User login
Dr. Jennifer Lee on VA Cancer Care
Federal Practitioner recently sat down with VA Deputy Under Secretary for Health for Policy and Services Jennifer Lee, MD, at the recent Launch Pad: Pathways to Cancer Innovation summit, November 29, 2016. In the interview, Dr. Lee discussed access to clinical trials for veterans, research, and the importance of partnering with other agencies, industry, and nonprofits to further veteran cancer care.
In the year since taking over for Madhulika Agarwal, MD, MPH, in the position, Dr. Lee has provided guidance to the Under Secretary for Health on matters related to health care policy, strategic objectives, and policy requirements for legislatively mandated health care delivery programs. She also directs research and other health policy and services programs within the VHA.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Federal Practitioner recently sat down with VA Deputy Under Secretary for Health for Policy and Services Jennifer Lee, MD, at the recent Launch Pad: Pathways to Cancer Innovation summit, November 29, 2016. In the interview, Dr. Lee discussed access to clinical trials for veterans, research, and the importance of partnering with other agencies, industry, and nonprofits to further veteran cancer care.
In the year since taking over for Madhulika Agarwal, MD, MPH, in the position, Dr. Lee has provided guidance to the Under Secretary for Health on matters related to health care policy, strategic objectives, and policy requirements for legislatively mandated health care delivery programs. She also directs research and other health policy and services programs within the VHA.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Federal Practitioner recently sat down with VA Deputy Under Secretary for Health for Policy and Services Jennifer Lee, MD, at the recent Launch Pad: Pathways to Cancer Innovation summit, November 29, 2016. In the interview, Dr. Lee discussed access to clinical trials for veterans, research, and the importance of partnering with other agencies, industry, and nonprofits to further veteran cancer care.
In the year since taking over for Madhulika Agarwal, MD, MPH, in the position, Dr. Lee has provided guidance to the Under Secretary for Health on matters related to health care policy, strategic objectives, and policy requirements for legislatively mandated health care delivery programs. She also directs research and other health policy and services programs within the VHA.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Study reveals CML patients likely to benefit from HSCT long-term
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.![]()
*Information presented at the meeting differs from the abstract.
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.![]()
*Information presented at the meeting differs from the abstract.
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.![]()
*Information presented at the meeting differs from the abstract.
Health Canada approves therapy for hemophilia A

Health Canada has approved the use of lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, in hemophilia A patients of all ages.
Lonoctocog alfa is indicated for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment to control bleeding episodes, and for perioperative management of bleeding (surgical prophylaxis).
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
Health Canada’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July.* ![]()
Health Canada has approved the use of lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, in hemophilia A patients of all ages.
Lonoctocog alfa is indicated for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment to control bleeding episodes, and for perioperative management of bleeding (surgical prophylaxis).
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
Health Canada’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July.* ![]()
Health Canada has approved the use of lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, in hemophilia A patients of all ages.
Lonoctocog alfa is indicated for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment to control bleeding episodes, and for perioperative management of bleeding (surgical prophylaxis).
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
Health Canada’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July.* ![]()
Characterizing FL transformation, progression

Patients with transformed follicular lymphoma (FL) and FL patients with early progression have “widely divergent patterns of clonal
dynamics,” according to researchers.
The team investigated the molecular events underlying transformation and early progression in FL and found that disparate evolutionary trajectories and mutational profiles drive these 2 distinct clinical endpoints.
Sohrab Shah, PhD, of the University of British Columbia in Vancouver, Canada, and his colleagues reported these findings in PLOS Medicine.
The researchers used whole-genome sequencing to analyze tumor specimens and matched normal specimens from 41 FL patients.
The team then classified the patients according to the following clinical endpoints:
- Patients who presented with transformation (n=15)
- Patients who experienced tumor progression within 2.5 years of starting treatment, without evidence of transformation (n=6)
- Patients who had neither transformation nor progression up to 5 years post-diagnosis (n=20).
The researchers also used targeted capture sequencing of known FL-associated genes in a larger cohort of 277 FL patients (395 samples) to investigate discrete genetic events that drive transformation and early progression.
Results showed that tumors that progress early evolve in different ways from those that transform.
The team found that, for tumors that transform, the cells or clones that constitute the majority of the aggressive tumor were extremely rare at diagnosis, if they were present at all.
In contrast, for early progressive FL, the clonal architecture remained similar from the time of diagnosis to relapse, indicating that the diagnostic tumor may already contain the properties that confer resistance to treatment.
Analysis of the larger cohort revealed genes and biological processes that were associated with transformation and progression.
The researchers identified 12 genes that were more commonly mutated at the time of transformation than the time of diagnosis—TP53, B2M, EZH2, MYC, CCND3, EBF1, PIM1, GNA13, ITPKB, CHD8, S1PR2, and P2RY8.
The team said their findings suggest that defective DNA damage response, increased proliferation, escape from immune surveillance, and loss of confinement within the germinal center are key features that drive histological transformation from indolent to aggressive lymphoma.
The researchers also identified 10 genes that were more commonly mutated in patients with early progression than in patients with late/no progression—B2M, BTG1, FAS, IKZF3, KMT2C, MKI67, MYD88, SOCS1, TP53, and XBP1.
The team noted that most patients with early progression (80%) had mutations in at least 1 of these 10 genes, but none of the genes were mutated at a frequency greater than 27%. This suggests that early progression is related to relatively infrequent genetic alterations.
The researchers said these findings provide a basis for future research on prognostic assay development and potential strategies for monitoring and treatment of patients with FL. ![]()
Patients with transformed follicular lymphoma (FL) and FL patients with early progression have “widely divergent patterns of clonal
dynamics,” according to researchers.
The team investigated the molecular events underlying transformation and early progression in FL and found that disparate evolutionary trajectories and mutational profiles drive these 2 distinct clinical endpoints.
Sohrab Shah, PhD, of the University of British Columbia in Vancouver, Canada, and his colleagues reported these findings in PLOS Medicine.
The researchers used whole-genome sequencing to analyze tumor specimens and matched normal specimens from 41 FL patients.
The team then classified the patients according to the following clinical endpoints:
- Patients who presented with transformation (n=15)
- Patients who experienced tumor progression within 2.5 years of starting treatment, without evidence of transformation (n=6)
- Patients who had neither transformation nor progression up to 5 years post-diagnosis (n=20).
The researchers also used targeted capture sequencing of known FL-associated genes in a larger cohort of 277 FL patients (395 samples) to investigate discrete genetic events that drive transformation and early progression.
Results showed that tumors that progress early evolve in different ways from those that transform.
The team found that, for tumors that transform, the cells or clones that constitute the majority of the aggressive tumor were extremely rare at diagnosis, if they were present at all.
In contrast, for early progressive FL, the clonal architecture remained similar from the time of diagnosis to relapse, indicating that the diagnostic tumor may already contain the properties that confer resistance to treatment.
Analysis of the larger cohort revealed genes and biological processes that were associated with transformation and progression.
The researchers identified 12 genes that were more commonly mutated at the time of transformation than the time of diagnosis—TP53, B2M, EZH2, MYC, CCND3, EBF1, PIM1, GNA13, ITPKB, CHD8, S1PR2, and P2RY8.
The team said their findings suggest that defective DNA damage response, increased proliferation, escape from immune surveillance, and loss of confinement within the germinal center are key features that drive histological transformation from indolent to aggressive lymphoma.
The researchers also identified 10 genes that were more commonly mutated in patients with early progression than in patients with late/no progression—B2M, BTG1, FAS, IKZF3, KMT2C, MKI67, MYD88, SOCS1, TP53, and XBP1.
The team noted that most patients with early progression (80%) had mutations in at least 1 of these 10 genes, but none of the genes were mutated at a frequency greater than 27%. This suggests that early progression is related to relatively infrequent genetic alterations.
The researchers said these findings provide a basis for future research on prognostic assay development and potential strategies for monitoring and treatment of patients with FL. ![]()
Patients with transformed follicular lymphoma (FL) and FL patients with early progression have “widely divergent patterns of clonal
dynamics,” according to researchers.
The team investigated the molecular events underlying transformation and early progression in FL and found that disparate evolutionary trajectories and mutational profiles drive these 2 distinct clinical endpoints.
Sohrab Shah, PhD, of the University of British Columbia in Vancouver, Canada, and his colleagues reported these findings in PLOS Medicine.
The researchers used whole-genome sequencing to analyze tumor specimens and matched normal specimens from 41 FL patients.
The team then classified the patients according to the following clinical endpoints:
- Patients who presented with transformation (n=15)
- Patients who experienced tumor progression within 2.5 years of starting treatment, without evidence of transformation (n=6)
- Patients who had neither transformation nor progression up to 5 years post-diagnosis (n=20).
The researchers also used targeted capture sequencing of known FL-associated genes in a larger cohort of 277 FL patients (395 samples) to investigate discrete genetic events that drive transformation and early progression.
Results showed that tumors that progress early evolve in different ways from those that transform.
The team found that, for tumors that transform, the cells or clones that constitute the majority of the aggressive tumor were extremely rare at diagnosis, if they were present at all.
In contrast, for early progressive FL, the clonal architecture remained similar from the time of diagnosis to relapse, indicating that the diagnostic tumor may already contain the properties that confer resistance to treatment.
Analysis of the larger cohort revealed genes and biological processes that were associated with transformation and progression.
The researchers identified 12 genes that were more commonly mutated at the time of transformation than the time of diagnosis—TP53, B2M, EZH2, MYC, CCND3, EBF1, PIM1, GNA13, ITPKB, CHD8, S1PR2, and P2RY8.
The team said their findings suggest that defective DNA damage response, increased proliferation, escape from immune surveillance, and loss of confinement within the germinal center are key features that drive histological transformation from indolent to aggressive lymphoma.
The researchers also identified 10 genes that were more commonly mutated in patients with early progression than in patients with late/no progression—B2M, BTG1, FAS, IKZF3, KMT2C, MKI67, MYD88, SOCS1, TP53, and XBP1.
The team noted that most patients with early progression (80%) had mutations in at least 1 of these 10 genes, but none of the genes were mutated at a frequency greater than 27%. This suggests that early progression is related to relatively infrequent genetic alterations.
The researchers said these findings provide a basis for future research on prognostic assay development and potential strategies for monitoring and treatment of patients with FL. ![]()
Platform could optimize treatment of ALL, other diseases

A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies. ![]()
A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies. ![]()
A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies. ![]()
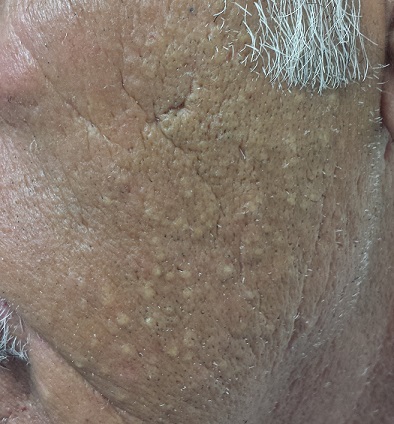
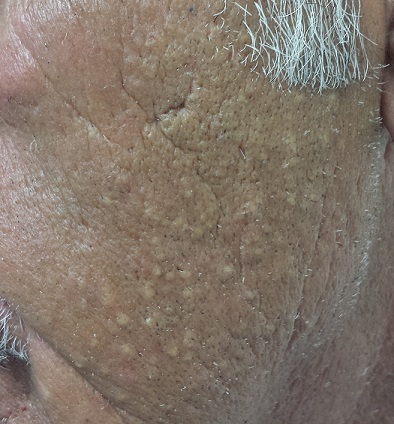
Dump Truck Dermis
Accompanied by family, a 68-year-old man presents for evaluation of changes on the left side of his face. His relatives, who have not seen him in years, find the changes quite alarming; the patient, however, is quite sure he has “looked this way for a while.” In any case, he has no associated symptoms.
He does have numerous other health problems, including hypertension, heart disease, and a 50–pack-year history of smoking. He has spent a great deal of time in the sun over the years, particularly when he drove a dump truck for work.
EXAMINATION
The left side of the patient’s face is covered with open and closed comedones superimposed on thickened, yellowed skin. Poikilodermatous changes and multiple telangiectasias can also be seen. The right side of his face demonstrates minor skin changes relative to the left.
What is the diagnosis?
DISCUSSION
These changes represent Favre-Racouchot syndrome (FRS). First described by Favre in 1932, the syndrome was refined in 1951 when Favre and Racouchot noticed its prevalence in truck drivers who also smoked. In France at that time, more than 80% of truck drivers were smokers—and they got the most sun exposure on the left sides of their faces. The condition was later identified in fishermen and other outdoor workers.
Studies in the US show that FRS affects nearly 6% of white adults (usually men) older than 60. Radiation therapy (eg, for squamous cell carcinoma) can produce similar effects.
Needless to say, smoking cessation is advisable. Beyond that, treatment involves preventing further sun damage and applying retinoid preparations, such as tretinoin or adapalene. Depending on the desired level of improvement, comedones may need to be surgically ablated.
TAKE-HOME LEARNING POINTS
• Favre-Racouchot syndrome (FRS) manifests with collections of open and closed comedones on sun-damaged facial skin.
• Most FRS patients are white male smokers older than 60.
• FRS is usually unilateral, resulting from overexposure to sunlight through a vehicle’s driver-side window; however, people working outdoors (eg, fishermen) can develop it over the whole face.
Accompanied by family, a 68-year-old man presents for evaluation of changes on the left side of his face. His relatives, who have not seen him in years, find the changes quite alarming; the patient, however, is quite sure he has “looked this way for a while.” In any case, he has no associated symptoms.
He does have numerous other health problems, including hypertension, heart disease, and a 50–pack-year history of smoking. He has spent a great deal of time in the sun over the years, particularly when he drove a dump truck for work.
EXAMINATION
The left side of the patient’s face is covered with open and closed comedones superimposed on thickened, yellowed skin. Poikilodermatous changes and multiple telangiectasias can also be seen. The right side of his face demonstrates minor skin changes relative to the left.
What is the diagnosis?
DISCUSSION
These changes represent Favre-Racouchot syndrome (FRS). First described by Favre in 1932, the syndrome was refined in 1951 when Favre and Racouchot noticed its prevalence in truck drivers who also smoked. In France at that time, more than 80% of truck drivers were smokers—and they got the most sun exposure on the left sides of their faces. The condition was later identified in fishermen and other outdoor workers.
Studies in the US show that FRS affects nearly 6% of white adults (usually men) older than 60. Radiation therapy (eg, for squamous cell carcinoma) can produce similar effects.
Needless to say, smoking cessation is advisable. Beyond that, treatment involves preventing further sun damage and applying retinoid preparations, such as tretinoin or adapalene. Depending on the desired level of improvement, comedones may need to be surgically ablated.
TAKE-HOME LEARNING POINTS
• Favre-Racouchot syndrome (FRS) manifests with collections of open and closed comedones on sun-damaged facial skin.
• Most FRS patients are white male smokers older than 60.
• FRS is usually unilateral, resulting from overexposure to sunlight through a vehicle’s driver-side window; however, people working outdoors (eg, fishermen) can develop it over the whole face.
Accompanied by family, a 68-year-old man presents for evaluation of changes on the left side of his face. His relatives, who have not seen him in years, find the changes quite alarming; the patient, however, is quite sure he has “looked this way for a while.” In any case, he has no associated symptoms.
He does have numerous other health problems, including hypertension, heart disease, and a 50–pack-year history of smoking. He has spent a great deal of time in the sun over the years, particularly when he drove a dump truck for work.
EXAMINATION
The left side of the patient’s face is covered with open and closed comedones superimposed on thickened, yellowed skin. Poikilodermatous changes and multiple telangiectasias can also be seen. The right side of his face demonstrates minor skin changes relative to the left.
What is the diagnosis?
DISCUSSION
These changes represent Favre-Racouchot syndrome (FRS). First described by Favre in 1932, the syndrome was refined in 1951 when Favre and Racouchot noticed its prevalence in truck drivers who also smoked. In France at that time, more than 80% of truck drivers were smokers—and they got the most sun exposure on the left sides of their faces. The condition was later identified in fishermen and other outdoor workers.
Studies in the US show that FRS affects nearly 6% of white adults (usually men) older than 60. Radiation therapy (eg, for squamous cell carcinoma) can produce similar effects.
Needless to say, smoking cessation is advisable. Beyond that, treatment involves preventing further sun damage and applying retinoid preparations, such as tretinoin or adapalene. Depending on the desired level of improvement, comedones may need to be surgically ablated.
TAKE-HOME LEARNING POINTS
• Favre-Racouchot syndrome (FRS) manifests with collections of open and closed comedones on sun-damaged facial skin.
• Most FRS patients are white male smokers older than 60.
• FRS is usually unilateral, resulting from overexposure to sunlight through a vehicle’s driver-side window; however, people working outdoors (eg, fishermen) can develop it over the whole face.
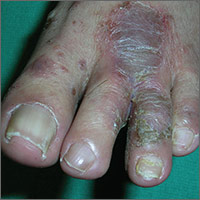
Painful rash on feet
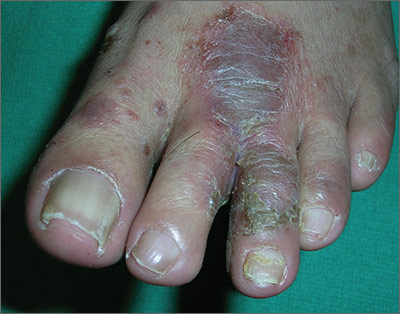
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP told the patient that he had ulcerative tinea pedis related to a bacterial superinfection. The FP performed a potassium hydroxide (KOH) preparation, which was positive for branching septate hyphae. (See a video on how to perform a KOH preparation.) The small ulcerations and crusts were due to a bacterial superinfection on top of the standard tinea pedis.
Ulcerative tinea pedis is uncommon and, thus, is not always mentioned when describing the standard types of tinea pedis (interdigital, moccasin distribution, and vesicular [vesiculobullous]). Ulcerative tinea pedis does not have to have large ulcers, but there is usually disruption in the skin barrier along with signs of bacterial superinfection (such as crusting and exudate).
This patient demonstrated these signs and the FP chose to treat the patient with an antibiotic that would cover typical skin organisms. There was no need for a culture, as it was likely to grow out many types of skin flora found on the foot. This case was unlikely to be the result of methicillin-resistant Staphylococcus aureus, so a first-generation cephalosporin was adequate.
The FP treated the patient with oral terbinafine 250 mg/d for 2 weeks, along with a course of oral cephalexin 500 mg 3 times daily for one week. Three weeks later, the skin on the patient’s feet was clear, except for some postinflammatory hyperpigmentation that was likely to fade over time.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Reppa R. Tinea pedis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:799-804.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Into the Wild: PA Edition
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.

Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.

Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.
Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.
Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
The island of Adak is Alaska’s last frontier. There are no stores, doctors, streetlights, traffic lights, or even a need for license plates. Just the lonely cry of a bald eagle and the howl of the arctic wind accompany you along the dark streets of this ghost town on the edge of the earth, where the North Pacific and the Bering Sea collide. The frigid Arctic front travels down the west coast, while the warmer Japanese current comes up the Pacific side. The low tundra does little to block the winds generated by that confluence, which often whip at 90 mph or more for days on end. Otherwise, the weather is generally gray and misty, and temperatures range from about 20°F to 65°F.
The weather matches the ambience and surroundings—decaying shambles of a once grand Navy base with facilities designed to make life as pleasant as possible for service members. But the Navy left in the late 1990s. By the time I arrived to practice here in 2013, only a few hundred people inhabited the island. There was virtually no economy, and almost all the roads and buildings were deserted. Man’s creation had fallen victim to vandals and nature.
My practice claimed a converted high school principal’s office and a two-bed emergency department (ED) that had some nice equipment—but with no medical aid, x-ray technician, phlebotomist, or lab technician, there was only me to operate it.
One of the oddities of the Alaskan bush is that medical providers often perform as All Species Providers; my first patient, Sadie, was a very well-behaved black Labrador retriever. Unable to anesthetize her, I was thankful for her even-tempered, patient breed (and for my experience as a veterinarian). Sadie lay calmly on the ED bed, her head in her owner’s hands, while I sutured her forepaw. In hindsight, she was one of my easier cases.
Adak’s isolated location (450 miles from the nearest settled outpost), rugged terrain, and vast wildlife come with an elevated risk for injuries and no shortage of challenges in treating them. During my first week, an autistic child presented with a foot laceration. There was no electricity when he arrived, as was often the case due to the wind; the only available light came through the open door. It was dusk and snow was gently falling, but we huddled in the doorway, as the patient’s mother and my husband held the boy while I sutured him. I just managed to get the last stitch in before the child entered his incoherent world.
Not surprisingly for an island, the surrounding seas brought many of my patients. Workers on commercial fishing vessels frequently sustained injuries that the conditions at sea would exacerbate. Hand injuries were common, as were slip-and-fall injuries such as dislocations and broken bones. It could take hours to get them into port. The most memorable was a fisherman who had lost the end of his thumb when it was crushed between the gunwale and a swinging, loaded crab pot being hoisted aboard. The electricity (surprise) was out, so I was forced to treat him by flashlight. He was a young man (early 30s) but especially nice. What I remember most is that he declined a digital block. I debrided the devitalized tissue via sharp dissection, but he never once cursed. So much for all those jokes about cussing like a sailor.
On a seemingly calm Saturday in my third week, one of the supervisors from the fish processing plant came in with two of his workers. One had bilateral ocular exposure to a cleaning solvent, while the other was stumbling and disoriented. My triage skills were put to the test as I anesthetized Worker 1’s eyes with proparacaine, then inserted Morgan lenses (which, thankfully, came with instructions in the packaging!). The supervisor, who was keeping Worker 2 calm simultaneously, held a basin below the patient’s eyes to catch runoff as I quickly attached a liter of saline to each lens and opened the valve full bore. With that situation under control for the moment, I switched gears to assess Worker 2, who was markedly incoherent and unable to give a history. What I did know was that he was ataxic and nauseous, with a temperature of 104°F. I flashed my penlight into his eyes, and he reacted like Dracula faced with a crucifix. In addition to his exquisite photophobia, he had marked neck stiffness. The diagnosis was my first case of human meningitis. I inserted an IV providing ceftriaxone, acyclovir, and ondansetron. He required 2.5 L of IV fluid, but he gradually recovered over the next two days.
Why, with all these challenges, did I go to Alaska? Because I felt that there I could be of most service; if ever there was an underserved community, Adak was it. Ironically, the other job offer I considered was on the Hawaiian island of Kauai. As idyllic as that might have been, it wasn’t quite the adventure that Adak turned out to be—and it wasn’t as desperately in need of a provider.
When I took the Adak post, I didn’t realize how much I would learn. Above all, I realized that a complete and meticulous physical exam is king. If I missed any life-threatening conditions, the ramifications would be extreme. There were no referrals or second opinions, just me with the patient, and I had to make the right decisions.
The bonds I formed with the people on this remote island, and the paths I trod through this unusual land, made for a unique experience. It was a thrill to look out toward the horizon and know that everything I could see had remained unchanged since the birth of these volcanic islands. In time, I came to recognize that I was not in charge. The planes carrying supplies in or patients out would come if the weather allowed. What was on the island was what I had to work with, and if I didn’t have what I wanted, it was only because I didn’t want what I had. The waves would come as they will. I could either surf them or be tossed by them. I chose to dive in.
Lack of natalizumab concentration increase before PML argues against extending dosing interval
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
Serum concentrations of natalizumab do not appear to rise before patients with relapsing-remitting multiple sclerosis are diagnosed with progressive multifocal leukoencephalopathy, contradicting the hypothesis that exposure to elevated concentrations of the drug is a risk factor for the disease, according to findings from a prospective, observational cohort study.
Zoé L.E. van Kempen, MD, and her colleagues at VU Medical Center Amsterdam noted that a small but increasing number of “neurologists are extending dose intervals of natalizumab [Tysabri] with the primary aim of reducing the risk of PML [progressive multifocal leukoencephalopathy] by lowering the natalizumab exposure per patient,” which also potentially has the benefit of decreasing drug costs, as well as clinic or hospital visits. However, there have been no prospective studies that confirm that extending natalizumab dose intervals does not affect efficacy.
Dr. van Kempen and her coinvestigators investigated this hypothesis by comparing serum concentrations of natalizumab in 5 patients with PML and 10 age- and sex-matched controls from a cohort of 219 relapsing-remitting multiple sclerosis patients taking natalizumab who had blood samples routinely drawn every 12 weeks before the infusion of natalizumab (Mult Scler. 2016 Dec 13. doi: 10.1177/1352458516684023). These 10 controls had a mean concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed before the diagnosis of PML in the 5 cases from the cohort. The five cases also did not show a rise in concentration before the diagnosis of PML and did not have concentrations fluctuate more than 11 mcg/mL during treatment. The patients with PML received a mean of 43 infusions, whereas controls received an average of 106 infusions. A median number of 5 pre-PML samples underwent testing, compared with yearly concentration measurements in the 10 controls who had at least 7 years’ treatment duration.
The increased risk of PML that has been documented in other studies with more than 24 months of natalizumab treatment also was not explained in this study by a rise in serum concentration over long-term follow-up.
“Although this cohort is too small to draw definite conclusions, our results do not support the hypothesis of high serum concentrations as a risk factor for developing PML,” the investigators wrote.
“With our small cohort in mind and insufficient data on this subject so far, neurologists should be careful in extending dose intervals and not overstate a possible decrease in the risk of developing PML. Obviously, if neurologists choose to extend dosing intervals of natalizumab in [John Cunningham virus]–positive patients, these patients should still receive stringent PML monitoring according to current recommendations,” they advised.
The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
FROM MULTIPLE SCLEROSIS JOURNAL
Key clinical point:
Major finding: A total of 10 control patients had a mean natalizumab serum concentration of 23.8 mcg/mL, which was in the same range as the 18.9 mcg/mL level observed in 5 patients before a diagnosis of PML.
Data source: A comparison of 5 patients with PML and 10 age- and sex-matched controls from a prospective cohort of 219 patients with relapsing-remitting MS who were treated with natalizumab at one center.
Disclosures: The study received support from the Brain Foundation Netherlands. Five of the authors reported financial ties to various pharmaceutical companies that market MS drugs, including Biogen, which markets natalizumab.
FDA gives nod to crisaborole for atopic dermatitis
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.

At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).