User login
Risankizumab impresses in phase 2 psoriatic arthritis trial
AMSTERDAM – Phase 2 data with the IL-23 inhibitor risankizumab at week 24 were even more impressive than the week 16 data, showing that without any further dosing after week 16, all doses provided protection against radiographic progression relative to placebo at 24 weeks, according to data presented at the European Congress of Rheumatology.
In a video interview, first author Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle, explained that it is not only the high rates of response to risankizumab but also the prolonged response that are attracting attention.
Risankizumab is among several monoclonal antibodies developed to target the p19 subunit of the proinflammatory cytokine IL-23. These drugs have already shown a high degree of efficacy for psoriasis, according to Dr. Mease. However, the new data with risankizumab confirm prolonged responses against a broad range of additional clinical targets specific to psoriatic arthritis, including bone destruction and enthesitis.
A prolonged response in patients treated with a single, relatively low dose of risankizumab is one of the intriguing findings. While three of the four active treatments arms received multiple infusions of 150 mg, the single-dose arm received only 75 mg of risankizumab once at baseline. At 16 weeks and 24 weeks, all arms, including the single-dose arm, met the primary endpoint of superiority to placebo for ACR20. At week 24, the single infusion of 75 mg was also providing significant benefit for several secondary endpoints, including radiographic progression.
However, the higher, more frequent doses did show greater efficacy overall. For example, patients in the arm with the most frequent dosing of risankizumab (every 4 weeks) and no dosing after week 16 continued to show significant improvement in enthesitis. A less frequent schedule of 150 mg risankizumab and the arm receiving a single dose of 75 mg risankizumab were not associated with a significant advantage over placebo for this endpoint.
Still, the prolonged responses at week 24 suggest that it may be possible to administer risankizumab at intervals that are less frequent than many other biologics.
So far, there “is nothing remarkable about safety,” Dr. Mease explained. A higher rate of infection relative to placebo was a treatment-emergent side effect in this study, but Dr. Mease said the drug is well tolerated.
Risankizumab is poised for evaluation in a phase 3 trial for psoriatic arthritis, and Dr. Mease was optimistic about its potential role, predicting that this, as well as other anti-IL23 p19 monoclonal antibodies, is likely to be an “important addition to our armamentarium.”
AbbVie and Boehringer Ingelheim funded the risankizumab study. Dr. Mease has received grant/research support from AbbVie and many other pharmaceutical companies. He also is a consultant to them and is on their speakers bureaus.
SOURCE: Mease P et al. Ann Rheum Dis. 2018;77(Suppl 2):200-1. Abstract OP0307
AMSTERDAM – Phase 2 data with the IL-23 inhibitor risankizumab at week 24 were even more impressive than the week 16 data, showing that without any further dosing after week 16, all doses provided protection against radiographic progression relative to placebo at 24 weeks, according to data presented at the European Congress of Rheumatology.
In a video interview, first author Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle, explained that it is not only the high rates of response to risankizumab but also the prolonged response that are attracting attention.
Risankizumab is among several monoclonal antibodies developed to target the p19 subunit of the proinflammatory cytokine IL-23. These drugs have already shown a high degree of efficacy for psoriasis, according to Dr. Mease. However, the new data with risankizumab confirm prolonged responses against a broad range of additional clinical targets specific to psoriatic arthritis, including bone destruction and enthesitis.
A prolonged response in patients treated with a single, relatively low dose of risankizumab is one of the intriguing findings. While three of the four active treatments arms received multiple infusions of 150 mg, the single-dose arm received only 75 mg of risankizumab once at baseline. At 16 weeks and 24 weeks, all arms, including the single-dose arm, met the primary endpoint of superiority to placebo for ACR20. At week 24, the single infusion of 75 mg was also providing significant benefit for several secondary endpoints, including radiographic progression.
However, the higher, more frequent doses did show greater efficacy overall. For example, patients in the arm with the most frequent dosing of risankizumab (every 4 weeks) and no dosing after week 16 continued to show significant improvement in enthesitis. A less frequent schedule of 150 mg risankizumab and the arm receiving a single dose of 75 mg risankizumab were not associated with a significant advantage over placebo for this endpoint.
Still, the prolonged responses at week 24 suggest that it may be possible to administer risankizumab at intervals that are less frequent than many other biologics.
So far, there “is nothing remarkable about safety,” Dr. Mease explained. A higher rate of infection relative to placebo was a treatment-emergent side effect in this study, but Dr. Mease said the drug is well tolerated.
Risankizumab is poised for evaluation in a phase 3 trial for psoriatic arthritis, and Dr. Mease was optimistic about its potential role, predicting that this, as well as other anti-IL23 p19 monoclonal antibodies, is likely to be an “important addition to our armamentarium.”
AbbVie and Boehringer Ingelheim funded the risankizumab study. Dr. Mease has received grant/research support from AbbVie and many other pharmaceutical companies. He also is a consultant to them and is on their speakers bureaus.
SOURCE: Mease P et al. Ann Rheum Dis. 2018;77(Suppl 2):200-1. Abstract OP0307
AMSTERDAM – Phase 2 data with the IL-23 inhibitor risankizumab at week 24 were even more impressive than the week 16 data, showing that without any further dosing after week 16, all doses provided protection against radiographic progression relative to placebo at 24 weeks, according to data presented at the European Congress of Rheumatology.
In a video interview, first author Philip J. Mease, MD, a rheumatologist at Swedish Medical Center in Seattle, explained that it is not only the high rates of response to risankizumab but also the prolonged response that are attracting attention.
Risankizumab is among several monoclonal antibodies developed to target the p19 subunit of the proinflammatory cytokine IL-23. These drugs have already shown a high degree of efficacy for psoriasis, according to Dr. Mease. However, the new data with risankizumab confirm prolonged responses against a broad range of additional clinical targets specific to psoriatic arthritis, including bone destruction and enthesitis.
A prolonged response in patients treated with a single, relatively low dose of risankizumab is one of the intriguing findings. While three of the four active treatments arms received multiple infusions of 150 mg, the single-dose arm received only 75 mg of risankizumab once at baseline. At 16 weeks and 24 weeks, all arms, including the single-dose arm, met the primary endpoint of superiority to placebo for ACR20. At week 24, the single infusion of 75 mg was also providing significant benefit for several secondary endpoints, including radiographic progression.
However, the higher, more frequent doses did show greater efficacy overall. For example, patients in the arm with the most frequent dosing of risankizumab (every 4 weeks) and no dosing after week 16 continued to show significant improvement in enthesitis. A less frequent schedule of 150 mg risankizumab and the arm receiving a single dose of 75 mg risankizumab were not associated with a significant advantage over placebo for this endpoint.
Still, the prolonged responses at week 24 suggest that it may be possible to administer risankizumab at intervals that are less frequent than many other biologics.
So far, there “is nothing remarkable about safety,” Dr. Mease explained. A higher rate of infection relative to placebo was a treatment-emergent side effect in this study, but Dr. Mease said the drug is well tolerated.
Risankizumab is poised for evaluation in a phase 3 trial for psoriatic arthritis, and Dr. Mease was optimistic about its potential role, predicting that this, as well as other anti-IL23 p19 monoclonal antibodies, is likely to be an “important addition to our armamentarium.”
AbbVie and Boehringer Ingelheim funded the risankizumab study. Dr. Mease has received grant/research support from AbbVie and many other pharmaceutical companies. He also is a consultant to them and is on their speakers bureaus.
SOURCE: Mease P et al. Ann Rheum Dis. 2018;77(Suppl 2):200-1. Abstract OP0307
REPORTING FROM the EULAR 2018 Congress

Diabetes patients pushed into high-deductible plans
ORLANDO – The proportion of diabetes patients enrolled in high-deductible health plans jumped from 10% in 2005 to about 50% in 2014, according to a review of insurance data for 63 million Americans under age 65 years.
Diabetes patients often don’t have a choice. To cut costs, high-deductible plans are increasingly the only ones employers offer.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While that may be adequate for healthy people, it’s quite another issue for people with chronic conditions, especially ones with low income. Out-of-pocket expenses can be thousands of dollars more than with traditional health plans, and the extra costs aren’t always offset by lower premiums.
The trend is concerning, said senior investigator J. Frank Wharam, MB, MPH, an associate professor of population medicine at Harvard Medical School, Boston. He explained the problem, and what’s being done about it, in an interview at the annual scientific sessions of the American Diabetes Association.
SOURCE: Garabedian LF et al. ADA 2018. Abstract 175-OR.
ORLANDO – The proportion of diabetes patients enrolled in high-deductible health plans jumped from 10% in 2005 to about 50% in 2014, according to a review of insurance data for 63 million Americans under age 65 years.
Diabetes patients often don’t have a choice. To cut costs, high-deductible plans are increasingly the only ones employers offer.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While that may be adequate for healthy people, it’s quite another issue for people with chronic conditions, especially ones with low income. Out-of-pocket expenses can be thousands of dollars more than with traditional health plans, and the extra costs aren’t always offset by lower premiums.
The trend is concerning, said senior investigator J. Frank Wharam, MB, MPH, an associate professor of population medicine at Harvard Medical School, Boston. He explained the problem, and what’s being done about it, in an interview at the annual scientific sessions of the American Diabetes Association.
SOURCE: Garabedian LF et al. ADA 2018. Abstract 175-OR.
ORLANDO – The proportion of diabetes patients enrolled in high-deductible health plans jumped from 10% in 2005 to about 50% in 2014, according to a review of insurance data for 63 million Americans under age 65 years.
Diabetes patients often don’t have a choice. To cut costs, high-deductible plans are increasingly the only ones employers offer.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While that may be adequate for healthy people, it’s quite another issue for people with chronic conditions, especially ones with low income. Out-of-pocket expenses can be thousands of dollars more than with traditional health plans, and the extra costs aren’t always offset by lower premiums.
The trend is concerning, said senior investigator J. Frank Wharam, MB, MPH, an associate professor of population medicine at Harvard Medical School, Boston. He explained the problem, and what’s being done about it, in an interview at the annual scientific sessions of the American Diabetes Association.
SOURCE: Garabedian LF et al. ADA 2018. Abstract 175-OR.
REPORTING FROM ADA 2018

KEYNOTE-427: Pembrolizumab monotherapy shows promise in accRCC
CHICAGO – (accRCC), according to findings from the phase 2 KEYNOTE-427 study.
At a median follow-up of 12 months, the overall response rate in 110 study participants with at least one post-baseline assessment was 38%. Three patients (2.7%) achieved a complete response and 39 (35.5%) achieved a partial response, David F. McDermott, MD, reported at the annual meeting of the American Society of Clinical Oncology.
“The disease control rate was 59%,” he said.
Overall, 67% of the patients experienced a reduction in tumor burden, 14% experienced at least an 80% reduction, and 7% experienced a 100% reduction of their target lesion, said Dr. McDermott of Beth Israel Deaconess Medical Center, Boston.
“Most tumor responses occurred early in the course of therapy,” he noted.
The median time to response was 2.8 months, and the median duration of response was not reached at data cutoff, but 74.8% of responders had a response lasting at least 6 months.
An analysis by International Metastatic Renal Cell Carcinoma Database Criteria (IMDC) category showed a confirmed overall response rate (ORR) of 32% among 41 patients with favorable risk, and 42% in 69 patients with intermediate or poor risk.
“Nine of 17 patients in the poor risk group achieved a major response,” Dr. McDermott noted. “Complete and durable responders were seen in all IMDC subgroups.”
In 46 patients with increased PD-L1 expression or a combined positive score of at least 1 the confirmed ORR was 50.0%, and in 53 patients with low PD-L1 expression and a combined positive score less than 1 it was 26%. The ORR was 45% in the remaining patients in whom PD-L1testing could not be performed.
“Of note, all of the complete responses were seen in the PD-L1-high or CPS-greater-than-1 group,” he said.
Median progression-free survival was 8.7 months, and median overall survival has not been reached.
Tolerability of pembrolizumab in this study was acceptable and consistent with that seen with pembrolizumab monotherapy in other tumor types. Although 80% of patients experienced a treatment-related adverse event, the events mainly included fatigue, pruritus, diarrhea, rash, and arthralgia, occurring in 12.7% to 27.3% of patients, he said.
Grade 3/4 events occurred in 21.8% of patients and one patient experienced a fatal grade 5 case of pneumonitis, he added, noting that 11% of patients discontinued treatment because of a treatment-related adverse event.
Overall, 61 patients discontinued therapy, and 33 of those discontinued because of disease progression.
Programmed death-1 (PD-1) inhibitor-based combination therapies have been shown to have clinical benefit when used first-line in accRCC, but data with respect to the clinical impact of first-line PD-1 inhibitor monotherapy are lacking, Dr. McDermott explained.
KEYNOTE-427 was a single-arm, open-label, two-cohort study evaluating the efficacy and safety of pembrolizumab as first-line monotherapy in accRCC and advanced non–clear cell RCC (anccRCC). Patients had accRCC or anccRCC, measurable disease, no prior systemic therapy and Karnofsky Performance Status score of 70% or greater. They were treated with intravenous pembrolizumab at a dose of 200 mg every 3 weeks, and response was assessed at week 12, then every 6 weeks thereafter until week 54, then every 12 weeks.
The current analysis focused on the accRCC cohort and showed that in treatment-naive patients with histologically confirmed accRCC and measurable disease, pembrolizumab shows promising antitumor activity across IMDC risk groups, he said.
“Encouraging activity was also observed in key subgroups, such as the IMDC intermediate/poor risk group ... and patients with [programmed death-ligand 1]-positive tumors,” he said. “The findings ... provide support for the exploration of pembrolizumab in the adjuvant setting and will allow investigators to put the benefit of anti-PD-1-based combination therapies in better context,” he concluded, noting that KEYNOTE-564, a study of pembrolizumab in the adjuvant setting is currently enrolling, and the current study (KEYNOTE-427) cohort B exploring pembrolizumab monotherapy in anccRCC patients is ongoing.
Merck sponsored the study. Dr. McDermott reported consulting or advisory roles with Array BioPharma, Bristol-Myers Squibb, Exelixis, Genentech/Roche, Merck, Novartis, Pfizer, and X4 Pharma. His institution has received research funding from Prometheus Laboratories.
SOURCE: McDermott DF et al., ASCO 2018 Abstract 4500.
CHICAGO – (accRCC), according to findings from the phase 2 KEYNOTE-427 study.
At a median follow-up of 12 months, the overall response rate in 110 study participants with at least one post-baseline assessment was 38%. Three patients (2.7%) achieved a complete response and 39 (35.5%) achieved a partial response, David F. McDermott, MD, reported at the annual meeting of the American Society of Clinical Oncology.
“The disease control rate was 59%,” he said.
Overall, 67% of the patients experienced a reduction in tumor burden, 14% experienced at least an 80% reduction, and 7% experienced a 100% reduction of their target lesion, said Dr. McDermott of Beth Israel Deaconess Medical Center, Boston.
“Most tumor responses occurred early in the course of therapy,” he noted.
The median time to response was 2.8 months, and the median duration of response was not reached at data cutoff, but 74.8% of responders had a response lasting at least 6 months.
An analysis by International Metastatic Renal Cell Carcinoma Database Criteria (IMDC) category showed a confirmed overall response rate (ORR) of 32% among 41 patients with favorable risk, and 42% in 69 patients with intermediate or poor risk.
“Nine of 17 patients in the poor risk group achieved a major response,” Dr. McDermott noted. “Complete and durable responders were seen in all IMDC subgroups.”
In 46 patients with increased PD-L1 expression or a combined positive score of at least 1 the confirmed ORR was 50.0%, and in 53 patients with low PD-L1 expression and a combined positive score less than 1 it was 26%. The ORR was 45% in the remaining patients in whom PD-L1testing could not be performed.
“Of note, all of the complete responses were seen in the PD-L1-high or CPS-greater-than-1 group,” he said.
Median progression-free survival was 8.7 months, and median overall survival has not been reached.
Tolerability of pembrolizumab in this study was acceptable and consistent with that seen with pembrolizumab monotherapy in other tumor types. Although 80% of patients experienced a treatment-related adverse event, the events mainly included fatigue, pruritus, diarrhea, rash, and arthralgia, occurring in 12.7% to 27.3% of patients, he said.
Grade 3/4 events occurred in 21.8% of patients and one patient experienced a fatal grade 5 case of pneumonitis, he added, noting that 11% of patients discontinued treatment because of a treatment-related adverse event.
Overall, 61 patients discontinued therapy, and 33 of those discontinued because of disease progression.
Programmed death-1 (PD-1) inhibitor-based combination therapies have been shown to have clinical benefit when used first-line in accRCC, but data with respect to the clinical impact of first-line PD-1 inhibitor monotherapy are lacking, Dr. McDermott explained.
KEYNOTE-427 was a single-arm, open-label, two-cohort study evaluating the efficacy and safety of pembrolizumab as first-line monotherapy in accRCC and advanced non–clear cell RCC (anccRCC). Patients had accRCC or anccRCC, measurable disease, no prior systemic therapy and Karnofsky Performance Status score of 70% or greater. They were treated with intravenous pembrolizumab at a dose of 200 mg every 3 weeks, and response was assessed at week 12, then every 6 weeks thereafter until week 54, then every 12 weeks.
The current analysis focused on the accRCC cohort and showed that in treatment-naive patients with histologically confirmed accRCC and measurable disease, pembrolizumab shows promising antitumor activity across IMDC risk groups, he said.
“Encouraging activity was also observed in key subgroups, such as the IMDC intermediate/poor risk group ... and patients with [programmed death-ligand 1]-positive tumors,” he said. “The findings ... provide support for the exploration of pembrolizumab in the adjuvant setting and will allow investigators to put the benefit of anti-PD-1-based combination therapies in better context,” he concluded, noting that KEYNOTE-564, a study of pembrolizumab in the adjuvant setting is currently enrolling, and the current study (KEYNOTE-427) cohort B exploring pembrolizumab monotherapy in anccRCC patients is ongoing.
Merck sponsored the study. Dr. McDermott reported consulting or advisory roles with Array BioPharma, Bristol-Myers Squibb, Exelixis, Genentech/Roche, Merck, Novartis, Pfizer, and X4 Pharma. His institution has received research funding from Prometheus Laboratories.
SOURCE: McDermott DF et al., ASCO 2018 Abstract 4500.
CHICAGO – (accRCC), according to findings from the phase 2 KEYNOTE-427 study.
At a median follow-up of 12 months, the overall response rate in 110 study participants with at least one post-baseline assessment was 38%. Three patients (2.7%) achieved a complete response and 39 (35.5%) achieved a partial response, David F. McDermott, MD, reported at the annual meeting of the American Society of Clinical Oncology.
“The disease control rate was 59%,” he said.
Overall, 67% of the patients experienced a reduction in tumor burden, 14% experienced at least an 80% reduction, and 7% experienced a 100% reduction of their target lesion, said Dr. McDermott of Beth Israel Deaconess Medical Center, Boston.
“Most tumor responses occurred early in the course of therapy,” he noted.
The median time to response was 2.8 months, and the median duration of response was not reached at data cutoff, but 74.8% of responders had a response lasting at least 6 months.
An analysis by International Metastatic Renal Cell Carcinoma Database Criteria (IMDC) category showed a confirmed overall response rate (ORR) of 32% among 41 patients with favorable risk, and 42% in 69 patients with intermediate or poor risk.
“Nine of 17 patients in the poor risk group achieved a major response,” Dr. McDermott noted. “Complete and durable responders were seen in all IMDC subgroups.”
In 46 patients with increased PD-L1 expression or a combined positive score of at least 1 the confirmed ORR was 50.0%, and in 53 patients with low PD-L1 expression and a combined positive score less than 1 it was 26%. The ORR was 45% in the remaining patients in whom PD-L1testing could not be performed.
“Of note, all of the complete responses were seen in the PD-L1-high or CPS-greater-than-1 group,” he said.
Median progression-free survival was 8.7 months, and median overall survival has not been reached.
Tolerability of pembrolizumab in this study was acceptable and consistent with that seen with pembrolizumab monotherapy in other tumor types. Although 80% of patients experienced a treatment-related adverse event, the events mainly included fatigue, pruritus, diarrhea, rash, and arthralgia, occurring in 12.7% to 27.3% of patients, he said.
Grade 3/4 events occurred in 21.8% of patients and one patient experienced a fatal grade 5 case of pneumonitis, he added, noting that 11% of patients discontinued treatment because of a treatment-related adverse event.
Overall, 61 patients discontinued therapy, and 33 of those discontinued because of disease progression.
Programmed death-1 (PD-1) inhibitor-based combination therapies have been shown to have clinical benefit when used first-line in accRCC, but data with respect to the clinical impact of first-line PD-1 inhibitor monotherapy are lacking, Dr. McDermott explained.
KEYNOTE-427 was a single-arm, open-label, two-cohort study evaluating the efficacy and safety of pembrolizumab as first-line monotherapy in accRCC and advanced non–clear cell RCC (anccRCC). Patients had accRCC or anccRCC, measurable disease, no prior systemic therapy and Karnofsky Performance Status score of 70% or greater. They were treated with intravenous pembrolizumab at a dose of 200 mg every 3 weeks, and response was assessed at week 12, then every 6 weeks thereafter until week 54, then every 12 weeks.
The current analysis focused on the accRCC cohort and showed that in treatment-naive patients with histologically confirmed accRCC and measurable disease, pembrolizumab shows promising antitumor activity across IMDC risk groups, he said.
“Encouraging activity was also observed in key subgroups, such as the IMDC intermediate/poor risk group ... and patients with [programmed death-ligand 1]-positive tumors,” he said. “The findings ... provide support for the exploration of pembrolizumab in the adjuvant setting and will allow investigators to put the benefit of anti-PD-1-based combination therapies in better context,” he concluded, noting that KEYNOTE-564, a study of pembrolizumab in the adjuvant setting is currently enrolling, and the current study (KEYNOTE-427) cohort B exploring pembrolizumab monotherapy in anccRCC patients is ongoing.
Merck sponsored the study. Dr. McDermott reported consulting or advisory roles with Array BioPharma, Bristol-Myers Squibb, Exelixis, Genentech/Roche, Merck, Novartis, Pfizer, and X4 Pharma. His institution has received research funding from Prometheus Laboratories.
SOURCE: McDermott DF et al., ASCO 2018 Abstract 4500.
REPORTING FROM ASCO 2018
Key clinical point: Pembrolizumab monotherapy shows promising efficacy and tolerability in accRCC.
Major finding: Overall response rate was 38%.
Study details: The phase 2 KEYNOTE-427 trial of 110 patients from one of two study cohorts.
Disclosures: Merck sponsored the study. Dr. McDermott reported consulting or advisory roles with Array BioPharma, Bristol-Myers Squibb, Exelixis, Genentech/Roche, Merck, Novartis, Pfizer, and X4 Pharma. His institution has received research funding from Prometheus Laboratories.
Source: McDermott DF et al. ASCO 2018, Abstract 4500.
The fragile gray mass between your ears
He’s almost 10 years younger than me.
He’d been in the hospital for 3 weeks. The ICU room had been decorated, as many families do, with pictures of his life. His wedding. His kids. He and his wife dressed as Darth Vader and Princess Leia for a Halloween party. A few religious items.
He was off sedation. EEG didn’t show any seizures. Head CT just showed the extensive damage from his head injury. The neurosurgeons can evacuate clots and decrease intracranial pressure, but they can’t repair brain tissue.
His wife was long past the point of shock when I met with her. After 3 weeks, she understood what the new normal was and how the lives of both herself and their kids would never be the same. She held his hand at the bedside as we talked, asked me a few pointed questions, and then thanked me for coming in to see him.
For me, it was just another day on call. I walked back to the nurses station, got some coffee from the galley, and sat down to dictate a note. There are always other patients to see on the coverage list.
But it still reminds you.
The brain doesn’t weigh much, just 2-3 pounds; it’s about the size of your fists put together.
But it’s everything that we are, both as individuals and as a species. All that humanity has achieved, good and bad, came from the brain.
The rest of him was in good shape. A healthy guy in his 40s. Probably in better condition than me. But with his brain irreparably damaged, none of that meant anything.

Even after almost 20 years of doing this work, this sort of thing still reminds me how lucky I, and most of us, are – and to be grateful for what I have.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
He’s almost 10 years younger than me.
He’d been in the hospital for 3 weeks. The ICU room had been decorated, as many families do, with pictures of his life. His wedding. His kids. He and his wife dressed as Darth Vader and Princess Leia for a Halloween party. A few religious items.
He was off sedation. EEG didn’t show any seizures. Head CT just showed the extensive damage from his head injury. The neurosurgeons can evacuate clots and decrease intracranial pressure, but they can’t repair brain tissue.
His wife was long past the point of shock when I met with her. After 3 weeks, she understood what the new normal was and how the lives of both herself and their kids would never be the same. She held his hand at the bedside as we talked, asked me a few pointed questions, and then thanked me for coming in to see him.
For me, it was just another day on call. I walked back to the nurses station, got some coffee from the galley, and sat down to dictate a note. There are always other patients to see on the coverage list.
But it still reminds you.
The brain doesn’t weigh much, just 2-3 pounds; it’s about the size of your fists put together.
But it’s everything that we are, both as individuals and as a species. All that humanity has achieved, good and bad, came from the brain.
The rest of him was in good shape. A healthy guy in his 40s. Probably in better condition than me. But with his brain irreparably damaged, none of that meant anything.
Even after almost 20 years of doing this work, this sort of thing still reminds me how lucky I, and most of us, are – and to be grateful for what I have.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
He’s almost 10 years younger than me.
He’d been in the hospital for 3 weeks. The ICU room had been decorated, as many families do, with pictures of his life. His wedding. His kids. He and his wife dressed as Darth Vader and Princess Leia for a Halloween party. A few religious items.
He was off sedation. EEG didn’t show any seizures. Head CT just showed the extensive damage from his head injury. The neurosurgeons can evacuate clots and decrease intracranial pressure, but they can’t repair brain tissue.
His wife was long past the point of shock when I met with her. After 3 weeks, she understood what the new normal was and how the lives of both herself and their kids would never be the same. She held his hand at the bedside as we talked, asked me a few pointed questions, and then thanked me for coming in to see him.
For me, it was just another day on call. I walked back to the nurses station, got some coffee from the galley, and sat down to dictate a note. There are always other patients to see on the coverage list.
But it still reminds you.
The brain doesn’t weigh much, just 2-3 pounds; it’s about the size of your fists put together.
But it’s everything that we are, both as individuals and as a species. All that humanity has achieved, good and bad, came from the brain.
The rest of him was in good shape. A healthy guy in his 40s. Probably in better condition than me. But with his brain irreparably damaged, none of that meant anything.
Even after almost 20 years of doing this work, this sort of thing still reminds me how lucky I, and most of us, are – and to be grateful for what I have.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Hints of nivolumab efficacy seen in biliary tract cancers
BARCELONA – The immune checkpoint inhibitor nivolumab (Opdivo) shows activity against biliary tract cancers (BTC) that have progressed on prior systemic therapies, investigators report.
Among 27 patients with intra- and extrahepatic cholangiocarcinoma and cancers of the gallbladder for whom at least one prior line of therapy had failed, the overall response rate with nivolumab monotherapy was 18.5%, reported Richard Kim, MD of Moffitt Cancer Center, in Tampa.
“Nivolumab demonstrated clinical efficacy in BTC patients. It was very well tolerated, with few grade 3 or 4 adverse events,” he said at the European Society of Medical Oncology World Congress on Gastrointestinal Cancer.
The worldwide incidence of biliary tract cancers has grown over the last 4 decades.
“It is a very aggressive disease, with 5-year overall survival rate of advance disease of less than 2%,” he said.
The standard of care for first-line treatment of advanced disease is gemcitabine and cisplatin, but there is no standard treatment available for patients for whom first-line therapy fails.
Median survival of patients with biliary tract cancers who are receiving second- or third-line therapies is approximately 6-7 months, Dr. Kim said.

The rationale for using nivolumab in this setting comes from evidence suggesting that cholangiocarcinoma is related to dysregulated immunity, with carcinogenesis linked to autoimmune conditions such as primary sclerosing cholangitis, and to chronic parasitic infections.
“Immune regulatory protein PD-1 is upregulated more in intrahepatic cholangiocarcinoma tissues than in adjacent normal tissue, and patients with memory CD8 T cells had longer relapse-free survival and overall survival in extrahepatic cholangiocarcinoma after resection,” he explained.
To see whether the use of an immune checkpoint inhibitor could provide clinically meaningful benefit in patients with advance biliary tract cancers, the investigators conducted a phase 2, two-stage study. They first accrued 18 patients with histologically confirmed, treatment-refractory biliary tract malignancies and treated them with nivolumab 240 mg IV every 2 weeks for 16 weeks, followed by 480 mg IV every 4 weeks.
According to the study protocol, if one or more patients had a complete or partial response, additional patients would be enrolled. As of May 2018, 34 patients had been treated.
The median patient age was 64.5 years. Two-thirds of the patients (64.7%) had intrahepatic cholangiocarcinoma, 2.9% had extrahepatic cholangiocarcinoma, and 32.4% had tumors of the gallbladder.
Twenty patients were failed by their first-line therapies, and 14 were failed by two or more lines of therapy. All 34 received at least one dose of nivolumab.
Of this group, 10 patients remained on study at the time of Dr. Kim’s presentation. Fifteen were withdrawn for progressive disease according to Response Evaluation Criteria in Solid Tumors (RECIST) revision 1.1, and 9 due to clinical progression.
Of 27 patients evaluable for investigator-assessed overall responses – the primary endpoint – 5 patients (18.5%) had a partial response, and 11 (40.7%) had stable disease, for a disease-control rate of 59.3%. The remaining 11 evaluable patients had progressive disease.
“Of interest, of our five patients who had a partial response, three had a diagnosis of intrahepatic cholangiocarcinoma, and two had a diagnosis of a gallbladder tumor,” Dr. Kim said.
All five patients remained on treatment at the time of the presentation, with response duration ranging from 24 to 64 weeks. The median duration of response in these patients has not been reached.
Median progression-free survival for all 34 patients treated with at least one dose was 3.5 months. Overall survival with a median follow-up of 9.9 months has not been reached. The 6-months overall survival rate was 73.5%.
Approximately 20% of patients experienced grade 3 or 4 treatment-related adverse events. There were no grade 4 events and no treatment-related deaths.
The most common grade 3 events were hyponatremia in three patients (8.8%), and lymphopenia, colitis, and hyperbilirubinemia in one patient each (2.9%).
The investigators have collected tissues from all patients and plan to present data from biomarker studies at future meetings. Based on the results of this study, they plan to add 20 more patients to the phase 2 trial to confirm efficacy of nivolumab in this setting.
BARCELONA – The immune checkpoint inhibitor nivolumab (Opdivo) shows activity against biliary tract cancers (BTC) that have progressed on prior systemic therapies, investigators report.
Among 27 patients with intra- and extrahepatic cholangiocarcinoma and cancers of the gallbladder for whom at least one prior line of therapy had failed, the overall response rate with nivolumab monotherapy was 18.5%, reported Richard Kim, MD of Moffitt Cancer Center, in Tampa.
“Nivolumab demonstrated clinical efficacy in BTC patients. It was very well tolerated, with few grade 3 or 4 adverse events,” he said at the European Society of Medical Oncology World Congress on Gastrointestinal Cancer.
The worldwide incidence of biliary tract cancers has grown over the last 4 decades.
“It is a very aggressive disease, with 5-year overall survival rate of advance disease of less than 2%,” he said.
The standard of care for first-line treatment of advanced disease is gemcitabine and cisplatin, but there is no standard treatment available for patients for whom first-line therapy fails.
Median survival of patients with biliary tract cancers who are receiving second- or third-line therapies is approximately 6-7 months, Dr. Kim said.
The rationale for using nivolumab in this setting comes from evidence suggesting that cholangiocarcinoma is related to dysregulated immunity, with carcinogenesis linked to autoimmune conditions such as primary sclerosing cholangitis, and to chronic parasitic infections.
“Immune regulatory protein PD-1 is upregulated more in intrahepatic cholangiocarcinoma tissues than in adjacent normal tissue, and patients with memory CD8 T cells had longer relapse-free survival and overall survival in extrahepatic cholangiocarcinoma after resection,” he explained.
To see whether the use of an immune checkpoint inhibitor could provide clinically meaningful benefit in patients with advance biliary tract cancers, the investigators conducted a phase 2, two-stage study. They first accrued 18 patients with histologically confirmed, treatment-refractory biliary tract malignancies and treated them with nivolumab 240 mg IV every 2 weeks for 16 weeks, followed by 480 mg IV every 4 weeks.
According to the study protocol, if one or more patients had a complete or partial response, additional patients would be enrolled. As of May 2018, 34 patients had been treated.
The median patient age was 64.5 years. Two-thirds of the patients (64.7%) had intrahepatic cholangiocarcinoma, 2.9% had extrahepatic cholangiocarcinoma, and 32.4% had tumors of the gallbladder.
Twenty patients were failed by their first-line therapies, and 14 were failed by two or more lines of therapy. All 34 received at least one dose of nivolumab.
Of this group, 10 patients remained on study at the time of Dr. Kim’s presentation. Fifteen were withdrawn for progressive disease according to Response Evaluation Criteria in Solid Tumors (RECIST) revision 1.1, and 9 due to clinical progression.
Of 27 patients evaluable for investigator-assessed overall responses – the primary endpoint – 5 patients (18.5%) had a partial response, and 11 (40.7%) had stable disease, for a disease-control rate of 59.3%. The remaining 11 evaluable patients had progressive disease.
“Of interest, of our five patients who had a partial response, three had a diagnosis of intrahepatic cholangiocarcinoma, and two had a diagnosis of a gallbladder tumor,” Dr. Kim said.
All five patients remained on treatment at the time of the presentation, with response duration ranging from 24 to 64 weeks. The median duration of response in these patients has not been reached.
Median progression-free survival for all 34 patients treated with at least one dose was 3.5 months. Overall survival with a median follow-up of 9.9 months has not been reached. The 6-months overall survival rate was 73.5%.
Approximately 20% of patients experienced grade 3 or 4 treatment-related adverse events. There were no grade 4 events and no treatment-related deaths.
The most common grade 3 events were hyponatremia in three patients (8.8%), and lymphopenia, colitis, and hyperbilirubinemia in one patient each (2.9%).
The investigators have collected tissues from all patients and plan to present data from biomarker studies at future meetings. Based on the results of this study, they plan to add 20 more patients to the phase 2 trial to confirm efficacy of nivolumab in this setting.
BARCELONA – The immune checkpoint inhibitor nivolumab (Opdivo) shows activity against biliary tract cancers (BTC) that have progressed on prior systemic therapies, investigators report.
Among 27 patients with intra- and extrahepatic cholangiocarcinoma and cancers of the gallbladder for whom at least one prior line of therapy had failed, the overall response rate with nivolumab monotherapy was 18.5%, reported Richard Kim, MD of Moffitt Cancer Center, in Tampa.
“Nivolumab demonstrated clinical efficacy in BTC patients. It was very well tolerated, with few grade 3 or 4 adverse events,” he said at the European Society of Medical Oncology World Congress on Gastrointestinal Cancer.
The worldwide incidence of biliary tract cancers has grown over the last 4 decades.
“It is a very aggressive disease, with 5-year overall survival rate of advance disease of less than 2%,” he said.
The standard of care for first-line treatment of advanced disease is gemcitabine and cisplatin, but there is no standard treatment available for patients for whom first-line therapy fails.
Median survival of patients with biliary tract cancers who are receiving second- or third-line therapies is approximately 6-7 months, Dr. Kim said.
The rationale for using nivolumab in this setting comes from evidence suggesting that cholangiocarcinoma is related to dysregulated immunity, with carcinogenesis linked to autoimmune conditions such as primary sclerosing cholangitis, and to chronic parasitic infections.
“Immune regulatory protein PD-1 is upregulated more in intrahepatic cholangiocarcinoma tissues than in adjacent normal tissue, and patients with memory CD8 T cells had longer relapse-free survival and overall survival in extrahepatic cholangiocarcinoma after resection,” he explained.
To see whether the use of an immune checkpoint inhibitor could provide clinically meaningful benefit in patients with advance biliary tract cancers, the investigators conducted a phase 2, two-stage study. They first accrued 18 patients with histologically confirmed, treatment-refractory biliary tract malignancies and treated them with nivolumab 240 mg IV every 2 weeks for 16 weeks, followed by 480 mg IV every 4 weeks.
According to the study protocol, if one or more patients had a complete or partial response, additional patients would be enrolled. As of May 2018, 34 patients had been treated.
The median patient age was 64.5 years. Two-thirds of the patients (64.7%) had intrahepatic cholangiocarcinoma, 2.9% had extrahepatic cholangiocarcinoma, and 32.4% had tumors of the gallbladder.
Twenty patients were failed by their first-line therapies, and 14 were failed by two or more lines of therapy. All 34 received at least one dose of nivolumab.
Of this group, 10 patients remained on study at the time of Dr. Kim’s presentation. Fifteen were withdrawn for progressive disease according to Response Evaluation Criteria in Solid Tumors (RECIST) revision 1.1, and 9 due to clinical progression.
Of 27 patients evaluable for investigator-assessed overall responses – the primary endpoint – 5 patients (18.5%) had a partial response, and 11 (40.7%) had stable disease, for a disease-control rate of 59.3%. The remaining 11 evaluable patients had progressive disease.
“Of interest, of our five patients who had a partial response, three had a diagnosis of intrahepatic cholangiocarcinoma, and two had a diagnosis of a gallbladder tumor,” Dr. Kim said.
All five patients remained on treatment at the time of the presentation, with response duration ranging from 24 to 64 weeks. The median duration of response in these patients has not been reached.
Median progression-free survival for all 34 patients treated with at least one dose was 3.5 months. Overall survival with a median follow-up of 9.9 months has not been reached. The 6-months overall survival rate was 73.5%.
Approximately 20% of patients experienced grade 3 or 4 treatment-related adverse events. There were no grade 4 events and no treatment-related deaths.
The most common grade 3 events were hyponatremia in three patients (8.8%), and lymphopenia, colitis, and hyperbilirubinemia in one patient each (2.9%).
The investigators have collected tissues from all patients and plan to present data from biomarker studies at future meetings. Based on the results of this study, they plan to add 20 more patients to the phase 2 trial to confirm efficacy of nivolumab in this setting.
REPORTING FROM ESMO GI 2018
Key clinical point: Nivolumab monotherapy appears to have activity in treatment-refractory biliary tract cancers.
Major finding: Five of 27 evaluable patients had partial responses to nivolumab.
Study details: Two-stage phase 2 trial of 34 patients with intrahepatic or extrahepatic cholangiocarcinomas or gallbladder tumors.
Disclosures: Bristol-Myers Squibb sponsored the study. Dr. Kim disclosed honoraria and institutional research funding from that company and others.
Source: Kim R et al. European Society of Medical Oncology World Congress on Gastrointestinal Cancer. Abstract O-009.
Clostridium difficile Colitis in a Patient With Abdominal Distention, Pain, and Severe Constipation
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L). 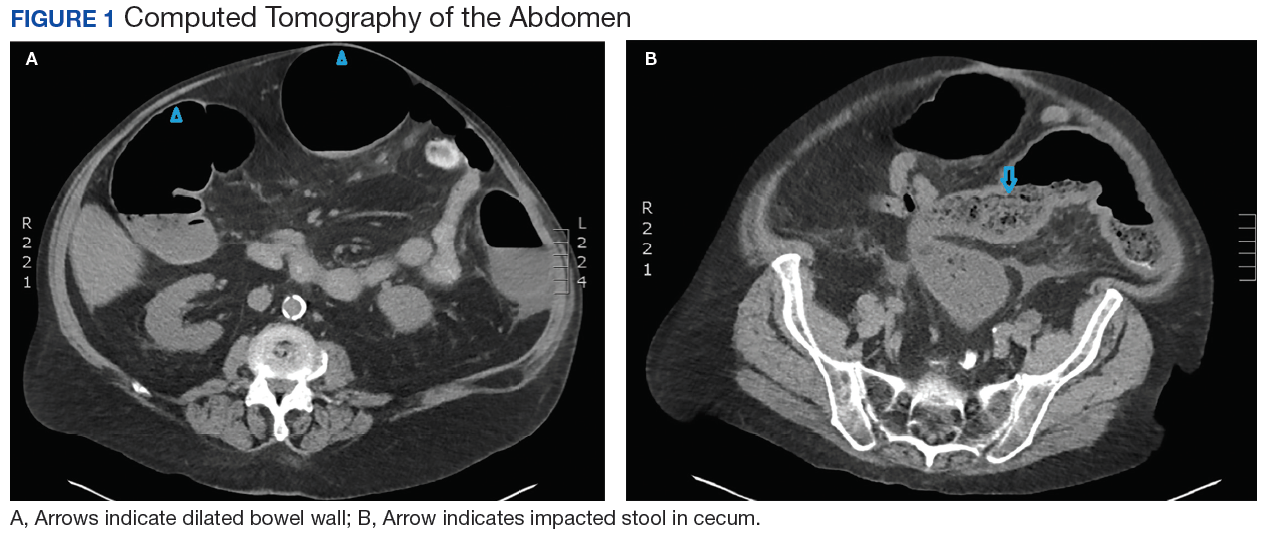
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy. 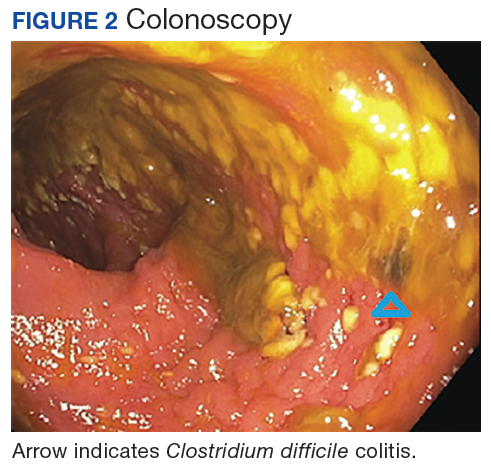
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L).
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy.
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L).
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy.
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
Avapritinib produces durable responses in SM

STOCKHOLM—The KIT/PDGFRA inhibitor avapritinib has produced durable responses in patients with systemic mastocytosis (SM).
In the phase 1 EXPLORER trial, avapritinib produced an overall response rate of 83%.
Responses have lasted up to 22 months, and 79% of responders remained on avapritinib as of the data cutoff.
The most common treatment-related adverse events (AEs) were periorbital edema, anemia, nausea, and fatigue.
These data were presented in a poster (abstract PF612) at the 23rd Congress of the European Hematology Association (EHA).
The trial was sponsored by Blueprint Medicines Corporation.
As of the data cutoff (April 30, 2018), 52 patients had been treated with avapritinib in the dose-escalation and expansion portions of the EXPLORER trial.
This included 25 patients with aggressive SM (ASM), 15 with advanced SM and an associated hematologic neoplasm (SM-AHN), 5 with mast cell leukemia (MCL), 5 pending central pathology diagnosis, and 2 with smoldering SM.
Thirty-five patients (67%) were previously treated, including 10 (19%) who previously received midostaurin. The patients’ median age was 63 (range, 34-83), and 52% were male.
Treatment
Thirty-two patients were treated in the dose-escalation portion of the study and received avapritinib at doses ranging from 30 mg to 400 mg daily. The 35 patients in the expansion portion received avapritinib at 300 mg daily.
Among all 52 enrolled patients, 42 remained on treatment as of the data cutoff date. Four patients discontinued treatment with avapritinib due to AEs. Three of these were treatment-related, and 1 was unrelated.
Three patients discontinued treatment due to clinical progression as determined by the investigator. None of the patients had documented disease progression by IWG-MRT-ECNM criteria.
Two patients discontinued due to investigator decision, and 1 withdrew consent.
Safety
All 52 patients were evaluable for safety.
Treatment-related AEs included periorbital edema (62%), anemia (33%), nausea (33%), fatigue (31%), peripheral edema (27%), diarrhea (25%), hair color changes (23%), thrombocytopenia (19%), cognitive effects (19%), vomiting (19%), and dizziness (12%).
Grade 3 or higher AEs, regardless of drug relationship, included thrombocytopenia (17%), anemia (15%), fatigue (6%), vomiting (6%), periorbital edema (4%), nausea (4%), diarrhea (2%), hair color changes (2%), and cognitive effects (2%).
Efficacy
As of the data cutoff, 23 patients were evaluable for response by IWG-MRT-ECNM criteria. This included 8 patients with ASM, 10 with SM-AHN, and 5 with MCL.
The overall response rate was 83% (n=19). All responses observed in the dose-escalation portion of the trial have been confirmed, and all responses in the dose-expansion portion of the trial are pending confirmation.
Four patients (17%) had a confirmed complete response with a full (n=1) or partial (n=3) recovery of peripheral blood counts. All of these responses occurred in patients with ASM.
Twelve patients (52%) had a partial response (7 confirmed, 5 pending confirmation). This included 6 patients with SM-AHN, 4 with MCL, and 2 with ASM.
Three patients (13%) had clinical improvement (2 confirmed, 1 pending confirmation), and 4 had stable disease. None of the patients progressed.
The duration of response ranged from 8 months to 22 months, and 79% of responders (15/19) remained on treatment at the data cutoff.
“As a clinician treating patients with this devastating and sometimes fatal rare disease, I’m excited to see that most patients with advanced systemic mastocytosis respond to treatment with avapritinib, and these responses deepen over time and are durable,” said study investigator Michael W. Deininger, MD, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City.
“These data further support avapritinib’s unique approach of selectively targeting D816V mutant KIT, the disease driver in most patients with systemic mastocytosis. If these results are confirmed in the planned phase 2 trial, avapritinib has the potential to become a new standard of care for patients with advanced forms of the disease.”
STOCKHOLM—The KIT/PDGFRA inhibitor avapritinib has produced durable responses in patients with systemic mastocytosis (SM).
In the phase 1 EXPLORER trial, avapritinib produced an overall response rate of 83%.
Responses have lasted up to 22 months, and 79% of responders remained on avapritinib as of the data cutoff.
The most common treatment-related adverse events (AEs) were periorbital edema, anemia, nausea, and fatigue.
These data were presented in a poster (abstract PF612) at the 23rd Congress of the European Hematology Association (EHA).
The trial was sponsored by Blueprint Medicines Corporation.
As of the data cutoff (April 30, 2018), 52 patients had been treated with avapritinib in the dose-escalation and expansion portions of the EXPLORER trial.
This included 25 patients with aggressive SM (ASM), 15 with advanced SM and an associated hematologic neoplasm (SM-AHN), 5 with mast cell leukemia (MCL), 5 pending central pathology diagnosis, and 2 with smoldering SM.
Thirty-five patients (67%) were previously treated, including 10 (19%) who previously received midostaurin. The patients’ median age was 63 (range, 34-83), and 52% were male.
Treatment
Thirty-two patients were treated in the dose-escalation portion of the study and received avapritinib at doses ranging from 30 mg to 400 mg daily. The 35 patients in the expansion portion received avapritinib at 300 mg daily.
Among all 52 enrolled patients, 42 remained on treatment as of the data cutoff date. Four patients discontinued treatment with avapritinib due to AEs. Three of these were treatment-related, and 1 was unrelated.
Three patients discontinued treatment due to clinical progression as determined by the investigator. None of the patients had documented disease progression by IWG-MRT-ECNM criteria.
Two patients discontinued due to investigator decision, and 1 withdrew consent.
Safety
All 52 patients were evaluable for safety.
Treatment-related AEs included periorbital edema (62%), anemia (33%), nausea (33%), fatigue (31%), peripheral edema (27%), diarrhea (25%), hair color changes (23%), thrombocytopenia (19%), cognitive effects (19%), vomiting (19%), and dizziness (12%).
Grade 3 or higher AEs, regardless of drug relationship, included thrombocytopenia (17%), anemia (15%), fatigue (6%), vomiting (6%), periorbital edema (4%), nausea (4%), diarrhea (2%), hair color changes (2%), and cognitive effects (2%).
Efficacy
As of the data cutoff, 23 patients were evaluable for response by IWG-MRT-ECNM criteria. This included 8 patients with ASM, 10 with SM-AHN, and 5 with MCL.
The overall response rate was 83% (n=19). All responses observed in the dose-escalation portion of the trial have been confirmed, and all responses in the dose-expansion portion of the trial are pending confirmation.
Four patients (17%) had a confirmed complete response with a full (n=1) or partial (n=3) recovery of peripheral blood counts. All of these responses occurred in patients with ASM.
Twelve patients (52%) had a partial response (7 confirmed, 5 pending confirmation). This included 6 patients with SM-AHN, 4 with MCL, and 2 with ASM.
Three patients (13%) had clinical improvement (2 confirmed, 1 pending confirmation), and 4 had stable disease. None of the patients progressed.
The duration of response ranged from 8 months to 22 months, and 79% of responders (15/19) remained on treatment at the data cutoff.
“As a clinician treating patients with this devastating and sometimes fatal rare disease, I’m excited to see that most patients with advanced systemic mastocytosis respond to treatment with avapritinib, and these responses deepen over time and are durable,” said study investigator Michael W. Deininger, MD, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City.
“These data further support avapritinib’s unique approach of selectively targeting D816V mutant KIT, the disease driver in most patients with systemic mastocytosis. If these results are confirmed in the planned phase 2 trial, avapritinib has the potential to become a new standard of care for patients with advanced forms of the disease.”
STOCKHOLM—The KIT/PDGFRA inhibitor avapritinib has produced durable responses in patients with systemic mastocytosis (SM).
In the phase 1 EXPLORER trial, avapritinib produced an overall response rate of 83%.
Responses have lasted up to 22 months, and 79% of responders remained on avapritinib as of the data cutoff.
The most common treatment-related adverse events (AEs) were periorbital edema, anemia, nausea, and fatigue.
These data were presented in a poster (abstract PF612) at the 23rd Congress of the European Hematology Association (EHA).
The trial was sponsored by Blueprint Medicines Corporation.
As of the data cutoff (April 30, 2018), 52 patients had been treated with avapritinib in the dose-escalation and expansion portions of the EXPLORER trial.
This included 25 patients with aggressive SM (ASM), 15 with advanced SM and an associated hematologic neoplasm (SM-AHN), 5 with mast cell leukemia (MCL), 5 pending central pathology diagnosis, and 2 with smoldering SM.
Thirty-five patients (67%) were previously treated, including 10 (19%) who previously received midostaurin. The patients’ median age was 63 (range, 34-83), and 52% were male.
Treatment
Thirty-two patients were treated in the dose-escalation portion of the study and received avapritinib at doses ranging from 30 mg to 400 mg daily. The 35 patients in the expansion portion received avapritinib at 300 mg daily.
Among all 52 enrolled patients, 42 remained on treatment as of the data cutoff date. Four patients discontinued treatment with avapritinib due to AEs. Three of these were treatment-related, and 1 was unrelated.
Three patients discontinued treatment due to clinical progression as determined by the investigator. None of the patients had documented disease progression by IWG-MRT-ECNM criteria.
Two patients discontinued due to investigator decision, and 1 withdrew consent.
Safety
All 52 patients were evaluable for safety.
Treatment-related AEs included periorbital edema (62%), anemia (33%), nausea (33%), fatigue (31%), peripheral edema (27%), diarrhea (25%), hair color changes (23%), thrombocytopenia (19%), cognitive effects (19%), vomiting (19%), and dizziness (12%).
Grade 3 or higher AEs, regardless of drug relationship, included thrombocytopenia (17%), anemia (15%), fatigue (6%), vomiting (6%), periorbital edema (4%), nausea (4%), diarrhea (2%), hair color changes (2%), and cognitive effects (2%).
Efficacy
As of the data cutoff, 23 patients were evaluable for response by IWG-MRT-ECNM criteria. This included 8 patients with ASM, 10 with SM-AHN, and 5 with MCL.
The overall response rate was 83% (n=19). All responses observed in the dose-escalation portion of the trial have been confirmed, and all responses in the dose-expansion portion of the trial are pending confirmation.
Four patients (17%) had a confirmed complete response with a full (n=1) or partial (n=3) recovery of peripheral blood counts. All of these responses occurred in patients with ASM.
Twelve patients (52%) had a partial response (7 confirmed, 5 pending confirmation). This included 6 patients with SM-AHN, 4 with MCL, and 2 with ASM.
Three patients (13%) had clinical improvement (2 confirmed, 1 pending confirmation), and 4 had stable disease. None of the patients progressed.
The duration of response ranged from 8 months to 22 months, and 79% of responders (15/19) remained on treatment at the data cutoff.
“As a clinician treating patients with this devastating and sometimes fatal rare disease, I’m excited to see that most patients with advanced systemic mastocytosis respond to treatment with avapritinib, and these responses deepen over time and are durable,” said study investigator Michael W. Deininger, MD, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City.
“These data further support avapritinib’s unique approach of selectively targeting D816V mutant KIT, the disease driver in most patients with systemic mastocytosis. If these results are confirmed in the planned phase 2 trial, avapritinib has the potential to become a new standard of care for patients with advanced forms of the disease.”
Urge expectant parents to have prenatal pediatrician visit
All parents-to-be, especially first-time parents, should visit a pediatrician during the third trimester of pregnancy to establish a relationship, according to an updated clinical report on the prenatal visit issued by the American Academy of Pediatrics. The report was published online June 25 and in the July issue of Pediatrics.
“It’s a chance to talk about how to keep a baby safe and thriving physically, but also ways to build strong parent-child bonds that promote resilience and help a child stay emotionally healthy,” Michael Yogman, MD, of Harvard Medical School, Boston, said in a statement. Dr. Yogman was the lead author of the report and chair of the AAP Committee on Psychosocial Aspects of Child and Family Health.
A comprehensive prenatal visit gives pediatricians the opportunity to meet four objectives: build a trusting relationship with parents, gather information about family history, provide advice and guidance on infant care and safety, and identify risk factors for psychosocial issues such as perinatal depression, according to the report in Pediatrics.
The prenatal visit allows families and clinicians to learn whether their philosophies align to start a relationship that may last for many years and this visit can include extended family members such as grandparents. In addition, pediatricians can use the prenatal visit as an opportunity to learn more about family history including past pregnancies, failed and successful, as well as pregnancy complications, chronic medical conditions in family members that may affect the home environment, and plans for child care if parents will be working outside the home.
The report also emphasizes “positive parenting” and the role of pediatricians at a prenatal visit in offering support and guidance to help prepare parents for infant care. This guidance may include advice on feeding, sleeping, diapering, and bathing, as well as acknowledging cultural practices.
The authors noted that a prime opporunity to schedule the prenatal visit is when an expectant parent seeking information about insurance, practice hours, and whether the practice is taking new patients.
The AAP advises clinicians to encourage same sex parents, parents expecting via surrogate, and parents who are adopting to schedule a prenatal visit to identify particular concerns they may have.
“This is the only routine child wellness visit recommended by the American Academy of Pediatrics that doesn’t actually require a child in the room,” coauthor Arthur Lavin, MD, also of Harvard Medical School, said in a statement.
The prenatal visit “gives parents an opportunity to really focus on any questions and concerns they may have. They can talk with a pediatrician before the fatigue of new parenthood sets in and there’s an adorably distracting little human in their arms who may be crying, spitting up, or in immediate need of feeding or a diaper change,” Dr. Lavin said.
“At its heart and soul,” Dr. Lavin noted, “this visit is about laying a foundation for a trusting, supportive relationship between the family and their pediatrician, who will work together to keep the child healthy for the next 18 or 20 years.”
The report recommends the Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents, Fourth Edition, as a resource for clinicians. The researchers had no financial conflicts to disclose.
SOURCE: Yogman M et al. Pediatrics. 2018; doi: 10.1542/peds. 2018-1218
All parents-to-be, especially first-time parents, should visit a pediatrician during the third trimester of pregnancy to establish a relationship, according to an updated clinical report on the prenatal visit issued by the American Academy of Pediatrics. The report was published online June 25 and in the July issue of Pediatrics.
“It’s a chance to talk about how to keep a baby safe and thriving physically, but also ways to build strong parent-child bonds that promote resilience and help a child stay emotionally healthy,” Michael Yogman, MD, of Harvard Medical School, Boston, said in a statement. Dr. Yogman was the lead author of the report and chair of the AAP Committee on Psychosocial Aspects of Child and Family Health.
A comprehensive prenatal visit gives pediatricians the opportunity to meet four objectives: build a trusting relationship with parents, gather information about family history, provide advice and guidance on infant care and safety, and identify risk factors for psychosocial issues such as perinatal depression, according to the report in Pediatrics.
The prenatal visit allows families and clinicians to learn whether their philosophies align to start a relationship that may last for many years and this visit can include extended family members such as grandparents. In addition, pediatricians can use the prenatal visit as an opportunity to learn more about family history including past pregnancies, failed and successful, as well as pregnancy complications, chronic medical conditions in family members that may affect the home environment, and plans for child care if parents will be working outside the home.
The report also emphasizes “positive parenting” and the role of pediatricians at a prenatal visit in offering support and guidance to help prepare parents for infant care. This guidance may include advice on feeding, sleeping, diapering, and bathing, as well as acknowledging cultural practices.
The authors noted that a prime opporunity to schedule the prenatal visit is when an expectant parent seeking information about insurance, practice hours, and whether the practice is taking new patients.
The AAP advises clinicians to encourage same sex parents, parents expecting via surrogate, and parents who are adopting to schedule a prenatal visit to identify particular concerns they may have.
“This is the only routine child wellness visit recommended by the American Academy of Pediatrics that doesn’t actually require a child in the room,” coauthor Arthur Lavin, MD, also of Harvard Medical School, said in a statement.
The prenatal visit “gives parents an opportunity to really focus on any questions and concerns they may have. They can talk with a pediatrician before the fatigue of new parenthood sets in and there’s an adorably distracting little human in their arms who may be crying, spitting up, or in immediate need of feeding or a diaper change,” Dr. Lavin said.
“At its heart and soul,” Dr. Lavin noted, “this visit is about laying a foundation for a trusting, supportive relationship between the family and their pediatrician, who will work together to keep the child healthy for the next 18 or 20 years.”
The report recommends the Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents, Fourth Edition, as a resource for clinicians. The researchers had no financial conflicts to disclose.
SOURCE: Yogman M et al. Pediatrics. 2018; doi: 10.1542/peds. 2018-1218
All parents-to-be, especially first-time parents, should visit a pediatrician during the third trimester of pregnancy to establish a relationship, according to an updated clinical report on the prenatal visit issued by the American Academy of Pediatrics. The report was published online June 25 and in the July issue of Pediatrics.
“It’s a chance to talk about how to keep a baby safe and thriving physically, but also ways to build strong parent-child bonds that promote resilience and help a child stay emotionally healthy,” Michael Yogman, MD, of Harvard Medical School, Boston, said in a statement. Dr. Yogman was the lead author of the report and chair of the AAP Committee on Psychosocial Aspects of Child and Family Health.
A comprehensive prenatal visit gives pediatricians the opportunity to meet four objectives: build a trusting relationship with parents, gather information about family history, provide advice and guidance on infant care and safety, and identify risk factors for psychosocial issues such as perinatal depression, according to the report in Pediatrics.
The prenatal visit allows families and clinicians to learn whether their philosophies align to start a relationship that may last for many years and this visit can include extended family members such as grandparents. In addition, pediatricians can use the prenatal visit as an opportunity to learn more about family history including past pregnancies, failed and successful, as well as pregnancy complications, chronic medical conditions in family members that may affect the home environment, and plans for child care if parents will be working outside the home.
The report also emphasizes “positive parenting” and the role of pediatricians at a prenatal visit in offering support and guidance to help prepare parents for infant care. This guidance may include advice on feeding, sleeping, diapering, and bathing, as well as acknowledging cultural practices.
The authors noted that a prime opporunity to schedule the prenatal visit is when an expectant parent seeking information about insurance, practice hours, and whether the practice is taking new patients.
The AAP advises clinicians to encourage same sex parents, parents expecting via surrogate, and parents who are adopting to schedule a prenatal visit to identify particular concerns they may have.
“This is the only routine child wellness visit recommended by the American Academy of Pediatrics that doesn’t actually require a child in the room,” coauthor Arthur Lavin, MD, also of Harvard Medical School, said in a statement.
The prenatal visit “gives parents an opportunity to really focus on any questions and concerns they may have. They can talk with a pediatrician before the fatigue of new parenthood sets in and there’s an adorably distracting little human in their arms who may be crying, spitting up, or in immediate need of feeding or a diaper change,” Dr. Lavin said.
“At its heart and soul,” Dr. Lavin noted, “this visit is about laying a foundation for a trusting, supportive relationship between the family and their pediatrician, who will work together to keep the child healthy for the next 18 or 20 years.”
The report recommends the Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents, Fourth Edition, as a resource for clinicians. The researchers had no financial conflicts to disclose.
SOURCE: Yogman M et al. Pediatrics. 2018; doi: 10.1542/peds. 2018-1218
FROM PEDIATRICS
Buprenorphine endangers lives and health of children
Eleven children died from exposure to buprenorphine – a drug used to treat opioid exposure – from 2007 to 2016, mostly very young children who accidentally ingested the drug.
Four deaths, however, were teens who took buprenorphine recreationally or used it in a suicide attempt, according to a new database review by Sara Post, MS, of the Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, and her associates.
“In 2016, the American Academy of Pediatrics issued a statement advocating for increased access to buprenorphine for opioid-addicted adolescents in primary care settings,” the authors noted. “This recommendation is warranted because of the high and increasing prevalence of opioid dependence among adolescents. However, caution should be used, because increased prescriptions among adolescents could lead to increased diversion and abuse and increased access to younger children in the home. Therefore, patient education for adolescents should include information about the dangers of misusing and/or abusing prescription drugs and the proper storage of medications.”
The deaths comprise a small fraction of the 11,275 children aged 19 years and younger whose buprenorphine ingestions were reported to a poison control center during that time, the investigators said. Nevertheless, almost half (45%) of the exposed children were admitted to a health care facility – with 22% needing treatment in a critical care unit.
The rate of exposures was highest during the years when only tablet formulations were available and fell after film was introduced, wrote Ms. Post, a medical student at the Northeast Ohio Medical University in Rootstown, and her colleagues. But after 2013, the rate held steady, at about 38 exposures per 1 million children per year.
Childproof packaging for all buprenorphine formulations could help protect younger children, and education could help protect older ones, she and her coinvestigators said. Manufacturers should use unit-dose packaging for all buprenorphine products to help prevent unintentional exposure among young children. Health care providers should inform caregivers of young children about the dangers of buprenorphine exposure and provide instructions on proper storage and disposal of medications. Adolescents should receive information regarding the risks of substance abuse and misuse.”
Ms. Post and her colleagues analyzed calls to poison control centers affiliated with the National Poison Data System from 2007 to 2016. During that time, the centers received 11,275 calls about buprenorphine exposure among children and adolescents 19 years and younger.
The mean age of exposure in children was about 4 years; children younger than 6 years comprised 86% of the exposures (9,709).
The investigators noted temporal trends in exposure rates in this group. From 2007 to 2013, the rate increased by 215%, peaking at 20 per 1 million in 2010. A decline followed, with exposure dropping to 12 per 1 million in 2013, before rising again to 13 in 2016.
The increase “was likely attributable to the increasing number of buprenorphine prescriptions dispensed since the Food and Drug Administration approved its use as a treatment of opioid dependence in 2002,” Ms. Post and her colleagues wrote.
The transient decrease may have been related to a shift in adult prescribing patterns, as the drug was prescribed less often to those in their 20s and gradually given more often to people aged 40-59 years.
The decrease also was probably related to the packaging shift from tablet to film. “In 2013, the leading brand-name tablets were voluntarily withdrawn from the U.S. market because of potential risk of unintentional pediatric exposures,” the team wrote. Unfortunately, the film packaging didn’t completely deter some children; from 2013 to 2016, there was a 30% increase in the frequency of exposures to buprenorphine film.
The bulk of exposures were unintentional (98%) and involved ingestion of a single buprenorphine product. However, the authors noted, even a single adult therapeutic dose can be extremely dangerous to a small child.
“Therapeutic doses of buprenorphine-naloxone for pediatric patients are 2 to 6 mcg/kg, so ingestion of a single 2-mg sublingual tablet in a 10-kg child can result in more than a 30-fold overdose. This is particularly dangerous, because children exposed to buprenorphine do not display the ‘ceiling effect’ reported in adults, in which escalating doses do not lead to additional increases in respiratory depression,” Ms. Post and her coauthors said.
This was reflected in the serious clinical effects experienced: respiratory depression, bradycardia, coma, cyanosis, respiratory arrest, seizure, and cardiac arrest. These youngest children experienced the most serious outcomes, with half requiring a hospital admittance and 21% experiencing a serious medical outcome. Seven died, six of whom were 2 years or younger.
There were 315 (3%) exposures in children aged 6-12 years; most of these (83%) were either unintentional or therapeutic errors (18%). About 30% of the group required hospital admission and about 12% experienced a serious medical outcome. There were no fatalities among this group, the investigators noted.
Among adolescents aged 13-19 years, there were 1,251 (11%) exposures and four deaths. The bulk of these (77%) was intentional, with suspected suicide accounting for 12%, and 30% involving more than one substance. The exposure rate followed the same general trends, rising to a peak of about 6 per 1 million in 2010 and the falling and leveling off at about 3 per 1 million in 2016, they said.
About 22% of teen exposures required hospital admission, with 11% needing treatment in a critical care unit. The four deaths, one of which was a suicide, all involved multiple substances (benzodiazepines, alcohol, and marijuana).
Ms. Post received a research stipend from the National Student Injury Research Training Program while she worked on the study. The coauthors had no relevant financial disclosures.
SOURCE: Post et al. Pediatrics. 2018;142:e20173652.
Eleven children died from exposure to buprenorphine – a drug used to treat opioid exposure – from 2007 to 2016, mostly very young children who accidentally ingested the drug.
Four deaths, however, were teens who took buprenorphine recreationally or used it in a suicide attempt, according to a new database review by Sara Post, MS, of the Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, and her associates.
“In 2016, the American Academy of Pediatrics issued a statement advocating for increased access to buprenorphine for opioid-addicted adolescents in primary care settings,” the authors noted. “This recommendation is warranted because of the high and increasing prevalence of opioid dependence among adolescents. However, caution should be used, because increased prescriptions among adolescents could lead to increased diversion and abuse and increased access to younger children in the home. Therefore, patient education for adolescents should include information about the dangers of misusing and/or abusing prescription drugs and the proper storage of medications.”
The deaths comprise a small fraction of the 11,275 children aged 19 years and younger whose buprenorphine ingestions were reported to a poison control center during that time, the investigators said. Nevertheless, almost half (45%) of the exposed children were admitted to a health care facility – with 22% needing treatment in a critical care unit.
The rate of exposures was highest during the years when only tablet formulations were available and fell after film was introduced, wrote Ms. Post, a medical student at the Northeast Ohio Medical University in Rootstown, and her colleagues. But after 2013, the rate held steady, at about 38 exposures per 1 million children per year.
Childproof packaging for all buprenorphine formulations could help protect younger children, and education could help protect older ones, she and her coinvestigators said. Manufacturers should use unit-dose packaging for all buprenorphine products to help prevent unintentional exposure among young children. Health care providers should inform caregivers of young children about the dangers of buprenorphine exposure and provide instructions on proper storage and disposal of medications. Adolescents should receive information regarding the risks of substance abuse and misuse.”
Ms. Post and her colleagues analyzed calls to poison control centers affiliated with the National Poison Data System from 2007 to 2016. During that time, the centers received 11,275 calls about buprenorphine exposure among children and adolescents 19 years and younger.
The mean age of exposure in children was about 4 years; children younger than 6 years comprised 86% of the exposures (9,709).
The investigators noted temporal trends in exposure rates in this group. From 2007 to 2013, the rate increased by 215%, peaking at 20 per 1 million in 2010. A decline followed, with exposure dropping to 12 per 1 million in 2013, before rising again to 13 in 2016.
The increase “was likely attributable to the increasing number of buprenorphine prescriptions dispensed since the Food and Drug Administration approved its use as a treatment of opioid dependence in 2002,” Ms. Post and her colleagues wrote.
The transient decrease may have been related to a shift in adult prescribing patterns, as the drug was prescribed less often to those in their 20s and gradually given more often to people aged 40-59 years.
The decrease also was probably related to the packaging shift from tablet to film. “In 2013, the leading brand-name tablets were voluntarily withdrawn from the U.S. market because of potential risk of unintentional pediatric exposures,” the team wrote. Unfortunately, the film packaging didn’t completely deter some children; from 2013 to 2016, there was a 30% increase in the frequency of exposures to buprenorphine film.
The bulk of exposures were unintentional (98%) and involved ingestion of a single buprenorphine product. However, the authors noted, even a single adult therapeutic dose can be extremely dangerous to a small child.
“Therapeutic doses of buprenorphine-naloxone for pediatric patients are 2 to 6 mcg/kg, so ingestion of a single 2-mg sublingual tablet in a 10-kg child can result in more than a 30-fold overdose. This is particularly dangerous, because children exposed to buprenorphine do not display the ‘ceiling effect’ reported in adults, in which escalating doses do not lead to additional increases in respiratory depression,” Ms. Post and her coauthors said.
This was reflected in the serious clinical effects experienced: respiratory depression, bradycardia, coma, cyanosis, respiratory arrest, seizure, and cardiac arrest. These youngest children experienced the most serious outcomes, with half requiring a hospital admittance and 21% experiencing a serious medical outcome. Seven died, six of whom were 2 years or younger.
There were 315 (3%) exposures in children aged 6-12 years; most of these (83%) were either unintentional or therapeutic errors (18%). About 30% of the group required hospital admission and about 12% experienced a serious medical outcome. There were no fatalities among this group, the investigators noted.
Among adolescents aged 13-19 years, there were 1,251 (11%) exposures and four deaths. The bulk of these (77%) was intentional, with suspected suicide accounting for 12%, and 30% involving more than one substance. The exposure rate followed the same general trends, rising to a peak of about 6 per 1 million in 2010 and the falling and leveling off at about 3 per 1 million in 2016, they said.
About 22% of teen exposures required hospital admission, with 11% needing treatment in a critical care unit. The four deaths, one of which was a suicide, all involved multiple substances (benzodiazepines, alcohol, and marijuana).
Ms. Post received a research stipend from the National Student Injury Research Training Program while she worked on the study. The coauthors had no relevant financial disclosures.
SOURCE: Post et al. Pediatrics. 2018;142:e20173652.
Eleven children died from exposure to buprenorphine – a drug used to treat opioid exposure – from 2007 to 2016, mostly very young children who accidentally ingested the drug.
Four deaths, however, were teens who took buprenorphine recreationally or used it in a suicide attempt, according to a new database review by Sara Post, MS, of the Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, and her associates.
“In 2016, the American Academy of Pediatrics issued a statement advocating for increased access to buprenorphine for opioid-addicted adolescents in primary care settings,” the authors noted. “This recommendation is warranted because of the high and increasing prevalence of opioid dependence among adolescents. However, caution should be used, because increased prescriptions among adolescents could lead to increased diversion and abuse and increased access to younger children in the home. Therefore, patient education for adolescents should include information about the dangers of misusing and/or abusing prescription drugs and the proper storage of medications.”
The deaths comprise a small fraction of the 11,275 children aged 19 years and younger whose buprenorphine ingestions were reported to a poison control center during that time, the investigators said. Nevertheless, almost half (45%) of the exposed children were admitted to a health care facility – with 22% needing treatment in a critical care unit.
The rate of exposures was highest during the years when only tablet formulations were available and fell after film was introduced, wrote Ms. Post, a medical student at the Northeast Ohio Medical University in Rootstown, and her colleagues. But after 2013, the rate held steady, at about 38 exposures per 1 million children per year.
Childproof packaging for all buprenorphine formulations could help protect younger children, and education could help protect older ones, she and her coinvestigators said. Manufacturers should use unit-dose packaging for all buprenorphine products to help prevent unintentional exposure among young children. Health care providers should inform caregivers of young children about the dangers of buprenorphine exposure and provide instructions on proper storage and disposal of medications. Adolescents should receive information regarding the risks of substance abuse and misuse.”
Ms. Post and her colleagues analyzed calls to poison control centers affiliated with the National Poison Data System from 2007 to 2016. During that time, the centers received 11,275 calls about buprenorphine exposure among children and adolescents 19 years and younger.
The mean age of exposure in children was about 4 years; children younger than 6 years comprised 86% of the exposures (9,709).
The investigators noted temporal trends in exposure rates in this group. From 2007 to 2013, the rate increased by 215%, peaking at 20 per 1 million in 2010. A decline followed, with exposure dropping to 12 per 1 million in 2013, before rising again to 13 in 2016.
The increase “was likely attributable to the increasing number of buprenorphine prescriptions dispensed since the Food and Drug Administration approved its use as a treatment of opioid dependence in 2002,” Ms. Post and her colleagues wrote.
The transient decrease may have been related to a shift in adult prescribing patterns, as the drug was prescribed less often to those in their 20s and gradually given more often to people aged 40-59 years.
The decrease also was probably related to the packaging shift from tablet to film. “In 2013, the leading brand-name tablets were voluntarily withdrawn from the U.S. market because of potential risk of unintentional pediatric exposures,” the team wrote. Unfortunately, the film packaging didn’t completely deter some children; from 2013 to 2016, there was a 30% increase in the frequency of exposures to buprenorphine film.
The bulk of exposures were unintentional (98%) and involved ingestion of a single buprenorphine product. However, the authors noted, even a single adult therapeutic dose can be extremely dangerous to a small child.
“Therapeutic doses of buprenorphine-naloxone for pediatric patients are 2 to 6 mcg/kg, so ingestion of a single 2-mg sublingual tablet in a 10-kg child can result in more than a 30-fold overdose. This is particularly dangerous, because children exposed to buprenorphine do not display the ‘ceiling effect’ reported in adults, in which escalating doses do not lead to additional increases in respiratory depression,” Ms. Post and her coauthors said.
This was reflected in the serious clinical effects experienced: respiratory depression, bradycardia, coma, cyanosis, respiratory arrest, seizure, and cardiac arrest. These youngest children experienced the most serious outcomes, with half requiring a hospital admittance and 21% experiencing a serious medical outcome. Seven died, six of whom were 2 years or younger.
There were 315 (3%) exposures in children aged 6-12 years; most of these (83%) were either unintentional or therapeutic errors (18%). About 30% of the group required hospital admission and about 12% experienced a serious medical outcome. There were no fatalities among this group, the investigators noted.
Among adolescents aged 13-19 years, there were 1,251 (11%) exposures and four deaths. The bulk of these (77%) was intentional, with suspected suicide accounting for 12%, and 30% involving more than one substance. The exposure rate followed the same general trends, rising to a peak of about 6 per 1 million in 2010 and the falling and leveling off at about 3 per 1 million in 2016, they said.
About 22% of teen exposures required hospital admission, with 11% needing treatment in a critical care unit. The four deaths, one of which was a suicide, all involved multiple substances (benzodiazepines, alcohol, and marijuana).
Ms. Post received a research stipend from the National Student Injury Research Training Program while she worked on the study. The coauthors had no relevant financial disclosures.
SOURCE: Post et al. Pediatrics. 2018;142:e20173652.
FROM PEDIATRICS
Key clinical point: Buprenorphine ingestion remains a threat to children, especially those younger than 6 years.
Major finding: From 2007 to 2016, 11,275 exposures were reported; 11 children died.
Study details: The database review looked at records from the National Poison Data System.
Disclosures: Ms. Post received a research stipend from the National Student Injury Research Training Program while she worked on the study. The coauthors had no relevant financial disclosures.
Source: Post et al. Pediatrics. 2018;142:e20173652.
Obesity didn’t just happen overnight
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.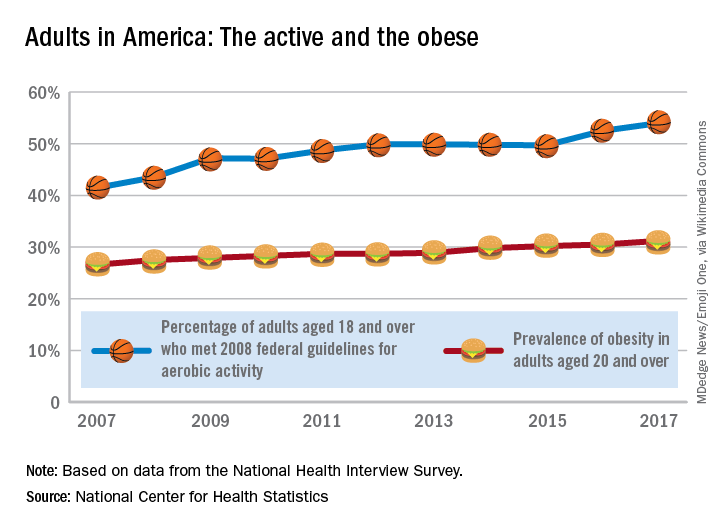
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.