User login
LABA achieves better asthma control when combined with FDC inhaler
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY
U.S. to jump-start antibiotic resistance research
The Centers for Disease Control and Prevention is providing $67 million to help U.S. health departments address antibiotic resistance and related patient safety concerns.
The new funding was made available through the CDC’s Epidemiology and Laboratory Capacity for Infectious Diseases Cooperative Agreement (ELC), according to a CDC statement, and will support seven new regional laboratories with specialized capabilities allowing rapid detection and identification of emerging antibiotic resistant threats.
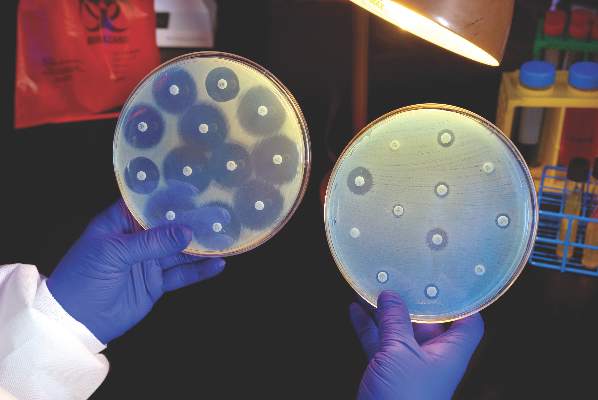
The CDC said it would distribute funds to all 50 state health departments, six local health departments (Chicago, the District of Columbia, Houston, Los Angeles County, New York City, and Philadelphia), and Puerto Rico, beginning Aug. 1, 2016. The agency said the grants would allow every state health department lab to test for carbapenem-resistant Enterobacteriaceae and ultimately perform whole genome sequencing on intestinal bacteria, including Salmonella, Shigella, and many Campylobacter strains.
The agency intends to provide support teams in nine state health departments for rapid response activities designed to “quickly identify and respond to the threat” of antibiotic-resistant gonorrhea in the United States, and will support high-level expertise to implement antimicrobial resistance activities in six states.
The CDC also said the promised funding would strengthen states’ ability to conduct foodborne disease tracking, investigation, and prevention, as it includes increased support for the PulseNet and OutbreakNet systems and for the Integrated Food Safety Centers of Excellence, as well as support for the National Antimicrobial Resistance Monitoring System (NARMS).
Global partnerships
Complementing the new CDC grants was an announcement from the U.S. Department of Health & Human Services that it would partner with the Wellcome Trust of London, the AMR Centre of Alderley Park (Cheshire, U.K.), and Boston University School of Law to create one of the world’s largest public-private partnerships focused on preclinical discovery and development of new antimicrobial products.
According to an HHS statement, the Combating Antibiotic Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) will bring together “multiple domestic and international partners and capabilities to find potential antibiotics and move them through preclinical testing to enable safety and efficacy testing in humans and greatly reducing the business risk,” to make antimicrobial development more attractive to private sector investment.
HHS said the federal Biomedical Advanced Research and Development Authority (BARDA) would provide $30 million during the first year of CARB-X, and up to $250 million during the 5-year project. CARB-X will provide funding for research and development, and technical assistance for companies with innovative and promising solutions to antibiotic resistance, HHS said.
“Our hope is that the combination of technical expertise and life science entrepreneurship experience within the CARB-X’s life science accelerators will remove barriers for companies pursuing the development of the next novel drug, diagnostic, or vaccine to combat this public health threat,” said Joe Larsen, PhD, acting BARDA deputy director, in the HHS statement.
On Twitter @richpizzi
The Centers for Disease Control and Prevention is providing $67 million to help U.S. health departments address antibiotic resistance and related patient safety concerns.
The new funding was made available through the CDC’s Epidemiology and Laboratory Capacity for Infectious Diseases Cooperative Agreement (ELC), according to a CDC statement, and will support seven new regional laboratories with specialized capabilities allowing rapid detection and identification of emerging antibiotic resistant threats.
The CDC said it would distribute funds to all 50 state health departments, six local health departments (Chicago, the District of Columbia, Houston, Los Angeles County, New York City, and Philadelphia), and Puerto Rico, beginning Aug. 1, 2016. The agency said the grants would allow every state health department lab to test for carbapenem-resistant Enterobacteriaceae and ultimately perform whole genome sequencing on intestinal bacteria, including Salmonella, Shigella, and many Campylobacter strains.
The agency intends to provide support teams in nine state health departments for rapid response activities designed to “quickly identify and respond to the threat” of antibiotic-resistant gonorrhea in the United States, and will support high-level expertise to implement antimicrobial resistance activities in six states.
The CDC also said the promised funding would strengthen states’ ability to conduct foodborne disease tracking, investigation, and prevention, as it includes increased support for the PulseNet and OutbreakNet systems and for the Integrated Food Safety Centers of Excellence, as well as support for the National Antimicrobial Resistance Monitoring System (NARMS).
Global partnerships
Complementing the new CDC grants was an announcement from the U.S. Department of Health & Human Services that it would partner with the Wellcome Trust of London, the AMR Centre of Alderley Park (Cheshire, U.K.), and Boston University School of Law to create one of the world’s largest public-private partnerships focused on preclinical discovery and development of new antimicrobial products.
According to an HHS statement, the Combating Antibiotic Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) will bring together “multiple domestic and international partners and capabilities to find potential antibiotics and move them through preclinical testing to enable safety and efficacy testing in humans and greatly reducing the business risk,” to make antimicrobial development more attractive to private sector investment.
HHS said the federal Biomedical Advanced Research and Development Authority (BARDA) would provide $30 million during the first year of CARB-X, and up to $250 million during the 5-year project. CARB-X will provide funding for research and development, and technical assistance for companies with innovative and promising solutions to antibiotic resistance, HHS said.
“Our hope is that the combination of technical expertise and life science entrepreneurship experience within the CARB-X’s life science accelerators will remove barriers for companies pursuing the development of the next novel drug, diagnostic, or vaccine to combat this public health threat,” said Joe Larsen, PhD, acting BARDA deputy director, in the HHS statement.
On Twitter @richpizzi
The Centers for Disease Control and Prevention is providing $67 million to help U.S. health departments address antibiotic resistance and related patient safety concerns.
The new funding was made available through the CDC’s Epidemiology and Laboratory Capacity for Infectious Diseases Cooperative Agreement (ELC), according to a CDC statement, and will support seven new regional laboratories with specialized capabilities allowing rapid detection and identification of emerging antibiotic resistant threats.
The CDC said it would distribute funds to all 50 state health departments, six local health departments (Chicago, the District of Columbia, Houston, Los Angeles County, New York City, and Philadelphia), and Puerto Rico, beginning Aug. 1, 2016. The agency said the grants would allow every state health department lab to test for carbapenem-resistant Enterobacteriaceae and ultimately perform whole genome sequencing on intestinal bacteria, including Salmonella, Shigella, and many Campylobacter strains.
The agency intends to provide support teams in nine state health departments for rapid response activities designed to “quickly identify and respond to the threat” of antibiotic-resistant gonorrhea in the United States, and will support high-level expertise to implement antimicrobial resistance activities in six states.
The CDC also said the promised funding would strengthen states’ ability to conduct foodborne disease tracking, investigation, and prevention, as it includes increased support for the PulseNet and OutbreakNet systems and for the Integrated Food Safety Centers of Excellence, as well as support for the National Antimicrobial Resistance Monitoring System (NARMS).
Global partnerships
Complementing the new CDC grants was an announcement from the U.S. Department of Health & Human Services that it would partner with the Wellcome Trust of London, the AMR Centre of Alderley Park (Cheshire, U.K.), and Boston University School of Law to create one of the world’s largest public-private partnerships focused on preclinical discovery and development of new antimicrobial products.
According to an HHS statement, the Combating Antibiotic Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) will bring together “multiple domestic and international partners and capabilities to find potential antibiotics and move them through preclinical testing to enable safety and efficacy testing in humans and greatly reducing the business risk,” to make antimicrobial development more attractive to private sector investment.
HHS said the federal Biomedical Advanced Research and Development Authority (BARDA) would provide $30 million during the first year of CARB-X, and up to $250 million during the 5-year project. CARB-X will provide funding for research and development, and technical assistance for companies with innovative and promising solutions to antibiotic resistance, HHS said.
“Our hope is that the combination of technical expertise and life science entrepreneurship experience within the CARB-X’s life science accelerators will remove barriers for companies pursuing the development of the next novel drug, diagnostic, or vaccine to combat this public health threat,” said Joe Larsen, PhD, acting BARDA deputy director, in the HHS statement.
On Twitter @richpizzi
Consistent Urine Screens Recommended for Patients on Opioids
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
EXPERT ANALYSIS FROM PAIN CARE FOR PRIMARY CARE
Consistent urine screens recommended for patients on opioids
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
LAKE BUENA VISTA, FLA. – Many clinicians dread discussing a screening test that suggests patients have not been compliant with their scheduled pain medication. Nevertheless, Edwin A. Salsitz, MD, said, those tests should be embraced for their value.
“Every single body that publishes guidelines regarding the use of opioids for chronic pain calls for the use of specimen testing. It is a standard of care,” reported Dr. Salsitz, medical director of office-based opioid therapy at Mount Sinai Beth Israel in New York.
“Urine drug testing is being performed for the patient, not to the patient, and it should increase, not decrease communication,” Dr. Salsitz said at the meeting. Most of all, “just because a urine test is positive, don’t dismiss the patient. That is not the point.”
Among biologic specimens used to monitor compliance with treatment plans, urine is the one most commonly performed, according to Dr. Salsitz, but it is not the only one. A growing number of centers are moving to saliva tests, he said, and those have several advantages. For example, collection of specimens is easier and opportunities for cheating are reduced.
Hair specimens pose an even lower risk of cheating, and they have a much longer drug detection window. Relative to blood and saliva specimens, which become positive almost immediately after drug exposures but lose validity within 24-48 hours, hair specimens do not become positive for days but still can prove positive for drugs months after exposure. Urine samples, which do not become positive for several hours after a drug exposure, typically remain reliable for several days.
Urine testing remains the most widely used screening tool and also the focus of most efforts to cheat, Dr. Salsitz said. He said an Internet search for strategies used to cheat on urine drug testing would generate pages of recommendations. For men, options include a prosthetic penis that permits cheating even for observed urine samples. Strapped to the body, the lifelike prosthetic includes a place to store a sample of urine that can be excreted by the prosthetic into a collection receptacle. This type of sophisticated effort to “game the system” can be a challenge when managing patients most intent on noncompliance.
For periodic drug screening at his own center, Dr. Salsitz reported that he often uses point-of-care urine testing. Although he conceded that those kits, which employ a dipstick technology similar to pregnancy tests, are not as reliable as laboratory analyses of urine samples, they are relatively inexpensive and provide immediate results. However, laboratory testing still may be needed if the patient denies drug use after a positive result or if a more comprehensive analysis of drug exposures is needed.
“I would not make a major clinical decision on the basis of point-of-care testing alone,” Dr. Salsitz reported. Indeed, he expressed caution about relying on specimen screening alone when other reasons exist to be concerned about noncompliance.
“It is unwise to accept at face value a urinary drug test report that seems to support an impression of clinical stability if, in fact, there is other clinical evidence to the contrary,” Dr. Salsitz said.
Successful screening strategies for noncompliance require an objective, nonjudgmental, and systematic approach, Dr. Salsitz said. Implementing a uniform policy common for all patients reduces the risk of conveying a sense of distrust. Indeed, uniform testing circumvents bias that could, for example, permit well-liked patients to avoid detection of noncompliance.
“In one study, reliance on aberrant behavior alone to trigger urine drug testing was estimated to miss almost half of those using drugs problematically,” Dr. Salsitz reported. However, he noted that the same study suggested that urine screening by itself also was insufficient. Rather, he said, “Monitoring urine and behavior identified more patients with inappropriate drug taking than either alone.”
Emphasizing that biologic specimen screening is “just a tool” in managing chronic pain patients on opioids, Dr. Salsitz reported several cases where he verified false-positive results with a point-of-care urine test using his own specimen, thereby validating claims made to him by patients. In one case, results were positive after he consumed a poppy bagel. “When the patient stopped eating poppy seed bagels, he stopped having positive tests,” Dr. Salsitz reported. In another case, a positive result occurred after consuming a commercially available tea made with coca leaves.
Dr. Salsitz reports that he has no relevant financial relationships to disclose. The meeting was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this news organization are owned the same company.
EXPERT ANALYSIS FROM PAIN CARE FOR PRIMARY CARE
Hepatitis outlook: July 2016
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
| ©Zerbor/Thinkstock |
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Visit http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c to learn more.
On Twitter @richpizzi
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
| ©Zerbor/Thinkstock |
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Visit http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c to learn more.
On Twitter @richpizzi
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
| ©Zerbor/Thinkstock |
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Visit http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c to learn more.
On Twitter @richpizzi
Hepatitis outlook: July 2016
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
On Twitter @richpizzi
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
On Twitter @richpizzi
If you work on the front lines of medical care treating patients with hepatitis, you may not have time to review all the hepatitis research that enters the medical literature every month. Here’s a quick look at some notable news items and journal articles published over the past month, covering a variety of the major hepatitis viruses.
Although hepatitis E virus infections are increasingly recognized as a global public health problem, there are “few methods for prevention and treatment that are widely available,” according to a recent analysis.
A “suboptimal plasma level of the antiviral drug daclatasvir allows the selection of resistance-associated variants” and fails to contribute to antiviral activity in HIV-hepatitis C virus (HCV) coinfected patients, according to a recent study, although no definite reason for the low daclatasvir level was found.
The new preservative-free inactivated hepatitis A vaccine (Healive) in two doses showed better persistence of antibody concentrations for 5 years after full-course immunization among children, compared with Havrix. The endurance of protective immunogenicity was estimated for at least 20 years.
Because of transplacental transfer of antihepatitis B virus antibodies (anti-HBVs), high levels of maternal anti-HBVs may suppress infants’ immune response to standard HBV vaccination, according to an analysis in the Journal of Viral Hepatitis.
The gamma-glutamyl transpeptidase-to-platelet ratio (GPR) is “a new serum model for the diagnosis” of liver fibrosis and cirrhosis, according to a recent study. Researchers said it shows advantages in Chinese hepatitis Be antigen (HBeAg)-positive patients with hepatitis B virus DNA greater than or equal to 5 log10 copies/mL and ALT less than or equal to two times ULN (upper limit of normal), compared with APRI (aspartate aminotransferase to platelet ratio index) and Fibrosis-4.
A baseline quantitative hepatitis B surface antigen (HBsAg) threshold of 3.141 log10 IU/mL and a baseline quantitative hepatitis B core-related antigen 3.450 log10 U/mL threshold, used separately or in combination, allow prediction of response to pegylated interferon-alpha-2a (PegIFN)-based “precision therapy” for hepatitis B virus infection, a new study found.
Male sex, age over 40 years, cirrhotic liver, and long length of stay are significant factors associated with death in hepatitis A virus-hospitalized cases, according to a study in the Journal of Viral Hepatitis.
Chronic kidney disease patients receiving three doses of hepatitis B adjuvanted vaccine were three times more likely to seroconvert than patients immunized with nonadjuvanted vaccines, according to results of a Spanish study. This meant fewer patients needed a second course of HBV vaccination and there were fewer outpatient visits.
Acute kidney injury is closely linked with increased short-term mortality in Chinese hepatitis B virus-related, acute-on-chronic liver failure patients, according to a study in the Journal of Viral Hepatitis.
Italian investigators attempted “to predict susceptibility of healthy patients to de novo HBV infection using a cultured IFN-gamma enzyme-linked immunospot (ELISPOT) assay.” Although the prognostic value of the assay was not demonstrated, data suggested that the subjects may be at risk for HBV infection.
Investigators demonstrated that treatment with sofosbuvir and simeprevir was effective in a real-life cohort of patients with hepatitis C virus genotype 4 infection and advanced liver fibrosis/cirrhosis. They said that adding ribavirin could be considered in treatment-experienced patients.
The presence of specific anti-envelope antibodies may be a factor that helps individuals at high risk of hepatitis C virus to resist infection, according to a study in the Journal of Viral Hepatitis.
A Chinese study determined that certain social network structural characteristics are related to hepatitis C virus infections in people who inject drugs, and used the data to identify the most susceptible individuals for HCV transmission in a network of people who inject drugs.
Drug resistance analyses of protease inhibitors that treat hepatitis C virus infection can be useful and essential in revealing the particular variants responsible for pretreatment natural resistance and also the particular mutations responsible for the viral breakthrough that may develop during the treatment, according to a study in the International Journal of Infectious Diseases.
Routine vaccination of toddlers against hepatitis A virus would be cost effective in Mexico using a single-dose vaccination strategy, according to a recent study, although the authors said the cost efficacy of a second dose depends on the assumptions of added safeguards by immune memory protection and the time horizon over which the analysis is enacted.
Hepatitis C virus-infected patients undergoing ribavirin-free sofosbuvir and velpatasvir regimens had significantly better patient-reported outcome scores during therapy, compared with those undergoing the ribavirin-containing regimen, a recent study found.
An analysis in Infectious Diseases in Clinical Practice reported the first case of visual hallucinations during chronic hepatitis C treatment with sofosbuvir and simeprevir. Investigators said hallucinations stopped upon starting antipsychotic medication, and the remainder of treatment was safe.
Sustained virologic response can be attained with pegylated interferon-alpha plus ribavirin combination therapy in hepatitis C virus–infected patients, but a relapse may occur in some patients, according to a recent study.
A quantitative HBsAg test can be used to ascertain high levels of hepatitis B viremia in women who might transmit the virus to their children, rather than a test for HBeAg or HBV DNA, according to a research letter in Hepatology.
A Chinese study found a robust relationship between Helicobacter pylori infection and chronic hepatitis B. This is especially true during hepatitis B virus progression.
The prevalence of antihepatitis E virus (HEV) antibodies was 49% (153/313) among blood donors in central Italy, according to a study published in Eurosurveillance. The authors said HEV infection is hyperendemic among blood donors (80% men, 18- to 64-years-old) from central Italy and associated with local dietary habits, such as eating raw dried pig liver sausage.
On Twitter @richpizzi
Cosmetic Corner: Dermatologists Weigh in on Lip Balms
To improve patient care and outcomes, leading dermatologists offered their recommendations on lip balms. Consideration must be given to:
- Aquaphor Lip Repair
Beiersdorf, Inc
Recommended by Gary Goldenberg, MD, New York, New York
- CeraVe Healing Ointment
Valeant Pharmaceuticals North America LLC
“Combining ceramides with hyaluronic acid in a waxy ointment base, it hydrates, protects, and repairs the damaged skin barrier.”—Joshua Zeichner, MD, New York, New York
- Lip Balm #1
Kiehl’s
“It is quite moisturizing with squalene, aloe vera, vitamin E, and wheat germ oil.”—Anthony M. Rossi, MD, New York, New York
- Lip Renewal SPF 50
June Jacobs Laboratories LLC
“This lip balm contains shea butter and vitamin E for hydration with a hint of pomegranate flavor. It also provides photoprotection for daily use.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Revitalizing Lip Balm SPF 20
Johnson & Johnson Consumer Inc
“It moisturizes the lips, protects from UV rays, and is available in a variety of shades. It should be reapplied frequently for UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Vanicream Lip Protectant SPF 30
Pharmaceutical Specialties, Inc
“It has titanium dioxide for good UV protection.”—Anthony M. Rossi, MD, New York, New York
- Xtend Your Youth Lip Filler & Volumizer
Dr. Brandt Skincare
“This unique formula restores moisture and smooths the lip surface, creating a canvas for longer-lasting lip color. Its potent blend of antioxidants including vitamin E, green tea, white tea, and grape seed extract protects lips from free radical damage, while also working to reduce the look of lines and wrinkles.”—Whitney Bowe, MD, New York, New York
Cutis invites readers to send us their recommendations. Self-tanners and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on lip balms. Consideration must be given to:
- Aquaphor Lip Repair
Beiersdorf, Inc
Recommended by Gary Goldenberg, MD, New York, New York
- CeraVe Healing Ointment
Valeant Pharmaceuticals North America LLC
“Combining ceramides with hyaluronic acid in a waxy ointment base, it hydrates, protects, and repairs the damaged skin barrier.”—Joshua Zeichner, MD, New York, New York
- Lip Balm #1
Kiehl’s
“It is quite moisturizing with squalene, aloe vera, vitamin E, and wheat germ oil.”—Anthony M. Rossi, MD, New York, New York
- Lip Renewal SPF 50
June Jacobs Laboratories LLC
“This lip balm contains shea butter and vitamin E for hydration with a hint of pomegranate flavor. It also provides photoprotection for daily use.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Revitalizing Lip Balm SPF 20
Johnson & Johnson Consumer Inc
“It moisturizes the lips, protects from UV rays, and is available in a variety of shades. It should be reapplied frequently for UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Vanicream Lip Protectant SPF 30
Pharmaceutical Specialties, Inc
“It has titanium dioxide for good UV protection.”—Anthony M. Rossi, MD, New York, New York
- Xtend Your Youth Lip Filler & Volumizer
Dr. Brandt Skincare
“This unique formula restores moisture and smooths the lip surface, creating a canvas for longer-lasting lip color. Its potent blend of antioxidants including vitamin E, green tea, white tea, and grape seed extract protects lips from free radical damage, while also working to reduce the look of lines and wrinkles.”—Whitney Bowe, MD, New York, New York
Cutis invites readers to send us their recommendations. Self-tanners and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on lip balms. Consideration must be given to:
- Aquaphor Lip Repair
Beiersdorf, Inc
Recommended by Gary Goldenberg, MD, New York, New York
- CeraVe Healing Ointment
Valeant Pharmaceuticals North America LLC
“Combining ceramides with hyaluronic acid in a waxy ointment base, it hydrates, protects, and repairs the damaged skin barrier.”—Joshua Zeichner, MD, New York, New York
- Lip Balm #1
Kiehl’s
“It is quite moisturizing with squalene, aloe vera, vitamin E, and wheat germ oil.”—Anthony M. Rossi, MD, New York, New York
- Lip Renewal SPF 50
June Jacobs Laboratories LLC
“This lip balm contains shea butter and vitamin E for hydration with a hint of pomegranate flavor. It also provides photoprotection for daily use.”—Cherise M. Levi, DO, New York, New York
- Neutrogena Revitalizing Lip Balm SPF 20
Johnson & Johnson Consumer Inc
“It moisturizes the lips, protects from UV rays, and is available in a variety of shades. It should be reapplied frequently for UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Vanicream Lip Protectant SPF 30
Pharmaceutical Specialties, Inc
“It has titanium dioxide for good UV protection.”—Anthony M. Rossi, MD, New York, New York
- Xtend Your Youth Lip Filler & Volumizer
Dr. Brandt Skincare
“This unique formula restores moisture and smooths the lip surface, creating a canvas for longer-lasting lip color. Its potent blend of antioxidants including vitamin E, green tea, white tea, and grape seed extract protects lips from free radical damage, while also working to reduce the look of lines and wrinkles.”—Whitney Bowe, MD, New York, New York
Cutis invites readers to send us their recommendations. Self-tanners and cleansing devices will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Understanding, Counsel Can Help to Navigate Payor Audits
Receiving notice from a payor that you are being audited can be alarming. Questions will inevitably run through your mind, such as, Why? How? How much will this cost?

Understanding the types of payor audits and how to navigate the process can make answering those questions easier. In addition, advanced preparation and knowing when to engage legal counsel can be critical to a successful audit outcome.
Audit Types
There are three general types of audits that providers face: Medicare audits, Medicaid audits, and private payor audits.
Medicare audits: The agency responsible for Medicare audits is the Centers for Medicare & Medicaid Services (CMS). There are three types of Medicare audits. Comprehensive Error Rate Testing (CERT) audits focus on providers who deliver high-cost items or services, have high volume, and/or have atypical billing or coding practices. Private contractors perform Recovery Audit Contractor (RAC) program audits; these contractors are paid a percentage of the amount of any improper payment discovered. Finally, Zone Program Integrity Contractor (ZPIC) audits are the most serious of the three audit types. ZPIC audits are performed by CMS contractors who mine the provider’s data for compliance with Medicare coverage and coding policies, investigate fraud, and may prepare cases for civil or criminal referral to CMS or law enforcement agencies.
Medicaid audits: Medicaid audits evaluate compliance with both CMS and applicable state regulations and investigate fraud. Evidence of fraud will be reported to the state attorney general for further review and prosecution.
Private payor audits: Private payor audits consist of informal reviews and formal audits. These audits can be triggered by actual allegations or evidence of noncompliance, or they can be random, in which general compliance is assessed. Audit procedures are typically determined by contract or the payor’s provider handbook and in accordance with applicable state law. The process can consist of prepayment reviews, in which the sufficiency of a claim and its supporting documentation is determined before payment is made to the provider, or post-payment reviews, during which claims are analyzed after the provider has been paid to determine if an overpayment was made and the amount of such overpayment. In the event an overpayment is discovered, a recoupment will be sought from the provider.
Focus
Consistent billing by a provider of high volumes of certain high-level services, high volumes of evaluation and management services, or consistently referring patients for certain testing can create suspicion in mayors.
In recent years, the primary focus of audits has been medical necessity due to payor concerns about specific fraud and abuse issues. Documentation of medical necessity is required during an audit. However, proving medical necessity can be difficult as the definition of “medical necessity” can vary by payor and within a payor depending on the underlying plan. In addition, private payors often have arbitrary and vague guidelines for defining and determining medical necessity, particularly when dealing with physicians or ordering clinicians. For this reason, it is critical that providers read their payor contracts and manuals carefully. If those materials are unclear, it is best to confirm requirements with the payor.
Regardless of the definition, medical necessity is a precondition to coverage for all payors. Proof is required that the services were reasonable and necessary to diagnosis or treat a patient’s medical condition. To satisfy this standard, providers should document the diagnosis for all procedures performed and all diagnostic tests ordered. In the case of repeat procedures, providers should clearly note the outcome of the previous procedure and the basis for reordering.
Responding to an Audit Request
All audit requests must be taken very seriously. Payors tend to copy what other payors are doing, and a problematic audit with one payor can cause other payors to initiate their own audits. Therefore, it is critical to respond appropriately to each audit request. Also, auditors often only check a few billing records. If errors are found, they will then extrapolate those findings, and the provider may be penalized.
Upon receipt of an audit request, it is important to immediately engage legal counsel well-versed in handling payor audits. Having an attorney who understands the audit process and has experience responding to audit requests can help ensure the best possible audit outcome. A negative outcome could result in recovery of overpayments, civil and/or criminal penalties, and exclusion from government programs.
Providers should work with such legal counsel to review the audit request and supply everything reasonably requested. A concerted effort should be made to submit all information to the auditor at one time. If information is missing, the auditor may determine that a significant error rate exists, which could cause the auditor to review all CPT codes to calculate the overpayment made to the provider. If it is not possible to gather the requested material before the auditor’s deadline, an extension should be requested. Any extensions granted should be documented.
It is important that an audit response and supporting documentation be thorough, clear, and concise. It should be submitted in a manner that allows the auditor to quickly review the information and understand the provider’s arguments. It should clearly state what measures the provider has already taken to terminate existing problems and prevent future issues. Competent legal counsel will be able to address procedural, legal, or factual flaws in the auditor’s position.
Advanced Preparation
The best way to ensure compliance and audit readiness is to develop and implement a compliance plan well in advance of any audit. Experienced legal counsel should play a pivotal role in development of such plan. As always, periodic self-audits or independent audits are necessary to proactively identify compliance issues and mitigate their impact.
Finally, regular and periodic training and education should be conducted regarding audit response obligations and responsibilities. Performing these tasks will help ensure a smooth audit experience with minimal infractions and penalties. TH
Receiving notice from a payor that you are being audited can be alarming. Questions will inevitably run through your mind, such as, Why? How? How much will this cost?
Understanding the types of payor audits and how to navigate the process can make answering those questions easier. In addition, advanced preparation and knowing when to engage legal counsel can be critical to a successful audit outcome.
Audit Types
There are three general types of audits that providers face: Medicare audits, Medicaid audits, and private payor audits.
Medicare audits: The agency responsible for Medicare audits is the Centers for Medicare & Medicaid Services (CMS). There are three types of Medicare audits. Comprehensive Error Rate Testing (CERT) audits focus on providers who deliver high-cost items or services, have high volume, and/or have atypical billing or coding practices. Private contractors perform Recovery Audit Contractor (RAC) program audits; these contractors are paid a percentage of the amount of any improper payment discovered. Finally, Zone Program Integrity Contractor (ZPIC) audits are the most serious of the three audit types. ZPIC audits are performed by CMS contractors who mine the provider’s data for compliance with Medicare coverage and coding policies, investigate fraud, and may prepare cases for civil or criminal referral to CMS or law enforcement agencies.
Medicaid audits: Medicaid audits evaluate compliance with both CMS and applicable state regulations and investigate fraud. Evidence of fraud will be reported to the state attorney general for further review and prosecution.
Private payor audits: Private payor audits consist of informal reviews and formal audits. These audits can be triggered by actual allegations or evidence of noncompliance, or they can be random, in which general compliance is assessed. Audit procedures are typically determined by contract or the payor’s provider handbook and in accordance with applicable state law. The process can consist of prepayment reviews, in which the sufficiency of a claim and its supporting documentation is determined before payment is made to the provider, or post-payment reviews, during which claims are analyzed after the provider has been paid to determine if an overpayment was made and the amount of such overpayment. In the event an overpayment is discovered, a recoupment will be sought from the provider.
Focus
Consistent billing by a provider of high volumes of certain high-level services, high volumes of evaluation and management services, or consistently referring patients for certain testing can create suspicion in mayors.
In recent years, the primary focus of audits has been medical necessity due to payor concerns about specific fraud and abuse issues. Documentation of medical necessity is required during an audit. However, proving medical necessity can be difficult as the definition of “medical necessity” can vary by payor and within a payor depending on the underlying plan. In addition, private payors often have arbitrary and vague guidelines for defining and determining medical necessity, particularly when dealing with physicians or ordering clinicians. For this reason, it is critical that providers read their payor contracts and manuals carefully. If those materials are unclear, it is best to confirm requirements with the payor.
Regardless of the definition, medical necessity is a precondition to coverage for all payors. Proof is required that the services were reasonable and necessary to diagnosis or treat a patient’s medical condition. To satisfy this standard, providers should document the diagnosis for all procedures performed and all diagnostic tests ordered. In the case of repeat procedures, providers should clearly note the outcome of the previous procedure and the basis for reordering.
Responding to an Audit Request
All audit requests must be taken very seriously. Payors tend to copy what other payors are doing, and a problematic audit with one payor can cause other payors to initiate their own audits. Therefore, it is critical to respond appropriately to each audit request. Also, auditors often only check a few billing records. If errors are found, they will then extrapolate those findings, and the provider may be penalized.
Upon receipt of an audit request, it is important to immediately engage legal counsel well-versed in handling payor audits. Having an attorney who understands the audit process and has experience responding to audit requests can help ensure the best possible audit outcome. A negative outcome could result in recovery of overpayments, civil and/or criminal penalties, and exclusion from government programs.
Providers should work with such legal counsel to review the audit request and supply everything reasonably requested. A concerted effort should be made to submit all information to the auditor at one time. If information is missing, the auditor may determine that a significant error rate exists, which could cause the auditor to review all CPT codes to calculate the overpayment made to the provider. If it is not possible to gather the requested material before the auditor’s deadline, an extension should be requested. Any extensions granted should be documented.
It is important that an audit response and supporting documentation be thorough, clear, and concise. It should be submitted in a manner that allows the auditor to quickly review the information and understand the provider’s arguments. It should clearly state what measures the provider has already taken to terminate existing problems and prevent future issues. Competent legal counsel will be able to address procedural, legal, or factual flaws in the auditor’s position.
Advanced Preparation
The best way to ensure compliance and audit readiness is to develop and implement a compliance plan well in advance of any audit. Experienced legal counsel should play a pivotal role in development of such plan. As always, periodic self-audits or independent audits are necessary to proactively identify compliance issues and mitigate their impact.
Finally, regular and periodic training and education should be conducted regarding audit response obligations and responsibilities. Performing these tasks will help ensure a smooth audit experience with minimal infractions and penalties. TH
Receiving notice from a payor that you are being audited can be alarming. Questions will inevitably run through your mind, such as, Why? How? How much will this cost?
Understanding the types of payor audits and how to navigate the process can make answering those questions easier. In addition, advanced preparation and knowing when to engage legal counsel can be critical to a successful audit outcome.
Audit Types
There are three general types of audits that providers face: Medicare audits, Medicaid audits, and private payor audits.
Medicare audits: The agency responsible for Medicare audits is the Centers for Medicare & Medicaid Services (CMS). There are three types of Medicare audits. Comprehensive Error Rate Testing (CERT) audits focus on providers who deliver high-cost items or services, have high volume, and/or have atypical billing or coding practices. Private contractors perform Recovery Audit Contractor (RAC) program audits; these contractors are paid a percentage of the amount of any improper payment discovered. Finally, Zone Program Integrity Contractor (ZPIC) audits are the most serious of the three audit types. ZPIC audits are performed by CMS contractors who mine the provider’s data for compliance with Medicare coverage and coding policies, investigate fraud, and may prepare cases for civil or criminal referral to CMS or law enforcement agencies.
Medicaid audits: Medicaid audits evaluate compliance with both CMS and applicable state regulations and investigate fraud. Evidence of fraud will be reported to the state attorney general for further review and prosecution.
Private payor audits: Private payor audits consist of informal reviews and formal audits. These audits can be triggered by actual allegations or evidence of noncompliance, or they can be random, in which general compliance is assessed. Audit procedures are typically determined by contract or the payor’s provider handbook and in accordance with applicable state law. The process can consist of prepayment reviews, in which the sufficiency of a claim and its supporting documentation is determined before payment is made to the provider, or post-payment reviews, during which claims are analyzed after the provider has been paid to determine if an overpayment was made and the amount of such overpayment. In the event an overpayment is discovered, a recoupment will be sought from the provider.
Focus
Consistent billing by a provider of high volumes of certain high-level services, high volumes of evaluation and management services, or consistently referring patients for certain testing can create suspicion in mayors.
In recent years, the primary focus of audits has been medical necessity due to payor concerns about specific fraud and abuse issues. Documentation of medical necessity is required during an audit. However, proving medical necessity can be difficult as the definition of “medical necessity” can vary by payor and within a payor depending on the underlying plan. In addition, private payors often have arbitrary and vague guidelines for defining and determining medical necessity, particularly when dealing with physicians or ordering clinicians. For this reason, it is critical that providers read their payor contracts and manuals carefully. If those materials are unclear, it is best to confirm requirements with the payor.
Regardless of the definition, medical necessity is a precondition to coverage for all payors. Proof is required that the services were reasonable and necessary to diagnosis or treat a patient’s medical condition. To satisfy this standard, providers should document the diagnosis for all procedures performed and all diagnostic tests ordered. In the case of repeat procedures, providers should clearly note the outcome of the previous procedure and the basis for reordering.
Responding to an Audit Request
All audit requests must be taken very seriously. Payors tend to copy what other payors are doing, and a problematic audit with one payor can cause other payors to initiate their own audits. Therefore, it is critical to respond appropriately to each audit request. Also, auditors often only check a few billing records. If errors are found, they will then extrapolate those findings, and the provider may be penalized.
Upon receipt of an audit request, it is important to immediately engage legal counsel well-versed in handling payor audits. Having an attorney who understands the audit process and has experience responding to audit requests can help ensure the best possible audit outcome. A negative outcome could result in recovery of overpayments, civil and/or criminal penalties, and exclusion from government programs.
Providers should work with such legal counsel to review the audit request and supply everything reasonably requested. A concerted effort should be made to submit all information to the auditor at one time. If information is missing, the auditor may determine that a significant error rate exists, which could cause the auditor to review all CPT codes to calculate the overpayment made to the provider. If it is not possible to gather the requested material before the auditor’s deadline, an extension should be requested. Any extensions granted should be documented.
It is important that an audit response and supporting documentation be thorough, clear, and concise. It should be submitted in a manner that allows the auditor to quickly review the information and understand the provider’s arguments. It should clearly state what measures the provider has already taken to terminate existing problems and prevent future issues. Competent legal counsel will be able to address procedural, legal, or factual flaws in the auditor’s position.
Advanced Preparation
The best way to ensure compliance and audit readiness is to develop and implement a compliance plan well in advance of any audit. Experienced legal counsel should play a pivotal role in development of such plan. As always, periodic self-audits or independent audits are necessary to proactively identify compliance issues and mitigate their impact.
Finally, regular and periodic training and education should be conducted regarding audit response obligations and responsibilities. Performing these tasks will help ensure a smooth audit experience with minimal infractions and penalties. TH
How iPSCs differentiate to blood cells

Image from Salk Institute
New research suggests the type of founder cell used to generate induced pluripotent stem cells (iPSCs) does not affect the iPSCs’ ability to differentiate into hematopoietic cells.
Instead, researchers found the expression of certain genes and DNA methylations were better indicators of the efficiency at which a cell line could be differentiated into the hematopoietic lineage.
The team reported these findings in Cell Stem Cell.
The researchers assessed the hematopoietic differentiation capacities of 35 iPSC lines derived from 4 types of somatic tissues—human dermal fibroblasts, hematopoietic cells such as cord blood and peripheral blood, dental pulp cells, and keratinocytes—from 15 donors.
The team also assessed 4 embryonic stem cell lines in early phase and late phase.
The researchers found that hematopoietic commitment capacity was associated with expression of IGF2 in undifferentiated iPSCs, but not with type of founder cell.
Higher expression of IFG2 was indicative of iPSCs initiating their conversion into hematopoietic cells. Even though IFG2 itself is not directly related to hematopoiesis, its uptake corresponded to an increase in the expression of genes that are.
Although IFG2 marked the beginnings of differentiation to hematopoietic lineage, the completion of differentiation was marked by the methylation profiles of the iPSC DNA.
“DNA methylation has an effect on a cell staying pluripotent or differentiating,” explained study author Yoshinori Yoshida, MD, PhD, of the Center for iPS Cell Research and Application at Kyoto University in Japan.
The completion of differentiation correlated with less aberrant methylation during the reprogramming process.
Hematopoietic founder cells showed a much lower propensity for aberrant methylation than did other founder cells, which could explain why, in the past, scientists attributed the founder cell to the effectiveness of differentiating iPSCs to the hematopoietic lineage.
Dr Yoshida and his colleagues said this research revealed molecular factors that can be used to evaluate the differentiation potential of different cell lines, which should expedite the progress of iPSCs to clinical use. ![]()
Image from Salk Institute
New research suggests the type of founder cell used to generate induced pluripotent stem cells (iPSCs) does not affect the iPSCs’ ability to differentiate into hematopoietic cells.
Instead, researchers found the expression of certain genes and DNA methylations were better indicators of the efficiency at which a cell line could be differentiated into the hematopoietic lineage.
The team reported these findings in Cell Stem Cell.
The researchers assessed the hematopoietic differentiation capacities of 35 iPSC lines derived from 4 types of somatic tissues—human dermal fibroblasts, hematopoietic cells such as cord blood and peripheral blood, dental pulp cells, and keratinocytes—from 15 donors.
The team also assessed 4 embryonic stem cell lines in early phase and late phase.
The researchers found that hematopoietic commitment capacity was associated with expression of IGF2 in undifferentiated iPSCs, but not with type of founder cell.
Higher expression of IFG2 was indicative of iPSCs initiating their conversion into hematopoietic cells. Even though IFG2 itself is not directly related to hematopoiesis, its uptake corresponded to an increase in the expression of genes that are.
Although IFG2 marked the beginnings of differentiation to hematopoietic lineage, the completion of differentiation was marked by the methylation profiles of the iPSC DNA.
“DNA methylation has an effect on a cell staying pluripotent or differentiating,” explained study author Yoshinori Yoshida, MD, PhD, of the Center for iPS Cell Research and Application at Kyoto University in Japan.
The completion of differentiation correlated with less aberrant methylation during the reprogramming process.
Hematopoietic founder cells showed a much lower propensity for aberrant methylation than did other founder cells, which could explain why, in the past, scientists attributed the founder cell to the effectiveness of differentiating iPSCs to the hematopoietic lineage.
Dr Yoshida and his colleagues said this research revealed molecular factors that can be used to evaluate the differentiation potential of different cell lines, which should expedite the progress of iPSCs to clinical use. ![]()
Image from Salk Institute
New research suggests the type of founder cell used to generate induced pluripotent stem cells (iPSCs) does not affect the iPSCs’ ability to differentiate into hematopoietic cells.
Instead, researchers found the expression of certain genes and DNA methylations were better indicators of the efficiency at which a cell line could be differentiated into the hematopoietic lineage.
The team reported these findings in Cell Stem Cell.
The researchers assessed the hematopoietic differentiation capacities of 35 iPSC lines derived from 4 types of somatic tissues—human dermal fibroblasts, hematopoietic cells such as cord blood and peripheral blood, dental pulp cells, and keratinocytes—from 15 donors.
The team also assessed 4 embryonic stem cell lines in early phase and late phase.
The researchers found that hematopoietic commitment capacity was associated with expression of IGF2 in undifferentiated iPSCs, but not with type of founder cell.
Higher expression of IFG2 was indicative of iPSCs initiating their conversion into hematopoietic cells. Even though IFG2 itself is not directly related to hematopoiesis, its uptake corresponded to an increase in the expression of genes that are.
Although IFG2 marked the beginnings of differentiation to hematopoietic lineage, the completion of differentiation was marked by the methylation profiles of the iPSC DNA.
“DNA methylation has an effect on a cell staying pluripotent or differentiating,” explained study author Yoshinori Yoshida, MD, PhD, of the Center for iPS Cell Research and Application at Kyoto University in Japan.
The completion of differentiation correlated with less aberrant methylation during the reprogramming process.
Hematopoietic founder cells showed a much lower propensity for aberrant methylation than did other founder cells, which could explain why, in the past, scientists attributed the founder cell to the effectiveness of differentiating iPSCs to the hematopoietic lineage.
Dr Yoshida and his colleagues said this research revealed molecular factors that can be used to evaluate the differentiation potential of different cell lines, which should expedite the progress of iPSCs to clinical use. ![]()
Method provides more accurate diagnosis of MDS, team says

Next-generation sequencing (NGS) of cell-free DNA should be the method of choice to confirm the diagnosis of myelodysplastic syndromes (MDS), according to researchers.
The team found that using NGS to analyze samples from MDS patients yielded more accurate results than Sanger sequencing.
And sequencing cell-free DNA rather than peripheral blood cell DNA increased the likelihood of detecting mutations associated with MDS.
The team reported these findings in Genetic Testing and Molecular Biomarkers. This research was funded by NeoGenomics Laboratories.
For this study, the researchers performed NGS on a panel of 14 target genes using total nucleic acid extracted from the plasma of 16 patients with early MDS (blasts <5%). The team also performed Sanger sequencing and NGS on peripheral blood cell DNA from the same patients.
The researchers found that NGS of cell-free DNA confirmed the diagnosis of MDS in all 16 patients.
In addition, NGS of cell-free DNA revealed abnormalities in 5 patients (31%) that were not detected by Sanger sequencing of peripheral blood cell DNA.
NGS of peripheral blood cell DNA produced the same results as NGS of cell-free DNA for 4 of the 5 patients. However, NGS of peripheral blood cell DNA did not detect a mutation in the RUNX1 gene that was evident in cell-free DNA from 1 patient.
Overall, the researchers found that mutant allele frequency was significantly higher in cell-free DNA than cellular DNA (P=0.008).
The team therefore concluded that cell-free DNA is more reliable than peripheral blood cell DNA for detecting molecular abnormalities in patients with MDS, and NGS is more accurate than Sanger sequencing. ![]()
Next-generation sequencing (NGS) of cell-free DNA should be the method of choice to confirm the diagnosis of myelodysplastic syndromes (MDS), according to researchers.
The team found that using NGS to analyze samples from MDS patients yielded more accurate results than Sanger sequencing.
And sequencing cell-free DNA rather than peripheral blood cell DNA increased the likelihood of detecting mutations associated with MDS.
The team reported these findings in Genetic Testing and Molecular Biomarkers. This research was funded by NeoGenomics Laboratories.
For this study, the researchers performed NGS on a panel of 14 target genes using total nucleic acid extracted from the plasma of 16 patients with early MDS (blasts <5%). The team also performed Sanger sequencing and NGS on peripheral blood cell DNA from the same patients.
The researchers found that NGS of cell-free DNA confirmed the diagnosis of MDS in all 16 patients.
In addition, NGS of cell-free DNA revealed abnormalities in 5 patients (31%) that were not detected by Sanger sequencing of peripheral blood cell DNA.
NGS of peripheral blood cell DNA produced the same results as NGS of cell-free DNA for 4 of the 5 patients. However, NGS of peripheral blood cell DNA did not detect a mutation in the RUNX1 gene that was evident in cell-free DNA from 1 patient.
Overall, the researchers found that mutant allele frequency was significantly higher in cell-free DNA than cellular DNA (P=0.008).
The team therefore concluded that cell-free DNA is more reliable than peripheral blood cell DNA for detecting molecular abnormalities in patients with MDS, and NGS is more accurate than Sanger sequencing. ![]()
Next-generation sequencing (NGS) of cell-free DNA should be the method of choice to confirm the diagnosis of myelodysplastic syndromes (MDS), according to researchers.
The team found that using NGS to analyze samples from MDS patients yielded more accurate results than Sanger sequencing.
And sequencing cell-free DNA rather than peripheral blood cell DNA increased the likelihood of detecting mutations associated with MDS.
The team reported these findings in Genetic Testing and Molecular Biomarkers. This research was funded by NeoGenomics Laboratories.
For this study, the researchers performed NGS on a panel of 14 target genes using total nucleic acid extracted from the plasma of 16 patients with early MDS (blasts <5%). The team also performed Sanger sequencing and NGS on peripheral blood cell DNA from the same patients.
The researchers found that NGS of cell-free DNA confirmed the diagnosis of MDS in all 16 patients.
In addition, NGS of cell-free DNA revealed abnormalities in 5 patients (31%) that were not detected by Sanger sequencing of peripheral blood cell DNA.
NGS of peripheral blood cell DNA produced the same results as NGS of cell-free DNA for 4 of the 5 patients. However, NGS of peripheral blood cell DNA did not detect a mutation in the RUNX1 gene that was evident in cell-free DNA from 1 patient.
Overall, the researchers found that mutant allele frequency was significantly higher in cell-free DNA than cellular DNA (P=0.008).
The team therefore concluded that cell-free DNA is more reliable than peripheral blood cell DNA for detecting molecular abnormalities in patients with MDS, and NGS is more accurate than Sanger sequencing. ![]()