 1.91) (P for interaction = .479). For the same comparison, there was no difference in outcomes among the three groups for the composite of death or hospitalization and death and ischemic events.
“The overall results of the AUGUSTUS trial are consistent across the three clinically important subgroups,” Dr. Windecker said. The reasons why patients received medical therapy remain unclear, “because it was at the physician’s discretion as to whether they were treated medically or underwent PCI,” he said. “The proportion very much reflects our clinical practice, where 20%-25% of patients are treated medically. What was surprising for me is that I would have anticipated there would be more elderly patients with comorbidities, but I did anticipate that there would be more female patients (in this subgroup).”
Robert A. Harrington, MD, an interventional cardiologist at Stanford (Calif.) University who served on the Data Safety and Monitoring Board for the trial, noted that the patients with atrial fibrillation represent 7%-10% of all ACS patients, “so it’s a big population,” he said. “What’s been disappointing is that none of the trials have been big enough to uncouple the bleeding vs. ischemic issue. We don’t know the answer for how long do you need the triple therapy versus when you can switch to the double therapy.”
Dr. Windecker said that the optimal duration of short-term aspirin remains unclear in this patient population. “Whether there is a benefit of giving aspirin for 2 weeks or 4 weeks remains unanswered,” he said. “Triple therapy is not the way to go, but we need to fine-tune, and probably individualize, which patients may benefit from a certain duration of aspirin.”
The study results were published online at the time of presentation (Circulation 2019 Sep 26. doi: 10.1161/CIRCULATIONAHA.119.043308.
AUGUSTUS was funded by Bristol-Myers Squibb and Pfizer Inc. Dr. Windecker reported having received institutional research and educational grants to Bern University Hospital from Abbott, Amgen, Bayer, BMS, CSL Behring, Boston Scientific, Biotronik, Edwards Lifesciences, Medtronic, Polares, and Sinomed. His coauthors reported having numerous financial ties to the pharmaceutical and device industries.
SOURCE: Windecker S. TCT 2019, Late-Breaking Trials 1 session.
SAN FRANCISCO – Anticoagulation with apixaban and a P2Y12 inhibitor without aspirin provides superior safety and similar efficacy in patients with atrial fibrillation who have an acute coronary syndrome, compared with regimens that include vitamin K antagonists, aspirin, or both.
The findings come from a prespecified analysis of data from the AUGUSTUS trial presented by Stephan Windecker, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting.
“This study adds very important information [to the notion] that triple therapy in the setting of atrial fibrillation and PCI [percutaneous coronary intervention] is really not the way to go,” Ori Ben-Yehuda, MD, FACC, executive director of the Cardiovascular Research Foundation’s Clinical Trials Center, said during a media briefing.
In the recent multicenter AUGUSTUS trial, Dr. Windecker, of the department of cardiology at Bern University Hospital, Switzerland, and colleagues found that among 4,614 patients with atrial fibrillation and a recent acute coronary syndrome or PCI treated with a P2Y12 inhibitor, apixaban without aspirin resulted in less bleeding, fewer hospitalizations, and no significant differences in ischemic events compared with regimens that included a vitamin K antagonist (VKA), aspirin, or both (N Engl J Med. 2019;380:1509-24). For this prespecified analysis, the researchers used a 2×2 factorial design to compare apixaban with VKA and aspirin with placebo in the AUGUSTUS trial participants with ACS treated medically (group 1; 1,097 patients, or 24%), those with ACS treated with PCI (group 2; 1,714 patients, or 37%), and those undergoing elective PCI (group 3; 1,784 patients, or 39%). The outcomes of interest were bleeding, death, and hospitalization as well as death and ischemic events by antithrombotic strategy in the study participants. This marks the only trial in the field that included patients with ACS managed medically, Dr. Windecker said.
At baseline, the median age of patients was 71 years, 30% were female, 36% had diabetes, and 45% had heart failure. Patients managed medically were younger (a median age of 70) and more frequently female; 57% presented with heart failure. The groups had identical CHA2DS2VASc scores (4), and very similar HAS-BLED scores (2 in groups 1 and 2, and 3 in group 3).
Apixaban compared with VKA showed lower International Society on Thrombosis and Haemostasis–defined major or clinically relevant nonmajor bleeding among patients in group 1 (HR, 0.44), group 2 (HR, 0.68), and group 3 (HR, 0.82) (P for interaction = .052). Apixaban compared with VKA reduced death or hospitalization among patients in group 1 (HR, 0.71), group 2 (HR 0.88), and group 3 (HR, 0.87) (P for interaction = .345). Compared with VKA, apixaban resulted in a similar effect on death and ischemic events among patients in all three treatment groups (P for interaction = .356).
Compared with placebo, aspirin had a higher rate of bleeding among patients in group 1 (HR, 1.49), group 2 (HR, 2.02) and group 3 (HR, 1.91) (P for interaction = .479). For the same comparison, there was no difference in outcomes among the three groups for the composite of death or hospitalization and death and ischemic events.
“The overall results of the AUGUSTUS trial are consistent across the three clinically important subgroups,” Dr. Windecker said. The reasons why patients received medical therapy remain unclear, “because it was at the physician’s discretion as to whether they were treated medically or underwent PCI,” he said. “The proportion very much reflects our clinical practice, where 20%-25% of patients are treated medically. What was surprising for me is that I would have anticipated there would be more elderly patients with comorbidities, but I did anticipate that there would be more female patients (in this subgroup).”
Robert A. Harrington, MD, an interventional cardiologist at Stanford (Calif.) University who served on the Data Safety and Monitoring Board for the trial, noted that the patients with atrial fibrillation represent 7%-10% of all ACS patients, “so it’s a big population,” he said. “What’s been disappointing is that none of the trials have been big enough to uncouple the bleeding vs. ischemic issue. We don’t know the answer for how long do you need the triple therapy versus when you can switch to the double therapy.”
Dr. Windecker said that the optimal duration of short-term aspirin remains unclear in this patient population. “Whether there is a benefit of giving aspirin for 2 weeks or 4 weeks remains unanswered,” he said. “Triple therapy is not the way to go, but we need to fine-tune, and probably individualize, which patients may benefit from a certain duration of aspirin.”
The study results were published online at the time of presentation (Circulation 2019 Sep 26. doi: 10.1161/CIRCULATIONAHA.119.043308.
AUGUSTUS was funded by Bristol-Myers Squibb and Pfizer Inc. Dr. Windecker reported having received institutional research and educational grants to Bern University Hospital from Abbott, Amgen, Bayer, BMS, CSL Behring, Boston Scientific, Biotronik, Edwards Lifesciences, Medtronic, Polares, and Sinomed. His coauthors reported having numerous financial ties to the pharmaceutical and device industries.
SOURCE: Windecker S. TCT 2019, Late-Breaking Trials 1 session.
SAN FRANCISCO – Anticoagulation with apixaban and a P2Y12 inhibitor without aspirin provides superior safety and similar efficacy in patients with atrial fibrillation who have an acute coronary syndrome, compared with regimens that include vitamin K antagonists, aspirin, or both.
The findings come from a prespecified analysis of data from the AUGUSTUS trial presented by Stephan Windecker, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting.
“This study adds very important information [to the notion] that triple therapy in the setting of atrial fibrillation and PCI [percutaneous coronary intervention] is really not the way to go,” Ori Ben-Yehuda, MD, FACC, executive director of the Cardiovascular Research Foundation’s Clinical Trials Center, said during a media briefing.
In the recent multicenter AUGUSTUS trial, Dr. Windecker, of the department of cardiology at Bern University Hospital, Switzerland, and colleagues found that among 4,614 patients with atrial fibrillation and a recent acute coronary syndrome or PCI treated with a P2Y12 inhibitor, apixaban without aspirin resulted in less bleeding, fewer hospitalizations, and no significant differences in ischemic events compared with regimens that included a vitamin K antagonist (VKA), aspirin, or both (N Engl J Med. 2019;380:1509-24). For this prespecified analysis, the researchers used a 2×2 factorial design to compare apixaban with VKA and aspirin with placebo in the AUGUSTUS trial participants with ACS treated medically (group 1; 1,097 patients, or 24%), those with ACS treated with PCI (group 2; 1,714 patients, or 37%), and those undergoing elective PCI (group 3; 1,784 patients, or 39%). The outcomes of interest were bleeding, death, and hospitalization as well as death and ischemic events by antithrombotic strategy in the study participants. This marks the only trial in the field that included patients with ACS managed medically, Dr. Windecker said.
At baseline, the median age of patients was 71 years, 30% were female, 36% had diabetes, and 45% had heart failure. Patients managed medically were younger (a median age of 70) and more frequently female; 57% presented with heart failure. The groups had identical CHA2DS2VASc scores (4), and very similar HAS-BLED scores (2 in groups 1 and 2, and 3 in group 3).
Apixaban compared with VKA showed lower International Society on Thrombosis and Haemostasis–defined major or clinically relevant nonmajor bleeding among patients in group 1 (HR, 0.44), group 2 (HR, 0.68), and group 3 (HR, 0.82) (P for interaction = .052). Apixaban compared with VKA reduced death or hospitalization among patients in group 1 (HR, 0.71), group 2 (HR 0.88), and group 3 (HR, 0.87) (P for interaction = .345). Compared with VKA, apixaban resulted in a similar effect on death and ischemic events among patients in all three treatment groups (P for interaction = .356).
Compared with placebo, aspirin had a higher rate of bleeding among patients in group 1 (HR, 1.49), group 2 (HR, 2.02) and group 3 (HR, 1.91) (P for interaction = .479). For the same comparison, there was no difference in outcomes among the three groups for the composite of death or hospitalization and death and ischemic events.
“The overall results of the AUGUSTUS trial are consistent across the three clinically important subgroups,” Dr. Windecker said. The reasons why patients received medical therapy remain unclear, “because it was at the physician’s discretion as to whether they were treated medically or underwent PCI,” he said. “The proportion very much reflects our clinical practice, where 20%-25% of patients are treated medically. What was surprising for me is that I would have anticipated there would be more elderly patients with comorbidities, but I did anticipate that there would be more female patients (in this subgroup).”
Robert A. Harrington, MD, an interventional cardiologist at Stanford (Calif.) University who served on the Data Safety and Monitoring Board for the trial, noted that the patients with atrial fibrillation represent 7%-10% of all ACS patients, “so it’s a big population,” he said. “What’s been disappointing is that none of the trials have been big enough to uncouple the bleeding vs. ischemic issue. We don’t know the answer for how long do you need the triple therapy versus when you can switch to the double therapy.”
Dr. Windecker said that the optimal duration of short-term aspirin remains unclear in this patient population. “Whether there is a benefit of giving aspirin for 2 weeks or 4 weeks remains unanswered,” he said. “Triple therapy is not the way to go, but we need to fine-tune, and probably individualize, which patients may benefit from a certain duration of aspirin.”
The study results were published online at the time of presentation (Circulation 2019 Sep 26. doi: 10.1161/CIRCULATIONAHA.119.043308.
AUGUSTUS was funded by Bristol-Myers Squibb and Pfizer Inc. Dr. Windecker reported having received institutional research and educational grants to Bern University Hospital from Abbott, Amgen, Bayer, BMS, CSL Behring, Boston Scientific, Biotronik, Edwards Lifesciences, Medtronic, Polares, and Sinomed. His coauthors reported having numerous financial ties to the pharmaceutical and device industries.
SOURCE: Windecker S. TCT 2019, Late-Breaking Trials 1 session.
AT TCT 2019