User login
Gene therapy for thalassemia normalizes hemoglobin
Most beta thalassemia patients were off transfusions at a median of 26 months after receiving gene therapy via a lentiviral vector, according to new results of two phase 1/2 studies regarding the use of LentiGlobin in transfusion-dependent beta thalassemia.
Of 13 patients who did not have the most severe beta0/beta0 genotype, all but 1 has become transfusion independent post transplant. Among patients who either had the beta0/beta0 genotype or had two copies of the IVS1-110 mutation, transfusions were down a median 73% annually, and three of these patients with more severe thalassemia became transfusion independent.
At the time of last data collection, hemoglobin levels in individual patients ranged from 8.2-13.7 g/dL.
“No clonal dominance related to vector integration was observed,” wrote Alexis Thompson, MD, and her collaborators, and replication-competent lentivirus had not been found in any patients.
Hematopoietic cell transplant (HCT) is an option primarily for younger beta thalassemia patients who have an HLA-matched sibling donor, the researchers wrote in the New England Journal of Medicine. Gene therapy represents an alternative to the current standard of care for patients who are not candidates for allogeneic HCT, which – without a good match – carries increased risk for rejection and graft-versus-host disease.
Patients with beta thalassemia aged 35 years or younger and without advanced organ damage were enrolled in the two studies, one conducted internationally and one conducted at a single site in France.
There were some protocol differences between the two studies; notably, the French study used enhanced red cell transfusion for 3 or more months before stem cell mobilization “to enrich for bona fide hematopoietic stem cells in the harvested CD34+ cell compartment by suppressing the erythroid lineage expansion and the skewing that is seen in beta thalassemia,” wrote Dr. Thompson, professor of pediatrics at Northwestern University, Chicago, and her colleagues.
In both studies, after mobilization, patients’ unmanipulated hematopoietic stem cells and progenitor cells were taken to a central processing facility, where CD34+ cells were enriched and then transduced with the lentiviral vector BB305, which encodes adult hemoglobin (HbA) with a T87Z amino acid substitution and thereby provides functioning Hb beta. Patients received the product via infusion after undergoing myeloablative conditioning with busulfan.
A total of 23 patients, 19 in the international study and 4 in the French study, went through mobilization and apheresis. One patient in the international study had apheresis failure, so a total of 22 patients received LentiGlobin, and all were followed for up to 2 years.
Patients were given the opportunity to participate in a follow-on open label study meant to continue for an additional 13 years after the initial 24-month period; 13 patients are currently enrolled in this long-term follow-up study.
When transfusion volume at baseline was assessed, patients in the international study were receiving a median annual red blood cell transfusion volume of 164 mL/kg per year, while the French study participants were receiving a median 182 mL/kg per year of red blood cell transfusion.
In both studies, blood HbAT87Q levels correlated with the vector copy numbers (R2, 0.75; P less than .001). Levels of HbAT87Q ranged from 3.4-10.0 g/dL.
“Other factors, such as age, genotype, and splenectomy status, did not appear to correlate with gene expression,” the researchers wrote.
An exploratory analysis looked at characteristics of patients who were able to stop transfusions after gene therapy. In this group, “the degree of hemolysis at first stabilized relative to pretransplantation levels and was fully corrected” in two patients by 36 months after treatment.
The researchers noted that the sponsor achieved “high-titer, large-scale, clinical-grade BB305 vector production and purification by ion-exchange chromatography” from a single site in the United States, which showed the feasibility of conducting this modality of gene therapy at scale.
The study was sponsored by bluebird bio, the National Institutes of Health, and by French national research organizations. Dr. Thompson reported research funding and fees from bluebird bio and other pharmaceutical companies.
SOURCE: Thompson A et al. N Engl J Med 2018;378:1479-93
Lentiviral vector hematopoietic stem cell (HSC) gene therapy represents a promising alternative to matched-sibling donor HSC transplants for treatment of beta thalassemia, with studies to date showing a safety profile that surpasses transplants from unrelated or alternative donors.
The prospect of a curative treatment raised by the work of Dr. Thompson and her colleagues also shows the feasibility of transfusion independence for beta0/betaE patients, who carry the most common beta thalassemia genotype, and a significant reduction in transfusions even for patients with the more severe beta0/beta0 genotype.
Beta thalassemia has greatest prevalence in North Africa, the Middle East, and Asia, where access to treatments is limited and patients’ prognoses are often grim. Gene therapy for beta thalassemia could thereby represent the first large-scale implementation of this intervention in developing countries.
Bringing HSC gene therapy to more patients will require not just the availability of autologous HSCs, but of high-quality vector and reliable, high-volume manufacturing of transduced cells.
Harnessing this still-evolving technology to bring a potentially curative treatment to patients in developing countries is an exciting, but challenging, frontier for physicians and researchers involved with gene therapy.
Alessandra Biffi, MD, is director of the gene therapy program at Dana Farber Cancer Institute/Boston Children’s Cancer and Blood Disorders Center, Boston. She serves on the board of directors of the American Society of Gene and Cell Therapy. These remarks were adapted from an accompanying editorial ( N Engl J Med. 2018;378[16]:1551-2 ).
Lentiviral vector hematopoietic stem cell (HSC) gene therapy represents a promising alternative to matched-sibling donor HSC transplants for treatment of beta thalassemia, with studies to date showing a safety profile that surpasses transplants from unrelated or alternative donors.
The prospect of a curative treatment raised by the work of Dr. Thompson and her colleagues also shows the feasibility of transfusion independence for beta0/betaE patients, who carry the most common beta thalassemia genotype, and a significant reduction in transfusions even for patients with the more severe beta0/beta0 genotype.
Beta thalassemia has greatest prevalence in North Africa, the Middle East, and Asia, where access to treatments is limited and patients’ prognoses are often grim. Gene therapy for beta thalassemia could thereby represent the first large-scale implementation of this intervention in developing countries.
Bringing HSC gene therapy to more patients will require not just the availability of autologous HSCs, but of high-quality vector and reliable, high-volume manufacturing of transduced cells.
Harnessing this still-evolving technology to bring a potentially curative treatment to patients in developing countries is an exciting, but challenging, frontier for physicians and researchers involved with gene therapy.
Alessandra Biffi, MD, is director of the gene therapy program at Dana Farber Cancer Institute/Boston Children’s Cancer and Blood Disorders Center, Boston. She serves on the board of directors of the American Society of Gene and Cell Therapy. These remarks were adapted from an accompanying editorial ( N Engl J Med. 2018;378[16]:1551-2 ).
Lentiviral vector hematopoietic stem cell (HSC) gene therapy represents a promising alternative to matched-sibling donor HSC transplants for treatment of beta thalassemia, with studies to date showing a safety profile that surpasses transplants from unrelated or alternative donors.
The prospect of a curative treatment raised by the work of Dr. Thompson and her colleagues also shows the feasibility of transfusion independence for beta0/betaE patients, who carry the most common beta thalassemia genotype, and a significant reduction in transfusions even for patients with the more severe beta0/beta0 genotype.
Beta thalassemia has greatest prevalence in North Africa, the Middle East, and Asia, where access to treatments is limited and patients’ prognoses are often grim. Gene therapy for beta thalassemia could thereby represent the first large-scale implementation of this intervention in developing countries.
Bringing HSC gene therapy to more patients will require not just the availability of autologous HSCs, but of high-quality vector and reliable, high-volume manufacturing of transduced cells.
Harnessing this still-evolving technology to bring a potentially curative treatment to patients in developing countries is an exciting, but challenging, frontier for physicians and researchers involved with gene therapy.
Alessandra Biffi, MD, is director of the gene therapy program at Dana Farber Cancer Institute/Boston Children’s Cancer and Blood Disorders Center, Boston. She serves on the board of directors of the American Society of Gene and Cell Therapy. These remarks were adapted from an accompanying editorial ( N Engl J Med. 2018;378[16]:1551-2 ).
Most beta thalassemia patients were off transfusions at a median of 26 months after receiving gene therapy via a lentiviral vector, according to new results of two phase 1/2 studies regarding the use of LentiGlobin in transfusion-dependent beta thalassemia.
Of 13 patients who did not have the most severe beta0/beta0 genotype, all but 1 has become transfusion independent post transplant. Among patients who either had the beta0/beta0 genotype or had two copies of the IVS1-110 mutation, transfusions were down a median 73% annually, and three of these patients with more severe thalassemia became transfusion independent.
At the time of last data collection, hemoglobin levels in individual patients ranged from 8.2-13.7 g/dL.
“No clonal dominance related to vector integration was observed,” wrote Alexis Thompson, MD, and her collaborators, and replication-competent lentivirus had not been found in any patients.
Hematopoietic cell transplant (HCT) is an option primarily for younger beta thalassemia patients who have an HLA-matched sibling donor, the researchers wrote in the New England Journal of Medicine. Gene therapy represents an alternative to the current standard of care for patients who are not candidates for allogeneic HCT, which – without a good match – carries increased risk for rejection and graft-versus-host disease.
Patients with beta thalassemia aged 35 years or younger and without advanced organ damage were enrolled in the two studies, one conducted internationally and one conducted at a single site in France.
There were some protocol differences between the two studies; notably, the French study used enhanced red cell transfusion for 3 or more months before stem cell mobilization “to enrich for bona fide hematopoietic stem cells in the harvested CD34+ cell compartment by suppressing the erythroid lineage expansion and the skewing that is seen in beta thalassemia,” wrote Dr. Thompson, professor of pediatrics at Northwestern University, Chicago, and her colleagues.
In both studies, after mobilization, patients’ unmanipulated hematopoietic stem cells and progenitor cells were taken to a central processing facility, where CD34+ cells were enriched and then transduced with the lentiviral vector BB305, which encodes adult hemoglobin (HbA) with a T87Z amino acid substitution and thereby provides functioning Hb beta. Patients received the product via infusion after undergoing myeloablative conditioning with busulfan.
A total of 23 patients, 19 in the international study and 4 in the French study, went through mobilization and apheresis. One patient in the international study had apheresis failure, so a total of 22 patients received LentiGlobin, and all were followed for up to 2 years.
Patients were given the opportunity to participate in a follow-on open label study meant to continue for an additional 13 years after the initial 24-month period; 13 patients are currently enrolled in this long-term follow-up study.
When transfusion volume at baseline was assessed, patients in the international study were receiving a median annual red blood cell transfusion volume of 164 mL/kg per year, while the French study participants were receiving a median 182 mL/kg per year of red blood cell transfusion.
In both studies, blood HbAT87Q levels correlated with the vector copy numbers (R2, 0.75; P less than .001). Levels of HbAT87Q ranged from 3.4-10.0 g/dL.
“Other factors, such as age, genotype, and splenectomy status, did not appear to correlate with gene expression,” the researchers wrote.
An exploratory analysis looked at characteristics of patients who were able to stop transfusions after gene therapy. In this group, “the degree of hemolysis at first stabilized relative to pretransplantation levels and was fully corrected” in two patients by 36 months after treatment.
The researchers noted that the sponsor achieved “high-titer, large-scale, clinical-grade BB305 vector production and purification by ion-exchange chromatography” from a single site in the United States, which showed the feasibility of conducting this modality of gene therapy at scale.
The study was sponsored by bluebird bio, the National Institutes of Health, and by French national research organizations. Dr. Thompson reported research funding and fees from bluebird bio and other pharmaceutical companies.
SOURCE: Thompson A et al. N Engl J Med 2018;378:1479-93
Most beta thalassemia patients were off transfusions at a median of 26 months after receiving gene therapy via a lentiviral vector, according to new results of two phase 1/2 studies regarding the use of LentiGlobin in transfusion-dependent beta thalassemia.
Of 13 patients who did not have the most severe beta0/beta0 genotype, all but 1 has become transfusion independent post transplant. Among patients who either had the beta0/beta0 genotype or had two copies of the IVS1-110 mutation, transfusions were down a median 73% annually, and three of these patients with more severe thalassemia became transfusion independent.
At the time of last data collection, hemoglobin levels in individual patients ranged from 8.2-13.7 g/dL.
“No clonal dominance related to vector integration was observed,” wrote Alexis Thompson, MD, and her collaborators, and replication-competent lentivirus had not been found in any patients.
Hematopoietic cell transplant (HCT) is an option primarily for younger beta thalassemia patients who have an HLA-matched sibling donor, the researchers wrote in the New England Journal of Medicine. Gene therapy represents an alternative to the current standard of care for patients who are not candidates for allogeneic HCT, which – without a good match – carries increased risk for rejection and graft-versus-host disease.
Patients with beta thalassemia aged 35 years or younger and without advanced organ damage were enrolled in the two studies, one conducted internationally and one conducted at a single site in France.
There were some protocol differences between the two studies; notably, the French study used enhanced red cell transfusion for 3 or more months before stem cell mobilization “to enrich for bona fide hematopoietic stem cells in the harvested CD34+ cell compartment by suppressing the erythroid lineage expansion and the skewing that is seen in beta thalassemia,” wrote Dr. Thompson, professor of pediatrics at Northwestern University, Chicago, and her colleagues.
In both studies, after mobilization, patients’ unmanipulated hematopoietic stem cells and progenitor cells were taken to a central processing facility, where CD34+ cells were enriched and then transduced with the lentiviral vector BB305, which encodes adult hemoglobin (HbA) with a T87Z amino acid substitution and thereby provides functioning Hb beta. Patients received the product via infusion after undergoing myeloablative conditioning with busulfan.
A total of 23 patients, 19 in the international study and 4 in the French study, went through mobilization and apheresis. One patient in the international study had apheresis failure, so a total of 22 patients received LentiGlobin, and all were followed for up to 2 years.
Patients were given the opportunity to participate in a follow-on open label study meant to continue for an additional 13 years after the initial 24-month period; 13 patients are currently enrolled in this long-term follow-up study.
When transfusion volume at baseline was assessed, patients in the international study were receiving a median annual red blood cell transfusion volume of 164 mL/kg per year, while the French study participants were receiving a median 182 mL/kg per year of red blood cell transfusion.
In both studies, blood HbAT87Q levels correlated with the vector copy numbers (R2, 0.75; P less than .001). Levels of HbAT87Q ranged from 3.4-10.0 g/dL.
“Other factors, such as age, genotype, and splenectomy status, did not appear to correlate with gene expression,” the researchers wrote.
An exploratory analysis looked at characteristics of patients who were able to stop transfusions after gene therapy. In this group, “the degree of hemolysis at first stabilized relative to pretransplantation levels and was fully corrected” in two patients by 36 months after treatment.
The researchers noted that the sponsor achieved “high-titer, large-scale, clinical-grade BB305 vector production and purification by ion-exchange chromatography” from a single site in the United States, which showed the feasibility of conducting this modality of gene therapy at scale.
The study was sponsored by bluebird bio, the National Institutes of Health, and by French national research organizations. Dr. Thompson reported research funding and fees from bluebird bio and other pharmaceutical companies.
SOURCE: Thompson A et al. N Engl J Med 2018;378:1479-93
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Transfusion requirements were down 73% annually in patients with the most severe thalassemia.
Study details: Data from 22 transfusion-dependent patients with beta thalassemia in ongoing phase 1/2 study of gene therapy delivered via lentiviral vector.
Disclosures: The study was sponsored by bluebird bio, the National Institutes of Health, and by French national research organizations. Dr. Thompson reported research funding and fees from bluebird bio and from other pharmaceutical companies.
Source: Thompson A et al. N Engl J Med. 2018;378:1479-93.
Impaired kidney function no problem for dabigatran reversal
ORLANDO – Idarucizumab, the reversal agent for the anticoagulant dabigatran, appeared as effective in quickly reversing dabigatran’s effects in patients with severe renal dysfunction as in patients with normally working kidneys, in a post hoc analysis of data collected in the drug’s pivotal trial.
A standard dose of idarucizumab “works just as well in patients with bad kidney function as it does in patients with preserved kidney function,” John W. Eikelboom, MD, said at the annual meeting of the American College of Cardiology. “The time to cessation of bleeding and the degree of normal hemostasis achieved was consistent” across the entire range of renal function examined, from severe renal dysfunction, with a creatinine clearance rate of less than 30 mL/min, to normal function, with an estimated rate of 80 mL/min or greater.

The ability of idarucizumab (Praxbind), conditionally approved by the Food and Drug Administration in 2015 and then fully approved in April 2018, to work in patients with impaired renal function has been an open question and concern because dabigatran (Pradaxa) is excreted renally, so it builds to unusually high levels in patients with poor kidney function. “Plasma dabigatran levels might be sky high, so a standard dose of idarucizumab might not work. That’s been a fear of clinicians,” explained Dr. Eikelboom, a hematologist at McMaster University in Hamilton, Ont.
To examine whether idarucizumab’s activity varied by renal function he used data from the patients enrolled in the RE-VERSE AD (Reversal Effects of Idarucizumab on Active Dabigatran) study, the pivotal dataset that led to idarucizumab’s U.S. approval. The new, post hoc analysis divided patients into four subgroups based on their kidney function, and focused on the 489 patients for whom renal data were available out of the 503 patients in the study (N Engl J Med. 2017 Aug 3;377[5]:431-41). The subgroups included 91 patients with severe dysfunction with a creatinine clearance rate of less than 30 mL/min; 127 with moderate dysfunction and a clearance rate of 30-49 mL/min; 163 with mild dysfunction and a clearance rate of 50-79 mL/min; and 108 with normal function and a creatinine clearance of at least 80 mL/min.
Patients in the subgroup with severe renal dysfunction had the worst clinical profile overall, and as predicted, had a markedly elevated average plasma level of dabigatran, 231 ng/mL, nearly five times higher than the 47-ng/mL average level in patients with normal renal function.
The ability of a single, standard dose of idarucizumab to reverse the anticoagulant effects of dabigatran were essentially identical across the four strata of renal activity, with 98% of patients in both the severely impaired subgroup and the normal subgroup having 100% reversal within 4 hours of treatment, Dr. Eikelboom reported. Every patient included in the analysis had more than 50% reversal.
The study followed patients to 12-24 hours after they received idarucizumab, and 55% of patients with severe renal dysfunction showed a plasma dabigatran level that crept back toward a clinically meaningful level and so might need a second idarucizumab dose. In contrast, this happened in 8% of patients with normal renal function.
In patients with severe renal dysfunction given idarucizumab, “be alert for a recurrent bleed,” which could require a second dose of idarucizumab, Dr. Eikelboom suggested.
SOURCE: Eikelboom JW et al. ACC 18, Abstract 1231M-11.
ORLANDO – Idarucizumab, the reversal agent for the anticoagulant dabigatran, appeared as effective in quickly reversing dabigatran’s effects in patients with severe renal dysfunction as in patients with normally working kidneys, in a post hoc analysis of data collected in the drug’s pivotal trial.
A standard dose of idarucizumab “works just as well in patients with bad kidney function as it does in patients with preserved kidney function,” John W. Eikelboom, MD, said at the annual meeting of the American College of Cardiology. “The time to cessation of bleeding and the degree of normal hemostasis achieved was consistent” across the entire range of renal function examined, from severe renal dysfunction, with a creatinine clearance rate of less than 30 mL/min, to normal function, with an estimated rate of 80 mL/min or greater.
The ability of idarucizumab (Praxbind), conditionally approved by the Food and Drug Administration in 2015 and then fully approved in April 2018, to work in patients with impaired renal function has been an open question and concern because dabigatran (Pradaxa) is excreted renally, so it builds to unusually high levels in patients with poor kidney function. “Plasma dabigatran levels might be sky high, so a standard dose of idarucizumab might not work. That’s been a fear of clinicians,” explained Dr. Eikelboom, a hematologist at McMaster University in Hamilton, Ont.
To examine whether idarucizumab’s activity varied by renal function he used data from the patients enrolled in the RE-VERSE AD (Reversal Effects of Idarucizumab on Active Dabigatran) study, the pivotal dataset that led to idarucizumab’s U.S. approval. The new, post hoc analysis divided patients into four subgroups based on their kidney function, and focused on the 489 patients for whom renal data were available out of the 503 patients in the study (N Engl J Med. 2017 Aug 3;377[5]:431-41). The subgroups included 91 patients with severe dysfunction with a creatinine clearance rate of less than 30 mL/min; 127 with moderate dysfunction and a clearance rate of 30-49 mL/min; 163 with mild dysfunction and a clearance rate of 50-79 mL/min; and 108 with normal function and a creatinine clearance of at least 80 mL/min.
Patients in the subgroup with severe renal dysfunction had the worst clinical profile overall, and as predicted, had a markedly elevated average plasma level of dabigatran, 231 ng/mL, nearly five times higher than the 47-ng/mL average level in patients with normal renal function.
The ability of a single, standard dose of idarucizumab to reverse the anticoagulant effects of dabigatran were essentially identical across the four strata of renal activity, with 98% of patients in both the severely impaired subgroup and the normal subgroup having 100% reversal within 4 hours of treatment, Dr. Eikelboom reported. Every patient included in the analysis had more than 50% reversal.
The study followed patients to 12-24 hours after they received idarucizumab, and 55% of patients with severe renal dysfunction showed a plasma dabigatran level that crept back toward a clinically meaningful level and so might need a second idarucizumab dose. In contrast, this happened in 8% of patients with normal renal function.
In patients with severe renal dysfunction given idarucizumab, “be alert for a recurrent bleed,” which could require a second dose of idarucizumab, Dr. Eikelboom suggested.
SOURCE: Eikelboom JW et al. ACC 18, Abstract 1231M-11.
ORLANDO – Idarucizumab, the reversal agent for the anticoagulant dabigatran, appeared as effective in quickly reversing dabigatran’s effects in patients with severe renal dysfunction as in patients with normally working kidneys, in a post hoc analysis of data collected in the drug’s pivotal trial.
A standard dose of idarucizumab “works just as well in patients with bad kidney function as it does in patients with preserved kidney function,” John W. Eikelboom, MD, said at the annual meeting of the American College of Cardiology. “The time to cessation of bleeding and the degree of normal hemostasis achieved was consistent” across the entire range of renal function examined, from severe renal dysfunction, with a creatinine clearance rate of less than 30 mL/min, to normal function, with an estimated rate of 80 mL/min or greater.
The ability of idarucizumab (Praxbind), conditionally approved by the Food and Drug Administration in 2015 and then fully approved in April 2018, to work in patients with impaired renal function has been an open question and concern because dabigatran (Pradaxa) is excreted renally, so it builds to unusually high levels in patients with poor kidney function. “Plasma dabigatran levels might be sky high, so a standard dose of idarucizumab might not work. That’s been a fear of clinicians,” explained Dr. Eikelboom, a hematologist at McMaster University in Hamilton, Ont.
To examine whether idarucizumab’s activity varied by renal function he used data from the patients enrolled in the RE-VERSE AD (Reversal Effects of Idarucizumab on Active Dabigatran) study, the pivotal dataset that led to idarucizumab’s U.S. approval. The new, post hoc analysis divided patients into four subgroups based on their kidney function, and focused on the 489 patients for whom renal data were available out of the 503 patients in the study (N Engl J Med. 2017 Aug 3;377[5]:431-41). The subgroups included 91 patients with severe dysfunction with a creatinine clearance rate of less than 30 mL/min; 127 with moderate dysfunction and a clearance rate of 30-49 mL/min; 163 with mild dysfunction and a clearance rate of 50-79 mL/min; and 108 with normal function and a creatinine clearance of at least 80 mL/min.
Patients in the subgroup with severe renal dysfunction had the worst clinical profile overall, and as predicted, had a markedly elevated average plasma level of dabigatran, 231 ng/mL, nearly five times higher than the 47-ng/mL average level in patients with normal renal function.
The ability of a single, standard dose of idarucizumab to reverse the anticoagulant effects of dabigatran were essentially identical across the four strata of renal activity, with 98% of patients in both the severely impaired subgroup and the normal subgroup having 100% reversal within 4 hours of treatment, Dr. Eikelboom reported. Every patient included in the analysis had more than 50% reversal.
The study followed patients to 12-24 hours after they received idarucizumab, and 55% of patients with severe renal dysfunction showed a plasma dabigatran level that crept back toward a clinically meaningful level and so might need a second idarucizumab dose. In contrast, this happened in 8% of patients with normal renal function.
In patients with severe renal dysfunction given idarucizumab, “be alert for a recurrent bleed,” which could require a second dose of idarucizumab, Dr. Eikelboom suggested.
SOURCE: Eikelboom JW et al. ACC 18, Abstract 1231M-11.
REPORTING FROM ACC 18
Key clinical point: Renal function had no impact on idarucizumab’s efficacy for dabigatran reversal.
Major finding: Complete dabigatran reversal occurred in 98% of patients with severe renal dysfunction who received idarucizumab.
Study details: Post hoc analysis of data from RE-VERSE AD, idarucizumab’s pivotal trial with 503 patients.
Disclosures: RE-VERSE AD was funded by Boehringer Ingelheim, the company that markets idarucizumab (Praxbind) and dabigatran (Pradaxa). Dr. Eikelboom has been a consultant to and has received research support from Boehringer Ingelheim, as well as from Bayer, Bristol-Myers Squibb, Daiichi-Sankyo, Janssen, and Pfizer.
Source: Eikelboom JW et al. ACC 18, Abstract 1231M-11.
Updates of ongoing clinical trials
Randomized Phase 3 Trial Evaluating the Addition of the IGF-1R Monoclonal Antibody Ganitumab (AMG 479, NSC# 750008) to Multiagent Chemotherapy for Patients With Newly Diagnosed Metastatic Ewing Sarcoma
NCT02306161
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Steven DuBois, Children’s Oncology Group and Dana-Farber Cancer Institute, Boston.
Study locations: Over 300 U.S. cancer centers
Study summary: This randomized phase 3 trial examines whether the monoclonal antibody ganitumab plus combination chemotherapy (vincristine sulfate, doxorubicin hydrochloride, cyclophosphamide, ifosfamide, and etoposide) improves event-free survival for patients with newly-diagnosed, metastatic Ewing sarcoma. Secondary outcomes include overall survival rate and comparative evaluations of toxicity.
Patients are randomized to induction and consolidation therapy with vincristine sulfate, doxorubicin hydrochloride and cyclophosphamide [VDC] and ifosfamide and etoposide [IE]) or to the same regimen plus ganitumab. Between weeks 13-18 of the trial, patients undergo surgery and/or radiation therapy for local control. Patients with lung metastases undergo definitive stereotactic body radiation therapy or external beam radiation therapy over 5 days.
Study inclusion summary: Patients up to 50 years old are eligible to participate in this trial if they have newly-diagnosed Ewing sarcoma or peripheral primitive neuroectodermal tumor (PNET) arising from bone or soft tissue and with metastatic disease involving lung, bone, bone marrow, or other metastatic site. Submission of pre-treatment serum, tumor tissue and whole blood is required. Patients should only have had a biopsy of the primary tumor without an attempt at complete or partial resection; patients will still be eligible if excision was attempted or accomplished as long as adequate anatomic imaging (MRI for most primary tumor sites) was obtained prior to surgery. Creatinine clearance or radioisotope glomerular filtration rate (GFR) must be at least 70 mL/min/1.73 m2 or greater. Total bilirubin must be less than 1.5 times the upper limit of normal, alanine aminotransferase must be less than 3 times the upper limit of normal, blood sugar must be normal, and heart ejection fraction must exceed 50%.
Induction therapy: Patients receive vincristine sulfate intravenously (IV) over 1 minute on day 1; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2; and cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 5, and 9; and ifosfamide IV over 1 hour on days 1 to 5 and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 7, and 11. Patients in the control group receive induction therapy and placebo and patients in the treatment group receive induction therapy and ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 1, 3, 5, 7, 9, and 11.
Consolidation therapy: Patients receive vincristine sulfate IV over 1 minute on day 1 of weeks 1, 7, 9, and 13; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2 of weeks 1 and 7; cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 7, 9, and 13; ifosfamide IV over 1 hour on days 1 to 5 of weeks 3, 5, 11, and 15; and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 5, 11, and 15. In addition to this standard consolidation therapy, pPatients in the active treatment group receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 7, 9, 11, 13, and 15.
Maintenance therapy: Patients receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 in weeks 1, 4, 7, 10, 13, 16, 19, and 22.
Follow up: After completion of study treatment, patients are followed for 10 years.
Combination Chemotherapy With or Without Temsirolimus in Treating Patients With Intermediate Risk Rhabdomyosarcoma
NCT02567435
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Abha Gupta, Children’s Oncology Group, The Hospital for Sick Children and Princess Margaret Cancer Centre.
Study locations: 293 cancer centers in the U.S. and Canada
Study summary: This randomized phase 3 trial compares standard combination chemotherapy with and without temsirolimus for patients with rhabdomyosarcoma that has an intermediate chance of recurrence after treatment. It is not yet known whether combination chemotherapy or combination chemotherapy plus temsirolimus is more effective in treating patients with intermediate-risk rhabdomyosarcoma.
Study inclusion summary: Patients up to age 40 with newly diagnosed RMS of any subtype, except adult-type pleomorphic, based upon institutional histopathologic classification, are eligible to enroll on the study. Lansky performance status score must be at least 50 for patients age 16 years and under; Karnofsky performance status score must be 50 or greater for patients over age 16. Peripheral absolute neutrophil count must be at least 750/uL and platelet count at least 75,000/uL. Creatinine clearance or radioisotope glomerular filtration rate must be at least 70 mL/min/1.73 m2. Total bilirubin must be no more than 1.5 times the upper limit of normal for patient age.
Treatment regimen: Patients are randomized to one of three study arms. One group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-13, 16, 17, 19, 20, 22-26, 28, 31-34, 37, 38, and 40, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, irinotecan hydrochloride IV over 90 minutes on days 1-5 of weeks 4, 10, 16, 19, 25, 31, and 37. The second group receives the same regimen plus temsirolimus IV over 30-60 minutes on day 1 of weeks 1-12 and 21-42. The third group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-10 and 13-22, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 4, 7, 10, 13, 16, 19, and 22, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 4, 7, and 10. Patients in all three groups also undergo radiation therapy beginning at week 13 for 6 weeks. Treatment continues in all three groups in the absence of disease progression or unacceptable toxicity.
Outcome Measures: The primary outcome measure is event-free survival (EFS) measured from study enrollment to the first occurrence of progression, relapse, second malignant neoplasm, or death as a first event. The secondary outcome measure is overall survival measured from study enrollment to death from any cause, assessed up to 10 years. TSJ
Randomized Phase 3 Trial Evaluating the Addition of the IGF-1R Monoclonal Antibody Ganitumab (AMG 479, NSC# 750008) to Multiagent Chemotherapy for Patients With Newly Diagnosed Metastatic Ewing Sarcoma
NCT02306161
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Steven DuBois, Children’s Oncology Group and Dana-Farber Cancer Institute, Boston.
Study locations: Over 300 U.S. cancer centers
Study summary: This randomized phase 3 trial examines whether the monoclonal antibody ganitumab plus combination chemotherapy (vincristine sulfate, doxorubicin hydrochloride, cyclophosphamide, ifosfamide, and etoposide) improves event-free survival for patients with newly-diagnosed, metastatic Ewing sarcoma. Secondary outcomes include overall survival rate and comparative evaluations of toxicity.
Patients are randomized to induction and consolidation therapy with vincristine sulfate, doxorubicin hydrochloride and cyclophosphamide [VDC] and ifosfamide and etoposide [IE]) or to the same regimen plus ganitumab. Between weeks 13-18 of the trial, patients undergo surgery and/or radiation therapy for local control. Patients with lung metastases undergo definitive stereotactic body radiation therapy or external beam radiation therapy over 5 days.
Study inclusion summary: Patients up to 50 years old are eligible to participate in this trial if they have newly-diagnosed Ewing sarcoma or peripheral primitive neuroectodermal tumor (PNET) arising from bone or soft tissue and with metastatic disease involving lung, bone, bone marrow, or other metastatic site. Submission of pre-treatment serum, tumor tissue and whole blood is required. Patients should only have had a biopsy of the primary tumor without an attempt at complete or partial resection; patients will still be eligible if excision was attempted or accomplished as long as adequate anatomic imaging (MRI for most primary tumor sites) was obtained prior to surgery. Creatinine clearance or radioisotope glomerular filtration rate (GFR) must be at least 70 mL/min/1.73 m2 or greater. Total bilirubin must be less than 1.5 times the upper limit of normal, alanine aminotransferase must be less than 3 times the upper limit of normal, blood sugar must be normal, and heart ejection fraction must exceed 50%.
Induction therapy: Patients receive vincristine sulfate intravenously (IV) over 1 minute on day 1; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2; and cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 5, and 9; and ifosfamide IV over 1 hour on days 1 to 5 and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 7, and 11. Patients in the control group receive induction therapy and placebo and patients in the treatment group receive induction therapy and ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 1, 3, 5, 7, 9, and 11.
Consolidation therapy: Patients receive vincristine sulfate IV over 1 minute on day 1 of weeks 1, 7, 9, and 13; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2 of weeks 1 and 7; cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 7, 9, and 13; ifosfamide IV over 1 hour on days 1 to 5 of weeks 3, 5, 11, and 15; and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 5, 11, and 15. In addition to this standard consolidation therapy, pPatients in the active treatment group receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 7, 9, 11, 13, and 15.
Maintenance therapy: Patients receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 in weeks 1, 4, 7, 10, 13, 16, 19, and 22.
Follow up: After completion of study treatment, patients are followed for 10 years.
Combination Chemotherapy With or Without Temsirolimus in Treating Patients With Intermediate Risk Rhabdomyosarcoma
NCT02567435
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Abha Gupta, Children’s Oncology Group, The Hospital for Sick Children and Princess Margaret Cancer Centre.
Study locations: 293 cancer centers in the U.S. and Canada
Study summary: This randomized phase 3 trial compares standard combination chemotherapy with and without temsirolimus for patients with rhabdomyosarcoma that has an intermediate chance of recurrence after treatment. It is not yet known whether combination chemotherapy or combination chemotherapy plus temsirolimus is more effective in treating patients with intermediate-risk rhabdomyosarcoma.
Study inclusion summary: Patients up to age 40 with newly diagnosed RMS of any subtype, except adult-type pleomorphic, based upon institutional histopathologic classification, are eligible to enroll on the study. Lansky performance status score must be at least 50 for patients age 16 years and under; Karnofsky performance status score must be 50 or greater for patients over age 16. Peripheral absolute neutrophil count must be at least 750/uL and platelet count at least 75,000/uL. Creatinine clearance or radioisotope glomerular filtration rate must be at least 70 mL/min/1.73 m2. Total bilirubin must be no more than 1.5 times the upper limit of normal for patient age.
Treatment regimen: Patients are randomized to one of three study arms. One group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-13, 16, 17, 19, 20, 22-26, 28, 31-34, 37, 38, and 40, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, irinotecan hydrochloride IV over 90 minutes on days 1-5 of weeks 4, 10, 16, 19, 25, 31, and 37. The second group receives the same regimen plus temsirolimus IV over 30-60 minutes on day 1 of weeks 1-12 and 21-42. The third group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-10 and 13-22, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 4, 7, 10, 13, 16, 19, and 22, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 4, 7, and 10. Patients in all three groups also undergo radiation therapy beginning at week 13 for 6 weeks. Treatment continues in all three groups in the absence of disease progression or unacceptable toxicity.
Outcome Measures: The primary outcome measure is event-free survival (EFS) measured from study enrollment to the first occurrence of progression, relapse, second malignant neoplasm, or death as a first event. The secondary outcome measure is overall survival measured from study enrollment to death from any cause, assessed up to 10 years. TSJ
Randomized Phase 3 Trial Evaluating the Addition of the IGF-1R Monoclonal Antibody Ganitumab (AMG 479, NSC# 750008) to Multiagent Chemotherapy for Patients With Newly Diagnosed Metastatic Ewing Sarcoma
NCT02306161
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Steven DuBois, Children’s Oncology Group and Dana-Farber Cancer Institute, Boston.
Study locations: Over 300 U.S. cancer centers
Study summary: This randomized phase 3 trial examines whether the monoclonal antibody ganitumab plus combination chemotherapy (vincristine sulfate, doxorubicin hydrochloride, cyclophosphamide, ifosfamide, and etoposide) improves event-free survival for patients with newly-diagnosed, metastatic Ewing sarcoma. Secondary outcomes include overall survival rate and comparative evaluations of toxicity.
Patients are randomized to induction and consolidation therapy with vincristine sulfate, doxorubicin hydrochloride and cyclophosphamide [VDC] and ifosfamide and etoposide [IE]) or to the same regimen plus ganitumab. Between weeks 13-18 of the trial, patients undergo surgery and/or radiation therapy for local control. Patients with lung metastases undergo definitive stereotactic body radiation therapy or external beam radiation therapy over 5 days.
Study inclusion summary: Patients up to 50 years old are eligible to participate in this trial if they have newly-diagnosed Ewing sarcoma or peripheral primitive neuroectodermal tumor (PNET) arising from bone or soft tissue and with metastatic disease involving lung, bone, bone marrow, or other metastatic site. Submission of pre-treatment serum, tumor tissue and whole blood is required. Patients should only have had a biopsy of the primary tumor without an attempt at complete or partial resection; patients will still be eligible if excision was attempted or accomplished as long as adequate anatomic imaging (MRI for most primary tumor sites) was obtained prior to surgery. Creatinine clearance or radioisotope glomerular filtration rate (GFR) must be at least 70 mL/min/1.73 m2 or greater. Total bilirubin must be less than 1.5 times the upper limit of normal, alanine aminotransferase must be less than 3 times the upper limit of normal, blood sugar must be normal, and heart ejection fraction must exceed 50%.
Induction therapy: Patients receive vincristine sulfate intravenously (IV) over 1 minute on day 1; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2; and cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 5, and 9; and ifosfamide IV over 1 hour on days 1 to 5 and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 7, and 11. Patients in the control group receive induction therapy and placebo and patients in the treatment group receive induction therapy and ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 1, 3, 5, 7, 9, and 11.
Consolidation therapy: Patients receive vincristine sulfate IV over 1 minute on day 1 of weeks 1, 7, 9, and 13; doxorubicin hydrochloride IV over 1-15 minutes on days 1 and 2 of weeks 1 and 7; cyclophosphamide IV over 30-60 minutes on day 1 of weeks 1, 7, 9, and 13; ifosfamide IV over 1 hour on days 1 to 5 of weeks 3, 5, 11, and 15; and etoposide IV over 1-2 hours on days 1 to 5 of weeks 3, 5, 11, and 15. In addition to this standard consolidation therapy, pPatients in the active treatment group receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 of weeks 7, 9, 11, 13, and 15.
Maintenance therapy: Patients receive ganitumab IV over 30-60 minutes or 60-120 minutes on day 1 in weeks 1, 4, 7, 10, 13, 16, 19, and 22.
Follow up: After completion of study treatment, patients are followed for 10 years.
Combination Chemotherapy With or Without Temsirolimus in Treating Patients With Intermediate Risk Rhabdomyosarcoma
NCT02567435
Sponsor: National Cancer Institute (NCI)
Principal Investigator: Abha Gupta, Children’s Oncology Group, The Hospital for Sick Children and Princess Margaret Cancer Centre.
Study locations: 293 cancer centers in the U.S. and Canada
Study summary: This randomized phase 3 trial compares standard combination chemotherapy with and without temsirolimus for patients with rhabdomyosarcoma that has an intermediate chance of recurrence after treatment. It is not yet known whether combination chemotherapy or combination chemotherapy plus temsirolimus is more effective in treating patients with intermediate-risk rhabdomyosarcoma.
Study inclusion summary: Patients up to age 40 with newly diagnosed RMS of any subtype, except adult-type pleomorphic, based upon institutional histopathologic classification, are eligible to enroll on the study. Lansky performance status score must be at least 50 for patients age 16 years and under; Karnofsky performance status score must be 50 or greater for patients over age 16. Peripheral absolute neutrophil count must be at least 750/uL and platelet count at least 75,000/uL. Creatinine clearance or radioisotope glomerular filtration rate must be at least 70 mL/min/1.73 m2. Total bilirubin must be no more than 1.5 times the upper limit of normal for patient age.
Treatment regimen: Patients are randomized to one of three study arms. One group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-13, 16, 17, 19, 20, 22-26, 28, 31-34, 37, 38, and 40, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 7, 13, 22, 28, 34, and 40, irinotecan hydrochloride IV over 90 minutes on days 1-5 of weeks 4, 10, 16, 19, 25, 31, and 37. The second group receives the same regimen plus temsirolimus IV over 30-60 minutes on day 1 of weeks 1-12 and 21-42. The third group receives vincristine sulfate IV over 1 minute on day 1 of weeks 1-10 and 13-22, dactinomycin IV over 1-5 minutes on day 1 of weeks 1, 4, 7, 10, 13, 16, 19, and 22, cyclophosphamide IV over 60 minutes on day 1 of weeks 1, 4, 7, and 10. Patients in all three groups also undergo radiation therapy beginning at week 13 for 6 weeks. Treatment continues in all three groups in the absence of disease progression or unacceptable toxicity.
Outcome Measures: The primary outcome measure is event-free survival (EFS) measured from study enrollment to the first occurrence of progression, relapse, second malignant neoplasm, or death as a first event. The secondary outcome measure is overall survival measured from study enrollment to death from any cause, assessed up to 10 years. TSJ
Picosecond 755-nm laser found effective for neck rejuvenation
DALLAS –
“It’s important to note that response was variable, but many patients were satisfied with the treatment,” Hana Jeon, MD said at the annual conference of the American Society for Laser Medicine and Surgery, Inc. “Further studies are needed to identify the clinical characteristics of neck laxity that would most benefit from this treatment.”
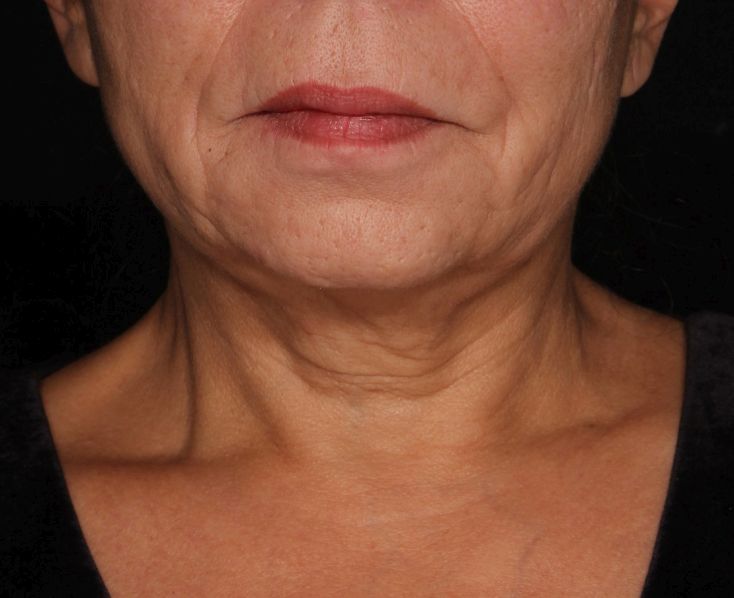
The researchers enrolled 25 patients with an average age of 58 years. The laser treatment settings were a 6-mm spot side-delivered at a fluence of 0.71 J/cm2 in a pulse width of 750 picoseconds. The patients were treated five times on the neck every 2-4 weeks, and follow-up visits were scheduled for 1 month and 3 months after the last treatment. Digital photos were taken at each visit. Formal assessment tools included patient and physician satisfaction scores and the Global Aesthetic Improvement Scale. In all, 21 women and 3 men completed the study. The majority (72%) had Fitzpatrick skin type II, while 16% had type III, 8% had type I, and 4% had type IV. An average of 5,042 pulses were delivered during each treatment session. The majority of patients (84%) required no anesthesia, while the rest used topical numbing medicine from 30 minutes to an hour prior to the procedure.
Dr. Jeon reported that the average pain score during the procedure was 4.7 on a 10-point scale. Forced-air cooling was used for comfort, and on average, mild redness following the treatment lasted less than 1 day (a mean of 0.6 days, with a range of 0-5 days). Mild pain also lasted less than 1 day (a mean of 0.1 days, with a range of 0-2 days). No swelling, crusting, bruising, bleeding, infection, blistering, scarring, burn, or dyspigmentation occurred.
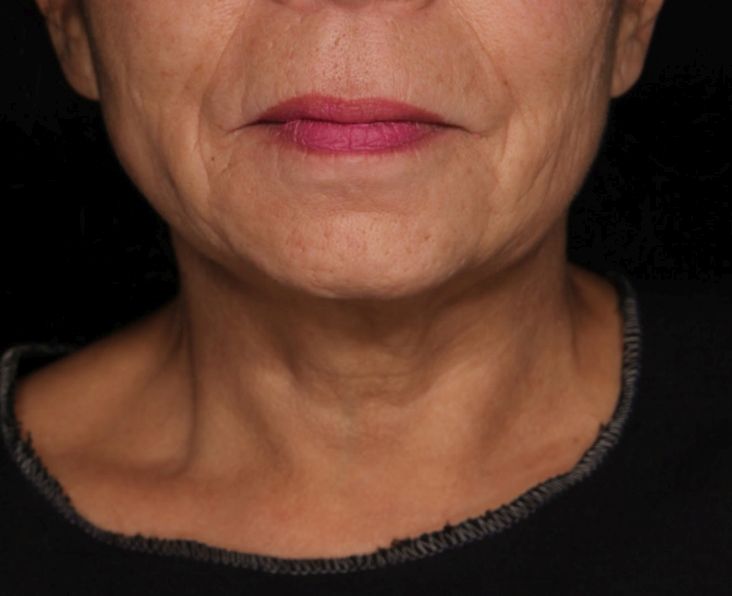
On the Global Aesthetic Improvement Scale at 1 and 3 months, physicians described 43% and 23% of cases, respectively, as “improved,” 17% and 18% of cases as “much improved,” and 4% and 9% of cases as “extremely improved.”
At 3 months, 35% of patients said they would be “somewhat likely” to recommend the procedure, and 30% said they would be “extremely likely” to recommend it.
Dr. Jeon reported having no financial disclosures.
DALLAS –
“It’s important to note that response was variable, but many patients were satisfied with the treatment,” Hana Jeon, MD said at the annual conference of the American Society for Laser Medicine and Surgery, Inc. “Further studies are needed to identify the clinical characteristics of neck laxity that would most benefit from this treatment.”
The researchers enrolled 25 patients with an average age of 58 years. The laser treatment settings were a 6-mm spot side-delivered at a fluence of 0.71 J/cm2 in a pulse width of 750 picoseconds. The patients were treated five times on the neck every 2-4 weeks, and follow-up visits were scheduled for 1 month and 3 months after the last treatment. Digital photos were taken at each visit. Formal assessment tools included patient and physician satisfaction scores and the Global Aesthetic Improvement Scale. In all, 21 women and 3 men completed the study. The majority (72%) had Fitzpatrick skin type II, while 16% had type III, 8% had type I, and 4% had type IV. An average of 5,042 pulses were delivered during each treatment session. The majority of patients (84%) required no anesthesia, while the rest used topical numbing medicine from 30 minutes to an hour prior to the procedure.
Dr. Jeon reported that the average pain score during the procedure was 4.7 on a 10-point scale. Forced-air cooling was used for comfort, and on average, mild redness following the treatment lasted less than 1 day (a mean of 0.6 days, with a range of 0-5 days). Mild pain also lasted less than 1 day (a mean of 0.1 days, with a range of 0-2 days). No swelling, crusting, bruising, bleeding, infection, blistering, scarring, burn, or dyspigmentation occurred.
On the Global Aesthetic Improvement Scale at 1 and 3 months, physicians described 43% and 23% of cases, respectively, as “improved,” 17% and 18% of cases as “much improved,” and 4% and 9% of cases as “extremely improved.”
At 3 months, 35% of patients said they would be “somewhat likely” to recommend the procedure, and 30% said they would be “extremely likely” to recommend it.
Dr. Jeon reported having no financial disclosures.
DALLAS –
“It’s important to note that response was variable, but many patients were satisfied with the treatment,” Hana Jeon, MD said at the annual conference of the American Society for Laser Medicine and Surgery, Inc. “Further studies are needed to identify the clinical characteristics of neck laxity that would most benefit from this treatment.”
The researchers enrolled 25 patients with an average age of 58 years. The laser treatment settings were a 6-mm spot side-delivered at a fluence of 0.71 J/cm2 in a pulse width of 750 picoseconds. The patients were treated five times on the neck every 2-4 weeks, and follow-up visits were scheduled for 1 month and 3 months after the last treatment. Digital photos were taken at each visit. Formal assessment tools included patient and physician satisfaction scores and the Global Aesthetic Improvement Scale. In all, 21 women and 3 men completed the study. The majority (72%) had Fitzpatrick skin type II, while 16% had type III, 8% had type I, and 4% had type IV. An average of 5,042 pulses were delivered during each treatment session. The majority of patients (84%) required no anesthesia, while the rest used topical numbing medicine from 30 minutes to an hour prior to the procedure.
Dr. Jeon reported that the average pain score during the procedure was 4.7 on a 10-point scale. Forced-air cooling was used for comfort, and on average, mild redness following the treatment lasted less than 1 day (a mean of 0.6 days, with a range of 0-5 days). Mild pain also lasted less than 1 day (a mean of 0.1 days, with a range of 0-2 days). No swelling, crusting, bruising, bleeding, infection, blistering, scarring, burn, or dyspigmentation occurred.
On the Global Aesthetic Improvement Scale at 1 and 3 months, physicians described 43% and 23% of cases, respectively, as “improved,” 17% and 18% of cases as “much improved,” and 4% and 9% of cases as “extremely improved.”
At 3 months, 35% of patients said they would be “somewhat likely” to recommend the procedure, and 30% said they would be “extremely likely” to recommend it.
Dr. Jeon reported having no financial disclosures.
REPORTING FROM ASLMS 2018
Key clinical point: Response to using a picosecond 755-nm laser with focus lens array for neck rejuvenation was variable.
Major finding: On the Global Aesthetic Improvement Scale at 1 and 3 months, physicians described 43% and 23% of cases, respectively, as “improved.”
Study details: A single-center study of 25 patients treated for neck laxity.
Disclosures: Dr. Jeon reported having no financial disclosures.
Infections predispose patients to developing Sjögren’s
WASHINGTON –
“We observed a consistent association between infections and the subsequent development of primary Sjögren’s syndrome,” said Johannes Mofors of the department of medicine at the Karolinska University Hospital, Stockholm, in his presentation. “Infections of certain anatomical sites have different associations to Sjögren’s.”
With risk measurements primarily reliant on detecting the presence of MHC genes, this knowledge could be helpful in identifying at-risk patients and give physicians the chance to act before the syndrome emerges, according to Mr. Mofors.
Investigators conducted a retrospective, multicenter, controlled cohort study of 9,993 Swedish individuals from the country’s national patient registry to observe the association between infections and Sjögren’s.
Patients were an average age of 55 years, with either an SSA or SSB infection, with an average observational period of 16 years before diagnosis.
Of the patients with Sjögren’s disease, 21% reported one or more infections prior to diagnosis, compared with 12% among the control group.
When assessing patients by their type of infection, Mr. Mofors and his colleagues found the likelihood of developing Sjögren’s varied depending on which infection was present.
“We looked at respiratory infections, with the SSA/Ro-, SSB/Ro-positive patients having a stronger association than the corresponding rate of SSA-, SSB-negative patients,” explained Mr. Mofors. “Interestingly, as we looked at patients with skin infections, we observed an association with the SSA-, SSB-positive patients having a stronger association than the negative patients.”
Investigators also tested gastrointestinal infections, but found no clear association to Sjögren’s.
Presence of more than one infection also appeared to increased disposition of patients to Sjögren’s syndrome, although it depended on the type of infection, Mr. Mofors said at the meeting, which was sponsored by Johns Hopkins University and the National Institutes of Health.
Patients with multiple respiratory infections showed a stronger association to Sjögren’s, patients with SSA- or SSB-positive infection displaying even stronger prevalence, and patients with skin infections showed a dose-response pattern.
Patients with SSA or SSB pattern showed no significant association.
It is possible, said Mr. Mofors, that patients became more susceptible to infection as their Sjögren’s manifested, so investigators extended the omission period of their study from 3 to 7 years.
“As the omission period was extended, in the aggregated group of cases we saw a less prominent association; however, for the respiratory infections the change in relationship was insignificant,” said Mr. Mofors.
For SSA- and SSB-negative patients, the association between infections and predisposition to Sögren’s was not significant when the omission period was extended.
Mr. Mofors reported no relevant financial disclosures.
WASHINGTON –
“We observed a consistent association between infections and the subsequent development of primary Sjögren’s syndrome,” said Johannes Mofors of the department of medicine at the Karolinska University Hospital, Stockholm, in his presentation. “Infections of certain anatomical sites have different associations to Sjögren’s.”
With risk measurements primarily reliant on detecting the presence of MHC genes, this knowledge could be helpful in identifying at-risk patients and give physicians the chance to act before the syndrome emerges, according to Mr. Mofors.
Investigators conducted a retrospective, multicenter, controlled cohort study of 9,993 Swedish individuals from the country’s national patient registry to observe the association between infections and Sjögren’s.
Patients were an average age of 55 years, with either an SSA or SSB infection, with an average observational period of 16 years before diagnosis.
Of the patients with Sjögren’s disease, 21% reported one or more infections prior to diagnosis, compared with 12% among the control group.
When assessing patients by their type of infection, Mr. Mofors and his colleagues found the likelihood of developing Sjögren’s varied depending on which infection was present.
“We looked at respiratory infections, with the SSA/Ro-, SSB/Ro-positive patients having a stronger association than the corresponding rate of SSA-, SSB-negative patients,” explained Mr. Mofors. “Interestingly, as we looked at patients with skin infections, we observed an association with the SSA-, SSB-positive patients having a stronger association than the negative patients.”
Investigators also tested gastrointestinal infections, but found no clear association to Sjögren’s.
Presence of more than one infection also appeared to increased disposition of patients to Sjögren’s syndrome, although it depended on the type of infection, Mr. Mofors said at the meeting, which was sponsored by Johns Hopkins University and the National Institutes of Health.
Patients with multiple respiratory infections showed a stronger association to Sjögren’s, patients with SSA- or SSB-positive infection displaying even stronger prevalence, and patients with skin infections showed a dose-response pattern.
Patients with SSA or SSB pattern showed no significant association.
It is possible, said Mr. Mofors, that patients became more susceptible to infection as their Sjögren’s manifested, so investigators extended the omission period of their study from 3 to 7 years.
“As the omission period was extended, in the aggregated group of cases we saw a less prominent association; however, for the respiratory infections the change in relationship was insignificant,” said Mr. Mofors.
For SSA- and SSB-negative patients, the association between infections and predisposition to Sögren’s was not significant when the omission period was extended.
Mr. Mofors reported no relevant financial disclosures.
WASHINGTON –
“We observed a consistent association between infections and the subsequent development of primary Sjögren’s syndrome,” said Johannes Mofors of the department of medicine at the Karolinska University Hospital, Stockholm, in his presentation. “Infections of certain anatomical sites have different associations to Sjögren’s.”
With risk measurements primarily reliant on detecting the presence of MHC genes, this knowledge could be helpful in identifying at-risk patients and give physicians the chance to act before the syndrome emerges, according to Mr. Mofors.
Investigators conducted a retrospective, multicenter, controlled cohort study of 9,993 Swedish individuals from the country’s national patient registry to observe the association between infections and Sjögren’s.
Patients were an average age of 55 years, with either an SSA or SSB infection, with an average observational period of 16 years before diagnosis.
Of the patients with Sjögren’s disease, 21% reported one or more infections prior to diagnosis, compared with 12% among the control group.
When assessing patients by their type of infection, Mr. Mofors and his colleagues found the likelihood of developing Sjögren’s varied depending on which infection was present.
“We looked at respiratory infections, with the SSA/Ro-, SSB/Ro-positive patients having a stronger association than the corresponding rate of SSA-, SSB-negative patients,” explained Mr. Mofors. “Interestingly, as we looked at patients with skin infections, we observed an association with the SSA-, SSB-positive patients having a stronger association than the negative patients.”
Investigators also tested gastrointestinal infections, but found no clear association to Sjögren’s.
Presence of more than one infection also appeared to increased disposition of patients to Sjögren’s syndrome, although it depended on the type of infection, Mr. Mofors said at the meeting, which was sponsored by Johns Hopkins University and the National Institutes of Health.
Patients with multiple respiratory infections showed a stronger association to Sjögren’s, patients with SSA- or SSB-positive infection displaying even stronger prevalence, and patients with skin infections showed a dose-response pattern.
Patients with SSA or SSB pattern showed no significant association.
It is possible, said Mr. Mofors, that patients became more susceptible to infection as their Sjögren’s manifested, so investigators extended the omission period of their study from 3 to 7 years.
“As the omission period was extended, in the aggregated group of cases we saw a less prominent association; however, for the respiratory infections the change in relationship was insignificant,” said Mr. Mofors.
For SSA- and SSB-negative patients, the association between infections and predisposition to Sögren’s was not significant when the omission period was extended.
Mr. Mofors reported no relevant financial disclosures.
REPORTING FROM ISSS
Key clinical point: Infections can be used to identify predisposition to Sjögren’s syndrome.
Major finding: Of the observed Sjögren’s syndrome patients, 21% had an infection prior to diagnosis, compared with 12% in the control group.
Study details: A controlled, multicenter, retrospective cohort study of 9,993 patients collected from the Swedish national patient database.
Disclosures: The investigators reported no relevant financial disclosures.
With these pearls, the med-tech space can be your oyster
BOSTON – Having a great idea is just the first step to landing a financial partner in device development – backers also scrutinize more intangible qualities.
The willingness to work as part of a team, the ability to project realistic expectations, the fortitude to take risks and persevere when circumstances get tough – these attributes are critical to forging a strong strategic alliance with a financial partner, Brian Tinkham said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.

Put your own skin in the game
“To me, ‘skin in the game’ is mainly money. Investing your own money, your family’s money, changes the way you behave at the board meeting and when you spend that money,” Mr. Tinkham said. “We like people who are all in on this. When I see an entrepreneur who’s showing a return that’s not good enough for his own investment, I can lose confidence and trust. We want people who won’t walk away from their money, their family’s money, or my money; when times get tough and challenging, decisions need to be made.”
Be realistic
There’s a difference between confidence and irrational confidence, Mr. Tinkham said. “If you come to me presenting a game plan that says you’ll have a commercially viable product in 1 year for a $500,000 investment, you’ll shoot your credibility right off. We know exactly how hard it is to build a $10 million business, never mind a $100 million business. When you obviously don’t understand what lies ahead of you, it hurts your credibility. Work with people who have experience and let them help you present your ideas and goals in a realistic way, and that will help with raising capital so you can execute your plan.”
Be capital efficient
“The key takeaway here is that raising $100 million doesn’t necessarily make for a strong return for investors. The strong return comes with $20-$40 million raised. Most likely businesses that have raised that much have built a commercial structure, provided proof of concept with some actual sales, and generated enough customer interest to attract strategic partners.”
Location, location, location
“This is so important when you’re developing technology: You need to know where the people with high levels of competency are, and where the money is. If you don’t live near these localities, get on a plane and get there – that’s where the business is being done.”
California and the Philadelphia-Boston-New York corridor are the two biggest med-tech and investor hot spots in the United States, Mr. Tinkham noted. Smaller centers of innovation are scattered around the country, including Seattle, Denver, Minneapolis, Chicago, Pittsburgh, Washington, Raleigh-Durham, Atlanta, Austin, and Phoenix.
Be patient
“Adopting a new technology takes time, and the more disruptive the idea, the longer it takes to achieve market adoption. Translating that into med-tech, the time from founding a company to exit will take more than 5 years. Only 10% of companies do it in less time than that,” Mr. Tinkham said. “And you have to remember that not all of the exits we see are good ones – they can be exits in which investors lose most of the capital they’ve brought into the company.”
De-risk
Be the entrepreneur who takes a vision to a viable product.
“Most physician entrepreneurs come up with an idea and protect it – but don’t move it further. We want to see an idea that’s been created and then de-risked. You protect it, you prototype it, go into preclinical studies, then clinically validate it or obtain regulatory approval. And then in the end, to us the best measure is your revenue. Are customers buying it? Do they see in it the same value that you, the entrepreneur, sees? If you can get it there, you’ve got something. The further you de-risk something, the more attractive you become.”
Are you a physician innovator?
If you have an idea for a new technology to improve gastroenterology, the AGA Center for GI Innovation and Technology can help you through the device development and adoption process.
Get in touch with us at [email protected].
BOSTON – Having a great idea is just the first step to landing a financial partner in device development – backers also scrutinize more intangible qualities.
The willingness to work as part of a team, the ability to project realistic expectations, the fortitude to take risks and persevere when circumstances get tough – these attributes are critical to forging a strong strategic alliance with a financial partner, Brian Tinkham said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
Put your own skin in the game
“To me, ‘skin in the game’ is mainly money. Investing your own money, your family’s money, changes the way you behave at the board meeting and when you spend that money,” Mr. Tinkham said. “We like people who are all in on this. When I see an entrepreneur who’s showing a return that’s not good enough for his own investment, I can lose confidence and trust. We want people who won’t walk away from their money, their family’s money, or my money; when times get tough and challenging, decisions need to be made.”
Be realistic
There’s a difference between confidence and irrational confidence, Mr. Tinkham said. “If you come to me presenting a game plan that says you’ll have a commercially viable product in 1 year for a $500,000 investment, you’ll shoot your credibility right off. We know exactly how hard it is to build a $10 million business, never mind a $100 million business. When you obviously don’t understand what lies ahead of you, it hurts your credibility. Work with people who have experience and let them help you present your ideas and goals in a realistic way, and that will help with raising capital so you can execute your plan.”
Be capital efficient
“The key takeaway here is that raising $100 million doesn’t necessarily make for a strong return for investors. The strong return comes with $20-$40 million raised. Most likely businesses that have raised that much have built a commercial structure, provided proof of concept with some actual sales, and generated enough customer interest to attract strategic partners.”
Location, location, location
“This is so important when you’re developing technology: You need to know where the people with high levels of competency are, and where the money is. If you don’t live near these localities, get on a plane and get there – that’s where the business is being done.”
California and the Philadelphia-Boston-New York corridor are the two biggest med-tech and investor hot spots in the United States, Mr. Tinkham noted. Smaller centers of innovation are scattered around the country, including Seattle, Denver, Minneapolis, Chicago, Pittsburgh, Washington, Raleigh-Durham, Atlanta, Austin, and Phoenix.
Be patient
“Adopting a new technology takes time, and the more disruptive the idea, the longer it takes to achieve market adoption. Translating that into med-tech, the time from founding a company to exit will take more than 5 years. Only 10% of companies do it in less time than that,” Mr. Tinkham said. “And you have to remember that not all of the exits we see are good ones – they can be exits in which investors lose most of the capital they’ve brought into the company.”
De-risk
Be the entrepreneur who takes a vision to a viable product.
“Most physician entrepreneurs come up with an idea and protect it – but don’t move it further. We want to see an idea that’s been created and then de-risked. You protect it, you prototype it, go into preclinical studies, then clinically validate it or obtain regulatory approval. And then in the end, to us the best measure is your revenue. Are customers buying it? Do they see in it the same value that you, the entrepreneur, sees? If you can get it there, you’ve got something. The further you de-risk something, the more attractive you become.”
Are you a physician innovator?
If you have an idea for a new technology to improve gastroenterology, the AGA Center for GI Innovation and Technology can help you through the device development and adoption process.
Get in touch with us at [email protected].
BOSTON – Having a great idea is just the first step to landing a financial partner in device development – backers also scrutinize more intangible qualities.
The willingness to work as part of a team, the ability to project realistic expectations, the fortitude to take risks and persevere when circumstances get tough – these attributes are critical to forging a strong strategic alliance with a financial partner, Brian Tinkham said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
Put your own skin in the game
“To me, ‘skin in the game’ is mainly money. Investing your own money, your family’s money, changes the way you behave at the board meeting and when you spend that money,” Mr. Tinkham said. “We like people who are all in on this. When I see an entrepreneur who’s showing a return that’s not good enough for his own investment, I can lose confidence and trust. We want people who won’t walk away from their money, their family’s money, or my money; when times get tough and challenging, decisions need to be made.”
Be realistic
There’s a difference between confidence and irrational confidence, Mr. Tinkham said. “If you come to me presenting a game plan that says you’ll have a commercially viable product in 1 year for a $500,000 investment, you’ll shoot your credibility right off. We know exactly how hard it is to build a $10 million business, never mind a $100 million business. When you obviously don’t understand what lies ahead of you, it hurts your credibility. Work with people who have experience and let them help you present your ideas and goals in a realistic way, and that will help with raising capital so you can execute your plan.”
Be capital efficient
“The key takeaway here is that raising $100 million doesn’t necessarily make for a strong return for investors. The strong return comes with $20-$40 million raised. Most likely businesses that have raised that much have built a commercial structure, provided proof of concept with some actual sales, and generated enough customer interest to attract strategic partners.”
Location, location, location
“This is so important when you’re developing technology: You need to know where the people with high levels of competency are, and where the money is. If you don’t live near these localities, get on a plane and get there – that’s where the business is being done.”
California and the Philadelphia-Boston-New York corridor are the two biggest med-tech and investor hot spots in the United States, Mr. Tinkham noted. Smaller centers of innovation are scattered around the country, including Seattle, Denver, Minneapolis, Chicago, Pittsburgh, Washington, Raleigh-Durham, Atlanta, Austin, and Phoenix.
Be patient
“Adopting a new technology takes time, and the more disruptive the idea, the longer it takes to achieve market adoption. Translating that into med-tech, the time from founding a company to exit will take more than 5 years. Only 10% of companies do it in less time than that,” Mr. Tinkham said. “And you have to remember that not all of the exits we see are good ones – they can be exits in which investors lose most of the capital they’ve brought into the company.”
De-risk
Be the entrepreneur who takes a vision to a viable product.
“Most physician entrepreneurs come up with an idea and protect it – but don’t move it further. We want to see an idea that’s been created and then de-risked. You protect it, you prototype it, go into preclinical studies, then clinically validate it or obtain regulatory approval. And then in the end, to us the best measure is your revenue. Are customers buying it? Do they see in it the same value that you, the entrepreneur, sees? If you can get it there, you’ve got something. The further you de-risk something, the more attractive you become.”
Are you a physician innovator?
If you have an idea for a new technology to improve gastroenterology, the AGA Center for GI Innovation and Technology can help you through the device development and adoption process.
Get in touch with us at [email protected].
EXPERT ANALYSIS FROM 2018 AGA TECH SUMMIT
Evidence is essential but not sufficient to move guidelines
BOSTON – For those considering how to navigate their innovative health care strategy into a position that will lead to an eventual guideline recommendation, it is important to think beyond demonstration of efficacy and safety in the design of randomized trials, according to an overview of how guideline committees currently function.
“In the old days, it was only the strength of the evidence. Now, in addition to the evidence, we have three other issues we look at to form the strength of a recommendation,” John M. Inadomi, MD, AGAF, head of the division of gastroenterology, University of Washington, Seattle, said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.

“I think the big thing is that we are trying to move away from is just-the-evidence [approach],” Dr. Inadomi explained to an audience that included physician entrepreneurs and investors with an interest in how to establish a new diagnostic tool or treatment device as a standard of care.
There is no doubt that randomized controlled trial data are critical for objectively establishing safety and efficacy, but there has been an evolutionary change. According to Dr. Inadomi, guideline committees are posing more pointed questions about the practical value of one strategy relative to others. They also have increased their scrutiny of the quality and consistency of the RCT data in relation to the specific indication being considered.
“The implication of a strong recommendation is that most people in the situation would want the recommended course of action and that only a small proportion would not,” Dr. Inadomi explained. On the basis of this criterion, an inconvenient, costly, or poorly accepted therapy may not receive a strong recommendation even if effective. Strong recommendations typically set a standard.
“For the health care provider, that means that most patients should receive that course of action,” Dr. Inadomi said. Conversely, “for a weak recommendation, it implies that the majority of people would want this, but many would not.”
Strong versus weak recommendations have an impact on health care policy, Dr. Inadomi added. Those measuring quality of care might, in some cases, evaluate the frequency with which patients receive guideline-based care that has been given a 1A rating, which identifies the strongest recommendation. Weak recommendations encourage a greater emphasis on shared decision making that recognizes alternative treatment strategies in the context of patient preferences and values.
A reorientation that considers the limits of objective data by itself is reflected in a less restrictive view on the source of the data used in guideline deliberations, according to Dr. Inadomi. “It was once thought that all RCTs are good and observational studies are bad,” he said, adding that this view has changed with greater appreciation of publication bias and RCT study limitations, such as enrollment of nonrepresentative patient populations. While RCT data are preferred, he contended that observational studies are influential to guideline committees when there is a large effect size and there is consistency of evidence.
The move away from evidence-only guidelines is driven by a greater appreciation of value, Dr. Inadomi suggested. For entrepreneurs who hope to shepherd their devices or tools into a central position in clinical medicine, safety and efficacy are critical but may no longer be sufficient.
Dr. Inadomi has no disclosures relevant to this topic.
BOSTON – For those considering how to navigate their innovative health care strategy into a position that will lead to an eventual guideline recommendation, it is important to think beyond demonstration of efficacy and safety in the design of randomized trials, according to an overview of how guideline committees currently function.
“In the old days, it was only the strength of the evidence. Now, in addition to the evidence, we have three other issues we look at to form the strength of a recommendation,” John M. Inadomi, MD, AGAF, head of the division of gastroenterology, University of Washington, Seattle, said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
“I think the big thing is that we are trying to move away from is just-the-evidence [approach],” Dr. Inadomi explained to an audience that included physician entrepreneurs and investors with an interest in how to establish a new diagnostic tool or treatment device as a standard of care.
There is no doubt that randomized controlled trial data are critical for objectively establishing safety and efficacy, but there has been an evolutionary change. According to Dr. Inadomi, guideline committees are posing more pointed questions about the practical value of one strategy relative to others. They also have increased their scrutiny of the quality and consistency of the RCT data in relation to the specific indication being considered.
“The implication of a strong recommendation is that most people in the situation would want the recommended course of action and that only a small proportion would not,” Dr. Inadomi explained. On the basis of this criterion, an inconvenient, costly, or poorly accepted therapy may not receive a strong recommendation even if effective. Strong recommendations typically set a standard.
“For the health care provider, that means that most patients should receive that course of action,” Dr. Inadomi said. Conversely, “for a weak recommendation, it implies that the majority of people would want this, but many would not.”
Strong versus weak recommendations have an impact on health care policy, Dr. Inadomi added. Those measuring quality of care might, in some cases, evaluate the frequency with which patients receive guideline-based care that has been given a 1A rating, which identifies the strongest recommendation. Weak recommendations encourage a greater emphasis on shared decision making that recognizes alternative treatment strategies in the context of patient preferences and values.
A reorientation that considers the limits of objective data by itself is reflected in a less restrictive view on the source of the data used in guideline deliberations, according to Dr. Inadomi. “It was once thought that all RCTs are good and observational studies are bad,” he said, adding that this view has changed with greater appreciation of publication bias and RCT study limitations, such as enrollment of nonrepresentative patient populations. While RCT data are preferred, he contended that observational studies are influential to guideline committees when there is a large effect size and there is consistency of evidence.
The move away from evidence-only guidelines is driven by a greater appreciation of value, Dr. Inadomi suggested. For entrepreneurs who hope to shepherd their devices or tools into a central position in clinical medicine, safety and efficacy are critical but may no longer be sufficient.
Dr. Inadomi has no disclosures relevant to this topic.
BOSTON – For those considering how to navigate their innovative health care strategy into a position that will lead to an eventual guideline recommendation, it is important to think beyond demonstration of efficacy and safety in the design of randomized trials, according to an overview of how guideline committees currently function.
“In the old days, it was only the strength of the evidence. Now, in addition to the evidence, we have three other issues we look at to form the strength of a recommendation,” John M. Inadomi, MD, AGAF, head of the division of gastroenterology, University of Washington, Seattle, said at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
“I think the big thing is that we are trying to move away from is just-the-evidence [approach],” Dr. Inadomi explained to an audience that included physician entrepreneurs and investors with an interest in how to establish a new diagnostic tool or treatment device as a standard of care.
There is no doubt that randomized controlled trial data are critical for objectively establishing safety and efficacy, but there has been an evolutionary change. According to Dr. Inadomi, guideline committees are posing more pointed questions about the practical value of one strategy relative to others. They also have increased their scrutiny of the quality and consistency of the RCT data in relation to the specific indication being considered.
“The implication of a strong recommendation is that most people in the situation would want the recommended course of action and that only a small proportion would not,” Dr. Inadomi explained. On the basis of this criterion, an inconvenient, costly, or poorly accepted therapy may not receive a strong recommendation even if effective. Strong recommendations typically set a standard.
“For the health care provider, that means that most patients should receive that course of action,” Dr. Inadomi said. Conversely, “for a weak recommendation, it implies that the majority of people would want this, but many would not.”
Strong versus weak recommendations have an impact on health care policy, Dr. Inadomi added. Those measuring quality of care might, in some cases, evaluate the frequency with which patients receive guideline-based care that has been given a 1A rating, which identifies the strongest recommendation. Weak recommendations encourage a greater emphasis on shared decision making that recognizes alternative treatment strategies in the context of patient preferences and values.
A reorientation that considers the limits of objective data by itself is reflected in a less restrictive view on the source of the data used in guideline deliberations, according to Dr. Inadomi. “It was once thought that all RCTs are good and observational studies are bad,” he said, adding that this view has changed with greater appreciation of publication bias and RCT study limitations, such as enrollment of nonrepresentative patient populations. While RCT data are preferred, he contended that observational studies are influential to guideline committees when there is a large effect size and there is consistency of evidence.
The move away from evidence-only guidelines is driven by a greater appreciation of value, Dr. Inadomi suggested. For entrepreneurs who hope to shepherd their devices or tools into a central position in clinical medicine, safety and efficacy are critical but may no longer be sufficient.
Dr. Inadomi has no disclosures relevant to this topic.
EXPERT ANALYSIS FROM 2018 AGA TECH SUMMIT
Physiology, not mechanics, explains benefit of bariatric procedures
boston – Rather than being a better strategy to block absorption of ingested calories, the future of bariatric surgery depends on treatment combinations that promote weight control through healthy physiology, according to three experts participating in a panel on this topic at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
“When we think about the mechanisms of surgery, the mechanical model is dead. There is no good supporting evidence for the mechanical model. The current model is all physiological, involving changes in signaling from the gut to the rest of the body,” asserted Lee Kaplan, MD, PhD, AGAF, director of the Weight Center at Massachusetts General Hospital, Boston.
The list of evidence suggesting that change in physiologic function is a far more important explanation for weight loss from bariatric interventions is long, according to Dr. Kaplan. Of his many examples, he noted that pregnant women gain weight normally after bariatric surgery.
“Now, if you cannot absorb food normally after bariatric surgery, how do you gain weight normally when pregnant?” Dr. Kaplan asked. The answer to this and other examples of a disconnect between a simple food-blocking mechanism and what is observed is that bariatric procedures favorably alter signals that control hunger, satiety, and metabolism.
The two other experts on the panel largely agreed. In discussing advances in small-bowel devices for the treatment of type 2 diabetes mellitus, Christopher Thompson, MD, AGAF, director of therapeutic endoscopy at Brigham and Women’s Hospital, Boston, also looked to physiologic effects of bariatric surgery. He placed particular emphasis on the foregut and hindgut hypotheses. These hypotheses are “not yet written in stone,” but they provide a conceptual basis for understanding metabolic changes observed after bariatric procedures.
“One way that gastric bypass might work is that it alters the incretins that drive insulin secretion and sensitivity,” Dr. Thompson said. The same principle has been proposed for a novel incisionless magnetic device developed by Dr. Thompson that is now in clinical trials. The device, which creates an anastomosis and a partial jejunal diversion, achieved a 40% excess weight loss and a significant reduction in hemoglobin A1c levels among patients with type 2 diabetes mellitus in an initial study. Dr. Thompson contended that this effect cannot be explained by a change in nutrient absorption.
A surgeon serving on the panel, Marina Kurian, MD, of New York University’s Langone Medical Center, New York, also referenced the evidence for physiologic effects when speaking about gastric bypass and sleeve gastrectomy. Although both involve a blocking function for food absorption, she agreed that there are several reasons why this may not account for benefits.
“Certainly with gastric bypass, we talk about foregut and hindgut theory in terms of incretin effect,” Dr. Kurian said. She also noted that even the procedures that produce the greatest restriction on food absorption are not typically effective as a single therapeutic approach. Rather, her major point was that no approach, whether surgical, endoscopic, or lifestyle, is generally sufficient to achieve and maintain weight loss indefinitely. In her own practice, she has been moving to a “one-stop shopping” approach to coordinate multiple options.
“Those of us working in obesity are very aware of its chronicity and how one intervention is not enough,” Dr. Kurian said. She suggested that coordinated care among surgeons, gastroenterologists, dietitians, behavioral therapists, and others will provide the road forward even if the next set of surgical procedures or endoscopic devices are incrementally more effective than current options for weight loss.
One reason that a single intervention may not be enough is that obesity is not a single disease but the product of multiple different pathological processes, according to Dr. Kaplan. This is supported by the varied response to current therapies. Producing a variety of examples, he showed that, although there are large weight reductions with the most successful therapies, some patients are exceptional responders, while a proportion of patients lose little or no weight and others actually gain weight. He expressed doubt that there will be a single solution applicable to all patients.
“Patients who respond to one therapy may not respond to another and vice versa, and so the goal is to match each patient with the therapy that is most appropriate and protective for them,” Dr. Kaplan said.
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The POWER (Practice Guide on Obesity and Weight Management, Education and Resources) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management.
Learn more at http://www.cghjournal.org/article/S1542-3565(16)309880/fulltext.
boston – Rather than being a better strategy to block absorption of ingested calories, the future of bariatric surgery depends on treatment combinations that promote weight control through healthy physiology, according to three experts participating in a panel on this topic at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
“When we think about the mechanisms of surgery, the mechanical model is dead. There is no good supporting evidence for the mechanical model. The current model is all physiological, involving changes in signaling from the gut to the rest of the body,” asserted Lee Kaplan, MD, PhD, AGAF, director of the Weight Center at Massachusetts General Hospital, Boston.
The list of evidence suggesting that change in physiologic function is a far more important explanation for weight loss from bariatric interventions is long, according to Dr. Kaplan. Of his many examples, he noted that pregnant women gain weight normally after bariatric surgery.
“Now, if you cannot absorb food normally after bariatric surgery, how do you gain weight normally when pregnant?” Dr. Kaplan asked. The answer to this and other examples of a disconnect between a simple food-blocking mechanism and what is observed is that bariatric procedures favorably alter signals that control hunger, satiety, and metabolism.
The two other experts on the panel largely agreed. In discussing advances in small-bowel devices for the treatment of type 2 diabetes mellitus, Christopher Thompson, MD, AGAF, director of therapeutic endoscopy at Brigham and Women’s Hospital, Boston, also looked to physiologic effects of bariatric surgery. He placed particular emphasis on the foregut and hindgut hypotheses. These hypotheses are “not yet written in stone,” but they provide a conceptual basis for understanding metabolic changes observed after bariatric procedures.
“One way that gastric bypass might work is that it alters the incretins that drive insulin secretion and sensitivity,” Dr. Thompson said. The same principle has been proposed for a novel incisionless magnetic device developed by Dr. Thompson that is now in clinical trials. The device, which creates an anastomosis and a partial jejunal diversion, achieved a 40% excess weight loss and a significant reduction in hemoglobin A1c levels among patients with type 2 diabetes mellitus in an initial study. Dr. Thompson contended that this effect cannot be explained by a change in nutrient absorption.
A surgeon serving on the panel, Marina Kurian, MD, of New York University’s Langone Medical Center, New York, also referenced the evidence for physiologic effects when speaking about gastric bypass and sleeve gastrectomy. Although both involve a blocking function for food absorption, she agreed that there are several reasons why this may not account for benefits.
“Certainly with gastric bypass, we talk about foregut and hindgut theory in terms of incretin effect,” Dr. Kurian said. She also noted that even the procedures that produce the greatest restriction on food absorption are not typically effective as a single therapeutic approach. Rather, her major point was that no approach, whether surgical, endoscopic, or lifestyle, is generally sufficient to achieve and maintain weight loss indefinitely. In her own practice, she has been moving to a “one-stop shopping” approach to coordinate multiple options.
“Those of us working in obesity are very aware of its chronicity and how one intervention is not enough,” Dr. Kurian said. She suggested that coordinated care among surgeons, gastroenterologists, dietitians, behavioral therapists, and others will provide the road forward even if the next set of surgical procedures or endoscopic devices are incrementally more effective than current options for weight loss.
One reason that a single intervention may not be enough is that obesity is not a single disease but the product of multiple different pathological processes, according to Dr. Kaplan. This is supported by the varied response to current therapies. Producing a variety of examples, he showed that, although there are large weight reductions with the most successful therapies, some patients are exceptional responders, while a proportion of patients lose little or no weight and others actually gain weight. He expressed doubt that there will be a single solution applicable to all patients.
“Patients who respond to one therapy may not respond to another and vice versa, and so the goal is to match each patient with the therapy that is most appropriate and protective for them,” Dr. Kaplan said.
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The POWER (Practice Guide on Obesity and Weight Management, Education and Resources) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management.
Learn more at http://www.cghjournal.org/article/S1542-3565(16)309880/fulltext.
boston – Rather than being a better strategy to block absorption of ingested calories, the future of bariatric surgery depends on treatment combinations that promote weight control through healthy physiology, according to three experts participating in a panel on this topic at the 2018 AGA Tech Summit, sponsored by the AGA Center for GI Innovation and Technology.
“When we think about the mechanisms of surgery, the mechanical model is dead. There is no good supporting evidence for the mechanical model. The current model is all physiological, involving changes in signaling from the gut to the rest of the body,” asserted Lee Kaplan, MD, PhD, AGAF, director of the Weight Center at Massachusetts General Hospital, Boston.
The list of evidence suggesting that change in physiologic function is a far more important explanation for weight loss from bariatric interventions is long, according to Dr. Kaplan. Of his many examples, he noted that pregnant women gain weight normally after bariatric surgery.
“Now, if you cannot absorb food normally after bariatric surgery, how do you gain weight normally when pregnant?” Dr. Kaplan asked. The answer to this and other examples of a disconnect between a simple food-blocking mechanism and what is observed is that bariatric procedures favorably alter signals that control hunger, satiety, and metabolism.
The two other experts on the panel largely agreed. In discussing advances in small-bowel devices for the treatment of type 2 diabetes mellitus, Christopher Thompson, MD, AGAF, director of therapeutic endoscopy at Brigham and Women’s Hospital, Boston, also looked to physiologic effects of bariatric surgery. He placed particular emphasis on the foregut and hindgut hypotheses. These hypotheses are “not yet written in stone,” but they provide a conceptual basis for understanding metabolic changes observed after bariatric procedures.
“One way that gastric bypass might work is that it alters the incretins that drive insulin secretion and sensitivity,” Dr. Thompson said. The same principle has been proposed for a novel incisionless magnetic device developed by Dr. Thompson that is now in clinical trials. The device, which creates an anastomosis and a partial jejunal diversion, achieved a 40% excess weight loss and a significant reduction in hemoglobin A1c levels among patients with type 2 diabetes mellitus in an initial study. Dr. Thompson contended that this effect cannot be explained by a change in nutrient absorption.
A surgeon serving on the panel, Marina Kurian, MD, of New York University’s Langone Medical Center, New York, also referenced the evidence for physiologic effects when speaking about gastric bypass and sleeve gastrectomy. Although both involve a blocking function for food absorption, she agreed that there are several reasons why this may not account for benefits.
“Certainly with gastric bypass, we talk about foregut and hindgut theory in terms of incretin effect,” Dr. Kurian said. She also noted that even the procedures that produce the greatest restriction on food absorption are not typically effective as a single therapeutic approach. Rather, her major point was that no approach, whether surgical, endoscopic, or lifestyle, is generally sufficient to achieve and maintain weight loss indefinitely. In her own practice, she has been moving to a “one-stop shopping” approach to coordinate multiple options.
“Those of us working in obesity are very aware of its chronicity and how one intervention is not enough,” Dr. Kurian said. She suggested that coordinated care among surgeons, gastroenterologists, dietitians, behavioral therapists, and others will provide the road forward even if the next set of surgical procedures or endoscopic devices are incrementally more effective than current options for weight loss.
One reason that a single intervention may not be enough is that obesity is not a single disease but the product of multiple different pathological processes, according to Dr. Kaplan. This is supported by the varied response to current therapies. Producing a variety of examples, he showed that, although there are large weight reductions with the most successful therapies, some patients are exceptional responders, while a proportion of patients lose little or no weight and others actually gain weight. He expressed doubt that there will be a single solution applicable to all patients.
“Patients who respond to one therapy may not respond to another and vice versa, and so the goal is to match each patient with the therapy that is most appropriate and protective for them,” Dr. Kaplan said.
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The POWER (Practice Guide on Obesity and Weight Management, Education and Resources) white paper provides physicians with a comprehensive, multidisciplinary process to guide and personalize innovative obesity care for safe and effective weight management.
Learn more at http://www.cghjournal.org/article/S1542-3565(16)309880/fulltext.
EXPERT ANALYSIS FROM 2018 AGA TECH SUMMIT
Eating Fish May Be Associated With a Reduced Risk of MS
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.
“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author Annette Langer-Gould, MD, PhD, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.

For this study, researchers examined the diets of 1,153 people (average age 36) from the MS Sunshine Study, a multi-ethnic matched case-control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
While the study suggests that omega-3 fatty acids, and how they are processed by the body, may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
The study was supported by the National Institute of Neurological Disorders and Stroke.
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.
“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author Annette Langer-Gould, MD, PhD, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.
For this study, researchers examined the diets of 1,153 people (average age 36) from the MS Sunshine Study, a multi-ethnic matched case-control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
While the study suggests that omega-3 fatty acids, and how they are processed by the body, may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
The study was supported by the National Institute of Neurological Disorders and Stroke.
LOS ANGELES—Eating fish at least once per week or eating fish one to three times per month, in addition to taking daily fish oil supplements, may be associated with a reduced risk of multiple sclerosis (MS), according to a preliminary study presented at the American Academy of Neurology’s 70th Annual Meeting. These findings suggest that the omega-3 fatty acids found in fish may be associated with lowering the risk of developing MS.
“Consuming fish that contain omega-3 fatty acids has been shown to have a variety of health benefits, so we wanted to see if this simple lifestyle modification, regularly eating fish and taking fish oil supplements, could reduce the risk of MS,” said lead study author Annette Langer-Gould, MD, PhD, Regional Lead for Clinical and Translational Neuroscience for the Southern California Permanente Medical Group in Pasadena, and Clinical Assistant Professor at the Keck School of Medicine of the University of Southern California in Los Angeles.
For this study, researchers examined the diets of 1,153 people (average age 36) from the MS Sunshine Study, a multi-ethnic matched case-control study of incident MS or clinically isolated syndrome (CIS), recruited from Kaiser Permanente Southern California.
Researchers queried participants about how much fish they consumed regularly. Investigators also examined 13 single nucleotide polymorphisms (SNPs) in FADS1, FADS2, and ELOV2, which regulate fatty acid biosynthesis.
High fish intake was defined as either eating one serving of fish per week or eating one to three servings per month in addition to taking daily fish oil supplements. Low intake was defined as less than one serving of fish per month and no fish oil supplements.
High fish intake was associated with a 45% reduced risk of MS or CIS, when compared with those who ate fish less than once a month and did not take fish oil supplements. A total of 180 of participants with MS had high fish intake compared with 251 of the healthy controls.
In addition, two SNPs, rs174611 and rs174618, in FADS2 were independently associated with a lower risk of MS, even after accounting for high fish intake. This suggests that some people may have a genetic advantage when it comes to regulating fatty acid levels, the researchers noted.
While the study suggests that omega-3 fatty acids, and how they are processed by the body, may play an important role in reducing MS risk. Dr. Langer-Gould and colleagues emphasized that their findings show an association, and not cause and effect. More research is needed to confirm the findings and to examine how omega-3 fatty acids may affect inflammation, metabolism, and nerve function.
The study was supported by the National Institute of Neurological Disorders and Stroke.
Hints of altered microRNA expression in women exposed to EDCs
Endocrine-disrupting chemicals (EDCs) are structurally similar to endogenous hormones and are therefore capable of mimicking these natural hormones, interfering with their biosynthesis, transport, binding action, and/or elimination. In animal studies and human clinical observational and epidemiologic studies of various EDCs, these chemicals have consistently been associated with diabetes mellitus, obesity, hormone-sensitive cancers, neurodevelopmental disorders in children exposed prenatally, and reproductive health.
In 2009, the Endocrine Society published a scientific statement in which it called EDCs a significant concern to human health (Endocr Rev. 2009;30[4]:293-342). Several years later, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a Committee Opinion on Exposure to Toxic Environmental Agents, warning that patient exposure to EDCs and other toxic environmental agents can have a “profound and lasting effect” on reproductive health outcomes across the life course and calling the reduction of exposure a “critical area of intervention” for ob.gyns. and other reproductive health care professionals (Obstet Gynecol. 2013;122[4]:931-5).
Despite strong calls by each of these organizations to not overlook EDCs in the clinical arena, as well as emerging evidence that EDCs may be a risk factor for gestational diabetes (GDM), EDC exposure may not be on the practicing ob.gyn.’s radar. Clinicians should know what these chemicals are and how to talk about them in preconception and prenatal visits. We should carefully consider their known – and potential – risks, and encourage our patients to identify and reduce exposure without being alarmist.
Low-dose effects
EDCs are used in the manufacture of pesticides, industrial chemicals, plastics and plasticizers, hand sanitizers, medical equipment, dental sealants, a variety of personal care products, cosmetics, and other common consumer and household products. They’re found, for example, in sunscreens, canned foods and beverages, food-packaging materials, baby bottles, flame-retardant furniture, stain-resistant carpet, and shoes. We are all ingesting and breathing them in to some degree.
Bisphenol A (BPA), one of the most extensively studied EDCs, is found in the thermal receipt paper routinely used by gas stations, supermarkets, and other stores. In a small study we conducted at Harvard, we found that urinary BPA concentrations increased after continual handling of receipts for 2 hours without gloves but did not increase significantly when gloves were used (JAMA. 2014 Feb 26;311[8]:859-60).

Informed consumers can then affect the market through their purchasing choices, but the removal of concerning chemicals from products takes a long time, and it’s not always immediately clear that replacement chemicals are safer. For instance, the BPA in “BPA-free” water bottles and canned foods has been replaced by bisphenol S (BPS), which has a very similar molecular structure to BPA. The potential adverse health effects of these replacement chemicals are now being examined in experimental and epidemiologic studies.
Through its National Health and Nutrition Examination Survey, the Centers for Disease Control and Prevention has reported detection rates of between 75% and 99% for different EDCs in urine samples collected from a representative sample of the U.S. population. In other human research, several EDCs have been shown to cross the placenta and have been measured in maternal blood and urine and in cord blood and amniotic fluid, as well as in placental tissue at birth.
It is interesting to note that BPA’s structure is similar to that of diethylstilbestrol (DES). BPA was first shown to have estrogenic activity in 1936 and was originally considered for use in pharmaceuticals to prevent miscarriages, spontaneous abortions, and premature labor but was put aside in favor of DES. (DES was eventually found to be carcinogenic and was taken off the market.) In the 1950s, the use of BPA was resuscitated though not in pharmaceuticals.

A better understanding about the mechanisms of action and dose-response patterns of EDCs has indicated that EDCs can act at low doses, and in many cases a nonmonotonic dose-response association has been demonstrated. This is a paradigm shift for traditional toxicology in which it is “the dose that makes the poison,” and some toxicologists have been critical of the claims of low-dose potency for EDCs.
A team of epidemiologists, toxicologists, and other scientists, including myself, critically analyzed in vitro, animal, and epidemiologic studies as part of a National Institute of Environmental Health Sciences working group on BPA to determine the strength of the evidence for low-dose effects (doses lower than those tested in traditional toxicology assessments) of BPA. We found that consistent, reproducible, and often adverse low-dose effects have been demonstrated for BPA in cell lines, primary cells and tissues, laboratory animals, and human populations. We also concluded that EDCs can pose the greatest threats when exposure occurs during early development, organogenesis, and during critical postnatal periods when tissues are differentiating (Endocr Disruptors [Austin, Tex.]. 2013 Sep;1:e25078-1-13).
A potential risk factor for GDM
Quite a lot of research has been done on EDCs and the risk of type 2 diabetes. A recent meta-analysis that included 41 cross-sectional and 8 prospective studies found that serum concentrations of dioxins, polychlorinated biphenyls, and chlorinated pesticides – and urine concentrations of BPA and phthalates – were significantly associated with type 2 diabetes risk. Comparing the highest and lowest concentration categories, the pooled relative risk was 1.45 for BPA and phthalates. EDC concentrations also were associated with indicators of impaired fasting glucose and insulin resistance (J Diabetes. 2016 Jul;8[4]:516-32).

Despite the mounting evidence for an association between BPA and type 2 diabetes, and despite the fact that the increased incidence of GDM in the past 20 years has mirrored the increasing use of EDCs, there has been a dearth of research examining the possible relationship between EDCs and GDM. The effects of BPA on GDM were identified as a knowledge gap by the National Institute of Environmental Health Sciences after a review of the literature from 2007 to 2013 (Environ Health Perspect. 2014 Aug:122[8]:775-86).
To understand the association between EDCs and GDM and the underlying mechanistic pathway of EDCs, we are conducting research that uses a growing body of evidence that suggests that environmental toxins are involved in the control of microRNA (miRNA) expression in trophoblast cells.
MiRNA, a single-stranded, short, noncoding RNA that is involved in posttranslational gene expression, can be packaged along with other signaling molecules inside extracellular vesicles in the placenta called exosomes. These exosomes appear to be shed from the placenta into the maternal circulation as early as 6-7 weeks into pregnancy. Once released into the maternal circulation, research has shown that the exosomes can target and reprogram other cells via the transfer of noncoding miRNAs, thereby changing the gene expression in these cells.
Such an exosome-mediated signaling pathway provides us with the opportunity to isolate exosomes, sequence the miRNAs, and look at whether women who are exposed to higher levels of EDCs (as indicated in urine concentration) have a particular miRNA signature that correlates with GDM. In other words, we’re working to determine whether particular EDCs and exposure levels affect the miRNA placental profiles, and if these profiles are predictive of GDM.
Thus far, in a pilot prospective cohort study of pregnant women, we are seeing hints of altered miRNA expression in relation to GDM. We have selected study participants who are at high risk of developing GDM (for example, prepregnancy body mass index greater than 30, past pregnancy with GDM, or macrosomia) because we suspect that, in many women, EDCs are a tipping point for the development of GDM rather than a sole causative factor. In addition to understanding the impact of EDCs on GDM, it is our hope that miRNAs in maternal circulation will serve as a noninvasive biomarker for early detection of GDM development or susceptibility.
Dr. Ehrlich is an assistant professor of pediatrics and environmental health at Cincinnati Children’s Hospital Medical Center.
Endocrine-disrupting chemicals (EDCs) are structurally similar to endogenous hormones and are therefore capable of mimicking these natural hormones, interfering with their biosynthesis, transport, binding action, and/or elimination. In animal studies and human clinical observational and epidemiologic studies of various EDCs, these chemicals have consistently been associated with diabetes mellitus, obesity, hormone-sensitive cancers, neurodevelopmental disorders in children exposed prenatally, and reproductive health.
In 2009, the Endocrine Society published a scientific statement in which it called EDCs a significant concern to human health (Endocr Rev. 2009;30[4]:293-342). Several years later, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a Committee Opinion on Exposure to Toxic Environmental Agents, warning that patient exposure to EDCs and other toxic environmental agents can have a “profound and lasting effect” on reproductive health outcomes across the life course and calling the reduction of exposure a “critical area of intervention” for ob.gyns. and other reproductive health care professionals (Obstet Gynecol. 2013;122[4]:931-5).
Despite strong calls by each of these organizations to not overlook EDCs in the clinical arena, as well as emerging evidence that EDCs may be a risk factor for gestational diabetes (GDM), EDC exposure may not be on the practicing ob.gyn.’s radar. Clinicians should know what these chemicals are and how to talk about them in preconception and prenatal visits. We should carefully consider their known – and potential – risks, and encourage our patients to identify and reduce exposure without being alarmist.
Low-dose effects
EDCs are used in the manufacture of pesticides, industrial chemicals, plastics and plasticizers, hand sanitizers, medical equipment, dental sealants, a variety of personal care products, cosmetics, and other common consumer and household products. They’re found, for example, in sunscreens, canned foods and beverages, food-packaging materials, baby bottles, flame-retardant furniture, stain-resistant carpet, and shoes. We are all ingesting and breathing them in to some degree.
Bisphenol A (BPA), one of the most extensively studied EDCs, is found in the thermal receipt paper routinely used by gas stations, supermarkets, and other stores. In a small study we conducted at Harvard, we found that urinary BPA concentrations increased after continual handling of receipts for 2 hours without gloves but did not increase significantly when gloves were used (JAMA. 2014 Feb 26;311[8]:859-60).
Informed consumers can then affect the market through their purchasing choices, but the removal of concerning chemicals from products takes a long time, and it’s not always immediately clear that replacement chemicals are safer. For instance, the BPA in “BPA-free” water bottles and canned foods has been replaced by bisphenol S (BPS), which has a very similar molecular structure to BPA. The potential adverse health effects of these replacement chemicals are now being examined in experimental and epidemiologic studies.
Through its National Health and Nutrition Examination Survey, the Centers for Disease Control and Prevention has reported detection rates of between 75% and 99% for different EDCs in urine samples collected from a representative sample of the U.S. population. In other human research, several EDCs have been shown to cross the placenta and have been measured in maternal blood and urine and in cord blood and amniotic fluid, as well as in placental tissue at birth.
It is interesting to note that BPA’s structure is similar to that of diethylstilbestrol (DES). BPA was first shown to have estrogenic activity in 1936 and was originally considered for use in pharmaceuticals to prevent miscarriages, spontaneous abortions, and premature labor but was put aside in favor of DES. (DES was eventually found to be carcinogenic and was taken off the market.) In the 1950s, the use of BPA was resuscitated though not in pharmaceuticals.
A better understanding about the mechanisms of action and dose-response patterns of EDCs has indicated that EDCs can act at low doses, and in many cases a nonmonotonic dose-response association has been demonstrated. This is a paradigm shift for traditional toxicology in which it is “the dose that makes the poison,” and some toxicologists have been critical of the claims of low-dose potency for EDCs.
A team of epidemiologists, toxicologists, and other scientists, including myself, critically analyzed in vitro, animal, and epidemiologic studies as part of a National Institute of Environmental Health Sciences working group on BPA to determine the strength of the evidence for low-dose effects (doses lower than those tested in traditional toxicology assessments) of BPA. We found that consistent, reproducible, and often adverse low-dose effects have been demonstrated for BPA in cell lines, primary cells and tissues, laboratory animals, and human populations. We also concluded that EDCs can pose the greatest threats when exposure occurs during early development, organogenesis, and during critical postnatal periods when tissues are differentiating (Endocr Disruptors [Austin, Tex.]. 2013 Sep;1:e25078-1-13).
A potential risk factor for GDM
Quite a lot of research has been done on EDCs and the risk of type 2 diabetes. A recent meta-analysis that included 41 cross-sectional and 8 prospective studies found that serum concentrations of dioxins, polychlorinated biphenyls, and chlorinated pesticides – and urine concentrations of BPA and phthalates – were significantly associated with type 2 diabetes risk. Comparing the highest and lowest concentration categories, the pooled relative risk was 1.45 for BPA and phthalates. EDC concentrations also were associated with indicators of impaired fasting glucose and insulin resistance (J Diabetes. 2016 Jul;8[4]:516-32).
Despite the mounting evidence for an association between BPA and type 2 diabetes, and despite the fact that the increased incidence of GDM in the past 20 years has mirrored the increasing use of EDCs, there has been a dearth of research examining the possible relationship between EDCs and GDM. The effects of BPA on GDM were identified as a knowledge gap by the National Institute of Environmental Health Sciences after a review of the literature from 2007 to 2013 (Environ Health Perspect. 2014 Aug:122[8]:775-86).
To understand the association between EDCs and GDM and the underlying mechanistic pathway of EDCs, we are conducting research that uses a growing body of evidence that suggests that environmental toxins are involved in the control of microRNA (miRNA) expression in trophoblast cells.
MiRNA, a single-stranded, short, noncoding RNA that is involved in posttranslational gene expression, can be packaged along with other signaling molecules inside extracellular vesicles in the placenta called exosomes. These exosomes appear to be shed from the placenta into the maternal circulation as early as 6-7 weeks into pregnancy. Once released into the maternal circulation, research has shown that the exosomes can target and reprogram other cells via the transfer of noncoding miRNAs, thereby changing the gene expression in these cells.
Such an exosome-mediated signaling pathway provides us with the opportunity to isolate exosomes, sequence the miRNAs, and look at whether women who are exposed to higher levels of EDCs (as indicated in urine concentration) have a particular miRNA signature that correlates with GDM. In other words, we’re working to determine whether particular EDCs and exposure levels affect the miRNA placental profiles, and if these profiles are predictive of GDM.
Thus far, in a pilot prospective cohort study of pregnant women, we are seeing hints of altered miRNA expression in relation to GDM. We have selected study participants who are at high risk of developing GDM (for example, prepregnancy body mass index greater than 30, past pregnancy with GDM, or macrosomia) because we suspect that, in many women, EDCs are a tipping point for the development of GDM rather than a sole causative factor. In addition to understanding the impact of EDCs on GDM, it is our hope that miRNAs in maternal circulation will serve as a noninvasive biomarker for early detection of GDM development or susceptibility.
Dr. Ehrlich is an assistant professor of pediatrics and environmental health at Cincinnati Children’s Hospital Medical Center.
Endocrine-disrupting chemicals (EDCs) are structurally similar to endogenous hormones and are therefore capable of mimicking these natural hormones, interfering with their biosynthesis, transport, binding action, and/or elimination. In animal studies and human clinical observational and epidemiologic studies of various EDCs, these chemicals have consistently been associated with diabetes mellitus, obesity, hormone-sensitive cancers, neurodevelopmental disorders in children exposed prenatally, and reproductive health.
In 2009, the Endocrine Society published a scientific statement in which it called EDCs a significant concern to human health (Endocr Rev. 2009;30[4]:293-342). Several years later, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a Committee Opinion on Exposure to Toxic Environmental Agents, warning that patient exposure to EDCs and other toxic environmental agents can have a “profound and lasting effect” on reproductive health outcomes across the life course and calling the reduction of exposure a “critical area of intervention” for ob.gyns. and other reproductive health care professionals (Obstet Gynecol. 2013;122[4]:931-5).
Despite strong calls by each of these organizations to not overlook EDCs in the clinical arena, as well as emerging evidence that EDCs may be a risk factor for gestational diabetes (GDM), EDC exposure may not be on the practicing ob.gyn.’s radar. Clinicians should know what these chemicals are and how to talk about them in preconception and prenatal visits. We should carefully consider their known – and potential – risks, and encourage our patients to identify and reduce exposure without being alarmist.
Low-dose effects
EDCs are used in the manufacture of pesticides, industrial chemicals, plastics and plasticizers, hand sanitizers, medical equipment, dental sealants, a variety of personal care products, cosmetics, and other common consumer and household products. They’re found, for example, in sunscreens, canned foods and beverages, food-packaging materials, baby bottles, flame-retardant furniture, stain-resistant carpet, and shoes. We are all ingesting and breathing them in to some degree.
Bisphenol A (BPA), one of the most extensively studied EDCs, is found in the thermal receipt paper routinely used by gas stations, supermarkets, and other stores. In a small study we conducted at Harvard, we found that urinary BPA concentrations increased after continual handling of receipts for 2 hours without gloves but did not increase significantly when gloves were used (JAMA. 2014 Feb 26;311[8]:859-60).
Informed consumers can then affect the market through their purchasing choices, but the removal of concerning chemicals from products takes a long time, and it’s not always immediately clear that replacement chemicals are safer. For instance, the BPA in “BPA-free” water bottles and canned foods has been replaced by bisphenol S (BPS), which has a very similar molecular structure to BPA. The potential adverse health effects of these replacement chemicals are now being examined in experimental and epidemiologic studies.
Through its National Health and Nutrition Examination Survey, the Centers for Disease Control and Prevention has reported detection rates of between 75% and 99% for different EDCs in urine samples collected from a representative sample of the U.S. population. In other human research, several EDCs have been shown to cross the placenta and have been measured in maternal blood and urine and in cord blood and amniotic fluid, as well as in placental tissue at birth.
It is interesting to note that BPA’s structure is similar to that of diethylstilbestrol (DES). BPA was first shown to have estrogenic activity in 1936 and was originally considered for use in pharmaceuticals to prevent miscarriages, spontaneous abortions, and premature labor but was put aside in favor of DES. (DES was eventually found to be carcinogenic and was taken off the market.) In the 1950s, the use of BPA was resuscitated though not in pharmaceuticals.
A better understanding about the mechanisms of action and dose-response patterns of EDCs has indicated that EDCs can act at low doses, and in many cases a nonmonotonic dose-response association has been demonstrated. This is a paradigm shift for traditional toxicology in which it is “the dose that makes the poison,” and some toxicologists have been critical of the claims of low-dose potency for EDCs.
A team of epidemiologists, toxicologists, and other scientists, including myself, critically analyzed in vitro, animal, and epidemiologic studies as part of a National Institute of Environmental Health Sciences working group on BPA to determine the strength of the evidence for low-dose effects (doses lower than those tested in traditional toxicology assessments) of BPA. We found that consistent, reproducible, and often adverse low-dose effects have been demonstrated for BPA in cell lines, primary cells and tissues, laboratory animals, and human populations. We also concluded that EDCs can pose the greatest threats when exposure occurs during early development, organogenesis, and during critical postnatal periods when tissues are differentiating (Endocr Disruptors [Austin, Tex.]. 2013 Sep;1:e25078-1-13).
A potential risk factor for GDM
Quite a lot of research has been done on EDCs and the risk of type 2 diabetes. A recent meta-analysis that included 41 cross-sectional and 8 prospective studies found that serum concentrations of dioxins, polychlorinated biphenyls, and chlorinated pesticides – and urine concentrations of BPA and phthalates – were significantly associated with type 2 diabetes risk. Comparing the highest and lowest concentration categories, the pooled relative risk was 1.45 for BPA and phthalates. EDC concentrations also were associated with indicators of impaired fasting glucose and insulin resistance (J Diabetes. 2016 Jul;8[4]:516-32).
Despite the mounting evidence for an association between BPA and type 2 diabetes, and despite the fact that the increased incidence of GDM in the past 20 years has mirrored the increasing use of EDCs, there has been a dearth of research examining the possible relationship between EDCs and GDM. The effects of BPA on GDM were identified as a knowledge gap by the National Institute of Environmental Health Sciences after a review of the literature from 2007 to 2013 (Environ Health Perspect. 2014 Aug:122[8]:775-86).
To understand the association between EDCs and GDM and the underlying mechanistic pathway of EDCs, we are conducting research that uses a growing body of evidence that suggests that environmental toxins are involved in the control of microRNA (miRNA) expression in trophoblast cells.
MiRNA, a single-stranded, short, noncoding RNA that is involved in posttranslational gene expression, can be packaged along with other signaling molecules inside extracellular vesicles in the placenta called exosomes. These exosomes appear to be shed from the placenta into the maternal circulation as early as 6-7 weeks into pregnancy. Once released into the maternal circulation, research has shown that the exosomes can target and reprogram other cells via the transfer of noncoding miRNAs, thereby changing the gene expression in these cells.
Such an exosome-mediated signaling pathway provides us with the opportunity to isolate exosomes, sequence the miRNAs, and look at whether women who are exposed to higher levels of EDCs (as indicated in urine concentration) have a particular miRNA signature that correlates with GDM. In other words, we’re working to determine whether particular EDCs and exposure levels affect the miRNA placental profiles, and if these profiles are predictive of GDM.
Thus far, in a pilot prospective cohort study of pregnant women, we are seeing hints of altered miRNA expression in relation to GDM. We have selected study participants who are at high risk of developing GDM (for example, prepregnancy body mass index greater than 30, past pregnancy with GDM, or macrosomia) because we suspect that, in many women, EDCs are a tipping point for the development of GDM rather than a sole causative factor. In addition to understanding the impact of EDCs on GDM, it is our hope that miRNAs in maternal circulation will serve as a noninvasive biomarker for early detection of GDM development or susceptibility.
Dr. Ehrlich is an assistant professor of pediatrics and environmental health at Cincinnati Children’s Hospital Medical Center.