User login
Oral prophylaxis and vaginal ring effective in adolescent HIV prevention
Dapivirine vaginal ring and oral pre-exposure prophylaxis (PrEP) are effective HIV prevention measures in adolescent girls, according to two studies presented at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
These solutions could be critical in lowering the rate of HIV and AIDS in girls between the ages of 15 and 25 years, a population that has proven to be particularly vulnerable to HIV infection, researchers say.
Females aged 15-24 years made up 20% of new HIV infections globally in 2015, even though they represented only 11% of the adult population, according to the National Institutes of Health (NIH).
“Adolescents and young people represent a growing share of people living with HIV worldwide,” Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases (NIAID) said in a statement released by the NIH. “Science has demonstrated that the HIV prevention needs of adolescents may be different than those of adults, which is why these new study findings are so important.”
In a phase II, double-blind, placebo-controlled trial, researchers administered a 25-mg dapivirine vaginal ring to 73 patients, with a placebo group of 23 patients, gathered from six sites across the United States once every 4 weeks over a period of 24 weeks.
Patients’ ages ranged from 15 to 17 years of age and a majority were African American, with a median of three sexual partners over the course of their lifetimes.
After the 24-week period, dapivirine residual drug levels indicated a 95% adherence rate, with a reported 42% of patients in the test group reporting never having removed the vaginal ring.
Among the patients given the ring, 63% reported never feeling the ring during intercourse, while 73% of those who said they did feel the ring reported not being bothered by it.
Overall, 93% of the study population reported not being bothered by the solution, which investigators interpreted as a positive sign for the dapivirine ring as an effective HIV prevention tool.
“We are encouraged by these results of the dapivirine ring in 15- to 17-year-old girls,” Sharon Hillier, PhD, of the NIH-funded Microbicide Trials Network (MTN), said in the NIH statement. “The study has demonstrated that the ring is safe in U.S. teens, and now we need data on the safety and acceptability of the ring in African adolescent girls. The REACH study, scheduled to launch later this year, will generate [these] data.”
In a second study presented at the conference, investigators tested the safety and acceptability of daily oral Truvada (emtricitabine/tenofovir) as a PrEP solution in 148 HIV-free adolescents aged 15-19 years from two study sites in South Africa over a span of 12 months. Truvada has not yet been approved by any national regulatory body for use as oral PrEP in adolescents.
Patients were majority female (99 girls/49 boys), with 74% of the population reporting having used a condom during their last sexual encounter.
At the start of the trial, sexually transmitted infections were present in 40% of participants, a level that remained constant throughout the study.
A total of 16 (11%) participants reported grade 2 adverse effects, including headaches, nausea, abdominal pain, and skin rashes, with another 2 patients reporting weight loss during the trial, according to Katherine Gill, MBBS, of the Desmond Tutu HIV Foundation, Cape Town, South Africa.
One instance of HIV infection was reported, although the patient in question dropped out of the program 24 weeks before diagnosis.
Overall, investigators found the Truvada PrEP program to be reasonably well tolerated, with plasma tenofovir levels detectable in 57% of participants after 12 weeks, 38% after 24 weeks and 38% at the end of the study, according to Dr. Gill and her colleagues.
Both studies were funded by NIH grants. Investigators of both studies reported no relevant financial conflicts.
[email protected]
On Twitter @eaztweets
Dapivirine vaginal ring and oral pre-exposure prophylaxis (PrEP) are effective HIV prevention measures in adolescent girls, according to two studies presented at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
These solutions could be critical in lowering the rate of HIV and AIDS in girls between the ages of 15 and 25 years, a population that has proven to be particularly vulnerable to HIV infection, researchers say.
Females aged 15-24 years made up 20% of new HIV infections globally in 2015, even though they represented only 11% of the adult population, according to the National Institutes of Health (NIH).
“Adolescents and young people represent a growing share of people living with HIV worldwide,” Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases (NIAID) said in a statement released by the NIH. “Science has demonstrated that the HIV prevention needs of adolescents may be different than those of adults, which is why these new study findings are so important.”
In a phase II, double-blind, placebo-controlled trial, researchers administered a 25-mg dapivirine vaginal ring to 73 patients, with a placebo group of 23 patients, gathered from six sites across the United States once every 4 weeks over a period of 24 weeks.
Patients’ ages ranged from 15 to 17 years of age and a majority were African American, with a median of three sexual partners over the course of their lifetimes.
After the 24-week period, dapivirine residual drug levels indicated a 95% adherence rate, with a reported 42% of patients in the test group reporting never having removed the vaginal ring.
Among the patients given the ring, 63% reported never feeling the ring during intercourse, while 73% of those who said they did feel the ring reported not being bothered by it.
Overall, 93% of the study population reported not being bothered by the solution, which investigators interpreted as a positive sign for the dapivirine ring as an effective HIV prevention tool.
“We are encouraged by these results of the dapivirine ring in 15- to 17-year-old girls,” Sharon Hillier, PhD, of the NIH-funded Microbicide Trials Network (MTN), said in the NIH statement. “The study has demonstrated that the ring is safe in U.S. teens, and now we need data on the safety and acceptability of the ring in African adolescent girls. The REACH study, scheduled to launch later this year, will generate [these] data.”
In a second study presented at the conference, investigators tested the safety and acceptability of daily oral Truvada (emtricitabine/tenofovir) as a PrEP solution in 148 HIV-free adolescents aged 15-19 years from two study sites in South Africa over a span of 12 months. Truvada has not yet been approved by any national regulatory body for use as oral PrEP in adolescents.
Patients were majority female (99 girls/49 boys), with 74% of the population reporting having used a condom during their last sexual encounter.
At the start of the trial, sexually transmitted infections were present in 40% of participants, a level that remained constant throughout the study.
A total of 16 (11%) participants reported grade 2 adverse effects, including headaches, nausea, abdominal pain, and skin rashes, with another 2 patients reporting weight loss during the trial, according to Katherine Gill, MBBS, of the Desmond Tutu HIV Foundation, Cape Town, South Africa.
One instance of HIV infection was reported, although the patient in question dropped out of the program 24 weeks before diagnosis.
Overall, investigators found the Truvada PrEP program to be reasonably well tolerated, with plasma tenofovir levels detectable in 57% of participants after 12 weeks, 38% after 24 weeks and 38% at the end of the study, according to Dr. Gill and her colleagues.
Both studies were funded by NIH grants. Investigators of both studies reported no relevant financial conflicts.
[email protected]
On Twitter @eaztweets
Dapivirine vaginal ring and oral pre-exposure prophylaxis (PrEP) are effective HIV prevention measures in adolescent girls, according to two studies presented at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
These solutions could be critical in lowering the rate of HIV and AIDS in girls between the ages of 15 and 25 years, a population that has proven to be particularly vulnerable to HIV infection, researchers say.
Females aged 15-24 years made up 20% of new HIV infections globally in 2015, even though they represented only 11% of the adult population, according to the National Institutes of Health (NIH).
“Adolescents and young people represent a growing share of people living with HIV worldwide,” Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases (NIAID) said in a statement released by the NIH. “Science has demonstrated that the HIV prevention needs of adolescents may be different than those of adults, which is why these new study findings are so important.”
In a phase II, double-blind, placebo-controlled trial, researchers administered a 25-mg dapivirine vaginal ring to 73 patients, with a placebo group of 23 patients, gathered from six sites across the United States once every 4 weeks over a period of 24 weeks.
Patients’ ages ranged from 15 to 17 years of age and a majority were African American, with a median of three sexual partners over the course of their lifetimes.
After the 24-week period, dapivirine residual drug levels indicated a 95% adherence rate, with a reported 42% of patients in the test group reporting never having removed the vaginal ring.
Among the patients given the ring, 63% reported never feeling the ring during intercourse, while 73% of those who said they did feel the ring reported not being bothered by it.
Overall, 93% of the study population reported not being bothered by the solution, which investigators interpreted as a positive sign for the dapivirine ring as an effective HIV prevention tool.
“We are encouraged by these results of the dapivirine ring in 15- to 17-year-old girls,” Sharon Hillier, PhD, of the NIH-funded Microbicide Trials Network (MTN), said in the NIH statement. “The study has demonstrated that the ring is safe in U.S. teens, and now we need data on the safety and acceptability of the ring in African adolescent girls. The REACH study, scheduled to launch later this year, will generate [these] data.”
In a second study presented at the conference, investigators tested the safety and acceptability of daily oral Truvada (emtricitabine/tenofovir) as a PrEP solution in 148 HIV-free adolescents aged 15-19 years from two study sites in South Africa over a span of 12 months. Truvada has not yet been approved by any national regulatory body for use as oral PrEP in adolescents.
Patients were majority female (99 girls/49 boys), with 74% of the population reporting having used a condom during their last sexual encounter.
At the start of the trial, sexually transmitted infections were present in 40% of participants, a level that remained constant throughout the study.
A total of 16 (11%) participants reported grade 2 adverse effects, including headaches, nausea, abdominal pain, and skin rashes, with another 2 patients reporting weight loss during the trial, according to Katherine Gill, MBBS, of the Desmond Tutu HIV Foundation, Cape Town, South Africa.
One instance of HIV infection was reported, although the patient in question dropped out of the program 24 weeks before diagnosis.
Overall, investigators found the Truvada PrEP program to be reasonably well tolerated, with plasma tenofovir levels detectable in 57% of participants after 12 weeks, 38% after 24 weeks and 38% at the end of the study, according to Dr. Gill and her colleagues.
Both studies were funded by NIH grants. Investigators of both studies reported no relevant financial conflicts.
[email protected]
On Twitter @eaztweets
FROM IAS 2017
FDA approves faster, pangenotypic cure for hep C virus
The first pangenotypic treatment for the hepatitis C virus, which also shaves 4 weeks off current regimens, has just been approved by the Food and Drug Administration.
Manufactured by AbbVie, glecaprevir/pibrentasvir (Mavyret) combines a nonstructural protein 3/4A protease inhibitor with a next-generation NS5A protein inhibitor for a once-daily, ribavirin-free treatment for adults with any of the major genotypes of chronic hepatitis C virus (HCV) infection.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” Edward Cox, MD, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in a statement.
The 8-week regimen is indicated in patients without cirrhosis or with compensated cirrhosis, who are new to treatment, and those with limited treatment options, such as patients with chronic kidney disease, including those on dialysis. The intervention also is indicated in adults with HCV genotype 1 who have been treated with either of the drugs in the combination, but not both. Glecaprevir/pibrentasvir is not recommended in patients with moderate cirrhosis and is contraindicated in patients with severe cirrhosis and in those taking the drugs atazanavir and rifampin.
The safety and efficacy of the treatment were evaluated in approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. In the clinical trials, between 92% and 100% of patients treated with glecaprevir/pibrentasvir for 8, 12, or 16 weeks had no detectable serum levels of the virus 12 weeks after finishing treatment. The most commonly reported adverse reactions were headache, fatigue, and nausea.
The FDA directs health care professionals to test all patients for current or prior hepatitis B virus (HBV) infection prior to starting this direct-acting antiviral drug combination since HBV reactivation has been reported in adult patients coinfected with both viruses who were undergoing or had completed treatment with HCV direct-acting antivirals and who were not receiving HBV antiviral therapy.
[email protected]
On Twitter @whitneymcknight
The first pangenotypic treatment for the hepatitis C virus, which also shaves 4 weeks off current regimens, has just been approved by the Food and Drug Administration.
Manufactured by AbbVie, glecaprevir/pibrentasvir (Mavyret) combines a nonstructural protein 3/4A protease inhibitor with a next-generation NS5A protein inhibitor for a once-daily, ribavirin-free treatment for adults with any of the major genotypes of chronic hepatitis C virus (HCV) infection.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” Edward Cox, MD, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in a statement.
The 8-week regimen is indicated in patients without cirrhosis or with compensated cirrhosis, who are new to treatment, and those with limited treatment options, such as patients with chronic kidney disease, including those on dialysis. The intervention also is indicated in adults with HCV genotype 1 who have been treated with either of the drugs in the combination, but not both. Glecaprevir/pibrentasvir is not recommended in patients with moderate cirrhosis and is contraindicated in patients with severe cirrhosis and in those taking the drugs atazanavir and rifampin.
The safety and efficacy of the treatment were evaluated in approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. In the clinical trials, between 92% and 100% of patients treated with glecaprevir/pibrentasvir for 8, 12, or 16 weeks had no detectable serum levels of the virus 12 weeks after finishing treatment. The most commonly reported adverse reactions were headache, fatigue, and nausea.
The FDA directs health care professionals to test all patients for current or prior hepatitis B virus (HBV) infection prior to starting this direct-acting antiviral drug combination since HBV reactivation has been reported in adult patients coinfected with both viruses who were undergoing or had completed treatment with HCV direct-acting antivirals and who were not receiving HBV antiviral therapy.
[email protected]
On Twitter @whitneymcknight
The first pangenotypic treatment for the hepatitis C virus, which also shaves 4 weeks off current regimens, has just been approved by the Food and Drug Administration.
Manufactured by AbbVie, glecaprevir/pibrentasvir (Mavyret) combines a nonstructural protein 3/4A protease inhibitor with a next-generation NS5A protein inhibitor for a once-daily, ribavirin-free treatment for adults with any of the major genotypes of chronic hepatitis C virus (HCV) infection.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” Edward Cox, MD, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, Silver Spring, Md., said in a statement.
The 8-week regimen is indicated in patients without cirrhosis or with compensated cirrhosis, who are new to treatment, and those with limited treatment options, such as patients with chronic kidney disease, including those on dialysis. The intervention also is indicated in adults with HCV genotype 1 who have been treated with either of the drugs in the combination, but not both. Glecaprevir/pibrentasvir is not recommended in patients with moderate cirrhosis and is contraindicated in patients with severe cirrhosis and in those taking the drugs atazanavir and rifampin.
The safety and efficacy of the treatment were evaluated in approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. In the clinical trials, between 92% and 100% of patients treated with glecaprevir/pibrentasvir for 8, 12, or 16 weeks had no detectable serum levels of the virus 12 weeks after finishing treatment. The most commonly reported adverse reactions were headache, fatigue, and nausea.
The FDA directs health care professionals to test all patients for current or prior hepatitis B virus (HBV) infection prior to starting this direct-acting antiviral drug combination since HBV reactivation has been reported in adult patients coinfected with both viruses who were undergoing or had completed treatment with HCV direct-acting antivirals and who were not receiving HBV antiviral therapy.
[email protected]
On Twitter @whitneymcknight
Diabetes’ social determinants: What they mean in our practices
More than many other pregnancy complications, diabetes exemplifies the impact of social determinants of health.
The medical management of diabetes during pregnancy involves major lifestyle changes. Diabetes care is largely a patient-driven social experience involving complex and demanding self-care behaviors and tasks.
The pregnant woman with diabetes is placed on a diet that is often novel to her and may be in conflict with the eating patterns of her family. She is advised to exercise, read nutrition labels, and purchase and cook healthy food. She often has to pick up prescriptions, check finger sticks and log results, accurately draw up insulin, and manage strict schedules.
Management requires a tremendous amount of daily engagement during a period of time that, in and of itself, is cognitively demanding.
Outcomes, in turn, are impacted by social context and social factors – by the patient’s economic stability and the safety and characteristics of her neighborhood, for instance, as well as her work schedule, her social support, and her level of health literacy. Each of these factors can influence behaviors and decision making, and ultimately glycemic control and perinatal outcomes.
The social determinants of diabetes-related health are so individualized and impactful that they must be realized and addressed throughout our care, from the way in which we communicate at the initial prenatal checkup to the support we offer for self-management.
Barriers to diabetes self-care
While the incidence of type 2 diabetes is increasing among all social, ethnic, and racial groups, its prevalence among nonpregnant U.S. adults is greatest among racial/ethnic minorities, as well as in individuals with a low-income status. Women who enter pregnancy with preexisting diabetes are more likely to be racial/ethnic minorities.
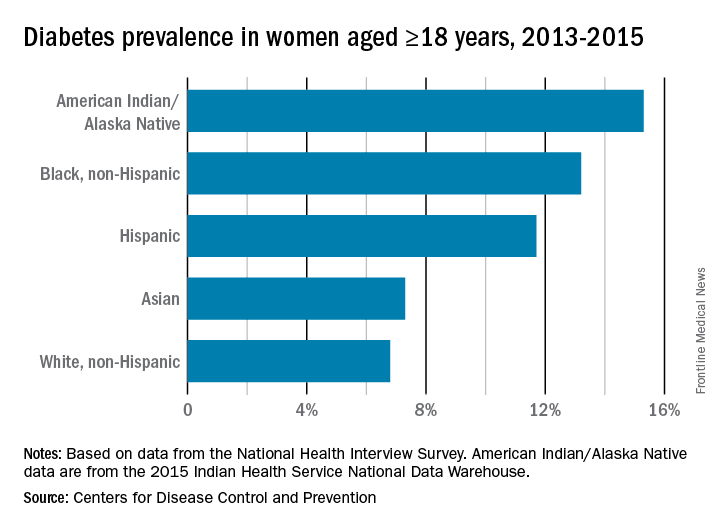
In pregnancy, minority women (especially Hispanic, but also Asian and non-Hispanic black women), and women with low-income status are similarly predisposed to developing gestational diabetes mellitus (GDM).
Social determinants of health are interwoven with inequities stemming from race/ethnicity, income, and other factors that affect outcomes. For example, not only do non-Hispanic black women experience a greater incidence of GDM than non-Hispanic white women, but when they have GDM, they also appear to experience worse pregnancy outcomes compared with white women who also have GDM. In addition, they have a greater likelihood of developing type 2 diabetes after a pregnancy with GDM.
I care for a population that consists largely of minority, low-income women with either gestational or pregestational diabetes. Despite their best intentions and efforts – and despite seemingly high motivation levels – these women struggle to achieve the levels of glycemic control necessary for preventing maternal and fetal complications.
Several years ago, I sought to better understand the barriers to diabetes self-care and behavioral change these women face. Through a series of in-depth, semi-structured interviews with 10 English-speaking women (half with pregestational diabetes) over the course of their pregnancies, we found that the barriers to self-care related to the following: disease novelty, social and economic instability, nutrition challenges, psychological stressors, a failure of outcome expectations, and the burden of disease management (J Health Care Poor Underserved. 2015 Aug;26[3]:926-40; J Nutr Educ Behav. 2016 Mar;48[3]:170-80.e1).
Some of these barriers, such as the lack of any prior experience with diabetes (through a family member, for instance) or the inability to believe that behavior change and other treatment could impact her diabetes and her fetus’ health, echoed other limited published data. However, women in our study also appeared to be affected by barriers driven by social instability (e.g., a lack of partner or family support, family conflict, or neighborhood violence), inadequate access to healthy food, and the psychological impact of diabetes.
They often felt isolated and overwhelmed by their diabetes; the condition amplified stresses they were already experiencing and contributed to worsening mental health in those who already had depression or anxiety. In the other direction, women also described how preexisting mental health challenges affected their ability to sustain recommended behavior changes.
However, we also identified factors that empowered women in this community to succeed with their diabetes during pregnancy – these included having prior familiarity and diabetes self-efficacy, being motivated by the health of the fetus or older children, having a supportive social and physical environment, and having the ability to self-regulate or set and achieve goals (J Perinatol. 2016 Jan;36[1]:13-8).
To address these barriers, my group has undertaken a series of projects aimed at improving care for pregnant women with diabetes. We developed a diabetes-specific text message support system, for instance, and are now transitioning this support to an advanced mobile health tool that can help patients beyond our site.
What we can do
Much of what we can do in our practices to identify and address social determinants and alleviate barriers to effective diabetes management is about finding the “sweet spot” – about being able to convey the right information in the right amount, with the right timing and the right delivery.
While we can’t improve a woman’s neighborhood or resolve food instability, I believe that we can still work to improve outcomes for women who experience these problems. Here are some key strategies for optimal support of our patients:
Inquire about social factors
Identify hurdles by asking questions such as: Where do you live? Is it safe to walk in your neighborhood? If not, where’s your closest mall? What kind of job do you have, and does your employer allow breaks to take care of your health? How are things going at home? Who is at home to help you? Are you having any trouble affording food? How can we help you learn to adapt your personal or cultural food preferences to healthier options?
Look for small actions to take. I often write letters to my patients’ employers requesting that they be given short, frequent breaks to accommodate their care regimens. I also work to ensure that diet recommendations and medication/insulin regimens are customized for patients with irregular meal and sleep schedules, such as those working night shifts.
Employ a social worker if possible, especially if your practice cares for large numbers of underserved women.
Serve as a resource center, and engage your team in doing so. Be prepared to refer women for social services support, food banks, intimate partner violence support services, and other local resources.
Take a low-health-literacy approach
Health literacy is the ability to obtain and utilize health information. It has been widely investigated outside of pregnancy (and to some extent during pregnancy), and has been found to be at the root of many disparities in health care and health outcomes. Numeracy, a type of health literacy, is the ability to understand numbers, perform basic calculations, and use simple math skills in a way that helps one’s health.
The barriers created by inadequate health literacy are distinct from language barriers. I’ve had patients who can read the labels on their insulin vials but cannot distinguish the short-acting from the long-acting formulation, or who can read the words on a nutrition label but don’t know how to interpret the amount of carbohydrates and determine if a food fits the diet plan.
Moreover, while health literacy is correlated with cognitive ability, it still is a distinct skill set. Studies have shown that patients educated in a traditional sense – college-educated professionals, for instance – will not necessarily understand health-related words and instructions.
Research similarly suggests that a low-health-literacy approach that uses focused, simple, and straightforward messages benefits everyone. This type of approach involves the following:
Simple language
Teach-back techniques (“tell me you what your understanding is of what I just told you”)
Diagrams, handouts, and brochures written at a sixth-grade level.
Teaching that is limited to five to eight key messages per session, and reinforcement of these messages over time.
Promote self-efficacy
Self-efficacy is the confidence in one’s ability to perform certain health behaviors. It involves motivation as well as knowledge of the disease, the rationale for treatment, and the specific behaviors that are required for effective self-care.
Help patients understand “why it matters” – that diabetes raises the risk of macrosomia, shoulder dystocia, hypertension, long-term diabetes, and other adverse maternal and neonatal outcomes. Explain basic physiologic concepts and provide background information. This builds self-efficacy.
Do not issue recommendations for exercising and eating well without asking: How can I help you do this? What do you need to be able to eat healthy? Do you need an appointment with a nutritionist? Do you need to see a social worker?
Inquire about and help patients identify supportive family members or other “champions.” Look for ways to incorporate these support people into the patient’s care. At a minimum, encourage the patient to ask her support person to eat healthy with her and/or to understand her daily tasks so that this individual can offer reminders and be a source of support when she feels exhausted or overwhelmed.
If possible, facilitate some type of “diabetes buddy” program to offer peer support and help patients stay engaged in their care, or use group education sessions.
Piggyback on your patients’ own motivating factors. Research has shown that women are extraordinarily motivated to stop smoking during pregnancy because of the health of the fetus. This should extend as well to the difficult lifestyle changes required for diabetes self-care.
View pregnancy as a “golden opportunity” to promote healthy life changes that endure because of the often-extraordinary levels of motivation that women feel or can be encouraged to feel.
Facilitate access
The ability to attend frequent appointments and to juggle the logistics of transportation, child care, and time off work (all part of the burden of disease management) is a social determinant of health. It’s something we should ask about, and it is often something we can positively impact by modifying our practice hours and/or using telehealth or mobile health techniques.

Coordinating newborn and pediatric care with the mother’s subsequent primary care is optimal. Women often prioritize their babies’ health over their own health and they rarely miss pediatric appointments. Coordinating care through medical homes or other mechanisms may help women remain engaged and may lessen the gaps between obstetrical and subsequent primary care.
For me, facilitating doctor-to-doctor transitions sometimes entails picking up the phone or sending communication to a primary care doctor to say, for instance, “I’m worried about my patient’s lifetime risk of type 2 diabetes, and I’d like to hand off her care to you.” This is one of many small but meaningful steps we can take.
Dr. Yee is an assistant professor in the division of maternal-fetal medicine at Northwestern University, Chicago. She reported having no relevant financial disclosures.
More than many other pregnancy complications, diabetes exemplifies the impact of social determinants of health.
The medical management of diabetes during pregnancy involves major lifestyle changes. Diabetes care is largely a patient-driven social experience involving complex and demanding self-care behaviors and tasks.
The pregnant woman with diabetes is placed on a diet that is often novel to her and may be in conflict with the eating patterns of her family. She is advised to exercise, read nutrition labels, and purchase and cook healthy food. She often has to pick up prescriptions, check finger sticks and log results, accurately draw up insulin, and manage strict schedules.
Management requires a tremendous amount of daily engagement during a period of time that, in and of itself, is cognitively demanding.
Outcomes, in turn, are impacted by social context and social factors – by the patient’s economic stability and the safety and characteristics of her neighborhood, for instance, as well as her work schedule, her social support, and her level of health literacy. Each of these factors can influence behaviors and decision making, and ultimately glycemic control and perinatal outcomes.
The social determinants of diabetes-related health are so individualized and impactful that they must be realized and addressed throughout our care, from the way in which we communicate at the initial prenatal checkup to the support we offer for self-management.
Barriers to diabetes self-care
While the incidence of type 2 diabetes is increasing among all social, ethnic, and racial groups, its prevalence among nonpregnant U.S. adults is greatest among racial/ethnic minorities, as well as in individuals with a low-income status. Women who enter pregnancy with preexisting diabetes are more likely to be racial/ethnic minorities.
In pregnancy, minority women (especially Hispanic, but also Asian and non-Hispanic black women), and women with low-income status are similarly predisposed to developing gestational diabetes mellitus (GDM).
Social determinants of health are interwoven with inequities stemming from race/ethnicity, income, and other factors that affect outcomes. For example, not only do non-Hispanic black women experience a greater incidence of GDM than non-Hispanic white women, but when they have GDM, they also appear to experience worse pregnancy outcomes compared with white women who also have GDM. In addition, they have a greater likelihood of developing type 2 diabetes after a pregnancy with GDM.
I care for a population that consists largely of minority, low-income women with either gestational or pregestational diabetes. Despite their best intentions and efforts – and despite seemingly high motivation levels – these women struggle to achieve the levels of glycemic control necessary for preventing maternal and fetal complications.
Several years ago, I sought to better understand the barriers to diabetes self-care and behavioral change these women face. Through a series of in-depth, semi-structured interviews with 10 English-speaking women (half with pregestational diabetes) over the course of their pregnancies, we found that the barriers to self-care related to the following: disease novelty, social and economic instability, nutrition challenges, psychological stressors, a failure of outcome expectations, and the burden of disease management (J Health Care Poor Underserved. 2015 Aug;26[3]:926-40; J Nutr Educ Behav. 2016 Mar;48[3]:170-80.e1).
Some of these barriers, such as the lack of any prior experience with diabetes (through a family member, for instance) or the inability to believe that behavior change and other treatment could impact her diabetes and her fetus’ health, echoed other limited published data. However, women in our study also appeared to be affected by barriers driven by social instability (e.g., a lack of partner or family support, family conflict, or neighborhood violence), inadequate access to healthy food, and the psychological impact of diabetes.
They often felt isolated and overwhelmed by their diabetes; the condition amplified stresses they were already experiencing and contributed to worsening mental health in those who already had depression or anxiety. In the other direction, women also described how preexisting mental health challenges affected their ability to sustain recommended behavior changes.
However, we also identified factors that empowered women in this community to succeed with their diabetes during pregnancy – these included having prior familiarity and diabetes self-efficacy, being motivated by the health of the fetus or older children, having a supportive social and physical environment, and having the ability to self-regulate or set and achieve goals (J Perinatol. 2016 Jan;36[1]:13-8).
To address these barriers, my group has undertaken a series of projects aimed at improving care for pregnant women with diabetes. We developed a diabetes-specific text message support system, for instance, and are now transitioning this support to an advanced mobile health tool that can help patients beyond our site.
What we can do
Much of what we can do in our practices to identify and address social determinants and alleviate barriers to effective diabetes management is about finding the “sweet spot” – about being able to convey the right information in the right amount, with the right timing and the right delivery.
While we can’t improve a woman’s neighborhood or resolve food instability, I believe that we can still work to improve outcomes for women who experience these problems. Here are some key strategies for optimal support of our patients:
Inquire about social factors
Identify hurdles by asking questions such as: Where do you live? Is it safe to walk in your neighborhood? If not, where’s your closest mall? What kind of job do you have, and does your employer allow breaks to take care of your health? How are things going at home? Who is at home to help you? Are you having any trouble affording food? How can we help you learn to adapt your personal or cultural food preferences to healthier options?
Look for small actions to take. I often write letters to my patients’ employers requesting that they be given short, frequent breaks to accommodate their care regimens. I also work to ensure that diet recommendations and medication/insulin regimens are customized for patients with irregular meal and sleep schedules, such as those working night shifts.
Employ a social worker if possible, especially if your practice cares for large numbers of underserved women.
Serve as a resource center, and engage your team in doing so. Be prepared to refer women for social services support, food banks, intimate partner violence support services, and other local resources.
Take a low-health-literacy approach
Health literacy is the ability to obtain and utilize health information. It has been widely investigated outside of pregnancy (and to some extent during pregnancy), and has been found to be at the root of many disparities in health care and health outcomes. Numeracy, a type of health literacy, is the ability to understand numbers, perform basic calculations, and use simple math skills in a way that helps one’s health.
The barriers created by inadequate health literacy are distinct from language barriers. I’ve had patients who can read the labels on their insulin vials but cannot distinguish the short-acting from the long-acting formulation, or who can read the words on a nutrition label but don’t know how to interpret the amount of carbohydrates and determine if a food fits the diet plan.
Moreover, while health literacy is correlated with cognitive ability, it still is a distinct skill set. Studies have shown that patients educated in a traditional sense – college-educated professionals, for instance – will not necessarily understand health-related words and instructions.
Research similarly suggests that a low-health-literacy approach that uses focused, simple, and straightforward messages benefits everyone. This type of approach involves the following:
Simple language
Teach-back techniques (“tell me you what your understanding is of what I just told you”)
Diagrams, handouts, and brochures written at a sixth-grade level.
Teaching that is limited to five to eight key messages per session, and reinforcement of these messages over time.
Promote self-efficacy
Self-efficacy is the confidence in one’s ability to perform certain health behaviors. It involves motivation as well as knowledge of the disease, the rationale for treatment, and the specific behaviors that are required for effective self-care.
Help patients understand “why it matters” – that diabetes raises the risk of macrosomia, shoulder dystocia, hypertension, long-term diabetes, and other adverse maternal and neonatal outcomes. Explain basic physiologic concepts and provide background information. This builds self-efficacy.
Do not issue recommendations for exercising and eating well without asking: How can I help you do this? What do you need to be able to eat healthy? Do you need an appointment with a nutritionist? Do you need to see a social worker?
Inquire about and help patients identify supportive family members or other “champions.” Look for ways to incorporate these support people into the patient’s care. At a minimum, encourage the patient to ask her support person to eat healthy with her and/or to understand her daily tasks so that this individual can offer reminders and be a source of support when she feels exhausted or overwhelmed.
If possible, facilitate some type of “diabetes buddy” program to offer peer support and help patients stay engaged in their care, or use group education sessions.
Piggyback on your patients’ own motivating factors. Research has shown that women are extraordinarily motivated to stop smoking during pregnancy because of the health of the fetus. This should extend as well to the difficult lifestyle changes required for diabetes self-care.
View pregnancy as a “golden opportunity” to promote healthy life changes that endure because of the often-extraordinary levels of motivation that women feel or can be encouraged to feel.
Facilitate access
The ability to attend frequent appointments and to juggle the logistics of transportation, child care, and time off work (all part of the burden of disease management) is a social determinant of health. It’s something we should ask about, and it is often something we can positively impact by modifying our practice hours and/or using telehealth or mobile health techniques.
Coordinating newborn and pediatric care with the mother’s subsequent primary care is optimal. Women often prioritize their babies’ health over their own health and they rarely miss pediatric appointments. Coordinating care through medical homes or other mechanisms may help women remain engaged and may lessen the gaps between obstetrical and subsequent primary care.
For me, facilitating doctor-to-doctor transitions sometimes entails picking up the phone or sending communication to a primary care doctor to say, for instance, “I’m worried about my patient’s lifetime risk of type 2 diabetes, and I’d like to hand off her care to you.” This is one of many small but meaningful steps we can take.
Dr. Yee is an assistant professor in the division of maternal-fetal medicine at Northwestern University, Chicago. She reported having no relevant financial disclosures.
More than many other pregnancy complications, diabetes exemplifies the impact of social determinants of health.
The medical management of diabetes during pregnancy involves major lifestyle changes. Diabetes care is largely a patient-driven social experience involving complex and demanding self-care behaviors and tasks.
The pregnant woman with diabetes is placed on a diet that is often novel to her and may be in conflict with the eating patterns of her family. She is advised to exercise, read nutrition labels, and purchase and cook healthy food. She often has to pick up prescriptions, check finger sticks and log results, accurately draw up insulin, and manage strict schedules.
Management requires a tremendous amount of daily engagement during a period of time that, in and of itself, is cognitively demanding.
Outcomes, in turn, are impacted by social context and social factors – by the patient’s economic stability and the safety and characteristics of her neighborhood, for instance, as well as her work schedule, her social support, and her level of health literacy. Each of these factors can influence behaviors and decision making, and ultimately glycemic control and perinatal outcomes.
The social determinants of diabetes-related health are so individualized and impactful that they must be realized and addressed throughout our care, from the way in which we communicate at the initial prenatal checkup to the support we offer for self-management.
Barriers to diabetes self-care
While the incidence of type 2 diabetes is increasing among all social, ethnic, and racial groups, its prevalence among nonpregnant U.S. adults is greatest among racial/ethnic minorities, as well as in individuals with a low-income status. Women who enter pregnancy with preexisting diabetes are more likely to be racial/ethnic minorities.
In pregnancy, minority women (especially Hispanic, but also Asian and non-Hispanic black women), and women with low-income status are similarly predisposed to developing gestational diabetes mellitus (GDM).
Social determinants of health are interwoven with inequities stemming from race/ethnicity, income, and other factors that affect outcomes. For example, not only do non-Hispanic black women experience a greater incidence of GDM than non-Hispanic white women, but when they have GDM, they also appear to experience worse pregnancy outcomes compared with white women who also have GDM. In addition, they have a greater likelihood of developing type 2 diabetes after a pregnancy with GDM.
I care for a population that consists largely of minority, low-income women with either gestational or pregestational diabetes. Despite their best intentions and efforts – and despite seemingly high motivation levels – these women struggle to achieve the levels of glycemic control necessary for preventing maternal and fetal complications.
Several years ago, I sought to better understand the barriers to diabetes self-care and behavioral change these women face. Through a series of in-depth, semi-structured interviews with 10 English-speaking women (half with pregestational diabetes) over the course of their pregnancies, we found that the barriers to self-care related to the following: disease novelty, social and economic instability, nutrition challenges, psychological stressors, a failure of outcome expectations, and the burden of disease management (J Health Care Poor Underserved. 2015 Aug;26[3]:926-40; J Nutr Educ Behav. 2016 Mar;48[3]:170-80.e1).
Some of these barriers, such as the lack of any prior experience with diabetes (through a family member, for instance) or the inability to believe that behavior change and other treatment could impact her diabetes and her fetus’ health, echoed other limited published data. However, women in our study also appeared to be affected by barriers driven by social instability (e.g., a lack of partner or family support, family conflict, or neighborhood violence), inadequate access to healthy food, and the psychological impact of diabetes.
They often felt isolated and overwhelmed by their diabetes; the condition amplified stresses they were already experiencing and contributed to worsening mental health in those who already had depression or anxiety. In the other direction, women also described how preexisting mental health challenges affected their ability to sustain recommended behavior changes.
However, we also identified factors that empowered women in this community to succeed with their diabetes during pregnancy – these included having prior familiarity and diabetes self-efficacy, being motivated by the health of the fetus or older children, having a supportive social and physical environment, and having the ability to self-regulate or set and achieve goals (J Perinatol. 2016 Jan;36[1]:13-8).
To address these barriers, my group has undertaken a series of projects aimed at improving care for pregnant women with diabetes. We developed a diabetes-specific text message support system, for instance, and are now transitioning this support to an advanced mobile health tool that can help patients beyond our site.
What we can do
Much of what we can do in our practices to identify and address social determinants and alleviate barriers to effective diabetes management is about finding the “sweet spot” – about being able to convey the right information in the right amount, with the right timing and the right delivery.
While we can’t improve a woman’s neighborhood or resolve food instability, I believe that we can still work to improve outcomes for women who experience these problems. Here are some key strategies for optimal support of our patients:
Inquire about social factors
Identify hurdles by asking questions such as: Where do you live? Is it safe to walk in your neighborhood? If not, where’s your closest mall? What kind of job do you have, and does your employer allow breaks to take care of your health? How are things going at home? Who is at home to help you? Are you having any trouble affording food? How can we help you learn to adapt your personal or cultural food preferences to healthier options?
Look for small actions to take. I often write letters to my patients’ employers requesting that they be given short, frequent breaks to accommodate their care regimens. I also work to ensure that diet recommendations and medication/insulin regimens are customized for patients with irregular meal and sleep schedules, such as those working night shifts.
Employ a social worker if possible, especially if your practice cares for large numbers of underserved women.
Serve as a resource center, and engage your team in doing so. Be prepared to refer women for social services support, food banks, intimate partner violence support services, and other local resources.
Take a low-health-literacy approach
Health literacy is the ability to obtain and utilize health information. It has been widely investigated outside of pregnancy (and to some extent during pregnancy), and has been found to be at the root of many disparities in health care and health outcomes. Numeracy, a type of health literacy, is the ability to understand numbers, perform basic calculations, and use simple math skills in a way that helps one’s health.
The barriers created by inadequate health literacy are distinct from language barriers. I’ve had patients who can read the labels on their insulin vials but cannot distinguish the short-acting from the long-acting formulation, or who can read the words on a nutrition label but don’t know how to interpret the amount of carbohydrates and determine if a food fits the diet plan.
Moreover, while health literacy is correlated with cognitive ability, it still is a distinct skill set. Studies have shown that patients educated in a traditional sense – college-educated professionals, for instance – will not necessarily understand health-related words and instructions.
Research similarly suggests that a low-health-literacy approach that uses focused, simple, and straightforward messages benefits everyone. This type of approach involves the following:
Simple language
Teach-back techniques (“tell me you what your understanding is of what I just told you”)
Diagrams, handouts, and brochures written at a sixth-grade level.
Teaching that is limited to five to eight key messages per session, and reinforcement of these messages over time.
Promote self-efficacy
Self-efficacy is the confidence in one’s ability to perform certain health behaviors. It involves motivation as well as knowledge of the disease, the rationale for treatment, and the specific behaviors that are required for effective self-care.
Help patients understand “why it matters” – that diabetes raises the risk of macrosomia, shoulder dystocia, hypertension, long-term diabetes, and other adverse maternal and neonatal outcomes. Explain basic physiologic concepts and provide background information. This builds self-efficacy.
Do not issue recommendations for exercising and eating well without asking: How can I help you do this? What do you need to be able to eat healthy? Do you need an appointment with a nutritionist? Do you need to see a social worker?
Inquire about and help patients identify supportive family members or other “champions.” Look for ways to incorporate these support people into the patient’s care. At a minimum, encourage the patient to ask her support person to eat healthy with her and/or to understand her daily tasks so that this individual can offer reminders and be a source of support when she feels exhausted or overwhelmed.
If possible, facilitate some type of “diabetes buddy” program to offer peer support and help patients stay engaged in their care, or use group education sessions.
Piggyback on your patients’ own motivating factors. Research has shown that women are extraordinarily motivated to stop smoking during pregnancy because of the health of the fetus. This should extend as well to the difficult lifestyle changes required for diabetes self-care.
View pregnancy as a “golden opportunity” to promote healthy life changes that endure because of the often-extraordinary levels of motivation that women feel or can be encouraged to feel.
Facilitate access
The ability to attend frequent appointments and to juggle the logistics of transportation, child care, and time off work (all part of the burden of disease management) is a social determinant of health. It’s something we should ask about, and it is often something we can positively impact by modifying our practice hours and/or using telehealth or mobile health techniques.
Coordinating newborn and pediatric care with the mother’s subsequent primary care is optimal. Women often prioritize their babies’ health over their own health and they rarely miss pediatric appointments. Coordinating care through medical homes or other mechanisms may help women remain engaged and may lessen the gaps between obstetrical and subsequent primary care.
For me, facilitating doctor-to-doctor transitions sometimes entails picking up the phone or sending communication to a primary care doctor to say, for instance, “I’m worried about my patient’s lifetime risk of type 2 diabetes, and I’d like to hand off her care to you.” This is one of many small but meaningful steps we can take.
Dr. Yee is an assistant professor in the division of maternal-fetal medicine at Northwestern University, Chicago. She reported having no relevant financial disclosures.
The moving target of gestational diabetes care
With the rise of obesity and diabetes, especially type 2 diabetes, in the general population, the likelihood of encountering a patient with diabetes in pregnancy also continues to increase. Women with diabetes who are pregnant require specialized medical guidance, additional monitoring, and a health care team well versed in the possible complications that can arise during pregnancy, delivery, and after birth.
Even with strict glycemic control, women with diabetes in pregnancy are much more likely to experience complications, such as preeclampsia, babies with major congenital defects, large-for-gestational-age fetuses, and children with a higher propensity for chronic diseases later in life, compared with women without diabetes.

Therefore, it has been an incredible honor for me to have taken part in the work of the Diabetes in Pregnancy Study Group of North America (DPSG-NA) for the last 20 years. The DPSG-NA meetings have served as a forum for the dissemination of data, gathered through collaboration between researchers and clinical care teams in the United States and abroad. This year, the DPSG-NA will meet in Washington, D.C., Oct. 26-28, to discuss a range of topics under the theme of managing and preventing diabetes and obesity in pregnancy.
I am delighted that one of the speakers at the DPSG-NA meeting is this month’s Master Class guest author, Lynn Yee, MD, assistant professor of obstetrics and gynecology at Northwestern University Feinberg School of Medicine, Chicago. Dr. Yee will address the need to reduce disparities in the quality and availability of care for patients with diabetes in pregnancy, an extension of the June Master Class column that discussed the critical role that ob.gyns. can play in improving health equity for all patients.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
With the rise of obesity and diabetes, especially type 2 diabetes, in the general population, the likelihood of encountering a patient with diabetes in pregnancy also continues to increase. Women with diabetes who are pregnant require specialized medical guidance, additional monitoring, and a health care team well versed in the possible complications that can arise during pregnancy, delivery, and after birth.
Even with strict glycemic control, women with diabetes in pregnancy are much more likely to experience complications, such as preeclampsia, babies with major congenital defects, large-for-gestational-age fetuses, and children with a higher propensity for chronic diseases later in life, compared with women without diabetes.
Therefore, it has been an incredible honor for me to have taken part in the work of the Diabetes in Pregnancy Study Group of North America (DPSG-NA) for the last 20 years. The DPSG-NA meetings have served as a forum for the dissemination of data, gathered through collaboration between researchers and clinical care teams in the United States and abroad. This year, the DPSG-NA will meet in Washington, D.C., Oct. 26-28, to discuss a range of topics under the theme of managing and preventing diabetes and obesity in pregnancy.
I am delighted that one of the speakers at the DPSG-NA meeting is this month’s Master Class guest author, Lynn Yee, MD, assistant professor of obstetrics and gynecology at Northwestern University Feinberg School of Medicine, Chicago. Dr. Yee will address the need to reduce disparities in the quality and availability of care for patients with diabetes in pregnancy, an extension of the June Master Class column that discussed the critical role that ob.gyns. can play in improving health equity for all patients.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
With the rise of obesity and diabetes, especially type 2 diabetes, in the general population, the likelihood of encountering a patient with diabetes in pregnancy also continues to increase. Women with diabetes who are pregnant require specialized medical guidance, additional monitoring, and a health care team well versed in the possible complications that can arise during pregnancy, delivery, and after birth.
Even with strict glycemic control, women with diabetes in pregnancy are much more likely to experience complications, such as preeclampsia, babies with major congenital defects, large-for-gestational-age fetuses, and children with a higher propensity for chronic diseases later in life, compared with women without diabetes.
Therefore, it has been an incredible honor for me to have taken part in the work of the Diabetes in Pregnancy Study Group of North America (DPSG-NA) for the last 20 years. The DPSG-NA meetings have served as a forum for the dissemination of data, gathered through collaboration between researchers and clinical care teams in the United States and abroad. This year, the DPSG-NA will meet in Washington, D.C., Oct. 26-28, to discuss a range of topics under the theme of managing and preventing diabetes and obesity in pregnancy.
I am delighted that one of the speakers at the DPSG-NA meeting is this month’s Master Class guest author, Lynn Yee, MD, assistant professor of obstetrics and gynecology at Northwestern University Feinberg School of Medicine, Chicago. Dr. Yee will address the need to reduce disparities in the quality and availability of care for patients with diabetes in pregnancy, an extension of the June Master Class column that discussed the critical role that ob.gyns. can play in improving health equity for all patients.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Safety of oral antidiabetic agents in pregnancy
The three most potent human teratogens, with the possible inclusion of some of the first antineoplastics, are isotretinoin, alcohol, and hyperglycemia.
As with all teratogens, the toxicity is dose related. For example, the risk of embryo-fetal harm from hyperglycemia increases markedly when the HbA1c is greater than 8%. Moreover, diabetes accounts for more than 90% of the harm caused by chronic diseases. Consequently, control of glucose levels in pregnancy is critical.

If these agents are used near term, there is a risk that they will cause hypoglycemia in the newborn. Changing from oral therapy to insulin is the safest course.
There are seven pharmacologic subclasses of oral antidiabetic agents: alpha-glucosidase inhibitors, biguanides, dipeptidyl peptidase-4 inhibitors, meglitinides, sulfonylureas, sodium-glucose cotransporter-2 inhibitors, and thiazolidinediones. Many of these drugs are available in combination with metformin. All of these agents are indicated as adjunct to diet and exercise for type 2 diabetes, but they also can be used for gestational diabetes. Although the human pregnancy data are very limited or nonexistent for most of these agents, none are known to cause structural defects in humans. Additional details of the exposures are available in the 11th edition of “Drugs in Pregnancy and Lactation” (2017: Wolters Kluwer).
Alpha-glucosidase inhibitors
The two agents is this subclass are acarbose (Precose) and miglitol (Glyset). The human pregnancy data with acarbose are limited, and no human pregnancy data have been found for miglitol. The animal data for both drugs suggest low risk.
Biguanides
There are substantial human pregnancy data for metformin in both type 2 and gestational diabetes. When combined with insulin, it is effective in significantly lowering the amount of insulin required to control hyperglycemia. It also may be effective when used alone. The risk of embryo-fetal harm with this drug appears to be very low or nonexistent. The animal data suggest low risk.
Dipeptidyl peptidase-4 inhibitors
There are four drugs in this subclass: alogliptin (Nesina), linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). No reports of the use of the first three drugs in human pregnancy have been found. However, the Merck Pregnancy Registries (2006-2009) described the outcomes of eight women who were exposed to sitagliptin or sitagliptin/metformin in the first trimester. The outcomes of these pregnancies were five healthy newborns, two spontaneous abortions, and one fetal death at 34 weeks’ gestation. In that case, the mother took sitagliptin and metformin separately during the first 5 weeks of gestation. The animal data for all four drugs suggest low risk.
Meglitinides
Nateglinide (Starlix) and repaglinide (Prandin) are the agents in this subclass. There is no human pregnancy data for nateglinide, but there is limited data (eight pregnancies) for repaglinide. No birth defects or other toxicity was noted in these cases. The animal data suggest low risk.
Sulfonylureas
Six drugs are included in this subclass: chlorpropamide, glimepiride (Amaryl), glipizide (Glucotrol), glyburide, tolazamide (Tolinase), and tolbutamide. These agents were among the first oral antidiabetic agents. As a result, they have the most human pregnancy data. Although birth defects were observed in newborns of mothers who had used one of these drugs, the defects were thought to be the result of uncontrolled diabetes. The animal data suggest low risk.
SGLT2 inhibitors
There are three drugs in this sodium-glucose cotransporter-2 inhibitor subclass: canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance). No reports describing the use of these drugs in human pregnancy have been located. The animal data suggest low risk.
Thiazolidinediones
Pioglitazone (Actos) and rosiglitazone (Avandia) form this subclass. There are limited human pregnancy data for both drugs. The animal data suggest moderate risk for embryo-fetal toxicity but not for structural defects.
Lactation
All of the above drugs will probably be excreted into breast milk, but the amounts are typically unknown. When they have been measured, the amounts were usually low. However, there is still a risk for hypoglycemia in a nursing infant. Combination products containing two antidiabetic agents are best avoided. The safest course is to use insulin, but, if this is not an option, then the lowest effective dose should be used. In addition, the infant’s blood glucose levels should be routinely monitored.
Mr. Briggs is a clinical professor of pharmacy at the University of California, San Francisco, and an adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. He coauthored “Drugs in Pregnancy and Lactation” and coedited “Diseases, Complications, and Drug Therapy in Obstetrics.” He reported having no relevant financial disclosures.
The three most potent human teratogens, with the possible inclusion of some of the first antineoplastics, are isotretinoin, alcohol, and hyperglycemia.
As with all teratogens, the toxicity is dose related. For example, the risk of embryo-fetal harm from hyperglycemia increases markedly when the HbA1c is greater than 8%. Moreover, diabetes accounts for more than 90% of the harm caused by chronic diseases. Consequently, control of glucose levels in pregnancy is critical.
If these agents are used near term, there is a risk that they will cause hypoglycemia in the newborn. Changing from oral therapy to insulin is the safest course.
There are seven pharmacologic subclasses of oral antidiabetic agents: alpha-glucosidase inhibitors, biguanides, dipeptidyl peptidase-4 inhibitors, meglitinides, sulfonylureas, sodium-glucose cotransporter-2 inhibitors, and thiazolidinediones. Many of these drugs are available in combination with metformin. All of these agents are indicated as adjunct to diet and exercise for type 2 diabetes, but they also can be used for gestational diabetes. Although the human pregnancy data are very limited or nonexistent for most of these agents, none are known to cause structural defects in humans. Additional details of the exposures are available in the 11th edition of “Drugs in Pregnancy and Lactation” (2017: Wolters Kluwer).
Alpha-glucosidase inhibitors
The two agents is this subclass are acarbose (Precose) and miglitol (Glyset). The human pregnancy data with acarbose are limited, and no human pregnancy data have been found for miglitol. The animal data for both drugs suggest low risk.
Biguanides
There are substantial human pregnancy data for metformin in both type 2 and gestational diabetes. When combined with insulin, it is effective in significantly lowering the amount of insulin required to control hyperglycemia. It also may be effective when used alone. The risk of embryo-fetal harm with this drug appears to be very low or nonexistent. The animal data suggest low risk.
Dipeptidyl peptidase-4 inhibitors
There are four drugs in this subclass: alogliptin (Nesina), linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). No reports of the use of the first three drugs in human pregnancy have been found. However, the Merck Pregnancy Registries (2006-2009) described the outcomes of eight women who were exposed to sitagliptin or sitagliptin/metformin in the first trimester. The outcomes of these pregnancies were five healthy newborns, two spontaneous abortions, and one fetal death at 34 weeks’ gestation. In that case, the mother took sitagliptin and metformin separately during the first 5 weeks of gestation. The animal data for all four drugs suggest low risk.
Meglitinides
Nateglinide (Starlix) and repaglinide (Prandin) are the agents in this subclass. There is no human pregnancy data for nateglinide, but there is limited data (eight pregnancies) for repaglinide. No birth defects or other toxicity was noted in these cases. The animal data suggest low risk.
Sulfonylureas
Six drugs are included in this subclass: chlorpropamide, glimepiride (Amaryl), glipizide (Glucotrol), glyburide, tolazamide (Tolinase), and tolbutamide. These agents were among the first oral antidiabetic agents. As a result, they have the most human pregnancy data. Although birth defects were observed in newborns of mothers who had used one of these drugs, the defects were thought to be the result of uncontrolled diabetes. The animal data suggest low risk.
SGLT2 inhibitors
There are three drugs in this sodium-glucose cotransporter-2 inhibitor subclass: canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance). No reports describing the use of these drugs in human pregnancy have been located. The animal data suggest low risk.
Thiazolidinediones
Pioglitazone (Actos) and rosiglitazone (Avandia) form this subclass. There are limited human pregnancy data for both drugs. The animal data suggest moderate risk for embryo-fetal toxicity but not for structural defects.
Lactation
All of the above drugs will probably be excreted into breast milk, but the amounts are typically unknown. When they have been measured, the amounts were usually low. However, there is still a risk for hypoglycemia in a nursing infant. Combination products containing two antidiabetic agents are best avoided. The safest course is to use insulin, but, if this is not an option, then the lowest effective dose should be used. In addition, the infant’s blood glucose levels should be routinely monitored.
Mr. Briggs is a clinical professor of pharmacy at the University of California, San Francisco, and an adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. He coauthored “Drugs in Pregnancy and Lactation” and coedited “Diseases, Complications, and Drug Therapy in Obstetrics.” He reported having no relevant financial disclosures.
The three most potent human teratogens, with the possible inclusion of some of the first antineoplastics, are isotretinoin, alcohol, and hyperglycemia.
As with all teratogens, the toxicity is dose related. For example, the risk of embryo-fetal harm from hyperglycemia increases markedly when the HbA1c is greater than 8%. Moreover, diabetes accounts for more than 90% of the harm caused by chronic diseases. Consequently, control of glucose levels in pregnancy is critical.
If these agents are used near term, there is a risk that they will cause hypoglycemia in the newborn. Changing from oral therapy to insulin is the safest course.
There are seven pharmacologic subclasses of oral antidiabetic agents: alpha-glucosidase inhibitors, biguanides, dipeptidyl peptidase-4 inhibitors, meglitinides, sulfonylureas, sodium-glucose cotransporter-2 inhibitors, and thiazolidinediones. Many of these drugs are available in combination with metformin. All of these agents are indicated as adjunct to diet and exercise for type 2 diabetes, but they also can be used for gestational diabetes. Although the human pregnancy data are very limited or nonexistent for most of these agents, none are known to cause structural defects in humans. Additional details of the exposures are available in the 11th edition of “Drugs in Pregnancy and Lactation” (2017: Wolters Kluwer).
Alpha-glucosidase inhibitors
The two agents is this subclass are acarbose (Precose) and miglitol (Glyset). The human pregnancy data with acarbose are limited, and no human pregnancy data have been found for miglitol. The animal data for both drugs suggest low risk.
Biguanides
There are substantial human pregnancy data for metformin in both type 2 and gestational diabetes. When combined with insulin, it is effective in significantly lowering the amount of insulin required to control hyperglycemia. It also may be effective when used alone. The risk of embryo-fetal harm with this drug appears to be very low or nonexistent. The animal data suggest low risk.
Dipeptidyl peptidase-4 inhibitors
There are four drugs in this subclass: alogliptin (Nesina), linagliptin (Tradjenta), saxagliptin (Onglyza), and sitagliptin (Januvia). No reports of the use of the first three drugs in human pregnancy have been found. However, the Merck Pregnancy Registries (2006-2009) described the outcomes of eight women who were exposed to sitagliptin or sitagliptin/metformin in the first trimester. The outcomes of these pregnancies were five healthy newborns, two spontaneous abortions, and one fetal death at 34 weeks’ gestation. In that case, the mother took sitagliptin and metformin separately during the first 5 weeks of gestation. The animal data for all four drugs suggest low risk.
Meglitinides
Nateglinide (Starlix) and repaglinide (Prandin) are the agents in this subclass. There is no human pregnancy data for nateglinide, but there is limited data (eight pregnancies) for repaglinide. No birth defects or other toxicity was noted in these cases. The animal data suggest low risk.
Sulfonylureas
Six drugs are included in this subclass: chlorpropamide, glimepiride (Amaryl), glipizide (Glucotrol), glyburide, tolazamide (Tolinase), and tolbutamide. These agents were among the first oral antidiabetic agents. As a result, they have the most human pregnancy data. Although birth defects were observed in newborns of mothers who had used one of these drugs, the defects were thought to be the result of uncontrolled diabetes. The animal data suggest low risk.
SGLT2 inhibitors
There are three drugs in this sodium-glucose cotransporter-2 inhibitor subclass: canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance). No reports describing the use of these drugs in human pregnancy have been located. The animal data suggest low risk.
Thiazolidinediones
Pioglitazone (Actos) and rosiglitazone (Avandia) form this subclass. There are limited human pregnancy data for both drugs. The animal data suggest moderate risk for embryo-fetal toxicity but not for structural defects.
Lactation
All of the above drugs will probably be excreted into breast milk, but the amounts are typically unknown. When they have been measured, the amounts were usually low. However, there is still a risk for hypoglycemia in a nursing infant. Combination products containing two antidiabetic agents are best avoided. The safest course is to use insulin, but, if this is not an option, then the lowest effective dose should be used. In addition, the infant’s blood glucose levels should be routinely monitored.
Mr. Briggs is a clinical professor of pharmacy at the University of California, San Francisco, and an adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. He coauthored “Drugs in Pregnancy and Lactation” and coedited “Diseases, Complications, and Drug Therapy in Obstetrics.” He reported having no relevant financial disclosures.
Optimizing HPV vaccination
Human papillomavirus (HPV) is the most common sexually transmitted infection. Exposure is widespread and most individuals clear the infection without symptoms or development of disease. However, a subset of individuals experience persistent infection, a state which can lead to carcinogenesis of lower genital tract malignancies, particularly cervical cancer.1
Vaccine coverage
Persistent infection with high-risk (oncogenic) HPV is well known to be the cause of cervical cancer. There are two HPV vaccines manufactured for the purposes of cervical cancer, anal cancer, and genital wart prevention (Cervarix and Gardasil). The Cervarix vaccine covers high-risk HPV subtypes 16 and 18 and the Gardasil vaccine prevents both low-risk HPV subtypes 6 and 11, which can cause genital warts, and high-risk HPV subtypes 16, 18, 31, 33, 45, 52 and 58, which cause cervical dysplasia and cancer.
High-risk HPV is also associated with head and neck, vulvar, vaginal, and penile cancers, though the vaccines are not approved by the Food and Drug Administration for prevention of these diseases.2
Vaccination indications
Since vaccination prevents multiple subtypes of HPV, an individual who has already been exposed will still benefit from protection from other subtypes of HPV through vaccination. HPV vaccination is not approved during pregnancy but can be initiated in the postpartum period when women are engaged in their health care and receiving other vaccinations, such as varicella or the MMR vaccine.
Recommended schedule
Until October 2016, the vaccination schedule was based on a three-dose series (0, 2, and 6 months). Currently, the CDC recommends that children aged under 15 years at the time of first dose may opt for a two-dose series (0 and 6-12 months). For those aged 15-26 years, the three-dose schedule remains the recommended course.
The benefits of two-dose schedule are convenience, cost, and increased likelihood of completion. Data presented at the 2017 Society of Gynecologic Oncology Annual Meeting on Women’s Cancer showed that rates of cervical dysplasia were equivalent for women who completed a two-dose schedule versus a three-dose schedule.4
Efficacy
A recent meta-analysis of clinical trials of the HPV vaccines describe efficacy of 95%-97% in prevention of CIN 1-3.5 While its greatest efficacy is in its ability to prevent primary HPV infection, there still is some benefit for individuals who already were exposed to HPV prior to vaccination. As stated previously, women with a history of prior HPV vaccination have lower rates of recurrence of cervical dysplasia after treatment. Additionally, recent research has shown that women who received HPV vaccinations after a LEEP procedure for CIN 2 or 3 experience significantly lower recurrence rates, compared with women who did not receive vaccinations after LEEP (2.5% vs. 8.5%).6 This raises the possibility of a therapeutic role for HPV vaccination in women infected with HPV. Prospective studies are currently evaluating this question.
Myths
The most common side effects of the HPV vaccine are pain, redness, or swelling at the injection site. Other known side effects include fever, headache or malaise, nausea, syncope, or muscle/joint pain – similar to other vaccinations. Anaphylaxis is a rare complication.
Some parents and pediatricians report concerns that vaccination could lead to earlier sexual activity. Multiple studies have shown that girls who receive HPV vaccination are no more likely to become pregnant or get a sexually transmitted infection (proxies for intercourse) than are girls who were not vaccinated.7,8
Maximizing vaccination rates
HPV vaccination rates in the United States lag significantly behind rates in countries with national vaccine programs, such as Australia and Denmark.9 Early data from Australia already have shown a decrease in genital warts and CIN 2+ incidence within the 10 years of starting its school-based vaccine program, with approximately 73% of 12- to 15-year-olds having completed the vaccine series.2 In contrast, just 40% and 22% of 13- to 17-year-old girls and boys in the United States, respectively, had completed the vaccine series in 2014, according to the CDC.10
Vaccination gaps between girls and boys are narrowing, and more teens will be able to complete the series with the new two-dose recommendation for those younger than 15 years. However, our current rates of vaccination are significantly lower for HPV than for other routinely recommended adolescent vaccines (such as Tdap and meningococcal) and more must be done to encourage vaccination.
Studies have shown that parents are more likely to vaccinate their children if providers recommend the vaccine.11 As women’s health care providers, we do not always see children during the time period that is ideal for vaccination. However, we take care of many women who are presenting for routine gynecologic care, pregnancy, or with abnormal Pap smear screenings. These are ideal opportunities to educate and offer HPV vaccination to women in the approved age groups, as well as to encourage parents to vaccinate their children.
As with other vaccines, the recommendation should be clear and focused on the cancer prevention benefit. Using methods in which the recommendation is “announced” in a brief statement assuming parents/patients are ready to vaccinate versus open-ended conversations, has been studied as a potentially successful method to increase uptake of HPV vaccination.12 Additionally, documentation of HPV vaccination status should be built into electronic medical record templates to prompt clinicians to ask and offer HPV vaccination at visits, including postpartum visits.

Dr. Rahangdale is an associate professor of ob.gyn. at the University of North Carolina, Chapel Hill, and is director of the North Carolina Women’s Hospital Cervical Dysplasia Clinic. Dr. Rossi is an assistant professor in the division of gynecologic oncology at UNC-Chapel Hill. They reported having no relevant financial disclosures.

References
1. Am J Epidemiol. 2008 Jul 15;168(2):123-37.
2. Int J Cancer. 2012 Nov 1;131(9):1969-82.
3. BMJ. 2012 Mar 27;344:e1401. doi: 10.1136/bmj.e1401.
4. Gynecol Oncol. 2017 Jun. doi. org/10.1016/j.ygyno.2017.03.031.
5. Int J Prev Med. 2017 Jun 1;8:44. doi: 10.4103/ijpvm.IJPVM_413_16.
6. Gynecol Oncol. 2013 Aug;130(2):264-8.
7. Pediatrics. 2012 Nov;130(5):798-805.
8. JAMA Intern Med. 2015 Apr;175(4):617-23.
9. Clin Pediatr (Phila). 2016 Sep;55(10):904-14.
10. MMWR Morb Mortal Wkly Rep. 2015 Jul 31;64(29):784-92.
11. Vaccine. 2016 Feb 24;34(9):1187-92.
12. Pediatrics. 2017 Jan;139(1). pii:e20161764. doi: 10.1542/peds.2016-1764.
Human papillomavirus (HPV) is the most common sexually transmitted infection. Exposure is widespread and most individuals clear the infection without symptoms or development of disease. However, a subset of individuals experience persistent infection, a state which can lead to carcinogenesis of lower genital tract malignancies, particularly cervical cancer.1
Vaccine coverage
Persistent infection with high-risk (oncogenic) HPV is well known to be the cause of cervical cancer. There are two HPV vaccines manufactured for the purposes of cervical cancer, anal cancer, and genital wart prevention (Cervarix and Gardasil). The Cervarix vaccine covers high-risk HPV subtypes 16 and 18 and the Gardasil vaccine prevents both low-risk HPV subtypes 6 and 11, which can cause genital warts, and high-risk HPV subtypes 16, 18, 31, 33, 45, 52 and 58, which cause cervical dysplasia and cancer.
High-risk HPV is also associated with head and neck, vulvar, vaginal, and penile cancers, though the vaccines are not approved by the Food and Drug Administration for prevention of these diseases.2
Vaccination indications
Since vaccination prevents multiple subtypes of HPV, an individual who has already been exposed will still benefit from protection from other subtypes of HPV through vaccination. HPV vaccination is not approved during pregnancy but can be initiated in the postpartum period when women are engaged in their health care and receiving other vaccinations, such as varicella or the MMR vaccine.
Recommended schedule
Until October 2016, the vaccination schedule was based on a three-dose series (0, 2, and 6 months). Currently, the CDC recommends that children aged under 15 years at the time of first dose may opt for a two-dose series (0 and 6-12 months). For those aged 15-26 years, the three-dose schedule remains the recommended course.
The benefits of two-dose schedule are convenience, cost, and increased likelihood of completion. Data presented at the 2017 Society of Gynecologic Oncology Annual Meeting on Women’s Cancer showed that rates of cervical dysplasia were equivalent for women who completed a two-dose schedule versus a three-dose schedule.4
Efficacy
A recent meta-analysis of clinical trials of the HPV vaccines describe efficacy of 95%-97% in prevention of CIN 1-3.5 While its greatest efficacy is in its ability to prevent primary HPV infection, there still is some benefit for individuals who already were exposed to HPV prior to vaccination. As stated previously, women with a history of prior HPV vaccination have lower rates of recurrence of cervical dysplasia after treatment. Additionally, recent research has shown that women who received HPV vaccinations after a LEEP procedure for CIN 2 or 3 experience significantly lower recurrence rates, compared with women who did not receive vaccinations after LEEP (2.5% vs. 8.5%).6 This raises the possibility of a therapeutic role for HPV vaccination in women infected with HPV. Prospective studies are currently evaluating this question.
Myths
The most common side effects of the HPV vaccine are pain, redness, or swelling at the injection site. Other known side effects include fever, headache or malaise, nausea, syncope, or muscle/joint pain – similar to other vaccinations. Anaphylaxis is a rare complication.
Some parents and pediatricians report concerns that vaccination could lead to earlier sexual activity. Multiple studies have shown that girls who receive HPV vaccination are no more likely to become pregnant or get a sexually transmitted infection (proxies for intercourse) than are girls who were not vaccinated.7,8
Maximizing vaccination rates
HPV vaccination rates in the United States lag significantly behind rates in countries with national vaccine programs, such as Australia and Denmark.9 Early data from Australia already have shown a decrease in genital warts and CIN 2+ incidence within the 10 years of starting its school-based vaccine program, with approximately 73% of 12- to 15-year-olds having completed the vaccine series.2 In contrast, just 40% and 22% of 13- to 17-year-old girls and boys in the United States, respectively, had completed the vaccine series in 2014, according to the CDC.10
Vaccination gaps between girls and boys are narrowing, and more teens will be able to complete the series with the new two-dose recommendation for those younger than 15 years. However, our current rates of vaccination are significantly lower for HPV than for other routinely recommended adolescent vaccines (such as Tdap and meningococcal) and more must be done to encourage vaccination.
Studies have shown that parents are more likely to vaccinate their children if providers recommend the vaccine.11 As women’s health care providers, we do not always see children during the time period that is ideal for vaccination. However, we take care of many women who are presenting for routine gynecologic care, pregnancy, or with abnormal Pap smear screenings. These are ideal opportunities to educate and offer HPV vaccination to women in the approved age groups, as well as to encourage parents to vaccinate their children.
As with other vaccines, the recommendation should be clear and focused on the cancer prevention benefit. Using methods in which the recommendation is “announced” in a brief statement assuming parents/patients are ready to vaccinate versus open-ended conversations, has been studied as a potentially successful method to increase uptake of HPV vaccination.12 Additionally, documentation of HPV vaccination status should be built into electronic medical record templates to prompt clinicians to ask and offer HPV vaccination at visits, including postpartum visits.
Dr. Rahangdale is an associate professor of ob.gyn. at the University of North Carolina, Chapel Hill, and is director of the North Carolina Women’s Hospital Cervical Dysplasia Clinic. Dr. Rossi is an assistant professor in the division of gynecologic oncology at UNC-Chapel Hill. They reported having no relevant financial disclosures.
References
1. Am J Epidemiol. 2008 Jul 15;168(2):123-37.
2. Int J Cancer. 2012 Nov 1;131(9):1969-82.
3. BMJ. 2012 Mar 27;344:e1401. doi: 10.1136/bmj.e1401.
4. Gynecol Oncol. 2017 Jun. doi. org/10.1016/j.ygyno.2017.03.031.
5. Int J Prev Med. 2017 Jun 1;8:44. doi: 10.4103/ijpvm.IJPVM_413_16.
6. Gynecol Oncol. 2013 Aug;130(2):264-8.
7. Pediatrics. 2012 Nov;130(5):798-805.
8. JAMA Intern Med. 2015 Apr;175(4):617-23.
9. Clin Pediatr (Phila). 2016 Sep;55(10):904-14.
10. MMWR Morb Mortal Wkly Rep. 2015 Jul 31;64(29):784-92.
11. Vaccine. 2016 Feb 24;34(9):1187-92.
12. Pediatrics. 2017 Jan;139(1). pii:e20161764. doi: 10.1542/peds.2016-1764.
Human papillomavirus (HPV) is the most common sexually transmitted infection. Exposure is widespread and most individuals clear the infection without symptoms or development of disease. However, a subset of individuals experience persistent infection, a state which can lead to carcinogenesis of lower genital tract malignancies, particularly cervical cancer.1
Vaccine coverage
Persistent infection with high-risk (oncogenic) HPV is well known to be the cause of cervical cancer. There are two HPV vaccines manufactured for the purposes of cervical cancer, anal cancer, and genital wart prevention (Cervarix and Gardasil). The Cervarix vaccine covers high-risk HPV subtypes 16 and 18 and the Gardasil vaccine prevents both low-risk HPV subtypes 6 and 11, which can cause genital warts, and high-risk HPV subtypes 16, 18, 31, 33, 45, 52 and 58, which cause cervical dysplasia and cancer.
High-risk HPV is also associated with head and neck, vulvar, vaginal, and penile cancers, though the vaccines are not approved by the Food and Drug Administration for prevention of these diseases.2
Vaccination indications
Since vaccination prevents multiple subtypes of HPV, an individual who has already been exposed will still benefit from protection from other subtypes of HPV through vaccination. HPV vaccination is not approved during pregnancy but can be initiated in the postpartum period when women are engaged in their health care and receiving other vaccinations, such as varicella or the MMR vaccine.
Recommended schedule
Until October 2016, the vaccination schedule was based on a three-dose series (0, 2, and 6 months). Currently, the CDC recommends that children aged under 15 years at the time of first dose may opt for a two-dose series (0 and 6-12 months). For those aged 15-26 years, the three-dose schedule remains the recommended course.
The benefits of two-dose schedule are convenience, cost, and increased likelihood of completion. Data presented at the 2017 Society of Gynecologic Oncology Annual Meeting on Women’s Cancer showed that rates of cervical dysplasia were equivalent for women who completed a two-dose schedule versus a three-dose schedule.4
Efficacy
A recent meta-analysis of clinical trials of the HPV vaccines describe efficacy of 95%-97% in prevention of CIN 1-3.5 While its greatest efficacy is in its ability to prevent primary HPV infection, there still is some benefit for individuals who already were exposed to HPV prior to vaccination. As stated previously, women with a history of prior HPV vaccination have lower rates of recurrence of cervical dysplasia after treatment. Additionally, recent research has shown that women who received HPV vaccinations after a LEEP procedure for CIN 2 or 3 experience significantly lower recurrence rates, compared with women who did not receive vaccinations after LEEP (2.5% vs. 8.5%).6 This raises the possibility of a therapeutic role for HPV vaccination in women infected with HPV. Prospective studies are currently evaluating this question.
Myths
The most common side effects of the HPV vaccine are pain, redness, or swelling at the injection site. Other known side effects include fever, headache or malaise, nausea, syncope, or muscle/joint pain – similar to other vaccinations. Anaphylaxis is a rare complication.
Some parents and pediatricians report concerns that vaccination could lead to earlier sexual activity. Multiple studies have shown that girls who receive HPV vaccination are no more likely to become pregnant or get a sexually transmitted infection (proxies for intercourse) than are girls who were not vaccinated.7,8
Maximizing vaccination rates
HPV vaccination rates in the United States lag significantly behind rates in countries with national vaccine programs, such as Australia and Denmark.9 Early data from Australia already have shown a decrease in genital warts and CIN 2+ incidence within the 10 years of starting its school-based vaccine program, with approximately 73% of 12- to 15-year-olds having completed the vaccine series.2 In contrast, just 40% and 22% of 13- to 17-year-old girls and boys in the United States, respectively, had completed the vaccine series in 2014, according to the CDC.10
Vaccination gaps between girls and boys are narrowing, and more teens will be able to complete the series with the new two-dose recommendation for those younger than 15 years. However, our current rates of vaccination are significantly lower for HPV than for other routinely recommended adolescent vaccines (such as Tdap and meningococcal) and more must be done to encourage vaccination.
Studies have shown that parents are more likely to vaccinate their children if providers recommend the vaccine.11 As women’s health care providers, we do not always see children during the time period that is ideal for vaccination. However, we take care of many women who are presenting for routine gynecologic care, pregnancy, or with abnormal Pap smear screenings. These are ideal opportunities to educate and offer HPV vaccination to women in the approved age groups, as well as to encourage parents to vaccinate their children.
As with other vaccines, the recommendation should be clear and focused on the cancer prevention benefit. Using methods in which the recommendation is “announced” in a brief statement assuming parents/patients are ready to vaccinate versus open-ended conversations, has been studied as a potentially successful method to increase uptake of HPV vaccination.12 Additionally, documentation of HPV vaccination status should be built into electronic medical record templates to prompt clinicians to ask and offer HPV vaccination at visits, including postpartum visits.
Dr. Rahangdale is an associate professor of ob.gyn. at the University of North Carolina, Chapel Hill, and is director of the North Carolina Women’s Hospital Cervical Dysplasia Clinic. Dr. Rossi is an assistant professor in the division of gynecologic oncology at UNC-Chapel Hill. They reported having no relevant financial disclosures.
References
1. Am J Epidemiol. 2008 Jul 15;168(2):123-37.
2. Int J Cancer. 2012 Nov 1;131(9):1969-82.
3. BMJ. 2012 Mar 27;344:e1401. doi: 10.1136/bmj.e1401.
4. Gynecol Oncol. 2017 Jun. doi. org/10.1016/j.ygyno.2017.03.031.
5. Int J Prev Med. 2017 Jun 1;8:44. doi: 10.4103/ijpvm.IJPVM_413_16.
6. Gynecol Oncol. 2013 Aug;130(2):264-8.
7. Pediatrics. 2012 Nov;130(5):798-805.
8. JAMA Intern Med. 2015 Apr;175(4):617-23.
9. Clin Pediatr (Phila). 2016 Sep;55(10):904-14.
10. MMWR Morb Mortal Wkly Rep. 2015 Jul 31;64(29):784-92.
11. Vaccine. 2016 Feb 24;34(9):1187-92.
12. Pediatrics. 2017 Jan;139(1). pii:e20161764. doi: 10.1542/peds.2016-1764.
Collaboration of the NIH and PHS Commissioned Corps in the International Ebola Clinical Research Response
The Ebola epidemic of 2014-2016 challenged many federal agencies to find creative ways to help address the vexing problems created by the spread of the disease.
The response from the U.S. and the global community took many forms: Not only was there a need for the typical medical care support, but also for basic public health systems to track the spread of disease, provide clean water, and dispose of infectious waste. Because no known preventive vaccines or therapeutics existed for those infected, the recognition of a research component to the response became abundantly clear as the epidemic continued. As a result, the National Institutes of Health (NIH) and the USPHS Commissioned Corps (Corps) serendipitously found themselves allied in a mutually beneficial relationship in the establishment of an Ebola clinical research program in West Africa.
This article describes the events that led to the NIH and Corps participation in the Ebola response, the roles filled by the Corps in supporting the NIH, and the lessons observed from that collaboration. Also presented are considerations regarding preparation of a clinical research response to future outbreaks.
NIH Clinical Research first Response
The 2014-2016 Ebola epidemic in West Africa demonstrated the need for federal agencies to reassess their capacity to respond to global threats to protect the health security of the U.S.1 The outbreak also challenged the U.S. government to mobilize unique resources that matched the need of this international (and domestic) response.
In 2014, President Barack Obama announced that the U.S. would launch a government response to the Ebola effort. Although a comprehensive research and development program already was in place to establish Ebola virus disease (EVD) countermeasures, no FDA-approved diagnostics, therapeutics, or preventive vaccines were readily available. Fortunately, FDA regulations regarding emergency use authorizations allowed for the use of several EVD diagnostics during this outbreak.2 However, the development of drugs and vaccines specific to Ebola had yet to make their way to phase 1 safety studies.
Two vaccine products went into phase 1 studies in the U.S. within months of the declaration of the emergency.3,4 Additionally, the NIH had organized a collaborative effort between the U.S. government and academic community to identify a research strategy for the evaluation of therapeutics.5 Regardless of the state of countermeasures and research proposals, the initial need was for disease control measures and care for Ebola patients. The CDC took the lead in working within the international community to establish an incident management system that could help the impacted countries enact mechanisms to bring the epidemic under control.6
As the epidemic progressed, leaders in the Corps and the NIH responded on pathways that eventually would intersect. One of the unfortunate outcomes of the early efforts of improperly protected health care providers was the unintentional transmission of Ebola.7 The Corps identified the need to provide high-level care to the health care worker community as one incentive to motivate health care workers to volunteer for hazardous duty inside Ebola treatment units (ETUs).8,9 Engulfed in the epidemic response, the U.S. government through the National Security Council and secretary of the Department of Health and Human Services (DHHS) evoked its statutory authority to deploy the Corps (42 U.S. Code 204a).
In the first week of October 2014, the Corps sent an advanced echelon team to assess the situation, partner with key host country and international stakeholders, and begin establishment of the U.S. government’s first ever ETU. With logistics, security, and resource support from the DoD and response coordination from the U.S. Agency for International Development, the Corps then deployed the first of four 70-person team rotations to staff the Monrovia Medical Unit (MMU), an ETU specifically dedicated to the treatment of Ebola-infected health care workers. At the time, it was the only ETU specifically dedicated to health care workers in all of Africa. The MMU operated until May 2015 and provided direct patient care for health care workers with Ebola, malaria, and other illnesses.8,10
In August 2014, representatives from the CDC met with Liberia’s Minister of Health and Social Welfare Walter T. Gwenigale, MD, to discuss the range of available options that could facilitate a better understanding of the prevention and treatment of the disease. This meeting resulted in a letter dated August 22, 2014, from Dr. Gwenigale to then DHHS Sylvia Burwell, requesting a research response. Secretary Burwell responded on October 2, 2014, describing the immediate dispatch of the deputy director for clinical research of the National Institute of Allergy and Infectious Diseases (NIAID) to Liberia to engage in initial discussions with the Liberian minister and other key Liberians involved in the response.
Representatives from the CDC and the commander of the Corps’ Ebola response (and acting deputy surgeon general) were included in those initial meetings, which led to a recognized need for a robust clinical research program of the highest ethical and scientific standards consistent with the expressed requirements of Liberia.11 A second and third trip to Liberia with larger U.S. teams resulted in an agreement signed on November 19, 2014 for the scientific investigation of strategies that tested interventions for treatment, control, and prevention of Ebola.12
The agreement led to the establishment of the Partnership for Research on Ebola Virus in Liberia (PREVAIL) to identify research priorities in a collaborative manner between Liberian and American scientists. The first protocol, a vaccine study, was launched in early February 2015.12 This monumental task involved the support of hundreds of Liberians and dozens of NIH staff who volunteered for rotations to Liberia. Of the 108 volunteers from within the NIH, 18 were PHS officers. Shortly after launching the vaccine study, the next priority was initiating the treatment study. This study was delayed primarily due to ZMapp (Mapp Biopharmaceatical, San Diego,CA) production limitations. ZMapp, a monoclonal antibody cocktail, was the first Ebola therapeutic product to be evaluated in a randomized trial.5,13
During the planning for the study, NIAID staff in Liberia met with Corps staff of the MMU to discuss the logistics associated with implementation of the ZMapp protocol at the MMU. During that meeting, the NIAID deputy director for clinical research expressed interest in obtaining Corps support from outside the NIH to sustain the research effort in West Africa. More specifically, additional pharmacy and laboratory staff were needed to augment NIH research operations. At the time, the MMU commander had recently transitioned from service as the acting surgeon general and was in a unique position to recommend additional Corps resources that could help in the research response.
The February 2015 discussion resulted in the establishment of an NIH/PHS research partnership that continues to exist. This new opportunity was not a significant stretch for the PHS as there was great interest from the Corps for responding to the Ebola crisis. The enthusiasm was consistent with the overall ethos of the Corps, which as a service was composed of highly qualified active-duty, deployable, uniformed, public health professionals who respond to public health crises at home and abroad. To date, 19 Corps officers from outside the NIH have deployed in support of the NIH Ebola clinical research program. An additional 18 Corps officers assigned within the NIH also volunteered for duty in West Africa. Of the 37 Corps officers supporting the NIH clinical research program, 7 served on more than 1 rotation.
Program Expansion
The Ebola clinical research program expanded over time from the initial PREVAIL vaccine study to include studies of therapeutic agents, natural history in Ebola survivors, and an additional vaccine study. The PHS officers have been integral in conducting these studies. The initial study implemented in Liberia, known as PREVAIL I, involved the evaluation of 2 vaccine strategies vs placebo.12,14 In addition to the NIH-based Corps officers supporting the study, the Readiness and Deployment Operations Group (RedDOG) initiated deployments for an additional 2 pharmacy and 7 laboratory officers to support this study. During the deployment, the pharmacists were asked to extend their reach to Sierra Leone and later to Guinea to help establish PREVAIL II, an evaluation of ZMapp in the treatment of Ebola.13 A total of 9 Corps pharmacists, 2 nurses, and 3 physicians deployed to Sierra Leone or Guinea to assist in the PREVAIL II study.
As the epidemic came to an end in Liberia in May 2015, the need for a long-term assessment of Ebola survivors was recognized, resulting in PREVAIL III.15 Noteworthy in the survivor study was an ophthalmic substudy led by a Corps officer assigned to the National Eye Institute.16,17 The survivor study also identified that the persistence of the Ebola virus was longer than previously known and that sexual transmission via semen from infected males remained a potential mode of transmission.18 To address the lingering viral load, a study of an antiviral drug was initiated in Liberia in the summer of 2016, PREVAIL IV.19
Four Corps pharmacists helped train Liberian pharmacists to establish and sustain this randomized, double-blind, placebo-controlled study. Most recently, Corps pharmacists were deployed to support the initiation of the Partnership for Research on Ebola Vaccines (PREVAC), a collaborative partnership with researchers from Liberia, Guinea, and Sierra Leone with cosponsors from the NIH, Institut national de la santé et de la recherche médicale (Inserm) in France, and the London School of Hygiene and Tropical Medicine in the United Kingdom.20
Deployment Procedures

Within a week of the February 2015 initial meeting in Liberia to establish the NIH/PHS collaboration, the NIH deployment team met by phone with the Corps’ RedDOG to discuss initial requirements (eg, number of officers needed, disciplines, time lines, and documentation needed for deployment). These initial discussions resulted in the establishment of more formal processes that evolved over time as the 2 organizations gained experience. Based on the identification of the numbers and types of officers needed, RedDOG used procedures similar to the process for staffing the MMU. A communication went out to the Corps seeking interested officers.
Deployment slots were filled based on the personal availability of the officer and coordination with their immediate supervisor and agency. Officers needed to meet medical clearance requirements and provide current health care provider licensure information. Additional training requirements needed to be completed (eg, U.S. State Department training and good clinical practice [GCP] if not already current). Corps officers also took part in the NIH orientation program for deploying personnel to familiarize them to the situation on the ground in West Africa and the specific clinical research protocols that they would encounter. Given that most of the Corps officers were coming from outside the NIH, the onboarding activities required significant attention to detail as procedures for arranging travel (eg, passport, visa, and airline reservations) and processes for reimbursement of travel/per diem pay differed from more traditional deployments directed through the Corps headquarters.
Commissioned Corps Roles in the Research Response
Whereas the establishment of the research program in Liberia was based primarily on relationships forged over a 2-month period by the NIAID deputy director for clinical research and staff, the extension of the research program into Sierra Leone (March 2015) and Guinea (June 2015) was on a substantially shorter time line. As a result, Corps officers were thrust into roles that immediately employed their leadership and diplomacy skills.
In Sierra Leone and Guinea, the NIAID deputy director for clinical research established initial relationships within the countries. However, Corps officers found themselves in regular interactions with regulators in the Ministry of Health to ensure that applications were complete and import permits for incoming shipments were cleared. Additionally, the research collaboration in Sierra Leone was coordinated through an investigator assigned to a military hospital converted into an ETU. The Corps officers were well suited to maintain and build on that relationship in expanding the protocol to other ETUs throughout Sierra Leone. A site established by the CDC within the Sierra Leone Ministry of Health coordinated ZMapp storage. The Corps officers formed working relationships with the CDC team to establish and improve cold-chain logistics and transportation of the ZMapp to the various ETUs around the country. Corps officers were integral in working with the in-country contract hiring agency. Activities included establishing criteria for clinical research positions, providing input on the interview of respective candidates, and training staff as the team formed. In Sierra Leone, local staff members were hired to work at specific facilities as research coordinators working with the health care delivery teams.
The U.S. team consisted of a physician, nurse research coordinator, and a pharmacist travelling to the sites with a logistics/operations staff member remaining in Freetown.
Fortunately, a Corps nurse on the team had been part of the initial MMU deployment and was trained to work in a special care unit at the NIH for patients with highly contagious infections. This practical experience was essential in the establishment of procedures in a hazardous environment for the administration of the IV ZMapp, monitoring of adverse effects (AEs), provision of medications to mitigate infusion-related reactions, and documentation of those AEs.
The U.S. research team regularly departed Freetown early in the morning 7 days a week with the various supplies needed as they visited up to 4 ETU sites to prepare the ZMapp at the site, await information on any AEs, and collect case report forms (Figures 1 and 2). The ETUs were spread out over a 90-mile radius and could be described as austere platforms for health care delivery.


An additional challenge was dealing with the multinational organizations that staffed the various ETUs. Relief organizations from Italy, the United Kingdom, China, as well as the Sierra Leone military provided the staffing for the 4 ETUs. Regardless of who operated the ETU, the concept of randomization to ZMapp or standard of care required significant tact and diplomacy in communicating the scientific necessity in order to appropriately answer the research question.
As the summer approached in Sierra Leone, the team worked through challenges in the IV administration of ZMapp as the protein structure of the monoclonal antibody had not previously been subjected to West African environmental extremes. A balance between speed of administration to prevent protein aggregation in the heat as opposed to the risk of infusion reactions from a foreign protein required the team to communicate frequently with, the manufacturer of ZMapp, to establish realistic infusion rate tables. Additionally, as the various deployment teams rotated in and out, procedures for establishing continuity of research operations were enacted and improved on with each rotation. Good documentation practices to adequately collect all required study information (eg, recording AEs, deviations, and signatures on various forms) proved critical to continuity of research operations.
In Guinea, not only was there the new wrinkle of working within a country where the primary language was French, but also a French cosponsor, Inserm. The NIAID clinical director capitalized on the research infrastructure established for a recently completed Inserm study of favipiravir in the treatment of Ebola to extend the ZMapp study to Guinea. Fortunately, many of the Inserm staff were bilingual and readily responded to the NIH training on the requirements of the ZMapp protocol. However, procedures for cold-chain storage and transportation needed to be established. In Guinea, the PHS officers were key in establishing access and temperature monitoring procedures for a secure room inside the U.S. embassy. The issues associated with cold-chain procedures in the infrastructure-limited environments of West Africa are substantial and warrant consideration of a stand-alone paper. Corps officers also took part in weekly country-focused team meetings with embassy staff to describe progress with the ZMapp study.
As the epidemic waned and NIH transitioned to the survivor and viral persistence studies, the operational tempo changed to allow Corps officers to take part in more definitive capacity building efforts. An initial PHS volunteer from the FDA accepted a position within NIAID as a clinical research oversight manager for pharmacy operations. This individual deployed on numerous occasions to the 3 affected West African countries to further establish cold-chain processes for pharmaceuticals and biologics. He also worked with a multidisciplinary team to renovate a clinical research facility in a rural setting in Guinea. In Liberia, he coordinated an effort with other Corps officers to provide educational seminars on clinical research principles and drug-specific topics with the University of Liberia School of Pharmacy.
Challenges
In each instance, the partnership experience was not without a few problems. The match of skills between the officers who wanted to help and those needed for the research program did not always coincide. While the Corps has more than 1,200 pharmacy officers on active duty, only a fraction of those have experience conducting FDA-regulated clinical research.
Communication problems and time pressures were also constant companions to both the Corps and the NIH. The Corps was going through the largest international deployment in its history to staff multiple missions (including the primary MMU mission in Liberia). The addition of the NIH partnership, while consistent with the MMU staffing mission, provided even more work for a very limited resource. Communicating to the many Corps officers who wanted to volunteer and keeping deployment time lines on track were a challenge. Complicating the matter was the addition of stray e-mails from well-intentioned NIH and Corps staff who communicated directly with colleagues to encourage participation, not fully understanding the policies and protocol governing the deployment process.
Time was always an issue as the rotation schedules were relatively short and the number of activities to make an officer deployment ready were numerous. Obtaining official passports and visas was a challenge as that activity required coordination with the U.S. Department of State. Airline schedules changed with little or no notice, complicating deployments and returns. As the NIH added additional research studies for which support was required, time lines for studies to start became difficult to predict with certainty due to factors outside the control of the NIH. Recently, additional security training requirements for government workers traveling abroad were instituted, further complicating the process of deploying an officer.
The Corps officers taking part in this research response (which was not consistent with customary deployments from Corps headquarters) necessarily were volunteers from full-time assignments within DHHS, and as such, required the permission of their supervisory chain to volunteer. Regardless of this limitation, there was widespread support for these additional and specific research deployments. Although the use of short-term rotations was not ideal, in the end, the rotation plans worked, and the NIH was able to fulfill its research mission with the support of the Corps.
Lessons Learned/Preparing for the Future
Many lessons have been learned and continue to be learned throughout this research response and NIH/Corps partnership. Effective and frequent communication between the organization requesting Corps officers and the Corps headquarters is crucial. In the initial deployments, officers were deployed from the FDA with the assumption that they would be familiar with FDA-regulated clinical research. This was not always the case. The NIH and Corps headquarters later collaborated to develop a survey to send to Corps officers that was used to identify specific skill sets needed by officers who would be deploying to conduct clinical research. NIH personnel prescreened survey responses to identify and prioritize officers for deployment consideration by the deployment authority. This process resulted in the selection of officers who generally needed less training and guidance.
Effective training in clinical research principles for deployed officers and other staff needs to be developed and made available to all deploying individuals. All clinical research staff are required to have training on GCP, but most GCP training programs focus primarily on the ethical principles of research as outlined by the Declaration of Helsinki, Nuremberg Code, and other documents. Few GCP training programs present adequate information on the hands-on conduct of clinical research, especially research regulated by the FDA and other government bodies and therefore subject to certain strict requirements. Examples of crucial but often overlooked topics are source document retention, good documentation practices, cold-chain principles, and other issues related to the creation and retention of adequate trial records.21
The handoff between returning and deploying officers is crucial. Due to various issues with changing time lines, flights, and administrative processes, it is imperative to plan adequate overlap between returning and deploying officers. Delays in obtaining passports or visas, flight cancellations, and other unforeseen issues may unexpectedly shorten any planned overlap periods. A full workweek is desirable for overlap so that the new officer may experience tasks that occur throughout the week, be introduced to the various team members, and have help if unexpected events occur. A regular staff member should check periodically that proper procedures are being followed, as some information may be missed during each handoff, and consecutive unchecked handoffs could result in large deviations of important procedures. Onboarding and offboarding checklists should be developed and updated regularly to guide the handoff process.
On a larger scale, the respective agencies and other stakeholders involved in planning clinical research for public health emergencies need to be included in regular tabletop training exercises to better understand how to coordinate a response when needed. Additionally, although many of the Corps officers who took part in this deployment served as mentors for others preparing for deployment, establishing a formal roster of experienced officers to support specific roles of this type of response would help serve as a resource center for future deployments. Finally, coordination between any operating division (or agency) and the Corps should be through the established Corps command infrastructure to eliminate miscommunication and complicating deployment processes.22
Conclusion
The increasing connectedness of this world, as demonstrated by the Ebola epidemic, requires that the HHS engage globally to provide international leadership and technical expertise in science, policy, and programs and work in concert with interagency partners.23 The missions of the PHS and NIH intersected in a synergistic manner in the research response to the Ebola epidemic of 2014-2016. The PHS Corps mission includes to “protect, promote, and advance the health and safety of the Nation...through rapid and effective response…and advancement of public health science.”24 The Corps mission directly supported the NIH mission to seek fundamental knowledge about the nature and behavior of living systems and the application of that knowledge to enhance health, lengthen life, and reduce illness and disability.25
The scope and scale of DHHS’s response to the Ebola epidemic was unprecedented. The NIH research program, although successful and an important component, was but a small part in bringing the Ebola crisis to an end. The CDC (including the many Corps officers assigned to that agency) worked successfully with the international community and the host countries to bring the disease under control. The Biological Advanced Research and Development Authority provided expert project management, making vaccines and therapeutics available for research.
The DoD was a partner in the development of countermeasures and phase 1 clinical research programs as well as establishing laboratory facilities in Liberia. The Department of State facilitated the many interactions required for the mobilization of resources into West Africa. The collective efforts of the U.S. government contributed immensely to the protection of U.S. borders and to the successful resolution of the Ebola outbreak of 2014-2016.
1. Bell BP, Damon IK, Jernigan DB, et al. Overview, control strategies, and lessons learned in the CDC response to the 2014-2016 Ebola epidemic. MMWR. 2016;65(suppl 3):4-11.
2. U.S. Food and Drug Administration. Emergency use authorization. https://www.fda.gov/EmergencyPreparedness/Counterterrorism/MedicalCountermeasures/MCMLegalRegulatoryandPolicyFramework/ucm182568.htm#ebola. Updated June 29, 2017. Accessed June 30, 2017.
3. Regules JA, Beigel JH, Paolino KM, et al; for the rVSVΔG-ZEBOV-GP Study Group. A recombinant vesicular stomatitis virus Ebola vaccine. N Engl J Med. 2017;376(4):330-341.
4. Tapia MD, Sow SO, Lyke KE, et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2016;16(1):31-42.
5. Dodd LE, Proschan MA, Neuhaus J, et al. Design of a randomized controlled trial for Ebola virus disease medical countermeasures: PREVAIL II, the Ebola MCM Study. J Infect Dis. 2016;213(12):1906-1913.
6. Brooks JC, Pinto M, Gill A, et al. Incident management systems and building emergency management capacity during the 2014-2016 Ebola epidemic—Liberia, Sierra Leone, and Guinea. MMWR. 2016;65(suppl 3):28-34.
7. Evans DK, Goldstein M, Popova A. Health-care worker mortality and the legacy of the Ebola epidemic. Lancet Glob Health. 2015;3(8):e439-e440.
8. Lushniak BD. The hope multipliers: the U.S. Public Health Service in Monrovia. Public Health Rep. 2015;130(6):562-565.
9. Lushniak BD. Update on the U.S. public health response to the Ebola outbreak. Public Health Rep. 2015;130(2):118-120.
10. Brown-Stephenson J. United States Public Health Service nurses: deployment in global crisis. Online J Issues Nurs. 2017;22(1):6.
11. Lane HC, Marston HD, Fauci AS. Conducting clinical trials in outbreak settings: points to consider. Clin Trials. 2016;13(1):92-95.
12. Kennedy SB, Neaton JD, Lane HC, et al. Implementation of an Ebola virus disease vaccine clinical trial during the Ebola epidemic in Liberia: design, procedures, and challenges. Clin Trials. 2016;13(1):49-56.
13. Davey RT. PREVAIL II: a randomized controlled trial of ZMappTM in acute Ebola virus infection. Paper presented at: Conference on Retroviruses and Opportunistic Infections; February 22-25, 2016; Boston, Massachusetts.
14. Doe-Anderson J, Baseler B, Driscoll P, et al. Beating the odds: successful establishment of a phase II/III clinical research trial in resource-poor Liberia during the largest-ever Ebola outbreak. Contemp Clin Trials Commun. 2016;4:68-73.
15. U.S. National Institutes of Health Clinical Center. Ebola virus disease survivors: clinical and immunologic follow-up. https://clinicaltrials.gov/ct2/show/NCT02431923. Updated June 30, 2017. Accessed July 5, 2017.
16. Jampol LM, Ferris FL III, Bishop RJ. Ebola and the eye. JAMA Ophthalmol. 2015;133(10):1105-1106.
17. Chertow DS, Nath A, Suffredini AF, et al. Severe meningoencephalitis in a case of Ebola virus disease: a case report. Ann Intern Med. 2016;165(4):301-304.
18. Pettitt J, Higgs ES, Fallah MP, Hensley LE. Assessment and optimization of the GeneXpert diagnostic platform for detection of Ebola virus RNA in seminal fluid. J Infect Dis. 2017;215(4):547-553.
19. U.S. National Institutes of Health Clinical Center. GS-5734 to assess the antiviral activity, longer-term clearance of Ebola virus, and safety in male Ebola survivors with evidence of Ebola virus persistence in semen. https://clinicaltrials.gov/show/NCT02818582. Updated June 30, 2017. Accessed July 5, 2017.
20. U.S. National Institutes of Health Clinical Center. Partnership for Research on Ebola VACcinations (PREVAC). https://clinicaltrials.gov/show/NCT02876328. Updated June 28, 2017. Accessed July 5, 2017.
21. Kirchoff MC, Pierson JF. Considerations for use of investigational drugs in public health emergencies. Ther Innov Regul Sci. 2017;51(2):146-152.
22. U.S. Department of Health and Human Services, Office of the Assistant Secretary for Preparedness and Response. U.S. Department of Health and Human Services Ebola response improvement plan (based on lessons learned from the 2014-2016 Ebola epidemic). https://www.phe.gov/Preparedness/respond ers/ebola/Documents/EbolaIP.pdf. Published June 2016. Accessed July 5, 2017.
23. U.S. Department of Health and Human Services, Office of Global Affairs. The Global Strategy of the U.S. Department of Health and Human Services. https://www.hhs.gov/sites/default/files/hhs-global -strategy.pdf. Accessed June 28, 2017.
24. U.S. Department of Health and Human Services. Mission and core values. Commissioned Corps of the U.S. Public Health Service website. https://www .usphs.gov/aboutus/mission.aspx. Updated February 3, 2014. Accessed June 28, 2017.
25. U.S. Department of Health and Human Services. Mission and Goals. National Institutes of Health website. https://www.nih.gov/about-nih/what-we -do/mission-goals. Accessed June 28, 2017.
The Ebola epidemic of 2014-2016 challenged many federal agencies to find creative ways to help address the vexing problems created by the spread of the disease.
The response from the U.S. and the global community took many forms: Not only was there a need for the typical medical care support, but also for basic public health systems to track the spread of disease, provide clean water, and dispose of infectious waste. Because no known preventive vaccines or therapeutics existed for those infected, the recognition of a research component to the response became abundantly clear as the epidemic continued. As a result, the National Institutes of Health (NIH) and the USPHS Commissioned Corps (Corps) serendipitously found themselves allied in a mutually beneficial relationship in the establishment of an Ebola clinical research program in West Africa.
This article describes the events that led to the NIH and Corps participation in the Ebola response, the roles filled by the Corps in supporting the NIH, and the lessons observed from that collaboration. Also presented are considerations regarding preparation of a clinical research response to future outbreaks.
NIH Clinical Research first Response
The 2014-2016 Ebola epidemic in West Africa demonstrated the need for federal agencies to reassess their capacity to respond to global threats to protect the health security of the U.S.1 The outbreak also challenged the U.S. government to mobilize unique resources that matched the need of this international (and domestic) response.
In 2014, President Barack Obama announced that the U.S. would launch a government response to the Ebola effort. Although a comprehensive research and development program already was in place to establish Ebola virus disease (EVD) countermeasures, no FDA-approved diagnostics, therapeutics, or preventive vaccines were readily available. Fortunately, FDA regulations regarding emergency use authorizations allowed for the use of several EVD diagnostics during this outbreak.2 However, the development of drugs and vaccines specific to Ebola had yet to make their way to phase 1 safety studies.
Two vaccine products went into phase 1 studies in the U.S. within months of the declaration of the emergency.3,4 Additionally, the NIH had organized a collaborative effort between the U.S. government and academic community to identify a research strategy for the evaluation of therapeutics.5 Regardless of the state of countermeasures and research proposals, the initial need was for disease control measures and care for Ebola patients. The CDC took the lead in working within the international community to establish an incident management system that could help the impacted countries enact mechanisms to bring the epidemic under control.6
As the epidemic progressed, leaders in the Corps and the NIH responded on pathways that eventually would intersect. One of the unfortunate outcomes of the early efforts of improperly protected health care providers was the unintentional transmission of Ebola.7 The Corps identified the need to provide high-level care to the health care worker community as one incentive to motivate health care workers to volunteer for hazardous duty inside Ebola treatment units (ETUs).8,9 Engulfed in the epidemic response, the U.S. government through the National Security Council and secretary of the Department of Health and Human Services (DHHS) evoked its statutory authority to deploy the Corps (42 U.S. Code 204a).
In the first week of October 2014, the Corps sent an advanced echelon team to assess the situation, partner with key host country and international stakeholders, and begin establishment of the U.S. government’s first ever ETU. With logistics, security, and resource support from the DoD and response coordination from the U.S. Agency for International Development, the Corps then deployed the first of four 70-person team rotations to staff the Monrovia Medical Unit (MMU), an ETU specifically dedicated to the treatment of Ebola-infected health care workers. At the time, it was the only ETU specifically dedicated to health care workers in all of Africa. The MMU operated until May 2015 and provided direct patient care for health care workers with Ebola, malaria, and other illnesses.8,10
In August 2014, representatives from the CDC met with Liberia’s Minister of Health and Social Welfare Walter T. Gwenigale, MD, to discuss the range of available options that could facilitate a better understanding of the prevention and treatment of the disease. This meeting resulted in a letter dated August 22, 2014, from Dr. Gwenigale to then DHHS Sylvia Burwell, requesting a research response. Secretary Burwell responded on October 2, 2014, describing the immediate dispatch of the deputy director for clinical research of the National Institute of Allergy and Infectious Diseases (NIAID) to Liberia to engage in initial discussions with the Liberian minister and other key Liberians involved in the response.
Representatives from the CDC and the commander of the Corps’ Ebola response (and acting deputy surgeon general) were included in those initial meetings, which led to a recognized need for a robust clinical research program of the highest ethical and scientific standards consistent with the expressed requirements of Liberia.11 A second and third trip to Liberia with larger U.S. teams resulted in an agreement signed on November 19, 2014 for the scientific investigation of strategies that tested interventions for treatment, control, and prevention of Ebola.12
The agreement led to the establishment of the Partnership for Research on Ebola Virus in Liberia (PREVAIL) to identify research priorities in a collaborative manner between Liberian and American scientists. The first protocol, a vaccine study, was launched in early February 2015.12 This monumental task involved the support of hundreds of Liberians and dozens of NIH staff who volunteered for rotations to Liberia. Of the 108 volunteers from within the NIH, 18 were PHS officers. Shortly after launching the vaccine study, the next priority was initiating the treatment study. This study was delayed primarily due to ZMapp (Mapp Biopharmaceatical, San Diego,CA) production limitations. ZMapp, a monoclonal antibody cocktail, was the first Ebola therapeutic product to be evaluated in a randomized trial.5,13
During the planning for the study, NIAID staff in Liberia met with Corps staff of the MMU to discuss the logistics associated with implementation of the ZMapp protocol at the MMU. During that meeting, the NIAID deputy director for clinical research expressed interest in obtaining Corps support from outside the NIH to sustain the research effort in West Africa. More specifically, additional pharmacy and laboratory staff were needed to augment NIH research operations. At the time, the MMU commander had recently transitioned from service as the acting surgeon general and was in a unique position to recommend additional Corps resources that could help in the research response.
The February 2015 discussion resulted in the establishment of an NIH/PHS research partnership that continues to exist. This new opportunity was not a significant stretch for the PHS as there was great interest from the Corps for responding to the Ebola crisis. The enthusiasm was consistent with the overall ethos of the Corps, which as a service was composed of highly qualified active-duty, deployable, uniformed, public health professionals who respond to public health crises at home and abroad. To date, 19 Corps officers from outside the NIH have deployed in support of the NIH Ebola clinical research program. An additional 18 Corps officers assigned within the NIH also volunteered for duty in West Africa. Of the 37 Corps officers supporting the NIH clinical research program, 7 served on more than 1 rotation.
Program Expansion
The Ebola clinical research program expanded over time from the initial PREVAIL vaccine study to include studies of therapeutic agents, natural history in Ebola survivors, and an additional vaccine study. The PHS officers have been integral in conducting these studies. The initial study implemented in Liberia, known as PREVAIL I, involved the evaluation of 2 vaccine strategies vs placebo.12,14 In addition to the NIH-based Corps officers supporting the study, the Readiness and Deployment Operations Group (RedDOG) initiated deployments for an additional 2 pharmacy and 7 laboratory officers to support this study. During the deployment, the pharmacists were asked to extend their reach to Sierra Leone and later to Guinea to help establish PREVAIL II, an evaluation of ZMapp in the treatment of Ebola.13 A total of 9 Corps pharmacists, 2 nurses, and 3 physicians deployed to Sierra Leone or Guinea to assist in the PREVAIL II study.
As the epidemic came to an end in Liberia in May 2015, the need for a long-term assessment of Ebola survivors was recognized, resulting in PREVAIL III.15 Noteworthy in the survivor study was an ophthalmic substudy led by a Corps officer assigned to the National Eye Institute.16,17 The survivor study also identified that the persistence of the Ebola virus was longer than previously known and that sexual transmission via semen from infected males remained a potential mode of transmission.18 To address the lingering viral load, a study of an antiviral drug was initiated in Liberia in the summer of 2016, PREVAIL IV.19
Four Corps pharmacists helped train Liberian pharmacists to establish and sustain this randomized, double-blind, placebo-controlled study. Most recently, Corps pharmacists were deployed to support the initiation of the Partnership for Research on Ebola Vaccines (PREVAC), a collaborative partnership with researchers from Liberia, Guinea, and Sierra Leone with cosponsors from the NIH, Institut national de la santé et de la recherche médicale (Inserm) in France, and the London School of Hygiene and Tropical Medicine in the United Kingdom.20
Deployment Procedures
Within a week of the February 2015 initial meeting in Liberia to establish the NIH/PHS collaboration, the NIH deployment team met by phone with the Corps’ RedDOG to discuss initial requirements (eg, number of officers needed, disciplines, time lines, and documentation needed for deployment). These initial discussions resulted in the establishment of more formal processes that evolved over time as the 2 organizations gained experience. Based on the identification of the numbers and types of officers needed, RedDOG used procedures similar to the process for staffing the MMU. A communication went out to the Corps seeking interested officers.
Deployment slots were filled based on the personal availability of the officer and coordination with their immediate supervisor and agency. Officers needed to meet medical clearance requirements and provide current health care provider licensure information. Additional training requirements needed to be completed (eg, U.S. State Department training and good clinical practice [GCP] if not already current). Corps officers also took part in the NIH orientation program for deploying personnel to familiarize them to the situation on the ground in West Africa and the specific clinical research protocols that they would encounter. Given that most of the Corps officers were coming from outside the NIH, the onboarding activities required significant attention to detail as procedures for arranging travel (eg, passport, visa, and airline reservations) and processes for reimbursement of travel/per diem pay differed from more traditional deployments directed through the Corps headquarters.
Commissioned Corps Roles in the Research Response
Whereas the establishment of the research program in Liberia was based primarily on relationships forged over a 2-month period by the NIAID deputy director for clinical research and staff, the extension of the research program into Sierra Leone (March 2015) and Guinea (June 2015) was on a substantially shorter time line. As a result, Corps officers were thrust into roles that immediately employed their leadership and diplomacy skills.
In Sierra Leone and Guinea, the NIAID deputy director for clinical research established initial relationships within the countries. However, Corps officers found themselves in regular interactions with regulators in the Ministry of Health to ensure that applications were complete and import permits for incoming shipments were cleared. Additionally, the research collaboration in Sierra Leone was coordinated through an investigator assigned to a military hospital converted into an ETU. The Corps officers were well suited to maintain and build on that relationship in expanding the protocol to other ETUs throughout Sierra Leone. A site established by the CDC within the Sierra Leone Ministry of Health coordinated ZMapp storage. The Corps officers formed working relationships with the CDC team to establish and improve cold-chain logistics and transportation of the ZMapp to the various ETUs around the country. Corps officers were integral in working with the in-country contract hiring agency. Activities included establishing criteria for clinical research positions, providing input on the interview of respective candidates, and training staff as the team formed. In Sierra Leone, local staff members were hired to work at specific facilities as research coordinators working with the health care delivery teams.
The U.S. team consisted of a physician, nurse research coordinator, and a pharmacist travelling to the sites with a logistics/operations staff member remaining in Freetown.
Fortunately, a Corps nurse on the team had been part of the initial MMU deployment and was trained to work in a special care unit at the NIH for patients with highly contagious infections. This practical experience was essential in the establishment of procedures in a hazardous environment for the administration of the IV ZMapp, monitoring of adverse effects (AEs), provision of medications to mitigate infusion-related reactions, and documentation of those AEs.
The U.S. research team regularly departed Freetown early in the morning 7 days a week with the various supplies needed as they visited up to 4 ETU sites to prepare the ZMapp at the site, await information on any AEs, and collect case report forms (Figures 1 and 2). The ETUs were spread out over a 90-mile radius and could be described as austere platforms for health care delivery.
An additional challenge was dealing with the multinational organizations that staffed the various ETUs. Relief organizations from Italy, the United Kingdom, China, as well as the Sierra Leone military provided the staffing for the 4 ETUs. Regardless of who operated the ETU, the concept of randomization to ZMapp or standard of care required significant tact and diplomacy in communicating the scientific necessity in order to appropriately answer the research question.
As the summer approached in Sierra Leone, the team worked through challenges in the IV administration of ZMapp as the protein structure of the monoclonal antibody had not previously been subjected to West African environmental extremes. A balance between speed of administration to prevent protein aggregation in the heat as opposed to the risk of infusion reactions from a foreign protein required the team to communicate frequently with, the manufacturer of ZMapp, to establish realistic infusion rate tables. Additionally, as the various deployment teams rotated in and out, procedures for establishing continuity of research operations were enacted and improved on with each rotation. Good documentation practices to adequately collect all required study information (eg, recording AEs, deviations, and signatures on various forms) proved critical to continuity of research operations.
In Guinea, not only was there the new wrinkle of working within a country where the primary language was French, but also a French cosponsor, Inserm. The NIAID clinical director capitalized on the research infrastructure established for a recently completed Inserm study of favipiravir in the treatment of Ebola to extend the ZMapp study to Guinea. Fortunately, many of the Inserm staff were bilingual and readily responded to the NIH training on the requirements of the ZMapp protocol. However, procedures for cold-chain storage and transportation needed to be established. In Guinea, the PHS officers were key in establishing access and temperature monitoring procedures for a secure room inside the U.S. embassy. The issues associated with cold-chain procedures in the infrastructure-limited environments of West Africa are substantial and warrant consideration of a stand-alone paper. Corps officers also took part in weekly country-focused team meetings with embassy staff to describe progress with the ZMapp study.
As the epidemic waned and NIH transitioned to the survivor and viral persistence studies, the operational tempo changed to allow Corps officers to take part in more definitive capacity building efforts. An initial PHS volunteer from the FDA accepted a position within NIAID as a clinical research oversight manager for pharmacy operations. This individual deployed on numerous occasions to the 3 affected West African countries to further establish cold-chain processes for pharmaceuticals and biologics. He also worked with a multidisciplinary team to renovate a clinical research facility in a rural setting in Guinea. In Liberia, he coordinated an effort with other Corps officers to provide educational seminars on clinical research principles and drug-specific topics with the University of Liberia School of Pharmacy.
Challenges
In each instance, the partnership experience was not without a few problems. The match of skills between the officers who wanted to help and those needed for the research program did not always coincide. While the Corps has more than 1,200 pharmacy officers on active duty, only a fraction of those have experience conducting FDA-regulated clinical research.
Communication problems and time pressures were also constant companions to both the Corps and the NIH. The Corps was going through the largest international deployment in its history to staff multiple missions (including the primary MMU mission in Liberia). The addition of the NIH partnership, while consistent with the MMU staffing mission, provided even more work for a very limited resource. Communicating to the many Corps officers who wanted to volunteer and keeping deployment time lines on track were a challenge. Complicating the matter was the addition of stray e-mails from well-intentioned NIH and Corps staff who communicated directly with colleagues to encourage participation, not fully understanding the policies and protocol governing the deployment process.
Time was always an issue as the rotation schedules were relatively short and the number of activities to make an officer deployment ready were numerous. Obtaining official passports and visas was a challenge as that activity required coordination with the U.S. Department of State. Airline schedules changed with little or no notice, complicating deployments and returns. As the NIH added additional research studies for which support was required, time lines for studies to start became difficult to predict with certainty due to factors outside the control of the NIH. Recently, additional security training requirements for government workers traveling abroad were instituted, further complicating the process of deploying an officer.
The Corps officers taking part in this research response (which was not consistent with customary deployments from Corps headquarters) necessarily were volunteers from full-time assignments within DHHS, and as such, required the permission of their supervisory chain to volunteer. Regardless of this limitation, there was widespread support for these additional and specific research deployments. Although the use of short-term rotations was not ideal, in the end, the rotation plans worked, and the NIH was able to fulfill its research mission with the support of the Corps.
Lessons Learned/Preparing for the Future
Many lessons have been learned and continue to be learned throughout this research response and NIH/Corps partnership. Effective and frequent communication between the organization requesting Corps officers and the Corps headquarters is crucial. In the initial deployments, officers were deployed from the FDA with the assumption that they would be familiar with FDA-regulated clinical research. This was not always the case. The NIH and Corps headquarters later collaborated to develop a survey to send to Corps officers that was used to identify specific skill sets needed by officers who would be deploying to conduct clinical research. NIH personnel prescreened survey responses to identify and prioritize officers for deployment consideration by the deployment authority. This process resulted in the selection of officers who generally needed less training and guidance.
Effective training in clinical research principles for deployed officers and other staff needs to be developed and made available to all deploying individuals. All clinical research staff are required to have training on GCP, but most GCP training programs focus primarily on the ethical principles of research as outlined by the Declaration of Helsinki, Nuremberg Code, and other documents. Few GCP training programs present adequate information on the hands-on conduct of clinical research, especially research regulated by the FDA and other government bodies and therefore subject to certain strict requirements. Examples of crucial but often overlooked topics are source document retention, good documentation practices, cold-chain principles, and other issues related to the creation and retention of adequate trial records.21
The handoff between returning and deploying officers is crucial. Due to various issues with changing time lines, flights, and administrative processes, it is imperative to plan adequate overlap between returning and deploying officers. Delays in obtaining passports or visas, flight cancellations, and other unforeseen issues may unexpectedly shorten any planned overlap periods. A full workweek is desirable for overlap so that the new officer may experience tasks that occur throughout the week, be introduced to the various team members, and have help if unexpected events occur. A regular staff member should check periodically that proper procedures are being followed, as some information may be missed during each handoff, and consecutive unchecked handoffs could result in large deviations of important procedures. Onboarding and offboarding checklists should be developed and updated regularly to guide the handoff process.
On a larger scale, the respective agencies and other stakeholders involved in planning clinical research for public health emergencies need to be included in regular tabletop training exercises to better understand how to coordinate a response when needed. Additionally, although many of the Corps officers who took part in this deployment served as mentors for others preparing for deployment, establishing a formal roster of experienced officers to support specific roles of this type of response would help serve as a resource center for future deployments. Finally, coordination between any operating division (or agency) and the Corps should be through the established Corps command infrastructure to eliminate miscommunication and complicating deployment processes.22
Conclusion
The increasing connectedness of this world, as demonstrated by the Ebola epidemic, requires that the HHS engage globally to provide international leadership and technical expertise in science, policy, and programs and work in concert with interagency partners.23 The missions of the PHS and NIH intersected in a synergistic manner in the research response to the Ebola epidemic of 2014-2016. The PHS Corps mission includes to “protect, promote, and advance the health and safety of the Nation...through rapid and effective response…and advancement of public health science.”24 The Corps mission directly supported the NIH mission to seek fundamental knowledge about the nature and behavior of living systems and the application of that knowledge to enhance health, lengthen life, and reduce illness and disability.25
The scope and scale of DHHS’s response to the Ebola epidemic was unprecedented. The NIH research program, although successful and an important component, was but a small part in bringing the Ebola crisis to an end. The CDC (including the many Corps officers assigned to that agency) worked successfully with the international community and the host countries to bring the disease under control. The Biological Advanced Research and Development Authority provided expert project management, making vaccines and therapeutics available for research.
The DoD was a partner in the development of countermeasures and phase 1 clinical research programs as well as establishing laboratory facilities in Liberia. The Department of State facilitated the many interactions required for the mobilization of resources into West Africa. The collective efforts of the U.S. government contributed immensely to the protection of U.S. borders and to the successful resolution of the Ebola outbreak of 2014-2016.
The Ebola epidemic of 2014-2016 challenged many federal agencies to find creative ways to help address the vexing problems created by the spread of the disease.
The response from the U.S. and the global community took many forms: Not only was there a need for the typical medical care support, but also for basic public health systems to track the spread of disease, provide clean water, and dispose of infectious waste. Because no known preventive vaccines or therapeutics existed for those infected, the recognition of a research component to the response became abundantly clear as the epidemic continued. As a result, the National Institutes of Health (NIH) and the USPHS Commissioned Corps (Corps) serendipitously found themselves allied in a mutually beneficial relationship in the establishment of an Ebola clinical research program in West Africa.
This article describes the events that led to the NIH and Corps participation in the Ebola response, the roles filled by the Corps in supporting the NIH, and the lessons observed from that collaboration. Also presented are considerations regarding preparation of a clinical research response to future outbreaks.
NIH Clinical Research first Response
The 2014-2016 Ebola epidemic in West Africa demonstrated the need for federal agencies to reassess their capacity to respond to global threats to protect the health security of the U.S.1 The outbreak also challenged the U.S. government to mobilize unique resources that matched the need of this international (and domestic) response.
In 2014, President Barack Obama announced that the U.S. would launch a government response to the Ebola effort. Although a comprehensive research and development program already was in place to establish Ebola virus disease (EVD) countermeasures, no FDA-approved diagnostics, therapeutics, or preventive vaccines were readily available. Fortunately, FDA regulations regarding emergency use authorizations allowed for the use of several EVD diagnostics during this outbreak.2 However, the development of drugs and vaccines specific to Ebola had yet to make their way to phase 1 safety studies.
Two vaccine products went into phase 1 studies in the U.S. within months of the declaration of the emergency.3,4 Additionally, the NIH had organized a collaborative effort between the U.S. government and academic community to identify a research strategy for the evaluation of therapeutics.5 Regardless of the state of countermeasures and research proposals, the initial need was for disease control measures and care for Ebola patients. The CDC took the lead in working within the international community to establish an incident management system that could help the impacted countries enact mechanisms to bring the epidemic under control.6
As the epidemic progressed, leaders in the Corps and the NIH responded on pathways that eventually would intersect. One of the unfortunate outcomes of the early efforts of improperly protected health care providers was the unintentional transmission of Ebola.7 The Corps identified the need to provide high-level care to the health care worker community as one incentive to motivate health care workers to volunteer for hazardous duty inside Ebola treatment units (ETUs).8,9 Engulfed in the epidemic response, the U.S. government through the National Security Council and secretary of the Department of Health and Human Services (DHHS) evoked its statutory authority to deploy the Corps (42 U.S. Code 204a).
In the first week of October 2014, the Corps sent an advanced echelon team to assess the situation, partner with key host country and international stakeholders, and begin establishment of the U.S. government’s first ever ETU. With logistics, security, and resource support from the DoD and response coordination from the U.S. Agency for International Development, the Corps then deployed the first of four 70-person team rotations to staff the Monrovia Medical Unit (MMU), an ETU specifically dedicated to the treatment of Ebola-infected health care workers. At the time, it was the only ETU specifically dedicated to health care workers in all of Africa. The MMU operated until May 2015 and provided direct patient care for health care workers with Ebola, malaria, and other illnesses.8,10
In August 2014, representatives from the CDC met with Liberia’s Minister of Health and Social Welfare Walter T. Gwenigale, MD, to discuss the range of available options that could facilitate a better understanding of the prevention and treatment of the disease. This meeting resulted in a letter dated August 22, 2014, from Dr. Gwenigale to then DHHS Sylvia Burwell, requesting a research response. Secretary Burwell responded on October 2, 2014, describing the immediate dispatch of the deputy director for clinical research of the National Institute of Allergy and Infectious Diseases (NIAID) to Liberia to engage in initial discussions with the Liberian minister and other key Liberians involved in the response.
Representatives from the CDC and the commander of the Corps’ Ebola response (and acting deputy surgeon general) were included in those initial meetings, which led to a recognized need for a robust clinical research program of the highest ethical and scientific standards consistent with the expressed requirements of Liberia.11 A second and third trip to Liberia with larger U.S. teams resulted in an agreement signed on November 19, 2014 for the scientific investigation of strategies that tested interventions for treatment, control, and prevention of Ebola.12
The agreement led to the establishment of the Partnership for Research on Ebola Virus in Liberia (PREVAIL) to identify research priorities in a collaborative manner between Liberian and American scientists. The first protocol, a vaccine study, was launched in early February 2015.12 This monumental task involved the support of hundreds of Liberians and dozens of NIH staff who volunteered for rotations to Liberia. Of the 108 volunteers from within the NIH, 18 were PHS officers. Shortly after launching the vaccine study, the next priority was initiating the treatment study. This study was delayed primarily due to ZMapp (Mapp Biopharmaceatical, San Diego,CA) production limitations. ZMapp, a monoclonal antibody cocktail, was the first Ebola therapeutic product to be evaluated in a randomized trial.5,13
During the planning for the study, NIAID staff in Liberia met with Corps staff of the MMU to discuss the logistics associated with implementation of the ZMapp protocol at the MMU. During that meeting, the NIAID deputy director for clinical research expressed interest in obtaining Corps support from outside the NIH to sustain the research effort in West Africa. More specifically, additional pharmacy and laboratory staff were needed to augment NIH research operations. At the time, the MMU commander had recently transitioned from service as the acting surgeon general and was in a unique position to recommend additional Corps resources that could help in the research response.
The February 2015 discussion resulted in the establishment of an NIH/PHS research partnership that continues to exist. This new opportunity was not a significant stretch for the PHS as there was great interest from the Corps for responding to the Ebola crisis. The enthusiasm was consistent with the overall ethos of the Corps, which as a service was composed of highly qualified active-duty, deployable, uniformed, public health professionals who respond to public health crises at home and abroad. To date, 19 Corps officers from outside the NIH have deployed in support of the NIH Ebola clinical research program. An additional 18 Corps officers assigned within the NIH also volunteered for duty in West Africa. Of the 37 Corps officers supporting the NIH clinical research program, 7 served on more than 1 rotation.
Program Expansion
The Ebola clinical research program expanded over time from the initial PREVAIL vaccine study to include studies of therapeutic agents, natural history in Ebola survivors, and an additional vaccine study. The PHS officers have been integral in conducting these studies. The initial study implemented in Liberia, known as PREVAIL I, involved the evaluation of 2 vaccine strategies vs placebo.12,14 In addition to the NIH-based Corps officers supporting the study, the Readiness and Deployment Operations Group (RedDOG) initiated deployments for an additional 2 pharmacy and 7 laboratory officers to support this study. During the deployment, the pharmacists were asked to extend their reach to Sierra Leone and later to Guinea to help establish PREVAIL II, an evaluation of ZMapp in the treatment of Ebola.13 A total of 9 Corps pharmacists, 2 nurses, and 3 physicians deployed to Sierra Leone or Guinea to assist in the PREVAIL II study.
As the epidemic came to an end in Liberia in May 2015, the need for a long-term assessment of Ebola survivors was recognized, resulting in PREVAIL III.15 Noteworthy in the survivor study was an ophthalmic substudy led by a Corps officer assigned to the National Eye Institute.16,17 The survivor study also identified that the persistence of the Ebola virus was longer than previously known and that sexual transmission via semen from infected males remained a potential mode of transmission.18 To address the lingering viral load, a study of an antiviral drug was initiated in Liberia in the summer of 2016, PREVAIL IV.19
Four Corps pharmacists helped train Liberian pharmacists to establish and sustain this randomized, double-blind, placebo-controlled study. Most recently, Corps pharmacists were deployed to support the initiation of the Partnership for Research on Ebola Vaccines (PREVAC), a collaborative partnership with researchers from Liberia, Guinea, and Sierra Leone with cosponsors from the NIH, Institut national de la santé et de la recherche médicale (Inserm) in France, and the London School of Hygiene and Tropical Medicine in the United Kingdom.20
Deployment Procedures
Within a week of the February 2015 initial meeting in Liberia to establish the NIH/PHS collaboration, the NIH deployment team met by phone with the Corps’ RedDOG to discuss initial requirements (eg, number of officers needed, disciplines, time lines, and documentation needed for deployment). These initial discussions resulted in the establishment of more formal processes that evolved over time as the 2 organizations gained experience. Based on the identification of the numbers and types of officers needed, RedDOG used procedures similar to the process for staffing the MMU. A communication went out to the Corps seeking interested officers.
Deployment slots were filled based on the personal availability of the officer and coordination with their immediate supervisor and agency. Officers needed to meet medical clearance requirements and provide current health care provider licensure information. Additional training requirements needed to be completed (eg, U.S. State Department training and good clinical practice [GCP] if not already current). Corps officers also took part in the NIH orientation program for deploying personnel to familiarize them to the situation on the ground in West Africa and the specific clinical research protocols that they would encounter. Given that most of the Corps officers were coming from outside the NIH, the onboarding activities required significant attention to detail as procedures for arranging travel (eg, passport, visa, and airline reservations) and processes for reimbursement of travel/per diem pay differed from more traditional deployments directed through the Corps headquarters.
Commissioned Corps Roles in the Research Response
Whereas the establishment of the research program in Liberia was based primarily on relationships forged over a 2-month period by the NIAID deputy director for clinical research and staff, the extension of the research program into Sierra Leone (March 2015) and Guinea (June 2015) was on a substantially shorter time line. As a result, Corps officers were thrust into roles that immediately employed their leadership and diplomacy skills.
In Sierra Leone and Guinea, the NIAID deputy director for clinical research established initial relationships within the countries. However, Corps officers found themselves in regular interactions with regulators in the Ministry of Health to ensure that applications were complete and import permits for incoming shipments were cleared. Additionally, the research collaboration in Sierra Leone was coordinated through an investigator assigned to a military hospital converted into an ETU. The Corps officers were well suited to maintain and build on that relationship in expanding the protocol to other ETUs throughout Sierra Leone. A site established by the CDC within the Sierra Leone Ministry of Health coordinated ZMapp storage. The Corps officers formed working relationships with the CDC team to establish and improve cold-chain logistics and transportation of the ZMapp to the various ETUs around the country. Corps officers were integral in working with the in-country contract hiring agency. Activities included establishing criteria for clinical research positions, providing input on the interview of respective candidates, and training staff as the team formed. In Sierra Leone, local staff members were hired to work at specific facilities as research coordinators working with the health care delivery teams.
The U.S. team consisted of a physician, nurse research coordinator, and a pharmacist travelling to the sites with a logistics/operations staff member remaining in Freetown.
Fortunately, a Corps nurse on the team had been part of the initial MMU deployment and was trained to work in a special care unit at the NIH for patients with highly contagious infections. This practical experience was essential in the establishment of procedures in a hazardous environment for the administration of the IV ZMapp, monitoring of adverse effects (AEs), provision of medications to mitigate infusion-related reactions, and documentation of those AEs.
The U.S. research team regularly departed Freetown early in the morning 7 days a week with the various supplies needed as they visited up to 4 ETU sites to prepare the ZMapp at the site, await information on any AEs, and collect case report forms (Figures 1 and 2). The ETUs were spread out over a 90-mile radius and could be described as austere platforms for health care delivery.
An additional challenge was dealing with the multinational organizations that staffed the various ETUs. Relief organizations from Italy, the United Kingdom, China, as well as the Sierra Leone military provided the staffing for the 4 ETUs. Regardless of who operated the ETU, the concept of randomization to ZMapp or standard of care required significant tact and diplomacy in communicating the scientific necessity in order to appropriately answer the research question.
As the summer approached in Sierra Leone, the team worked through challenges in the IV administration of ZMapp as the protein structure of the monoclonal antibody had not previously been subjected to West African environmental extremes. A balance between speed of administration to prevent protein aggregation in the heat as opposed to the risk of infusion reactions from a foreign protein required the team to communicate frequently with, the manufacturer of ZMapp, to establish realistic infusion rate tables. Additionally, as the various deployment teams rotated in and out, procedures for establishing continuity of research operations were enacted and improved on with each rotation. Good documentation practices to adequately collect all required study information (eg, recording AEs, deviations, and signatures on various forms) proved critical to continuity of research operations.
In Guinea, not only was there the new wrinkle of working within a country where the primary language was French, but also a French cosponsor, Inserm. The NIAID clinical director capitalized on the research infrastructure established for a recently completed Inserm study of favipiravir in the treatment of Ebola to extend the ZMapp study to Guinea. Fortunately, many of the Inserm staff were bilingual and readily responded to the NIH training on the requirements of the ZMapp protocol. However, procedures for cold-chain storage and transportation needed to be established. In Guinea, the PHS officers were key in establishing access and temperature monitoring procedures for a secure room inside the U.S. embassy. The issues associated with cold-chain procedures in the infrastructure-limited environments of West Africa are substantial and warrant consideration of a stand-alone paper. Corps officers also took part in weekly country-focused team meetings with embassy staff to describe progress with the ZMapp study.
As the epidemic waned and NIH transitioned to the survivor and viral persistence studies, the operational tempo changed to allow Corps officers to take part in more definitive capacity building efforts. An initial PHS volunteer from the FDA accepted a position within NIAID as a clinical research oversight manager for pharmacy operations. This individual deployed on numerous occasions to the 3 affected West African countries to further establish cold-chain processes for pharmaceuticals and biologics. He also worked with a multidisciplinary team to renovate a clinical research facility in a rural setting in Guinea. In Liberia, he coordinated an effort with other Corps officers to provide educational seminars on clinical research principles and drug-specific topics with the University of Liberia School of Pharmacy.
Challenges
In each instance, the partnership experience was not without a few problems. The match of skills between the officers who wanted to help and those needed for the research program did not always coincide. While the Corps has more than 1,200 pharmacy officers on active duty, only a fraction of those have experience conducting FDA-regulated clinical research.
Communication problems and time pressures were also constant companions to both the Corps and the NIH. The Corps was going through the largest international deployment in its history to staff multiple missions (including the primary MMU mission in Liberia). The addition of the NIH partnership, while consistent with the MMU staffing mission, provided even more work for a very limited resource. Communicating to the many Corps officers who wanted to volunteer and keeping deployment time lines on track were a challenge. Complicating the matter was the addition of stray e-mails from well-intentioned NIH and Corps staff who communicated directly with colleagues to encourage participation, not fully understanding the policies and protocol governing the deployment process.
Time was always an issue as the rotation schedules were relatively short and the number of activities to make an officer deployment ready were numerous. Obtaining official passports and visas was a challenge as that activity required coordination with the U.S. Department of State. Airline schedules changed with little or no notice, complicating deployments and returns. As the NIH added additional research studies for which support was required, time lines for studies to start became difficult to predict with certainty due to factors outside the control of the NIH. Recently, additional security training requirements for government workers traveling abroad were instituted, further complicating the process of deploying an officer.
The Corps officers taking part in this research response (which was not consistent with customary deployments from Corps headquarters) necessarily were volunteers from full-time assignments within DHHS, and as such, required the permission of their supervisory chain to volunteer. Regardless of this limitation, there was widespread support for these additional and specific research deployments. Although the use of short-term rotations was not ideal, in the end, the rotation plans worked, and the NIH was able to fulfill its research mission with the support of the Corps.
Lessons Learned/Preparing for the Future
Many lessons have been learned and continue to be learned throughout this research response and NIH/Corps partnership. Effective and frequent communication between the organization requesting Corps officers and the Corps headquarters is crucial. In the initial deployments, officers were deployed from the FDA with the assumption that they would be familiar with FDA-regulated clinical research. This was not always the case. The NIH and Corps headquarters later collaborated to develop a survey to send to Corps officers that was used to identify specific skill sets needed by officers who would be deploying to conduct clinical research. NIH personnel prescreened survey responses to identify and prioritize officers for deployment consideration by the deployment authority. This process resulted in the selection of officers who generally needed less training and guidance.
Effective training in clinical research principles for deployed officers and other staff needs to be developed and made available to all deploying individuals. All clinical research staff are required to have training on GCP, but most GCP training programs focus primarily on the ethical principles of research as outlined by the Declaration of Helsinki, Nuremberg Code, and other documents. Few GCP training programs present adequate information on the hands-on conduct of clinical research, especially research regulated by the FDA and other government bodies and therefore subject to certain strict requirements. Examples of crucial but often overlooked topics are source document retention, good documentation practices, cold-chain principles, and other issues related to the creation and retention of adequate trial records.21
The handoff between returning and deploying officers is crucial. Due to various issues with changing time lines, flights, and administrative processes, it is imperative to plan adequate overlap between returning and deploying officers. Delays in obtaining passports or visas, flight cancellations, and other unforeseen issues may unexpectedly shorten any planned overlap periods. A full workweek is desirable for overlap so that the new officer may experience tasks that occur throughout the week, be introduced to the various team members, and have help if unexpected events occur. A regular staff member should check periodically that proper procedures are being followed, as some information may be missed during each handoff, and consecutive unchecked handoffs could result in large deviations of important procedures. Onboarding and offboarding checklists should be developed and updated regularly to guide the handoff process.
On a larger scale, the respective agencies and other stakeholders involved in planning clinical research for public health emergencies need to be included in regular tabletop training exercises to better understand how to coordinate a response when needed. Additionally, although many of the Corps officers who took part in this deployment served as mentors for others preparing for deployment, establishing a formal roster of experienced officers to support specific roles of this type of response would help serve as a resource center for future deployments. Finally, coordination between any operating division (or agency) and the Corps should be through the established Corps command infrastructure to eliminate miscommunication and complicating deployment processes.22
Conclusion
The increasing connectedness of this world, as demonstrated by the Ebola epidemic, requires that the HHS engage globally to provide international leadership and technical expertise in science, policy, and programs and work in concert with interagency partners.23 The missions of the PHS and NIH intersected in a synergistic manner in the research response to the Ebola epidemic of 2014-2016. The PHS Corps mission includes to “protect, promote, and advance the health and safety of the Nation...through rapid and effective response…and advancement of public health science.”24 The Corps mission directly supported the NIH mission to seek fundamental knowledge about the nature and behavior of living systems and the application of that knowledge to enhance health, lengthen life, and reduce illness and disability.25
The scope and scale of DHHS’s response to the Ebola epidemic was unprecedented. The NIH research program, although successful and an important component, was but a small part in bringing the Ebola crisis to an end. The CDC (including the many Corps officers assigned to that agency) worked successfully with the international community and the host countries to bring the disease under control. The Biological Advanced Research and Development Authority provided expert project management, making vaccines and therapeutics available for research.
The DoD was a partner in the development of countermeasures and phase 1 clinical research programs as well as establishing laboratory facilities in Liberia. The Department of State facilitated the many interactions required for the mobilization of resources into West Africa. The collective efforts of the U.S. government contributed immensely to the protection of U.S. borders and to the successful resolution of the Ebola outbreak of 2014-2016.
1. Bell BP, Damon IK, Jernigan DB, et al. Overview, control strategies, and lessons learned in the CDC response to the 2014-2016 Ebola epidemic. MMWR. 2016;65(suppl 3):4-11.
2. U.S. Food and Drug Administration. Emergency use authorization. https://www.fda.gov/EmergencyPreparedness/Counterterrorism/MedicalCountermeasures/MCMLegalRegulatoryandPolicyFramework/ucm182568.htm#ebola. Updated June 29, 2017. Accessed June 30, 2017.
3. Regules JA, Beigel JH, Paolino KM, et al; for the rVSVΔG-ZEBOV-GP Study Group. A recombinant vesicular stomatitis virus Ebola vaccine. N Engl J Med. 2017;376(4):330-341.
4. Tapia MD, Sow SO, Lyke KE, et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2016;16(1):31-42.
5. Dodd LE, Proschan MA, Neuhaus J, et al. Design of a randomized controlled trial for Ebola virus disease medical countermeasures: PREVAIL II, the Ebola MCM Study. J Infect Dis. 2016;213(12):1906-1913.
6. Brooks JC, Pinto M, Gill A, et al. Incident management systems and building emergency management capacity during the 2014-2016 Ebola epidemic—Liberia, Sierra Leone, and Guinea. MMWR. 2016;65(suppl 3):28-34.
7. Evans DK, Goldstein M, Popova A. Health-care worker mortality and the legacy of the Ebola epidemic. Lancet Glob Health. 2015;3(8):e439-e440.
8. Lushniak BD. The hope multipliers: the U.S. Public Health Service in Monrovia. Public Health Rep. 2015;130(6):562-565.
9. Lushniak BD. Update on the U.S. public health response to the Ebola outbreak. Public Health Rep. 2015;130(2):118-120.
10. Brown-Stephenson J. United States Public Health Service nurses: deployment in global crisis. Online J Issues Nurs. 2017;22(1):6.
11. Lane HC, Marston HD, Fauci AS. Conducting clinical trials in outbreak settings: points to consider. Clin Trials. 2016;13(1):92-95.
12. Kennedy SB, Neaton JD, Lane HC, et al. Implementation of an Ebola virus disease vaccine clinical trial during the Ebola epidemic in Liberia: design, procedures, and challenges. Clin Trials. 2016;13(1):49-56.
13. Davey RT. PREVAIL II: a randomized controlled trial of ZMappTM in acute Ebola virus infection. Paper presented at: Conference on Retroviruses and Opportunistic Infections; February 22-25, 2016; Boston, Massachusetts.
14. Doe-Anderson J, Baseler B, Driscoll P, et al. Beating the odds: successful establishment of a phase II/III clinical research trial in resource-poor Liberia during the largest-ever Ebola outbreak. Contemp Clin Trials Commun. 2016;4:68-73.
15. U.S. National Institutes of Health Clinical Center. Ebola virus disease survivors: clinical and immunologic follow-up. https://clinicaltrials.gov/ct2/show/NCT02431923. Updated June 30, 2017. Accessed July 5, 2017.
16. Jampol LM, Ferris FL III, Bishop RJ. Ebola and the eye. JAMA Ophthalmol. 2015;133(10):1105-1106.
17. Chertow DS, Nath A, Suffredini AF, et al. Severe meningoencephalitis in a case of Ebola virus disease: a case report. Ann Intern Med. 2016;165(4):301-304.
18. Pettitt J, Higgs ES, Fallah MP, Hensley LE. Assessment and optimization of the GeneXpert diagnostic platform for detection of Ebola virus RNA in seminal fluid. J Infect Dis. 2017;215(4):547-553.
19. U.S. National Institutes of Health Clinical Center. GS-5734 to assess the antiviral activity, longer-term clearance of Ebola virus, and safety in male Ebola survivors with evidence of Ebola virus persistence in semen. https://clinicaltrials.gov/show/NCT02818582. Updated June 30, 2017. Accessed July 5, 2017.
20. U.S. National Institutes of Health Clinical Center. Partnership for Research on Ebola VACcinations (PREVAC). https://clinicaltrials.gov/show/NCT02876328. Updated June 28, 2017. Accessed July 5, 2017.
21. Kirchoff MC, Pierson JF. Considerations for use of investigational drugs in public health emergencies. Ther Innov Regul Sci. 2017;51(2):146-152.
22. U.S. Department of Health and Human Services, Office of the Assistant Secretary for Preparedness and Response. U.S. Department of Health and Human Services Ebola response improvement plan (based on lessons learned from the 2014-2016 Ebola epidemic). https://www.phe.gov/Preparedness/respond ers/ebola/Documents/EbolaIP.pdf. Published June 2016. Accessed July 5, 2017.
23. U.S. Department of Health and Human Services, Office of Global Affairs. The Global Strategy of the U.S. Department of Health and Human Services. https://www.hhs.gov/sites/default/files/hhs-global -strategy.pdf. Accessed June 28, 2017.
24. U.S. Department of Health and Human Services. Mission and core values. Commissioned Corps of the U.S. Public Health Service website. https://www .usphs.gov/aboutus/mission.aspx. Updated February 3, 2014. Accessed June 28, 2017.
25. U.S. Department of Health and Human Services. Mission and Goals. National Institutes of Health website. https://www.nih.gov/about-nih/what-we -do/mission-goals. Accessed June 28, 2017.
1. Bell BP, Damon IK, Jernigan DB, et al. Overview, control strategies, and lessons learned in the CDC response to the 2014-2016 Ebola epidemic. MMWR. 2016;65(suppl 3):4-11.
2. U.S. Food and Drug Administration. Emergency use authorization. https://www.fda.gov/EmergencyPreparedness/Counterterrorism/MedicalCountermeasures/MCMLegalRegulatoryandPolicyFramework/ucm182568.htm#ebola. Updated June 29, 2017. Accessed June 30, 2017.
3. Regules JA, Beigel JH, Paolino KM, et al; for the rVSVΔG-ZEBOV-GP Study Group. A recombinant vesicular stomatitis virus Ebola vaccine. N Engl J Med. 2017;376(4):330-341.
4. Tapia MD, Sow SO, Lyke KE, et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2016;16(1):31-42.
5. Dodd LE, Proschan MA, Neuhaus J, et al. Design of a randomized controlled trial for Ebola virus disease medical countermeasures: PREVAIL II, the Ebola MCM Study. J Infect Dis. 2016;213(12):1906-1913.
6. Brooks JC, Pinto M, Gill A, et al. Incident management systems and building emergency management capacity during the 2014-2016 Ebola epidemic—Liberia, Sierra Leone, and Guinea. MMWR. 2016;65(suppl 3):28-34.
7. Evans DK, Goldstein M, Popova A. Health-care worker mortality and the legacy of the Ebola epidemic. Lancet Glob Health. 2015;3(8):e439-e440.
8. Lushniak BD. The hope multipliers: the U.S. Public Health Service in Monrovia. Public Health Rep. 2015;130(6):562-565.
9. Lushniak BD. Update on the U.S. public health response to the Ebola outbreak. Public Health Rep. 2015;130(2):118-120.
10. Brown-Stephenson J. United States Public Health Service nurses: deployment in global crisis. Online J Issues Nurs. 2017;22(1):6.
11. Lane HC, Marston HD, Fauci AS. Conducting clinical trials in outbreak settings: points to consider. Clin Trials. 2016;13(1):92-95.
12. Kennedy SB, Neaton JD, Lane HC, et al. Implementation of an Ebola virus disease vaccine clinical trial during the Ebola epidemic in Liberia: design, procedures, and challenges. Clin Trials. 2016;13(1):49-56.
13. Davey RT. PREVAIL II: a randomized controlled trial of ZMappTM in acute Ebola virus infection. Paper presented at: Conference on Retroviruses and Opportunistic Infections; February 22-25, 2016; Boston, Massachusetts.
14. Doe-Anderson J, Baseler B, Driscoll P, et al. Beating the odds: successful establishment of a phase II/III clinical research trial in resource-poor Liberia during the largest-ever Ebola outbreak. Contemp Clin Trials Commun. 2016;4:68-73.
15. U.S. National Institutes of Health Clinical Center. Ebola virus disease survivors: clinical and immunologic follow-up. https://clinicaltrials.gov/ct2/show/NCT02431923. Updated June 30, 2017. Accessed July 5, 2017.
16. Jampol LM, Ferris FL III, Bishop RJ. Ebola and the eye. JAMA Ophthalmol. 2015;133(10):1105-1106.
17. Chertow DS, Nath A, Suffredini AF, et al. Severe meningoencephalitis in a case of Ebola virus disease: a case report. Ann Intern Med. 2016;165(4):301-304.
18. Pettitt J, Higgs ES, Fallah MP, Hensley LE. Assessment and optimization of the GeneXpert diagnostic platform for detection of Ebola virus RNA in seminal fluid. J Infect Dis. 2017;215(4):547-553.
19. U.S. National Institutes of Health Clinical Center. GS-5734 to assess the antiviral activity, longer-term clearance of Ebola virus, and safety in male Ebola survivors with evidence of Ebola virus persistence in semen. https://clinicaltrials.gov/show/NCT02818582. Updated June 30, 2017. Accessed July 5, 2017.
20. U.S. National Institutes of Health Clinical Center. Partnership for Research on Ebola VACcinations (PREVAC). https://clinicaltrials.gov/show/NCT02876328. Updated June 28, 2017. Accessed July 5, 2017.
21. Kirchoff MC, Pierson JF. Considerations for use of investigational drugs in public health emergencies. Ther Innov Regul Sci. 2017;51(2):146-152.
22. U.S. Department of Health and Human Services, Office of the Assistant Secretary for Preparedness and Response. U.S. Department of Health and Human Services Ebola response improvement plan (based on lessons learned from the 2014-2016 Ebola epidemic). https://www.phe.gov/Preparedness/respond ers/ebola/Documents/EbolaIP.pdf. Published June 2016. Accessed July 5, 2017.
23. U.S. Department of Health and Human Services, Office of Global Affairs. The Global Strategy of the U.S. Department of Health and Human Services. https://www.hhs.gov/sites/default/files/hhs-global -strategy.pdf. Accessed June 28, 2017.
24. U.S. Department of Health and Human Services. Mission and core values. Commissioned Corps of the U.S. Public Health Service website. https://www .usphs.gov/aboutus/mission.aspx. Updated February 3, 2014. Accessed June 28, 2017.
25. U.S. Department of Health and Human Services. Mission and Goals. National Institutes of Health website. https://www.nih.gov/about-nih/what-we -do/mission-goals. Accessed June 28, 2017.

VIDEO: What role does autoimmune dysfunction play in schizophrenia?
SAN DIEGO – Researchers are looking at the potential role that autoimmune reactions, such as inflammation, play in the pathogenesis of schizophrenia, both with and without comorbid substance use.
“What we see is evidence of autoimmune involvement through the lifespan in schizophrenia,” explained Brian Miller, MD, PhD, of the department of psychiatry at Augusta (Georgia) University. “We know there is a bidirectional association between schizophrenia and autoimmune disorders, [where] starting with either increases the chances of the other occurring comorbidly.”
Previous studies have shown that adding anti-inflammatory agents to second-generation antipsychotics has been associated with greater symptomatic improvement in schizophrenia than with second-generation antipsychotics alone. Now, Dr. Miller and his colleagues are conducting a randomized, controlled study using the systemic immunotherapy siltuximab to see if adjunct anti-inflammatory treatments can improve cognition in schizophrenia. They are using the monoclonal antibody instead of nonsteroidal anti-inflammatory drugs because of its precise ability to target specific autoimmune pathways, according to Dr. Miller.
In addition, Dr. Miller and his colleagues conducted a post-hoc analysis of data from the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) study to determine if there was a difference between those patients with comorbid substance in schizophrenia, compared with those without. He and his coinvestigators found in a post-hoc analysis that the higher the level of certain inflammatory markers, the higher the Positive and Negative Syndrome Scale (PANSS) psychopathology score in people who’d tested positive for marijuana use at baseline.
The results, he said, beg the question about whether trial inclusion criteria for schizophrenia research need revisiting.
“What we really need to do is investigate, in a well-controlled, well-designed, prospective sample of patients who have this substance use comorbidity, [whether] we see alterations in these immune markers, and then how they vary over the course of illness,” Dr. Miller explained in a video interview at the International Congress on Schizophrenia Research. “It’s an important selection bias that as a field we’ve ignored. Maybe that’s a population that is enriched for some kind of immune dysfunction that might be targeted by some kind of anti-inflammatory or other immunotherapy strategy.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
SAN DIEGO – Researchers are looking at the potential role that autoimmune reactions, such as inflammation, play in the pathogenesis of schizophrenia, both with and without comorbid substance use.
“What we see is evidence of autoimmune involvement through the lifespan in schizophrenia,” explained Brian Miller, MD, PhD, of the department of psychiatry at Augusta (Georgia) University. “We know there is a bidirectional association between schizophrenia and autoimmune disorders, [where] starting with either increases the chances of the other occurring comorbidly.”
Previous studies have shown that adding anti-inflammatory agents to second-generation antipsychotics has been associated with greater symptomatic improvement in schizophrenia than with second-generation antipsychotics alone. Now, Dr. Miller and his colleagues are conducting a randomized, controlled study using the systemic immunotherapy siltuximab to see if adjunct anti-inflammatory treatments can improve cognition in schizophrenia. They are using the monoclonal antibody instead of nonsteroidal anti-inflammatory drugs because of its precise ability to target specific autoimmune pathways, according to Dr. Miller.
In addition, Dr. Miller and his colleagues conducted a post-hoc analysis of data from the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) study to determine if there was a difference between those patients with comorbid substance in schizophrenia, compared with those without. He and his coinvestigators found in a post-hoc analysis that the higher the level of certain inflammatory markers, the higher the Positive and Negative Syndrome Scale (PANSS) psychopathology score in people who’d tested positive for marijuana use at baseline.
The results, he said, beg the question about whether trial inclusion criteria for schizophrenia research need revisiting.
“What we really need to do is investigate, in a well-controlled, well-designed, prospective sample of patients who have this substance use comorbidity, [whether] we see alterations in these immune markers, and then how they vary over the course of illness,” Dr. Miller explained in a video interview at the International Congress on Schizophrenia Research. “It’s an important selection bias that as a field we’ve ignored. Maybe that’s a population that is enriched for some kind of immune dysfunction that might be targeted by some kind of anti-inflammatory or other immunotherapy strategy.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
SAN DIEGO – Researchers are looking at the potential role that autoimmune reactions, such as inflammation, play in the pathogenesis of schizophrenia, both with and without comorbid substance use.
“What we see is evidence of autoimmune involvement through the lifespan in schizophrenia,” explained Brian Miller, MD, PhD, of the department of psychiatry at Augusta (Georgia) University. “We know there is a bidirectional association between schizophrenia and autoimmune disorders, [where] starting with either increases the chances of the other occurring comorbidly.”
Previous studies have shown that adding anti-inflammatory agents to second-generation antipsychotics has been associated with greater symptomatic improvement in schizophrenia than with second-generation antipsychotics alone. Now, Dr. Miller and his colleagues are conducting a randomized, controlled study using the systemic immunotherapy siltuximab to see if adjunct anti-inflammatory treatments can improve cognition in schizophrenia. They are using the monoclonal antibody instead of nonsteroidal anti-inflammatory drugs because of its precise ability to target specific autoimmune pathways, according to Dr. Miller.
In addition, Dr. Miller and his colleagues conducted a post-hoc analysis of data from the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) study to determine if there was a difference between those patients with comorbid substance in schizophrenia, compared with those without. He and his coinvestigators found in a post-hoc analysis that the higher the level of certain inflammatory markers, the higher the Positive and Negative Syndrome Scale (PANSS) psychopathology score in people who’d tested positive for marijuana use at baseline.
The results, he said, beg the question about whether trial inclusion criteria for schizophrenia research need revisiting.
“What we really need to do is investigate, in a well-controlled, well-designed, prospective sample of patients who have this substance use comorbidity, [whether] we see alterations in these immune markers, and then how they vary over the course of illness,” Dr. Miller explained in a video interview at the International Congress on Schizophrenia Research. “It’s an important selection bias that as a field we’ve ignored. Maybe that’s a population that is enriched for some kind of immune dysfunction that might be targeted by some kind of anti-inflammatory or other immunotherapy strategy.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
AT THE ICSR BIENNIAL MEETING

FDA approves drug to treat 2 types of AML

The US Food and Drug Administration (FDA) has granted approval for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
CPX-351 is approved to treat adults with 2 types of acute myeloid leukemia (AML)—AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML.
The FDA granted the approval of CPX-351 to Jazz Pharmaceuticals.
The company says the drug will be commercially available within a week.
The FDA approval of CPX-351 is based on data from a phase 3 trial in which researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage.
For more information on CPX-351, visit http://www.vyxeos.com. ![]()
The US Food and Drug Administration (FDA) has granted approval for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
CPX-351 is approved to treat adults with 2 types of acute myeloid leukemia (AML)—AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML.
The FDA granted the approval of CPX-351 to Jazz Pharmaceuticals.
The company says the drug will be commercially available within a week.
The FDA approval of CPX-351 is based on data from a phase 3 trial in which researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage.
For more information on CPX-351, visit http://www.vyxeos.com. ![]()
The US Food and Drug Administration (FDA) has granted approval for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
CPX-351 is approved to treat adults with 2 types of acute myeloid leukemia (AML)—AML with myelodysplasia-related changes and newly diagnosed, therapy-related AML.
The FDA granted the approval of CPX-351 to Jazz Pharmaceuticals.
The company says the drug will be commercially available within a week.
The FDA approval of CPX-351 is based on data from a phase 3 trial in which researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage.
For more information on CPX-351, visit http://www.vyxeos.com. ![]()
Mast cell inhibitors might be effective against DVT
Preclinical research suggests common anti-allergy medicines might be able to treat deep vein thrombosis (DVT).
Researchers discovered that mice genetically depleted of mast cells were protected from developing DVT but still had normal hemostasis.
The team therefore theorized that mast cell inhibitors, which are already approved to treat some allergic diseases, might be effective against DVT.
Alex Brill, MD, PhD, of the University of Birmingham in the UK, and his colleagues reported these findings in Circulation Research.
“These findings offer new hope for the treatment of deep vein thrombosis without a risk of bleeding,” Dr Brill said. “If further human studies support our findings in mice, drugs to block mast cell production could be used in the future alongside lower doses of anticoagulants such as warfarin, significantly reducing bleeding risk.”
“This is particularly exciting because this is a group of drugs which already exists, and some forms are approved for the treatment of allergies such as hay fever and asthma, meaning that this discovery could help people with DVT sooner rather than later.” ![]()
Preclinical research suggests common anti-allergy medicines might be able to treat deep vein thrombosis (DVT).
Researchers discovered that mice genetically depleted of mast cells were protected from developing DVT but still had normal hemostasis.
The team therefore theorized that mast cell inhibitors, which are already approved to treat some allergic diseases, might be effective against DVT.
Alex Brill, MD, PhD, of the University of Birmingham in the UK, and his colleagues reported these findings in Circulation Research.
“These findings offer new hope for the treatment of deep vein thrombosis without a risk of bleeding,” Dr Brill said. “If further human studies support our findings in mice, drugs to block mast cell production could be used in the future alongside lower doses of anticoagulants such as warfarin, significantly reducing bleeding risk.”
“This is particularly exciting because this is a group of drugs which already exists, and some forms are approved for the treatment of allergies such as hay fever and asthma, meaning that this discovery could help people with DVT sooner rather than later.” ![]()
Preclinical research suggests common anti-allergy medicines might be able to treat deep vein thrombosis (DVT).
Researchers discovered that mice genetically depleted of mast cells were protected from developing DVT but still had normal hemostasis.
The team therefore theorized that mast cell inhibitors, which are already approved to treat some allergic diseases, might be effective against DVT.
Alex Brill, MD, PhD, of the University of Birmingham in the UK, and his colleagues reported these findings in Circulation Research.
“These findings offer new hope for the treatment of deep vein thrombosis without a risk of bleeding,” Dr Brill said. “If further human studies support our findings in mice, drugs to block mast cell production could be used in the future alongside lower doses of anticoagulants such as warfarin, significantly reducing bleeding risk.”
“This is particularly exciting because this is a group of drugs which already exists, and some forms are approved for the treatment of allergies such as hay fever and asthma, meaning that this discovery could help people with DVT sooner rather than later.” ![]()