User login
Liquid Levodopa Formulation May Simplify Continuous Drug Delivery
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.

Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
VANCOUVER—A liquid formulation of levodopa and carbidopa delivered continuously and subcutaneously is generally well tolerated and may reduce motor fluctuations in patients with Parkinson’s disease, according to research described at the 68th Annual Meeting of the American Academy of Neurology.
Levodopa has a short half-life, and long-term oral levodopa treatment is associated with motor fluctuations and dyskinesia. Continuous levodopa delivery may be the best way to overcome those limitations of the drug, said Sheila Oren, MD, MBA, Vice President of Clinical and Regulatory Affairs at NeuroDerm in Rehovot, Israel. Current continuous infusion systems, however, must be surgically implanted to permit direct infusion of the drug in gel form into the jejunum.
Sheila Oren, MD, MBA
Until recently, poor levodopa solubility had prevented the development of a liquid formulation of the drug that could be delivered subcutaneously. ND0612L, a novel, proprietary formulation of levodopa and carbidopa, “is the first time that this [drug] is a real liquid,” Dr. Oren said. “Not a suspension or a gel, but a liquid that can be administered parenterally.… In this way, we hope to be able to deliver levodopa continuously in a much simpler way.”
Safety and Tolerability Trial
To study the pharmacokinetic and clinical profile of ND0612L in patients with Parkinson’s disease with motor fluctuations, Dr. Oren and colleagues conducted a phase II randomized, placebo-controlled, double-blind study in 30 patients with Parkinson’s disease.
Patients were randomized two-to-one to receive a low-dose formulation of ND0612L (ie, 60 mg of levodopa and 14 mg of carbidopa per milliliter) or placebo in addition to their standard of care oral treatment for two weeks. A belt pump system delivered ND0612L or placebo subcutaneously at a nightly rate of 0.08 mL/h for eight hours (ie, from 6 pm to 2 am) and then at a higher daily rate of 0.24 mL/h for the next 16 hours.
Investigators enrolled patients with idiopathic Parkinson’s disease who had more than two waking hours of off time per day and took optimized doses of levodopa three or more times per day. The researchers excluded people taking controlled-release levodopa formulations, people who had undergone neurosurgical intervention for Parkinson’s disease, and people with severe, disabling dyskinesia.
Participants had a mean age of approximately 64 and mean disease duration of about eight years. Participants had received levodopa therapy for about seven years and took 700 mg of levodopa daily, on average. Participants had experienced motor fluctuations and dyskinesias for several years, and their average total daily off time was almost six hours.ND0612L was generally well tolerated and safe. Cutaneous side effects included mild and transient edema and erythema at the injection site. In addition, most patients developed at least one small, painless nodule, Dr. Oren said. Nodules typically resolved within two months. Researchers did not observe systemic adverse events, dyskinesia, or psychiatric symptoms.
Compared with the placebo group, patients who received adjunctive ND0612L had a reduction in plasma levodopa concentration fluctuations. Their plasma levodopa concentrations consistently remained above a mean of 800 ng/mL, thus avoiding the low trough levels that occurred in the placebo group. The ND0612L group had a lower peak-to-trough ratio and fluctuation index, compared with the placebo group.
Preliminary Evidence of Efficacy
In an exploratory efficacy analysis, researchers observed that ND0612L treatment reduced off time in clinic by 2.42 hours, compared with a 0.41-hour reduction with placebo.
Treatment also improved sleep quality (Parkinson’s Disease Sleep Scale score improvement from baseline of 17.1 with ND0612L vs 0.5 with placebo), quality of life (Parkinson’s Disease Questionnaire-39 score improvement of 6.6 with ND0612L vs 1.78 with placebo), and global impression (90% of patients treated with ND0612L had improved Global Impression of Change scores, compared with 36% of patients treated with placebo).
Investigators are developing a way to deliver ND0612L via a patch pump. Investigators also are investigating a high-dose formulation of the drug, ND0612H, developed for advanced patients as an alternative to surgical interventions. Researchers plan to conduct further studies of the drug’s safety, pharmacokinetics, and potential clinical benefits, Dr. Oren said.
—Jake Remaly
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.
Suggested Reading
Senek M, Nyholm D. Continuous drug delivery in Parkinson’s disease. CNS Drugs. 2014;28(1):19-27.

Using a Combination of Therapies to Manage Rosacea

What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.
What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.
What do your patients need to know at the first visit?
Patients need to understand that rosacea has no gender, age, or race predilection. It is caused by a personal and genetic proinflammatory predisposition. Rosacea patients seem to have a genetic predisposition to overproduce cathelicidins, a small antimicrobial peptide that is produced by the action of stratum corneum tryptic enzyme. They have more production and abnormal forms of cathelicidins produced by high levels of stratum corneum tryptic enzyme. This upregulation of inflammatory cathelicidins on the dermis is associated with vascular instability and exacerbated by triggers such as sunlight, hot drinks, spicy foods, stress, and rapid changing weather.
I divide the condition into proinflammatory predisposition, vascular instability with redness, Demodex infection of the hair follicle, and sebaceous gland overgrowth. More recently, the bacterium Bacillus oleronius was isolated from inside a Demodex mite and was found to produce molecules provoking an immune reaction in rosacea patients (Erbaguci and Ozgöztaşi). Other studies have shown that patients with varying types of rosacea react to the molecules produced by this bacterium, exposing it as a likely trigger for the condition (Li et al). What’s more, this bacterium is sensitive to the antibiotics used to treat rosacea.
What are your go-to treatments? What are the side effects?
For inflammation I prescribe anti-inflammatory (low dose) or antibacterial (high dose) doses of doxycycline and/or anti-inflammatory azelaic acid gel 15% twice daily after application of barrier repair topical hyaluronic acid. For the vascular component I use the temporary relief from the application of brimonidine gel 0.33% in the morning in addition to the topical given for inflammatory rosacea, and the more durable excel V (532 and 1064 nm) laser. Ultimately, topical ivermectin is prescribed for those patients who do not respond to previously mentioned treatments for coverage of Demodex infestation. For rhinophyma I offer a surgical approach and laser treatments; surgical removal of the excess glandular growth is followed by fractional ablative and nonablative treatments for scar reduction after surgery.
All patients should apply an inorganic sun protection factor 50+ sunblock with titanium dioxide and zinc oxide to prevent sunlight from being a trigger. All patients are encouraged to avoid triggers.
I try to prevent the potential side effects associated with rosacea treatments. For example, applying barrier repair hyaluronic acid before azelaic acid to prevent irritation and telling patients they might have vascular rebound phenomena with more redness after brimonidine application wears off. I also explain to patients that laser treatments induce temporary erythema and swelling that may last 3 days.
How do you keep patients compliant with treatment?
In general, my patients are compliant with their treatments, which I ascribe to the simplicity of a twice-daily regimen that is written for them. They understand that I design a treatment regimen for each individual patient based on his/her presentation.
What resources do you recommend to patients for more information?
I recommend web-based resources that can provide further assistance and information, such as the American Academy of Dermatology website (https://www.aad.org/public/diseases/acne-and-rosacea/rosacea), National Rosacea Society (www.rosacea.org), and specific disease foundations (eg, International Rosacea Foundation [www.internationalrosaceafoundation.org]).Suggested Readings
- Erbaguci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425.
- Li J, O’Reilly N, Sheha H, et al. Correlation between ocular Demodex infestation and serum immunoreactivity to Bacillus proteins in patients with facial rosacea. Ophthalmology. 2010;117:870-877.

NSAID plus TNFi linked to less ankylosing spondylitis progression
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.

These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Ankylosing spondylitis patients treated with a TNF inhibitor plus an NSAID had the lowest level of new bone formation during treatment for more than 2 years.
Major finding: Patients on a TNF inhibitor and an NSAID had 67% less new bone formation, compared with patients not on an NSAID.
Data source: Cross-sectional cohort study of 527 patients with ankylosing spondylitis enrolled in the Prospective Study of Outcomes in Ankylosing Spondylitis.
Disclosures: Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.

NSAID plus TNFi linked to less ankylosing spondylitis progression
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
LONDON – Patients with ankylosing spondylitis who remained on long-term treatment with a nonsteroidal anti-inflammatory drug and a tumor necrosis factor inhibitor had significantly less new spinal-bone formation in a cross-sectional analysis of a multicenter cohort of 527 U.S. patients.
A related analysis of the same cohort also showed significantly less ankylosing spondylitis (AS) progression as measured by radiographic progression among patients who received treatment with a tumor necrosis factor–alpha (TNF) inhibitor for 2.1-3.5 years regardless of their treatment with a nonsteroidal anti-inflammatory drug (NSAID), although this link trended to a stronger effect among the patients taking both, Lianne S. Gensler, MD, reported in a pair of posters at the European Congress of Rheumatology.
These finding suggest “there may be synergy between NSAIDs and TNF inhibitors [for slowing or preventing progression] in a select group of AS patients at high risk for progression,” said Dr. Gensler, a rheumatologist and director of the Ankylosing Spondylitis Clinic at the University of California, San Francisco.
But Dr. Gensler also cautioned that these findings are merely “hypothesis generating” and should not be used as a rationale to place or maintain AS patients on long-term treatment with an NSAID, a TNF inhibitor, or both drugs.
“You treat the disease burden. The message is absolutely not to always put AS patients on both types of drugs. When an AS patient is well controlled on a TNF inhibitor alone, I would not tell them to also take a NSAID,” she said in an interview. “This is only relevant for patients who require treatment with both drug classes because of their clinical status.”
As the list of treatment options for patients with AS grows – it now includes NSAIDs, TNF inhibitors, and the interleukin-17 inhibitor secukinumab (Cosentyx) – the impact of these agents on disease progression as assessed by radiography and new bone formation becomes a new dimension to start to consider in addition to the standard criterion of immediate clinical response, Dr. Gensler explained. AS progression “is another factor to think about as we decide on treatment strategies. There is growing evidence that long-term treatment with a tumor necrosis factor inhibitor and with a NSAID have potential roles in disease modification.” But the evidence is indirect, from cohort studies that make cause and effect assessments difficult because of possible unidentified confounding factors, she noted.
“There has never been a randomized, controlled trial examining the disease-modifying effects of a TNF inhibitor because you can’t keep patients on placebo for a long enough time to see this benefit,” Dr. Gensler said. It takes a long time to see progression in AS patients, she noted.
A prior cohort analysis run by Dr. Gensler and her associates found evidence for an effect of long-term treatment with a TNF inhibitor and reduced AS progression measured using the modified Stoke AS Spine Score (mSASSS), compared with AS patients not on a TNF inhibitor in a propensity-score matched analysis of 334 patients. A link between TNF inhibitor use and a discernible difference in mSASSS only occurred when patients were on TNF inhibitor treatment for at least 3.9 years (Arthritis Rheum. 2013 Oct;65[10]:2645-54). In addition, a separate report at the EULAR congress on 168 AS patients maintained on treatment with secukinumab for 2 years showed evidence for slowed progression of mSASSS scores, compared with historical controls as well as with similar patients who were not on secukinumab treatment for as long a period of time.
The new analysis reported by Dr. Gensler looked at 527 AS patients in the multicenter Prospective Study of Outcomes in AS cohort followed for a median of 3.7 years. Clinicians participating in this cohort saw patients every 6 months, and radiographic assessments by mSASSS and for new bone formation occurred every 2 years. At entry into the registry, about 57% of patients received a TNF inhibitor and about 63% received an NSAID, with a third on an NSAID only, 27% on a TNF inhibitor only, 30% on both drugs, and 10% receiving neither drug.
The analysis showed that among patients followed for 2.1-3.5 years, the fraction of patients on a TNF inhibitor who showed progression of their mSASSS was 77% lower than patients not on a TNF inhibitor, a statistically significant difference, Dr. Gensler reported. The researchers saw no statistically significant difference in mSASSS progression rates between the patients on a TNF inhibitor at baseline and those not on a TNF inhibitor at baseline among patients followed for 2 years and among those followed for more than 3.5 years, although the analysis did show nominally higher levels of response among TNF-inhibitor users followed for more than 3.5 years. Dr. Gensler speculated that one reason for the loss of a statistically significant difference during longer follow-up could be that fewer patients reached these levels of prolonged follow-up, making statistically significant differences harder to see. This analysis also showed a strong trend for less progression among the patients treated with an NSAID, a 51% relative reduction in mSASSS progression, compared with patients not taking an NSAID, but this relationship just missed statistical significance.
A second analysis by Dr. Gensler and her associates used data from the same cohort but focused on new bone formation during follow-up. This analysis again showed a statistically significant, 72% reduction in this outcome among patients taking a TNF inhibitor at baseline, compared with those not on a TNF inhibitor, when followed for 2.1-3.5 years, with no statistically significant relationship seen among patients followed for less or more time, Dr. Gensler reported. However, the results from this analysis also showed a statistically significant impact from NSAID treatment: Patients on a TNF inhibitor and an NSAID at baseline had 67% less new bone formation, compared with those who received a TNF inhibitor but were not on an NSAID at baseline.
Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Ankylosing spondylitis patients treated with a TNF inhibitor plus an NSAID had the lowest level of new bone formation during treatment for more than 2 years.
Major finding: Patients on a TNF inhibitor and an NSAID had 67% less new bone formation, compared with patients not on an NSAID.
Data source: Cross-sectional cohort study of 527 patients with ankylosing spondylitis enrolled in the Prospective Study of Outcomes in Ankylosing Spondylitis.
Disclosures: Dr. Gensler has been a consultant to or investigator funded by AbbVie, Amgen, Janssen, Novartis, and UCB. The Prospective Study of Outcomes in Ankylosing Spondylitis receives no commercial support.

Should Medical Marijuana and Cannabinoids Be Used to Treat Epilepsy?
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.

“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.
“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes
VANCOUVER—“Neurologists are often asked to ‘certify’ patients, but should you recommend or support medical marijuana use for patients with epilepsy?”
Daniel Friedman, MD, MSc, Assistant Professor of Neurology at New York University Langone School of Medicine in New York, addressed this question in his presentation at the 68th Annual Meeting of the American Academy of Neurology.
“There is a disconnect between what the patient, families, and the public understand about cannabinoid therapies for epilepsy and what we as a neurologic community know,” he said. “Much of human evidence for the efficacy of cannabinoids for the treatment of epilepsy is anecdotal” and, of the “few controlled trials, most are small and methodologically flawed.”
A Brief History of Medical Marijuana
Cannabis sativa was first cultivated in approximately 8,000 BCE in China for rope, then medicinally for conditions ranging from menstruation to absentmindedness and, eventually, more than 100 ailments. In the early 19th century, W.B. O’Shaughnessy introduced cannabis to England, where Gowers and other Victorian neurologists used the “Indian hemp” to treat epilepsy.
Medicinal use of cannabis dwindled in the 1930s due to a move to synthetic medicine and Prohibition, which also “coincided with the beginning of the era of rigorous scientific investigation of drug claims,” said Dr. Friedman. Now, “the discovery of the endocannabinoid system in the brain has sparked new research into the therapeutic potential of cannabinoids.”
The Chemicals in Cannabis
The genus Cannabis includes C. sativa, C. indica, and C. ruderalis. A total of 85 phytocannabinoids are found in Cannabis species plants, and cannabidiol (CBD) and delta-9 tetrahydrocannabinol (THC) are the most abundant neuroactive chemicals; others include the terpenes and flavonoids.
CBD-rich oil is a concentrate made from cannabis bred to have a low level of THC and a high level of CBD. GW Pharma manufactures a solution with equal parts purified plant-derived THC and CBD (Sativex). The company also makes an investigational agent, pure CBD (Epidiolex), a 98% purified plant-derived CBD. INSYS Therapeutics is developing a synthetic CBD that is structurally identical to the plant-derived compound.
What Is the Evidence?
Experimental and human studies have provided evidence for the anticonvulsant properties of CBD and THC, and antiseizure effects have also been observed with cannabidivarin (CBDV).
Since 2014, more than 10 US centers have been using Epidiolex under an FDA expanded-access program to treat patients between ages 1 and 30 (median age, 10.5) with severe, treatment-resistant, childhood-onset epilepsy. Patients’ baseline motor seizure rate was 29.5 per month (range, 11.0–96.0 per month).
As of January 2015, 162 patients had completed 12 weeks of observation. Preliminary results showed median motor seizure reduction to be 36.5%. Patients with Dravet syndrome had a 49.8% reduction in seizures, and patients with Lennox-Gastaut syndrome had a 36.8% reduction. Serious adverse events, primarily status epilepticus, were observed in 12% of participants. Only 3% of patients discontinued treatment, however. Commonly reported adverse events included somnolence (25.3%), decreased appetite (19.1%), diarrhea (19.1%), fatigue (13.0%), and convulsion (11.1%).
In addition, children with Dravet syndrome in a phase III trial of Epidiolex had a median reduction in motor seizures of 39% versus 13% for placebo.
“Much of the safety data for chronic cannabinoid use” has come from studies of recreational use, which is “inherently confounded,” Dr. Friedman said. Acute affects include impaired memory, judgment, and motor performance. Chronic use leads to addiction in approximately 9% of users, as well as to cognitive impairment, decreased motivation, and increased risk for psychotic disorders.
Researchers examined pooled data from adults with multiple sclerosis who used cannabinoids for spasticity, pain, and dyskinesias for less than six months. The data encompassed approximately 1,600 exposures to the drugs. About 7% of participants discontinued treatment due to adverse events that included nausea, behavioral and mood changes, suicidality, hallucinations, dizziness, and weakness.
CBD and THC are inhibitors of P450 isozymes, primarily CYP2C and CYP3A, but drug–drug interaction effects are not typically seen in doses used in human studies, said Dr. Friedman. At low micromolar concentrations, CBD inhibits CYP2C19, an enzyme involved in the metabolism of N-desmethylclobazam, phenytoin, diazepam, citalopram, and some tricyclic antidepressants. The CYP2B family may also be induced, thus affecting valproate and clobazam metabolism.
Drugs that affect CBD metabolism include the CYP3A4 inducers, such as carbamazepine and phenytoin, and the CYP3A4 inhibitors such as ketoconazole.
Randomized controlled trials of CBD and CBDV are in progress in patients with Dravet syndrome, Lennox-Gastaut syndrome, and refractory focal epilepsy.
Is Cannabis Right for My Patient?
Since New York State approved the medical use of cannabis, Dr. Friedman has discussed the drug in one of every four of his clinic visits. Deciding whether to recommend medical marijuana for a patient can be difficult, however.
“Would you authorize its use for a 22-year-old patient with treatment-resistant epilepsy who is not a candidate for epilepsy surgery, is having two complex partial seizures a week and one tonic-clonic seizure per month, and is currently on three antiepileptic drugs [AEDs] at high doses,” asked Dr. Friedman. “How about for a 63-year-old woman with focal motor seizures following a meningioma resection who has not been able to tolerate adequate doses of four prior AEDs?”
The answer depends on myriad factors, not the least of which is the legal status of medical marijuana in the neurologist’s state. Although “23 states and the District of Columbia have approved medical marijuana for certain conditions, including epilepsy,” the US Drug Enforcement Administration still considers cannabis and its derivatives schedule I compounds, which means that they have “no accepted medical use in the US,” a high abuse potential, and cannot be prescribed—only “recommended”—by a physician, saidDr. Friedman.
Patients should be evaluated at a comprehensive epilepsy center to determine whether they have been unable to achieve control with conventional therapies due to lack of efficacy or side effects, and whether other proven effective therapies, such as vagus nerve stimulation, a ketogenic diet, or surgery, have been considered, he added.
For patients who do initiate treatment with medical marijuana, the risks and benefits should be carefully weighed, and a treatment plan that includes a timeline for discontinuation should be developed. Laboratory values and clinical status should be monitored regularly.
“Perhaps one day we’ll have [cannabis] in the pharmacy,” Dr. Freidman concluded. Until then, patients should be cautioned against buying cannabinoids on the Internet because, as one FDA study showed, products bought from such sources may have no detectable cannabinoid levels.
—Debra Hughes

Maternal Exposure to Pregabalin May Cause Birth Defects
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
First-trimester exposure to pregabalin may increase the risk of major birth defects, according to a study published online ahead of print May 18 in Neurology.
Pregabalin is an FDA-approved treatment for seizures and neuropathic pain. It is also a common off-label treatment for restless legs syndrome, cyclic mood disorders, and generalized anxiety disorder.
Ursula Winterfeld, PhD, of the Swiss Teratogen Information Service and Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland, and colleagues conducted a multicenter, observational cohort study in which they compared pregnancy outcomes of 164 women exposed to pregabalin with 656 controls who were not exposed to any known teratogenic medications or antiepileptic drugs. Data for this study were collected from 2004 to 2013 and included data from France, the United Kingdom, Italy, Finland, Switzerland, the Netherlands, and Turkey.
Of the women on the medication, 77% started taking pregabalin before they became pregnant and stopped taking the drug at a median of six weeks into their pregnancies. Of the women taking pregabalin, 13% were also taking another antiepileptic drug.
Pregnancies of the women who took pregabalin during the first trimester of pregnancy were three times more likely to result in major birth defects than those of women who did not take the drug—6.0% versus 2.1%, respectively. The major birth defects included heart defects and structural problems with the CNS or other organ systems. The study also revealed a lower rate of live births in the pregabalin group due to elective and medically indicated pregnancy terminations.
“We can’t draw any definitive conclusions from this study, since many of the women were taking other drugs that could have played a role in the birth defects and because the study was small and the results need to be confirmed with larger studies, but these results do signal that there may be an increased risk for major birth defects after taking pregabalin during the first trimester of pregnancy,” said Dr. Winterfeld.
She suggested that before a woman is prescribed pregabalin, it is important to make sure the benefits outweigh the risks and that she is carefully informed about the use of effective birth control.
—Adaeze Stephanie Onyechi
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].
Suggested Reading
Winterfeld U, Merlob P, Baud D, et al. Pregnancy outcome following maternal exposure to pregabalin may call for concern. Neurology. 2016 May 18 [Epub ahead of print].
Pennell PB, Meador KJ. A common medication for neuropsychiatric illness may cause common problems in pregnancy. Neurology. 2016 May 18 [Epub ahead of print].
Primary care management of sepsis survivors does not improve mental health quality of life
Patients who have survived sepsis or septic shock do not receive any significant benefit in the quality of their mental health by receiving primary care management intervention, according to a new study published by JAMA.
“Many survivors of sepsis have multiple medical comorbidities that are typically managed in primary care [but] interventions for managing sepsis sequelae in primary care have not been developed,” states the study, which was led by Jochen Gensichen, MD, of the Institute of General Practice & Family Medicine at Jena (Germany) University Hospital.
“To our knowledge, this is the first large-scale, randomized controlled clinical trial of an intervention to improve outcomes in survivors of sepsis in primary care,” Dr. Gensichen and his coinvestigators added.
The study recruited sepsis and septic shock survivors from nine ICUs across Germany between February 2011 and December 2014, excluding any patients with cognitive impairment, defined as a Telephone Interview of Cognitive Status score no greater than 27. Ultimately, 291 patients aged 18 years or older (mean age of 61.6 years) were selected for inclusion and randomized into cohorts receiving either primary care–based intervention (n = 148) or usual care (n = 143) (JAMA. 2016;315:2703-11. doi: 10.1001/jama.2016.7207).
Those assigned to the usual care cohort received the standard care that their primary care providers would normally carry out, which included “periodic contacts, referrals to specialists, and prescription of medication and therapeutic aids at quantities comparable with those for other populations with multiple chronic conditions.” Those in the other cohort were given active monitoring of symptoms from providers who had been given evidence-based care training and clinical decision support from nurses who underwent training to become case managers. Case managers would take patients through an hour-long face-to-face training on sepsis sequelae within 2-20 days of ICU discharge, along with subsequent follow-up conversations over the phone.
“Case managers monitored patients’ symptoms using validated screening tools to assess critical illness polyneuropathy/myopathy, wasting, neurocognitive deficits, [posttraumatic stress disorder], depressive and pain symptoms, as well as patient self-management behaviors focusing on physical activity and individual self-management goals,” the authors said, noting that case managers would report their results to a consulting physician who “supervised the case managers and provided clinical decision support to the [primary care physicians].”
Baseline Mental Component Summary (MCS) scores were taken for subjects in both cohorts to determine mental health-based quality of life, which averaged 49.1 for the intervention cohort and 49.3 for the control. MCS scores at 6 months’ follow-up were 52.9 for the intervention group (95% confidence interval, 1.05-6.54) and 51.0 for the control group (95% CI, –1.22-4.51), for a mean change of 3.8 in the intervention cohort and 1.6 for the control group. The mean treatment effect was 2.15 (95% CI, –1.79-6.09; P = .28), indicating no significant difference between the two.
“There was no evidence for a differential treatment effect on the study’s primary outcome, postsepsis MCS scores,” the authors concluded. “This finding is similar to those from previous trials of care management interventions following critical illness.”
The authors added that “further research is needed to determine if modified approaches to primary care management may be more effective.”
The study was funded by the Center for Sepsis Control and Care, the German Federal Ministry of Education and Research, and the German Sepsis Society. Dr. Gensichen reported receiving personal fees from the Primary Health Care Foundation and receiving a grant from the German Federal Ministry of Education and Research.
Patients who have survived sepsis or septic shock do not receive any significant benefit in the quality of their mental health by receiving primary care management intervention, according to a new study published by JAMA.
“Many survivors of sepsis have multiple medical comorbidities that are typically managed in primary care [but] interventions for managing sepsis sequelae in primary care have not been developed,” states the study, which was led by Jochen Gensichen, MD, of the Institute of General Practice & Family Medicine at Jena (Germany) University Hospital.
“To our knowledge, this is the first large-scale, randomized controlled clinical trial of an intervention to improve outcomes in survivors of sepsis in primary care,” Dr. Gensichen and his coinvestigators added.
The study recruited sepsis and septic shock survivors from nine ICUs across Germany between February 2011 and December 2014, excluding any patients with cognitive impairment, defined as a Telephone Interview of Cognitive Status score no greater than 27. Ultimately, 291 patients aged 18 years or older (mean age of 61.6 years) were selected for inclusion and randomized into cohorts receiving either primary care–based intervention (n = 148) or usual care (n = 143) (JAMA. 2016;315:2703-11. doi: 10.1001/jama.2016.7207).
Those assigned to the usual care cohort received the standard care that their primary care providers would normally carry out, which included “periodic contacts, referrals to specialists, and prescription of medication and therapeutic aids at quantities comparable with those for other populations with multiple chronic conditions.” Those in the other cohort were given active monitoring of symptoms from providers who had been given evidence-based care training and clinical decision support from nurses who underwent training to become case managers. Case managers would take patients through an hour-long face-to-face training on sepsis sequelae within 2-20 days of ICU discharge, along with subsequent follow-up conversations over the phone.
“Case managers monitored patients’ symptoms using validated screening tools to assess critical illness polyneuropathy/myopathy, wasting, neurocognitive deficits, [posttraumatic stress disorder], depressive and pain symptoms, as well as patient self-management behaviors focusing on physical activity and individual self-management goals,” the authors said, noting that case managers would report their results to a consulting physician who “supervised the case managers and provided clinical decision support to the [primary care physicians].”
Baseline Mental Component Summary (MCS) scores were taken for subjects in both cohorts to determine mental health-based quality of life, which averaged 49.1 for the intervention cohort and 49.3 for the control. MCS scores at 6 months’ follow-up were 52.9 for the intervention group (95% confidence interval, 1.05-6.54) and 51.0 for the control group (95% CI, –1.22-4.51), for a mean change of 3.8 in the intervention cohort and 1.6 for the control group. The mean treatment effect was 2.15 (95% CI, –1.79-6.09; P = .28), indicating no significant difference between the two.
“There was no evidence for a differential treatment effect on the study’s primary outcome, postsepsis MCS scores,” the authors concluded. “This finding is similar to those from previous trials of care management interventions following critical illness.”
The authors added that “further research is needed to determine if modified approaches to primary care management may be more effective.”
The study was funded by the Center for Sepsis Control and Care, the German Federal Ministry of Education and Research, and the German Sepsis Society. Dr. Gensichen reported receiving personal fees from the Primary Health Care Foundation and receiving a grant from the German Federal Ministry of Education and Research.
Patients who have survived sepsis or septic shock do not receive any significant benefit in the quality of their mental health by receiving primary care management intervention, according to a new study published by JAMA.
“Many survivors of sepsis have multiple medical comorbidities that are typically managed in primary care [but] interventions for managing sepsis sequelae in primary care have not been developed,” states the study, which was led by Jochen Gensichen, MD, of the Institute of General Practice & Family Medicine at Jena (Germany) University Hospital.
“To our knowledge, this is the first large-scale, randomized controlled clinical trial of an intervention to improve outcomes in survivors of sepsis in primary care,” Dr. Gensichen and his coinvestigators added.
The study recruited sepsis and septic shock survivors from nine ICUs across Germany between February 2011 and December 2014, excluding any patients with cognitive impairment, defined as a Telephone Interview of Cognitive Status score no greater than 27. Ultimately, 291 patients aged 18 years or older (mean age of 61.6 years) were selected for inclusion and randomized into cohorts receiving either primary care–based intervention (n = 148) or usual care (n = 143) (JAMA. 2016;315:2703-11. doi: 10.1001/jama.2016.7207).
Those assigned to the usual care cohort received the standard care that their primary care providers would normally carry out, which included “periodic contacts, referrals to specialists, and prescription of medication and therapeutic aids at quantities comparable with those for other populations with multiple chronic conditions.” Those in the other cohort were given active monitoring of symptoms from providers who had been given evidence-based care training and clinical decision support from nurses who underwent training to become case managers. Case managers would take patients through an hour-long face-to-face training on sepsis sequelae within 2-20 days of ICU discharge, along with subsequent follow-up conversations over the phone.
“Case managers monitored patients’ symptoms using validated screening tools to assess critical illness polyneuropathy/myopathy, wasting, neurocognitive deficits, [posttraumatic stress disorder], depressive and pain symptoms, as well as patient self-management behaviors focusing on physical activity and individual self-management goals,” the authors said, noting that case managers would report their results to a consulting physician who “supervised the case managers and provided clinical decision support to the [primary care physicians].”
Baseline Mental Component Summary (MCS) scores were taken for subjects in both cohorts to determine mental health-based quality of life, which averaged 49.1 for the intervention cohort and 49.3 for the control. MCS scores at 6 months’ follow-up were 52.9 for the intervention group (95% confidence interval, 1.05-6.54) and 51.0 for the control group (95% CI, –1.22-4.51), for a mean change of 3.8 in the intervention cohort and 1.6 for the control group. The mean treatment effect was 2.15 (95% CI, –1.79-6.09; P = .28), indicating no significant difference between the two.
“There was no evidence for a differential treatment effect on the study’s primary outcome, postsepsis MCS scores,” the authors concluded. “This finding is similar to those from previous trials of care management interventions following critical illness.”
The authors added that “further research is needed to determine if modified approaches to primary care management may be more effective.”
The study was funded by the Center for Sepsis Control and Care, the German Federal Ministry of Education and Research, and the German Sepsis Society. Dr. Gensichen reported receiving personal fees from the Primary Health Care Foundation and receiving a grant from the German Federal Ministry of Education and Research.
FROM JAMA
Key clinical point: Primary care intervention does not improve mental health–related quality of life in survivors of sepsis or septic shock.
Major finding: Mean Mental Component Summary (MCS) scores showed no significant change between the time of ICU discharge (49.1) versus at 6 months postdischarge (52.9) (95% CI, 1.05-6.54), compared with the control group: 49.3 at baseline vs. 51.0 at 6 months follow-up (95% CI, –1.22-4.51).
Data source: A multicenter, unblinded, two-group randomized clinical trial of 291 adult sepsis or septic shock survivors recruited from nine German ICUs from February 2011 through December 2014.
Disclosures: Study funded by the Center for Sepsis Control and Care, the German Federal Ministry of Education and Research, and the German Sepsis Society. Dr. Gensichen reported receiving personal fees from The Primary Health Care Foundation and receiving a grant from the German Federal Ministry of Education and Research.
ICU-based therapy fails to shorten hospital stay
Standardized rehabilitation therapy did not reduce hospital length of stay in patients with acute respiratory failure, based on data from a randomized trial of 300 adults published online in JAMA.
Hospital length of stay averaged 10 days for patients in the standardized rehabilitation therapy group (SRT) and 10 days in the control group that received usual ICU care, wrote Dr. Peter E. Morris of the division of pulmonary, critical care and sleep medicine at the University of Kentucky, Lexington, and his colleagues (JAMA. 2016 Jun;315:2694-702. doi: 10.1001/jama.2016.7201).

The patients were followed for 6 months; 84 patients in the SRT group and 81 in the usual group completed the study.
Patients in the SRT group received daily therapy including passive range of motion, physical therapy, and progressive-resistance exercises. The usual care group received weekday physical therapy as determined by the clinical team.
The researchers also assessed secondary outcomes related to physical function and quality of life, including ventilator days, Short Physical Performance Battery (SPPB) score, handgrip, Mini-Mental State Examination, and Functional Performance Inventory (FPI).
Overall, there was no difference in duration of ventilation or ICU care between the two groups, and score of handgrip strength and mental health also were similar at 6 months’ follow up. However, the SF-36 physical function scores were significantly higher in the SRT group (difference, 12.2; 95% confidence interval, 3.8-20.7; P = .001), and the FPI scores and SPPB scores were higher, compared with the usual care group at 6 months.
“These findings from the exploratory analysis may highlight the emerging role of placing long-term outcomes within critical care clinical trial design not only as a secondary outcome, but possibly as the primary outcome,” the researchers noted. “In view of the SPPB, SF-36 PFS, and FPI data at 6 months, the SRT group demonstrated a potential signal of improvement compared with the usual care group that was not evident at hospital discharge,” they wrote.
The study was supported by the National Institutes of Health, National Institute of Nursing Research, and National Heart, Lung, and Blood Institute. Lead author, Dr. Morris, had no financial conflicts to disclose.
Standardized rehabilitation therapy did not reduce hospital length of stay in patients with acute respiratory failure, based on data from a randomized trial of 300 adults published online in JAMA.
Hospital length of stay averaged 10 days for patients in the standardized rehabilitation therapy group (SRT) and 10 days in the control group that received usual ICU care, wrote Dr. Peter E. Morris of the division of pulmonary, critical care and sleep medicine at the University of Kentucky, Lexington, and his colleagues (JAMA. 2016 Jun;315:2694-702. doi: 10.1001/jama.2016.7201).
The patients were followed for 6 months; 84 patients in the SRT group and 81 in the usual group completed the study.
Patients in the SRT group received daily therapy including passive range of motion, physical therapy, and progressive-resistance exercises. The usual care group received weekday physical therapy as determined by the clinical team.
The researchers also assessed secondary outcomes related to physical function and quality of life, including ventilator days, Short Physical Performance Battery (SPPB) score, handgrip, Mini-Mental State Examination, and Functional Performance Inventory (FPI).
Overall, there was no difference in duration of ventilation or ICU care between the two groups, and score of handgrip strength and mental health also were similar at 6 months’ follow up. However, the SF-36 physical function scores were significantly higher in the SRT group (difference, 12.2; 95% confidence interval, 3.8-20.7; P = .001), and the FPI scores and SPPB scores were higher, compared with the usual care group at 6 months.
“These findings from the exploratory analysis may highlight the emerging role of placing long-term outcomes within critical care clinical trial design not only as a secondary outcome, but possibly as the primary outcome,” the researchers noted. “In view of the SPPB, SF-36 PFS, and FPI data at 6 months, the SRT group demonstrated a potential signal of improvement compared with the usual care group that was not evident at hospital discharge,” they wrote.
The study was supported by the National Institutes of Health, National Institute of Nursing Research, and National Heart, Lung, and Blood Institute. Lead author, Dr. Morris, had no financial conflicts to disclose.
Standardized rehabilitation therapy did not reduce hospital length of stay in patients with acute respiratory failure, based on data from a randomized trial of 300 adults published online in JAMA.
Hospital length of stay averaged 10 days for patients in the standardized rehabilitation therapy group (SRT) and 10 days in the control group that received usual ICU care, wrote Dr. Peter E. Morris of the division of pulmonary, critical care and sleep medicine at the University of Kentucky, Lexington, and his colleagues (JAMA. 2016 Jun;315:2694-702. doi: 10.1001/jama.2016.7201).
The patients were followed for 6 months; 84 patients in the SRT group and 81 in the usual group completed the study.
Patients in the SRT group received daily therapy including passive range of motion, physical therapy, and progressive-resistance exercises. The usual care group received weekday physical therapy as determined by the clinical team.
The researchers also assessed secondary outcomes related to physical function and quality of life, including ventilator days, Short Physical Performance Battery (SPPB) score, handgrip, Mini-Mental State Examination, and Functional Performance Inventory (FPI).
Overall, there was no difference in duration of ventilation or ICU care between the two groups, and score of handgrip strength and mental health also were similar at 6 months’ follow up. However, the SF-36 physical function scores were significantly higher in the SRT group (difference, 12.2; 95% confidence interval, 3.8-20.7; P = .001), and the FPI scores and SPPB scores were higher, compared with the usual care group at 6 months.
“These findings from the exploratory analysis may highlight the emerging role of placing long-term outcomes within critical care clinical trial design not only as a secondary outcome, but possibly as the primary outcome,” the researchers noted. “In view of the SPPB, SF-36 PFS, and FPI data at 6 months, the SRT group demonstrated a potential signal of improvement compared with the usual care group that was not evident at hospital discharge,” they wrote.
The study was supported by the National Institutes of Health, National Institute of Nursing Research, and National Heart, Lung, and Blood Institute. Lead author, Dr. Morris, had no financial conflicts to disclose.
FROM JAMA
Key clinical point: Rehabilitation therapy in the ICU did not reduce hospital stay in patients with acute respiratory failure.
Major finding: The average length of stay was 10 days in both the therapy and control groups.
Data source: A randomized, single-center study including 300 adults with acute respiratory failure.
Disclosures: The study was supported by the National Institutes of Health, National Institute of Nursing Research, and National Heart, Lung, and Blood Institute. Lead author Dr. Morris had no financial conflicts to disclose.

Using lipid guidelines to manage metabolic syndrome for patients taking an antipsychotic
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?

Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
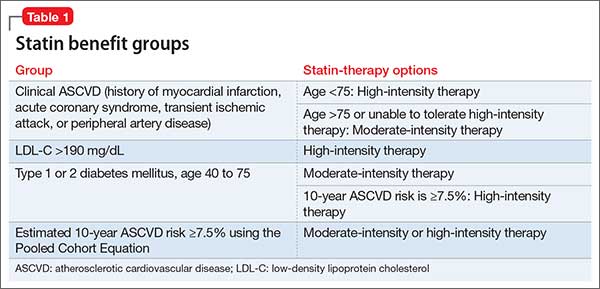
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
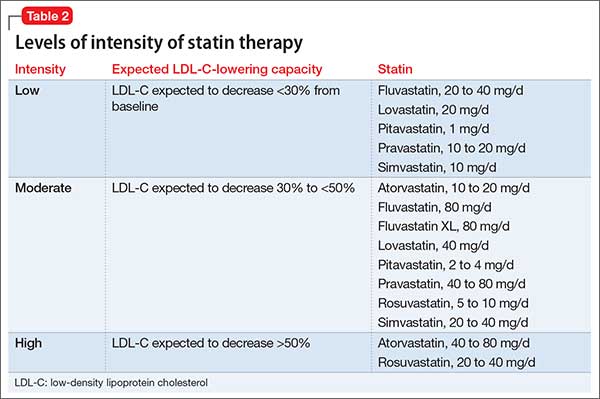
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
How to talk to patients and families about brain stimulation
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).

Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.