User login
Team makes ‘fundamental’ AML discovery

A newly identified pathway plays a “fundamental” role in acute myeloid leukemia (AML), according to researchers.
The team discovered that AML cells have a secretory pathway that leads to the production and release of the immune receptor Tim-3 and its ligand galectin-9, both of which prevent natural killer (NK) and other cytotoxic cells from killing the AML cells.
Vadim Sumbayev, PhD, of the University of Kent in the UK, and his colleagues recounted these findings in EBioMedicine.
The researchers found that AML cells—but not healthy blood cells—express a receptor called latrophilin 1 (LPHN1). LPHN1 induces activation of PKCα, which triggers the translation and secretion of Tim-3 and galectin-9.
Soluble Tim-3 prevents the secretion of interleukin 2, which is required for the activation of NK cells and cytotoxic T cells. Galectin-9 impairs the AML-cell-killing ability of NK cells and other cytotoxic lymphocytes.
The researchers said their work revealed both biomarkers for AML diagnostics and potential targets for AML treatment.
“Targeting this pathway will crucially enhance patients’ own immune defenses, helping them to eliminate leukemia cells,” Dr Sumbayev said.
He added that his group’s discovery might be applied to the treatment of other cancers as well. ![]()
A newly identified pathway plays a “fundamental” role in acute myeloid leukemia (AML), according to researchers.
The team discovered that AML cells have a secretory pathway that leads to the production and release of the immune receptor Tim-3 and its ligand galectin-9, both of which prevent natural killer (NK) and other cytotoxic cells from killing the AML cells.
Vadim Sumbayev, PhD, of the University of Kent in the UK, and his colleagues recounted these findings in EBioMedicine.
The researchers found that AML cells—but not healthy blood cells—express a receptor called latrophilin 1 (LPHN1). LPHN1 induces activation of PKCα, which triggers the translation and secretion of Tim-3 and galectin-9.
Soluble Tim-3 prevents the secretion of interleukin 2, which is required for the activation of NK cells and cytotoxic T cells. Galectin-9 impairs the AML-cell-killing ability of NK cells and other cytotoxic lymphocytes.
The researchers said their work revealed both biomarkers for AML diagnostics and potential targets for AML treatment.
“Targeting this pathway will crucially enhance patients’ own immune defenses, helping them to eliminate leukemia cells,” Dr Sumbayev said.
He added that his group’s discovery might be applied to the treatment of other cancers as well. ![]()
A newly identified pathway plays a “fundamental” role in acute myeloid leukemia (AML), according to researchers.
The team discovered that AML cells have a secretory pathway that leads to the production and release of the immune receptor Tim-3 and its ligand galectin-9, both of which prevent natural killer (NK) and other cytotoxic cells from killing the AML cells.
Vadim Sumbayev, PhD, of the University of Kent in the UK, and his colleagues recounted these findings in EBioMedicine.
The researchers found that AML cells—but not healthy blood cells—express a receptor called latrophilin 1 (LPHN1). LPHN1 induces activation of PKCα, which triggers the translation and secretion of Tim-3 and galectin-9.
Soluble Tim-3 prevents the secretion of interleukin 2, which is required for the activation of NK cells and cytotoxic T cells. Galectin-9 impairs the AML-cell-killing ability of NK cells and other cytotoxic lymphocytes.
The researchers said their work revealed both biomarkers for AML diagnostics and potential targets for AML treatment.
“Targeting this pathway will crucially enhance patients’ own immune defenses, helping them to eliminate leukemia cells,” Dr Sumbayev said.
He added that his group’s discovery might be applied to the treatment of other cancers as well. ![]()
PANS and PANDAS – A step forward?
In the Journal of Child and Adolescent Psychopharmacology’s July 2017 issue, a group of respected individuals representing diverse expertise published “guidelines” to support clinical management of pediatric acute-onset neuropsychiatric syndrome (PANS) and its subclass PANDAS (those associated with streptococcal infection). PANS represents an enigmatic clinical syndrome that includes abrupt onset of obsessive-compulsive disorder (OCD) or eating restriction in combination with anxiety, attention deficit, hyperkinesia, emotional lability, irritability, aggressive or oppositional behavior, or academic decline. Neurologic findings also may be present; these are most often motor or vocal tics, but choreiform movements of the finger (repetitive motions that are rapid, jerky, and involuntary), deteriorating penmanship, sleep disruptions, or urinary frequency also may be present. The clinical course most often is relapsing and remitting with overall improvement over months or years.
(J Child Adolesc Psychopharmacol. 2017 Apr 7. doi: 10.1089/cap.2016.0151).
Specific recommendations include:
1. Searching for a coexisting infectious etiology with history, exam, and appropriate laboratory testing (including ASO and ADB antibodies), and, when present, treating accordingly. Even in the absence of definitive evidence of GAS infection, they recommend an initial course of antimicrobial therapy such as that given to patients with rheumatic fever.
2. For children with PANDAS (PANS with either culture or serologic evidence of GAS), consider instituting long-term streptococcal prophylaxis. The data on its value is mixed; however, most studies find more than 40% (and as many as 75%) of exacerbations are associated with GAS, and at least one study reports a reduction in neuropsychiatric exacerbations in children on penicillin or azithromycin prophylaxis for a 1-year period. Such decisions should be individualized: In children with strong evidence of exacerbations linked to GAS, there was thought to be greater likelihood of benefit, while, in those with no evidence for prior GAS infection, the potential for benefit was thought to be insufficient to justify prophylaxis. Furthermore, the optimal duration of prophylaxis is unknown. The guidelines recommend up to 2 years, but individualization is appropriate since severe cases may warrant prolonged prophylaxis.
3. In children who present with PANDAS and a positive throat culture for GAS, follow-up should be the same as that given for rheumatic fever, with reculture at 2-7 days and retreatment if there is persistence of GAS.
4. Vigilance for GAS infection in family members is appropriate, including obtaining throat cultures from persons with pharyngitis and treating them promptly when results are positive.
5. When GAS infection is not identified, the clinician should search for alternative infectious agents, such as Mycoplasma pneumoniae (using polymerase chain reaction on throat or nasopharyngeal swab), influenza virus, or alternative infections such as sinusitis, and treat accordingly.
6. Children with PANS and PANDAS should be immunized according to Advisory Committee of Immunization Practices recommendations, which includes annual influenza immunization. The committee reported that symptom flares after immunization were uncommon, brief, and manageable with NSAIDs.
7. The committee suggested that optimization of serum vitamin D levels among children with PANS and PANDAS could be of benefit, despite limited evidence. The committee members recommended treating children with PANS/PANDAS with vitamin D3 as needed to maintain serum 25-hydroxy vitamin D levels above 30 ng/mL. No benefit for adenotonsillectomy was identified. The committee recommended that tonsillectomy and/or adenoidectomy should limited to those with traditional indications (sleep apnea, failure to thrive, and abnormally large tonsils, etc.). The committee also found no evidence to suggest that probiotics modulate this condition.
These guidelines come with an important caveat. They represent a practical clinical approach for the management of infection in the context of PANS or PANDAS and rely heavily on the clinical experience of the members of the PANS/PANDAS Consortium. They provide criteria for the retrospective diagnosis of GAS infection and recommend treatment of GAS in all patients with newly diagnosed PANS. The suggested guidelines are supported by limited data and recognize that further prospective study of the mechanistic link between infection and PANS, clarification of the risk factors for development of PANS, and definitive study of the risks and benefits of antimicrobial prophylaxis are needed.
The consortium also has published two accompanying guidelines that address psychiatric (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0145) and immunomodulatory management (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0148) in the same issue of the Journal of Child and Adolescent Psychopharmacology.
Proposed criteria for documenting GAS infection in PANS pediatric patients

- A rise in serial antibody level, regardless of rapid test or culture result. This definition does not require clinical pharyngitis.
- Acute pharyngitis with a positive GAS throat culture, with or without a rising antibody level.
- Pharyngitis with characteristic palatal petechiae.
- Pharyngitis with a characteristic scarlatiniform rash.
- Pharyngitis without a throat swab or serology, but intimate (usually household) exposure to a proven GAS case.
- Asymptomatic pharyngeal colonization documented after an intimate exposure.
- Asymptomatic pharyngeal colonization after a negative throat swab documented within the prior 3-4 months.
- Single ASO or ADB antibody level within 6 months after the initial onset of neuropsychiatric symptoms may be accepted as positive if it is more than 95th percentile, using the laboratory’s normal standard for children of comparable age, or provisionally ASO greater than or equal to 1:480 or ADB greater than or equal to 1:1280.
- Both ASO and ADB are elevated at more than 80% percentile for age in the same serum sample within 6 months after the initial onset of neuropsychiatric symptoms.
- Culture-documented streptococcal dermatitis.
Source: J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0151.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at [email protected].
In the Journal of Child and Adolescent Psychopharmacology’s July 2017 issue, a group of respected individuals representing diverse expertise published “guidelines” to support clinical management of pediatric acute-onset neuropsychiatric syndrome (PANS) and its subclass PANDAS (those associated with streptococcal infection). PANS represents an enigmatic clinical syndrome that includes abrupt onset of obsessive-compulsive disorder (OCD) or eating restriction in combination with anxiety, attention deficit, hyperkinesia, emotional lability, irritability, aggressive or oppositional behavior, or academic decline. Neurologic findings also may be present; these are most often motor or vocal tics, but choreiform movements of the finger (repetitive motions that are rapid, jerky, and involuntary), deteriorating penmanship, sleep disruptions, or urinary frequency also may be present. The clinical course most often is relapsing and remitting with overall improvement over months or years.
(J Child Adolesc Psychopharmacol. 2017 Apr 7. doi: 10.1089/cap.2016.0151).
Specific recommendations include:
1. Searching for a coexisting infectious etiology with history, exam, and appropriate laboratory testing (including ASO and ADB antibodies), and, when present, treating accordingly. Even in the absence of definitive evidence of GAS infection, they recommend an initial course of antimicrobial therapy such as that given to patients with rheumatic fever.
2. For children with PANDAS (PANS with either culture or serologic evidence of GAS), consider instituting long-term streptococcal prophylaxis. The data on its value is mixed; however, most studies find more than 40% (and as many as 75%) of exacerbations are associated with GAS, and at least one study reports a reduction in neuropsychiatric exacerbations in children on penicillin or azithromycin prophylaxis for a 1-year period. Such decisions should be individualized: In children with strong evidence of exacerbations linked to GAS, there was thought to be greater likelihood of benefit, while, in those with no evidence for prior GAS infection, the potential for benefit was thought to be insufficient to justify prophylaxis. Furthermore, the optimal duration of prophylaxis is unknown. The guidelines recommend up to 2 years, but individualization is appropriate since severe cases may warrant prolonged prophylaxis.
3. In children who present with PANDAS and a positive throat culture for GAS, follow-up should be the same as that given for rheumatic fever, with reculture at 2-7 days and retreatment if there is persistence of GAS.
4. Vigilance for GAS infection in family members is appropriate, including obtaining throat cultures from persons with pharyngitis and treating them promptly when results are positive.
5. When GAS infection is not identified, the clinician should search for alternative infectious agents, such as Mycoplasma pneumoniae (using polymerase chain reaction on throat or nasopharyngeal swab), influenza virus, or alternative infections such as sinusitis, and treat accordingly.
6. Children with PANS and PANDAS should be immunized according to Advisory Committee of Immunization Practices recommendations, which includes annual influenza immunization. The committee reported that symptom flares after immunization were uncommon, brief, and manageable with NSAIDs.
7. The committee suggested that optimization of serum vitamin D levels among children with PANS and PANDAS could be of benefit, despite limited evidence. The committee members recommended treating children with PANS/PANDAS with vitamin D3 as needed to maintain serum 25-hydroxy vitamin D levels above 30 ng/mL. No benefit for adenotonsillectomy was identified. The committee recommended that tonsillectomy and/or adenoidectomy should limited to those with traditional indications (sleep apnea, failure to thrive, and abnormally large tonsils, etc.). The committee also found no evidence to suggest that probiotics modulate this condition.
These guidelines come with an important caveat. They represent a practical clinical approach for the management of infection in the context of PANS or PANDAS and rely heavily on the clinical experience of the members of the PANS/PANDAS Consortium. They provide criteria for the retrospective diagnosis of GAS infection and recommend treatment of GAS in all patients with newly diagnosed PANS. The suggested guidelines are supported by limited data and recognize that further prospective study of the mechanistic link between infection and PANS, clarification of the risk factors for development of PANS, and definitive study of the risks and benefits of antimicrobial prophylaxis are needed.
The consortium also has published two accompanying guidelines that address psychiatric (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0145) and immunomodulatory management (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0148) in the same issue of the Journal of Child and Adolescent Psychopharmacology.
Proposed criteria for documenting GAS infection in PANS pediatric patients
- A rise in serial antibody level, regardless of rapid test or culture result. This definition does not require clinical pharyngitis.
- Acute pharyngitis with a positive GAS throat culture, with or without a rising antibody level.
- Pharyngitis with characteristic palatal petechiae.
- Pharyngitis with a characteristic scarlatiniform rash.
- Pharyngitis without a throat swab or serology, but intimate (usually household) exposure to a proven GAS case.
- Asymptomatic pharyngeal colonization documented after an intimate exposure.
- Asymptomatic pharyngeal colonization after a negative throat swab documented within the prior 3-4 months.
- Single ASO or ADB antibody level within 6 months after the initial onset of neuropsychiatric symptoms may be accepted as positive if it is more than 95th percentile, using the laboratory’s normal standard for children of comparable age, or provisionally ASO greater than or equal to 1:480 or ADB greater than or equal to 1:1280.
- Both ASO and ADB are elevated at more than 80% percentile for age in the same serum sample within 6 months after the initial onset of neuropsychiatric symptoms.
- Culture-documented streptococcal dermatitis.
Source: J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0151.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at [email protected].
In the Journal of Child and Adolescent Psychopharmacology’s July 2017 issue, a group of respected individuals representing diverse expertise published “guidelines” to support clinical management of pediatric acute-onset neuropsychiatric syndrome (PANS) and its subclass PANDAS (those associated with streptococcal infection). PANS represents an enigmatic clinical syndrome that includes abrupt onset of obsessive-compulsive disorder (OCD) or eating restriction in combination with anxiety, attention deficit, hyperkinesia, emotional lability, irritability, aggressive or oppositional behavior, or academic decline. Neurologic findings also may be present; these are most often motor or vocal tics, but choreiform movements of the finger (repetitive motions that are rapid, jerky, and involuntary), deteriorating penmanship, sleep disruptions, or urinary frequency also may be present. The clinical course most often is relapsing and remitting with overall improvement over months or years.
(J Child Adolesc Psychopharmacol. 2017 Apr 7. doi: 10.1089/cap.2016.0151).
Specific recommendations include:
1. Searching for a coexisting infectious etiology with history, exam, and appropriate laboratory testing (including ASO and ADB antibodies), and, when present, treating accordingly. Even in the absence of definitive evidence of GAS infection, they recommend an initial course of antimicrobial therapy such as that given to patients with rheumatic fever.
2. For children with PANDAS (PANS with either culture or serologic evidence of GAS), consider instituting long-term streptococcal prophylaxis. The data on its value is mixed; however, most studies find more than 40% (and as many as 75%) of exacerbations are associated with GAS, and at least one study reports a reduction in neuropsychiatric exacerbations in children on penicillin or azithromycin prophylaxis for a 1-year period. Such decisions should be individualized: In children with strong evidence of exacerbations linked to GAS, there was thought to be greater likelihood of benefit, while, in those with no evidence for prior GAS infection, the potential for benefit was thought to be insufficient to justify prophylaxis. Furthermore, the optimal duration of prophylaxis is unknown. The guidelines recommend up to 2 years, but individualization is appropriate since severe cases may warrant prolonged prophylaxis.
3. In children who present with PANDAS and a positive throat culture for GAS, follow-up should be the same as that given for rheumatic fever, with reculture at 2-7 days and retreatment if there is persistence of GAS.
4. Vigilance for GAS infection in family members is appropriate, including obtaining throat cultures from persons with pharyngitis and treating them promptly when results are positive.
5. When GAS infection is not identified, the clinician should search for alternative infectious agents, such as Mycoplasma pneumoniae (using polymerase chain reaction on throat or nasopharyngeal swab), influenza virus, or alternative infections such as sinusitis, and treat accordingly.
6. Children with PANS and PANDAS should be immunized according to Advisory Committee of Immunization Practices recommendations, which includes annual influenza immunization. The committee reported that symptom flares after immunization were uncommon, brief, and manageable with NSAIDs.
7. The committee suggested that optimization of serum vitamin D levels among children with PANS and PANDAS could be of benefit, despite limited evidence. The committee members recommended treating children with PANS/PANDAS with vitamin D3 as needed to maintain serum 25-hydroxy vitamin D levels above 30 ng/mL. No benefit for adenotonsillectomy was identified. The committee recommended that tonsillectomy and/or adenoidectomy should limited to those with traditional indications (sleep apnea, failure to thrive, and abnormally large tonsils, etc.). The committee also found no evidence to suggest that probiotics modulate this condition.
These guidelines come with an important caveat. They represent a practical clinical approach for the management of infection in the context of PANS or PANDAS and rely heavily on the clinical experience of the members of the PANS/PANDAS Consortium. They provide criteria for the retrospective diagnosis of GAS infection and recommend treatment of GAS in all patients with newly diagnosed PANS. The suggested guidelines are supported by limited data and recognize that further prospective study of the mechanistic link between infection and PANS, clarification of the risk factors for development of PANS, and definitive study of the risks and benefits of antimicrobial prophylaxis are needed.
The consortium also has published two accompanying guidelines that address psychiatric (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0145) and immunomodulatory management (J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0148) in the same issue of the Journal of Child and Adolescent Psychopharmacology.
Proposed criteria for documenting GAS infection in PANS pediatric patients
- A rise in serial antibody level, regardless of rapid test or culture result. This definition does not require clinical pharyngitis.
- Acute pharyngitis with a positive GAS throat culture, with or without a rising antibody level.
- Pharyngitis with characteristic palatal petechiae.
- Pharyngitis with a characteristic scarlatiniform rash.
- Pharyngitis without a throat swab or serology, but intimate (usually household) exposure to a proven GAS case.
- Asymptomatic pharyngeal colonization documented after an intimate exposure.
- Asymptomatic pharyngeal colonization after a negative throat swab documented within the prior 3-4 months.
- Single ASO or ADB antibody level within 6 months after the initial onset of neuropsychiatric symptoms may be accepted as positive if it is more than 95th percentile, using the laboratory’s normal standard for children of comparable age, or provisionally ASO greater than or equal to 1:480 or ADB greater than or equal to 1:1280.
- Both ASO and ADB are elevated at more than 80% percentile for age in the same serum sample within 6 months after the initial onset of neuropsychiatric symptoms.
- Culture-documented streptococcal dermatitis.
Source: J Child Adolesc Psychopharmacol. 2017. doi: 10.1089/cap.2016.0151.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at [email protected].
Disease-Modifying Drug Treatment Before, During, and After Pregnancy in Women With MS
NEW ORLEANS—In a population of women with multiple sclerosis (MS) and a live birth, the rate of disease-modifying drug treatment decreased before and during pregnancy and increased steadily post partum, according to a report presented at the 31st Annual Meeting of the Consortium of MS Centers. In a separate analysis by the same researchers, they reported that less than one-third of women with MS and a live birth initiated a disease-modifying treatment within one year after delivery. “The rate of disease-modifying drug initiation increased with the number of relapses the patient experienced prior to pregnancy,” said Maria K. Houtchens, MD, on behalf of her research collaborators. Dr. Houtchens is an Assistant Professor in the Department of Neurology at Harvard Medical School and Director of the Women’s Health Program at the Partners MS Center at Brigham and Women’s Hospital in Boston.

Treatment Before, During, and After Pregnancy
To evaluate treatment patterns before, during, and after pregnancy in women with MS and a live birth, Dr. Houtchens and colleagues used a US administrative claims database to conduct a retrospective analysis of women ages 18 to 65 with MS, a claim indicative of a live birth, and one-year continuous eligibility before and after pregnancy in the IMS Health Real World Data Adjudicated Claims US database from January 1, 2006, to June 30, 2015. Disease-modifying drug treatment was evaluated during the year prior to pregnancy (at three-month intervals), the three trimesters of pregnancy, puerperium (six weeks post pregnancy), and one year post pregnancy (seven to 12 weeks post pregnancy and three to six, six to nine, and nine to 12 months post pregnancy). The researchers evaluated the proportion of women exposed to disease-modifying drug treatment during the 12 time periods. Results were also stratified by the number of relapses women experienced in the year prior to pregnancy.
Of 190,475 women with MS, 2,158 met eligibility criteria. Mean age was 30.26. Most women had commercial health insurance (98%) and were from the Midwest (32%), South (30%), or Northeast (29%) regions of the US.
The proportion of women with MS and a live birth treated with any disease-modifying drug was 20.48% at nine to 12 months pre-pregnancy, 21.46% at six to nine months pre-pregnancy, 20.62% at three to six months pre-pregnancy, and 17.75% at three months pre-pregnancy. During pregnancy, the proportion of women treated with a disease-modifying drug decreased to 12.05% during the first trimester and 1.90% during the second trimester, and then increased slightly to 2.97% during the third trimester. The proportion of women treated with disease-modifying drugs increased to 8.34% during puerperium, 12.93% during seven to 12 weeks post partum, 21.97% during three to six months post partum, 24.47% during six to nine months post partum, and 25.49% during nine to 12 months post partum. The majority of women (81.9%) had received disease-modifying drug treatment by six to nine months post partum. The proportion of women with disease-modifying drug treatment before and after pregnancy increased numerically with the number of relapses experienced before pregnancy.
Treatment After a Live Birth
In a separate analysis using the same cohort, Dr. Houtchens and colleagues looked closer at the time to initiation of disease-modifying drug treatment after a live birth in women with MS. Of the 2,094 women included in this analysis, the proportion with a live birth initiating a disease-modifying drug treatment within one year was 28.46%, and the proportion with no disease-modifying treatment within one year was 71.54%.
For those initiating a disease-modifying treatment within one year, mean time from live birth to first treatment was 118.98 days, and median time to first treatment was 93.50 days. A total of 16.11% received a disease-modifying drug less than 30 days after live birth, approximately half initiated a treatment within 90 days (47.82%), and three-quarters initiated a disease-modifying drug within six months (75.5%). The proportion of patients initiating treatment within one year after live birth increased with higher numbers of pre-pregnancy relapses (zero relapses, n = 441, 24.53%; one relapse, n = 108, 50.94%; two relapses, n = 33, 54.10%; three or more relapses, n = 14, 60.87%). The mean number of days until disease-modifying drug initiation for those receiving
This study was supported by EMD Serono.
NEW ORLEANS—In a population of women with multiple sclerosis (MS) and a live birth, the rate of disease-modifying drug treatment decreased before and during pregnancy and increased steadily post partum, according to a report presented at the 31st Annual Meeting of the Consortium of MS Centers. In a separate analysis by the same researchers, they reported that less than one-third of women with MS and a live birth initiated a disease-modifying treatment within one year after delivery. “The rate of disease-modifying drug initiation increased with the number of relapses the patient experienced prior to pregnancy,” said Maria K. Houtchens, MD, on behalf of her research collaborators. Dr. Houtchens is an Assistant Professor in the Department of Neurology at Harvard Medical School and Director of the Women’s Health Program at the Partners MS Center at Brigham and Women’s Hospital in Boston.
Treatment Before, During, and After Pregnancy
To evaluate treatment patterns before, during, and after pregnancy in women with MS and a live birth, Dr. Houtchens and colleagues used a US administrative claims database to conduct a retrospective analysis of women ages 18 to 65 with MS, a claim indicative of a live birth, and one-year continuous eligibility before and after pregnancy in the IMS Health Real World Data Adjudicated Claims US database from January 1, 2006, to June 30, 2015. Disease-modifying drug treatment was evaluated during the year prior to pregnancy (at three-month intervals), the three trimesters of pregnancy, puerperium (six weeks post pregnancy), and one year post pregnancy (seven to 12 weeks post pregnancy and three to six, six to nine, and nine to 12 months post pregnancy). The researchers evaluated the proportion of women exposed to disease-modifying drug treatment during the 12 time periods. Results were also stratified by the number of relapses women experienced in the year prior to pregnancy.
Of 190,475 women with MS, 2,158 met eligibility criteria. Mean age was 30.26. Most women had commercial health insurance (98%) and were from the Midwest (32%), South (30%), or Northeast (29%) regions of the US.
The proportion of women with MS and a live birth treated with any disease-modifying drug was 20.48% at nine to 12 months pre-pregnancy, 21.46% at six to nine months pre-pregnancy, 20.62% at three to six months pre-pregnancy, and 17.75% at three months pre-pregnancy. During pregnancy, the proportion of women treated with a disease-modifying drug decreased to 12.05% during the first trimester and 1.90% during the second trimester, and then increased slightly to 2.97% during the third trimester. The proportion of women treated with disease-modifying drugs increased to 8.34% during puerperium, 12.93% during seven to 12 weeks post partum, 21.97% during three to six months post partum, 24.47% during six to nine months post partum, and 25.49% during nine to 12 months post partum. The majority of women (81.9%) had received disease-modifying drug treatment by six to nine months post partum. The proportion of women with disease-modifying drug treatment before and after pregnancy increased numerically with the number of relapses experienced before pregnancy.
Treatment After a Live Birth
In a separate analysis using the same cohort, Dr. Houtchens and colleagues looked closer at the time to initiation of disease-modifying drug treatment after a live birth in women with MS. Of the 2,094 women included in this analysis, the proportion with a live birth initiating a disease-modifying drug treatment within one year was 28.46%, and the proportion with no disease-modifying treatment within one year was 71.54%.
For those initiating a disease-modifying treatment within one year, mean time from live birth to first treatment was 118.98 days, and median time to first treatment was 93.50 days. A total of 16.11% received a disease-modifying drug less than 30 days after live birth, approximately half initiated a treatment within 90 days (47.82%), and three-quarters initiated a disease-modifying drug within six months (75.5%). The proportion of patients initiating treatment within one year after live birth increased with higher numbers of pre-pregnancy relapses (zero relapses, n = 441, 24.53%; one relapse, n = 108, 50.94%; two relapses, n = 33, 54.10%; three or more relapses, n = 14, 60.87%). The mean number of days until disease-modifying drug initiation for those receiving
This study was supported by EMD Serono.
NEW ORLEANS—In a population of women with multiple sclerosis (MS) and a live birth, the rate of disease-modifying drug treatment decreased before and during pregnancy and increased steadily post partum, according to a report presented at the 31st Annual Meeting of the Consortium of MS Centers. In a separate analysis by the same researchers, they reported that less than one-third of women with MS and a live birth initiated a disease-modifying treatment within one year after delivery. “The rate of disease-modifying drug initiation increased with the number of relapses the patient experienced prior to pregnancy,” said Maria K. Houtchens, MD, on behalf of her research collaborators. Dr. Houtchens is an Assistant Professor in the Department of Neurology at Harvard Medical School and Director of the Women’s Health Program at the Partners MS Center at Brigham and Women’s Hospital in Boston.
Treatment Before, During, and After Pregnancy
To evaluate treatment patterns before, during, and after pregnancy in women with MS and a live birth, Dr. Houtchens and colleagues used a US administrative claims database to conduct a retrospective analysis of women ages 18 to 65 with MS, a claim indicative of a live birth, and one-year continuous eligibility before and after pregnancy in the IMS Health Real World Data Adjudicated Claims US database from January 1, 2006, to June 30, 2015. Disease-modifying drug treatment was evaluated during the year prior to pregnancy (at three-month intervals), the three trimesters of pregnancy, puerperium (six weeks post pregnancy), and one year post pregnancy (seven to 12 weeks post pregnancy and three to six, six to nine, and nine to 12 months post pregnancy). The researchers evaluated the proportion of women exposed to disease-modifying drug treatment during the 12 time periods. Results were also stratified by the number of relapses women experienced in the year prior to pregnancy.
Of 190,475 women with MS, 2,158 met eligibility criteria. Mean age was 30.26. Most women had commercial health insurance (98%) and were from the Midwest (32%), South (30%), or Northeast (29%) regions of the US.
The proportion of women with MS and a live birth treated with any disease-modifying drug was 20.48% at nine to 12 months pre-pregnancy, 21.46% at six to nine months pre-pregnancy, 20.62% at three to six months pre-pregnancy, and 17.75% at three months pre-pregnancy. During pregnancy, the proportion of women treated with a disease-modifying drug decreased to 12.05% during the first trimester and 1.90% during the second trimester, and then increased slightly to 2.97% during the third trimester. The proportion of women treated with disease-modifying drugs increased to 8.34% during puerperium, 12.93% during seven to 12 weeks post partum, 21.97% during three to six months post partum, 24.47% during six to nine months post partum, and 25.49% during nine to 12 months post partum. The majority of women (81.9%) had received disease-modifying drug treatment by six to nine months post partum. The proportion of women with disease-modifying drug treatment before and after pregnancy increased numerically with the number of relapses experienced before pregnancy.
Treatment After a Live Birth
In a separate analysis using the same cohort, Dr. Houtchens and colleagues looked closer at the time to initiation of disease-modifying drug treatment after a live birth in women with MS. Of the 2,094 women included in this analysis, the proportion with a live birth initiating a disease-modifying drug treatment within one year was 28.46%, and the proportion with no disease-modifying treatment within one year was 71.54%.
For those initiating a disease-modifying treatment within one year, mean time from live birth to first treatment was 118.98 days, and median time to first treatment was 93.50 days. A total of 16.11% received a disease-modifying drug less than 30 days after live birth, approximately half initiated a treatment within 90 days (47.82%), and three-quarters initiated a disease-modifying drug within six months (75.5%). The proportion of patients initiating treatment within one year after live birth increased with higher numbers of pre-pregnancy relapses (zero relapses, n = 441, 24.53%; one relapse, n = 108, 50.94%; two relapses, n = 33, 54.10%; three or more relapses, n = 14, 60.87%). The mean number of days until disease-modifying drug initiation for those receiving
This study was supported by EMD Serono.
A shot in the arm: Boost your knowledge about immunizations for psychiatric patients
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.

We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.

Risk of exposure

Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.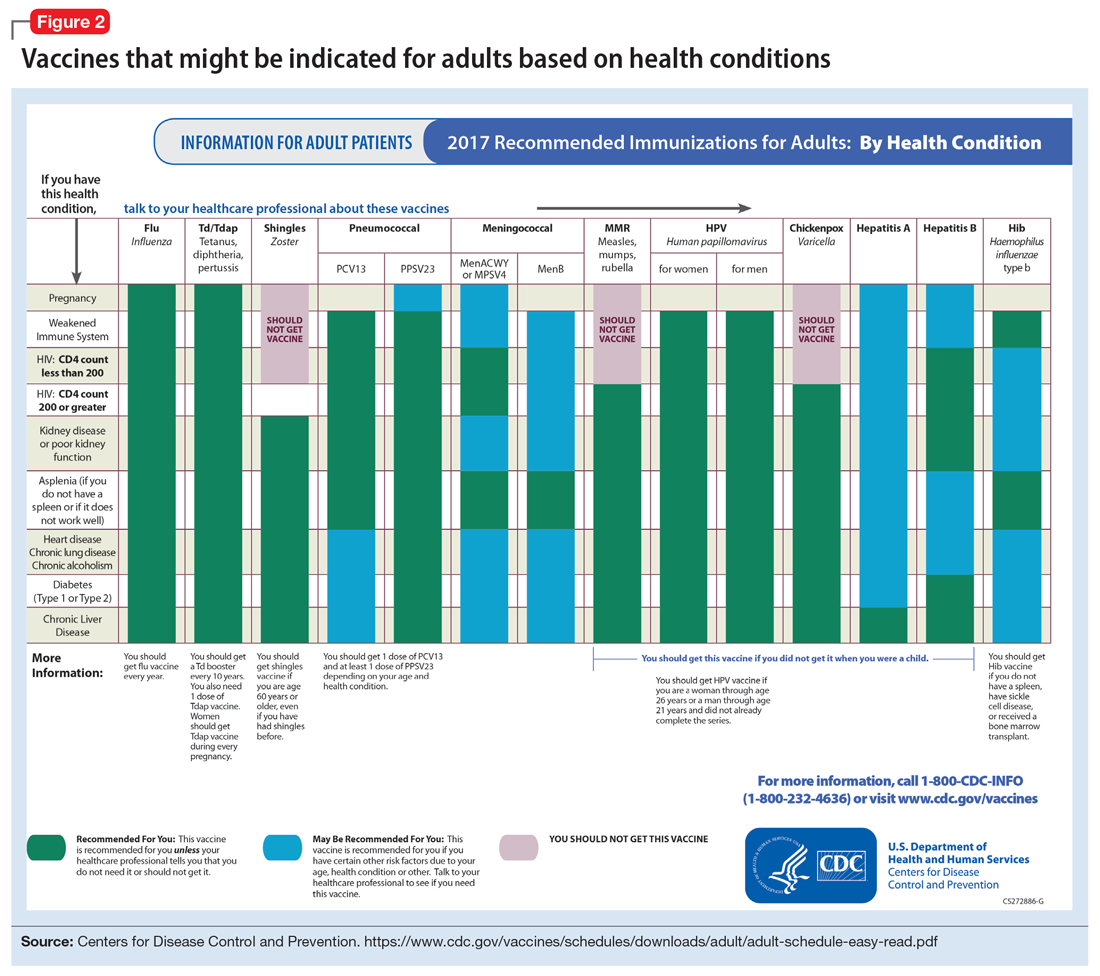
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.
We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.
Risk of exposure
Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.
We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.
Risk of exposure
Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.

Patient Satisfaction With Prophylactic Migraine Medications
BOSTON—Effectiveness and tolerability were the key factors influencing patients’ satisfaction with prophylactic migraine medications, according to a study presented at the 59th Annual Scientific Meeting of the American Headache Society. “There is an unmet need for treatments that have better efficacy and tolerability profiles than existing prophylactic migraine medications,” said lead author Marci Clark, PharmD, Director of Patient-Centered Outcomes Assessment at RTI Health Solutions in Ann Arbor, Michigan, on behalf of her study collaborators.

To gain insight into reasons for patient satisfaction or dissatisfaction with prophylactic migraine medications and identify medication characteristics that would increase patient satisfaction with future prophylactic migraine medications, the researchers recruited study participants from research facilities in Charlotte, North Carolina; St. Louis, Missouri; Southfield, Michigan; and Tampa, Florida. Individuals were eligible to participate if they met the following self-reported criteria: age 18 to 55, physician diagnosis of migraine for 12 or more months, current or prior experience taking one or more prescription prophylactic migraine medication in the past six months, experiencing four or more migraine headache days per month, and further classification as having episodic or chronic migraine (based on the headache/migraine days reported). Individual discussions with all participants were conducted by two experienced interviewers.
Forty participants were recruited and interviewed; 36 met all eligibility criteria. Twenty-one participants met the criteria for episodic migraine, 15 met the criteria for chronic migraine, and four participants, who were current migraineurs, were unable to be classified as having episodic or chronic migraine. Participants provided feedback on 20 prescription prophylactic migraine medications. The most commonly used current or prior migraine prophylactics were topiramate (n = 27), amitriptyline (n = 6), propranolol (n = 6), onabotulinumtoxinA (n = 5), and zonisamide (n = 3).
In general, medication effectiveness and tolerability were the most influential factors on patient satisfaction or dissatisfaction with their prophylactic migraine medications. Lack of efficacy, cited by 56% of respondents, was the most frequently reported reason for participants’ dissatisfaction with their medications. Regarding tolerabilit
Among those who were satisfied with their prophylactic migraine medications, decreased migraine frequency was the most common reason for their satisfaction (65%), followed by decreased migraine severity or intensity, which was reported by 28% of participants.
The most frequently reported desirable characteristics of a new prophylactic migraine medication that would increase participants’ satisfaction were related to the medication’s ability to decrease the frequency and severity or intensity of migraines. Nausea (36%), weight gain (26%), dizziness/light-headedness (23%), drowsiness/grogginess (15%), and memory loss, including the ability to remember words and speak clearly (13%), are side effects that patients reported that they do not want to see in a new or future prophylactic migraine medication.
BOSTON—Effectiveness and tolerability were the key factors influencing patients’ satisfaction with prophylactic migraine medications, according to a study presented at the 59th Annual Scientific Meeting of the American Headache Society. “There is an unmet need for treatments that have better efficacy and tolerability profiles than existing prophylactic migraine medications,” said lead author Marci Clark, PharmD, Director of Patient-Centered Outcomes Assessment at RTI Health Solutions in Ann Arbor, Michigan, on behalf of her study collaborators.
To gain insight into reasons for patient satisfaction or dissatisfaction with prophylactic migraine medications and identify medication characteristics that would increase patient satisfaction with future prophylactic migraine medications, the researchers recruited study participants from research facilities in Charlotte, North Carolina; St. Louis, Missouri; Southfield, Michigan; and Tampa, Florida. Individuals were eligible to participate if they met the following self-reported criteria: age 18 to 55, physician diagnosis of migraine for 12 or more months, current or prior experience taking one or more prescription prophylactic migraine medication in the past six months, experiencing four or more migraine headache days per month, and further classification as having episodic or chronic migraine (based on the headache/migraine days reported). Individual discussions with all participants were conducted by two experienced interviewers.
Forty participants were recruited and interviewed; 36 met all eligibility criteria. Twenty-one participants met the criteria for episodic migraine, 15 met the criteria for chronic migraine, and four participants, who were current migraineurs, were unable to be classified as having episodic or chronic migraine. Participants provided feedback on 20 prescription prophylactic migraine medications. The most commonly used current or prior migraine prophylactics were topiramate (n = 27), amitriptyline (n = 6), propranolol (n = 6), onabotulinumtoxinA (n = 5), and zonisamide (n = 3).
In general, medication effectiveness and tolerability were the most influential factors on patient satisfaction or dissatisfaction with their prophylactic migraine medications. Lack of efficacy, cited by 56% of respondents, was the most frequently reported reason for participants’ dissatisfaction with their medications. Regarding tolerabilit
Among those who were satisfied with their prophylactic migraine medications, decreased migraine frequency was the most common reason for their satisfaction (65%), followed by decreased migraine severity or intensity, which was reported by 28% of participants.
The most frequently reported desirable characteristics of a new prophylactic migraine medication that would increase participants’ satisfaction were related to the medication’s ability to decrease the frequency and severity or intensity of migraines. Nausea (36%), weight gain (26%), dizziness/light-headedness (23%), drowsiness/grogginess (15%), and memory loss, including the ability to remember words and speak clearly (13%), are side effects that patients reported that they do not want to see in a new or future prophylactic migraine medication.
BOSTON—Effectiveness and tolerability were the key factors influencing patients’ satisfaction with prophylactic migraine medications, according to a study presented at the 59th Annual Scientific Meeting of the American Headache Society. “There is an unmet need for treatments that have better efficacy and tolerability profiles than existing prophylactic migraine medications,” said lead author Marci Clark, PharmD, Director of Patient-Centered Outcomes Assessment at RTI Health Solutions in Ann Arbor, Michigan, on behalf of her study collaborators.
To gain insight into reasons for patient satisfaction or dissatisfaction with prophylactic migraine medications and identify medication characteristics that would increase patient satisfaction with future prophylactic migraine medications, the researchers recruited study participants from research facilities in Charlotte, North Carolina; St. Louis, Missouri; Southfield, Michigan; and Tampa, Florida. Individuals were eligible to participate if they met the following self-reported criteria: age 18 to 55, physician diagnosis of migraine for 12 or more months, current or prior experience taking one or more prescription prophylactic migraine medication in the past six months, experiencing four or more migraine headache days per month, and further classification as having episodic or chronic migraine (based on the headache/migraine days reported). Individual discussions with all participants were conducted by two experienced interviewers.
Forty participants were recruited and interviewed; 36 met all eligibility criteria. Twenty-one participants met the criteria for episodic migraine, 15 met the criteria for chronic migraine, and four participants, who were current migraineurs, were unable to be classified as having episodic or chronic migraine. Participants provided feedback on 20 prescription prophylactic migraine medications. The most commonly used current or prior migraine prophylactics were topiramate (n = 27), amitriptyline (n = 6), propranolol (n = 6), onabotulinumtoxinA (n = 5), and zonisamide (n = 3).
In general, medication effectiveness and tolerability were the most influential factors on patient satisfaction or dissatisfaction with their prophylactic migraine medications. Lack of efficacy, cited by 56% of respondents, was the most frequently reported reason for participants’ dissatisfaction with their medications. Regarding tolerabilit
Among those who were satisfied with their prophylactic migraine medications, decreased migraine frequency was the most common reason for their satisfaction (65%), followed by decreased migraine severity or intensity, which was reported by 28% of participants.
The most frequently reported desirable characteristics of a new prophylactic migraine medication that would increase participants’ satisfaction were related to the medication’s ability to decrease the frequency and severity or intensity of migraines. Nausea (36%), weight gain (26%), dizziness/light-headedness (23%), drowsiness/grogginess (15%), and memory loss, including the ability to remember words and speak clearly (13%), are side effects that patients reported that they do not want to see in a new or future prophylactic migraine medication.
Sulindac-erlotinib as chemoprevention for FAP
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited disorder caused by germline mutations in the APC (adenomatous polyposis coli) gene. The disease is characterized by the formation of hundreds to thousands of adenomatous polyps in the colorectum and a nearly 100% lifetime risk of colorectal cancer, if left untreated.

Six randomized clinical trials explore chemoprevention in FAP, including the use of sulindac, celecoxib, low-dose aspirin, eicosapentaenoic acid, and one ongoing trial using sulindac and difluoromethylornithine. These trials have shown at most a 30% decrease in colorectal adenoma burden over short-term treatment, compared with placebo.
Preclinical studies have shown that inactivation of the APC gene and epidermal growth factor receptor (EGFR) signaling promotes cyclooxygenase (COX)-2 expression and subsequent development of intestinal neoplasia. Our group has previously shown that the combination of COX and EGFR inhibition with sulindac and erlotinib led to a 71% reduction in duodenal polyp burden in patients with FAP.
The hypothesis of this study was that a combination of COX and EGFR inhibition would inhibit colorectal adenoma formation in patient with FAP. We designed a phase 2 double-blind, placebo-controlled randomized trial in FAP patients who received combination therapy with 150 mg sulindac b.i.d. and 75 mg erlotinib daily, or placebo tablets for 6 months and assessed the number of polyps in the colorectum, rectum, or ileal pouch at baseline and at 6 months.
The total colorectal polyp count was significantly different between the placebo and sulindac-erlotinib groups at 6 months in FAP patients with an intact colon (P less than .0001), with a net percentage change of 89.3% between the two groups. Similar reductions were found in the ileal pouch anal anastomosis and ileorectal anastomosis groups.
Further research is necessary to evaluate these preliminary findings in a larger study population with longer follow-up to determine whether the observed effects will result in improved long-term clinical outcomes. A new phase 2 multicenter clinical trial (sponsored by the National Cancer Institute) of erlotinib therapy in patients with FAP will be activating this summer across seven U.S. cancer centers and will add further evidence for this chemoprevention strategy.
Dr. Samadder is in the division of gastroenterology and hepatology, Mayo Clinic, Scottsdale, Ariz. His comments were made during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited disorder caused by germline mutations in the APC (adenomatous polyposis coli) gene. The disease is characterized by the formation of hundreds to thousands of adenomatous polyps in the colorectum and a nearly 100% lifetime risk of colorectal cancer, if left untreated.
Six randomized clinical trials explore chemoprevention in FAP, including the use of sulindac, celecoxib, low-dose aspirin, eicosapentaenoic acid, and one ongoing trial using sulindac and difluoromethylornithine. These trials have shown at most a 30% decrease in colorectal adenoma burden over short-term treatment, compared with placebo.
Preclinical studies have shown that inactivation of the APC gene and epidermal growth factor receptor (EGFR) signaling promotes cyclooxygenase (COX)-2 expression and subsequent development of intestinal neoplasia. Our group has previously shown that the combination of COX and EGFR inhibition with sulindac and erlotinib led to a 71% reduction in duodenal polyp burden in patients with FAP.
The hypothesis of this study was that a combination of COX and EGFR inhibition would inhibit colorectal adenoma formation in patient with FAP. We designed a phase 2 double-blind, placebo-controlled randomized trial in FAP patients who received combination therapy with 150 mg sulindac b.i.d. and 75 mg erlotinib daily, or placebo tablets for 6 months and assessed the number of polyps in the colorectum, rectum, or ileal pouch at baseline and at 6 months.
The total colorectal polyp count was significantly different between the placebo and sulindac-erlotinib groups at 6 months in FAP patients with an intact colon (P less than .0001), with a net percentage change of 89.3% between the two groups. Similar reductions were found in the ileal pouch anal anastomosis and ileorectal anastomosis groups.
Further research is necessary to evaluate these preliminary findings in a larger study population with longer follow-up to determine whether the observed effects will result in improved long-term clinical outcomes. A new phase 2 multicenter clinical trial (sponsored by the National Cancer Institute) of erlotinib therapy in patients with FAP will be activating this summer across seven U.S. cancer centers and will add further evidence for this chemoprevention strategy.
Dr. Samadder is in the division of gastroenterology and hepatology, Mayo Clinic, Scottsdale, Ariz. His comments were made during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited disorder caused by germline mutations in the APC (adenomatous polyposis coli) gene. The disease is characterized by the formation of hundreds to thousands of adenomatous polyps in the colorectum and a nearly 100% lifetime risk of colorectal cancer, if left untreated.
Six randomized clinical trials explore chemoprevention in FAP, including the use of sulindac, celecoxib, low-dose aspirin, eicosapentaenoic acid, and one ongoing trial using sulindac and difluoromethylornithine. These trials have shown at most a 30% decrease in colorectal adenoma burden over short-term treatment, compared with placebo.
Preclinical studies have shown that inactivation of the APC gene and epidermal growth factor receptor (EGFR) signaling promotes cyclooxygenase (COX)-2 expression and subsequent development of intestinal neoplasia. Our group has previously shown that the combination of COX and EGFR inhibition with sulindac and erlotinib led to a 71% reduction in duodenal polyp burden in patients with FAP.
The hypothesis of this study was that a combination of COX and EGFR inhibition would inhibit colorectal adenoma formation in patient with FAP. We designed a phase 2 double-blind, placebo-controlled randomized trial in FAP patients who received combination therapy with 150 mg sulindac b.i.d. and 75 mg erlotinib daily, or placebo tablets for 6 months and assessed the number of polyps in the colorectum, rectum, or ileal pouch at baseline and at 6 months.
The total colorectal polyp count was significantly different between the placebo and sulindac-erlotinib groups at 6 months in FAP patients with an intact colon (P less than .0001), with a net percentage change of 89.3% between the two groups. Similar reductions were found in the ileal pouch anal anastomosis and ileorectal anastomosis groups.
Further research is necessary to evaluate these preliminary findings in a larger study population with longer follow-up to determine whether the observed effects will result in improved long-term clinical outcomes. A new phase 2 multicenter clinical trial (sponsored by the National Cancer Institute) of erlotinib therapy in patients with FAP will be activating this summer across seven U.S. cancer centers and will add further evidence for this chemoprevention strategy.
Dr. Samadder is in the division of gastroenterology and hepatology, Mayo Clinic, Scottsdale, Ariz. His comments were made during the AGA Institute Presidential Plenary at the Annual Digestive Disease Week.
Novel drug-eluting coronary stent looks good in DESSOLVE III
PARIS – A unique drug-eluting coronary stent showed positive interim results in the DESSOLVE III trial presented at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
In DESSOLVE III, 1,398 patients undergoing percutaneous coronary intervention were randomized to the widely used Xience everolimus-eluting stent or to MiStent, a novel thin-strut stent with a polymer coating that is quickly absorbed after delivering microcrystalline sirolimus into the vessel wall for prolonged release at a near-linear rate.
At an interim analysis at 12 months of follow-up, the primary endpoint – a composite of cardiac death, target vessel MI, and clinically indicated target lesion revascularization – had occurred in 5.8% of the MiStent group and 6.5% of Xience recipients in the study, which was designed as a noninferiority trial, reported Robbert J. de Winter, MD, professor of clinical cardiology at the Academic Medical Center in Amsterdam.
“The data support the hypothesis that long-term cytostatic inhibition of early neointima could prevent the late neointimal growth seen at medium and long term with conventional drug-eluting stents,” he said.
MiStent is designed to overcome a shortcoming of conventional drug-eluting stents: namely, their durable polymer coating sticks around after the cytostatic drug is finished being released. It is believed that this residual polymer, which may not disappear for 6-9 months, induces inflammation in the vessel wall, eventually leading to intimal growth, restenosis, and new atherosclerosis.
“The unique feature of the MiStent is that the polymer is bioabsorbable and is fully absorbed at 3 months, whereas the drug is present in the vessel wall out to 9 months, well after the coating has disappeared. So in theory you would expect that any inflammatory response caused by the polymer coating will be counteracted by the drug. This was seen in animal models. And in the DESSOLVE I and II studies, angiographic follow-up showed that late luminal loss was flat after 6 months, in contrast to other drug-eluting stents, which show accrual of late neointimal growth after 6 months to a year,” according to the cardiologist.
DESSOLVE III is planned as a 3-year study. Already by 6 months the curves for target lesion revascularization started to separate, with a 12-month rate of 2.6% in the MiStent group versus 3.8% for Xience. And while this difference is not yet statistically significant, Dr. de Winter and his coinvestigators expect that by 3 years the separation will have grown to the point that the difference becomes statistically significant and clinically meaningful.
The MiStent platform is composed of a cobalt/chromium alloy. The stent strut thickness is only 64 microns, in contrast to 81 microns for the Xience stent. Thinner stent struts have previously been shown to be less injurious to the vessel wall.
DESSOLVE III is an all-comers trial conducted at 20 sites in four European countries. Participants had to have a reference vessel diameter of 2.5-3.5 mm. Roughly 60% of patients had an acute coronary syndrome as their indication for PCI. The study population included, among others, patients with left main coronary artery lesions, restenotic lesions, and failed saphenous vein grafts. Dual-antiplatelet therapy was given for 6 months in patients with stable angina and 12 months for those with ACS, in accordance with European Society of Cardiology guidelines.
Discussant Robert A. Byrne, MD, of the German Heart Center in Munich, declared: “For me, this is a potentially interesting device. It’s the only device where we have a drug elution that’s more prolonged than the polymer, and this does offer the potential for later benefit.”
Dr. Byrne was a member of a European Commission–backed task force that developed European regulatory guidance for the evaluation of new coronary stents. “This MiStent trial program ticks off a lot of boxes: We had a successful first human use study, then we had a modest-size angiographic endpoint study where the late lumen loss looked good, and now we have a clinical endpoint study. This is what we want to see in the regulatory space.”
Another discussant, Chaim Lotan, MD, pronounced the MiStent “definitely another good stent on the shelf.”
It’s impossible to say whether the excellent 1-year outcomes seen with MiStent in DESSOLVE III were due to the prolonged-release microcrystalline sirolimus, the ultrathin stent struts, or both. In any case, the major adverse cardiovascular event rates seen in DESSOLVE III are so low by historical standards that it will become extremely difficult to show superiority for one contemporary drug-eluting stent over another, predicted Dr. Lotan of Hadassah Medical Center in Jerusalem.
Dr. de Winter concurred.
“I think we can now say that the benchmark for present day drug-eluting stents is a target lesion failure rate of about 6% at 12 months and a stent thrombosis rate below 1% at 12 months. It’s going to be increasingly more difficult to improve on that,” he said.
The MiStent, manufactured by Micell Technologies, is commercially available in Europe but investigational in the United States.
DESSOLVE III was sponsored by the European Cardiovascular Research Institute and supported by grants from Micell Technologies and Stentys.
Dr. de Winter reported receiving research grants from OrbusNeich, Abbott Vascular, AstraZeneca, Stentys, and Tryton.
PARIS – A unique drug-eluting coronary stent showed positive interim results in the DESSOLVE III trial presented at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
In DESSOLVE III, 1,398 patients undergoing percutaneous coronary intervention were randomized to the widely used Xience everolimus-eluting stent or to MiStent, a novel thin-strut stent with a polymer coating that is quickly absorbed after delivering microcrystalline sirolimus into the vessel wall for prolonged release at a near-linear rate.
At an interim analysis at 12 months of follow-up, the primary endpoint – a composite of cardiac death, target vessel MI, and clinically indicated target lesion revascularization – had occurred in 5.8% of the MiStent group and 6.5% of Xience recipients in the study, which was designed as a noninferiority trial, reported Robbert J. de Winter, MD, professor of clinical cardiology at the Academic Medical Center in Amsterdam.
“The data support the hypothesis that long-term cytostatic inhibition of early neointima could prevent the late neointimal growth seen at medium and long term with conventional drug-eluting stents,” he said.
MiStent is designed to overcome a shortcoming of conventional drug-eluting stents: namely, their durable polymer coating sticks around after the cytostatic drug is finished being released. It is believed that this residual polymer, which may not disappear for 6-9 months, induces inflammation in the vessel wall, eventually leading to intimal growth, restenosis, and new atherosclerosis.
“The unique feature of the MiStent is that the polymer is bioabsorbable and is fully absorbed at 3 months, whereas the drug is present in the vessel wall out to 9 months, well after the coating has disappeared. So in theory you would expect that any inflammatory response caused by the polymer coating will be counteracted by the drug. This was seen in animal models. And in the DESSOLVE I and II studies, angiographic follow-up showed that late luminal loss was flat after 6 months, in contrast to other drug-eluting stents, which show accrual of late neointimal growth after 6 months to a year,” according to the cardiologist.
DESSOLVE III is planned as a 3-year study. Already by 6 months the curves for target lesion revascularization started to separate, with a 12-month rate of 2.6% in the MiStent group versus 3.8% for Xience. And while this difference is not yet statistically significant, Dr. de Winter and his coinvestigators expect that by 3 years the separation will have grown to the point that the difference becomes statistically significant and clinically meaningful.
The MiStent platform is composed of a cobalt/chromium alloy. The stent strut thickness is only 64 microns, in contrast to 81 microns for the Xience stent. Thinner stent struts have previously been shown to be less injurious to the vessel wall.
DESSOLVE III is an all-comers trial conducted at 20 sites in four European countries. Participants had to have a reference vessel diameter of 2.5-3.5 mm. Roughly 60% of patients had an acute coronary syndrome as their indication for PCI. The study population included, among others, patients with left main coronary artery lesions, restenotic lesions, and failed saphenous vein grafts. Dual-antiplatelet therapy was given for 6 months in patients with stable angina and 12 months for those with ACS, in accordance with European Society of Cardiology guidelines.
Discussant Robert A. Byrne, MD, of the German Heart Center in Munich, declared: “For me, this is a potentially interesting device. It’s the only device where we have a drug elution that’s more prolonged than the polymer, and this does offer the potential for later benefit.”
Dr. Byrne was a member of a European Commission–backed task force that developed European regulatory guidance for the evaluation of new coronary stents. “This MiStent trial program ticks off a lot of boxes: We had a successful first human use study, then we had a modest-size angiographic endpoint study where the late lumen loss looked good, and now we have a clinical endpoint study. This is what we want to see in the regulatory space.”
Another discussant, Chaim Lotan, MD, pronounced the MiStent “definitely another good stent on the shelf.”
It’s impossible to say whether the excellent 1-year outcomes seen with MiStent in DESSOLVE III were due to the prolonged-release microcrystalline sirolimus, the ultrathin stent struts, or both. In any case, the major adverse cardiovascular event rates seen in DESSOLVE III are so low by historical standards that it will become extremely difficult to show superiority for one contemporary drug-eluting stent over another, predicted Dr. Lotan of Hadassah Medical Center in Jerusalem.
Dr. de Winter concurred.
“I think we can now say that the benchmark for present day drug-eluting stents is a target lesion failure rate of about 6% at 12 months and a stent thrombosis rate below 1% at 12 months. It’s going to be increasingly more difficult to improve on that,” he said.
The MiStent, manufactured by Micell Technologies, is commercially available in Europe but investigational in the United States.
DESSOLVE III was sponsored by the European Cardiovascular Research Institute and supported by grants from Micell Technologies and Stentys.
Dr. de Winter reported receiving research grants from OrbusNeich, Abbott Vascular, AstraZeneca, Stentys, and Tryton.
PARIS – A unique drug-eluting coronary stent showed positive interim results in the DESSOLVE III trial presented at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
In DESSOLVE III, 1,398 patients undergoing percutaneous coronary intervention were randomized to the widely used Xience everolimus-eluting stent or to MiStent, a novel thin-strut stent with a polymer coating that is quickly absorbed after delivering microcrystalline sirolimus into the vessel wall for prolonged release at a near-linear rate.
At an interim analysis at 12 months of follow-up, the primary endpoint – a composite of cardiac death, target vessel MI, and clinically indicated target lesion revascularization – had occurred in 5.8% of the MiStent group and 6.5% of Xience recipients in the study, which was designed as a noninferiority trial, reported Robbert J. de Winter, MD, professor of clinical cardiology at the Academic Medical Center in Amsterdam.
“The data support the hypothesis that long-term cytostatic inhibition of early neointima could prevent the late neointimal growth seen at medium and long term with conventional drug-eluting stents,” he said.
MiStent is designed to overcome a shortcoming of conventional drug-eluting stents: namely, their durable polymer coating sticks around after the cytostatic drug is finished being released. It is believed that this residual polymer, which may not disappear for 6-9 months, induces inflammation in the vessel wall, eventually leading to intimal growth, restenosis, and new atherosclerosis.
“The unique feature of the MiStent is that the polymer is bioabsorbable and is fully absorbed at 3 months, whereas the drug is present in the vessel wall out to 9 months, well after the coating has disappeared. So in theory you would expect that any inflammatory response caused by the polymer coating will be counteracted by the drug. This was seen in animal models. And in the DESSOLVE I and II studies, angiographic follow-up showed that late luminal loss was flat after 6 months, in contrast to other drug-eluting stents, which show accrual of late neointimal growth after 6 months to a year,” according to the cardiologist.
DESSOLVE III is planned as a 3-year study. Already by 6 months the curves for target lesion revascularization started to separate, with a 12-month rate of 2.6% in the MiStent group versus 3.8% for Xience. And while this difference is not yet statistically significant, Dr. de Winter and his coinvestigators expect that by 3 years the separation will have grown to the point that the difference becomes statistically significant and clinically meaningful.
The MiStent platform is composed of a cobalt/chromium alloy. The stent strut thickness is only 64 microns, in contrast to 81 microns for the Xience stent. Thinner stent struts have previously been shown to be less injurious to the vessel wall.
DESSOLVE III is an all-comers trial conducted at 20 sites in four European countries. Participants had to have a reference vessel diameter of 2.5-3.5 mm. Roughly 60% of patients had an acute coronary syndrome as their indication for PCI. The study population included, among others, patients with left main coronary artery lesions, restenotic lesions, and failed saphenous vein grafts. Dual-antiplatelet therapy was given for 6 months in patients with stable angina and 12 months for those with ACS, in accordance with European Society of Cardiology guidelines.
Discussant Robert A. Byrne, MD, of the German Heart Center in Munich, declared: “For me, this is a potentially interesting device. It’s the only device where we have a drug elution that’s more prolonged than the polymer, and this does offer the potential for later benefit.”
Dr. Byrne was a member of a European Commission–backed task force that developed European regulatory guidance for the evaluation of new coronary stents. “This MiStent trial program ticks off a lot of boxes: We had a successful first human use study, then we had a modest-size angiographic endpoint study where the late lumen loss looked good, and now we have a clinical endpoint study. This is what we want to see in the regulatory space.”
Another discussant, Chaim Lotan, MD, pronounced the MiStent “definitely another good stent on the shelf.”
It’s impossible to say whether the excellent 1-year outcomes seen with MiStent in DESSOLVE III were due to the prolonged-release microcrystalline sirolimus, the ultrathin stent struts, or both. In any case, the major adverse cardiovascular event rates seen in DESSOLVE III are so low by historical standards that it will become extremely difficult to show superiority for one contemporary drug-eluting stent over another, predicted Dr. Lotan of Hadassah Medical Center in Jerusalem.
Dr. de Winter concurred.
“I think we can now say that the benchmark for present day drug-eluting stents is a target lesion failure rate of about 6% at 12 months and a stent thrombosis rate below 1% at 12 months. It’s going to be increasingly more difficult to improve on that,” he said.
The MiStent, manufactured by Micell Technologies, is commercially available in Europe but investigational in the United States.
DESSOLVE III was sponsored by the European Cardiovascular Research Institute and supported by grants from Micell Technologies and Stentys.
Dr. de Winter reported receiving research grants from OrbusNeich, Abbott Vascular, AstraZeneca, Stentys, and Tryton.
AT EUROPCR
Key clinical point:
Major finding: At an interim 12-month analysis, the composite rate of cardiac death, target vessel MI, and clinically indicated target lesion revascularization was 6.5% in recipients of a Xience everolimus-eluting coronary stent and 5.8% in those randomized to the novel MiStent.
Data source: DESSOLVE III, a prospective, randomized, international trial that randomized 1,398 real-world-type all-comers undergoing PCI to the Xience device or the MiStent.
Disclosures: DESSOLVE III was sponsored by the European Cardiovascular Research Institute and supported by grants from Micell Technologies and Stentys. The presenter reported receiving research grants from both companies.

Integrated health system builds collegial network of rural surgeons
Recruiting and retaining general surgeons is a longstanding problem for rural hospitals. A regional medical system in the Upper Midwest tackles this challenge by integrating small-town general surgeons into the network and emphasizing professional development, fair compensation, and a sustainable call and leave schedule.
The Gundersen Health System (GHS) is a physician-led, nonprofit health care network that operates in 19 rural counties in parts of Wisconsin, Minnesota, and Iowa, with its main campus in La Crosse, Wis. The network, established over a period of almost 40 years, is a mix of larger GHS medical centers, community hospitals, medical clinics, and GHS-managed and independent critical access hospitals (CAHs).

A sustainable model
“It is clear that the older paradigm of a single rural surgeon providing care 24/7 to an isolated community is vanishing. Design of a more sustainable model involves small groups of rural surgeons working together to provide general surgery and some subspecialty care locally, but who are also part of a larger network for administrative and clinical support,” said Dr. Cogbill in an interview.

Lessons learned
Dr. Cogbill said, “Our 38-year experience with rural surgery in our region has taught us many lessons. The strategy of trying to place a solo general surgeon in every small town with a CAH within our service area was not sustainable nor practical. The development of several rural centers of care within our region has allowed us to be more successful in the recruitment and retention of rural general surgeons who are hired to be part of a small group (optimally three) who provide care to their home community as well as outreach surgical care to several outlying CAHs near their home CAH. This has made it possible to offer a reasonable call schedule, mutual assistance, and the chance to build adequate case volumes. Connectivity to the health system should not mean ‘send all the great cases to the main campus,’ but instead should support the rural surgeons in performing appropriate cases locally.”
The survey respondents were aged 36-55 years, five were male, and all were graduates of U.S. medical schools. Eight are board certified and seven are either fellows or associate members of the American College of Surgeons. Their tenure in the GHS system averaged at least 7 years, ranging from 2 years to more than 20. Their surgical logs for a recent 1-year period show a case mix of endoscopy (63.8%), general surgery (26.7%), and obstetrics (6.1%). Mean annual relative value units for the group were 3,627 (range 2,456-5,846).
One goal of the confidential survey was to explore the reasons behind these surgeons’ choice of a rural practice. Their primary motivations were a preference for a rural lifestyle and a desire for a broad scope of practice. Loan forgiveness motivated some (37.5%), and the influence of a mentor was important for others (25%). The opportunity to join an integrated health system such as GHS was deemed extremely important to seven of the respondents.
Retention of rural surgeons
The most important factors mentioned by survey respondents for remaining in their positions were lifestyle (87.5%), family (75.0%), relationship with patients and colleagues, and scope of practice (75.0%), and compensation (62.6%).
Reasons to consider leaving were call burden (37.5%), relationship with the local hospital (25.0%), and compensation (25.0%).
The survey also looked at potential retention of these general surgeons in the coming 5 years: 37.5% said they were somewhat likely to remain, 25% said they were very likely to remain, and 37.5% said they were extremely likely to stay.
Two successful strategies have been promoting a satisfactory case mix and comanagement of patients who are referred to the main campus. The surgeons from the small towns are encouraged to come to La Crosse to assist in procedures on referred patients, to teach in the surgical residency and the Transition to Practice General Surgery fellowship programs at Gundersen, to participate in clinical research activities, and to engage in a variety of professional activities that strengthen the bonds between GHS and rural surgeons. These interactions help minimize professional isolation, a serious problem for surgeons working on their own in small communities.
Communication is maintained electronically. “Our system includes the use of a common EMR across the entire system allowing mutual access to both inpatient and outpatient records, including full access to digitized diagnostic imaging. GHS has established a number of distance-learning telemedicine links between the main campus and the rural communities that permit real-time patient consultations as well as participation in teaching conferences including Morbidity and Mortality Conferences.”
Reducing burnout in rural surgeons
The GHS model may have some impact on burnout among the rural surgeons in the system, said Dr. Cogbill. “Rural surgeon employment as part of a fully integrated regional network has the potential to reduce the magnitude of burnout by providing administrative assistance to help navigate bureaucratic complexities, easy access for subspecialty consults with colleagues who are known entities, and a model of rural surgery involving pods of three colleagues who can share call, mutual assistance, and case volumes.” Fair and competitive compensation and some degree of loan forgiveness have been in the mix of factors that have helped with recruitment. Administrative assistance from the main campus eases the clerical burden the surgeons face. Guaranteed free time for vacations and educational meetings, as well as a reasonable call schedule, are all built into contracts; this has had a big impact on recruitment. GHS has concluded that three general surgeons in a community is the optimal number to maintain call coverage and mutual assistance. Dr. Cogbill said, “The call schedule is managed by each “pod” of rural general surgeons themselves. With a full complement of three rural surgeons in a pod, they maintain an every third night call schedule. In towns in which there are fewer than three surgeons, the GHS surgeons often share call with surgeons who are not part of GHS to maintain a reasonable/sustainable call schedule.”
The retention track record at GHS is impressive. Since 1978, 19 rural general surgeons have been employed by GHS. Four (21%) rural general surgeons have retired 10 (53%) continue to practice in the network; only 5 (26%) left prior to retirement. Six rural general surgeons practiced in one location for over 20 years.
[email protected]
On Twitter @ThereseBorden
The rural surgeon needs some financial assurance, a reasonable call schedule that allows him/her time away from the job without compromising the care of patients in his/her town, regulatory relief, and a relationship with a “mother ship” referral center that is a true two-way street. The days of the independent private solo practitioner are numbered, and the statistics bear this out. The convergence of declining reimbursement, increasing burden of scrutiny and documentation placed by payers and the government, and an emerging workforce that values work-life balance all contribute to the need to develop programs like this one at Gunderson to maintain the surgical workforce in our small towns. Rural surgery comes with a great deal of intrinsic reward, which makes it an excellent career, if these obstacles can be overcome.
Mark Savarise, MD, FACS, is a general surgeon practicing in South Jordan, Utah, and is clinical associate professor of surgery at the University of Utah, Salt Lake City. He has no disclosures.
The rural surgeon needs some financial assurance, a reasonable call schedule that allows him/her time away from the job without compromising the care of patients in his/her town, regulatory relief, and a relationship with a “mother ship” referral center that is a true two-way street. The days of the independent private solo practitioner are numbered, and the statistics bear this out. The convergence of declining reimbursement, increasing burden of scrutiny and documentation placed by payers and the government, and an emerging workforce that values work-life balance all contribute to the need to develop programs like this one at Gunderson to maintain the surgical workforce in our small towns. Rural surgery comes with a great deal of intrinsic reward, which makes it an excellent career, if these obstacles can be overcome.
Mark Savarise, MD, FACS, is a general surgeon practicing in South Jordan, Utah, and is clinical associate professor of surgery at the University of Utah, Salt Lake City. He has no disclosures.
The rural surgeon needs some financial assurance, a reasonable call schedule that allows him/her time away from the job without compromising the care of patients in his/her town, regulatory relief, and a relationship with a “mother ship” referral center that is a true two-way street. The days of the independent private solo practitioner are numbered, and the statistics bear this out. The convergence of declining reimbursement, increasing burden of scrutiny and documentation placed by payers and the government, and an emerging workforce that values work-life balance all contribute to the need to develop programs like this one at Gunderson to maintain the surgical workforce in our small towns. Rural surgery comes with a great deal of intrinsic reward, which makes it an excellent career, if these obstacles can be overcome.
Mark Savarise, MD, FACS, is a general surgeon practicing in South Jordan, Utah, and is clinical associate professor of surgery at the University of Utah, Salt Lake City. He has no disclosures.
Recruiting and retaining general surgeons is a longstanding problem for rural hospitals. A regional medical system in the Upper Midwest tackles this challenge by integrating small-town general surgeons into the network and emphasizing professional development, fair compensation, and a sustainable call and leave schedule.
The Gundersen Health System (GHS) is a physician-led, nonprofit health care network that operates in 19 rural counties in parts of Wisconsin, Minnesota, and Iowa, with its main campus in La Crosse, Wis. The network, established over a period of almost 40 years, is a mix of larger GHS medical centers, community hospitals, medical clinics, and GHS-managed and independent critical access hospitals (CAHs).
A sustainable model
“It is clear that the older paradigm of a single rural surgeon providing care 24/7 to an isolated community is vanishing. Design of a more sustainable model involves small groups of rural surgeons working together to provide general surgery and some subspecialty care locally, but who are also part of a larger network for administrative and clinical support,” said Dr. Cogbill in an interview.
Lessons learned
Dr. Cogbill said, “Our 38-year experience with rural surgery in our region has taught us many lessons. The strategy of trying to place a solo general surgeon in every small town with a CAH within our service area was not sustainable nor practical. The development of several rural centers of care within our region has allowed us to be more successful in the recruitment and retention of rural general surgeons who are hired to be part of a small group (optimally three) who provide care to their home community as well as outreach surgical care to several outlying CAHs near their home CAH. This has made it possible to offer a reasonable call schedule, mutual assistance, and the chance to build adequate case volumes. Connectivity to the health system should not mean ‘send all the great cases to the main campus,’ but instead should support the rural surgeons in performing appropriate cases locally.”
The survey respondents were aged 36-55 years, five were male, and all were graduates of U.S. medical schools. Eight are board certified and seven are either fellows or associate members of the American College of Surgeons. Their tenure in the GHS system averaged at least 7 years, ranging from 2 years to more than 20. Their surgical logs for a recent 1-year period show a case mix of endoscopy (63.8%), general surgery (26.7%), and obstetrics (6.1%). Mean annual relative value units for the group were 3,627 (range 2,456-5,846).
One goal of the confidential survey was to explore the reasons behind these surgeons’ choice of a rural practice. Their primary motivations were a preference for a rural lifestyle and a desire for a broad scope of practice. Loan forgiveness motivated some (37.5%), and the influence of a mentor was important for others (25%). The opportunity to join an integrated health system such as GHS was deemed extremely important to seven of the respondents.
Retention of rural surgeons
The most important factors mentioned by survey respondents for remaining in their positions were lifestyle (87.5%), family (75.0%), relationship with patients and colleagues, and scope of practice (75.0%), and compensation (62.6%).
Reasons to consider leaving were call burden (37.5%), relationship with the local hospital (25.0%), and compensation (25.0%).
The survey also looked at potential retention of these general surgeons in the coming 5 years: 37.5% said they were somewhat likely to remain, 25% said they were very likely to remain, and 37.5% said they were extremely likely to stay.
Two successful strategies have been promoting a satisfactory case mix and comanagement of patients who are referred to the main campus. The surgeons from the small towns are encouraged to come to La Crosse to assist in procedures on referred patients, to teach in the surgical residency and the Transition to Practice General Surgery fellowship programs at Gundersen, to participate in clinical research activities, and to engage in a variety of professional activities that strengthen the bonds between GHS and rural surgeons. These interactions help minimize professional isolation, a serious problem for surgeons working on their own in small communities.
Communication is maintained electronically. “Our system includes the use of a common EMR across the entire system allowing mutual access to both inpatient and outpatient records, including full access to digitized diagnostic imaging. GHS has established a number of distance-learning telemedicine links between the main campus and the rural communities that permit real-time patient consultations as well as participation in teaching conferences including Morbidity and Mortality Conferences.”
Reducing burnout in rural surgeons
The GHS model may have some impact on burnout among the rural surgeons in the system, said Dr. Cogbill. “Rural surgeon employment as part of a fully integrated regional network has the potential to reduce the magnitude of burnout by providing administrative assistance to help navigate bureaucratic complexities, easy access for subspecialty consults with colleagues who are known entities, and a model of rural surgery involving pods of three colleagues who can share call, mutual assistance, and case volumes.” Fair and competitive compensation and some degree of loan forgiveness have been in the mix of factors that have helped with recruitment. Administrative assistance from the main campus eases the clerical burden the surgeons face. Guaranteed free time for vacations and educational meetings, as well as a reasonable call schedule, are all built into contracts; this has had a big impact on recruitment. GHS has concluded that three general surgeons in a community is the optimal number to maintain call coverage and mutual assistance. Dr. Cogbill said, “The call schedule is managed by each “pod” of rural general surgeons themselves. With a full complement of three rural surgeons in a pod, they maintain an every third night call schedule. In towns in which there are fewer than three surgeons, the GHS surgeons often share call with surgeons who are not part of GHS to maintain a reasonable/sustainable call schedule.”
The retention track record at GHS is impressive. Since 1978, 19 rural general surgeons have been employed by GHS. Four (21%) rural general surgeons have retired 10 (53%) continue to practice in the network; only 5 (26%) left prior to retirement. Six rural general surgeons practiced in one location for over 20 years.
[email protected]
On Twitter @ThereseBorden
Recruiting and retaining general surgeons is a longstanding problem for rural hospitals. A regional medical system in the Upper Midwest tackles this challenge by integrating small-town general surgeons into the network and emphasizing professional development, fair compensation, and a sustainable call and leave schedule.
The Gundersen Health System (GHS) is a physician-led, nonprofit health care network that operates in 19 rural counties in parts of Wisconsin, Minnesota, and Iowa, with its main campus in La Crosse, Wis. The network, established over a period of almost 40 years, is a mix of larger GHS medical centers, community hospitals, medical clinics, and GHS-managed and independent critical access hospitals (CAHs).
A sustainable model
“It is clear that the older paradigm of a single rural surgeon providing care 24/7 to an isolated community is vanishing. Design of a more sustainable model involves small groups of rural surgeons working together to provide general surgery and some subspecialty care locally, but who are also part of a larger network for administrative and clinical support,” said Dr. Cogbill in an interview.
Lessons learned
Dr. Cogbill said, “Our 38-year experience with rural surgery in our region has taught us many lessons. The strategy of trying to place a solo general surgeon in every small town with a CAH within our service area was not sustainable nor practical. The development of several rural centers of care within our region has allowed us to be more successful in the recruitment and retention of rural general surgeons who are hired to be part of a small group (optimally three) who provide care to their home community as well as outreach surgical care to several outlying CAHs near their home CAH. This has made it possible to offer a reasonable call schedule, mutual assistance, and the chance to build adequate case volumes. Connectivity to the health system should not mean ‘send all the great cases to the main campus,’ but instead should support the rural surgeons in performing appropriate cases locally.”
The survey respondents were aged 36-55 years, five were male, and all were graduates of U.S. medical schools. Eight are board certified and seven are either fellows or associate members of the American College of Surgeons. Their tenure in the GHS system averaged at least 7 years, ranging from 2 years to more than 20. Their surgical logs for a recent 1-year period show a case mix of endoscopy (63.8%), general surgery (26.7%), and obstetrics (6.1%). Mean annual relative value units for the group were 3,627 (range 2,456-5,846).
One goal of the confidential survey was to explore the reasons behind these surgeons’ choice of a rural practice. Their primary motivations were a preference for a rural lifestyle and a desire for a broad scope of practice. Loan forgiveness motivated some (37.5%), and the influence of a mentor was important for others (25%). The opportunity to join an integrated health system such as GHS was deemed extremely important to seven of the respondents.
Retention of rural surgeons
The most important factors mentioned by survey respondents for remaining in their positions were lifestyle (87.5%), family (75.0%), relationship with patients and colleagues, and scope of practice (75.0%), and compensation (62.6%).
Reasons to consider leaving were call burden (37.5%), relationship with the local hospital (25.0%), and compensation (25.0%).
The survey also looked at potential retention of these general surgeons in the coming 5 years: 37.5% said they were somewhat likely to remain, 25% said they were very likely to remain, and 37.5% said they were extremely likely to stay.
Two successful strategies have been promoting a satisfactory case mix and comanagement of patients who are referred to the main campus. The surgeons from the small towns are encouraged to come to La Crosse to assist in procedures on referred patients, to teach in the surgical residency and the Transition to Practice General Surgery fellowship programs at Gundersen, to participate in clinical research activities, and to engage in a variety of professional activities that strengthen the bonds between GHS and rural surgeons. These interactions help minimize professional isolation, a serious problem for surgeons working on their own in small communities.
Communication is maintained electronically. “Our system includes the use of a common EMR across the entire system allowing mutual access to both inpatient and outpatient records, including full access to digitized diagnostic imaging. GHS has established a number of distance-learning telemedicine links between the main campus and the rural communities that permit real-time patient consultations as well as participation in teaching conferences including Morbidity and Mortality Conferences.”
Reducing burnout in rural surgeons
The GHS model may have some impact on burnout among the rural surgeons in the system, said Dr. Cogbill. “Rural surgeon employment as part of a fully integrated regional network has the potential to reduce the magnitude of burnout by providing administrative assistance to help navigate bureaucratic complexities, easy access for subspecialty consults with colleagues who are known entities, and a model of rural surgery involving pods of three colleagues who can share call, mutual assistance, and case volumes.” Fair and competitive compensation and some degree of loan forgiveness have been in the mix of factors that have helped with recruitment. Administrative assistance from the main campus eases the clerical burden the surgeons face. Guaranteed free time for vacations and educational meetings, as well as a reasonable call schedule, are all built into contracts; this has had a big impact on recruitment. GHS has concluded that three general surgeons in a community is the optimal number to maintain call coverage and mutual assistance. Dr. Cogbill said, “The call schedule is managed by each “pod” of rural general surgeons themselves. With a full complement of three rural surgeons in a pod, they maintain an every third night call schedule. In towns in which there are fewer than three surgeons, the GHS surgeons often share call with surgeons who are not part of GHS to maintain a reasonable/sustainable call schedule.”
The retention track record at GHS is impressive. Since 1978, 19 rural general surgeons have been employed by GHS. Four (21%) rural general surgeons have retired 10 (53%) continue to practice in the network; only 5 (26%) left prior to retirement. Six rural general surgeons practiced in one location for over 20 years.
[email protected]
On Twitter @ThereseBorden
Comprehensive guidelines released for enhanced colorectal surgery recovery
New guidelines for enhanced recovery from colon and rectal surgery highlight the small steps that can add up to big improvements in patient outcomes.
“I think one of the most surprising aspects” of the guidelines – a joint effort from the American Society of Colon and Rectal Surgeons (ASCRS) and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES) – “is how enhanced recovery in many ways involves all the little things,” said senior author Scott Steele, MD, FACS, chairman of the department of colorectal surgery at the Cleveland Clinic (Dis Colon Rectum. 2017 Aug;60[8]:761-84. doi: 10.1097/DCR.0000000000000883). The guideline includes 24 literature-based recommendations covering everything from preoperative stoma counseling to postop chewing gum, all rated by quality of evidence.
“Many are easy to incorporate into day-to-day practice: getting [patients] out of bed, avoiding nasogastric tubes, not giving as much IV fluid as we used to, having patients take oral food and drink right after surgery, and having nursing/anesthesia/surgeons all on the same page and understanding that ... multidisciplinary, multisetting care leads to the best outcomes,” he said.

ASCRS and SAGES joined forces after noting that previous guidelines for enhanced recovery – perhaps better known as enhanced recovery after surgery, or ERAS, protocols – are dated, including studies only up to 2012; much has been published since then.
Preop measures
Some of the new recommendations encourage closer patient involvement with care. For instance, the groups strongly recommend discussing goals and discharge criteria with patients before surgery. Recent work has found that compliance and success go up when patients understand what’s going on, and length of stay and complications go down. For similar reasons, stoma education, stoma marking, and counseling on avoiding dehydration should happen preoperatively.
Meanwhile, “although there appear to be no meaningful benefits of [mechanical bowel prep (MBP)] alone in terms of complications,” the groups made a weak recommendation for MBP plus oral antibiotics before surgery. “A meta-analysis of seven RCTs comparing MBP with [antibiotics] versus MBP alone showed a reduction in total surgical site infection and incisional site infection,” they noted.
ASCRS and SAGES strongly recommended that patients drink clear fluids in the 2 hours before surgery, and also recommended carbohydrate loading – specifically drinks high in complex carbohydrates – in nondiabetic patients to attenuate insulin resistance induced by surgery and starvation.
The groups also recommended preset orders to standardize care, and care bundles to reduce surgical site infections. Measures could include preop chlorhexidine showers, ertapenem (Invanz) within an hour of incision, gown and glove changes before fascial closure, and washing incisions with chlorhexidine during recovery.
Pain control
“A multimodal, opioid-sparing, pain management plan should be ... implemented before the induction of anesthesia” for earlier return of bowel function and shorter lengths of stay, they said in a strong recommendation. “One of the simplest techniques to limit opioid intake is to schedule narcotic alternatives, such as oral acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and gabapentin, rather than giving them on an as-needed basis.” The risk of anastomotic leaks with NSAIDs appears to be most pronounced when patients are on them for more than 3 days.
Wound infiltration and abdominal trunk blocks with liposomal bupivacaine have shown promising results, as well. “Limited data demonstrate that the (TAP) block with a local anesthetic [is] associated with decreased length of stay ... TAP blocks performed before surgery appear to provide better analgesia than TAP blocks performed at the end,” the groups said.
ASCRS and SAGES strongly recommended thoracic epidural analgesia for open colorectal cases, but not for routine use in laparoscopic cases. “The modest analgesic benefits provided by TEA do not support a faster recovery in laparoscopic surgery,” they said, noting that at least in open cases, infusion of a local anesthetic and a lipophilic opioid seems to work better than either option alone.
They also strongly recommended that surgery teams preempt postop nausea and vomiting. Dexamethasone at anesthesia induction and ondansetron at emergence is a common option for patients at risk. Others include total intravenous anesthesia, intravenous acetaminophen, and gabapentin.
Fluid management
Intraoperative crystalloids have to be managed to avoid volume overload and its bad effects. “A maintenance infusion of 1.5-2 mL/kg/h of balanced crystalloid solution is sufficient to cover the needs derived from salt water homeostasis during major abdominal surgery,” ASCRS and SAGES said in a strong recommendation.
“The neuroendocrine response induced by surgical trauma leads to a physiologic reduction of urine output that, in the absence of other signs of hypovolemia, should not trigger additional fluid administration.” Also, “crystalloid or colloid preloading does not prevent hypotension induced by neuraxial blockade ... hypotension induced by epidural analgesia should be managed by reducing the epidural infusion rate and with small doses of vasopressors” – not IV fluids – “so long at the patient is normovolemic,” they noted.
Intravenous fluids should be stopped after recovery room discharge, and clear fluids encouraged as soon as patients can tolerate them.
Postop care
ASCRS and SAGES made strong recommendations for minimally invasive surgery when possible, and for avoiding intra-abdominal drains and nasogastric tubes, both recommendations that support current practice in many places. NG tubes can push oral intake back 2 days, and there’s no evidence that abdominal drains prevent anastomotic leaks, plus there can be complications with both.
The groups also strongly recommended early and progressive patient mobilization to shorten length of stay, and a regular diet immediately after surgery.
As for the chewing gum, “sham feeding (i.e., chewing sugar-free gum for [at least] 10 minutes 3-4 times per day) after colorectal surgery is safe, results in small improvements in GI recovery” – flatus and bowel moments happen sooner – “and may be associated with a reduction in the length of hospital stay.” The groups strongly recommended it based on high-quality evidence
Alvimopan was also a strong recommendation to reverse increased GI transit time and constipation from opioids after open cases. “Several RCTs and pooled post hoc analyses showed accelerated time to recovery of GI function with 6- and 12-mg doses compared with placebo and a significantly shorter hospital length of stay in the alvimopan 12-mg group.” It’s unclear at this point, however, if alvimopan has a role in laparoscopic cases, the groups said.
To reduce the risk of urinary tract infections, they said urinary catheters should be pulled within 24 hours of elective colonic or upper rectal resection not involving a vesicular fistula, and within 48 hours of midrectal/lower rectal resections, which carry a greater risk of urinary retention.
ASCRS and SAGES funded the work. Seven of the 10 authors, including Dr. Steele, had no financial disclosures. One author is a speaker for Pacira Pharmaceuticals, and her institution has received unrestricted educational grants from the company. Another author reported grant support from Medtronic and Merck, maker of alvimopan and ertapenem, and a third reported collaborations with Medtronic and Johnson & Johnson.
New guidelines for enhanced recovery from colon and rectal surgery highlight the small steps that can add up to big improvements in patient outcomes.
“I think one of the most surprising aspects” of the guidelines – a joint effort from the American Society of Colon and Rectal Surgeons (ASCRS) and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES) – “is how enhanced recovery in many ways involves all the little things,” said senior author Scott Steele, MD, FACS, chairman of the department of colorectal surgery at the Cleveland Clinic (Dis Colon Rectum. 2017 Aug;60[8]:761-84. doi: 10.1097/DCR.0000000000000883). The guideline includes 24 literature-based recommendations covering everything from preoperative stoma counseling to postop chewing gum, all rated by quality of evidence.
“Many are easy to incorporate into day-to-day practice: getting [patients] out of bed, avoiding nasogastric tubes, not giving as much IV fluid as we used to, having patients take oral food and drink right after surgery, and having nursing/anesthesia/surgeons all on the same page and understanding that ... multidisciplinary, multisetting care leads to the best outcomes,” he said.
ASCRS and SAGES joined forces after noting that previous guidelines for enhanced recovery – perhaps better known as enhanced recovery after surgery, or ERAS, protocols – are dated, including studies only up to 2012; much has been published since then.
Preop measures
Some of the new recommendations encourage closer patient involvement with care. For instance, the groups strongly recommend discussing goals and discharge criteria with patients before surgery. Recent work has found that compliance and success go up when patients understand what’s going on, and length of stay and complications go down. For similar reasons, stoma education, stoma marking, and counseling on avoiding dehydration should happen preoperatively.
Meanwhile, “although there appear to be no meaningful benefits of [mechanical bowel prep (MBP)] alone in terms of complications,” the groups made a weak recommendation for MBP plus oral antibiotics before surgery. “A meta-analysis of seven RCTs comparing MBP with [antibiotics] versus MBP alone showed a reduction in total surgical site infection and incisional site infection,” they noted.
ASCRS and SAGES strongly recommended that patients drink clear fluids in the 2 hours before surgery, and also recommended carbohydrate loading – specifically drinks high in complex carbohydrates – in nondiabetic patients to attenuate insulin resistance induced by surgery and starvation.
The groups also recommended preset orders to standardize care, and care bundles to reduce surgical site infections. Measures could include preop chlorhexidine showers, ertapenem (Invanz) within an hour of incision, gown and glove changes before fascial closure, and washing incisions with chlorhexidine during recovery.
Pain control
“A multimodal, opioid-sparing, pain management plan should be ... implemented before the induction of anesthesia” for earlier return of bowel function and shorter lengths of stay, they said in a strong recommendation. “One of the simplest techniques to limit opioid intake is to schedule narcotic alternatives, such as oral acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and gabapentin, rather than giving them on an as-needed basis.” The risk of anastomotic leaks with NSAIDs appears to be most pronounced when patients are on them for more than 3 days.
Wound infiltration and abdominal trunk blocks with liposomal bupivacaine have shown promising results, as well. “Limited data demonstrate that the (TAP) block with a local anesthetic [is] associated with decreased length of stay ... TAP blocks performed before surgery appear to provide better analgesia than TAP blocks performed at the end,” the groups said.
ASCRS and SAGES strongly recommended thoracic epidural analgesia for open colorectal cases, but not for routine use in laparoscopic cases. “The modest analgesic benefits provided by TEA do not support a faster recovery in laparoscopic surgery,” they said, noting that at least in open cases, infusion of a local anesthetic and a lipophilic opioid seems to work better than either option alone.
They also strongly recommended that surgery teams preempt postop nausea and vomiting. Dexamethasone at anesthesia induction and ondansetron at emergence is a common option for patients at risk. Others include total intravenous anesthesia, intravenous acetaminophen, and gabapentin.
Fluid management
Intraoperative crystalloids have to be managed to avoid volume overload and its bad effects. “A maintenance infusion of 1.5-2 mL/kg/h of balanced crystalloid solution is sufficient to cover the needs derived from salt water homeostasis during major abdominal surgery,” ASCRS and SAGES said in a strong recommendation.
“The neuroendocrine response induced by surgical trauma leads to a physiologic reduction of urine output that, in the absence of other signs of hypovolemia, should not trigger additional fluid administration.” Also, “crystalloid or colloid preloading does not prevent hypotension induced by neuraxial blockade ... hypotension induced by epidural analgesia should be managed by reducing the epidural infusion rate and with small doses of vasopressors” – not IV fluids – “so long at the patient is normovolemic,” they noted.
Intravenous fluids should be stopped after recovery room discharge, and clear fluids encouraged as soon as patients can tolerate them.
Postop care
ASCRS and SAGES made strong recommendations for minimally invasive surgery when possible, and for avoiding intra-abdominal drains and nasogastric tubes, both recommendations that support current practice in many places. NG tubes can push oral intake back 2 days, and there’s no evidence that abdominal drains prevent anastomotic leaks, plus there can be complications with both.
The groups also strongly recommended early and progressive patient mobilization to shorten length of stay, and a regular diet immediately after surgery.
As for the chewing gum, “sham feeding (i.e., chewing sugar-free gum for [at least] 10 minutes 3-4 times per day) after colorectal surgery is safe, results in small improvements in GI recovery” – flatus and bowel moments happen sooner – “and may be associated with a reduction in the length of hospital stay.” The groups strongly recommended it based on high-quality evidence
Alvimopan was also a strong recommendation to reverse increased GI transit time and constipation from opioids after open cases. “Several RCTs and pooled post hoc analyses showed accelerated time to recovery of GI function with 6- and 12-mg doses compared with placebo and a significantly shorter hospital length of stay in the alvimopan 12-mg group.” It’s unclear at this point, however, if alvimopan has a role in laparoscopic cases, the groups said.
To reduce the risk of urinary tract infections, they said urinary catheters should be pulled within 24 hours of elective colonic or upper rectal resection not involving a vesicular fistula, and within 48 hours of midrectal/lower rectal resections, which carry a greater risk of urinary retention.
ASCRS and SAGES funded the work. Seven of the 10 authors, including Dr. Steele, had no financial disclosures. One author is a speaker for Pacira Pharmaceuticals, and her institution has received unrestricted educational grants from the company. Another author reported grant support from Medtronic and Merck, maker of alvimopan and ertapenem, and a third reported collaborations with Medtronic and Johnson & Johnson.
New guidelines for enhanced recovery from colon and rectal surgery highlight the small steps that can add up to big improvements in patient outcomes.
“I think one of the most surprising aspects” of the guidelines – a joint effort from the American Society of Colon and Rectal Surgeons (ASCRS) and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES) – “is how enhanced recovery in many ways involves all the little things,” said senior author Scott Steele, MD, FACS, chairman of the department of colorectal surgery at the Cleveland Clinic (Dis Colon Rectum. 2017 Aug;60[8]:761-84. doi: 10.1097/DCR.0000000000000883). The guideline includes 24 literature-based recommendations covering everything from preoperative stoma counseling to postop chewing gum, all rated by quality of evidence.
“Many are easy to incorporate into day-to-day practice: getting [patients] out of bed, avoiding nasogastric tubes, not giving as much IV fluid as we used to, having patients take oral food and drink right after surgery, and having nursing/anesthesia/surgeons all on the same page and understanding that ... multidisciplinary, multisetting care leads to the best outcomes,” he said.
ASCRS and SAGES joined forces after noting that previous guidelines for enhanced recovery – perhaps better known as enhanced recovery after surgery, or ERAS, protocols – are dated, including studies only up to 2012; much has been published since then.
Preop measures
Some of the new recommendations encourage closer patient involvement with care. For instance, the groups strongly recommend discussing goals and discharge criteria with patients before surgery. Recent work has found that compliance and success go up when patients understand what’s going on, and length of stay and complications go down. For similar reasons, stoma education, stoma marking, and counseling on avoiding dehydration should happen preoperatively.
Meanwhile, “although there appear to be no meaningful benefits of [mechanical bowel prep (MBP)] alone in terms of complications,” the groups made a weak recommendation for MBP plus oral antibiotics before surgery. “A meta-analysis of seven RCTs comparing MBP with [antibiotics] versus MBP alone showed a reduction in total surgical site infection and incisional site infection,” they noted.
ASCRS and SAGES strongly recommended that patients drink clear fluids in the 2 hours before surgery, and also recommended carbohydrate loading – specifically drinks high in complex carbohydrates – in nondiabetic patients to attenuate insulin resistance induced by surgery and starvation.
The groups also recommended preset orders to standardize care, and care bundles to reduce surgical site infections. Measures could include preop chlorhexidine showers, ertapenem (Invanz) within an hour of incision, gown and glove changes before fascial closure, and washing incisions with chlorhexidine during recovery.
Pain control
“A multimodal, opioid-sparing, pain management plan should be ... implemented before the induction of anesthesia” for earlier return of bowel function and shorter lengths of stay, they said in a strong recommendation. “One of the simplest techniques to limit opioid intake is to schedule narcotic alternatives, such as oral acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and gabapentin, rather than giving them on an as-needed basis.” The risk of anastomotic leaks with NSAIDs appears to be most pronounced when patients are on them for more than 3 days.
Wound infiltration and abdominal trunk blocks with liposomal bupivacaine have shown promising results, as well. “Limited data demonstrate that the (TAP) block with a local anesthetic [is] associated with decreased length of stay ... TAP blocks performed before surgery appear to provide better analgesia than TAP blocks performed at the end,” the groups said.
ASCRS and SAGES strongly recommended thoracic epidural analgesia for open colorectal cases, but not for routine use in laparoscopic cases. “The modest analgesic benefits provided by TEA do not support a faster recovery in laparoscopic surgery,” they said, noting that at least in open cases, infusion of a local anesthetic and a lipophilic opioid seems to work better than either option alone.
They also strongly recommended that surgery teams preempt postop nausea and vomiting. Dexamethasone at anesthesia induction and ondansetron at emergence is a common option for patients at risk. Others include total intravenous anesthesia, intravenous acetaminophen, and gabapentin.
Fluid management
Intraoperative crystalloids have to be managed to avoid volume overload and its bad effects. “A maintenance infusion of 1.5-2 mL/kg/h of balanced crystalloid solution is sufficient to cover the needs derived from salt water homeostasis during major abdominal surgery,” ASCRS and SAGES said in a strong recommendation.
“The neuroendocrine response induced by surgical trauma leads to a physiologic reduction of urine output that, in the absence of other signs of hypovolemia, should not trigger additional fluid administration.” Also, “crystalloid or colloid preloading does not prevent hypotension induced by neuraxial blockade ... hypotension induced by epidural analgesia should be managed by reducing the epidural infusion rate and with small doses of vasopressors” – not IV fluids – “so long at the patient is normovolemic,” they noted.
Intravenous fluids should be stopped after recovery room discharge, and clear fluids encouraged as soon as patients can tolerate them.
Postop care
ASCRS and SAGES made strong recommendations for minimally invasive surgery when possible, and for avoiding intra-abdominal drains and nasogastric tubes, both recommendations that support current practice in many places. NG tubes can push oral intake back 2 days, and there’s no evidence that abdominal drains prevent anastomotic leaks, plus there can be complications with both.
The groups also strongly recommended early and progressive patient mobilization to shorten length of stay, and a regular diet immediately after surgery.
As for the chewing gum, “sham feeding (i.e., chewing sugar-free gum for [at least] 10 minutes 3-4 times per day) after colorectal surgery is safe, results in small improvements in GI recovery” – flatus and bowel moments happen sooner – “and may be associated with a reduction in the length of hospital stay.” The groups strongly recommended it based on high-quality evidence
Alvimopan was also a strong recommendation to reverse increased GI transit time and constipation from opioids after open cases. “Several RCTs and pooled post hoc analyses showed accelerated time to recovery of GI function with 6- and 12-mg doses compared with placebo and a significantly shorter hospital length of stay in the alvimopan 12-mg group.” It’s unclear at this point, however, if alvimopan has a role in laparoscopic cases, the groups said.
To reduce the risk of urinary tract infections, they said urinary catheters should be pulled within 24 hours of elective colonic or upper rectal resection not involving a vesicular fistula, and within 48 hours of midrectal/lower rectal resections, which carry a greater risk of urinary retention.
ASCRS and SAGES funded the work. Seven of the 10 authors, including Dr. Steele, had no financial disclosures. One author is a speaker for Pacira Pharmaceuticals, and her institution has received unrestricted educational grants from the company. Another author reported grant support from Medtronic and Merck, maker of alvimopan and ertapenem, and a third reported collaborations with Medtronic and Johnson & Johnson.
FROM DISEASES OF THE COLON AND RECTUM
Molecular subtypes predicted outcomes in gastric cancer
Recurrence-free and overall survival in gastric cancer was highest among patients with the Epstein-Barr virus molecular subtype, investigators reported online July 27 in Clinical Cancer Research.
In contrast, the genomically stable subtype had the worst prognosis and was least likely to benefit from adjuvant chemotherapy, Bo Hwa Sohn, PhD, of the University of Texas MD Anderson Cancer Center, Houston, reported with her associates. Their prediction model “successfully stratified patients by survival and adjuvant chemotherapy outcomes,” they concluded.
The MSI subtype was tied to a moderate prognosis. Prognosis for the CIN subtype also was moderate, but was worse in the cohort from South Korea than in the MD Anderson cohort. These results might reflect more heterogeneity within the CIN subtype, the researchers said.
Importantly, adjuvant chemotherapy most benefited CIN patients – 59% were alive without recurrence at 3 years, compared with only 34% of those who received no adjuvant therapy (hazard ratio, 0.39; 95% confidence interval, 0.16-0.94; P = .03). In contrast, adjuvant chemotherapy produced only modest benefits in MSI (HR, 0.55; P = .18), and did not benefit GS patients (P = .66).
Analyses of 24 gastric cancer cell lines from the Genomics of Drug Sensitivity in Cancer project supported that finding, said the researchers. Lines of GS cells had the highest 5-fluorouracil IC50 values, indicating they are resistant to 5-fluorouracil. The activated transcription regulator NUPR1 has been linked to chemoresistance in other cancers and is often altered in the GS subtype, they noted. They only studied adjuvant chemotherapy in the MD Anderson cohort because most South Korean patients did not receive it. Also, all EBV patients received adjuvant chemotherapy, so the researchers could not examine outcomes for this subtype.
The researchers also created a predictive model by pooling probabilities of recurrence. On a scale of 0 to 100, patients who scored under 20 had a 67% chance of recurrence-free survival at 5 years, compared with 52% for patients scoring between 20 and 30 and 38% for patients scoring above 30. Differences between these probabilities were highly statistically significant, and results for overall survival were similar. Score independently predicted recurrence-free survival after other clinicopathologic variables were controlled for in the pooled cohorts and in patients with more heterogeneous stage II cancers.
This prediction model will need to be whittled down to a few genes that adequately represent each subtype before it is useful in the clinic, the researchers acknowledged. “Nevertheless, the validation of our prediction model in two independent patient cohorts and the fact that the model reflects the biological characteristics associated with each subtype indicate that this prediction model could be used to develop rational therapy recommendations,” they concluded.
Funders included the National Institutes of Health, The University of Texas MD Anderson Cancer Center, the Korea National Research Foundation, the Scientific Research Center Program, and the Korean Research Institute of Bioscience and Biotechnology. No investigator declared potential conflicts of interest.
Recurrence-free and overall survival in gastric cancer was highest among patients with the Epstein-Barr virus molecular subtype, investigators reported online July 27 in Clinical Cancer Research.
In contrast, the genomically stable subtype had the worst prognosis and was least likely to benefit from adjuvant chemotherapy, Bo Hwa Sohn, PhD, of the University of Texas MD Anderson Cancer Center, Houston, reported with her associates. Their prediction model “successfully stratified patients by survival and adjuvant chemotherapy outcomes,” they concluded.
The MSI subtype was tied to a moderate prognosis. Prognosis for the CIN subtype also was moderate, but was worse in the cohort from South Korea than in the MD Anderson cohort. These results might reflect more heterogeneity within the CIN subtype, the researchers said.
Importantly, adjuvant chemotherapy most benefited CIN patients – 59% were alive without recurrence at 3 years, compared with only 34% of those who received no adjuvant therapy (hazard ratio, 0.39; 95% confidence interval, 0.16-0.94; P = .03). In contrast, adjuvant chemotherapy produced only modest benefits in MSI (HR, 0.55; P = .18), and did not benefit GS patients (P = .66).
Analyses of 24 gastric cancer cell lines from the Genomics of Drug Sensitivity in Cancer project supported that finding, said the researchers. Lines of GS cells had the highest 5-fluorouracil IC50 values, indicating they are resistant to 5-fluorouracil. The activated transcription regulator NUPR1 has been linked to chemoresistance in other cancers and is often altered in the GS subtype, they noted. They only studied adjuvant chemotherapy in the MD Anderson cohort because most South Korean patients did not receive it. Also, all EBV patients received adjuvant chemotherapy, so the researchers could not examine outcomes for this subtype.
The researchers also created a predictive model by pooling probabilities of recurrence. On a scale of 0 to 100, patients who scored under 20 had a 67% chance of recurrence-free survival at 5 years, compared with 52% for patients scoring between 20 and 30 and 38% for patients scoring above 30. Differences between these probabilities were highly statistically significant, and results for overall survival were similar. Score independently predicted recurrence-free survival after other clinicopathologic variables were controlled for in the pooled cohorts and in patients with more heterogeneous stage II cancers.
This prediction model will need to be whittled down to a few genes that adequately represent each subtype before it is useful in the clinic, the researchers acknowledged. “Nevertheless, the validation of our prediction model in two independent patient cohorts and the fact that the model reflects the biological characteristics associated with each subtype indicate that this prediction model could be used to develop rational therapy recommendations,” they concluded.
Funders included the National Institutes of Health, The University of Texas MD Anderson Cancer Center, the Korea National Research Foundation, the Scientific Research Center Program, and the Korean Research Institute of Bioscience and Biotechnology. No investigator declared potential conflicts of interest.
Recurrence-free and overall survival in gastric cancer was highest among patients with the Epstein-Barr virus molecular subtype, investigators reported online July 27 in Clinical Cancer Research.
In contrast, the genomically stable subtype had the worst prognosis and was least likely to benefit from adjuvant chemotherapy, Bo Hwa Sohn, PhD, of the University of Texas MD Anderson Cancer Center, Houston, reported with her associates. Their prediction model “successfully stratified patients by survival and adjuvant chemotherapy outcomes,” they concluded.
The MSI subtype was tied to a moderate prognosis. Prognosis for the CIN subtype also was moderate, but was worse in the cohort from South Korea than in the MD Anderson cohort. These results might reflect more heterogeneity within the CIN subtype, the researchers said.
Importantly, adjuvant chemotherapy most benefited CIN patients – 59% were alive without recurrence at 3 years, compared with only 34% of those who received no adjuvant therapy (hazard ratio, 0.39; 95% confidence interval, 0.16-0.94; P = .03). In contrast, adjuvant chemotherapy produced only modest benefits in MSI (HR, 0.55; P = .18), and did not benefit GS patients (P = .66).
Analyses of 24 gastric cancer cell lines from the Genomics of Drug Sensitivity in Cancer project supported that finding, said the researchers. Lines of GS cells had the highest 5-fluorouracil IC50 values, indicating they are resistant to 5-fluorouracil. The activated transcription regulator NUPR1 has been linked to chemoresistance in other cancers and is often altered in the GS subtype, they noted. They only studied adjuvant chemotherapy in the MD Anderson cohort because most South Korean patients did not receive it. Also, all EBV patients received adjuvant chemotherapy, so the researchers could not examine outcomes for this subtype.
The researchers also created a predictive model by pooling probabilities of recurrence. On a scale of 0 to 100, patients who scored under 20 had a 67% chance of recurrence-free survival at 5 years, compared with 52% for patients scoring between 20 and 30 and 38% for patients scoring above 30. Differences between these probabilities were highly statistically significant, and results for overall survival were similar. Score independently predicted recurrence-free survival after other clinicopathologic variables were controlled for in the pooled cohorts and in patients with more heterogeneous stage II cancers.
This prediction model will need to be whittled down to a few genes that adequately represent each subtype before it is useful in the clinic, the researchers acknowledged. “Nevertheless, the validation of our prediction model in two independent patient cohorts and the fact that the model reflects the biological characteristics associated with each subtype indicate that this prediction model could be used to develop rational therapy recommendations,” they concluded.
Funders included the National Institutes of Health, The University of Texas MD Anderson Cancer Center, the Korea National Research Foundation, the Scientific Research Center Program, and the Korean Research Institute of Bioscience and Biotechnology. No investigator declared potential conflicts of interest.
FROM CLINICAL CANCER RESEARCH
Key clinical point: In gastric cancer, molecular subtypes predicted survival and response to adjuvant chemotherapy.
Major finding: Epstein-Barr virus subtype was associated with the highest probability of recurrence-free and overall survival. The genomically stable subtype had the worst prognosis and was least likely to benefit from adjuvant chemotherapy.
Data source: A retrospective study of 267 patients from South Korea and 432 patients from MD Anderson with gastric cancer.
Disclosures: Funders included the National Institutes of Health, The University of Texas MD Anderson Cancer Center, the Korea National Research Foundation, the Scientific Research Center Program, and the Korean Research Institute of Bioscience and Biotechnology. No investigator declared potential conflicts of interest.