User login
The surprising way to fight asthma symptoms
Asthma is a sneaky foe.
a professor of nursing at Columbia University and a spokesperson for the Asthma and Allergy Foundation of America.
But that doesn’t mean exercise should be avoided, she said.
Exercise, in fact, is one of the best ways to reduce asthma symptoms. Research over the past 2 decades has shown that physical activity can help improve lung function and boost quality of life for someone with asthma.
As their fitness improves, asthma patients report better sleep, reduced stress, improved weight control, and more days without symptoms. In some cases, they’re able to cut down their medication doses.
Exercise reduces inflammatory cytokines and increases anti-inflammatory cytokines, according to a 2023 review by researchers in the United Kingdom. That could help calm chronic airway inflammation, easing symptoms of asthma.
A few simple guidelines can help patients reap those benefits while staying safe.
Make sure the first steps aren’t the last steps
For someone who’s new to exercise, there’s only one way to begin: Carefully.
The Global Initiative for Asthma recommends twice-weekly cardio and strength training.
“You always start low and slow,” said Spencer Nadolsky, DO, a board-certified obesity and lipid specialist and medical director of Sequence, a comprehensive weight management program.
“Low” means light loads in the weight room. “Slow” means short, easy walks.
Many have been put “through the wringer” when starting out, discouraging them from continuing, Dr. Nadolsky said. “They were too sore, and it felt more like punishment.”
An even bigger concern is triggering an asthma attack. Patients should take steps to lower the risk by carrying their rescue inhalers and keeping up on medications, he added.
“A health care professional should be consulted” before the start of a new activity or ramping up a program, or anytime asthma interferes with a workout, Dr. George said.
Those who exercise outside need to be aware of the air quality, especially at a time when smoke and particulates from a wildfires in Canada can trigger asthma symptoms in people thousands of miles away.
The harder one works, the higher one’s “ventilation” – taking more air into the lungs, and potentially more allergens and pollutants.
Temperature and humidity also become risky at the extremes. Cold, dry air can dehydrate and constrict the airways, making it hard to breathe.
How to choose the best type of exercise
Step 1: Be realistic. People with asthma often have less exercise capacity than those who don’t – understandable when shortness of breath is the default setting.
Second, allow for plenty of time to warm up. A solid warm-up routine – particularly one with a mix of lower- and higher-intensity exercises – may help prevent exercise-induced bronchoconstriction causing shortness of breath and wheezing.
For example, warming up on a treadmill or exercise bike could be mixed with a few short bursts of faster running or cycling, with a couple of minutes of recovery at a slower pace in between.
That concept can be expanded into a full-blown workout.
High-intensity interval training (HIIT) is a promising option for people with asthma. A 2021 study showed that three 20-minute interval workouts per week significantly improved asthma control.
“The benefit of HIIT is that ventilation is able to recover intermittently,” said Carley O’Neill, PhD, an exercise scientist at Acadia University in Nova Scotia and the study’s lead author.
That’s a key difference from conventional cardio, where the constant exertion can evaporate water from the lungs faster than your body can replenish it. “Dehydrating of the airways can, in some, trigger exercise-induced asthma,” Dr. O’Neill said.
HIIT, conversely, allows the airways to recover and rehydrate between exercise bouts.
Another recent study found that people with asthma who did HIIT workouts had fewer breathing problems and felt less fatigued, compared with a matched group who did cardio training at a constant pace. (Both types of cardio led to similar improvements in aerobic fitness.)
Individuals can choose other types of intermittent exercise as well. Strength training, for example, requires relatively short periods of exertion, with plenty of rest in between.
The one choice you don’t want to make
While there are lots of good exercise options for someone with asthma, there’s one clearly bad choice, according to Dr. George: “Avoiding exercise.”
Being inactive puts one at higher risk for obesity and all the health problems that go with it. And allowing one’s fitness level to decline makes it much harder to move when one needs or wants to.
Any choice is better than that one.
A version of this article first appeared on WebMD.com.
Asthma is a sneaky foe.
a professor of nursing at Columbia University and a spokesperson for the Asthma and Allergy Foundation of America.
But that doesn’t mean exercise should be avoided, she said.
Exercise, in fact, is one of the best ways to reduce asthma symptoms. Research over the past 2 decades has shown that physical activity can help improve lung function and boost quality of life for someone with asthma.
As their fitness improves, asthma patients report better sleep, reduced stress, improved weight control, and more days without symptoms. In some cases, they’re able to cut down their medication doses.
Exercise reduces inflammatory cytokines and increases anti-inflammatory cytokines, according to a 2023 review by researchers in the United Kingdom. That could help calm chronic airway inflammation, easing symptoms of asthma.
A few simple guidelines can help patients reap those benefits while staying safe.
Make sure the first steps aren’t the last steps
For someone who’s new to exercise, there’s only one way to begin: Carefully.
The Global Initiative for Asthma recommends twice-weekly cardio and strength training.
“You always start low and slow,” said Spencer Nadolsky, DO, a board-certified obesity and lipid specialist and medical director of Sequence, a comprehensive weight management program.
“Low” means light loads in the weight room. “Slow” means short, easy walks.
Many have been put “through the wringer” when starting out, discouraging them from continuing, Dr. Nadolsky said. “They were too sore, and it felt more like punishment.”
An even bigger concern is triggering an asthma attack. Patients should take steps to lower the risk by carrying their rescue inhalers and keeping up on medications, he added.
“A health care professional should be consulted” before the start of a new activity or ramping up a program, or anytime asthma interferes with a workout, Dr. George said.
Those who exercise outside need to be aware of the air quality, especially at a time when smoke and particulates from a wildfires in Canada can trigger asthma symptoms in people thousands of miles away.
The harder one works, the higher one’s “ventilation” – taking more air into the lungs, and potentially more allergens and pollutants.
Temperature and humidity also become risky at the extremes. Cold, dry air can dehydrate and constrict the airways, making it hard to breathe.
How to choose the best type of exercise
Step 1: Be realistic. People with asthma often have less exercise capacity than those who don’t – understandable when shortness of breath is the default setting.
Second, allow for plenty of time to warm up. A solid warm-up routine – particularly one with a mix of lower- and higher-intensity exercises – may help prevent exercise-induced bronchoconstriction causing shortness of breath and wheezing.
For example, warming up on a treadmill or exercise bike could be mixed with a few short bursts of faster running or cycling, with a couple of minutes of recovery at a slower pace in between.
That concept can be expanded into a full-blown workout.
High-intensity interval training (HIIT) is a promising option for people with asthma. A 2021 study showed that three 20-minute interval workouts per week significantly improved asthma control.
“The benefit of HIIT is that ventilation is able to recover intermittently,” said Carley O’Neill, PhD, an exercise scientist at Acadia University in Nova Scotia and the study’s lead author.
That’s a key difference from conventional cardio, where the constant exertion can evaporate water from the lungs faster than your body can replenish it. “Dehydrating of the airways can, in some, trigger exercise-induced asthma,” Dr. O’Neill said.
HIIT, conversely, allows the airways to recover and rehydrate between exercise bouts.
Another recent study found that people with asthma who did HIIT workouts had fewer breathing problems and felt less fatigued, compared with a matched group who did cardio training at a constant pace. (Both types of cardio led to similar improvements in aerobic fitness.)
Individuals can choose other types of intermittent exercise as well. Strength training, for example, requires relatively short periods of exertion, with plenty of rest in between.
The one choice you don’t want to make
While there are lots of good exercise options for someone with asthma, there’s one clearly bad choice, according to Dr. George: “Avoiding exercise.”
Being inactive puts one at higher risk for obesity and all the health problems that go with it. And allowing one’s fitness level to decline makes it much harder to move when one needs or wants to.
Any choice is better than that one.
A version of this article first appeared on WebMD.com.
Asthma is a sneaky foe.
a professor of nursing at Columbia University and a spokesperson for the Asthma and Allergy Foundation of America.
But that doesn’t mean exercise should be avoided, she said.
Exercise, in fact, is one of the best ways to reduce asthma symptoms. Research over the past 2 decades has shown that physical activity can help improve lung function and boost quality of life for someone with asthma.
As their fitness improves, asthma patients report better sleep, reduced stress, improved weight control, and more days without symptoms. In some cases, they’re able to cut down their medication doses.
Exercise reduces inflammatory cytokines and increases anti-inflammatory cytokines, according to a 2023 review by researchers in the United Kingdom. That could help calm chronic airway inflammation, easing symptoms of asthma.
A few simple guidelines can help patients reap those benefits while staying safe.
Make sure the first steps aren’t the last steps
For someone who’s new to exercise, there’s only one way to begin: Carefully.
The Global Initiative for Asthma recommends twice-weekly cardio and strength training.
“You always start low and slow,” said Spencer Nadolsky, DO, a board-certified obesity and lipid specialist and medical director of Sequence, a comprehensive weight management program.
“Low” means light loads in the weight room. “Slow” means short, easy walks.
Many have been put “through the wringer” when starting out, discouraging them from continuing, Dr. Nadolsky said. “They were too sore, and it felt more like punishment.”
An even bigger concern is triggering an asthma attack. Patients should take steps to lower the risk by carrying their rescue inhalers and keeping up on medications, he added.
“A health care professional should be consulted” before the start of a new activity or ramping up a program, or anytime asthma interferes with a workout, Dr. George said.
Those who exercise outside need to be aware of the air quality, especially at a time when smoke and particulates from a wildfires in Canada can trigger asthma symptoms in people thousands of miles away.
The harder one works, the higher one’s “ventilation” – taking more air into the lungs, and potentially more allergens and pollutants.
Temperature and humidity also become risky at the extremes. Cold, dry air can dehydrate and constrict the airways, making it hard to breathe.
How to choose the best type of exercise
Step 1: Be realistic. People with asthma often have less exercise capacity than those who don’t – understandable when shortness of breath is the default setting.
Second, allow for plenty of time to warm up. A solid warm-up routine – particularly one with a mix of lower- and higher-intensity exercises – may help prevent exercise-induced bronchoconstriction causing shortness of breath and wheezing.
For example, warming up on a treadmill or exercise bike could be mixed with a few short bursts of faster running or cycling, with a couple of minutes of recovery at a slower pace in between.
That concept can be expanded into a full-blown workout.
High-intensity interval training (HIIT) is a promising option for people with asthma. A 2021 study showed that three 20-minute interval workouts per week significantly improved asthma control.
“The benefit of HIIT is that ventilation is able to recover intermittently,” said Carley O’Neill, PhD, an exercise scientist at Acadia University in Nova Scotia and the study’s lead author.
That’s a key difference from conventional cardio, where the constant exertion can evaporate water from the lungs faster than your body can replenish it. “Dehydrating of the airways can, in some, trigger exercise-induced asthma,” Dr. O’Neill said.
HIIT, conversely, allows the airways to recover and rehydrate between exercise bouts.
Another recent study found that people with asthma who did HIIT workouts had fewer breathing problems and felt less fatigued, compared with a matched group who did cardio training at a constant pace. (Both types of cardio led to similar improvements in aerobic fitness.)
Individuals can choose other types of intermittent exercise as well. Strength training, for example, requires relatively short periods of exertion, with plenty of rest in between.
The one choice you don’t want to make
While there are lots of good exercise options for someone with asthma, there’s one clearly bad choice, according to Dr. George: “Avoiding exercise.”
Being inactive puts one at higher risk for obesity and all the health problems that go with it. And allowing one’s fitness level to decline makes it much harder to move when one needs or wants to.
Any choice is better than that one.
A version of this article first appeared on WebMD.com.
Psilocybin reduces symptoms, disability in major depression
The randomized, phase 2 trial was conducted at 11 sites across the United States and is the latest to demonstrate the psychedelic drug’s potential as a treatment for depression.
The project was funded by Usona Institute, a nonprofit medical research organization based in Madison, Wisc. The institute issued a press statement, but researchers did not comment further on the findings.
“As the largest and most rigorous study conducted across a wide spectrum of individuals with major depressive disorder, the results show promise for all people struggling with this condition,” lead author Charles Raison, MD, director of clinical and translational research at Usona, said in the statement.
The 34 coauthors on the study are affiliated with public universities, research centers, and private companies. Eight of the investigators are identified as employees of Usona Institute.
Declining further comment, an institute spokesperson told this news organization that, “Usona has chosen the approach of no interviews, and this applies for all coauthors.”
The findings were published online in JAMA.
Largest study to date
Usona’s investigational psilocybin drug has been granted a breakthrough designation by the Food and Drug Administration, a process designed to speed drug development and review.
Previous smaller studies have suggested a rapid antidepressant response with psilocybin, but they have been small, unblinded, and have had short duration of follow-up, they write. This randomized, double-blind, phase 2 clinical trial is the largest study of psilocybin for depression to date, the researchers note.
It included 104 adults aged 21-65 years with MDD who had a current depressive episode of at least 60 days and a Montgomery-Åsberg Depression Rating Scale (MADRS) total score of 28 or more at baseline.
Participants had to be free of psychedelic drugs for at least 5 years, have had no active suicidal ideation or suicidal behavior in the prior 12 months, no personal or first-degree family history of psychosis or mania, and no history of moderate/severe alcohol or drug use disorder.
Before the study, participants had a 7- to 35-day screening period for psychiatric medication tapering, underwent baseline assessments, and received 6-8 hours of preparation with two facilitators who would be with them during dosing.
Dosing occurred within 7 days of baseline assessments. During the 6- to 8-hour session, participants received either a single 25-mg oral dose of psilocybin or 100-mg dose of niacin. One participant randomly assigned to receive psilocybin received the incorrect treatment, resulting in 50 participants receiving psilocybin and 54 receiving niacin.
Participants returned the next day, the next week, and then every 2 weeks for assessments, for a follow-up of 6 weeks.
Psychosocial support
Participants who received psilocybin reported significantly greater improvements in MDD symptoms, compared with those who received niacin. MADRS scores – a scale from 0 to 60 where higher scores indicate more severe depression – showed greater reductions with treatment vs. placebo at 8 days (mean difference, −12.0; 95% confidence interval, −16.6 to −7.4; P < .001), and at day 43 (mean difference, −12.3; 95% CI, −17.5 to −7.2; P < .001).
More participants receiving psilocybin had sustained depressive symptom response (42% vs. 11%; P = .002) and more improvement in the Sheehan Disability Scale score, which measures functional disability, 43 days after treatment (P < .001).
The effects persisted through the end of the study, although the differences between groups were no longer significant by week 6.

“This is another exciting piece of evidence that adds to the current literature regarding the potential efficacy of psilocybin for the treatment of mental health conditions, particularly depression,” said Greg Fonzo, MD, codirector of the Center for Psychedelic Research and Therapy at the University of Texas at Austin, who commented on the findings.
Significantly more people in the psilocybin group reported at least one treatment-related adverse event (AE, 82% vs. 44%), although most were mild to moderate. Headache and nausea were the most common side effects and most resolved within 1 day of dosing.
While those numbers are high, Dr. Fonzo said they’re not out of line with AEs reported in other studies.
“Particularly with the types of adverse events reported here, like headache and nausea, those are things you would typically expect to see in this treatment,” said Dr. Fonzo, who was not part of the research.
“But it is high, and it underscores that this is not a treatment without certain risks, even though it was good that they were primarily mild in severity,” he added.
A ‘stepping stone’ to FDA approval?
The use of tools to measure disability in addition to symptoms of depression severity is a strength of the study, Dr. Fonzo added. The use of an active comparator and the 6-week follow-up also offer something new over previous studies.
Despite the longer follow up, questions remain about the durability of response, something only a longer study could answer, Dr. Fonzo said. The small and homogeneous sample-size are also a concern. Nearly 90% of participants were White, and more than half had an income of $75,000 a year or higher.
“It’s another stepping stone in the process to FDA approval, but the next step in that process would be much larger phase 3 trials that would have much larger samples, a longer follow-up, and hopefully have a more inclusive swath of the population,” Dr. Fonzo said.
But perhaps one of the most significant limitations is the use of niacin as an active comparator, said Caleb Alexander, MD, codirector of the Center for Drug Safety and Effectiveness at Johns Hopkins University in Baltimore.

The use of an agent that doesn’t produce effects similar to those expected from a psychedelic introduced the potential for functional unblinding, Dr. Alexander said. Investigators did not ask participants to guess whether they received psilocybin or niacin, so the quality of the blinding was not assessed in the study.
“We’d like to see the use of [an] active comparator that might have a chance of obscuring to people as to whether they’ve been randomized to the treatment arm or control arm,” said Dr. Alexander, who wasn’t involved in the study. “Why not use a benzodiazepine or another drug that produces a transient euphoria that would better obscure whether or not people were receiving the psilocybin?”
The authors of an accompanying editorial shared these concerns, also noting that the study included “a significant number of patients who did not respond to therapy.”
“It is important to analyze and understand adverse outcomes in psychedelic trials and conduct longitudinal studies to determine how sustained the effects will be and what may initiate a recrudescence of symptoms,” write Rachel Yehuda, PhD, and Amy Lehrner, PhD, both of the Peters VA Medical Center and Icahn School of Medicine at Mount Sinai, New York.

“Future studies will help identify who is most likely to benefit from psychedelics, whether booster or repeated treatment is safe and beneficial, and what the optimal dose and therapeutic frameworks are.”
A long-term follow-up of the current trial was terminated last year because of low enrollment. The spokesperson with Usona Institute did not respond to questions about that study, and the institute’s statement only added that preparations are underway to launch another study that “will provide additional safety and efficacy data to support submission of a new drug application to the FDA.”
Usona published its manufacturing process that it used to synthesize psilocybin in an open-access journal and signed a statement on “open science and open praxis” with psilocybin and similar substances, which appears on their website. That statement was signed by 31 research and service organizations around the world and nearly 150 scientists, scholars, and practitioners.
The study was funded by Usona Institute. Dr. Raison reported receiving personal fees from Usona Institute and grants to Usona Institute from Dr. Bronner’s All-One, Fournier Family Foundation, Good Ventures, Steven and Alexandra Cohen Foundation, Tiny Blue Dot Foundation, Turnbull Family Foundation, and William A. Linton during the conduct of the study; and personal fees from Novartis, Sage/Biogen, Emory Healthcare, and Vail Health outside the submitted work. Dr. Fonzo and Dr. Alexander report no relevant financial relationships. Dr. Yehuda reports receiving nonfinancial support from the Multidisciplinary Association for Psychedelic Studies Public Benefit (MAPS PBC) and grants from COMPASS Pathways. Dr. Lehrner is an investigator on trials sponsored by MAPS PBC and COMPASS Pathways.
A version of this article first appeared on Medscape.com.
The randomized, phase 2 trial was conducted at 11 sites across the United States and is the latest to demonstrate the psychedelic drug’s potential as a treatment for depression.
The project was funded by Usona Institute, a nonprofit medical research organization based in Madison, Wisc. The institute issued a press statement, but researchers did not comment further on the findings.
“As the largest and most rigorous study conducted across a wide spectrum of individuals with major depressive disorder, the results show promise for all people struggling with this condition,” lead author Charles Raison, MD, director of clinical and translational research at Usona, said in the statement.
The 34 coauthors on the study are affiliated with public universities, research centers, and private companies. Eight of the investigators are identified as employees of Usona Institute.
Declining further comment, an institute spokesperson told this news organization that, “Usona has chosen the approach of no interviews, and this applies for all coauthors.”
The findings were published online in JAMA.
Largest study to date
Usona’s investigational psilocybin drug has been granted a breakthrough designation by the Food and Drug Administration, a process designed to speed drug development and review.
Previous smaller studies have suggested a rapid antidepressant response with psilocybin, but they have been small, unblinded, and have had short duration of follow-up, they write. This randomized, double-blind, phase 2 clinical trial is the largest study of psilocybin for depression to date, the researchers note.
It included 104 adults aged 21-65 years with MDD who had a current depressive episode of at least 60 days and a Montgomery-Åsberg Depression Rating Scale (MADRS) total score of 28 or more at baseline.
Participants had to be free of psychedelic drugs for at least 5 years, have had no active suicidal ideation or suicidal behavior in the prior 12 months, no personal or first-degree family history of psychosis or mania, and no history of moderate/severe alcohol or drug use disorder.
Before the study, participants had a 7- to 35-day screening period for psychiatric medication tapering, underwent baseline assessments, and received 6-8 hours of preparation with two facilitators who would be with them during dosing.
Dosing occurred within 7 days of baseline assessments. During the 6- to 8-hour session, participants received either a single 25-mg oral dose of psilocybin or 100-mg dose of niacin. One participant randomly assigned to receive psilocybin received the incorrect treatment, resulting in 50 participants receiving psilocybin and 54 receiving niacin.
Participants returned the next day, the next week, and then every 2 weeks for assessments, for a follow-up of 6 weeks.
Psychosocial support
Participants who received psilocybin reported significantly greater improvements in MDD symptoms, compared with those who received niacin. MADRS scores – a scale from 0 to 60 where higher scores indicate more severe depression – showed greater reductions with treatment vs. placebo at 8 days (mean difference, −12.0; 95% confidence interval, −16.6 to −7.4; P < .001), and at day 43 (mean difference, −12.3; 95% CI, −17.5 to −7.2; P < .001).
More participants receiving psilocybin had sustained depressive symptom response (42% vs. 11%; P = .002) and more improvement in the Sheehan Disability Scale score, which measures functional disability, 43 days after treatment (P < .001).
The effects persisted through the end of the study, although the differences between groups were no longer significant by week 6.
“This is another exciting piece of evidence that adds to the current literature regarding the potential efficacy of psilocybin for the treatment of mental health conditions, particularly depression,” said Greg Fonzo, MD, codirector of the Center for Psychedelic Research and Therapy at the University of Texas at Austin, who commented on the findings.
Significantly more people in the psilocybin group reported at least one treatment-related adverse event (AE, 82% vs. 44%), although most were mild to moderate. Headache and nausea were the most common side effects and most resolved within 1 day of dosing.
While those numbers are high, Dr. Fonzo said they’re not out of line with AEs reported in other studies.
“Particularly with the types of adverse events reported here, like headache and nausea, those are things you would typically expect to see in this treatment,” said Dr. Fonzo, who was not part of the research.
“But it is high, and it underscores that this is not a treatment without certain risks, even though it was good that they were primarily mild in severity,” he added.
A ‘stepping stone’ to FDA approval?
The use of tools to measure disability in addition to symptoms of depression severity is a strength of the study, Dr. Fonzo added. The use of an active comparator and the 6-week follow-up also offer something new over previous studies.
Despite the longer follow up, questions remain about the durability of response, something only a longer study could answer, Dr. Fonzo said. The small and homogeneous sample-size are also a concern. Nearly 90% of participants were White, and more than half had an income of $75,000 a year or higher.
“It’s another stepping stone in the process to FDA approval, but the next step in that process would be much larger phase 3 trials that would have much larger samples, a longer follow-up, and hopefully have a more inclusive swath of the population,” Dr. Fonzo said.
But perhaps one of the most significant limitations is the use of niacin as an active comparator, said Caleb Alexander, MD, codirector of the Center for Drug Safety and Effectiveness at Johns Hopkins University in Baltimore.
The use of an agent that doesn’t produce effects similar to those expected from a psychedelic introduced the potential for functional unblinding, Dr. Alexander said. Investigators did not ask participants to guess whether they received psilocybin or niacin, so the quality of the blinding was not assessed in the study.
“We’d like to see the use of [an] active comparator that might have a chance of obscuring to people as to whether they’ve been randomized to the treatment arm or control arm,” said Dr. Alexander, who wasn’t involved in the study. “Why not use a benzodiazepine or another drug that produces a transient euphoria that would better obscure whether or not people were receiving the psilocybin?”
The authors of an accompanying editorial shared these concerns, also noting that the study included “a significant number of patients who did not respond to therapy.”
“It is important to analyze and understand adverse outcomes in psychedelic trials and conduct longitudinal studies to determine how sustained the effects will be and what may initiate a recrudescence of symptoms,” write Rachel Yehuda, PhD, and Amy Lehrner, PhD, both of the Peters VA Medical Center and Icahn School of Medicine at Mount Sinai, New York.
“Future studies will help identify who is most likely to benefit from psychedelics, whether booster or repeated treatment is safe and beneficial, and what the optimal dose and therapeutic frameworks are.”
A long-term follow-up of the current trial was terminated last year because of low enrollment. The spokesperson with Usona Institute did not respond to questions about that study, and the institute’s statement only added that preparations are underway to launch another study that “will provide additional safety and efficacy data to support submission of a new drug application to the FDA.”
Usona published its manufacturing process that it used to synthesize psilocybin in an open-access journal and signed a statement on “open science and open praxis” with psilocybin and similar substances, which appears on their website. That statement was signed by 31 research and service organizations around the world and nearly 150 scientists, scholars, and practitioners.
The study was funded by Usona Institute. Dr. Raison reported receiving personal fees from Usona Institute and grants to Usona Institute from Dr. Bronner’s All-One, Fournier Family Foundation, Good Ventures, Steven and Alexandra Cohen Foundation, Tiny Blue Dot Foundation, Turnbull Family Foundation, and William A. Linton during the conduct of the study; and personal fees from Novartis, Sage/Biogen, Emory Healthcare, and Vail Health outside the submitted work. Dr. Fonzo and Dr. Alexander report no relevant financial relationships. Dr. Yehuda reports receiving nonfinancial support from the Multidisciplinary Association for Psychedelic Studies Public Benefit (MAPS PBC) and grants from COMPASS Pathways. Dr. Lehrner is an investigator on trials sponsored by MAPS PBC and COMPASS Pathways.
A version of this article first appeared on Medscape.com.
The randomized, phase 2 trial was conducted at 11 sites across the United States and is the latest to demonstrate the psychedelic drug’s potential as a treatment for depression.
The project was funded by Usona Institute, a nonprofit medical research organization based in Madison, Wisc. The institute issued a press statement, but researchers did not comment further on the findings.
“As the largest and most rigorous study conducted across a wide spectrum of individuals with major depressive disorder, the results show promise for all people struggling with this condition,” lead author Charles Raison, MD, director of clinical and translational research at Usona, said in the statement.
The 34 coauthors on the study are affiliated with public universities, research centers, and private companies. Eight of the investigators are identified as employees of Usona Institute.
Declining further comment, an institute spokesperson told this news organization that, “Usona has chosen the approach of no interviews, and this applies for all coauthors.”
The findings were published online in JAMA.
Largest study to date
Usona’s investigational psilocybin drug has been granted a breakthrough designation by the Food and Drug Administration, a process designed to speed drug development and review.
Previous smaller studies have suggested a rapid antidepressant response with psilocybin, but they have been small, unblinded, and have had short duration of follow-up, they write. This randomized, double-blind, phase 2 clinical trial is the largest study of psilocybin for depression to date, the researchers note.
It included 104 adults aged 21-65 years with MDD who had a current depressive episode of at least 60 days and a Montgomery-Åsberg Depression Rating Scale (MADRS) total score of 28 or more at baseline.
Participants had to be free of psychedelic drugs for at least 5 years, have had no active suicidal ideation or suicidal behavior in the prior 12 months, no personal or first-degree family history of psychosis or mania, and no history of moderate/severe alcohol or drug use disorder.
Before the study, participants had a 7- to 35-day screening period for psychiatric medication tapering, underwent baseline assessments, and received 6-8 hours of preparation with two facilitators who would be with them during dosing.
Dosing occurred within 7 days of baseline assessments. During the 6- to 8-hour session, participants received either a single 25-mg oral dose of psilocybin or 100-mg dose of niacin. One participant randomly assigned to receive psilocybin received the incorrect treatment, resulting in 50 participants receiving psilocybin and 54 receiving niacin.
Participants returned the next day, the next week, and then every 2 weeks for assessments, for a follow-up of 6 weeks.
Psychosocial support
Participants who received psilocybin reported significantly greater improvements in MDD symptoms, compared with those who received niacin. MADRS scores – a scale from 0 to 60 where higher scores indicate more severe depression – showed greater reductions with treatment vs. placebo at 8 days (mean difference, −12.0; 95% confidence interval, −16.6 to −7.4; P < .001), and at day 43 (mean difference, −12.3; 95% CI, −17.5 to −7.2; P < .001).
More participants receiving psilocybin had sustained depressive symptom response (42% vs. 11%; P = .002) and more improvement in the Sheehan Disability Scale score, which measures functional disability, 43 days after treatment (P < .001).
The effects persisted through the end of the study, although the differences between groups were no longer significant by week 6.
“This is another exciting piece of evidence that adds to the current literature regarding the potential efficacy of psilocybin for the treatment of mental health conditions, particularly depression,” said Greg Fonzo, MD, codirector of the Center for Psychedelic Research and Therapy at the University of Texas at Austin, who commented on the findings.
Significantly more people in the psilocybin group reported at least one treatment-related adverse event (AE, 82% vs. 44%), although most were mild to moderate. Headache and nausea were the most common side effects and most resolved within 1 day of dosing.
While those numbers are high, Dr. Fonzo said they’re not out of line with AEs reported in other studies.
“Particularly with the types of adverse events reported here, like headache and nausea, those are things you would typically expect to see in this treatment,” said Dr. Fonzo, who was not part of the research.
“But it is high, and it underscores that this is not a treatment without certain risks, even though it was good that they were primarily mild in severity,” he added.
A ‘stepping stone’ to FDA approval?
The use of tools to measure disability in addition to symptoms of depression severity is a strength of the study, Dr. Fonzo added. The use of an active comparator and the 6-week follow-up also offer something new over previous studies.
Despite the longer follow up, questions remain about the durability of response, something only a longer study could answer, Dr. Fonzo said. The small and homogeneous sample-size are also a concern. Nearly 90% of participants were White, and more than half had an income of $75,000 a year or higher.
“It’s another stepping stone in the process to FDA approval, but the next step in that process would be much larger phase 3 trials that would have much larger samples, a longer follow-up, and hopefully have a more inclusive swath of the population,” Dr. Fonzo said.
But perhaps one of the most significant limitations is the use of niacin as an active comparator, said Caleb Alexander, MD, codirector of the Center for Drug Safety and Effectiveness at Johns Hopkins University in Baltimore.
The use of an agent that doesn’t produce effects similar to those expected from a psychedelic introduced the potential for functional unblinding, Dr. Alexander said. Investigators did not ask participants to guess whether they received psilocybin or niacin, so the quality of the blinding was not assessed in the study.
“We’d like to see the use of [an] active comparator that might have a chance of obscuring to people as to whether they’ve been randomized to the treatment arm or control arm,” said Dr. Alexander, who wasn’t involved in the study. “Why not use a benzodiazepine or another drug that produces a transient euphoria that would better obscure whether or not people were receiving the psilocybin?”
The authors of an accompanying editorial shared these concerns, also noting that the study included “a significant number of patients who did not respond to therapy.”
“It is important to analyze and understand adverse outcomes in psychedelic trials and conduct longitudinal studies to determine how sustained the effects will be and what may initiate a recrudescence of symptoms,” write Rachel Yehuda, PhD, and Amy Lehrner, PhD, both of the Peters VA Medical Center and Icahn School of Medicine at Mount Sinai, New York.
“Future studies will help identify who is most likely to benefit from psychedelics, whether booster or repeated treatment is safe and beneficial, and what the optimal dose and therapeutic frameworks are.”
A long-term follow-up of the current trial was terminated last year because of low enrollment. The spokesperson with Usona Institute did not respond to questions about that study, and the institute’s statement only added that preparations are underway to launch another study that “will provide additional safety and efficacy data to support submission of a new drug application to the FDA.”
Usona published its manufacturing process that it used to synthesize psilocybin in an open-access journal and signed a statement on “open science and open praxis” with psilocybin and similar substances, which appears on their website. That statement was signed by 31 research and service organizations around the world and nearly 150 scientists, scholars, and practitioners.
The study was funded by Usona Institute. Dr. Raison reported receiving personal fees from Usona Institute and grants to Usona Institute from Dr. Bronner’s All-One, Fournier Family Foundation, Good Ventures, Steven and Alexandra Cohen Foundation, Tiny Blue Dot Foundation, Turnbull Family Foundation, and William A. Linton during the conduct of the study; and personal fees from Novartis, Sage/Biogen, Emory Healthcare, and Vail Health outside the submitted work. Dr. Fonzo and Dr. Alexander report no relevant financial relationships. Dr. Yehuda reports receiving nonfinancial support from the Multidisciplinary Association for Psychedelic Studies Public Benefit (MAPS PBC) and grants from COMPASS Pathways. Dr. Lehrner is an investigator on trials sponsored by MAPS PBC and COMPASS Pathways.
A version of this article first appeared on Medscape.com.
FROM JAMA
Drug price alerts convince 12% of clinicians to change prescriptions
, according to findings from a study published in JAMA Internal Medicine.
The findings suggest that incorporating the alerts into electronic health record software could be useful for reducing patient expenses, said lead author Anna D. Sinaiko, PhD, assistant professor of health economics and policy at Harvard T. H. Chan School of Public Health, Boston. Showing clinicians the actual prices of medications their patient would pay led to changes in one in six orders when the potential cost savings to the patient was $20 or more, she said.
“This suggests that clinicians are taking medication out-of-pocket prices into account when they are most meaningful for patients.”
Such “real-time benefit tools” provide more meaningful information about patient drug prices in clinical settings than has previously been available, Dr. Sinaiko said. They provide out-of-pocket price estimates specific to individual patients and account for their health plans as opposed to symbols or colors indicating drugs that are more or less expensive, which has been the status quo.
Also, she said, Medicare has promoted the use of these tools by health systems and health plans.
Dr. Sinaiko and colleagues examined EHR data for 103,953 primary care clinic encounters with 72,420 patients in the University of Colorado Health system (81.5% White; 59.5% female; 51.4% aged 65 years or older; 51.9% on Medicare). The patients were treated from July 2019 to July 2022 by 889 clinicians (physicians, nurse practitioners, and physician assistants), who wrote nearly 1.9 million medication orders. Of those orders, 181,887 (9.7%) included a price estimate.
For each prescription, the EHR displayed out-of-pocket costs for patients and offered alternative drugs if those drugs were at least 15 cents cheaper or if they were available at an on-site pharmacy.
Clinicians changed prescriptions 12.3% of the time after they saw price information. The percentage went up to 14% when possible cost savings were $5 or more.
Researchers also found that, while there was the option for clinicians to click a button in the EHR and learn a patient’s specific medication price before ordering a drug, very few clinicians requested price estimates directly, Dr. Sinaiko said. Fewer than 1% (0.9%) did so. The other 99.1% did not, meaning they received information about prices via alerts only after ordering prescriptions.
Researchers also found that clinicians weren’t more likely to change psychiatric medications when the cost savings for the patient was higher. The demographics of patients – such as whether they were poorer or richer – didn’t affect the willingness of clinicians to change prescriptions after receiving price information.
In the big picture, Dr. Sinaiko said, “The fact that medication orders were changed more often when the potential cost savings for patients were larger suggests to me that clinicians were taking out-of-pocket cost into account when it was most salient for the patient.”
It’s not clear, however, why clinicians did not revise more prescriptions to help patients save money.
One theory is that they may ignore the alerts because of “alert fatigue,” she said. “I’d like to know if clinicians discount or ignore the price estimate because they don’t know where it comes from or whether it is accurate. It’s also possible that clinicians discuss the option to change a medication order with their patient, and for reasons other than cost, they decide to keep the original selection. This suggests that clinicians might be using price information to guide – not dictate – their clinical decisions.”
The study had limitations. The researchers did not assess whether the cheaper alternative medications were appropriate in individual cases. Also, they did not take into account other factors, such as patient preferences, that affect how clinicians make prescription decisions.
Clinicians may also not know whether their patients worry about drug costs.
“There isn’t really good data on who wants to talk to their physician about costs, but it is definitely nowhere near 100%,” said health services researcher Alyna Chien, MD, a pediatrician at Boston Children’s Hospital. “For physicians, there is also good reason to keep cost out of the picture until asked so that patients don’t feel like they’re getting suboptimal choices.”
University of Washington, Seattle, graduate student Shiven Bhardwaj, PharmD, who studies health policy, said in an interview that the new study “suggests that physicians are not frequently selecting less costly agents suggested by the real-time benefit tool, and they may not even be considering these alternatives.”
According to Dr. Bhardwaj, previous research has found that physicians “are unable to estimate what their patients’ out-of-pocket costs may be, which is not surprising, given wide variation in health insurance benefit designs.”
Why aren’t more clinicians choosing cheaper alternatives, even when they’re directly told about them? Dr. Bhardwaj suggests that many health systems may be implementing electronic drug cost alerts in the absence of official notification or training.
“Health systems should be making providers aware of the system and its potential to reduce patients’ out-of-pocket costs.”
What’s next for research in this area?
Lead author Dr. Sinaiko said she and her team will interview clinicians and patients in practices at University of Colorado Health to understand how these price estimates are used in clinical encounters and how they affect clinician practice and patient experiences
“We are interested in learning about the cost-savings thresholds that are important to patients,” she said.
The researchers will also examine whether cost information helps to boost access to medications for chronic conditions among Black and Hispanic patients and patients who live in rural areas, she said.
The study was funded by the Harvard School of Public Health Dean’s Fund and the National Institute on Minority Health and Health Disparities. Dr. Sinaiko, Dr. Bhardwaj, and Dr. Chien have no relevant disclosures. Two study authors report having received a grant from the National Institute on Aging and consulting fees from Dispatch Health and Credo Health.
A version of this article first appeared on Medscape.com.
, according to findings from a study published in JAMA Internal Medicine.
The findings suggest that incorporating the alerts into electronic health record software could be useful for reducing patient expenses, said lead author Anna D. Sinaiko, PhD, assistant professor of health economics and policy at Harvard T. H. Chan School of Public Health, Boston. Showing clinicians the actual prices of medications their patient would pay led to changes in one in six orders when the potential cost savings to the patient was $20 or more, she said.
“This suggests that clinicians are taking medication out-of-pocket prices into account when they are most meaningful for patients.”
Such “real-time benefit tools” provide more meaningful information about patient drug prices in clinical settings than has previously been available, Dr. Sinaiko said. They provide out-of-pocket price estimates specific to individual patients and account for their health plans as opposed to symbols or colors indicating drugs that are more or less expensive, which has been the status quo.
Also, she said, Medicare has promoted the use of these tools by health systems and health plans.
Dr. Sinaiko and colleagues examined EHR data for 103,953 primary care clinic encounters with 72,420 patients in the University of Colorado Health system (81.5% White; 59.5% female; 51.4% aged 65 years or older; 51.9% on Medicare). The patients were treated from July 2019 to July 2022 by 889 clinicians (physicians, nurse practitioners, and physician assistants), who wrote nearly 1.9 million medication orders. Of those orders, 181,887 (9.7%) included a price estimate.
For each prescription, the EHR displayed out-of-pocket costs for patients and offered alternative drugs if those drugs were at least 15 cents cheaper or if they were available at an on-site pharmacy.
Clinicians changed prescriptions 12.3% of the time after they saw price information. The percentage went up to 14% when possible cost savings were $5 or more.
Researchers also found that, while there was the option for clinicians to click a button in the EHR and learn a patient’s specific medication price before ordering a drug, very few clinicians requested price estimates directly, Dr. Sinaiko said. Fewer than 1% (0.9%) did so. The other 99.1% did not, meaning they received information about prices via alerts only after ordering prescriptions.
Researchers also found that clinicians weren’t more likely to change psychiatric medications when the cost savings for the patient was higher. The demographics of patients – such as whether they were poorer or richer – didn’t affect the willingness of clinicians to change prescriptions after receiving price information.
In the big picture, Dr. Sinaiko said, “The fact that medication orders were changed more often when the potential cost savings for patients were larger suggests to me that clinicians were taking out-of-pocket cost into account when it was most salient for the patient.”
It’s not clear, however, why clinicians did not revise more prescriptions to help patients save money.
One theory is that they may ignore the alerts because of “alert fatigue,” she said. “I’d like to know if clinicians discount or ignore the price estimate because they don’t know where it comes from or whether it is accurate. It’s also possible that clinicians discuss the option to change a medication order with their patient, and for reasons other than cost, they decide to keep the original selection. This suggests that clinicians might be using price information to guide – not dictate – their clinical decisions.”
The study had limitations. The researchers did not assess whether the cheaper alternative medications were appropriate in individual cases. Also, they did not take into account other factors, such as patient preferences, that affect how clinicians make prescription decisions.
Clinicians may also not know whether their patients worry about drug costs.
“There isn’t really good data on who wants to talk to their physician about costs, but it is definitely nowhere near 100%,” said health services researcher Alyna Chien, MD, a pediatrician at Boston Children’s Hospital. “For physicians, there is also good reason to keep cost out of the picture until asked so that patients don’t feel like they’re getting suboptimal choices.”
University of Washington, Seattle, graduate student Shiven Bhardwaj, PharmD, who studies health policy, said in an interview that the new study “suggests that physicians are not frequently selecting less costly agents suggested by the real-time benefit tool, and they may not even be considering these alternatives.”
According to Dr. Bhardwaj, previous research has found that physicians “are unable to estimate what their patients’ out-of-pocket costs may be, which is not surprising, given wide variation in health insurance benefit designs.”
Why aren’t more clinicians choosing cheaper alternatives, even when they’re directly told about them? Dr. Bhardwaj suggests that many health systems may be implementing electronic drug cost alerts in the absence of official notification or training.
“Health systems should be making providers aware of the system and its potential to reduce patients’ out-of-pocket costs.”
What’s next for research in this area?
Lead author Dr. Sinaiko said she and her team will interview clinicians and patients in practices at University of Colorado Health to understand how these price estimates are used in clinical encounters and how they affect clinician practice and patient experiences
“We are interested in learning about the cost-savings thresholds that are important to patients,” she said.
The researchers will also examine whether cost information helps to boost access to medications for chronic conditions among Black and Hispanic patients and patients who live in rural areas, she said.
The study was funded by the Harvard School of Public Health Dean’s Fund and the National Institute on Minority Health and Health Disparities. Dr. Sinaiko, Dr. Bhardwaj, and Dr. Chien have no relevant disclosures. Two study authors report having received a grant from the National Institute on Aging and consulting fees from Dispatch Health and Credo Health.
A version of this article first appeared on Medscape.com.
, according to findings from a study published in JAMA Internal Medicine.
The findings suggest that incorporating the alerts into electronic health record software could be useful for reducing patient expenses, said lead author Anna D. Sinaiko, PhD, assistant professor of health economics and policy at Harvard T. H. Chan School of Public Health, Boston. Showing clinicians the actual prices of medications their patient would pay led to changes in one in six orders when the potential cost savings to the patient was $20 or more, she said.
“This suggests that clinicians are taking medication out-of-pocket prices into account when they are most meaningful for patients.”
Such “real-time benefit tools” provide more meaningful information about patient drug prices in clinical settings than has previously been available, Dr. Sinaiko said. They provide out-of-pocket price estimates specific to individual patients and account for their health plans as opposed to symbols or colors indicating drugs that are more or less expensive, which has been the status quo.
Also, she said, Medicare has promoted the use of these tools by health systems and health plans.
Dr. Sinaiko and colleagues examined EHR data for 103,953 primary care clinic encounters with 72,420 patients in the University of Colorado Health system (81.5% White; 59.5% female; 51.4% aged 65 years or older; 51.9% on Medicare). The patients were treated from July 2019 to July 2022 by 889 clinicians (physicians, nurse practitioners, and physician assistants), who wrote nearly 1.9 million medication orders. Of those orders, 181,887 (9.7%) included a price estimate.
For each prescription, the EHR displayed out-of-pocket costs for patients and offered alternative drugs if those drugs were at least 15 cents cheaper or if they were available at an on-site pharmacy.
Clinicians changed prescriptions 12.3% of the time after they saw price information. The percentage went up to 14% when possible cost savings were $5 or more.
Researchers also found that, while there was the option for clinicians to click a button in the EHR and learn a patient’s specific medication price before ordering a drug, very few clinicians requested price estimates directly, Dr. Sinaiko said. Fewer than 1% (0.9%) did so. The other 99.1% did not, meaning they received information about prices via alerts only after ordering prescriptions.
Researchers also found that clinicians weren’t more likely to change psychiatric medications when the cost savings for the patient was higher. The demographics of patients – such as whether they were poorer or richer – didn’t affect the willingness of clinicians to change prescriptions after receiving price information.
In the big picture, Dr. Sinaiko said, “The fact that medication orders were changed more often when the potential cost savings for patients were larger suggests to me that clinicians were taking out-of-pocket cost into account when it was most salient for the patient.”
It’s not clear, however, why clinicians did not revise more prescriptions to help patients save money.
One theory is that they may ignore the alerts because of “alert fatigue,” she said. “I’d like to know if clinicians discount or ignore the price estimate because they don’t know where it comes from or whether it is accurate. It’s also possible that clinicians discuss the option to change a medication order with their patient, and for reasons other than cost, they decide to keep the original selection. This suggests that clinicians might be using price information to guide – not dictate – their clinical decisions.”
The study had limitations. The researchers did not assess whether the cheaper alternative medications were appropriate in individual cases. Also, they did not take into account other factors, such as patient preferences, that affect how clinicians make prescription decisions.
Clinicians may also not know whether their patients worry about drug costs.
“There isn’t really good data on who wants to talk to their physician about costs, but it is definitely nowhere near 100%,” said health services researcher Alyna Chien, MD, a pediatrician at Boston Children’s Hospital. “For physicians, there is also good reason to keep cost out of the picture until asked so that patients don’t feel like they’re getting suboptimal choices.”
University of Washington, Seattle, graduate student Shiven Bhardwaj, PharmD, who studies health policy, said in an interview that the new study “suggests that physicians are not frequently selecting less costly agents suggested by the real-time benefit tool, and they may not even be considering these alternatives.”
According to Dr. Bhardwaj, previous research has found that physicians “are unable to estimate what their patients’ out-of-pocket costs may be, which is not surprising, given wide variation in health insurance benefit designs.”
Why aren’t more clinicians choosing cheaper alternatives, even when they’re directly told about them? Dr. Bhardwaj suggests that many health systems may be implementing electronic drug cost alerts in the absence of official notification or training.
“Health systems should be making providers aware of the system and its potential to reduce patients’ out-of-pocket costs.”
What’s next for research in this area?
Lead author Dr. Sinaiko said she and her team will interview clinicians and patients in practices at University of Colorado Health to understand how these price estimates are used in clinical encounters and how they affect clinician practice and patient experiences
“We are interested in learning about the cost-savings thresholds that are important to patients,” she said.
The researchers will also examine whether cost information helps to boost access to medications for chronic conditions among Black and Hispanic patients and patients who live in rural areas, she said.
The study was funded by the Harvard School of Public Health Dean’s Fund and the National Institute on Minority Health and Health Disparities. Dr. Sinaiko, Dr. Bhardwaj, and Dr. Chien have no relevant disclosures. Two study authors report having received a grant from the National Institute on Aging and consulting fees from Dispatch Health and Credo Health.
A version of this article first appeared on Medscape.com.
FROM JAMA INTERNAL MEDICINE
Suicidal behavior tied to increased all-cause mortality in MDD
Investigators studied close to 143,000 patients, encompassing more than 150,000 MDD episodes. Episodes of depression with suicidal behavior (MDD-SB) were compared to MDD episodes without suicidal behavior (MDD-non-SB).
Suicidal behavior was associated with a 2.6-fold higher rate of all-cause mortality, as well as considerably higher health care resource utilization (HCRU) and work loss, compared with matched controls.
Patients with depression who had attempted suicide were younger and more commonly suffering from other psychiatric comorbidities, such as anxiety and addiction. Important risk factors for suicidal acts within a year after the onset of a depressive episode were previous suicide attempts, substance use disorder, anxiety, and sleeping disorders.
“The findings tell us that the care provided for this particular group needs to be developed,” lead author Johan Lundberg, MD, PhD, adjunct professor in psychiatry and senior physician in psychiatry, Karolinska Institute, Stockholm, told this news organization.
“The take-home message is that, when treating patients with increased risk of suicidal behavior, one should offer treatments with this in mind,” said Dr. Lundberg, also the head of the section of mood disorders, Northern Stockholm Psychiatry Clinic. “One possible option is lithium augmentation.”
The study was published online in JAMA Psychiatry.
Identifying subgroups
Depression is associated with increased all-cause mortality, the authors write. Suicidal behavior and previous suicide attempts are known to increase the risk of suicide-associated mortality, with up to 13% of patients with nonfatal suicide attempts dying of suicide at a later time.
Previous studies investigating the association between suicidal behavior and mortality have been limited by nonrandom sampling due to “nonuniversal access to health care and/or exclusion of primary care data,” they state.
For this reason, it’s not established to what extent these estimates actually represent patients with MDD as a whole, or to what extent suicidal behavior is a risk factor for all-cause mortality.
“We think there is a need to identify subgroups within the very large group of individuals with MDD in order to improve treatment outcomes,” Dr. Lundberg said.
To do so, the researchers turned to data from the Stockholm MDD Cohort (SMC), which comprises all patients diagnosed with MDD in any health care setting in the regions of Stockholm from 2010 to 2018. They identified 5 years of recorded MDD episodes (n = 158,169) in patients aged 18 years and older (n = 145,577). A single patient could contribute more than one episode.
At index, MDD-SB patients (n = 2,219; mean age, 41 years) were matched with MDD-non-SB patients (9,574; mean age, 41 years) based on age, sex, year of MDD diagnosis, and socioeconomic status. In total, 2,219 episodes (63.2% in women, 36.8% in men) were compared to 11,109 episodes (63.4% in women, 36.6% in men), respectively.
Enhanced monitoring, optimized treatment
The median time from the start of the episode until the first suicidal behavior was 165 days.
The all-cause mortality rate in the MDD-SB and MDD-non-SB groups was 2.5 per 100 person-years vs. 1 per 100 person-years, respectively (based on 466 deaths), corresponding to a hazard ratio of 2.62 (95% confidence interval, 2.15-3.20).
Patients in the MDD-SB group were younger, were more frequently diagnosed while in specialized care, and had sustained more work loss than their counterparts in the MDD-non-SB group. They also showed a gradual increase in the prevalence of comorbid conditions from about 12 months before index, with this increase being “most pronounced” for anxiety, stress, substance use, and personality disorders.
MDD-SB episodes were associated with higher HCRU and more work loss, compared with MDD-non-SB episodes.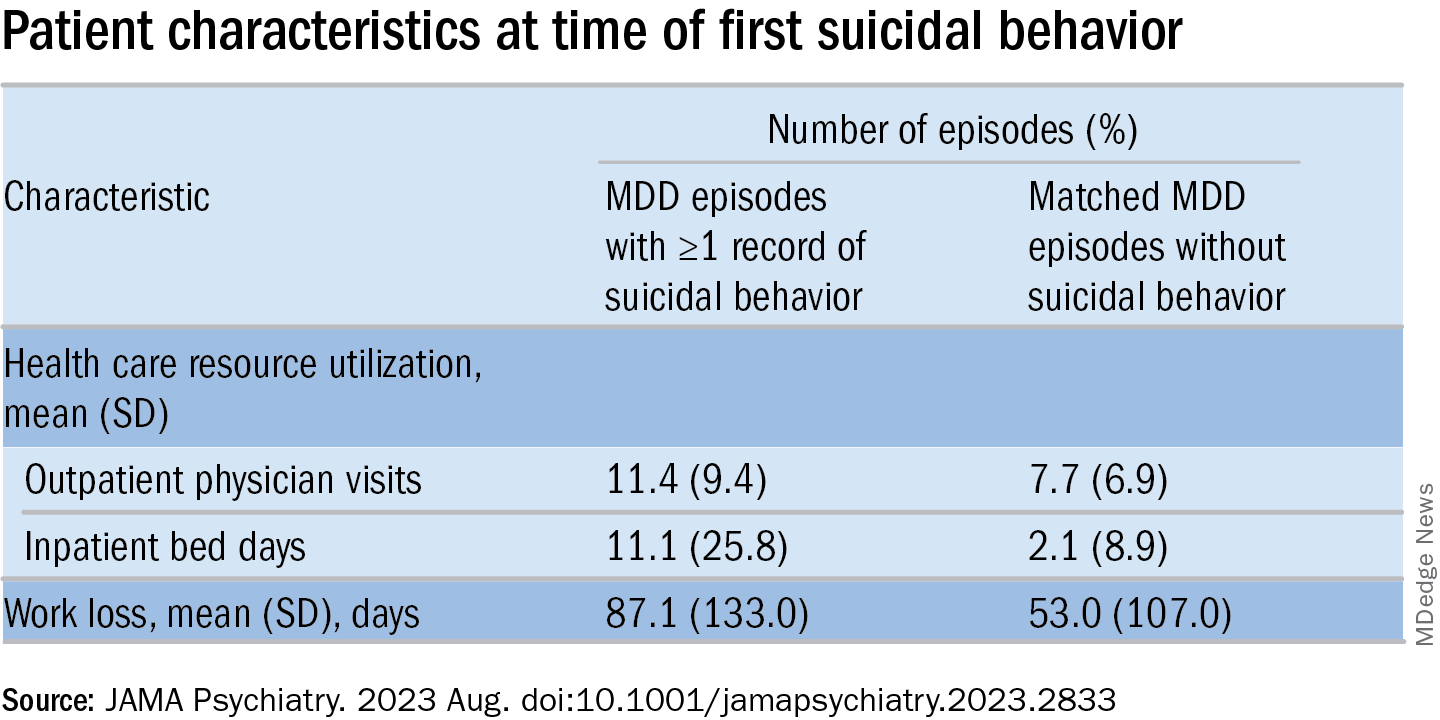
The researchers calculated a risk score for factors associated with suicidal behavior within 1 year after the start of an MDD episode (outcome). The two most important risk factors for suicidal behavior were a history of suicidal behavior together with age, which had a “U-shaped association” with the outcome, they write, with individuals younger than age 20 and older than age 70 having the highest risks.
The final risk score included additional factors that increased the risk of the outcome (in descending order): history of substance use, history of sleep disorders, health care level in which MDD was diagnosed, history of antidepressant use, and history of anxiety disorders.
These results “indicate that patients at risk for suicidal behavior can be identified at an early stage to allow for enhanced monitoring and optimized treatment with the goal of preventing suicidal behavior and reducing mortality,” the authors state.
The specific causes of death weren’t analyzed in this particular paper, Dr. Lundberg noted. A previous study conducted by the same group found the risk of death was doubled in MDD patients, compared with controls.
“We don’t speculate about which causes other than suicide might explain the difference” and account for the increased mortality risk, he said. “This should be studied in future projects.”
Complicated family of destructive behaviors
In a comment, Russell Copelan, MD, a former emergency department psychiatrist at the University of Colorado Affiliated Hospital and currently an expert consultant to the American Association of Suicidology, said a take-home message of the study is that suicide is “a complex and complicated family of destructive behaviors.”
The findings “should not suggest a wait-and-see clinical approach,” warned Dr. Copelan, who wasn’t involved with the study.
Underrecognized or misdiagnosed anxiety, agitation, and insomnia may be “barriers to remission and treatment response,” he noted.
Dr. Copelan, who is also the founder and CEO of eMed Logic, which offers assessment tools for suicide and violence, encouraged clinicians “not to minimize the proportion of patients who experience anxiety, agitation, and insomnia in response to what some may consider a personal misfortune, such as interpersonal, employment, or financial crisis.”
A version of this article first appeared on Medscape.com.
Investigators studied close to 143,000 patients, encompassing more than 150,000 MDD episodes. Episodes of depression with suicidal behavior (MDD-SB) were compared to MDD episodes without suicidal behavior (MDD-non-SB).
Suicidal behavior was associated with a 2.6-fold higher rate of all-cause mortality, as well as considerably higher health care resource utilization (HCRU) and work loss, compared with matched controls.
Patients with depression who had attempted suicide were younger and more commonly suffering from other psychiatric comorbidities, such as anxiety and addiction. Important risk factors for suicidal acts within a year after the onset of a depressive episode were previous suicide attempts, substance use disorder, anxiety, and sleeping disorders.
“The findings tell us that the care provided for this particular group needs to be developed,” lead author Johan Lundberg, MD, PhD, adjunct professor in psychiatry and senior physician in psychiatry, Karolinska Institute, Stockholm, told this news organization.
“The take-home message is that, when treating patients with increased risk of suicidal behavior, one should offer treatments with this in mind,” said Dr. Lundberg, also the head of the section of mood disorders, Northern Stockholm Psychiatry Clinic. “One possible option is lithium augmentation.”
The study was published online in JAMA Psychiatry.
Identifying subgroups
Depression is associated with increased all-cause mortality, the authors write. Suicidal behavior and previous suicide attempts are known to increase the risk of suicide-associated mortality, with up to 13% of patients with nonfatal suicide attempts dying of suicide at a later time.
Previous studies investigating the association between suicidal behavior and mortality have been limited by nonrandom sampling due to “nonuniversal access to health care and/or exclusion of primary care data,” they state.
For this reason, it’s not established to what extent these estimates actually represent patients with MDD as a whole, or to what extent suicidal behavior is a risk factor for all-cause mortality.
“We think there is a need to identify subgroups within the very large group of individuals with MDD in order to improve treatment outcomes,” Dr. Lundberg said.
To do so, the researchers turned to data from the Stockholm MDD Cohort (SMC), which comprises all patients diagnosed with MDD in any health care setting in the regions of Stockholm from 2010 to 2018. They identified 5 years of recorded MDD episodes (n = 158,169) in patients aged 18 years and older (n = 145,577). A single patient could contribute more than one episode.
At index, MDD-SB patients (n = 2,219; mean age, 41 years) were matched with MDD-non-SB patients (9,574; mean age, 41 years) based on age, sex, year of MDD diagnosis, and socioeconomic status. In total, 2,219 episodes (63.2% in women, 36.8% in men) were compared to 11,109 episodes (63.4% in women, 36.6% in men), respectively.
Enhanced monitoring, optimized treatment
The median time from the start of the episode until the first suicidal behavior was 165 days.
The all-cause mortality rate in the MDD-SB and MDD-non-SB groups was 2.5 per 100 person-years vs. 1 per 100 person-years, respectively (based on 466 deaths), corresponding to a hazard ratio of 2.62 (95% confidence interval, 2.15-3.20).
Patients in the MDD-SB group were younger, were more frequently diagnosed while in specialized care, and had sustained more work loss than their counterparts in the MDD-non-SB group. They also showed a gradual increase in the prevalence of comorbid conditions from about 12 months before index, with this increase being “most pronounced” for anxiety, stress, substance use, and personality disorders.
MDD-SB episodes were associated with higher HCRU and more work loss, compared with MDD-non-SB episodes.
The researchers calculated a risk score for factors associated with suicidal behavior within 1 year after the start of an MDD episode (outcome). The two most important risk factors for suicidal behavior were a history of suicidal behavior together with age, which had a “U-shaped association” with the outcome, they write, with individuals younger than age 20 and older than age 70 having the highest risks.
The final risk score included additional factors that increased the risk of the outcome (in descending order): history of substance use, history of sleep disorders, health care level in which MDD was diagnosed, history of antidepressant use, and history of anxiety disorders.
These results “indicate that patients at risk for suicidal behavior can be identified at an early stage to allow for enhanced monitoring and optimized treatment with the goal of preventing suicidal behavior and reducing mortality,” the authors state.
The specific causes of death weren’t analyzed in this particular paper, Dr. Lundberg noted. A previous study conducted by the same group found the risk of death was doubled in MDD patients, compared with controls.
“We don’t speculate about which causes other than suicide might explain the difference” and account for the increased mortality risk, he said. “This should be studied in future projects.”
Complicated family of destructive behaviors
In a comment, Russell Copelan, MD, a former emergency department psychiatrist at the University of Colorado Affiliated Hospital and currently an expert consultant to the American Association of Suicidology, said a take-home message of the study is that suicide is “a complex and complicated family of destructive behaviors.”
The findings “should not suggest a wait-and-see clinical approach,” warned Dr. Copelan, who wasn’t involved with the study.
Underrecognized or misdiagnosed anxiety, agitation, and insomnia may be “barriers to remission and treatment response,” he noted.
Dr. Copelan, who is also the founder and CEO of eMed Logic, which offers assessment tools for suicide and violence, encouraged clinicians “not to minimize the proportion of patients who experience anxiety, agitation, and insomnia in response to what some may consider a personal misfortune, such as interpersonal, employment, or financial crisis.”
A version of this article first appeared on Medscape.com.
Investigators studied close to 143,000 patients, encompassing more than 150,000 MDD episodes. Episodes of depression with suicidal behavior (MDD-SB) were compared to MDD episodes without suicidal behavior (MDD-non-SB).
Suicidal behavior was associated with a 2.6-fold higher rate of all-cause mortality, as well as considerably higher health care resource utilization (HCRU) and work loss, compared with matched controls.
Patients with depression who had attempted suicide were younger and more commonly suffering from other psychiatric comorbidities, such as anxiety and addiction. Important risk factors for suicidal acts within a year after the onset of a depressive episode were previous suicide attempts, substance use disorder, anxiety, and sleeping disorders.
“The findings tell us that the care provided for this particular group needs to be developed,” lead author Johan Lundberg, MD, PhD, adjunct professor in psychiatry and senior physician in psychiatry, Karolinska Institute, Stockholm, told this news organization.
“The take-home message is that, when treating patients with increased risk of suicidal behavior, one should offer treatments with this in mind,” said Dr. Lundberg, also the head of the section of mood disorders, Northern Stockholm Psychiatry Clinic. “One possible option is lithium augmentation.”
The study was published online in JAMA Psychiatry.
Identifying subgroups
Depression is associated with increased all-cause mortality, the authors write. Suicidal behavior and previous suicide attempts are known to increase the risk of suicide-associated mortality, with up to 13% of patients with nonfatal suicide attempts dying of suicide at a later time.
Previous studies investigating the association between suicidal behavior and mortality have been limited by nonrandom sampling due to “nonuniversal access to health care and/or exclusion of primary care data,” they state.
For this reason, it’s not established to what extent these estimates actually represent patients with MDD as a whole, or to what extent suicidal behavior is a risk factor for all-cause mortality.
“We think there is a need to identify subgroups within the very large group of individuals with MDD in order to improve treatment outcomes,” Dr. Lundberg said.
To do so, the researchers turned to data from the Stockholm MDD Cohort (SMC), which comprises all patients diagnosed with MDD in any health care setting in the regions of Stockholm from 2010 to 2018. They identified 5 years of recorded MDD episodes (n = 158,169) in patients aged 18 years and older (n = 145,577). A single patient could contribute more than one episode.
At index, MDD-SB patients (n = 2,219; mean age, 41 years) were matched with MDD-non-SB patients (9,574; mean age, 41 years) based on age, sex, year of MDD diagnosis, and socioeconomic status. In total, 2,219 episodes (63.2% in women, 36.8% in men) were compared to 11,109 episodes (63.4% in women, 36.6% in men), respectively.
Enhanced monitoring, optimized treatment
The median time from the start of the episode until the first suicidal behavior was 165 days.
The all-cause mortality rate in the MDD-SB and MDD-non-SB groups was 2.5 per 100 person-years vs. 1 per 100 person-years, respectively (based on 466 deaths), corresponding to a hazard ratio of 2.62 (95% confidence interval, 2.15-3.20).
Patients in the MDD-SB group were younger, were more frequently diagnosed while in specialized care, and had sustained more work loss than their counterparts in the MDD-non-SB group. They also showed a gradual increase in the prevalence of comorbid conditions from about 12 months before index, with this increase being “most pronounced” for anxiety, stress, substance use, and personality disorders.
MDD-SB episodes were associated with higher HCRU and more work loss, compared with MDD-non-SB episodes.
The researchers calculated a risk score for factors associated with suicidal behavior within 1 year after the start of an MDD episode (outcome). The two most important risk factors for suicidal behavior were a history of suicidal behavior together with age, which had a “U-shaped association” with the outcome, they write, with individuals younger than age 20 and older than age 70 having the highest risks.
The final risk score included additional factors that increased the risk of the outcome (in descending order): history of substance use, history of sleep disorders, health care level in which MDD was diagnosed, history of antidepressant use, and history of anxiety disorders.
These results “indicate that patients at risk for suicidal behavior can be identified at an early stage to allow for enhanced monitoring and optimized treatment with the goal of preventing suicidal behavior and reducing mortality,” the authors state.
The specific causes of death weren’t analyzed in this particular paper, Dr. Lundberg noted. A previous study conducted by the same group found the risk of death was doubled in MDD patients, compared with controls.
“We don’t speculate about which causes other than suicide might explain the difference” and account for the increased mortality risk, he said. “This should be studied in future projects.”
Complicated family of destructive behaviors
In a comment, Russell Copelan, MD, a former emergency department psychiatrist at the University of Colorado Affiliated Hospital and currently an expert consultant to the American Association of Suicidology, said a take-home message of the study is that suicide is “a complex and complicated family of destructive behaviors.”
The findings “should not suggest a wait-and-see clinical approach,” warned Dr. Copelan, who wasn’t involved with the study.
Underrecognized or misdiagnosed anxiety, agitation, and insomnia may be “barriers to remission and treatment response,” he noted.
Dr. Copelan, who is also the founder and CEO of eMed Logic, which offers assessment tools for suicide and violence, encouraged clinicians “not to minimize the proportion of patients who experience anxiety, agitation, and insomnia in response to what some may consider a personal misfortune, such as interpersonal, employment, or financial crisis.”
A version of this article first appeared on Medscape.com.
FROM JAMA PSYCHIATRY
Predicting prostate cancer risk: Are polygenic risk scores ready for prime time?
DNA testing for prostate cancer – of the patients’ inherited DNA and their tumors’ somatic DNA – is increasingly used in the U.S. to determine whether and how to treat low-grade, localized prostate cancers.
Another genetic approach, known as the polygenic risk score (PRS), is emerging as a third genetic approach for sorting out prostate cancer risks.
PRS aims to stratify a person’s disease risk by going beyond rare variants in genes, such as BRCA2, and compiling a weighted score that integrates thousands of common variants whose role in cancer may be unknown but are found more frequently in men with prostate cancer. Traditional germline testing, by contrast, looks for about 30 specific genes directly linked to prostate cancer.
Essentially, “a polygenic risk score estimates your risk by adding together the number of bad cards you were dealt by the impact of each card, such as an ace versus a deuce,” said William Catalona, MD, a urologist at Northwestern University Feinberg School of Medicine, Chicago, known as the father of prostate-specific antigen (PSA) screening.
In combination, these variants can have powerful predictive value.
Having a tool that can mine the depths of a person’s genetic makeup and help doctors devise a nuanced risk assessment for prostate cancer seems like a winning proposition.
Despite its promise, PRS testing is not yet used routinely in practice. The central uncertainty regarding its use lies in whether the risk score can accurately predict who will develop aggressive prostate cancer that needs to be treated and who won’t. The research to date has been mixed, and experts remain polarized.
“PRS absolutely, irrefutably can distinguish between the probability of somebody developing prostate cancer or not. Nobody could look at the data and argue with that,” said Todd Morgan, MD, a genomics researcher from the University of Michigan, Ann Arbor. “What [the data] so far haven’t really been able to do is distinguish whether somebody is likely to have clinically significant prostate cancer versus lower-risk prostate cancer.”
The promise of PRS in prostate cancer?
, according to Burcu Darst, PhD, a genetic epidemiologist at Fred Hutchinson Cancer Center, Seattle.
Research in the area has intensified in recent years as genome-wide association studies (GWAS) have become more affordable and the genetic information from these studies has been increasingly aggregated in biobanks.
“Because the sample sizes now are so much bigger than they used to be for GWAS studies, we’re able to develop much better polygenic risk scores than we were before,” said Dr. Darst.
Dr. Darst is lead author on the largest, most diverse prostate GWAS analysis, which led to the development of a PRS that is highly predictive of prostate cancer risk across diverse populations.
In the 2021 meta-analysis, which included 107,247 case patients and 127,006 control patients, Dr. Darst and colleagues identified 86 new genetic risk variants independently associated with prostate cancer risk, bringing the total to 269 known risk variants.
Compared with men at average genetic risk for prostate cancer – those in the 40%-60% genetic risk score category – men in the top 10% of the risk score (90%-100%) had between a 3.74-fold to fivefold higher risk for prostate cancer. However, the team did not find evidence that the genetic risk score could differentiate a person’s risk for aggressive versus nonaggressive disease.
As Dr. Darst’s team continues to improve the PRS, Dr. Darst says it will get better at predicting aggressive disease. One recent analysis from Dr. Darst and colleagues found that “although the PRS generally did not differentiate aggressive versus nonaggressive prostate cancer,” about 40% of men who will develop aggressive disease have a PRS in the top 20%, whereas only about 7% of men who will develop aggressive tumors have a PRS in the bottom 20%. Another recent study from Dr. Darst and colleagues found that PRS can distinguish between aggressive and nonaggressive disease in men of African ancestry.
These findings highlight “the potential clinical utility of the polygenic risk score,” Dr. Darst said.
Although the growing body of research makes Dr. Catalona, Dr. Darst, and others optimistic about PRS, the landscape is also littered with critics and studies showcasing its limitations.
An analysis, published in JAMA Internal Medicine, found that, compared with a contemporary clinical risk predictor, PRS did not improve prediction of aggressive prostate cancers. Another recent study, which used a 6.6 million–variant PRS to predict the risk of incident prostate cancer among 5,701 healthy men of European descent older than age 69, found that men in the top 20% of the PRS distribution “had an almost three times higher risk of prostate cancer,” compared with men in the lowest quintile; however, a higher PRS was not associated with a higher Gleason grade group, indicative of more aggressive disease.
“While a PRS for prostate cancer is strongly associated with incident risk” in the cohort, “the clinical utility of the PRS as a biomarker is currently limited by its inability to select for clinically significant disease,” the authors concluded.
Utility in practice?
Although PRS has been billed as a predictive test, Dr. Catalona believes PRS could have a range of uses both before and after diagnosis.
PRS may, for instance, guide treatment choices for men diagnosed with prostate cancer, Dr. Catalona noted. For men with a PRS that signals a higher risk for aggressive disease, a positive prostate biopsy result could help them decide whether to seek active treatment with surgery or radiation or go on active surveillance.
PRS could also help inform cancer screening. If a PRS test found a patient’s risk for prostate cancer was high, that person could decide to seek PSA screening before age 50 – the recommended age for average-risk men.
However, Aroon Hingorani, MD, a professor of genetic epidemiology at the University College London, expressed concern over using PRS to inform cancer screenings.
Part of the issue, Dr. Hingorani and colleagues explained in a recent article in the BMJ, is that “risk is notoriously difficult to communicate.”
PRS estimates a person’s relative risk for a disease but does not factor in the underlying population risk. Risk prediction should include both, Dr. Hingorani said.
People with high-risk scores may, for instance, discuss earlier screening with their clinician, even if their absolute risk for the disease – which accounts for both relative risk and underlying population disease risk – is still small, Dr. Hingorani and colleagues said. “Conversely, people who do not have ‘high risk’ polygenic scores might be less likely to seek medical attention for concerning symptoms, or their clinicians might be less inclined to investigate.”
Given this, Dr. Hingorani and colleagues believe polygenic scores “will always be limited in their ability to predict disease” and “will always remain one of many risk factors,” such as environmental influences.
Another caveat is that PRS generally is based on data collected from European populations, said Eric Klein, MD, chairman emeritus of urology at the Cleveland Clinic and now a scientist at the biotechnology company Grail, which developed the Galleri blood test that screens for 50 types of cancer. While a valid concern, “that’s easy to fix ultimately,” he said, as the diversity of inputs from various ethnicities increases over time.
Although several companies offer PRS products, moving PRS into the clinic would require an infrastructure for testing which does not yet exist in the U.S., said Dr. Catalona.
Giordano Botta, PhD, CEO of New York–based PRS software start-up Alleica, which bills itself as the Polygenic Risk Score Company, said “test demand is growing rapidly.” His company offers PRS scores that integrate up to 700,000 markers for prostate cancer depending on ancestry and charges patients $250 out of pocket for testing.
Dr. Botta noted that thousands of American patients have undergone PRS testing through his company. Several health systems, including Penn Medicine, Brigham and Women’s Hospital, and the University of Alabama at Birmingham, have been using the test to help “see beyond what traditional risk factors allow,” he said.
However, this and other PRS tests are not yet widely used in the primary care setting.
A major barrier to wider adoption is that experts remain divided on its clinical utility. “People either say it’s ready, and it should be implemented, or they say it’s never going to work,” said Sowmiya Moorthie, PhD, a senior policy analyst with the PHG Foundation, a Cambridge University–associated think tank.
Dr. Klein sits in the optimistic camp. He envisions a day soon when patients will undergo whole-genome testing to collect data on risk scores and incorporate the full genome into the electronic record. At a certain age, primary care physicians would then query the data to determine the patient’s germline risk for a variety of diseases.
“At age 45, if I were a primary care physician seeing a male, I would query the PRS for prostate cancer, and if the risks were low, I would say, ‘You don’t need your first PSA probably until you’re 50,’ ” Dr. Klein said. “If your risk is high, I’d say, ‘Let’s do a baseline PSA now.’ ”
We would then have the data to watch these patients a little more closely, he said.
Dr. Moorthie, however, remains more reserved about the future of PRS. “I take the middle ground and say, I think there is some value because it’s an additional data point,” Dr. Moorthie said. “And I can see it having value in certain scenarios, but we still don’t have a clear picture of what these are and how best to use and communicate this information.”
A version of this article first appeared on Medscape.com.
DNA testing for prostate cancer – of the patients’ inherited DNA and their tumors’ somatic DNA – is increasingly used in the U.S. to determine whether and how to treat low-grade, localized prostate cancers.
Another genetic approach, known as the polygenic risk score (PRS), is emerging as a third genetic approach for sorting out prostate cancer risks.
PRS aims to stratify a person’s disease risk by going beyond rare variants in genes, such as BRCA2, and compiling a weighted score that integrates thousands of common variants whose role in cancer may be unknown but are found more frequently in men with prostate cancer. Traditional germline testing, by contrast, looks for about 30 specific genes directly linked to prostate cancer.
Essentially, “a polygenic risk score estimates your risk by adding together the number of bad cards you were dealt by the impact of each card, such as an ace versus a deuce,” said William Catalona, MD, a urologist at Northwestern University Feinberg School of Medicine, Chicago, known as the father of prostate-specific antigen (PSA) screening.
In combination, these variants can have powerful predictive value.
Having a tool that can mine the depths of a person’s genetic makeup and help doctors devise a nuanced risk assessment for prostate cancer seems like a winning proposition.
Despite its promise, PRS testing is not yet used routinely in practice. The central uncertainty regarding its use lies in whether the risk score can accurately predict who will develop aggressive prostate cancer that needs to be treated and who won’t. The research to date has been mixed, and experts remain polarized.
“PRS absolutely, irrefutably can distinguish between the probability of somebody developing prostate cancer or not. Nobody could look at the data and argue with that,” said Todd Morgan, MD, a genomics researcher from the University of Michigan, Ann Arbor. “What [the data] so far haven’t really been able to do is distinguish whether somebody is likely to have clinically significant prostate cancer versus lower-risk prostate cancer.”
The promise of PRS in prostate cancer?
, according to Burcu Darst, PhD, a genetic epidemiologist at Fred Hutchinson Cancer Center, Seattle.
Research in the area has intensified in recent years as genome-wide association studies (GWAS) have become more affordable and the genetic information from these studies has been increasingly aggregated in biobanks.
“Because the sample sizes now are so much bigger than they used to be for GWAS studies, we’re able to develop much better polygenic risk scores than we were before,” said Dr. Darst.
Dr. Darst is lead author on the largest, most diverse prostate GWAS analysis, which led to the development of a PRS that is highly predictive of prostate cancer risk across diverse populations.
In the 2021 meta-analysis, which included 107,247 case patients and 127,006 control patients, Dr. Darst and colleagues identified 86 new genetic risk variants independently associated with prostate cancer risk, bringing the total to 269 known risk variants.
Compared with men at average genetic risk for prostate cancer – those in the 40%-60% genetic risk score category – men in the top 10% of the risk score (90%-100%) had between a 3.74-fold to fivefold higher risk for prostate cancer. However, the team did not find evidence that the genetic risk score could differentiate a person’s risk for aggressive versus nonaggressive disease.
As Dr. Darst’s team continues to improve the PRS, Dr. Darst says it will get better at predicting aggressive disease. One recent analysis from Dr. Darst and colleagues found that “although the PRS generally did not differentiate aggressive versus nonaggressive prostate cancer,” about 40% of men who will develop aggressive disease have a PRS in the top 20%, whereas only about 7% of men who will develop aggressive tumors have a PRS in the bottom 20%. Another recent study from Dr. Darst and colleagues found that PRS can distinguish between aggressive and nonaggressive disease in men of African ancestry.
These findings highlight “the potential clinical utility of the polygenic risk score,” Dr. Darst said.
Although the growing body of research makes Dr. Catalona, Dr. Darst, and others optimistic about PRS, the landscape is also littered with critics and studies showcasing its limitations.
An analysis, published in JAMA Internal Medicine, found that, compared with a contemporary clinical risk predictor, PRS did not improve prediction of aggressive prostate cancers. Another recent study, which used a 6.6 million–variant PRS to predict the risk of incident prostate cancer among 5,701 healthy men of European descent older than age 69, found that men in the top 20% of the PRS distribution “had an almost three times higher risk of prostate cancer,” compared with men in the lowest quintile; however, a higher PRS was not associated with a higher Gleason grade group, indicative of more aggressive disease.
“While a PRS for prostate cancer is strongly associated with incident risk” in the cohort, “the clinical utility of the PRS as a biomarker is currently limited by its inability to select for clinically significant disease,” the authors concluded.
Utility in practice?
Although PRS has been billed as a predictive test, Dr. Catalona believes PRS could have a range of uses both before and after diagnosis.
PRS may, for instance, guide treatment choices for men diagnosed with prostate cancer, Dr. Catalona noted. For men with a PRS that signals a higher risk for aggressive disease, a positive prostate biopsy result could help them decide whether to seek active treatment with surgery or radiation or go on active surveillance.
PRS could also help inform cancer screening. If a PRS test found a patient’s risk for prostate cancer was high, that person could decide to seek PSA screening before age 50 – the recommended age for average-risk men.
However, Aroon Hingorani, MD, a professor of genetic epidemiology at the University College London, expressed concern over using PRS to inform cancer screenings.
Part of the issue, Dr. Hingorani and colleagues explained in a recent article in the BMJ, is that “risk is notoriously difficult to communicate.”
PRS estimates a person’s relative risk for a disease but does not factor in the underlying population risk. Risk prediction should include both, Dr. Hingorani said.
People with high-risk scores may, for instance, discuss earlier screening with their clinician, even if their absolute risk for the disease – which accounts for both relative risk and underlying population disease risk – is still small, Dr. Hingorani and colleagues said. “Conversely, people who do not have ‘high risk’ polygenic scores might be less likely to seek medical attention for concerning symptoms, or their clinicians might be less inclined to investigate.”
Given this, Dr. Hingorani and colleagues believe polygenic scores “will always be limited in their ability to predict disease” and “will always remain one of many risk factors,” such as environmental influences.
Another caveat is that PRS generally is based on data collected from European populations, said Eric Klein, MD, chairman emeritus of urology at the Cleveland Clinic and now a scientist at the biotechnology company Grail, which developed the Galleri blood test that screens for 50 types of cancer. While a valid concern, “that’s easy to fix ultimately,” he said, as the diversity of inputs from various ethnicities increases over time.
Although several companies offer PRS products, moving PRS into the clinic would require an infrastructure for testing which does not yet exist in the U.S., said Dr. Catalona.
Giordano Botta, PhD, CEO of New York–based PRS software start-up Alleica, which bills itself as the Polygenic Risk Score Company, said “test demand is growing rapidly.” His company offers PRS scores that integrate up to 700,000 markers for prostate cancer depending on ancestry and charges patients $250 out of pocket for testing.
Dr. Botta noted that thousands of American patients have undergone PRS testing through his company. Several health systems, including Penn Medicine, Brigham and Women’s Hospital, and the University of Alabama at Birmingham, have been using the test to help “see beyond what traditional risk factors allow,” he said.
However, this and other PRS tests are not yet widely used in the primary care setting.
A major barrier to wider adoption is that experts remain divided on its clinical utility. “People either say it’s ready, and it should be implemented, or they say it’s never going to work,” said Sowmiya Moorthie, PhD, a senior policy analyst with the PHG Foundation, a Cambridge University–associated think tank.
Dr. Klein sits in the optimistic camp. He envisions a day soon when patients will undergo whole-genome testing to collect data on risk scores and incorporate the full genome into the electronic record. At a certain age, primary care physicians would then query the data to determine the patient’s germline risk for a variety of diseases.
“At age 45, if I were a primary care physician seeing a male, I would query the PRS for prostate cancer, and if the risks were low, I would say, ‘You don’t need your first PSA probably until you’re 50,’ ” Dr. Klein said. “If your risk is high, I’d say, ‘Let’s do a baseline PSA now.’ ”
We would then have the data to watch these patients a little more closely, he said.
Dr. Moorthie, however, remains more reserved about the future of PRS. “I take the middle ground and say, I think there is some value because it’s an additional data point,” Dr. Moorthie said. “And I can see it having value in certain scenarios, but we still don’t have a clear picture of what these are and how best to use and communicate this information.”
A version of this article first appeared on Medscape.com.
DNA testing for prostate cancer – of the patients’ inherited DNA and their tumors’ somatic DNA – is increasingly used in the U.S. to determine whether and how to treat low-grade, localized prostate cancers.
Another genetic approach, known as the polygenic risk score (PRS), is emerging as a third genetic approach for sorting out prostate cancer risks.
PRS aims to stratify a person’s disease risk by going beyond rare variants in genes, such as BRCA2, and compiling a weighted score that integrates thousands of common variants whose role in cancer may be unknown but are found more frequently in men with prostate cancer. Traditional germline testing, by contrast, looks for about 30 specific genes directly linked to prostate cancer.
Essentially, “a polygenic risk score estimates your risk by adding together the number of bad cards you were dealt by the impact of each card, such as an ace versus a deuce,” said William Catalona, MD, a urologist at Northwestern University Feinberg School of Medicine, Chicago, known as the father of prostate-specific antigen (PSA) screening.
In combination, these variants can have powerful predictive value.
Having a tool that can mine the depths of a person’s genetic makeup and help doctors devise a nuanced risk assessment for prostate cancer seems like a winning proposition.
Despite its promise, PRS testing is not yet used routinely in practice. The central uncertainty regarding its use lies in whether the risk score can accurately predict who will develop aggressive prostate cancer that needs to be treated and who won’t. The research to date has been mixed, and experts remain polarized.
“PRS absolutely, irrefutably can distinguish between the probability of somebody developing prostate cancer or not. Nobody could look at the data and argue with that,” said Todd Morgan, MD, a genomics researcher from the University of Michigan, Ann Arbor. “What [the data] so far haven’t really been able to do is distinguish whether somebody is likely to have clinically significant prostate cancer versus lower-risk prostate cancer.”
The promise of PRS in prostate cancer?
, according to Burcu Darst, PhD, a genetic epidemiologist at Fred Hutchinson Cancer Center, Seattle.
Research in the area has intensified in recent years as genome-wide association studies (GWAS) have become more affordable and the genetic information from these studies has been increasingly aggregated in biobanks.
“Because the sample sizes now are so much bigger than they used to be for GWAS studies, we’re able to develop much better polygenic risk scores than we were before,” said Dr. Darst.
Dr. Darst is lead author on the largest, most diverse prostate GWAS analysis, which led to the development of a PRS that is highly predictive of prostate cancer risk across diverse populations.
In the 2021 meta-analysis, which included 107,247 case patients and 127,006 control patients, Dr. Darst and colleagues identified 86 new genetic risk variants independently associated with prostate cancer risk, bringing the total to 269 known risk variants.
Compared with men at average genetic risk for prostate cancer – those in the 40%-60% genetic risk score category – men in the top 10% of the risk score (90%-100%) had between a 3.74-fold to fivefold higher risk for prostate cancer. However, the team did not find evidence that the genetic risk score could differentiate a person’s risk for aggressive versus nonaggressive disease.
As Dr. Darst’s team continues to improve the PRS, Dr. Darst says it will get better at predicting aggressive disease. One recent analysis from Dr. Darst and colleagues found that “although the PRS generally did not differentiate aggressive versus nonaggressive prostate cancer,” about 40% of men who will develop aggressive disease have a PRS in the top 20%, whereas only about 7% of men who will develop aggressive tumors have a PRS in the bottom 20%. Another recent study from Dr. Darst and colleagues found that PRS can distinguish between aggressive and nonaggressive disease in men of African ancestry.
These findings highlight “the potential clinical utility of the polygenic risk score,” Dr. Darst said.
Although the growing body of research makes Dr. Catalona, Dr. Darst, and others optimistic about PRS, the landscape is also littered with critics and studies showcasing its limitations.
An analysis, published in JAMA Internal Medicine, found that, compared with a contemporary clinical risk predictor, PRS did not improve prediction of aggressive prostate cancers. Another recent study, which used a 6.6 million–variant PRS to predict the risk of incident prostate cancer among 5,701 healthy men of European descent older than age 69, found that men in the top 20% of the PRS distribution “had an almost three times higher risk of prostate cancer,” compared with men in the lowest quintile; however, a higher PRS was not associated with a higher Gleason grade group, indicative of more aggressive disease.
“While a PRS for prostate cancer is strongly associated with incident risk” in the cohort, “the clinical utility of the PRS as a biomarker is currently limited by its inability to select for clinically significant disease,” the authors concluded.
Utility in practice?
Although PRS has been billed as a predictive test, Dr. Catalona believes PRS could have a range of uses both before and after diagnosis.
PRS may, for instance, guide treatment choices for men diagnosed with prostate cancer, Dr. Catalona noted. For men with a PRS that signals a higher risk for aggressive disease, a positive prostate biopsy result could help them decide whether to seek active treatment with surgery or radiation or go on active surveillance.
PRS could also help inform cancer screening. If a PRS test found a patient’s risk for prostate cancer was high, that person could decide to seek PSA screening before age 50 – the recommended age for average-risk men.
However, Aroon Hingorani, MD, a professor of genetic epidemiology at the University College London, expressed concern over using PRS to inform cancer screenings.
Part of the issue, Dr. Hingorani and colleagues explained in a recent article in the BMJ, is that “risk is notoriously difficult to communicate.”
PRS estimates a person’s relative risk for a disease but does not factor in the underlying population risk. Risk prediction should include both, Dr. Hingorani said.
People with high-risk scores may, for instance, discuss earlier screening with their clinician, even if their absolute risk for the disease – which accounts for both relative risk and underlying population disease risk – is still small, Dr. Hingorani and colleagues said. “Conversely, people who do not have ‘high risk’ polygenic scores might be less likely to seek medical attention for concerning symptoms, or their clinicians might be less inclined to investigate.”
Given this, Dr. Hingorani and colleagues believe polygenic scores “will always be limited in their ability to predict disease” and “will always remain one of many risk factors,” such as environmental influences.
Another caveat is that PRS generally is based on data collected from European populations, said Eric Klein, MD, chairman emeritus of urology at the Cleveland Clinic and now a scientist at the biotechnology company Grail, which developed the Galleri blood test that screens for 50 types of cancer. While a valid concern, “that’s easy to fix ultimately,” he said, as the diversity of inputs from various ethnicities increases over time.
Although several companies offer PRS products, moving PRS into the clinic would require an infrastructure for testing which does not yet exist in the U.S., said Dr. Catalona.
Giordano Botta, PhD, CEO of New York–based PRS software start-up Alleica, which bills itself as the Polygenic Risk Score Company, said “test demand is growing rapidly.” His company offers PRS scores that integrate up to 700,000 markers for prostate cancer depending on ancestry and charges patients $250 out of pocket for testing.
Dr. Botta noted that thousands of American patients have undergone PRS testing through his company. Several health systems, including Penn Medicine, Brigham and Women’s Hospital, and the University of Alabama at Birmingham, have been using the test to help “see beyond what traditional risk factors allow,” he said.
However, this and other PRS tests are not yet widely used in the primary care setting.
A major barrier to wider adoption is that experts remain divided on its clinical utility. “People either say it’s ready, and it should be implemented, or they say it’s never going to work,” said Sowmiya Moorthie, PhD, a senior policy analyst with the PHG Foundation, a Cambridge University–associated think tank.
Dr. Klein sits in the optimistic camp. He envisions a day soon when patients will undergo whole-genome testing to collect data on risk scores and incorporate the full genome into the electronic record. At a certain age, primary care physicians would then query the data to determine the patient’s germline risk for a variety of diseases.
“At age 45, if I were a primary care physician seeing a male, I would query the PRS for prostate cancer, and if the risks were low, I would say, ‘You don’t need your first PSA probably until you’re 50,’ ” Dr. Klein said. “If your risk is high, I’d say, ‘Let’s do a baseline PSA now.’ ”
We would then have the data to watch these patients a little more closely, he said.
Dr. Moorthie, however, remains more reserved about the future of PRS. “I take the middle ground and say, I think there is some value because it’s an additional data point,” Dr. Moorthie said. “And I can see it having value in certain scenarios, but we still don’t have a clear picture of what these are and how best to use and communicate this information.”
A version of this article first appeared on Medscape.com.
Screen bipolar patients for eating disorders
Previous research of bipolar disorder (BD) shows a high rate of comorbidities with other psychiatric disorders, including eating disorders (EDs), Valentin Flaudias, PhD, of Nantes (France) University and colleagues wrote.

“There is growing evidence that, compared with individuals with BD alone, individuals with both BD and EDs have a more severe clinical profile, including increased mood instability, alcohol use disorders, anxiety disorders, more depressive episodes, more rapid cycling, increased suicidality, and poorer response to medication,” but studies of BD type-specific ED prevalence have been inconsistent, they said.
In a study published in the Journal of Affective Disorders, the researchers reviewed data from 2,929 outpatients who underwent assessments for BD at 1 of 12 psychiatric centers in France. Of these, 1,505 met criteria for type I and 1,424 met criteria for type II. The post hoc analysis included identification of lifetime prevalence of ED. Diagnosis was based on the DSM-4-TR and the researchers considered three ED types: anorexia nervosa (AN), bulimia nervosa (BN), and binge-eating disorder (BED). Subtypes of BD were type I and type II. DSM not otherwise specified diagnoses for BD and EDs were excluded. The mean age of the participants was 40.5 years, and 61% were women.
A total of 479 individuals met criteria for comorbid EDs (16.4%). ED prevalence was significantly higher in BD type II patients than in BD type I patients (20.6 % vs. 12.4 %, P < .001). The overall breakdown according to ED subtype was 30% for AN, 13% for BN, and 56% for BED. The researchers found no significant differences in patients with AN, BN, or BED according to BD subtype.
In a multivariate analysis, BD patients with ED were more likely than those without ED to be women (77% vs. 55%), especially those with AN (95% vs. 82%).
BD patients with ED also tended to be younger than those without ED (37 years vs. 41 years) and reported more frequent suicide attempts (50% vs. 35%). Younger age and more frequent suicide attempts were further significant among BD patients with AN, compared with those with BED, but BD patients with BED reported higher levels of childhood trauma.
BD patients with ED also reported higher levels of depressive symptoms than those without ED, although history of psychosis was less frequent among BD patients with AN and BED compared with BD patients without EDs.
Overall, “after controlling for other variables, the independent factors differentiating BD patients with versus without ED were primarily younger age, female gender, abnormal BMI, increased affective lability and higher comorbidity with anxiety disorders,” the researchers wrote. In addition, presence of EDs except for AN was associated with decreased current functioning.
The findings were limited by several factors including the cross-sectional design, lack of a control group of non-BD individuals, and the consideration of ED over a lifetime, and small number of BN cases, the researchers noted.
However, the results suggest a high prevalence of ED in BD patients and highlight the need to screen BD patients for ED and provide integrated care. More research is needed to explore the evolution of the two conditions as comorbidities and to examine subtypes and of both conditions and their interactions, they concluded.
The study was supported by the FondaMental Foundation, French National Institute for Health and Medical Research, Public Hospitals of Paris, and the French National Research Agency’s Investment for the Future program. The researchers had no financial conflicts to disclose.
Previous research of bipolar disorder (BD) shows a high rate of comorbidities with other psychiatric disorders, including eating disorders (EDs), Valentin Flaudias, PhD, of Nantes (France) University and colleagues wrote.
“There is growing evidence that, compared with individuals with BD alone, individuals with both BD and EDs have a more severe clinical profile, including increased mood instability, alcohol use disorders, anxiety disorders, more depressive episodes, more rapid cycling, increased suicidality, and poorer response to medication,” but studies of BD type-specific ED prevalence have been inconsistent, they said.
In a study published in the Journal of Affective Disorders, the researchers reviewed data from 2,929 outpatients who underwent assessments for BD at 1 of 12 psychiatric centers in France. Of these, 1,505 met criteria for type I and 1,424 met criteria for type II. The post hoc analysis included identification of lifetime prevalence of ED. Diagnosis was based on the DSM-4-TR and the researchers considered three ED types: anorexia nervosa (AN), bulimia nervosa (BN), and binge-eating disorder (BED). Subtypes of BD were type I and type II. DSM not otherwise specified diagnoses for BD and EDs were excluded. The mean age of the participants was 40.5 years, and 61% were women.
A total of 479 individuals met criteria for comorbid EDs (16.4%). ED prevalence was significantly higher in BD type II patients than in BD type I patients (20.6 % vs. 12.4 %, P < .001). The overall breakdown according to ED subtype was 30% for AN, 13% for BN, and 56% for BED. The researchers found no significant differences in patients with AN, BN, or BED according to BD subtype.
In a multivariate analysis, BD patients with ED were more likely than those without ED to be women (77% vs. 55%), especially those with AN (95% vs. 82%).
BD patients with ED also tended to be younger than those without ED (37 years vs. 41 years) and reported more frequent suicide attempts (50% vs. 35%). Younger age and more frequent suicide attempts were further significant among BD patients with AN, compared with those with BED, but BD patients with BED reported higher levels of childhood trauma.
BD patients with ED also reported higher levels of depressive symptoms than those without ED, although history of psychosis was less frequent among BD patients with AN and BED compared with BD patients without EDs.
Overall, “after controlling for other variables, the independent factors differentiating BD patients with versus without ED were primarily younger age, female gender, abnormal BMI, increased affective lability and higher comorbidity with anxiety disorders,” the researchers wrote. In addition, presence of EDs except for AN was associated with decreased current functioning.
The findings were limited by several factors including the cross-sectional design, lack of a control group of non-BD individuals, and the consideration of ED over a lifetime, and small number of BN cases, the researchers noted.
However, the results suggest a high prevalence of ED in BD patients and highlight the need to screen BD patients for ED and provide integrated care. More research is needed to explore the evolution of the two conditions as comorbidities and to examine subtypes and of both conditions and their interactions, they concluded.
The study was supported by the FondaMental Foundation, French National Institute for Health and Medical Research, Public Hospitals of Paris, and the French National Research Agency’s Investment for the Future program. The researchers had no financial conflicts to disclose.
Previous research of bipolar disorder (BD) shows a high rate of comorbidities with other psychiatric disorders, including eating disorders (EDs), Valentin Flaudias, PhD, of Nantes (France) University and colleagues wrote.
“There is growing evidence that, compared with individuals with BD alone, individuals with both BD and EDs have a more severe clinical profile, including increased mood instability, alcohol use disorders, anxiety disorders, more depressive episodes, more rapid cycling, increased suicidality, and poorer response to medication,” but studies of BD type-specific ED prevalence have been inconsistent, they said.
In a study published in the Journal of Affective Disorders, the researchers reviewed data from 2,929 outpatients who underwent assessments for BD at 1 of 12 psychiatric centers in France. Of these, 1,505 met criteria for type I and 1,424 met criteria for type II. The post hoc analysis included identification of lifetime prevalence of ED. Diagnosis was based on the DSM-4-TR and the researchers considered three ED types: anorexia nervosa (AN), bulimia nervosa (BN), and binge-eating disorder (BED). Subtypes of BD were type I and type II. DSM not otherwise specified diagnoses for BD and EDs were excluded. The mean age of the participants was 40.5 years, and 61% were women.
A total of 479 individuals met criteria for comorbid EDs (16.4%). ED prevalence was significantly higher in BD type II patients than in BD type I patients (20.6 % vs. 12.4 %, P < .001). The overall breakdown according to ED subtype was 30% for AN, 13% for BN, and 56% for BED. The researchers found no significant differences in patients with AN, BN, or BED according to BD subtype.
In a multivariate analysis, BD patients with ED were more likely than those without ED to be women (77% vs. 55%), especially those with AN (95% vs. 82%).
BD patients with ED also tended to be younger than those without ED (37 years vs. 41 years) and reported more frequent suicide attempts (50% vs. 35%). Younger age and more frequent suicide attempts were further significant among BD patients with AN, compared with those with BED, but BD patients with BED reported higher levels of childhood trauma.
BD patients with ED also reported higher levels of depressive symptoms than those without ED, although history of psychosis was less frequent among BD patients with AN and BED compared with BD patients without EDs.
Overall, “after controlling for other variables, the independent factors differentiating BD patients with versus without ED were primarily younger age, female gender, abnormal BMI, increased affective lability and higher comorbidity with anxiety disorders,” the researchers wrote. In addition, presence of EDs except for AN was associated with decreased current functioning.
The findings were limited by several factors including the cross-sectional design, lack of a control group of non-BD individuals, and the consideration of ED over a lifetime, and small number of BN cases, the researchers noted.
However, the results suggest a high prevalence of ED in BD patients and highlight the need to screen BD patients for ED and provide integrated care. More research is needed to explore the evolution of the two conditions as comorbidities and to examine subtypes and of both conditions and their interactions, they concluded.
The study was supported by the FondaMental Foundation, French National Institute for Health and Medical Research, Public Hospitals of Paris, and the French National Research Agency’s Investment for the Future program. The researchers had no financial conflicts to disclose.
FROM THE JOURNAL OF AFFECTIVE DISORDERS
Your patient bequeathed money to you: Can you accept it?
Michael Victoroff, MD, described the phone call he received from an attorney asking a thorny ethics question involving a patient’s gift to another physician. Dr. Victoroff, a past member of the ethics committee of the American Academy of Family Physicians, had definite thoughts about it.
“The attorney was representing the daughters of an elderly gentleman who had moved from the East Coast to Colorado to be closer to them,” said Dr. Victoroff, who teaches bioethics in the MBA program at the University of Denver and also practices at the University of Colorado School of Medicine.
“The father visited his new primary care physician frequently because he had multiple health issues.”
The patient was happy with the doctor’s medical care and over time that they developed a friendship. Dr. Victoroff emphasized that no sexual or romantic impropriety ever took place between the patient and his physician.
“But the social relationship went beyond the ordinary doctor-patient boundaries. The patient ultimately named the doctor as his health care proxy in the event that he became unable to make decisions regarding his care. He also mentioned he was going to leave her $100,000 in his will,” says Dr. Victoroff.
The physician did accept the role of proxy, “which raises a whole host of ethical issues,” says Dr. Victoroff. As it happened, she was never called upon to exercise that decision-making authority, since the patient died suddenly and was mentally competent at the time.
for her to accept such a substantial bequest from a patient, and they hired an attorney to contest the will.
No law against it
Dennis Hursh, attorney and managing partner of Physician Agreements Health Law, a Pennsylvania-based law firm that represents physicians, noted in an interview that, “the problem isn’t legal per se. Rather, the problem is an ethical one.”
Legally speaking, there’s no prohibition against receiving a bequest or other form of gift from a patient. “People are free to dispose of their estates in whatever way they see fit, and no law technically precludes a physician from accepting a bequest,” says Dr. Victoroff. “But this presupposes there is nothing improper going on, such as extortion, deception, coercion, or exercising undue influence.”
The issue of bequeathing money to their physician gained attention in a recent case that took place in Australia. Peter Alexakis, MD, received a whopping bequest of $24 million from a patient. The elderly patient had changed his will to name Dr. Alexakis as the sole beneficiary – after Dr. Alexakis had visited him at home 92 times during the preceding months. The original heirs filed a lawsuit in Australia’s Supreme Court against Dr. Alexakis, contesting the will.
The lawsuit was unsuccessful in court, but Dr. Alexakis was found guilty of malpractice by Australia’s Health Care Complaints Commission after being reported to the HCCC by the palliative care physicians who were treating the patient. They alleged that Dr. Alexakis had interfered with their care of the patient. The more serious allegation was that the doctor had engaged in a deliberate strategy to exploit the relationship for financial gain.
Dr. Alexakis was chastised by the HCCC for engaging in “obtuse” and “suspicious” behavior and for “blurring the boundaries of the doctor-patient relationship.”
There are three domains – legal, ethical, and practical – when it comes to accepting bequests or any gifts from patients, says Dr. Victoroff.
“[In] the legal domain, for example, if you receive a bequest from anyone, patient or otherwise, you have to know your local laws about estates and taxes and so forth and obey them,” he said.
Attorney Hursh pointed out that the Australian doctor wasn’t found guilty of wrongdoing in a court of law but rather of unethical conduct by the Australian medical licensing entity.
Patients giving gifts is often a part of a physician’s life
When Ian Schorr, MD, first started out in practice, he was surprised that patients began bringing him gifts of food to express gratitude for his care.
“I thought it was unethical to accept their gifts, so I turned them down and wouldn’t accept so much as a cookie,” Dr. Schorr, a now-retired ophthalmologist, told this news organization. “But that changed because my office staff told me that some patients were feeling disappointed and insulted. I realized that some people want to express appreciation in ways that go beyond a monetary payment.”
The next time he received a gift from a patient, he “accepted it gracefully.” And he wrote a thank you note, which he continued to do any time he received a gift from a patient.
Kenneth Prager, MD, professor of clinical medicine, director of clinical ethics and chairman of the Medical Ethics Committee at Columbia University Medical Center, New York, says, “I have literally received hundreds of gifts, the vast majority being tokens of patients’ appreciation,” he said. “I’ll get boxes of chocolate or cakes, or sometimes articles of clothing.”
Occasionally, Dr. Prager receives a “somewhat larger gift” – for example, two tickets to a baseball game. “To reject these gifts would be a slap in the face to the patient,” he says, but “where it gets more ethically cloudy is when a gift is very substantial.”
Dr. Prager has never been offered a “substantial” gift or bequest personally. “But a patient whose brother I cared for has indicated that she has left instructions in her will to endow an associate chair of ethics in my honor, and I didn’t decline that,” he said.
The AMA Code of Ethics confirms that accepting gifts offered “as an expression of gratitude or a reflection of the patient’s cultural tradition” can “enhance the patient-physician relationship.” But sometimes gifts “may signal psychological needs that require the physician’s attention.” Accepting such gifts is “likely to damage the patient-physician relationship.”
Potential damage to the therapeutic relationship applies to all physicians but especially for psychiatrists and mental health professionals. “There are more stringent ethical requirements when it comes to these disciplines, where gift-giving gets into the territory of transference or may have particular psychological meaning, and accepting the gift may muddy the therapeutic waters,” Dr. Victoroff said.
Impact on the patient’s family and on other patients
The AMA statement encourages physicians to be “sensitive to the gift’s value, relative to the patient’s or physician’s means.” Physicians should decline gifts that are “disproportionately or inappropriately large, or when the physician would be uncomfortable to have colleagues know the gift had been accepted.”
They should also decline a bequest from a patient if they have reason to believe that to accept it “would present an emotional or financial hardship to the patient’s family.”
“If Bill Gates were leaving $100,000 to his doctor, I imagine Melinda would be just fine,” Mr. Hursh said. “But under ordinary circumstances, if the patient’s family might feel the impact of the bequest, it would be unethical to accept it and could be grounds for revocation of the doctor’s license.”
The AMA statement also warns physicians that by offering a gift, some patients may be seeking to “secure or influence care or to secure preferential treatment,” which can “undermine physicians’ obligation to provide services fairly to all patients.”
For this reason, bequests are “sticky,” said Laurel Lyckholm, MD, professor of hematology and oncology at West Virginia University School of Medicine. In the case of institutions where patients or community members donate money, “we know whose names are on the plaques that hang on the hospital walls, so it’s a delicate balance. What if there’s only one bed or one ventilator? Will the wife of the donor get preferential treatment?”
Follow institutional policy
A “very small gift, such as a fruitcake, is fine,” says Dr. Lyckholm, author of an essay on accepting gifts from patients. She said there’s a dollar amount ($15) that her institution mandates, above which a gift – even food – is considered too expensive to accept. “I was a nurse before I became a physician, and people always tried to give us gifts because we were so close to the minute-by-minute care of the patients,” she said. “We were not allowed to accept money or anything lavish.”
But in the case of small gifts, “the risk-benefit analysis is that there’s much more risk not to take it and to hurt the patient’s feelings.”
Gifts above $15 are given to charity. “I explain to patients that I’m not allowed to take such a large gift, but I’d love to give it to the hospital’s Rosenbaum Family House that provides patients and their relatives with lodging, or to the homeless shelter in Morgantown.”
Dr. Lyckholm, who serves on the ethics committee at J.W. Ruby Memorial Hospital, once was offered expensive tickets and said to the patient, “This is so incredibly thoughtful and kind, but I can’t accept them. I would like to give the tickets to a charity that can auction them off.”
She advises physicians to find out their institution’s policies. Many institutions have policies about what gifts their staff – whether physicians, nurses, or other health care professionals – can accept.
Passing the ‘smell test’
Accepting a large gift from a patient could potentially make it look like you might have exercised undue influence.
“That concern brings us to the third domain, which is very practical and all about appearances and perceptions,” Dr. Victoroff said.
He noted that there is “an inherent power differential between a physician and a patient. The very nature of the relationship can create a risk of ‘undue influence’ on the doctor’s part, even if it’s not apparent to the doctor.” For this reason, it’s necessary to be utterly transparent about how the bequest came about.
He suggests that if a patient informs you that he or she would like to leave money to you, it might be wise to suggest a meeting with the patient’s family, thus establishing some transparency.
It may not be possible to meet with the patient’s family for logistical reasons or because the patient would prefer not to involve their family in their estate planning. But in any case, it’s advisable to document any conversation in the patient’s chart, Dr. Victoroff advised.
“You should make a contemporaneous note that the patient initiated the suggestion and that you counseled them about the implications, no differently than you would with an interaction of a clinical nature,” he suggests. That way, if money has been left to you and is disputed, there’s a clear record that you didn’t solicit it or use any undue influence to bring it about.
He also recommended getting advice from a trusted colleague or a member of your institution’s ethics committee. “Taking time to get a second opinion about an ethical question is a safeguard, like having a chaperone in the room during an examination.”
Ultimately, “there is no human relationship without potential conflicts of interest. Our job is to manage those as best as we can, and sunlight is the best antidote to bad appearances,” Dr. Victoroff said.
A version of this article appeared on Medscape.com.
Michael Victoroff, MD, described the phone call he received from an attorney asking a thorny ethics question involving a patient’s gift to another physician. Dr. Victoroff, a past member of the ethics committee of the American Academy of Family Physicians, had definite thoughts about it.
“The attorney was representing the daughters of an elderly gentleman who had moved from the East Coast to Colorado to be closer to them,” said Dr. Victoroff, who teaches bioethics in the MBA program at the University of Denver and also practices at the University of Colorado School of Medicine.
“The father visited his new primary care physician frequently because he had multiple health issues.”
The patient was happy with the doctor’s medical care and over time that they developed a friendship. Dr. Victoroff emphasized that no sexual or romantic impropriety ever took place between the patient and his physician.
“But the social relationship went beyond the ordinary doctor-patient boundaries. The patient ultimately named the doctor as his health care proxy in the event that he became unable to make decisions regarding his care. He also mentioned he was going to leave her $100,000 in his will,” says Dr. Victoroff.
The physician did accept the role of proxy, “which raises a whole host of ethical issues,” says Dr. Victoroff. As it happened, she was never called upon to exercise that decision-making authority, since the patient died suddenly and was mentally competent at the time.
for her to accept such a substantial bequest from a patient, and they hired an attorney to contest the will.
No law against it
Dennis Hursh, attorney and managing partner of Physician Agreements Health Law, a Pennsylvania-based law firm that represents physicians, noted in an interview that, “the problem isn’t legal per se. Rather, the problem is an ethical one.”
Legally speaking, there’s no prohibition against receiving a bequest or other form of gift from a patient. “People are free to dispose of their estates in whatever way they see fit, and no law technically precludes a physician from accepting a bequest,” says Dr. Victoroff. “But this presupposes there is nothing improper going on, such as extortion, deception, coercion, or exercising undue influence.”
The issue of bequeathing money to their physician gained attention in a recent case that took place in Australia. Peter Alexakis, MD, received a whopping bequest of $24 million from a patient. The elderly patient had changed his will to name Dr. Alexakis as the sole beneficiary – after Dr. Alexakis had visited him at home 92 times during the preceding months. The original heirs filed a lawsuit in Australia’s Supreme Court against Dr. Alexakis, contesting the will.
The lawsuit was unsuccessful in court, but Dr. Alexakis was found guilty of malpractice by Australia’s Health Care Complaints Commission after being reported to the HCCC by the palliative care physicians who were treating the patient. They alleged that Dr. Alexakis had interfered with their care of the patient. The more serious allegation was that the doctor had engaged in a deliberate strategy to exploit the relationship for financial gain.
Dr. Alexakis was chastised by the HCCC for engaging in “obtuse” and “suspicious” behavior and for “blurring the boundaries of the doctor-patient relationship.”
There are three domains – legal, ethical, and practical – when it comes to accepting bequests or any gifts from patients, says Dr. Victoroff.
“[In] the legal domain, for example, if you receive a bequest from anyone, patient or otherwise, you have to know your local laws about estates and taxes and so forth and obey them,” he said.
Attorney Hursh pointed out that the Australian doctor wasn’t found guilty of wrongdoing in a court of law but rather of unethical conduct by the Australian medical licensing entity.
Patients giving gifts is often a part of a physician’s life
When Ian Schorr, MD, first started out in practice, he was surprised that patients began bringing him gifts of food to express gratitude for his care.
“I thought it was unethical to accept their gifts, so I turned them down and wouldn’t accept so much as a cookie,” Dr. Schorr, a now-retired ophthalmologist, told this news organization. “But that changed because my office staff told me that some patients were feeling disappointed and insulted. I realized that some people want to express appreciation in ways that go beyond a monetary payment.”
The next time he received a gift from a patient, he “accepted it gracefully.” And he wrote a thank you note, which he continued to do any time he received a gift from a patient.
Kenneth Prager, MD, professor of clinical medicine, director of clinical ethics and chairman of the Medical Ethics Committee at Columbia University Medical Center, New York, says, “I have literally received hundreds of gifts, the vast majority being tokens of patients’ appreciation,” he said. “I’ll get boxes of chocolate or cakes, or sometimes articles of clothing.”
Occasionally, Dr. Prager receives a “somewhat larger gift” – for example, two tickets to a baseball game. “To reject these gifts would be a slap in the face to the patient,” he says, but “where it gets more ethically cloudy is when a gift is very substantial.”
Dr. Prager has never been offered a “substantial” gift or bequest personally. “But a patient whose brother I cared for has indicated that she has left instructions in her will to endow an associate chair of ethics in my honor, and I didn’t decline that,” he said.
The AMA Code of Ethics confirms that accepting gifts offered “as an expression of gratitude or a reflection of the patient’s cultural tradition” can “enhance the patient-physician relationship.” But sometimes gifts “may signal psychological needs that require the physician’s attention.” Accepting such gifts is “likely to damage the patient-physician relationship.”
Potential damage to the therapeutic relationship applies to all physicians but especially for psychiatrists and mental health professionals. “There are more stringent ethical requirements when it comes to these disciplines, where gift-giving gets into the territory of transference or may have particular psychological meaning, and accepting the gift may muddy the therapeutic waters,” Dr. Victoroff said.
Impact on the patient’s family and on other patients
The AMA statement encourages physicians to be “sensitive to the gift’s value, relative to the patient’s or physician’s means.” Physicians should decline gifts that are “disproportionately or inappropriately large, or when the physician would be uncomfortable to have colleagues know the gift had been accepted.”
They should also decline a bequest from a patient if they have reason to believe that to accept it “would present an emotional or financial hardship to the patient’s family.”
“If Bill Gates were leaving $100,000 to his doctor, I imagine Melinda would be just fine,” Mr. Hursh said. “But under ordinary circumstances, if the patient’s family might feel the impact of the bequest, it would be unethical to accept it and could be grounds for revocation of the doctor’s license.”
The AMA statement also warns physicians that by offering a gift, some patients may be seeking to “secure or influence care or to secure preferential treatment,” which can “undermine physicians’ obligation to provide services fairly to all patients.”
For this reason, bequests are “sticky,” said Laurel Lyckholm, MD, professor of hematology and oncology at West Virginia University School of Medicine. In the case of institutions where patients or community members donate money, “we know whose names are on the plaques that hang on the hospital walls, so it’s a delicate balance. What if there’s only one bed or one ventilator? Will the wife of the donor get preferential treatment?”
Follow institutional policy
A “very small gift, such as a fruitcake, is fine,” says Dr. Lyckholm, author of an essay on accepting gifts from patients. She said there’s a dollar amount ($15) that her institution mandates, above which a gift – even food – is considered too expensive to accept. “I was a nurse before I became a physician, and people always tried to give us gifts because we were so close to the minute-by-minute care of the patients,” she said. “We were not allowed to accept money or anything lavish.”
But in the case of small gifts, “the risk-benefit analysis is that there’s much more risk not to take it and to hurt the patient’s feelings.”
Gifts above $15 are given to charity. “I explain to patients that I’m not allowed to take such a large gift, but I’d love to give it to the hospital’s Rosenbaum Family House that provides patients and their relatives with lodging, or to the homeless shelter in Morgantown.”
Dr. Lyckholm, who serves on the ethics committee at J.W. Ruby Memorial Hospital, once was offered expensive tickets and said to the patient, “This is so incredibly thoughtful and kind, but I can’t accept them. I would like to give the tickets to a charity that can auction them off.”
She advises physicians to find out their institution’s policies. Many institutions have policies about what gifts their staff – whether physicians, nurses, or other health care professionals – can accept.
Passing the ‘smell test’
Accepting a large gift from a patient could potentially make it look like you might have exercised undue influence.
“That concern brings us to the third domain, which is very practical and all about appearances and perceptions,” Dr. Victoroff said.
He noted that there is “an inherent power differential between a physician and a patient. The very nature of the relationship can create a risk of ‘undue influence’ on the doctor’s part, even if it’s not apparent to the doctor.” For this reason, it’s necessary to be utterly transparent about how the bequest came about.
He suggests that if a patient informs you that he or she would like to leave money to you, it might be wise to suggest a meeting with the patient’s family, thus establishing some transparency.
It may not be possible to meet with the patient’s family for logistical reasons or because the patient would prefer not to involve their family in their estate planning. But in any case, it’s advisable to document any conversation in the patient’s chart, Dr. Victoroff advised.
“You should make a contemporaneous note that the patient initiated the suggestion and that you counseled them about the implications, no differently than you would with an interaction of a clinical nature,” he suggests. That way, if money has been left to you and is disputed, there’s a clear record that you didn’t solicit it or use any undue influence to bring it about.
He also recommended getting advice from a trusted colleague or a member of your institution’s ethics committee. “Taking time to get a second opinion about an ethical question is a safeguard, like having a chaperone in the room during an examination.”
Ultimately, “there is no human relationship without potential conflicts of interest. Our job is to manage those as best as we can, and sunlight is the best antidote to bad appearances,” Dr. Victoroff said.
A version of this article appeared on Medscape.com.
Michael Victoroff, MD, described the phone call he received from an attorney asking a thorny ethics question involving a patient’s gift to another physician. Dr. Victoroff, a past member of the ethics committee of the American Academy of Family Physicians, had definite thoughts about it.
“The attorney was representing the daughters of an elderly gentleman who had moved from the East Coast to Colorado to be closer to them,” said Dr. Victoroff, who teaches bioethics in the MBA program at the University of Denver and also practices at the University of Colorado School of Medicine.
“The father visited his new primary care physician frequently because he had multiple health issues.”
The patient was happy with the doctor’s medical care and over time that they developed a friendship. Dr. Victoroff emphasized that no sexual or romantic impropriety ever took place between the patient and his physician.
“But the social relationship went beyond the ordinary doctor-patient boundaries. The patient ultimately named the doctor as his health care proxy in the event that he became unable to make decisions regarding his care. He also mentioned he was going to leave her $100,000 in his will,” says Dr. Victoroff.
The physician did accept the role of proxy, “which raises a whole host of ethical issues,” says Dr. Victoroff. As it happened, she was never called upon to exercise that decision-making authority, since the patient died suddenly and was mentally competent at the time.
for her to accept such a substantial bequest from a patient, and they hired an attorney to contest the will.
No law against it
Dennis Hursh, attorney and managing partner of Physician Agreements Health Law, a Pennsylvania-based law firm that represents physicians, noted in an interview that, “the problem isn’t legal per se. Rather, the problem is an ethical one.”
Legally speaking, there’s no prohibition against receiving a bequest or other form of gift from a patient. “People are free to dispose of their estates in whatever way they see fit, and no law technically precludes a physician from accepting a bequest,” says Dr. Victoroff. “But this presupposes there is nothing improper going on, such as extortion, deception, coercion, or exercising undue influence.”
The issue of bequeathing money to their physician gained attention in a recent case that took place in Australia. Peter Alexakis, MD, received a whopping bequest of $24 million from a patient. The elderly patient had changed his will to name Dr. Alexakis as the sole beneficiary – after Dr. Alexakis had visited him at home 92 times during the preceding months. The original heirs filed a lawsuit in Australia’s Supreme Court against Dr. Alexakis, contesting the will.
The lawsuit was unsuccessful in court, but Dr. Alexakis was found guilty of malpractice by Australia’s Health Care Complaints Commission after being reported to the HCCC by the palliative care physicians who were treating the patient. They alleged that Dr. Alexakis had interfered with their care of the patient. The more serious allegation was that the doctor had engaged in a deliberate strategy to exploit the relationship for financial gain.
Dr. Alexakis was chastised by the HCCC for engaging in “obtuse” and “suspicious” behavior and for “blurring the boundaries of the doctor-patient relationship.”
There are three domains – legal, ethical, and practical – when it comes to accepting bequests or any gifts from patients, says Dr. Victoroff.
“[In] the legal domain, for example, if you receive a bequest from anyone, patient or otherwise, you have to know your local laws about estates and taxes and so forth and obey them,” he said.
Attorney Hursh pointed out that the Australian doctor wasn’t found guilty of wrongdoing in a court of law but rather of unethical conduct by the Australian medical licensing entity.
Patients giving gifts is often a part of a physician’s life
When Ian Schorr, MD, first started out in practice, he was surprised that patients began bringing him gifts of food to express gratitude for his care.
“I thought it was unethical to accept their gifts, so I turned them down and wouldn’t accept so much as a cookie,” Dr. Schorr, a now-retired ophthalmologist, told this news organization. “But that changed because my office staff told me that some patients were feeling disappointed and insulted. I realized that some people want to express appreciation in ways that go beyond a monetary payment.”
The next time he received a gift from a patient, he “accepted it gracefully.” And he wrote a thank you note, which he continued to do any time he received a gift from a patient.
Kenneth Prager, MD, professor of clinical medicine, director of clinical ethics and chairman of the Medical Ethics Committee at Columbia University Medical Center, New York, says, “I have literally received hundreds of gifts, the vast majority being tokens of patients’ appreciation,” he said. “I’ll get boxes of chocolate or cakes, or sometimes articles of clothing.”
Occasionally, Dr. Prager receives a “somewhat larger gift” – for example, two tickets to a baseball game. “To reject these gifts would be a slap in the face to the patient,” he says, but “where it gets more ethically cloudy is when a gift is very substantial.”
Dr. Prager has never been offered a “substantial” gift or bequest personally. “But a patient whose brother I cared for has indicated that she has left instructions in her will to endow an associate chair of ethics in my honor, and I didn’t decline that,” he said.
The AMA Code of Ethics confirms that accepting gifts offered “as an expression of gratitude or a reflection of the patient’s cultural tradition” can “enhance the patient-physician relationship.” But sometimes gifts “may signal psychological needs that require the physician’s attention.” Accepting such gifts is “likely to damage the patient-physician relationship.”
Potential damage to the therapeutic relationship applies to all physicians but especially for psychiatrists and mental health professionals. “There are more stringent ethical requirements when it comes to these disciplines, where gift-giving gets into the territory of transference or may have particular psychological meaning, and accepting the gift may muddy the therapeutic waters,” Dr. Victoroff said.
Impact on the patient’s family and on other patients
The AMA statement encourages physicians to be “sensitive to the gift’s value, relative to the patient’s or physician’s means.” Physicians should decline gifts that are “disproportionately or inappropriately large, or when the physician would be uncomfortable to have colleagues know the gift had been accepted.”
They should also decline a bequest from a patient if they have reason to believe that to accept it “would present an emotional or financial hardship to the patient’s family.”
“If Bill Gates were leaving $100,000 to his doctor, I imagine Melinda would be just fine,” Mr. Hursh said. “But under ordinary circumstances, if the patient’s family might feel the impact of the bequest, it would be unethical to accept it and could be grounds for revocation of the doctor’s license.”
The AMA statement also warns physicians that by offering a gift, some patients may be seeking to “secure or influence care or to secure preferential treatment,” which can “undermine physicians’ obligation to provide services fairly to all patients.”
For this reason, bequests are “sticky,” said Laurel Lyckholm, MD, professor of hematology and oncology at West Virginia University School of Medicine. In the case of institutions where patients or community members donate money, “we know whose names are on the plaques that hang on the hospital walls, so it’s a delicate balance. What if there’s only one bed or one ventilator? Will the wife of the donor get preferential treatment?”
Follow institutional policy
A “very small gift, such as a fruitcake, is fine,” says Dr. Lyckholm, author of an essay on accepting gifts from patients. She said there’s a dollar amount ($15) that her institution mandates, above which a gift – even food – is considered too expensive to accept. “I was a nurse before I became a physician, and people always tried to give us gifts because we were so close to the minute-by-minute care of the patients,” she said. “We were not allowed to accept money or anything lavish.”
But in the case of small gifts, “the risk-benefit analysis is that there’s much more risk not to take it and to hurt the patient’s feelings.”
Gifts above $15 are given to charity. “I explain to patients that I’m not allowed to take such a large gift, but I’d love to give it to the hospital’s Rosenbaum Family House that provides patients and their relatives with lodging, or to the homeless shelter in Morgantown.”
Dr. Lyckholm, who serves on the ethics committee at J.W. Ruby Memorial Hospital, once was offered expensive tickets and said to the patient, “This is so incredibly thoughtful and kind, but I can’t accept them. I would like to give the tickets to a charity that can auction them off.”
She advises physicians to find out their institution’s policies. Many institutions have policies about what gifts their staff – whether physicians, nurses, or other health care professionals – can accept.
Passing the ‘smell test’
Accepting a large gift from a patient could potentially make it look like you might have exercised undue influence.
“That concern brings us to the third domain, which is very practical and all about appearances and perceptions,” Dr. Victoroff said.
He noted that there is “an inherent power differential between a physician and a patient. The very nature of the relationship can create a risk of ‘undue influence’ on the doctor’s part, even if it’s not apparent to the doctor.” For this reason, it’s necessary to be utterly transparent about how the bequest came about.
He suggests that if a patient informs you that he or she would like to leave money to you, it might be wise to suggest a meeting with the patient’s family, thus establishing some transparency.
It may not be possible to meet with the patient’s family for logistical reasons or because the patient would prefer not to involve their family in their estate planning. But in any case, it’s advisable to document any conversation in the patient’s chart, Dr. Victoroff advised.
“You should make a contemporaneous note that the patient initiated the suggestion and that you counseled them about the implications, no differently than you would with an interaction of a clinical nature,” he suggests. That way, if money has been left to you and is disputed, there’s a clear record that you didn’t solicit it or use any undue influence to bring it about.
He also recommended getting advice from a trusted colleague or a member of your institution’s ethics committee. “Taking time to get a second opinion about an ethical question is a safeguard, like having a chaperone in the room during an examination.”
Ultimately, “there is no human relationship without potential conflicts of interest. Our job is to manage those as best as we can, and sunlight is the best antidote to bad appearances,” Dr. Victoroff said.
A version of this article appeared on Medscape.com.
Aspirin still needed in first month after PCI: STOPDAPT-3
AMSTERDAM – Dropping aspirin and using low-dose prasugrel (Effient) alone in the initial month of treatment after percutaneous coronary intervention (PCI) failed to lower bleeding risk, compared with dual antiplatelet therapy (DAPT), and there was a signal of possible harm in terms of increased subacute stent thrombosis, in the STOPDAPT-3 trial.
“Therefore, dual antiplatelet therapy with aspirin and a P2Y12 inhibitor should still remain the standard strategy at least for 1 month after PCI,” said the trial’s lead investigator Masahiro Natsuaki, MD, Saga (Japan) University.
The STOPDAPT-3 trial was presented at the recent annual congress of the European Society of Cardiology.
Designated discussant Marco Valgimigli, MD, Cardiocentro Ticino Foundation, Lugano, Switzerland, explained that the current wisdom before this study was that aspirin withdrawal in the postacute phase after PCI (after 1 month of DAPT onwards) is associated with lower bleeding risk without affecting ischemic risk, but this STOPDAPT-3 trial is the first to look at the idea of not giving aspirin at all.
“This study is a well-designed, well-conducted trial, and the results are very clear: Dr. Valgimigli said.
He pointed out that the possible harm was not related to the coprimary cardiovascular composite endpoint, which did fulfill noninferiority, although he acknowledged the “generous” noninferiority margin.
Rather, the possible harm was related to an increase in subacute stent thrombosis, which was three times higher in the nonaspirin group (0.58% vs. 0.17%).
“While these absolute event rates are extremely low, they are unquestionably higher in the nonaspirin group,” he added.
In his presentation, Dr. Natsuaki explained that very short durations (1-3 months) of DAPT followed by P2Y12 inhibitor monotherapy has been shown to reduce bleeding events without increasing cardiovascular events, compared with longer durations of DAPT after PCI using drug-eluting stents.
However, the incidence of major bleeding events within the 1-month mandatory DAPT period after PCI remains high in clinical practice, particularly in patients with ACS or high bleeding risk.
In single-arm studies, use of prasugrel or ticagrelor (Brilinta) alone following new-generation drug-eluting stent implantation was not associated with any stent thrombosis in selected low-risk patients with or without ACS, and it is thought that removing aspirin from the DAPT regimen might reduce bleeding events early after PCI without compromising the risk of cardiovascular events. However, the efficacy and safety of this strategy has not been proven in randomized trials.
STOPDAPT-3 trial
STOPDAPT-3 investigated the efficacy and safety of prasugrel monotherapy compared with 1-month DAPT with aspirin and prasugrel in Japanese patients with ACS or high bleeding risk undergoing PCI with cobalt-chromium everolimus-eluting stents.
The study enrolled 6,002 patients with ACS or high bleeding risk who were randomly assigned to prasugrel monotherapy (3.75 mg/day; the licensed dose in Japan) or to DAPT with aspirin (81-100 mg/day) and prasugrel after a loading dose of prasugrel 20 mg in both groups.
There were two primary endpoints: major bleeding events (defined as BARC type 3 or 5) at 1 month for superiority and cardiovascular events (a composite of cardiovascular death, myocardial infarction, definite stent thrombosis, or stroke) at 1 month for noninferiority.
The major secondary endpoint was a composite of the coprimary bleeding and cardiovascular endpoints (cardiovascular death, myocardial infarction, definite stent thrombosis, stroke, or major bleeding) at 1 month representing net clinical benefit.
Results showed that, at 1 month, the no-aspirin strategy was not superior to DAPT for the coprimary bleeding endpoint, with major bleeding events occurring in 4.47% of the prasugrel monotherapy group versus 4.71% of those on DAPT (hazard ratio, 0.95; 95% confidence interval, 0.75-1.20).
The prasugrel monotherapy strategy was noninferior to DAPT, although there was a relative 50% margin for the coprimary cardiovascular endpoint. Cardiovascular endpoints occurred in 4.12% of prasugrel monotherapy group versus 3.69% of the DAPT patients (HR, 1.12; 95% CI, 0.87-1.45; P for noninferiority = .01).
The major secondary net clinical benefit endpoint occurred in 7.14% patients in the prasugrel monotherapy group and 7.38% patients in the DAPT group, with no between-group difference, indicating a similar effect on net clinical benefit for both groups.
However, there was an excess of any coronary revascularization (1.15% vs. 0.57%) and definite or probable stent thrombosis (0.71% vs. 0.44%) in the prasugrel monotherapy group compared with the DAPT group, while definite stent thrombosis was not different between the two groups (0.47% vs. 0.37%).
In a subgroup analysis stratified by ACS and non-ACS, the excess risk for cardiovascular events in the no-aspirin group, compared with the DAPT group, was seen in patients with ACS, but not in those without ACS.
Future: Focus on dose and timing
In his discussion, Dr. Valgimigli said the implications of this trial for clinical practice were very clear: “Aspirin remains a cornerstone treatment in the periprocedural and acute phase of PCI in patients without indications for oral anticoagulation.”
However, he added that the study opens several important points for subsequent discussion.
These include the role of type and dose of P2Y12 inhibitor therapy used; specifically, he questioned whether the 3.75-mg dose of prasugrel was enough.
Dr. Valgimigli also pointed out that this study did not include a purely high bleeding risk population, and he said there was still potential to investigate periprocedure versus postprocedure aspirin administration.
The STOPDAPT-3 trial was funded by Abbott Medical Japan. Dr. Natsuaki reported receiving honoraria from Abbott Medical Japan, Daiichi Sankyo, and Bayer.
A version of this article first appeared on Medscape.com.
AMSTERDAM – Dropping aspirin and using low-dose prasugrel (Effient) alone in the initial month of treatment after percutaneous coronary intervention (PCI) failed to lower bleeding risk, compared with dual antiplatelet therapy (DAPT), and there was a signal of possible harm in terms of increased subacute stent thrombosis, in the STOPDAPT-3 trial.
“Therefore, dual antiplatelet therapy with aspirin and a P2Y12 inhibitor should still remain the standard strategy at least for 1 month after PCI,” said the trial’s lead investigator Masahiro Natsuaki, MD, Saga (Japan) University.
The STOPDAPT-3 trial was presented at the recent annual congress of the European Society of Cardiology.
Designated discussant Marco Valgimigli, MD, Cardiocentro Ticino Foundation, Lugano, Switzerland, explained that the current wisdom before this study was that aspirin withdrawal in the postacute phase after PCI (after 1 month of DAPT onwards) is associated with lower bleeding risk without affecting ischemic risk, but this STOPDAPT-3 trial is the first to look at the idea of not giving aspirin at all.
“This study is a well-designed, well-conducted trial, and the results are very clear: Dr. Valgimigli said.
He pointed out that the possible harm was not related to the coprimary cardiovascular composite endpoint, which did fulfill noninferiority, although he acknowledged the “generous” noninferiority margin.
Rather, the possible harm was related to an increase in subacute stent thrombosis, which was three times higher in the nonaspirin group (0.58% vs. 0.17%).
“While these absolute event rates are extremely low, they are unquestionably higher in the nonaspirin group,” he added.
In his presentation, Dr. Natsuaki explained that very short durations (1-3 months) of DAPT followed by P2Y12 inhibitor monotherapy has been shown to reduce bleeding events without increasing cardiovascular events, compared with longer durations of DAPT after PCI using drug-eluting stents.
However, the incidence of major bleeding events within the 1-month mandatory DAPT period after PCI remains high in clinical practice, particularly in patients with ACS or high bleeding risk.
In single-arm studies, use of prasugrel or ticagrelor (Brilinta) alone following new-generation drug-eluting stent implantation was not associated with any stent thrombosis in selected low-risk patients with or without ACS, and it is thought that removing aspirin from the DAPT regimen might reduce bleeding events early after PCI without compromising the risk of cardiovascular events. However, the efficacy and safety of this strategy has not been proven in randomized trials.
STOPDAPT-3 trial
STOPDAPT-3 investigated the efficacy and safety of prasugrel monotherapy compared with 1-month DAPT with aspirin and prasugrel in Japanese patients with ACS or high bleeding risk undergoing PCI with cobalt-chromium everolimus-eluting stents.
The study enrolled 6,002 patients with ACS or high bleeding risk who were randomly assigned to prasugrel monotherapy (3.75 mg/day; the licensed dose in Japan) or to DAPT with aspirin (81-100 mg/day) and prasugrel after a loading dose of prasugrel 20 mg in both groups.
There were two primary endpoints: major bleeding events (defined as BARC type 3 or 5) at 1 month for superiority and cardiovascular events (a composite of cardiovascular death, myocardial infarction, definite stent thrombosis, or stroke) at 1 month for noninferiority.
The major secondary endpoint was a composite of the coprimary bleeding and cardiovascular endpoints (cardiovascular death, myocardial infarction, definite stent thrombosis, stroke, or major bleeding) at 1 month representing net clinical benefit.
Results showed that, at 1 month, the no-aspirin strategy was not superior to DAPT for the coprimary bleeding endpoint, with major bleeding events occurring in 4.47% of the prasugrel monotherapy group versus 4.71% of those on DAPT (hazard ratio, 0.95; 95% confidence interval, 0.75-1.20).
The prasugrel monotherapy strategy was noninferior to DAPT, although there was a relative 50% margin for the coprimary cardiovascular endpoint. Cardiovascular endpoints occurred in 4.12% of prasugrel monotherapy group versus 3.69% of the DAPT patients (HR, 1.12; 95% CI, 0.87-1.45; P for noninferiority = .01).
The major secondary net clinical benefit endpoint occurred in 7.14% patients in the prasugrel monotherapy group and 7.38% patients in the DAPT group, with no between-group difference, indicating a similar effect on net clinical benefit for both groups.
However, there was an excess of any coronary revascularization (1.15% vs. 0.57%) and definite or probable stent thrombosis (0.71% vs. 0.44%) in the prasugrel monotherapy group compared with the DAPT group, while definite stent thrombosis was not different between the two groups (0.47% vs. 0.37%).
In a subgroup analysis stratified by ACS and non-ACS, the excess risk for cardiovascular events in the no-aspirin group, compared with the DAPT group, was seen in patients with ACS, but not in those without ACS.
Future: Focus on dose and timing
In his discussion, Dr. Valgimigli said the implications of this trial for clinical practice were very clear: “Aspirin remains a cornerstone treatment in the periprocedural and acute phase of PCI in patients without indications for oral anticoagulation.”
However, he added that the study opens several important points for subsequent discussion.
These include the role of type and dose of P2Y12 inhibitor therapy used; specifically, he questioned whether the 3.75-mg dose of prasugrel was enough.
Dr. Valgimigli also pointed out that this study did not include a purely high bleeding risk population, and he said there was still potential to investigate periprocedure versus postprocedure aspirin administration.
The STOPDAPT-3 trial was funded by Abbott Medical Japan. Dr. Natsuaki reported receiving honoraria from Abbott Medical Japan, Daiichi Sankyo, and Bayer.
A version of this article first appeared on Medscape.com.
AMSTERDAM – Dropping aspirin and using low-dose prasugrel (Effient) alone in the initial month of treatment after percutaneous coronary intervention (PCI) failed to lower bleeding risk, compared with dual antiplatelet therapy (DAPT), and there was a signal of possible harm in terms of increased subacute stent thrombosis, in the STOPDAPT-3 trial.
“Therefore, dual antiplatelet therapy with aspirin and a P2Y12 inhibitor should still remain the standard strategy at least for 1 month after PCI,” said the trial’s lead investigator Masahiro Natsuaki, MD, Saga (Japan) University.
The STOPDAPT-3 trial was presented at the recent annual congress of the European Society of Cardiology.
Designated discussant Marco Valgimigli, MD, Cardiocentro Ticino Foundation, Lugano, Switzerland, explained that the current wisdom before this study was that aspirin withdrawal in the postacute phase after PCI (after 1 month of DAPT onwards) is associated with lower bleeding risk without affecting ischemic risk, but this STOPDAPT-3 trial is the first to look at the idea of not giving aspirin at all.
“This study is a well-designed, well-conducted trial, and the results are very clear: Dr. Valgimigli said.
He pointed out that the possible harm was not related to the coprimary cardiovascular composite endpoint, which did fulfill noninferiority, although he acknowledged the “generous” noninferiority margin.
Rather, the possible harm was related to an increase in subacute stent thrombosis, which was three times higher in the nonaspirin group (0.58% vs. 0.17%).
“While these absolute event rates are extremely low, they are unquestionably higher in the nonaspirin group,” he added.
In his presentation, Dr. Natsuaki explained that very short durations (1-3 months) of DAPT followed by P2Y12 inhibitor monotherapy has been shown to reduce bleeding events without increasing cardiovascular events, compared with longer durations of DAPT after PCI using drug-eluting stents.
However, the incidence of major bleeding events within the 1-month mandatory DAPT period after PCI remains high in clinical practice, particularly in patients with ACS or high bleeding risk.
In single-arm studies, use of prasugrel or ticagrelor (Brilinta) alone following new-generation drug-eluting stent implantation was not associated with any stent thrombosis in selected low-risk patients with or without ACS, and it is thought that removing aspirin from the DAPT regimen might reduce bleeding events early after PCI without compromising the risk of cardiovascular events. However, the efficacy and safety of this strategy has not been proven in randomized trials.
STOPDAPT-3 trial
STOPDAPT-3 investigated the efficacy and safety of prasugrel monotherapy compared with 1-month DAPT with aspirin and prasugrel in Japanese patients with ACS or high bleeding risk undergoing PCI with cobalt-chromium everolimus-eluting stents.
The study enrolled 6,002 patients with ACS or high bleeding risk who were randomly assigned to prasugrel monotherapy (3.75 mg/day; the licensed dose in Japan) or to DAPT with aspirin (81-100 mg/day) and prasugrel after a loading dose of prasugrel 20 mg in both groups.
There were two primary endpoints: major bleeding events (defined as BARC type 3 or 5) at 1 month for superiority and cardiovascular events (a composite of cardiovascular death, myocardial infarction, definite stent thrombosis, or stroke) at 1 month for noninferiority.
The major secondary endpoint was a composite of the coprimary bleeding and cardiovascular endpoints (cardiovascular death, myocardial infarction, definite stent thrombosis, stroke, or major bleeding) at 1 month representing net clinical benefit.
Results showed that, at 1 month, the no-aspirin strategy was not superior to DAPT for the coprimary bleeding endpoint, with major bleeding events occurring in 4.47% of the prasugrel monotherapy group versus 4.71% of those on DAPT (hazard ratio, 0.95; 95% confidence interval, 0.75-1.20).
The prasugrel monotherapy strategy was noninferior to DAPT, although there was a relative 50% margin for the coprimary cardiovascular endpoint. Cardiovascular endpoints occurred in 4.12% of prasugrel monotherapy group versus 3.69% of the DAPT patients (HR, 1.12; 95% CI, 0.87-1.45; P for noninferiority = .01).
The major secondary net clinical benefit endpoint occurred in 7.14% patients in the prasugrel monotherapy group and 7.38% patients in the DAPT group, with no between-group difference, indicating a similar effect on net clinical benefit for both groups.
However, there was an excess of any coronary revascularization (1.15% vs. 0.57%) and definite or probable stent thrombosis (0.71% vs. 0.44%) in the prasugrel monotherapy group compared with the DAPT group, while definite stent thrombosis was not different between the two groups (0.47% vs. 0.37%).
In a subgroup analysis stratified by ACS and non-ACS, the excess risk for cardiovascular events in the no-aspirin group, compared with the DAPT group, was seen in patients with ACS, but not in those without ACS.
Future: Focus on dose and timing
In his discussion, Dr. Valgimigli said the implications of this trial for clinical practice were very clear: “Aspirin remains a cornerstone treatment in the periprocedural and acute phase of PCI in patients without indications for oral anticoagulation.”
However, he added that the study opens several important points for subsequent discussion.
These include the role of type and dose of P2Y12 inhibitor therapy used; specifically, he questioned whether the 3.75-mg dose of prasugrel was enough.
Dr. Valgimigli also pointed out that this study did not include a purely high bleeding risk population, and he said there was still potential to investigate periprocedure versus postprocedure aspirin administration.
The STOPDAPT-3 trial was funded by Abbott Medical Japan. Dr. Natsuaki reported receiving honoraria from Abbott Medical Japan, Daiichi Sankyo, and Bayer.
A version of this article first appeared on Medscape.com.
AT THE ESC CONGRESS 2023
‘Missed opportunities’ for accurate diagnosing of women with vaginitis

Although the standard of care of diagnosing vaginitis is clinical evaluation, many practices do not perform accurate and comprehensive clinical examinations for a variety for reasons, and the Centers for Disease Control and Prevention currently recommends molecular testing, wrote Casey N. Pinto, PhD, of Penn State University, Hershey, and colleagues. The CDC also recommends testing women with vaginitis for Chlamydia trachomatis (CT) and Neissaria gonorrhoeae (NG) given the high rate of coinfections between vaginitis and these sexually transmitted infections, but data on cotesting in clinical practice are limited, they said.
In a study published in Sexually Transmitted Diseases, the researchers reviewed data from a commercial administrative claims database for 1,359,289 women aged 18-50 years who were diagnosed with vaginitis between 2012 and 2017.
The women were categorized into groups based on type of vaginitis diagnosis: nucleic amplification test (NAAT), DNA probe test, traditional lab test, and those diagnosed clinically at an index visit but with no CPT code for further testing.
Overall, nearly half of the women (49.2%) had no CPT code for further vaginitis testing beyond clinical diagnosis. Of those with CPT codes for testing, 50.9% underwent traditional point-of-care testing, wet mount, or culture, 23.5% had a DNA probe, and 20.6% had NAAT testing.
Approximately one-third (34%) of women were cotested for CT/NG. Testing rates varied widely across the type of vaginitis test, from 70.8% of women who received NAAT to 22.8% of women with no CPT code. In multivariate analysis including age, region, and the Charlson Comorbidity Index (CCI), those tested with NAAT were eight times more likely to be cotested for CT/NG than those with no CPT code (odds ratio, 8.77; P < .0001).
Women who received a traditional test or DNA probe test for vaginitis also were more likely to have CT/NG testing than women with no CPT code, but only 1.8-2.5 times as likely.
“Our data suggest that most clinicians are not engaging the standard of care for testing and diagnosing vaginitis, or not engaging in comprehensive care by cotesting for vaginitis and CT/NG when patients may be at risk, resulting in missed opportunities for accurate diagnosis and potential associated coinfections,” the researchers wrote in their discussion. The higher rates for CT/NG testing among women receiving either NAAT or DNA probe vaginitis testing could be attributed to bundled testing, they noted, and the lower rate of CT/NG testing for patients with no CPT code could stem from limited access to microscopy or clinician preference for clinical diagnosis only, they said.
The findings were limited by several factors, including the lack of data on testing and diagnoses prior to the study period and not billed to insurance, and by the inability to account for variables including race, ethnicity, and socioeconomic status, the researchers noted.
However, the results highlight the need for more comprehensive care in vaginitis testing to take advantage of opportunities to identify CT or NG in women diagnosed with vaginitis, they concluded.
The study was supported by Becton, Dickinson and Company. Lead author Dr. Pinto disclosed consulting for Becton, Dickinson and Company, and receiving an honorarium from Roche.
Although the standard of care of diagnosing vaginitis is clinical evaluation, many practices do not perform accurate and comprehensive clinical examinations for a variety for reasons, and the Centers for Disease Control and Prevention currently recommends molecular testing, wrote Casey N. Pinto, PhD, of Penn State University, Hershey, and colleagues. The CDC also recommends testing women with vaginitis for Chlamydia trachomatis (CT) and Neissaria gonorrhoeae (NG) given the high rate of coinfections between vaginitis and these sexually transmitted infections, but data on cotesting in clinical practice are limited, they said.
In a study published in Sexually Transmitted Diseases, the researchers reviewed data from a commercial administrative claims database for 1,359,289 women aged 18-50 years who were diagnosed with vaginitis between 2012 and 2017.
The women were categorized into groups based on type of vaginitis diagnosis: nucleic amplification test (NAAT), DNA probe test, traditional lab test, and those diagnosed clinically at an index visit but with no CPT code for further testing.
Overall, nearly half of the women (49.2%) had no CPT code for further vaginitis testing beyond clinical diagnosis. Of those with CPT codes for testing, 50.9% underwent traditional point-of-care testing, wet mount, or culture, 23.5% had a DNA probe, and 20.6% had NAAT testing.
Approximately one-third (34%) of women were cotested for CT/NG. Testing rates varied widely across the type of vaginitis test, from 70.8% of women who received NAAT to 22.8% of women with no CPT code. In multivariate analysis including age, region, and the Charlson Comorbidity Index (CCI), those tested with NAAT were eight times more likely to be cotested for CT/NG than those with no CPT code (odds ratio, 8.77; P < .0001).
Women who received a traditional test or DNA probe test for vaginitis also were more likely to have CT/NG testing than women with no CPT code, but only 1.8-2.5 times as likely.
“Our data suggest that most clinicians are not engaging the standard of care for testing and diagnosing vaginitis, or not engaging in comprehensive care by cotesting for vaginitis and CT/NG when patients may be at risk, resulting in missed opportunities for accurate diagnosis and potential associated coinfections,” the researchers wrote in their discussion. The higher rates for CT/NG testing among women receiving either NAAT or DNA probe vaginitis testing could be attributed to bundled testing, they noted, and the lower rate of CT/NG testing for patients with no CPT code could stem from limited access to microscopy or clinician preference for clinical diagnosis only, they said.
The findings were limited by several factors, including the lack of data on testing and diagnoses prior to the study period and not billed to insurance, and by the inability to account for variables including race, ethnicity, and socioeconomic status, the researchers noted.
However, the results highlight the need for more comprehensive care in vaginitis testing to take advantage of opportunities to identify CT or NG in women diagnosed with vaginitis, they concluded.
The study was supported by Becton, Dickinson and Company. Lead author Dr. Pinto disclosed consulting for Becton, Dickinson and Company, and receiving an honorarium from Roche.
Although the standard of care of diagnosing vaginitis is clinical evaluation, many practices do not perform accurate and comprehensive clinical examinations for a variety for reasons, and the Centers for Disease Control and Prevention currently recommends molecular testing, wrote Casey N. Pinto, PhD, of Penn State University, Hershey, and colleagues. The CDC also recommends testing women with vaginitis for Chlamydia trachomatis (CT) and Neissaria gonorrhoeae (NG) given the high rate of coinfections between vaginitis and these sexually transmitted infections, but data on cotesting in clinical practice are limited, they said.
In a study published in Sexually Transmitted Diseases, the researchers reviewed data from a commercial administrative claims database for 1,359,289 women aged 18-50 years who were diagnosed with vaginitis between 2012 and 2017.
The women were categorized into groups based on type of vaginitis diagnosis: nucleic amplification test (NAAT), DNA probe test, traditional lab test, and those diagnosed clinically at an index visit but with no CPT code for further testing.
Overall, nearly half of the women (49.2%) had no CPT code for further vaginitis testing beyond clinical diagnosis. Of those with CPT codes for testing, 50.9% underwent traditional point-of-care testing, wet mount, or culture, 23.5% had a DNA probe, and 20.6% had NAAT testing.
Approximately one-third (34%) of women were cotested for CT/NG. Testing rates varied widely across the type of vaginitis test, from 70.8% of women who received NAAT to 22.8% of women with no CPT code. In multivariate analysis including age, region, and the Charlson Comorbidity Index (CCI), those tested with NAAT were eight times more likely to be cotested for CT/NG than those with no CPT code (odds ratio, 8.77; P < .0001).
Women who received a traditional test or DNA probe test for vaginitis also were more likely to have CT/NG testing than women with no CPT code, but only 1.8-2.5 times as likely.
“Our data suggest that most clinicians are not engaging the standard of care for testing and diagnosing vaginitis, or not engaging in comprehensive care by cotesting for vaginitis and CT/NG when patients may be at risk, resulting in missed opportunities for accurate diagnosis and potential associated coinfections,” the researchers wrote in their discussion. The higher rates for CT/NG testing among women receiving either NAAT or DNA probe vaginitis testing could be attributed to bundled testing, they noted, and the lower rate of CT/NG testing for patients with no CPT code could stem from limited access to microscopy or clinician preference for clinical diagnosis only, they said.
The findings were limited by several factors, including the lack of data on testing and diagnoses prior to the study period and not billed to insurance, and by the inability to account for variables including race, ethnicity, and socioeconomic status, the researchers noted.
However, the results highlight the need for more comprehensive care in vaginitis testing to take advantage of opportunities to identify CT or NG in women diagnosed with vaginitis, they concluded.
The study was supported by Becton, Dickinson and Company. Lead author Dr. Pinto disclosed consulting for Becton, Dickinson and Company, and receiving an honorarium from Roche.
FROM SEXUALLY TRANSMITTED DISEASES
Are you ready for RSV season? There’s a new preventive option
There is now an additional option for the prevention of respiratory syncytial virus (RSV), the most common cause of hospitalization among infants and children in the United States. In July, the US Food and Drug Administration (FDA) approved nirsevimab, an RSV preventive monoclonal antibody, for use in neonates and infants born during or entering their first RSV season and in children up to 24 months of age who remain vulnerable to RSV during their second season.1 The Advisory Committee on Immunization Practices (ACIP) subsequently made 2 recommendations regarding use of nirsevimab, which I’ll detail in a moment.2
First, a word about RSV. The Centers for Disease Control and Prevention estimates that each year in children younger than 5 years, RSV is responsible for 1.5 million outpatient clinic visits, 520,000 emergency department visits, 58,000 to 80,000 hospitalizations, and 100 to 200 deaths.2 The risk for hospitalization from RSV is highest in the second and third months of life and decreases with increasing age.
There are some racial disparities in RSV severity, likely reflecting social drivers of health: ICU admission rates are 1.2 to 1.6 times higher among non-Hispanic Black infants younger than 6 months than among non-Hispanic White infants, and hospitalization rates are up to 5 times higher in American Indian and Alaska Native populations.2
What nirsevimab adds to the toolbox. Until recently, there was only 1 RSV preventive agent available: palivizumab, also a monoclonal antibody. The American Academy of Pediatrics has recommended palivizumab be used only for infants at high risk for RSV infection, due to its high cost and the need for monthly injections for the duration of an RSV season. In addition, the Academy has noted that palivizumab has limited effect on RSV hospitalizations on a population basis and does not appear to affect mortality.3
Nirsevimab has a longer half-life than palivizumab, and only 1 injection is needed for the RSV season. Early studies on nirsevimab demonstrate 79% effectiveness in preventing medical-attended lower respiratory tract infection, 80.6% effectiveness in preventing hospitalization, and 90% effectiveness in preventing ICU admission. The number needed to immunize with nirsevimab to prevent an outpatient visit is estimated to be 17; to prevent an ED visit, 48; and to prevent an inpatient admission, 128. Due to the low RSV death rate, the studies were not able to demonstrate reduced mortality.2
What the ACIP recommends. At a special meeting in July, the ACIP recommended 1 dose of nirsevimab for2:
- all infants younger than 8 months who are born during or entering their first RSV season
- children ages 8 to 19 months who are at increased risk for severe RSV disease and entering their second RSV season.
Those at risk include children with chronic lung disease of prematurity who required medical support any time during the 6-month period before the start of their second RSV season; those with severe immunocompromise; those with cystic fibrosis who have manifestations of severe lung disease or weight-for-length < 10th percentile; and American Indian and Alaska Native children.2
How to administer nirsevimab. The dose of nirsevimab is 50 mg IM for those weighing < 5 kg, 100 mg for those weighing ≥ 5 kg, and 200 mg for high-risk children entering their second RSV season.2 Nirsevimab can be co-administered with other recommended vaccines; however, both nirsevimab and palivizumab should not be used in the same child in the same RSV season.
Nirsevimab should be administered in the first week of life for infants born shortly before or during RSV season, and shortly before the season for infants younger than 8 months and those ages 8 to 19 months who are at high risk.4 The months of highest RSV transmission in most locations are December through February, but this can vary. Local epidemiology and advice from state and local health departments are the best source of information about when RSV season starts and ends in your area.
On the horizon. Nirsevimab will be included in the Vaccines for Children program and covered by commercial health plans with no cost sharing.5 A maternal vaccine to prevent RSV in newborns is likely to be approved by the FDA in the near future.
1. FDA. FDA approves new drug to prevent RSV in babies and toddlers [press release]. Published July 17, 2023. Accessed August 29, 2023. www.fda.gov/news-events/press-announcements/fda-approves-new-drug-prevent-rsv-babies-and-toddlers
2. Jones J. Evidence to recommendation framework: nirsevimab updates. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131586
3. American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014;134:e620–e638. doi: 10.1542/peds.2014-1666
4. Jones J. Proposed clinical consideration updates for nirsevimab. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2023-08-3/04-rsv-jones-508.pdf
5. Peacock G. Nirsevimab: implementation considerations. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131587
There is now an additional option for the prevention of respiratory syncytial virus (RSV), the most common cause of hospitalization among infants and children in the United States. In July, the US Food and Drug Administration (FDA) approved nirsevimab, an RSV preventive monoclonal antibody, for use in neonates and infants born during or entering their first RSV season and in children up to 24 months of age who remain vulnerable to RSV during their second season.1 The Advisory Committee on Immunization Practices (ACIP) subsequently made 2 recommendations regarding use of nirsevimab, which I’ll detail in a moment.2
First, a word about RSV. The Centers for Disease Control and Prevention estimates that each year in children younger than 5 years, RSV is responsible for 1.5 million outpatient clinic visits, 520,000 emergency department visits, 58,000 to 80,000 hospitalizations, and 100 to 200 deaths.2 The risk for hospitalization from RSV is highest in the second and third months of life and decreases with increasing age.
There are some racial disparities in RSV severity, likely reflecting social drivers of health: ICU admission rates are 1.2 to 1.6 times higher among non-Hispanic Black infants younger than 6 months than among non-Hispanic White infants, and hospitalization rates are up to 5 times higher in American Indian and Alaska Native populations.2
What nirsevimab adds to the toolbox. Until recently, there was only 1 RSV preventive agent available: palivizumab, also a monoclonal antibody. The American Academy of Pediatrics has recommended palivizumab be used only for infants at high risk for RSV infection, due to its high cost and the need for monthly injections for the duration of an RSV season. In addition, the Academy has noted that palivizumab has limited effect on RSV hospitalizations on a population basis and does not appear to affect mortality.3
Nirsevimab has a longer half-life than palivizumab, and only 1 injection is needed for the RSV season. Early studies on nirsevimab demonstrate 79% effectiveness in preventing medical-attended lower respiratory tract infection, 80.6% effectiveness in preventing hospitalization, and 90% effectiveness in preventing ICU admission. The number needed to immunize with nirsevimab to prevent an outpatient visit is estimated to be 17; to prevent an ED visit, 48; and to prevent an inpatient admission, 128. Due to the low RSV death rate, the studies were not able to demonstrate reduced mortality.2
What the ACIP recommends. At a special meeting in July, the ACIP recommended 1 dose of nirsevimab for2:
- all infants younger than 8 months who are born during or entering their first RSV season
- children ages 8 to 19 months who are at increased risk for severe RSV disease and entering their second RSV season.
Those at risk include children with chronic lung disease of prematurity who required medical support any time during the 6-month period before the start of their second RSV season; those with severe immunocompromise; those with cystic fibrosis who have manifestations of severe lung disease or weight-for-length < 10th percentile; and American Indian and Alaska Native children.2
How to administer nirsevimab. The dose of nirsevimab is 50 mg IM for those weighing < 5 kg, 100 mg for those weighing ≥ 5 kg, and 200 mg for high-risk children entering their second RSV season.2 Nirsevimab can be co-administered with other recommended vaccines; however, both nirsevimab and palivizumab should not be used in the same child in the same RSV season.
Nirsevimab should be administered in the first week of life for infants born shortly before or during RSV season, and shortly before the season for infants younger than 8 months and those ages 8 to 19 months who are at high risk.4 The months of highest RSV transmission in most locations are December through February, but this can vary. Local epidemiology and advice from state and local health departments are the best source of information about when RSV season starts and ends in your area.
On the horizon. Nirsevimab will be included in the Vaccines for Children program and covered by commercial health plans with no cost sharing.5 A maternal vaccine to prevent RSV in newborns is likely to be approved by the FDA in the near future.
There is now an additional option for the prevention of respiratory syncytial virus (RSV), the most common cause of hospitalization among infants and children in the United States. In July, the US Food and Drug Administration (FDA) approved nirsevimab, an RSV preventive monoclonal antibody, for use in neonates and infants born during or entering their first RSV season and in children up to 24 months of age who remain vulnerable to RSV during their second season.1 The Advisory Committee on Immunization Practices (ACIP) subsequently made 2 recommendations regarding use of nirsevimab, which I’ll detail in a moment.2
First, a word about RSV. The Centers for Disease Control and Prevention estimates that each year in children younger than 5 years, RSV is responsible for 1.5 million outpatient clinic visits, 520,000 emergency department visits, 58,000 to 80,000 hospitalizations, and 100 to 200 deaths.2 The risk for hospitalization from RSV is highest in the second and third months of life and decreases with increasing age.
There are some racial disparities in RSV severity, likely reflecting social drivers of health: ICU admission rates are 1.2 to 1.6 times higher among non-Hispanic Black infants younger than 6 months than among non-Hispanic White infants, and hospitalization rates are up to 5 times higher in American Indian and Alaska Native populations.2
What nirsevimab adds to the toolbox. Until recently, there was only 1 RSV preventive agent available: palivizumab, also a monoclonal antibody. The American Academy of Pediatrics has recommended palivizumab be used only for infants at high risk for RSV infection, due to its high cost and the need for monthly injections for the duration of an RSV season. In addition, the Academy has noted that palivizumab has limited effect on RSV hospitalizations on a population basis and does not appear to affect mortality.3
Nirsevimab has a longer half-life than palivizumab, and only 1 injection is needed for the RSV season. Early studies on nirsevimab demonstrate 79% effectiveness in preventing medical-attended lower respiratory tract infection, 80.6% effectiveness in preventing hospitalization, and 90% effectiveness in preventing ICU admission. The number needed to immunize with nirsevimab to prevent an outpatient visit is estimated to be 17; to prevent an ED visit, 48; and to prevent an inpatient admission, 128. Due to the low RSV death rate, the studies were not able to demonstrate reduced mortality.2
What the ACIP recommends. At a special meeting in July, the ACIP recommended 1 dose of nirsevimab for2:
- all infants younger than 8 months who are born during or entering their first RSV season
- children ages 8 to 19 months who are at increased risk for severe RSV disease and entering their second RSV season.
Those at risk include children with chronic lung disease of prematurity who required medical support any time during the 6-month period before the start of their second RSV season; those with severe immunocompromise; those with cystic fibrosis who have manifestations of severe lung disease or weight-for-length < 10th percentile; and American Indian and Alaska Native children.2
How to administer nirsevimab. The dose of nirsevimab is 50 mg IM for those weighing < 5 kg, 100 mg for those weighing ≥ 5 kg, and 200 mg for high-risk children entering their second RSV season.2 Nirsevimab can be co-administered with other recommended vaccines; however, both nirsevimab and palivizumab should not be used in the same child in the same RSV season.
Nirsevimab should be administered in the first week of life for infants born shortly before or during RSV season, and shortly before the season for infants younger than 8 months and those ages 8 to 19 months who are at high risk.4 The months of highest RSV transmission in most locations are December through February, but this can vary. Local epidemiology and advice from state and local health departments are the best source of information about when RSV season starts and ends in your area.
On the horizon. Nirsevimab will be included in the Vaccines for Children program and covered by commercial health plans with no cost sharing.5 A maternal vaccine to prevent RSV in newborns is likely to be approved by the FDA in the near future.
1. FDA. FDA approves new drug to prevent RSV in babies and toddlers [press release]. Published July 17, 2023. Accessed August 29, 2023. www.fda.gov/news-events/press-announcements/fda-approves-new-drug-prevent-rsv-babies-and-toddlers
2. Jones J. Evidence to recommendation framework: nirsevimab updates. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131586
3. American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014;134:e620–e638. doi: 10.1542/peds.2014-1666
4. Jones J. Proposed clinical consideration updates for nirsevimab. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2023-08-3/04-rsv-jones-508.pdf
5. Peacock G. Nirsevimab: implementation considerations. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131587
1. FDA. FDA approves new drug to prevent RSV in babies and toddlers [press release]. Published July 17, 2023. Accessed August 29, 2023. www.fda.gov/news-events/press-announcements/fda-approves-new-drug-prevent-rsv-babies-and-toddlers
2. Jones J. Evidence to recommendation framework: nirsevimab updates. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131586
3. American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014;134:e620–e638. doi: 10.1542/peds.2014-1666
4. Jones J. Proposed clinical consideration updates for nirsevimab. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2023-08-3/04-rsv-jones-508.pdf
5. Peacock G. Nirsevimab: implementation considerations. Presented to the ACIP on August 3, 2023. Accessed August 23, 2023. https://stacks.cdc.gov/view/cdc/131587
