User login
Docs prescribe drugs despite possible interaction

Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Overcoming drug resistance in malaria
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
Serum Vitamin D Levels, Atopy Not Significantly Linked
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.

Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
AT THE 2016 SID ANNUAL MEETING

Serum vitamin D levels, atopy not significantly linked
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.

Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
AT THE 2016 SID ANNUAL MEETING
Key clinical point: Serum vitamin D was not a significant marker for pediatric atopic dermatitis or disease severity.
Major finding: The average serum vitamin D level was lower among patients with atopic dermatitis than healthy children, but the difference did not reach statistical significance.
Data source: A single-center retrospective review of electronic medical records from 655 patients aged 21 years and younger (average age, 10 years).
Disclosures: Ms. Darji reported no funding sources and had no disclosures.

MCIs and the Orlando Nightclub Shooting
While most Americans were still reacting in horror and disbelief to news of a mass shooting at the Pulse nightclub in Orlando, Florida in the early morning hours of June 12, 2016, the thoughts of most emergency physicians (EPs) were probably focused on the ongoing efforts to save as many victims as possible: What was the closest Level 1 trauma center, and how deep was the ED staffing there that night? Was there a need for additional resources, and perhaps even, could they get there in time to help? In this issue of Emergency Medicine (EM), Residency Program Director Salvatore Silvestri, MD, and his emergency medicine colleagues masterfully recount events from that night as they unfolded, both at the scene and two blocks away at the Orlando Regional Medical Center (ORMC) ED, in the minutes and hours following the first reported shootings.
Being prepared for a mass-casualty incident (MCI) is an extraordinarily expensive requirement of a hospital: It must acquire and maintain adequate resources and communication capabilities; periodically perform unannounced drills that disrupt other hospital activities; and ensure that all EPs and staff are not only able to perform their own day-to-day roles as attending physicians, residents, nurses, etc, but are also capable of taking on even greater responsibilities during an MCI—depending on the day and time it occurs. As you will read in the pages that follow, in the early morning hours of June 12, many of the practiced exercises and rehearsed procedures proved useful, even life-saving, while others had to be discarded or ignored in favor of improvised solutions to rapidly transport and treat the large number of victims with unanticipated needs, under unique conditions.
In any MCI, saving the largest number of victims invariably depends on rapidly instituting some deviations from standard operating procedures (SOPs) and standards of care (SOCs). As described by the ORMC EP authors, “...law enforcement vehicles and ambulances would make the two-block drive from the scene to ORMC carrying as many patients as they safely could, and return immediately after offload….minimal interventions were performed and unlike standard procedure, EMS could offer no prearrival report to the hospital.”
Deviations from SOPs and SOCs during an MCI are not confined to prehospital care, but often extend into the ED and beyond. Most difficult for an EP participating in an MCI is the moment of realization that the sheer number of seriously injured and dying patients arriving en masse mandates a change from “ED triage” to “battlefield triage.” As Dr Ponder recalled, “One of the first few patients I saw was pulseless, and as I went to start chest compressions, I was stopped by a trauma surgeon who said, ‘He’s gone, focus on the ones we can help.’”
In EDs, dying patients are attended to first with extensive staff and resources, while, as a result, less seriously ill patients must sometimes wait longer for care. The opposite is true on the battlefield, where near-death victims of lethal injuries are provided only with comfort care, at most, in order to save those who have a chance of surviving their extensive or serious injuries.
For hospitals and staff, the costs incurred by disaster preparedness are great and invariably exceed government funding provided for these efforts, but how much greater would be the human toll from an MCI without such efforts? All EPs should be proud of what our colleagues at ORMC accomplished on June 12. Though most EPs may never have to deal directly with an MCI, the Orlando nightclub shooting incident was only one of the latest MCIs, certainly not one of the last.
Previous editorials on MCIs may be found in EM September 2006 (9/11), June 2011 (first responders), September 2011 (9/11), November 2011 (surge capacity), and September 2012 (surge capacity/Hurricane Sandy).
While most Americans were still reacting in horror and disbelief to news of a mass shooting at the Pulse nightclub in Orlando, Florida in the early morning hours of June 12, 2016, the thoughts of most emergency physicians (EPs) were probably focused on the ongoing efforts to save as many victims as possible: What was the closest Level 1 trauma center, and how deep was the ED staffing there that night? Was there a need for additional resources, and perhaps even, could they get there in time to help? In this issue of Emergency Medicine (EM), Residency Program Director Salvatore Silvestri, MD, and his emergency medicine colleagues masterfully recount events from that night as they unfolded, both at the scene and two blocks away at the Orlando Regional Medical Center (ORMC) ED, in the minutes and hours following the first reported shootings.
Being prepared for a mass-casualty incident (MCI) is an extraordinarily expensive requirement of a hospital: It must acquire and maintain adequate resources and communication capabilities; periodically perform unannounced drills that disrupt other hospital activities; and ensure that all EPs and staff are not only able to perform their own day-to-day roles as attending physicians, residents, nurses, etc, but are also capable of taking on even greater responsibilities during an MCI—depending on the day and time it occurs. As you will read in the pages that follow, in the early morning hours of June 12, many of the practiced exercises and rehearsed procedures proved useful, even life-saving, while others had to be discarded or ignored in favor of improvised solutions to rapidly transport and treat the large number of victims with unanticipated needs, under unique conditions.
In any MCI, saving the largest number of victims invariably depends on rapidly instituting some deviations from standard operating procedures (SOPs) and standards of care (SOCs). As described by the ORMC EP authors, “...law enforcement vehicles and ambulances would make the two-block drive from the scene to ORMC carrying as many patients as they safely could, and return immediately after offload….minimal interventions were performed and unlike standard procedure, EMS could offer no prearrival report to the hospital.”
Deviations from SOPs and SOCs during an MCI are not confined to prehospital care, but often extend into the ED and beyond. Most difficult for an EP participating in an MCI is the moment of realization that the sheer number of seriously injured and dying patients arriving en masse mandates a change from “ED triage” to “battlefield triage.” As Dr Ponder recalled, “One of the first few patients I saw was pulseless, and as I went to start chest compressions, I was stopped by a trauma surgeon who said, ‘He’s gone, focus on the ones we can help.’”
In EDs, dying patients are attended to first with extensive staff and resources, while, as a result, less seriously ill patients must sometimes wait longer for care. The opposite is true on the battlefield, where near-death victims of lethal injuries are provided only with comfort care, at most, in order to save those who have a chance of surviving their extensive or serious injuries.
For hospitals and staff, the costs incurred by disaster preparedness are great and invariably exceed government funding provided for these efforts, but how much greater would be the human toll from an MCI without such efforts? All EPs should be proud of what our colleagues at ORMC accomplished on June 12. Though most EPs may never have to deal directly with an MCI, the Orlando nightclub shooting incident was only one of the latest MCIs, certainly not one of the last.
Previous editorials on MCIs may be found in EM September 2006 (9/11), June 2011 (first responders), September 2011 (9/11), November 2011 (surge capacity), and September 2012 (surge capacity/Hurricane Sandy).
While most Americans were still reacting in horror and disbelief to news of a mass shooting at the Pulse nightclub in Orlando, Florida in the early morning hours of June 12, 2016, the thoughts of most emergency physicians (EPs) were probably focused on the ongoing efforts to save as many victims as possible: What was the closest Level 1 trauma center, and how deep was the ED staffing there that night? Was there a need for additional resources, and perhaps even, could they get there in time to help? In this issue of Emergency Medicine (EM), Residency Program Director Salvatore Silvestri, MD, and his emergency medicine colleagues masterfully recount events from that night as they unfolded, both at the scene and two blocks away at the Orlando Regional Medical Center (ORMC) ED, in the minutes and hours following the first reported shootings.
Being prepared for a mass-casualty incident (MCI) is an extraordinarily expensive requirement of a hospital: It must acquire and maintain adequate resources and communication capabilities; periodically perform unannounced drills that disrupt other hospital activities; and ensure that all EPs and staff are not only able to perform their own day-to-day roles as attending physicians, residents, nurses, etc, but are also capable of taking on even greater responsibilities during an MCI—depending on the day and time it occurs. As you will read in the pages that follow, in the early morning hours of June 12, many of the practiced exercises and rehearsed procedures proved useful, even life-saving, while others had to be discarded or ignored in favor of improvised solutions to rapidly transport and treat the large number of victims with unanticipated needs, under unique conditions.
In any MCI, saving the largest number of victims invariably depends on rapidly instituting some deviations from standard operating procedures (SOPs) and standards of care (SOCs). As described by the ORMC EP authors, “...law enforcement vehicles and ambulances would make the two-block drive from the scene to ORMC carrying as many patients as they safely could, and return immediately after offload….minimal interventions were performed and unlike standard procedure, EMS could offer no prearrival report to the hospital.”
Deviations from SOPs and SOCs during an MCI are not confined to prehospital care, but often extend into the ED and beyond. Most difficult for an EP participating in an MCI is the moment of realization that the sheer number of seriously injured and dying patients arriving en masse mandates a change from “ED triage” to “battlefield triage.” As Dr Ponder recalled, “One of the first few patients I saw was pulseless, and as I went to start chest compressions, I was stopped by a trauma surgeon who said, ‘He’s gone, focus on the ones we can help.’”
In EDs, dying patients are attended to first with extensive staff and resources, while, as a result, less seriously ill patients must sometimes wait longer for care. The opposite is true on the battlefield, where near-death victims of lethal injuries are provided only with comfort care, at most, in order to save those who have a chance of surviving their extensive or serious injuries.
For hospitals and staff, the costs incurred by disaster preparedness are great and invariably exceed government funding provided for these efforts, but how much greater would be the human toll from an MCI without such efforts? All EPs should be proud of what our colleagues at ORMC accomplished on June 12. Though most EPs may never have to deal directly with an MCI, the Orlando nightclub shooting incident was only one of the latest MCIs, certainly not one of the last.
Previous editorials on MCIs may be found in EM September 2006 (9/11), June 2011 (first responders), September 2011 (9/11), November 2011 (surge capacity), and September 2012 (surge capacity/Hurricane Sandy).

Pediatric Dermatology Consult - August 2016
Dr. Catalina Matiz and David Ginsberg describe the diagnosis and treatment of discoid lupus erythematosus in children.
Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is a relatively common form of chronic cutaneous lupus, although its presentation in children is rare. The typical presentation of DLE is well-circumscribed, indurated, sometimes scaly round or oval plaques with pigmentary change, often red to purple in color. DLE also is clinically associated with telangiectasia, scarring, and follicular plugging, which has a characteristic appearance of “carpet tacking” beneath the scale.
When left untreated, these lesions may result in areas of long-term hypo- or hyperpigmentation, as well as atrophy and scarring.1 These cutaneous manifestations often are exacerbated by UV light exposure. This is particularly problematic because DLE most often affects the face, although lesions also can be found on the scalp, ears, trunk, extremities, and in the mouth as well.
Currently, there are few studies looking specifically at DLE in children and, based on the studies that have been done, there appear to be several important differences between the adult and pediatric populations. DLE affects women more than twice as much as men in the adult population, but reports vary as to whether this female predominance carries over to affected children.2,3 Adults with DLE rarely have a family history of systemic lupus erythematosus (SLE), with rates reported between 1% and 4.4%. In children, however, the reported rates of family history increase tenfold to 11%-40%.2
One study showed that children with DLE progress to SLE at a rate of 23.5%-26%, which is higher than reported rates in adults of 5%-20%.2,4 For this reason, repeated laboratory studies are essential in the follow-up of children diagnosed with DLE, given the possibility of a transition to systemic disease, particularly within the first year of diagnosis. Disseminated lesions can be a red flag for future progression to SLE.2
Differential diagnosis
The differential diagnosis of infiltrated annular lesions on the face and ears should include conditions such as tinea faciae, seborrheic dermatitis, granuloma annulare, cutaneous lymphoma, sarcoidosis, and leishmaniasis. When the lesions are present on the ear or nose, as in the case of this patient, relapsing polychonritis also may be considered.
Although histology plays a large part in diagnosing DLE, clinical presentation and recognizing the need for a biopsy are important.5 Tinea faciei, a fungal infection of the face, can present as erythematous scaly plaques.6 A thorough history and physical exam are important in differentiating tinea faciei from DLE because the former often begins as a small scaly papule that annularly expands outward to form larger plaque with scale around the outer rim, as opposed to DLE, which may have an adherent scale across the entire plaque.5 A potassium hydroxide (KOH) preparation of scraped scale from one of the lesions or a fungal culture can confirm the diagnosis of tinea faciei.6
Seborrheic dermatitis can be localized to the face and scalp and presents with greasy yellow scales. Early lesions of DLE can be difficult to differentiate from seborrheic dermatitis.
Infiltrated annular lesions on the face may represent granulomatous conditions such as granuloma annulare or sarcoidosis, but these lesions usually lack the presence of scale that can be seen in DLE.
Relapsing polychondritis presents as intermittent episodes of cartilage inflammation, usually affecting the cartilage of the ear, nose, and respiratory tract. Areas affected do not show changes on the surface of the skin as it occurs in DLE lesions.
As mentioned above, family history of SLE could indicate a potential for DLE in a small percentage of patients, but the clinical feature of the scaly plaques with the carpet tacking underneath the scale, caused by follicular plugging, is helpful in making the diagnosis clinically. Ultimately, the best way to differentiate anything resembling DLE is through histology and direct immunofluorescence (DIF). Histologic findings in DLE include epidermal atrophy, basal membrane cell vacuolization, hyperkeratosis, parakeratosis, corneal plugs, pseudoscysts, and acanthosis.3 The lupus band test is done using DIF and is a widely used tool for making the diagnosis of DLE based on the distribution of immunoglobulin deposition in the basement membrane zone.7
Treatment
Without timely diagnosis and treatment of DLE, the lesions can progress to scarring and atrophy, leading to a decreased quality of life. UV light exposure and smoking can exacerbate DLE, so sun protection and smoking cessation are both recommended in patients with DLE, although, admittedly, the latter is less relevant in the pediatric population.1 Topical, intralesional, or systemic corticosteroids, with or without antimalarials, are the first line therapy for the management of DLE.1 For refractory cases, some reports document the use of topical calcineurin inhibitors, dapsone, methotrexate, and topical or systemic retinoids.1 For severe cases, intravenous immunoglobulin, ustekinumab, and rituximab also may be used.1
References
- Dermatol Ther. 2016 Apr 12 Epub.
- Pediatr Dermatol. 2008 Mar-Apr;25(2):163-7.
- Pediatr Dermatol. 2003 Mar-Apr;20(2):103-7.
- J Am Acad Dermatol. 2015 Apr;72(4):628-33.
- Pediatr Dermatol. 2016 Mar-Apr;33(2):200-8.
- Pediatr Clin North Am. 2014 Apr;61(2):443-55.
- Am J Dermatopathol. 2016 Feb;38(2):121-3.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
Dr. Catalina Matiz and David Ginsberg describe the diagnosis and treatment of discoid lupus erythematosus in children.
Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is a relatively common form of chronic cutaneous lupus, although its presentation in children is rare. The typical presentation of DLE is well-circumscribed, indurated, sometimes scaly round or oval plaques with pigmentary change, often red to purple in color. DLE also is clinically associated with telangiectasia, scarring, and follicular plugging, which has a characteristic appearance of “carpet tacking” beneath the scale.
When left untreated, these lesions may result in areas of long-term hypo- or hyperpigmentation, as well as atrophy and scarring.1 These cutaneous manifestations often are exacerbated by UV light exposure. This is particularly problematic because DLE most often affects the face, although lesions also can be found on the scalp, ears, trunk, extremities, and in the mouth as well.
Currently, there are few studies looking specifically at DLE in children and, based on the studies that have been done, there appear to be several important differences between the adult and pediatric populations. DLE affects women more than twice as much as men in the adult population, but reports vary as to whether this female predominance carries over to affected children.2,3 Adults with DLE rarely have a family history of systemic lupus erythematosus (SLE), with rates reported between 1% and 4.4%. In children, however, the reported rates of family history increase tenfold to 11%-40%.2
One study showed that children with DLE progress to SLE at a rate of 23.5%-26%, which is higher than reported rates in adults of 5%-20%.2,4 For this reason, repeated laboratory studies are essential in the follow-up of children diagnosed with DLE, given the possibility of a transition to systemic disease, particularly within the first year of diagnosis. Disseminated lesions can be a red flag for future progression to SLE.2
Differential diagnosis
The differential diagnosis of infiltrated annular lesions on the face and ears should include conditions such as tinea faciae, seborrheic dermatitis, granuloma annulare, cutaneous lymphoma, sarcoidosis, and leishmaniasis. When the lesions are present on the ear or nose, as in the case of this patient, relapsing polychonritis also may be considered.
Although histology plays a large part in diagnosing DLE, clinical presentation and recognizing the need for a biopsy are important.5 Tinea faciei, a fungal infection of the face, can present as erythematous scaly plaques.6 A thorough history and physical exam are important in differentiating tinea faciei from DLE because the former often begins as a small scaly papule that annularly expands outward to form larger plaque with scale around the outer rim, as opposed to DLE, which may have an adherent scale across the entire plaque.5 A potassium hydroxide (KOH) preparation of scraped scale from one of the lesions or a fungal culture can confirm the diagnosis of tinea faciei.6
Seborrheic dermatitis can be localized to the face and scalp and presents with greasy yellow scales. Early lesions of DLE can be difficult to differentiate from seborrheic dermatitis.
Infiltrated annular lesions on the face may represent granulomatous conditions such as granuloma annulare or sarcoidosis, but these lesions usually lack the presence of scale that can be seen in DLE.
Relapsing polychondritis presents as intermittent episodes of cartilage inflammation, usually affecting the cartilage of the ear, nose, and respiratory tract. Areas affected do not show changes on the surface of the skin as it occurs in DLE lesions.
As mentioned above, family history of SLE could indicate a potential for DLE in a small percentage of patients, but the clinical feature of the scaly plaques with the carpet tacking underneath the scale, caused by follicular plugging, is helpful in making the diagnosis clinically. Ultimately, the best way to differentiate anything resembling DLE is through histology and direct immunofluorescence (DIF). Histologic findings in DLE include epidermal atrophy, basal membrane cell vacuolization, hyperkeratosis, parakeratosis, corneal plugs, pseudoscysts, and acanthosis.3 The lupus band test is done using DIF and is a widely used tool for making the diagnosis of DLE based on the distribution of immunoglobulin deposition in the basement membrane zone.7
Treatment
Without timely diagnosis and treatment of DLE, the lesions can progress to scarring and atrophy, leading to a decreased quality of life. UV light exposure and smoking can exacerbate DLE, so sun protection and smoking cessation are both recommended in patients with DLE, although, admittedly, the latter is less relevant in the pediatric population.1 Topical, intralesional, or systemic corticosteroids, with or without antimalarials, are the first line therapy for the management of DLE.1 For refractory cases, some reports document the use of topical calcineurin inhibitors, dapsone, methotrexate, and topical or systemic retinoids.1 For severe cases, intravenous immunoglobulin, ustekinumab, and rituximab also may be used.1
References
- Dermatol Ther. 2016 Apr 12 Epub.
- Pediatr Dermatol. 2008 Mar-Apr;25(2):163-7.
- Pediatr Dermatol. 2003 Mar-Apr;20(2):103-7.
- J Am Acad Dermatol. 2015 Apr;72(4):628-33.
- Pediatr Dermatol. 2016 Mar-Apr;33(2):200-8.
- Pediatr Clin North Am. 2014 Apr;61(2):443-55.
- Am J Dermatopathol. 2016 Feb;38(2):121-3.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
Dr. Catalina Matiz and David Ginsberg describe the diagnosis and treatment of discoid lupus erythematosus in children.
Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is a relatively common form of chronic cutaneous lupus, although its presentation in children is rare. The typical presentation of DLE is well-circumscribed, indurated, sometimes scaly round or oval plaques with pigmentary change, often red to purple in color. DLE also is clinically associated with telangiectasia, scarring, and follicular plugging, which has a characteristic appearance of “carpet tacking” beneath the scale.
When left untreated, these lesions may result in areas of long-term hypo- or hyperpigmentation, as well as atrophy and scarring.1 These cutaneous manifestations often are exacerbated by UV light exposure. This is particularly problematic because DLE most often affects the face, although lesions also can be found on the scalp, ears, trunk, extremities, and in the mouth as well.
Currently, there are few studies looking specifically at DLE in children and, based on the studies that have been done, there appear to be several important differences between the adult and pediatric populations. DLE affects women more than twice as much as men in the adult population, but reports vary as to whether this female predominance carries over to affected children.2,3 Adults with DLE rarely have a family history of systemic lupus erythematosus (SLE), with rates reported between 1% and 4.4%. In children, however, the reported rates of family history increase tenfold to 11%-40%.2
One study showed that children with DLE progress to SLE at a rate of 23.5%-26%, which is higher than reported rates in adults of 5%-20%.2,4 For this reason, repeated laboratory studies are essential in the follow-up of children diagnosed with DLE, given the possibility of a transition to systemic disease, particularly within the first year of diagnosis. Disseminated lesions can be a red flag for future progression to SLE.2
Differential diagnosis
The differential diagnosis of infiltrated annular lesions on the face and ears should include conditions such as tinea faciae, seborrheic dermatitis, granuloma annulare, cutaneous lymphoma, sarcoidosis, and leishmaniasis. When the lesions are present on the ear or nose, as in the case of this patient, relapsing polychonritis also may be considered.
Although histology plays a large part in diagnosing DLE, clinical presentation and recognizing the need for a biopsy are important.5 Tinea faciei, a fungal infection of the face, can present as erythematous scaly plaques.6 A thorough history and physical exam are important in differentiating tinea faciei from DLE because the former often begins as a small scaly papule that annularly expands outward to form larger plaque with scale around the outer rim, as opposed to DLE, which may have an adherent scale across the entire plaque.5 A potassium hydroxide (KOH) preparation of scraped scale from one of the lesions or a fungal culture can confirm the diagnosis of tinea faciei.6
Seborrheic dermatitis can be localized to the face and scalp and presents with greasy yellow scales. Early lesions of DLE can be difficult to differentiate from seborrheic dermatitis.
Infiltrated annular lesions on the face may represent granulomatous conditions such as granuloma annulare or sarcoidosis, but these lesions usually lack the presence of scale that can be seen in DLE.
Relapsing polychondritis presents as intermittent episodes of cartilage inflammation, usually affecting the cartilage of the ear, nose, and respiratory tract. Areas affected do not show changes on the surface of the skin as it occurs in DLE lesions.
As mentioned above, family history of SLE could indicate a potential for DLE in a small percentage of patients, but the clinical feature of the scaly plaques with the carpet tacking underneath the scale, caused by follicular plugging, is helpful in making the diagnosis clinically. Ultimately, the best way to differentiate anything resembling DLE is through histology and direct immunofluorescence (DIF). Histologic findings in DLE include epidermal atrophy, basal membrane cell vacuolization, hyperkeratosis, parakeratosis, corneal plugs, pseudoscysts, and acanthosis.3 The lupus band test is done using DIF and is a widely used tool for making the diagnosis of DLE based on the distribution of immunoglobulin deposition in the basement membrane zone.7
Treatment
Without timely diagnosis and treatment of DLE, the lesions can progress to scarring and atrophy, leading to a decreased quality of life. UV light exposure and smoking can exacerbate DLE, so sun protection and smoking cessation are both recommended in patients with DLE, although, admittedly, the latter is less relevant in the pediatric population.1 Topical, intralesional, or systemic corticosteroids, with or without antimalarials, are the first line therapy for the management of DLE.1 For refractory cases, some reports document the use of topical calcineurin inhibitors, dapsone, methotrexate, and topical or systemic retinoids.1 For severe cases, intravenous immunoglobulin, ustekinumab, and rituximab also may be used.1
References
- Dermatol Ther. 2016 Apr 12 Epub.
- Pediatr Dermatol. 2008 Mar-Apr;25(2):163-7.
- Pediatr Dermatol. 2003 Mar-Apr;20(2):103-7.
- J Am Acad Dermatol. 2015 Apr;72(4):628-33.
- Pediatr Dermatol. 2016 Mar-Apr;33(2):200-8.
- Pediatr Clin North Am. 2014 Apr;61(2):443-55.
- Am J Dermatopathol. 2016 Feb;38(2):121-3.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California, San Diego, and Mr. Ginsberg is a research associate at the hospital. Dr. Matiz and Mr. Ginsberg said they have no relevant financial disclosures.
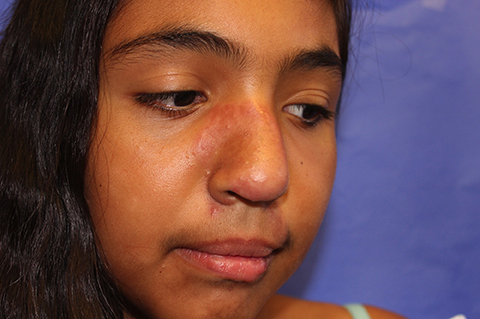
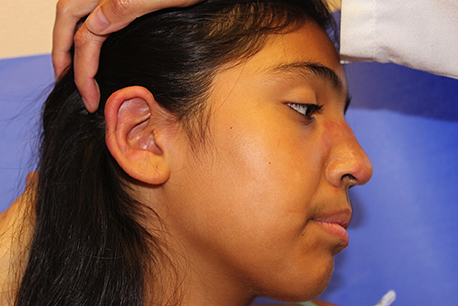
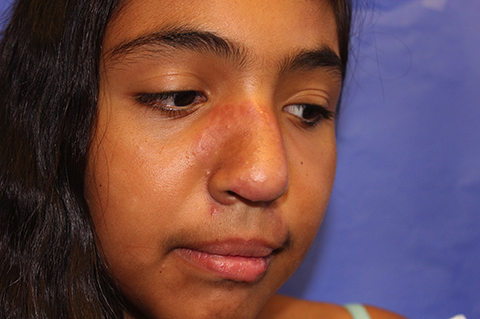
The patient is a well-appearing adolescent in no acute distress, but complaining of continued mild pain of her ear. Upon inspection, she has pink and violaceous indurated annular plaques on her right nasal sidewall and cheek. There is a pink edematous plaque on her right helix and an indurated plaque on the left upper cutaneous lip. The lesions are limited to her face, with no scalp or oral mucosal involvement. Her nails and hair are unaffected.
Testosterone might counteract chemotherapy heart damage
SEATTLE – Adjunct testosterone improved short-term cardiac function in head, neck, and cervical cancer patients undergoing standard treatment in a small randomized trial from the University of Texas Medical Branch, Galveston.
The finding suggests that testosterone might counteract the cardiotoxic effects of chemotherapy, reducing “the incidence of chemotherapy-induced remodeling. It might also have rehabilitation implications and make patients better surgical candidates. Further investigation is warranted,” said investigator Albert Chamberlain, MD, an endocrine research fellow at the university. Although the results were positive, follow-up was short; years-long data are needed to know if testosterone really protects the heart from chemotherapy damage.

Dr. Chamberlain’s team looked into the issue because “many current chemotherapy drug classes have cardiotoxicity that progresses subclinically for a long time” before problems emerge. “Testosterone is known to cause vasodilation in both large and resistance arteries,” which might help prevent damage. “With that in mind, we decided to” investigate testosterone’s impact on cardiac performance during chemotherapy, he said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
Five women and one man were randomized to weekly intramuscular 100 mg testosterone injections for 7 weeks; six men and four women were randomized to placebo injections. They were all recently diagnosed with stage IIIB, IV, or recurrent head and neck cancer, or cervical cancer, and were undergoing concomitant standard-of-care chemotherapy or chemoradiation. Cardiac function was measured blindly by transthoracic echocardiogram at baseline and the end of the study.
The testosterone group had significantly improved stroke volumes (+18.2% versus –2.6%, P = 0.01), ejection fractions (+6.2% versus –1.8%, P = 0.02), and cardiac output (+1402.2 mL/min versus –16.8mL/min, P = 0.011). Heart rate, arterial pressure, end-diastolic volume, and end-systolic volume remained unchanged in both groups, so the improved systolic function was attributed to reduced vascular resistance in the testosterone group (–26.5% versus +3.9% in the placebo group, P = 0.001).
Systolic improvements remained as cardiac index increased 27.6% in the testosterone group versus 2.8% in the placebo group. Testosterone didn’t seem to have any negative impacts on diastolic function. A placebo patient had a stroke, but there were no other adverse events in the study.
Although improved stroke volume is likely due to the reduced afterload, “increased contractility cannot be eliminated as a potential contributing factor. End diastolic volume remained unchanged in both groups, [suggesting] that preload is unlikely to be the mechanism for increased stroke volume,” Dr. Chamberlain said.
This study was funded by the National Cancer Institute. Dr. Chamberlain reported having no relevant disclosures.

|
Dr. Benjamin Judson |
The data are promising but preliminary for a problem we see a lot, chemotherapy-induced cardiotoxicity that presents years after treatment. We have to be really careful before we give testosterone to anyone who is under active treatment for cancer, because I don’t think we really know if it’s safe.
Benjamin Judson, MD, is an associate professor of otolaryngologic surgery at Yale Medical School in New Haven, Conn. He moderated Dr. Chamberlain’s talk and was not involved in the study.
|
|
Dr. Benjamin Judson |
The data are promising but preliminary for a problem we see a lot, chemotherapy-induced cardiotoxicity that presents years after treatment. We have to be really careful before we give testosterone to anyone who is under active treatment for cancer, because I don’t think we really know if it’s safe.
Benjamin Judson, MD, is an associate professor of otolaryngologic surgery at Yale Medical School in New Haven, Conn. He moderated Dr. Chamberlain’s talk and was not involved in the study.
|
|
Dr. Benjamin Judson |
The data are promising but preliminary for a problem we see a lot, chemotherapy-induced cardiotoxicity that presents years after treatment. We have to be really careful before we give testosterone to anyone who is under active treatment for cancer, because I don’t think we really know if it’s safe.
Benjamin Judson, MD, is an associate professor of otolaryngologic surgery at Yale Medical School in New Haven, Conn. He moderated Dr. Chamberlain’s talk and was not involved in the study.
SEATTLE – Adjunct testosterone improved short-term cardiac function in head, neck, and cervical cancer patients undergoing standard treatment in a small randomized trial from the University of Texas Medical Branch, Galveston.
The finding suggests that testosterone might counteract the cardiotoxic effects of chemotherapy, reducing “the incidence of chemotherapy-induced remodeling. It might also have rehabilitation implications and make patients better surgical candidates. Further investigation is warranted,” said investigator Albert Chamberlain, MD, an endocrine research fellow at the university. Although the results were positive, follow-up was short; years-long data are needed to know if testosterone really protects the heart from chemotherapy damage.
Dr. Chamberlain’s team looked into the issue because “many current chemotherapy drug classes have cardiotoxicity that progresses subclinically for a long time” before problems emerge. “Testosterone is known to cause vasodilation in both large and resistance arteries,” which might help prevent damage. “With that in mind, we decided to” investigate testosterone’s impact on cardiac performance during chemotherapy, he said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
Five women and one man were randomized to weekly intramuscular 100 mg testosterone injections for 7 weeks; six men and four women were randomized to placebo injections. They were all recently diagnosed with stage IIIB, IV, or recurrent head and neck cancer, or cervical cancer, and were undergoing concomitant standard-of-care chemotherapy or chemoradiation. Cardiac function was measured blindly by transthoracic echocardiogram at baseline and the end of the study.
The testosterone group had significantly improved stroke volumes (+18.2% versus –2.6%, P = 0.01), ejection fractions (+6.2% versus –1.8%, P = 0.02), and cardiac output (+1402.2 mL/min versus –16.8mL/min, P = 0.011). Heart rate, arterial pressure, end-diastolic volume, and end-systolic volume remained unchanged in both groups, so the improved systolic function was attributed to reduced vascular resistance in the testosterone group (–26.5% versus +3.9% in the placebo group, P = 0.001).
Systolic improvements remained as cardiac index increased 27.6% in the testosterone group versus 2.8% in the placebo group. Testosterone didn’t seem to have any negative impacts on diastolic function. A placebo patient had a stroke, but there were no other adverse events in the study.
Although improved stroke volume is likely due to the reduced afterload, “increased contractility cannot be eliminated as a potential contributing factor. End diastolic volume remained unchanged in both groups, [suggesting] that preload is unlikely to be the mechanism for increased stroke volume,” Dr. Chamberlain said.
This study was funded by the National Cancer Institute. Dr. Chamberlain reported having no relevant disclosures.
SEATTLE – Adjunct testosterone improved short-term cardiac function in head, neck, and cervical cancer patients undergoing standard treatment in a small randomized trial from the University of Texas Medical Branch, Galveston.
The finding suggests that testosterone might counteract the cardiotoxic effects of chemotherapy, reducing “the incidence of chemotherapy-induced remodeling. It might also have rehabilitation implications and make patients better surgical candidates. Further investigation is warranted,” said investigator Albert Chamberlain, MD, an endocrine research fellow at the university. Although the results were positive, follow-up was short; years-long data are needed to know if testosterone really protects the heart from chemotherapy damage.
Dr. Chamberlain’s team looked into the issue because “many current chemotherapy drug classes have cardiotoxicity that progresses subclinically for a long time” before problems emerge. “Testosterone is known to cause vasodilation in both large and resistance arteries,” which might help prevent damage. “With that in mind, we decided to” investigate testosterone’s impact on cardiac performance during chemotherapy, he said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
Five women and one man were randomized to weekly intramuscular 100 mg testosterone injections for 7 weeks; six men and four women were randomized to placebo injections. They were all recently diagnosed with stage IIIB, IV, or recurrent head and neck cancer, or cervical cancer, and were undergoing concomitant standard-of-care chemotherapy or chemoradiation. Cardiac function was measured blindly by transthoracic echocardiogram at baseline and the end of the study.
The testosterone group had significantly improved stroke volumes (+18.2% versus –2.6%, P = 0.01), ejection fractions (+6.2% versus –1.8%, P = 0.02), and cardiac output (+1402.2 mL/min versus –16.8mL/min, P = 0.011). Heart rate, arterial pressure, end-diastolic volume, and end-systolic volume remained unchanged in both groups, so the improved systolic function was attributed to reduced vascular resistance in the testosterone group (–26.5% versus +3.9% in the placebo group, P = 0.001).
Systolic improvements remained as cardiac index increased 27.6% in the testosterone group versus 2.8% in the placebo group. Testosterone didn’t seem to have any negative impacts on diastolic function. A placebo patient had a stroke, but there were no other adverse events in the study.
Although improved stroke volume is likely due to the reduced afterload, “increased contractility cannot be eliminated as a potential contributing factor. End diastolic volume remained unchanged in both groups, [suggesting] that preload is unlikely to be the mechanism for increased stroke volume,” Dr. Chamberlain said.
This study was funded by the National Cancer Institute. Dr. Chamberlain reported having no relevant disclosures.
AT AHNS 2016
Key clinical point: Adjunct testosterone improved cardiac function in head, neck, and cervical cancer patients undergoing standard treatment in a small randomized trial.
Major finding: The testosterone group had significantly improved stroke volumes (+18.2% versus –2.6%, P = 0.01), ejection fractions (+6.2% versus –1.8%, P = 0.02), and cardiac output (+1402.2 mL/min versus –16.8mL/min, P = 0.011), but there was no years-long follow-up to show lasting cardiac benefit.
Data source: Randomized trial with 16 patients.
Disclosures: The National Cancer Institute funded the work. The lead investigator had no disclosures.

PHM16: Tips on Meeting Needs of Children with a Medical Complexity
Presenters: Mary L Ehlenbach, MD, FAAP; Megan Z Cardoso, MD, FAAP; and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex care program. Presenters opened with a discussion of how to define children with medical complexity going through a variety of different methods including some research based aggregation of ICD-10 codes, referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about 1/3 of healthcare spending and due to advances in technology and medicine this group of children is growing in numbers. This group makes up about 10% of all pediatric admissions.
The session then went on to break down into four small groups which focused on details about how to create a complex care program and how to evaluate effectiveness of the program. Group 1 discussed methods of identifying patients that the program will serve. This included setting guidelines if a certain group or diagnosis should be excluded from the program. Different models of what services were also discussed which ranged from providing a comprehensive medical home to inpatient consults or care coordination services. The second group focused on what team members it may be beneficial to have involved. Team composition varied widely usually including MDs, NPs, social workers, RNs and at times a documentation expert who could aid with proper billing to boost revenue. The third group focused on how to measure quality services including family surveys of quantitative impact and satisfaction, PCP satisfaction. The final session consisted of the business and financial considerations of beginning a complex care program.
Key Takeaways:
- Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
- When working to initiate a complex care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are non-clinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children's National Medical Center in Washington D.C.

Presenters: Mary L Ehlenbach, MD, FAAP; Megan Z Cardoso, MD, FAAP; and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex care program. Presenters opened with a discussion of how to define children with medical complexity going through a variety of different methods including some research based aggregation of ICD-10 codes, referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about 1/3 of healthcare spending and due to advances in technology and medicine this group of children is growing in numbers. This group makes up about 10% of all pediatric admissions.
The session then went on to break down into four small groups which focused on details about how to create a complex care program and how to evaluate effectiveness of the program. Group 1 discussed methods of identifying patients that the program will serve. This included setting guidelines if a certain group or diagnosis should be excluded from the program. Different models of what services were also discussed which ranged from providing a comprehensive medical home to inpatient consults or care coordination services. The second group focused on what team members it may be beneficial to have involved. Team composition varied widely usually including MDs, NPs, social workers, RNs and at times a documentation expert who could aid with proper billing to boost revenue. The third group focused on how to measure quality services including family surveys of quantitative impact and satisfaction, PCP satisfaction. The final session consisted of the business and financial considerations of beginning a complex care program.
Key Takeaways:
- Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
- When working to initiate a complex care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are non-clinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children's National Medical Center in Washington D.C.
Presenters: Mary L Ehlenbach, MD, FAAP; Megan Z Cardoso, MD, FAAP; and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex care program. Presenters opened with a discussion of how to define children with medical complexity going through a variety of different methods including some research based aggregation of ICD-10 codes, referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about 1/3 of healthcare spending and due to advances in technology and medicine this group of children is growing in numbers. This group makes up about 10% of all pediatric admissions.
The session then went on to break down into four small groups which focused on details about how to create a complex care program and how to evaluate effectiveness of the program. Group 1 discussed methods of identifying patients that the program will serve. This included setting guidelines if a certain group or diagnosis should be excluded from the program. Different models of what services were also discussed which ranged from providing a comprehensive medical home to inpatient consults or care coordination services. The second group focused on what team members it may be beneficial to have involved. Team composition varied widely usually including MDs, NPs, social workers, RNs and at times a documentation expert who could aid with proper billing to boost revenue. The third group focused on how to measure quality services including family surveys of quantitative impact and satisfaction, PCP satisfaction. The final session consisted of the business and financial considerations of beginning a complex care program.
Key Takeaways:
- Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
- When working to initiate a complex care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are non-clinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children's National Medical Center in Washington D.C.

Zika virus update: A rapidly moving target
We recently reviewed the most current information on the epidemiology, clinical manifestations, and diagnosis of maternal and congenital Zika virus (ZV) infection.1 We also offered tentative recommendations for reducing the risk of infection and for managing the treatment of women exposed to the virus.
In this update, we present new information on the broadened spectrum of anomalies now known to be causally related to congenital ZV infection and on the increasing number of serious neurologic complications directly related to ZV infection in adults. We also update recommendations for diagnosing maternal, fetal, and neonatal infection and present guidelines for preventing sexual transmission of ZV infection.
CASE Woman from Brazil gives birth to stillborn baby with microcephaly
A 23-year-old woman (G2P1) recently emigrated from Pernambuco in Brazil to the United States and now presents to the hospital in advanced labor. Based on results of first-trimester ultrasonography performed in Brazil, it is determined that she is at 39 weeks’ gestation. The patient has not had any prenatal care since early in the second trimester because of low income and lack of medical insurance. She reports no serious illness before or during the pregnancy.
In the labor and delivery suite, she rapidly delivers a stillborn female infant—5 pounds 3 ounces, growth restricted, with multiple congenital anomalies. Postmortem examination reveals microcephaly, ventriculomegaly, extensive brain atrophy, intracranial calcifications, cerebellar agenesis, cataracts, ocular calcifications, redundant scalp tissue, and multiple joint contractures.
What is the most likely cause of these multiple anomalies?
The patient’s findings are most consistent with a diagnosis of severe intrauterine infection. Possible pathogenic organisms include rubella virus, cytomegalovirus, lymphocytic choriomeningitis virus, toxoplasmosis, and ZV.2 Given the patient’s recent move from Pernambuco in northeastern Brazil, the epicenter of the ZV epidemic in the Americas, the most likely diagnosis is congenital ZV infection.
The initial reports of congenital anomalies associated with ZV infection focused on microcephaly, usually defined as head circumference less than 3 standard deviations below the mean, or less than the third or fifth percentile for gestational age. Subsequent reports have linked many other serious central nervous system (CNS) anomalies to the virus. In a retrospective case series, de Fatima Vasco Aragao and colleagues3 described neuroimaging findings in 23 infants with presumed congenital ZV infection. Of the 22 with computed tomography scans, all had calcifications at the junction of cortical and subcortical white matter, 21 (95%) had disordered cortical development, 20 (91%) had a significant decrease in brain volume, 19 (86%) had ventriculomegaly, and half had distinct hypoplasia of either cerebellum or brainstem. In addition, of the 8 infants with magnetic resonance imaging (MRI) studies, 7 (88%) had an enlarged cisterna magna, 7 (88%) had delayed myelination, 6 (75%) had a simplified gyral pattern, and 3 (38%) had hypoplasia of corpus callosum.
De Paula Freitas and colleagues4 recently found congenital ZV infection associated with severe ocular abnormalities. Comprehensive ophthalmologic examination of 29 infants with microcephaly, presumed caused by congenital ZV infection, revealed 10 (35%) had abnormalities, which included focal pigment mottling, chorioretinal atrophy, hypoplasia and cupping of optic disk, loss of foveal reflex, macular atrophy, lens subluxation, and coloboma of iris.
Other conditions linked to congenital ZV infection include intrauterine growth restriction, redundant scalp tissue, contractures of multiple joints, and clubfoot.2
Bottom line. Although the ocular abnormalities are undetectable by prenatal ultrasound, many of the CNS and skeletal anomalies can be identified antenatally. Therefore, serial ultrasound examinations should be performed on adults who have a clinical illness consistent with ZV infection or who have traveled to an endemic area or have a sexual partner who has been in an endemic area. Patients should be assessed for possible microcephaly, ventriculomegaly, agenesis of corpus callosum, hypoplasia of cerebellum, and skeletal deformities.
Zika virus has been shown to be a direct cause of microcephaly
To make the determination that Zika virus (ZV) causes microcephaly, Rasmussen and colleagues1 very recently evaluated Shepard’s 7 criteria,2 published in 1994, for establishing a cause between a microorganism and a specific clinical condition. These 7 criteria are:
- There must be a proven exposure at one or more critical times during prenatal development.
Rasmussen and colleagues1 pointed to case reports, case series, and epidemiologic studies showing a clear association between ZV exposure and microcephaly. Although exposure at any time during pregnancy may cause congenital infection, exposure in the late first and early second trimesters seems to pose the most risk for severe central nervous system (CNS) injury. - There must be consistent findings in 2 or more high-quality epidemiologic studies.
The studies must control for important confounding variables and include an appropriate number of patients to clearly identify an association between a given exposure and specific fetal anomalies. Rasmussen and colleagues1 cited 2 important epidemiologic studies. The first, a prospective cohort investigation of women in Brazil, found that 29% of those with ZV infection had abnormalities on prenatal ultrasound.3
In the second investigation, a retrospective study of 8 infants in French Polynesia, the mathematical modeling performed by the authors4 suggested microcephaly occurred in 1% of infants born to women with first-trimester ZV infection. Using a different mathematical model, Johansson and colleagues5 found that the risk of fetal microcephaly associated with first-trimester infection may range from as low as 1% to as high as 13%.
Although these studies are helpful in quantifying the risk of congenital infection, they only partially satisfy Shepard’s second criterion. - The suspected microorganism must produce a specific defect or clearly delineated syndrome.
Rasmussen and colleagues1 argued that this criterion has been fulfilled. Zika virus infection causes a distinct phenotype that includes microcephaly, multiple other CNS anomalies, redundant scalp skin, ocular abnormalities, joint contractures (arthrogryposis), and clubfoot.6,7 - The observed birth defect must be associated with a rare environmental exposure.
This criterion also has been met, Rasmussen and colleagues1 reported. They noted that congenital microcephaly is rare in the United States (only about 6 cases in 10,000 liveborn infants) but that the number of cases in Brazil and French Polynesia is much in excess of what would be predicted in the absence of the ZV epidemic. - Teratogenicity should be demonstrated in laboratory animals.
Shepard indicated that this criterion is important but not essential to prove causation. As there is yet no animal model for ZV infection, this criterion has not been fulfilled. - The association between the exposure and the observed anomaly or spectrum of anomalies should be biologically plausible.
Rasmussen and colleagues1 demonstrated that the findings linked to maternal ZV infection are similar to those described for at least 2 other viral pathogens, rubella virus and cytomegalovirus. Animal models also have clearly shown that the ZV is neurotropic. Moreover, ZV has been clearly identified in the brains of infants with microcephaly.8 - Shepard’s seventh criterion relates to a medication or chemical exposure and is not relevant to a microorganism.
References
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Brasil P, Pereira JP Jr, Raja Gabaglia C, et al. Zika virus infection in pregnant women in Rio de Janeiro—preliminary report [published online ahead of print March 4, 2016]. N Engl J Med. doi:10.1056/NEJMoa1602412.
- Cauchemez S, Besnard M, Bompard P, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet. 2016;387(10033):2125–2132.
- Johansson MA, Mier-Y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly [published online ahead of print May 25, 2016; updated June 9, 2016]. N Engl J Med. 2016;375:1–4. doi:10.1056/NEJMp1605367.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika virus. N Engl J Med. 2016;374(16):1552–1563.
- Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374(10):951–958.
Did ZV cause these anomalies?
How certain can we be that the anomalies present in the case patient’s baby were caused by ZV? In the past, and for many years, scientists relied on Koch’s 4 postulates (TABLE 1) to answer this question and establish a causal relationship between a microorganism and a specific clinical disease.5 Koch’s postulates have not been satisfied for the relationship between maternal ZV infection and congenital anomalies. Today’s more relevant standards for determining causality of a teratogen were published in 1994 by Shepard.6 In 2016, Rasmussen and colleagues7 found that the critical components of these criteria are fulfilled and concluded that there is little doubt ZV is a proven and extremely dangerous teratogen. See “Zika virus has been shown to be a direct cause of microcephaly”.

Rasmussen and colleagues7 also used Hill’s criteria to assess the evidence for causation. Hill’s systematic assessment is based on 9 factors (TABLE 2)8, and Rasmussen and colleagues7 concluded that the necessary 7 of these 9 criteria have been met (the experimental animal model criterion was not satisfied, and the biological gradient criterion was not applicable). Given their assessment of Shepard’s criteria,6 the authors argued that the link between maternal ZV infection and severe congenital anomalies has risen from association to well-defined causation.

How should ZV infection be confirmed in adults and newborns?
After our first review was published in March 2016,1 the testing algorithm recommended by the US Centers for Disease Control and Prevention (CDC) was revised.9 Now, according to the CDC, if a patient has had symptoms of ZV infection for less than 5 days, serum and urine should be obtained for reverse transcriptase–polymerase chain reaction (RT-PCR) testing. If symptoms have been present for 5 to 14 days, urine should be tested by RT-PCR because urine samples appear to remain positive for virus longer than serum samples do. If RT-PCR is performed within the appropriate period and the result is negative, ZV infection is excluded; if the result is positive, acute ZV infection is confirmed, and additional testing is not indicated. RT-PCR can be performed by 2 commercial laboratories (Quest Diagnostics and LabCorp), state health departments, and the CDC.
If serum or urine is collected more than 5 days after symptom onset and the RT-PCR result is negative, the patient should have an immunoglobulin M (IgM) assay for ZV. If the assay result is negative, infection is excluded; if the result is positive or equivocal, additional testing is needed to ensure that the presence of the antibody does not reflect a cross-reaction to dengue or chikungunya virus. The confirmatory plaque reduction neutralization test (PRNT) is performed only by the CDC. To be considered positive, the PRNT result must be at least 4-fold higher than the dengue virus neutralizing antibody titer.
In patients with suspected Guillain-Barré syndrome (GBS), RT-PCR can be performed on cerebrospinal fluid. For suspected fetal or neonatal infection, RT-PCR can be performed on amniotic fluid, umbilical cord blood, and fetal and placental tissue.
CASE 2 Nonpregnant woman with possible Zika virus exposure presents to ED with neurologic symptoms
A 31-year-old nulligravid woman presents to the emergency department (ED) for evaluation of numbness, tingling, and weakness in the lower extremities and difficulty walking. She reports having had a low-grade fever and a fine disseminated macular rash 1 week earlier. She denies recent travel and exposure to friends or relatives with illness, but she says her husband travels extensively and was living and working in Puerto Rico. The patient has no other neurologic symptoms.
Serum and cerebrospinal fluid chemistries and MRI findings are normal. However, the ZV IgM assay is positive, and nerve conduction study results are consistent with GBS. The patient is admitted to the hospital, treated with intravenous immunoglobulin and given supportive care. Over 10 days, her neurologic condition gradually improves.
What is the link between ZV infection and serious neurologic complications in adults?
ZV infection has been associated with serious neurologic complications in adults. Investigators in several countries have reported dramatic increases in GBS cases during the ZV outbreak.10
GBS is an acute, immune-mediated, demyelinating peripheral neuropathy that can vary in presentation but most commonly manifests as a rapidly ascending paralysis. The disorder often is preceded by an immunization or live viral infection. In some patients, paralysis severely weakens the respiratory muscles and even the cranial nerves, and affected individuals may require intubation, ventilator support, and parenteral or enteral alimentation.
In a case-control study conducted duringthe 2013–2014 outbreak in French Polynesia, the association between ZV infection and GBS was evaluated in 3 groups of patients: 42 patients with GBS, 98 control patients, and 70 patients with ZV infection but no neurologic complications.11 Symptoms of ZV infection were present in about 88% of the patients with GBS, and the median interval from viral infection to onset of neurologic symptoms was 6 days. The ZV IgM assay was positive in 93% of GBS cases. Nerve conduction study results were consistent with the acute motor axonal neuropathy of GBS. All patients were treated with intravenous immunoglobulin; 38% of patients had to be admitted to the intensive care unit, and 29% needed respiratory support. There were no fatalities. The overall incidence of GBS was 2.4 cases per 10,000 ZV infections.
Other neurologic complications that have been associated with ZV infection are meningoencephalitis,12 brain ischemia,13 and myelitis.14
Bottom line. ZV infection may cause serious neurologic complications in adults. The most devastating complication is GBS, which can result in respiratory muscle paralysis and cranial nerve palsies.
How can patients prevent sexual transmission of ZV infection?
The ZV can be transmitted by sexual contact, including vaginal, anal, and oral sex.15 It is known to persist longer in semen than in blood or urine, though the exact duration remains unknown. Atkinson and colleagues16 reported RT-PCR detection of ZV RNA in semen about 62 days after onset of febrile illness—long after the virus became undetectable in blood.15
Mansuy and colleagues17 found that the viral load in semen was more than 100,000 times that in blood and urine more than 2 weeks after symptom onset.16 The ZV has been detected in saliva, urine, and breast milk. Although it has not been identified in vaginal secretions in humans, it has been detected in the vaginal secretions of nonhuman primates up to 7 days after subcutaneous inoculation of virus.18 In addition, the first case of female-to-male sexual transmission of ZV infection was just reported.19 In this report, transmission seems to have occurred on day 3 of the woman’s symptomatic illness, when she had unprotected vaginal intercourse with her partner. The partner became symptomatic 7 days after sexual exposure. To date, there is no evidence that infection is spread through kissing or breastfeeding.
The most recent recommendations from the CDC are that a man with symptomatic ZV infection wait at least 6 months before having unprotected sexual contact. In addition, a man who is asymptomatic after ZV exposure should wait at least 8 weeks before having unprotected sexual contact.17
A woman planning a pregnancy should know there is no evidence that prior ZV infection increases the risk of birth defects. However, a woman with a proven ZV infection should wait at least 8 weeks after symptom onset before trying to conceive. Even an asymptomatic woman with possible exposure should wait at least 8 weeks after the last exposure before attempting conception. In addition, given the risks associated with maternal and fetal infection, a man who has been exposed to the virus and who has a pregnant partner should abstain from unprotected sexual contact for the duration of the pregnancy.20
Key takeaways
- Zika virus has now been clearly established as the cause of severe fetal malformations, particularly microcephaly.
- The risk of fetal injury appears to be greater when maternal infection occurs in the first trimester of pregnancy.
- Zika virus has now been established as the cause of Guillain-Barré syndrome in adults.
- Although most cases of Zika virus infection are transmitted as the result of mosquito bites, patients can acquire the infection through sexual contact. Both male-to-female and female-to-male transmission have been documented.
- If symptoms have been present for 5 to 14 days, only the urine RT-PCR test should be performed.
- If symptoms have been present for more than 14 days, the patient should have an immunoglobulin M assay for Zika virus. If this test is equivocal or positive, a plaque reduction neutralization test should be performed to exclude infection caused by dengue or chikungunya virus.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28–34.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- de Fatima Vasco Aragao M, van der Linden V, Brainer-Lima AM, et al. Clinical features and neuroimaging (CT and MRI) findings in presumed Zika virus related congenital infection and microcephaly: retrospective case series study. BMJ. 2016;353:i1901.
- de Paula Freitas B, de Oliveira Dias JR, Prazeres J, et al. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil [published online ahead of print February 9, 2016]. JAMA Ophthalmol. doi:10.1001/jamaophthalmol.2016.0267.
- Segen JC. Concise Dictionary of Modern Medicine. New York, NY: McGraw-Hill; 2002.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Hill AB. The environment and disease: association or causation? 1965. J R Soc Med. 2015;108(1):32–37.
- Florida Department of Health. Zika fever: sample submission guidance for county health departments (CHDs). Version 2.0. http://www.floridahealth.gov/diseases-and-conditions/disease-reporting-and-management/disease-reporting-and-surveillance/_documents/zika-fever-sample-submission-guidance-for-chds.pdf. Published June 7, 2016. Accessed July 8, 2016.
- European Centre for Disease Prevention and Control. Zika virus disease epidemic: potential association with microcephaly and Guillain-Barré syndrome (first update). http://ecdc.europa.eu/en/publications/Publications/rapid-risk-assessment-zika-virus-first-update-jan-2016.pdf. Published January 21, 2016. Accessed January 25, 2016.
- Cao-Lormeau VM, Blake A, Mons S, et al. Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet. 2016;387(10027):1531–1539.
- Carteaux G, Maquart M, Bedet A, et al. Zika virus associated with meningoencephalitis. N Engl J Med. 2016;374(16):1595–1596.
- Baud D, Van Mieghem T, Musso D, Truttmann AC, Panchaud A, Vouga M. Clinical management of pregnant women exposed to Zika virus [published online ahead of print April 4, 2016]. Lancet Infect Dis. 2016;16(5):523. doi:10.1016/S1473-3099(16)30008-1.
- Mécharles S, Herrmann C, Poullain P, et al. Acute myelitis due to Zika virus infection. Lancet. 2016;387(10026):1481.
- Oster AM, Russell K, Stryker JE, et al. Update: interim guidance for prevention of sexual transmission of Zika virus—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):323–325.
- Atkinson B, Hearn P, Afrough B, et al. Detection of Zika virus in semen. Emerg Infect Dis. 2016;22(5):940.
- Mansuy JM, Dutertre M, Mengelle C, et al. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis. 2016;16(4):405.
- Dudley DM, Aliota MT, Mohr EL, et al. A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun. 2016;7:12204.
- Davidson A, Slavinski S, Komoto K, Rakeman J, Weiss D. Suspected female-to-male sexual transmission of Zika virus-New York City, 2016. MMWR Morb Mortal Wkly Rep. 2016; 65(28):716-717.
- Petersen EE, Polen KN, Meaney-Delman D, et al. Update: interim guidance for health care providers caring for women of reproductive age with possible Zika virus exposure—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):315–322.
Anushka Chelliah, MD, and Patrick Duff, MD

Dr. Chelliah is a Maternal Fetal Medicine Fellow in the Division of Maternal Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.

The authors report no financial relationships relevant to this article.
Anushka Chelliah, MD, and Patrick Duff, MD
Dr. Chelliah is a Maternal Fetal Medicine Fellow in the Division of Maternal Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Anushka Chelliah, MD, and Patrick Duff, MD
Dr. Chelliah is a Maternal Fetal Medicine Fellow in the Division of Maternal Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
We recently reviewed the most current information on the epidemiology, clinical manifestations, and diagnosis of maternal and congenital Zika virus (ZV) infection.1 We also offered tentative recommendations for reducing the risk of infection and for managing the treatment of women exposed to the virus.
In this update, we present new information on the broadened spectrum of anomalies now known to be causally related to congenital ZV infection and on the increasing number of serious neurologic complications directly related to ZV infection in adults. We also update recommendations for diagnosing maternal, fetal, and neonatal infection and present guidelines for preventing sexual transmission of ZV infection.
CASE Woman from Brazil gives birth to stillborn baby with microcephaly
A 23-year-old woman (G2P1) recently emigrated from Pernambuco in Brazil to the United States and now presents to the hospital in advanced labor. Based on results of first-trimester ultrasonography performed in Brazil, it is determined that she is at 39 weeks’ gestation. The patient has not had any prenatal care since early in the second trimester because of low income and lack of medical insurance. She reports no serious illness before or during the pregnancy.
In the labor and delivery suite, she rapidly delivers a stillborn female infant—5 pounds 3 ounces, growth restricted, with multiple congenital anomalies. Postmortem examination reveals microcephaly, ventriculomegaly, extensive brain atrophy, intracranial calcifications, cerebellar agenesis, cataracts, ocular calcifications, redundant scalp tissue, and multiple joint contractures.
What is the most likely cause of these multiple anomalies?
The patient’s findings are most consistent with a diagnosis of severe intrauterine infection. Possible pathogenic organisms include rubella virus, cytomegalovirus, lymphocytic choriomeningitis virus, toxoplasmosis, and ZV.2 Given the patient’s recent move from Pernambuco in northeastern Brazil, the epicenter of the ZV epidemic in the Americas, the most likely diagnosis is congenital ZV infection.
The initial reports of congenital anomalies associated with ZV infection focused on microcephaly, usually defined as head circumference less than 3 standard deviations below the mean, or less than the third or fifth percentile for gestational age. Subsequent reports have linked many other serious central nervous system (CNS) anomalies to the virus. In a retrospective case series, de Fatima Vasco Aragao and colleagues3 described neuroimaging findings in 23 infants with presumed congenital ZV infection. Of the 22 with computed tomography scans, all had calcifications at the junction of cortical and subcortical white matter, 21 (95%) had disordered cortical development, 20 (91%) had a significant decrease in brain volume, 19 (86%) had ventriculomegaly, and half had distinct hypoplasia of either cerebellum or brainstem. In addition, of the 8 infants with magnetic resonance imaging (MRI) studies, 7 (88%) had an enlarged cisterna magna, 7 (88%) had delayed myelination, 6 (75%) had a simplified gyral pattern, and 3 (38%) had hypoplasia of corpus callosum.
De Paula Freitas and colleagues4 recently found congenital ZV infection associated with severe ocular abnormalities. Comprehensive ophthalmologic examination of 29 infants with microcephaly, presumed caused by congenital ZV infection, revealed 10 (35%) had abnormalities, which included focal pigment mottling, chorioretinal atrophy, hypoplasia and cupping of optic disk, loss of foveal reflex, macular atrophy, lens subluxation, and coloboma of iris.
Other conditions linked to congenital ZV infection include intrauterine growth restriction, redundant scalp tissue, contractures of multiple joints, and clubfoot.2
Bottom line. Although the ocular abnormalities are undetectable by prenatal ultrasound, many of the CNS and skeletal anomalies can be identified antenatally. Therefore, serial ultrasound examinations should be performed on adults who have a clinical illness consistent with ZV infection or who have traveled to an endemic area or have a sexual partner who has been in an endemic area. Patients should be assessed for possible microcephaly, ventriculomegaly, agenesis of corpus callosum, hypoplasia of cerebellum, and skeletal deformities.
Zika virus has been shown to be a direct cause of microcephaly
To make the determination that Zika virus (ZV) causes microcephaly, Rasmussen and colleagues1 very recently evaluated Shepard’s 7 criteria,2 published in 1994, for establishing a cause between a microorganism and a specific clinical condition. These 7 criteria are:
- There must be a proven exposure at one or more critical times during prenatal development.
Rasmussen and colleagues1 pointed to case reports, case series, and epidemiologic studies showing a clear association between ZV exposure and microcephaly. Although exposure at any time during pregnancy may cause congenital infection, exposure in the late first and early second trimesters seems to pose the most risk for severe central nervous system (CNS) injury. - There must be consistent findings in 2 or more high-quality epidemiologic studies.
The studies must control for important confounding variables and include an appropriate number of patients to clearly identify an association between a given exposure and specific fetal anomalies. Rasmussen and colleagues1 cited 2 important epidemiologic studies. The first, a prospective cohort investigation of women in Brazil, found that 29% of those with ZV infection had abnormalities on prenatal ultrasound.3
In the second investigation, a retrospective study of 8 infants in French Polynesia, the mathematical modeling performed by the authors4 suggested microcephaly occurred in 1% of infants born to women with first-trimester ZV infection. Using a different mathematical model, Johansson and colleagues5 found that the risk of fetal microcephaly associated with first-trimester infection may range from as low as 1% to as high as 13%.
Although these studies are helpful in quantifying the risk of congenital infection, they only partially satisfy Shepard’s second criterion. - The suspected microorganism must produce a specific defect or clearly delineated syndrome.
Rasmussen and colleagues1 argued that this criterion has been fulfilled. Zika virus infection causes a distinct phenotype that includes microcephaly, multiple other CNS anomalies, redundant scalp skin, ocular abnormalities, joint contractures (arthrogryposis), and clubfoot.6,7 - The observed birth defect must be associated with a rare environmental exposure.
This criterion also has been met, Rasmussen and colleagues1 reported. They noted that congenital microcephaly is rare in the United States (only about 6 cases in 10,000 liveborn infants) but that the number of cases in Brazil and French Polynesia is much in excess of what would be predicted in the absence of the ZV epidemic. - Teratogenicity should be demonstrated in laboratory animals.
Shepard indicated that this criterion is important but not essential to prove causation. As there is yet no animal model for ZV infection, this criterion has not been fulfilled. - The association between the exposure and the observed anomaly or spectrum of anomalies should be biologically plausible.
Rasmussen and colleagues1 demonstrated that the findings linked to maternal ZV infection are similar to those described for at least 2 other viral pathogens, rubella virus and cytomegalovirus. Animal models also have clearly shown that the ZV is neurotropic. Moreover, ZV has been clearly identified in the brains of infants with microcephaly.8 - Shepard’s seventh criterion relates to a medication or chemical exposure and is not relevant to a microorganism.
References
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Brasil P, Pereira JP Jr, Raja Gabaglia C, et al. Zika virus infection in pregnant women in Rio de Janeiro—preliminary report [published online ahead of print March 4, 2016]. N Engl J Med. doi:10.1056/NEJMoa1602412.
- Cauchemez S, Besnard M, Bompard P, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet. 2016;387(10033):2125–2132.
- Johansson MA, Mier-Y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly [published online ahead of print May 25, 2016; updated June 9, 2016]. N Engl J Med. 2016;375:1–4. doi:10.1056/NEJMp1605367.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika virus. N Engl J Med. 2016;374(16):1552–1563.
- Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374(10):951–958.
Did ZV cause these anomalies?
How certain can we be that the anomalies present in the case patient’s baby were caused by ZV? In the past, and for many years, scientists relied on Koch’s 4 postulates (TABLE 1) to answer this question and establish a causal relationship between a microorganism and a specific clinical disease.5 Koch’s postulates have not been satisfied for the relationship between maternal ZV infection and congenital anomalies. Today’s more relevant standards for determining causality of a teratogen were published in 1994 by Shepard.6 In 2016, Rasmussen and colleagues7 found that the critical components of these criteria are fulfilled and concluded that there is little doubt ZV is a proven and extremely dangerous teratogen. See “Zika virus has been shown to be a direct cause of microcephaly”.
Rasmussen and colleagues7 also used Hill’s criteria to assess the evidence for causation. Hill’s systematic assessment is based on 9 factors (TABLE 2)8, and Rasmussen and colleagues7 concluded that the necessary 7 of these 9 criteria have been met (the experimental animal model criterion was not satisfied, and the biological gradient criterion was not applicable). Given their assessment of Shepard’s criteria,6 the authors argued that the link between maternal ZV infection and severe congenital anomalies has risen from association to well-defined causation.
How should ZV infection be confirmed in adults and newborns?
After our first review was published in March 2016,1 the testing algorithm recommended by the US Centers for Disease Control and Prevention (CDC) was revised.9 Now, according to the CDC, if a patient has had symptoms of ZV infection for less than 5 days, serum and urine should be obtained for reverse transcriptase–polymerase chain reaction (RT-PCR) testing. If symptoms have been present for 5 to 14 days, urine should be tested by RT-PCR because urine samples appear to remain positive for virus longer than serum samples do. If RT-PCR is performed within the appropriate period and the result is negative, ZV infection is excluded; if the result is positive, acute ZV infection is confirmed, and additional testing is not indicated. RT-PCR can be performed by 2 commercial laboratories (Quest Diagnostics and LabCorp), state health departments, and the CDC.
If serum or urine is collected more than 5 days after symptom onset and the RT-PCR result is negative, the patient should have an immunoglobulin M (IgM) assay for ZV. If the assay result is negative, infection is excluded; if the result is positive or equivocal, additional testing is needed to ensure that the presence of the antibody does not reflect a cross-reaction to dengue or chikungunya virus. The confirmatory plaque reduction neutralization test (PRNT) is performed only by the CDC. To be considered positive, the PRNT result must be at least 4-fold higher than the dengue virus neutralizing antibody titer.
In patients with suspected Guillain-Barré syndrome (GBS), RT-PCR can be performed on cerebrospinal fluid. For suspected fetal or neonatal infection, RT-PCR can be performed on amniotic fluid, umbilical cord blood, and fetal and placental tissue.
CASE 2 Nonpregnant woman with possible Zika virus exposure presents to ED with neurologic symptoms
A 31-year-old nulligravid woman presents to the emergency department (ED) for evaluation of numbness, tingling, and weakness in the lower extremities and difficulty walking. She reports having had a low-grade fever and a fine disseminated macular rash 1 week earlier. She denies recent travel and exposure to friends or relatives with illness, but she says her husband travels extensively and was living and working in Puerto Rico. The patient has no other neurologic symptoms.
Serum and cerebrospinal fluid chemistries and MRI findings are normal. However, the ZV IgM assay is positive, and nerve conduction study results are consistent with GBS. The patient is admitted to the hospital, treated with intravenous immunoglobulin and given supportive care. Over 10 days, her neurologic condition gradually improves.
What is the link between ZV infection and serious neurologic complications in adults?
ZV infection has been associated with serious neurologic complications in adults. Investigators in several countries have reported dramatic increases in GBS cases during the ZV outbreak.10
GBS is an acute, immune-mediated, demyelinating peripheral neuropathy that can vary in presentation but most commonly manifests as a rapidly ascending paralysis. The disorder often is preceded by an immunization or live viral infection. In some patients, paralysis severely weakens the respiratory muscles and even the cranial nerves, and affected individuals may require intubation, ventilator support, and parenteral or enteral alimentation.
In a case-control study conducted duringthe 2013–2014 outbreak in French Polynesia, the association between ZV infection and GBS was evaluated in 3 groups of patients: 42 patients with GBS, 98 control patients, and 70 patients with ZV infection but no neurologic complications.11 Symptoms of ZV infection were present in about 88% of the patients with GBS, and the median interval from viral infection to onset of neurologic symptoms was 6 days. The ZV IgM assay was positive in 93% of GBS cases. Nerve conduction study results were consistent with the acute motor axonal neuropathy of GBS. All patients were treated with intravenous immunoglobulin; 38% of patients had to be admitted to the intensive care unit, and 29% needed respiratory support. There were no fatalities. The overall incidence of GBS was 2.4 cases per 10,000 ZV infections.
Other neurologic complications that have been associated with ZV infection are meningoencephalitis,12 brain ischemia,13 and myelitis.14
Bottom line. ZV infection may cause serious neurologic complications in adults. The most devastating complication is GBS, which can result in respiratory muscle paralysis and cranial nerve palsies.
How can patients prevent sexual transmission of ZV infection?
The ZV can be transmitted by sexual contact, including vaginal, anal, and oral sex.15 It is known to persist longer in semen than in blood or urine, though the exact duration remains unknown. Atkinson and colleagues16 reported RT-PCR detection of ZV RNA in semen about 62 days after onset of febrile illness—long after the virus became undetectable in blood.15
Mansuy and colleagues17 found that the viral load in semen was more than 100,000 times that in blood and urine more than 2 weeks after symptom onset.16 The ZV has been detected in saliva, urine, and breast milk. Although it has not been identified in vaginal secretions in humans, it has been detected in the vaginal secretions of nonhuman primates up to 7 days after subcutaneous inoculation of virus.18 In addition, the first case of female-to-male sexual transmission of ZV infection was just reported.19 In this report, transmission seems to have occurred on day 3 of the woman’s symptomatic illness, when she had unprotected vaginal intercourse with her partner. The partner became symptomatic 7 days after sexual exposure. To date, there is no evidence that infection is spread through kissing or breastfeeding.
The most recent recommendations from the CDC are that a man with symptomatic ZV infection wait at least 6 months before having unprotected sexual contact. In addition, a man who is asymptomatic after ZV exposure should wait at least 8 weeks before having unprotected sexual contact.17
A woman planning a pregnancy should know there is no evidence that prior ZV infection increases the risk of birth defects. However, a woman with a proven ZV infection should wait at least 8 weeks after symptom onset before trying to conceive. Even an asymptomatic woman with possible exposure should wait at least 8 weeks after the last exposure before attempting conception. In addition, given the risks associated with maternal and fetal infection, a man who has been exposed to the virus and who has a pregnant partner should abstain from unprotected sexual contact for the duration of the pregnancy.20
Key takeaways
- Zika virus has now been clearly established as the cause of severe fetal malformations, particularly microcephaly.
- The risk of fetal injury appears to be greater when maternal infection occurs in the first trimester of pregnancy.
- Zika virus has now been established as the cause of Guillain-Barré syndrome in adults.
- Although most cases of Zika virus infection are transmitted as the result of mosquito bites, patients can acquire the infection through sexual contact. Both male-to-female and female-to-male transmission have been documented.
- If symptoms have been present for 5 to 14 days, only the urine RT-PCR test should be performed.
- If symptoms have been present for more than 14 days, the patient should have an immunoglobulin M assay for Zika virus. If this test is equivocal or positive, a plaque reduction neutralization test should be performed to exclude infection caused by dengue or chikungunya virus.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
We recently reviewed the most current information on the epidemiology, clinical manifestations, and diagnosis of maternal and congenital Zika virus (ZV) infection.1 We also offered tentative recommendations for reducing the risk of infection and for managing the treatment of women exposed to the virus.
In this update, we present new information on the broadened spectrum of anomalies now known to be causally related to congenital ZV infection and on the increasing number of serious neurologic complications directly related to ZV infection in adults. We also update recommendations for diagnosing maternal, fetal, and neonatal infection and present guidelines for preventing sexual transmission of ZV infection.
CASE Woman from Brazil gives birth to stillborn baby with microcephaly
A 23-year-old woman (G2P1) recently emigrated from Pernambuco in Brazil to the United States and now presents to the hospital in advanced labor. Based on results of first-trimester ultrasonography performed in Brazil, it is determined that she is at 39 weeks’ gestation. The patient has not had any prenatal care since early in the second trimester because of low income and lack of medical insurance. She reports no serious illness before or during the pregnancy.
In the labor and delivery suite, she rapidly delivers a stillborn female infant—5 pounds 3 ounces, growth restricted, with multiple congenital anomalies. Postmortem examination reveals microcephaly, ventriculomegaly, extensive brain atrophy, intracranial calcifications, cerebellar agenesis, cataracts, ocular calcifications, redundant scalp tissue, and multiple joint contractures.
What is the most likely cause of these multiple anomalies?
The patient’s findings are most consistent with a diagnosis of severe intrauterine infection. Possible pathogenic organisms include rubella virus, cytomegalovirus, lymphocytic choriomeningitis virus, toxoplasmosis, and ZV.2 Given the patient’s recent move from Pernambuco in northeastern Brazil, the epicenter of the ZV epidemic in the Americas, the most likely diagnosis is congenital ZV infection.
The initial reports of congenital anomalies associated with ZV infection focused on microcephaly, usually defined as head circumference less than 3 standard deviations below the mean, or less than the third or fifth percentile for gestational age. Subsequent reports have linked many other serious central nervous system (CNS) anomalies to the virus. In a retrospective case series, de Fatima Vasco Aragao and colleagues3 described neuroimaging findings in 23 infants with presumed congenital ZV infection. Of the 22 with computed tomography scans, all had calcifications at the junction of cortical and subcortical white matter, 21 (95%) had disordered cortical development, 20 (91%) had a significant decrease in brain volume, 19 (86%) had ventriculomegaly, and half had distinct hypoplasia of either cerebellum or brainstem. In addition, of the 8 infants with magnetic resonance imaging (MRI) studies, 7 (88%) had an enlarged cisterna magna, 7 (88%) had delayed myelination, 6 (75%) had a simplified gyral pattern, and 3 (38%) had hypoplasia of corpus callosum.
De Paula Freitas and colleagues4 recently found congenital ZV infection associated with severe ocular abnormalities. Comprehensive ophthalmologic examination of 29 infants with microcephaly, presumed caused by congenital ZV infection, revealed 10 (35%) had abnormalities, which included focal pigment mottling, chorioretinal atrophy, hypoplasia and cupping of optic disk, loss of foveal reflex, macular atrophy, lens subluxation, and coloboma of iris.
Other conditions linked to congenital ZV infection include intrauterine growth restriction, redundant scalp tissue, contractures of multiple joints, and clubfoot.2
Bottom line. Although the ocular abnormalities are undetectable by prenatal ultrasound, many of the CNS and skeletal anomalies can be identified antenatally. Therefore, serial ultrasound examinations should be performed on adults who have a clinical illness consistent with ZV infection or who have traveled to an endemic area or have a sexual partner who has been in an endemic area. Patients should be assessed for possible microcephaly, ventriculomegaly, agenesis of corpus callosum, hypoplasia of cerebellum, and skeletal deformities.
Zika virus has been shown to be a direct cause of microcephaly
To make the determination that Zika virus (ZV) causes microcephaly, Rasmussen and colleagues1 very recently evaluated Shepard’s 7 criteria,2 published in 1994, for establishing a cause between a microorganism and a specific clinical condition. These 7 criteria are:
- There must be a proven exposure at one or more critical times during prenatal development.
Rasmussen and colleagues1 pointed to case reports, case series, and epidemiologic studies showing a clear association between ZV exposure and microcephaly. Although exposure at any time during pregnancy may cause congenital infection, exposure in the late first and early second trimesters seems to pose the most risk for severe central nervous system (CNS) injury. - There must be consistent findings in 2 or more high-quality epidemiologic studies.
The studies must control for important confounding variables and include an appropriate number of patients to clearly identify an association between a given exposure and specific fetal anomalies. Rasmussen and colleagues1 cited 2 important epidemiologic studies. The first, a prospective cohort investigation of women in Brazil, found that 29% of those with ZV infection had abnormalities on prenatal ultrasound.3
In the second investigation, a retrospective study of 8 infants in French Polynesia, the mathematical modeling performed by the authors4 suggested microcephaly occurred in 1% of infants born to women with first-trimester ZV infection. Using a different mathematical model, Johansson and colleagues5 found that the risk of fetal microcephaly associated with first-trimester infection may range from as low as 1% to as high as 13%.
Although these studies are helpful in quantifying the risk of congenital infection, they only partially satisfy Shepard’s second criterion. - The suspected microorganism must produce a specific defect or clearly delineated syndrome.
Rasmussen and colleagues1 argued that this criterion has been fulfilled. Zika virus infection causes a distinct phenotype that includes microcephaly, multiple other CNS anomalies, redundant scalp skin, ocular abnormalities, joint contractures (arthrogryposis), and clubfoot.6,7 - The observed birth defect must be associated with a rare environmental exposure.
This criterion also has been met, Rasmussen and colleagues1 reported. They noted that congenital microcephaly is rare in the United States (only about 6 cases in 10,000 liveborn infants) but that the number of cases in Brazil and French Polynesia is much in excess of what would be predicted in the absence of the ZV epidemic. - Teratogenicity should be demonstrated in laboratory animals.
Shepard indicated that this criterion is important but not essential to prove causation. As there is yet no animal model for ZV infection, this criterion has not been fulfilled. - The association between the exposure and the observed anomaly or spectrum of anomalies should be biologically plausible.
Rasmussen and colleagues1 demonstrated that the findings linked to maternal ZV infection are similar to those described for at least 2 other viral pathogens, rubella virus and cytomegalovirus. Animal models also have clearly shown that the ZV is neurotropic. Moreover, ZV has been clearly identified in the brains of infants with microcephaly.8 - Shepard’s seventh criterion relates to a medication or chemical exposure and is not relevant to a microorganism.
References
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Brasil P, Pereira JP Jr, Raja Gabaglia C, et al. Zika virus infection in pregnant women in Rio de Janeiro—preliminary report [published online ahead of print March 4, 2016]. N Engl J Med. doi:10.1056/NEJMoa1602412.
- Cauchemez S, Besnard M, Bompard P, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet. 2016;387(10033):2125–2132.
- Johansson MA, Mier-Y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly [published online ahead of print May 25, 2016; updated June 9, 2016]. N Engl J Med. 2016;375:1–4. doi:10.1056/NEJMp1605367.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika virus. N Engl J Med. 2016;374(16):1552–1563.
- Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374(10):951–958.
Did ZV cause these anomalies?
How certain can we be that the anomalies present in the case patient’s baby were caused by ZV? In the past, and for many years, scientists relied on Koch’s 4 postulates (TABLE 1) to answer this question and establish a causal relationship between a microorganism and a specific clinical disease.5 Koch’s postulates have not been satisfied for the relationship between maternal ZV infection and congenital anomalies. Today’s more relevant standards for determining causality of a teratogen were published in 1994 by Shepard.6 In 2016, Rasmussen and colleagues7 found that the critical components of these criteria are fulfilled and concluded that there is little doubt ZV is a proven and extremely dangerous teratogen. See “Zika virus has been shown to be a direct cause of microcephaly”.
Rasmussen and colleagues7 also used Hill’s criteria to assess the evidence for causation. Hill’s systematic assessment is based on 9 factors (TABLE 2)8, and Rasmussen and colleagues7 concluded that the necessary 7 of these 9 criteria have been met (the experimental animal model criterion was not satisfied, and the biological gradient criterion was not applicable). Given their assessment of Shepard’s criteria,6 the authors argued that the link between maternal ZV infection and severe congenital anomalies has risen from association to well-defined causation.
How should ZV infection be confirmed in adults and newborns?
After our first review was published in March 2016,1 the testing algorithm recommended by the US Centers for Disease Control and Prevention (CDC) was revised.9 Now, according to the CDC, if a patient has had symptoms of ZV infection for less than 5 days, serum and urine should be obtained for reverse transcriptase–polymerase chain reaction (RT-PCR) testing. If symptoms have been present for 5 to 14 days, urine should be tested by RT-PCR because urine samples appear to remain positive for virus longer than serum samples do. If RT-PCR is performed within the appropriate period and the result is negative, ZV infection is excluded; if the result is positive, acute ZV infection is confirmed, and additional testing is not indicated. RT-PCR can be performed by 2 commercial laboratories (Quest Diagnostics and LabCorp), state health departments, and the CDC.
If serum or urine is collected more than 5 days after symptom onset and the RT-PCR result is negative, the patient should have an immunoglobulin M (IgM) assay for ZV. If the assay result is negative, infection is excluded; if the result is positive or equivocal, additional testing is needed to ensure that the presence of the antibody does not reflect a cross-reaction to dengue or chikungunya virus. The confirmatory plaque reduction neutralization test (PRNT) is performed only by the CDC. To be considered positive, the PRNT result must be at least 4-fold higher than the dengue virus neutralizing antibody titer.
In patients with suspected Guillain-Barré syndrome (GBS), RT-PCR can be performed on cerebrospinal fluid. For suspected fetal or neonatal infection, RT-PCR can be performed on amniotic fluid, umbilical cord blood, and fetal and placental tissue.
CASE 2 Nonpregnant woman with possible Zika virus exposure presents to ED with neurologic symptoms
A 31-year-old nulligravid woman presents to the emergency department (ED) for evaluation of numbness, tingling, and weakness in the lower extremities and difficulty walking. She reports having had a low-grade fever and a fine disseminated macular rash 1 week earlier. She denies recent travel and exposure to friends or relatives with illness, but she says her husband travels extensively and was living and working in Puerto Rico. The patient has no other neurologic symptoms.
Serum and cerebrospinal fluid chemistries and MRI findings are normal. However, the ZV IgM assay is positive, and nerve conduction study results are consistent with GBS. The patient is admitted to the hospital, treated with intravenous immunoglobulin and given supportive care. Over 10 days, her neurologic condition gradually improves.
What is the link between ZV infection and serious neurologic complications in adults?
ZV infection has been associated with serious neurologic complications in adults. Investigators in several countries have reported dramatic increases in GBS cases during the ZV outbreak.10
GBS is an acute, immune-mediated, demyelinating peripheral neuropathy that can vary in presentation but most commonly manifests as a rapidly ascending paralysis. The disorder often is preceded by an immunization or live viral infection. In some patients, paralysis severely weakens the respiratory muscles and even the cranial nerves, and affected individuals may require intubation, ventilator support, and parenteral or enteral alimentation.
In a case-control study conducted duringthe 2013–2014 outbreak in French Polynesia, the association between ZV infection and GBS was evaluated in 3 groups of patients: 42 patients with GBS, 98 control patients, and 70 patients with ZV infection but no neurologic complications.11 Symptoms of ZV infection were present in about 88% of the patients with GBS, and the median interval from viral infection to onset of neurologic symptoms was 6 days. The ZV IgM assay was positive in 93% of GBS cases. Nerve conduction study results were consistent with the acute motor axonal neuropathy of GBS. All patients were treated with intravenous immunoglobulin; 38% of patients had to be admitted to the intensive care unit, and 29% needed respiratory support. There were no fatalities. The overall incidence of GBS was 2.4 cases per 10,000 ZV infections.
Other neurologic complications that have been associated with ZV infection are meningoencephalitis,12 brain ischemia,13 and myelitis.14
Bottom line. ZV infection may cause serious neurologic complications in adults. The most devastating complication is GBS, which can result in respiratory muscle paralysis and cranial nerve palsies.
How can patients prevent sexual transmission of ZV infection?
The ZV can be transmitted by sexual contact, including vaginal, anal, and oral sex.15 It is known to persist longer in semen than in blood or urine, though the exact duration remains unknown. Atkinson and colleagues16 reported RT-PCR detection of ZV RNA in semen about 62 days after onset of febrile illness—long after the virus became undetectable in blood.15
Mansuy and colleagues17 found that the viral load in semen was more than 100,000 times that in blood and urine more than 2 weeks after symptom onset.16 The ZV has been detected in saliva, urine, and breast milk. Although it has not been identified in vaginal secretions in humans, it has been detected in the vaginal secretions of nonhuman primates up to 7 days after subcutaneous inoculation of virus.18 In addition, the first case of female-to-male sexual transmission of ZV infection was just reported.19 In this report, transmission seems to have occurred on day 3 of the woman’s symptomatic illness, when she had unprotected vaginal intercourse with her partner. The partner became symptomatic 7 days after sexual exposure. To date, there is no evidence that infection is spread through kissing or breastfeeding.
The most recent recommendations from the CDC are that a man with symptomatic ZV infection wait at least 6 months before having unprotected sexual contact. In addition, a man who is asymptomatic after ZV exposure should wait at least 8 weeks before having unprotected sexual contact.17
A woman planning a pregnancy should know there is no evidence that prior ZV infection increases the risk of birth defects. However, a woman with a proven ZV infection should wait at least 8 weeks after symptom onset before trying to conceive. Even an asymptomatic woman with possible exposure should wait at least 8 weeks after the last exposure before attempting conception. In addition, given the risks associated with maternal and fetal infection, a man who has been exposed to the virus and who has a pregnant partner should abstain from unprotected sexual contact for the duration of the pregnancy.20
Key takeaways
- Zika virus has now been clearly established as the cause of severe fetal malformations, particularly microcephaly.
- The risk of fetal injury appears to be greater when maternal infection occurs in the first trimester of pregnancy.
- Zika virus has now been established as the cause of Guillain-Barré syndrome in adults.
- Although most cases of Zika virus infection are transmitted as the result of mosquito bites, patients can acquire the infection through sexual contact. Both male-to-female and female-to-male transmission have been documented.
- If symptoms have been present for 5 to 14 days, only the urine RT-PCR test should be performed.
- If symptoms have been present for more than 14 days, the patient should have an immunoglobulin M assay for Zika virus. If this test is equivocal or positive, a plaque reduction neutralization test should be performed to exclude infection caused by dengue or chikungunya virus.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28–34.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- de Fatima Vasco Aragao M, van der Linden V, Brainer-Lima AM, et al. Clinical features and neuroimaging (CT and MRI) findings in presumed Zika virus related congenital infection and microcephaly: retrospective case series study. BMJ. 2016;353:i1901.
- de Paula Freitas B, de Oliveira Dias JR, Prazeres J, et al. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil [published online ahead of print February 9, 2016]. JAMA Ophthalmol. doi:10.1001/jamaophthalmol.2016.0267.
- Segen JC. Concise Dictionary of Modern Medicine. New York, NY: McGraw-Hill; 2002.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Hill AB. The environment and disease: association or causation? 1965. J R Soc Med. 2015;108(1):32–37.
- Florida Department of Health. Zika fever: sample submission guidance for county health departments (CHDs). Version 2.0. http://www.floridahealth.gov/diseases-and-conditions/disease-reporting-and-management/disease-reporting-and-surveillance/_documents/zika-fever-sample-submission-guidance-for-chds.pdf. Published June 7, 2016. Accessed July 8, 2016.
- European Centre for Disease Prevention and Control. Zika virus disease epidemic: potential association with microcephaly and Guillain-Barré syndrome (first update). http://ecdc.europa.eu/en/publications/Publications/rapid-risk-assessment-zika-virus-first-update-jan-2016.pdf. Published January 21, 2016. Accessed January 25, 2016.
- Cao-Lormeau VM, Blake A, Mons S, et al. Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet. 2016;387(10027):1531–1539.
- Carteaux G, Maquart M, Bedet A, et al. Zika virus associated with meningoencephalitis. N Engl J Med. 2016;374(16):1595–1596.
- Baud D, Van Mieghem T, Musso D, Truttmann AC, Panchaud A, Vouga M. Clinical management of pregnant women exposed to Zika virus [published online ahead of print April 4, 2016]. Lancet Infect Dis. 2016;16(5):523. doi:10.1016/S1473-3099(16)30008-1.
- Mécharles S, Herrmann C, Poullain P, et al. Acute myelitis due to Zika virus infection. Lancet. 2016;387(10026):1481.
- Oster AM, Russell K, Stryker JE, et al. Update: interim guidance for prevention of sexual transmission of Zika virus—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):323–325.
- Atkinson B, Hearn P, Afrough B, et al. Detection of Zika virus in semen. Emerg Infect Dis. 2016;22(5):940.
- Mansuy JM, Dutertre M, Mengelle C, et al. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis. 2016;16(4):405.
- Dudley DM, Aliota MT, Mohr EL, et al. A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun. 2016;7:12204.
- Davidson A, Slavinski S, Komoto K, Rakeman J, Weiss D. Suspected female-to-male sexual transmission of Zika virus-New York City, 2016. MMWR Morb Mortal Wkly Rep. 2016; 65(28):716-717.
- Petersen EE, Polen KN, Meaney-Delman D, et al. Update: interim guidance for health care providers caring for women of reproductive age with possible Zika virus exposure—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):315–322.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28–34.
- Meaney-Delman D, Rasmussen SA, Staples JE, et al. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol. 2016;127(4):642–648.
- de Fatima Vasco Aragao M, van der Linden V, Brainer-Lima AM, et al. Clinical features and neuroimaging (CT and MRI) findings in presumed Zika virus related congenital infection and microcephaly: retrospective case series study. BMJ. 2016;353:i1901.
- de Paula Freitas B, de Oliveira Dias JR, Prazeres J, et al. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil [published online ahead of print February 9, 2016]. JAMA Ophthalmol. doi:10.1001/jamaophthalmol.2016.0267.
- Segen JC. Concise Dictionary of Modern Medicine. New York, NY: McGraw-Hill; 2002.
- Shepard TH. “Proof” of human teratogenicity. Teratology. 1994;50(2):97–98.
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374(20):1981–1987.
- Hill AB. The environment and disease: association or causation? 1965. J R Soc Med. 2015;108(1):32–37.
- Florida Department of Health. Zika fever: sample submission guidance for county health departments (CHDs). Version 2.0. http://www.floridahealth.gov/diseases-and-conditions/disease-reporting-and-management/disease-reporting-and-surveillance/_documents/zika-fever-sample-submission-guidance-for-chds.pdf. Published June 7, 2016. Accessed July 8, 2016.
- European Centre for Disease Prevention and Control. Zika virus disease epidemic: potential association with microcephaly and Guillain-Barré syndrome (first update). http://ecdc.europa.eu/en/publications/Publications/rapid-risk-assessment-zika-virus-first-update-jan-2016.pdf. Published January 21, 2016. Accessed January 25, 2016.
- Cao-Lormeau VM, Blake A, Mons S, et al. Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet. 2016;387(10027):1531–1539.
- Carteaux G, Maquart M, Bedet A, et al. Zika virus associated with meningoencephalitis. N Engl J Med. 2016;374(16):1595–1596.
- Baud D, Van Mieghem T, Musso D, Truttmann AC, Panchaud A, Vouga M. Clinical management of pregnant women exposed to Zika virus [published online ahead of print April 4, 2016]. Lancet Infect Dis. 2016;16(5):523. doi:10.1016/S1473-3099(16)30008-1.
- Mécharles S, Herrmann C, Poullain P, et al. Acute myelitis due to Zika virus infection. Lancet. 2016;387(10026):1481.
- Oster AM, Russell K, Stryker JE, et al. Update: interim guidance for prevention of sexual transmission of Zika virus—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):323–325.
- Atkinson B, Hearn P, Afrough B, et al. Detection of Zika virus in semen. Emerg Infect Dis. 2016;22(5):940.
- Mansuy JM, Dutertre M, Mengelle C, et al. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis. 2016;16(4):405.
- Dudley DM, Aliota MT, Mohr EL, et al. A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun. 2016;7:12204.
- Davidson A, Slavinski S, Komoto K, Rakeman J, Weiss D. Suspected female-to-male sexual transmission of Zika virus-New York City, 2016. MMWR Morb Mortal Wkly Rep. 2016; 65(28):716-717.
- Petersen EE, Polen KN, Meaney-Delman D, et al. Update: interim guidance for health care providers caring for women of reproductive age with possible Zika virus exposure—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(12):315–322.
In this Article
- Confirming Zika virus infection
- Zika virus and Guillain-Barré syndrome
- Preventing sexual transmission
2016 Update on contraception
Contraception is an important tool that allows patients to carry out their reproductive-life plans. In the United States, the average woman desires 2 children.1 To achieve this goal, she will spend more than 30 years of her reproductive life avoiding pregnancy.1 The most effective reversible contraceptive methods, the intrauterine device (IUD) and the contraceptive implant, offer an efficient way to cover this significant period. Currently, American women more commonly choose an IUD than an implant by a factor 8 to 1.2 Between 2002 and 2012, the percentage of US contraceptive users aged 15 to 44 using the IUD rose from 2% to 10%.2
Significant barriers to contraceptive access still exist, however. Although widespread reports have lauded the decrease in unintended pregnancies in the United States, improvement only has been marginal for women who live below the poverty level. In fact, for unintended pregnancy the gap between women above and below the poverty level has increased from a 2.6-fold difference in 1994 to a 5.6-fold difference in 2011 (FIGURE).3−5 Since the decrease in the unintended pregnancy rate is most likely related to an increase in contraceptive use, particularly the IUD, we are not providing equal contraceptive access to all women.5
Both the copper IUD and the 3 available levonorgestrel (LNG)-releasing intrauterine system (IUS) products provide safe and effective contraception. As IUD research expands, it is imperative for providers to stay up to date so patients can have full access to these devices.
In this article, we present important updates regarding IUD use that will help break down some continuing barriers to contraceptive access, including:
- clinical trial data demonstrating efficacy of LNG 52-mg IUS for 7 years
- a novel emergency contraception (EC) regimen of same-day oral LNG and the LNG 52-mg IUS
- a large prospective trial demonstrating that women can safely have IUS placement without known sexually transmitted infection (STI) screening results and that pelvic infection rates are not higher in the time shortly after IUS placement.

WHO study demonstrates LNG 52-mg IUS is highly effective for up to 7 years of use
Rowe P, Farley T, Peregoudov A, et al; IUD Research Group of the UNDP/UNFPA/WHO/World Bank Special Programme of Research; Development and Research Training in Human Reproduction. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93(6):498−506.
Currently, the 2 LNG 52-mg IUS products (Liletta, Mirena) approved by the US Food and Drug Administration (FDA) are available for use for 3 and 5 years, respectively. The pivotal approval trial for Liletta is still ongoing and is planned to continue for up to 7 years.6 The TCu380A (ParaGard) copper IUD is FDA approved for up to 10 years of use; however, this product initially was approved for only 4 years. The duration of use was expanded to 10 years based on continued clinical trials.
Based on current data, we will not need to wait for the Liletta pivotal trial to have clinical evidence of a longer duration of efficacy for the LNG 52-mg IUS. A collaborative group as part of the UNDP/UNFPA/WHO/World Bank Special Programme of Research, Development and Research Training in Human Reproduction performed a multicenter, open-label randomized controlled trial to evaluate outcomes through 7 years of use of the LNG 52-mg IUS and the TCu380A IUD.
Details of the study
A total of 3,836 women were enrolled at 20 centers in Europe, Asia, South America, and China and were randomly assigned to one of the 2 products. Eligible women were aged 16 to 40 years, parous, and without known leiomyoma or recent pelvic infection. After excluding 15 failed IUD insertions, 1,910 women received an LNG 52-mg IUS and 1,911 received a TCu380A. Ultimately, 398 women in the LNG 52-mg IUS group and 682 in the TCu380A group completed 7-year follow-up with the IUD in place. Women were surveyed regarding pregnancy and method discontinuation.
Lower pregnancy rate, higher discontinuation with LNG IUS
The cumulative 7-year pregnancy rate among LNG 52-mg IUS users was significantly lower than among TCu380A users (0.53 per 100 women vs 2.45 per 100 women, respectively). All pregnancies in the LNG 52-mg IUS group occurred in the first 5 years of study follow-up--with no pregnancies in years 6 through 7 (TABLE). The cumulative pregnancy rate in the TCu380A group in this study is consistent with that in a previous long-term trial of this IUD.7

Early removal was significantly higher in the LNG 52-mg IUS group, with a cumulative discontinuation rate of 70.6 per 100 women, compared with 40.8 per 100 women in the TCu380A group. Significant cultural variation existed when it came to both rate and reason for discontinuation. Most women at Chinese centers cited amenorrhea and decreased bleeding as the primary reason for discontinuation, and they did so at twice the rate of women at non-Chinese centers.
The patterns of method discontinuation in this study were different from those found among US women. By comparison, a recent study in the United States had lower overall discontinuation rates and did not find decreased bleeding to be among the main reasons for LNG 52-mg IUS removal.7 In fact, most women who discontinued the LNG 52-mg IUS cited concerns about upcoming expiration as their reason for removal.
The results of this large study also generally corroborated the low-risk profile of IUDs. Only 1 reported IUD perforation occurred, for a rate of 0.03 per 1,000 women. Device expulsion rates were similar between the IUDs and were uncommon overall with 7-year rates of 8 to 9 per 100 women. Pelvic infection was cited as reason for removal in only 7 women (0.18 per 100 women) over 7 years.
What this evidence means for practiceThis exciting study is the first large-scale clinical trial demonstrating continued high efficacy with LNG 52-mg IUS use through 7 years. This information affords women extended contraceptive coverage. While additional research will be welcome, particularly in younger women who will maintain greater fertility across the IUS’s 7-year life span, we are confident in extending the 7-year duration for this IUS to our patients.
Additionally, the method discontinuation findings in this study highlight the importance of discussing the expected menstrual changes of hormonal IUS use with women prior to insertion so they can determine if the potential changes would be satisfactory. As the acceptability of medical menstrual suppression may be new to many women, providers should frame the adverse effects in this context. Providers can use this opportunity to review the noncontraceptive benefits of the hormonal IUS as well.
Novel combination of LNG 52-mg IUS and oral LNG 1.5 mg is promising for emergency contraception
Turok DK, Sanders JN, Thompson IS, Royer PA, Eggebroten J, Gawron LM. Preference for and efficacy of oral levonorgestrel for emergency contraception with concomitant placement of a levonorgestrel IUD: a prospective cohort study. Contraception. 2016;93(6):526−532.
The copper IUD is superior for EC relative to oral agents and has the added benefit of providing ongoing highly effective contraception after placement.8 Despite this strong evidence, the copper IUD remains underutilized for this indication. Turok and colleagues noted that women in their clinic seeking IUDs for non-EC purposes preferred the LNG 52-mg IUS over the copper IUD. It is understandable that women might carry these preferences into EC encounters as well. Thus, the investigators devised a novel combination of LNG 52-mg IUS and oral LNG 1.5 mg, which provided both known EC benefit and same-day access to a more popular contraceptive device.
Details of the study
Women presenting for EC who desired same-day IUD placement were enrolled in the prospective cohort study. Eligible women had a negative urine pregnancy test, known last menstrual period (LMP), regular menstrual cycle, and reported unprotected intercourse within 120 hours prior to presentation. Importantly, women with multiple episodes of unprotected intercourse in the weeks prior to presentation were also included to provide a population more comparable to that encountered clinically. The women were then offered the choice of a TCu380A copper IUD or oral LNG EC with LNG 52-mg IUS placement. They were counseled on the potential increased risk of pregnancy with the novel oral LNG EC plus LNG IUS combination compared with the copper IUD. Participants were given a home pregnancy test that they were to complete in 2 weeks and then report the results to the clinic.
Of the 1,004 women presenting to the clinic for EC over the 16-month study period, 188 (18%) desired same-day IUD insertion. Of these, more opted for the oral LNG EC plus LNG IUS combination (n = 121, 64%) than the copper IUD (n = 67, 36%), demonstrating that women were often willing to accept a possible decrease in EC efficacy with the goal of obtaining their preferred lUD type.
Excluding failed insertion, undiagnosed uterine didelphys, and patient withdrawal, 110 women received the oral LNG EC plus LNG IUS and 66 received the copper IUD. Demographics were comparable between groups except for body mass index (BMI). Of note, more than half (61%) of the women who opted for the oral LNG EC plus LNG IUS combination were overweight or obese.
Both EC methods are effective, broadening options
All women who received the copper IUD followed up at 2 weeks, and no pregnancies were reported. Of the women who received oral LNG EC plus the LNG IUS, 107 (97%) had follow-up at 2 weeks. In this group, there was 1 reported ectopic pregnancy that ultimately required surgical management. However, further review of the patient's coital history suggested that conception occurred prior to IUD insertion, and the case was not classified by the investigators as an EC failure.
Although this study was not powered to detect pregnancy rate differences between the traditional copper IUD and the oral LNG EC plus LNG IUS combination, the results are promising. An important strength of this study is the presence of 2 high-risk groups for EC failure in the oral LNG EC plus LNG IUS group (elevated BMI and multiple episodes of unprotected intercourse).
What this evidence means for practiceEncounters for EC are important opportunities in which to discuss a woman’s reproductive goals. For women who are interested in an IUD, this study opens up the option of same-day placement of the LNG 52-mg IUS. While larger trials need to be conducted to obtain more information regarding pregnancy rates with an oral EC plus LNG IUS combination versus the TCu380A, offering such a combination is reasonable. Given that ulipristal acetate (UPA) is a more effective EC product, especially for women who are overweight or obese,9 offering a UPA EC plus LNG IUS combination may be a better alternative.
It is time to remove the STI screening barrier to same-day IUD insertion
Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement [published online ahead of print May 12, 2016]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2016.05017.
Provider concerns regarding the presence of pelvic infection remain a significant barrier to same-day IUD insertion. Older studies suggested a higher risk of pelvic infection in the first 20 days after IUD placement and extrapolated that pelvic infection was related to inserting an IUD in a woman at risk for STI.10 As a result, patients may be restricted from same-day IUD insertion by providers who think that obtaining results of STI testing is required prior to placement. Recently, a systematic review suggested, based on limited evidence, that IUD placement does not increase the risk of pelvic infection in asymptomatic women compared with those without an IUD.11
Turok and colleagues (including M.D.C., coauthor of this article) reported results from a planned secondary analysis of A Comprehensive Contraceptive Efficacy and Safety Study of an IUS (ACCESS IUS), a component of the regulatory approval of the Liletta LNG 52-mg IUS. This analysis represents the first large-scale prospective investigation of pelvic infection rates during the first 2 years after IUD placement in US women.
Details of the study
Of the 1,751 women enrolled in the study, 1,714 had successful IUS insertions. Infection was assessed via baseline pelvic visual and bimanual examination and Chlamydia testing in all women. Gonorrhea testing was also performed in women who had not been tested with their current sexual partner. STI test results were not required for IUS insertion. Participants were assessed in person at 1, 3, 6, 12, and 24 months after insertion. Additional pelvic exams were performed as needed based on reported symptoms. At 6 months, 1 year, and 2 years, IUS continuation was reported in 1,553 (90.6%), 1,401 (81.7%), and 1,157 (67.3%) women, respectively.
Nearly all women received baseline STI testing (98.4%); however, results were not available prior to same-day IUS insertion for 79.6% of participants. Twenty-nine (1.7%) women had positive baseline STI tests (25 for Chlamydia, 3 for gonorrhea, 1 for both). Of these, only 6 women had results available prior to IUS placement. All women with a positive STI test were treated, and the IUS was left in place. Importantly, none of these women developed pelvic infection in the subsequent 2 years of follow-up and none requested IUS removal.
Infection risk is low with IUD placement
Among women with negative baseline STI tests, there were only 9 (0.5%) clinical diagnoses of pelvic infections over the first 2 years of follow-up. Diagnosis was typically made based on physical examination findings. Most women underwent repeat Chlamydia and gonorrhea testing at the time of pelvic infection diagnosis, and none had positive results. There were no medically recommended IUS removals; 2 women with pelvic infection requested IUS removal per their preference.
Three of the 9 women with pelvic infections were diagnosed within 1 week of IUS placement, 1 at 39 days after placement, and the remaining 5 more than 6 months after placement, suggesting that pelvic infection is not temporally related to IUS placement. Most women were successfully treated as outpatients.
What this evidence means for practiceThis study provides further reassurance regarding the low risk of pelvic infection among women with an LNG IUS. Insertion of an IUS should not be delayed to await results of Chlamydia or gonorrhea testing in a woman without clinical evidence of pelvic infection. Risk-based, as opposed to universal testing, is imperative.12 These recommendations are in agreement with current recommendations of the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists.13,14 Practices that employ 2-visit protocols unnecessarily limit women’s access to the IUS, as research has shown that nearly half of women desiring an IUD do not return for device placement if a second encounter is required.15
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Office of Population Affairs, Department of Health and Human Services. Family planning program, FY 1999 service program grants by state. Bethesda, MD: Office of Population Affairs; 1999.
- Kavanaugh ML, Jerman J, Finer LB. Changes in use of long-acting reversible contraceptive methods among US women, 2009-2012. Obstet Gynecol. 2015;126(5):917–927.
- Ventura SJ, Abma JC, Mosher WD, Henshaw S. Revised pregnancy rates, 1990-97, and new rates for 1998-99: United States. Natl Vital Stat Rep. 2003;52(7):1−14.
- Finer LB, Zolna MR. Shifts in intended and unintended pregnancies in the United States, 2001-2008. Am J Public Health. 2014;104(suppl 1):S43–S48.
- Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008-2011. N Engl J Med. 2016;374(9):843–852.
- Eisenberg DL, Schreiber CA, Turok DK, Teal SB, Westhoff CL, Creinin MD; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92(1):10–16.
- United Nations Development Programme, United Nations Population Fund, World Health Organization, World Bank, Special Programme of Research, Development, and Research Training in Human Reproduction. Long-term reversible contraception: twelve years of experience with the TCu380A and TCu220C. Contraception. 1997;56(6):341–352.
- Cleland K, Zhu H, Goldstuck N, Cheng L, Trussell J. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27(7):1994–2000.
- Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84(4):363–367.
- Farley TM, Rosenberg MJ, Rowe PJ, Chen JH, Meirik O. Intrauterine devices and pelvic inflammatory disease: an international perspective. Lancet. 1992;339(8796):785–788.
- Jatlaoui TC, Simmons KB, Curtis KM. The safety of intrauterine contraception initiation among women with current asymptomatic cervical infections or at increased risk of sexually transmitted infections [published online ahead of print June 1, 2016]. Contraception. doi:10.1016/j.contracep tion.2016.05.013.
- Grentzer JM, Peipert JF, Zhao Q, McNicholas C, Secura GM, Madden T. Risk-based screening for Chlamydia trachomatis and Neisseria gonorrhoeae prior to intrauterine device insertion. Contraception. 2015;92(4):313–318.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 121: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2011;118(1):184–196.
- Centers for Disease Control and Prevention. US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd ed. MMWR Recomm Rep. 2013;62(RR05):1–46.
- Bergin A, Tristan S, Terplan M, Gilliam ML, Whitaker AK. A missed opportunity for care: two-visit IUD insertion protocols inhibit placement. Contraception. 2012;86(6):694–697.
Contraception is an important tool that allows patients to carry out their reproductive-life plans. In the United States, the average woman desires 2 children.1 To achieve this goal, she will spend more than 30 years of her reproductive life avoiding pregnancy.1 The most effective reversible contraceptive methods, the intrauterine device (IUD) and the contraceptive implant, offer an efficient way to cover this significant period. Currently, American women more commonly choose an IUD than an implant by a factor 8 to 1.2 Between 2002 and 2012, the percentage of US contraceptive users aged 15 to 44 using the IUD rose from 2% to 10%.2
Significant barriers to contraceptive access still exist, however. Although widespread reports have lauded the decrease in unintended pregnancies in the United States, improvement only has been marginal for women who live below the poverty level. In fact, for unintended pregnancy the gap between women above and below the poverty level has increased from a 2.6-fold difference in 1994 to a 5.6-fold difference in 2011 (FIGURE).3−5 Since the decrease in the unintended pregnancy rate is most likely related to an increase in contraceptive use, particularly the IUD, we are not providing equal contraceptive access to all women.5
Both the copper IUD and the 3 available levonorgestrel (LNG)-releasing intrauterine system (IUS) products provide safe and effective contraception. As IUD research expands, it is imperative for providers to stay up to date so patients can have full access to these devices.
In this article, we present important updates regarding IUD use that will help break down some continuing barriers to contraceptive access, including:
- clinical trial data demonstrating efficacy of LNG 52-mg IUS for 7 years
- a novel emergency contraception (EC) regimen of same-day oral LNG and the LNG 52-mg IUS
- a large prospective trial demonstrating that women can safely have IUS placement without known sexually transmitted infection (STI) screening results and that pelvic infection rates are not higher in the time shortly after IUS placement.
WHO study demonstrates LNG 52-mg IUS is highly effective for up to 7 years of use
Rowe P, Farley T, Peregoudov A, et al; IUD Research Group of the UNDP/UNFPA/WHO/World Bank Special Programme of Research; Development and Research Training in Human Reproduction. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93(6):498−506.
Currently, the 2 LNG 52-mg IUS products (Liletta, Mirena) approved by the US Food and Drug Administration (FDA) are available for use for 3 and 5 years, respectively. The pivotal approval trial for Liletta is still ongoing and is planned to continue for up to 7 years.6 The TCu380A (ParaGard) copper IUD is FDA approved for up to 10 years of use; however, this product initially was approved for only 4 years. The duration of use was expanded to 10 years based on continued clinical trials.
Based on current data, we will not need to wait for the Liletta pivotal trial to have clinical evidence of a longer duration of efficacy for the LNG 52-mg IUS. A collaborative group as part of the UNDP/UNFPA/WHO/World Bank Special Programme of Research, Development and Research Training in Human Reproduction performed a multicenter, open-label randomized controlled trial to evaluate outcomes through 7 years of use of the LNG 52-mg IUS and the TCu380A IUD.
Details of the study
A total of 3,836 women were enrolled at 20 centers in Europe, Asia, South America, and China and were randomly assigned to one of the 2 products. Eligible women were aged 16 to 40 years, parous, and without known leiomyoma or recent pelvic infection. After excluding 15 failed IUD insertions, 1,910 women received an LNG 52-mg IUS and 1,911 received a TCu380A. Ultimately, 398 women in the LNG 52-mg IUS group and 682 in the TCu380A group completed 7-year follow-up with the IUD in place. Women were surveyed regarding pregnancy and method discontinuation.
Lower pregnancy rate, higher discontinuation with LNG IUS
The cumulative 7-year pregnancy rate among LNG 52-mg IUS users was significantly lower than among TCu380A users (0.53 per 100 women vs 2.45 per 100 women, respectively). All pregnancies in the LNG 52-mg IUS group occurred in the first 5 years of study follow-up--with no pregnancies in years 6 through 7 (TABLE). The cumulative pregnancy rate in the TCu380A group in this study is consistent with that in a previous long-term trial of this IUD.7
Early removal was significantly higher in the LNG 52-mg IUS group, with a cumulative discontinuation rate of 70.6 per 100 women, compared with 40.8 per 100 women in the TCu380A group. Significant cultural variation existed when it came to both rate and reason for discontinuation. Most women at Chinese centers cited amenorrhea and decreased bleeding as the primary reason for discontinuation, and they did so at twice the rate of women at non-Chinese centers.
The patterns of method discontinuation in this study were different from those found among US women. By comparison, a recent study in the United States had lower overall discontinuation rates and did not find decreased bleeding to be among the main reasons for LNG 52-mg IUS removal.7 In fact, most women who discontinued the LNG 52-mg IUS cited concerns about upcoming expiration as their reason for removal.
The results of this large study also generally corroborated the low-risk profile of IUDs. Only 1 reported IUD perforation occurred, for a rate of 0.03 per 1,000 women. Device expulsion rates were similar between the IUDs and were uncommon overall with 7-year rates of 8 to 9 per 100 women. Pelvic infection was cited as reason for removal in only 7 women (0.18 per 100 women) over 7 years.
What this evidence means for practiceThis exciting study is the first large-scale clinical trial demonstrating continued high efficacy with LNG 52-mg IUS use through 7 years. This information affords women extended contraceptive coverage. While additional research will be welcome, particularly in younger women who will maintain greater fertility across the IUS’s 7-year life span, we are confident in extending the 7-year duration for this IUS to our patients.
Additionally, the method discontinuation findings in this study highlight the importance of discussing the expected menstrual changes of hormonal IUS use with women prior to insertion so they can determine if the potential changes would be satisfactory. As the acceptability of medical menstrual suppression may be new to many women, providers should frame the adverse effects in this context. Providers can use this opportunity to review the noncontraceptive benefits of the hormonal IUS as well.
Novel combination of LNG 52-mg IUS and oral LNG 1.5 mg is promising for emergency contraception
Turok DK, Sanders JN, Thompson IS, Royer PA, Eggebroten J, Gawron LM. Preference for and efficacy of oral levonorgestrel for emergency contraception with concomitant placement of a levonorgestrel IUD: a prospective cohort study. Contraception. 2016;93(6):526−532.
The copper IUD is superior for EC relative to oral agents and has the added benefit of providing ongoing highly effective contraception after placement.8 Despite this strong evidence, the copper IUD remains underutilized for this indication. Turok and colleagues noted that women in their clinic seeking IUDs for non-EC purposes preferred the LNG 52-mg IUS over the copper IUD. It is understandable that women might carry these preferences into EC encounters as well. Thus, the investigators devised a novel combination of LNG 52-mg IUS and oral LNG 1.5 mg, which provided both known EC benefit and same-day access to a more popular contraceptive device.
Details of the study
Women presenting for EC who desired same-day IUD placement were enrolled in the prospective cohort study. Eligible women had a negative urine pregnancy test, known last menstrual period (LMP), regular menstrual cycle, and reported unprotected intercourse within 120 hours prior to presentation. Importantly, women with multiple episodes of unprotected intercourse in the weeks prior to presentation were also included to provide a population more comparable to that encountered clinically. The women were then offered the choice of a TCu380A copper IUD or oral LNG EC with LNG 52-mg IUS placement. They were counseled on the potential increased risk of pregnancy with the novel oral LNG EC plus LNG IUS combination compared with the copper IUD. Participants were given a home pregnancy test that they were to complete in 2 weeks and then report the results to the clinic.
Of the 1,004 women presenting to the clinic for EC over the 16-month study period, 188 (18%) desired same-day IUD insertion. Of these, more opted for the oral LNG EC plus LNG IUS combination (n = 121, 64%) than the copper IUD (n = 67, 36%), demonstrating that women were often willing to accept a possible decrease in EC efficacy with the goal of obtaining their preferred lUD type.
Excluding failed insertion, undiagnosed uterine didelphys, and patient withdrawal, 110 women received the oral LNG EC plus LNG IUS and 66 received the copper IUD. Demographics were comparable between groups except for body mass index (BMI). Of note, more than half (61%) of the women who opted for the oral LNG EC plus LNG IUS combination were overweight or obese.
Both EC methods are effective, broadening options
All women who received the copper IUD followed up at 2 weeks, and no pregnancies were reported. Of the women who received oral LNG EC plus the LNG IUS, 107 (97%) had follow-up at 2 weeks. In this group, there was 1 reported ectopic pregnancy that ultimately required surgical management. However, further review of the patient's coital history suggested that conception occurred prior to IUD insertion, and the case was not classified by the investigators as an EC failure.
Although this study was not powered to detect pregnancy rate differences between the traditional copper IUD and the oral LNG EC plus LNG IUS combination, the results are promising. An important strength of this study is the presence of 2 high-risk groups for EC failure in the oral LNG EC plus LNG IUS group (elevated BMI and multiple episodes of unprotected intercourse).
What this evidence means for practiceEncounters for EC are important opportunities in which to discuss a woman’s reproductive goals. For women who are interested in an IUD, this study opens up the option of same-day placement of the LNG 52-mg IUS. While larger trials need to be conducted to obtain more information regarding pregnancy rates with an oral EC plus LNG IUS combination versus the TCu380A, offering such a combination is reasonable. Given that ulipristal acetate (UPA) is a more effective EC product, especially for women who are overweight or obese,9 offering a UPA EC plus LNG IUS combination may be a better alternative.
It is time to remove the STI screening barrier to same-day IUD insertion
Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement [published online ahead of print May 12, 2016]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2016.05017.
Provider concerns regarding the presence of pelvic infection remain a significant barrier to same-day IUD insertion. Older studies suggested a higher risk of pelvic infection in the first 20 days after IUD placement and extrapolated that pelvic infection was related to inserting an IUD in a woman at risk for STI.10 As a result, patients may be restricted from same-day IUD insertion by providers who think that obtaining results of STI testing is required prior to placement. Recently, a systematic review suggested, based on limited evidence, that IUD placement does not increase the risk of pelvic infection in asymptomatic women compared with those without an IUD.11
Turok and colleagues (including M.D.C., coauthor of this article) reported results from a planned secondary analysis of A Comprehensive Contraceptive Efficacy and Safety Study of an IUS (ACCESS IUS), a component of the regulatory approval of the Liletta LNG 52-mg IUS. This analysis represents the first large-scale prospective investigation of pelvic infection rates during the first 2 years after IUD placement in US women.
Details of the study
Of the 1,751 women enrolled in the study, 1,714 had successful IUS insertions. Infection was assessed via baseline pelvic visual and bimanual examination and Chlamydia testing in all women. Gonorrhea testing was also performed in women who had not been tested with their current sexual partner. STI test results were not required for IUS insertion. Participants were assessed in person at 1, 3, 6, 12, and 24 months after insertion. Additional pelvic exams were performed as needed based on reported symptoms. At 6 months, 1 year, and 2 years, IUS continuation was reported in 1,553 (90.6%), 1,401 (81.7%), and 1,157 (67.3%) women, respectively.
Nearly all women received baseline STI testing (98.4%); however, results were not available prior to same-day IUS insertion for 79.6% of participants. Twenty-nine (1.7%) women had positive baseline STI tests (25 for Chlamydia, 3 for gonorrhea, 1 for both). Of these, only 6 women had results available prior to IUS placement. All women with a positive STI test were treated, and the IUS was left in place. Importantly, none of these women developed pelvic infection in the subsequent 2 years of follow-up and none requested IUS removal.
Infection risk is low with IUD placement
Among women with negative baseline STI tests, there were only 9 (0.5%) clinical diagnoses of pelvic infections over the first 2 years of follow-up. Diagnosis was typically made based on physical examination findings. Most women underwent repeat Chlamydia and gonorrhea testing at the time of pelvic infection diagnosis, and none had positive results. There were no medically recommended IUS removals; 2 women with pelvic infection requested IUS removal per their preference.
Three of the 9 women with pelvic infections were diagnosed within 1 week of IUS placement, 1 at 39 days after placement, and the remaining 5 more than 6 months after placement, suggesting that pelvic infection is not temporally related to IUS placement. Most women were successfully treated as outpatients.
What this evidence means for practiceThis study provides further reassurance regarding the low risk of pelvic infection among women with an LNG IUS. Insertion of an IUS should not be delayed to await results of Chlamydia or gonorrhea testing in a woman without clinical evidence of pelvic infection. Risk-based, as opposed to universal testing, is imperative.12 These recommendations are in agreement with current recommendations of the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists.13,14 Practices that employ 2-visit protocols unnecessarily limit women’s access to the IUS, as research has shown that nearly half of women desiring an IUD do not return for device placement if a second encounter is required.15
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Contraception is an important tool that allows patients to carry out their reproductive-life plans. In the United States, the average woman desires 2 children.1 To achieve this goal, she will spend more than 30 years of her reproductive life avoiding pregnancy.1 The most effective reversible contraceptive methods, the intrauterine device (IUD) and the contraceptive implant, offer an efficient way to cover this significant period. Currently, American women more commonly choose an IUD than an implant by a factor 8 to 1.2 Between 2002 and 2012, the percentage of US contraceptive users aged 15 to 44 using the IUD rose from 2% to 10%.2
Significant barriers to contraceptive access still exist, however. Although widespread reports have lauded the decrease in unintended pregnancies in the United States, improvement only has been marginal for women who live below the poverty level. In fact, for unintended pregnancy the gap between women above and below the poverty level has increased from a 2.6-fold difference in 1994 to a 5.6-fold difference in 2011 (FIGURE).3−5 Since the decrease in the unintended pregnancy rate is most likely related to an increase in contraceptive use, particularly the IUD, we are not providing equal contraceptive access to all women.5
Both the copper IUD and the 3 available levonorgestrel (LNG)-releasing intrauterine system (IUS) products provide safe and effective contraception. As IUD research expands, it is imperative for providers to stay up to date so patients can have full access to these devices.
In this article, we present important updates regarding IUD use that will help break down some continuing barriers to contraceptive access, including:
- clinical trial data demonstrating efficacy of LNG 52-mg IUS for 7 years
- a novel emergency contraception (EC) regimen of same-day oral LNG and the LNG 52-mg IUS
- a large prospective trial demonstrating that women can safely have IUS placement without known sexually transmitted infection (STI) screening results and that pelvic infection rates are not higher in the time shortly after IUS placement.
WHO study demonstrates LNG 52-mg IUS is highly effective for up to 7 years of use
Rowe P, Farley T, Peregoudov A, et al; IUD Research Group of the UNDP/UNFPA/WHO/World Bank Special Programme of Research; Development and Research Training in Human Reproduction. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93(6):498−506.
Currently, the 2 LNG 52-mg IUS products (Liletta, Mirena) approved by the US Food and Drug Administration (FDA) are available for use for 3 and 5 years, respectively. The pivotal approval trial for Liletta is still ongoing and is planned to continue for up to 7 years.6 The TCu380A (ParaGard) copper IUD is FDA approved for up to 10 years of use; however, this product initially was approved for only 4 years. The duration of use was expanded to 10 years based on continued clinical trials.
Based on current data, we will not need to wait for the Liletta pivotal trial to have clinical evidence of a longer duration of efficacy for the LNG 52-mg IUS. A collaborative group as part of the UNDP/UNFPA/WHO/World Bank Special Programme of Research, Development and Research Training in Human Reproduction performed a multicenter, open-label randomized controlled trial to evaluate outcomes through 7 years of use of the LNG 52-mg IUS and the TCu380A IUD.
Details of the study
A total of 3,836 women were enrolled at 20 centers in Europe, Asia, South America, and China and were randomly assigned to one of the 2 products. Eligible women were aged 16 to 40 years, parous, and without known leiomyoma or recent pelvic infection. After excluding 15 failed IUD insertions, 1,910 women received an LNG 52-mg IUS and 1,911 received a TCu380A. Ultimately, 398 women in the LNG 52-mg IUS group and 682 in the TCu380A group completed 7-year follow-up with the IUD in place. Women were surveyed regarding pregnancy and method discontinuation.
Lower pregnancy rate, higher discontinuation with LNG IUS
The cumulative 7-year pregnancy rate among LNG 52-mg IUS users was significantly lower than among TCu380A users (0.53 per 100 women vs 2.45 per 100 women, respectively). All pregnancies in the LNG 52-mg IUS group occurred in the first 5 years of study follow-up--with no pregnancies in years 6 through 7 (TABLE). The cumulative pregnancy rate in the TCu380A group in this study is consistent with that in a previous long-term trial of this IUD.7
Early removal was significantly higher in the LNG 52-mg IUS group, with a cumulative discontinuation rate of 70.6 per 100 women, compared with 40.8 per 100 women in the TCu380A group. Significant cultural variation existed when it came to both rate and reason for discontinuation. Most women at Chinese centers cited amenorrhea and decreased bleeding as the primary reason for discontinuation, and they did so at twice the rate of women at non-Chinese centers.
The patterns of method discontinuation in this study were different from those found among US women. By comparison, a recent study in the United States had lower overall discontinuation rates and did not find decreased bleeding to be among the main reasons for LNG 52-mg IUS removal.7 In fact, most women who discontinued the LNG 52-mg IUS cited concerns about upcoming expiration as their reason for removal.
The results of this large study also generally corroborated the low-risk profile of IUDs. Only 1 reported IUD perforation occurred, for a rate of 0.03 per 1,000 women. Device expulsion rates were similar between the IUDs and were uncommon overall with 7-year rates of 8 to 9 per 100 women. Pelvic infection was cited as reason for removal in only 7 women (0.18 per 100 women) over 7 years.
What this evidence means for practiceThis exciting study is the first large-scale clinical trial demonstrating continued high efficacy with LNG 52-mg IUS use through 7 years. This information affords women extended contraceptive coverage. While additional research will be welcome, particularly in younger women who will maintain greater fertility across the IUS’s 7-year life span, we are confident in extending the 7-year duration for this IUS to our patients.
Additionally, the method discontinuation findings in this study highlight the importance of discussing the expected menstrual changes of hormonal IUS use with women prior to insertion so they can determine if the potential changes would be satisfactory. As the acceptability of medical menstrual suppression may be new to many women, providers should frame the adverse effects in this context. Providers can use this opportunity to review the noncontraceptive benefits of the hormonal IUS as well.
Novel combination of LNG 52-mg IUS and oral LNG 1.5 mg is promising for emergency contraception
Turok DK, Sanders JN, Thompson IS, Royer PA, Eggebroten J, Gawron LM. Preference for and efficacy of oral levonorgestrel for emergency contraception with concomitant placement of a levonorgestrel IUD: a prospective cohort study. Contraception. 2016;93(6):526−532.
The copper IUD is superior for EC relative to oral agents and has the added benefit of providing ongoing highly effective contraception after placement.8 Despite this strong evidence, the copper IUD remains underutilized for this indication. Turok and colleagues noted that women in their clinic seeking IUDs for non-EC purposes preferred the LNG 52-mg IUS over the copper IUD. It is understandable that women might carry these preferences into EC encounters as well. Thus, the investigators devised a novel combination of LNG 52-mg IUS and oral LNG 1.5 mg, which provided both known EC benefit and same-day access to a more popular contraceptive device.
Details of the study
Women presenting for EC who desired same-day IUD placement were enrolled in the prospective cohort study. Eligible women had a negative urine pregnancy test, known last menstrual period (LMP), regular menstrual cycle, and reported unprotected intercourse within 120 hours prior to presentation. Importantly, women with multiple episodes of unprotected intercourse in the weeks prior to presentation were also included to provide a population more comparable to that encountered clinically. The women were then offered the choice of a TCu380A copper IUD or oral LNG EC with LNG 52-mg IUS placement. They were counseled on the potential increased risk of pregnancy with the novel oral LNG EC plus LNG IUS combination compared with the copper IUD. Participants were given a home pregnancy test that they were to complete in 2 weeks and then report the results to the clinic.
Of the 1,004 women presenting to the clinic for EC over the 16-month study period, 188 (18%) desired same-day IUD insertion. Of these, more opted for the oral LNG EC plus LNG IUS combination (n = 121, 64%) than the copper IUD (n = 67, 36%), demonstrating that women were often willing to accept a possible decrease in EC efficacy with the goal of obtaining their preferred lUD type.
Excluding failed insertion, undiagnosed uterine didelphys, and patient withdrawal, 110 women received the oral LNG EC plus LNG IUS and 66 received the copper IUD. Demographics were comparable between groups except for body mass index (BMI). Of note, more than half (61%) of the women who opted for the oral LNG EC plus LNG IUS combination were overweight or obese.
Both EC methods are effective, broadening options
All women who received the copper IUD followed up at 2 weeks, and no pregnancies were reported. Of the women who received oral LNG EC plus the LNG IUS, 107 (97%) had follow-up at 2 weeks. In this group, there was 1 reported ectopic pregnancy that ultimately required surgical management. However, further review of the patient's coital history suggested that conception occurred prior to IUD insertion, and the case was not classified by the investigators as an EC failure.
Although this study was not powered to detect pregnancy rate differences between the traditional copper IUD and the oral LNG EC plus LNG IUS combination, the results are promising. An important strength of this study is the presence of 2 high-risk groups for EC failure in the oral LNG EC plus LNG IUS group (elevated BMI and multiple episodes of unprotected intercourse).
What this evidence means for practiceEncounters for EC are important opportunities in which to discuss a woman’s reproductive goals. For women who are interested in an IUD, this study opens up the option of same-day placement of the LNG 52-mg IUS. While larger trials need to be conducted to obtain more information regarding pregnancy rates with an oral EC plus LNG IUS combination versus the TCu380A, offering such a combination is reasonable. Given that ulipristal acetate (UPA) is a more effective EC product, especially for women who are overweight or obese,9 offering a UPA EC plus LNG IUS combination may be a better alternative.
It is time to remove the STI screening barrier to same-day IUD insertion
Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement [published online ahead of print May 12, 2016]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2016.05017.
Provider concerns regarding the presence of pelvic infection remain a significant barrier to same-day IUD insertion. Older studies suggested a higher risk of pelvic infection in the first 20 days after IUD placement and extrapolated that pelvic infection was related to inserting an IUD in a woman at risk for STI.10 As a result, patients may be restricted from same-day IUD insertion by providers who think that obtaining results of STI testing is required prior to placement. Recently, a systematic review suggested, based on limited evidence, that IUD placement does not increase the risk of pelvic infection in asymptomatic women compared with those without an IUD.11
Turok and colleagues (including M.D.C., coauthor of this article) reported results from a planned secondary analysis of A Comprehensive Contraceptive Efficacy and Safety Study of an IUS (ACCESS IUS), a component of the regulatory approval of the Liletta LNG 52-mg IUS. This analysis represents the first large-scale prospective investigation of pelvic infection rates during the first 2 years after IUD placement in US women.
Details of the study
Of the 1,751 women enrolled in the study, 1,714 had successful IUS insertions. Infection was assessed via baseline pelvic visual and bimanual examination and Chlamydia testing in all women. Gonorrhea testing was also performed in women who had not been tested with their current sexual partner. STI test results were not required for IUS insertion. Participants were assessed in person at 1, 3, 6, 12, and 24 months after insertion. Additional pelvic exams were performed as needed based on reported symptoms. At 6 months, 1 year, and 2 years, IUS continuation was reported in 1,553 (90.6%), 1,401 (81.7%), and 1,157 (67.3%) women, respectively.
Nearly all women received baseline STI testing (98.4%); however, results were not available prior to same-day IUS insertion for 79.6% of participants. Twenty-nine (1.7%) women had positive baseline STI tests (25 for Chlamydia, 3 for gonorrhea, 1 for both). Of these, only 6 women had results available prior to IUS placement. All women with a positive STI test were treated, and the IUS was left in place. Importantly, none of these women developed pelvic infection in the subsequent 2 years of follow-up and none requested IUS removal.
Infection risk is low with IUD placement
Among women with negative baseline STI tests, there were only 9 (0.5%) clinical diagnoses of pelvic infections over the first 2 years of follow-up. Diagnosis was typically made based on physical examination findings. Most women underwent repeat Chlamydia and gonorrhea testing at the time of pelvic infection diagnosis, and none had positive results. There were no medically recommended IUS removals; 2 women with pelvic infection requested IUS removal per their preference.
Three of the 9 women with pelvic infections were diagnosed within 1 week of IUS placement, 1 at 39 days after placement, and the remaining 5 more than 6 months after placement, suggesting that pelvic infection is not temporally related to IUS placement. Most women were successfully treated as outpatients.
What this evidence means for practiceThis study provides further reassurance regarding the low risk of pelvic infection among women with an LNG IUS. Insertion of an IUS should not be delayed to await results of Chlamydia or gonorrhea testing in a woman without clinical evidence of pelvic infection. Risk-based, as opposed to universal testing, is imperative.12 These recommendations are in agreement with current recommendations of the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists.13,14 Practices that employ 2-visit protocols unnecessarily limit women’s access to the IUS, as research has shown that nearly half of women desiring an IUD do not return for device placement if a second encounter is required.15
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Office of Population Affairs, Department of Health and Human Services. Family planning program, FY 1999 service program grants by state. Bethesda, MD: Office of Population Affairs; 1999.
- Kavanaugh ML, Jerman J, Finer LB. Changes in use of long-acting reversible contraceptive methods among US women, 2009-2012. Obstet Gynecol. 2015;126(5):917–927.
- Ventura SJ, Abma JC, Mosher WD, Henshaw S. Revised pregnancy rates, 1990-97, and new rates for 1998-99: United States. Natl Vital Stat Rep. 2003;52(7):1−14.
- Finer LB, Zolna MR. Shifts in intended and unintended pregnancies in the United States, 2001-2008. Am J Public Health. 2014;104(suppl 1):S43–S48.
- Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008-2011. N Engl J Med. 2016;374(9):843–852.
- Eisenberg DL, Schreiber CA, Turok DK, Teal SB, Westhoff CL, Creinin MD; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92(1):10–16.
- United Nations Development Programme, United Nations Population Fund, World Health Organization, World Bank, Special Programme of Research, Development, and Research Training in Human Reproduction. Long-term reversible contraception: twelve years of experience with the TCu380A and TCu220C. Contraception. 1997;56(6):341–352.
- Cleland K, Zhu H, Goldstuck N, Cheng L, Trussell J. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27(7):1994–2000.
- Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84(4):363–367.
- Farley TM, Rosenberg MJ, Rowe PJ, Chen JH, Meirik O. Intrauterine devices and pelvic inflammatory disease: an international perspective. Lancet. 1992;339(8796):785–788.
- Jatlaoui TC, Simmons KB, Curtis KM. The safety of intrauterine contraception initiation among women with current asymptomatic cervical infections or at increased risk of sexually transmitted infections [published online ahead of print June 1, 2016]. Contraception. doi:10.1016/j.contracep tion.2016.05.013.
- Grentzer JM, Peipert JF, Zhao Q, McNicholas C, Secura GM, Madden T. Risk-based screening for Chlamydia trachomatis and Neisseria gonorrhoeae prior to intrauterine device insertion. Contraception. 2015;92(4):313–318.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 121: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2011;118(1):184–196.
- Centers for Disease Control and Prevention. US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd ed. MMWR Recomm Rep. 2013;62(RR05):1–46.
- Bergin A, Tristan S, Terplan M, Gilliam ML, Whitaker AK. A missed opportunity for care: two-visit IUD insertion protocols inhibit placement. Contraception. 2012;86(6):694–697.
- Office of Population Affairs, Department of Health and Human Services. Family planning program, FY 1999 service program grants by state. Bethesda, MD: Office of Population Affairs; 1999.
- Kavanaugh ML, Jerman J, Finer LB. Changes in use of long-acting reversible contraceptive methods among US women, 2009-2012. Obstet Gynecol. 2015;126(5):917–927.
- Ventura SJ, Abma JC, Mosher WD, Henshaw S. Revised pregnancy rates, 1990-97, and new rates for 1998-99: United States. Natl Vital Stat Rep. 2003;52(7):1−14.
- Finer LB, Zolna MR. Shifts in intended and unintended pregnancies in the United States, 2001-2008. Am J Public Health. 2014;104(suppl 1):S43–S48.
- Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008-2011. N Engl J Med. 2016;374(9):843–852.
- Eisenberg DL, Schreiber CA, Turok DK, Teal SB, Westhoff CL, Creinin MD; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92(1):10–16.
- United Nations Development Programme, United Nations Population Fund, World Health Organization, World Bank, Special Programme of Research, Development, and Research Training in Human Reproduction. Long-term reversible contraception: twelve years of experience with the TCu380A and TCu220C. Contraception. 1997;56(6):341–352.
- Cleland K, Zhu H, Goldstuck N, Cheng L, Trussell J. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27(7):1994–2000.
- Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84(4):363–367.
- Farley TM, Rosenberg MJ, Rowe PJ, Chen JH, Meirik O. Intrauterine devices and pelvic inflammatory disease: an international perspective. Lancet. 1992;339(8796):785–788.
- Jatlaoui TC, Simmons KB, Curtis KM. The safety of intrauterine contraception initiation among women with current asymptomatic cervical infections or at increased risk of sexually transmitted infections [published online ahead of print June 1, 2016]. Contraception. doi:10.1016/j.contracep tion.2016.05.013.
- Grentzer JM, Peipert JF, Zhao Q, McNicholas C, Secura GM, Madden T. Risk-based screening for Chlamydia trachomatis and Neisseria gonorrhoeae prior to intrauterine device insertion. Contraception. 2015;92(4):313–318.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 121: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2011;118(1):184–196.
- Centers for Disease Control and Prevention. US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd ed. MMWR Recomm Rep. 2013;62(RR05):1–46.
- Bergin A, Tristan S, Terplan M, Gilliam ML, Whitaker AK. A missed opportunity for care: two-visit IUD insertion protocols inhibit placement. Contraception. 2012;86(6):694–697.
In this article
- Extended use of LNG IUS
- Oral LNG and LNG IUS combo for emergency contraception
- STI screening and same-day IUD placement