User login
R-CHOP matched more intense regimens for overall survival in high-risk, untreated DLBCL
High-dose chemotherapy with upfront autologous stem cell transplantation did not meaningfully improve survival at 5 years in a phase 3 trial of 399 patients under age 66 years with intermediate to high– or high-risk untreated diffuse large B-cell lymphoma.
Rates of treatment failure-free survival at 2 years were 71% for patients who received rituximab dose–dense chemotherapy followed by autologous stem cell transplantation (ASCT) and 62% for patients who received only the rituximab regimen (hazard ratio, 0.65; 95% confidence interval, 0.47-0.91; P = .01), Annalisa Chiappella, MD, of Azienda Ospedaliero Universitaria Città della Salute e della Scienza di Torino (Italy) reported with her associates in Lancet Oncology.

Patients with diffuse large B-cell lymphoma, whose age-adjusted International Prognostic Index (aa-IPI) scores are 2 or 3 (intermediate to high or high risk), have a poor prognosis despite standard treatment with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone). Given a historic lack of late-phase studies of this population, the researchers designed a multicenter, open-label, randomized phase 3 trial comparing rituximab dose–dense chemotherapy without ASCT with a shorter rituximab regimen followed by consolidation with R-MAD (rituximab plus high-dose cytarabine plus mitoxantrone plus dexamethasone) and high-dose BEAM chemotherapy (carmustine, etoposide, cytarabine, and melphalan) plus ASCT in untreated patients aged 18-65 years.
Rituximab-based chemotherapy consisted of 14-day cycles of either standard R-CHOP or an intensified R-CHOP–like regimen that increased the doses of cyclophosphamide (1200 mg/m2) and doxorubicin (70 mg/m2), the investigators noted (Lancet Oncol. 2017 Jun 28. doi: 10.1016/S1470-2045[17]30444-8).
In all, 96 patients were assigned to receive only standard R-CHOP (eight cycles), 100 patients were assigned to high-dose R-CHOP (six cycles), 103 patients were assigned to standard R-CHOP (four cycles) plus R-MAD (2 cycles) plus BEAM plus ASCT, and 96 patients were assigned to high-dose R-CHOP (four cycles) plus the same sequence of R-MAD, BEAM, and ASCT. A total of 325 (81%) patients completed treatment, and the median follow-up period was 72 months (interquartile range, 57 to 88 months). There was no evidence that treatment efficacy varied by age, sex, bone marrow involvement, or other histology results, but only the intermediate-high risk patients experienced a benefit in terms of failure-free survival.
Grade 3 or higher adverse hematologic events affected 183 (92%) ASCT recipients and 135 (68%) patients who received only R-CHOP or high-dose R-CHOP. Nonhematologic adverse events affected 45% and 16% of patients, respectively, and were mostly gastrointestinal in nature. Of recipients of ASCT, 12 stopped treatment because of infections, prolonged neutropenia, gastrointestinal symptoms, or cardiac abnormalities, and two patients who did not undergo ASCT stopped treatment because of infections or prolonged neutropenia. Three (13%) patients died from treatment-related causes, including eight patients in the transplantation groups and five of the other patients.
Previous studies have reported benefits from intensified R-CHOP–like regimens in diffuse large B-cell lymphoma, but these trials included low-risk patients.
“Our study enrolled only intermediate to high–risk or high-risk patients, and the findings do not support the hypothesis that increasing the dose of R-CHOP improves outcomes in patients with diffuse large B-cell lymphoma who are at high risk,” they wrote. “The addition of novel drugs, such as lenalidomide, ibrutinib, bortezomib, and others, to standard R-CHOP regimens has been reported in phase 1 or 2 studies with promising results in high-risk patients, leading to ongoing phase 3 randomized trials to assess the efficacy of these strategies. While awaiting the results of these randomized studies, the standard treatment in patients with diffuse large B-cell lymphoma at intermediate to high and high risk remains chemoimmunotherapy based on the standard R-CHOP regimen.”
Fondazione Italiana Linfomi funded the study. Dr. Chiappelli and several coinvestigators disclosed ties to Amgen, Celgene, Janssen, Nanostring, Pfizer, and other companies.
High-dose chemotherapy with upfront autologous stem cell transplantation did not meaningfully improve survival at 5 years in a phase 3 trial of 399 patients under age 66 years with intermediate to high– or high-risk untreated diffuse large B-cell lymphoma.
Rates of treatment failure-free survival at 2 years were 71% for patients who received rituximab dose–dense chemotherapy followed by autologous stem cell transplantation (ASCT) and 62% for patients who received only the rituximab regimen (hazard ratio, 0.65; 95% confidence interval, 0.47-0.91; P = .01), Annalisa Chiappella, MD, of Azienda Ospedaliero Universitaria Città della Salute e della Scienza di Torino (Italy) reported with her associates in Lancet Oncology.
Patients with diffuse large B-cell lymphoma, whose age-adjusted International Prognostic Index (aa-IPI) scores are 2 or 3 (intermediate to high or high risk), have a poor prognosis despite standard treatment with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone). Given a historic lack of late-phase studies of this population, the researchers designed a multicenter, open-label, randomized phase 3 trial comparing rituximab dose–dense chemotherapy without ASCT with a shorter rituximab regimen followed by consolidation with R-MAD (rituximab plus high-dose cytarabine plus mitoxantrone plus dexamethasone) and high-dose BEAM chemotherapy (carmustine, etoposide, cytarabine, and melphalan) plus ASCT in untreated patients aged 18-65 years.
Rituximab-based chemotherapy consisted of 14-day cycles of either standard R-CHOP or an intensified R-CHOP–like regimen that increased the doses of cyclophosphamide (1200 mg/m2) and doxorubicin (70 mg/m2), the investigators noted (Lancet Oncol. 2017 Jun 28. doi: 10.1016/S1470-2045[17]30444-8).
In all, 96 patients were assigned to receive only standard R-CHOP (eight cycles), 100 patients were assigned to high-dose R-CHOP (six cycles), 103 patients were assigned to standard R-CHOP (four cycles) plus R-MAD (2 cycles) plus BEAM plus ASCT, and 96 patients were assigned to high-dose R-CHOP (four cycles) plus the same sequence of R-MAD, BEAM, and ASCT. A total of 325 (81%) patients completed treatment, and the median follow-up period was 72 months (interquartile range, 57 to 88 months). There was no evidence that treatment efficacy varied by age, sex, bone marrow involvement, or other histology results, but only the intermediate-high risk patients experienced a benefit in terms of failure-free survival.
Grade 3 or higher adverse hematologic events affected 183 (92%) ASCT recipients and 135 (68%) patients who received only R-CHOP or high-dose R-CHOP. Nonhematologic adverse events affected 45% and 16% of patients, respectively, and were mostly gastrointestinal in nature. Of recipients of ASCT, 12 stopped treatment because of infections, prolonged neutropenia, gastrointestinal symptoms, or cardiac abnormalities, and two patients who did not undergo ASCT stopped treatment because of infections or prolonged neutropenia. Three (13%) patients died from treatment-related causes, including eight patients in the transplantation groups and five of the other patients.
Previous studies have reported benefits from intensified R-CHOP–like regimens in diffuse large B-cell lymphoma, but these trials included low-risk patients.
“Our study enrolled only intermediate to high–risk or high-risk patients, and the findings do not support the hypothesis that increasing the dose of R-CHOP improves outcomes in patients with diffuse large B-cell lymphoma who are at high risk,” they wrote. “The addition of novel drugs, such as lenalidomide, ibrutinib, bortezomib, and others, to standard R-CHOP regimens has been reported in phase 1 or 2 studies with promising results in high-risk patients, leading to ongoing phase 3 randomized trials to assess the efficacy of these strategies. While awaiting the results of these randomized studies, the standard treatment in patients with diffuse large B-cell lymphoma at intermediate to high and high risk remains chemoimmunotherapy based on the standard R-CHOP regimen.”
Fondazione Italiana Linfomi funded the study. Dr. Chiappelli and several coinvestigators disclosed ties to Amgen, Celgene, Janssen, Nanostring, Pfizer, and other companies.
High-dose chemotherapy with upfront autologous stem cell transplantation did not meaningfully improve survival at 5 years in a phase 3 trial of 399 patients under age 66 years with intermediate to high– or high-risk untreated diffuse large B-cell lymphoma.
Rates of treatment failure-free survival at 2 years were 71% for patients who received rituximab dose–dense chemotherapy followed by autologous stem cell transplantation (ASCT) and 62% for patients who received only the rituximab regimen (hazard ratio, 0.65; 95% confidence interval, 0.47-0.91; P = .01), Annalisa Chiappella, MD, of Azienda Ospedaliero Universitaria Città della Salute e della Scienza di Torino (Italy) reported with her associates in Lancet Oncology.
Patients with diffuse large B-cell lymphoma, whose age-adjusted International Prognostic Index (aa-IPI) scores are 2 or 3 (intermediate to high or high risk), have a poor prognosis despite standard treatment with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone). Given a historic lack of late-phase studies of this population, the researchers designed a multicenter, open-label, randomized phase 3 trial comparing rituximab dose–dense chemotherapy without ASCT with a shorter rituximab regimen followed by consolidation with R-MAD (rituximab plus high-dose cytarabine plus mitoxantrone plus dexamethasone) and high-dose BEAM chemotherapy (carmustine, etoposide, cytarabine, and melphalan) plus ASCT in untreated patients aged 18-65 years.
Rituximab-based chemotherapy consisted of 14-day cycles of either standard R-CHOP or an intensified R-CHOP–like regimen that increased the doses of cyclophosphamide (1200 mg/m2) and doxorubicin (70 mg/m2), the investigators noted (Lancet Oncol. 2017 Jun 28. doi: 10.1016/S1470-2045[17]30444-8).
In all, 96 patients were assigned to receive only standard R-CHOP (eight cycles), 100 patients were assigned to high-dose R-CHOP (six cycles), 103 patients were assigned to standard R-CHOP (four cycles) plus R-MAD (2 cycles) plus BEAM plus ASCT, and 96 patients were assigned to high-dose R-CHOP (four cycles) plus the same sequence of R-MAD, BEAM, and ASCT. A total of 325 (81%) patients completed treatment, and the median follow-up period was 72 months (interquartile range, 57 to 88 months). There was no evidence that treatment efficacy varied by age, sex, bone marrow involvement, or other histology results, but only the intermediate-high risk patients experienced a benefit in terms of failure-free survival.
Grade 3 or higher adverse hematologic events affected 183 (92%) ASCT recipients and 135 (68%) patients who received only R-CHOP or high-dose R-CHOP. Nonhematologic adverse events affected 45% and 16% of patients, respectively, and were mostly gastrointestinal in nature. Of recipients of ASCT, 12 stopped treatment because of infections, prolonged neutropenia, gastrointestinal symptoms, or cardiac abnormalities, and two patients who did not undergo ASCT stopped treatment because of infections or prolonged neutropenia. Three (13%) patients died from treatment-related causes, including eight patients in the transplantation groups and five of the other patients.
Previous studies have reported benefits from intensified R-CHOP–like regimens in diffuse large B-cell lymphoma, but these trials included low-risk patients.
“Our study enrolled only intermediate to high–risk or high-risk patients, and the findings do not support the hypothesis that increasing the dose of R-CHOP improves outcomes in patients with diffuse large B-cell lymphoma who are at high risk,” they wrote. “The addition of novel drugs, such as lenalidomide, ibrutinib, bortezomib, and others, to standard R-CHOP regimens has been reported in phase 1 or 2 studies with promising results in high-risk patients, leading to ongoing phase 3 randomized trials to assess the efficacy of these strategies. While awaiting the results of these randomized studies, the standard treatment in patients with diffuse large B-cell lymphoma at intermediate to high and high risk remains chemoimmunotherapy based on the standard R-CHOP regimen.”
Fondazione Italiana Linfomi funded the study. Dr. Chiappelli and several coinvestigators disclosed ties to Amgen, Celgene, Janssen, Nanostring, Pfizer, and other companies.
FROM LANCET ONCOLOGY
Key clinical point: Pending phase 3 trial results for novel drugs, R-CHOP should remain the standard treatment for high-risk diffuse large B-cell lymphoma.
Major finding: For patients given rituximab dose-dense chemotherapy followed by chemoimmunotherapy consolidation and autologous stem cell transplantation, versus rituximab dose-dense chemotherapy alone, 5-year rates of overall survival were 78% and 77%, respectively,
Data source: A randomized, open-label, phase 3 study of 399 patients, aged 18-65 years.
Disclosures: Fondazione Italiana Linfomi funded the study. Dr. Chiappelli and several coinvestigators disclosed ties to Amgen, Celgene, Janssen, Nanostring, Pfizer, and other companies.
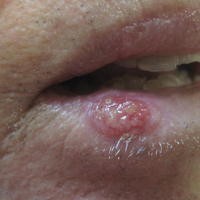
Prior mycobacterial infection linked to Sjögren’s syndrome
MADRID – in a large population-based study reported at the European Congress of Rheumatology.
Study investigator Hsin-Hua Chen, MD, and his colleagues at the Taichung (Taiwan) Veterans Hospital found that the adjusted odds ratio for having Sjögren’s syndrome after nontuberculous mycobacteria infection (NTM) was 11.24, with a 95% confidence interval of 2.37-53.24.
The risk for having Sjögren’s syndrome was found to be highest in those aged 40-65 years, versus those older than 65 (aOR, 39.24; P = .09) and in those with no prior history of bronchiectasis (aOR, 37.98; P = .09). Although, in both analyses, the 95% CIs were very wide (3.97-387.75 and 3.83-376.92, respectively), and the P values were not significant.

Dr. Chen and his colleagues decided to look at the association between tuberculous or nontuberculous mycobacteria with Sjögren’s syndrome for several reasons. First, mycobacterial infections have been linked to the development of autoimmunity. Second, there has been an increased incidence of tuberculosis reported in patients with Sjögren’s syndrome. Third, both Sjögren’s and infection with NTM occurred predominantly in middle-aged women, suggesting a shared potential mechanism.
To investigate a possible association, a matched case-control study was conducted, with data obtained from the Taiwan National Health Insurance Database. There were 5,751 new cases of Sjögren’s syndrome that were identified and validated by at least two qualified rheumatologists and matched to 86,265 controls from the general population according to age, gender, and year of diagnosis. Patients with rheumatoid arthritis and systemic lupus erythematosus were excluded. International Classification of Disease codes were used to identify individuals who had prior TB or NTM infections.
The mean age of patients in both groups was 55 years, and approximately 87% of participants in both groups were female. There was a significant difference in baseline Charlson Comorbidity Index scores between cases and controls (0.5 vs. 0.4; P less than .001), and more cases than controls had bronchiectasis (4.1% vs. 1.3%; P less than .001). Results were adjusted accordingly.
While there was an association between NTM infection and Sjögren’s syndrome, there was no association with tuberculous mycobacteria infection.
Of course, it is not clear if infection with NTM actually causes the condition, and reverse causality cannot be ruled out, Dr. Chen said, so further mechanistic studies would be needed to investigate NTM’s possible role in the development of Sjögren’s syndrome.
Dr. Chen and coauthors had nothing to disclose.
MADRID – in a large population-based study reported at the European Congress of Rheumatology.
Study investigator Hsin-Hua Chen, MD, and his colleagues at the Taichung (Taiwan) Veterans Hospital found that the adjusted odds ratio for having Sjögren’s syndrome after nontuberculous mycobacteria infection (NTM) was 11.24, with a 95% confidence interval of 2.37-53.24.
The risk for having Sjögren’s syndrome was found to be highest in those aged 40-65 years, versus those older than 65 (aOR, 39.24; P = .09) and in those with no prior history of bronchiectasis (aOR, 37.98; P = .09). Although, in both analyses, the 95% CIs were very wide (3.97-387.75 and 3.83-376.92, respectively), and the P values were not significant.
Dr. Chen and his colleagues decided to look at the association between tuberculous or nontuberculous mycobacteria with Sjögren’s syndrome for several reasons. First, mycobacterial infections have been linked to the development of autoimmunity. Second, there has been an increased incidence of tuberculosis reported in patients with Sjögren’s syndrome. Third, both Sjögren’s and infection with NTM occurred predominantly in middle-aged women, suggesting a shared potential mechanism.
To investigate a possible association, a matched case-control study was conducted, with data obtained from the Taiwan National Health Insurance Database. There were 5,751 new cases of Sjögren’s syndrome that were identified and validated by at least two qualified rheumatologists and matched to 86,265 controls from the general population according to age, gender, and year of diagnosis. Patients with rheumatoid arthritis and systemic lupus erythematosus were excluded. International Classification of Disease codes were used to identify individuals who had prior TB or NTM infections.
The mean age of patients in both groups was 55 years, and approximately 87% of participants in both groups were female. There was a significant difference in baseline Charlson Comorbidity Index scores between cases and controls (0.5 vs. 0.4; P less than .001), and more cases than controls had bronchiectasis (4.1% vs. 1.3%; P less than .001). Results were adjusted accordingly.
While there was an association between NTM infection and Sjögren’s syndrome, there was no association with tuberculous mycobacteria infection.
Of course, it is not clear if infection with NTM actually causes the condition, and reverse causality cannot be ruled out, Dr. Chen said, so further mechanistic studies would be needed to investigate NTM’s possible role in the development of Sjögren’s syndrome.
Dr. Chen and coauthors had nothing to disclose.
MADRID – in a large population-based study reported at the European Congress of Rheumatology.
Study investigator Hsin-Hua Chen, MD, and his colleagues at the Taichung (Taiwan) Veterans Hospital found that the adjusted odds ratio for having Sjögren’s syndrome after nontuberculous mycobacteria infection (NTM) was 11.24, with a 95% confidence interval of 2.37-53.24.
The risk for having Sjögren’s syndrome was found to be highest in those aged 40-65 years, versus those older than 65 (aOR, 39.24; P = .09) and in those with no prior history of bronchiectasis (aOR, 37.98; P = .09). Although, in both analyses, the 95% CIs were very wide (3.97-387.75 and 3.83-376.92, respectively), and the P values were not significant.
Dr. Chen and his colleagues decided to look at the association between tuberculous or nontuberculous mycobacteria with Sjögren’s syndrome for several reasons. First, mycobacterial infections have been linked to the development of autoimmunity. Second, there has been an increased incidence of tuberculosis reported in patients with Sjögren’s syndrome. Third, both Sjögren’s and infection with NTM occurred predominantly in middle-aged women, suggesting a shared potential mechanism.
To investigate a possible association, a matched case-control study was conducted, with data obtained from the Taiwan National Health Insurance Database. There were 5,751 new cases of Sjögren’s syndrome that were identified and validated by at least two qualified rheumatologists and matched to 86,265 controls from the general population according to age, gender, and year of diagnosis. Patients with rheumatoid arthritis and systemic lupus erythematosus were excluded. International Classification of Disease codes were used to identify individuals who had prior TB or NTM infections.
The mean age of patients in both groups was 55 years, and approximately 87% of participants in both groups were female. There was a significant difference in baseline Charlson Comorbidity Index scores between cases and controls (0.5 vs. 0.4; P less than .001), and more cases than controls had bronchiectasis (4.1% vs. 1.3%; P less than .001). Results were adjusted accordingly.
While there was an association between NTM infection and Sjögren’s syndrome, there was no association with tuberculous mycobacteria infection.
Of course, it is not clear if infection with NTM actually causes the condition, and reverse causality cannot be ruled out, Dr. Chen said, so further mechanistic studies would be needed to investigate NTM’s possible role in the development of Sjögren’s syndrome.
Dr. Chen and coauthors had nothing to disclose.
AT THE EULAR 2017 CONGRESS
Key clinical point: Screening for Sjögren’s syndrome might be needed among people infected with nontuberculous mycobacteria if these data are confirmed.
Major finding: A strong association between prior infection with NTM and the development of Sjögren’s syndrome was found.
Data source: A retrospective, population-based study involving 5,751 newly diagnosed cases of Sjögren’s syndrome and 86,265 control subjects.
Disclosures: The author had no disclosures.
Determinants of Suboptimal Migraine Diagnosis and Treatment in the Primary Care Setting
From the Mayo Clinic, Scottsdale, AZ.
Abstract
- Objective: To review the impact of migraine and explore the barriers to optimal migraine diagnosis and treatment.
- Methods: Review of the literature.
- Results: Several factors may play a role in the inadequate care of migraine patients, including issues related to poor access to care, diagnostic insight, misdiagnosis, adherence to treatment, and management of comorbidities. Both patient and physician factors play an important role and many be modifiable.
- Conclusions: A focus on education of both patients and physicians is of paramount importance to improve the care provided to migraine patients. Patient evaluations should be multisystemic and include addressing comorbid conditions as well as a discussion about appropriate use of prevention and avoidance of medication overuse.
Key words: migraine; triptans; medication overuse headache; medication adherence; primary care.
Migraine is a common, debilitating condition that is a significant source of reduced productivity and increased disability [1]. According to the latest government statistics, 14.2% of US adults have reported having migraine or severe headaches in the previous 3 months, with an overall age-adjusted 3-month prevalence of 19.1% in females and 9.0% in males [2]. In a self-administered headache questionnaire mailed to 120,000 representative US households, the 1-year period prevalence for migraine was 11.7% (17.1% in women and 5.6% in men). Prevalence peaked in middle life and was lower in adolescents and those older than age 60 years [3]. Migraine is an important cause of reduced health-related quality of life and has a very high economic burden [4]. This effect is even more marked in those with chronic migraine, who are even more likely to have professional and social absenteeism and experience more severe disability [4].
Migraine and headache are a common reason for primary care physician (PCP) visits. Some estimates suggest that as many as 10% of primary care consultations are due to headache [5]. Approximately 75% of all patients complaining of headache in primary care will eventually be diagnosed with migraine [6]. Of these, as many as 1% to 5% will have chronic migraine [6].
Despite the high frequency and social and economic impact of migraine, migraine is underrecognized and undertreated. A survey of US households revealed that only 13% of migraineurs were currently using a preventive thrapy while 43.3% had never used one [3]. This is despite the fact that 32.4% met expert criteria for consideration of a preventive medication [3]. The reasons for underrecognition and undertreatment are multifactorial and include both patient and physician factors.
Physician Factors
Although migraine and headache are a leading cause of physicians visits, most physicians have had little formal training in headache. In the United States, medical students spend an average of 1 hour of preclinical and 2 hours of clinical education in headache [7]. Furthermore, primary care physicians receive little formal training in headache during residency [8]. In addition to the lack of formal training, there is also a lack of substantial clinic time available to fully evaluate and treat a new headache patient in the primary care setting [8]. Headache consultations can often be timely and detail-driven in order to determine the correct diagnosis and treatment [9].
Misdiagnosis
Evidence suggests that misdiagnosis plays a large role in the suboptimal management of migraineurs. Studies have shown that as many as 59.7% of migraineurs were not given a diagnosis of migraine by their primary care provider [10]. Common mistaken diagnoses include tension-type headache [11], “sinus headache” [12], cervical pain syndrome or cervicogenic headache [13], and “stress headache” [14].
The reasons for these misdiagnoses is not certain. It may be that the patient and practitioner assume that location of the pain is suggestive of the cause [13]. This is even though more than half of those with migraine have associated neck pain [15]. A recent study suggests that 60% of migraineurs who self-reported a diagnosis of cervical pain have been subsequently diagnosed with cervicalgia by a physician [13]. If patients endorse stress as a precipitant or the presence of cervical pain, they are more likely to obtain a diagnosis other than migraine. The presence of aura in association with the headache appears to be protective against misdiagnosis [13].
Similarly, patients are often given a diagnosis of “sinus headache.” This diagnosis is often made without radiologic evidence of sinusitis and even in those with a more typical migraine headache [16]. In one survey, 40% of patients meeting criteria for migraine were given this diagnosis. Many of these patients did have nasal symptoms or facial pain without clear evidence or rhinosinusitis, and in some cases these symptoms would respond to migraine treatments [16]. This is a particularly important misdiagnosis to highlight, as attributing symptoms to sinus disease may lead to unnecessary consultations and even sinus instrumentation.
In addition to common misdiagnoses, many PCPs are unfamiliar with the “red flags” that may indicate a secondary headache disorder and are also unfamiliar with appropriate use of neuroimaging in headache patients [17].
Misuse of As-Needed Medications
Studies have suggested that a large proportion of PCPs will prescribe nonspecific analgesics for migraine rather than migraine-specific medications [18]. These treatments may include NSAIDs, acetaminophen, barbiturates, and even opiates. This appears to be the pattern even for those with severe attacks [18], suggesting that migraine-specific medications such as triptans may be underused in the primary care setting. Postulated reasons for this pattern include lack of physician knowledge regarding the specific recommendations for managing migraine, the cost of medications, as well as lack of insurance coverage for these medications [19]. Misuse of as-needed medications can lead to medication overuse headache (MOH), which is an underrecognized problem in the primary care setting [20]. In a survey of PCPs in Boston, only 54% of PCPs were aware that barbiturates can cause MOH and only 34% were aware that opiates can cause MOH [17]. The same survey revealed that approximately 20% of PCPs had never made the diagnosis of MOH [17].
Underuse of Preventive Medications
As many as 40% of migraineurs need preventive therapy, but only approximately 13% are currently receiving it [3]. Additionally, the average time from diagnosis of migraine to instituting preventive treatment is 4.3 years, and often there is only a single preventive medication trial if one is instituted [21]. The reasons for this appear to be complex. The physician factors contributing to the underuse of preventive medications include inadequate education, discomfort and inadequate time for assessments. Only 27.8% of surveyed PCPs were aware of the American Academy of Neurology guidelines for prescribing preventive medications [17].
There may be an underestimate of the disability experienced by migraineurs, which can explain some of the underuse of preventive medications. While many PCPs endorse inquiring about headache-related disability, many do not used validated scales such as the Migraine Disability Assessment Score (MIDAS) or the Headache Impact Test (HIT) [17]. In addition, patients often underreport their headache days and report only their severe exacerbations unless clearly asked about a daily headache [22]. This may be part of the reason why only 20% of migraineurs who meet criteria for chronic migraine are diagnosed as such and why preventatives may not be offered [23].
After preventatives are started, less than 25% of patients will be adherent to oral migraine preventive agents at 1 year [24]. Common reasons for discontinuing preventives include adverse effects and perceived inefficacy [22]. Preventive medications may need a 6- to 8-week trial before efficacy is determined, but in practice medications may be stopped before this threshold is reached. Inadequate follow-up and lack of detail with regard to medication trials may result in the perception of an intractable patient prematurely. It has been suggested that a systematic approach to documenting and choosing preventive agents is helpful in the treatment of migraine [25], although this is not always practical in the primary care setting.
Another contributor to underuse of effective prophylaxis is related to access. Treatment with onabotulinumtoxin A, an efficacious prophylactic treatment approved for select chronic migraine patients [26], will usually require referral to a headache specialist, which is not always available to PCPs in a timely manner [7].
Nonpharmacologic Approaches
Effective nonpharmacologic treatment modalities for migraine, such as cognitive-behavioral therapy and biofeedback [27], are not commonly recommended by PCPs [17]. Instead, there appears to be more focus on avoidance of triggers and referral to non–evidence-based resources, such as special diets and massage therapy [17]. While these methods are not always inappropriate, it should be noted that they often have little or no evidence for efficacy.
Patients often wish for non-medication approaches to migraine management, but for those with significant and severe disability, these are probably insufficient. In these patients, non-medication approaches may best be used as a supplement to pharmacological treatment, with education on pharmacologic prevention given. Neuromodulation is a promising, novel approach that is emerging as a new treatment for migraine, but likely will require referral to a headache specialist.
Suboptimal Management of Migraine Comorbidities
There are several disorders that are commonly comorbid with migraine. Among the most common are anxiety, depression, medication (and caffeine) overuse, obesity, and sleep disorders [22]. A survey of PCPs reveals that only 50.6% of PCPs screen for anxiety, 60.2% for depression, and 73.5% for sleep disorders [17]. They are, for the most part, modifiable or treatable conditions and their proper management may help ease migraine disability.
In addition, the presence of these comorbidities may alter choice of treatment, for example, favoring the use of an serotonin and norepinephrine reuptake inhibitor such as venlafaxine for treatment in those with comorbid anxiety and depression. It is also worthwhile to have a high index of suspicion for obstructive sleep apnea in patients with headache, particularly in the obese and in those who endorse nonrestorative sleep or excessive daytime somnolence. It appears that patients who are adherent to the treatment of sleep apnea are more likely to report improvement in their headache [28].
Given the time constraints that often exist in the PCP office setting, addressing these comorbidities thoroughly is not always possible. It is reasonable, however, to have patients use screening tools while in the waiting room or prior to an appointment, to better identify those with modifiable comorbidities. Depression, anxiety, and excessive daytime sleepiness can all be screened for relatively easily with tools such as the PHQ-9 [29], GAD-7 [30] and Epworth Sleepiness Scale [31], respectively. A positive screen on any of these could lead the PCP to further investigate these entities as a possible contributor to migraine.
Patient Factors
In addition to the physician factors identified above, patient factors can contribute to the suboptimal management of migraine as well. These factors include a lack insight into diagnosis, poor compliance with treatment of migraine or its comorbidities, and overuse of abortive medications. There are also less modifiable patient factors such as socioeconomic status and the stigma that may be associated with migraine.
Poor Insight Into Diagnosis
Despite the high prevalence and burden of migraine in the general population, there is a staggering lack of awareness among migraineurs. Some estimates state that as many as 54% of patients were unaware that their headaches represented migraine [32]. The most common self-reported diagnoses in migraineurs are sinus headache (39%), tension-type headache (31%) and stress headache (29%) [14]. In addition, many patients believe they are suffering from cervical spine–related pain [13]. This is likely due to the common presence of posteriorly located pain, attacks triggered by poor sleep, or attacks associated with weather changes [13]. Patients presenting with aura are more likely to report and to receive a physician diagnosis of migraine [14]. Women are more likely to receive and report a diagnosis of migraine compared with men [32].
There are many factors that play a role in poor insight. Many patients appear to believe that the location of the pain is suggestive of the cause [13]. Many patients never seek out consultation for their headaches, and thus never receive a proper diagnosis [33]. Some patients may seek out medical care for their headaches, but fail to remember their diagnosis or receive an improper diagnosis [34].
Poor Adherence
The body of literature examining adherence with headache treatment is growing, but remains small [35]. In a recent systematic review of treatment adherence in pediatric and adult patients with headache, adherence rates in adults with headache ranged from 25% to 94% [35]. In this review, prescription claims data analyses found poor persistence in patients prescribed triptans for migraine treatment. In one large claims-based study, 53.8% of patients receiving a new triptan prescription did not persistently refill their index triptan [36]. Although some of these patients switched to an alternative triptan, the majority switched to a non-triptan migraine medication, including opioids and nonsteroidal anti-inflammatory drugs [36].
Cady and colleagues’ study of lapsed and sustained triptan users found that sustained users were significantly more satisfied with their medication, confident in the medication’s ability to control headache, and reported control of migraine with fewer doses of medication [37]. The authors concluded that the findings suggest that lapsed users may not be receiving optimal treatment. In a review by Rains et al [38], the authors found that headache treatment adherence declines “with more frequent and complex dosing regimens, side effects, and costs, and is subject to a wide range of psychosocial influences.”
Adherence issues also exist for migraine prevention. Less than 25% of chronic migraine patients continue to take oral preventive therapies at 1 year [24]. The reasons for this nonadherence are not completely clear, but are likely multifactorial. Preventives may take several weeks to months to become effective, which may contribute to noncompliance. In addition, migraineurs appears to have inadequate follow-up for migraine. Studies from France suggest that only 18% of those aware of their migraine diagnosis received medical follow-up [39].
Medication Overuse
While the data is not entirely clear, it is likely that overuse of as-needed medication plays a role in migraine chronification [40]. The reasons for medication overuse in the migraine population include some of the issues already highlighted above, including inadequate patient education, poor insight into diagnosis, not seeking care, misdiagnosis, and treatment nonadherence. Patients should be educated on the proper use of as-needed medication. Limits to medication use should be set during the physician-patient encounter. Patients should be counselled to limit their as-needed medication to no more than 10 days per month to reduce the risk of medication overuse headache. Ideally, opiates and barbiturates should be avoided, and never used as first-line therapy in patients who lack contraindications to NSAIDs and triptans. If their use in unavoidable for other reasons, they should be used sparingly, as use on as few as 5 to 8 days per month can be problematic [41]. Furthermore it is important to note that if patients are using several different acute analgesics, the combined total use of all as-needed pain medications needs to be less than 10 days per month to reduce the potential for medication overuse headache.
Socioeconomic Factors
Low socioeconomic status has been associated with an increased prevalence for all headache forms and an increased migraine attack frequency [42], but there appear to be few studies looking at the impact of low socioeconomic status and treatment. Lipton et al found that health insurance status was an important predictor of persons with migraine consulting a health care professional [43]. Among consulters, women were far more likely to be diagnosed than men, suggesting that gender bias in diagnosis may be an important barrier for men. Higher household income appeared to be a predictor for receiving a correct diagnosis of migraine. These researchers also found economic barriers related to use of appropriate prescription medications [43]. Differences in diagnosis and treatment may indicate racial and ethnic disparities in access and quality of care for minority patients [44].
Stigma
At least 1 study has reported that migraine patients experience stigma. In Young et al’s study of 123 episodic migraine patients, 123 chronic migraine patients, and 62 epilepsy patients, adjusted stigma was similar for chronic migraine and epilepsy, which were greater than for episodic migraine [45]. Stigma correlated most strongly with inability to work. Migraine patients reported equally high stigma scores across age, income, and education. The stigma of migraine may pose a barrier to seeking consultation and treatment. Further, the perception that migraine is “just a headache” may lead to stigmatizing attitudes on the part of friends, family, and coworkers of patients with migraine.
Conclusions and Recommendations
Migraine is a prevalent and frequently disabling condition that is underrecognized and undertreated in the primary care setting. Both physician and patient factors pose barriers to the optimal diagnosis and treatment of migraine. Remedies to address these barriers include education of both patients and physicians first and foremost. Targeting physician education in medical school and during residency training, including in primary care subspecialties, could include additional didactic teaching, but also clinical encounters in headache subspecialty clinics to increase exposure. Patient advocacy groups and public campaigns to improve understanding of migraine in the community may be a means for improving patient education and reducing stigma. Patients should be encouraged to seek out consultations for headache to reduce long-term headache disability. Management of comorbidities is paramount, and screening tools for migraine-associated disability, anxiety, depression, and medication use may be helpful to implement in the primary care setting as they are easy to use and time saving.
Recent surveys of PCPs suggest that the resource that is most desired is ready access to subspecialists for advice and “curb-side” consultation [17]. While this solution is not always practical, it may be worthwhile exploring closer relationships between primary care and subspecialty headache clinics, or perhaps more access to e-consultation or telephone consultation for more rural areas. Recently, Minen et al examined education strategies for PCPs. While in-person education sessions with PCPs were poorly attended, multiple possibilities for further education were identified. It was suggested that PCPs having real-time access to resources during the patient encounter would improve their comfort in managing patients. This includes online databases, simple algorithms for treatment, and directions for when to refer to a neurologist [46]. In addition, it may be worthwhile to train not only PCPs but also nursing and allied health staff so that they can provide headache education to patients. This may help ease some of the time burden on PCPs as well as provide a collaborative environment in which headache can be managed [46].
Corresponding author: William S. Kingston, MD, Mayo Clinic, 13400 E. Shea Blvd., Scottsdale, AZ 85259.
Financial disclosures: None.
1. Stewart WF, Schechter A, Lipton RB. Migraine heterogeneity. Disability, pain intensity and attack frequency and duration. Neurology 1994; 44(suppl 4):S24–S39
2. Burch RC, Loder S, Loder E, Smitherman TA. The prevalence of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache 2015;55:21–34.
3. Lipton RB, Bigal ME, Diamond M, et al. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 2007;68:343–9.
4. Blumenfeld AM, Varon SF, Wilcox TK, et al. Disability, HRQoL and resource use amoung chronic and episodic migraineurs: results from the International Burden of Migraine Study (IBMS). Cephalalgia 2011;31:301–15.
5. Ahmed F. Headache disorders: differentiating and managing the common subtypes. Br J Pain 2012;6:124–32.
6. Natoli JL, Manack A, Dean B, et al. Global prevalence of chronic migraine: a systemic review. Cephalalgia 2010;30: 599–609.
7. Finkel AG. American academic headache specialists in neurology: Practice characteristics and culture. Cephalalgia 2004; 24:522–7.
8. Sheftell FD, Cady RK, Borchert LD, et al. Optimizing the diagnosis and treatment of migraine. J Am Acad Nurse Pract 2005;17:309–17.
9. Lipton RB, Scher AI, Steiner TJ, et al. Patterns of health care utilization for migraine in England and in the United States. Neurology 2003;60:441–8.
10. De Diego EV, Lanteri-Minet M. Recognition and management of migraine in primary care: Influence of functional impact measures by the Headache Impact Test (HIT). Cephalalgia 2005;25:184–90.
11. Miller S, Matharu MS. Migraine is underdiagnosed and undertreated. Practitioner 2014;258:19–24.
12. Al-Hashel JY, Ahmed SF, Alroughani R, et al. Migraine misdiagnosis as sinusitis, a delay that can last for many years. J Headache Pain 2013;14:97.
13. Viana M, Sances G, Terrazzino S, et al. When cervical pain is actually migraine: an observational study in 207 patients. Cephalalgia 2016. Epub ahead of print.
14. Diamond MD, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: Results from the American Migraine Prevalence and Prevention Study. Headache 2007;47:355–63.
15. Aguila MR, Rebbeck T, Mendoza KG, et al. Definitions and participant characteristics of frequent recurrent headache types in clinical trials: A systematic review. Cephalalgia 2017. Epub ahead of print.
16. Senbil N, Yavus Gurer YK, Uner C, Barut Y. Sinusitis in children and adolescents with chronic or recurrent headache: A case-control study. J Headache Pain 2008;9:33–6.
17. Minen MT, Loder E, Tishler L, Silbersweig D. Migraine diagnosis and treatment: A knowledge and needs assessment amoung primary care providers. Cephalalgia 2016; 36:358–70.
18. MacGregor EA, Brandes J, Eikerman A. Migraine prevalence and treatment patterns: The global migraine and zolmitriptan evaluation survey. Headache 2003;33:19–26.
19. Khan S, Mascarenhas A, Moore JE, et al. Access to triptans for acute episodic migraine: a qualitative study. Headache 2015; 44(suppl 4):199–211.
20. Tepper SJ. Medication-overuse headache. Continuum 2012;18:807–22.
21. Dekker F, Dielemann J, Neven AK, et al. Preventive treatment for migraine in primary care, a population based study in the Netherlands. Cephalalgia 2013;33:1170–8.
22. Starling AJ, Dodick DW. Best practices for patients with chronic migraine: burden, diagnosis and management in primary care. Mayo Clin Proc 2015;90:408–14.
23. Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology 2008;71:559–66.
24. Hepp Z, Dodick D, Varon S, et al. Adherence to oral migraine preventive-medications among patients with chronic migraine. Cephalalgia 2015;35:478–88.
25. Smith JH, Schwedt TJ. What constitutes an “adequate” trial in migraine prevention? Curr Pain Headache Rep 2015;19:52.
26. Dodick DW, Turkel CC, DeGryse RE, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache 2010;50:921–36.
27. Silberstein SD. Practice parameter: evidence based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000;55: 754–62.
28. Johnson KG, Ziemba AM, Garb JL. Improvement in headaches with continuous positive airway pressure for obstructive sleep apnea: a retrospective analysis. Headache 2013;53:333–43.
29. Altura KC, Patten SB, FIest KM, et al. Suicidal ideation in persons with neurological conditions: prevalence, associations and validation of the PHQ-9 for suicidal ideation. Gen Hosp Psychiatry 2016;42:22–6.
30. Seo JG, Park SP. Validation of the Generalized Anxiety Disorder-7 (GAD-7) and GAD-2 in patients with migraine. J Headache Pain 2015;16:97.
31. Corlateanu A, Pylchenko S, DIrcu V, Botnaru V. Predictors of daytime sleepiness in patients with obstructive sleep apnea. Pneumologia 2015;64:21–5.
32. Linde M, Dahlof C. Attitudes and burden of disease among self-considered migraineurs – a nation-wide population-based survey in Sweden. Cephalalgia 2004;24:455–65.
33. Osterhaus JT, Gutterman DL, Plachetka JR. Health care resources and lost labor costs of migraine headaches in the United States. Pharmacoeconomics 1992;36:69–76.
34. Tepper SJ, Dahlof CG, Dowson A et al. Prevalence and diagnosis of migraine in patients consulting their physician with a complaint of headache: Data from the Landmark Study. Headache 2004;44:856–64.
35. Ramsey RR, Ryan JL, Hershey AD, et al. Treatment adherence in patients with headache: a systematic review. Headache 2014;54:795–816.
36. Katic BJ, Rajagopalan S, Ho TW, et al. Triptan persistency among newly initiated users in a pharmacy claims database. Cephalalgia 2011;31:488–500.
37. Cady RK, Maizels M, Reeves DL, Levinson DM, Evans JK. Predictors of adherence to triptans: factors of sustained vs lapsed users. Headache 2009;49:386–94.
38. Rains JC, Lipchik GL, Penzien DB. Behavioral facilitation of medical treatment for headache--part I: Review of headache treatment compliance. Headache 2006;46:1387–94.
39. Lucas C, Chaffaut C, Artaz MA, Lanteri-Minet M. FRAMIG 2000: Medical and therapeutic management of migraine in France. Cephalalgia 2005;25:267–79.
40. Bigal ME, Serrano D, Buse D et al. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal population-based study. Headache 2008;48:1157–68.
41. Diener HC, Limmroth V. Medication-overuse headache: a worldwide problem. Lancet Neurol 2004;3:475–83.
42. Winter AC, Berger K, Buring JE, Kurth T. Associations of socioeconomic status with migraine and non-migraine headache. Cephalalgia 2012;32:159–70.
43. Lipton, RB, Serrano D, Holland S et al. Barriers to the diagnosis and treatment of migraine: effects of sex, income, and headache features. Headache 2013;53: 81–92.
44. Loder S, Sheikh HU, Loder E. The prevalence, burden, and treatment of severe, frequent, and migraine headaches in US minority populations: statistics from National Survey studies. Headache 2015;55:214–28.
45. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8:e54074.
46. Minen A, Shome A, Hapern A, et al. A migraine training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and consideration for further research. Headache 2016;56:725–40.
From the Mayo Clinic, Scottsdale, AZ.
Abstract
- Objective: To review the impact of migraine and explore the barriers to optimal migraine diagnosis and treatment.
- Methods: Review of the literature.
- Results: Several factors may play a role in the inadequate care of migraine patients, including issues related to poor access to care, diagnostic insight, misdiagnosis, adherence to treatment, and management of comorbidities. Both patient and physician factors play an important role and many be modifiable.
- Conclusions: A focus on education of both patients and physicians is of paramount importance to improve the care provided to migraine patients. Patient evaluations should be multisystemic and include addressing comorbid conditions as well as a discussion about appropriate use of prevention and avoidance of medication overuse.
Key words: migraine; triptans; medication overuse headache; medication adherence; primary care.
Migraine is a common, debilitating condition that is a significant source of reduced productivity and increased disability [1]. According to the latest government statistics, 14.2% of US adults have reported having migraine or severe headaches in the previous 3 months, with an overall age-adjusted 3-month prevalence of 19.1% in females and 9.0% in males [2]. In a self-administered headache questionnaire mailed to 120,000 representative US households, the 1-year period prevalence for migraine was 11.7% (17.1% in women and 5.6% in men). Prevalence peaked in middle life and was lower in adolescents and those older than age 60 years [3]. Migraine is an important cause of reduced health-related quality of life and has a very high economic burden [4]. This effect is even more marked in those with chronic migraine, who are even more likely to have professional and social absenteeism and experience more severe disability [4].
Migraine and headache are a common reason for primary care physician (PCP) visits. Some estimates suggest that as many as 10% of primary care consultations are due to headache [5]. Approximately 75% of all patients complaining of headache in primary care will eventually be diagnosed with migraine [6]. Of these, as many as 1% to 5% will have chronic migraine [6].
Despite the high frequency and social and economic impact of migraine, migraine is underrecognized and undertreated. A survey of US households revealed that only 13% of migraineurs were currently using a preventive thrapy while 43.3% had never used one [3]. This is despite the fact that 32.4% met expert criteria for consideration of a preventive medication [3]. The reasons for underrecognition and undertreatment are multifactorial and include both patient and physician factors.
Physician Factors
Although migraine and headache are a leading cause of physicians visits, most physicians have had little formal training in headache. In the United States, medical students spend an average of 1 hour of preclinical and 2 hours of clinical education in headache [7]. Furthermore, primary care physicians receive little formal training in headache during residency [8]. In addition to the lack of formal training, there is also a lack of substantial clinic time available to fully evaluate and treat a new headache patient in the primary care setting [8]. Headache consultations can often be timely and detail-driven in order to determine the correct diagnosis and treatment [9].
Misdiagnosis
Evidence suggests that misdiagnosis plays a large role in the suboptimal management of migraineurs. Studies have shown that as many as 59.7% of migraineurs were not given a diagnosis of migraine by their primary care provider [10]. Common mistaken diagnoses include tension-type headache [11], “sinus headache” [12], cervical pain syndrome or cervicogenic headache [13], and “stress headache” [14].
The reasons for these misdiagnoses is not certain. It may be that the patient and practitioner assume that location of the pain is suggestive of the cause [13]. This is even though more than half of those with migraine have associated neck pain [15]. A recent study suggests that 60% of migraineurs who self-reported a diagnosis of cervical pain have been subsequently diagnosed with cervicalgia by a physician [13]. If patients endorse stress as a precipitant or the presence of cervical pain, they are more likely to obtain a diagnosis other than migraine. The presence of aura in association with the headache appears to be protective against misdiagnosis [13].
Similarly, patients are often given a diagnosis of “sinus headache.” This diagnosis is often made without radiologic evidence of sinusitis and even in those with a more typical migraine headache [16]. In one survey, 40% of patients meeting criteria for migraine were given this diagnosis. Many of these patients did have nasal symptoms or facial pain without clear evidence or rhinosinusitis, and in some cases these symptoms would respond to migraine treatments [16]. This is a particularly important misdiagnosis to highlight, as attributing symptoms to sinus disease may lead to unnecessary consultations and even sinus instrumentation.
In addition to common misdiagnoses, many PCPs are unfamiliar with the “red flags” that may indicate a secondary headache disorder and are also unfamiliar with appropriate use of neuroimaging in headache patients [17].
Misuse of As-Needed Medications
Studies have suggested that a large proportion of PCPs will prescribe nonspecific analgesics for migraine rather than migraine-specific medications [18]. These treatments may include NSAIDs, acetaminophen, barbiturates, and even opiates. This appears to be the pattern even for those with severe attacks [18], suggesting that migraine-specific medications such as triptans may be underused in the primary care setting. Postulated reasons for this pattern include lack of physician knowledge regarding the specific recommendations for managing migraine, the cost of medications, as well as lack of insurance coverage for these medications [19]. Misuse of as-needed medications can lead to medication overuse headache (MOH), which is an underrecognized problem in the primary care setting [20]. In a survey of PCPs in Boston, only 54% of PCPs were aware that barbiturates can cause MOH and only 34% were aware that opiates can cause MOH [17]. The same survey revealed that approximately 20% of PCPs had never made the diagnosis of MOH [17].
Underuse of Preventive Medications
As many as 40% of migraineurs need preventive therapy, but only approximately 13% are currently receiving it [3]. Additionally, the average time from diagnosis of migraine to instituting preventive treatment is 4.3 years, and often there is only a single preventive medication trial if one is instituted [21]. The reasons for this appear to be complex. The physician factors contributing to the underuse of preventive medications include inadequate education, discomfort and inadequate time for assessments. Only 27.8% of surveyed PCPs were aware of the American Academy of Neurology guidelines for prescribing preventive medications [17].
There may be an underestimate of the disability experienced by migraineurs, which can explain some of the underuse of preventive medications. While many PCPs endorse inquiring about headache-related disability, many do not used validated scales such as the Migraine Disability Assessment Score (MIDAS) or the Headache Impact Test (HIT) [17]. In addition, patients often underreport their headache days and report only their severe exacerbations unless clearly asked about a daily headache [22]. This may be part of the reason why only 20% of migraineurs who meet criteria for chronic migraine are diagnosed as such and why preventatives may not be offered [23].
After preventatives are started, less than 25% of patients will be adherent to oral migraine preventive agents at 1 year [24]. Common reasons for discontinuing preventives include adverse effects and perceived inefficacy [22]. Preventive medications may need a 6- to 8-week trial before efficacy is determined, but in practice medications may be stopped before this threshold is reached. Inadequate follow-up and lack of detail with regard to medication trials may result in the perception of an intractable patient prematurely. It has been suggested that a systematic approach to documenting and choosing preventive agents is helpful in the treatment of migraine [25], although this is not always practical in the primary care setting.
Another contributor to underuse of effective prophylaxis is related to access. Treatment with onabotulinumtoxin A, an efficacious prophylactic treatment approved for select chronic migraine patients [26], will usually require referral to a headache specialist, which is not always available to PCPs in a timely manner [7].
Nonpharmacologic Approaches
Effective nonpharmacologic treatment modalities for migraine, such as cognitive-behavioral therapy and biofeedback [27], are not commonly recommended by PCPs [17]. Instead, there appears to be more focus on avoidance of triggers and referral to non–evidence-based resources, such as special diets and massage therapy [17]. While these methods are not always inappropriate, it should be noted that they often have little or no evidence for efficacy.
Patients often wish for non-medication approaches to migraine management, but for those with significant and severe disability, these are probably insufficient. In these patients, non-medication approaches may best be used as a supplement to pharmacological treatment, with education on pharmacologic prevention given. Neuromodulation is a promising, novel approach that is emerging as a new treatment for migraine, but likely will require referral to a headache specialist.
Suboptimal Management of Migraine Comorbidities
There are several disorders that are commonly comorbid with migraine. Among the most common are anxiety, depression, medication (and caffeine) overuse, obesity, and sleep disorders [22]. A survey of PCPs reveals that only 50.6% of PCPs screen for anxiety, 60.2% for depression, and 73.5% for sleep disorders [17]. They are, for the most part, modifiable or treatable conditions and their proper management may help ease migraine disability.
In addition, the presence of these comorbidities may alter choice of treatment, for example, favoring the use of an serotonin and norepinephrine reuptake inhibitor such as venlafaxine for treatment in those with comorbid anxiety and depression. It is also worthwhile to have a high index of suspicion for obstructive sleep apnea in patients with headache, particularly in the obese and in those who endorse nonrestorative sleep or excessive daytime somnolence. It appears that patients who are adherent to the treatment of sleep apnea are more likely to report improvement in their headache [28].
Given the time constraints that often exist in the PCP office setting, addressing these comorbidities thoroughly is not always possible. It is reasonable, however, to have patients use screening tools while in the waiting room or prior to an appointment, to better identify those with modifiable comorbidities. Depression, anxiety, and excessive daytime sleepiness can all be screened for relatively easily with tools such as the PHQ-9 [29], GAD-7 [30] and Epworth Sleepiness Scale [31], respectively. A positive screen on any of these could lead the PCP to further investigate these entities as a possible contributor to migraine.
Patient Factors
In addition to the physician factors identified above, patient factors can contribute to the suboptimal management of migraine as well. These factors include a lack insight into diagnosis, poor compliance with treatment of migraine or its comorbidities, and overuse of abortive medications. There are also less modifiable patient factors such as socioeconomic status and the stigma that may be associated with migraine.
Poor Insight Into Diagnosis
Despite the high prevalence and burden of migraine in the general population, there is a staggering lack of awareness among migraineurs. Some estimates state that as many as 54% of patients were unaware that their headaches represented migraine [32]. The most common self-reported diagnoses in migraineurs are sinus headache (39%), tension-type headache (31%) and stress headache (29%) [14]. In addition, many patients believe they are suffering from cervical spine–related pain [13]. This is likely due to the common presence of posteriorly located pain, attacks triggered by poor sleep, or attacks associated with weather changes [13]. Patients presenting with aura are more likely to report and to receive a physician diagnosis of migraine [14]. Women are more likely to receive and report a diagnosis of migraine compared with men [32].
There are many factors that play a role in poor insight. Many patients appear to believe that the location of the pain is suggestive of the cause [13]. Many patients never seek out consultation for their headaches, and thus never receive a proper diagnosis [33]. Some patients may seek out medical care for their headaches, but fail to remember their diagnosis or receive an improper diagnosis [34].
Poor Adherence
The body of literature examining adherence with headache treatment is growing, but remains small [35]. In a recent systematic review of treatment adherence in pediatric and adult patients with headache, adherence rates in adults with headache ranged from 25% to 94% [35]. In this review, prescription claims data analyses found poor persistence in patients prescribed triptans for migraine treatment. In one large claims-based study, 53.8% of patients receiving a new triptan prescription did not persistently refill their index triptan [36]. Although some of these patients switched to an alternative triptan, the majority switched to a non-triptan migraine medication, including opioids and nonsteroidal anti-inflammatory drugs [36].
Cady and colleagues’ study of lapsed and sustained triptan users found that sustained users were significantly more satisfied with their medication, confident in the medication’s ability to control headache, and reported control of migraine with fewer doses of medication [37]. The authors concluded that the findings suggest that lapsed users may not be receiving optimal treatment. In a review by Rains et al [38], the authors found that headache treatment adherence declines “with more frequent and complex dosing regimens, side effects, and costs, and is subject to a wide range of psychosocial influences.”
Adherence issues also exist for migraine prevention. Less than 25% of chronic migraine patients continue to take oral preventive therapies at 1 year [24]. The reasons for this nonadherence are not completely clear, but are likely multifactorial. Preventives may take several weeks to months to become effective, which may contribute to noncompliance. In addition, migraineurs appears to have inadequate follow-up for migraine. Studies from France suggest that only 18% of those aware of their migraine diagnosis received medical follow-up [39].
Medication Overuse
While the data is not entirely clear, it is likely that overuse of as-needed medication plays a role in migraine chronification [40]. The reasons for medication overuse in the migraine population include some of the issues already highlighted above, including inadequate patient education, poor insight into diagnosis, not seeking care, misdiagnosis, and treatment nonadherence. Patients should be educated on the proper use of as-needed medication. Limits to medication use should be set during the physician-patient encounter. Patients should be counselled to limit their as-needed medication to no more than 10 days per month to reduce the risk of medication overuse headache. Ideally, opiates and barbiturates should be avoided, and never used as first-line therapy in patients who lack contraindications to NSAIDs and triptans. If their use in unavoidable for other reasons, they should be used sparingly, as use on as few as 5 to 8 days per month can be problematic [41]. Furthermore it is important to note that if patients are using several different acute analgesics, the combined total use of all as-needed pain medications needs to be less than 10 days per month to reduce the potential for medication overuse headache.
Socioeconomic Factors
Low socioeconomic status has been associated with an increased prevalence for all headache forms and an increased migraine attack frequency [42], but there appear to be few studies looking at the impact of low socioeconomic status and treatment. Lipton et al found that health insurance status was an important predictor of persons with migraine consulting a health care professional [43]. Among consulters, women were far more likely to be diagnosed than men, suggesting that gender bias in diagnosis may be an important barrier for men. Higher household income appeared to be a predictor for receiving a correct diagnosis of migraine. These researchers also found economic barriers related to use of appropriate prescription medications [43]. Differences in diagnosis and treatment may indicate racial and ethnic disparities in access and quality of care for minority patients [44].
Stigma
At least 1 study has reported that migraine patients experience stigma. In Young et al’s study of 123 episodic migraine patients, 123 chronic migraine patients, and 62 epilepsy patients, adjusted stigma was similar for chronic migraine and epilepsy, which were greater than for episodic migraine [45]. Stigma correlated most strongly with inability to work. Migraine patients reported equally high stigma scores across age, income, and education. The stigma of migraine may pose a barrier to seeking consultation and treatment. Further, the perception that migraine is “just a headache” may lead to stigmatizing attitudes on the part of friends, family, and coworkers of patients with migraine.
Conclusions and Recommendations
Migraine is a prevalent and frequently disabling condition that is underrecognized and undertreated in the primary care setting. Both physician and patient factors pose barriers to the optimal diagnosis and treatment of migraine. Remedies to address these barriers include education of both patients and physicians first and foremost. Targeting physician education in medical school and during residency training, including in primary care subspecialties, could include additional didactic teaching, but also clinical encounters in headache subspecialty clinics to increase exposure. Patient advocacy groups and public campaigns to improve understanding of migraine in the community may be a means for improving patient education and reducing stigma. Patients should be encouraged to seek out consultations for headache to reduce long-term headache disability. Management of comorbidities is paramount, and screening tools for migraine-associated disability, anxiety, depression, and medication use may be helpful to implement in the primary care setting as they are easy to use and time saving.
Recent surveys of PCPs suggest that the resource that is most desired is ready access to subspecialists for advice and “curb-side” consultation [17]. While this solution is not always practical, it may be worthwhile exploring closer relationships between primary care and subspecialty headache clinics, or perhaps more access to e-consultation or telephone consultation for more rural areas. Recently, Minen et al examined education strategies for PCPs. While in-person education sessions with PCPs were poorly attended, multiple possibilities for further education were identified. It was suggested that PCPs having real-time access to resources during the patient encounter would improve their comfort in managing patients. This includes online databases, simple algorithms for treatment, and directions for when to refer to a neurologist [46]. In addition, it may be worthwhile to train not only PCPs but also nursing and allied health staff so that they can provide headache education to patients. This may help ease some of the time burden on PCPs as well as provide a collaborative environment in which headache can be managed [46].
Corresponding author: William S. Kingston, MD, Mayo Clinic, 13400 E. Shea Blvd., Scottsdale, AZ 85259.
Financial disclosures: None.
From the Mayo Clinic, Scottsdale, AZ.
Abstract
- Objective: To review the impact of migraine and explore the barriers to optimal migraine diagnosis and treatment.
- Methods: Review of the literature.
- Results: Several factors may play a role in the inadequate care of migraine patients, including issues related to poor access to care, diagnostic insight, misdiagnosis, adherence to treatment, and management of comorbidities. Both patient and physician factors play an important role and many be modifiable.
- Conclusions: A focus on education of both patients and physicians is of paramount importance to improve the care provided to migraine patients. Patient evaluations should be multisystemic and include addressing comorbid conditions as well as a discussion about appropriate use of prevention and avoidance of medication overuse.
Key words: migraine; triptans; medication overuse headache; medication adherence; primary care.
Migraine is a common, debilitating condition that is a significant source of reduced productivity and increased disability [1]. According to the latest government statistics, 14.2% of US adults have reported having migraine or severe headaches in the previous 3 months, with an overall age-adjusted 3-month prevalence of 19.1% in females and 9.0% in males [2]. In a self-administered headache questionnaire mailed to 120,000 representative US households, the 1-year period prevalence for migraine was 11.7% (17.1% in women and 5.6% in men). Prevalence peaked in middle life and was lower in adolescents and those older than age 60 years [3]. Migraine is an important cause of reduced health-related quality of life and has a very high economic burden [4]. This effect is even more marked in those with chronic migraine, who are even more likely to have professional and social absenteeism and experience more severe disability [4].
Migraine and headache are a common reason for primary care physician (PCP) visits. Some estimates suggest that as many as 10% of primary care consultations are due to headache [5]. Approximately 75% of all patients complaining of headache in primary care will eventually be diagnosed with migraine [6]. Of these, as many as 1% to 5% will have chronic migraine [6].
Despite the high frequency and social and economic impact of migraine, migraine is underrecognized and undertreated. A survey of US households revealed that only 13% of migraineurs were currently using a preventive thrapy while 43.3% had never used one [3]. This is despite the fact that 32.4% met expert criteria for consideration of a preventive medication [3]. The reasons for underrecognition and undertreatment are multifactorial and include both patient and physician factors.
Physician Factors
Although migraine and headache are a leading cause of physicians visits, most physicians have had little formal training in headache. In the United States, medical students spend an average of 1 hour of preclinical and 2 hours of clinical education in headache [7]. Furthermore, primary care physicians receive little formal training in headache during residency [8]. In addition to the lack of formal training, there is also a lack of substantial clinic time available to fully evaluate and treat a new headache patient in the primary care setting [8]. Headache consultations can often be timely and detail-driven in order to determine the correct diagnosis and treatment [9].
Misdiagnosis
Evidence suggests that misdiagnosis plays a large role in the suboptimal management of migraineurs. Studies have shown that as many as 59.7% of migraineurs were not given a diagnosis of migraine by their primary care provider [10]. Common mistaken diagnoses include tension-type headache [11], “sinus headache” [12], cervical pain syndrome or cervicogenic headache [13], and “stress headache” [14].
The reasons for these misdiagnoses is not certain. It may be that the patient and practitioner assume that location of the pain is suggestive of the cause [13]. This is even though more than half of those with migraine have associated neck pain [15]. A recent study suggests that 60% of migraineurs who self-reported a diagnosis of cervical pain have been subsequently diagnosed with cervicalgia by a physician [13]. If patients endorse stress as a precipitant or the presence of cervical pain, they are more likely to obtain a diagnosis other than migraine. The presence of aura in association with the headache appears to be protective against misdiagnosis [13].
Similarly, patients are often given a diagnosis of “sinus headache.” This diagnosis is often made without radiologic evidence of sinusitis and even in those with a more typical migraine headache [16]. In one survey, 40% of patients meeting criteria for migraine were given this diagnosis. Many of these patients did have nasal symptoms or facial pain without clear evidence or rhinosinusitis, and in some cases these symptoms would respond to migraine treatments [16]. This is a particularly important misdiagnosis to highlight, as attributing symptoms to sinus disease may lead to unnecessary consultations and even sinus instrumentation.
In addition to common misdiagnoses, many PCPs are unfamiliar with the “red flags” that may indicate a secondary headache disorder and are also unfamiliar with appropriate use of neuroimaging in headache patients [17].
Misuse of As-Needed Medications
Studies have suggested that a large proportion of PCPs will prescribe nonspecific analgesics for migraine rather than migraine-specific medications [18]. These treatments may include NSAIDs, acetaminophen, barbiturates, and even opiates. This appears to be the pattern even for those with severe attacks [18], suggesting that migraine-specific medications such as triptans may be underused in the primary care setting. Postulated reasons for this pattern include lack of physician knowledge regarding the specific recommendations for managing migraine, the cost of medications, as well as lack of insurance coverage for these medications [19]. Misuse of as-needed medications can lead to medication overuse headache (MOH), which is an underrecognized problem in the primary care setting [20]. In a survey of PCPs in Boston, only 54% of PCPs were aware that barbiturates can cause MOH and only 34% were aware that opiates can cause MOH [17]. The same survey revealed that approximately 20% of PCPs had never made the diagnosis of MOH [17].
Underuse of Preventive Medications
As many as 40% of migraineurs need preventive therapy, but only approximately 13% are currently receiving it [3]. Additionally, the average time from diagnosis of migraine to instituting preventive treatment is 4.3 years, and often there is only a single preventive medication trial if one is instituted [21]. The reasons for this appear to be complex. The physician factors contributing to the underuse of preventive medications include inadequate education, discomfort and inadequate time for assessments. Only 27.8% of surveyed PCPs were aware of the American Academy of Neurology guidelines for prescribing preventive medications [17].
There may be an underestimate of the disability experienced by migraineurs, which can explain some of the underuse of preventive medications. While many PCPs endorse inquiring about headache-related disability, many do not used validated scales such as the Migraine Disability Assessment Score (MIDAS) or the Headache Impact Test (HIT) [17]. In addition, patients often underreport their headache days and report only their severe exacerbations unless clearly asked about a daily headache [22]. This may be part of the reason why only 20% of migraineurs who meet criteria for chronic migraine are diagnosed as such and why preventatives may not be offered [23].
After preventatives are started, less than 25% of patients will be adherent to oral migraine preventive agents at 1 year [24]. Common reasons for discontinuing preventives include adverse effects and perceived inefficacy [22]. Preventive medications may need a 6- to 8-week trial before efficacy is determined, but in practice medications may be stopped before this threshold is reached. Inadequate follow-up and lack of detail with regard to medication trials may result in the perception of an intractable patient prematurely. It has been suggested that a systematic approach to documenting and choosing preventive agents is helpful in the treatment of migraine [25], although this is not always practical in the primary care setting.
Another contributor to underuse of effective prophylaxis is related to access. Treatment with onabotulinumtoxin A, an efficacious prophylactic treatment approved for select chronic migraine patients [26], will usually require referral to a headache specialist, which is not always available to PCPs in a timely manner [7].
Nonpharmacologic Approaches
Effective nonpharmacologic treatment modalities for migraine, such as cognitive-behavioral therapy and biofeedback [27], are not commonly recommended by PCPs [17]. Instead, there appears to be more focus on avoidance of triggers and referral to non–evidence-based resources, such as special diets and massage therapy [17]. While these methods are not always inappropriate, it should be noted that they often have little or no evidence for efficacy.
Patients often wish for non-medication approaches to migraine management, but for those with significant and severe disability, these are probably insufficient. In these patients, non-medication approaches may best be used as a supplement to pharmacological treatment, with education on pharmacologic prevention given. Neuromodulation is a promising, novel approach that is emerging as a new treatment for migraine, but likely will require referral to a headache specialist.
Suboptimal Management of Migraine Comorbidities
There are several disorders that are commonly comorbid with migraine. Among the most common are anxiety, depression, medication (and caffeine) overuse, obesity, and sleep disorders [22]. A survey of PCPs reveals that only 50.6% of PCPs screen for anxiety, 60.2% for depression, and 73.5% for sleep disorders [17]. They are, for the most part, modifiable or treatable conditions and their proper management may help ease migraine disability.
In addition, the presence of these comorbidities may alter choice of treatment, for example, favoring the use of an serotonin and norepinephrine reuptake inhibitor such as venlafaxine for treatment in those with comorbid anxiety and depression. It is also worthwhile to have a high index of suspicion for obstructive sleep apnea in patients with headache, particularly in the obese and in those who endorse nonrestorative sleep or excessive daytime somnolence. It appears that patients who are adherent to the treatment of sleep apnea are more likely to report improvement in their headache [28].
Given the time constraints that often exist in the PCP office setting, addressing these comorbidities thoroughly is not always possible. It is reasonable, however, to have patients use screening tools while in the waiting room or prior to an appointment, to better identify those with modifiable comorbidities. Depression, anxiety, and excessive daytime sleepiness can all be screened for relatively easily with tools such as the PHQ-9 [29], GAD-7 [30] and Epworth Sleepiness Scale [31], respectively. A positive screen on any of these could lead the PCP to further investigate these entities as a possible contributor to migraine.
Patient Factors
In addition to the physician factors identified above, patient factors can contribute to the suboptimal management of migraine as well. These factors include a lack insight into diagnosis, poor compliance with treatment of migraine or its comorbidities, and overuse of abortive medications. There are also less modifiable patient factors such as socioeconomic status and the stigma that may be associated with migraine.
Poor Insight Into Diagnosis
Despite the high prevalence and burden of migraine in the general population, there is a staggering lack of awareness among migraineurs. Some estimates state that as many as 54% of patients were unaware that their headaches represented migraine [32]. The most common self-reported diagnoses in migraineurs are sinus headache (39%), tension-type headache (31%) and stress headache (29%) [14]. In addition, many patients believe they are suffering from cervical spine–related pain [13]. This is likely due to the common presence of posteriorly located pain, attacks triggered by poor sleep, or attacks associated with weather changes [13]. Patients presenting with aura are more likely to report and to receive a physician diagnosis of migraine [14]. Women are more likely to receive and report a diagnosis of migraine compared with men [32].
There are many factors that play a role in poor insight. Many patients appear to believe that the location of the pain is suggestive of the cause [13]. Many patients never seek out consultation for their headaches, and thus never receive a proper diagnosis [33]. Some patients may seek out medical care for their headaches, but fail to remember their diagnosis or receive an improper diagnosis [34].
Poor Adherence
The body of literature examining adherence with headache treatment is growing, but remains small [35]. In a recent systematic review of treatment adherence in pediatric and adult patients with headache, adherence rates in adults with headache ranged from 25% to 94% [35]. In this review, prescription claims data analyses found poor persistence in patients prescribed triptans for migraine treatment. In one large claims-based study, 53.8% of patients receiving a new triptan prescription did not persistently refill their index triptan [36]. Although some of these patients switched to an alternative triptan, the majority switched to a non-triptan migraine medication, including opioids and nonsteroidal anti-inflammatory drugs [36].
Cady and colleagues’ study of lapsed and sustained triptan users found that sustained users were significantly more satisfied with their medication, confident in the medication’s ability to control headache, and reported control of migraine with fewer doses of medication [37]. The authors concluded that the findings suggest that lapsed users may not be receiving optimal treatment. In a review by Rains et al [38], the authors found that headache treatment adherence declines “with more frequent and complex dosing regimens, side effects, and costs, and is subject to a wide range of psychosocial influences.”
Adherence issues also exist for migraine prevention. Less than 25% of chronic migraine patients continue to take oral preventive therapies at 1 year [24]. The reasons for this nonadherence are not completely clear, but are likely multifactorial. Preventives may take several weeks to months to become effective, which may contribute to noncompliance. In addition, migraineurs appears to have inadequate follow-up for migraine. Studies from France suggest that only 18% of those aware of their migraine diagnosis received medical follow-up [39].
Medication Overuse
While the data is not entirely clear, it is likely that overuse of as-needed medication plays a role in migraine chronification [40]. The reasons for medication overuse in the migraine population include some of the issues already highlighted above, including inadequate patient education, poor insight into diagnosis, not seeking care, misdiagnosis, and treatment nonadherence. Patients should be educated on the proper use of as-needed medication. Limits to medication use should be set during the physician-patient encounter. Patients should be counselled to limit their as-needed medication to no more than 10 days per month to reduce the risk of medication overuse headache. Ideally, opiates and barbiturates should be avoided, and never used as first-line therapy in patients who lack contraindications to NSAIDs and triptans. If their use in unavoidable for other reasons, they should be used sparingly, as use on as few as 5 to 8 days per month can be problematic [41]. Furthermore it is important to note that if patients are using several different acute analgesics, the combined total use of all as-needed pain medications needs to be less than 10 days per month to reduce the potential for medication overuse headache.
Socioeconomic Factors
Low socioeconomic status has been associated with an increased prevalence for all headache forms and an increased migraine attack frequency [42], but there appear to be few studies looking at the impact of low socioeconomic status and treatment. Lipton et al found that health insurance status was an important predictor of persons with migraine consulting a health care professional [43]. Among consulters, women were far more likely to be diagnosed than men, suggesting that gender bias in diagnosis may be an important barrier for men. Higher household income appeared to be a predictor for receiving a correct diagnosis of migraine. These researchers also found economic barriers related to use of appropriate prescription medications [43]. Differences in diagnosis and treatment may indicate racial and ethnic disparities in access and quality of care for minority patients [44].
Stigma
At least 1 study has reported that migraine patients experience stigma. In Young et al’s study of 123 episodic migraine patients, 123 chronic migraine patients, and 62 epilepsy patients, adjusted stigma was similar for chronic migraine and epilepsy, which were greater than for episodic migraine [45]. Stigma correlated most strongly with inability to work. Migraine patients reported equally high stigma scores across age, income, and education. The stigma of migraine may pose a barrier to seeking consultation and treatment. Further, the perception that migraine is “just a headache” may lead to stigmatizing attitudes on the part of friends, family, and coworkers of patients with migraine.
Conclusions and Recommendations
Migraine is a prevalent and frequently disabling condition that is underrecognized and undertreated in the primary care setting. Both physician and patient factors pose barriers to the optimal diagnosis and treatment of migraine. Remedies to address these barriers include education of both patients and physicians first and foremost. Targeting physician education in medical school and during residency training, including in primary care subspecialties, could include additional didactic teaching, but also clinical encounters in headache subspecialty clinics to increase exposure. Patient advocacy groups and public campaigns to improve understanding of migraine in the community may be a means for improving patient education and reducing stigma. Patients should be encouraged to seek out consultations for headache to reduce long-term headache disability. Management of comorbidities is paramount, and screening tools for migraine-associated disability, anxiety, depression, and medication use may be helpful to implement in the primary care setting as they are easy to use and time saving.
Recent surveys of PCPs suggest that the resource that is most desired is ready access to subspecialists for advice and “curb-side” consultation [17]. While this solution is not always practical, it may be worthwhile exploring closer relationships between primary care and subspecialty headache clinics, or perhaps more access to e-consultation or telephone consultation for more rural areas. Recently, Minen et al examined education strategies for PCPs. While in-person education sessions with PCPs were poorly attended, multiple possibilities for further education were identified. It was suggested that PCPs having real-time access to resources during the patient encounter would improve their comfort in managing patients. This includes online databases, simple algorithms for treatment, and directions for when to refer to a neurologist [46]. In addition, it may be worthwhile to train not only PCPs but also nursing and allied health staff so that they can provide headache education to patients. This may help ease some of the time burden on PCPs as well as provide a collaborative environment in which headache can be managed [46].
Corresponding author: William S. Kingston, MD, Mayo Clinic, 13400 E. Shea Blvd., Scottsdale, AZ 85259.
Financial disclosures: None.
1. Stewart WF, Schechter A, Lipton RB. Migraine heterogeneity. Disability, pain intensity and attack frequency and duration. Neurology 1994; 44(suppl 4):S24–S39
2. Burch RC, Loder S, Loder E, Smitherman TA. The prevalence of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache 2015;55:21–34.
3. Lipton RB, Bigal ME, Diamond M, et al. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 2007;68:343–9.
4. Blumenfeld AM, Varon SF, Wilcox TK, et al. Disability, HRQoL and resource use amoung chronic and episodic migraineurs: results from the International Burden of Migraine Study (IBMS). Cephalalgia 2011;31:301–15.
5. Ahmed F. Headache disorders: differentiating and managing the common subtypes. Br J Pain 2012;6:124–32.
6. Natoli JL, Manack A, Dean B, et al. Global prevalence of chronic migraine: a systemic review. Cephalalgia 2010;30: 599–609.
7. Finkel AG. American academic headache specialists in neurology: Practice characteristics and culture. Cephalalgia 2004; 24:522–7.
8. Sheftell FD, Cady RK, Borchert LD, et al. Optimizing the diagnosis and treatment of migraine. J Am Acad Nurse Pract 2005;17:309–17.
9. Lipton RB, Scher AI, Steiner TJ, et al. Patterns of health care utilization for migraine in England and in the United States. Neurology 2003;60:441–8.
10. De Diego EV, Lanteri-Minet M. Recognition and management of migraine in primary care: Influence of functional impact measures by the Headache Impact Test (HIT). Cephalalgia 2005;25:184–90.
11. Miller S, Matharu MS. Migraine is underdiagnosed and undertreated. Practitioner 2014;258:19–24.
12. Al-Hashel JY, Ahmed SF, Alroughani R, et al. Migraine misdiagnosis as sinusitis, a delay that can last for many years. J Headache Pain 2013;14:97.
13. Viana M, Sances G, Terrazzino S, et al. When cervical pain is actually migraine: an observational study in 207 patients. Cephalalgia 2016. Epub ahead of print.
14. Diamond MD, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: Results from the American Migraine Prevalence and Prevention Study. Headache 2007;47:355–63.
15. Aguila MR, Rebbeck T, Mendoza KG, et al. Definitions and participant characteristics of frequent recurrent headache types in clinical trials: A systematic review. Cephalalgia 2017. Epub ahead of print.
16. Senbil N, Yavus Gurer YK, Uner C, Barut Y. Sinusitis in children and adolescents with chronic or recurrent headache: A case-control study. J Headache Pain 2008;9:33–6.
17. Minen MT, Loder E, Tishler L, Silbersweig D. Migraine diagnosis and treatment: A knowledge and needs assessment amoung primary care providers. Cephalalgia 2016; 36:358–70.
18. MacGregor EA, Brandes J, Eikerman A. Migraine prevalence and treatment patterns: The global migraine and zolmitriptan evaluation survey. Headache 2003;33:19–26.
19. Khan S, Mascarenhas A, Moore JE, et al. Access to triptans for acute episodic migraine: a qualitative study. Headache 2015; 44(suppl 4):199–211.
20. Tepper SJ. Medication-overuse headache. Continuum 2012;18:807–22.
21. Dekker F, Dielemann J, Neven AK, et al. Preventive treatment for migraine in primary care, a population based study in the Netherlands. Cephalalgia 2013;33:1170–8.
22. Starling AJ, Dodick DW. Best practices for patients with chronic migraine: burden, diagnosis and management in primary care. Mayo Clin Proc 2015;90:408–14.
23. Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology 2008;71:559–66.
24. Hepp Z, Dodick D, Varon S, et al. Adherence to oral migraine preventive-medications among patients with chronic migraine. Cephalalgia 2015;35:478–88.
25. Smith JH, Schwedt TJ. What constitutes an “adequate” trial in migraine prevention? Curr Pain Headache Rep 2015;19:52.
26. Dodick DW, Turkel CC, DeGryse RE, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache 2010;50:921–36.
27. Silberstein SD. Practice parameter: evidence based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000;55: 754–62.
28. Johnson KG, Ziemba AM, Garb JL. Improvement in headaches with continuous positive airway pressure for obstructive sleep apnea: a retrospective analysis. Headache 2013;53:333–43.
29. Altura KC, Patten SB, FIest KM, et al. Suicidal ideation in persons with neurological conditions: prevalence, associations and validation of the PHQ-9 for suicidal ideation. Gen Hosp Psychiatry 2016;42:22–6.
30. Seo JG, Park SP. Validation of the Generalized Anxiety Disorder-7 (GAD-7) and GAD-2 in patients with migraine. J Headache Pain 2015;16:97.
31. Corlateanu A, Pylchenko S, DIrcu V, Botnaru V. Predictors of daytime sleepiness in patients with obstructive sleep apnea. Pneumologia 2015;64:21–5.
32. Linde M, Dahlof C. Attitudes and burden of disease among self-considered migraineurs – a nation-wide population-based survey in Sweden. Cephalalgia 2004;24:455–65.
33. Osterhaus JT, Gutterman DL, Plachetka JR. Health care resources and lost labor costs of migraine headaches in the United States. Pharmacoeconomics 1992;36:69–76.
34. Tepper SJ, Dahlof CG, Dowson A et al. Prevalence and diagnosis of migraine in patients consulting their physician with a complaint of headache: Data from the Landmark Study. Headache 2004;44:856–64.
35. Ramsey RR, Ryan JL, Hershey AD, et al. Treatment adherence in patients with headache: a systematic review. Headache 2014;54:795–816.
36. Katic BJ, Rajagopalan S, Ho TW, et al. Triptan persistency among newly initiated users in a pharmacy claims database. Cephalalgia 2011;31:488–500.
37. Cady RK, Maizels M, Reeves DL, Levinson DM, Evans JK. Predictors of adherence to triptans: factors of sustained vs lapsed users. Headache 2009;49:386–94.
38. Rains JC, Lipchik GL, Penzien DB. Behavioral facilitation of medical treatment for headache--part I: Review of headache treatment compliance. Headache 2006;46:1387–94.
39. Lucas C, Chaffaut C, Artaz MA, Lanteri-Minet M. FRAMIG 2000: Medical and therapeutic management of migraine in France. Cephalalgia 2005;25:267–79.
40. Bigal ME, Serrano D, Buse D et al. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal population-based study. Headache 2008;48:1157–68.
41. Diener HC, Limmroth V. Medication-overuse headache: a worldwide problem. Lancet Neurol 2004;3:475–83.
42. Winter AC, Berger K, Buring JE, Kurth T. Associations of socioeconomic status with migraine and non-migraine headache. Cephalalgia 2012;32:159–70.
43. Lipton, RB, Serrano D, Holland S et al. Barriers to the diagnosis and treatment of migraine: effects of sex, income, and headache features. Headache 2013;53: 81–92.
44. Loder S, Sheikh HU, Loder E. The prevalence, burden, and treatment of severe, frequent, and migraine headaches in US minority populations: statistics from National Survey studies. Headache 2015;55:214–28.
45. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8:e54074.
46. Minen A, Shome A, Hapern A, et al. A migraine training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and consideration for further research. Headache 2016;56:725–40.
1. Stewart WF, Schechter A, Lipton RB. Migraine heterogeneity. Disability, pain intensity and attack frequency and duration. Neurology 1994; 44(suppl 4):S24–S39
2. Burch RC, Loder S, Loder E, Smitherman TA. The prevalence of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache 2015;55:21–34.
3. Lipton RB, Bigal ME, Diamond M, et al. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 2007;68:343–9.
4. Blumenfeld AM, Varon SF, Wilcox TK, et al. Disability, HRQoL and resource use amoung chronic and episodic migraineurs: results from the International Burden of Migraine Study (IBMS). Cephalalgia 2011;31:301–15.
5. Ahmed F. Headache disorders: differentiating and managing the common subtypes. Br J Pain 2012;6:124–32.
6. Natoli JL, Manack A, Dean B, et al. Global prevalence of chronic migraine: a systemic review. Cephalalgia 2010;30: 599–609.
7. Finkel AG. American academic headache specialists in neurology: Practice characteristics and culture. Cephalalgia 2004; 24:522–7.
8. Sheftell FD, Cady RK, Borchert LD, et al. Optimizing the diagnosis and treatment of migraine. J Am Acad Nurse Pract 2005;17:309–17.
9. Lipton RB, Scher AI, Steiner TJ, et al. Patterns of health care utilization for migraine in England and in the United States. Neurology 2003;60:441–8.
10. De Diego EV, Lanteri-Minet M. Recognition and management of migraine in primary care: Influence of functional impact measures by the Headache Impact Test (HIT). Cephalalgia 2005;25:184–90.
11. Miller S, Matharu MS. Migraine is underdiagnosed and undertreated. Practitioner 2014;258:19–24.
12. Al-Hashel JY, Ahmed SF, Alroughani R, et al. Migraine misdiagnosis as sinusitis, a delay that can last for many years. J Headache Pain 2013;14:97.
13. Viana M, Sances G, Terrazzino S, et al. When cervical pain is actually migraine: an observational study in 207 patients. Cephalalgia 2016. Epub ahead of print.
14. Diamond MD, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: Results from the American Migraine Prevalence and Prevention Study. Headache 2007;47:355–63.
15. Aguila MR, Rebbeck T, Mendoza KG, et al. Definitions and participant characteristics of frequent recurrent headache types in clinical trials: A systematic review. Cephalalgia 2017. Epub ahead of print.
16. Senbil N, Yavus Gurer YK, Uner C, Barut Y. Sinusitis in children and adolescents with chronic or recurrent headache: A case-control study. J Headache Pain 2008;9:33–6.
17. Minen MT, Loder E, Tishler L, Silbersweig D. Migraine diagnosis and treatment: A knowledge and needs assessment amoung primary care providers. Cephalalgia 2016; 36:358–70.
18. MacGregor EA, Brandes J, Eikerman A. Migraine prevalence and treatment patterns: The global migraine and zolmitriptan evaluation survey. Headache 2003;33:19–26.
19. Khan S, Mascarenhas A, Moore JE, et al. Access to triptans for acute episodic migraine: a qualitative study. Headache 2015; 44(suppl 4):199–211.
20. Tepper SJ. Medication-overuse headache. Continuum 2012;18:807–22.
21. Dekker F, Dielemann J, Neven AK, et al. Preventive treatment for migraine in primary care, a population based study in the Netherlands. Cephalalgia 2013;33:1170–8.
22. Starling AJ, Dodick DW. Best practices for patients with chronic migraine: burden, diagnosis and management in primary care. Mayo Clin Proc 2015;90:408–14.
23. Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology 2008;71:559–66.
24. Hepp Z, Dodick D, Varon S, et al. Adherence to oral migraine preventive-medications among patients with chronic migraine. Cephalalgia 2015;35:478–88.
25. Smith JH, Schwedt TJ. What constitutes an “adequate” trial in migraine prevention? Curr Pain Headache Rep 2015;19:52.
26. Dodick DW, Turkel CC, DeGryse RE, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache 2010;50:921–36.
27. Silberstein SD. Practice parameter: evidence based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000;55: 754–62.
28. Johnson KG, Ziemba AM, Garb JL. Improvement in headaches with continuous positive airway pressure for obstructive sleep apnea: a retrospective analysis. Headache 2013;53:333–43.
29. Altura KC, Patten SB, FIest KM, et al. Suicidal ideation in persons with neurological conditions: prevalence, associations and validation of the PHQ-9 for suicidal ideation. Gen Hosp Psychiatry 2016;42:22–6.
30. Seo JG, Park SP. Validation of the Generalized Anxiety Disorder-7 (GAD-7) and GAD-2 in patients with migraine. J Headache Pain 2015;16:97.
31. Corlateanu A, Pylchenko S, DIrcu V, Botnaru V. Predictors of daytime sleepiness in patients with obstructive sleep apnea. Pneumologia 2015;64:21–5.
32. Linde M, Dahlof C. Attitudes and burden of disease among self-considered migraineurs – a nation-wide population-based survey in Sweden. Cephalalgia 2004;24:455–65.
33. Osterhaus JT, Gutterman DL, Plachetka JR. Health care resources and lost labor costs of migraine headaches in the United States. Pharmacoeconomics 1992;36:69–76.
34. Tepper SJ, Dahlof CG, Dowson A et al. Prevalence and diagnosis of migraine in patients consulting their physician with a complaint of headache: Data from the Landmark Study. Headache 2004;44:856–64.
35. Ramsey RR, Ryan JL, Hershey AD, et al. Treatment adherence in patients with headache: a systematic review. Headache 2014;54:795–816.
36. Katic BJ, Rajagopalan S, Ho TW, et al. Triptan persistency among newly initiated users in a pharmacy claims database. Cephalalgia 2011;31:488–500.
37. Cady RK, Maizels M, Reeves DL, Levinson DM, Evans JK. Predictors of adherence to triptans: factors of sustained vs lapsed users. Headache 2009;49:386–94.
38. Rains JC, Lipchik GL, Penzien DB. Behavioral facilitation of medical treatment for headache--part I: Review of headache treatment compliance. Headache 2006;46:1387–94.
39. Lucas C, Chaffaut C, Artaz MA, Lanteri-Minet M. FRAMIG 2000: Medical and therapeutic management of migraine in France. Cephalalgia 2005;25:267–79.
40. Bigal ME, Serrano D, Buse D et al. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal population-based study. Headache 2008;48:1157–68.
41. Diener HC, Limmroth V. Medication-overuse headache: a worldwide problem. Lancet Neurol 2004;3:475–83.
42. Winter AC, Berger K, Buring JE, Kurth T. Associations of socioeconomic status with migraine and non-migraine headache. Cephalalgia 2012;32:159–70.
43. Lipton, RB, Serrano D, Holland S et al. Barriers to the diagnosis and treatment of migraine: effects of sex, income, and headache features. Headache 2013;53: 81–92.
44. Loder S, Sheikh HU, Loder E. The prevalence, burden, and treatment of severe, frequent, and migraine headaches in US minority populations: statistics from National Survey studies. Headache 2015;55:214–28.
45. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8:e54074.
46. Minen A, Shome A, Hapern A, et al. A migraine training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and consideration for further research. Headache 2016;56:725–40.
Patients’ profile deemed best criteria for LAIs in bipolar disorder
MIAMI – Expert clinicians endorsed long-acting injectables as a preferred treatment for bipolar I disorder on the basis of patient characteristics and treatment history, rather than on an assumed level of treatment adherence, according to a small survey.
“Just over three-quarters of the experts we surveyed said they were ‘somewhat’ or ‘not very’ confident about their ability to assess their patients’ adherence,” said Martha Sajatovic, MD, who presented the data during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly the New Clinical Drug Evaluation Unit meeting.
The finding reflects a shift, according to Dr. Sajatovic, professor of psychiatry and neurology, and the Willard Brown Chair in Neurological Outcomes Research, at Case Western University in Cleveland.

The traditional view of using long-acting injectable antipsychotics (LAIs) for patients with bipolar I disorder is that they are appropriate only in certain cohorts, such as patients with very severe illness or at the more extreme spectrum in terms of risk, and those who are homeless or pose a risk to themselves or others, Dr. Sajatovic said. Also, when it comes to the use of LAIs, there is a lack of guidance – which might contribute to clinicians’ reluctance to prescribe or recommend them, she said.
In the survey, of the 42 experts contacted by Dr. Sajatovic and her colleagues, 34 responded. According to those respondents, 11% of their patients with bipolar disorder were being treated with LAIs, compared with one-third of all patients with schizophrenia/schizoaffective disorder.
Using a scale of 1-9, with 1 being “extremely inappropriate,” 2-3 being “usually inappropriate,” 4-6 being “sometimes appropriate,” 7-8 being “usually appropriate,” and 9 being “extremely appropriate,” all tended to favor patient characteristics and treatment history over adherence when rating criteria for treatment selection. This was true regardless of whether patients were newly diagnosed with bipolar disorder, or whether their diagnosis was established and treated with an antipsychotic for 2 or more years.
For comparison, Dr. Sajatovic and her colleagues also surveyed the expert panel members on their use of LAIs in established schizophrenia and schizoaffective disorder.
Patients with a history of two or more hospitalizations for bipolar relapses and those who were either homeless or had unstable housing were rated by most respondents as usually appropriate for LAIs as first-line treatment. For those with dubious treatment adherence, the profile was similar: LAIs were considered by a majority of respondents as usually appropriate if there was a history of two or more hospitalizations for bipolar relapses, as well as homelessness or an unstable housing situation. LAIs also were considered by a majority as usually appropriate in this cohort if there was a history of violence to others, and if patients had poor insight into their illness.
Spotty treatment adherence to medications was the most common treatment history characteristic cited for first-line prescription of LAIs in patients with an established bipolar disorder diagnosis. Other first-line LAI criteria cited by most respondents for this cohort were if they previously had done well on an LAI, and if they frequently missed clinic appointments.
Virtually all the criteria above applied to patients with established illness and questionable adherence, although the expert clinicians also largely cited a failure to respond to lithium or an anticonvulsant mood stabilizer, a predominant history of manic relapse, and a strong therapeutic alliance as additional reasons to view LAIs as usually appropriate.
Regardless of the assumption of adherence or nonadherence, in most cases in which patients had an established bipolar diagnosis, more than half of the expert panel said use of an LAI was extremely appropriate.
In patients with an established diagnosis of bipolar disorder with questionable treatment adherence, respondents strongly endorsed the idea that it was usually appropriate to use LAIs as first-line treatment if the patients had a history of two or more hospitalizations for bipolar relapses, homelessness or an otherwise unstable living arrangement, violence toward others, and poor insight into their illness.
The panel members were blinded to the study’s sponsor, which was Otsuka. All respondents had an average of 25 years of clinical experience and an average of 22 years of research experience, and all had extensive expertise in the use of two or more LAIs, although no specific antipsychotic brand names were included in the survey.
Just more than one-third of respondents reported spending all or most of their professional time seeing patients, and one-fifth reported that they saw patients half of the time. The average age of patients seen by the respondents was 35-65 years.
Dr. Sajatovic disclosed receiving research grants from the National Institutes of Health, Alkermes, Janssen, Merck, and several other pharmaceutical companies and foundations; serving as a consultant for numerous entities, including Otsuka; and receiving royalties from UpToDate, and several publishing companies.
[email protected]
On Twitter @whitneymcknight
MIAMI – Expert clinicians endorsed long-acting injectables as a preferred treatment for bipolar I disorder on the basis of patient characteristics and treatment history, rather than on an assumed level of treatment adherence, according to a small survey.
“Just over three-quarters of the experts we surveyed said they were ‘somewhat’ or ‘not very’ confident about their ability to assess their patients’ adherence,” said Martha Sajatovic, MD, who presented the data during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly the New Clinical Drug Evaluation Unit meeting.
The finding reflects a shift, according to Dr. Sajatovic, professor of psychiatry and neurology, and the Willard Brown Chair in Neurological Outcomes Research, at Case Western University in Cleveland.
The traditional view of using long-acting injectable antipsychotics (LAIs) for patients with bipolar I disorder is that they are appropriate only in certain cohorts, such as patients with very severe illness or at the more extreme spectrum in terms of risk, and those who are homeless or pose a risk to themselves or others, Dr. Sajatovic said. Also, when it comes to the use of LAIs, there is a lack of guidance – which might contribute to clinicians’ reluctance to prescribe or recommend them, she said.
In the survey, of the 42 experts contacted by Dr. Sajatovic and her colleagues, 34 responded. According to those respondents, 11% of their patients with bipolar disorder were being treated with LAIs, compared with one-third of all patients with schizophrenia/schizoaffective disorder.
Using a scale of 1-9, with 1 being “extremely inappropriate,” 2-3 being “usually inappropriate,” 4-6 being “sometimes appropriate,” 7-8 being “usually appropriate,” and 9 being “extremely appropriate,” all tended to favor patient characteristics and treatment history over adherence when rating criteria for treatment selection. This was true regardless of whether patients were newly diagnosed with bipolar disorder, or whether their diagnosis was established and treated with an antipsychotic for 2 or more years.
For comparison, Dr. Sajatovic and her colleagues also surveyed the expert panel members on their use of LAIs in established schizophrenia and schizoaffective disorder.
Patients with a history of two or more hospitalizations for bipolar relapses and those who were either homeless or had unstable housing were rated by most respondents as usually appropriate for LAIs as first-line treatment. For those with dubious treatment adherence, the profile was similar: LAIs were considered by a majority of respondents as usually appropriate if there was a history of two or more hospitalizations for bipolar relapses, as well as homelessness or an unstable housing situation. LAIs also were considered by a majority as usually appropriate in this cohort if there was a history of violence to others, and if patients had poor insight into their illness.
Spotty treatment adherence to medications was the most common treatment history characteristic cited for first-line prescription of LAIs in patients with an established bipolar disorder diagnosis. Other first-line LAI criteria cited by most respondents for this cohort were if they previously had done well on an LAI, and if they frequently missed clinic appointments.
Virtually all the criteria above applied to patients with established illness and questionable adherence, although the expert clinicians also largely cited a failure to respond to lithium or an anticonvulsant mood stabilizer, a predominant history of manic relapse, and a strong therapeutic alliance as additional reasons to view LAIs as usually appropriate.
Regardless of the assumption of adherence or nonadherence, in most cases in which patients had an established bipolar diagnosis, more than half of the expert panel said use of an LAI was extremely appropriate.
In patients with an established diagnosis of bipolar disorder with questionable treatment adherence, respondents strongly endorsed the idea that it was usually appropriate to use LAIs as first-line treatment if the patients had a history of two or more hospitalizations for bipolar relapses, homelessness or an otherwise unstable living arrangement, violence toward others, and poor insight into their illness.
The panel members were blinded to the study’s sponsor, which was Otsuka. All respondents had an average of 25 years of clinical experience and an average of 22 years of research experience, and all had extensive expertise in the use of two or more LAIs, although no specific antipsychotic brand names were included in the survey.
Just more than one-third of respondents reported spending all or most of their professional time seeing patients, and one-fifth reported that they saw patients half of the time. The average age of patients seen by the respondents was 35-65 years.
Dr. Sajatovic disclosed receiving research grants from the National Institutes of Health, Alkermes, Janssen, Merck, and several other pharmaceutical companies and foundations; serving as a consultant for numerous entities, including Otsuka; and receiving royalties from UpToDate, and several publishing companies.
[email protected]
On Twitter @whitneymcknight
MIAMI – Expert clinicians endorsed long-acting injectables as a preferred treatment for bipolar I disorder on the basis of patient characteristics and treatment history, rather than on an assumed level of treatment adherence, according to a small survey.
“Just over three-quarters of the experts we surveyed said they were ‘somewhat’ or ‘not very’ confident about their ability to assess their patients’ adherence,” said Martha Sajatovic, MD, who presented the data during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly the New Clinical Drug Evaluation Unit meeting.
The finding reflects a shift, according to Dr. Sajatovic, professor of psychiatry and neurology, and the Willard Brown Chair in Neurological Outcomes Research, at Case Western University in Cleveland.
The traditional view of using long-acting injectable antipsychotics (LAIs) for patients with bipolar I disorder is that they are appropriate only in certain cohorts, such as patients with very severe illness or at the more extreme spectrum in terms of risk, and those who are homeless or pose a risk to themselves or others, Dr. Sajatovic said. Also, when it comes to the use of LAIs, there is a lack of guidance – which might contribute to clinicians’ reluctance to prescribe or recommend them, she said.
In the survey, of the 42 experts contacted by Dr. Sajatovic and her colleagues, 34 responded. According to those respondents, 11% of their patients with bipolar disorder were being treated with LAIs, compared with one-third of all patients with schizophrenia/schizoaffective disorder.
Using a scale of 1-9, with 1 being “extremely inappropriate,” 2-3 being “usually inappropriate,” 4-6 being “sometimes appropriate,” 7-8 being “usually appropriate,” and 9 being “extremely appropriate,” all tended to favor patient characteristics and treatment history over adherence when rating criteria for treatment selection. This was true regardless of whether patients were newly diagnosed with bipolar disorder, or whether their diagnosis was established and treated with an antipsychotic for 2 or more years.
For comparison, Dr. Sajatovic and her colleagues also surveyed the expert panel members on their use of LAIs in established schizophrenia and schizoaffective disorder.
Patients with a history of two or more hospitalizations for bipolar relapses and those who were either homeless or had unstable housing were rated by most respondents as usually appropriate for LAIs as first-line treatment. For those with dubious treatment adherence, the profile was similar: LAIs were considered by a majority of respondents as usually appropriate if there was a history of two or more hospitalizations for bipolar relapses, as well as homelessness or an unstable housing situation. LAIs also were considered by a majority as usually appropriate in this cohort if there was a history of violence to others, and if patients had poor insight into their illness.
Spotty treatment adherence to medications was the most common treatment history characteristic cited for first-line prescription of LAIs in patients with an established bipolar disorder diagnosis. Other first-line LAI criteria cited by most respondents for this cohort were if they previously had done well on an LAI, and if they frequently missed clinic appointments.
Virtually all the criteria above applied to patients with established illness and questionable adherence, although the expert clinicians also largely cited a failure to respond to lithium or an anticonvulsant mood stabilizer, a predominant history of manic relapse, and a strong therapeutic alliance as additional reasons to view LAIs as usually appropriate.
Regardless of the assumption of adherence or nonadherence, in most cases in which patients had an established bipolar diagnosis, more than half of the expert panel said use of an LAI was extremely appropriate.
In patients with an established diagnosis of bipolar disorder with questionable treatment adherence, respondents strongly endorsed the idea that it was usually appropriate to use LAIs as first-line treatment if the patients had a history of two or more hospitalizations for bipolar relapses, homelessness or an otherwise unstable living arrangement, violence toward others, and poor insight into their illness.
The panel members were blinded to the study’s sponsor, which was Otsuka. All respondents had an average of 25 years of clinical experience and an average of 22 years of research experience, and all had extensive expertise in the use of two or more LAIs, although no specific antipsychotic brand names were included in the survey.
Just more than one-third of respondents reported spending all or most of their professional time seeing patients, and one-fifth reported that they saw patients half of the time. The average age of patients seen by the respondents was 35-65 years.
Dr. Sajatovic disclosed receiving research grants from the National Institutes of Health, Alkermes, Janssen, Merck, and several other pharmaceutical companies and foundations; serving as a consultant for numerous entities, including Otsuka; and receiving royalties from UpToDate, and several publishing companies.
[email protected]
On Twitter @whitneymcknight
AT THE ASCP ANNUAL MEETING
Key clinical point:
Major finding: Clinicians very strongly endorsed the use long-acting injectables as first-line treatment in patients with bipolar disorder who met several criteria, including having a history of two or more hospitalizations for bipolar relapses.
Data source: A blinded, consensus survey of 34 high-prescribing clinicians experienced in treating mood and psychotic disorders.
Disclosures: Dr. Sajatovic disclosed receiving research grants from the National Institutes of Health, Alkermes, Janssen, Merck, and several other pharmaceutical companies and foundations; serving as a consultant for numerous entities, including Otsuka, which sponsored the study; and receiving royalties from UpToDate, and several publishing companies.
Association Between Ventilator Strategy and Neurocognitive Outcomes in Out-of-Hospital Cardiac Arrest Patients
Study Overview
Objective. To determine if there is an association between low tidal volume (VT) ventilation and neurocognitive outcomes in patients after out-of-hospital cardiac arrest (OHCA).
Design. Retrospective cohort study.
Setting and participants. Data was obtained from retrospective review of all adults admitted between 2008 and 2014 to one of 2 centers (A or B) with nontraumatic OHCA requiring mechanical ventilation for greater than 48 hours. The study physicians screened records primarily using chart review with secondary confirmation of the diagnosis of OHCA and eligibility criteria. Patients with an outside hospital stay greater than 24 hours, intracranial hemorrhage, use of extracorporeal membranous oxygenation (ECMO), use of airway pressure release mode of ventilation, chronic dependence on mechanical ventilation, or missing data were excluded. Of the 579 patients with OHCA, 256 (44.2%) met the inclusion criteria and were included in the main analysis. A total of 97 patients were identified as having high VT (defined as > 8 mL/kg of predicted body weight [PBW]) and were matched to 97 of the 159 patients identified as having a low VT as part of the propensity-matched subgroup analysis using 1:1 optimal caliper matching.
Main outcome measure. The primary outcome was a favorable neurocognitive outcome at hospital discharge (Cerebral Performance Category score [CPC] of 1 or 2). A CPC of 1 or 2 corresponds to normal life or life that is disabled but independent, respectively. A CPC of 3 is disabled and dependent, and a CPC of 5 is alive but brain dead. Two physicians blinded to VT and other measures of illness severity calculated the CPC via chart review. Discordant scores were resolved by consensus, and a Kappa statistic was calculated to quantify agreement between investigators. Secondary outcomes included ventilator-free days, hospital-free days, ICU-free days, shock-free days, and extrapulmonary organ failure–free days. Logistic regression with backward elimination was used to identify predictors of receiving VT ≤ 8 mL/kg PBW to be used in the propensity-matched analysis, along with relevant predictors identified from the literature. The odds ratio for the primary outcome was calculated using both logistic regression analysis and propensity-matched analysis. Other methods of sensitivity analysis (propensity quintile adjustment, inverse-probability-of-treatment weighting) were used to confirm the robustness of the initial analysis to different statistical methods. A P value of < 0.05 was considered significant.
Main results. Of the study patients, approximately half (49% in high VT, 52% in low VT) had an initial rhythm of ventricular tachycardia or ventricular fibrillation. Patients with low VT were significantly younger (mean age 59 yr vs. 66 yr), taller (mean height 177 cm vs. 165 cm), and heavier (mean weight 88 kg vs. 81 kg). There were also significantly fewer females in the low VT group (19% vs. 46%). There were no significant differences between baseline comorbidities, arrest characteristics, or illness severity between the 2 groups with the exception of significantly more patients in the low VT underwent therapeutic hypothermia (87% vs. 76%) and were admitted to hospital A (69% vs. 55%). There were no significant differences between the groups across ventilator parameters aside from tidal volume. The average VT in mL/kg PBW was 9.3 in the high VT group and 7.1 in the low VT group over the first 48 hours.
In the multivariate regression analysis, significant independent predictors of receiving high VT included height, weight, and hospital of admission. The final propensity model to predict VT included age, height, weight, sex, illness severity measures (APACHE-II score and presence of circulatory shock in the first 24 hours of admission), arrest characteristics, and respiratory characteristics (initial pH, initial PaCO2, PaO2:FiO2 ratio, and initial peak inspiratory pressure) as covariates. The use of low VT was significantly associated with a favorable neurocognitive outcome in the multivariate regression analysis (odds ratio [OR] 1.65, 95% confidence interval [CI] 1.18–2.29). This association held in both the propensity matched analysis (OR 1.68, 95% CI 1.11–2.55) as well as conditional logistic regression analysis using propensity score as a covariate (OR 1.61, 95% CI 1.13–2.28).
In the propensity-adjusted conditional logistic regression analysis, a lower VT (1 mL/kg of PBW decrease) was significantly associated with ventilator-free days (OR 1.78, 95% CI 0.39–3.16), shock-free days (OR 1.31, 95% CI 0.10–2.51), ICU-free days (OR 1.38, 95% CI 0.13–2.63), and hospital-free days (OR 1.07, 95% CI 0.04–2.09). There was a nonsignificant trend towards improved survival to hospital discharge (OR 1.23, 95% CI 0.95–1.60, P = 0.115). After propensity score adjustment, lower VT was not associated with therapeutic hypothermia (OR 0.14, 95% CI −0.19 to 0.47), and in the multivariate regression analysis there was no association between favorable neurocognitive outcome and therapeutic hypothermia (P = 0.516). While there was a significant association between lower VT and site of admission (Hospital A: OR 1.50, 95% CI 1.04–2.17 per 1 mL/kg of PBW decrease), there was no association between favorable neurocognitive outcome and hospital site of admission in the final adjusted regression analysis (P = 0.588).
Conclusion. In this retrospective cohort study, lower VT in the first 48 hours of admission following OHCA was independently associated with favorable neurocognitive outcomes as measured by the CPC score, as well as more ventilator-free, shock-free, ICU-free, and hospital-free days.
Commentary
Neurocognitive impairment following nontraumatic OHCA is common, estimated to occur in roughly half of all survivors [1]. Similar to the acute respiratory distress syndrome (ARDS), the post–cardiac arrest syndrome (PCAS) is recognized as a systemic process with multi-organ effects thought to be mediated in part by inflammatory cytokines [2]. While the beneficial role of low VT in patients with ARDS is well established, currently there are no recommendations for specific VT targets in post–cardiac arrest care, and the effect of VT on outcomes following cardiac arrest is unknown [3].
In this study, Beitler and colleagues suggest a possible association between VT and neurocognitive outcomes following OHCA. Using retrospective data drawn from 2 centers, and employing both regression analysis and propensity matching, the authors identified a significant beneficial effect of lower VT on neurocognitive outcomes in their cohort. This benefit held regardless of the statistical analytic method employed and was present even when correcting for the difference between groups in hospital admission site and use of therapeutic hypothermia in the original cohort. The authors also demonstrated a lower VT was associated with a number of secondary outcomes including fewer hospital, ICU, and ventilator days. While the statistical methods employed by the authors are robust and attempt to account for the limitations inherent to observational studies, a number of questions remain.
First, as the authors appropriately note, causality cannot be proven from a retrospective study. While the analytic methods employed by the authors serve to limit the effect of residual confounding, they do not eliminate it. Although unlikely, it is possible low VT may be a marker for an unmeasured variable that leads to more favorable neurocognitive outcomes. Further research into a possible casual association between VT and neurocognitive outcomes is needed.
The authors also suggest a number of inflammatory-related mechanisms for the association between lower VT and improved neurocognitive outcomes, which they collectively name “brain-lung communication.” While this is a physiologically attractive hypothesis in light of what is known regarding PCAS, the retrospective nature of the study prevents measurement of any inflammatory markers or cytokine levels that might strengthen this hypothesis. As it stands, further exploration of the mechanisms that might link lower VT to improved neurocognitive outcomes will be required before a more definitive statement regarding brain-lung communication can be accepted.
Although the authors identified an association between lower VT and a number of secondary outcomes, their results show there were no significant associations between lower VT and fewer days of extrapulmonary organ failure or improved survival. Given the contradictory nature of some of these secondary outcomes (such as an association with fewer shock-free days but no association with less extrapulmonary organ failure, a known consequence of hemodynamic shock), the true impact of low VT on these outcomes is unclear. While it is logical that the association between lower VT and some secondary outcomes (such as fewer ICU days and fewer ventilator-dependent days) is a result of improved neurocognitive outcomes, further work is required to elucidate the true clinical significance of these secondary outcomes.
Finally, while there was no significant difference between groups in terms of initial pH or PCO2, and these variables were included in the propensity matching analysis, both groups had mean initial PCO2 levels that were elevated (47 mm Hg and 49 mm Hg in the high and low VT groups, respectively). These values are above the physiological range (35–45 mm Hg) recommended by the 2015 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care [3]. The authors suggest that the recommended eucapnic targets can be met in a low VT strategy by increasing the respiratory rate. However, current literature suggests that patients with ARDS exposed to higher respiratory rates may have more frequent exposure to ventilator-induced lung injury (VILI) stresses and an increased rate of lung injury [4]. While there are no clinical trials proving the benefit of a low vs. high respiratory rate strategy, current recommendations for reducing the risk of VILI include limiting the respiratory rate. It is unclear at this time if an increase in the respiratory rate would increase the incidence of VILI and negate any potential benefit provided by low VT in these patients, but this would be an important cost to account for when employing a low VT strategy.
Applications for Clinical Practice
In this study, Beitler and colleagues found that using a low VT ventilation strategy in OHCA patients was associated with improved neurocognitive outcomes. This study is primarily useful as a hypothesis generator. Further research into the effects of ventilator parameters such as VT on the inflammatory cascade, neurocognitive outcomes in other groups of patients (such as those with ARDS), and the existence of a “brain-lung communication” pathway is warranted. From a practical standpoint, evidence continues to mount that lower VT is associated with a number of beneficial effects that are not limited to patients with ARDS [5]. This study would support the current practice of many intensivists to utilize a low VT strategy unless a compelling contraindication exists, as the potential benefits are substantial and the risks minimal. However, this practice will have to be balanced with the need to avoid hypercapnia, and the elevated respiratory rates used to achieve eucapnia may have unforeseen consequences.
—Arun Jose, MD, The George Washington University, Washington, DC
1. Moulaert VR, Verbunt JA, van Heugten CM, Wade DT. Cognitive impairments in survivors of out-of-hospital cardiac arrest: a systematic review. Resuscitation 2009;80:297–305.
2. Peberdy MA, Andersen LW, Abbate A, et al. Inflammatory markers following resuscitation from out-of-hospital cardiac arrest – A prospective multicenter observational study. Resuscitation 2016;103:117–24.
3. Callaway CW, Soar J, Aibiki M, et al. Advanced life support chapter collaborators. Part 4: Advanced life support: 2015 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation 2015;132:S84–S145.
4. Beitler JR, Malhotra A, Thompson BT. Ventilator-induced lung injury. Clin Chest Med 2016;37:633–46.
5. Serpa Neto A, Cardoso SO, Manetta JA, et al. Association between use of lung-protective ventilation with lower tidal volumes and clinical outcomes among patients without acute respiratory distress syndrome: a meta-analysis. JAMA 2012;
308:1651–9.
Study Overview
Objective. To determine if there is an association between low tidal volume (VT) ventilation and neurocognitive outcomes in patients after out-of-hospital cardiac arrest (OHCA).
Design. Retrospective cohort study.
Setting and participants. Data was obtained from retrospective review of all adults admitted between 2008 and 2014 to one of 2 centers (A or B) with nontraumatic OHCA requiring mechanical ventilation for greater than 48 hours. The study physicians screened records primarily using chart review with secondary confirmation of the diagnosis of OHCA and eligibility criteria. Patients with an outside hospital stay greater than 24 hours, intracranial hemorrhage, use of extracorporeal membranous oxygenation (ECMO), use of airway pressure release mode of ventilation, chronic dependence on mechanical ventilation, or missing data were excluded. Of the 579 patients with OHCA, 256 (44.2%) met the inclusion criteria and were included in the main analysis. A total of 97 patients were identified as having high VT (defined as > 8 mL/kg of predicted body weight [PBW]) and were matched to 97 of the 159 patients identified as having a low VT as part of the propensity-matched subgroup analysis using 1:1 optimal caliper matching.
Main outcome measure. The primary outcome was a favorable neurocognitive outcome at hospital discharge (Cerebral Performance Category score [CPC] of 1 or 2). A CPC of 1 or 2 corresponds to normal life or life that is disabled but independent, respectively. A CPC of 3 is disabled and dependent, and a CPC of 5 is alive but brain dead. Two physicians blinded to VT and other measures of illness severity calculated the CPC via chart review. Discordant scores were resolved by consensus, and a Kappa statistic was calculated to quantify agreement between investigators. Secondary outcomes included ventilator-free days, hospital-free days, ICU-free days, shock-free days, and extrapulmonary organ failure–free days. Logistic regression with backward elimination was used to identify predictors of receiving VT ≤ 8 mL/kg PBW to be used in the propensity-matched analysis, along with relevant predictors identified from the literature. The odds ratio for the primary outcome was calculated using both logistic regression analysis and propensity-matched analysis. Other methods of sensitivity analysis (propensity quintile adjustment, inverse-probability-of-treatment weighting) were used to confirm the robustness of the initial analysis to different statistical methods. A P value of < 0.05 was considered significant.
Main results. Of the study patients, approximately half (49% in high VT, 52% in low VT) had an initial rhythm of ventricular tachycardia or ventricular fibrillation. Patients with low VT were significantly younger (mean age 59 yr vs. 66 yr), taller (mean height 177 cm vs. 165 cm), and heavier (mean weight 88 kg vs. 81 kg). There were also significantly fewer females in the low VT group (19% vs. 46%). There were no significant differences between baseline comorbidities, arrest characteristics, or illness severity between the 2 groups with the exception of significantly more patients in the low VT underwent therapeutic hypothermia (87% vs. 76%) and were admitted to hospital A (69% vs. 55%). There were no significant differences between the groups across ventilator parameters aside from tidal volume. The average VT in mL/kg PBW was 9.3 in the high VT group and 7.1 in the low VT group over the first 48 hours.
In the multivariate regression analysis, significant independent predictors of receiving high VT included height, weight, and hospital of admission. The final propensity model to predict VT included age, height, weight, sex, illness severity measures (APACHE-II score and presence of circulatory shock in the first 24 hours of admission), arrest characteristics, and respiratory characteristics (initial pH, initial PaCO2, PaO2:FiO2 ratio, and initial peak inspiratory pressure) as covariates. The use of low VT was significantly associated with a favorable neurocognitive outcome in the multivariate regression analysis (odds ratio [OR] 1.65, 95% confidence interval [CI] 1.18–2.29). This association held in both the propensity matched analysis (OR 1.68, 95% CI 1.11–2.55) as well as conditional logistic regression analysis using propensity score as a covariate (OR 1.61, 95% CI 1.13–2.28).
In the propensity-adjusted conditional logistic regression analysis, a lower VT (1 mL/kg of PBW decrease) was significantly associated with ventilator-free days (OR 1.78, 95% CI 0.39–3.16), shock-free days (OR 1.31, 95% CI 0.10–2.51), ICU-free days (OR 1.38, 95% CI 0.13–2.63), and hospital-free days (OR 1.07, 95% CI 0.04–2.09). There was a nonsignificant trend towards improved survival to hospital discharge (OR 1.23, 95% CI 0.95–1.60, P = 0.115). After propensity score adjustment, lower VT was not associated with therapeutic hypothermia (OR 0.14, 95% CI −0.19 to 0.47), and in the multivariate regression analysis there was no association between favorable neurocognitive outcome and therapeutic hypothermia (P = 0.516). While there was a significant association between lower VT and site of admission (Hospital A: OR 1.50, 95% CI 1.04–2.17 per 1 mL/kg of PBW decrease), there was no association between favorable neurocognitive outcome and hospital site of admission in the final adjusted regression analysis (P = 0.588).
Conclusion. In this retrospective cohort study, lower VT in the first 48 hours of admission following OHCA was independently associated with favorable neurocognitive outcomes as measured by the CPC score, as well as more ventilator-free, shock-free, ICU-free, and hospital-free days.
Commentary
Neurocognitive impairment following nontraumatic OHCA is common, estimated to occur in roughly half of all survivors [1]. Similar to the acute respiratory distress syndrome (ARDS), the post–cardiac arrest syndrome (PCAS) is recognized as a systemic process with multi-organ effects thought to be mediated in part by inflammatory cytokines [2]. While the beneficial role of low VT in patients with ARDS is well established, currently there are no recommendations for specific VT targets in post–cardiac arrest care, and the effect of VT on outcomes following cardiac arrest is unknown [3].
In this study, Beitler and colleagues suggest a possible association between VT and neurocognitive outcomes following OHCA. Using retrospective data drawn from 2 centers, and employing both regression analysis and propensity matching, the authors identified a significant beneficial effect of lower VT on neurocognitive outcomes in their cohort. This benefit held regardless of the statistical analytic method employed and was present even when correcting for the difference between groups in hospital admission site and use of therapeutic hypothermia in the original cohort. The authors also demonstrated a lower VT was associated with a number of secondary outcomes including fewer hospital, ICU, and ventilator days. While the statistical methods employed by the authors are robust and attempt to account for the limitations inherent to observational studies, a number of questions remain.
First, as the authors appropriately note, causality cannot be proven from a retrospective study. While the analytic methods employed by the authors serve to limit the effect of residual confounding, they do not eliminate it. Although unlikely, it is possible low VT may be a marker for an unmeasured variable that leads to more favorable neurocognitive outcomes. Further research into a possible casual association between VT and neurocognitive outcomes is needed.
The authors also suggest a number of inflammatory-related mechanisms for the association between lower VT and improved neurocognitive outcomes, which they collectively name “brain-lung communication.” While this is a physiologically attractive hypothesis in light of what is known regarding PCAS, the retrospective nature of the study prevents measurement of any inflammatory markers or cytokine levels that might strengthen this hypothesis. As it stands, further exploration of the mechanisms that might link lower VT to improved neurocognitive outcomes will be required before a more definitive statement regarding brain-lung communication can be accepted.
Although the authors identified an association between lower VT and a number of secondary outcomes, their results show there were no significant associations between lower VT and fewer days of extrapulmonary organ failure or improved survival. Given the contradictory nature of some of these secondary outcomes (such as an association with fewer shock-free days but no association with less extrapulmonary organ failure, a known consequence of hemodynamic shock), the true impact of low VT on these outcomes is unclear. While it is logical that the association between lower VT and some secondary outcomes (such as fewer ICU days and fewer ventilator-dependent days) is a result of improved neurocognitive outcomes, further work is required to elucidate the true clinical significance of these secondary outcomes.
Finally, while there was no significant difference between groups in terms of initial pH or PCO2, and these variables were included in the propensity matching analysis, both groups had mean initial PCO2 levels that were elevated (47 mm Hg and 49 mm Hg in the high and low VT groups, respectively). These values are above the physiological range (35–45 mm Hg) recommended by the 2015 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care [3]. The authors suggest that the recommended eucapnic targets can be met in a low VT strategy by increasing the respiratory rate. However, current literature suggests that patients with ARDS exposed to higher respiratory rates may have more frequent exposure to ventilator-induced lung injury (VILI) stresses and an increased rate of lung injury [4]. While there are no clinical trials proving the benefit of a low vs. high respiratory rate strategy, current recommendations for reducing the risk of VILI include limiting the respiratory rate. It is unclear at this time if an increase in the respiratory rate would increase the incidence of VILI and negate any potential benefit provided by low VT in these patients, but this would be an important cost to account for when employing a low VT strategy.
Applications for Clinical Practice
In this study, Beitler and colleagues found that using a low VT ventilation strategy in OHCA patients was associated with improved neurocognitive outcomes. This study is primarily useful as a hypothesis generator. Further research into the effects of ventilator parameters such as VT on the inflammatory cascade, neurocognitive outcomes in other groups of patients (such as those with ARDS), and the existence of a “brain-lung communication” pathway is warranted. From a practical standpoint, evidence continues to mount that lower VT is associated with a number of beneficial effects that are not limited to patients with ARDS [5]. This study would support the current practice of many intensivists to utilize a low VT strategy unless a compelling contraindication exists, as the potential benefits are substantial and the risks minimal. However, this practice will have to be balanced with the need to avoid hypercapnia, and the elevated respiratory rates used to achieve eucapnia may have unforeseen consequences.
—Arun Jose, MD, The George Washington University, Washington, DC
Study Overview
Objective. To determine if there is an association between low tidal volume (VT) ventilation and neurocognitive outcomes in patients after out-of-hospital cardiac arrest (OHCA).
Design. Retrospective cohort study.
Setting and participants. Data was obtained from retrospective review of all adults admitted between 2008 and 2014 to one of 2 centers (A or B) with nontraumatic OHCA requiring mechanical ventilation for greater than 48 hours. The study physicians screened records primarily using chart review with secondary confirmation of the diagnosis of OHCA and eligibility criteria. Patients with an outside hospital stay greater than 24 hours, intracranial hemorrhage, use of extracorporeal membranous oxygenation (ECMO), use of airway pressure release mode of ventilation, chronic dependence on mechanical ventilation, or missing data were excluded. Of the 579 patients with OHCA, 256 (44.2%) met the inclusion criteria and were included in the main analysis. A total of 97 patients were identified as having high VT (defined as > 8 mL/kg of predicted body weight [PBW]) and were matched to 97 of the 159 patients identified as having a low VT as part of the propensity-matched subgroup analysis using 1:1 optimal caliper matching.
Main outcome measure. The primary outcome was a favorable neurocognitive outcome at hospital discharge (Cerebral Performance Category score [CPC] of 1 or 2). A CPC of 1 or 2 corresponds to normal life or life that is disabled but independent, respectively. A CPC of 3 is disabled and dependent, and a CPC of 5 is alive but brain dead. Two physicians blinded to VT and other measures of illness severity calculated the CPC via chart review. Discordant scores were resolved by consensus, and a Kappa statistic was calculated to quantify agreement between investigators. Secondary outcomes included ventilator-free days, hospital-free days, ICU-free days, shock-free days, and extrapulmonary organ failure–free days. Logistic regression with backward elimination was used to identify predictors of receiving VT ≤ 8 mL/kg PBW to be used in the propensity-matched analysis, along with relevant predictors identified from the literature. The odds ratio for the primary outcome was calculated using both logistic regression analysis and propensity-matched analysis. Other methods of sensitivity analysis (propensity quintile adjustment, inverse-probability-of-treatment weighting) were used to confirm the robustness of the initial analysis to different statistical methods. A P value of < 0.05 was considered significant.
Main results. Of the study patients, approximately half (49% in high VT, 52% in low VT) had an initial rhythm of ventricular tachycardia or ventricular fibrillation. Patients with low VT were significantly younger (mean age 59 yr vs. 66 yr), taller (mean height 177 cm vs. 165 cm), and heavier (mean weight 88 kg vs. 81 kg). There were also significantly fewer females in the low VT group (19% vs. 46%). There were no significant differences between baseline comorbidities, arrest characteristics, or illness severity between the 2 groups with the exception of significantly more patients in the low VT underwent therapeutic hypothermia (87% vs. 76%) and were admitted to hospital A (69% vs. 55%). There were no significant differences between the groups across ventilator parameters aside from tidal volume. The average VT in mL/kg PBW was 9.3 in the high VT group and 7.1 in the low VT group over the first 48 hours.
In the multivariate regression analysis, significant independent predictors of receiving high VT included height, weight, and hospital of admission. The final propensity model to predict VT included age, height, weight, sex, illness severity measures (APACHE-II score and presence of circulatory shock in the first 24 hours of admission), arrest characteristics, and respiratory characteristics (initial pH, initial PaCO2, PaO2:FiO2 ratio, and initial peak inspiratory pressure) as covariates. The use of low VT was significantly associated with a favorable neurocognitive outcome in the multivariate regression analysis (odds ratio [OR] 1.65, 95% confidence interval [CI] 1.18–2.29). This association held in both the propensity matched analysis (OR 1.68, 95% CI 1.11–2.55) as well as conditional logistic regression analysis using propensity score as a covariate (OR 1.61, 95% CI 1.13–2.28).
In the propensity-adjusted conditional logistic regression analysis, a lower VT (1 mL/kg of PBW decrease) was significantly associated with ventilator-free days (OR 1.78, 95% CI 0.39–3.16), shock-free days (OR 1.31, 95% CI 0.10–2.51), ICU-free days (OR 1.38, 95% CI 0.13–2.63), and hospital-free days (OR 1.07, 95% CI 0.04–2.09). There was a nonsignificant trend towards improved survival to hospital discharge (OR 1.23, 95% CI 0.95–1.60, P = 0.115). After propensity score adjustment, lower VT was not associated with therapeutic hypothermia (OR 0.14, 95% CI −0.19 to 0.47), and in the multivariate regression analysis there was no association between favorable neurocognitive outcome and therapeutic hypothermia (P = 0.516). While there was a significant association between lower VT and site of admission (Hospital A: OR 1.50, 95% CI 1.04–2.17 per 1 mL/kg of PBW decrease), there was no association between favorable neurocognitive outcome and hospital site of admission in the final adjusted regression analysis (P = 0.588).
Conclusion. In this retrospective cohort study, lower VT in the first 48 hours of admission following OHCA was independently associated with favorable neurocognitive outcomes as measured by the CPC score, as well as more ventilator-free, shock-free, ICU-free, and hospital-free days.
Commentary
Neurocognitive impairment following nontraumatic OHCA is common, estimated to occur in roughly half of all survivors [1]. Similar to the acute respiratory distress syndrome (ARDS), the post–cardiac arrest syndrome (PCAS) is recognized as a systemic process with multi-organ effects thought to be mediated in part by inflammatory cytokines [2]. While the beneficial role of low VT in patients with ARDS is well established, currently there are no recommendations for specific VT targets in post–cardiac arrest care, and the effect of VT on outcomes following cardiac arrest is unknown [3].
In this study, Beitler and colleagues suggest a possible association between VT and neurocognitive outcomes following OHCA. Using retrospective data drawn from 2 centers, and employing both regression analysis and propensity matching, the authors identified a significant beneficial effect of lower VT on neurocognitive outcomes in their cohort. This benefit held regardless of the statistical analytic method employed and was present even when correcting for the difference between groups in hospital admission site and use of therapeutic hypothermia in the original cohort. The authors also demonstrated a lower VT was associated with a number of secondary outcomes including fewer hospital, ICU, and ventilator days. While the statistical methods employed by the authors are robust and attempt to account for the limitations inherent to observational studies, a number of questions remain.
First, as the authors appropriately note, causality cannot be proven from a retrospective study. While the analytic methods employed by the authors serve to limit the effect of residual confounding, they do not eliminate it. Although unlikely, it is possible low VT may be a marker for an unmeasured variable that leads to more favorable neurocognitive outcomes. Further research into a possible casual association between VT and neurocognitive outcomes is needed.
The authors also suggest a number of inflammatory-related mechanisms for the association between lower VT and improved neurocognitive outcomes, which they collectively name “brain-lung communication.” While this is a physiologically attractive hypothesis in light of what is known regarding PCAS, the retrospective nature of the study prevents measurement of any inflammatory markers or cytokine levels that might strengthen this hypothesis. As it stands, further exploration of the mechanisms that might link lower VT to improved neurocognitive outcomes will be required before a more definitive statement regarding brain-lung communication can be accepted.
Although the authors identified an association between lower VT and a number of secondary outcomes, their results show there were no significant associations between lower VT and fewer days of extrapulmonary organ failure or improved survival. Given the contradictory nature of some of these secondary outcomes (such as an association with fewer shock-free days but no association with less extrapulmonary organ failure, a known consequence of hemodynamic shock), the true impact of low VT on these outcomes is unclear. While it is logical that the association between lower VT and some secondary outcomes (such as fewer ICU days and fewer ventilator-dependent days) is a result of improved neurocognitive outcomes, further work is required to elucidate the true clinical significance of these secondary outcomes.
Finally, while there was no significant difference between groups in terms of initial pH or PCO2, and these variables were included in the propensity matching analysis, both groups had mean initial PCO2 levels that were elevated (47 mm Hg and 49 mm Hg in the high and low VT groups, respectively). These values are above the physiological range (35–45 mm Hg) recommended by the 2015 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care [3]. The authors suggest that the recommended eucapnic targets can be met in a low VT strategy by increasing the respiratory rate. However, current literature suggests that patients with ARDS exposed to higher respiratory rates may have more frequent exposure to ventilator-induced lung injury (VILI) stresses and an increased rate of lung injury [4]. While there are no clinical trials proving the benefit of a low vs. high respiratory rate strategy, current recommendations for reducing the risk of VILI include limiting the respiratory rate. It is unclear at this time if an increase in the respiratory rate would increase the incidence of VILI and negate any potential benefit provided by low VT in these patients, but this would be an important cost to account for when employing a low VT strategy.
Applications for Clinical Practice
In this study, Beitler and colleagues found that using a low VT ventilation strategy in OHCA patients was associated with improved neurocognitive outcomes. This study is primarily useful as a hypothesis generator. Further research into the effects of ventilator parameters such as VT on the inflammatory cascade, neurocognitive outcomes in other groups of patients (such as those with ARDS), and the existence of a “brain-lung communication” pathway is warranted. From a practical standpoint, evidence continues to mount that lower VT is associated with a number of beneficial effects that are not limited to patients with ARDS [5]. This study would support the current practice of many intensivists to utilize a low VT strategy unless a compelling contraindication exists, as the potential benefits are substantial and the risks minimal. However, this practice will have to be balanced with the need to avoid hypercapnia, and the elevated respiratory rates used to achieve eucapnia may have unforeseen consequences.
—Arun Jose, MD, The George Washington University, Washington, DC
1. Moulaert VR, Verbunt JA, van Heugten CM, Wade DT. Cognitive impairments in survivors of out-of-hospital cardiac arrest: a systematic review. Resuscitation 2009;80:297–305.
2. Peberdy MA, Andersen LW, Abbate A, et al. Inflammatory markers following resuscitation from out-of-hospital cardiac arrest – A prospective multicenter observational study. Resuscitation 2016;103:117–24.
3. Callaway CW, Soar J, Aibiki M, et al. Advanced life support chapter collaborators. Part 4: Advanced life support: 2015 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation 2015;132:S84–S145.
4. Beitler JR, Malhotra A, Thompson BT. Ventilator-induced lung injury. Clin Chest Med 2016;37:633–46.
5. Serpa Neto A, Cardoso SO, Manetta JA, et al. Association between use of lung-protective ventilation with lower tidal volumes and clinical outcomes among patients without acute respiratory distress syndrome: a meta-analysis. JAMA 2012;
308:1651–9.
1. Moulaert VR, Verbunt JA, van Heugten CM, Wade DT. Cognitive impairments in survivors of out-of-hospital cardiac arrest: a systematic review. Resuscitation 2009;80:297–305.
2. Peberdy MA, Andersen LW, Abbate A, et al. Inflammatory markers following resuscitation from out-of-hospital cardiac arrest – A prospective multicenter observational study. Resuscitation 2016;103:117–24.
3. Callaway CW, Soar J, Aibiki M, et al. Advanced life support chapter collaborators. Part 4: Advanced life support: 2015 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation 2015;132:S84–S145.
4. Beitler JR, Malhotra A, Thompson BT. Ventilator-induced lung injury. Clin Chest Med 2016;37:633–46.
5. Serpa Neto A, Cardoso SO, Manetta JA, et al. Association between use of lung-protective ventilation with lower tidal volumes and clinical outcomes among patients without acute respiratory distress syndrome: a meta-analysis. JAMA 2012;
308:1651–9.
Use of Lay Navigation Is Associated with Reduction in Health Care Use and Medicare Costs Among Older Adults with Cancer
Study Overview
Objective. To determine the effect of navigation for older cancer patients by lay persons on health care use and Medicare costs.
Design. Observational cohort study using propensity score–matched controls.
Setting and participants. The study was conducted at the University of Alabama at Birmingham Health System Cancer Community Network, which consists of 2 academic and 10 community cancer centers across Alabama, Georgia, Florida, Mississippi and Tennessee. Participants were Medicare beneficiaries who received care at these facilities from 1 Jan 2012 to 31 Dec 2015. The patient population includes Medicare beneficiaries age 65 years or older with primary Medicare Part A and B insurance and a cancer diagnosis from 2012 to 2015, and excludes those with Medicare health maintenance organization coverage. Only patients with 1 quarter of observation prior to receiving patient navigation and those with 2 quarters of observation after initiation of navigation services were included in the sample. Propensity score matching method was used to establish a matched group of patients without patient navigation. Covariates included in the propensity score matching include age at diagnosis, race, sex, cancer acuity, phase of care, comorbidity score, cost of care, treatment with chemotherapy and emergency department (ED) and intensive care unit use at baseline.
Patients were identified to receive patient navigation through review of patient census in the ED and hospital, clinical referral, and self-referral. High-risk patients were prioritized to receive navigation; this included patients with metastatic disease, cancers that have high morbidity, chronic diseases that have high morbidity, or a history of recent acute care utilization.
Navigation program. The patient navigation program was started in 2013 as an innovation project funded by the Center for Medicare and Medicaid Services. Implementation began in March 2013 and all sites started to enroll patients for patient navigation by October 2013. Patient navigation was conducted by lay navigators who were hired from within the community. Navigators were required to have a bachelor’s degree but were not licensed clinicians such as a nurse or social worker. Patient navigators aimed to proactively identify patient needs, connect patient with resources, coordinate patient care, and empower patients to take an active role in their own care. Navigators performed distress screenings that assessed patients’ practical, information, financial, familial, emotional, spiritual and physical concerns. Assistance was given to patients at patients’ request and algorithms were used to guide frequency of contact.
Main outcome measures. The main outcome measure was Medicare costs per beneficiary per quarter; these costs include all amounts paid by Medicare for all care received but exclude Medicare Part D prescription drug costs. Other outcomes included source of costs and resource utilization defined by number of ED visits, hospitalizations, and intensive care unit admissions per quarter. The return on investment was also examined with the investment defined by salaries for the patient navigators, including fringe benefits; each navigator had a mean caseload of 152 patients per quarter. The return on investment was calculated as reduced Medicare costs of navigated patients compared with non-navigated patients multiplied by the number of patients served.
Main results. A total of 6214 matched pairs of navigated and non-navigated patients were included in the analysis. The mean age of the patients was 75 (SD ± 7) and 12% were African American. Medicare costs declined faster among those with navigation compared to the control group by $781 more per quarter per navigated patient. Inpatient and outpatient costs had the largest decline at $294 and $275 respectively. Resource use decreased in the navigated group more so than the non-navigated group, with a 6% more decrease per quarter in ED visits, 8% more decrease in hospitalization, and 11% more decrease in intensive care unit admissions. The return on investment was estimated at 1:10 with a $475,024 reduction in Medicare cost per navigator annually.
Conclusion. Navigation by lay persons among older Medicare beneficiaries with cancer is associated with a decrease in Medicare costs and resource utilization. There is substantial return on investment when considering the salaries of navigator staff against the Medicare savings associated with care navigation. Lay navigation appears to have benefits of reducing health care costs and resource use.
Commentary
The U.S. health care system is often difficult to navigate for older persons, particularly those with complex health care needs. Older adults with chronic disease, including cancer, may utilize care in multiple settings and with multiple providers and teams, making it challenging to organize and obtain needed care. In this study, the authors examined the impact of a patient navigation program on older patients with cancer and found that the use of lay navigation is associated with reduced costs and resource use for the health care system. Prior studies have examined the use of care navigation in complex chronic disease management, such as HIV infection and cancer [1,2] and have often found positive impacts on patient satisfaction, adherence to treatments, and reducing care disparities in vulnerable populations [3]. Care navigation has been performed using clinical staff, such as nursing, but with lay persons as well [4]. Lay persons offer the benefit of reduced costs, and if they are able to perform the care navigation tasks well with training, clinical staff use may not be necessary.
This study uses alternative methods to randomization to generate a balanced non-navigated control group for comparison to determine the impact of care navigation. The limitation is that the propensity score method used may balance potential confounders that are specified but unmeasured confounders were not accounted for in the analysis [5]. Another limitation is that the comparison group may not be concurrent, because non-navigated patients prior to the initiation of the navigation program were included in the control group. As care delivery often changes over time, non-concurrent comparison may introduce bias in the study. Nonetheless, the effect that is found on costs and resource utilization appear to be a strong one and is consistent with prior studies on the effects of care navigation.
Although this study spanned several clinical settings across a large geographic area, it is unclear if the program will offer similar benefits at other institutions. Additional studies that examine the impact of lay navigation on other patient outcomes such as satisfaction will be useful, as will studying the model at other institutions and in other settings to examine whether the program’s effects can be generalized.
Applications for Clinical Practice
In general, evidence suggests that patient navigation is an effective intervention for use in health care. Clinicians should consider assigning team members to help their patients with cancer navigate the health care system.
—William W. Hung, MD, MPH
1. Shacham E, López JD, Brown TM, et al. Enhancing adherence to care in the HIV care continuum: the Barrier Elimination and Care Navigation (BEACON) project evaluation. AIDS Behav 2017.
2. Ali-Faisal SF, Colella TJ, Medina-Jaudes N, Benz Scott L. The effectiveness of patient navigation to improve healthcare utilization outcomes: A meta-analysis of randomized controlled trials. Patient Educ Couns 2017;100:436–48.
3. Steinberg ML, Fremont A, Khan DC, et al. Lay patient navigator program implementation for equal access to cancer care and clinical trials: essential steps and initial challenges. Cancer 2006;107:2669–77.
4. Kim K, Choi JS, Choi E, et al. Effects of community-based health worker interventions to improve chronic disease management and care among vulnerable populations: a systematic review. Am J Public Health 2016;106:e3–e28.
5. Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med 2007;26:734–53.
Study Overview
Objective. To determine the effect of navigation for older cancer patients by lay persons on health care use and Medicare costs.
Design. Observational cohort study using propensity score–matched controls.
Setting and participants. The study was conducted at the University of Alabama at Birmingham Health System Cancer Community Network, which consists of 2 academic and 10 community cancer centers across Alabama, Georgia, Florida, Mississippi and Tennessee. Participants were Medicare beneficiaries who received care at these facilities from 1 Jan 2012 to 31 Dec 2015. The patient population includes Medicare beneficiaries age 65 years or older with primary Medicare Part A and B insurance and a cancer diagnosis from 2012 to 2015, and excludes those with Medicare health maintenance organization coverage. Only patients with 1 quarter of observation prior to receiving patient navigation and those with 2 quarters of observation after initiation of navigation services were included in the sample. Propensity score matching method was used to establish a matched group of patients without patient navigation. Covariates included in the propensity score matching include age at diagnosis, race, sex, cancer acuity, phase of care, comorbidity score, cost of care, treatment with chemotherapy and emergency department (ED) and intensive care unit use at baseline.
Patients were identified to receive patient navigation through review of patient census in the ED and hospital, clinical referral, and self-referral. High-risk patients were prioritized to receive navigation; this included patients with metastatic disease, cancers that have high morbidity, chronic diseases that have high morbidity, or a history of recent acute care utilization.
Navigation program. The patient navigation program was started in 2013 as an innovation project funded by the Center for Medicare and Medicaid Services. Implementation began in March 2013 and all sites started to enroll patients for patient navigation by October 2013. Patient navigation was conducted by lay navigators who were hired from within the community. Navigators were required to have a bachelor’s degree but were not licensed clinicians such as a nurse or social worker. Patient navigators aimed to proactively identify patient needs, connect patient with resources, coordinate patient care, and empower patients to take an active role in their own care. Navigators performed distress screenings that assessed patients’ practical, information, financial, familial, emotional, spiritual and physical concerns. Assistance was given to patients at patients’ request and algorithms were used to guide frequency of contact.
Main outcome measures. The main outcome measure was Medicare costs per beneficiary per quarter; these costs include all amounts paid by Medicare for all care received but exclude Medicare Part D prescription drug costs. Other outcomes included source of costs and resource utilization defined by number of ED visits, hospitalizations, and intensive care unit admissions per quarter. The return on investment was also examined with the investment defined by salaries for the patient navigators, including fringe benefits; each navigator had a mean caseload of 152 patients per quarter. The return on investment was calculated as reduced Medicare costs of navigated patients compared with non-navigated patients multiplied by the number of patients served.
Main results. A total of 6214 matched pairs of navigated and non-navigated patients were included in the analysis. The mean age of the patients was 75 (SD ± 7) and 12% were African American. Medicare costs declined faster among those with navigation compared to the control group by $781 more per quarter per navigated patient. Inpatient and outpatient costs had the largest decline at $294 and $275 respectively. Resource use decreased in the navigated group more so than the non-navigated group, with a 6% more decrease per quarter in ED visits, 8% more decrease in hospitalization, and 11% more decrease in intensive care unit admissions. The return on investment was estimated at 1:10 with a $475,024 reduction in Medicare cost per navigator annually.
Conclusion. Navigation by lay persons among older Medicare beneficiaries with cancer is associated with a decrease in Medicare costs and resource utilization. There is substantial return on investment when considering the salaries of navigator staff against the Medicare savings associated with care navigation. Lay navigation appears to have benefits of reducing health care costs and resource use.
Commentary
The U.S. health care system is often difficult to navigate for older persons, particularly those with complex health care needs. Older adults with chronic disease, including cancer, may utilize care in multiple settings and with multiple providers and teams, making it challenging to organize and obtain needed care. In this study, the authors examined the impact of a patient navigation program on older patients with cancer and found that the use of lay navigation is associated with reduced costs and resource use for the health care system. Prior studies have examined the use of care navigation in complex chronic disease management, such as HIV infection and cancer [1,2] and have often found positive impacts on patient satisfaction, adherence to treatments, and reducing care disparities in vulnerable populations [3]. Care navigation has been performed using clinical staff, such as nursing, but with lay persons as well [4]. Lay persons offer the benefit of reduced costs, and if they are able to perform the care navigation tasks well with training, clinical staff use may not be necessary.
This study uses alternative methods to randomization to generate a balanced non-navigated control group for comparison to determine the impact of care navigation. The limitation is that the propensity score method used may balance potential confounders that are specified but unmeasured confounders were not accounted for in the analysis [5]. Another limitation is that the comparison group may not be concurrent, because non-navigated patients prior to the initiation of the navigation program were included in the control group. As care delivery often changes over time, non-concurrent comparison may introduce bias in the study. Nonetheless, the effect that is found on costs and resource utilization appear to be a strong one and is consistent with prior studies on the effects of care navigation.
Although this study spanned several clinical settings across a large geographic area, it is unclear if the program will offer similar benefits at other institutions. Additional studies that examine the impact of lay navigation on other patient outcomes such as satisfaction will be useful, as will studying the model at other institutions and in other settings to examine whether the program’s effects can be generalized.
Applications for Clinical Practice
In general, evidence suggests that patient navigation is an effective intervention for use in health care. Clinicians should consider assigning team members to help their patients with cancer navigate the health care system.
—William W. Hung, MD, MPH
Study Overview
Objective. To determine the effect of navigation for older cancer patients by lay persons on health care use and Medicare costs.
Design. Observational cohort study using propensity score–matched controls.
Setting and participants. The study was conducted at the University of Alabama at Birmingham Health System Cancer Community Network, which consists of 2 academic and 10 community cancer centers across Alabama, Georgia, Florida, Mississippi and Tennessee. Participants were Medicare beneficiaries who received care at these facilities from 1 Jan 2012 to 31 Dec 2015. The patient population includes Medicare beneficiaries age 65 years or older with primary Medicare Part A and B insurance and a cancer diagnosis from 2012 to 2015, and excludes those with Medicare health maintenance organization coverage. Only patients with 1 quarter of observation prior to receiving patient navigation and those with 2 quarters of observation after initiation of navigation services were included in the sample. Propensity score matching method was used to establish a matched group of patients without patient navigation. Covariates included in the propensity score matching include age at diagnosis, race, sex, cancer acuity, phase of care, comorbidity score, cost of care, treatment with chemotherapy and emergency department (ED) and intensive care unit use at baseline.
Patients were identified to receive patient navigation through review of patient census in the ED and hospital, clinical referral, and self-referral. High-risk patients were prioritized to receive navigation; this included patients with metastatic disease, cancers that have high morbidity, chronic diseases that have high morbidity, or a history of recent acute care utilization.
Navigation program. The patient navigation program was started in 2013 as an innovation project funded by the Center for Medicare and Medicaid Services. Implementation began in March 2013 and all sites started to enroll patients for patient navigation by October 2013. Patient navigation was conducted by lay navigators who were hired from within the community. Navigators were required to have a bachelor’s degree but were not licensed clinicians such as a nurse or social worker. Patient navigators aimed to proactively identify patient needs, connect patient with resources, coordinate patient care, and empower patients to take an active role in their own care. Navigators performed distress screenings that assessed patients’ practical, information, financial, familial, emotional, spiritual and physical concerns. Assistance was given to patients at patients’ request and algorithms were used to guide frequency of contact.
Main outcome measures. The main outcome measure was Medicare costs per beneficiary per quarter; these costs include all amounts paid by Medicare for all care received but exclude Medicare Part D prescription drug costs. Other outcomes included source of costs and resource utilization defined by number of ED visits, hospitalizations, and intensive care unit admissions per quarter. The return on investment was also examined with the investment defined by salaries for the patient navigators, including fringe benefits; each navigator had a mean caseload of 152 patients per quarter. The return on investment was calculated as reduced Medicare costs of navigated patients compared with non-navigated patients multiplied by the number of patients served.
Main results. A total of 6214 matched pairs of navigated and non-navigated patients were included in the analysis. The mean age of the patients was 75 (SD ± 7) and 12% were African American. Medicare costs declined faster among those with navigation compared to the control group by $781 more per quarter per navigated patient. Inpatient and outpatient costs had the largest decline at $294 and $275 respectively. Resource use decreased in the navigated group more so than the non-navigated group, with a 6% more decrease per quarter in ED visits, 8% more decrease in hospitalization, and 11% more decrease in intensive care unit admissions. The return on investment was estimated at 1:10 with a $475,024 reduction in Medicare cost per navigator annually.
Conclusion. Navigation by lay persons among older Medicare beneficiaries with cancer is associated with a decrease in Medicare costs and resource utilization. There is substantial return on investment when considering the salaries of navigator staff against the Medicare savings associated with care navigation. Lay navigation appears to have benefits of reducing health care costs and resource use.
Commentary
The U.S. health care system is often difficult to navigate for older persons, particularly those with complex health care needs. Older adults with chronic disease, including cancer, may utilize care in multiple settings and with multiple providers and teams, making it challenging to organize and obtain needed care. In this study, the authors examined the impact of a patient navigation program on older patients with cancer and found that the use of lay navigation is associated with reduced costs and resource use for the health care system. Prior studies have examined the use of care navigation in complex chronic disease management, such as HIV infection and cancer [1,2] and have often found positive impacts on patient satisfaction, adherence to treatments, and reducing care disparities in vulnerable populations [3]. Care navigation has been performed using clinical staff, such as nursing, but with lay persons as well [4]. Lay persons offer the benefit of reduced costs, and if they are able to perform the care navigation tasks well with training, clinical staff use may not be necessary.
This study uses alternative methods to randomization to generate a balanced non-navigated control group for comparison to determine the impact of care navigation. The limitation is that the propensity score method used may balance potential confounders that are specified but unmeasured confounders were not accounted for in the analysis [5]. Another limitation is that the comparison group may not be concurrent, because non-navigated patients prior to the initiation of the navigation program were included in the control group. As care delivery often changes over time, non-concurrent comparison may introduce bias in the study. Nonetheless, the effect that is found on costs and resource utilization appear to be a strong one and is consistent with prior studies on the effects of care navigation.
Although this study spanned several clinical settings across a large geographic area, it is unclear if the program will offer similar benefits at other institutions. Additional studies that examine the impact of lay navigation on other patient outcomes such as satisfaction will be useful, as will studying the model at other institutions and in other settings to examine whether the program’s effects can be generalized.
Applications for Clinical Practice
In general, evidence suggests that patient navigation is an effective intervention for use in health care. Clinicians should consider assigning team members to help their patients with cancer navigate the health care system.
—William W. Hung, MD, MPH
1. Shacham E, López JD, Brown TM, et al. Enhancing adherence to care in the HIV care continuum: the Barrier Elimination and Care Navigation (BEACON) project evaluation. AIDS Behav 2017.
2. Ali-Faisal SF, Colella TJ, Medina-Jaudes N, Benz Scott L. The effectiveness of patient navigation to improve healthcare utilization outcomes: A meta-analysis of randomized controlled trials. Patient Educ Couns 2017;100:436–48.
3. Steinberg ML, Fremont A, Khan DC, et al. Lay patient navigator program implementation for equal access to cancer care and clinical trials: essential steps and initial challenges. Cancer 2006;107:2669–77.
4. Kim K, Choi JS, Choi E, et al. Effects of community-based health worker interventions to improve chronic disease management and care among vulnerable populations: a systematic review. Am J Public Health 2016;106:e3–e28.
5. Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med 2007;26:734–53.
1. Shacham E, López JD, Brown TM, et al. Enhancing adherence to care in the HIV care continuum: the Barrier Elimination and Care Navigation (BEACON) project evaluation. AIDS Behav 2017.
2. Ali-Faisal SF, Colella TJ, Medina-Jaudes N, Benz Scott L. The effectiveness of patient navigation to improve healthcare utilization outcomes: A meta-analysis of randomized controlled trials. Patient Educ Couns 2017;100:436–48.
3. Steinberg ML, Fremont A, Khan DC, et al. Lay patient navigator program implementation for equal access to cancer care and clinical trials: essential steps and initial challenges. Cancer 2006;107:2669–77.
4. Kim K, Choi JS, Choi E, et al. Effects of community-based health worker interventions to improve chronic disease management and care among vulnerable populations: a systematic review. Am J Public Health 2016;106:e3–e28.
5. Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med 2007;26:734–53.
Vaccination does not eliminate risk for meningococcal disease in eculizumab recipients
Patients taking eculizumab are at a significant risk for meningococcal disease even if they have received the quadrivalent meningococcal conjugate (MenACWY) and serogroup B (MenB) meningococcal vaccines, according to the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report, released July 7.
Between 2008 and 2016, 16 cases of meningococcal disease were reported in eculizumab users in 10 jurisdictions within the United States. Of those infected, 14 had received MenACWY and MenB vaccines as recommended by the Advisory Committee on Immunization Practices, according to the CDC report.
Required vaccination plus antimicrobial prophylaxis for the duration of eculizumab treatment might reduce the risk for meningococcal disease in these patients, but the addition of antibiotic prophylaxis is no guarantee that all cases of meningococcal disease would be prevented, wrote Lucy A. McNamara, PhD, of the division of bacterial diseases, National Center for Immunization and Respiratory Diseases, CDC, and her colleagues.
They advised physician and patient vigilance regarding meningococcal disease symptoms and urged that patients be advised to seek immediate care and be rapidly treated, regardless of meningococcal vaccination or antimicrobial prophylaxis status.
Health organizations in Europe, including France and the United Kingdom, are recommending eculizumab users receive penicillin during eculizumab treatment. A recent study of invasive meningococcal isolates in the United States found most were susceptible to penicillin, according to the report.
In the 16 U.S. cases reported, nongroupable Neisseria meningitidis caused meningococcal disease in 11 of the patients, serogroup Y was the cause in 4 patients, and the cause was not identified in 1 patient.
Ten patients had meningococcemia without meningitis, the researchers noted. “Initial symptoms of meningococcemia are often relatively mild and nonspecific and might include fever, chills, fatigue, vomiting, diarrhea, and aches or pains in the muscles, joints, chest, or abdomen; however, these symptoms can progress to severe illness and death within hours.”
Eculizumab (Soliris, Alexion Pharmaceuticals) is licensed in the United States for treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, two diseases that are rare and can be fatal.
Eculizumab is associated with a 1,000-fold to 2,000-fold increased incidence of meningococcal disease among persons receiving the drug. The Food and Drug Administration–approved prescribing information includes a boxed warning regarding increased risk for meningococcal disease.
The CDC is collecting reports from state health departments for further analysis of the risk among eculizumab recipients.
The researchers reported having no conflicts of interest.
[email protected]
On Twitter @eaztweets
Patients taking eculizumab are at a significant risk for meningococcal disease even if they have received the quadrivalent meningococcal conjugate (MenACWY) and serogroup B (MenB) meningococcal vaccines, according to the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report, released July 7.
Between 2008 and 2016, 16 cases of meningococcal disease were reported in eculizumab users in 10 jurisdictions within the United States. Of those infected, 14 had received MenACWY and MenB vaccines as recommended by the Advisory Committee on Immunization Practices, according to the CDC report.
Required vaccination plus antimicrobial prophylaxis for the duration of eculizumab treatment might reduce the risk for meningococcal disease in these patients, but the addition of antibiotic prophylaxis is no guarantee that all cases of meningococcal disease would be prevented, wrote Lucy A. McNamara, PhD, of the division of bacterial diseases, National Center for Immunization and Respiratory Diseases, CDC, and her colleagues.
They advised physician and patient vigilance regarding meningococcal disease symptoms and urged that patients be advised to seek immediate care and be rapidly treated, regardless of meningococcal vaccination or antimicrobial prophylaxis status.
Health organizations in Europe, including France and the United Kingdom, are recommending eculizumab users receive penicillin during eculizumab treatment. A recent study of invasive meningococcal isolates in the United States found most were susceptible to penicillin, according to the report.
In the 16 U.S. cases reported, nongroupable Neisseria meningitidis caused meningococcal disease in 11 of the patients, serogroup Y was the cause in 4 patients, and the cause was not identified in 1 patient.
Ten patients had meningococcemia without meningitis, the researchers noted. “Initial symptoms of meningococcemia are often relatively mild and nonspecific and might include fever, chills, fatigue, vomiting, diarrhea, and aches or pains in the muscles, joints, chest, or abdomen; however, these symptoms can progress to severe illness and death within hours.”
Eculizumab (Soliris, Alexion Pharmaceuticals) is licensed in the United States for treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, two diseases that are rare and can be fatal.
Eculizumab is associated with a 1,000-fold to 2,000-fold increased incidence of meningococcal disease among persons receiving the drug. The Food and Drug Administration–approved prescribing information includes a boxed warning regarding increased risk for meningococcal disease.
The CDC is collecting reports from state health departments for further analysis of the risk among eculizumab recipients.
The researchers reported having no conflicts of interest.
[email protected]
On Twitter @eaztweets
Patients taking eculizumab are at a significant risk for meningococcal disease even if they have received the quadrivalent meningococcal conjugate (MenACWY) and serogroup B (MenB) meningococcal vaccines, according to the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report, released July 7.
Between 2008 and 2016, 16 cases of meningococcal disease were reported in eculizumab users in 10 jurisdictions within the United States. Of those infected, 14 had received MenACWY and MenB vaccines as recommended by the Advisory Committee on Immunization Practices, according to the CDC report.
Required vaccination plus antimicrobial prophylaxis for the duration of eculizumab treatment might reduce the risk for meningococcal disease in these patients, but the addition of antibiotic prophylaxis is no guarantee that all cases of meningococcal disease would be prevented, wrote Lucy A. McNamara, PhD, of the division of bacterial diseases, National Center for Immunization and Respiratory Diseases, CDC, and her colleagues.
They advised physician and patient vigilance regarding meningococcal disease symptoms and urged that patients be advised to seek immediate care and be rapidly treated, regardless of meningococcal vaccination or antimicrobial prophylaxis status.
Health organizations in Europe, including France and the United Kingdom, are recommending eculizumab users receive penicillin during eculizumab treatment. A recent study of invasive meningococcal isolates in the United States found most were susceptible to penicillin, according to the report.
In the 16 U.S. cases reported, nongroupable Neisseria meningitidis caused meningococcal disease in 11 of the patients, serogroup Y was the cause in 4 patients, and the cause was not identified in 1 patient.
Ten patients had meningococcemia without meningitis, the researchers noted. “Initial symptoms of meningococcemia are often relatively mild and nonspecific and might include fever, chills, fatigue, vomiting, diarrhea, and aches or pains in the muscles, joints, chest, or abdomen; however, these symptoms can progress to severe illness and death within hours.”
Eculizumab (Soliris, Alexion Pharmaceuticals) is licensed in the United States for treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, two diseases that are rare and can be fatal.
Eculizumab is associated with a 1,000-fold to 2,000-fold increased incidence of meningococcal disease among persons receiving the drug. The Food and Drug Administration–approved prescribing information includes a boxed warning regarding increased risk for meningococcal disease.
The CDC is collecting reports from state health departments for further analysis of the risk among eculizumab recipients.
The researchers reported having no conflicts of interest.
[email protected]
On Twitter @eaztweets
FROM MMWR
Standardization lacking in pediatric trials of atopic dermatitis
CHICAGO – There is considerable variability and poor documentation of severity assessments used for inclusion criteria and baseline severity evaluations in randomized, controlled pediatric atopic dermatitis (AD) trials, results from a systematic review showed.
“It is important for clinicians and investigators to recognize that these differences may limit our ability to reproduce trials, interpret individual studies, and compare results between studies of similar target populations for severity,” lead study author Rishi Chopra, MS, said in an interview in advance of the World Congress of Pediatric Dermatology. “Moreover, this heterogeneity should be considered when retroactively pooling results for meta-analyses of pediatric atopic dermatitis randomized, controlled trials.”

In an effort to evaluate the documentation and characterize the severity assessments used in inclusion criteria and baseline evaluations for randomized, controlled trials of pediatric AD internationally, the researchers performed a systematic review of relevant studies contained in the Cochrane Library, Embase, LILACS, GREAT, MEDLINE, and Scopus databases during 2007-2016. Inclusion criteria were RCT with a pharmacological intervention and any comparison with a control group, children, and males or females. In all, 89 studies met the inclusion/exclusion criteria. Most (70.8%) were studies of pediatric populations aged 0-17 years, and almost 17% were studies of infants aged 0-1 years. The most common target populations were mild-moderate AD (31.5%), moderate-severe AD (18.0%), or undefined (36.0%).
Mr. Chopra and his associates found that the most commonly used severity indices were Scoring AD (SCORAD) in 29.2%, Body Surface Area (BSA) in 16.9%, and global assessments in 13.4%, while the most common assessments of baseline severity were SCORAD in 43.8%, global assessments in 20.2%, Eczema Area and Severity Index in 17.9%, BSA in 14.6%, and visual itch in 13.5%. Only 85.4% of studies recorded the severity assessments used for recruiting the predefined target population and only 76.4% of studies documented baseline severity.
There was considerable heterogeneity across studies, as 16 unique assessments were used as inclusion criteria and 34 assessments were used to evaluate baseline severity. “In addition, even within an individual study, there was substantial discordance in their use as only 71.2% of studies used the same assessments for inclusion and documenting baseline disease severity,” Mr. Chopra said. “Altogether, this multidimensional lack of documentation and heterogeneity of inclusion criteria and baseline severity assessments limits our ability to assess whether the recruitment methods for patients were adequate and confirm whether the intended target population for severity was successfully enrolled.”
He acknowledged certain limitations of the study, including the fact that it may not be generalizable to nonpharmacological interventional trials or noninterventional studies. “In addition, we could only conduct an analysis for studies that provided adequate documentation of inclusion criteria and baseline severity,” Mr. Chopra said. “Thus, those studies that did not provide this information were left out. Nevertheless, across all studies, the uniformity and concordance between assessments likely are even more negatively impacted. It should also be noted that lack of documentation of assessments for inclusion criteria and baseline severity does not imply lack of their utilization. Finally, it is important to acknowledge that AD’s diverse phenotype and relapsing and remitting course may result in the unavoidable heterogeneity of severity assessment use. This may actually help to capture a broader range of disease and improve the external validity of results.”
He reported having no financial disclosures.
CHICAGO – There is considerable variability and poor documentation of severity assessments used for inclusion criteria and baseline severity evaluations in randomized, controlled pediatric atopic dermatitis (AD) trials, results from a systematic review showed.
“It is important for clinicians and investigators to recognize that these differences may limit our ability to reproduce trials, interpret individual studies, and compare results between studies of similar target populations for severity,” lead study author Rishi Chopra, MS, said in an interview in advance of the World Congress of Pediatric Dermatology. “Moreover, this heterogeneity should be considered when retroactively pooling results for meta-analyses of pediatric atopic dermatitis randomized, controlled trials.”
In an effort to evaluate the documentation and characterize the severity assessments used in inclusion criteria and baseline evaluations for randomized, controlled trials of pediatric AD internationally, the researchers performed a systematic review of relevant studies contained in the Cochrane Library, Embase, LILACS, GREAT, MEDLINE, and Scopus databases during 2007-2016. Inclusion criteria were RCT with a pharmacological intervention and any comparison with a control group, children, and males or females. In all, 89 studies met the inclusion/exclusion criteria. Most (70.8%) were studies of pediatric populations aged 0-17 years, and almost 17% were studies of infants aged 0-1 years. The most common target populations were mild-moderate AD (31.5%), moderate-severe AD (18.0%), or undefined (36.0%).
Mr. Chopra and his associates found that the most commonly used severity indices were Scoring AD (SCORAD) in 29.2%, Body Surface Area (BSA) in 16.9%, and global assessments in 13.4%, while the most common assessments of baseline severity were SCORAD in 43.8%, global assessments in 20.2%, Eczema Area and Severity Index in 17.9%, BSA in 14.6%, and visual itch in 13.5%. Only 85.4% of studies recorded the severity assessments used for recruiting the predefined target population and only 76.4% of studies documented baseline severity.
There was considerable heterogeneity across studies, as 16 unique assessments were used as inclusion criteria and 34 assessments were used to evaluate baseline severity. “In addition, even within an individual study, there was substantial discordance in their use as only 71.2% of studies used the same assessments for inclusion and documenting baseline disease severity,” Mr. Chopra said. “Altogether, this multidimensional lack of documentation and heterogeneity of inclusion criteria and baseline severity assessments limits our ability to assess whether the recruitment methods for patients were adequate and confirm whether the intended target population for severity was successfully enrolled.”
He acknowledged certain limitations of the study, including the fact that it may not be generalizable to nonpharmacological interventional trials or noninterventional studies. “In addition, we could only conduct an analysis for studies that provided adequate documentation of inclusion criteria and baseline severity,” Mr. Chopra said. “Thus, those studies that did not provide this information were left out. Nevertheless, across all studies, the uniformity and concordance between assessments likely are even more negatively impacted. It should also be noted that lack of documentation of assessments for inclusion criteria and baseline severity does not imply lack of their utilization. Finally, it is important to acknowledge that AD’s diverse phenotype and relapsing and remitting course may result in the unavoidable heterogeneity of severity assessment use. This may actually help to capture a broader range of disease and improve the external validity of results.”
He reported having no financial disclosures.
CHICAGO – There is considerable variability and poor documentation of severity assessments used for inclusion criteria and baseline severity evaluations in randomized, controlled pediatric atopic dermatitis (AD) trials, results from a systematic review showed.
“It is important for clinicians and investigators to recognize that these differences may limit our ability to reproduce trials, interpret individual studies, and compare results between studies of similar target populations for severity,” lead study author Rishi Chopra, MS, said in an interview in advance of the World Congress of Pediatric Dermatology. “Moreover, this heterogeneity should be considered when retroactively pooling results for meta-analyses of pediatric atopic dermatitis randomized, controlled trials.”
In an effort to evaluate the documentation and characterize the severity assessments used in inclusion criteria and baseline evaluations for randomized, controlled trials of pediatric AD internationally, the researchers performed a systematic review of relevant studies contained in the Cochrane Library, Embase, LILACS, GREAT, MEDLINE, and Scopus databases during 2007-2016. Inclusion criteria were RCT with a pharmacological intervention and any comparison with a control group, children, and males or females. In all, 89 studies met the inclusion/exclusion criteria. Most (70.8%) were studies of pediatric populations aged 0-17 years, and almost 17% were studies of infants aged 0-1 years. The most common target populations were mild-moderate AD (31.5%), moderate-severe AD (18.0%), or undefined (36.0%).
Mr. Chopra and his associates found that the most commonly used severity indices were Scoring AD (SCORAD) in 29.2%, Body Surface Area (BSA) in 16.9%, and global assessments in 13.4%, while the most common assessments of baseline severity were SCORAD in 43.8%, global assessments in 20.2%, Eczema Area and Severity Index in 17.9%, BSA in 14.6%, and visual itch in 13.5%. Only 85.4% of studies recorded the severity assessments used for recruiting the predefined target population and only 76.4% of studies documented baseline severity.
There was considerable heterogeneity across studies, as 16 unique assessments were used as inclusion criteria and 34 assessments were used to evaluate baseline severity. “In addition, even within an individual study, there was substantial discordance in their use as only 71.2% of studies used the same assessments for inclusion and documenting baseline disease severity,” Mr. Chopra said. “Altogether, this multidimensional lack of documentation and heterogeneity of inclusion criteria and baseline severity assessments limits our ability to assess whether the recruitment methods for patients were adequate and confirm whether the intended target population for severity was successfully enrolled.”
He acknowledged certain limitations of the study, including the fact that it may not be generalizable to nonpharmacological interventional trials or noninterventional studies. “In addition, we could only conduct an analysis for studies that provided adequate documentation of inclusion criteria and baseline severity,” Mr. Chopra said. “Thus, those studies that did not provide this information were left out. Nevertheless, across all studies, the uniformity and concordance between assessments likely are even more negatively impacted. It should also be noted that lack of documentation of assessments for inclusion criteria and baseline severity does not imply lack of their utilization. Finally, it is important to acknowledge that AD’s diverse phenotype and relapsing and remitting course may result in the unavoidable heterogeneity of severity assessment use. This may actually help to capture a broader range of disease and improve the external validity of results.”
He reported having no financial disclosures.
AT WCPD 2017
Key clinical point:
Major finding: Only 85.4% of studies recorded the severity assessments used for recruiting the predefined target population, and only 76.4% of studies documented baseline severity.
Data source: A systematic review of 89 pediatric atopic dermatitis randomized, controlled trials published during 2007-2016.
Disclosures: Mr. Chopra reported having no financial disclosures.
Subset of pediatric herpes simplex virus entails frequent episodes
There’s a newly identified, atypical, but substantial subset of patients: Young children who have frequent episodes of herpes simplex virus (HSV) that may not affect mucosal tissue, often are multifocal, and may require suppressive therapy, according to a report published in Pediatric Dermatology.
“Most of our knowledge of the clinical manifestations of HSV infections in children derives from cases of common mucosal HSV that present to and are treated by primary care providers,” said Julia K. Gittler, MD, and her associates at New York University. 
These cases comprise primary herpetic gingivostomatitis or the well-known orolabial HSV. To characterize the more atypical presentations in the pediatric population, the investigators reviewed the charts of all 48 patients referred to their pediatric dermatology clinic in a 10-year period after receiving a diagnosis of HSV. Only 19% of these patients were older than 11 years of age, and more than 40% were younger than 2 years of age at presentation.
Dr. Gittler and her associates found a “substantial” subset of 19 patients (approximately 40% of the study sample) who had six or more outbreaks per year. Only four patients (8.3%) had one or fewer outbreaks per year. The mean frequency of outbreaks in their study population was seven per year. “Reported triggers for outbreaks included fever, viral infections, sun, cold, stress, vaccinations, and courses of oral corticosteroids for asthma flares,” the investigators said.
Given this high frequency of outbreaks, antiviral therapy was initiated in 16 patients (33%). The average age of this treatment initiation was only 6 years. “In the general population, only approximately 15%-40% of patients with serologic evidence of HSV-1 have recurrent HSV; most of these will have an average of 2 outbreaks per year, and only 5%-10% have more than 6 outbreaks per year,” the investigators wrote (Pediatr Dermatol. 2017. doi: 10.1111/pde.13190).
The majority of the study participants (29 patients) had no labial or mucosal involvement. Approximately 23% had cutaneous involvement of the cheek, “which may have been because the primary infection was acquired through the kiss of a caregiver,” they said. Other sites of cutaneous lesions that were frequent in this population but are considered atypical in general were the ear, forehead, chest, and knees.
Most commonly, the physical examination findings were vesicles or papulovesicles (52%), but crusting (33%), pustules (6%), and erosions (23%) also occurred.
Fully half of the study population had multifocal rather than unifocal presentations.
Atypical presentation of HSV is frequently misdiagnosed as impetigo or herpes zoster, according to reports in the literature. Fifteen (31%) patients in this study had a previous misdiagnosis of impetigo. “These less typical cutaneous presentations of HSV may evade accurate diagnosis and delay initiation of appropriate treatment,” Dr. Gittler and her associates noted.
No financial disclosures were provided for Dr. Gittler and her associates.
There’s a newly identified, atypical, but substantial subset of patients: Young children who have frequent episodes of herpes simplex virus (HSV) that may not affect mucosal tissue, often are multifocal, and may require suppressive therapy, according to a report published in Pediatric Dermatology.
“Most of our knowledge of the clinical manifestations of HSV infections in children derives from cases of common mucosal HSV that present to and are treated by primary care providers,” said Julia K. Gittler, MD, and her associates at New York University.
These cases comprise primary herpetic gingivostomatitis or the well-known orolabial HSV. To characterize the more atypical presentations in the pediatric population, the investigators reviewed the charts of all 48 patients referred to their pediatric dermatology clinic in a 10-year period after receiving a diagnosis of HSV. Only 19% of these patients were older than 11 years of age, and more than 40% were younger than 2 years of age at presentation.
Dr. Gittler and her associates found a “substantial” subset of 19 patients (approximately 40% of the study sample) who had six or more outbreaks per year. Only four patients (8.3%) had one or fewer outbreaks per year. The mean frequency of outbreaks in their study population was seven per year. “Reported triggers for outbreaks included fever, viral infections, sun, cold, stress, vaccinations, and courses of oral corticosteroids for asthma flares,” the investigators said.
Given this high frequency of outbreaks, antiviral therapy was initiated in 16 patients (33%). The average age of this treatment initiation was only 6 years. “In the general population, only approximately 15%-40% of patients with serologic evidence of HSV-1 have recurrent HSV; most of these will have an average of 2 outbreaks per year, and only 5%-10% have more than 6 outbreaks per year,” the investigators wrote (Pediatr Dermatol. 2017. doi: 10.1111/pde.13190).
The majority of the study participants (29 patients) had no labial or mucosal involvement. Approximately 23% had cutaneous involvement of the cheek, “which may have been because the primary infection was acquired through the kiss of a caregiver,” they said. Other sites of cutaneous lesions that were frequent in this population but are considered atypical in general were the ear, forehead, chest, and knees.
Most commonly, the physical examination findings were vesicles or papulovesicles (52%), but crusting (33%), pustules (6%), and erosions (23%) also occurred.
Fully half of the study population had multifocal rather than unifocal presentations.
Atypical presentation of HSV is frequently misdiagnosed as impetigo or herpes zoster, according to reports in the literature. Fifteen (31%) patients in this study had a previous misdiagnosis of impetigo. “These less typical cutaneous presentations of HSV may evade accurate diagnosis and delay initiation of appropriate treatment,” Dr. Gittler and her associates noted.
No financial disclosures were provided for Dr. Gittler and her associates.
There’s a newly identified, atypical, but substantial subset of patients: Young children who have frequent episodes of herpes simplex virus (HSV) that may not affect mucosal tissue, often are multifocal, and may require suppressive therapy, according to a report published in Pediatric Dermatology.
“Most of our knowledge of the clinical manifestations of HSV infections in children derives from cases of common mucosal HSV that present to and are treated by primary care providers,” said Julia K. Gittler, MD, and her associates at New York University.
These cases comprise primary herpetic gingivostomatitis or the well-known orolabial HSV. To characterize the more atypical presentations in the pediatric population, the investigators reviewed the charts of all 48 patients referred to their pediatric dermatology clinic in a 10-year period after receiving a diagnosis of HSV. Only 19% of these patients were older than 11 years of age, and more than 40% were younger than 2 years of age at presentation.
Dr. Gittler and her associates found a “substantial” subset of 19 patients (approximately 40% of the study sample) who had six or more outbreaks per year. Only four patients (8.3%) had one or fewer outbreaks per year. The mean frequency of outbreaks in their study population was seven per year. “Reported triggers for outbreaks included fever, viral infections, sun, cold, stress, vaccinations, and courses of oral corticosteroids for asthma flares,” the investigators said.
Given this high frequency of outbreaks, antiviral therapy was initiated in 16 patients (33%). The average age of this treatment initiation was only 6 years. “In the general population, only approximately 15%-40% of patients with serologic evidence of HSV-1 have recurrent HSV; most of these will have an average of 2 outbreaks per year, and only 5%-10% have more than 6 outbreaks per year,” the investigators wrote (Pediatr Dermatol. 2017. doi: 10.1111/pde.13190).
The majority of the study participants (29 patients) had no labial or mucosal involvement. Approximately 23% had cutaneous involvement of the cheek, “which may have been because the primary infection was acquired through the kiss of a caregiver,” they said. Other sites of cutaneous lesions that were frequent in this population but are considered atypical in general were the ear, forehead, chest, and knees.
Most commonly, the physical examination findings were vesicles or papulovesicles (52%), but crusting (33%), pustules (6%), and erosions (23%) also occurred.
Fully half of the study population had multifocal rather than unifocal presentations.
Atypical presentation of HSV is frequently misdiagnosed as impetigo or herpes zoster, according to reports in the literature. Fifteen (31%) patients in this study had a previous misdiagnosis of impetigo. “These less typical cutaneous presentations of HSV may evade accurate diagnosis and delay initiation of appropriate treatment,” Dr. Gittler and her associates noted.
No financial disclosures were provided for Dr. Gittler and her associates.
FROM PEDIATRIC DERMATOLOGY
Key clinical point:
Major finding: 19 of 48 (40%) patients had six or more HSV outbreaks per year, and the mean frequency of outbreaks in this study population was seven per year.
Data source: Chart review of 48 cases referred to a single pediatric dermatology clinic in a 10-year period.
Disclosures: No financial disclosures were provided for Dr. Gittler and her associates.
Eroded Plaque on the Lower Lip
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.

The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
An 83-year-old man presented with a new-onset 1.2-cm eroded plaque on the vermilion border of the right lower lip that reportedly developed 2 weeks prior and was increasing in size. The plaque was moist and was composed of confluent glistening papules. Medical history was notable for the presence of both basal cell and squamous cell carcinomas.