User login
Anti-Remicade antibodies also cross-react with infliximab biosimilar
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).

In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Rheumatology patients with positive antibodies to Remicade should not be switched to infliximab biosimilar (Remsima, Inflectra).
Major finding: Antibodies to infliximab in Remicade-treated rheumatology patients showed identical reactivity towards the biosimilar CT-P13.
Data source: A retrospective study of 250 consecutive patients with RA and SpA taking Remicade and 77 infliximab-naive controls.
Disclosures: Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.

Temsirolimus results in good but short-duration responses in primary CNS lymphoma
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Relapsed/refractory primary CNS lymphoma has a poor prognosis and no standard treatment option.
Major finding: The overall response rate to once-weekly temsirolimus was 54%; most responses were short lived.
Data source: Open-label phase 2 study in 37 adults with relapsed/refractory primary CNS lymphoma.
Disclosures: The study was supported by Pfizer Germany. Dr. Korfel and several colleagues disclosed research support from or consulting/advising for the company.
IMWG issues renal impairment recommendations for myeloma patients
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: All patients diagnosed with multiple myeloma should be evaluated for renal impairment.
Major finding: Bortezomib-based regimens are the standard of care for patients with multiple myeloma.
Data source: Evidence-based clinical recommendations.
Disclosures: Many coauthors disclosed multiple relationships with companies that make antimyeloma therapies and other medications.
Progressive Cardiomyopathy in a Patient With Elevated Cobalt Ion Levels and Bilateral Metal-on-Metal Hip Arthroplasties
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
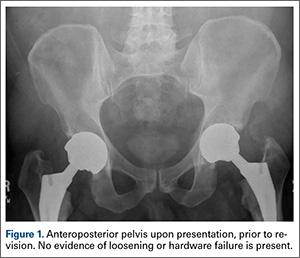
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
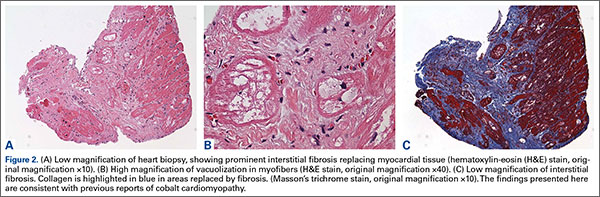
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
Partial Flexor Tendon Laceration Assessment: Interobserver and Intraobserver Reliability
How to manage complete flexor tendon lacerations in the hand is well documented and a subject of relative agreement among authors. However, treatment of partial flexor tendon lacerations is controversial and lacking clear consensus in the literature. Managing these injuries can be challenging, as clinicians must weigh the diminished tensile strength in the injured tendon and the potential for later complications (eg, entrapment, triggering, rupture) against the negative effects of tenorrhaphy.1 Several studies have found impaired tendon gliding on the basis of bulk and inflammatory reaction secondary to suture material within the flexor sheath as well as decreased tendon strength after tenorrhaphy.2-6 This finding led the investigators to recommend nonsurgical management for partial lacerations up to as much as 95% of the cross-sectional area (CSA) of the tendon. According to a survey by McCarthy and colleagues,7 45% of 591 members of the American Society for Surgery of the Hand (ASSH) indicated they would perform tenorrhaphy for a laceration that involved more than 50% of the tendon.
However, accurate assessment of partial-thickness flexor tendon lacerations is difficult owing to the subjectivity of evaluation. In the survey just mentioned,7 the majority of surgeons used the naked eye to make assessments, and only 14% used other means, such as a ruler, a pair of calipers, or loupe magnification. In addition, flexor tendon injuries are often evaluated under less than ideal circumstances—a dirty or bloody field, poor lighting, an uncomfortable patient.
We conducted a study to determine the interobserver and intraobserver reliability of surgeons assessing the percentage of CSA injured in partially lacerated digital flexor tendons. We hypothesized that participants’ accuracy and agreement would be poor.
Materials and Methods
Eight 1-cm transverse, volar skin incisions were made over the midportions of the middle and proximal phalanges of the index, middle, ring, and small fingers of a fresh-frozen human cadaver hand (Figure 1). The tendon sheaths were incised, and the flexor digitorum profundus tendons to each digit were delivered through the wound. With use of a method described previously by Manning and colleagues,8 the tendon was then placed over a flat metal post to be used as a cutting board, and the proposed laceration site was marked with ink. Under loupe magnification, a No. 15 blade was used to create a partial transverse, volar-to-dorsal laceration in each tendon.8 The goal was to create lacerations of about 30%, 50%, and 70% of the total CSA of the tendon. The tendons were then returned to the wound, and visibility of the marked laceration within the wound was ensured. A similar exercise was performed at the level of the proximal palmar crease. Four flexor digitorum superficialis tendons were exposed through 1-cm transverse incisions, and partial lacerations were made in the volar substance of the tendons. The tendons were then returned to the wound, resulting in 12 partially lacerated tendons (8 flexor digitorum profundus, 4 flexor digitorum superficialis).
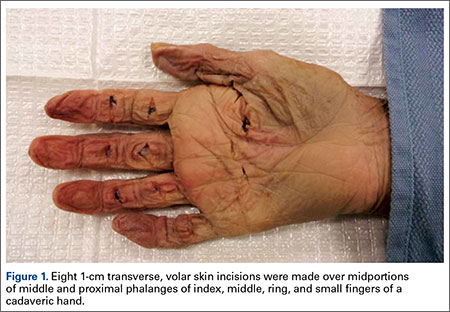
Six orthopedic surgery residents (2 postgraduate year 1 [PGY-1], 2 PGY-3, 2 PGY-5) and 4 fellowship-trained hand surgeons participated in our study. Each was asked to evaluate the tendons and determine the percentage of total CSA lacerated. Loupe magnification and measuring tools were not permitted, but participants were allowed to handle the tendons. In addition, they were asked if they would perform tenorrhaphy on the injured tendons, given only the amount of injury. The participants repeated this exercise 4 weeks later.
After all measurements were made, a longitudinal incision was made down each of the digits, and the flexor tendons were exposed within the flexor sheath. The transverse incisions in the palm were connected to expose the flexor digitorum superficialis tendons. Under an operating microscope, a pair of digital microcalipers (Kobalt 0.5-ft Metric and SAE Caliper; Figure 2) accurate to 0.01 mm was used to measure the external width (a) and height (b + bˈ) of the tendons just proximal to the lacerations. Measurements were made with the caliper blades just touching the edges of the lacerated tendon, thus minimizing deformation of the tendon. Other measurements made at the laceration site were width of the remaining tendon (c) and height of the remaining tendon (bˈ). CSA of the tendon was calculated assuming a regular ellipsoid shape and using the equation:
Area = 1/2π(b+b')
The area of the tendon injured was determined by calculating the area under a parabola and using the equation:
Area = 2/3c[(b+b')-b']
Last, the percentage of total CSA lacerated was calculated using the equation:
Area (total area)
Statistical analysis was performed to determine accuracy and interobserver and intraobserver reliability. Paired t tests were used in the assessment of accuracy to determine if there were differences between estimated and calibrated measurements.

Results
The 10 participants’ estimates differed significantly (P < .0006) from the calibrated measurements, as did residents’ estimates (P < .0025) and fellowship-trained hand surgeons’ estimates (P < .0002). Estimates were scored 1 to 5 on the basis of proximity to calibrated measurements (Table 1). Thus, more accurate estimates received lower scores. Individual estimates were then scored and stratified into groups for comparison. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. These results are listed in Table 2. Once overall and grouped accuracy was analyzed, κ statistics were calculated to compare interobserver and intraobserver reliability. Overall interobserver agreement was poor for both initial readings (κ = 0.16) and secondary readings (κ = 0.16), indicating poor strength of agreement between individuals both initially and secondarily. Table 3 presents the κ interpretations. There was moderate overall intraobserver agreement (45.83%), indicating participants’ secondary estimates agreed with their primary estimates 46% of the time. Fellowship-trained hand surgeons and first-year residents had the highest intraobserver agreement (50.0%). These results are listed in Table 4.




Discussion
Accurate assessment of partial flexor tendon lacerations is difficult and subjective. There is no standardized method for determining the extent of injury, regardless of whether the evaluation is performed in an emergency department or in the operating room. As McCarthy and colleagues7 noted in their survey of ASSH members, naked eye assessment was by far the most popular means of estimating percentage injured in partial lacerations, and only 10% of the survey respondents used intraoperative measuring devices. Our study showed that participants agreed with one another less than 50% of the time when evaluating injuries without the aid of measuring devices. In addition, interobserver agreement in this study was about 50%, highlighting the difficulty in making an accurate and reproducible assessment.
In a study of canine flexor tendons, McCarthy and colleagues9 found calipers are inaccurate as well and do not provide a reliable means of assessing partial flexor tendon lacerations. They compared caliper measurements with laser micrometer measurements, and the differences averaged 29.3%. They suggested that methods for calculating loss of CSA and for creating precise lacerations must be developed in order to evaluate treatments. One such method is the “tenotome,” devised by Hitchcock and colleagues10: A device with standard scalpel blades is used to make uniform lacerations in tendons by leaving a constant area of the tendon intact, regardless of the size or shape of the original tendon. Measurements made with calipers or rulers assume the tendon has a regular ellipsoid shape, but in reality the shape is a double-ellipse, particularly within the flexor sheath.
Dobyns and colleagues11 observed that changes in CSA size can be related to changes in the size of the bundle pattern of the tendon. They found that, on average, the radial bundle comprised about 60% of the total CSA of the tendon. This finding was clarified by Grewal and colleagues.12 Using histologic sections of tendons plus photomicrographs, they determined that, in zone II of the index and small fingers, the ulnar bundle had an area consistently larger than 50% and the radial bundle less than 50% of the total tendon area. In the ring and middle fingers, the areas of both bundles were almost 50% of the total tendon area. The authors suggested that, using this bundle pattern theory of injury, surgeons could more accurately evaluate the extent of injury with the naked eye.
One of the questions that prompted our study is how reliable is the information a surgeon receives regarding a partial flexor tendon injury evaluated by someone else in another setting. What is done with this information is another question. The scenario can be considered in 2 settings: emergency department and operating room.
Given the poor accuracy and interobserver agreement found in our study, along with the inaccuracy of caliper and ruler measurements, it seems decisions to perform tenorrhaphy based on reported percentages lacerated are unreliable. Our results showed that the ability to accurately assess partial tendon injuries does not improve with surgeon experience, as fellowship-trained hand surgeons were not statistically more accurate or consistent than residents. To this effect, one institution treats all its partial flexor tendon lacerations with wound inspection and irrigation in the emergency department, under digital block and after neurovascular injury has been excluded.8 If the patient is able to actively flex and extend the digit without triggering, then the wound is closed without closing the tendon sheath, a dorsal blocking splint is applied, and motion is begun early, 48 hours later, regardless of laceration severity.
Once the decision has been made to go to the operating room and the injury is being evaluated, what should be done with the information from the measurement, whether made with loupe magnification, calipers, rulers, or the naked eye? Surgeons must weigh the risks for triggering, entrapment, and rupture of untreated partial tendon lacerations1 with the added bulk and potential for adhesions, along with the tensile strength reduction that accompanies tendon repair. Both Reynolds and colleagues13 and Ollinger and colleagues14 found tensile strength significantly diminished in sutured tendons. Ollinger and colleagues14 showed a decrease in tendon gliding after surgical exposure and tenorrhaphy for partial tendon lacerations. Reynolds and colleagues13 concluded that surgical repair leads to poorer results than nonsurgical treatment.
Clinical studies have demonstrated excellent results with nonintervention, and in vivo and in vitro studies have indicated that early motion can be initiated in partial lacerations of up to 95% of total CSA. Wray and Weeks6 treated 26 patients with partial lacerations varying from 25% to 95% of total CSA and noted 1 incidence of trigger finger (which resolved) and no late ruptures. They advocated treatment with early motion and excision or repair of beveled partial lacerations with simple sutures. Stahl and colleagues2 reported comparable outcomes in children with partial lacerations up to 75% of total CSA treated with and without surgery and noted no complications in either group. In a biomechanical study, Hariharan and colleagues4 found lacerations up to 75% can withstand forces associated with active unresisted mobilization.
Conversely, how many patients or surgeons want to return to the operating room to fix a late rupture when it could have been repaired in the primary setting? Schlenker and colleagues,1 reporting on a late flexor pollicus tendon rupture that required tendon grafting, recommended exploration and primary repair of all partial flexor tendon lacerations. Often, it is difficult to determine whether surgical repair is necessary to ensure the best outcome for the patient.
Our study results showed that, in the evaluation of flexor tendon lacerations, both accuracy and interobserver agreement were poor among residents and fellowship-trained hand surgeons, and intraobserver agreement was moderate. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. Our results highlight the difficulty in making accurate assessments of flexor tendon lacerations owing to the subjectivity of evaluation, which appear not to improve with surgeon experience.
1. Schlenker JD, Lister GD, Kleinert HE. Three complications of untreated partial laceration of flexor tendon—entrapment, rupture, and triggering. J Hand Surg Am. 1981;6(4):392-398.
2. Stahl S, Kaufman T, Bialik V. Partial lacerations of flexor tendons in children. Primary repair versus conservative treatment. J Hand Surg Br. 1997;22(3):377-380.
3. Al-Qattan MM. Conservative management of zone II partial flexor tendon lacerations greater than half the width of the tendon. J Hand Surg Am. 2000;25(6):1118-1121.
4. Hariharan JS, Diao E, Soejima O, Lotz JC. Partial lacerations of human digital flexor tendons: a biomechanical analysis. J Hand Surg Am. 1997;22(6):1011-1015.
5. Bishop AT, Cooney WP 3rd, Wood MB. Treatment of partial flexor tendon lacerations: the effect of tenorrhaphy and early protected mobilization. J Trauma. 1986;26(4):301-312.
6. Wray RC Jr, Weeks PM. Treatment of partial tendon lacerations. Hand. 1980;12(2):163-166.
7. McCarthy DM, Boardman ND 3rd, Tramaglini DM, Sotereanos DG, Herndon JH. Clinical management of partially lacerated digital flexor tendons: a survey of hand surgeons. J Hand Surg Am. 1995;20(2):273-275.
8. Manning DW, Spiguel AR, Mass DP. Biomechanical analysis of partial flexor tendon lacerations in zone II of human cadavers. J Hand Surg Am. 2010;35(1):11-18.
9. McCarthy DM, Tramaglini DM, Chan SS, Schmidt CC, Sotereanos DG, Herndon JH. Effect of partial laceration on the structural properties of the canine FDP tendon: an in vitro study. J Hand Surg Am. 1995;20(5):795-800.
10. Hitchcock TF, Candel AG, Light TR, Blevens AD. New technique for producing uniform partial lacerations of tendons. J Orthop Res. 1989;7(3):451-455.
11. Dobyns RC, Cooney WC, Wood MB. Effect of partial lacerations on canine flexor tendons. Minn Med. 1982;65(1):27-32.
12. Grewal R, Sotereanos DG, Rao U, Herndon JH, Woo SL. Bundle pattern of the flexor digitorum profundus tendon in zone II of the hand: a quantitative assessment of the size of a laceration. J Hand Surg Am. 1996;21(6):978-983.
13. Reynolds B, Wray RC Jr, Weeks PM. Should an incompletely severed tendon be sutured? Plast Reconstr Surg. 1976;57(1):36-38.
14. Ollinger H, Wray RC Jr, Weeks PM. Effects of suture on tensile strength gain of partially and completely severed tendons. Surg Forum. 1975;26:63-64.
How to manage complete flexor tendon lacerations in the hand is well documented and a subject of relative agreement among authors. However, treatment of partial flexor tendon lacerations is controversial and lacking clear consensus in the literature. Managing these injuries can be challenging, as clinicians must weigh the diminished tensile strength in the injured tendon and the potential for later complications (eg, entrapment, triggering, rupture) against the negative effects of tenorrhaphy.1 Several studies have found impaired tendon gliding on the basis of bulk and inflammatory reaction secondary to suture material within the flexor sheath as well as decreased tendon strength after tenorrhaphy.2-6 This finding led the investigators to recommend nonsurgical management for partial lacerations up to as much as 95% of the cross-sectional area (CSA) of the tendon. According to a survey by McCarthy and colleagues,7 45% of 591 members of the American Society for Surgery of the Hand (ASSH) indicated they would perform tenorrhaphy for a laceration that involved more than 50% of the tendon.
However, accurate assessment of partial-thickness flexor tendon lacerations is difficult owing to the subjectivity of evaluation. In the survey just mentioned,7 the majority of surgeons used the naked eye to make assessments, and only 14% used other means, such as a ruler, a pair of calipers, or loupe magnification. In addition, flexor tendon injuries are often evaluated under less than ideal circumstances—a dirty or bloody field, poor lighting, an uncomfortable patient.
We conducted a study to determine the interobserver and intraobserver reliability of surgeons assessing the percentage of CSA injured in partially lacerated digital flexor tendons. We hypothesized that participants’ accuracy and agreement would be poor.
Materials and Methods
Eight 1-cm transverse, volar skin incisions were made over the midportions of the middle and proximal phalanges of the index, middle, ring, and small fingers of a fresh-frozen human cadaver hand (Figure 1). The tendon sheaths were incised, and the flexor digitorum profundus tendons to each digit were delivered through the wound. With use of a method described previously by Manning and colleagues,8 the tendon was then placed over a flat metal post to be used as a cutting board, and the proposed laceration site was marked with ink. Under loupe magnification, a No. 15 blade was used to create a partial transverse, volar-to-dorsal laceration in each tendon.8 The goal was to create lacerations of about 30%, 50%, and 70% of the total CSA of the tendon. The tendons were then returned to the wound, and visibility of the marked laceration within the wound was ensured. A similar exercise was performed at the level of the proximal palmar crease. Four flexor digitorum superficialis tendons were exposed through 1-cm transverse incisions, and partial lacerations were made in the volar substance of the tendons. The tendons were then returned to the wound, resulting in 12 partially lacerated tendons (8 flexor digitorum profundus, 4 flexor digitorum superficialis).
Six orthopedic surgery residents (2 postgraduate year 1 [PGY-1], 2 PGY-3, 2 PGY-5) and 4 fellowship-trained hand surgeons participated in our study. Each was asked to evaluate the tendons and determine the percentage of total CSA lacerated. Loupe magnification and measuring tools were not permitted, but participants were allowed to handle the tendons. In addition, they were asked if they would perform tenorrhaphy on the injured tendons, given only the amount of injury. The participants repeated this exercise 4 weeks later.
After all measurements were made, a longitudinal incision was made down each of the digits, and the flexor tendons were exposed within the flexor sheath. The transverse incisions in the palm were connected to expose the flexor digitorum superficialis tendons. Under an operating microscope, a pair of digital microcalipers (Kobalt 0.5-ft Metric and SAE Caliper; Figure 2) accurate to 0.01 mm was used to measure the external width (a) and height (b + bˈ) of the tendons just proximal to the lacerations. Measurements were made with the caliper blades just touching the edges of the lacerated tendon, thus minimizing deformation of the tendon. Other measurements made at the laceration site were width of the remaining tendon (c) and height of the remaining tendon (bˈ). CSA of the tendon was calculated assuming a regular ellipsoid shape and using the equation:
Area = 1/2π(b+b')
The area of the tendon injured was determined by calculating the area under a parabola and using the equation:
Area = 2/3c[(b+b')-b']
Last, the percentage of total CSA lacerated was calculated using the equation:
Area (total area)
Statistical analysis was performed to determine accuracy and interobserver and intraobserver reliability. Paired t tests were used in the assessment of accuracy to determine if there were differences between estimated and calibrated measurements.
Results
The 10 participants’ estimates differed significantly (P < .0006) from the calibrated measurements, as did residents’ estimates (P < .0025) and fellowship-trained hand surgeons’ estimates (P < .0002). Estimates were scored 1 to 5 on the basis of proximity to calibrated measurements (Table 1). Thus, more accurate estimates received lower scores. Individual estimates were then scored and stratified into groups for comparison. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. These results are listed in Table 2. Once overall and grouped accuracy was analyzed, κ statistics were calculated to compare interobserver and intraobserver reliability. Overall interobserver agreement was poor for both initial readings (κ = 0.16) and secondary readings (κ = 0.16), indicating poor strength of agreement between individuals both initially and secondarily. Table 3 presents the κ interpretations. There was moderate overall intraobserver agreement (45.83%), indicating participants’ secondary estimates agreed with their primary estimates 46% of the time. Fellowship-trained hand surgeons and first-year residents had the highest intraobserver agreement (50.0%). These results are listed in Table 4.
Discussion
Accurate assessment of partial flexor tendon lacerations is difficult and subjective. There is no standardized method for determining the extent of injury, regardless of whether the evaluation is performed in an emergency department or in the operating room. As McCarthy and colleagues7 noted in their survey of ASSH members, naked eye assessment was by far the most popular means of estimating percentage injured in partial lacerations, and only 10% of the survey respondents used intraoperative measuring devices. Our study showed that participants agreed with one another less than 50% of the time when evaluating injuries without the aid of measuring devices. In addition, interobserver agreement in this study was about 50%, highlighting the difficulty in making an accurate and reproducible assessment.
In a study of canine flexor tendons, McCarthy and colleagues9 found calipers are inaccurate as well and do not provide a reliable means of assessing partial flexor tendon lacerations. They compared caliper measurements with laser micrometer measurements, and the differences averaged 29.3%. They suggested that methods for calculating loss of CSA and for creating precise lacerations must be developed in order to evaluate treatments. One such method is the “tenotome,” devised by Hitchcock and colleagues10: A device with standard scalpel blades is used to make uniform lacerations in tendons by leaving a constant area of the tendon intact, regardless of the size or shape of the original tendon. Measurements made with calipers or rulers assume the tendon has a regular ellipsoid shape, but in reality the shape is a double-ellipse, particularly within the flexor sheath.
Dobyns and colleagues11 observed that changes in CSA size can be related to changes in the size of the bundle pattern of the tendon. They found that, on average, the radial bundle comprised about 60% of the total CSA of the tendon. This finding was clarified by Grewal and colleagues.12 Using histologic sections of tendons plus photomicrographs, they determined that, in zone II of the index and small fingers, the ulnar bundle had an area consistently larger than 50% and the radial bundle less than 50% of the total tendon area. In the ring and middle fingers, the areas of both bundles were almost 50% of the total tendon area. The authors suggested that, using this bundle pattern theory of injury, surgeons could more accurately evaluate the extent of injury with the naked eye.
One of the questions that prompted our study is how reliable is the information a surgeon receives regarding a partial flexor tendon injury evaluated by someone else in another setting. What is done with this information is another question. The scenario can be considered in 2 settings: emergency department and operating room.
Given the poor accuracy and interobserver agreement found in our study, along with the inaccuracy of caliper and ruler measurements, it seems decisions to perform tenorrhaphy based on reported percentages lacerated are unreliable. Our results showed that the ability to accurately assess partial tendon injuries does not improve with surgeon experience, as fellowship-trained hand surgeons were not statistically more accurate or consistent than residents. To this effect, one institution treats all its partial flexor tendon lacerations with wound inspection and irrigation in the emergency department, under digital block and after neurovascular injury has been excluded.8 If the patient is able to actively flex and extend the digit without triggering, then the wound is closed without closing the tendon sheath, a dorsal blocking splint is applied, and motion is begun early, 48 hours later, regardless of laceration severity.
Once the decision has been made to go to the operating room and the injury is being evaluated, what should be done with the information from the measurement, whether made with loupe magnification, calipers, rulers, or the naked eye? Surgeons must weigh the risks for triggering, entrapment, and rupture of untreated partial tendon lacerations1 with the added bulk and potential for adhesions, along with the tensile strength reduction that accompanies tendon repair. Both Reynolds and colleagues13 and Ollinger and colleagues14 found tensile strength significantly diminished in sutured tendons. Ollinger and colleagues14 showed a decrease in tendon gliding after surgical exposure and tenorrhaphy for partial tendon lacerations. Reynolds and colleagues13 concluded that surgical repair leads to poorer results than nonsurgical treatment.
Clinical studies have demonstrated excellent results with nonintervention, and in vivo and in vitro studies have indicated that early motion can be initiated in partial lacerations of up to 95% of total CSA. Wray and Weeks6 treated 26 patients with partial lacerations varying from 25% to 95% of total CSA and noted 1 incidence of trigger finger (which resolved) and no late ruptures. They advocated treatment with early motion and excision or repair of beveled partial lacerations with simple sutures. Stahl and colleagues2 reported comparable outcomes in children with partial lacerations up to 75% of total CSA treated with and without surgery and noted no complications in either group. In a biomechanical study, Hariharan and colleagues4 found lacerations up to 75% can withstand forces associated with active unresisted mobilization.
Conversely, how many patients or surgeons want to return to the operating room to fix a late rupture when it could have been repaired in the primary setting? Schlenker and colleagues,1 reporting on a late flexor pollicus tendon rupture that required tendon grafting, recommended exploration and primary repair of all partial flexor tendon lacerations. Often, it is difficult to determine whether surgical repair is necessary to ensure the best outcome for the patient.
Our study results showed that, in the evaluation of flexor tendon lacerations, both accuracy and interobserver agreement were poor among residents and fellowship-trained hand surgeons, and intraobserver agreement was moderate. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. Our results highlight the difficulty in making accurate assessments of flexor tendon lacerations owing to the subjectivity of evaluation, which appear not to improve with surgeon experience.
How to manage complete flexor tendon lacerations in the hand is well documented and a subject of relative agreement among authors. However, treatment of partial flexor tendon lacerations is controversial and lacking clear consensus in the literature. Managing these injuries can be challenging, as clinicians must weigh the diminished tensile strength in the injured tendon and the potential for later complications (eg, entrapment, triggering, rupture) against the negative effects of tenorrhaphy.1 Several studies have found impaired tendon gliding on the basis of bulk and inflammatory reaction secondary to suture material within the flexor sheath as well as decreased tendon strength after tenorrhaphy.2-6 This finding led the investigators to recommend nonsurgical management for partial lacerations up to as much as 95% of the cross-sectional area (CSA) of the tendon. According to a survey by McCarthy and colleagues,7 45% of 591 members of the American Society for Surgery of the Hand (ASSH) indicated they would perform tenorrhaphy for a laceration that involved more than 50% of the tendon.
However, accurate assessment of partial-thickness flexor tendon lacerations is difficult owing to the subjectivity of evaluation. In the survey just mentioned,7 the majority of surgeons used the naked eye to make assessments, and only 14% used other means, such as a ruler, a pair of calipers, or loupe magnification. In addition, flexor tendon injuries are often evaluated under less than ideal circumstances—a dirty or bloody field, poor lighting, an uncomfortable patient.
We conducted a study to determine the interobserver and intraobserver reliability of surgeons assessing the percentage of CSA injured in partially lacerated digital flexor tendons. We hypothesized that participants’ accuracy and agreement would be poor.
Materials and Methods
Eight 1-cm transverse, volar skin incisions were made over the midportions of the middle and proximal phalanges of the index, middle, ring, and small fingers of a fresh-frozen human cadaver hand (Figure 1). The tendon sheaths were incised, and the flexor digitorum profundus tendons to each digit were delivered through the wound. With use of a method described previously by Manning and colleagues,8 the tendon was then placed over a flat metal post to be used as a cutting board, and the proposed laceration site was marked with ink. Under loupe magnification, a No. 15 blade was used to create a partial transverse, volar-to-dorsal laceration in each tendon.8 The goal was to create lacerations of about 30%, 50%, and 70% of the total CSA of the tendon. The tendons were then returned to the wound, and visibility of the marked laceration within the wound was ensured. A similar exercise was performed at the level of the proximal palmar crease. Four flexor digitorum superficialis tendons were exposed through 1-cm transverse incisions, and partial lacerations were made in the volar substance of the tendons. The tendons were then returned to the wound, resulting in 12 partially lacerated tendons (8 flexor digitorum profundus, 4 flexor digitorum superficialis).
Six orthopedic surgery residents (2 postgraduate year 1 [PGY-1], 2 PGY-3, 2 PGY-5) and 4 fellowship-trained hand surgeons participated in our study. Each was asked to evaluate the tendons and determine the percentage of total CSA lacerated. Loupe magnification and measuring tools were not permitted, but participants were allowed to handle the tendons. In addition, they were asked if they would perform tenorrhaphy on the injured tendons, given only the amount of injury. The participants repeated this exercise 4 weeks later.
After all measurements were made, a longitudinal incision was made down each of the digits, and the flexor tendons were exposed within the flexor sheath. The transverse incisions in the palm were connected to expose the flexor digitorum superficialis tendons. Under an operating microscope, a pair of digital microcalipers (Kobalt 0.5-ft Metric and SAE Caliper; Figure 2) accurate to 0.01 mm was used to measure the external width (a) and height (b + bˈ) of the tendons just proximal to the lacerations. Measurements were made with the caliper blades just touching the edges of the lacerated tendon, thus minimizing deformation of the tendon. Other measurements made at the laceration site were width of the remaining tendon (c) and height of the remaining tendon (bˈ). CSA of the tendon was calculated assuming a regular ellipsoid shape and using the equation:
Area = 1/2π(b+b')
The area of the tendon injured was determined by calculating the area under a parabola and using the equation:
Area = 2/3c[(b+b')-b']
Last, the percentage of total CSA lacerated was calculated using the equation:
Area (total area)
Statistical analysis was performed to determine accuracy and interobserver and intraobserver reliability. Paired t tests were used in the assessment of accuracy to determine if there were differences between estimated and calibrated measurements.
Results
The 10 participants’ estimates differed significantly (P < .0006) from the calibrated measurements, as did residents’ estimates (P < .0025) and fellowship-trained hand surgeons’ estimates (P < .0002). Estimates were scored 1 to 5 on the basis of proximity to calibrated measurements (Table 1). Thus, more accurate estimates received lower scores. Individual estimates were then scored and stratified into groups for comparison. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. These results are listed in Table 2. Once overall and grouped accuracy was analyzed, κ statistics were calculated to compare interobserver and intraobserver reliability. Overall interobserver agreement was poor for both initial readings (κ = 0.16) and secondary readings (κ = 0.16), indicating poor strength of agreement between individuals both initially and secondarily. Table 3 presents the κ interpretations. There was moderate overall intraobserver agreement (45.83%), indicating participants’ secondary estimates agreed with their primary estimates 46% of the time. Fellowship-trained hand surgeons and first-year residents had the highest intraobserver agreement (50.0%). These results are listed in Table 4.
Discussion
Accurate assessment of partial flexor tendon lacerations is difficult and subjective. There is no standardized method for determining the extent of injury, regardless of whether the evaluation is performed in an emergency department or in the operating room. As McCarthy and colleagues7 noted in their survey of ASSH members, naked eye assessment was by far the most popular means of estimating percentage injured in partial lacerations, and only 10% of the survey respondents used intraoperative measuring devices. Our study showed that participants agreed with one another less than 50% of the time when evaluating injuries without the aid of measuring devices. In addition, interobserver agreement in this study was about 50%, highlighting the difficulty in making an accurate and reproducible assessment.
In a study of canine flexor tendons, McCarthy and colleagues9 found calipers are inaccurate as well and do not provide a reliable means of assessing partial flexor tendon lacerations. They compared caliper measurements with laser micrometer measurements, and the differences averaged 29.3%. They suggested that methods for calculating loss of CSA and for creating precise lacerations must be developed in order to evaluate treatments. One such method is the “tenotome,” devised by Hitchcock and colleagues10: A device with standard scalpel blades is used to make uniform lacerations in tendons by leaving a constant area of the tendon intact, regardless of the size or shape of the original tendon. Measurements made with calipers or rulers assume the tendon has a regular ellipsoid shape, but in reality the shape is a double-ellipse, particularly within the flexor sheath.
Dobyns and colleagues11 observed that changes in CSA size can be related to changes in the size of the bundle pattern of the tendon. They found that, on average, the radial bundle comprised about 60% of the total CSA of the tendon. This finding was clarified by Grewal and colleagues.12 Using histologic sections of tendons plus photomicrographs, they determined that, in zone II of the index and small fingers, the ulnar bundle had an area consistently larger than 50% and the radial bundle less than 50% of the total tendon area. In the ring and middle fingers, the areas of both bundles were almost 50% of the total tendon area. The authors suggested that, using this bundle pattern theory of injury, surgeons could more accurately evaluate the extent of injury with the naked eye.
One of the questions that prompted our study is how reliable is the information a surgeon receives regarding a partial flexor tendon injury evaluated by someone else in another setting. What is done with this information is another question. The scenario can be considered in 2 settings: emergency department and operating room.
Given the poor accuracy and interobserver agreement found in our study, along with the inaccuracy of caliper and ruler measurements, it seems decisions to perform tenorrhaphy based on reported percentages lacerated are unreliable. Our results showed that the ability to accurately assess partial tendon injuries does not improve with surgeon experience, as fellowship-trained hand surgeons were not statistically more accurate or consistent than residents. To this effect, one institution treats all its partial flexor tendon lacerations with wound inspection and irrigation in the emergency department, under digital block and after neurovascular injury has been excluded.8 If the patient is able to actively flex and extend the digit without triggering, then the wound is closed without closing the tendon sheath, a dorsal blocking splint is applied, and motion is begun early, 48 hours later, regardless of laceration severity.
Once the decision has been made to go to the operating room and the injury is being evaluated, what should be done with the information from the measurement, whether made with loupe magnification, calipers, rulers, or the naked eye? Surgeons must weigh the risks for triggering, entrapment, and rupture of untreated partial tendon lacerations1 with the added bulk and potential for adhesions, along with the tensile strength reduction that accompanies tendon repair. Both Reynolds and colleagues13 and Ollinger and colleagues14 found tensile strength significantly diminished in sutured tendons. Ollinger and colleagues14 showed a decrease in tendon gliding after surgical exposure and tenorrhaphy for partial tendon lacerations. Reynolds and colleagues13 concluded that surgical repair leads to poorer results than nonsurgical treatment.
Clinical studies have demonstrated excellent results with nonintervention, and in vivo and in vitro studies have indicated that early motion can be initiated in partial lacerations of up to 95% of total CSA. Wray and Weeks6 treated 26 patients with partial lacerations varying from 25% to 95% of total CSA and noted 1 incidence of trigger finger (which resolved) and no late ruptures. They advocated treatment with early motion and excision or repair of beveled partial lacerations with simple sutures. Stahl and colleagues2 reported comparable outcomes in children with partial lacerations up to 75% of total CSA treated with and without surgery and noted no complications in either group. In a biomechanical study, Hariharan and colleagues4 found lacerations up to 75% can withstand forces associated with active unresisted mobilization.
Conversely, how many patients or surgeons want to return to the operating room to fix a late rupture when it could have been repaired in the primary setting? Schlenker and colleagues,1 reporting on a late flexor pollicus tendon rupture that required tendon grafting, recommended exploration and primary repair of all partial flexor tendon lacerations. Often, it is difficult to determine whether surgical repair is necessary to ensure the best outcome for the patient.
Our study results showed that, in the evaluation of flexor tendon lacerations, both accuracy and interobserver agreement were poor among residents and fellowship-trained hand surgeons, and intraobserver agreement was moderate. Third-year residents were the most accurate residents, and there was no difference in accuracy between residents and fellowship-trained hand surgeons. Our results highlight the difficulty in making accurate assessments of flexor tendon lacerations owing to the subjectivity of evaluation, which appear not to improve with surgeon experience.
1. Schlenker JD, Lister GD, Kleinert HE. Three complications of untreated partial laceration of flexor tendon—entrapment, rupture, and triggering. J Hand Surg Am. 1981;6(4):392-398.
2. Stahl S, Kaufman T, Bialik V. Partial lacerations of flexor tendons in children. Primary repair versus conservative treatment. J Hand Surg Br. 1997;22(3):377-380.
3. Al-Qattan MM. Conservative management of zone II partial flexor tendon lacerations greater than half the width of the tendon. J Hand Surg Am. 2000;25(6):1118-1121.
4. Hariharan JS, Diao E, Soejima O, Lotz JC. Partial lacerations of human digital flexor tendons: a biomechanical analysis. J Hand Surg Am. 1997;22(6):1011-1015.
5. Bishop AT, Cooney WP 3rd, Wood MB. Treatment of partial flexor tendon lacerations: the effect of tenorrhaphy and early protected mobilization. J Trauma. 1986;26(4):301-312.
6. Wray RC Jr, Weeks PM. Treatment of partial tendon lacerations. Hand. 1980;12(2):163-166.
7. McCarthy DM, Boardman ND 3rd, Tramaglini DM, Sotereanos DG, Herndon JH. Clinical management of partially lacerated digital flexor tendons: a survey of hand surgeons. J Hand Surg Am. 1995;20(2):273-275.
8. Manning DW, Spiguel AR, Mass DP. Biomechanical analysis of partial flexor tendon lacerations in zone II of human cadavers. J Hand Surg Am. 2010;35(1):11-18.
9. McCarthy DM, Tramaglini DM, Chan SS, Schmidt CC, Sotereanos DG, Herndon JH. Effect of partial laceration on the structural properties of the canine FDP tendon: an in vitro study. J Hand Surg Am. 1995;20(5):795-800.
10. Hitchcock TF, Candel AG, Light TR, Blevens AD. New technique for producing uniform partial lacerations of tendons. J Orthop Res. 1989;7(3):451-455.
11. Dobyns RC, Cooney WC, Wood MB. Effect of partial lacerations on canine flexor tendons. Minn Med. 1982;65(1):27-32.
12. Grewal R, Sotereanos DG, Rao U, Herndon JH, Woo SL. Bundle pattern of the flexor digitorum profundus tendon in zone II of the hand: a quantitative assessment of the size of a laceration. J Hand Surg Am. 1996;21(6):978-983.
13. Reynolds B, Wray RC Jr, Weeks PM. Should an incompletely severed tendon be sutured? Plast Reconstr Surg. 1976;57(1):36-38.
14. Ollinger H, Wray RC Jr, Weeks PM. Effects of suture on tensile strength gain of partially and completely severed tendons. Surg Forum. 1975;26:63-64.
1. Schlenker JD, Lister GD, Kleinert HE. Three complications of untreated partial laceration of flexor tendon—entrapment, rupture, and triggering. J Hand Surg Am. 1981;6(4):392-398.
2. Stahl S, Kaufman T, Bialik V. Partial lacerations of flexor tendons in children. Primary repair versus conservative treatment. J Hand Surg Br. 1997;22(3):377-380.
3. Al-Qattan MM. Conservative management of zone II partial flexor tendon lacerations greater than half the width of the tendon. J Hand Surg Am. 2000;25(6):1118-1121.
4. Hariharan JS, Diao E, Soejima O, Lotz JC. Partial lacerations of human digital flexor tendons: a biomechanical analysis. J Hand Surg Am. 1997;22(6):1011-1015.
5. Bishop AT, Cooney WP 3rd, Wood MB. Treatment of partial flexor tendon lacerations: the effect of tenorrhaphy and early protected mobilization. J Trauma. 1986;26(4):301-312.
6. Wray RC Jr, Weeks PM. Treatment of partial tendon lacerations. Hand. 1980;12(2):163-166.
7. McCarthy DM, Boardman ND 3rd, Tramaglini DM, Sotereanos DG, Herndon JH. Clinical management of partially lacerated digital flexor tendons: a survey of hand surgeons. J Hand Surg Am. 1995;20(2):273-275.
8. Manning DW, Spiguel AR, Mass DP. Biomechanical analysis of partial flexor tendon lacerations in zone II of human cadavers. J Hand Surg Am. 2010;35(1):11-18.
9. McCarthy DM, Tramaglini DM, Chan SS, Schmidt CC, Sotereanos DG, Herndon JH. Effect of partial laceration on the structural properties of the canine FDP tendon: an in vitro study. J Hand Surg Am. 1995;20(5):795-800.
10. Hitchcock TF, Candel AG, Light TR, Blevens AD. New technique for producing uniform partial lacerations of tendons. J Orthop Res. 1989;7(3):451-455.
11. Dobyns RC, Cooney WC, Wood MB. Effect of partial lacerations on canine flexor tendons. Minn Med. 1982;65(1):27-32.
12. Grewal R, Sotereanos DG, Rao U, Herndon JH, Woo SL. Bundle pattern of the flexor digitorum profundus tendon in zone II of the hand: a quantitative assessment of the size of a laceration. J Hand Surg Am. 1996;21(6):978-983.
13. Reynolds B, Wray RC Jr, Weeks PM. Should an incompletely severed tendon be sutured? Plast Reconstr Surg. 1976;57(1):36-38.
14. Ollinger H, Wray RC Jr, Weeks PM. Effects of suture on tensile strength gain of partially and completely severed tendons. Surg Forum. 1975;26:63-64.

Technical Errors May Affect Accuracy of Torque Limiter in Locking Plate Osteosynthesis
Proper surgical technique must be used to ensure that surgical fracture management is long-lasting. Plate implantation and screw implantation are among the most common orthopedic procedures performed. Plate and screw osteosynthesis can be done with nonlocking or locking plate and screw constructs or with hybrid fixation that incorporates both methods.
Nonlocking plate and screw osteosynthesis uses friction-fit for fixation. In osteoporotic bone, less torque is generated because of poor bone quality, and thus less friction force between plate and bone.1,2 Locked plating has dramatically changed fracture management, especially in frail and comminuted osteoporotic bone, with significant advantages over conventional plating.3-7
Development of locked plating systems, including the Less Invasive Stabilization System (LISS; DePuy Synthes) with its soft-tissue and fracture-fragment preservation, has changed treatment of distal femur and proximal tibia fractures. Cole and colleagues8 reported stable fixation and union in 97% of their patients. The LISS system proved to be stable, but there were cases of implant removal difficulty with this titanium construct. In 1 of the 10 cases in which the LISS plate was removed, 4 of the 11 locking screws were welded to the plate.8
Cold welding, in which similar metals are chemically bonded together under extreme pressure, is a complication associated with use of titanium-only plates and screws.9 This process, which is more likely to happen if cross-threading occurs within the screw–plate interface, can make screw removal extremely difficult. Screw removal difficulty strips screw heads, and often the surgeon must use either metal cutting instruments or trephines to remove screw remnants, which often results in retained implant or debris and damage or necrosis to surrounding bone.9,10
Locking screws are often inserted under power with a torque-limiting device attached to the drill mechanism to reduce the risk of lock screw overtightening and to try to prevent difficult implant removal. Although standard practice is to insert the screw and stop just before screw head engagement, with final tightening with a torque limiter and hand power, final tightening is often inadvertently done under power.3 Most technique guides instruct surgeons how to insert screws under power while using a torque limiter, but the exact technique is not emphasized.
We conducted a study to determine if rotational speed of screw insertion affects maximum torque on screw with use of a torque limiter. We describe proper use of a torque limiter as well as possible pitfalls. We hypothesized that improper use would result in substantially higher than expected insertion torque.
Materials and Methods
Torque-Limiting Attachments, Torque Wrench, and Drill
The Small Fragment Locking Compression Plate System (Synthes) includes a 1.5-Nm torque-limiting attachment and quick-coupling wooden handles and Star Drive attachments. All devices in this study were in active use at 6 urban institutions (3 level I trauma centers, 2 level II trauma centers, 1 level III hospital). Permission to obtain and test each device was granted by each institution.
A 0.25-inch dial torque wrench (751LDIN; CDI Torque Products) was purchased through an established distributor. The manufacturer includes a traceable certificate of accuracy to verify correct calibration. The torque wrench has a torque range of 0 to 9 Nm with visual increment demarcations of 0.2 Nm and a memory needle to retain maximum torque measurement. The same torque wrench was used in each experiment in order to maintain consistent measurements between devices. It was reset to zero after each use.
This study used a 0.5-inch, 19.2-V lithium drill (Craftsman C3) with 2 speed options: 0 to 440 rpm high torque and 0 to 1600 rpm high speed. This device provides variable torque output with a maximum output of 38.4 Nm. For this study, all measurements were done with the device on its high torque setting.
Maximum Torque Determination for Different Scenarios
Each torque limiter was evaluated for variations in maximum torque under 4 different scenarios. In each scenario, the torque limiter was coupled to the Star Drive attachment and then to that scenario’s rotating force. The completed system was then inserted into the torque wrench, which was secured to a flat working surface and rotated in accordance with each scenario; maximum torque was measured and recorded (Figures 1, 2). A torque-limiting event was defined as a single audible click on the torque limiter.


In scenario 1, each torque-limiting attachment system was attached to a quick-coupling wooden handle. The completed system was then rotated at controlled low velocity under hand power until 1 torque-limiting event occurred. This scenario was also used as an internal control to verify that the torque limiters were calibrated correctly.
In Scenario 2, the device was again attached to a quick-coupling wooden handle. The completed system was rotated at high velocity under hand power until multiple torque-limiting events occurred in a row. High velocity was defined as the operator freely rotating the wooden handle in a single action with full power resulting in multiple torque-limiting events.
In Scenario 3, the device was attached to a power drill braced to the flat working surface and rotated at low velocity under power until 1 torque-limiting event occurred.
In Scenario 4, the device was again attached to a power drill braced to the flat working surface. The completed system was rotated at high velocity under power until multiple torque-limiting events occurred.
After each trial, we recorded maximum torque achieved before each device’s torque-limiting event. Either an orthopedic surgery resident or a qualified medical student tested each torque-limiting device in each standardized testing scenario.
Statistical Analysis
Experiments for each torque limiter were repeated for 3 trials of each of the 4 different scenarios. For comparative statistics between experiments, maximum torque measurements were expressed as means and SDs; 95% confidence interval (95% CI) was calculated and reported to determine extent of variation within a single group. One-way analysis of variance (ANOVA) and Tukey post hoc tests were performed between groups for comparison of the normally distributed data. Significance was set at P ≤ .05.
Results
During simulation, we successfully measured maximum torque achieved with each torque limiter under the 4 different scenarios. All testing was done by 2 operators. ANOVA demonstrated significant (P ≤ .001) differences in torque among the scenarios.
In scenario 1, mean (SD) maximum torque under hand power at low velocity was 1.49 (0.15) Nm (95% CI, 1.43-1.55), near the advertised maximum torque of 1.5 Nm, with relatively minimal variation between devices. This scenario confirmed proper calibration of properly used torque limiters. Mean maximum torque ranged from 1.25 to 1.93 Nm.
In scenario 2, mean (SD) maximum torque under hand power at high velocity was 3.73 (0.79) Nm (95% CI, 3.33-4.13), a 2.5-fold increase compared with scenario 1 (P < .0001) (Figure 3). There also was an increase in variation of maximum torque between trials of individual devices and between different devices. Mean maximum torque ranged from 2.27 to 5.53 Nm.
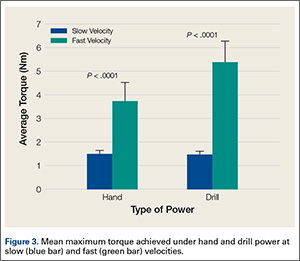
In scenario 3, mean (SD) maximum torque under drill power at controlled low velocity was 1.47 (0.14) Nm (95% CI, 1.37-1.56), again near the advertised maximum torque of 1.5 Nm, with relatively minimal variation. Mean maximum torque ranged from 1.10 to 1.73 Nm.
In scenario 4, mean (SD) maximum torque under drill power at full power/high velocity was 5.37 (0.90) Nm (95% CI, 4.92-5.83), a 3.65-fold increase compared with scenario 3 (P < .0001) (Figure 3). Mean maximum torque measured in 3 tests ranged from 3.40 to 6.92 Nm.
There was no significant difference in mean maximum torque between the scenarios of hand power at low velocity and drill power at low velocities (P = .999) (Figure 4). Highest maximum torque from any device was 9.0 Nm (drill at full power). Results are summarized in the Table. There was no statistical significance in the test between the 2 test operators.
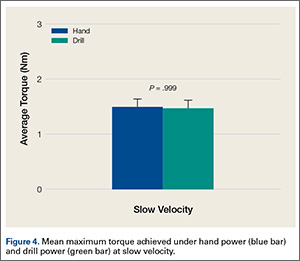
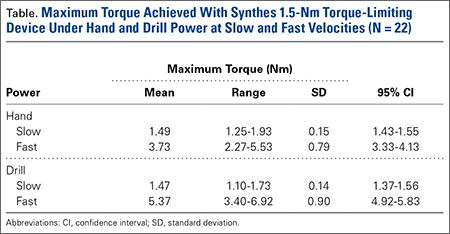
Discussion
Maximum torque was measured using a torque-limiting attachment under 4 different simulated scenarios. Our goals were to determine if varying practice and rotational velocity would affect maximum insertional torque and to measure consistency among torque limiters. We designed the scenarios to mimic practice patterns, including hand insertion and power insertion of locking screws. Results demonstrated that misuse of a torque-limiting device may inadvertently produce insertional torque substantially higher than recommended. Highest maximum torque was 9.0 Nm, which is 6.0-fold higher than expected for a locking screw using a 1.5-Nm torque limiter.
Our study results showed that insertion under controlled hand power (and low-velocity drill power) until 1 torque-limiting event occurred produced the most consistent and predictable results. Insertion under drill power or high-velocity hand power produced multiple sequential torque-limiting events, yielding inaccurate insertion torque. Low-velocity insertion under hand power, or carefully controlled drill power, consistently produced torque similar to advertised values.
Manufacturers’ technique guides are available for proximal humerus locking compression plate (LCP) systems, small-fragment LCP systems, the Proximal Humeral Interlocking System (PHILOS; DePuy Synthes), and the LISS. These technique guides clearly state that insertion can be performed under power. Only the PHILOS and LISS guides state that insertion should be performed under power until a single click is heard or that final tightening should be completed under hand power. The proximal humerus LCP guide states that surgeons should insert the locking screw under power until the torque-limiting device clicks. The small-fragment LCP guide states that insertion under power should always be completed with the torque-limiting attachment; there is no mention of reducing power or a single click (this may give the surgeon a false sense of security).
Screw overtightening and head/thread stripping can make screw removal challenging.10 Removal rates for LISS plates range from 8% to 26%, and removal is often reported as taking longer than the index procedure, with complication rates as high as 47%.11-13 Bae and colleagues3 reported significant difficulty in removing 24 of 279 self-tapping locking screws (3.5 mm).
It is important to note that these complications, most notably cold welding, are mostly associated with titanium locking plate and screw constructs. Although stainless steel constructs have gained favor, titanium constructs are still widely used around the world.14,15
In 10% of cases in a laboratory setting, insertion of a 3.5-mm locking screw at 4 to 6 Nm damaged the screw.9 Removal of 3.5-mm locking screws had a stripping rate of 8.6%, and use of the torque limiter did not make removal easy all the time.3 Torque limiters are set specific to each screw diameter to reduce the risk of damage/stripping or even overtightening. Even when a surgeon intends to stop a drill before locking, final tightening often inadvertently occurs under power.3
Cold welding is often described as a cause of difficult implant removal.3,12 According to a newer definition, this process is independent of temperature and can occur when 2 metallic surfaces are in direct contact.16 High contact pressures between 2 similar metals can lead to this solid state welding.17 Theoretically, improper use of torque limiters can increase the risk of welding; however, it appears to be associated only with titanium locking plate and screw constructs.
Locked plating osteosynthesis is a valuable tool for fracture management, but improper use can have significant consequences, including morbid implant removal procedures, which are more difficult and time-consuming than the index surgery. We determined that proper use of torque limiters involves insertion under hand or power control at slow velocity until 1 torque-limiting event occurs. Many orthopedic surgeons may assume that torque limiters are accurate no matter how screws are inserted into locking plates. In addition, they may be unaware guidelines exist, as these are often deeply embedded within text. Therefore, we must emphasize that torque limiters can be inaccurate when used improperly.
One limitation of this study is that it tested only the Synthes 1.5-Nm torque-limiting attachment, though we can speculate that torque limiters designed for larger screws and limiters manufactured by different companies will behave similarly. Another limitation is that we did not obtain the hospitals’ service records for the tested equipment and assumed the equipment was properly checked for accuracy by the providing company. However, we hypothesized that, if maintenance were an issue, then our results would not be similar across all sites tested.
These tests involved a torque limiter linked to a torque-measuring device and may not perfectly represent actual torque measured at the locked screw–thread interface. However, we think our construct accurately determines the torque produced at the level of the driver tip. Also, we can speculate that the torque produced with improper use will lead to the complications mentioned and demonstrated in previous studies. Welding of the screw–plate interface may simply be a result of improper trajectory and cross-threading. However, if we assume that torque limiters prevent excessive torque no matter how they are used, high insertion speeds may compound the effect of welding. Additional biomechanical studies with full locked plate osteosynthesis constructs on bone specimens are planned to further characterize the potential complications of this issue.
1. Sommer C, Babst R, Müller M, Hanson B. Locking compression plate loosening and plate breakage: a report of four cases. J Orthop Trauma. 2004;18(8):571-577.
2. Schütz M, Südkamp NP. Revolution in plate osteosynthesis: new internal fixator systems. J Orthop Sci. 2003;8(2):252-258.
3. Bae JH, Oh JK, Oh CW, Hur CR. Technical difficulties of removal of locking screw after locking compression plating. Arch Orthop Trauma Surg. 2009;129(1):91-95.
4. Frigg R. Locking compression plate (LCP). An osteosynthesis plate based on the dynamic compression plate and the point contact fixator (PC-Fix). Injury. 2001;32(suppl 2):63-66.
5. Frigg R. Development of the locking compression plate. Injury. 2003;34(suppl 2):B6-B10.
6. Korner J, Lill H, Müller LP, Rommens PM, Schneider E, Linke B. The LCP-concept in the operative treatment of distal humerus fractures—biological, biomechanical and surgical aspects. Injury. 2003;34(suppl 2):B20-B30.
7. Egol KA, Kubiak EN, Fulkerson E, Kummer FJ, Koval KJ. Biomechanics of locked plates and screws. J Orthop Trauma. 2004;18(8):488-493.
8. Cole PA, Zlowodzki M, Kregor PJ. Treatment of proximal tibia fractures using the Less Invasive Stabilization System: surgical experience and early clinical results in 77 fractures. J Orthop Trauma. 2004;18(8):528-535.
9. Ehlinger M, Adam P, Simon P, Bonnomet F. Technical difficulties in hardware removal in titanium compression plates with locking screws. Orthop Traumatol Surg Res. 2009;95(5):373-376.
10. Gopinathan NR, Dhillon MS, Kumar R. Surgical technique: simple technique for removing a locking recon plate with damaged screw heads. Clin Orthop Relat Res. 2013;471(5):1572-1575.
11. Pattison G, Reynolds J, Hardy J. Salvaging a stripped drive connection when removing screws. Injury. 1999;30(1):74-75.
12. Raja S, Imbuldeniya AM, Garg S, Groom G. Difficulties encountered removing locked plates. Ann R Coll Surg Engl. 2012;94(7):502-505.
13. Kumar G, Dunlop C. Case report: a technique to remove a jammed locking screw from a locking plate. Clin Orthop Relat Res. 2011;469(2):613-616.
14. Disegi JA. Titanium alloys for fracture fixation implants. Injury. 2000;31(suppl 4):14-17.
15. El-Zayat BF, Ruchholtz S, Efe T, Paletta J, Kreslo D, Zettl R. Results of titanium locking plate and stainless steel cerclage wire combination in femoral fractures. Indian J Orthop. 2013;47(5):454-458.
16. Van Nortwick SS, Yao J, Ladd AL. Titanium integration with bone, welding, and screw head destruction complicating hardware removal of the distal radius: report of 2 cases. J Hand Surg. 2012;37(7):1388-1392.
17. Ferguson GS, Chaudhury MK, Sigal GB, Whitesides GM. Contact adhesion of thin gold films on elastomeric supports: cold welding under ambient conditions. Science. 1991;253(5021):776-778.
Proper surgical technique must be used to ensure that surgical fracture management is long-lasting. Plate implantation and screw implantation are among the most common orthopedic procedures performed. Plate and screw osteosynthesis can be done with nonlocking or locking plate and screw constructs or with hybrid fixation that incorporates both methods.
Nonlocking plate and screw osteosynthesis uses friction-fit for fixation. In osteoporotic bone, less torque is generated because of poor bone quality, and thus less friction force between plate and bone.1,2 Locked plating has dramatically changed fracture management, especially in frail and comminuted osteoporotic bone, with significant advantages over conventional plating.3-7
Development of locked plating systems, including the Less Invasive Stabilization System (LISS; DePuy Synthes) with its soft-tissue and fracture-fragment preservation, has changed treatment of distal femur and proximal tibia fractures. Cole and colleagues8 reported stable fixation and union in 97% of their patients. The LISS system proved to be stable, but there were cases of implant removal difficulty with this titanium construct. In 1 of the 10 cases in which the LISS plate was removed, 4 of the 11 locking screws were welded to the plate.8
Cold welding, in which similar metals are chemically bonded together under extreme pressure, is a complication associated with use of titanium-only plates and screws.9 This process, which is more likely to happen if cross-threading occurs within the screw–plate interface, can make screw removal extremely difficult. Screw removal difficulty strips screw heads, and often the surgeon must use either metal cutting instruments or trephines to remove screw remnants, which often results in retained implant or debris and damage or necrosis to surrounding bone.9,10
Locking screws are often inserted under power with a torque-limiting device attached to the drill mechanism to reduce the risk of lock screw overtightening and to try to prevent difficult implant removal. Although standard practice is to insert the screw and stop just before screw head engagement, with final tightening with a torque limiter and hand power, final tightening is often inadvertently done under power.3 Most technique guides instruct surgeons how to insert screws under power while using a torque limiter, but the exact technique is not emphasized.
We conducted a study to determine if rotational speed of screw insertion affects maximum torque on screw with use of a torque limiter. We describe proper use of a torque limiter as well as possible pitfalls. We hypothesized that improper use would result in substantially higher than expected insertion torque.
Materials and Methods
Torque-Limiting Attachments, Torque Wrench, and Drill
The Small Fragment Locking Compression Plate System (Synthes) includes a 1.5-Nm torque-limiting attachment and quick-coupling wooden handles and Star Drive attachments. All devices in this study were in active use at 6 urban institutions (3 level I trauma centers, 2 level II trauma centers, 1 level III hospital). Permission to obtain and test each device was granted by each institution.
A 0.25-inch dial torque wrench (751LDIN; CDI Torque Products) was purchased through an established distributor. The manufacturer includes a traceable certificate of accuracy to verify correct calibration. The torque wrench has a torque range of 0 to 9 Nm with visual increment demarcations of 0.2 Nm and a memory needle to retain maximum torque measurement. The same torque wrench was used in each experiment in order to maintain consistent measurements between devices. It was reset to zero after each use.
This study used a 0.5-inch, 19.2-V lithium drill (Craftsman C3) with 2 speed options: 0 to 440 rpm high torque and 0 to 1600 rpm high speed. This device provides variable torque output with a maximum output of 38.4 Nm. For this study, all measurements were done with the device on its high torque setting.
Maximum Torque Determination for Different Scenarios
Each torque limiter was evaluated for variations in maximum torque under 4 different scenarios. In each scenario, the torque limiter was coupled to the Star Drive attachment and then to that scenario’s rotating force. The completed system was then inserted into the torque wrench, which was secured to a flat working surface and rotated in accordance with each scenario; maximum torque was measured and recorded (Figures 1, 2). A torque-limiting event was defined as a single audible click on the torque limiter.
In scenario 1, each torque-limiting attachment system was attached to a quick-coupling wooden handle. The completed system was then rotated at controlled low velocity under hand power until 1 torque-limiting event occurred. This scenario was also used as an internal control to verify that the torque limiters were calibrated correctly.
In Scenario 2, the device was again attached to a quick-coupling wooden handle. The completed system was rotated at high velocity under hand power until multiple torque-limiting events occurred in a row. High velocity was defined as the operator freely rotating the wooden handle in a single action with full power resulting in multiple torque-limiting events.
In Scenario 3, the device was attached to a power drill braced to the flat working surface and rotated at low velocity under power until 1 torque-limiting event occurred.
In Scenario 4, the device was again attached to a power drill braced to the flat working surface. The completed system was rotated at high velocity under power until multiple torque-limiting events occurred.
After each trial, we recorded maximum torque achieved before each device’s torque-limiting event. Either an orthopedic surgery resident or a qualified medical student tested each torque-limiting device in each standardized testing scenario.
Statistical Analysis
Experiments for each torque limiter were repeated for 3 trials of each of the 4 different scenarios. For comparative statistics between experiments, maximum torque measurements were expressed as means and SDs; 95% confidence interval (95% CI) was calculated and reported to determine extent of variation within a single group. One-way analysis of variance (ANOVA) and Tukey post hoc tests were performed between groups for comparison of the normally distributed data. Significance was set at P ≤ .05.
Results
During simulation, we successfully measured maximum torque achieved with each torque limiter under the 4 different scenarios. All testing was done by 2 operators. ANOVA demonstrated significant (P ≤ .001) differences in torque among the scenarios.
In scenario 1, mean (SD) maximum torque under hand power at low velocity was 1.49 (0.15) Nm (95% CI, 1.43-1.55), near the advertised maximum torque of 1.5 Nm, with relatively minimal variation between devices. This scenario confirmed proper calibration of properly used torque limiters. Mean maximum torque ranged from 1.25 to 1.93 Nm.
In scenario 2, mean (SD) maximum torque under hand power at high velocity was 3.73 (0.79) Nm (95% CI, 3.33-4.13), a 2.5-fold increase compared with scenario 1 (P < .0001) (Figure 3). There also was an increase in variation of maximum torque between trials of individual devices and between different devices. Mean maximum torque ranged from 2.27 to 5.53 Nm.
In scenario 3, mean (SD) maximum torque under drill power at controlled low velocity was 1.47 (0.14) Nm (95% CI, 1.37-1.56), again near the advertised maximum torque of 1.5 Nm, with relatively minimal variation. Mean maximum torque ranged from 1.10 to 1.73 Nm.
In scenario 4, mean (SD) maximum torque under drill power at full power/high velocity was 5.37 (0.90) Nm (95% CI, 4.92-5.83), a 3.65-fold increase compared with scenario 3 (P < .0001) (Figure 3). Mean maximum torque measured in 3 tests ranged from 3.40 to 6.92 Nm.
There was no significant difference in mean maximum torque between the scenarios of hand power at low velocity and drill power at low velocities (P = .999) (Figure 4). Highest maximum torque from any device was 9.0 Nm (drill at full power). Results are summarized in the Table. There was no statistical significance in the test between the 2 test operators.
Discussion
Maximum torque was measured using a torque-limiting attachment under 4 different simulated scenarios. Our goals were to determine if varying practice and rotational velocity would affect maximum insertional torque and to measure consistency among torque limiters. We designed the scenarios to mimic practice patterns, including hand insertion and power insertion of locking screws. Results demonstrated that misuse of a torque-limiting device may inadvertently produce insertional torque substantially higher than recommended. Highest maximum torque was 9.0 Nm, which is 6.0-fold higher than expected for a locking screw using a 1.5-Nm torque limiter.
Our study results showed that insertion under controlled hand power (and low-velocity drill power) until 1 torque-limiting event occurred produced the most consistent and predictable results. Insertion under drill power or high-velocity hand power produced multiple sequential torque-limiting events, yielding inaccurate insertion torque. Low-velocity insertion under hand power, or carefully controlled drill power, consistently produced torque similar to advertised values.
Manufacturers’ technique guides are available for proximal humerus locking compression plate (LCP) systems, small-fragment LCP systems, the Proximal Humeral Interlocking System (PHILOS; DePuy Synthes), and the LISS. These technique guides clearly state that insertion can be performed under power. Only the PHILOS and LISS guides state that insertion should be performed under power until a single click is heard or that final tightening should be completed under hand power. The proximal humerus LCP guide states that surgeons should insert the locking screw under power until the torque-limiting device clicks. The small-fragment LCP guide states that insertion under power should always be completed with the torque-limiting attachment; there is no mention of reducing power or a single click (this may give the surgeon a false sense of security).
Screw overtightening and head/thread stripping can make screw removal challenging.10 Removal rates for LISS plates range from 8% to 26%, and removal is often reported as taking longer than the index procedure, with complication rates as high as 47%.11-13 Bae and colleagues3 reported significant difficulty in removing 24 of 279 self-tapping locking screws (3.5 mm).
It is important to note that these complications, most notably cold welding, are mostly associated with titanium locking plate and screw constructs. Although stainless steel constructs have gained favor, titanium constructs are still widely used around the world.14,15
In 10% of cases in a laboratory setting, insertion of a 3.5-mm locking screw at 4 to 6 Nm damaged the screw.9 Removal of 3.5-mm locking screws had a stripping rate of 8.6%, and use of the torque limiter did not make removal easy all the time.3 Torque limiters are set specific to each screw diameter to reduce the risk of damage/stripping or even overtightening. Even when a surgeon intends to stop a drill before locking, final tightening often inadvertently occurs under power.3
Cold welding is often described as a cause of difficult implant removal.3,12 According to a newer definition, this process is independent of temperature and can occur when 2 metallic surfaces are in direct contact.16 High contact pressures between 2 similar metals can lead to this solid state welding.17 Theoretically, improper use of torque limiters can increase the risk of welding; however, it appears to be associated only with titanium locking plate and screw constructs.
Locked plating osteosynthesis is a valuable tool for fracture management, but improper use can have significant consequences, including morbid implant removal procedures, which are more difficult and time-consuming than the index surgery. We determined that proper use of torque limiters involves insertion under hand or power control at slow velocity until 1 torque-limiting event occurs. Many orthopedic surgeons may assume that torque limiters are accurate no matter how screws are inserted into locking plates. In addition, they may be unaware guidelines exist, as these are often deeply embedded within text. Therefore, we must emphasize that torque limiters can be inaccurate when used improperly.
One limitation of this study is that it tested only the Synthes 1.5-Nm torque-limiting attachment, though we can speculate that torque limiters designed for larger screws and limiters manufactured by different companies will behave similarly. Another limitation is that we did not obtain the hospitals’ service records for the tested equipment and assumed the equipment was properly checked for accuracy by the providing company. However, we hypothesized that, if maintenance were an issue, then our results would not be similar across all sites tested.
These tests involved a torque limiter linked to a torque-measuring device and may not perfectly represent actual torque measured at the locked screw–thread interface. However, we think our construct accurately determines the torque produced at the level of the driver tip. Also, we can speculate that the torque produced with improper use will lead to the complications mentioned and demonstrated in previous studies. Welding of the screw–plate interface may simply be a result of improper trajectory and cross-threading. However, if we assume that torque limiters prevent excessive torque no matter how they are used, high insertion speeds may compound the effect of welding. Additional biomechanical studies with full locked plate osteosynthesis constructs on bone specimens are planned to further characterize the potential complications of this issue.
Proper surgical technique must be used to ensure that surgical fracture management is long-lasting. Plate implantation and screw implantation are among the most common orthopedic procedures performed. Plate and screw osteosynthesis can be done with nonlocking or locking plate and screw constructs or with hybrid fixation that incorporates both methods.
Nonlocking plate and screw osteosynthesis uses friction-fit for fixation. In osteoporotic bone, less torque is generated because of poor bone quality, and thus less friction force between plate and bone.1,2 Locked plating has dramatically changed fracture management, especially in frail and comminuted osteoporotic bone, with significant advantages over conventional plating.3-7
Development of locked plating systems, including the Less Invasive Stabilization System (LISS; DePuy Synthes) with its soft-tissue and fracture-fragment preservation, has changed treatment of distal femur and proximal tibia fractures. Cole and colleagues8 reported stable fixation and union in 97% of their patients. The LISS system proved to be stable, but there were cases of implant removal difficulty with this titanium construct. In 1 of the 10 cases in which the LISS plate was removed, 4 of the 11 locking screws were welded to the plate.8
Cold welding, in which similar metals are chemically bonded together under extreme pressure, is a complication associated with use of titanium-only plates and screws.9 This process, which is more likely to happen if cross-threading occurs within the screw–plate interface, can make screw removal extremely difficult. Screw removal difficulty strips screw heads, and often the surgeon must use either metal cutting instruments or trephines to remove screw remnants, which often results in retained implant or debris and damage or necrosis to surrounding bone.9,10
Locking screws are often inserted under power with a torque-limiting device attached to the drill mechanism to reduce the risk of lock screw overtightening and to try to prevent difficult implant removal. Although standard practice is to insert the screw and stop just before screw head engagement, with final tightening with a torque limiter and hand power, final tightening is often inadvertently done under power.3 Most technique guides instruct surgeons how to insert screws under power while using a torque limiter, but the exact technique is not emphasized.
We conducted a study to determine if rotational speed of screw insertion affects maximum torque on screw with use of a torque limiter. We describe proper use of a torque limiter as well as possible pitfalls. We hypothesized that improper use would result in substantially higher than expected insertion torque.
Materials and Methods
Torque-Limiting Attachments, Torque Wrench, and Drill
The Small Fragment Locking Compression Plate System (Synthes) includes a 1.5-Nm torque-limiting attachment and quick-coupling wooden handles and Star Drive attachments. All devices in this study were in active use at 6 urban institutions (3 level I trauma centers, 2 level II trauma centers, 1 level III hospital). Permission to obtain and test each device was granted by each institution.
A 0.25-inch dial torque wrench (751LDIN; CDI Torque Products) was purchased through an established distributor. The manufacturer includes a traceable certificate of accuracy to verify correct calibration. The torque wrench has a torque range of 0 to 9 Nm with visual increment demarcations of 0.2 Nm and a memory needle to retain maximum torque measurement. The same torque wrench was used in each experiment in order to maintain consistent measurements between devices. It was reset to zero after each use.
This study used a 0.5-inch, 19.2-V lithium drill (Craftsman C3) with 2 speed options: 0 to 440 rpm high torque and 0 to 1600 rpm high speed. This device provides variable torque output with a maximum output of 38.4 Nm. For this study, all measurements were done with the device on its high torque setting.
Maximum Torque Determination for Different Scenarios
Each torque limiter was evaluated for variations in maximum torque under 4 different scenarios. In each scenario, the torque limiter was coupled to the Star Drive attachment and then to that scenario’s rotating force. The completed system was then inserted into the torque wrench, which was secured to a flat working surface and rotated in accordance with each scenario; maximum torque was measured and recorded (Figures 1, 2). A torque-limiting event was defined as a single audible click on the torque limiter.
In scenario 1, each torque-limiting attachment system was attached to a quick-coupling wooden handle. The completed system was then rotated at controlled low velocity under hand power until 1 torque-limiting event occurred. This scenario was also used as an internal control to verify that the torque limiters were calibrated correctly.
In Scenario 2, the device was again attached to a quick-coupling wooden handle. The completed system was rotated at high velocity under hand power until multiple torque-limiting events occurred in a row. High velocity was defined as the operator freely rotating the wooden handle in a single action with full power resulting in multiple torque-limiting events.
In Scenario 3, the device was attached to a power drill braced to the flat working surface and rotated at low velocity under power until 1 torque-limiting event occurred.
In Scenario 4, the device was again attached to a power drill braced to the flat working surface. The completed system was rotated at high velocity under power until multiple torque-limiting events occurred.
After each trial, we recorded maximum torque achieved before each device’s torque-limiting event. Either an orthopedic surgery resident or a qualified medical student tested each torque-limiting device in each standardized testing scenario.
Statistical Analysis
Experiments for each torque limiter were repeated for 3 trials of each of the 4 different scenarios. For comparative statistics between experiments, maximum torque measurements were expressed as means and SDs; 95% confidence interval (95% CI) was calculated and reported to determine extent of variation within a single group. One-way analysis of variance (ANOVA) and Tukey post hoc tests were performed between groups for comparison of the normally distributed data. Significance was set at P ≤ .05.
Results
During simulation, we successfully measured maximum torque achieved with each torque limiter under the 4 different scenarios. All testing was done by 2 operators. ANOVA demonstrated significant (P ≤ .001) differences in torque among the scenarios.
In scenario 1, mean (SD) maximum torque under hand power at low velocity was 1.49 (0.15) Nm (95% CI, 1.43-1.55), near the advertised maximum torque of 1.5 Nm, with relatively minimal variation between devices. This scenario confirmed proper calibration of properly used torque limiters. Mean maximum torque ranged from 1.25 to 1.93 Nm.
In scenario 2, mean (SD) maximum torque under hand power at high velocity was 3.73 (0.79) Nm (95% CI, 3.33-4.13), a 2.5-fold increase compared with scenario 1 (P < .0001) (Figure 3). There also was an increase in variation of maximum torque between trials of individual devices and between different devices. Mean maximum torque ranged from 2.27 to 5.53 Nm.
In scenario 3, mean (SD) maximum torque under drill power at controlled low velocity was 1.47 (0.14) Nm (95% CI, 1.37-1.56), again near the advertised maximum torque of 1.5 Nm, with relatively minimal variation. Mean maximum torque ranged from 1.10 to 1.73 Nm.
In scenario 4, mean (SD) maximum torque under drill power at full power/high velocity was 5.37 (0.90) Nm (95% CI, 4.92-5.83), a 3.65-fold increase compared with scenario 3 (P < .0001) (Figure 3). Mean maximum torque measured in 3 tests ranged from 3.40 to 6.92 Nm.
There was no significant difference in mean maximum torque between the scenarios of hand power at low velocity and drill power at low velocities (P = .999) (Figure 4). Highest maximum torque from any device was 9.0 Nm (drill at full power). Results are summarized in the Table. There was no statistical significance in the test between the 2 test operators.
Discussion
Maximum torque was measured using a torque-limiting attachment under 4 different simulated scenarios. Our goals were to determine if varying practice and rotational velocity would affect maximum insertional torque and to measure consistency among torque limiters. We designed the scenarios to mimic practice patterns, including hand insertion and power insertion of locking screws. Results demonstrated that misuse of a torque-limiting device may inadvertently produce insertional torque substantially higher than recommended. Highest maximum torque was 9.0 Nm, which is 6.0-fold higher than expected for a locking screw using a 1.5-Nm torque limiter.
Our study results showed that insertion under controlled hand power (and low-velocity drill power) until 1 torque-limiting event occurred produced the most consistent and predictable results. Insertion under drill power or high-velocity hand power produced multiple sequential torque-limiting events, yielding inaccurate insertion torque. Low-velocity insertion under hand power, or carefully controlled drill power, consistently produced torque similar to advertised values.
Manufacturers’ technique guides are available for proximal humerus locking compression plate (LCP) systems, small-fragment LCP systems, the Proximal Humeral Interlocking System (PHILOS; DePuy Synthes), and the LISS. These technique guides clearly state that insertion can be performed under power. Only the PHILOS and LISS guides state that insertion should be performed under power until a single click is heard or that final tightening should be completed under hand power. The proximal humerus LCP guide states that surgeons should insert the locking screw under power until the torque-limiting device clicks. The small-fragment LCP guide states that insertion under power should always be completed with the torque-limiting attachment; there is no mention of reducing power or a single click (this may give the surgeon a false sense of security).
Screw overtightening and head/thread stripping can make screw removal challenging.10 Removal rates for LISS plates range from 8% to 26%, and removal is often reported as taking longer than the index procedure, with complication rates as high as 47%.11-13 Bae and colleagues3 reported significant difficulty in removing 24 of 279 self-tapping locking screws (3.5 mm).
It is important to note that these complications, most notably cold welding, are mostly associated with titanium locking plate and screw constructs. Although stainless steel constructs have gained favor, titanium constructs are still widely used around the world.14,15
In 10% of cases in a laboratory setting, insertion of a 3.5-mm locking screw at 4 to 6 Nm damaged the screw.9 Removal of 3.5-mm locking screws had a stripping rate of 8.6%, and use of the torque limiter did not make removal easy all the time.3 Torque limiters are set specific to each screw diameter to reduce the risk of damage/stripping or even overtightening. Even when a surgeon intends to stop a drill before locking, final tightening often inadvertently occurs under power.3
Cold welding is often described as a cause of difficult implant removal.3,12 According to a newer definition, this process is independent of temperature and can occur when 2 metallic surfaces are in direct contact.16 High contact pressures between 2 similar metals can lead to this solid state welding.17 Theoretically, improper use of torque limiters can increase the risk of welding; however, it appears to be associated only with titanium locking plate and screw constructs.
Locked plating osteosynthesis is a valuable tool for fracture management, but improper use can have significant consequences, including morbid implant removal procedures, which are more difficult and time-consuming than the index surgery. We determined that proper use of torque limiters involves insertion under hand or power control at slow velocity until 1 torque-limiting event occurs. Many orthopedic surgeons may assume that torque limiters are accurate no matter how screws are inserted into locking plates. In addition, they may be unaware guidelines exist, as these are often deeply embedded within text. Therefore, we must emphasize that torque limiters can be inaccurate when used improperly.
One limitation of this study is that it tested only the Synthes 1.5-Nm torque-limiting attachment, though we can speculate that torque limiters designed for larger screws and limiters manufactured by different companies will behave similarly. Another limitation is that we did not obtain the hospitals’ service records for the tested equipment and assumed the equipment was properly checked for accuracy by the providing company. However, we hypothesized that, if maintenance were an issue, then our results would not be similar across all sites tested.
These tests involved a torque limiter linked to a torque-measuring device and may not perfectly represent actual torque measured at the locked screw–thread interface. However, we think our construct accurately determines the torque produced at the level of the driver tip. Also, we can speculate that the torque produced with improper use will lead to the complications mentioned and demonstrated in previous studies. Welding of the screw–plate interface may simply be a result of improper trajectory and cross-threading. However, if we assume that torque limiters prevent excessive torque no matter how they are used, high insertion speeds may compound the effect of welding. Additional biomechanical studies with full locked plate osteosynthesis constructs on bone specimens are planned to further characterize the potential complications of this issue.
1. Sommer C, Babst R, Müller M, Hanson B. Locking compression plate loosening and plate breakage: a report of four cases. J Orthop Trauma. 2004;18(8):571-577.
2. Schütz M, Südkamp NP. Revolution in plate osteosynthesis: new internal fixator systems. J Orthop Sci. 2003;8(2):252-258.
3. Bae JH, Oh JK, Oh CW, Hur CR. Technical difficulties of removal of locking screw after locking compression plating. Arch Orthop Trauma Surg. 2009;129(1):91-95.
4. Frigg R. Locking compression plate (LCP). An osteosynthesis plate based on the dynamic compression plate and the point contact fixator (PC-Fix). Injury. 2001;32(suppl 2):63-66.
5. Frigg R. Development of the locking compression plate. Injury. 2003;34(suppl 2):B6-B10.
6. Korner J, Lill H, Müller LP, Rommens PM, Schneider E, Linke B. The LCP-concept in the operative treatment of distal humerus fractures—biological, biomechanical and surgical aspects. Injury. 2003;34(suppl 2):B20-B30.
7. Egol KA, Kubiak EN, Fulkerson E, Kummer FJ, Koval KJ. Biomechanics of locked plates and screws. J Orthop Trauma. 2004;18(8):488-493.
8. Cole PA, Zlowodzki M, Kregor PJ. Treatment of proximal tibia fractures using the Less Invasive Stabilization System: surgical experience and early clinical results in 77 fractures. J Orthop Trauma. 2004;18(8):528-535.
9. Ehlinger M, Adam P, Simon P, Bonnomet F. Technical difficulties in hardware removal in titanium compression plates with locking screws. Orthop Traumatol Surg Res. 2009;95(5):373-376.
10. Gopinathan NR, Dhillon MS, Kumar R. Surgical technique: simple technique for removing a locking recon plate with damaged screw heads. Clin Orthop Relat Res. 2013;471(5):1572-1575.
11. Pattison G, Reynolds J, Hardy J. Salvaging a stripped drive connection when removing screws. Injury. 1999;30(1):74-75.
12. Raja S, Imbuldeniya AM, Garg S, Groom G. Difficulties encountered removing locked plates. Ann R Coll Surg Engl. 2012;94(7):502-505.
13. Kumar G, Dunlop C. Case report: a technique to remove a jammed locking screw from a locking plate. Clin Orthop Relat Res. 2011;469(2):613-616.
14. Disegi JA. Titanium alloys for fracture fixation implants. Injury. 2000;31(suppl 4):14-17.
15. El-Zayat BF, Ruchholtz S, Efe T, Paletta J, Kreslo D, Zettl R. Results of titanium locking plate and stainless steel cerclage wire combination in femoral fractures. Indian J Orthop. 2013;47(5):454-458.
16. Van Nortwick SS, Yao J, Ladd AL. Titanium integration with bone, welding, and screw head destruction complicating hardware removal of the distal radius: report of 2 cases. J Hand Surg. 2012;37(7):1388-1392.
17. Ferguson GS, Chaudhury MK, Sigal GB, Whitesides GM. Contact adhesion of thin gold films on elastomeric supports: cold welding under ambient conditions. Science. 1991;253(5021):776-778.
1. Sommer C, Babst R, Müller M, Hanson B. Locking compression plate loosening and plate breakage: a report of four cases. J Orthop Trauma. 2004;18(8):571-577.
2. Schütz M, Südkamp NP. Revolution in plate osteosynthesis: new internal fixator systems. J Orthop Sci. 2003;8(2):252-258.
3. Bae JH, Oh JK, Oh CW, Hur CR. Technical difficulties of removal of locking screw after locking compression plating. Arch Orthop Trauma Surg. 2009;129(1):91-95.
4. Frigg R. Locking compression plate (LCP). An osteosynthesis plate based on the dynamic compression plate and the point contact fixator (PC-Fix). Injury. 2001;32(suppl 2):63-66.
5. Frigg R. Development of the locking compression plate. Injury. 2003;34(suppl 2):B6-B10.
6. Korner J, Lill H, Müller LP, Rommens PM, Schneider E, Linke B. The LCP-concept in the operative treatment of distal humerus fractures—biological, biomechanical and surgical aspects. Injury. 2003;34(suppl 2):B20-B30.
7. Egol KA, Kubiak EN, Fulkerson E, Kummer FJ, Koval KJ. Biomechanics of locked plates and screws. J Orthop Trauma. 2004;18(8):488-493.
8. Cole PA, Zlowodzki M, Kregor PJ. Treatment of proximal tibia fractures using the Less Invasive Stabilization System: surgical experience and early clinical results in 77 fractures. J Orthop Trauma. 2004;18(8):528-535.
9. Ehlinger M, Adam P, Simon P, Bonnomet F. Technical difficulties in hardware removal in titanium compression plates with locking screws. Orthop Traumatol Surg Res. 2009;95(5):373-376.
10. Gopinathan NR, Dhillon MS, Kumar R. Surgical technique: simple technique for removing a locking recon plate with damaged screw heads. Clin Orthop Relat Res. 2013;471(5):1572-1575.
11. Pattison G, Reynolds J, Hardy J. Salvaging a stripped drive connection when removing screws. Injury. 1999;30(1):74-75.
12. Raja S, Imbuldeniya AM, Garg S, Groom G. Difficulties encountered removing locked plates. Ann R Coll Surg Engl. 2012;94(7):502-505.
13. Kumar G, Dunlop C. Case report: a technique to remove a jammed locking screw from a locking plate. Clin Orthop Relat Res. 2011;469(2):613-616.
14. Disegi JA. Titanium alloys for fracture fixation implants. Injury. 2000;31(suppl 4):14-17.
15. El-Zayat BF, Ruchholtz S, Efe T, Paletta J, Kreslo D, Zettl R. Results of titanium locking plate and stainless steel cerclage wire combination in femoral fractures. Indian J Orthop. 2013;47(5):454-458.
16. Van Nortwick SS, Yao J, Ladd AL. Titanium integration with bone, welding, and screw head destruction complicating hardware removal of the distal radius: report of 2 cases. J Hand Surg. 2012;37(7):1388-1392.
17. Ferguson GS, Chaudhury MK, Sigal GB, Whitesides GM. Contact adhesion of thin gold films on elastomeric supports: cold welding under ambient conditions. Science. 1991;253(5021):776-778.

Poor Sun Protection Practices in Hispanic Patients Increases Risk for Skin Cancer
The most common type of skin cancer in Hispanic patients is basal cell carcinoma, according to Maritza Perez, MD, Associate Clinical Professor of Dermatology at the Icahn School of Medicine at Mount Sinai, New York, New York. Dr. Perez reviewed skin cancer in Hispanics as part of the “Skin Issues in Latino Patients” forum at the 74th Annual Meeting of the American Academy of Dermatology (AAD)(March 4-8, 2016) in Washington, DC. The head and neck region is the most common location of BCC in Hispanics, and it is more common among Hispanic males. Squamous cell carcinomas are the second most common, and melanomas are third most common.
The mortality rate from squamous cell carcinoma is higher in Hispanic patients compared to white patients. Dr. Perez indicated that UV light exposure is a predisposing factor for these types of skin cancer, and poor sun protection practices in this population are to blame.
“Patients [in this population] do not perceive that they are prone to skin cancer,” said Dr. Perez. This erroneous perception has led to delayed diagnosis and treatment of patients in this group, which has caused increased mortality and decreased survival.
Marta Rendon, MD, director of the session at the AAD and Medical Director of The Rendon Center for Dermatology & Aesthetic Medicine, Boca Raton, Florida, noted that Latinos are the fastest growing minority population in the United States and it is estimated that they will be approximately 30% of the population by the year 2050. As a result, dermatologists must emphasize sun protection, avoidance of midday sun, and avoidance of UV radiation.
In his Cutis practical pearls, “Patient Compliance With Photoprotection,” Vincent A. DeLeo, MD, provided tips on sunscreen use. “Patients need a realistic approach to photoprotection based on their genetics, including Fitzpatrick skin type and family history . . . and lifestyle history, which should include location of residence as well as occupation and recreational pursuits,” he said. “These inquiries should lead to a frank discussion of the patient’s risk for developing photodamage and skin cancer.”
Encourage routine use of a sunscreen with a sun protection factor of 30 or higher that carries a “broad spectrum” label. “There is evidence that sunscreens prevent squamous cell carcinoma, actinic keratosis, and photoaging. Early evidence, less strong but positive, also suggests protection against basal cell carcinoma and melanoma,” Dr. DeLeo reported.
The most common type of skin cancer in Hispanic patients is basal cell carcinoma, according to Maritza Perez, MD, Associate Clinical Professor of Dermatology at the Icahn School of Medicine at Mount Sinai, New York, New York. Dr. Perez reviewed skin cancer in Hispanics as part of the “Skin Issues in Latino Patients” forum at the 74th Annual Meeting of the American Academy of Dermatology (AAD)(March 4-8, 2016) in Washington, DC. The head and neck region is the most common location of BCC in Hispanics, and it is more common among Hispanic males. Squamous cell carcinomas are the second most common, and melanomas are third most common.
The mortality rate from squamous cell carcinoma is higher in Hispanic patients compared to white patients. Dr. Perez indicated that UV light exposure is a predisposing factor for these types of skin cancer, and poor sun protection practices in this population are to blame.
“Patients [in this population] do not perceive that they are prone to skin cancer,” said Dr. Perez. This erroneous perception has led to delayed diagnosis and treatment of patients in this group, which has caused increased mortality and decreased survival.
Marta Rendon, MD, director of the session at the AAD and Medical Director of The Rendon Center for Dermatology & Aesthetic Medicine, Boca Raton, Florida, noted that Latinos are the fastest growing minority population in the United States and it is estimated that they will be approximately 30% of the population by the year 2050. As a result, dermatologists must emphasize sun protection, avoidance of midday sun, and avoidance of UV radiation.
In his Cutis practical pearls, “Patient Compliance With Photoprotection,” Vincent A. DeLeo, MD, provided tips on sunscreen use. “Patients need a realistic approach to photoprotection based on their genetics, including Fitzpatrick skin type and family history . . . and lifestyle history, which should include location of residence as well as occupation and recreational pursuits,” he said. “These inquiries should lead to a frank discussion of the patient’s risk for developing photodamage and skin cancer.”
Encourage routine use of a sunscreen with a sun protection factor of 30 or higher that carries a “broad spectrum” label. “There is evidence that sunscreens prevent squamous cell carcinoma, actinic keratosis, and photoaging. Early evidence, less strong but positive, also suggests protection against basal cell carcinoma and melanoma,” Dr. DeLeo reported.
The most common type of skin cancer in Hispanic patients is basal cell carcinoma, according to Maritza Perez, MD, Associate Clinical Professor of Dermatology at the Icahn School of Medicine at Mount Sinai, New York, New York. Dr. Perez reviewed skin cancer in Hispanics as part of the “Skin Issues in Latino Patients” forum at the 74th Annual Meeting of the American Academy of Dermatology (AAD)(March 4-8, 2016) in Washington, DC. The head and neck region is the most common location of BCC in Hispanics, and it is more common among Hispanic males. Squamous cell carcinomas are the second most common, and melanomas are third most common.
The mortality rate from squamous cell carcinoma is higher in Hispanic patients compared to white patients. Dr. Perez indicated that UV light exposure is a predisposing factor for these types of skin cancer, and poor sun protection practices in this population are to blame.
“Patients [in this population] do not perceive that they are prone to skin cancer,” said Dr. Perez. This erroneous perception has led to delayed diagnosis and treatment of patients in this group, which has caused increased mortality and decreased survival.
Marta Rendon, MD, director of the session at the AAD and Medical Director of The Rendon Center for Dermatology & Aesthetic Medicine, Boca Raton, Florida, noted that Latinos are the fastest growing minority population in the United States and it is estimated that they will be approximately 30% of the population by the year 2050. As a result, dermatologists must emphasize sun protection, avoidance of midday sun, and avoidance of UV radiation.
In his Cutis practical pearls, “Patient Compliance With Photoprotection,” Vincent A. DeLeo, MD, provided tips on sunscreen use. “Patients need a realistic approach to photoprotection based on their genetics, including Fitzpatrick skin type and family history . . . and lifestyle history, which should include location of residence as well as occupation and recreational pursuits,” he said. “These inquiries should lead to a frank discussion of the patient’s risk for developing photodamage and skin cancer.”
Encourage routine use of a sunscreen with a sun protection factor of 30 or higher that carries a “broad spectrum” label. “There is evidence that sunscreens prevent squamous cell carcinoma, actinic keratosis, and photoaging. Early evidence, less strong but positive, also suggests protection against basal cell carcinoma and melanoma,” Dr. DeLeo reported.

Obesity, oral contraceptive use are risk factors for cerebral venous thrombosis in women
Obese women taking oral contraceptives had a 30-fold increased odds for cerebral venous thrombosis (CVT), compared with normal-weight women, in an unmatched case-control study of 186 CVT cases and 6,134 controls.
The association between a body-mass index of 30 kg/m2 or higher and increased odds of CVT “appears to be fully attributable to a strongly increased risk in women who use oral contraceptives [OCs],” said Dr. Susanna M. Zuurbier of the Academic Medical Centre in Amsterdam, and her colleagues.
“Although the relative risks are increased substantially, the absolute risks of CVT are still small,” the researchers said. “Moreover, withholding oral contraceptives may lead to an increase in unintended pregnancies and thus the number of pregnancy-related thrombosis cases.”
After controlling for multiple variables, CVT was 2.63 times more likely to occur in obese individuals, compared with those of normal weight. When the findings were stratified by sex, a BMI greater than 30 kg/m2 had no significant impact on CVT in men, but the odds rose 3.5-fold in women, the investigators reported online March 14 in JAMA Neurology (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2016.000100).
On further analysis, overweight and obesity had a dose-dependent impact on the odds of CVT among women who used OCs. Adjusted odds ratios were 11.87 for those with BMIs ranging from 25 kg/m2 to 29 kg/m2, and 29.26 for those with BMIs of 30 kg/m2 or higher when compared against normal-weight women who did not use OCs. No association was found between obesity and CVT risk in women who did not use OCs, the researchers noted.
Patients with CVT were recruited in 2006-2014 from two medical centers, one in the Netherlands and the other in Switzerland. The controls were part of the Dutch Multiple Environmental and Genetic Assessment of Risk Factors for Venous Thrombosis (MEGA) study. Overall, patients with CVT were more likely to be younger than controls (median age 40 years vs. 48 years). They also were more likely than controls to be female (72% vs. 53%), users of oral contraceptives (73% vs. 24%) and have a history of cancer (9% vs. 4%).
The study was limited by several factors, including the small number of CVT cases, the lack of complete BMI data for the study population, and time difference in recruitment of cases and controls, the researchers noted, but they advised clinicians to keep the risk in mind when counseling obese women about OC use.
“Alternative methods of contraception that are not associated with thrombosis, such as an intrauterine device, might be offered to these women,” they wrote.
The study was funded by grants from the Netherlands Organisation for Scientific Research, the Dutch Thrombosis Society, the Remmert Adriaan Laan Foundation, and the Swiss Heart Foundation. Study coauthor Dr. Marcel Arnold disclosed receiving honoraria for giving lectures and serving on advisory boards for Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer, and Covidien.
“The discovery of increased rates of asymptomatic atherosclerosis in patients with VTE led to the systematic study of cardiovascular risk factors, such as obesity, in VTE,” but the possible association between obesity and CVT has not been explored, Dr. Chirantan Banerjee wrote in an accompanying editorial.
The current study results are “novel and in concordance with prior studies on obesity and OC use as risk factors for VTE,” Dr. Banerjee noted. The study’s strengths lie in the inclusion of appropriate subgroups, controls, and confounding variables. In addition to further research to validate the findings, “studies investigating the effect of inherited thrombophilia on the association between obesity and CVT would also be important because the current study did not have this data,” he noted. “Other potential confounders, such as obstructive sleep apnea and anemia, should be included in future studies.
In the meantime, “the authors correctly point out that despite the manifold increased relative risk, the absolute risk of CVT in obese women taking OCs still remains low and should not preclude OC use among them,” Dr. Banerjee said.
Dr. Banerjee is a member of the department of neurology at the Medical University of Charleston, S.C., and had no financial conflicts to disclose. These comments were taken from his editorial accompanying Dr. Zuurbier and colleagues’ report (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2015.5107).
“The discovery of increased rates of asymptomatic atherosclerosis in patients with VTE led to the systematic study of cardiovascular risk factors, such as obesity, in VTE,” but the possible association between obesity and CVT has not been explored, Dr. Chirantan Banerjee wrote in an accompanying editorial.
The current study results are “novel and in concordance with prior studies on obesity and OC use as risk factors for VTE,” Dr. Banerjee noted. The study’s strengths lie in the inclusion of appropriate subgroups, controls, and confounding variables. In addition to further research to validate the findings, “studies investigating the effect of inherited thrombophilia on the association between obesity and CVT would also be important because the current study did not have this data,” he noted. “Other potential confounders, such as obstructive sleep apnea and anemia, should be included in future studies.
In the meantime, “the authors correctly point out that despite the manifold increased relative risk, the absolute risk of CVT in obese women taking OCs still remains low and should not preclude OC use among them,” Dr. Banerjee said.
Dr. Banerjee is a member of the department of neurology at the Medical University of Charleston, S.C., and had no financial conflicts to disclose. These comments were taken from his editorial accompanying Dr. Zuurbier and colleagues’ report (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2015.5107).
“The discovery of increased rates of asymptomatic atherosclerosis in patients with VTE led to the systematic study of cardiovascular risk factors, such as obesity, in VTE,” but the possible association between obesity and CVT has not been explored, Dr. Chirantan Banerjee wrote in an accompanying editorial.
The current study results are “novel and in concordance with prior studies on obesity and OC use as risk factors for VTE,” Dr. Banerjee noted. The study’s strengths lie in the inclusion of appropriate subgroups, controls, and confounding variables. In addition to further research to validate the findings, “studies investigating the effect of inherited thrombophilia on the association between obesity and CVT would also be important because the current study did not have this data,” he noted. “Other potential confounders, such as obstructive sleep apnea and anemia, should be included in future studies.
In the meantime, “the authors correctly point out that despite the manifold increased relative risk, the absolute risk of CVT in obese women taking OCs still remains low and should not preclude OC use among them,” Dr. Banerjee said.
Dr. Banerjee is a member of the department of neurology at the Medical University of Charleston, S.C., and had no financial conflicts to disclose. These comments were taken from his editorial accompanying Dr. Zuurbier and colleagues’ report (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2015.5107).
Obese women taking oral contraceptives had a 30-fold increased odds for cerebral venous thrombosis (CVT), compared with normal-weight women, in an unmatched case-control study of 186 CVT cases and 6,134 controls.
The association between a body-mass index of 30 kg/m2 or higher and increased odds of CVT “appears to be fully attributable to a strongly increased risk in women who use oral contraceptives [OCs],” said Dr. Susanna M. Zuurbier of the Academic Medical Centre in Amsterdam, and her colleagues.
“Although the relative risks are increased substantially, the absolute risks of CVT are still small,” the researchers said. “Moreover, withholding oral contraceptives may lead to an increase in unintended pregnancies and thus the number of pregnancy-related thrombosis cases.”
After controlling for multiple variables, CVT was 2.63 times more likely to occur in obese individuals, compared with those of normal weight. When the findings were stratified by sex, a BMI greater than 30 kg/m2 had no significant impact on CVT in men, but the odds rose 3.5-fold in women, the investigators reported online March 14 in JAMA Neurology (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2016.000100).
On further analysis, overweight and obesity had a dose-dependent impact on the odds of CVT among women who used OCs. Adjusted odds ratios were 11.87 for those with BMIs ranging from 25 kg/m2 to 29 kg/m2, and 29.26 for those with BMIs of 30 kg/m2 or higher when compared against normal-weight women who did not use OCs. No association was found between obesity and CVT risk in women who did not use OCs, the researchers noted.
Patients with CVT were recruited in 2006-2014 from two medical centers, one in the Netherlands and the other in Switzerland. The controls were part of the Dutch Multiple Environmental and Genetic Assessment of Risk Factors for Venous Thrombosis (MEGA) study. Overall, patients with CVT were more likely to be younger than controls (median age 40 years vs. 48 years). They also were more likely than controls to be female (72% vs. 53%), users of oral contraceptives (73% vs. 24%) and have a history of cancer (9% vs. 4%).
The study was limited by several factors, including the small number of CVT cases, the lack of complete BMI data for the study population, and time difference in recruitment of cases and controls, the researchers noted, but they advised clinicians to keep the risk in mind when counseling obese women about OC use.
“Alternative methods of contraception that are not associated with thrombosis, such as an intrauterine device, might be offered to these women,” they wrote.
The study was funded by grants from the Netherlands Organisation for Scientific Research, the Dutch Thrombosis Society, the Remmert Adriaan Laan Foundation, and the Swiss Heart Foundation. Study coauthor Dr. Marcel Arnold disclosed receiving honoraria for giving lectures and serving on advisory boards for Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer, and Covidien.
Obese women taking oral contraceptives had a 30-fold increased odds for cerebral venous thrombosis (CVT), compared with normal-weight women, in an unmatched case-control study of 186 CVT cases and 6,134 controls.
The association between a body-mass index of 30 kg/m2 or higher and increased odds of CVT “appears to be fully attributable to a strongly increased risk in women who use oral contraceptives [OCs],” said Dr. Susanna M. Zuurbier of the Academic Medical Centre in Amsterdam, and her colleagues.
“Although the relative risks are increased substantially, the absolute risks of CVT are still small,” the researchers said. “Moreover, withholding oral contraceptives may lead to an increase in unintended pregnancies and thus the number of pregnancy-related thrombosis cases.”
After controlling for multiple variables, CVT was 2.63 times more likely to occur in obese individuals, compared with those of normal weight. When the findings were stratified by sex, a BMI greater than 30 kg/m2 had no significant impact on CVT in men, but the odds rose 3.5-fold in women, the investigators reported online March 14 in JAMA Neurology (JAMA Neurol. 2016 Mar 14. doi: 10.1001/jamaneurol.2016.000100).
On further analysis, overweight and obesity had a dose-dependent impact on the odds of CVT among women who used OCs. Adjusted odds ratios were 11.87 for those with BMIs ranging from 25 kg/m2 to 29 kg/m2, and 29.26 for those with BMIs of 30 kg/m2 or higher when compared against normal-weight women who did not use OCs. No association was found between obesity and CVT risk in women who did not use OCs, the researchers noted.
Patients with CVT were recruited in 2006-2014 from two medical centers, one in the Netherlands and the other in Switzerland. The controls were part of the Dutch Multiple Environmental and Genetic Assessment of Risk Factors for Venous Thrombosis (MEGA) study. Overall, patients with CVT were more likely to be younger than controls (median age 40 years vs. 48 years). They also were more likely than controls to be female (72% vs. 53%), users of oral contraceptives (73% vs. 24%) and have a history of cancer (9% vs. 4%).
The study was limited by several factors, including the small number of CVT cases, the lack of complete BMI data for the study population, and time difference in recruitment of cases and controls, the researchers noted, but they advised clinicians to keep the risk in mind when counseling obese women about OC use.
“Alternative methods of contraception that are not associated with thrombosis, such as an intrauterine device, might be offered to these women,” they wrote.
The study was funded by grants from the Netherlands Organisation for Scientific Research, the Dutch Thrombosis Society, the Remmert Adriaan Laan Foundation, and the Swiss Heart Foundation. Study coauthor Dr. Marcel Arnold disclosed receiving honoraria for giving lectures and serving on advisory boards for Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer, and Covidien.
FROM JAMA NEUROLOGY
Key clinical point: Obesity and oral contraceptive use significantly increased the odds for cerebral venous thrombosis in adults.
Major finding: The risk of CVT was 2.63 times more likely among obese women compared to nonobese women, and nearly 30 times more likely in women taking oral contraceptives.
Data source: An unmatched, case-control study of 186 CVT patients and 6,134 healthy controls.
Disclosures: The study was funded by grants from the Netherlands Organisation for Scientific Research, the Dutch Thrombosis Society, the Remmert Adriaan Laan Foundation, and the Swiss Heart Foundation. Study coauthor Dr. Marcel Arnold disclosed receiving honoraria for giving lectures and serving on advisory boards for Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer, and Covidien.
High false positives found for Medtronic’s implantable AF detectors
LOS ANGELES – Medtronic’s Reveal LINQ implantable loop recorders misidentified 84% of rhythm anomalies as atrial fibrillation in 52 stroke patients at Emory University in Atlanta, according to a presentation at the International Stroke Conference.
Two electrophysiologists reviewed a random sample of 166 rhythm strips from those patients that were identified by Reveal as atrial fibrillation (AF) over a 2-month period; 140 (84%) were false positives. Eighty (57%) of the false positives were premature atrial complexes, 31 (22%) were due to T wave over-sensing, 14 (10%) to noise, 7 (5%) to premature ventricular complexes, 4 (2.9%) to under-sensing, and 4 (2.9%) to sinus arrhythmias.
There wasn’t a mix-and-match of true and false positives in the same patient; false and true positives were consistent in patients over the study period.
The take-home message from the study is that the high sensitivity of Medtronic’s implantable loop recorders means that they are good at detecting possible AF, but their findings must be reviewed and confirmed before being acted upon.

“There are high rates of false positives, but the results can be easily adjudicated by electrophysiologists as evidenced by our observer agreement,” which was 100%. “These strips need to be adjudicated by somebody, and not taken at face value,” said investigator Dr. Spencer Maddox, an Emory resident.
The devices look mainly at RR intervals and the presence or absence of P waves. Runs of 2 minutes are required for AF. One of the issues in the study was that T waves were identified as QRS complexes, which changed the RR interval and trigged the device to report AF, he said.
Medtronic could reduce the false positive rate by, for instance, extending the run required for AF, but it would be a bad idea. “The goal here is to not miss atrial fibrillation,” Dr. Maddox said. “There are false positives, but as long as you go back and reread, I think that’s fine. I wouldn’t mess with the sensitivity of the device.”
The company said the same thing when asked for comment on the study.
“The AF detection algorithm ... is tuned to favor sensitivity because manual review can subsequently rule out any false positives. This is one reason it is important to have trained cardiologists or electrophysiologists involved in reviewing reports of irregular heart rhythms to determine a diagnosis of AF,” said Medtronic spokesman Ryan Mathre, who also noted that company data suggest a lower false positive rate (Cerebrovasc Dis. 2015;40:175-81).

Even so, confirmation doesn’t always happen, “and that’s the scary part. In practice almost all the time,” the report comes in “and it’s acted on,” said Dr. Robert Hart, a neurology professor at McMaster University in Hamilton, Ont., and comoderator of the study presentation.
The Emory patients were all recovering from ischemic strokes or TIAs. They were about 70 years old on average, about 60% were men, and almost all were white. A total of 38 had implantable loop recorders for cryptogenic strokes; the devices detected true AF in 4 (11%) after a mean of 92 days. Early Holter monitoring missed it.
Emory generally prescribes anticoagulants if AF is confirmed, but, as several audience members noted, the burden of AF that requires anticoagulation is unclear. “It’s an area with a lot of thought now, but no good answers,” Dr. Maddox said at the conference, sponsored by the American Heart Association.
The investigators had no disclosures, and there was no outside funding for the work.
LOS ANGELES – Medtronic’s Reveal LINQ implantable loop recorders misidentified 84% of rhythm anomalies as atrial fibrillation in 52 stroke patients at Emory University in Atlanta, according to a presentation at the International Stroke Conference.
Two electrophysiologists reviewed a random sample of 166 rhythm strips from those patients that were identified by Reveal as atrial fibrillation (AF) over a 2-month period; 140 (84%) were false positives. Eighty (57%) of the false positives were premature atrial complexes, 31 (22%) were due to T wave over-sensing, 14 (10%) to noise, 7 (5%) to premature ventricular complexes, 4 (2.9%) to under-sensing, and 4 (2.9%) to sinus arrhythmias.
There wasn’t a mix-and-match of true and false positives in the same patient; false and true positives were consistent in patients over the study period.
The take-home message from the study is that the high sensitivity of Medtronic’s implantable loop recorders means that they are good at detecting possible AF, but their findings must be reviewed and confirmed before being acted upon.
“There are high rates of false positives, but the results can be easily adjudicated by electrophysiologists as evidenced by our observer agreement,” which was 100%. “These strips need to be adjudicated by somebody, and not taken at face value,” said investigator Dr. Spencer Maddox, an Emory resident.
The devices look mainly at RR intervals and the presence or absence of P waves. Runs of 2 minutes are required for AF. One of the issues in the study was that T waves were identified as QRS complexes, which changed the RR interval and trigged the device to report AF, he said.
Medtronic could reduce the false positive rate by, for instance, extending the run required for AF, but it would be a bad idea. “The goal here is to not miss atrial fibrillation,” Dr. Maddox said. “There are false positives, but as long as you go back and reread, I think that’s fine. I wouldn’t mess with the sensitivity of the device.”
The company said the same thing when asked for comment on the study.
“The AF detection algorithm ... is tuned to favor sensitivity because manual review can subsequently rule out any false positives. This is one reason it is important to have trained cardiologists or electrophysiologists involved in reviewing reports of irregular heart rhythms to determine a diagnosis of AF,” said Medtronic spokesman Ryan Mathre, who also noted that company data suggest a lower false positive rate (Cerebrovasc Dis. 2015;40:175-81).
Even so, confirmation doesn’t always happen, “and that’s the scary part. In practice almost all the time,” the report comes in “and it’s acted on,” said Dr. Robert Hart, a neurology professor at McMaster University in Hamilton, Ont., and comoderator of the study presentation.
The Emory patients were all recovering from ischemic strokes or TIAs. They were about 70 years old on average, about 60% were men, and almost all were white. A total of 38 had implantable loop recorders for cryptogenic strokes; the devices detected true AF in 4 (11%) after a mean of 92 days. Early Holter monitoring missed it.
Emory generally prescribes anticoagulants if AF is confirmed, but, as several audience members noted, the burden of AF that requires anticoagulation is unclear. “It’s an area with a lot of thought now, but no good answers,” Dr. Maddox said at the conference, sponsored by the American Heart Association.
The investigators had no disclosures, and there was no outside funding for the work.
LOS ANGELES – Medtronic’s Reveal LINQ implantable loop recorders misidentified 84% of rhythm anomalies as atrial fibrillation in 52 stroke patients at Emory University in Atlanta, according to a presentation at the International Stroke Conference.
Two electrophysiologists reviewed a random sample of 166 rhythm strips from those patients that were identified by Reveal as atrial fibrillation (AF) over a 2-month period; 140 (84%) were false positives. Eighty (57%) of the false positives were premature atrial complexes, 31 (22%) were due to T wave over-sensing, 14 (10%) to noise, 7 (5%) to premature ventricular complexes, 4 (2.9%) to under-sensing, and 4 (2.9%) to sinus arrhythmias.
There wasn’t a mix-and-match of true and false positives in the same patient; false and true positives were consistent in patients over the study period.
The take-home message from the study is that the high sensitivity of Medtronic’s implantable loop recorders means that they are good at detecting possible AF, but their findings must be reviewed and confirmed before being acted upon.
“There are high rates of false positives, but the results can be easily adjudicated by electrophysiologists as evidenced by our observer agreement,” which was 100%. “These strips need to be adjudicated by somebody, and not taken at face value,” said investigator Dr. Spencer Maddox, an Emory resident.
The devices look mainly at RR intervals and the presence or absence of P waves. Runs of 2 minutes are required for AF. One of the issues in the study was that T waves were identified as QRS complexes, which changed the RR interval and trigged the device to report AF, he said.
Medtronic could reduce the false positive rate by, for instance, extending the run required for AF, but it would be a bad idea. “The goal here is to not miss atrial fibrillation,” Dr. Maddox said. “There are false positives, but as long as you go back and reread, I think that’s fine. I wouldn’t mess with the sensitivity of the device.”
The company said the same thing when asked for comment on the study.
“The AF detection algorithm ... is tuned to favor sensitivity because manual review can subsequently rule out any false positives. This is one reason it is important to have trained cardiologists or electrophysiologists involved in reviewing reports of irregular heart rhythms to determine a diagnosis of AF,” said Medtronic spokesman Ryan Mathre, who also noted that company data suggest a lower false positive rate (Cerebrovasc Dis. 2015;40:175-81).
Even so, confirmation doesn’t always happen, “and that’s the scary part. In practice almost all the time,” the report comes in “and it’s acted on,” said Dr. Robert Hart, a neurology professor at McMaster University in Hamilton, Ont., and comoderator of the study presentation.
The Emory patients were all recovering from ischemic strokes or TIAs. They were about 70 years old on average, about 60% were men, and almost all were white. A total of 38 had implantable loop recorders for cryptogenic strokes; the devices detected true AF in 4 (11%) after a mean of 92 days. Early Holter monitoring missed it.
Emory generally prescribes anticoagulants if AF is confirmed, but, as several audience members noted, the burden of AF that requires anticoagulation is unclear. “It’s an area with a lot of thought now, but no good answers,” Dr. Maddox said at the conference, sponsored by the American Heart Association.
The investigators had no disclosures, and there was no outside funding for the work.
AT The INTERNATIONAL STROKE CONFERENCE
Key clinical point: The high sensitivity of Medtronic’s implantable loop recorders means that they’re good at detecting possible AF, but their findings must be reviewed and confirmed before being acted upon.
Major finding: Two electrophysiologists reviewed a random sample of 166 rhythm strips identified by Reveal as atrial fibrillation; 140 (84%) were false positives.
Data source: Fifty-two ischemic stroke and TIA patients.
Disclosures: The investigators had no disclosures. There was no outside funding for the work.

Lenalidomide, thalidomide in melphalan regimen yields similar progression-free survival
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
FROM BLOOD
Key clinical point: Lenalidomide or thalidomide added to a melphalan backbone regimen yielded similar outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma.
Major finding: At 36 months median follow-up, median PFS was 20 months for patients treated with thalidomide, compared with 23 months for those treated with lenalidomide.
Data source: Randomized controlled trial of 637 patients with multiple myeloma who were not candidates for autologous stem cell transplantation.
Disclosures: The Dutch Cancer Society, Norwegian Cancer Society, and Celgene supported the study. Dr. Zweegman and other colleagues reported financial relationships with Celgene.