User login
Type 2 diabetes peer-led intervention in primary care tied to improved depression symptoms
BETHESDA, MD. – A novel, peer- and nurse-led intervention in a primary care setting for type 2 diabetes in people with serious mental illness was associated with improvements in depression symptoms, global psychopathology, and overall health, a study has shown.
“The intervention really is patient self-management. It could be a nice complement to team-based, multidisciplinary care,” said Martha Sajatovic, MD, who presented the data in a poster at a National Institute of Mental Health conference on mental health services research. Dr. Sajatovic is the Willard W. Brown Chair and director of the Neurological & Behavioral Outcomes Center at University Hospitals Neurological Institute in Cleveland.

People with serious mental illness (SMI) have a significantly higher risk of premature death than do those in the general population, in part because this cohort experiences higher rates of metabolic disease, often exacerbated by higher rates of smoking, poor diet, substance abuse, and lack of exercise. However, in a 60-week randomized controlled trial of 200 people with SMI and comorbid type 2 diabetes, which was conducted in a primary care setting, those who were taught better self-care fared better than did those who received treatment as usual.
The group-based, psychosocial intervention – called “targeted training in illness” – blended psychoeducation, problem identification, goal setting, behavioral modeling, and care coordination around SMI and diabetes. In the first 12 weeks, groups of 6-10 people met in weekly, hour-long sessions co-led by a peer and nurse educator. Group discussions focused on self-management of diabetes through proper eating habits, regular exercise, tobacco cessation, and other forms of behavior modification.
Meeting as a group helped to “combat some of the social isolation that you see in this population,” Dr. Sajatovic said in an interview. “The peer leadership is really critical, too, because it empowers [the participants]. I believe peer support gives resilience ... and helps [the group] see you don’t have to be perfect to make progress.” In the study, the 3 months of group sessions were followed by weekly telephone maintenance sessions with either the peer or nurse educator for 48 weeks.
Half of the study’s participants – two-thirds of whom were women, and just over half of whom were black – had had a diagnosis of diabetes for at least 10 years; half of all participants used insulin. All had either schizophrenia, schizoaffective disorder, bipolar disorder, or major depressive disorder. Baseline rates of depression were high, and psychotic symptoms were minimal.
After assessments at baseline, 13, 30, and 60 weeks, the study arm was found to have improvements in depression, global psychopathology, and functional status, which Dr. Sajatovic said could be attributable to the group’s significantly improved knowledge about diabetes (P less than .01).
Glycemic control improved generally, a surprising finding that Dr. Sajatovic said could have been tied to the expansion of Medicaid in Ohio, where the study was done, and a “real concerted effort” to provide treatment by medical homes at this time.
While no significant difference between the groups was found overall, a post hoc analysis showed a difference in the 53% of the entire sample who had good to fair glycemic control (hemoglobin A1c equal to or less than 7.5) at baseline: At 60 weeks, those in the treatment arm achieved stable, long-term control compared with controls, whose values had worsened slightly (P = .024). Those people tended to be older, more likely to have schizophrenia, and less likely to be on insulin, and to have a shorter history of diabetes, said Dr. Sajatovic, professor of psychiatry and of neurology at Case Western Reserve University, Cleveland.
Compared with controls, the study arm had greater improvement at 60 weeks in Clinical Global Impression scores (P = .0008); Montgomery-Åsberg Depression Rating Scale scores (P = .0156); Global Assessment of Functioning scores (P = .0031); and knowledge of diabetes (P less than .0002), as well as an improvement trend in Sheehan Disability Scale scores (P = .0863). There was no difference between the groups on the Brief Psychiatric Rating Scale, the Short Form–36 or HbA1c values. By study’s end, Dr. Sajatovic said about a quarter had been lost to follow-up.
The intervention meets three important criteria, she said. “First, people need to know what to do. Then, they need to have confidence, or self-efficacy. The third thing is that the person has to believe in a given outcome based on a given behavior.”
Dr. Sajatovic did not have any relevant disclosures. The National Institutes of Health funded the study.
On Twitter @whitneymcknight
BETHESDA, MD. – A novel, peer- and nurse-led intervention in a primary care setting for type 2 diabetes in people with serious mental illness was associated with improvements in depression symptoms, global psychopathology, and overall health, a study has shown.
“The intervention really is patient self-management. It could be a nice complement to team-based, multidisciplinary care,” said Martha Sajatovic, MD, who presented the data in a poster at a National Institute of Mental Health conference on mental health services research. Dr. Sajatovic is the Willard W. Brown Chair and director of the Neurological & Behavioral Outcomes Center at University Hospitals Neurological Institute in Cleveland.
People with serious mental illness (SMI) have a significantly higher risk of premature death than do those in the general population, in part because this cohort experiences higher rates of metabolic disease, often exacerbated by higher rates of smoking, poor diet, substance abuse, and lack of exercise. However, in a 60-week randomized controlled trial of 200 people with SMI and comorbid type 2 diabetes, which was conducted in a primary care setting, those who were taught better self-care fared better than did those who received treatment as usual.
The group-based, psychosocial intervention – called “targeted training in illness” – blended psychoeducation, problem identification, goal setting, behavioral modeling, and care coordination around SMI and diabetes. In the first 12 weeks, groups of 6-10 people met in weekly, hour-long sessions co-led by a peer and nurse educator. Group discussions focused on self-management of diabetes through proper eating habits, regular exercise, tobacco cessation, and other forms of behavior modification.
Meeting as a group helped to “combat some of the social isolation that you see in this population,” Dr. Sajatovic said in an interview. “The peer leadership is really critical, too, because it empowers [the participants]. I believe peer support gives resilience ... and helps [the group] see you don’t have to be perfect to make progress.” In the study, the 3 months of group sessions were followed by weekly telephone maintenance sessions with either the peer or nurse educator for 48 weeks.
Half of the study’s participants – two-thirds of whom were women, and just over half of whom were black – had had a diagnosis of diabetes for at least 10 years; half of all participants used insulin. All had either schizophrenia, schizoaffective disorder, bipolar disorder, or major depressive disorder. Baseline rates of depression were high, and psychotic symptoms were minimal.
After assessments at baseline, 13, 30, and 60 weeks, the study arm was found to have improvements in depression, global psychopathology, and functional status, which Dr. Sajatovic said could be attributable to the group’s significantly improved knowledge about diabetes (P less than .01).
Glycemic control improved generally, a surprising finding that Dr. Sajatovic said could have been tied to the expansion of Medicaid in Ohio, where the study was done, and a “real concerted effort” to provide treatment by medical homes at this time.
While no significant difference between the groups was found overall, a post hoc analysis showed a difference in the 53% of the entire sample who had good to fair glycemic control (hemoglobin A1c equal to or less than 7.5) at baseline: At 60 weeks, those in the treatment arm achieved stable, long-term control compared with controls, whose values had worsened slightly (P = .024). Those people tended to be older, more likely to have schizophrenia, and less likely to be on insulin, and to have a shorter history of diabetes, said Dr. Sajatovic, professor of psychiatry and of neurology at Case Western Reserve University, Cleveland.
Compared with controls, the study arm had greater improvement at 60 weeks in Clinical Global Impression scores (P = .0008); Montgomery-Åsberg Depression Rating Scale scores (P = .0156); Global Assessment of Functioning scores (P = .0031); and knowledge of diabetes (P less than .0002), as well as an improvement trend in Sheehan Disability Scale scores (P = .0863). There was no difference between the groups on the Brief Psychiatric Rating Scale, the Short Form–36 or HbA1c values. By study’s end, Dr. Sajatovic said about a quarter had been lost to follow-up.
The intervention meets three important criteria, she said. “First, people need to know what to do. Then, they need to have confidence, or self-efficacy. The third thing is that the person has to believe in a given outcome based on a given behavior.”
Dr. Sajatovic did not have any relevant disclosures. The National Institutes of Health funded the study.
On Twitter @whitneymcknight
BETHESDA, MD. – A novel, peer- and nurse-led intervention in a primary care setting for type 2 diabetes in people with serious mental illness was associated with improvements in depression symptoms, global psychopathology, and overall health, a study has shown.
“The intervention really is patient self-management. It could be a nice complement to team-based, multidisciplinary care,” said Martha Sajatovic, MD, who presented the data in a poster at a National Institute of Mental Health conference on mental health services research. Dr. Sajatovic is the Willard W. Brown Chair and director of the Neurological & Behavioral Outcomes Center at University Hospitals Neurological Institute in Cleveland.
People with serious mental illness (SMI) have a significantly higher risk of premature death than do those in the general population, in part because this cohort experiences higher rates of metabolic disease, often exacerbated by higher rates of smoking, poor diet, substance abuse, and lack of exercise. However, in a 60-week randomized controlled trial of 200 people with SMI and comorbid type 2 diabetes, which was conducted in a primary care setting, those who were taught better self-care fared better than did those who received treatment as usual.
The group-based, psychosocial intervention – called “targeted training in illness” – blended psychoeducation, problem identification, goal setting, behavioral modeling, and care coordination around SMI and diabetes. In the first 12 weeks, groups of 6-10 people met in weekly, hour-long sessions co-led by a peer and nurse educator. Group discussions focused on self-management of diabetes through proper eating habits, regular exercise, tobacco cessation, and other forms of behavior modification.
Meeting as a group helped to “combat some of the social isolation that you see in this population,” Dr. Sajatovic said in an interview. “The peer leadership is really critical, too, because it empowers [the participants]. I believe peer support gives resilience ... and helps [the group] see you don’t have to be perfect to make progress.” In the study, the 3 months of group sessions were followed by weekly telephone maintenance sessions with either the peer or nurse educator for 48 weeks.
Half of the study’s participants – two-thirds of whom were women, and just over half of whom were black – had had a diagnosis of diabetes for at least 10 years; half of all participants used insulin. All had either schizophrenia, schizoaffective disorder, bipolar disorder, or major depressive disorder. Baseline rates of depression were high, and psychotic symptoms were minimal.
After assessments at baseline, 13, 30, and 60 weeks, the study arm was found to have improvements in depression, global psychopathology, and functional status, which Dr. Sajatovic said could be attributable to the group’s significantly improved knowledge about diabetes (P less than .01).
Glycemic control improved generally, a surprising finding that Dr. Sajatovic said could have been tied to the expansion of Medicaid in Ohio, where the study was done, and a “real concerted effort” to provide treatment by medical homes at this time.
While no significant difference between the groups was found overall, a post hoc analysis showed a difference in the 53% of the entire sample who had good to fair glycemic control (hemoglobin A1c equal to or less than 7.5) at baseline: At 60 weeks, those in the treatment arm achieved stable, long-term control compared with controls, whose values had worsened slightly (P = .024). Those people tended to be older, more likely to have schizophrenia, and less likely to be on insulin, and to have a shorter history of diabetes, said Dr. Sajatovic, professor of psychiatry and of neurology at Case Western Reserve University, Cleveland.
Compared with controls, the study arm had greater improvement at 60 weeks in Clinical Global Impression scores (P = .0008); Montgomery-Åsberg Depression Rating Scale scores (P = .0156); Global Assessment of Functioning scores (P = .0031); and knowledge of diabetes (P less than .0002), as well as an improvement trend in Sheehan Disability Scale scores (P = .0863). There was no difference between the groups on the Brief Psychiatric Rating Scale, the Short Form–36 or HbA1c values. By study’s end, Dr. Sajatovic said about a quarter had been lost to follow-up.
The intervention meets three important criteria, she said. “First, people need to know what to do. Then, they need to have confidence, or self-efficacy. The third thing is that the person has to believe in a given outcome based on a given behavior.”
Dr. Sajatovic did not have any relevant disclosures. The National Institutes of Health funded the study.
On Twitter @whitneymcknight
AT AN NIMH CONFERENCE
Key clinical point: Targeted training in illness management effectively improves overall health outcomes in people with serious mental illness and comorbid type 2 diabetes.
Major finding: Compared with treatment as usual, peer-led intervention improved depression, overall health, and knowledge of diabetes at 60 weeks.
Data source: Randomized, controlled study of 200 people with serious mental illness and comorbid type 2 diabetes seen in primary care.
Disclosures: Dr. Sajatovic did not have any relevant disclosures. The National Institutes of Health funded the study.

Data-based Recommendations for CKD Screening
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Clinician Reviews in partnership with
![]()
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Rebecca V. Rokosky, MSN, APRN, FNP, who practices at the Renal Associates Clinical Advancement Center in San Antonio, Texas, and Tricia A. Howard, MHS, PA-C, DFAAPA, Associate Professor and Assistant Program Director in the PA Program at South University in Savannah, Georgia.
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
Q)
I’ve received mixed messages about whom to screen for chronic kidney disease (CKD). The US Preventive Services Task Force (USPSTF) recommends screening only patients at high risk, but kidney experts advise screening everyone. Who is right? What does the data show?
In 2012, the USPSTF stated that there was insufficient evidence to assess the benefit, or harm, of regularly screening asymptomatic adults for CKD.1 Other expert medical panels have come to this conclusion as well, and therefore only recommend screening highrisk patients.2
The National Kidney Foundation (NKF) encourages clinicians to assess all patients for risk factors of CKD. Diabetes and hypertension are strongly established risk factors for kidney disease; others include family history of kidney disease; cardiovascular disease; obesity; and older age.
If a patient is at risk for CKD, the NKF recommends testing serum creatinine levels to estimate glomerular filtration rate and testing urine for protein (microalbuminuria or macroalbuminuria). These tests are readily accessible in a primary care setting. It should be noted that one-time testing of serum creatinine and/or urine has not been studied for sensitivity or specificity in the diagnosis of CKD. Diagnosis should be based on decreased renal function or kidney damage occurring over a three-month span.3
In May 2016, Canadian researchers published results from the See Kidney Disease Targeted Screening Program for CKD, comparing CKD screening in the general population with a targeted, at-risk individual population.4 The study, which included more than 6,000 participants, revealed a higher rate of unrecognized CKD in the at-risk population than in the general population (21.9% and 14.7%, respectively).
These findings support the idea that screening at-risk patients identifies more cases of CKD than screening the general patient population does.4 Early diagnosis of CKD, through recognition of risk factors, provides an opportunity to decrease complications and manage conditions that contribute to the progression of renal disease.2,3 —RVR
Rebecca V. Rokosky, MSN, APRN, FNP
Renal Associates Clinical Advancement Center in San Antonio, Texas
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
1. Moyer VA. Screening for chronic kidney disease: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157(8):567-570.
2. Vassalotti JA, Centor R, Turner BJ, et al. Practical approach to detection and management of chronic kidney disease for the primary care clinician. Am J Med. 2016;129(2):153-162.
3. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313(8):837-846.
NIH Launches Study for New Zika Vaccine
An early-stage study to evaluate the safety and efficacy of an experimental Zika vaccine in humans is being launched by the National Institute of Allergy and Infectious Diseases (NIAID). “Results in animal testing have been very encouraging,” said NIAID Director Anthony Fauci, MD. “Although it will take some time before a vaccine against Zika is commercially available, the launch of this study is an important step forward.”
The NIAID scientists developed the investigational vaccine earlier this year. The approach is similar to that taken for another NIAID investigational vaccine developed for West Nile virus, which was found to be safe and effective in a phase 1 clinical trial. The vaccine includes a plasmid (small piece of DNA) engineered to contain genes that code for proteins of the Zika virus. The body reads the genes and makes Zika virus proteins, which cause an immune response. The DNA vaccines do not contain infectious material and cannot cause a vaccinated person to become infected with Zika.
The phase 1 clinical trial (VRC 319) will involve 4 groups of 20 people. The participants will be vaccinated at the first visit, and then half will receive another vaccination 8 weeks or 12 weeks later. The remaining participants will receive 2 additional vaccines, 1 group at week 4 and week 8; the other group, at week 4 and week 20. Participants will be followed for 44 weeks.
The study will be conducted at the NIH Clinical Center in Bethesda, the Center for Vaccine Development at the University of Maryland, and Emory University, Atlanta. Initial safety and immunogenicity data from the trial are expected by January 2017. In early 2017, If the results are favorable, NIAID plans a phase 2 trial in Zika-endemic countries.
An early-stage study to evaluate the safety and efficacy of an experimental Zika vaccine in humans is being launched by the National Institute of Allergy and Infectious Diseases (NIAID). “Results in animal testing have been very encouraging,” said NIAID Director Anthony Fauci, MD. “Although it will take some time before a vaccine against Zika is commercially available, the launch of this study is an important step forward.”
The NIAID scientists developed the investigational vaccine earlier this year. The approach is similar to that taken for another NIAID investigational vaccine developed for West Nile virus, which was found to be safe and effective in a phase 1 clinical trial. The vaccine includes a plasmid (small piece of DNA) engineered to contain genes that code for proteins of the Zika virus. The body reads the genes and makes Zika virus proteins, which cause an immune response. The DNA vaccines do not contain infectious material and cannot cause a vaccinated person to become infected with Zika.
The phase 1 clinical trial (VRC 319) will involve 4 groups of 20 people. The participants will be vaccinated at the first visit, and then half will receive another vaccination 8 weeks or 12 weeks later. The remaining participants will receive 2 additional vaccines, 1 group at week 4 and week 8; the other group, at week 4 and week 20. Participants will be followed for 44 weeks.
The study will be conducted at the NIH Clinical Center in Bethesda, the Center for Vaccine Development at the University of Maryland, and Emory University, Atlanta. Initial safety and immunogenicity data from the trial are expected by January 2017. In early 2017, If the results are favorable, NIAID plans a phase 2 trial in Zika-endemic countries.
An early-stage study to evaluate the safety and efficacy of an experimental Zika vaccine in humans is being launched by the National Institute of Allergy and Infectious Diseases (NIAID). “Results in animal testing have been very encouraging,” said NIAID Director Anthony Fauci, MD. “Although it will take some time before a vaccine against Zika is commercially available, the launch of this study is an important step forward.”
The NIAID scientists developed the investigational vaccine earlier this year. The approach is similar to that taken for another NIAID investigational vaccine developed for West Nile virus, which was found to be safe and effective in a phase 1 clinical trial. The vaccine includes a plasmid (small piece of DNA) engineered to contain genes that code for proteins of the Zika virus. The body reads the genes and makes Zika virus proteins, which cause an immune response. The DNA vaccines do not contain infectious material and cannot cause a vaccinated person to become infected with Zika.
The phase 1 clinical trial (VRC 319) will involve 4 groups of 20 people. The participants will be vaccinated at the first visit, and then half will receive another vaccination 8 weeks or 12 weeks later. The remaining participants will receive 2 additional vaccines, 1 group at week 4 and week 8; the other group, at week 4 and week 20. Participants will be followed for 44 weeks.
The study will be conducted at the NIH Clinical Center in Bethesda, the Center for Vaccine Development at the University of Maryland, and Emory University, Atlanta. Initial safety and immunogenicity data from the trial are expected by January 2017. In early 2017, If the results are favorable, NIAID plans a phase 2 trial in Zika-endemic countries.
An Evolutionary Perspective on Basal Insulin in Diabetes Treatment

- Role of Insulin Therapy in Diabetes
- Basal Insulin in Primary Care
- Innovations in Insulin: Insulin Degludec U-100 and U-200
- Innovations in Insulin: Insulin Glargine U-300
- Role of Insulin Therapy in Diabetes
- Basal Insulin in Primary Care
- Innovations in Insulin: Insulin Degludec U-100 and U-200
- Innovations in Insulin: Insulin Glargine U-300
- Role of Insulin Therapy in Diabetes
- Basal Insulin in Primary Care
- Innovations in Insulin: Insulin Degludec U-100 and U-200
- Innovations in Insulin: Insulin Glargine U-300

Physical/functional limitations top risk factor for late-life depression
New findings show there are several major risk factors that influence late-life depression (LLD), with physical/functional limitations the most prevalent, according to Shun-Chiao Chang, ScD, of Brigham and Women’s Hospital, Boston, and her associates.
They examined 21,728 women aged older than 65 years who had no prior depression. During a 10-year follow-up, 3,945 incident LLD cases were identified. In those cases, social factors and lifestyle/behavioral factors did affect LLD, but the categories with the largest effect magnitudes for higher LLD risk were severe/very severe bodily pain (hazard ratio, 2.22; 95% confidence interval, 1.88-2.62), difficulty sleeping most/all the time (HR, 2.04; 95% CI, 1.77-2.36), and daily sleep of 10 hours or more (HR, 1.96; 95% CI, 1.56-2.46). The most prevalent risk factor, physical/functional limitations, was associated with a 42% increase in risk

Sleep difficulty some to all of the time, no/very little exercise, and moderate to very severe bodily pain also were factors, with population attributable fraction (PAF) values of 10% or higher. The factor with the largest PAF was physical/functional limitation (26.4%).
Overall, the behavioral factors appeared to contribute relatively equally to LLD among women with and without physical/functional limitations; however, health factors had much bigger contributions to risk among women with limitations.
“Together, model predictors accounted for almost 60% of all new LLD cases in this population, and physical/functional limitation is the largest single contributor to total risk,” the researchers concluded. “A substantial proportion of LLD cases may be preventable by increasing exercise and intervening or preventing sleep difficulties and pain.”
Find the full study in Preventive Medicine (doi: 10.1016/j.ypmed.2016.08.014).
New findings show there are several major risk factors that influence late-life depression (LLD), with physical/functional limitations the most prevalent, according to Shun-Chiao Chang, ScD, of Brigham and Women’s Hospital, Boston, and her associates.
They examined 21,728 women aged older than 65 years who had no prior depression. During a 10-year follow-up, 3,945 incident LLD cases were identified. In those cases, social factors and lifestyle/behavioral factors did affect LLD, but the categories with the largest effect magnitudes for higher LLD risk were severe/very severe bodily pain (hazard ratio, 2.22; 95% confidence interval, 1.88-2.62), difficulty sleeping most/all the time (HR, 2.04; 95% CI, 1.77-2.36), and daily sleep of 10 hours or more (HR, 1.96; 95% CI, 1.56-2.46). The most prevalent risk factor, physical/functional limitations, was associated with a 42% increase in risk
Sleep difficulty some to all of the time, no/very little exercise, and moderate to very severe bodily pain also were factors, with population attributable fraction (PAF) values of 10% or higher. The factor with the largest PAF was physical/functional limitation (26.4%).
Overall, the behavioral factors appeared to contribute relatively equally to LLD among women with and without physical/functional limitations; however, health factors had much bigger contributions to risk among women with limitations.
“Together, model predictors accounted for almost 60% of all new LLD cases in this population, and physical/functional limitation is the largest single contributor to total risk,” the researchers concluded. “A substantial proportion of LLD cases may be preventable by increasing exercise and intervening or preventing sleep difficulties and pain.”
Find the full study in Preventive Medicine (doi: 10.1016/j.ypmed.2016.08.014).
New findings show there are several major risk factors that influence late-life depression (LLD), with physical/functional limitations the most prevalent, according to Shun-Chiao Chang, ScD, of Brigham and Women’s Hospital, Boston, and her associates.
They examined 21,728 women aged older than 65 years who had no prior depression. During a 10-year follow-up, 3,945 incident LLD cases were identified. In those cases, social factors and lifestyle/behavioral factors did affect LLD, but the categories with the largest effect magnitudes for higher LLD risk were severe/very severe bodily pain (hazard ratio, 2.22; 95% confidence interval, 1.88-2.62), difficulty sleeping most/all the time (HR, 2.04; 95% CI, 1.77-2.36), and daily sleep of 10 hours or more (HR, 1.96; 95% CI, 1.56-2.46). The most prevalent risk factor, physical/functional limitations, was associated with a 42% increase in risk
Sleep difficulty some to all of the time, no/very little exercise, and moderate to very severe bodily pain also were factors, with population attributable fraction (PAF) values of 10% or higher. The factor with the largest PAF was physical/functional limitation (26.4%).
Overall, the behavioral factors appeared to contribute relatively equally to LLD among women with and without physical/functional limitations; however, health factors had much bigger contributions to risk among women with limitations.
“Together, model predictors accounted for almost 60% of all new LLD cases in this population, and physical/functional limitation is the largest single contributor to total risk,” the researchers concluded. “A substantial proportion of LLD cases may be preventable by increasing exercise and intervening or preventing sleep difficulties and pain.”
Find the full study in Preventive Medicine (doi: 10.1016/j.ypmed.2016.08.014).
FROM PREVENTIVE MEDICINE

Painful ulcers in mouth
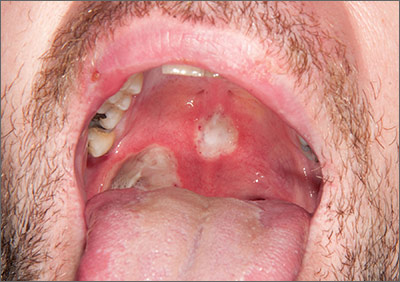
The patient was diagnosed with Behçet’s disease (BD) based on his clinical presentation. BD is a rare multisystem inflammatory disorder of unknown cause. The development of ulceration at the site of superficial skin injury (pathergy) is typical of BD. (The patient underwent multiple venipunctures while being investigated for a presumed infective illness prior to this presentation.)
There are no diagnostic laboratory tests for BD; laboratory findings usually reflect systemic inflammation. The International Study Group for BD, however, has derived classification criteria for use in clinical research studies. Their criteria include oral ulceration (that has recurred at least 3 times in a 12-month period), plus 2 of the following: recurrent genital ulceration, eye lesions, cutaneous lesions, or a positive pathergy test.
Recurrent mouth ulcers frequently involve the soft palate and oropharynx. Genital ulceration is the second most common manifestation of BD and is present in 57% to 93% of patients. The scrotum is most commonly involved, although the shaft and glans penis may also be affected. Ocular involvement is also seen in 30% to 70% of patients and is more frequent and severe in men.
There is no curative treatment for BD. The goals of treatment are to prevent organ damage and alleviate symptoms. Mucocutaneous disease is treated with potent topical corticosteroids. Severe attacks are treated with oral corticosteroids—1 mg/kg of prednisolone. The drug is tapered and discontinued once the disease is under control. Colchicine or dapsone is also an option. In refractory cases, consider thalidomide (50 mg once a day) or azathioprine (1-3 mg/kg). An anti-tumor necrosis factor agent may also be considered.
In this case, the patient was prescribed prednisolone 60 mg once a day, but relapsed once he was weaned off of it. He was then given thalidomide 50 mg once a day for 4 months and the disease resolved completely. The thalidomide was then reduced to 50 mg 3 times a week for 4 weeks, and then stopped completely. Nearly 2 years later, the patient has remained disease free.
Adapted from: Aslam A, Chalmers R. Mucocutaneous ulceration in a previously healthy man. J Fam Pract. 2014;63:97-100.
The patient was diagnosed with Behçet’s disease (BD) based on his clinical presentation. BD is a rare multisystem inflammatory disorder of unknown cause. The development of ulceration at the site of superficial skin injury (pathergy) is typical of BD. (The patient underwent multiple venipunctures while being investigated for a presumed infective illness prior to this presentation.)
There are no diagnostic laboratory tests for BD; laboratory findings usually reflect systemic inflammation. The International Study Group for BD, however, has derived classification criteria for use in clinical research studies. Their criteria include oral ulceration (that has recurred at least 3 times in a 12-month period), plus 2 of the following: recurrent genital ulceration, eye lesions, cutaneous lesions, or a positive pathergy test.
Recurrent mouth ulcers frequently involve the soft palate and oropharynx. Genital ulceration is the second most common manifestation of BD and is present in 57% to 93% of patients. The scrotum is most commonly involved, although the shaft and glans penis may also be affected. Ocular involvement is also seen in 30% to 70% of patients and is more frequent and severe in men.
There is no curative treatment for BD. The goals of treatment are to prevent organ damage and alleviate symptoms. Mucocutaneous disease is treated with potent topical corticosteroids. Severe attacks are treated with oral corticosteroids—1 mg/kg of prednisolone. The drug is tapered and discontinued once the disease is under control. Colchicine or dapsone is also an option. In refractory cases, consider thalidomide (50 mg once a day) or azathioprine (1-3 mg/kg). An anti-tumor necrosis factor agent may also be considered.
In this case, the patient was prescribed prednisolone 60 mg once a day, but relapsed once he was weaned off of it. He was then given thalidomide 50 mg once a day for 4 months and the disease resolved completely. The thalidomide was then reduced to 50 mg 3 times a week for 4 weeks, and then stopped completely. Nearly 2 years later, the patient has remained disease free.
Adapted from: Aslam A, Chalmers R. Mucocutaneous ulceration in a previously healthy man. J Fam Pract. 2014;63:97-100.
The patient was diagnosed with Behçet’s disease (BD) based on his clinical presentation. BD is a rare multisystem inflammatory disorder of unknown cause. The development of ulceration at the site of superficial skin injury (pathergy) is typical of BD. (The patient underwent multiple venipunctures while being investigated for a presumed infective illness prior to this presentation.)
There are no diagnostic laboratory tests for BD; laboratory findings usually reflect systemic inflammation. The International Study Group for BD, however, has derived classification criteria for use in clinical research studies. Their criteria include oral ulceration (that has recurred at least 3 times in a 12-month period), plus 2 of the following: recurrent genital ulceration, eye lesions, cutaneous lesions, or a positive pathergy test.
Recurrent mouth ulcers frequently involve the soft palate and oropharynx. Genital ulceration is the second most common manifestation of BD and is present in 57% to 93% of patients. The scrotum is most commonly involved, although the shaft and glans penis may also be affected. Ocular involvement is also seen in 30% to 70% of patients and is more frequent and severe in men.
There is no curative treatment for BD. The goals of treatment are to prevent organ damage and alleviate symptoms. Mucocutaneous disease is treated with potent topical corticosteroids. Severe attacks are treated with oral corticosteroids—1 mg/kg of prednisolone. The drug is tapered and discontinued once the disease is under control. Colchicine or dapsone is also an option. In refractory cases, consider thalidomide (50 mg once a day) or azathioprine (1-3 mg/kg). An anti-tumor necrosis factor agent may also be considered.
In this case, the patient was prescribed prednisolone 60 mg once a day, but relapsed once he was weaned off of it. He was then given thalidomide 50 mg once a day for 4 months and the disease resolved completely. The thalidomide was then reduced to 50 mg 3 times a week for 4 weeks, and then stopped completely. Nearly 2 years later, the patient has remained disease free.
Adapted from: Aslam A, Chalmers R. Mucocutaneous ulceration in a previously healthy man. J Fam Pract. 2014;63:97-100.
Prostate cancer incidence continues to decrease after recommendation against screening
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.

From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.
From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.
From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
FROM JAMA ONCOLOGY
Key clinical point: Incidence rates for localized- and regional-stage prostate cancer continue to decline.
Major finding: The incidence rates for localized- and regional-stage prostate cancer per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older.
Data source: Meta-analysis from 18 SEER registries.
Disclosures: The American Cancer Society funded the study. The authors had no relevant disclosures to report.

Itch, Scratch, Ad Infinitum, Part 2
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
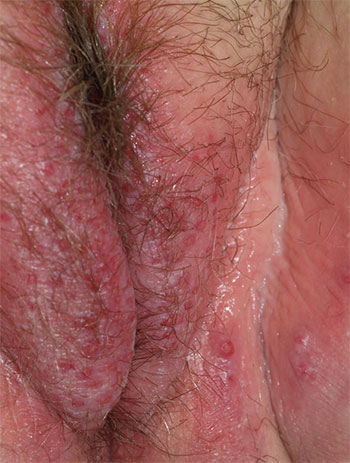
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
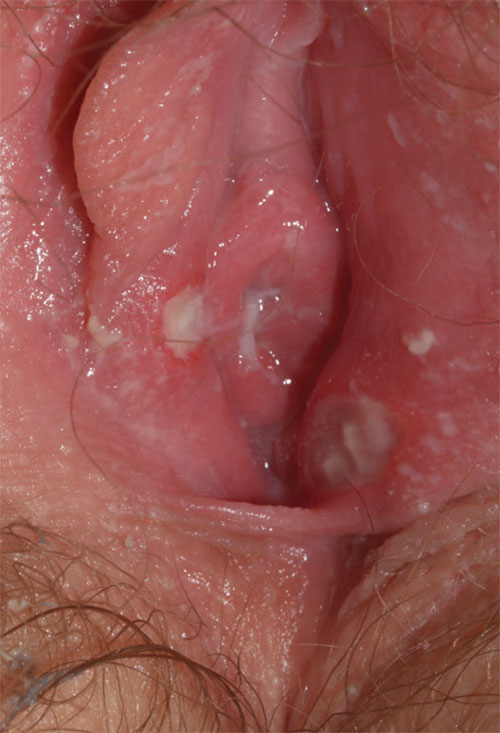
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
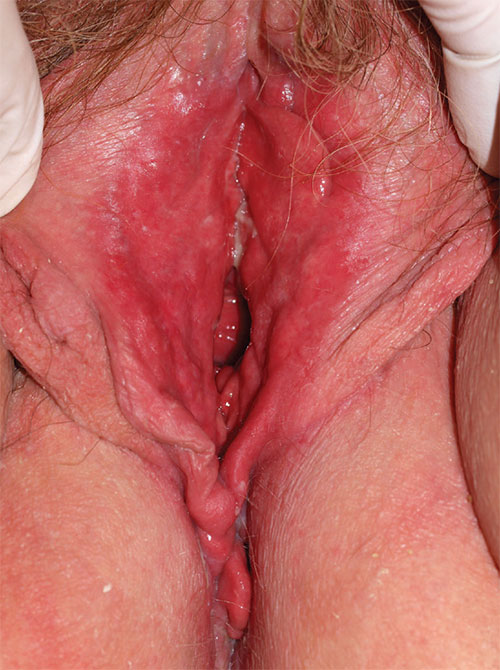
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.

Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.
Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.
Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
AATS Week 2017 Call for Abstracts & Videos
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST

“Honoring Our Mentor” Fellowships Open for Submission
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016
