User login
What’s Eating You? Tropical Rat Mite (Ornithonyssus bacoti)
The tropical rat mite (Ornithonyssus bacoti) belongs to the family Macronyssidae. Theses mites are commonly mistaken for red bird mites or Nordic bird mites because they belong to the same family and have similar characteristics.1 Although O bacoti is called the tropical rat mite, it also can be found in moderate climates.2,3
Characteristics
The life cycle of a tropical rat mite lasts 11 to 13 days and includes 5 stages: egg, larva, protonymph, deutonymph, and adult.1,2 The length of the mite (0.3–0.7 mm) varies with the stage of development.1 Adults can reach 0.75 to 1.40 mm, with females larger than males and possibly visible with the naked eye.1,2
Two or 3 days after a blood meal, the female mite lays approximately 100 eggs in its nest but not on the surface of a host. The eggs hatch into larvae after 1 to 4 days and go on to complete their life cyle.1 During developmental stages, mites occupy their hosts for blood meals. Mites search for their hosts at night and prefer wild or pet rodents for blood meals but are not host specific and can be found on many mammals including birds, cats, racoons, and squirrels.4
Although tropical rat mites prefer rodent hosts, they can infest humans when their preferred host is unavailable. In the United States, the first case of human dermatitis due to a tropical rate mite occurred in 1923. In Europe, rat mite dermatitis was first reported in a human in 1931, possibly due to contamination of sailing vessels.4
Infestation and Transmission
Tropical rat mites prefer wild and pet rodents as hosts because the mites are able to feed on their blood over long periods.4 During the day, the mite spends most of its time hiding in dark dry spaces; it is most active during the night, traveling to find a host for meals.3-5 If a preferred host is not present, the mite may choose to infest a human.5
Human infestation occurs most often upon close bodily contact with an infected animal or pet rodent that was sold without parasites having been eliminated.3-5 Mites are able to survive without a host for as long as 6 months; they may travel after a meal.1,2 Therefore, individuals who do not have a pet rodent can be infested if an infected wild rodent has infested their living space.1,3-5
Clinical Presentation of Infestation
Patients infested with tropical rat mites present with pruritic cutaneous lesions, most often on unclothed parts of the body that are easily exposed to mites; lesions rarely occur on the scalp.5 People of any age or gender can be infested. Rat mite bites can present as single or grouped, pruritic, erythematous papules ranging in size from 4 to 10 mm in diameter.5-7 Excoriations may be present due to excessive scratching. Although rare, vesicles or nodules have been reported.5,7
Diagnosis of the underlying cause of the cutaneous manifestations often is difficult because mites are not visible during the day, as they are less active then.2 Lesions often are misdiagnosed as an allergic response, a bacterial infection, or various forms of dermatitis.1 A parasitic cause often is not considered unless the physician or patient detects a mite or many trials of therapies fail to provide relief.1,3-5 Eliciting a thorough history may disclose that the patient has had close contact with rodents or lives in a community center, shelter, or shared space. If any of the patient’s close contacts have a similar presentation, infestation with mites should be considered.
Treatment and Prevention
Patients should be educated about treatment options and measures that need to be taken to prevent reinfection. It has been reported that tropical rat mites can survive without a blood meal for as long as 6 months; therefore, meticulous inspection and decontamination of all living spaces is required.1,4 Once identified, physicians may prescribe an antiparasitic such as permethrin or pyriproxyfen to prevent further infestation and eliminate mites on the host.5 Lindane and benzyl benzoate previously were reported to be effective but should be prescribed only in correctly diagnosed cases due to the potential adverse effects of both therapies.4,7-10 For effective treatment, physicians should thoroughly review the proper application of topical treatments with patients. Topical creams should be massaged into the skin from the head to the soles of the feet, covering all creases of the skin and between the fingers and toes. Antiparasitic creams should be left on the skin for 8 to 14 hours, and all members of the household should be examined and treated, if necessary, by a physician. A thorough bath removes tropical rat mites, but preventive measures should be taken to prevent reinfestation.4 Antihistamines or glucocorticoids also can be used as symptomatic treatment.6,8
Avoiding Reinfestation—Preventive measures should be taken to prevent reinfestation, including evaluation by an exterminator for any wild rodents to remove nests and treat the living space with an acaricide.5 Insecticides administered by exterminators, including malathion, methyl carbamate, and lindane, also have been reported to be effective for preventing reinfestation.5,7-9 A veterinarian should be consulted if the patient owns any pets to ensure proper identification of any potential tropical rat mites and treatments that may be necessary for any household pets.1
Case Report
A 68-year-old man presented to the dermatology outpatient clinic with diffuse pruritus of the skin and scalp. He reported no other symptoms and had never had a total-body skin examination. His primary care physician recently prescribed a dose pack of methylprednisolone 4-mg tablets, which relieved the symptoms except for a mild scalp itch. His wife did not experience itching, and he denied noticing mites or fleas on his pet dog. Physical examination did not reveal any contributory findings, such as erythema or rash. Ketoconazole shampoo 2% and fluocinolone solution 0.01% were prescribed for scalp pruritus; however, he could not afford those medications and therefore did not take them.
Two weeks later, the patient presented with diffuse itching that involved the scalp, trunk, and extremities. He denied groin pruritus. He reported that the itching was worse at night. His wife continued to be asymptomatic. The patient reported that his health screening was up-to-date, and he had no interval health changes. A complete blood cell count, thyroid studies, and a comprehensive metabolic panel performed recently were within reference range. He denied recent travel or taking new medications. Physical examination revealed a somewhat linear distribution of erythematous urticarial papules on the right side of the abdomen. Red dermatographic excoriations were noted on the back. No burrows were visualized. He was given intramuscular triamcinolone 60 mg and was advised to have his house evaluated for bed bugs and his pet dog evaluated by a veterinarian for mites. During the triamcinolone injection, the medical assistant observed a 1- to 2-mm red insect, which fell into his clothing and could not be further evaluated.
After 1 month, the patient had no improvement of the pruritus; instead, it became worse. During this time, his wife developed intermittent urticarial-like eruptions. He was taking oral diphenhydramine nightly and applying triamcinolone cream 0.5% that he had at home from an earlier skin problem as needed. Physical examination findings correlated with worsening symptoms. He had multiple erythematous urticarial papules—many of which were excoriated—across the chest, abdomen, buttocks, and back. The arms had multiple excoriations. The urticarial papules coalesced in the anterior axillary folds, yet no burrows were visualized. In the left anterior axillary fold adjacent to one of the urticarial papules, a 1-mm mobile mite was identified on dermoscopy. Further evaluation by microscopy showed morphologic characteristics of a tropical rat mite (Figure). The patient admitted that his house had a mouse infestation that he was struggling to eliminate. Permethrin cream 5% was prescribed. Because the patient could not afford the prescription, he was advised to use the triamcinolone cream 0.5%and oral diphenhydramine that he had at home nightly for symptomatic relief. He was advised to hire an exterminator to eradicate the mouse and mite infestation to prevent reinfestation.
 under microscopy")
Identification of Rate Mite Dermatitis
The characteristics of tropical rat mite dermatitis can be confused with many other conditions, such as infection. Even when a mite is identified, it can be difficult to classify it as a tropical rat mite. To confirm the diagnosis of tropical rat mite dermatitis, the parasite needs to be identified. Skin scrapings can be collected from pruritic lesions and examined microscopically in the hope of revealing the rat mites. The recommendation is to collect skin scrapings from the dorsal aspect of the hands or from the neck.5 Patients may report finding mites in their living space or on their bedding or clothing.
Although the tropical rat mite was reported as a vector for endemic typhus between humans, no other cases of transmission between humans have been reported since.11,12 Studies reporting non–human subject research and case reports have shown that O bacoti is a vector for Rickettsia akari, Coxiella burnetii, Francisella tularensis, Yersinia pestis, Eastern equine encephalitis virus (Alphavirus), Enterovirus (Picornaviridae), Langat virus (Flavivirus), and Hantaan orthohantavirus.5,11-17 However, no cases of these infectious diseases being transmitted naturally have been reported.5
Confirmation of O bacoti as a vector for human pathogens is difficult because it relies on identification of the mite in the clinic.5 The epidemiologic importance of the mite in transmitting infectious disease is unknown; reports of human cases of mite infestation are rare. We present this information to increase awareness and help dermatologists and other health care providers identify O bacoti.
- Beck W, Fölster-Holst R. Tropical rat mites (Ornithonyssus bacoti)—serious ectoparasites. J Dtsch Dermatol Ges. 2009;7:667-670. doi:10.1111/j.1610-0387.2009.07140.x
- Baumstark J, Beck W, Hofmann H. Outbreak of tropical rat mite (Ornithonyssus bacoti) dermatitis in a home for disabled persons. Dermatology. 2007;215:66-68. doi:10.1159/000102037
- Beck W. Occurrence of a house-infesting tropical rat mite (Ornithonyssus bacoti) on murides and human beings. Travel Med Infect Dis. 2008;6:245-249. doi:10.1016/j.tmaid.2008.01.002
- Beck W. Tropical rat mites as newly emerging disease pathogens in rodents and man. Trav Med Infect Dis. 2007;5:403. doi:10.1016/j.tmaid.2007.09.016
- Engel PM, Welzel J, Maass M, et al. Tropical rat mite dermatitis: case report and review. Clin Infect Dis. 1998;27:1465-1469. doi:10.1086/515016
- Hetherington GW, Holder WR, Smith EB. Rat mite dermatitis. JAMA. 1971;215:1499-1500.
- Fox JG. Outbreak of tropical rat mite dermatitis in laboratory personnel. Arch Dermatol. 1982;118:676-678. doi:10.1001/archderm.1982.01650210056019
- Fishman HC. Rat mite dermatitis. Cutis. 1988;42:414-416.
- Ram SM, Satija KC, Kaushik RK. Ornithonyssus bacoti infestation in laboratory personnel and veterinary students. Int J Zoonoses. 1986;13:138-140.
- Brown S, Becher J, Brady W. Treatment of ectoparasitic infections: review of the English-language literature, 1982-1992. Clin Infect Dis. 1995;20(suppl 1):S104-S109. doi:10.1093/clinids/20.supplement_1.s104
- Reeves WK, Loftis AD, Szumlas DE, et al. Rickettsial pathogens in the tropical rat mite Ornithonyssus bacoti (Acari: Macronyssidae) from Egyptian rats (Rattus spp.). Exp Appl Acarol. 2007;41:101-107. doi:10.1007/s10493-006-9040-3
- Philip CB, Hughes LE. The tropical rat mite; Liponyssus bacoti, as an experimental vector of rickettsialpox. Am J Trop Med Hyg. 1948;28:697-705. doi:10.4269/ajtmh.1948.s1-28.697
- Zemskaia AA, Pchelkina AA. Experimental infection of ticks Dermanyssus gallinae Redi Bdellonyssus bacoti Hirst with Q fever. Dokl Akad Nauk SSSR. 1955;101:391-392.
- Hopla CE. Experimental transmission of tularemia by the tropical rat mite. Am J Trop Med Hyg. 1951;31:768-783. doi:10.4269/ajtmh.1951.s1-31.768
- Clark GM, Lutz AE, Fadnessl. Observations on the ability of Haemogamasus liponyssoides Ewing and Ornithonyssus bacoti (Hirst) (Acarina, Gamasina) to retain eastern equine encephalitis virus: preliminary report. Am J Trop Med Hyg. 1966;15:107-112. doi:10.4269/ajtmh.1966.15.107
- Schwab M, Allen R, Sulkin SE. The tropical rat mite (Liponyssus bacoti) as an experimental vector of Coxsackie virus. Am J Trop Med Hyg. 1952;1:982-986. doi:10.4269/ajtmh.1952.1.982
- Durden LA, Turell MJ. Inefficient mechanical transmission of Langat (tick-borne encephalitis virus complex) virus by blood-feeding mites (Acari) to laboratory mice. J Med Entomol. 1993;30:639-641. doi:10.1093/jmedent/30.3.639
The tropical rat mite (Ornithonyssus bacoti) belongs to the family Macronyssidae. Theses mites are commonly mistaken for red bird mites or Nordic bird mites because they belong to the same family and have similar characteristics.1 Although O bacoti is called the tropical rat mite, it also can be found in moderate climates.2,3
Characteristics
The life cycle of a tropical rat mite lasts 11 to 13 days and includes 5 stages: egg, larva, protonymph, deutonymph, and adult.1,2 The length of the mite (0.3–0.7 mm) varies with the stage of development.1 Adults can reach 0.75 to 1.40 mm, with females larger than males and possibly visible with the naked eye.1,2
Two or 3 days after a blood meal, the female mite lays approximately 100 eggs in its nest but not on the surface of a host. The eggs hatch into larvae after 1 to 4 days and go on to complete their life cyle.1 During developmental stages, mites occupy their hosts for blood meals. Mites search for their hosts at night and prefer wild or pet rodents for blood meals but are not host specific and can be found on many mammals including birds, cats, racoons, and squirrels.4
Although tropical rat mites prefer rodent hosts, they can infest humans when their preferred host is unavailable. In the United States, the first case of human dermatitis due to a tropical rate mite occurred in 1923. In Europe, rat mite dermatitis was first reported in a human in 1931, possibly due to contamination of sailing vessels.4
Infestation and Transmission
Tropical rat mites prefer wild and pet rodents as hosts because the mites are able to feed on their blood over long periods.4 During the day, the mite spends most of its time hiding in dark dry spaces; it is most active during the night, traveling to find a host for meals.3-5 If a preferred host is not present, the mite may choose to infest a human.5
Human infestation occurs most often upon close bodily contact with an infected animal or pet rodent that was sold without parasites having been eliminated.3-5 Mites are able to survive without a host for as long as 6 months; they may travel after a meal.1,2 Therefore, individuals who do not have a pet rodent can be infested if an infected wild rodent has infested their living space.1,3-5
Clinical Presentation of Infestation
Patients infested with tropical rat mites present with pruritic cutaneous lesions, most often on unclothed parts of the body that are easily exposed to mites; lesions rarely occur on the scalp.5 People of any age or gender can be infested. Rat mite bites can present as single or grouped, pruritic, erythematous papules ranging in size from 4 to 10 mm in diameter.5-7 Excoriations may be present due to excessive scratching. Although rare, vesicles or nodules have been reported.5,7
Diagnosis of the underlying cause of the cutaneous manifestations often is difficult because mites are not visible during the day, as they are less active then.2 Lesions often are misdiagnosed as an allergic response, a bacterial infection, or various forms of dermatitis.1 A parasitic cause often is not considered unless the physician or patient detects a mite or many trials of therapies fail to provide relief.1,3-5 Eliciting a thorough history may disclose that the patient has had close contact with rodents or lives in a community center, shelter, or shared space. If any of the patient’s close contacts have a similar presentation, infestation with mites should be considered.
Treatment and Prevention
Patients should be educated about treatment options and measures that need to be taken to prevent reinfection. It has been reported that tropical rat mites can survive without a blood meal for as long as 6 months; therefore, meticulous inspection and decontamination of all living spaces is required.1,4 Once identified, physicians may prescribe an antiparasitic such as permethrin or pyriproxyfen to prevent further infestation and eliminate mites on the host.5 Lindane and benzyl benzoate previously were reported to be effective but should be prescribed only in correctly diagnosed cases due to the potential adverse effects of both therapies.4,7-10 For effective treatment, physicians should thoroughly review the proper application of topical treatments with patients. Topical creams should be massaged into the skin from the head to the soles of the feet, covering all creases of the skin and between the fingers and toes. Antiparasitic creams should be left on the skin for 8 to 14 hours, and all members of the household should be examined and treated, if necessary, by a physician. A thorough bath removes tropical rat mites, but preventive measures should be taken to prevent reinfestation.4 Antihistamines or glucocorticoids also can be used as symptomatic treatment.6,8
Avoiding Reinfestation—Preventive measures should be taken to prevent reinfestation, including evaluation by an exterminator for any wild rodents to remove nests and treat the living space with an acaricide.5 Insecticides administered by exterminators, including malathion, methyl carbamate, and lindane, also have been reported to be effective for preventing reinfestation.5,7-9 A veterinarian should be consulted if the patient owns any pets to ensure proper identification of any potential tropical rat mites and treatments that may be necessary for any household pets.1
Case Report
A 68-year-old man presented to the dermatology outpatient clinic with diffuse pruritus of the skin and scalp. He reported no other symptoms and had never had a total-body skin examination. His primary care physician recently prescribed a dose pack of methylprednisolone 4-mg tablets, which relieved the symptoms except for a mild scalp itch. His wife did not experience itching, and he denied noticing mites or fleas on his pet dog. Physical examination did not reveal any contributory findings, such as erythema or rash. Ketoconazole shampoo 2% and fluocinolone solution 0.01% were prescribed for scalp pruritus; however, he could not afford those medications and therefore did not take them.
Two weeks later, the patient presented with diffuse itching that involved the scalp, trunk, and extremities. He denied groin pruritus. He reported that the itching was worse at night. His wife continued to be asymptomatic. The patient reported that his health screening was up-to-date, and he had no interval health changes. A complete blood cell count, thyroid studies, and a comprehensive metabolic panel performed recently were within reference range. He denied recent travel or taking new medications. Physical examination revealed a somewhat linear distribution of erythematous urticarial papules on the right side of the abdomen. Red dermatographic excoriations were noted on the back. No burrows were visualized. He was given intramuscular triamcinolone 60 mg and was advised to have his house evaluated for bed bugs and his pet dog evaluated by a veterinarian for mites. During the triamcinolone injection, the medical assistant observed a 1- to 2-mm red insect, which fell into his clothing and could not be further evaluated.
After 1 month, the patient had no improvement of the pruritus; instead, it became worse. During this time, his wife developed intermittent urticarial-like eruptions. He was taking oral diphenhydramine nightly and applying triamcinolone cream 0.5% that he had at home from an earlier skin problem as needed. Physical examination findings correlated with worsening symptoms. He had multiple erythematous urticarial papules—many of which were excoriated—across the chest, abdomen, buttocks, and back. The arms had multiple excoriations. The urticarial papules coalesced in the anterior axillary folds, yet no burrows were visualized. In the left anterior axillary fold adjacent to one of the urticarial papules, a 1-mm mobile mite was identified on dermoscopy. Further evaluation by microscopy showed morphologic characteristics of a tropical rat mite (Figure). The patient admitted that his house had a mouse infestation that he was struggling to eliminate. Permethrin cream 5% was prescribed. Because the patient could not afford the prescription, he was advised to use the triamcinolone cream 0.5%and oral diphenhydramine that he had at home nightly for symptomatic relief. He was advised to hire an exterminator to eradicate the mouse and mite infestation to prevent reinfestation.
Identification of Rate Mite Dermatitis
The characteristics of tropical rat mite dermatitis can be confused with many other conditions, such as infection. Even when a mite is identified, it can be difficult to classify it as a tropical rat mite. To confirm the diagnosis of tropical rat mite dermatitis, the parasite needs to be identified. Skin scrapings can be collected from pruritic lesions and examined microscopically in the hope of revealing the rat mites. The recommendation is to collect skin scrapings from the dorsal aspect of the hands or from the neck.5 Patients may report finding mites in their living space or on their bedding or clothing.
Although the tropical rat mite was reported as a vector for endemic typhus between humans, no other cases of transmission between humans have been reported since.11,12 Studies reporting non–human subject research and case reports have shown that O bacoti is a vector for Rickettsia akari, Coxiella burnetii, Francisella tularensis, Yersinia pestis, Eastern equine encephalitis virus (Alphavirus), Enterovirus (Picornaviridae), Langat virus (Flavivirus), and Hantaan orthohantavirus.5,11-17 However, no cases of these infectious diseases being transmitted naturally have been reported.5
Confirmation of O bacoti as a vector for human pathogens is difficult because it relies on identification of the mite in the clinic.5 The epidemiologic importance of the mite in transmitting infectious disease is unknown; reports of human cases of mite infestation are rare. We present this information to increase awareness and help dermatologists and other health care providers identify O bacoti.
The tropical rat mite (Ornithonyssus bacoti) belongs to the family Macronyssidae. Theses mites are commonly mistaken for red bird mites or Nordic bird mites because they belong to the same family and have similar characteristics.1 Although O bacoti is called the tropical rat mite, it also can be found in moderate climates.2,3
Characteristics
The life cycle of a tropical rat mite lasts 11 to 13 days and includes 5 stages: egg, larva, protonymph, deutonymph, and adult.1,2 The length of the mite (0.3–0.7 mm) varies with the stage of development.1 Adults can reach 0.75 to 1.40 mm, with females larger than males and possibly visible with the naked eye.1,2
Two or 3 days after a blood meal, the female mite lays approximately 100 eggs in its nest but not on the surface of a host. The eggs hatch into larvae after 1 to 4 days and go on to complete their life cyle.1 During developmental stages, mites occupy their hosts for blood meals. Mites search for their hosts at night and prefer wild or pet rodents for blood meals but are not host specific and can be found on many mammals including birds, cats, racoons, and squirrels.4
Although tropical rat mites prefer rodent hosts, they can infest humans when their preferred host is unavailable. In the United States, the first case of human dermatitis due to a tropical rate mite occurred in 1923. In Europe, rat mite dermatitis was first reported in a human in 1931, possibly due to contamination of sailing vessels.4
Infestation and Transmission
Tropical rat mites prefer wild and pet rodents as hosts because the mites are able to feed on their blood over long periods.4 During the day, the mite spends most of its time hiding in dark dry spaces; it is most active during the night, traveling to find a host for meals.3-5 If a preferred host is not present, the mite may choose to infest a human.5
Human infestation occurs most often upon close bodily contact with an infected animal or pet rodent that was sold without parasites having been eliminated.3-5 Mites are able to survive without a host for as long as 6 months; they may travel after a meal.1,2 Therefore, individuals who do not have a pet rodent can be infested if an infected wild rodent has infested their living space.1,3-5
Clinical Presentation of Infestation
Patients infested with tropical rat mites present with pruritic cutaneous lesions, most often on unclothed parts of the body that are easily exposed to mites; lesions rarely occur on the scalp.5 People of any age or gender can be infested. Rat mite bites can present as single or grouped, pruritic, erythematous papules ranging in size from 4 to 10 mm in diameter.5-7 Excoriations may be present due to excessive scratching. Although rare, vesicles or nodules have been reported.5,7
Diagnosis of the underlying cause of the cutaneous manifestations often is difficult because mites are not visible during the day, as they are less active then.2 Lesions often are misdiagnosed as an allergic response, a bacterial infection, or various forms of dermatitis.1 A parasitic cause often is not considered unless the physician or patient detects a mite or many trials of therapies fail to provide relief.1,3-5 Eliciting a thorough history may disclose that the patient has had close contact with rodents or lives in a community center, shelter, or shared space. If any of the patient’s close contacts have a similar presentation, infestation with mites should be considered.
Treatment and Prevention
Patients should be educated about treatment options and measures that need to be taken to prevent reinfection. It has been reported that tropical rat mites can survive without a blood meal for as long as 6 months; therefore, meticulous inspection and decontamination of all living spaces is required.1,4 Once identified, physicians may prescribe an antiparasitic such as permethrin or pyriproxyfen to prevent further infestation and eliminate mites on the host.5 Lindane and benzyl benzoate previously were reported to be effective but should be prescribed only in correctly diagnosed cases due to the potential adverse effects of both therapies.4,7-10 For effective treatment, physicians should thoroughly review the proper application of topical treatments with patients. Topical creams should be massaged into the skin from the head to the soles of the feet, covering all creases of the skin and between the fingers and toes. Antiparasitic creams should be left on the skin for 8 to 14 hours, and all members of the household should be examined and treated, if necessary, by a physician. A thorough bath removes tropical rat mites, but preventive measures should be taken to prevent reinfestation.4 Antihistamines or glucocorticoids also can be used as symptomatic treatment.6,8
Avoiding Reinfestation—Preventive measures should be taken to prevent reinfestation, including evaluation by an exterminator for any wild rodents to remove nests and treat the living space with an acaricide.5 Insecticides administered by exterminators, including malathion, methyl carbamate, and lindane, also have been reported to be effective for preventing reinfestation.5,7-9 A veterinarian should be consulted if the patient owns any pets to ensure proper identification of any potential tropical rat mites and treatments that may be necessary for any household pets.1
Case Report
A 68-year-old man presented to the dermatology outpatient clinic with diffuse pruritus of the skin and scalp. He reported no other symptoms and had never had a total-body skin examination. His primary care physician recently prescribed a dose pack of methylprednisolone 4-mg tablets, which relieved the symptoms except for a mild scalp itch. His wife did not experience itching, and he denied noticing mites or fleas on his pet dog. Physical examination did not reveal any contributory findings, such as erythema or rash. Ketoconazole shampoo 2% and fluocinolone solution 0.01% were prescribed for scalp pruritus; however, he could not afford those medications and therefore did not take them.
Two weeks later, the patient presented with diffuse itching that involved the scalp, trunk, and extremities. He denied groin pruritus. He reported that the itching was worse at night. His wife continued to be asymptomatic. The patient reported that his health screening was up-to-date, and he had no interval health changes. A complete blood cell count, thyroid studies, and a comprehensive metabolic panel performed recently were within reference range. He denied recent travel or taking new medications. Physical examination revealed a somewhat linear distribution of erythematous urticarial papules on the right side of the abdomen. Red dermatographic excoriations were noted on the back. No burrows were visualized. He was given intramuscular triamcinolone 60 mg and was advised to have his house evaluated for bed bugs and his pet dog evaluated by a veterinarian for mites. During the triamcinolone injection, the medical assistant observed a 1- to 2-mm red insect, which fell into his clothing and could not be further evaluated.
After 1 month, the patient had no improvement of the pruritus; instead, it became worse. During this time, his wife developed intermittent urticarial-like eruptions. He was taking oral diphenhydramine nightly and applying triamcinolone cream 0.5% that he had at home from an earlier skin problem as needed. Physical examination findings correlated with worsening symptoms. He had multiple erythematous urticarial papules—many of which were excoriated—across the chest, abdomen, buttocks, and back. The arms had multiple excoriations. The urticarial papules coalesced in the anterior axillary folds, yet no burrows were visualized. In the left anterior axillary fold adjacent to one of the urticarial papules, a 1-mm mobile mite was identified on dermoscopy. Further evaluation by microscopy showed morphologic characteristics of a tropical rat mite (Figure). The patient admitted that his house had a mouse infestation that he was struggling to eliminate. Permethrin cream 5% was prescribed. Because the patient could not afford the prescription, he was advised to use the triamcinolone cream 0.5%and oral diphenhydramine that he had at home nightly for symptomatic relief. He was advised to hire an exterminator to eradicate the mouse and mite infestation to prevent reinfestation.
Identification of Rate Mite Dermatitis
The characteristics of tropical rat mite dermatitis can be confused with many other conditions, such as infection. Even when a mite is identified, it can be difficult to classify it as a tropical rat mite. To confirm the diagnosis of tropical rat mite dermatitis, the parasite needs to be identified. Skin scrapings can be collected from pruritic lesions and examined microscopically in the hope of revealing the rat mites. The recommendation is to collect skin scrapings from the dorsal aspect of the hands or from the neck.5 Patients may report finding mites in their living space or on their bedding or clothing.
Although the tropical rat mite was reported as a vector for endemic typhus between humans, no other cases of transmission between humans have been reported since.11,12 Studies reporting non–human subject research and case reports have shown that O bacoti is a vector for Rickettsia akari, Coxiella burnetii, Francisella tularensis, Yersinia pestis, Eastern equine encephalitis virus (Alphavirus), Enterovirus (Picornaviridae), Langat virus (Flavivirus), and Hantaan orthohantavirus.5,11-17 However, no cases of these infectious diseases being transmitted naturally have been reported.5
Confirmation of O bacoti as a vector for human pathogens is difficult because it relies on identification of the mite in the clinic.5 The epidemiologic importance of the mite in transmitting infectious disease is unknown; reports of human cases of mite infestation are rare. We present this information to increase awareness and help dermatologists and other health care providers identify O bacoti.
- Beck W, Fölster-Holst R. Tropical rat mites (Ornithonyssus bacoti)—serious ectoparasites. J Dtsch Dermatol Ges. 2009;7:667-670. doi:10.1111/j.1610-0387.2009.07140.x
- Baumstark J, Beck W, Hofmann H. Outbreak of tropical rat mite (Ornithonyssus bacoti) dermatitis in a home for disabled persons. Dermatology. 2007;215:66-68. doi:10.1159/000102037
- Beck W. Occurrence of a house-infesting tropical rat mite (Ornithonyssus bacoti) on murides and human beings. Travel Med Infect Dis. 2008;6:245-249. doi:10.1016/j.tmaid.2008.01.002
- Beck W. Tropical rat mites as newly emerging disease pathogens in rodents and man. Trav Med Infect Dis. 2007;5:403. doi:10.1016/j.tmaid.2007.09.016
- Engel PM, Welzel J, Maass M, et al. Tropical rat mite dermatitis: case report and review. Clin Infect Dis. 1998;27:1465-1469. doi:10.1086/515016
- Hetherington GW, Holder WR, Smith EB. Rat mite dermatitis. JAMA. 1971;215:1499-1500.
- Fox JG. Outbreak of tropical rat mite dermatitis in laboratory personnel. Arch Dermatol. 1982;118:676-678. doi:10.1001/archderm.1982.01650210056019
- Fishman HC. Rat mite dermatitis. Cutis. 1988;42:414-416.
- Ram SM, Satija KC, Kaushik RK. Ornithonyssus bacoti infestation in laboratory personnel and veterinary students. Int J Zoonoses. 1986;13:138-140.
- Brown S, Becher J, Brady W. Treatment of ectoparasitic infections: review of the English-language literature, 1982-1992. Clin Infect Dis. 1995;20(suppl 1):S104-S109. doi:10.1093/clinids/20.supplement_1.s104
- Reeves WK, Loftis AD, Szumlas DE, et al. Rickettsial pathogens in the tropical rat mite Ornithonyssus bacoti (Acari: Macronyssidae) from Egyptian rats (Rattus spp.). Exp Appl Acarol. 2007;41:101-107. doi:10.1007/s10493-006-9040-3
- Philip CB, Hughes LE. The tropical rat mite; Liponyssus bacoti, as an experimental vector of rickettsialpox. Am J Trop Med Hyg. 1948;28:697-705. doi:10.4269/ajtmh.1948.s1-28.697
- Zemskaia AA, Pchelkina AA. Experimental infection of ticks Dermanyssus gallinae Redi Bdellonyssus bacoti Hirst with Q fever. Dokl Akad Nauk SSSR. 1955;101:391-392.
- Hopla CE. Experimental transmission of tularemia by the tropical rat mite. Am J Trop Med Hyg. 1951;31:768-783. doi:10.4269/ajtmh.1951.s1-31.768
- Clark GM, Lutz AE, Fadnessl. Observations on the ability of Haemogamasus liponyssoides Ewing and Ornithonyssus bacoti (Hirst) (Acarina, Gamasina) to retain eastern equine encephalitis virus: preliminary report. Am J Trop Med Hyg. 1966;15:107-112. doi:10.4269/ajtmh.1966.15.107
- Schwab M, Allen R, Sulkin SE. The tropical rat mite (Liponyssus bacoti) as an experimental vector of Coxsackie virus. Am J Trop Med Hyg. 1952;1:982-986. doi:10.4269/ajtmh.1952.1.982
- Durden LA, Turell MJ. Inefficient mechanical transmission of Langat (tick-borne encephalitis virus complex) virus by blood-feeding mites (Acari) to laboratory mice. J Med Entomol. 1993;30:639-641. doi:10.1093/jmedent/30.3.639
- Beck W, Fölster-Holst R. Tropical rat mites (Ornithonyssus bacoti)—serious ectoparasites. J Dtsch Dermatol Ges. 2009;7:667-670. doi:10.1111/j.1610-0387.2009.07140.x
- Baumstark J, Beck W, Hofmann H. Outbreak of tropical rat mite (Ornithonyssus bacoti) dermatitis in a home for disabled persons. Dermatology. 2007;215:66-68. doi:10.1159/000102037
- Beck W. Occurrence of a house-infesting tropical rat mite (Ornithonyssus bacoti) on murides and human beings. Travel Med Infect Dis. 2008;6:245-249. doi:10.1016/j.tmaid.2008.01.002
- Beck W. Tropical rat mites as newly emerging disease pathogens in rodents and man. Trav Med Infect Dis. 2007;5:403. doi:10.1016/j.tmaid.2007.09.016
- Engel PM, Welzel J, Maass M, et al. Tropical rat mite dermatitis: case report and review. Clin Infect Dis. 1998;27:1465-1469. doi:10.1086/515016
- Hetherington GW, Holder WR, Smith EB. Rat mite dermatitis. JAMA. 1971;215:1499-1500.
- Fox JG. Outbreak of tropical rat mite dermatitis in laboratory personnel. Arch Dermatol. 1982;118:676-678. doi:10.1001/archderm.1982.01650210056019
- Fishman HC. Rat mite dermatitis. Cutis. 1988;42:414-416.
- Ram SM, Satija KC, Kaushik RK. Ornithonyssus bacoti infestation in laboratory personnel and veterinary students. Int J Zoonoses. 1986;13:138-140.
- Brown S, Becher J, Brady W. Treatment of ectoparasitic infections: review of the English-language literature, 1982-1992. Clin Infect Dis. 1995;20(suppl 1):S104-S109. doi:10.1093/clinids/20.supplement_1.s104
- Reeves WK, Loftis AD, Szumlas DE, et al. Rickettsial pathogens in the tropical rat mite Ornithonyssus bacoti (Acari: Macronyssidae) from Egyptian rats (Rattus spp.). Exp Appl Acarol. 2007;41:101-107. doi:10.1007/s10493-006-9040-3
- Philip CB, Hughes LE. The tropical rat mite; Liponyssus bacoti, as an experimental vector of rickettsialpox. Am J Trop Med Hyg. 1948;28:697-705. doi:10.4269/ajtmh.1948.s1-28.697
- Zemskaia AA, Pchelkina AA. Experimental infection of ticks Dermanyssus gallinae Redi Bdellonyssus bacoti Hirst with Q fever. Dokl Akad Nauk SSSR. 1955;101:391-392.
- Hopla CE. Experimental transmission of tularemia by the tropical rat mite. Am J Trop Med Hyg. 1951;31:768-783. doi:10.4269/ajtmh.1951.s1-31.768
- Clark GM, Lutz AE, Fadnessl. Observations on the ability of Haemogamasus liponyssoides Ewing and Ornithonyssus bacoti (Hirst) (Acarina, Gamasina) to retain eastern equine encephalitis virus: preliminary report. Am J Trop Med Hyg. 1966;15:107-112. doi:10.4269/ajtmh.1966.15.107
- Schwab M, Allen R, Sulkin SE. The tropical rat mite (Liponyssus bacoti) as an experimental vector of Coxsackie virus. Am J Trop Med Hyg. 1952;1:982-986. doi:10.4269/ajtmh.1952.1.982
- Durden LA, Turell MJ. Inefficient mechanical transmission of Langat (tick-borne encephalitis virus complex) virus by blood-feeding mites (Acari) to laboratory mice. J Med Entomol. 1993;30:639-641. doi:10.1093/jmedent/30.3.639
Practice Points
- The tropical rat mite (Ornithonyssus bacoti) can infest humans who make bodily contact with a rodent, reside in living spaces infested with rodents, or own any pets.
- Patients infested with rat mites may present with pruritic, erythematous, cutaneous lesions with secondary excoriations that can be mistaken for an infection or dermatitis.
- The recommended treatment of rate mite infestation includes antiparasitic medications such as permethrin or pyriproxyfen. Preventive measures include proper disinfestation of living spaces.
 under microscopy")
Sarcoidosis in Post–9/11 Military Veterans
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.

Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18

When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).

An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.
Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18
When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).
An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.
Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18
When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).
An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
Practice Points
- Cutaneous sarcoidosis is the most common extrapulmonary manifestation of the disease.
- Cutaneous sarcoidosis can precede systemic manifestations of the disease and should prompt further workup.
- Sarcoidosis is a presumptive diagnosis under the PACT Act and may be a service-connected condition. Veterans with presumptive exposures should be referred to the US Department of Veterans Affairs.

Treatments for Hidradenitis Suppurativa Comorbidities Help With Pain Management
Hidradenitis suppurativa (HS) has an unpredictable disease course and poses substantial therapeutic challenges. It carries an increased risk for adverse cardiovascular outcomes and all-cause mortality. It also is associated with comorbidities including mood disorders, tobacco smoking, obesity, diabetes mellitus, sleep disorders, sexual dysfunction, and autoimmune diseases, which can complicate its management and considerably affect patients’ quality of life (QOL).1 Hidradenitis suppurativa also disproportionately affects minority groups and has far-reaching inequities; for example, the condition has a notable economic impact on patients, including higher unemployment and disability rates, lower-paying jobs, less paid time off, and other indirect costs.2,3 Race can impact how pain itself is treated. In one study (N = 217), Black patients with extremity fractures presenting to anemergency department were significantly less likely to receive analgesia compared to White patients despite reporting similar pain (57% vs 74%, respectively; P = .01).4 In another study, Hispanic patients were 7-times less likely to be treated with opioids compared to non-Hispanic patients with long-bone fractures.5 Herein, we highlight pain management disparities in HS patients.
Treating HS Comorbidities Helps Improve Pain
Pain is reported by almost all HS patients and is the symptom most associated with QOL impairment.6,7 Pain in HS is multifactorial, with other symptoms and comorbidities affecting its severity. Treatment of acute flares often is painful and procedural, including intralesional steroid injections or incision and drainage.8 Algorithms for addressing pain through the treatment of comorbidities also have been developed.6 Although there are few studies on the medications that treat related comorbidities in HS, there is evidence of their benefits in similar diseases; for example, treating depression in patients with irritable bowel disease (IBD) improved pain perception, cognitive function, and sexual dysfunction.9
Depression exacerbates pain, and higher levels of depression have been observed in severe HS.10,11 Additionally, more than 80% of individuals with HS report tobacco smoking.1 Nicotine not only increases pain sensitivity and decreases pain tolerance but also worsens neuropathic, nociceptive, and psychosocial pain, as well as mood disorders and sleep disturbances.12 Given the higher prevalence of depression and smoking in HS patients and the impact on pain, addressing these comorbidities is crucial. Additionally, poor sleep amplifies pain sensitivity and affects neurologic pain modulation.13 Chronic pain also is associated with obesity and sleep dysfunction.14
Treatments Targeting Pain and Comorbidities
Treatments that target comorbidities and other symptoms of HS also may improve pain. Bupropion is a well-studied antidepressant and first-line option to aid in smoking cessation. It provides acute and chronic pain relief associated with IBD and may perform similarly in patients with HS.15-18 Bupropion also demonstrated dose-dependent weight reduction in obese and overweight individuals.19,20 Additionally, varenicline is a first-line option to aid in smoking cessation and can be combined with bupropion to increase long-term efficacy.21,22
Other antidepressants may alleviate HS pain. The selective norepinephrine reuptake inhibitors duloxetine and venlafaxine are recommended for chronic pain in HS.6 Selective serotonin reuptake inhibitors such as citalopram, escitalopram, and paroxetine are inexpensive and widely available antidepressants. Citalopram is as efficacious as duloxetine for chronic pain with fewer side effects.23 Paroxetine has been shown to improve pain and pruritus, QOL, and depression in patients with IBD.24 Benefits such as improved weight and sexual dysfunction also have been reported.25
Metformin is well studied in Black patients, and greater glycemic response supports its efficacy for diabetes as well as HS, which disproportionately affects individuals with skin of color.26 Metformin also targets other comorbidities of HS, such as improving insulin resistance, polycystic ovary syndrome, acne vulgaris, weight loss, hyperlipidemia, cardiovascular risk, and neuropsychologic conditions.27 Growing evidence supports the use of metformin as a new agent in chronic pain management, specifically for patients with HS.28,29
Final Thoughts
Hidradenitis suppurativa is a complex medical condition seen disproportionately in minority groups. Understanding common comorbidities as well as the biases associated with pain management will allow providers to treat HS patients more effectively. Dermatologists who see many HS patients should become more familiar with treating these associated comorbidities to provide patient care that is more holistic and effective.
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Yang H, Mu F, et al. Impact of hidradenitis suppurativa on work loss, indirect costs and income. Br J Dermatol. 2019;181:147-154. doi:10.1111/bjd.17101
- Udechukwu NS, Fleischer AB. Higher risk of care for hidradenitis suppurativa in African American and non-Hispanic patients in the United States. J Natl Med Assoc. 2017;109:44-48. doi:10.1016/j.jnma.2016.09.002
- Todd KH, Deaton C, D’Adamo AP, et al. Ethnicity and analgesic practice. Ann Emerg Med. 2000;35:11-16. doi:10.1016/s0196-0644(00)70099-0
- Todd KH, Samaroo N, Hoffman JR. Ethnicity as a risk factor for inadequate emergency department analgesia. JAMA. 1993;269:1537-1539.
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Matusiak Ł, Szcze˛ch J, Kaaz K, et al. Clinical characteristics of pruritus and pain in patients with hidradenitis suppurativa. Acta Derm Venereol. 2018;98:191-194. doi:10.2340/00015555-2815
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. Gen Hosp Psychiatry. 1996;18:220-229. doi:10.1016/0163-8343(96)00036-9
- Phan K, Huo YR, Smith SD. Hidradenitis suppurativa and psychiatric comorbidities, suicides and substance abuse: systematic review and meta-analysis. Ann Transl Med. 2020;8:821. doi:10.21037/atm-20-1028
- Woo AK. Depression and anxiety in pain. Rev Pain. 2010;4:8-12. doi:10.1177/204946371000400103
- Iida H, Yamaguchi S, Goyagi T, et al. Consensus statement on smoking cessation in patients with pain. J Anesth. 2022;36:671-687. doi:10.1007/s00540-022-03097-w
- Krause AJ, Prather AA, Wager TD, et al. The pain of sleep loss: a brain characterization in humans. J Neurosci. 2019;39:2291-2300. doi:10.1523/JNEUROSCI.2408-18.2018
- Mundal I, Gråwe RW, Bjørngaard JH, et al. Prevalence and long-term predictors of persistent chronic widespread pain in the general population in an 11-year prospective study: the HUNT study. BMC Musculoskelet Disord. 2014;15:213. doi:10.1186/1471-2474-15-213
- Aubin H-J. Tolerability and safety of sustained-release bupropion in the management of smoking cessation. Drugs. 2002;(62 suppl 2):45-52. doi:10.2165/00003495-200262002-00005
- Shah TH, Moradimehr A. Bupropion for the treatment of neuropathic pain. Am J Hosp Palliat Care. 2010;27:333-336. doi:10.1177/1049909110361229
- Baune BT, Renger L. Pharmacological and non-pharmacological interventions to improve cognitive dysfunction and functional ability in clinical depression—a systematic review. Psychiatry Res. 2014;219:25-50. doi:10.1016/j.psychres.2014.05.013
- Walker PW, Cole JO, Gardner EA, et al. Improvement in fluoxetine-associated sexual dysfunction in patients switched to bupropion. J Clin Psychiatry. 1993;54:459-465.
- Sherman MM, Ungureanu S, Rey JA. Naltrexone/bupropion ER (contrave): newly approved treatment option for chronic weight management in obese adults. P T. 2016;41:164-172.
- Anderson JW, Greenway FL, Fujioka K, et al. Bupropion SR enhances weight loss: a 48-week double-blind, placebo-controlled trial. Obes Res. 2002;10:633-641. doi:10.1038/oby.2002.86
- Kalkhoran S, Benowitz NL, Rigotti NA. Prevention and treatment of tobacco use: JACC health promotion series. J Am Coll Cardiol. 2018;72:1030-1045. doi:10.1016/j.jacc.2018.06.036
- Singh D, Saadabadi A. Varenicline. StatPearls Publishing; 2023.
- Mazza M, Mazza O, Pazzaglia C, et al. Escitalopram 20 mg versus duloxetine 60 mg for the treatment of chronic low back pain. Expert Opin Pharmacother. 2010;11:1049-1052. doi:10.1517/14656561003730413
- Docherty MJ, Jones RCW, Wallace MS. Managing pain in inflammatory bowel disease. Gastroenterol Hepatol (N Y). 2011;7:592-601.
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. StatPearls Publishing; 2022.
- Williams LK, Padhukasahasram B, Ahmedani BK, et al. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab. 2014;99:3160-3168. doi:10.1210/jc.2014-1539
- Sharma S, Mathur DK, Paliwal V, et al. Efficacy of metformin in the treatment of acne in women with polycystic ovarian syndrome: a newer approach to acne therapy. J Clin Aesthet Dermatol. 2019;12:34-38.
- Scheinfeld N. Hidradenitis suppurativa: a practical review of possible medical treatments based on over 350 hidradenitis patients. Dermatol Online J. 2013;19:1. doi:10.5070/D35VW402NF
- Baeza-Flores GDC, Guzmán-Priego CG, Parra-Flores LI, et al. Metformin: a prospective alternative for the treatment of chronic pain. Front Pharmacol. 2020;11:558474. doi:10.3389/fphar.2020.558474
Hidradenitis suppurativa (HS) has an unpredictable disease course and poses substantial therapeutic challenges. It carries an increased risk for adverse cardiovascular outcomes and all-cause mortality. It also is associated with comorbidities including mood disorders, tobacco smoking, obesity, diabetes mellitus, sleep disorders, sexual dysfunction, and autoimmune diseases, which can complicate its management and considerably affect patients’ quality of life (QOL).1 Hidradenitis suppurativa also disproportionately affects minority groups and has far-reaching inequities; for example, the condition has a notable economic impact on patients, including higher unemployment and disability rates, lower-paying jobs, less paid time off, and other indirect costs.2,3 Race can impact how pain itself is treated. In one study (N = 217), Black patients with extremity fractures presenting to anemergency department were significantly less likely to receive analgesia compared to White patients despite reporting similar pain (57% vs 74%, respectively; P = .01).4 In another study, Hispanic patients were 7-times less likely to be treated with opioids compared to non-Hispanic patients with long-bone fractures.5 Herein, we highlight pain management disparities in HS patients.
Treating HS Comorbidities Helps Improve Pain
Pain is reported by almost all HS patients and is the symptom most associated with QOL impairment.6,7 Pain in HS is multifactorial, with other symptoms and comorbidities affecting its severity. Treatment of acute flares often is painful and procedural, including intralesional steroid injections or incision and drainage.8 Algorithms for addressing pain through the treatment of comorbidities also have been developed.6 Although there are few studies on the medications that treat related comorbidities in HS, there is evidence of their benefits in similar diseases; for example, treating depression in patients with irritable bowel disease (IBD) improved pain perception, cognitive function, and sexual dysfunction.9
Depression exacerbates pain, and higher levels of depression have been observed in severe HS.10,11 Additionally, more than 80% of individuals with HS report tobacco smoking.1 Nicotine not only increases pain sensitivity and decreases pain tolerance but also worsens neuropathic, nociceptive, and psychosocial pain, as well as mood disorders and sleep disturbances.12 Given the higher prevalence of depression and smoking in HS patients and the impact on pain, addressing these comorbidities is crucial. Additionally, poor sleep amplifies pain sensitivity and affects neurologic pain modulation.13 Chronic pain also is associated with obesity and sleep dysfunction.14
Treatments Targeting Pain and Comorbidities
Treatments that target comorbidities and other symptoms of HS also may improve pain. Bupropion is a well-studied antidepressant and first-line option to aid in smoking cessation. It provides acute and chronic pain relief associated with IBD and may perform similarly in patients with HS.15-18 Bupropion also demonstrated dose-dependent weight reduction in obese and overweight individuals.19,20 Additionally, varenicline is a first-line option to aid in smoking cessation and can be combined with bupropion to increase long-term efficacy.21,22
Other antidepressants may alleviate HS pain. The selective norepinephrine reuptake inhibitors duloxetine and venlafaxine are recommended for chronic pain in HS.6 Selective serotonin reuptake inhibitors such as citalopram, escitalopram, and paroxetine are inexpensive and widely available antidepressants. Citalopram is as efficacious as duloxetine for chronic pain with fewer side effects.23 Paroxetine has been shown to improve pain and pruritus, QOL, and depression in patients with IBD.24 Benefits such as improved weight and sexual dysfunction also have been reported.25
Metformin is well studied in Black patients, and greater glycemic response supports its efficacy for diabetes as well as HS, which disproportionately affects individuals with skin of color.26 Metformin also targets other comorbidities of HS, such as improving insulin resistance, polycystic ovary syndrome, acne vulgaris, weight loss, hyperlipidemia, cardiovascular risk, and neuropsychologic conditions.27 Growing evidence supports the use of metformin as a new agent in chronic pain management, specifically for patients with HS.28,29
Final Thoughts
Hidradenitis suppurativa is a complex medical condition seen disproportionately in minority groups. Understanding common comorbidities as well as the biases associated with pain management will allow providers to treat HS patients more effectively. Dermatologists who see many HS patients should become more familiar with treating these associated comorbidities to provide patient care that is more holistic and effective.
Hidradenitis suppurativa (HS) has an unpredictable disease course and poses substantial therapeutic challenges. It carries an increased risk for adverse cardiovascular outcomes and all-cause mortality. It also is associated with comorbidities including mood disorders, tobacco smoking, obesity, diabetes mellitus, sleep disorders, sexual dysfunction, and autoimmune diseases, which can complicate its management and considerably affect patients’ quality of life (QOL).1 Hidradenitis suppurativa also disproportionately affects minority groups and has far-reaching inequities; for example, the condition has a notable economic impact on patients, including higher unemployment and disability rates, lower-paying jobs, less paid time off, and other indirect costs.2,3 Race can impact how pain itself is treated. In one study (N = 217), Black patients with extremity fractures presenting to anemergency department were significantly less likely to receive analgesia compared to White patients despite reporting similar pain (57% vs 74%, respectively; P = .01).4 In another study, Hispanic patients were 7-times less likely to be treated with opioids compared to non-Hispanic patients with long-bone fractures.5 Herein, we highlight pain management disparities in HS patients.
Treating HS Comorbidities Helps Improve Pain
Pain is reported by almost all HS patients and is the symptom most associated with QOL impairment.6,7 Pain in HS is multifactorial, with other symptoms and comorbidities affecting its severity. Treatment of acute flares often is painful and procedural, including intralesional steroid injections or incision and drainage.8 Algorithms for addressing pain through the treatment of comorbidities also have been developed.6 Although there are few studies on the medications that treat related comorbidities in HS, there is evidence of their benefits in similar diseases; for example, treating depression in patients with irritable bowel disease (IBD) improved pain perception, cognitive function, and sexual dysfunction.9
Depression exacerbates pain, and higher levels of depression have been observed in severe HS.10,11 Additionally, more than 80% of individuals with HS report tobacco smoking.1 Nicotine not only increases pain sensitivity and decreases pain tolerance but also worsens neuropathic, nociceptive, and psychosocial pain, as well as mood disorders and sleep disturbances.12 Given the higher prevalence of depression and smoking in HS patients and the impact on pain, addressing these comorbidities is crucial. Additionally, poor sleep amplifies pain sensitivity and affects neurologic pain modulation.13 Chronic pain also is associated with obesity and sleep dysfunction.14
Treatments Targeting Pain and Comorbidities
Treatments that target comorbidities and other symptoms of HS also may improve pain. Bupropion is a well-studied antidepressant and first-line option to aid in smoking cessation. It provides acute and chronic pain relief associated with IBD and may perform similarly in patients with HS.15-18 Bupropion also demonstrated dose-dependent weight reduction in obese and overweight individuals.19,20 Additionally, varenicline is a first-line option to aid in smoking cessation and can be combined with bupropion to increase long-term efficacy.21,22
Other antidepressants may alleviate HS pain. The selective norepinephrine reuptake inhibitors duloxetine and venlafaxine are recommended for chronic pain in HS.6 Selective serotonin reuptake inhibitors such as citalopram, escitalopram, and paroxetine are inexpensive and widely available antidepressants. Citalopram is as efficacious as duloxetine for chronic pain with fewer side effects.23 Paroxetine has been shown to improve pain and pruritus, QOL, and depression in patients with IBD.24 Benefits such as improved weight and sexual dysfunction also have been reported.25
Metformin is well studied in Black patients, and greater glycemic response supports its efficacy for diabetes as well as HS, which disproportionately affects individuals with skin of color.26 Metformin also targets other comorbidities of HS, such as improving insulin resistance, polycystic ovary syndrome, acne vulgaris, weight loss, hyperlipidemia, cardiovascular risk, and neuropsychologic conditions.27 Growing evidence supports the use of metformin as a new agent in chronic pain management, specifically for patients with HS.28,29
Final Thoughts
Hidradenitis suppurativa is a complex medical condition seen disproportionately in minority groups. Understanding common comorbidities as well as the biases associated with pain management will allow providers to treat HS patients more effectively. Dermatologists who see many HS patients should become more familiar with treating these associated comorbidities to provide patient care that is more holistic and effective.
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Yang H, Mu F, et al. Impact of hidradenitis suppurativa on work loss, indirect costs and income. Br J Dermatol. 2019;181:147-154. doi:10.1111/bjd.17101
- Udechukwu NS, Fleischer AB. Higher risk of care for hidradenitis suppurativa in African American and non-Hispanic patients in the United States. J Natl Med Assoc. 2017;109:44-48. doi:10.1016/j.jnma.2016.09.002
- Todd KH, Deaton C, D’Adamo AP, et al. Ethnicity and analgesic practice. Ann Emerg Med. 2000;35:11-16. doi:10.1016/s0196-0644(00)70099-0
- Todd KH, Samaroo N, Hoffman JR. Ethnicity as a risk factor for inadequate emergency department analgesia. JAMA. 1993;269:1537-1539.
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Matusiak Ł, Szcze˛ch J, Kaaz K, et al. Clinical characteristics of pruritus and pain in patients with hidradenitis suppurativa. Acta Derm Venereol. 2018;98:191-194. doi:10.2340/00015555-2815
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. Gen Hosp Psychiatry. 1996;18:220-229. doi:10.1016/0163-8343(96)00036-9
- Phan K, Huo YR, Smith SD. Hidradenitis suppurativa and psychiatric comorbidities, suicides and substance abuse: systematic review and meta-analysis. Ann Transl Med. 2020;8:821. doi:10.21037/atm-20-1028
- Woo AK. Depression and anxiety in pain. Rev Pain. 2010;4:8-12. doi:10.1177/204946371000400103
- Iida H, Yamaguchi S, Goyagi T, et al. Consensus statement on smoking cessation in patients with pain. J Anesth. 2022;36:671-687. doi:10.1007/s00540-022-03097-w
- Krause AJ, Prather AA, Wager TD, et al. The pain of sleep loss: a brain characterization in humans. J Neurosci. 2019;39:2291-2300. doi:10.1523/JNEUROSCI.2408-18.2018
- Mundal I, Gråwe RW, Bjørngaard JH, et al. Prevalence and long-term predictors of persistent chronic widespread pain in the general population in an 11-year prospective study: the HUNT study. BMC Musculoskelet Disord. 2014;15:213. doi:10.1186/1471-2474-15-213
- Aubin H-J. Tolerability and safety of sustained-release bupropion in the management of smoking cessation. Drugs. 2002;(62 suppl 2):45-52. doi:10.2165/00003495-200262002-00005
- Shah TH, Moradimehr A. Bupropion for the treatment of neuropathic pain. Am J Hosp Palliat Care. 2010;27:333-336. doi:10.1177/1049909110361229
- Baune BT, Renger L. Pharmacological and non-pharmacological interventions to improve cognitive dysfunction and functional ability in clinical depression—a systematic review. Psychiatry Res. 2014;219:25-50. doi:10.1016/j.psychres.2014.05.013
- Walker PW, Cole JO, Gardner EA, et al. Improvement in fluoxetine-associated sexual dysfunction in patients switched to bupropion. J Clin Psychiatry. 1993;54:459-465.
- Sherman MM, Ungureanu S, Rey JA. Naltrexone/bupropion ER (contrave): newly approved treatment option for chronic weight management in obese adults. P T. 2016;41:164-172.
- Anderson JW, Greenway FL, Fujioka K, et al. Bupropion SR enhances weight loss: a 48-week double-blind, placebo-controlled trial. Obes Res. 2002;10:633-641. doi:10.1038/oby.2002.86
- Kalkhoran S, Benowitz NL, Rigotti NA. Prevention and treatment of tobacco use: JACC health promotion series. J Am Coll Cardiol. 2018;72:1030-1045. doi:10.1016/j.jacc.2018.06.036
- Singh D, Saadabadi A. Varenicline. StatPearls Publishing; 2023.
- Mazza M, Mazza O, Pazzaglia C, et al. Escitalopram 20 mg versus duloxetine 60 mg for the treatment of chronic low back pain. Expert Opin Pharmacother. 2010;11:1049-1052. doi:10.1517/14656561003730413
- Docherty MJ, Jones RCW, Wallace MS. Managing pain in inflammatory bowel disease. Gastroenterol Hepatol (N Y). 2011;7:592-601.
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. StatPearls Publishing; 2022.
- Williams LK, Padhukasahasram B, Ahmedani BK, et al. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab. 2014;99:3160-3168. doi:10.1210/jc.2014-1539
- Sharma S, Mathur DK, Paliwal V, et al. Efficacy of metformin in the treatment of acne in women with polycystic ovarian syndrome: a newer approach to acne therapy. J Clin Aesthet Dermatol. 2019;12:34-38.
- Scheinfeld N. Hidradenitis suppurativa: a practical review of possible medical treatments based on over 350 hidradenitis patients. Dermatol Online J. 2013;19:1. doi:10.5070/D35VW402NF
- Baeza-Flores GDC, Guzmán-Priego CG, Parra-Flores LI, et al. Metformin: a prospective alternative for the treatment of chronic pain. Front Pharmacol. 2020;11:558474. doi:10.3389/fphar.2020.558474
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Yang H, Mu F, et al. Impact of hidradenitis suppurativa on work loss, indirect costs and income. Br J Dermatol. 2019;181:147-154. doi:10.1111/bjd.17101
- Udechukwu NS, Fleischer AB. Higher risk of care for hidradenitis suppurativa in African American and non-Hispanic patients in the United States. J Natl Med Assoc. 2017;109:44-48. doi:10.1016/j.jnma.2016.09.002
- Todd KH, Deaton C, D’Adamo AP, et al. Ethnicity and analgesic practice. Ann Emerg Med. 2000;35:11-16. doi:10.1016/s0196-0644(00)70099-0
- Todd KH, Samaroo N, Hoffman JR. Ethnicity as a risk factor for inadequate emergency department analgesia. JAMA. 1993;269:1537-1539.
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Matusiak Ł, Szcze˛ch J, Kaaz K, et al. Clinical characteristics of pruritus and pain in patients with hidradenitis suppurativa. Acta Derm Venereol. 2018;98:191-194. doi:10.2340/00015555-2815
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. Gen Hosp Psychiatry. 1996;18:220-229. doi:10.1016/0163-8343(96)00036-9
- Phan K, Huo YR, Smith SD. Hidradenitis suppurativa and psychiatric comorbidities, suicides and substance abuse: systematic review and meta-analysis. Ann Transl Med. 2020;8:821. doi:10.21037/atm-20-1028
- Woo AK. Depression and anxiety in pain. Rev Pain. 2010;4:8-12. doi:10.1177/204946371000400103
- Iida H, Yamaguchi S, Goyagi T, et al. Consensus statement on smoking cessation in patients with pain. J Anesth. 2022;36:671-687. doi:10.1007/s00540-022-03097-w
- Krause AJ, Prather AA, Wager TD, et al. The pain of sleep loss: a brain characterization in humans. J Neurosci. 2019;39:2291-2300. doi:10.1523/JNEUROSCI.2408-18.2018
- Mundal I, Gråwe RW, Bjørngaard JH, et al. Prevalence and long-term predictors of persistent chronic widespread pain in the general population in an 11-year prospective study: the HUNT study. BMC Musculoskelet Disord. 2014;15:213. doi:10.1186/1471-2474-15-213
- Aubin H-J. Tolerability and safety of sustained-release bupropion in the management of smoking cessation. Drugs. 2002;(62 suppl 2):45-52. doi:10.2165/00003495-200262002-00005
- Shah TH, Moradimehr A. Bupropion for the treatment of neuropathic pain. Am J Hosp Palliat Care. 2010;27:333-336. doi:10.1177/1049909110361229
- Baune BT, Renger L. Pharmacological and non-pharmacological interventions to improve cognitive dysfunction and functional ability in clinical depression—a systematic review. Psychiatry Res. 2014;219:25-50. doi:10.1016/j.psychres.2014.05.013
- Walker PW, Cole JO, Gardner EA, et al. Improvement in fluoxetine-associated sexual dysfunction in patients switched to bupropion. J Clin Psychiatry. 1993;54:459-465.
- Sherman MM, Ungureanu S, Rey JA. Naltrexone/bupropion ER (contrave): newly approved treatment option for chronic weight management in obese adults. P T. 2016;41:164-172.
- Anderson JW, Greenway FL, Fujioka K, et al. Bupropion SR enhances weight loss: a 48-week double-blind, placebo-controlled trial. Obes Res. 2002;10:633-641. doi:10.1038/oby.2002.86
- Kalkhoran S, Benowitz NL, Rigotti NA. Prevention and treatment of tobacco use: JACC health promotion series. J Am Coll Cardiol. 2018;72:1030-1045. doi:10.1016/j.jacc.2018.06.036
- Singh D, Saadabadi A. Varenicline. StatPearls Publishing; 2023.
- Mazza M, Mazza O, Pazzaglia C, et al. Escitalopram 20 mg versus duloxetine 60 mg for the treatment of chronic low back pain. Expert Opin Pharmacother. 2010;11:1049-1052. doi:10.1517/14656561003730413
- Docherty MJ, Jones RCW, Wallace MS. Managing pain in inflammatory bowel disease. Gastroenterol Hepatol (N Y). 2011;7:592-601.
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. StatPearls Publishing; 2022.
- Williams LK, Padhukasahasram B, Ahmedani BK, et al. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab. 2014;99:3160-3168. doi:10.1210/jc.2014-1539
- Sharma S, Mathur DK, Paliwal V, et al. Efficacy of metformin in the treatment of acne in women with polycystic ovarian syndrome: a newer approach to acne therapy. J Clin Aesthet Dermatol. 2019;12:34-38.
- Scheinfeld N. Hidradenitis suppurativa: a practical review of possible medical treatments based on over 350 hidradenitis patients. Dermatol Online J. 2013;19:1. doi:10.5070/D35VW402NF
- Baeza-Flores GDC, Guzmán-Priego CG, Parra-Flores LI, et al. Metformin: a prospective alternative for the treatment of chronic pain. Front Pharmacol. 2020;11:558474. doi:10.3389/fphar.2020.558474

Cadaveric Split-Thickness Skin Graft With Partial Guiding Closure for Scalp Defects Extending to the Periosteum
Practice Gap
Scalp defects that extend to or below the periosteum often pose a reconstructive conundrum. Secondary-intention healing is challenging without an intact periosteum, and complex rotational flaps are required in these scenarios.1 For a tumor that is at high risk for recurrence or when adjuvant therapy is necessary, tissue distortion of flaps can make monitoring for recurrence difficult. Similarly, for patients in poor health or who are elderly and have substantial skin atrophy, extensive closure may be undesirable or more technically challenging with a higher risk for adverse events. In these scenarios, additional strategies are necessary to optimize wound healing and cosmesis. A cadaveric split-thickness skin graft (STSG) consisting of biologically active tissue can be used to expedite granulation.2

Technique
Following tumor clearance on the scalp (Figure 1), wide undermining is performed and 3-0 polyglactin 910 epidermal pulley sutures are placed to partially close the defect. A cadaveric STSG is placed over the remaining exposed periosteum and secured under the pulley sutures (Figure 2). The cadaveric STSG is replaced at 1-week intervals. At 4 weeks, sutures typically are removed. The cadaveric STSG is used until the exposed periosteum is fully granulated and the surgeon decides that granulation arrest is unlikely. The wound then heals by unassisted granulation. This approach provides an excellent final cosmetic outcome while avoiding extensive reconstruction (Figure 3).
 decrease the size of the defect and secure a cadaveric split-thickness skin graft over the remaining exposed periosteum.")
Practice Implications
Scalp defects requiring closure are common for dermatologic surgeons. Several techniques to promote tissue granulation in defects that involve exposed periosteum have been reported, including (1) creation of small holes with a scalpel or chisel to access cortical circulation and (2) using laser modalities to stimulate granulation (eg, an erbium:YAG or CO2 laser).3,4 Although direct comparative studies are needed, the cadaveric STSG provides an approach that increases tissue granulation but does not require more invasive techniques or equipment.

Autologous STSGs need a wound bed and can fail with an exposed periosteum. Furthermore, an autologous STSG that survives may leave an unsightly, hypopigmented, depressed defect. When a defect involves the periosteum and a primary closure or flap is not ideal, a skin substitute may be an option.
Skin substitutes, including cadaveric STSG, generally are classified as bioengineered skin equivalents, amniotic tissue, or cadaveric bioproducts (Table). Unlike autologous grafts, these skin substitutes can provide rapid coverage of the defect and do not require a highly vascularized wound bed.6 They also minimize the inflammatory response and potentially improve the final cosmetic outcome by improving granulation rather than immediate STSG closure creating a step-off in deep wounds.6
Cadaveric STSGs also have been used in nonhealing ulcerations; diabetic foot ulcers; and ulcerations in which muscle, tendon, or bone are exposed, demonstrating induction of wound healing with superior scar quality and skin function.2,7,8 The utility of the cadaveric STSG is further highlighted by its potential to reduce costs9 compared to bioengineered skin substitutes, though considerable variability exists in pricing (Table).

Consider using a cadaveric STSG with a guiding closure in cases in which there is concern for delayed or absent tissue granulation or when monitoring for recurrence is essential.
- Jibbe A, Tolkachjov SN. An efficient single-layer suture technique for large scalp flaps. J Am Acad Dermatol. 2020;83:E395-E396. doi:10.1016/j.jaad.2019.07.062
- Mosti G, Mattaliano V, Magliaro A, et al. Cadaveric skin grafts may greatly increase the healing rate of recalcitrant ulcers when used both alone and in combination with split-thickness skin grafts. Dermatol Surg. 2020;46:169-179. doi:10.1097/dss.0000000000001990
- Valesky EM, Vogl T, Kaufmann R, et al. Trepanation or complete removal of the outer table of the calvarium for granulation induction: the erbium:YAG laser as an alternative to the rose head burr. Dermatology. 2015;230:276-281. doi:10.1159/000368749
- Drosou A, Trieu D, Goldberg LH. Scalpel-made holes on exposed scalp bone to promote second intention healing. J Am Acad Dermatol. 2014;71:387-388. doi:10.1016/j.jaad.2014.04.020
- Centers for Medicare & Medicaid Services. April 2023 ASP Pricing. Accessed August 25, 2023. https://www.cms.gov/medicare/medicare-part-b-drug-average-sales-price/asp-pricing-files
- Shores JT, Gabriel A, Gupta S. Skin substitutes and alternatives: a review. Adv Skin Wound Care. 2007;20(9 Pt 1):493-508. doi:10.1097/01.ASW.0000288217.83128.f3
- Li X, Meng X, Wang X, et al. Human acellular dermal matrix allograft: a randomized, controlled human trial for the long-term evaluation of patients with extensive burns. Burns. 2015;41:689-699. doi:10.1016/j.burns.2014.12.007
- Juhasz I, Kiss B, Lukacs L, et al. Long-term followup of dermal substitution with acellular dermal implant in burns and postburn scar corrections. Dermatol Res Pract. 2010;2010:210150. doi:10.1155/2010/210150
- Towler MA, Rush EW, Richardson MK, et al. Randomized, prospective, blinded-enrollment, head-to-head venous leg ulcer healing trial comparing living, bioengineered skin graft substitute (Apligraf) with living, cryopreserved, human skin allograft (TheraSkin). Clin Podiatr Med Surg. 2018;35:357-365. doi:10.1016/j.cpm.2018.02.006
Practice Gap
Scalp defects that extend to or below the periosteum often pose a reconstructive conundrum. Secondary-intention healing is challenging without an intact periosteum, and complex rotational flaps are required in these scenarios.1 For a tumor that is at high risk for recurrence or when adjuvant therapy is necessary, tissue distortion of flaps can make monitoring for recurrence difficult. Similarly, for patients in poor health or who are elderly and have substantial skin atrophy, extensive closure may be undesirable or more technically challenging with a higher risk for adverse events. In these scenarios, additional strategies are necessary to optimize wound healing and cosmesis. A cadaveric split-thickness skin graft (STSG) consisting of biologically active tissue can be used to expedite granulation.2
Technique
Following tumor clearance on the scalp (Figure 1), wide undermining is performed and 3-0 polyglactin 910 epidermal pulley sutures are placed to partially close the defect. A cadaveric STSG is placed over the remaining exposed periosteum and secured under the pulley sutures (Figure 2). The cadaveric STSG is replaced at 1-week intervals. At 4 weeks, sutures typically are removed. The cadaveric STSG is used until the exposed periosteum is fully granulated and the surgeon decides that granulation arrest is unlikely. The wound then heals by unassisted granulation. This approach provides an excellent final cosmetic outcome while avoiding extensive reconstruction (Figure 3).
Practice Implications
Scalp defects requiring closure are common for dermatologic surgeons. Several techniques to promote tissue granulation in defects that involve exposed periosteum have been reported, including (1) creation of small holes with a scalpel or chisel to access cortical circulation and (2) using laser modalities to stimulate granulation (eg, an erbium:YAG or CO2 laser).3,4 Although direct comparative studies are needed, the cadaveric STSG provides an approach that increases tissue granulation but does not require more invasive techniques or equipment.
Autologous STSGs need a wound bed and can fail with an exposed periosteum. Furthermore, an autologous STSG that survives may leave an unsightly, hypopigmented, depressed defect. When a defect involves the periosteum and a primary closure or flap is not ideal, a skin substitute may be an option.
Skin substitutes, including cadaveric STSG, generally are classified as bioengineered skin equivalents, amniotic tissue, or cadaveric bioproducts (Table). Unlike autologous grafts, these skin substitutes can provide rapid coverage of the defect and do not require a highly vascularized wound bed.6 They also minimize the inflammatory response and potentially improve the final cosmetic outcome by improving granulation rather than immediate STSG closure creating a step-off in deep wounds.6
Cadaveric STSGs also have been used in nonhealing ulcerations; diabetic foot ulcers; and ulcerations in which muscle, tendon, or bone are exposed, demonstrating induction of wound healing with superior scar quality and skin function.2,7,8 The utility of the cadaveric STSG is further highlighted by its potential to reduce costs9 compared to bioengineered skin substitutes, though considerable variability exists in pricing (Table).
Consider using a cadaveric STSG with a guiding closure in cases in which there is concern for delayed or absent tissue granulation or when monitoring for recurrence is essential.
Practice Gap
Scalp defects that extend to or below the periosteum often pose a reconstructive conundrum. Secondary-intention healing is challenging without an intact periosteum, and complex rotational flaps are required in these scenarios.1 For a tumor that is at high risk for recurrence or when adjuvant therapy is necessary, tissue distortion of flaps can make monitoring for recurrence difficult. Similarly, for patients in poor health or who are elderly and have substantial skin atrophy, extensive closure may be undesirable or more technically challenging with a higher risk for adverse events. In these scenarios, additional strategies are necessary to optimize wound healing and cosmesis. A cadaveric split-thickness skin graft (STSG) consisting of biologically active tissue can be used to expedite granulation.2
Technique
Following tumor clearance on the scalp (Figure 1), wide undermining is performed and 3-0 polyglactin 910 epidermal pulley sutures are placed to partially close the defect. A cadaveric STSG is placed over the remaining exposed periosteum and secured under the pulley sutures (Figure 2). The cadaveric STSG is replaced at 1-week intervals. At 4 weeks, sutures typically are removed. The cadaveric STSG is used until the exposed periosteum is fully granulated and the surgeon decides that granulation arrest is unlikely. The wound then heals by unassisted granulation. This approach provides an excellent final cosmetic outcome while avoiding extensive reconstruction (Figure 3).
Practice Implications
Scalp defects requiring closure are common for dermatologic surgeons. Several techniques to promote tissue granulation in defects that involve exposed periosteum have been reported, including (1) creation of small holes with a scalpel or chisel to access cortical circulation and (2) using laser modalities to stimulate granulation (eg, an erbium:YAG or CO2 laser).3,4 Although direct comparative studies are needed, the cadaveric STSG provides an approach that increases tissue granulation but does not require more invasive techniques or equipment.
Autologous STSGs need a wound bed and can fail with an exposed periosteum. Furthermore, an autologous STSG that survives may leave an unsightly, hypopigmented, depressed defect. When a defect involves the periosteum and a primary closure or flap is not ideal, a skin substitute may be an option.
Skin substitutes, including cadaveric STSG, generally are classified as bioengineered skin equivalents, amniotic tissue, or cadaveric bioproducts (Table). Unlike autologous grafts, these skin substitutes can provide rapid coverage of the defect and do not require a highly vascularized wound bed.6 They also minimize the inflammatory response and potentially improve the final cosmetic outcome by improving granulation rather than immediate STSG closure creating a step-off in deep wounds.6
Cadaveric STSGs also have been used in nonhealing ulcerations; diabetic foot ulcers; and ulcerations in which muscle, tendon, or bone are exposed, demonstrating induction of wound healing with superior scar quality and skin function.2,7,8 The utility of the cadaveric STSG is further highlighted by its potential to reduce costs9 compared to bioengineered skin substitutes, though considerable variability exists in pricing (Table).
Consider using a cadaveric STSG with a guiding closure in cases in which there is concern for delayed or absent tissue granulation or when monitoring for recurrence is essential.
- Jibbe A, Tolkachjov SN. An efficient single-layer suture technique for large scalp flaps. J Am Acad Dermatol. 2020;83:E395-E396. doi:10.1016/j.jaad.2019.07.062
- Mosti G, Mattaliano V, Magliaro A, et al. Cadaveric skin grafts may greatly increase the healing rate of recalcitrant ulcers when used both alone and in combination with split-thickness skin grafts. Dermatol Surg. 2020;46:169-179. doi:10.1097/dss.0000000000001990
- Valesky EM, Vogl T, Kaufmann R, et al. Trepanation or complete removal of the outer table of the calvarium for granulation induction: the erbium:YAG laser as an alternative to the rose head burr. Dermatology. 2015;230:276-281. doi:10.1159/000368749
- Drosou A, Trieu D, Goldberg LH. Scalpel-made holes on exposed scalp bone to promote second intention healing. J Am Acad Dermatol. 2014;71:387-388. doi:10.1016/j.jaad.2014.04.020
- Centers for Medicare & Medicaid Services. April 2023 ASP Pricing. Accessed August 25, 2023. https://www.cms.gov/medicare/medicare-part-b-drug-average-sales-price/asp-pricing-files
- Shores JT, Gabriel A, Gupta S. Skin substitutes and alternatives: a review. Adv Skin Wound Care. 2007;20(9 Pt 1):493-508. doi:10.1097/01.ASW.0000288217.83128.f3
- Li X, Meng X, Wang X, et al. Human acellular dermal matrix allograft: a randomized, controlled human trial for the long-term evaluation of patients with extensive burns. Burns. 2015;41:689-699. doi:10.1016/j.burns.2014.12.007
- Juhasz I, Kiss B, Lukacs L, et al. Long-term followup of dermal substitution with acellular dermal implant in burns and postburn scar corrections. Dermatol Res Pract. 2010;2010:210150. doi:10.1155/2010/210150
- Towler MA, Rush EW, Richardson MK, et al. Randomized, prospective, blinded-enrollment, head-to-head venous leg ulcer healing trial comparing living, bioengineered skin graft substitute (Apligraf) with living, cryopreserved, human skin allograft (TheraSkin). Clin Podiatr Med Surg. 2018;35:357-365. doi:10.1016/j.cpm.2018.02.006
- Jibbe A, Tolkachjov SN. An efficient single-layer suture technique for large scalp flaps. J Am Acad Dermatol. 2020;83:E395-E396. doi:10.1016/j.jaad.2019.07.062
- Mosti G, Mattaliano V, Magliaro A, et al. Cadaveric skin grafts may greatly increase the healing rate of recalcitrant ulcers when used both alone and in combination with split-thickness skin grafts. Dermatol Surg. 2020;46:169-179. doi:10.1097/dss.0000000000001990
- Valesky EM, Vogl T, Kaufmann R, et al. Trepanation or complete removal of the outer table of the calvarium for granulation induction: the erbium:YAG laser as an alternative to the rose head burr. Dermatology. 2015;230:276-281. doi:10.1159/000368749
- Drosou A, Trieu D, Goldberg LH. Scalpel-made holes on exposed scalp bone to promote second intention healing. J Am Acad Dermatol. 2014;71:387-388. doi:10.1016/j.jaad.2014.04.020
- Centers for Medicare & Medicaid Services. April 2023 ASP Pricing. Accessed August 25, 2023. https://www.cms.gov/medicare/medicare-part-b-drug-average-sales-price/asp-pricing-files
- Shores JT, Gabriel A, Gupta S. Skin substitutes and alternatives: a review. Adv Skin Wound Care. 2007;20(9 Pt 1):493-508. doi:10.1097/01.ASW.0000288217.83128.f3
- Li X, Meng X, Wang X, et al. Human acellular dermal matrix allograft: a randomized, controlled human trial for the long-term evaluation of patients with extensive burns. Burns. 2015;41:689-699. doi:10.1016/j.burns.2014.12.007
- Juhasz I, Kiss B, Lukacs L, et al. Long-term followup of dermal substitution with acellular dermal implant in burns and postburn scar corrections. Dermatol Res Pract. 2010;2010:210150. doi:10.1155/2010/210150
- Towler MA, Rush EW, Richardson MK, et al. Randomized, prospective, blinded-enrollment, head-to-head venous leg ulcer healing trial comparing living, bioengineered skin graft substitute (Apligraf) with living, cryopreserved, human skin allograft (TheraSkin). Clin Podiatr Med Surg. 2018;35:357-365. doi:10.1016/j.cpm.2018.02.006
 decrease the size of the defect and secure a cadaveric split-thickness skin graft over the remaining exposed periosteum.")
Complications of Body Piercings: A Systematic Review
The practice of body piercing has been present in cultures around the world for centuries. Piercings may be performed for religious or spiritual reasons or as a form of self-expression. In recent years, body piercings have become increasingly popular in all genders, with the most common sites being the ears, mouth, nose, eyebrows, nipples, navel, and genitals.1 The prevalence of body piercing in the general population is estimated to be as high as 50%.2 With the rising popularity of piercings, there also has been an increase in their associated complications, with one study noting that up to 35% of individuals with pierced ears and 30% of all pierced sites developed a complication.3 Common problems following piercing include infections, keloid formation, allergic contact dermatitis, site deformation, and tooth fractures.4 It is of utmost importance that health care professionals are aware of the potential complications associated with such a common practice. A comprehensive review of complications associated with cutaneous and mucosal piercings is lacking. We conducted a systematic review to summarize the clinical characteristics, complication types and frequency, and treatments reported for cutaneous and mucosal piercings.
METHODS
We conducted a systematic review of the literature adhering to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analyses) reporting guidelines.5
Search Strategy, Study Eligibility Criteria, and Study Selection
A literature search of the Embase, MEDLINE, and PubMed databases was performed on June 20, 2022, using search terms related to body piercing and possible piercing-induced complications (Supplemental Information online). All studies reporting complications following body piercing were included. In vitro and animal studies were excluded. Title and abstract screening were completed by 6 independent researchers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) using Covidence online systematic review software (www.covidence.org). Six reviewers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) independently evaluated titles, abstracts, and full texts to identify relevant studies. Conflicts were resolved by the senior reviewer (I.M.M.).
Data Extraction and Synthesis
Five reviewers (S.C., K.K., M.M-B., K.A., T.S.) independently extracted data from eligible studies using a standardized extraction form that included title; authors; year of publication; sample size; and key findings, including mean age, sex, piercing location, complication type, and treatment received.
Treatment type was placed into the following categories: surgical treatments, antimicrobials, medical treatments, direct-target therapy, oral procedures, avoidance, miscellaneous therapies, and no treatment. (Data regarding treatments can be found in the Supplemental Information online.)
RESULTS
The combined search yielded 2679 studies, 617 of which underwent full-text review; 319 studies were included (Figure). Studies were published from 1950 to June 2022 and included both adult and pediatric populations.
 diagram of study selection process.")
Patient Characteristics
In total, our pooled analysis included data on 30,090 complications across 36,803 pierced sites in 30,231 patients (Table 1). Demographic data are available for 55% (n=30,231) of patients. Overall, 74% (22,247/30,231) of the individuals included in our analysis were female. The mean age was 27.8 years (range, 0–76 years).

Piercing Location
Overall, 36,803 pierced sites had a reported complication. The oral cavity, location not otherwise specified, was the most common site associated with a complication, accounting for 67% (n=24,478) of complications (Table 1). Other reported sites included (in decreasing frequency) the ears (21%, n=7551), tongue (5%, n=1669), lip (3%, n=998), navel (2%, n=605), nose (1%, n=540), nipple (1%, n=344), face/body (1%, n=269), genitals/groin (0%, n=183), eyebrow (0%, n=161), hand (0%, n=4), and eyelid (0%, n=1). Piercing complications were more commonly reported among females across all piercing locations except for the eyebrow, which was equal in both sexes.
Complications
Local Infections—Local infections accounted for 36% of reported complication types (n=10,872/30,090): perichondritis (1%, n=85); abscesses (0%, n=25); bacterial colonization (1%, n=106); and local infections, not otherwise specified (98%, n=10,648)(Table 2). The majority of local infections were found to be secondary to piercings of the ear and oral cavity. The nipple was found to be a common site for abscesses (40%, n=10), whereas the tongue was found to be the most common site for bacterial colonization (69%, n=73).


Immune-Mediated Issues—Immune-mediated issues encompassed 5% of the total reported complications (n=1561/30,090). The most commonly reported immune-mediated complications included allergies (31%, n=482), edema and swelling (21%, n=331), dermatitis (18%, n=282), and inflammatory lesions (17%, n=270). The majority were found to occur secondary to ear piercings, with the exception of edema, which mainly occurred secondary to tongue piercings (45%, n=150), and allergy, which primarily was associated with oral piercings (51%, n=245)(Table 2).
Tissue Damage—Tissue damage accounted for 43% of all complications (n=13,036/30,090). The most common forms of tissue damage were trauma (55%, n=7182), dysesthesia (22%, n=2883), bleeding and bruising (18%, n=2376), and pain (3%, n=370)(Table 2). Trauma was mainly found to be a complication in the context of oral piercings (99%, n=7121). Similarly, 94% (n=2242) of bleeding and bruising occurred secondary to oral piercings. Embedded piercings (92%, n=127), deformity (91%, n=29), and necrosis (75%, n=3) mostly occurred following ear piercings. Lip piercings were found to be the most common cause of damage to surrounding structures (98%, n=50).
Oral—Overall, 3193 intraoral complications were reported, constituting 11% of the total complications (Table 2). Oral complications included dental damage (86%, n=2732), gum recession (14%, n=459), and gingivitis (0%, n=2). Dental damage was mostly reported following oral piercings (90%, n=2453), whereas gum recession was mostly reported following lip piercings (59%, n=272).
Proliferations—Proliferations accounted for 795 (3%) of reported piercing complications. The majority (97%, n=772) were keloids, 2% (n=16) were other benign growths, and 1% (n=7) were malignancies. These complications mostly occurred secondary to ear piercings, which resulted in 741 (96%) keloids, 6 (38%) benign growths, and 4 (57%) malignancies.
Systemic—Overall, 2% (n=633) of the total complications were classified as systemic issues, including functional impairment (45%, n=282), secondary organ involvement (24%, n=150), cardiac issues (3%, n=21), and aspiration/inhalation (1%, n=8). Nonlocalized infections such as hepatitis or an increased risk thereof (17%, n=107), tetanus (8%, n=52), chlamydia (1%, n=9), HIV (0%, n=1), herpes simplex virus (0%, n=1), gonorrhea (0%, n=1), and bacterial vaginosis (0%, n=1) also were included in this category. The tongue, ear, and genitals were the locations most involved in these complications (Table 2). Secondary organ involvement mostly occurred after ear (36%, n=54) and genital piercings (27%, n=41). A total of 8 cases of piercing aspiration and/or inhalation were reported in association with piercings of the head and neck (Table 2).
COMMENT
Piercing Complications
Overall, the ear, tongue, and oral cavity were found to be the sites with the most associated complications recorded in the literature, and local infection and tissue damage were found to be the most prevalent types of complications. A plethora of treatments were used to manage piercing-induced complications, including surgical or medical treatments and avoidance (Supplemental Information). Reports by Metts6 and Escudero-Castaño et al7 provide detailed protocols and photographs of piercings.
Infections
Our review found that local infections were commonly reported complications associated with body piercings, which is consistent with other studies.1 The initial trauma inherent in the piercing process followed by the presence of an ongoing foreign body lends itself to an increased risk for developing these complications. Wound healing after piercing also varies based on the piercing location.
The rate and severity of the infection are influenced by the anatomic location of the piercing, hygiene, method of piercing, types of materials used, and aftercare.8 Piercing cartilage sites, such as the helix, concha, or nose, increases susceptibility to infections and permanent deformities. Cartilage is particularly at risk because of its avascular nature.9 Other studies have reported similar incidences of superficial localized infections; infectious complications were seen in 10% to 30% of body piercings in one study,3 while 45% of American and Australian college students reported infection at a piercing site in a second study.10
Systemic Issues
Systemic issues are potentially the most dangerous piercing-induced complications, though they were rarer in our analysis. Some serious complications included septic emboli, fatal staphylococcal toxic shock syndrome, and death. Although some systemic issues, such as staphylococcal toxic shock syndrome and septic sacroiliitis, required extensive hospital stays and complex treatment, others had lifelong repercussions, such as hepatitis and HIV. One report showed an increased incidence of endocarditis associated with body piercing, including staphylococcal endocarditis following nasal piercings, Neisseria endocarditis following tongue piercings, and Staphylococcus epidermidis endocarditis following nipple piercings.11 Moreover, Mariano et al12—who noted a case of endocarditis and meningitis associated with a nape piercing in a young female in 2015—reinforced the notion that information pertaining to the risks associated with body piercing must be better disseminated, given the potential for morbid or fatal outcomes. Finally, nonsterile piercing techniques and poor hygiene were found to contribute substantially to the increased risk for infection, so it is of utmost importance to reinforce proper practices in piercing salons.4
Immune-Mediated Issues
Because piercings are foreign bodies, they are susceptible to both acute and chronic immune responses. Our study found that allergies and dermatitis made up almost half of the immune-mediated piercing complications. It is especially important to emphasize that costume jewelry exposes our skin to a variety of contact allergens, most prominently nickel, heightening the risk for developing allergic contact dermatitis.13 Moreover, a study conducted by Brandão et al14 found that patients with pierced ears were significantly more likely to react to nickel than those without pierced ears (P=.031). Although other studies have found that allergy to metals ranges from 8.3% to 20% in the general population,15 we were not able to quantify the incidence in our study due to a lack of reporting of common benign complications, such as contact dermatitis.
Tissue Damage and Local Problems
Our review found that tissue and oral damage also were commonly reported piercing complications, with the most common pathologies being trauma, dysesthesia, bleeding/bruising, and dental damage. Laumann and Derick16 reported that bleeding, tissue trauma, and local problems were common physical health problems associated with body piercing. Severe complications, such as abscesses, toxic shock syndrome, and endocarditis, also have been reported in association with intraoral piercings.17 Moreover, other studies have shown that oral piercings are associated with several adverse oral and systemic conditions. A meta-analysis of individuals with oral piercings found a similar prevalence of dental fracture, gingival recession, and tooth wear (34%), as well as unspecified dental damage (27%) and tooth chipping (22%). Additionally, this meta-analysis reported a 3-fold increased risk for dental fracture and 7-fold increased risk for gingival recession with oral piercings.18 Another meta-analysis of oral piercing complications found a similar prevalence of dental fracture (34%), tooth wear (34%), gingival recession (33%), unspecified dental damage (27%), and tooth chipping (22%).19 Considering the extensive amount of cumulative damage, wearers of oral jewelry require periodic periodontal evaluations to monitor for dental damage and gingival recession.20 There are limited data on treatments for complications of oral piercings, and further research in this area is warranted.
Proliferations and Scars
Although proliferations and scarring were among the least common complications reported in the literature, they are some of the most cosmetically disfiguring for patients. Keloids, the most common type of growth associated with piercings, do not naturally regress and thus require some form of intervention. Given the multimodal approach used to treat keloids, as described by the evidence-based algorithm by Ogawa,21 it is not surprising that keloids also represented the complication most treated with medical therapies, such as steroids, and also with direct-target therapy, such as liquid nitrogen therapy (Supplemental Information).
Other proliferations reported in the literature include benign pyogenic granulomas22 and much less commonly malignant neoplasms such as basal cell carcinoma23 and squamous cell carcinoma.24 Although rare, treatment of piercing-associated malignancies include surgical removal, chemotherapy, and radiation therapy (Supplemental Information).
Limitations
There are several limitations to our systematic review. First, heterogeneity in study designs, patient populations, treatment interventions, and outcome measures of included studies may have affected the quality and generalizability of our results. Moreover, because the studies included in this systematic review focused on specific complications, we could not compare our results to the literature that analyzes incidence rates of piercing complications. Furthermore, not all studies included the data that we hoped to extract, and thus only available data were reported in these instances. Finally, the articles we reviewed may have included publication bias, with positive findings being more frequently published, potentially inflating certain types and sites of complications and treatment choices. Despite these limitations, our review provides essential information that must be interpreted in a clinical context.
CONCLUSION
Given that cutaneous and mucosal piercing has become more prevalent in recent years, along with an increase in the variety of piercing-induced complications, it is of utmost importance that piercing salons have proper hygiene practices in place and that patients are aware of the multitude of potential complications that can arise—whether common and benign or rare but life-threatening.
- Preslar D, Borger J. Body piercing infections. In: StatPearls. StatPearls Publishing; 2022.
- Antoszewski B, Jedrzejczak M, Kruk-Jeromin J. Complications after body piercing in patient suffering from type 1 diabetes mellitus. Int J Dermatol. 2007;46:1250-1252.
- Simplot TC, Hoffman HT. Comparison between cartilage and soft tissue ear piercing complications. Am J Otolaryngol. 1998;19:305-310.
- Meltzer DI. Complications of body piercing. Am Fam Physician. 2005;72:2029-2034.
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
- Metts J. Common complications of body piercing. West J Med. 2002;176:85-86.
- Escudero-Castaño N, Perea-García MA, Campo-Trapero J, et al. Oral and perioral piercing complications. Open Dent J. 2008;2:133-136.
- Tweeten SS, Rickman LS. Infectious complications of body piercing. Clin Infect Dis. 1998;26:735-740.
- Gabriel OT, Anthony OO, Paul EA, et al. Trends and complications of ear piercing among selected Nigerian population. J Family Med Prim Care. 2017;6:517-521.
- Armstrong ML, Koch JR, Saunders JC, et al. The hole picture: risks, decision making, purpose, regulations, and the future of body piercing. Clin Dermatol. 2007;25:398-406.
- Millar BC, Moore JE. Antibiotic prophylaxis, body piercing and infective endocarditis. J Antimicrob Chemother. 2004;53:123-126.
- Mariano A, Pisapia R, Abdeddaim A, et al. Endocarditis and meningitis associated to nape piercing in a young female: a case report. Infez Med. 2015;23:275-279.
- Ivey LA, Limone BA, Jacob SE. Approach to the jewelry aficionado. Pediatr Dermatol. 2018;35:274-275.
- Brandão MH, Gontijo B, Girundi MA, et al. Ear piercing as a risk factor for contact allergy to nickel. J Pediatr (Rio J). 2010;86:149-154.
- Schuttelaar MLA, Ofenloch RF, Bruze M, et al. Prevalence of contact allergy to metals in the European general population with a focus on nickel and piercings: The EDEN Fragrance Study. Contact Dermatitis. 2018;79:1-9.
- Laumann AE, Derick AJ. Tattoos and body piercings in the United States: a national data set. J Am Acad Dermatol. 2006;55:413-421.
- De Moor RJ, De Witte AM, Delmé KI, et al. Dental and oral complications of lip and tongue piercings. Br Dent J. 2005;199:506-509.
- Offen E, Allison JR. Do oral piercings cause problems in the mouth? Evid Based Dent. 2022;23:126-127.
- Passos PF, Pintor AVB, Marañón-Vásquez GA, et al. Oral manifestations arising from oral piercings: A systematic review and meta-analyses. Oral Surg Oral Med Oral Pathol Oral Radiol. 2022;134:327-341.
- Covello F, Salerno C, Giovannini V, et al. Piercing and oral health: a study on the knowledge of risks and complications. Int J Environ Res Public Health. 2020;17:613.
- Ogawa R. The most current algorithms for the treatment and prevention of hypertrophic scars and keloids: a 2020 update of the algorithms published 10 years ago. Plast Reconstr Surg. 2022;149:E79-E94.
- Kumar Ghosh S, Bandyopadhyay D. Granuloma pyogenicum as a complication of decorative nose piercing: report of eight cases from eastern India. J Cutan Med Surg. 2012;16:197-200.
- Dreher K, Kern M, Rush L, et al. Basal cell carcinoma invasion of an ear piercing. Dermatol Online J. 2022;28.
- Stanko P, Poruban D, Mracna J, et al. Squamous cell carcinoma and piercing of the tongue—a case report. J Craniomaxillofac Surg. 2012;40:329-331.
The practice of body piercing has been present in cultures around the world for centuries. Piercings may be performed for religious or spiritual reasons or as a form of self-expression. In recent years, body piercings have become increasingly popular in all genders, with the most common sites being the ears, mouth, nose, eyebrows, nipples, navel, and genitals.1 The prevalence of body piercing in the general population is estimated to be as high as 50%.2 With the rising popularity of piercings, there also has been an increase in their associated complications, with one study noting that up to 35% of individuals with pierced ears and 30% of all pierced sites developed a complication.3 Common problems following piercing include infections, keloid formation, allergic contact dermatitis, site deformation, and tooth fractures.4 It is of utmost importance that health care professionals are aware of the potential complications associated with such a common practice. A comprehensive review of complications associated with cutaneous and mucosal piercings is lacking. We conducted a systematic review to summarize the clinical characteristics, complication types and frequency, and treatments reported for cutaneous and mucosal piercings.
METHODS
We conducted a systematic review of the literature adhering to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analyses) reporting guidelines.5
Search Strategy, Study Eligibility Criteria, and Study Selection
A literature search of the Embase, MEDLINE, and PubMed databases was performed on June 20, 2022, using search terms related to body piercing and possible piercing-induced complications (Supplemental Information online). All studies reporting complications following body piercing were included. In vitro and animal studies were excluded. Title and abstract screening were completed by 6 independent researchers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) using Covidence online systematic review software (www.covidence.org). Six reviewers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) independently evaluated titles, abstracts, and full texts to identify relevant studies. Conflicts were resolved by the senior reviewer (I.M.M.).
Data Extraction and Synthesis
Five reviewers (S.C., K.K., M.M-B., K.A., T.S.) independently extracted data from eligible studies using a standardized extraction form that included title; authors; year of publication; sample size; and key findings, including mean age, sex, piercing location, complication type, and treatment received.
Treatment type was placed into the following categories: surgical treatments, antimicrobials, medical treatments, direct-target therapy, oral procedures, avoidance, miscellaneous therapies, and no treatment. (Data regarding treatments can be found in the Supplemental Information online.)
RESULTS
The combined search yielded 2679 studies, 617 of which underwent full-text review; 319 studies were included (Figure). Studies were published from 1950 to June 2022 and included both adult and pediatric populations.
Patient Characteristics
In total, our pooled analysis included data on 30,090 complications across 36,803 pierced sites in 30,231 patients (Table 1). Demographic data are available for 55% (n=30,231) of patients. Overall, 74% (22,247/30,231) of the individuals included in our analysis were female. The mean age was 27.8 years (range, 0–76 years).
Piercing Location
Overall, 36,803 pierced sites had a reported complication. The oral cavity, location not otherwise specified, was the most common site associated with a complication, accounting for 67% (n=24,478) of complications (Table 1). Other reported sites included (in decreasing frequency) the ears (21%, n=7551), tongue (5%, n=1669), lip (3%, n=998), navel (2%, n=605), nose (1%, n=540), nipple (1%, n=344), face/body (1%, n=269), genitals/groin (0%, n=183), eyebrow (0%, n=161), hand (0%, n=4), and eyelid (0%, n=1). Piercing complications were more commonly reported among females across all piercing locations except for the eyebrow, which was equal in both sexes.
Complications
Local Infections—Local infections accounted for 36% of reported complication types (n=10,872/30,090): perichondritis (1%, n=85); abscesses (0%, n=25); bacterial colonization (1%, n=106); and local infections, not otherwise specified (98%, n=10,648)(Table 2). The majority of local infections were found to be secondary to piercings of the ear and oral cavity. The nipple was found to be a common site for abscesses (40%, n=10), whereas the tongue was found to be the most common site for bacterial colonization (69%, n=73).
Immune-Mediated Issues—Immune-mediated issues encompassed 5% of the total reported complications (n=1561/30,090). The most commonly reported immune-mediated complications included allergies (31%, n=482), edema and swelling (21%, n=331), dermatitis (18%, n=282), and inflammatory lesions (17%, n=270). The majority were found to occur secondary to ear piercings, with the exception of edema, which mainly occurred secondary to tongue piercings (45%, n=150), and allergy, which primarily was associated with oral piercings (51%, n=245)(Table 2).
Tissue Damage—Tissue damage accounted for 43% of all complications (n=13,036/30,090). The most common forms of tissue damage were trauma (55%, n=7182), dysesthesia (22%, n=2883), bleeding and bruising (18%, n=2376), and pain (3%, n=370)(Table 2). Trauma was mainly found to be a complication in the context of oral piercings (99%, n=7121). Similarly, 94% (n=2242) of bleeding and bruising occurred secondary to oral piercings. Embedded piercings (92%, n=127), deformity (91%, n=29), and necrosis (75%, n=3) mostly occurred following ear piercings. Lip piercings were found to be the most common cause of damage to surrounding structures (98%, n=50).
Oral—Overall, 3193 intraoral complications were reported, constituting 11% of the total complications (Table 2). Oral complications included dental damage (86%, n=2732), gum recession (14%, n=459), and gingivitis (0%, n=2). Dental damage was mostly reported following oral piercings (90%, n=2453), whereas gum recession was mostly reported following lip piercings (59%, n=272).
Proliferations—Proliferations accounted for 795 (3%) of reported piercing complications. The majority (97%, n=772) were keloids, 2% (n=16) were other benign growths, and 1% (n=7) were malignancies. These complications mostly occurred secondary to ear piercings, which resulted in 741 (96%) keloids, 6 (38%) benign growths, and 4 (57%) malignancies.
Systemic—Overall, 2% (n=633) of the total complications were classified as systemic issues, including functional impairment (45%, n=282), secondary organ involvement (24%, n=150), cardiac issues (3%, n=21), and aspiration/inhalation (1%, n=8). Nonlocalized infections such as hepatitis or an increased risk thereof (17%, n=107), tetanus (8%, n=52), chlamydia (1%, n=9), HIV (0%, n=1), herpes simplex virus (0%, n=1), gonorrhea (0%, n=1), and bacterial vaginosis (0%, n=1) also were included in this category. The tongue, ear, and genitals were the locations most involved in these complications (Table 2). Secondary organ involvement mostly occurred after ear (36%, n=54) and genital piercings (27%, n=41). A total of 8 cases of piercing aspiration and/or inhalation were reported in association with piercings of the head and neck (Table 2).
COMMENT
Piercing Complications
Overall, the ear, tongue, and oral cavity were found to be the sites with the most associated complications recorded in the literature, and local infection and tissue damage were found to be the most prevalent types of complications. A plethora of treatments were used to manage piercing-induced complications, including surgical or medical treatments and avoidance (Supplemental Information). Reports by Metts6 and Escudero-Castaño et al7 provide detailed protocols and photographs of piercings.
Infections
Our review found that local infections were commonly reported complications associated with body piercings, which is consistent with other studies.1 The initial trauma inherent in the piercing process followed by the presence of an ongoing foreign body lends itself to an increased risk for developing these complications. Wound healing after piercing also varies based on the piercing location.
The rate and severity of the infection are influenced by the anatomic location of the piercing, hygiene, method of piercing, types of materials used, and aftercare.8 Piercing cartilage sites, such as the helix, concha, or nose, increases susceptibility to infections and permanent deformities. Cartilage is particularly at risk because of its avascular nature.9 Other studies have reported similar incidences of superficial localized infections; infectious complications were seen in 10% to 30% of body piercings in one study,3 while 45% of American and Australian college students reported infection at a piercing site in a second study.10
Systemic Issues
Systemic issues are potentially the most dangerous piercing-induced complications, though they were rarer in our analysis. Some serious complications included septic emboli, fatal staphylococcal toxic shock syndrome, and death. Although some systemic issues, such as staphylococcal toxic shock syndrome and septic sacroiliitis, required extensive hospital stays and complex treatment, others had lifelong repercussions, such as hepatitis and HIV. One report showed an increased incidence of endocarditis associated with body piercing, including staphylococcal endocarditis following nasal piercings, Neisseria endocarditis following tongue piercings, and Staphylococcus epidermidis endocarditis following nipple piercings.11 Moreover, Mariano et al12—who noted a case of endocarditis and meningitis associated with a nape piercing in a young female in 2015—reinforced the notion that information pertaining to the risks associated with body piercing must be better disseminated, given the potential for morbid or fatal outcomes. Finally, nonsterile piercing techniques and poor hygiene were found to contribute substantially to the increased risk for infection, so it is of utmost importance to reinforce proper practices in piercing salons.4
Immune-Mediated Issues
Because piercings are foreign bodies, they are susceptible to both acute and chronic immune responses. Our study found that allergies and dermatitis made up almost half of the immune-mediated piercing complications. It is especially important to emphasize that costume jewelry exposes our skin to a variety of contact allergens, most prominently nickel, heightening the risk for developing allergic contact dermatitis.13 Moreover, a study conducted by Brandão et al14 found that patients with pierced ears were significantly more likely to react to nickel than those without pierced ears (P=.031). Although other studies have found that allergy to metals ranges from 8.3% to 20% in the general population,15 we were not able to quantify the incidence in our study due to a lack of reporting of common benign complications, such as contact dermatitis.
Tissue Damage and Local Problems
Our review found that tissue and oral damage also were commonly reported piercing complications, with the most common pathologies being trauma, dysesthesia, bleeding/bruising, and dental damage. Laumann and Derick16 reported that bleeding, tissue trauma, and local problems were common physical health problems associated with body piercing. Severe complications, such as abscesses, toxic shock syndrome, and endocarditis, also have been reported in association with intraoral piercings.17 Moreover, other studies have shown that oral piercings are associated with several adverse oral and systemic conditions. A meta-analysis of individuals with oral piercings found a similar prevalence of dental fracture, gingival recession, and tooth wear (34%), as well as unspecified dental damage (27%) and tooth chipping (22%). Additionally, this meta-analysis reported a 3-fold increased risk for dental fracture and 7-fold increased risk for gingival recession with oral piercings.18 Another meta-analysis of oral piercing complications found a similar prevalence of dental fracture (34%), tooth wear (34%), gingival recession (33%), unspecified dental damage (27%), and tooth chipping (22%).19 Considering the extensive amount of cumulative damage, wearers of oral jewelry require periodic periodontal evaluations to monitor for dental damage and gingival recession.20 There are limited data on treatments for complications of oral piercings, and further research in this area is warranted.
Proliferations and Scars
Although proliferations and scarring were among the least common complications reported in the literature, they are some of the most cosmetically disfiguring for patients. Keloids, the most common type of growth associated with piercings, do not naturally regress and thus require some form of intervention. Given the multimodal approach used to treat keloids, as described by the evidence-based algorithm by Ogawa,21 it is not surprising that keloids also represented the complication most treated with medical therapies, such as steroids, and also with direct-target therapy, such as liquid nitrogen therapy (Supplemental Information).
Other proliferations reported in the literature include benign pyogenic granulomas22 and much less commonly malignant neoplasms such as basal cell carcinoma23 and squamous cell carcinoma.24 Although rare, treatment of piercing-associated malignancies include surgical removal, chemotherapy, and radiation therapy (Supplemental Information).
Limitations
There are several limitations to our systematic review. First, heterogeneity in study designs, patient populations, treatment interventions, and outcome measures of included studies may have affected the quality and generalizability of our results. Moreover, because the studies included in this systematic review focused on specific complications, we could not compare our results to the literature that analyzes incidence rates of piercing complications. Furthermore, not all studies included the data that we hoped to extract, and thus only available data were reported in these instances. Finally, the articles we reviewed may have included publication bias, with positive findings being more frequently published, potentially inflating certain types and sites of complications and treatment choices. Despite these limitations, our review provides essential information that must be interpreted in a clinical context.
CONCLUSION
Given that cutaneous and mucosal piercing has become more prevalent in recent years, along with an increase in the variety of piercing-induced complications, it is of utmost importance that piercing salons have proper hygiene practices in place and that patients are aware of the multitude of potential complications that can arise—whether common and benign or rare but life-threatening.
The practice of body piercing has been present in cultures around the world for centuries. Piercings may be performed for religious or spiritual reasons or as a form of self-expression. In recent years, body piercings have become increasingly popular in all genders, with the most common sites being the ears, mouth, nose, eyebrows, nipples, navel, and genitals.1 The prevalence of body piercing in the general population is estimated to be as high as 50%.2 With the rising popularity of piercings, there also has been an increase in their associated complications, with one study noting that up to 35% of individuals with pierced ears and 30% of all pierced sites developed a complication.3 Common problems following piercing include infections, keloid formation, allergic contact dermatitis, site deformation, and tooth fractures.4 It is of utmost importance that health care professionals are aware of the potential complications associated with such a common practice. A comprehensive review of complications associated with cutaneous and mucosal piercings is lacking. We conducted a systematic review to summarize the clinical characteristics, complication types and frequency, and treatments reported for cutaneous and mucosal piercings.
METHODS
We conducted a systematic review of the literature adhering to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analyses) reporting guidelines.5
Search Strategy, Study Eligibility Criteria, and Study Selection
A literature search of the Embase, MEDLINE, and PubMed databases was performed on June 20, 2022, using search terms related to body piercing and possible piercing-induced complications (Supplemental Information online). All studies reporting complications following body piercing were included. In vitro and animal studies were excluded. Title and abstract screening were completed by 6 independent researchers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) using Covidence online systematic review software (www.covidence.org). Six reviewers (S.C., K.K., M.M-B., K.A., T.S., I.M.M.) independently evaluated titles, abstracts, and full texts to identify relevant studies. Conflicts were resolved by the senior reviewer (I.M.M.).
Data Extraction and Synthesis
Five reviewers (S.C., K.K., M.M-B., K.A., T.S.) independently extracted data from eligible studies using a standardized extraction form that included title; authors; year of publication; sample size; and key findings, including mean age, sex, piercing location, complication type, and treatment received.
Treatment type was placed into the following categories: surgical treatments, antimicrobials, medical treatments, direct-target therapy, oral procedures, avoidance, miscellaneous therapies, and no treatment. (Data regarding treatments can be found in the Supplemental Information online.)
RESULTS
The combined search yielded 2679 studies, 617 of which underwent full-text review; 319 studies were included (Figure). Studies were published from 1950 to June 2022 and included both adult and pediatric populations.
Patient Characteristics
In total, our pooled analysis included data on 30,090 complications across 36,803 pierced sites in 30,231 patients (Table 1). Demographic data are available for 55% (n=30,231) of patients. Overall, 74% (22,247/30,231) of the individuals included in our analysis were female. The mean age was 27.8 years (range, 0–76 years).
Piercing Location
Overall, 36,803 pierced sites had a reported complication. The oral cavity, location not otherwise specified, was the most common site associated with a complication, accounting for 67% (n=24,478) of complications (Table 1). Other reported sites included (in decreasing frequency) the ears (21%, n=7551), tongue (5%, n=1669), lip (3%, n=998), navel (2%, n=605), nose (1%, n=540), nipple (1%, n=344), face/body (1%, n=269), genitals/groin (0%, n=183), eyebrow (0%, n=161), hand (0%, n=4), and eyelid (0%, n=1). Piercing complications were more commonly reported among females across all piercing locations except for the eyebrow, which was equal in both sexes.
Complications
Local Infections—Local infections accounted for 36% of reported complication types (n=10,872/30,090): perichondritis (1%, n=85); abscesses (0%, n=25); bacterial colonization (1%, n=106); and local infections, not otherwise specified (98%, n=10,648)(Table 2). The majority of local infections were found to be secondary to piercings of the ear and oral cavity. The nipple was found to be a common site for abscesses (40%, n=10), whereas the tongue was found to be the most common site for bacterial colonization (69%, n=73).
Immune-Mediated Issues—Immune-mediated issues encompassed 5% of the total reported complications (n=1561/30,090). The most commonly reported immune-mediated complications included allergies (31%, n=482), edema and swelling (21%, n=331), dermatitis (18%, n=282), and inflammatory lesions (17%, n=270). The majority were found to occur secondary to ear piercings, with the exception of edema, which mainly occurred secondary to tongue piercings (45%, n=150), and allergy, which primarily was associated with oral piercings (51%, n=245)(Table 2).
Tissue Damage—Tissue damage accounted for 43% of all complications (n=13,036/30,090). The most common forms of tissue damage were trauma (55%, n=7182), dysesthesia (22%, n=2883), bleeding and bruising (18%, n=2376), and pain (3%, n=370)(Table 2). Trauma was mainly found to be a complication in the context of oral piercings (99%, n=7121). Similarly, 94% (n=2242) of bleeding and bruising occurred secondary to oral piercings. Embedded piercings (92%, n=127), deformity (91%, n=29), and necrosis (75%, n=3) mostly occurred following ear piercings. Lip piercings were found to be the most common cause of damage to surrounding structures (98%, n=50).
Oral—Overall, 3193 intraoral complications were reported, constituting 11% of the total complications (Table 2). Oral complications included dental damage (86%, n=2732), gum recession (14%, n=459), and gingivitis (0%, n=2). Dental damage was mostly reported following oral piercings (90%, n=2453), whereas gum recession was mostly reported following lip piercings (59%, n=272).
Proliferations—Proliferations accounted for 795 (3%) of reported piercing complications. The majority (97%, n=772) were keloids, 2% (n=16) were other benign growths, and 1% (n=7) were malignancies. These complications mostly occurred secondary to ear piercings, which resulted in 741 (96%) keloids, 6 (38%) benign growths, and 4 (57%) malignancies.
Systemic—Overall, 2% (n=633) of the total complications were classified as systemic issues, including functional impairment (45%, n=282), secondary organ involvement (24%, n=150), cardiac issues (3%, n=21), and aspiration/inhalation (1%, n=8). Nonlocalized infections such as hepatitis or an increased risk thereof (17%, n=107), tetanus (8%, n=52), chlamydia (1%, n=9), HIV (0%, n=1), herpes simplex virus (0%, n=1), gonorrhea (0%, n=1), and bacterial vaginosis (0%, n=1) also were included in this category. The tongue, ear, and genitals were the locations most involved in these complications (Table 2). Secondary organ involvement mostly occurred after ear (36%, n=54) and genital piercings (27%, n=41). A total of 8 cases of piercing aspiration and/or inhalation were reported in association with piercings of the head and neck (Table 2).
COMMENT
Piercing Complications
Overall, the ear, tongue, and oral cavity were found to be the sites with the most associated complications recorded in the literature, and local infection and tissue damage were found to be the most prevalent types of complications. A plethora of treatments were used to manage piercing-induced complications, including surgical or medical treatments and avoidance (Supplemental Information). Reports by Metts6 and Escudero-Castaño et al7 provide detailed protocols and photographs of piercings.
Infections
Our review found that local infections were commonly reported complications associated with body piercings, which is consistent with other studies.1 The initial trauma inherent in the piercing process followed by the presence of an ongoing foreign body lends itself to an increased risk for developing these complications. Wound healing after piercing also varies based on the piercing location.
The rate and severity of the infection are influenced by the anatomic location of the piercing, hygiene, method of piercing, types of materials used, and aftercare.8 Piercing cartilage sites, such as the helix, concha, or nose, increases susceptibility to infections and permanent deformities. Cartilage is particularly at risk because of its avascular nature.9 Other studies have reported similar incidences of superficial localized infections; infectious complications were seen in 10% to 30% of body piercings in one study,3 while 45% of American and Australian college students reported infection at a piercing site in a second study.10
Systemic Issues
Systemic issues are potentially the most dangerous piercing-induced complications, though they were rarer in our analysis. Some serious complications included septic emboli, fatal staphylococcal toxic shock syndrome, and death. Although some systemic issues, such as staphylococcal toxic shock syndrome and septic sacroiliitis, required extensive hospital stays and complex treatment, others had lifelong repercussions, such as hepatitis and HIV. One report showed an increased incidence of endocarditis associated with body piercing, including staphylococcal endocarditis following nasal piercings, Neisseria endocarditis following tongue piercings, and Staphylococcus epidermidis endocarditis following nipple piercings.11 Moreover, Mariano et al12—who noted a case of endocarditis and meningitis associated with a nape piercing in a young female in 2015—reinforced the notion that information pertaining to the risks associated with body piercing must be better disseminated, given the potential for morbid or fatal outcomes. Finally, nonsterile piercing techniques and poor hygiene were found to contribute substantially to the increased risk for infection, so it is of utmost importance to reinforce proper practices in piercing salons.4
Immune-Mediated Issues
Because piercings are foreign bodies, they are susceptible to both acute and chronic immune responses. Our study found that allergies and dermatitis made up almost half of the immune-mediated piercing complications. It is especially important to emphasize that costume jewelry exposes our skin to a variety of contact allergens, most prominently nickel, heightening the risk for developing allergic contact dermatitis.13 Moreover, a study conducted by Brandão et al14 found that patients with pierced ears were significantly more likely to react to nickel than those without pierced ears (P=.031). Although other studies have found that allergy to metals ranges from 8.3% to 20% in the general population,15 we were not able to quantify the incidence in our study due to a lack of reporting of common benign complications, such as contact dermatitis.
Tissue Damage and Local Problems
Our review found that tissue and oral damage also were commonly reported piercing complications, with the most common pathologies being trauma, dysesthesia, bleeding/bruising, and dental damage. Laumann and Derick16 reported that bleeding, tissue trauma, and local problems were common physical health problems associated with body piercing. Severe complications, such as abscesses, toxic shock syndrome, and endocarditis, also have been reported in association with intraoral piercings.17 Moreover, other studies have shown that oral piercings are associated with several adverse oral and systemic conditions. A meta-analysis of individuals with oral piercings found a similar prevalence of dental fracture, gingival recession, and tooth wear (34%), as well as unspecified dental damage (27%) and tooth chipping (22%). Additionally, this meta-analysis reported a 3-fold increased risk for dental fracture and 7-fold increased risk for gingival recession with oral piercings.18 Another meta-analysis of oral piercing complications found a similar prevalence of dental fracture (34%), tooth wear (34%), gingival recession (33%), unspecified dental damage (27%), and tooth chipping (22%).19 Considering the extensive amount of cumulative damage, wearers of oral jewelry require periodic periodontal evaluations to monitor for dental damage and gingival recession.20 There are limited data on treatments for complications of oral piercings, and further research in this area is warranted.
Proliferations and Scars
Although proliferations and scarring were among the least common complications reported in the literature, they are some of the most cosmetically disfiguring for patients. Keloids, the most common type of growth associated with piercings, do not naturally regress and thus require some form of intervention. Given the multimodal approach used to treat keloids, as described by the evidence-based algorithm by Ogawa,21 it is not surprising that keloids also represented the complication most treated with medical therapies, such as steroids, and also with direct-target therapy, such as liquid nitrogen therapy (Supplemental Information).
Other proliferations reported in the literature include benign pyogenic granulomas22 and much less commonly malignant neoplasms such as basal cell carcinoma23 and squamous cell carcinoma.24 Although rare, treatment of piercing-associated malignancies include surgical removal, chemotherapy, and radiation therapy (Supplemental Information).
Limitations
There are several limitations to our systematic review. First, heterogeneity in study designs, patient populations, treatment interventions, and outcome measures of included studies may have affected the quality and generalizability of our results. Moreover, because the studies included in this systematic review focused on specific complications, we could not compare our results to the literature that analyzes incidence rates of piercing complications. Furthermore, not all studies included the data that we hoped to extract, and thus only available data were reported in these instances. Finally, the articles we reviewed may have included publication bias, with positive findings being more frequently published, potentially inflating certain types and sites of complications and treatment choices. Despite these limitations, our review provides essential information that must be interpreted in a clinical context.
CONCLUSION
Given that cutaneous and mucosal piercing has become more prevalent in recent years, along with an increase in the variety of piercing-induced complications, it is of utmost importance that piercing salons have proper hygiene practices in place and that patients are aware of the multitude of potential complications that can arise—whether common and benign or rare but life-threatening.
- Preslar D, Borger J. Body piercing infections. In: StatPearls. StatPearls Publishing; 2022.
- Antoszewski B, Jedrzejczak M, Kruk-Jeromin J. Complications after body piercing in patient suffering from type 1 diabetes mellitus. Int J Dermatol. 2007;46:1250-1252.
- Simplot TC, Hoffman HT. Comparison between cartilage and soft tissue ear piercing complications. Am J Otolaryngol. 1998;19:305-310.
- Meltzer DI. Complications of body piercing. Am Fam Physician. 2005;72:2029-2034.
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
- Metts J. Common complications of body piercing. West J Med. 2002;176:85-86.
- Escudero-Castaño N, Perea-García MA, Campo-Trapero J, et al. Oral and perioral piercing complications. Open Dent J. 2008;2:133-136.
- Tweeten SS, Rickman LS. Infectious complications of body piercing. Clin Infect Dis. 1998;26:735-740.
- Gabriel OT, Anthony OO, Paul EA, et al. Trends and complications of ear piercing among selected Nigerian population. J Family Med Prim Care. 2017;6:517-521.
- Armstrong ML, Koch JR, Saunders JC, et al. The hole picture: risks, decision making, purpose, regulations, and the future of body piercing. Clin Dermatol. 2007;25:398-406.
- Millar BC, Moore JE. Antibiotic prophylaxis, body piercing and infective endocarditis. J Antimicrob Chemother. 2004;53:123-126.
- Mariano A, Pisapia R, Abdeddaim A, et al. Endocarditis and meningitis associated to nape piercing in a young female: a case report. Infez Med. 2015;23:275-279.
- Ivey LA, Limone BA, Jacob SE. Approach to the jewelry aficionado. Pediatr Dermatol. 2018;35:274-275.
- Brandão MH, Gontijo B, Girundi MA, et al. Ear piercing as a risk factor for contact allergy to nickel. J Pediatr (Rio J). 2010;86:149-154.
- Schuttelaar MLA, Ofenloch RF, Bruze M, et al. Prevalence of contact allergy to metals in the European general population with a focus on nickel and piercings: The EDEN Fragrance Study. Contact Dermatitis. 2018;79:1-9.
- Laumann AE, Derick AJ. Tattoos and body piercings in the United States: a national data set. J Am Acad Dermatol. 2006;55:413-421.
- De Moor RJ, De Witte AM, Delmé KI, et al. Dental and oral complications of lip and tongue piercings. Br Dent J. 2005;199:506-509.
- Offen E, Allison JR. Do oral piercings cause problems in the mouth? Evid Based Dent. 2022;23:126-127.
- Passos PF, Pintor AVB, Marañón-Vásquez GA, et al. Oral manifestations arising from oral piercings: A systematic review and meta-analyses. Oral Surg Oral Med Oral Pathol Oral Radiol. 2022;134:327-341.
- Covello F, Salerno C, Giovannini V, et al. Piercing and oral health: a study on the knowledge of risks and complications. Int J Environ Res Public Health. 2020;17:613.
- Ogawa R. The most current algorithms for the treatment and prevention of hypertrophic scars and keloids: a 2020 update of the algorithms published 10 years ago. Plast Reconstr Surg. 2022;149:E79-E94.
- Kumar Ghosh S, Bandyopadhyay D. Granuloma pyogenicum as a complication of decorative nose piercing: report of eight cases from eastern India. J Cutan Med Surg. 2012;16:197-200.
- Dreher K, Kern M, Rush L, et al. Basal cell carcinoma invasion of an ear piercing. Dermatol Online J. 2022;28.
- Stanko P, Poruban D, Mracna J, et al. Squamous cell carcinoma and piercing of the tongue—a case report. J Craniomaxillofac Surg. 2012;40:329-331.
- Preslar D, Borger J. Body piercing infections. In: StatPearls. StatPearls Publishing; 2022.
- Antoszewski B, Jedrzejczak M, Kruk-Jeromin J. Complications after body piercing in patient suffering from type 1 diabetes mellitus. Int J Dermatol. 2007;46:1250-1252.
- Simplot TC, Hoffman HT. Comparison between cartilage and soft tissue ear piercing complications. Am J Otolaryngol. 1998;19:305-310.
- Meltzer DI. Complications of body piercing. Am Fam Physician. 2005;72:2029-2034.
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
- Metts J. Common complications of body piercing. West J Med. 2002;176:85-86.
- Escudero-Castaño N, Perea-García MA, Campo-Trapero J, et al. Oral and perioral piercing complications. Open Dent J. 2008;2:133-136.
- Tweeten SS, Rickman LS. Infectious complications of body piercing. Clin Infect Dis. 1998;26:735-740.
- Gabriel OT, Anthony OO, Paul EA, et al. Trends and complications of ear piercing among selected Nigerian population. J Family Med Prim Care. 2017;6:517-521.
- Armstrong ML, Koch JR, Saunders JC, et al. The hole picture: risks, decision making, purpose, regulations, and the future of body piercing. Clin Dermatol. 2007;25:398-406.
- Millar BC, Moore JE. Antibiotic prophylaxis, body piercing and infective endocarditis. J Antimicrob Chemother. 2004;53:123-126.
- Mariano A, Pisapia R, Abdeddaim A, et al. Endocarditis and meningitis associated to nape piercing in a young female: a case report. Infez Med. 2015;23:275-279.
- Ivey LA, Limone BA, Jacob SE. Approach to the jewelry aficionado. Pediatr Dermatol. 2018;35:274-275.
- Brandão MH, Gontijo B, Girundi MA, et al. Ear piercing as a risk factor for contact allergy to nickel. J Pediatr (Rio J). 2010;86:149-154.
- Schuttelaar MLA, Ofenloch RF, Bruze M, et al. Prevalence of contact allergy to metals in the European general population with a focus on nickel and piercings: The EDEN Fragrance Study. Contact Dermatitis. 2018;79:1-9.
- Laumann AE, Derick AJ. Tattoos and body piercings in the United States: a national data set. J Am Acad Dermatol. 2006;55:413-421.
- De Moor RJ, De Witte AM, Delmé KI, et al. Dental and oral complications of lip and tongue piercings. Br Dent J. 2005;199:506-509.
- Offen E, Allison JR. Do oral piercings cause problems in the mouth? Evid Based Dent. 2022;23:126-127.
- Passos PF, Pintor AVB, Marañón-Vásquez GA, et al. Oral manifestations arising from oral piercings: A systematic review and meta-analyses. Oral Surg Oral Med Oral Pathol Oral Radiol. 2022;134:327-341.
- Covello F, Salerno C, Giovannini V, et al. Piercing and oral health: a study on the knowledge of risks and complications. Int J Environ Res Public Health. 2020;17:613.
- Ogawa R. The most current algorithms for the treatment and prevention of hypertrophic scars and keloids: a 2020 update of the algorithms published 10 years ago. Plast Reconstr Surg. 2022;149:E79-E94.
- Kumar Ghosh S, Bandyopadhyay D. Granuloma pyogenicum as a complication of decorative nose piercing: report of eight cases from eastern India. J Cutan Med Surg. 2012;16:197-200.
- Dreher K, Kern M, Rush L, et al. Basal cell carcinoma invasion of an ear piercing. Dermatol Online J. 2022;28.
- Stanko P, Poruban D, Mracna J, et al. Squamous cell carcinoma and piercing of the tongue—a case report. J Craniomaxillofac Surg. 2012;40:329-331.
Practice Points
- Intraoral piercings of the tongue, lip, gingiva, or mucosa are associated with the most acute and chronic complications.
- Tissue damage is a common complication associated with cutaneous and mucocutaneous piercings, including trauma, bleeding and bruising, or dysesthesia.
- Given the rapid rise in the popularity of piercings, general practitioners and dermatologists should be aware of the multitude of acute or chronic complications associated with body piercings as well as effective treatment modalities.

Disseminated Papules and Nodules on the Skin and Oral Mucosa in an Infant
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
.")
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.

Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
A 38-week-old infant boy presented at birth with disseminated, nonblanching, purple to dark red papules and nodules on the skin and oral mucosa. He was born spontaneously after an uncomplicated pregnancy. The mother experienced an episode of oral herpes simplex virus during pregnancy. The infant was otherwise healthy. Laboratory tests including a complete blood cell count and routine serum biochemical analyses were within reference range; however, an infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies. Ophthalmologic and auditory screenings were normal.


Domestic violence in health care is real and underreported
To protect survivors’ identities, some names have been changed or shortened.
Natasha Abadilla, MD, met the man who would become her abuser while working abroad for a public health nonprofit. When he began emotionally and physically abusing her, she did everything she could to hide it.
“My coworkers knew nothing of the abuse. I became an expert in applying makeup to hide the bruises,” recalls Dr. Abadilla, now a second-year resident and pediatric neurologist at Lucile Packard Children’s Hospital at Stanford.
Dr. Abadilla says she strongly identifies as a hard worker and – to this day – hopes her work did not falter despite her partner’s constant drain on her. But the impact of the abuse continued to affect her for years. Like many survivors of domestic violence, she struggled with PTSD and depression.
Health care workers are often the first point of contact for survivors of domestic violence. Experts and advocates continue to push for more training for clinicians to identify and respond to signs among their patients. Often missing from this conversation is the reality that those tasked with screening can also be victims of intimate partner violence themselves.
What’s more: The very strengths that medical professionals often pride themselves on – perfectionism, empathy, grit – can make it harder for them to identify abuse in their own relationships and push through humiliation and shame to seek help.
Dr. Abadilla is exceptional among survivors in the medical field. Rather than keep her experience quiet, she has shared it publicly.
Awareness, she believes, can save lives.
An understudied problem in an underserved group
The majority of research on health care workers in this area has focused on workplace violence, which 62% experience worldwide. But intimate partner violence remains understudied and underdiscussed. Some medical professionals are even saddled with a “double burden,” facing trauma at work and at home, note the authors of a 2022 meta-analysis published in the journal Trauma, Violence, & Abuse.
The problem has had dire consequences. In recent years, many health care workers have been killed by their abusers:
- In 2016, Casey M. Drawert, MD, a Texas-based critical care anesthesiologist, was fatally shot by her husband in a murder-suicide.
- In 2018, Tamara O’Neal, MD, an ER physician, and Dayna Less, a first-year pharmacy resident, were killed by Dr. O’Neal’s ex-fiancé at Mercy Hospital in Chicago.
- In 2019, Sarah Hawley, MD, a first-year University of Utah resident, was fatally shot by her boyfriend in a murder-suicide.
- In 2021, Moria Kinsey, a nurse practitioner in Tahlequah, Okla., was murdered by a physician.
- In July of 2023, Gwendolyn Lavonne Riddick, DO, an ob.gyn. in North Carolina, was fatally shot by the father of her 3-year-old son.
There are others.
In the wake of these tragedies, calls for health care workers to screen each other as well as patients have grown. But for an untold number of survivors, breaking the silence is still not possible due to concerns about their reputation, professional consequences, the threat of harassment from abusers who are often in the same field, a medical culture of selfless endurance, and a lack of appropriate resources.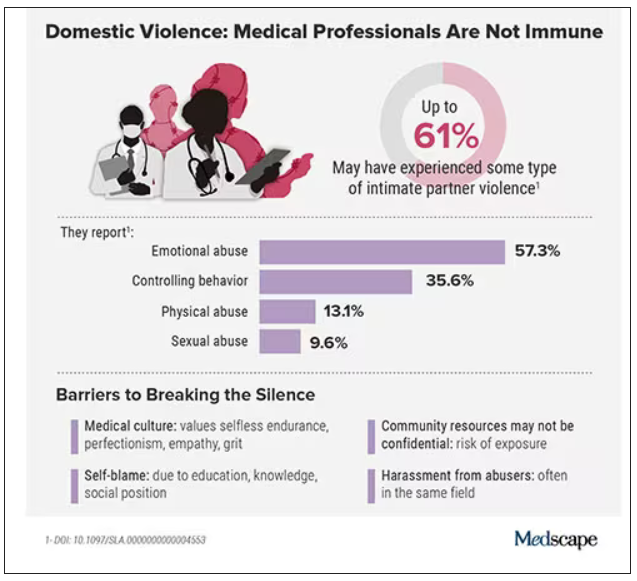
While the vast majority have stayed silent, those who have spoken out say there’s a need for targeted interventions to educate medical professionals as well as more supportive policies throughout the health care system.
Are health care workers more at risk?
Although more studies are needed, research indicates health care workers experience domestic violence at rates comparable to those of other populations, whereas some data suggest rates may be higher.
In the United States, more than one in three women and one in four men experience some form of intimate partner violence in their lifetime. Similarly, a 2020 study found that 24% of 400 physicians responding to a survey reported a history of domestic violence, with 15% reporting verbal abuse, 8% reporting physical violence, 4% reporting sexual abuse, and 4% reporting stalking.
Meanwhile, in an anonymous survey completed by 882 practicing surgeons and trainees in the United States from late 2018 to early 2019, more than 60% reported experiencing some type of intimate partner violence, most commonly emotional abuse.
Recent studies in the United Kingdom, Australia, and elsewhere show that significant numbers of medical professionals are fighting this battle. A 2019 study of more than 2,000 nurses, midwives, and health care assistants in the United Kingdom found that nurses were three times more likely to experience domestic violence than the average person.
What would help solve this problem: More study of health care worker-survivors as a unique group with unique risk factors. In general, domestic violence is most prevalent among women and people in marginalized groups. But young adults, such as medical students and trainees, can face an increased risk due to economic strain. Major life changes, such as relocating for residency, can also drive up stress and fray social connections, further isolating victims.
Why it’s so much harder for medical professionals to reveal abuse
For medical professionals accustomed to being strong and forging on, identifying as a victim of abuse can seem like a personal contradiction. It can feel easier to separate their personal and professional lives rather than face a complex reality.
In a personal essay on KevinMD.com, medical student Chloe N. L. Lee describes this emotional turmoil. “As an aspiring psychiatrist, I questioned my character judgment (how did I end up with a misogynistic abuser?) and wondered if I ought to have known better. I worried that my colleagues would deem me unfit to care for patients. And I thought that this was not supposed to happen to women like me,” Ms. Lee writes.
Kimberly, a licensed therapist, experienced a similar pattern of self-blame when her partner began exhibiting violent behavior. “For a long time, I felt guilty because I said to myself, You’re a therapist. You’re supposed to know this,” she recalls. At the same time, she felt driven to help him and sought couples therapy as his violence escalated.
Whitney, a pharmacist, recognized the “hallmarks” of abuse in her relationship, but she coped by compartmentalizing. Whitney says she was vulnerable to her abuser as a young college student who struggled financially. As he showered her with gifts, she found herself waving away red flags like aggressiveness or overprotectiveness.
After Whitney graduated, her partner’s emotional manipulation escalated into frequent physical assaults. When he gave her a black eye, she could not bring herself to go into work. She quit her job without notice. Despite a spotless record, none of her coworkers ever reached out to investigate her sudden departure.
It would take 8 years for Whitney to acknowledge the abuse and seize a moment to escape. She fled with just her purse and started over in a new city, rebuilding her life in the midst of harassment and threats from her ex. She says she’s grateful to be alive.
An imperfect system doesn’t help
Health care workers rarely ask for support or disclose abuse at work. Some have cited stigma, a lack of confidentiality (especially when the abuser is also in health care), fears about colleagues’ judgment, and a culture that doesn’t prioritize self-care.
Sometimes policies get in the way: In a 2021 qualitative study of interviews with 21 female physician-survivors in the United Kingdom, many said that despite the intense stress of abuse and recovery, they were unable to take any time off.
Of 180 UK-based midwife-survivors interviewed in a 2018 study, only 60 sought support at work and 30 received it. Many said their supervisors pressured them to report the abuse and get back to work, called social services behind their back, or reported them to their professional regulator. “I was treated like the perpetrator,” one said. Barbara Hernandez, PhD, a researcher who studies physician-survivors and director of physician vitality at Loma Linda University in southern California, says workplace violence and mistreatment from patients or colleagues – and a poor institutional response – can make those in health care feel like they have to “shut up and put up,” priming them to also tolerate abuse at home.
When survivors do reach out, there can be a disconnect between the resources they need and those they’re offered, Dr. Hernandez adds. In a recent survey of 400 physicians she conducted, respondents typically said they would advise a physician-survivor to “get to a shelter quickly.” But when roles were reversed, they admitted going to a shelter was the least feasible option. Support groups can also be problematic in smaller communities where physicians might be recognized or see their own patients.
Complicating matters further, the violence often comes from within the medical community. This can lead to particularly malicious abuse tactics like sending false accusations to a victim’s regulatory college or board; prolonged court and custody battles to drain them of all resources and their ability to hold a job; or even sabotage, harassment, or violence at work. The sheen of the abuser’s public persona, on the other hand, can guard them from any accountability.
For example, one physician-survivor said her ex-partner, a psychiatrist, coerced her into believing she was mentally ill, claimed she was “psychotic” in order to take back their children after she left, and had numerous colleagues serve as character witnesses in court for him, “saying he couldn’t have done any of these things, how great he is, and what a wonderful father he is.”
Slow progress is still progress
After Sherilyn M. Gordon-Burroughs, MD, a Texas-based transplant surgeon, mother, and educator, was killed by her husband in a murder-suicide in 2017, her friends Barbara Lee Bass, MD, president of the American College of Surgeons, and Patricia L. Turner, MD, were spurred into action. Together, they founded the ACS Intimate Partner Violence Task Force. Their mission is to educate surgeons to identify the signs of intimate partner violence (IPV) in themselves and their colleagues and connect them with resources.
“There is a concerted effort to close that gap,” says D’Andrea K. Joseph, MD, cochair of the task force and chief of trauma and acute care surgery at NYU Langone in New York. In the future, Dr. Joseph predicts, “making this a part of the curriculum, that it’s standardized for residents and trainees, that there is a safe place for victims ... and that we can band together and really recognize and assist our colleagues who are in trouble.”
Resources created by the ACS IPV task force, such as the toolkit and curriculum, provide a model for other health care leaders. But there have been few similar initiatives aimed at increasing IPV intervention within the medical system.
What you can do in your workplace
In her essay, Ms. Lee explains that a major turning point came when a physician friend explicitly asked if she was experiencing abuse. He then gently confirmed she was, and asked without judgment how he could support her, an approach that mirrors advice from the National Domestic Violence Hotline.
“Having a physician validate that this was, indeed, an abusive situation helped enormously ... I believe it may have saved my life,” she writes.
That validation can be crucial, and Dr. Abadilla urges other physicians to regularly check in with colleagues, especially those who seem particularly positive with a go-getter attitude and yet may not seem themselves. That was how she presented when she was struggling the most.
Supporting systemic changes within your organization and beyond is also important. The authors of the 2022 meta-analysis stress the need for domestic violence training, legislative changes, paid leave, and union support.
Finding strength in recovery
Over a decade after escaping her abuser, Whitney says she’s only just begun to share her experience, but what she’s learned has made her a better pharmacist. She says she’s more attuned to subtle signs something could be off with patients and coworkers. When someone makes comments about feeling anxious or that they can’t do anything right, it’s important to ask why, she says.
Recently, Kimberly has opened up to her mentor and other therapists, many of whom have shared that they’re also survivors.
“The last thing I said to [my abuser] is you think you’ve won and you’re hurting me, but what you’ve done to me – I’m going to utilize this and I’m going to help other people,” Kimberly says. “This pain that I have will go away, and I’m going to save the lives of others.”
A version of this article first appeared on Medscape.com.
To protect survivors’ identities, some names have been changed or shortened.
Natasha Abadilla, MD, met the man who would become her abuser while working abroad for a public health nonprofit. When he began emotionally and physically abusing her, she did everything she could to hide it.
“My coworkers knew nothing of the abuse. I became an expert in applying makeup to hide the bruises,” recalls Dr. Abadilla, now a second-year resident and pediatric neurologist at Lucile Packard Children’s Hospital at Stanford.
Dr. Abadilla says she strongly identifies as a hard worker and – to this day – hopes her work did not falter despite her partner’s constant drain on her. But the impact of the abuse continued to affect her for years. Like many survivors of domestic violence, she struggled with PTSD and depression.
Health care workers are often the first point of contact for survivors of domestic violence. Experts and advocates continue to push for more training for clinicians to identify and respond to signs among their patients. Often missing from this conversation is the reality that those tasked with screening can also be victims of intimate partner violence themselves.
What’s more: The very strengths that medical professionals often pride themselves on – perfectionism, empathy, grit – can make it harder for them to identify abuse in their own relationships and push through humiliation and shame to seek help.
Dr. Abadilla is exceptional among survivors in the medical field. Rather than keep her experience quiet, she has shared it publicly.
Awareness, she believes, can save lives.
An understudied problem in an underserved group
The majority of research on health care workers in this area has focused on workplace violence, which 62% experience worldwide. But intimate partner violence remains understudied and underdiscussed. Some medical professionals are even saddled with a “double burden,” facing trauma at work and at home, note the authors of a 2022 meta-analysis published in the journal Trauma, Violence, & Abuse.
The problem has had dire consequences. In recent years, many health care workers have been killed by their abusers:
- In 2016, Casey M. Drawert, MD, a Texas-based critical care anesthesiologist, was fatally shot by her husband in a murder-suicide.
- In 2018, Tamara O’Neal, MD, an ER physician, and Dayna Less, a first-year pharmacy resident, were killed by Dr. O’Neal’s ex-fiancé at Mercy Hospital in Chicago.
- In 2019, Sarah Hawley, MD, a first-year University of Utah resident, was fatally shot by her boyfriend in a murder-suicide.
- In 2021, Moria Kinsey, a nurse practitioner in Tahlequah, Okla., was murdered by a physician.
- In July of 2023, Gwendolyn Lavonne Riddick, DO, an ob.gyn. in North Carolina, was fatally shot by the father of her 3-year-old son.
There are others.
In the wake of these tragedies, calls for health care workers to screen each other as well as patients have grown. But for an untold number of survivors, breaking the silence is still not possible due to concerns about their reputation, professional consequences, the threat of harassment from abusers who are often in the same field, a medical culture of selfless endurance, and a lack of appropriate resources.
While the vast majority have stayed silent, those who have spoken out say there’s a need for targeted interventions to educate medical professionals as well as more supportive policies throughout the health care system.
Are health care workers more at risk?
Although more studies are needed, research indicates health care workers experience domestic violence at rates comparable to those of other populations, whereas some data suggest rates may be higher.
In the United States, more than one in three women and one in four men experience some form of intimate partner violence in their lifetime. Similarly, a 2020 study found that 24% of 400 physicians responding to a survey reported a history of domestic violence, with 15% reporting verbal abuse, 8% reporting physical violence, 4% reporting sexual abuse, and 4% reporting stalking.
Meanwhile, in an anonymous survey completed by 882 practicing surgeons and trainees in the United States from late 2018 to early 2019, more than 60% reported experiencing some type of intimate partner violence, most commonly emotional abuse.
Recent studies in the United Kingdom, Australia, and elsewhere show that significant numbers of medical professionals are fighting this battle. A 2019 study of more than 2,000 nurses, midwives, and health care assistants in the United Kingdom found that nurses were three times more likely to experience domestic violence than the average person.
What would help solve this problem: More study of health care worker-survivors as a unique group with unique risk factors. In general, domestic violence is most prevalent among women and people in marginalized groups. But young adults, such as medical students and trainees, can face an increased risk due to economic strain. Major life changes, such as relocating for residency, can also drive up stress and fray social connections, further isolating victims.
Why it’s so much harder for medical professionals to reveal abuse
For medical professionals accustomed to being strong and forging on, identifying as a victim of abuse can seem like a personal contradiction. It can feel easier to separate their personal and professional lives rather than face a complex reality.
In a personal essay on KevinMD.com, medical student Chloe N. L. Lee describes this emotional turmoil. “As an aspiring psychiatrist, I questioned my character judgment (how did I end up with a misogynistic abuser?) and wondered if I ought to have known better. I worried that my colleagues would deem me unfit to care for patients. And I thought that this was not supposed to happen to women like me,” Ms. Lee writes.
Kimberly, a licensed therapist, experienced a similar pattern of self-blame when her partner began exhibiting violent behavior. “For a long time, I felt guilty because I said to myself, You’re a therapist. You’re supposed to know this,” she recalls. At the same time, she felt driven to help him and sought couples therapy as his violence escalated.
Whitney, a pharmacist, recognized the “hallmarks” of abuse in her relationship, but she coped by compartmentalizing. Whitney says she was vulnerable to her abuser as a young college student who struggled financially. As he showered her with gifts, she found herself waving away red flags like aggressiveness or overprotectiveness.
After Whitney graduated, her partner’s emotional manipulation escalated into frequent physical assaults. When he gave her a black eye, she could not bring herself to go into work. She quit her job without notice. Despite a spotless record, none of her coworkers ever reached out to investigate her sudden departure.
It would take 8 years for Whitney to acknowledge the abuse and seize a moment to escape. She fled with just her purse and started over in a new city, rebuilding her life in the midst of harassment and threats from her ex. She says she’s grateful to be alive.
An imperfect system doesn’t help
Health care workers rarely ask for support or disclose abuse at work. Some have cited stigma, a lack of confidentiality (especially when the abuser is also in health care), fears about colleagues’ judgment, and a culture that doesn’t prioritize self-care.
Sometimes policies get in the way: In a 2021 qualitative study of interviews with 21 female physician-survivors in the United Kingdom, many said that despite the intense stress of abuse and recovery, they were unable to take any time off.
Of 180 UK-based midwife-survivors interviewed in a 2018 study, only 60 sought support at work and 30 received it. Many said their supervisors pressured them to report the abuse and get back to work, called social services behind their back, or reported them to their professional regulator. “I was treated like the perpetrator,” one said. Barbara Hernandez, PhD, a researcher who studies physician-survivors and director of physician vitality at Loma Linda University in southern California, says workplace violence and mistreatment from patients or colleagues – and a poor institutional response – can make those in health care feel like they have to “shut up and put up,” priming them to also tolerate abuse at home.
When survivors do reach out, there can be a disconnect between the resources they need and those they’re offered, Dr. Hernandez adds. In a recent survey of 400 physicians she conducted, respondents typically said they would advise a physician-survivor to “get to a shelter quickly.” But when roles were reversed, they admitted going to a shelter was the least feasible option. Support groups can also be problematic in smaller communities where physicians might be recognized or see their own patients.
Complicating matters further, the violence often comes from within the medical community. This can lead to particularly malicious abuse tactics like sending false accusations to a victim’s regulatory college or board; prolonged court and custody battles to drain them of all resources and their ability to hold a job; or even sabotage, harassment, or violence at work. The sheen of the abuser’s public persona, on the other hand, can guard them from any accountability.
For example, one physician-survivor said her ex-partner, a psychiatrist, coerced her into believing she was mentally ill, claimed she was “psychotic” in order to take back their children after she left, and had numerous colleagues serve as character witnesses in court for him, “saying he couldn’t have done any of these things, how great he is, and what a wonderful father he is.”
Slow progress is still progress
After Sherilyn M. Gordon-Burroughs, MD, a Texas-based transplant surgeon, mother, and educator, was killed by her husband in a murder-suicide in 2017, her friends Barbara Lee Bass, MD, president of the American College of Surgeons, and Patricia L. Turner, MD, were spurred into action. Together, they founded the ACS Intimate Partner Violence Task Force. Their mission is to educate surgeons to identify the signs of intimate partner violence (IPV) in themselves and their colleagues and connect them with resources.
“There is a concerted effort to close that gap,” says D’Andrea K. Joseph, MD, cochair of the task force and chief of trauma and acute care surgery at NYU Langone in New York. In the future, Dr. Joseph predicts, “making this a part of the curriculum, that it’s standardized for residents and trainees, that there is a safe place for victims ... and that we can band together and really recognize and assist our colleagues who are in trouble.”
Resources created by the ACS IPV task force, such as the toolkit and curriculum, provide a model for other health care leaders. But there have been few similar initiatives aimed at increasing IPV intervention within the medical system.
What you can do in your workplace
In her essay, Ms. Lee explains that a major turning point came when a physician friend explicitly asked if she was experiencing abuse. He then gently confirmed she was, and asked without judgment how he could support her, an approach that mirrors advice from the National Domestic Violence Hotline.
“Having a physician validate that this was, indeed, an abusive situation helped enormously ... I believe it may have saved my life,” she writes.
That validation can be crucial, and Dr. Abadilla urges other physicians to regularly check in with colleagues, especially those who seem particularly positive with a go-getter attitude and yet may not seem themselves. That was how she presented when she was struggling the most.
Supporting systemic changes within your organization and beyond is also important. The authors of the 2022 meta-analysis stress the need for domestic violence training, legislative changes, paid leave, and union support.
Finding strength in recovery
Over a decade after escaping her abuser, Whitney says she’s only just begun to share her experience, but what she’s learned has made her a better pharmacist. She says she’s more attuned to subtle signs something could be off with patients and coworkers. When someone makes comments about feeling anxious or that they can’t do anything right, it’s important to ask why, she says.
Recently, Kimberly has opened up to her mentor and other therapists, many of whom have shared that they’re also survivors.
“The last thing I said to [my abuser] is you think you’ve won and you’re hurting me, but what you’ve done to me – I’m going to utilize this and I’m going to help other people,” Kimberly says. “This pain that I have will go away, and I’m going to save the lives of others.”
A version of this article first appeared on Medscape.com.
To protect survivors’ identities, some names have been changed or shortened.
Natasha Abadilla, MD, met the man who would become her abuser while working abroad for a public health nonprofit. When he began emotionally and physically abusing her, she did everything she could to hide it.
“My coworkers knew nothing of the abuse. I became an expert in applying makeup to hide the bruises,” recalls Dr. Abadilla, now a second-year resident and pediatric neurologist at Lucile Packard Children’s Hospital at Stanford.
Dr. Abadilla says she strongly identifies as a hard worker and – to this day – hopes her work did not falter despite her partner’s constant drain on her. But the impact of the abuse continued to affect her for years. Like many survivors of domestic violence, she struggled with PTSD and depression.
Health care workers are often the first point of contact for survivors of domestic violence. Experts and advocates continue to push for more training for clinicians to identify and respond to signs among their patients. Often missing from this conversation is the reality that those tasked with screening can also be victims of intimate partner violence themselves.
What’s more: The very strengths that medical professionals often pride themselves on – perfectionism, empathy, grit – can make it harder for them to identify abuse in their own relationships and push through humiliation and shame to seek help.
Dr. Abadilla is exceptional among survivors in the medical field. Rather than keep her experience quiet, she has shared it publicly.
Awareness, she believes, can save lives.
An understudied problem in an underserved group
The majority of research on health care workers in this area has focused on workplace violence, which 62% experience worldwide. But intimate partner violence remains understudied and underdiscussed. Some medical professionals are even saddled with a “double burden,” facing trauma at work and at home, note the authors of a 2022 meta-analysis published in the journal Trauma, Violence, & Abuse.
The problem has had dire consequences. In recent years, many health care workers have been killed by their abusers:
- In 2016, Casey M. Drawert, MD, a Texas-based critical care anesthesiologist, was fatally shot by her husband in a murder-suicide.
- In 2018, Tamara O’Neal, MD, an ER physician, and Dayna Less, a first-year pharmacy resident, were killed by Dr. O’Neal’s ex-fiancé at Mercy Hospital in Chicago.
- In 2019, Sarah Hawley, MD, a first-year University of Utah resident, was fatally shot by her boyfriend in a murder-suicide.
- In 2021, Moria Kinsey, a nurse practitioner in Tahlequah, Okla., was murdered by a physician.
- In July of 2023, Gwendolyn Lavonne Riddick, DO, an ob.gyn. in North Carolina, was fatally shot by the father of her 3-year-old son.
There are others.
In the wake of these tragedies, calls for health care workers to screen each other as well as patients have grown. But for an untold number of survivors, breaking the silence is still not possible due to concerns about their reputation, professional consequences, the threat of harassment from abusers who are often in the same field, a medical culture of selfless endurance, and a lack of appropriate resources.
While the vast majority have stayed silent, those who have spoken out say there’s a need for targeted interventions to educate medical professionals as well as more supportive policies throughout the health care system.
Are health care workers more at risk?
Although more studies are needed, research indicates health care workers experience domestic violence at rates comparable to those of other populations, whereas some data suggest rates may be higher.
In the United States, more than one in three women and one in four men experience some form of intimate partner violence in their lifetime. Similarly, a 2020 study found that 24% of 400 physicians responding to a survey reported a history of domestic violence, with 15% reporting verbal abuse, 8% reporting physical violence, 4% reporting sexual abuse, and 4% reporting stalking.
Meanwhile, in an anonymous survey completed by 882 practicing surgeons and trainees in the United States from late 2018 to early 2019, more than 60% reported experiencing some type of intimate partner violence, most commonly emotional abuse.
Recent studies in the United Kingdom, Australia, and elsewhere show that significant numbers of medical professionals are fighting this battle. A 2019 study of more than 2,000 nurses, midwives, and health care assistants in the United Kingdom found that nurses were three times more likely to experience domestic violence than the average person.
What would help solve this problem: More study of health care worker-survivors as a unique group with unique risk factors. In general, domestic violence is most prevalent among women and people in marginalized groups. But young adults, such as medical students and trainees, can face an increased risk due to economic strain. Major life changes, such as relocating for residency, can also drive up stress and fray social connections, further isolating victims.
Why it’s so much harder for medical professionals to reveal abuse
For medical professionals accustomed to being strong and forging on, identifying as a victim of abuse can seem like a personal contradiction. It can feel easier to separate their personal and professional lives rather than face a complex reality.
In a personal essay on KevinMD.com, medical student Chloe N. L. Lee describes this emotional turmoil. “As an aspiring psychiatrist, I questioned my character judgment (how did I end up with a misogynistic abuser?) and wondered if I ought to have known better. I worried that my colleagues would deem me unfit to care for patients. And I thought that this was not supposed to happen to women like me,” Ms. Lee writes.
Kimberly, a licensed therapist, experienced a similar pattern of self-blame when her partner began exhibiting violent behavior. “For a long time, I felt guilty because I said to myself, You’re a therapist. You’re supposed to know this,” she recalls. At the same time, she felt driven to help him and sought couples therapy as his violence escalated.
Whitney, a pharmacist, recognized the “hallmarks” of abuse in her relationship, but she coped by compartmentalizing. Whitney says she was vulnerable to her abuser as a young college student who struggled financially. As he showered her with gifts, she found herself waving away red flags like aggressiveness or overprotectiveness.
After Whitney graduated, her partner’s emotional manipulation escalated into frequent physical assaults. When he gave her a black eye, she could not bring herself to go into work. She quit her job without notice. Despite a spotless record, none of her coworkers ever reached out to investigate her sudden departure.
It would take 8 years for Whitney to acknowledge the abuse and seize a moment to escape. She fled with just her purse and started over in a new city, rebuilding her life in the midst of harassment and threats from her ex. She says she’s grateful to be alive.
An imperfect system doesn’t help
Health care workers rarely ask for support or disclose abuse at work. Some have cited stigma, a lack of confidentiality (especially when the abuser is also in health care), fears about colleagues’ judgment, and a culture that doesn’t prioritize self-care.
Sometimes policies get in the way: In a 2021 qualitative study of interviews with 21 female physician-survivors in the United Kingdom, many said that despite the intense stress of abuse and recovery, they were unable to take any time off.
Of 180 UK-based midwife-survivors interviewed in a 2018 study, only 60 sought support at work and 30 received it. Many said their supervisors pressured them to report the abuse and get back to work, called social services behind their back, or reported them to their professional regulator. “I was treated like the perpetrator,” one said. Barbara Hernandez, PhD, a researcher who studies physician-survivors and director of physician vitality at Loma Linda University in southern California, says workplace violence and mistreatment from patients or colleagues – and a poor institutional response – can make those in health care feel like they have to “shut up and put up,” priming them to also tolerate abuse at home.
When survivors do reach out, there can be a disconnect between the resources they need and those they’re offered, Dr. Hernandez adds. In a recent survey of 400 physicians she conducted, respondents typically said they would advise a physician-survivor to “get to a shelter quickly.” But when roles were reversed, they admitted going to a shelter was the least feasible option. Support groups can also be problematic in smaller communities where physicians might be recognized or see their own patients.
Complicating matters further, the violence often comes from within the medical community. This can lead to particularly malicious abuse tactics like sending false accusations to a victim’s regulatory college or board; prolonged court and custody battles to drain them of all resources and their ability to hold a job; or even sabotage, harassment, or violence at work. The sheen of the abuser’s public persona, on the other hand, can guard them from any accountability.
For example, one physician-survivor said her ex-partner, a psychiatrist, coerced her into believing she was mentally ill, claimed she was “psychotic” in order to take back their children after she left, and had numerous colleagues serve as character witnesses in court for him, “saying he couldn’t have done any of these things, how great he is, and what a wonderful father he is.”
Slow progress is still progress
After Sherilyn M. Gordon-Burroughs, MD, a Texas-based transplant surgeon, mother, and educator, was killed by her husband in a murder-suicide in 2017, her friends Barbara Lee Bass, MD, president of the American College of Surgeons, and Patricia L. Turner, MD, were spurred into action. Together, they founded the ACS Intimate Partner Violence Task Force. Their mission is to educate surgeons to identify the signs of intimate partner violence (IPV) in themselves and their colleagues and connect them with resources.
“There is a concerted effort to close that gap,” says D’Andrea K. Joseph, MD, cochair of the task force and chief of trauma and acute care surgery at NYU Langone in New York. In the future, Dr. Joseph predicts, “making this a part of the curriculum, that it’s standardized for residents and trainees, that there is a safe place for victims ... and that we can band together and really recognize and assist our colleagues who are in trouble.”
Resources created by the ACS IPV task force, such as the toolkit and curriculum, provide a model for other health care leaders. But there have been few similar initiatives aimed at increasing IPV intervention within the medical system.
What you can do in your workplace
In her essay, Ms. Lee explains that a major turning point came when a physician friend explicitly asked if she was experiencing abuse. He then gently confirmed she was, and asked without judgment how he could support her, an approach that mirrors advice from the National Domestic Violence Hotline.
“Having a physician validate that this was, indeed, an abusive situation helped enormously ... I believe it may have saved my life,” she writes.
That validation can be crucial, and Dr. Abadilla urges other physicians to regularly check in with colleagues, especially those who seem particularly positive with a go-getter attitude and yet may not seem themselves. That was how she presented when she was struggling the most.
Supporting systemic changes within your organization and beyond is also important. The authors of the 2022 meta-analysis stress the need for domestic violence training, legislative changes, paid leave, and union support.
Finding strength in recovery
Over a decade after escaping her abuser, Whitney says she’s only just begun to share her experience, but what she’s learned has made her a better pharmacist. She says she’s more attuned to subtle signs something could be off with patients and coworkers. When someone makes comments about feeling anxious or that they can’t do anything right, it’s important to ask why, she says.
Recently, Kimberly has opened up to her mentor and other therapists, many of whom have shared that they’re also survivors.
“The last thing I said to [my abuser] is you think you’ve won and you’re hurting me, but what you’ve done to me – I’m going to utilize this and I’m going to help other people,” Kimberly says. “This pain that I have will go away, and I’m going to save the lives of others.”
A version of this article first appeared on Medscape.com.
One in five doctors with long COVID can no longer work: Survey
Crippling symptoms, lost careers, and eroded incomes: This is the harsh reality for doctors suffering with long COVID, according to the first major survey of physicians with the condition.
The survey, conducted by the British Medical Association and the Long COVID Doctors for Action support group, sheds light on the lingering effects of long COVID on more than 600 chronically ill and disabled doctors with the condition. It also spotlights what they describe as a lack of medical and financial support from their government and employers at the National Health Service.
“We feel betrayed and abandoned,” said Kelly Fearnley, MBChB, chair and cofounder of Long COVID Doctors for Action. “At a time of national crisis, when health care workers were asked to step up, we did. When the nation needed us, we stepped up. We put our lives on the line. We put our families’ lives on the line. And now that we are injured after knowingly being unprotected and deliberately and repeatedly exposed to a level 3 biohazard, we now find ourselves in this position.”
Dr. Fearnley fell ill while working in a hospital’s COVID ward in November 2020. She is one of an estimated 2 million people in the United Kingdom – including thousands of NHS employees – with long COVID. She hasn’t been able to return to work in nearly 3 years.
Long COVID affects more than 65 million people worldwide. It is estimated that 1 in 10 people infected with the virus develop long-term symptoms. In the United Kingdom, health care and social care workers are seven times more likely to have had severe COVID-19 than other types of employees.
Doctors responding to the BMA survey reported a wide range of long COVID symptoms, including fatigue, headaches, muscular pain, nerve damage, joint pain, and respiratory problems.
Among the survey’s key findings, 60% of doctors said long COVID has affected their ability to carry out day-to-day tasks on a regular basis. Almost one in five (18%) said they were no longer able to work, while fewer than one in three (31%) were working full time. This compares with more than half (57%) of respondents working full time before the onset of their COVID illness – a decline of 46%.
Nearly half (48%) of respondents said they have experienced some form of loss of earnings as a result of long COVID, and almost half of the doctors were never referred to an NHS long COVID clinic. The survey included the following first-person accounts from doctors living with the condition.
- One doctor said: “I nearly lost my life, my home, my partner and my career. I have received little support to help keep these. The impact on my mental health nearly cost [me] my life again.”
- A senior consulting physician commented: “Life is absolutely miserable. Every day is a struggle. I wake up exhausted, the insomnia and night terrors are horrendous as I live through my worst fears every night. Any activity such as eating meals, washing, etc., will mean I have to go to bed for a few hours. I am unable to look after myself or my child, exercise or maintain social relationships. I have no financial security. Long COVID has totally destroyed my life.”
- A salaried general practitioner said: “I can no longer work, finances are ruined. I didn’t have employment protection so am now unemployed and penniless.”
Calls for action from the BMA include the following:
- Financial support for doctors and health care staff with long COVID.
- The recognition of long COVID as an occupational disease among health care workers, along with a definition of the condition that covers all of the debilitating disease’s symptoms.
- Improved access to physical and mental health services to help comprehensive assessment, investigations, and treatment.
- Greater workplace protection for health care staff who risk their lives for others.
- Better support for long COVID sufferers to return to work safely if they can, including a flexible approach to the use of workplace adjustments.
“One would think, given the circumstances under which we fell ill and current workforce shortages, NHS employers would be eager to do everything to facilitate the return to work of people with long COVID,” said Dr. Fearnley. “However, NHS employers are legally required to implement only ‘reasonable adjustments,’ and so things such as extended phased return or adjustments to shift patterns are not always being facilitated. Instead, an increasing number of employers are choosing to terminate contracts.”
Raymond Agius, the BMA’s occupational medicine committee cochair, also put the blame on inadequate safety measures for doctors. Those inadequate measures persist to this day, inasmuch as U.K. hospitals have dropped masking requirements.
“During the COVID-19 pandemic, doctors were left exposed and unprotected at work,” he said in a BMA press release. “They often did not have access to the right PPE. ... Too many risk assessments of workplaces and especially of vulnerable doctors were not undertaken.”
A small minority of doctors who were surveyed said they had access to respiratory protective equipment about the time they contracted COVID-19. Only 11% had access to an FFP2 respirator (the equivalent of an N95 mask); 16% had an FFP3 respirator (the equivalent of an N99 mask).
To date, the British government hasn’t issued much of a response to the survey, saying only that it has invested more than ₤50 million to better understand long COVID.
A version of this article first appeared on Medscape.com.
Crippling symptoms, lost careers, and eroded incomes: This is the harsh reality for doctors suffering with long COVID, according to the first major survey of physicians with the condition.
The survey, conducted by the British Medical Association and the Long COVID Doctors for Action support group, sheds light on the lingering effects of long COVID on more than 600 chronically ill and disabled doctors with the condition. It also spotlights what they describe as a lack of medical and financial support from their government and employers at the National Health Service.
“We feel betrayed and abandoned,” said Kelly Fearnley, MBChB, chair and cofounder of Long COVID Doctors for Action. “At a time of national crisis, when health care workers were asked to step up, we did. When the nation needed us, we stepped up. We put our lives on the line. We put our families’ lives on the line. And now that we are injured after knowingly being unprotected and deliberately and repeatedly exposed to a level 3 biohazard, we now find ourselves in this position.”
Dr. Fearnley fell ill while working in a hospital’s COVID ward in November 2020. She is one of an estimated 2 million people in the United Kingdom – including thousands of NHS employees – with long COVID. She hasn’t been able to return to work in nearly 3 years.
Long COVID affects more than 65 million people worldwide. It is estimated that 1 in 10 people infected with the virus develop long-term symptoms. In the United Kingdom, health care and social care workers are seven times more likely to have had severe COVID-19 than other types of employees.
Doctors responding to the BMA survey reported a wide range of long COVID symptoms, including fatigue, headaches, muscular pain, nerve damage, joint pain, and respiratory problems.
Among the survey’s key findings, 60% of doctors said long COVID has affected their ability to carry out day-to-day tasks on a regular basis. Almost one in five (18%) said they were no longer able to work, while fewer than one in three (31%) were working full time. This compares with more than half (57%) of respondents working full time before the onset of their COVID illness – a decline of 46%.
Nearly half (48%) of respondents said they have experienced some form of loss of earnings as a result of long COVID, and almost half of the doctors were never referred to an NHS long COVID clinic. The survey included the following first-person accounts from doctors living with the condition.
- One doctor said: “I nearly lost my life, my home, my partner and my career. I have received little support to help keep these. The impact on my mental health nearly cost [me] my life again.”
- A senior consulting physician commented: “Life is absolutely miserable. Every day is a struggle. I wake up exhausted, the insomnia and night terrors are horrendous as I live through my worst fears every night. Any activity such as eating meals, washing, etc., will mean I have to go to bed for a few hours. I am unable to look after myself or my child, exercise or maintain social relationships. I have no financial security. Long COVID has totally destroyed my life.”
- A salaried general practitioner said: “I can no longer work, finances are ruined. I didn’t have employment protection so am now unemployed and penniless.”
Calls for action from the BMA include the following:
- Financial support for doctors and health care staff with long COVID.
- The recognition of long COVID as an occupational disease among health care workers, along with a definition of the condition that covers all of the debilitating disease’s symptoms.
- Improved access to physical and mental health services to help comprehensive assessment, investigations, and treatment.
- Greater workplace protection for health care staff who risk their lives for others.
- Better support for long COVID sufferers to return to work safely if they can, including a flexible approach to the use of workplace adjustments.
“One would think, given the circumstances under which we fell ill and current workforce shortages, NHS employers would be eager to do everything to facilitate the return to work of people with long COVID,” said Dr. Fearnley. “However, NHS employers are legally required to implement only ‘reasonable adjustments,’ and so things such as extended phased return or adjustments to shift patterns are not always being facilitated. Instead, an increasing number of employers are choosing to terminate contracts.”
Raymond Agius, the BMA’s occupational medicine committee cochair, also put the blame on inadequate safety measures for doctors. Those inadequate measures persist to this day, inasmuch as U.K. hospitals have dropped masking requirements.
“During the COVID-19 pandemic, doctors were left exposed and unprotected at work,” he said in a BMA press release. “They often did not have access to the right PPE. ... Too many risk assessments of workplaces and especially of vulnerable doctors were not undertaken.”
A small minority of doctors who were surveyed said they had access to respiratory protective equipment about the time they contracted COVID-19. Only 11% had access to an FFP2 respirator (the equivalent of an N95 mask); 16% had an FFP3 respirator (the equivalent of an N99 mask).
To date, the British government hasn’t issued much of a response to the survey, saying only that it has invested more than ₤50 million to better understand long COVID.
A version of this article first appeared on Medscape.com.
Crippling symptoms, lost careers, and eroded incomes: This is the harsh reality for doctors suffering with long COVID, according to the first major survey of physicians with the condition.
The survey, conducted by the British Medical Association and the Long COVID Doctors for Action support group, sheds light on the lingering effects of long COVID on more than 600 chronically ill and disabled doctors with the condition. It also spotlights what they describe as a lack of medical and financial support from their government and employers at the National Health Service.
“We feel betrayed and abandoned,” said Kelly Fearnley, MBChB, chair and cofounder of Long COVID Doctors for Action. “At a time of national crisis, when health care workers were asked to step up, we did. When the nation needed us, we stepped up. We put our lives on the line. We put our families’ lives on the line. And now that we are injured after knowingly being unprotected and deliberately and repeatedly exposed to a level 3 biohazard, we now find ourselves in this position.”
Dr. Fearnley fell ill while working in a hospital’s COVID ward in November 2020. She is one of an estimated 2 million people in the United Kingdom – including thousands of NHS employees – with long COVID. She hasn’t been able to return to work in nearly 3 years.
Long COVID affects more than 65 million people worldwide. It is estimated that 1 in 10 people infected with the virus develop long-term symptoms. In the United Kingdom, health care and social care workers are seven times more likely to have had severe COVID-19 than other types of employees.
Doctors responding to the BMA survey reported a wide range of long COVID symptoms, including fatigue, headaches, muscular pain, nerve damage, joint pain, and respiratory problems.
Among the survey’s key findings, 60% of doctors said long COVID has affected their ability to carry out day-to-day tasks on a regular basis. Almost one in five (18%) said they were no longer able to work, while fewer than one in three (31%) were working full time. This compares with more than half (57%) of respondents working full time before the onset of their COVID illness – a decline of 46%.
Nearly half (48%) of respondents said they have experienced some form of loss of earnings as a result of long COVID, and almost half of the doctors were never referred to an NHS long COVID clinic. The survey included the following first-person accounts from doctors living with the condition.
- One doctor said: “I nearly lost my life, my home, my partner and my career. I have received little support to help keep these. The impact on my mental health nearly cost [me] my life again.”
- A senior consulting physician commented: “Life is absolutely miserable. Every day is a struggle. I wake up exhausted, the insomnia and night terrors are horrendous as I live through my worst fears every night. Any activity such as eating meals, washing, etc., will mean I have to go to bed for a few hours. I am unable to look after myself or my child, exercise or maintain social relationships. I have no financial security. Long COVID has totally destroyed my life.”
- A salaried general practitioner said: “I can no longer work, finances are ruined. I didn’t have employment protection so am now unemployed and penniless.”
Calls for action from the BMA include the following:
- Financial support for doctors and health care staff with long COVID.
- The recognition of long COVID as an occupational disease among health care workers, along with a definition of the condition that covers all of the debilitating disease’s symptoms.
- Improved access to physical and mental health services to help comprehensive assessment, investigations, and treatment.
- Greater workplace protection for health care staff who risk their lives for others.
- Better support for long COVID sufferers to return to work safely if they can, including a flexible approach to the use of workplace adjustments.
“One would think, given the circumstances under which we fell ill and current workforce shortages, NHS employers would be eager to do everything to facilitate the return to work of people with long COVID,” said Dr. Fearnley. “However, NHS employers are legally required to implement only ‘reasonable adjustments,’ and so things such as extended phased return or adjustments to shift patterns are not always being facilitated. Instead, an increasing number of employers are choosing to terminate contracts.”
Raymond Agius, the BMA’s occupational medicine committee cochair, also put the blame on inadequate safety measures for doctors. Those inadequate measures persist to this day, inasmuch as U.K. hospitals have dropped masking requirements.
“During the COVID-19 pandemic, doctors were left exposed and unprotected at work,” he said in a BMA press release. “They often did not have access to the right PPE. ... Too many risk assessments of workplaces and especially of vulnerable doctors were not undertaken.”
A small minority of doctors who were surveyed said they had access to respiratory protective equipment about the time they contracted COVID-19. Only 11% had access to an FFP2 respirator (the equivalent of an N95 mask); 16% had an FFP3 respirator (the equivalent of an N99 mask).
To date, the British government hasn’t issued much of a response to the survey, saying only that it has invested more than ₤50 million to better understand long COVID.
A version of this article first appeared on Medscape.com.
Resident creates AI alternative to U.S. News med school ranking
For decades, pre-med students depended on the annual medical school rankings by U.S. News and World Report to decide where to apply for physician education. But after several prominent med schools pulled out of the rankings, one resident began experimenting with artificial intelligence (AI) to create an alternative.
Brandon Turner MD, MSc, a radiation oncology resident at Massachusetts General Hospital in Boston, developed a free do-it-yourself tool using AI that allows prospective students to rank medical schools based on considerations that are most important to them. His research was published online in JAMA Network Open.
“One of the flaws with conventional ranking systems is that the metrics used in these tools are weighted based on the preferences and views of the people who developed these rankings, but those may not work for everyone,” Dr. Turner told this news organization.
He explained that there are different types of metrics used in the U.S. News ranking: one for research and the other for primary care. “The research rankings carry the most prestige and are the ones that most people know about,” he explained. These metrics take into account factors such as how many grant dollars the medical school receives and the average size of those grants per faculty member, Dr. Turner said.
Admission metrics are also included – for example, the median grade point average or MCAT scores of students who have been accepted. “These don’t tell you anything about the research output of the school, only about how selective the school is,” he said.
Primary care metrics might focus on how many graduates of a given school go into primary care, or how other schools rate the quality of primary care training at a given school – a process called peer assessment, Dr. Turner said.
But even though these might be helpful, students may be more interested in the cost of attendance, average debt, representation of minorities, and how many graduates pass their boards, he said. “U.S. News metrics don’t capture these things, but I included them in my algorithm.”
A U.S. News spokesperson said that the publication continues to help students and their families make decisions about their future education. The spokesperson cited U.S. News’ explanation of how it calculates its rankings. “A school’s overall Best Medical Schools rank should be one consideration and not the lone determinant in where a student applies and accepts,” the article states.
Dr. Turner agreed ranking systems are a good starting point when researching med schools, “but the values reflected in the ranking may not reflect an individual’s goals.”
Tyra-Lee Brett, a premed student at the University of South Florida, Tampa, believes an additional tool for students to evaluate medical schools is needed – and she could potentially see herself using Dr. Turner’s creation.
Still, Ms. Brett, a premed trustee of the American Medical Student Association, doesn’t regard any ranking tool as the “be all and end all.” Rather, she feels that the most effective tool would be based on students’ lived experiences. The AMSA is developing a scorecard in which students grade schools based on their opinions about such issues as housing, family planning, and environmental health, she said.
No prior judgments
To develop his algorithm, Dr. Turner used a branch of AI called “unsupervised learning.” It doesn’t make a prior judgment about what the data should look like, Dr. Turner explained.
“You’re just analyzing natural trends within the data.”
The algorithm tries to find and discover clusters or patterns within the data. “It’s like saying to the algorithm: ‘I want you to tell me what schools you think should be grouped together based on the data I feed you,’ which is the data that the user selects based on his or her personal preferences.”
U.S. News has been transparent about the metrics it uses, Dr. Turner notes. “When I started looking into how rankings are developed, I saw that there was transparency, and the reasoning for choosing the metrics used to develop the ranking was pretty sound,” he said.
“But I didn’t see any justification as to why they chose the particular metrics and weighted them in the way that they did.”
Dr. Turner extracted data from the 2023 U.S. News report, which ranked 109 allopathic medical schools, and applied several scenarios to the results to create his alternative ranking system.
In one scenario, he used the same research metrics used by U.S. News, such as a peer research assessment, median federal research activity per full-time faculty member, median GPA, median MCAT, acceptance rate, and faculty-student ratio.
In another scenario, he included four additional metrics: debt, in-state cost of attendance, USMLE Step 1 passing rate, and percentage of underrepresented students with minority race or ethnicity at the school.
For example, a user can rank the importance of the diversity of the class, amount of debt students expect to incur, and amount of research funding the medical school receives. After selecting those factors, the tool generates tiered results displayed in a circle, a shape chosen to avoid the appearance of the hierarchy associated with traditional rankings, Dr. Turner said.
“A prospective student might not care about acceptance rates and MCAT scores, and instead cares about diversity and debt,” Dr. Turner said. He looks forward to extending this approach to the ranking of colleges as well.
‘Imperfect measures’
“The model and interesting online tool that Dr. Turner created allows a premed [student] to generate custom rankings that are in line with their own priorities,” said Christopher Worsham, MD, MPH, a critical care physician in Mass General’s division of pulmonary and critical care medicine.
But Dr. Worsham, also a teaching associate at Harvard Medical School’s department of health care policy, expressed concern that factors figuring into the rankings by U.S. News and Dr. Turner’s alternative “are imperfect measures of medical school quality.”
For example, a student interested in research might favor federal research funding in their customized rankings with Dr. Turner’s model. “But higher research funding doesn’t necessarily translate into a better education for students, particularly when differentiating between two major research systems,” Dr. Worsham noted.
Dr. Worsham added that neither ranking system accurately predicts the quality of doctors graduating from the schools. Instead, he’d like to see ranking systems based on which schools’ graduates deliver the best patient outcomes, whether that’s through direct patient care, impactful research, or leadership within the health care system.
Michael Sauder, PhD, professor of sociology at the University of Iowa, Iowa City, said the model could offer a valuable alternative to the U.S. News ranking system. It might help users develop their own criteria for determining the ranking of medical schools, which is a big improvement over a “one-size-fits-all” approach, Dr. Sauder said.
And Hanna Stotland, an admission consultant based in Chicago, noted that most students rely on rankings because they “don’t have the luxury of advisers who know the ins and outs of different medical schools.” Given the role that rankings play, Ms. Stotland expects that every new ranking tool will have some influence on students.
This tool in particular “has the potential to be useful for students who have identified values they want their medical school to share.” For example, students who care about racial diversity “could use it to easily identify schools that are successful on that metric,” Ms. Stotland said.
Sujay Ratna, a 2nd-year med student at Icahn School of Medicine at Mount Sinai in New York, said he considered the U.S. News ranking his “go-to tool” when he was applying to med school.
But after reading Dr. Turner’s article, the AMSA membership vice president tried the algorithm. “I definitely would have used it had it existed when I was thinking of what schools to apply to and what [schools] to attend.”
The study had no specific funding. Dr. Turner, Dr. Worsham, Dr. Sauder, Ms. Stotland, Ms. Brett, and Mr. Ratna report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For decades, pre-med students depended on the annual medical school rankings by U.S. News and World Report to decide where to apply for physician education. But after several prominent med schools pulled out of the rankings, one resident began experimenting with artificial intelligence (AI) to create an alternative.
Brandon Turner MD, MSc, a radiation oncology resident at Massachusetts General Hospital in Boston, developed a free do-it-yourself tool using AI that allows prospective students to rank medical schools based on considerations that are most important to them. His research was published online in JAMA Network Open.
“One of the flaws with conventional ranking systems is that the metrics used in these tools are weighted based on the preferences and views of the people who developed these rankings, but those may not work for everyone,” Dr. Turner told this news organization.
He explained that there are different types of metrics used in the U.S. News ranking: one for research and the other for primary care. “The research rankings carry the most prestige and are the ones that most people know about,” he explained. These metrics take into account factors such as how many grant dollars the medical school receives and the average size of those grants per faculty member, Dr. Turner said.
Admission metrics are also included – for example, the median grade point average or MCAT scores of students who have been accepted. “These don’t tell you anything about the research output of the school, only about how selective the school is,” he said.
Primary care metrics might focus on how many graduates of a given school go into primary care, or how other schools rate the quality of primary care training at a given school – a process called peer assessment, Dr. Turner said.
But even though these might be helpful, students may be more interested in the cost of attendance, average debt, representation of minorities, and how many graduates pass their boards, he said. “U.S. News metrics don’t capture these things, but I included them in my algorithm.”
A U.S. News spokesperson said that the publication continues to help students and their families make decisions about their future education. The spokesperson cited U.S. News’ explanation of how it calculates its rankings. “A school’s overall Best Medical Schools rank should be one consideration and not the lone determinant in where a student applies and accepts,” the article states.
Dr. Turner agreed ranking systems are a good starting point when researching med schools, “but the values reflected in the ranking may not reflect an individual’s goals.”
Tyra-Lee Brett, a premed student at the University of South Florida, Tampa, believes an additional tool for students to evaluate medical schools is needed – and she could potentially see herself using Dr. Turner’s creation.
Still, Ms. Brett, a premed trustee of the American Medical Student Association, doesn’t regard any ranking tool as the “be all and end all.” Rather, she feels that the most effective tool would be based on students’ lived experiences. The AMSA is developing a scorecard in which students grade schools based on their opinions about such issues as housing, family planning, and environmental health, she said.
No prior judgments
To develop his algorithm, Dr. Turner used a branch of AI called “unsupervised learning.” It doesn’t make a prior judgment about what the data should look like, Dr. Turner explained.
“You’re just analyzing natural trends within the data.”
The algorithm tries to find and discover clusters or patterns within the data. “It’s like saying to the algorithm: ‘I want you to tell me what schools you think should be grouped together based on the data I feed you,’ which is the data that the user selects based on his or her personal preferences.”
U.S. News has been transparent about the metrics it uses, Dr. Turner notes. “When I started looking into how rankings are developed, I saw that there was transparency, and the reasoning for choosing the metrics used to develop the ranking was pretty sound,” he said.
“But I didn’t see any justification as to why they chose the particular metrics and weighted them in the way that they did.”
Dr. Turner extracted data from the 2023 U.S. News report, which ranked 109 allopathic medical schools, and applied several scenarios to the results to create his alternative ranking system.
In one scenario, he used the same research metrics used by U.S. News, such as a peer research assessment, median federal research activity per full-time faculty member, median GPA, median MCAT, acceptance rate, and faculty-student ratio.
In another scenario, he included four additional metrics: debt, in-state cost of attendance, USMLE Step 1 passing rate, and percentage of underrepresented students with minority race or ethnicity at the school.
For example, a user can rank the importance of the diversity of the class, amount of debt students expect to incur, and amount of research funding the medical school receives. After selecting those factors, the tool generates tiered results displayed in a circle, a shape chosen to avoid the appearance of the hierarchy associated with traditional rankings, Dr. Turner said.
“A prospective student might not care about acceptance rates and MCAT scores, and instead cares about diversity and debt,” Dr. Turner said. He looks forward to extending this approach to the ranking of colleges as well.
‘Imperfect measures’
“The model and interesting online tool that Dr. Turner created allows a premed [student] to generate custom rankings that are in line with their own priorities,” said Christopher Worsham, MD, MPH, a critical care physician in Mass General’s division of pulmonary and critical care medicine.
But Dr. Worsham, also a teaching associate at Harvard Medical School’s department of health care policy, expressed concern that factors figuring into the rankings by U.S. News and Dr. Turner’s alternative “are imperfect measures of medical school quality.”
For example, a student interested in research might favor federal research funding in their customized rankings with Dr. Turner’s model. “But higher research funding doesn’t necessarily translate into a better education for students, particularly when differentiating between two major research systems,” Dr. Worsham noted.
Dr. Worsham added that neither ranking system accurately predicts the quality of doctors graduating from the schools. Instead, he’d like to see ranking systems based on which schools’ graduates deliver the best patient outcomes, whether that’s through direct patient care, impactful research, or leadership within the health care system.
Michael Sauder, PhD, professor of sociology at the University of Iowa, Iowa City, said the model could offer a valuable alternative to the U.S. News ranking system. It might help users develop their own criteria for determining the ranking of medical schools, which is a big improvement over a “one-size-fits-all” approach, Dr. Sauder said.
And Hanna Stotland, an admission consultant based in Chicago, noted that most students rely on rankings because they “don’t have the luxury of advisers who know the ins and outs of different medical schools.” Given the role that rankings play, Ms. Stotland expects that every new ranking tool will have some influence on students.
This tool in particular “has the potential to be useful for students who have identified values they want their medical school to share.” For example, students who care about racial diversity “could use it to easily identify schools that are successful on that metric,” Ms. Stotland said.
Sujay Ratna, a 2nd-year med student at Icahn School of Medicine at Mount Sinai in New York, said he considered the U.S. News ranking his “go-to tool” when he was applying to med school.
But after reading Dr. Turner’s article, the AMSA membership vice president tried the algorithm. “I definitely would have used it had it existed when I was thinking of what schools to apply to and what [schools] to attend.”
The study had no specific funding. Dr. Turner, Dr. Worsham, Dr. Sauder, Ms. Stotland, Ms. Brett, and Mr. Ratna report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For decades, pre-med students depended on the annual medical school rankings by U.S. News and World Report to decide where to apply for physician education. But after several prominent med schools pulled out of the rankings, one resident began experimenting with artificial intelligence (AI) to create an alternative.
Brandon Turner MD, MSc, a radiation oncology resident at Massachusetts General Hospital in Boston, developed a free do-it-yourself tool using AI that allows prospective students to rank medical schools based on considerations that are most important to them. His research was published online in JAMA Network Open.
“One of the flaws with conventional ranking systems is that the metrics used in these tools are weighted based on the preferences and views of the people who developed these rankings, but those may not work for everyone,” Dr. Turner told this news organization.
He explained that there are different types of metrics used in the U.S. News ranking: one for research and the other for primary care. “The research rankings carry the most prestige and are the ones that most people know about,” he explained. These metrics take into account factors such as how many grant dollars the medical school receives and the average size of those grants per faculty member, Dr. Turner said.
Admission metrics are also included – for example, the median grade point average or MCAT scores of students who have been accepted. “These don’t tell you anything about the research output of the school, only about how selective the school is,” he said.
Primary care metrics might focus on how many graduates of a given school go into primary care, or how other schools rate the quality of primary care training at a given school – a process called peer assessment, Dr. Turner said.
But even though these might be helpful, students may be more interested in the cost of attendance, average debt, representation of minorities, and how many graduates pass their boards, he said. “U.S. News metrics don’t capture these things, but I included them in my algorithm.”
A U.S. News spokesperson said that the publication continues to help students and their families make decisions about their future education. The spokesperson cited U.S. News’ explanation of how it calculates its rankings. “A school’s overall Best Medical Schools rank should be one consideration and not the lone determinant in where a student applies and accepts,” the article states.
Dr. Turner agreed ranking systems are a good starting point when researching med schools, “but the values reflected in the ranking may not reflect an individual’s goals.”
Tyra-Lee Brett, a premed student at the University of South Florida, Tampa, believes an additional tool for students to evaluate medical schools is needed – and she could potentially see herself using Dr. Turner’s creation.
Still, Ms. Brett, a premed trustee of the American Medical Student Association, doesn’t regard any ranking tool as the “be all and end all.” Rather, she feels that the most effective tool would be based on students’ lived experiences. The AMSA is developing a scorecard in which students grade schools based on their opinions about such issues as housing, family planning, and environmental health, she said.
No prior judgments
To develop his algorithm, Dr. Turner used a branch of AI called “unsupervised learning.” It doesn’t make a prior judgment about what the data should look like, Dr. Turner explained.
“You’re just analyzing natural trends within the data.”
The algorithm tries to find and discover clusters or patterns within the data. “It’s like saying to the algorithm: ‘I want you to tell me what schools you think should be grouped together based on the data I feed you,’ which is the data that the user selects based on his or her personal preferences.”
U.S. News has been transparent about the metrics it uses, Dr. Turner notes. “When I started looking into how rankings are developed, I saw that there was transparency, and the reasoning for choosing the metrics used to develop the ranking was pretty sound,” he said.
“But I didn’t see any justification as to why they chose the particular metrics and weighted them in the way that they did.”
Dr. Turner extracted data from the 2023 U.S. News report, which ranked 109 allopathic medical schools, and applied several scenarios to the results to create his alternative ranking system.
In one scenario, he used the same research metrics used by U.S. News, such as a peer research assessment, median federal research activity per full-time faculty member, median GPA, median MCAT, acceptance rate, and faculty-student ratio.
In another scenario, he included four additional metrics: debt, in-state cost of attendance, USMLE Step 1 passing rate, and percentage of underrepresented students with minority race or ethnicity at the school.
For example, a user can rank the importance of the diversity of the class, amount of debt students expect to incur, and amount of research funding the medical school receives. After selecting those factors, the tool generates tiered results displayed in a circle, a shape chosen to avoid the appearance of the hierarchy associated with traditional rankings, Dr. Turner said.
“A prospective student might not care about acceptance rates and MCAT scores, and instead cares about diversity and debt,” Dr. Turner said. He looks forward to extending this approach to the ranking of colleges as well.
‘Imperfect measures’
“The model and interesting online tool that Dr. Turner created allows a premed [student] to generate custom rankings that are in line with their own priorities,” said Christopher Worsham, MD, MPH, a critical care physician in Mass General’s division of pulmonary and critical care medicine.
But Dr. Worsham, also a teaching associate at Harvard Medical School’s department of health care policy, expressed concern that factors figuring into the rankings by U.S. News and Dr. Turner’s alternative “are imperfect measures of medical school quality.”
For example, a student interested in research might favor federal research funding in their customized rankings with Dr. Turner’s model. “But higher research funding doesn’t necessarily translate into a better education for students, particularly when differentiating between two major research systems,” Dr. Worsham noted.
Dr. Worsham added that neither ranking system accurately predicts the quality of doctors graduating from the schools. Instead, he’d like to see ranking systems based on which schools’ graduates deliver the best patient outcomes, whether that’s through direct patient care, impactful research, or leadership within the health care system.
Michael Sauder, PhD, professor of sociology at the University of Iowa, Iowa City, said the model could offer a valuable alternative to the U.S. News ranking system. It might help users develop their own criteria for determining the ranking of medical schools, which is a big improvement over a “one-size-fits-all” approach, Dr. Sauder said.
And Hanna Stotland, an admission consultant based in Chicago, noted that most students rely on rankings because they “don’t have the luxury of advisers who know the ins and outs of different medical schools.” Given the role that rankings play, Ms. Stotland expects that every new ranking tool will have some influence on students.
This tool in particular “has the potential to be useful for students who have identified values they want their medical school to share.” For example, students who care about racial diversity “could use it to easily identify schools that are successful on that metric,” Ms. Stotland said.
Sujay Ratna, a 2nd-year med student at Icahn School of Medicine at Mount Sinai in New York, said he considered the U.S. News ranking his “go-to tool” when he was applying to med school.
But after reading Dr. Turner’s article, the AMSA membership vice president tried the algorithm. “I definitely would have used it had it existed when I was thinking of what schools to apply to and what [schools] to attend.”
The study had no specific funding. Dr. Turner, Dr. Worsham, Dr. Sauder, Ms. Stotland, Ms. Brett, and Mr. Ratna report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Rise in number of unclaimed dead bodies used in medical schools
An increasing number of unclaimed dead bodies went to help train medical students in Texas between 2017 and 2021, new research reveals.
Investigators did not expect to see such an increase, said lead author Eli Shupe, PhD, assistant professor in the department of philosophy and humanities at the University of Texas at Arlington.
The numbers jumped from 64 unclaimed bodies to 446 bodies annually over those 5 years. “People are usually under the impression that this is something that either doesn’t happen anymore or it’s on the decline as more people step up to be willed body donors,” said Dr. Shupe, who is also codirector of the medical humanities and bioethics program at UTA.
The study findings were published in JAMA as a research letter. Researchers said that the number of unclaimed bodies – those not claimed by next of kin for burial or cremation – has dropped significantly across the United States since the middle of the 20th century.
Some people don’t want to discuss the practice because it is controversial, said Matthew DeCamp, MD, PhD, associate professor at the Center for Bioethics and Humanities and Division of General Internal Medicine, University of Colorado Anschutz Medical Campus, Aurora. “But ‘sweeping it under the rug’ means we miss the opportunity for dialogues about respect, consent, social justice, and so on – as well as the opportunity to change policy.”
The study included all medical schools in Texas, and researchers say it’s likely happening elsewhere in the United States and abroad. The practice is legal in most counties and states. One exception is New York, which passed a law in 2016 that does not allow unclaimed bodies to go to medical schools without prior written consent from the deceased.
“Although limited to one state, these findings suggest that use of unclaimed bodies may be both more common than we thought and increasing,” added Dr. DeCamp, who was not affiliated with the current study.
Even doctors can be split on the value to medical training versus the rights of the dead. “I know that medical professionals are divided on the role of dissection and anatomy learning and its necessity,” Dr. Shupe said. She predicted working with cadavers in medical schools will probably continue for the foreseeable future.
The marginalized and the vulnerable
So who are the unclaimed? They can include those who are unhoused and those who do not leave enough money to cover cost of burial or cremation. In some cases, they don’t have a next of kin or their next of kin is unwilling or unable to pay for their burial or cremation.
“Predominantly, these are going to be people who are poor or members of marginalized or vulnerable populations,” Dr. Shupe said. She estimated that about 80% of the people who die in poverty in her region, the Dallas–Fort Worth area, are Black or Hispanic individuals.
“It is alarming that we are going in the wrong direction when it comes to the increasing utility of unclaimed bodies,” said Joy Balta, PhD, associate professor of anatomy and founding director of Anatomy Learning Institute at Point Loma Nazarene University, San Diego, when asked to comment on the study. The hope is to rely solely on donated human bodies to ensure that donors have provided informed consent for their use in education, research, and clinical training.
“These unclaimed bodies did not provide any consent, [which] raises ethical questions,” Dr. Balta said.
Key findings
In Texas in 2021, 43% of the cadavers in 14 medical schools studied came from unclaimed bodies. A total 14% of schools reported that they accepted unclaimed bodies, 28% possibly accepted them because they were transferred from institutions that use them, and the remaining 57% do not accept unclaimed bodies.
The total number and proportion of unclaimed bodies going to medical education in the study increased during the study. The 14% in 2021 was a jump from 2% in 2017, for example.
The 14 medical schools studied included both public and private institutions. The investigators also looked at data from the Texas State Anatomical Board, which tracks how cadavers are attained and distributed in the state, including how many began as unclaimed bodies.
Legal in most jurisdictions
Dr. Shupe first learned about what can happen to unclaimed bodies as a hospice volunteer. She was accompanying the hospice chaplain one day who said: “Poor Mr. Smith [not his real name] doesn’t have long, and then he’s off to the medical school.” Dr. Shupe asked what the chaplain meant because she was unaware of the practice.
“I stumbled on this by chance, and it ended up being a really fruitful research area,” she added.
The bigger picture
Greater awareness is needed and there is not a lot of research out there, Dr. Shupe said. One exception is a 2018 study of medical schools nationwide that found 12.4% reported possible use of unclaimed bodies.
Dr. DeCamp, an author of that previous research, said: “Knowing this practice continues is the most important thing for doctors and medical students to know.”
It remains unclear whether the COVID pandemic or the opioid epidemic contributed to the rise of unclaimed bodies going to medical training. That is a question for future study, Dr. Shupe said.
Most bodies willingly donated
The majority of cadavers that go to medical training in the United States are ‘full body donors,’ people or relatives who agree to voluntarily send a body to medical schools. “We are fortunate to have a lot of people who are willing to become whole body donors,” she said.
Greater awareness about how donated cadavers could make a difference to further increase willful donations, Dr. Shupe said. “Honoring those gifts by allowing them to help train the next generation of doctors is a wonderful thing.”
A May 2023 study from Dr. Balta and colleagues on body donation programs in the United States “found that the number of whole-body donations have decreased in some states and the numbers are not enough to meet the needs for education, research and clinical training,” Dr. Balta said. This could explain the increasing use of unclaimed bodies.
“Some medical schools have explicit educational interventions on this topic, and formally recognize the unclaimed at anatomical gift ceremonies,” Dr. DeCamp said. “More should.”
Research support was provided by the UTA. Dr. Shupe, Dr. Balta, and Dr. DeCamp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
An increasing number of unclaimed dead bodies went to help train medical students in Texas between 2017 and 2021, new research reveals.
Investigators did not expect to see such an increase, said lead author Eli Shupe, PhD, assistant professor in the department of philosophy and humanities at the University of Texas at Arlington.
The numbers jumped from 64 unclaimed bodies to 446 bodies annually over those 5 years. “People are usually under the impression that this is something that either doesn’t happen anymore or it’s on the decline as more people step up to be willed body donors,” said Dr. Shupe, who is also codirector of the medical humanities and bioethics program at UTA.
The study findings were published in JAMA as a research letter. Researchers said that the number of unclaimed bodies – those not claimed by next of kin for burial or cremation – has dropped significantly across the United States since the middle of the 20th century.
Some people don’t want to discuss the practice because it is controversial, said Matthew DeCamp, MD, PhD, associate professor at the Center for Bioethics and Humanities and Division of General Internal Medicine, University of Colorado Anschutz Medical Campus, Aurora. “But ‘sweeping it under the rug’ means we miss the opportunity for dialogues about respect, consent, social justice, and so on – as well as the opportunity to change policy.”
The study included all medical schools in Texas, and researchers say it’s likely happening elsewhere in the United States and abroad. The practice is legal in most counties and states. One exception is New York, which passed a law in 2016 that does not allow unclaimed bodies to go to medical schools without prior written consent from the deceased.
“Although limited to one state, these findings suggest that use of unclaimed bodies may be both more common than we thought and increasing,” added Dr. DeCamp, who was not affiliated with the current study.
Even doctors can be split on the value to medical training versus the rights of the dead. “I know that medical professionals are divided on the role of dissection and anatomy learning and its necessity,” Dr. Shupe said. She predicted working with cadavers in medical schools will probably continue for the foreseeable future.
The marginalized and the vulnerable
So who are the unclaimed? They can include those who are unhoused and those who do not leave enough money to cover cost of burial or cremation. In some cases, they don’t have a next of kin or their next of kin is unwilling or unable to pay for their burial or cremation.
“Predominantly, these are going to be people who are poor or members of marginalized or vulnerable populations,” Dr. Shupe said. She estimated that about 80% of the people who die in poverty in her region, the Dallas–Fort Worth area, are Black or Hispanic individuals.
“It is alarming that we are going in the wrong direction when it comes to the increasing utility of unclaimed bodies,” said Joy Balta, PhD, associate professor of anatomy and founding director of Anatomy Learning Institute at Point Loma Nazarene University, San Diego, when asked to comment on the study. The hope is to rely solely on donated human bodies to ensure that donors have provided informed consent for their use in education, research, and clinical training.
“These unclaimed bodies did not provide any consent, [which] raises ethical questions,” Dr. Balta said.
Key findings
In Texas in 2021, 43% of the cadavers in 14 medical schools studied came from unclaimed bodies. A total 14% of schools reported that they accepted unclaimed bodies, 28% possibly accepted them because they were transferred from institutions that use them, and the remaining 57% do not accept unclaimed bodies.
The total number and proportion of unclaimed bodies going to medical education in the study increased during the study. The 14% in 2021 was a jump from 2% in 2017, for example.
The 14 medical schools studied included both public and private institutions. The investigators also looked at data from the Texas State Anatomical Board, which tracks how cadavers are attained and distributed in the state, including how many began as unclaimed bodies.
Legal in most jurisdictions
Dr. Shupe first learned about what can happen to unclaimed bodies as a hospice volunteer. She was accompanying the hospice chaplain one day who said: “Poor Mr. Smith [not his real name] doesn’t have long, and then he’s off to the medical school.” Dr. Shupe asked what the chaplain meant because she was unaware of the practice.
“I stumbled on this by chance, and it ended up being a really fruitful research area,” she added.
The bigger picture
Greater awareness is needed and there is not a lot of research out there, Dr. Shupe said. One exception is a 2018 study of medical schools nationwide that found 12.4% reported possible use of unclaimed bodies.
Dr. DeCamp, an author of that previous research, said: “Knowing this practice continues is the most important thing for doctors and medical students to know.”
It remains unclear whether the COVID pandemic or the opioid epidemic contributed to the rise of unclaimed bodies going to medical training. That is a question for future study, Dr. Shupe said.
Most bodies willingly donated
The majority of cadavers that go to medical training in the United States are ‘full body donors,’ people or relatives who agree to voluntarily send a body to medical schools. “We are fortunate to have a lot of people who are willing to become whole body donors,” she said.
Greater awareness about how donated cadavers could make a difference to further increase willful donations, Dr. Shupe said. “Honoring those gifts by allowing them to help train the next generation of doctors is a wonderful thing.”
A May 2023 study from Dr. Balta and colleagues on body donation programs in the United States “found that the number of whole-body donations have decreased in some states and the numbers are not enough to meet the needs for education, research and clinical training,” Dr. Balta said. This could explain the increasing use of unclaimed bodies.
“Some medical schools have explicit educational interventions on this topic, and formally recognize the unclaimed at anatomical gift ceremonies,” Dr. DeCamp said. “More should.”
Research support was provided by the UTA. Dr. Shupe, Dr. Balta, and Dr. DeCamp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
An increasing number of unclaimed dead bodies went to help train medical students in Texas between 2017 and 2021, new research reveals.
Investigators did not expect to see such an increase, said lead author Eli Shupe, PhD, assistant professor in the department of philosophy and humanities at the University of Texas at Arlington.
The numbers jumped from 64 unclaimed bodies to 446 bodies annually over those 5 years. “People are usually under the impression that this is something that either doesn’t happen anymore or it’s on the decline as more people step up to be willed body donors,” said Dr. Shupe, who is also codirector of the medical humanities and bioethics program at UTA.
The study findings were published in JAMA as a research letter. Researchers said that the number of unclaimed bodies – those not claimed by next of kin for burial or cremation – has dropped significantly across the United States since the middle of the 20th century.
Some people don’t want to discuss the practice because it is controversial, said Matthew DeCamp, MD, PhD, associate professor at the Center for Bioethics and Humanities and Division of General Internal Medicine, University of Colorado Anschutz Medical Campus, Aurora. “But ‘sweeping it under the rug’ means we miss the opportunity for dialogues about respect, consent, social justice, and so on – as well as the opportunity to change policy.”
The study included all medical schools in Texas, and researchers say it’s likely happening elsewhere in the United States and abroad. The practice is legal in most counties and states. One exception is New York, which passed a law in 2016 that does not allow unclaimed bodies to go to medical schools without prior written consent from the deceased.
“Although limited to one state, these findings suggest that use of unclaimed bodies may be both more common than we thought and increasing,” added Dr. DeCamp, who was not affiliated with the current study.
Even doctors can be split on the value to medical training versus the rights of the dead. “I know that medical professionals are divided on the role of dissection and anatomy learning and its necessity,” Dr. Shupe said. She predicted working with cadavers in medical schools will probably continue for the foreseeable future.
The marginalized and the vulnerable
So who are the unclaimed? They can include those who are unhoused and those who do not leave enough money to cover cost of burial or cremation. In some cases, they don’t have a next of kin or their next of kin is unwilling or unable to pay for their burial or cremation.
“Predominantly, these are going to be people who are poor or members of marginalized or vulnerable populations,” Dr. Shupe said. She estimated that about 80% of the people who die in poverty in her region, the Dallas–Fort Worth area, are Black or Hispanic individuals.
“It is alarming that we are going in the wrong direction when it comes to the increasing utility of unclaimed bodies,” said Joy Balta, PhD, associate professor of anatomy and founding director of Anatomy Learning Institute at Point Loma Nazarene University, San Diego, when asked to comment on the study. The hope is to rely solely on donated human bodies to ensure that donors have provided informed consent for their use in education, research, and clinical training.
“These unclaimed bodies did not provide any consent, [which] raises ethical questions,” Dr. Balta said.
Key findings
In Texas in 2021, 43% of the cadavers in 14 medical schools studied came from unclaimed bodies. A total 14% of schools reported that they accepted unclaimed bodies, 28% possibly accepted them because they were transferred from institutions that use them, and the remaining 57% do not accept unclaimed bodies.
The total number and proportion of unclaimed bodies going to medical education in the study increased during the study. The 14% in 2021 was a jump from 2% in 2017, for example.
The 14 medical schools studied included both public and private institutions. The investigators also looked at data from the Texas State Anatomical Board, which tracks how cadavers are attained and distributed in the state, including how many began as unclaimed bodies.
Legal in most jurisdictions
Dr. Shupe first learned about what can happen to unclaimed bodies as a hospice volunteer. She was accompanying the hospice chaplain one day who said: “Poor Mr. Smith [not his real name] doesn’t have long, and then he’s off to the medical school.” Dr. Shupe asked what the chaplain meant because she was unaware of the practice.
“I stumbled on this by chance, and it ended up being a really fruitful research area,” she added.
The bigger picture
Greater awareness is needed and there is not a lot of research out there, Dr. Shupe said. One exception is a 2018 study of medical schools nationwide that found 12.4% reported possible use of unclaimed bodies.
Dr. DeCamp, an author of that previous research, said: “Knowing this practice continues is the most important thing for doctors and medical students to know.”
It remains unclear whether the COVID pandemic or the opioid epidemic contributed to the rise of unclaimed bodies going to medical training. That is a question for future study, Dr. Shupe said.
Most bodies willingly donated
The majority of cadavers that go to medical training in the United States are ‘full body donors,’ people or relatives who agree to voluntarily send a body to medical schools. “We are fortunate to have a lot of people who are willing to become whole body donors,” she said.
Greater awareness about how donated cadavers could make a difference to further increase willful donations, Dr. Shupe said. “Honoring those gifts by allowing them to help train the next generation of doctors is a wonderful thing.”
A May 2023 study from Dr. Balta and colleagues on body donation programs in the United States “found that the number of whole-body donations have decreased in some states and the numbers are not enough to meet the needs for education, research and clinical training,” Dr. Balta said. This could explain the increasing use of unclaimed bodies.
“Some medical schools have explicit educational interventions on this topic, and formally recognize the unclaimed at anatomical gift ceremonies,” Dr. DeCamp said. “More should.”
Research support was provided by the UTA. Dr. Shupe, Dr. Balta, and Dr. DeCamp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA