User login
Perioperative ACE‐Inhibitor Management
The management of perioperative medications is a tale of progressive scientific enquiry. Although long‐term use of agents such as aspirin or statins improves clinical outcomes, use during surgery ranges from problematic to protective. The delicate balance between proven long‐term benefits in the nonoperative setting versus short‐term uncertainty in the perioperative setting must be assessed using a lens that incorporates patient risk, surgical process, and pharmacodynamic principles. Advances in our understanding of perioperative physiology, coupled with robust clinical and outcome data, have led to new knowledge and insights regarding how best to manage these medications. As well illustrated by the near 180 change in the use of perioperative ‐blockers to prevent adverse cardiovascular outcomes, these new data have led to substantial progress in surgical safety and patient outcomes.
In this issue of the Journal of Hospital Medicine, 2 retrospective cohort studies add to this growing body of evidence by examining risks associated with use of angiotensin‐converting enzyme (ACE) inhibitors in the perioperative setting. In the first study, conducted at a single academic medical center, Nielson and colleagues evaluate the association of preoperative ACE‐inhibitor use with hypotension and acute kidney injury in patients undergoing major elective orthopedic surgery.[1] The authors report that patients receiving ACE‐inhibitors were not only more likely to experience hypotension after induction of anesthesia (12.2% vs 6.7%, P = 0.005), but also were more likely to develop postoperative acute kidney injury (odds ratio [OR]: 2.68, 95% confidence interval [CI]: 1.25‐1.99). In the second study which focused solely on patients who were receiving preoperative ACE‐inhibitor therapy, Mudumbai and colleagues used a national Veterans Affairs database to examine the association between failure to resume ACE‐inhibitor treatment after surgery and outcomes at 30 days.[2] The authors found that failure to resume treatment 14 days after surgery was not only common (affecting 1 in 4 patients), but was also associated with increased 30‐day mortality (hazard ratio: 3.44, 95% CI: 3.30‐3.60).[2] Taken together, these 2 studies shed new light on clinical practice and policy implications for the use of these agents in the surgical period. Both sets of authors should be congratulated on moving the needle forward in this enquiry.
ACE‐inhibitors physiologically mediate their effects by preventing the formation of the potent vasoconstrictor angiotensin‐II from its precursor angiotensin‐I. In doing so, they decrease arterial resistance, increase venous capacitance, decrease glomerular filtration pressure, and promote natriuresis. These vascular effects have key benefits for the management of a number of chronic diseases including hypertension, congestive heart failure, and diabetic nephropathy. However, these clinical alterations are often problematic in the perioperative setting. For example, ACE‐inhibitors may cause vasoplegia during anesthetic administration, commonly manifested as hypotension during induction.[3] This hemodynamic alteration has been viewed as being so precarious, that some authors recommend withholding ACE‐inhibitors prior to major cardiovascular procedures such as coronary artery bypass grafting.[4] Although hypotension leads to management challenges (often increasing vasoconstrictor requirements), several studies report that ACE‐inhibitor use during surgery may also be associated with increased risks of acute kidney injury and mortality.[5, 6] However, despite these data, existing literature has not shown a consistent association between ACE‐inhibitor use and adverse postoperative outcomes. For example, a propensity‐matched cohort study of 79,228 patients at the Cleveland Clinic found no difference in hemodynamic characteristics, vasopressor requirements, or cardiorespiratory complications among patients who were or were not using ACE‐inhibitors during noncardiac surgery.[7] Furthermore, some research has also found that withdrawal of ACE‐inhibitors may itself lead to harm. For instance, a contemporary study reported that withdrawal of ACE‐inhibitor therapy in patients undergoing coronary artery bypass was associated with increased in‐hospital cardiovascular events.[8] Given this uncertainty, it is not surprising that management of ACE‐inhibitors in the perioperative period remains a subject of ongoing controversy with clinical reviews often recommending consideration of the risks and benefits associated with use of these agents.[9]
It is important to note that driver of this ambiguity is the very design of relevant studies. For instance, studies that focus on patients undergoing coronary artery bypass grafting surgery have limited external validity, as these patients are very different from those who undergo elective hip or knee replacement (such as those included by Nielsen and colleagues). Additionally, many studies suffer from methodological constraints or biases. For instance, retrospective observational studies often suffer from selection bias and residual confounding; that is, the individuals chosen for inclusion in the study and the variables available for analysis are often limited by available data, curtailing the conclusions that can be generated. Although the use of multivariable regression or propensity score techniques helps address these limitations, residual confounding by unmeasured variables always remains a threat to statistical inference. The potential influence for this bias is particularly relevant when examining the results of the study by Nielsen and colleagues. Another important limitation is survivor bias, a problem inherent in the study by Mudumbai and colleagues. Put simply, for a patient to resume ACE‐inhibitors after surgery, this same patient, must also survive for at least 15 to 30 days after surgery. Thus, for some patients, failure to resume this treatment may simply be a marker of early mortality rather than failure to resume the ACE itself. The potential influence of this bias is supported by an included sensitivity analysis, where a large change in the adjusted OR was observed when patients suffering early mortality were excluded. This swing in effect size suggests that biases related to comparing patients who survived to those who did not with respect to ACE use may, in part, account for the results of the study.
These limitations aside, the studies brought forth by both authors help inform practice with respect to the use of these agents during surgery. In this context, 3 paradigms are relevant for practicing hospitalists.
First, if a patient is maintained on an ACE‐inhibitor before surgery, should the medication be temporarily held before surgery to minimize hypotension during anesthesia? The study by Nielsen and colleagues (comparing those on ACE‐inhibitor treatment to those without), in addition to the evidence generated from other studies in this area[10, 11, 12] suggest that this is a rational decision. Although the existence of a withdrawal state from abrupt cessation of ACE‐inhibitor use is theoretically plausible, this has yet to be reliably reported in the literature. Given the short half‐life of most ACE‐inhibitors, cessation 24 hours before surgery appears to be the most pragmatic clinical approach.
Second, if a patient is on an ACE‐inhibitor before surgery, when should the medication be resumed after surgery? The findings from the study by Mudumbai and colleagues, in addition to contemporary evidence,[7, 8, 13] support the resumption of these agents as soon as possible following operative intervention. Once hemodynamic stability and volume status have been assured, risks associated with postoperative ACE‐inhibitor use appear to be outweighed by benefits, though specific care is likely necessary in those with preexisting renal dysfunction.[14] A program that ensures reconciliation of medications in the postoperative setting may be valuable in ensuring that such treatment is restarted.
Third, if a patient is not on ACE‐inhibitor therapy, should this be started before surgery for perioperative or long‐term benefit? Although neither study examines this issue, the potential for significant risk make this an unattractive option. Future interventional studies with thoughtfully weighed safety parameters may be necessary to assess whether such a paradigm may be valuable.
The studies included in this issue of the Journal of Hospital Medicine suggest that the use of ACE‐inhibitors during the perioperative period may be considered a function of time and place. Resuming ACE‐inhibitors and cessation of treatment at specific intervals in relation to surgery can help ensure positive outcomes. Hospitalists have an important role in this regard, as they are ideally situated to manage these agents in the many patients undergoing surgery across the United States.
Disclosures: Dr. Wijeysundera is supported by a Clinician‐Scientist Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto. Dr. Chopra is supported by a Career‐Development Award (1K08HS022835‐01) from the Agency of Healthcare Research and Quality.
- , , , . Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283–288.
- , , , , , . Thirty‐day mortality risk associated with postoperative nonresumption of angiotensin‐converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289–296.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , , . Angiotensin‐converting enzyme inhibitors increase vasoconstrictor requirements after cardiopulmonary bypass. Anesth Analg. 1995;80(3):473–479.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57(8):736–744.
- , , , , , . Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552–560.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87(1029):472–481.
- , , , et al; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , , , . Effects of renin‐angiotensin system inhibitors on the occurrence of acute kidney injury following off‐pump coronary artery bypass grafting. Circulation J. 2010;74(9):1852–1858.
- , , , , , . Prophylactic vasopressin in patients receiving the angiotensin‐converting enzyme inhibitor ramipril undergoing coronary artery bypass graft surgery. J Cardiothorac Vasc Anesth. 2010;24(2):230–238.
- , , , , . Neither diabetes nor glucose‐lowering drugs are associated with mortality after noncardiac surgery in patients with coronary artery disease or heart failure. Can J Cardiol. 2013;29(4):423–428.
- , , , , , . Interventions for protecting renal function in the perioperative period. Cochrane Database Syst Rev. 2008;(4):CD003590.
The management of perioperative medications is a tale of progressive scientific enquiry. Although long‐term use of agents such as aspirin or statins improves clinical outcomes, use during surgery ranges from problematic to protective. The delicate balance between proven long‐term benefits in the nonoperative setting versus short‐term uncertainty in the perioperative setting must be assessed using a lens that incorporates patient risk, surgical process, and pharmacodynamic principles. Advances in our understanding of perioperative physiology, coupled with robust clinical and outcome data, have led to new knowledge and insights regarding how best to manage these medications. As well illustrated by the near 180 change in the use of perioperative ‐blockers to prevent adverse cardiovascular outcomes, these new data have led to substantial progress in surgical safety and patient outcomes.
In this issue of the Journal of Hospital Medicine, 2 retrospective cohort studies add to this growing body of evidence by examining risks associated with use of angiotensin‐converting enzyme (ACE) inhibitors in the perioperative setting. In the first study, conducted at a single academic medical center, Nielson and colleagues evaluate the association of preoperative ACE‐inhibitor use with hypotension and acute kidney injury in patients undergoing major elective orthopedic surgery.[1] The authors report that patients receiving ACE‐inhibitors were not only more likely to experience hypotension after induction of anesthesia (12.2% vs 6.7%, P = 0.005), but also were more likely to develop postoperative acute kidney injury (odds ratio [OR]: 2.68, 95% confidence interval [CI]: 1.25‐1.99). In the second study which focused solely on patients who were receiving preoperative ACE‐inhibitor therapy, Mudumbai and colleagues used a national Veterans Affairs database to examine the association between failure to resume ACE‐inhibitor treatment after surgery and outcomes at 30 days.[2] The authors found that failure to resume treatment 14 days after surgery was not only common (affecting 1 in 4 patients), but was also associated with increased 30‐day mortality (hazard ratio: 3.44, 95% CI: 3.30‐3.60).[2] Taken together, these 2 studies shed new light on clinical practice and policy implications for the use of these agents in the surgical period. Both sets of authors should be congratulated on moving the needle forward in this enquiry.
ACE‐inhibitors physiologically mediate their effects by preventing the formation of the potent vasoconstrictor angiotensin‐II from its precursor angiotensin‐I. In doing so, they decrease arterial resistance, increase venous capacitance, decrease glomerular filtration pressure, and promote natriuresis. These vascular effects have key benefits for the management of a number of chronic diseases including hypertension, congestive heart failure, and diabetic nephropathy. However, these clinical alterations are often problematic in the perioperative setting. For example, ACE‐inhibitors may cause vasoplegia during anesthetic administration, commonly manifested as hypotension during induction.[3] This hemodynamic alteration has been viewed as being so precarious, that some authors recommend withholding ACE‐inhibitors prior to major cardiovascular procedures such as coronary artery bypass grafting.[4] Although hypotension leads to management challenges (often increasing vasoconstrictor requirements), several studies report that ACE‐inhibitor use during surgery may also be associated with increased risks of acute kidney injury and mortality.[5, 6] However, despite these data, existing literature has not shown a consistent association between ACE‐inhibitor use and adverse postoperative outcomes. For example, a propensity‐matched cohort study of 79,228 patients at the Cleveland Clinic found no difference in hemodynamic characteristics, vasopressor requirements, or cardiorespiratory complications among patients who were or were not using ACE‐inhibitors during noncardiac surgery.[7] Furthermore, some research has also found that withdrawal of ACE‐inhibitors may itself lead to harm. For instance, a contemporary study reported that withdrawal of ACE‐inhibitor therapy in patients undergoing coronary artery bypass was associated with increased in‐hospital cardiovascular events.[8] Given this uncertainty, it is not surprising that management of ACE‐inhibitors in the perioperative period remains a subject of ongoing controversy with clinical reviews often recommending consideration of the risks and benefits associated with use of these agents.[9]
It is important to note that driver of this ambiguity is the very design of relevant studies. For instance, studies that focus on patients undergoing coronary artery bypass grafting surgery have limited external validity, as these patients are very different from those who undergo elective hip or knee replacement (such as those included by Nielsen and colleagues). Additionally, many studies suffer from methodological constraints or biases. For instance, retrospective observational studies often suffer from selection bias and residual confounding; that is, the individuals chosen for inclusion in the study and the variables available for analysis are often limited by available data, curtailing the conclusions that can be generated. Although the use of multivariable regression or propensity score techniques helps address these limitations, residual confounding by unmeasured variables always remains a threat to statistical inference. The potential influence for this bias is particularly relevant when examining the results of the study by Nielsen and colleagues. Another important limitation is survivor bias, a problem inherent in the study by Mudumbai and colleagues. Put simply, for a patient to resume ACE‐inhibitors after surgery, this same patient, must also survive for at least 15 to 30 days after surgery. Thus, for some patients, failure to resume this treatment may simply be a marker of early mortality rather than failure to resume the ACE itself. The potential influence of this bias is supported by an included sensitivity analysis, where a large change in the adjusted OR was observed when patients suffering early mortality were excluded. This swing in effect size suggests that biases related to comparing patients who survived to those who did not with respect to ACE use may, in part, account for the results of the study.
These limitations aside, the studies brought forth by both authors help inform practice with respect to the use of these agents during surgery. In this context, 3 paradigms are relevant for practicing hospitalists.
First, if a patient is maintained on an ACE‐inhibitor before surgery, should the medication be temporarily held before surgery to minimize hypotension during anesthesia? The study by Nielsen and colleagues (comparing those on ACE‐inhibitor treatment to those without), in addition to the evidence generated from other studies in this area[10, 11, 12] suggest that this is a rational decision. Although the existence of a withdrawal state from abrupt cessation of ACE‐inhibitor use is theoretically plausible, this has yet to be reliably reported in the literature. Given the short half‐life of most ACE‐inhibitors, cessation 24 hours before surgery appears to be the most pragmatic clinical approach.
Second, if a patient is on an ACE‐inhibitor before surgery, when should the medication be resumed after surgery? The findings from the study by Mudumbai and colleagues, in addition to contemporary evidence,[7, 8, 13] support the resumption of these agents as soon as possible following operative intervention. Once hemodynamic stability and volume status have been assured, risks associated with postoperative ACE‐inhibitor use appear to be outweighed by benefits, though specific care is likely necessary in those with preexisting renal dysfunction.[14] A program that ensures reconciliation of medications in the postoperative setting may be valuable in ensuring that such treatment is restarted.
Third, if a patient is not on ACE‐inhibitor therapy, should this be started before surgery for perioperative or long‐term benefit? Although neither study examines this issue, the potential for significant risk make this an unattractive option. Future interventional studies with thoughtfully weighed safety parameters may be necessary to assess whether such a paradigm may be valuable.
The studies included in this issue of the Journal of Hospital Medicine suggest that the use of ACE‐inhibitors during the perioperative period may be considered a function of time and place. Resuming ACE‐inhibitors and cessation of treatment at specific intervals in relation to surgery can help ensure positive outcomes. Hospitalists have an important role in this regard, as they are ideally situated to manage these agents in the many patients undergoing surgery across the United States.
Disclosures: Dr. Wijeysundera is supported by a Clinician‐Scientist Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto. Dr. Chopra is supported by a Career‐Development Award (1K08HS022835‐01) from the Agency of Healthcare Research and Quality.
The management of perioperative medications is a tale of progressive scientific enquiry. Although long‐term use of agents such as aspirin or statins improves clinical outcomes, use during surgery ranges from problematic to protective. The delicate balance between proven long‐term benefits in the nonoperative setting versus short‐term uncertainty in the perioperative setting must be assessed using a lens that incorporates patient risk, surgical process, and pharmacodynamic principles. Advances in our understanding of perioperative physiology, coupled with robust clinical and outcome data, have led to new knowledge and insights regarding how best to manage these medications. As well illustrated by the near 180 change in the use of perioperative ‐blockers to prevent adverse cardiovascular outcomes, these new data have led to substantial progress in surgical safety and patient outcomes.
In this issue of the Journal of Hospital Medicine, 2 retrospective cohort studies add to this growing body of evidence by examining risks associated with use of angiotensin‐converting enzyme (ACE) inhibitors in the perioperative setting. In the first study, conducted at a single academic medical center, Nielson and colleagues evaluate the association of preoperative ACE‐inhibitor use with hypotension and acute kidney injury in patients undergoing major elective orthopedic surgery.[1] The authors report that patients receiving ACE‐inhibitors were not only more likely to experience hypotension after induction of anesthesia (12.2% vs 6.7%, P = 0.005), but also were more likely to develop postoperative acute kidney injury (odds ratio [OR]: 2.68, 95% confidence interval [CI]: 1.25‐1.99). In the second study which focused solely on patients who were receiving preoperative ACE‐inhibitor therapy, Mudumbai and colleagues used a national Veterans Affairs database to examine the association between failure to resume ACE‐inhibitor treatment after surgery and outcomes at 30 days.[2] The authors found that failure to resume treatment 14 days after surgery was not only common (affecting 1 in 4 patients), but was also associated with increased 30‐day mortality (hazard ratio: 3.44, 95% CI: 3.30‐3.60).[2] Taken together, these 2 studies shed new light on clinical practice and policy implications for the use of these agents in the surgical period. Both sets of authors should be congratulated on moving the needle forward in this enquiry.
ACE‐inhibitors physiologically mediate their effects by preventing the formation of the potent vasoconstrictor angiotensin‐II from its precursor angiotensin‐I. In doing so, they decrease arterial resistance, increase venous capacitance, decrease glomerular filtration pressure, and promote natriuresis. These vascular effects have key benefits for the management of a number of chronic diseases including hypertension, congestive heart failure, and diabetic nephropathy. However, these clinical alterations are often problematic in the perioperative setting. For example, ACE‐inhibitors may cause vasoplegia during anesthetic administration, commonly manifested as hypotension during induction.[3] This hemodynamic alteration has been viewed as being so precarious, that some authors recommend withholding ACE‐inhibitors prior to major cardiovascular procedures such as coronary artery bypass grafting.[4] Although hypotension leads to management challenges (often increasing vasoconstrictor requirements), several studies report that ACE‐inhibitor use during surgery may also be associated with increased risks of acute kidney injury and mortality.[5, 6] However, despite these data, existing literature has not shown a consistent association between ACE‐inhibitor use and adverse postoperative outcomes. For example, a propensity‐matched cohort study of 79,228 patients at the Cleveland Clinic found no difference in hemodynamic characteristics, vasopressor requirements, or cardiorespiratory complications among patients who were or were not using ACE‐inhibitors during noncardiac surgery.[7] Furthermore, some research has also found that withdrawal of ACE‐inhibitors may itself lead to harm. For instance, a contemporary study reported that withdrawal of ACE‐inhibitor therapy in patients undergoing coronary artery bypass was associated with increased in‐hospital cardiovascular events.[8] Given this uncertainty, it is not surprising that management of ACE‐inhibitors in the perioperative period remains a subject of ongoing controversy with clinical reviews often recommending consideration of the risks and benefits associated with use of these agents.[9]
It is important to note that driver of this ambiguity is the very design of relevant studies. For instance, studies that focus on patients undergoing coronary artery bypass grafting surgery have limited external validity, as these patients are very different from those who undergo elective hip or knee replacement (such as those included by Nielsen and colleagues). Additionally, many studies suffer from methodological constraints or biases. For instance, retrospective observational studies often suffer from selection bias and residual confounding; that is, the individuals chosen for inclusion in the study and the variables available for analysis are often limited by available data, curtailing the conclusions that can be generated. Although the use of multivariable regression or propensity score techniques helps address these limitations, residual confounding by unmeasured variables always remains a threat to statistical inference. The potential influence for this bias is particularly relevant when examining the results of the study by Nielsen and colleagues. Another important limitation is survivor bias, a problem inherent in the study by Mudumbai and colleagues. Put simply, for a patient to resume ACE‐inhibitors after surgery, this same patient, must also survive for at least 15 to 30 days after surgery. Thus, for some patients, failure to resume this treatment may simply be a marker of early mortality rather than failure to resume the ACE itself. The potential influence of this bias is supported by an included sensitivity analysis, where a large change in the adjusted OR was observed when patients suffering early mortality were excluded. This swing in effect size suggests that biases related to comparing patients who survived to those who did not with respect to ACE use may, in part, account for the results of the study.
These limitations aside, the studies brought forth by both authors help inform practice with respect to the use of these agents during surgery. In this context, 3 paradigms are relevant for practicing hospitalists.
First, if a patient is maintained on an ACE‐inhibitor before surgery, should the medication be temporarily held before surgery to minimize hypotension during anesthesia? The study by Nielsen and colleagues (comparing those on ACE‐inhibitor treatment to those without), in addition to the evidence generated from other studies in this area[10, 11, 12] suggest that this is a rational decision. Although the existence of a withdrawal state from abrupt cessation of ACE‐inhibitor use is theoretically plausible, this has yet to be reliably reported in the literature. Given the short half‐life of most ACE‐inhibitors, cessation 24 hours before surgery appears to be the most pragmatic clinical approach.
Second, if a patient is on an ACE‐inhibitor before surgery, when should the medication be resumed after surgery? The findings from the study by Mudumbai and colleagues, in addition to contemporary evidence,[7, 8, 13] support the resumption of these agents as soon as possible following operative intervention. Once hemodynamic stability and volume status have been assured, risks associated with postoperative ACE‐inhibitor use appear to be outweighed by benefits, though specific care is likely necessary in those with preexisting renal dysfunction.[14] A program that ensures reconciliation of medications in the postoperative setting may be valuable in ensuring that such treatment is restarted.
Third, if a patient is not on ACE‐inhibitor therapy, should this be started before surgery for perioperative or long‐term benefit? Although neither study examines this issue, the potential for significant risk make this an unattractive option. Future interventional studies with thoughtfully weighed safety parameters may be necessary to assess whether such a paradigm may be valuable.
The studies included in this issue of the Journal of Hospital Medicine suggest that the use of ACE‐inhibitors during the perioperative period may be considered a function of time and place. Resuming ACE‐inhibitors and cessation of treatment at specific intervals in relation to surgery can help ensure positive outcomes. Hospitalists have an important role in this regard, as they are ideally situated to manage these agents in the many patients undergoing surgery across the United States.
Disclosures: Dr. Wijeysundera is supported by a Clinician‐Scientist Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto. Dr. Chopra is supported by a Career‐Development Award (1K08HS022835‐01) from the Agency of Healthcare Research and Quality.
- , , , . Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283–288.
- , , , , , . Thirty‐day mortality risk associated with postoperative nonresumption of angiotensin‐converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289–296.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , , . Angiotensin‐converting enzyme inhibitors increase vasoconstrictor requirements after cardiopulmonary bypass. Anesth Analg. 1995;80(3):473–479.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57(8):736–744.
- , , , , , . Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552–560.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87(1029):472–481.
- , , , et al; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , , , . Effects of renin‐angiotensin system inhibitors on the occurrence of acute kidney injury following off‐pump coronary artery bypass grafting. Circulation J. 2010;74(9):1852–1858.
- , , , , , . Prophylactic vasopressin in patients receiving the angiotensin‐converting enzyme inhibitor ramipril undergoing coronary artery bypass graft surgery. J Cardiothorac Vasc Anesth. 2010;24(2):230–238.
- , , , , . Neither diabetes nor glucose‐lowering drugs are associated with mortality after noncardiac surgery in patients with coronary artery disease or heart failure. Can J Cardiol. 2013;29(4):423–428.
- , , , , , . Interventions for protecting renal function in the perioperative period. Cochrane Database Syst Rev. 2008;(4):CD003590.
- , , , . Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283–288.
- , , , , , . Thirty‐day mortality risk associated with postoperative nonresumption of angiotensin‐converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289–296.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , , . Angiotensin‐converting enzyme inhibitors increase vasoconstrictor requirements after cardiopulmonary bypass. Anesth Analg. 1995;80(3):473–479.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57(8):736–744.
- , , , , , . Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552–560.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87(1029):472–481.
- , , , et al; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , , , . Effects of renin‐angiotensin system inhibitors on the occurrence of acute kidney injury following off‐pump coronary artery bypass grafting. Circulation J. 2010;74(9):1852–1858.
- , , , , , . Prophylactic vasopressin in patients receiving the angiotensin‐converting enzyme inhibitor ramipril undergoing coronary artery bypass graft surgery. J Cardiothorac Vasc Anesth. 2010;24(2):230–238.
- , , , , . Neither diabetes nor glucose‐lowering drugs are associated with mortality after noncardiac surgery in patients with coronary artery disease or heart failure. Can J Cardiol. 2013;29(4):423–428.
- , , , , , . Interventions for protecting renal function in the perioperative period. Cochrane Database Syst Rev. 2008;(4):CD003590.
Nonresumption of an ACE‐I
Perioperative medication management requires careful consideration, because surgical patients, especially older ones, may be receiving multiple medications for the treatment of acute or chronic comorbidities.[1] Because patients often present to surgery stabilized on their drug regimens, nonresumption of medications for chronic conditions may be problematic in controlling underlying diseases.[2] For example, nonresumption of cardiovascular medications such as ‐blockers postoperatively has been shown to lead to increased longer‐term mortality.[3] Little data, however, exist to guide practitioners on the postoperative management risks for another widely used class of cardiovascular medication: angiotensin‐converting enzyme inhibitors (ACE‐Is).[4]
About 170 million prescriptions for an ACE‐I are dispensed in the United States annually, which reflects a multiple criteria for their use including hypertension, heart failure, ischemic heart disease, coronary disease risk, diabetes mellitus, chronic kidney disease, recurrent stroke prevention, and vascular disease.[5, 6, 7] ACE‐Is have been shown to improve outcomes in patients with ischemic heart disease and heart failure.[8, 9] An observational study found that perioperative use of an ACE‐I in coronary artery bypass grafting (CABG) patients was associated with increased mortality, use of vasopressors, and postoperative acute renal failure.[10] Data also indicate that patients who continue the use of an ACE‐I perioperatively can experience severe hypotension.[11] As a result, some have recommended that consideration be given to not restarting the ACE‐I perioperatively, especially with hypertensive patients undergoing noncardiac surgery.[12] However, little evidence exists to document benefits and risks of not restarting an ACE‐I in surgical patients for various intervals. To evaluate these risks, we tested the hypothesis that postoperative nonresumption of an ACE‐I occurs frequently for broad cohorts of Veterans Affairs (VA) surgery patients within the first 14 days and is associated with increased 30‐day mortality.
MATERIALS AND METHODS
After institutional review board approval (University of California, San Francisco), we examined surgeries conducted at hospitals at 120 stations within the VA Health Care System (VAHCS). The VAHCS is the largest integrated healthcare system in the United States, with long‐standing electronic medical records capturing detailed demographic, pharmacy, and mortality information.[13] Data were extracted from Medical Statistical Analysis System (SAS) and Corporate Data Warehouse (CDW) files in the VA Informatics and Computing Infrastructure.[14]
Development of the Study Population
To identify surgery patients who were consistently prescribed an ACE‐I preoperatively (Figure 1), we first located 1,213,086 surgical admissions in 846,454 patients from 1999 to 2012 using Medical SAS files and classified them by specialty of the surgeon (eg, neurosurgery, orthopedic, urology, cardiothoracic, general, vascular, plastic, and other [such as gynecology]). We identified comorbidities and cardiovascular risk factors from inpatient/outpatient diagnosis files in the CDW using International Classification of Diseases (ICD‐9) diagnosis codes (see Supporting Information, Tables 1 and 2, in the online version of this article). To ensure chronic preoperative ACE‐I use, we included surgeries with 3 outpatient prescription fills of an ACE‐I and <180‐day gap. ACE‐Is included benazepril, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, and ramipril. We excluded cases with a surgery in the prior 90 days and missing diagnosis codes. Our final population was comprised of 294,505 surgical admissions in 240,978 patients.
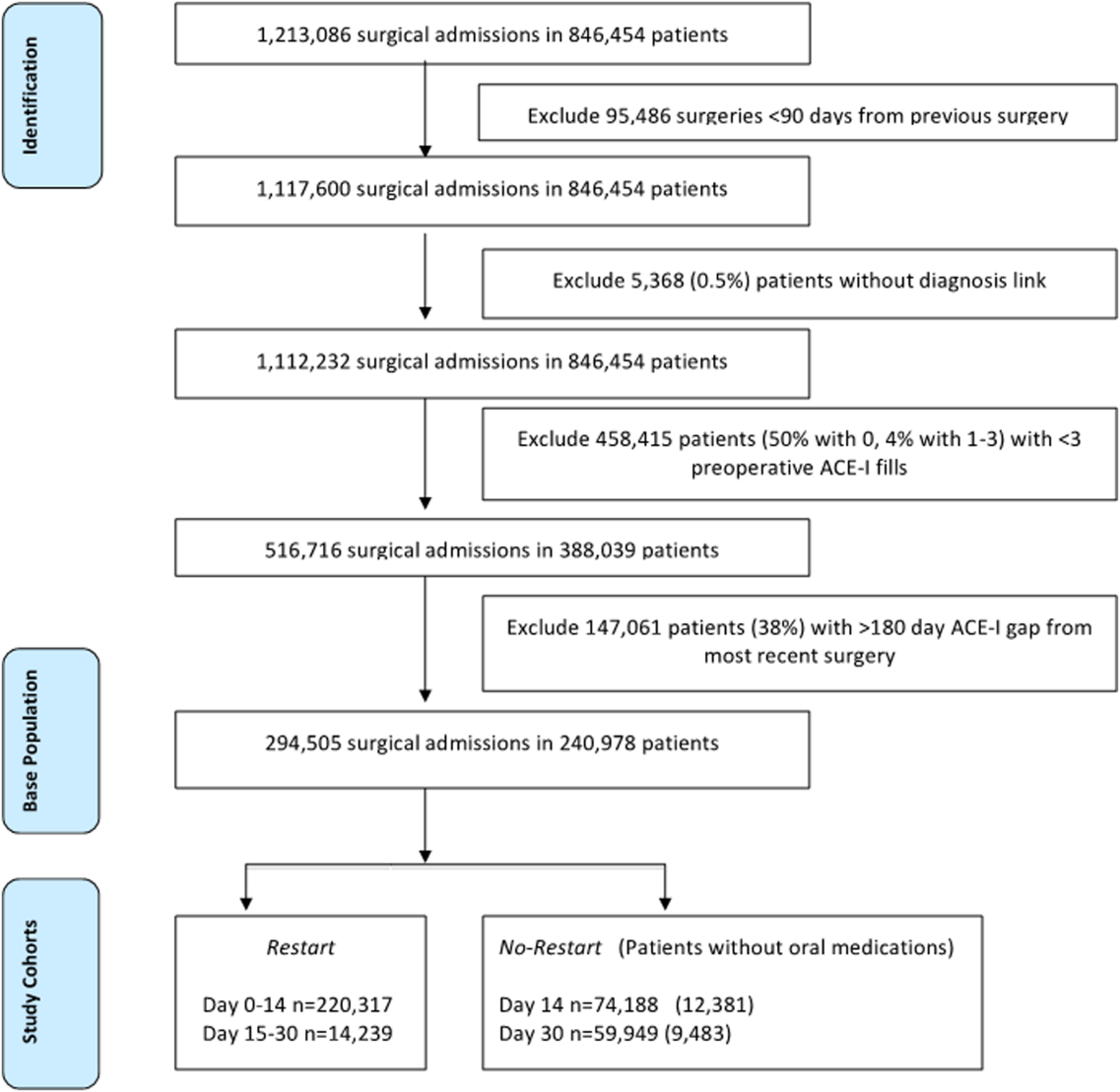
| Parameter | Surgeries, No. (%), Total=294,505 | Died by 30‐Days, Total=9,227 | P Value |
|---|---|---|---|
| |||
| No restart, 014 daysa | 59,949 (20%) | 7.3% | <0.001 |
| Restart, 014 daysb | 220,317 (75%) | 2.1% | |
| Restart, 1530 daysc | 14,239 (5%) | 1.7% | |
| Age, y | |||
| <60 | 74,326 (14%) | 1.7% | <0.001 |
| 6170 | 97,731 (24%) | 2.3% | |
| 7190 | 119,775 (60%) | 4.6% | |
| >90 | 2,673 (1%) | 6.9% | |
| Gender | |||
| Female | 7,186 (2%) | 1.6% | <0.001 |
| Male | 287,319 (98%) | 3.2% | |
| Indications for use of ACE‐I | |||
| Hypertension | 270,486 (92%) | 2.8% | <0.001 |
| Ischemic heart disease | 129,212 (44%) | 3.8% | <0.001 |
| Vascular disease | 75,410 (26%) | 3.7% | <0.001 |
| Heart failure | 59,809 (20%) | 5.7% | <0.001 |
| Chronic kidney disease | 8,804 (3%) | 4.9% | <0.001 |
| Diabetes mellitus | 170,320 (58%) | 3.0% | <0.001 |
| Coronary disease riskd | 280,958 (95%) | 3.1% | <0.001 |
| Stroke | 22,285 (8%) | 5.2% | <0.001 |
| Comorbidity scoree | |||
| 0 | 72,126 (24%) | 1.4% | <0.001 |
| 1 | 59,609 (20%) | 1.5% | |
| 2 4 | 116,914 (40%) | 3.5% | |
| >4 | 45,856 (16%) | 7.0% | |
| Preoperative ACE‐I gap, daysf | |||
| 045 | 21,383 (7%) | 3.7% | <0.001 |
| 4690 | 30,237 (10%) | 3.8% | |
| 91180 | 242,885 (83%) | 3.0% | |
| Surgical specialty | |||
| General | 98,210 (33%) | 4.6% | <0.001 |
| Neurosurgery | 15,423 (5%) | 2.3% | |
| Orthopedic | 51,600 (18%) | 1.9% | |
| Plastic | 12,547 (4%) | 3.8% | |
| Thoracic | 44,728 (15%) | 3.2% | |
| Urology | 34,595 (12%) | 1.5% | |
| Vascular | 34,228 (12%) | 2.8% | |
| Other (gynecology) | 3,174 (1%) | 1.4% | |
| Year of surgery | |||
| 19992002 | 66,689 (23%) | 4.2% | <0.001 |
| 20032005 | 75,420 (26%) | 3.4% | |
| 20062008 | 76,563 (26%) | 2.8% | |
| 20092012 | 75,833 (26%) | 2.2% | |
| No. of prior surgeries | |||
| 0 | 215,443 (74%) | 3.2% | 0.413 |
| 1 | 56,419 (19%) | 3.1% | |
| 2 | 22,643 (7%) | 3.1% | |
| Length of stay, d | |||
| 1 | 40,538 (14%) | 1.4% | <0.001 |
| 23 | 59,817 (20%) | 1.4% | |
| 47 | 83,366 (28%) | 2.0% | |
| 821 | 83,379 (28%) | 4.7% | |
| >21 | 27,405 (9%) | 8.0% | |
| Center surgical volume quartileg | |||
| 0%25% | 74,846 (25%) | 3.7% | <0.001 |
| 25%50% | 74,569 (25%) | 3.1% | |
| 50%75% | 69,947 (24%) | 2.8% | |
| 75%100% | 75,143 (26%) | 2.8% | |
| Center restart quartileh | |||
| 0%25% | 73,750 (25%) | 3.1% | 0.014 |
| 25%50% | 81,071 (28%) | 3.0% | |
| 50%75% | 83,952 (29%) | 3.3% | |
| 75%100% | 55,732 (19%) | 3.2% | |
| No complication | 80,700 (27%) | 1.3% | <0.001 |
| Minor complicationi | 181,924 (62%) | 4.2% | <0.001 |
| Major complicationj | 46,977 (16%) | 8.3% | <0.001 |
| Complications | |||
| Arrhythmia | 3,037 (1%) | 2.0% | <0.001 |
| Bleeding | 12,887 (4%) | 4.8% | <0.001 |
| Deep venous thrombosis | 6,075 (2%) | 3.6% | <0.001 |
| Myocardial infarction | 9,114 (3%) | 7.7% | <0.001 |
| Pneumonia | 109,660 (37%) | 5.1% | <0.001 |
| Pulmonary embolism | 5,064 (2%) | 6.2% | <0.001 |
| Renal failure | 25,513 (9%) | 11.0% | <0.001 |
| Sepsis | 5,846 (2%) | 16.5% | <0.001 |
| Stroke | 19,546 (7%) | 5.0% | <0.001 |
| Urinary tract infection | 32,548 (11%) | 4.9% | <0.001 |
| Unadjusted Hazard for 30‐Day Mortality (OR [95% CI]) | Adjusted hazard for 30 day mortality (OR [95% CI]) | ||||
|---|---|---|---|---|---|
| Restart (014 Days) (Referent)a | No Restart, 014 Daysb | Restart, 1530 Daysc | Restart, 014 Days (Referent) | No Restart, 014 Days | Restart, 1530 Days |
| |||||
| 1 | 3.44 (3.303.60)d | 0.23 (0.200.26)d | 1 | 2.79 (2.672.92)d | 0.24 (0.210.28)d |
| Restart, 014 Days (Referent) | No Restart, 014 Days | NA | Restart, 014 Days (Referent) | No Restart, 014 Days | NA |
| 1 | 2.92 (2.803.05)d | NA27 | 1 | 2.39 (2.292.50)d | NA27 |
Postoperative Medication Use
We defined patients as postoperative restart (014 days) if an ACE‐I was administered in‐hospital (oral or intravenous) or a postdischarge outpatient ACE‐I prescription was filled in the 14 days following surgery. In absence of ACE‐I administration or prescription during postoperative days 0 to 14, patients were classified as no restart (014 days). Intraclass changes from one ACE‐I to another were considered a restart if they occurred within 0 to 14 days of surgery. We also tracked ACE‐I prescription fills through postoperative day 15 to 30 (ie, restart [1530 days]) and noted administration or filling of oral medications. Oral medications were classified as tablets or caplets in formularies.
Patient Characteristics
We categorized patients by age strata: <60, 61 to 70, 71 to 90, and >90 years old; gender; and epochs (every 34 years starting from calendar year 1999). We tracked prior surgery admissions and length of stay.
Hospital Factors
To account for clustering of surgeries and hospital‐related factors affecting ACE‐I use practices, we divided hospitals into quartiles of (1) total surgical volume based on total number of surgeries done at a hospital from 1999 to 2012 (0%25%, n<2378; 50%, n=3498; 75%, n=4531; highest surgical volume, 8162); and (2) percent of cases restarted on ACE‐I at 14 days (71%, 76%, 79%, and 100%).
Indications, Patient Illness Severity, and Complications
We determined probable indications for ACE‐I usage (ie, heart failure) and comorbidities using ICD‐9 codes in medical records prior to surgical admissions (see Supporting Information, Tables 1 and 2, in the online version of this article). Comorbidities were aggregated using algorithms developed by Gagne aggregating comorbidity conditions (defined by Elixhauser) into scores similar to Charlson scores.[15] The Gagne score has higher correlation with 30‐day, 90‐day, 180‐day, and 1‐year mortality than Charlson scores.[15]
After evaluating secondary diagnosis codes in the clinic or hospital visits prior to surgery date, complications were defined using codes newly incident after surgery and up to 90 days following discharge. We organized complications into major and minor. Major complications were myocardial infarction, renal failure, and stroke; minor complications included arrhythmia, postoperative bleeding, deep venous thrombosis, pneumonia, pulmonary embolism, sepsis, and urinary tract infection.
Mortality
Deaths were ascertained from VA Vital Status files.
Statistical Analysis
The unit of analysis was surgical episode; surgeries were stratified by 30‐day mortality. We evaluated differences between the 2 groups using 2 tests accounting for restarting of an ACE‐I through day 30, risk factors, patient, and hospital‐stay characteristics. We also compared those who did not restart from postoperative day 0 to 14 and 15 to 30 to all others who did not restart at any point up to 90 days. Independent variables included age, gender, indications for ACE‐I, comorbidity burden, type and year of surgery, previous hospitalizations, length of stay, and complications. To account for site‐related effects and clustering of observations (ie, surgeries within hospitals), we included quartiles of hospital volume and hospital rates of ACE‐I restart in models and used cluster command in Stata (StataCorp, College Station, TX).
Risk of Mortality
We developed Cox regression models to examine 30‐day mortality risks between restart (015 days) and restart (1530 days) groups to a reference group of patients who did not restart in the first 14 days after surgery (ie, no restart [014 days]). We considered those who had restarted their ACE‐I beyond day 14 and excluded these from comparisons to the no restart group. Independent variables included age, gender, indications for ACE‐I usage, comorbidity, type and year of surgery, previous hospitalizations, length of stay, quartiles of hospital surgical volume and rates of restarting an ACE‐I, and complications.
Sensitivity Analyses
Using Cox regression, we tested robustness of results regarding no restart (014 days) versus restart (014 days) in subsets after excluding patients who died postoperative day 0 to 2 and those with no oral medications on postoperative day 0 to 14, those with low comorbidity burden, within subtypes of surgery, and by surgical episode. To evaluate confounding by indication, we examined subsets without major complications and after excluding patients who died postoperative day 0 to 14. We then developed a propensity score model using quintiles to estimate average treatment effects associated with no restart (014 days).[16] A propensity score reflecting the probability of ACE‐I administration at 14 days was developed using logistic regression accounting for all independent variables. For analyses, we considered a 2‐tailed P value of 0.05 as statistically significant. Stata 12.1 software (Stata Corp.) was used.
RESULTS
Table 1 describes the characteristics and 30‐day mortality rates for our cohort. By postoperative day 14, 75% of the study sample (n=220,317) had restarted an ACE‐I (Figure 1). Our sample consisted primarily of older men with a substantial comorbidity burden and multiple indications for an ACE‐I. Most patients had 1 surgical episode, with the largest fraction undergoing general surgery overall. A third of the cases had lengths of stay >1 week, and surgeries occurred throughout the study period. The largest number of surgeries was noted for centers in 75% to 100% surgical volume and 50% to 75% restart quartiles. Most surgeries had no or minor complications.
The no restart (014 days) group had a higher 30‐day mortality rate (7.3%) compared to those who restarted by postoperative day 14 (2.1%) or 30 (1.7%). The highest mortality rates were found in patients aged >90 years, with a >4 comorbidity index or hospital stays >3 weeks, and those experiencing major postoperative complications.
30‐Day Mortality
Table 2 indicates that nonresumption of an ACE‐I from postoperative day 0 to 14 was independently associated with an approximately 2.5‐fold increased risk of 30‐day mortality (hazard ratio [HR]: 3.44; 95% confidence interval [CI]: 3.30‐3.60; P<0.001). Lower hazard ratios were noted when patients who restarted postoperative days 15 to 30 were included in models (HR: 2.79; 95% CI: 2.67‐2.92; P<0.001).
The sensitivity analyses illustrate the durability of treatment effects (Table 3). After excluding patients who died during days 0 to 2 and without a record of receiving an oral medication by postoperative day 14, ACE‐I nonresumption was associated with an 88% increase in 30‐day mortality risk (HR: 1.88; 95% CI: 1.79‐1.98; P<0.001). Similar increased risks were seen in patients with less comorbidity for each specialty and for those who did not experience a major complication. In data not shown, adjusting by propensity score did not modulate treatment effects (HR for no restart [014 days]: 3.03; 95% CI: 2.78‐3.30; P<0.001).
| Population | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|
| ||
| Exclude patients who died day 02 or no record of oral medications days 014 | 2.29 (2.182.40) | 1.88 (1.791.98) |
| Cases with 02 comorbidity scoreb | 1.92 (1.742.12) | 1.72 (1.551.90) |
| Only cardiothoracic surgery casesb | 2.07 (1.832.35) | 1.94 (1.702.21) |
| Only neurosurgery casesb | 1.49 (1.102.02) | 1.46 (1.072.00) |
| Only orthopedic surgery casesb | 2.48 (2.122.91) | 2.17 (1.842.55) |
| Only urologic surgery casesb | 1.92 (1.582.34) | 1.37 (1.121.68) |
| Only first surgery casesb | 2.22 (2.092.35) | 1.86 (1.751.97) |
| Subsequent surgery casesb | 2.49 (2.272.73) | 1.96 (1.782.16) |
| Cases with no major complicationsb | 2.49 (2.362.64) | 2.25 (2.122.38) |
| Exclude patients who died within the first 14 days after surgeryc | 2.26 (2.112.41) | 1.66 (1.551.78) |
Other factors associated with increased 30‐day mortality are displayed in Table 4. The risk associated with not restarting an ACE‐I was similar to effect of age >90years and a >4 comorbidity index.
| Parameter | Reference Group | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|---|
| |||
| No restart (014 days)b | Restart (014 days)c | 2.92 (2.803.05) | 2.39 (2.292.50) |
| Age, y | |||
| 6170 | Age <60 years | 1.33 (1.241.43) | 1.36 (1.261.46) |
| 7190 | 2.72 (2.552.90) | 2.01 (1.892.30) | |
| >90 | 4.05 (3.454.76) | 2.70 (2.183.74) | |
| Male | Female | 2.11 (1.742.57) | 1.54 (1.271.88) |
| Comorbidity score | |||
| 24 | 1 | 2.19 (2.062.33) | 1.36 (1.271.45) |
| >4 | 4.57 (4.294.87) | 1.97 (1.822.13) | |
| Center surgical volume quartile | |||
| 025th percentile | 76th100th percentile | 1.35 (1.281.43) | 1.21 (1.141.29) |
| 26th50th percentile | 1.11 (1.041.18) | 1.05 (0.991.12) | |
| Indications | |||
| Heart failure | No heart failure | 2.23 (2.142.34) | 1.19 (1.121.26) |
| Year of surgery | |||
| 19992002 | 20062008 | 1.49 (1.411.58) | 1.07 (1.011.13) |
| 20032005 | 1.21 (1.451.29) | 1.13 (1.061.20) | |
DISCUSSION
The results from this national retrospective study confirm our hypothesis that nonresumption of an ACE‐I for 14 or more postoperative days occurs frequently for VA surgery patients. However, we found that nonresumption of an ACE‐I during the first 2 weeks after surgery is independently associated with increased 30‐day mortality. Our study is one of the first to examine the patterns and risks of postoperative ACE‐I management across a large and varied surgical population.[11, 17]
The lack of inpatient and outpatient ACE‐I prescription use by postoperative day 14 across multiple surgery classes suggests that surgical patients may be prone to short‐term nonresumption of an ACE‐I. Our intention in using a 14‐day window to evaluate restarting strategies was to account for immediate postoperative management. After surgery, careful appraisal of whether medications should be restarted is often necessary in the face of substantially deranged physiology, hypercoagulability, and blood loss.[18] After physiologic stabilization over several days, cardiovascular drugs are usually restarted thereafter to help manage chronic comorbidities.[19] One immediate conclusion from our findings is that ACE‐I are commonly discontinued perioperatively (potentially due to concerns for hypotension), and are often not restarted.[20, 21, 22, 23, 24, 25]
Our rates of ACE‐I nonresumption are comparable to rates of nonresumption reported postoperatively for other medications and raise concerns for inadequate medication reconciliation in surgical cohorts. Bell et al. conducted a population‐based cohort study of patients undergoing elective surgery and found that 11.4% of 45,220 patients chronically prescribed warfarin were not restarted by postoperative day 180.[22] A subsequent study showed intensive care unit (ICU) admission was associated with increased rates of not restarting 4 of 5 medication groups (range, 4.5%19.4%; statins, antiplatelet/anticoagulant agents, levothyroxine, respiratory inhalers, and gastric acid‐suppressing drugs).[21] One‐year follow‐up showed elevated odds for the secondary composite outcome of death in the statins group (odds ratio [OR]: 1.07; 95% CI: 1.03‐1.11) and antiplatelet/anticoagulant agents group (OR: 1.10; 95% CI: 1.03‐1.16). Drenger et al. noted a 50% rate for no restart of ACE‐I after CABG surgery; restarting was associated with a decreased composite outcome of cardiac, cerebral, and renal events and in‐hospital mortality (OR: 0.50; 95% CI: 0.38‐0.66).[26] Because medication management has been noted to be problematic at care transitions, the inpatient medication reconciliation recommendations articulated in recent Joint Commission National Patient Safety Goals may be particularly relevant for high‐risk surgical patients who experience multiple transitions of care (ie, operating room to ICU to surgical ward to rehabilitation unit to discharge).[19, 24, 27]
In examining the crucial interval for the surgical patientthe postoperative period when medication changes are commonwe found a nearly 2.5‐fold increase in risk for 30‐day mortality associated with nonresumption of an ACE‐I.[4, 19, 28] We also noted that those who were restarted later on day 15 to 30 fared better than those not restarted (Table 2). Similar effect sizes have been found with postoperative nonresumption of other cardiovascular medications. Not restarting chronic ‐blocker treatment after surgery is associated with a significant 1‐year mortality risk (HR: 2.7; 95% CI: 1.25.9).[29] Postoperative statin withdrawal (>4 days) is an independent predictor of postoperative myonecrosis (OR: 2.9; 95% CI: 1.6‐5.5).[30, 31] Biologic mechanisms contributing to mortality after a temporary failure to restart an ACE‐I are speculative and were not addressed in this study. Potential mechanisms may lie with hypertensive rebound and associated cardiac decompensation. Withdrawing an ACE‐I can cause rapid increases in blood pressure within 48 hours on home self‐measured blood pressure in hypertensive patients and in diabetic patients with chronic renal failure.[32, 33] Patients with heart failure or coronary artery disease may then experience myocardial ischemia in the context of elevated blood pressure. Not restarting an ACE‐I may also lead to compromised microcirculatory flow with renal complications and mortality.[34, 35]
Alternative explanations for the magnitude of our findings may lie with unmeasured confounders. Our analysis did not evaluate potential interactions arising from the failure to restart of all other medications (eg, ‐blockers) or evaluate changes to angiotensin receptor blockers (ARBs). In addition, our study lacked data on health system variations or emergent versus elective surgeries. However, a key starting point of our analysis was distinguishing between purposeful versus potentially unintentional nonresumption of an ACE‐I. To accomplish this, we included patients who had at least 3 prescription ACE‐I fills prior to surgery, evaluated the preoperative indications for an ACE‐I and the ability to take postoperative oral medications (eg, immortal time bias), and accounted for minor and major postoperative complications.
To address bias from unmeasured confounders, we conducted sensitivity analyses in more homogeneous subpopulations. With each sensitivity analysis, we found consistently strong associations between increased 30‐day mortality and nonresumption of an ACE‐I (Table 3). Strong effects were observed in patients without major complications and with low comorbidity burdens, patients in whom we would not expect an effect. Because deaths in postoperative day 0 to 2 could be attributed to surgical factors (ie, hemorrhage) or that patients who did not restart an ACE‐I in postoperative day 0 to 14 were too sick to tolerate oral medications, we excluded these patients along with patients who died before postoperative day 14. Both sensitivity analyses maintained our primary finding. Somewhat attenuated risks were found when we examined ACE‐I nonresumption by individual surgery types, perhaps reflective of differences in comorbidity burden.
Finally, although this study did not examine predictors of nonresumption, our models showed that in the context of postoperative ACE‐I management, factors including increasing age, being male, those with heart failure, and surgeries conducted in centers with low surgical volume were associated with increased 30‐day mortality (Table 4). Future research might consider how reinstitution of an ACE‐I occurs in these subpopulations to identify potential mechanisms for nonresumption.
Our study has several strengths. We examined patients over a decade, considered all major types of surgery, and studied patients across a healthcare system. Moreover, we used computerized prescription data and medical records (eg, discharge diagnosis, ICD‐9 codes) to derive risk factors. VA prescription data are standardized and accurate because of intensive efforts to contain costs.[36] Within VA data, the estimated sensitivity of computerized diagnoses exceeds 80% in the administrative files, with specificity of 91% to 100% for common diagnoses such as coronary artery disease.[37] These records also carefully and accurately identify death.[38]
We also identified potential limitations to our study. First, a retrospective, observational, cohort study may be prone to selection bias, and therefore we report associations that are not necessarily causal relationships. However, our methods are supported by the fact that we developed a large study sample consisting of consecutive surgical patients over a decade and noted large effect sizes across multiple subpopulations. Second, for group assignment, we used prescription records rather than medication administration data. Nevertheless, a cohort analysis focusing on exposure is standard for epidemiologic studies and shows outcomes of care resulting from daily clinical practice.[39] Third, we did not study the cause of death, data that may help to identify potential causal pathways between not restarting an ACE‐I and mortality. Fourth, our results come from VA medical centers and so may not be generalizable to non‐VA institutions. However, the length of observation under conditions of routine clinical practice at multiple medical centers and a diverse set of surgical procedures support the external validity of our study results. Fifth, we did not have clinical data accounting for surgeon‐level effects potentially affecting rates of nonresumption of an ACE‐I, American Society of Anesthesiology physical status, information on perioperative hypotension or vasopressors, or the presence of a postoperative primary care visit.
In conclusion, in the VA Healthcare System, temporary nonresumption of an ACE‐I is common. Postoperative nonresumption of an ACE‐I, although sometimes indicated and appropriate, is associated with increased risk of mortality. Careful attention to the issue of eventual reinstitution of medications for chronic conditions, such as an ACE‐I, is indicated to avoid unnecessary mortality. Because early experience showed that dose titration was a key for successful application of an ACE‐I, practitioners may also need to consider dose modification rather than simply continuation or not restarting.[40] Future research is needed to confirm our results in other healthcare systems and to define mechanisms that link postoperative nonresumption of an ACE‐I to mortality.
Acknowledgements
The authors acknowledge Dr. Edward R. Mariano, Chief Anesthesia Service, VA Palo Alto Health Care System, and Associate Professor, Stanford Department of Anesthesiology for general support of this research and critical review of the manuscript. We would also like to thank Dr. Ronald Pearl, Chair, Department of Anesthesiology, Perioperative, and Pain Medicine, Stanford University, for his support of our research. This material is the result of work supported with resources and the use of facilities at the Veterans Affairs Medical Center, San Francisco and Veterans Affairs Palo Alto Healthcare System.
Disclosure: The Northern California Institute for Research and Education and the Veterans Affairs Medical Center, San Francisco, California supported this work. This work was presented at the American Society of Anesthesiologists Annual Meeting, Chicago, Illinois, October 1519, 2011, and the Veterans Affairs National Health Services Research and Development National Conference, National Harbor, Maryland, July 1619, 2012.
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
- , , , . The development of polypharmacy. A longitudinal study. Fam Pract. 2000;17(3):261–267.
- , , , , . Polypharmacy in a general surgical unit and consequences of drug withdrawal. Br J Clin Pharmacol. 2000;49(4):353–362.
- , , , , , . Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients. Am Heart J. 2001;141(1):148–153.
- . Perioperative medication management: general principles and practical applications. Cleve Clin J Med. 2009;76(suppl 4):S126–S132.
- , , , et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572.
- IMS Health. Top therapeutic classes by U.S. dispensed prescriptions. April 7, 2011. Available at: http://www.imshealth.com/deployedfiles/imshealth/Global/Content/StaticFile/Top_Line_Data/2010_Top_Therap eutic_Classes_by_RX.pdf. Accessed September 5, 2011.
- , , , et al. The consistency of the treatment effect of an ACE‐inhibitor based treatment regimen in patients with vascular disease or high risk of vascular disease: a combined analysis of individual data of ADVANCE, EUROPA, and PROGRESS trials. Eur Heart J. 2009;30(11):1385–1394.
- , , , , , . Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153.
- , , , , , . Effects of the early administration of enalapril on mortality in patients with acute myocardial infarction. Results of the Cooperative New Scandinavian Enalapril Survival Study II (CONSENSUS II). N Engl J Med. 1992;327(10):678–684.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636–644.
- , , . Guidelines for pre‐operative cardiac risk assessment and perioperative cardiac management in non‐cardiac surgery: The Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non‐cardiac Surgery of the European Society of Cardiology (ESC) and endorsed by the European Society of Anaesthesiology (ESA). Eur Heart J. 2009;30(22):2769–2812.
- , , , . Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218–2227.
- VA Information Resource Center; VIReC Research User Guide: VHA Decision support system clinical national data extracts. 2nd ed. Hines, IL: U.S. Department of VA, Health Services Research and Development Service, VA Information Resource Center, 2009. Available at: http://www.virec.research.va.gov/RUGs/RUGs-Index.htm. Accessed February 27, 2013.
- , , , , . A combined comorbidity score predicted mortality in elderly patients better than existing scores. J Clin Epidemiol. 2011;64(7):749–759.
- . A tutorial and case study in propensity score analysis: an application to estimating the effect of in‐hospital smoking cessation counseling on mortality. Multi Behav Res. 2011;46(1):119–151.
- , . Stopping and restarting medications in the perioperative period. J Gen Intern Med. 1987;2(4):270–283.
- , , . Perioperative management of drug therapy, clinical considerations. Drugs. 1996;51(2):238–259.
- , , , et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation. 2007;116(17):1971–1996.
- , , , et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med. 2008;23(9):1414–1422.
- , , , et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA. 2011;306(8):840–847.
- , , , , , . Potentially unintended discontinuation of long‐term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525–2531.
- , , , . Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314–323.
- , . Discontinuation and reinstitution of medications during the perioperative period. Am J Health Syst Pharm. 2004;61(9):899–912.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med. 2010;5(8):477–485.
- , , . Guidelines for the management of chronic medication in the perioperative period: systematic review and formal consensus. J Clin Pharm Therap. 2011;36(4):446–467.
- , , , et al. Increase of 1‐year mortality after perioperative beta‐blocker withdrawal in endovascular and vascular surgery patients. Eur J Vasc Endovasc Surg. 2007;33(1):13–19.
- , , , et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg. 2007;104(6):1326–1333.
- , , , et al. Effect of statin withdrawal on frequency of cardiac events after vascular surgery. Am J Cardiol. 2007;100(2):316–320.
- , , , et al. Short‐term effects of withdrawing angiotensin converting enzyme inhibitor therapy on home self‐measured blood pressure in hypertensive patients. Am J Hypertens. 1998;11(2):165–173.
- , , , et al. Hypertensive rebound after angiotensin converting enzyme inhibitor withdrawal in diabetic patients with chronic renal failure. Nephrol Dial Trans. 2001;16(5):1084–1085.
- , , . Vascular protective effects of angiotensin converting enzyme inhibitors and their relation to clinical events. J Cardiovasc Pharmacol. 2001;37(suppl 1):S21–S30.
- , , , et al. Angiotensin‐converting enzyme inhibitor withdrawal and ACE gene polymorphism. Clin Nephrol. 2003;60(4):225–232.
- , . Pharmacy data in the VA health care system. Med Care Res Rev. 2003;60(3 suppl):92S–123S.
- , , , , . Accuracy of computerized outpatient diagnoses in a Veterans Affairs general medicine clinic. Am J Manag Care. 2002;8(1):37–43.
- , , , . Mortality ascertainment in the veteran population: alternatives to the National Death Index. Am J Epidemiol. 1995;141(3):242–250.
- . Statistical considerations in the intent‐to‐treat principle. Control Clin Trials. 2000;21(3):167–189.
- , , . ACE inhibitors in cardiac surgery: current studies and controversies. Hypertens Res. 2010;34(1):15–22.
Perioperative medication management requires careful consideration, because surgical patients, especially older ones, may be receiving multiple medications for the treatment of acute or chronic comorbidities.[1] Because patients often present to surgery stabilized on their drug regimens, nonresumption of medications for chronic conditions may be problematic in controlling underlying diseases.[2] For example, nonresumption of cardiovascular medications such as ‐blockers postoperatively has been shown to lead to increased longer‐term mortality.[3] Little data, however, exist to guide practitioners on the postoperative management risks for another widely used class of cardiovascular medication: angiotensin‐converting enzyme inhibitors (ACE‐Is).[4]
About 170 million prescriptions for an ACE‐I are dispensed in the United States annually, which reflects a multiple criteria for their use including hypertension, heart failure, ischemic heart disease, coronary disease risk, diabetes mellitus, chronic kidney disease, recurrent stroke prevention, and vascular disease.[5, 6, 7] ACE‐Is have been shown to improve outcomes in patients with ischemic heart disease and heart failure.[8, 9] An observational study found that perioperative use of an ACE‐I in coronary artery bypass grafting (CABG) patients was associated with increased mortality, use of vasopressors, and postoperative acute renal failure.[10] Data also indicate that patients who continue the use of an ACE‐I perioperatively can experience severe hypotension.[11] As a result, some have recommended that consideration be given to not restarting the ACE‐I perioperatively, especially with hypertensive patients undergoing noncardiac surgery.[12] However, little evidence exists to document benefits and risks of not restarting an ACE‐I in surgical patients for various intervals. To evaluate these risks, we tested the hypothesis that postoperative nonresumption of an ACE‐I occurs frequently for broad cohorts of Veterans Affairs (VA) surgery patients within the first 14 days and is associated with increased 30‐day mortality.
MATERIALS AND METHODS
After institutional review board approval (University of California, San Francisco), we examined surgeries conducted at hospitals at 120 stations within the VA Health Care System (VAHCS). The VAHCS is the largest integrated healthcare system in the United States, with long‐standing electronic medical records capturing detailed demographic, pharmacy, and mortality information.[13] Data were extracted from Medical Statistical Analysis System (SAS) and Corporate Data Warehouse (CDW) files in the VA Informatics and Computing Infrastructure.[14]
Development of the Study Population
To identify surgery patients who were consistently prescribed an ACE‐I preoperatively (Figure 1), we first located 1,213,086 surgical admissions in 846,454 patients from 1999 to 2012 using Medical SAS files and classified them by specialty of the surgeon (eg, neurosurgery, orthopedic, urology, cardiothoracic, general, vascular, plastic, and other [such as gynecology]). We identified comorbidities and cardiovascular risk factors from inpatient/outpatient diagnosis files in the CDW using International Classification of Diseases (ICD‐9) diagnosis codes (see Supporting Information, Tables 1 and 2, in the online version of this article). To ensure chronic preoperative ACE‐I use, we included surgeries with 3 outpatient prescription fills of an ACE‐I and <180‐day gap. ACE‐Is included benazepril, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, and ramipril. We excluded cases with a surgery in the prior 90 days and missing diagnosis codes. Our final population was comprised of 294,505 surgical admissions in 240,978 patients.
| Parameter | Surgeries, No. (%), Total=294,505 | Died by 30‐Days, Total=9,227 | P Value |
|---|---|---|---|
| |||
| No restart, 014 daysa | 59,949 (20%) | 7.3% | <0.001 |
| Restart, 014 daysb | 220,317 (75%) | 2.1% | |
| Restart, 1530 daysc | 14,239 (5%) | 1.7% | |
| Age, y | |||
| <60 | 74,326 (14%) | 1.7% | <0.001 |
| 6170 | 97,731 (24%) | 2.3% | |
| 7190 | 119,775 (60%) | 4.6% | |
| >90 | 2,673 (1%) | 6.9% | |
| Gender | |||
| Female | 7,186 (2%) | 1.6% | <0.001 |
| Male | 287,319 (98%) | 3.2% | |
| Indications for use of ACE‐I | |||
| Hypertension | 270,486 (92%) | 2.8% | <0.001 |
| Ischemic heart disease | 129,212 (44%) | 3.8% | <0.001 |
| Vascular disease | 75,410 (26%) | 3.7% | <0.001 |
| Heart failure | 59,809 (20%) | 5.7% | <0.001 |
| Chronic kidney disease | 8,804 (3%) | 4.9% | <0.001 |
| Diabetes mellitus | 170,320 (58%) | 3.0% | <0.001 |
| Coronary disease riskd | 280,958 (95%) | 3.1% | <0.001 |
| Stroke | 22,285 (8%) | 5.2% | <0.001 |
| Comorbidity scoree | |||
| 0 | 72,126 (24%) | 1.4% | <0.001 |
| 1 | 59,609 (20%) | 1.5% | |
| 2 4 | 116,914 (40%) | 3.5% | |
| >4 | 45,856 (16%) | 7.0% | |
| Preoperative ACE‐I gap, daysf | |||
| 045 | 21,383 (7%) | 3.7% | <0.001 |
| 4690 | 30,237 (10%) | 3.8% | |
| 91180 | 242,885 (83%) | 3.0% | |
| Surgical specialty | |||
| General | 98,210 (33%) | 4.6% | <0.001 |
| Neurosurgery | 15,423 (5%) | 2.3% | |
| Orthopedic | 51,600 (18%) | 1.9% | |
| Plastic | 12,547 (4%) | 3.8% | |
| Thoracic | 44,728 (15%) | 3.2% | |
| Urology | 34,595 (12%) | 1.5% | |
| Vascular | 34,228 (12%) | 2.8% | |
| Other (gynecology) | 3,174 (1%) | 1.4% | |
| Year of surgery | |||
| 19992002 | 66,689 (23%) | 4.2% | <0.001 |
| 20032005 | 75,420 (26%) | 3.4% | |
| 20062008 | 76,563 (26%) | 2.8% | |
| 20092012 | 75,833 (26%) | 2.2% | |
| No. of prior surgeries | |||
| 0 | 215,443 (74%) | 3.2% | 0.413 |
| 1 | 56,419 (19%) | 3.1% | |
| 2 | 22,643 (7%) | 3.1% | |
| Length of stay, d | |||
| 1 | 40,538 (14%) | 1.4% | <0.001 |
| 23 | 59,817 (20%) | 1.4% | |
| 47 | 83,366 (28%) | 2.0% | |
| 821 | 83,379 (28%) | 4.7% | |
| >21 | 27,405 (9%) | 8.0% | |
| Center surgical volume quartileg | |||
| 0%25% | 74,846 (25%) | 3.7% | <0.001 |
| 25%50% | 74,569 (25%) | 3.1% | |
| 50%75% | 69,947 (24%) | 2.8% | |
| 75%100% | 75,143 (26%) | 2.8% | |
| Center restart quartileh | |||
| 0%25% | 73,750 (25%) | 3.1% | 0.014 |
| 25%50% | 81,071 (28%) | 3.0% | |
| 50%75% | 83,952 (29%) | 3.3% | |
| 75%100% | 55,732 (19%) | 3.2% | |
| No complication | 80,700 (27%) | 1.3% | <0.001 |
| Minor complicationi | 181,924 (62%) | 4.2% | <0.001 |
| Major complicationj | 46,977 (16%) | 8.3% | <0.001 |
| Complications | |||
| Arrhythmia | 3,037 (1%) | 2.0% | <0.001 |
| Bleeding | 12,887 (4%) | 4.8% | <0.001 |
| Deep venous thrombosis | 6,075 (2%) | 3.6% | <0.001 |
| Myocardial infarction | 9,114 (3%) | 7.7% | <0.001 |
| Pneumonia | 109,660 (37%) | 5.1% | <0.001 |
| Pulmonary embolism | 5,064 (2%) | 6.2% | <0.001 |
| Renal failure | 25,513 (9%) | 11.0% | <0.001 |
| Sepsis | 5,846 (2%) | 16.5% | <0.001 |
| Stroke | 19,546 (7%) | 5.0% | <0.001 |
| Urinary tract infection | 32,548 (11%) | 4.9% | <0.001 |
| Unadjusted Hazard for 30‐Day Mortality (OR [95% CI]) | Adjusted hazard for 30 day mortality (OR [95% CI]) | ||||
|---|---|---|---|---|---|
| Restart (014 Days) (Referent)a | No Restart, 014 Daysb | Restart, 1530 Daysc | Restart, 014 Days (Referent) | No Restart, 014 Days | Restart, 1530 Days |
| |||||
| 1 | 3.44 (3.303.60)d | 0.23 (0.200.26)d | 1 | 2.79 (2.672.92)d | 0.24 (0.210.28)d |
| Restart, 014 Days (Referent) | No Restart, 014 Days | NA | Restart, 014 Days (Referent) | No Restart, 014 Days | NA |
| 1 | 2.92 (2.803.05)d | NA27 | 1 | 2.39 (2.292.50)d | NA27 |
Postoperative Medication Use
We defined patients as postoperative restart (014 days) if an ACE‐I was administered in‐hospital (oral or intravenous) or a postdischarge outpatient ACE‐I prescription was filled in the 14 days following surgery. In absence of ACE‐I administration or prescription during postoperative days 0 to 14, patients were classified as no restart (014 days). Intraclass changes from one ACE‐I to another were considered a restart if they occurred within 0 to 14 days of surgery. We also tracked ACE‐I prescription fills through postoperative day 15 to 30 (ie, restart [1530 days]) and noted administration or filling of oral medications. Oral medications were classified as tablets or caplets in formularies.
Patient Characteristics
We categorized patients by age strata: <60, 61 to 70, 71 to 90, and >90 years old; gender; and epochs (every 34 years starting from calendar year 1999). We tracked prior surgery admissions and length of stay.
Hospital Factors
To account for clustering of surgeries and hospital‐related factors affecting ACE‐I use practices, we divided hospitals into quartiles of (1) total surgical volume based on total number of surgeries done at a hospital from 1999 to 2012 (0%25%, n<2378; 50%, n=3498; 75%, n=4531; highest surgical volume, 8162); and (2) percent of cases restarted on ACE‐I at 14 days (71%, 76%, 79%, and 100%).
Indications, Patient Illness Severity, and Complications
We determined probable indications for ACE‐I usage (ie, heart failure) and comorbidities using ICD‐9 codes in medical records prior to surgical admissions (see Supporting Information, Tables 1 and 2, in the online version of this article). Comorbidities were aggregated using algorithms developed by Gagne aggregating comorbidity conditions (defined by Elixhauser) into scores similar to Charlson scores.[15] The Gagne score has higher correlation with 30‐day, 90‐day, 180‐day, and 1‐year mortality than Charlson scores.[15]
After evaluating secondary diagnosis codes in the clinic or hospital visits prior to surgery date, complications were defined using codes newly incident after surgery and up to 90 days following discharge. We organized complications into major and minor. Major complications were myocardial infarction, renal failure, and stroke; minor complications included arrhythmia, postoperative bleeding, deep venous thrombosis, pneumonia, pulmonary embolism, sepsis, and urinary tract infection.
Mortality
Deaths were ascertained from VA Vital Status files.
Statistical Analysis
The unit of analysis was surgical episode; surgeries were stratified by 30‐day mortality. We evaluated differences between the 2 groups using 2 tests accounting for restarting of an ACE‐I through day 30, risk factors, patient, and hospital‐stay characteristics. We also compared those who did not restart from postoperative day 0 to 14 and 15 to 30 to all others who did not restart at any point up to 90 days. Independent variables included age, gender, indications for ACE‐I, comorbidity burden, type and year of surgery, previous hospitalizations, length of stay, and complications. To account for site‐related effects and clustering of observations (ie, surgeries within hospitals), we included quartiles of hospital volume and hospital rates of ACE‐I restart in models and used cluster command in Stata (StataCorp, College Station, TX).
Risk of Mortality
We developed Cox regression models to examine 30‐day mortality risks between restart (015 days) and restart (1530 days) groups to a reference group of patients who did not restart in the first 14 days after surgery (ie, no restart [014 days]). We considered those who had restarted their ACE‐I beyond day 14 and excluded these from comparisons to the no restart group. Independent variables included age, gender, indications for ACE‐I usage, comorbidity, type and year of surgery, previous hospitalizations, length of stay, quartiles of hospital surgical volume and rates of restarting an ACE‐I, and complications.
Sensitivity Analyses
Using Cox regression, we tested robustness of results regarding no restart (014 days) versus restart (014 days) in subsets after excluding patients who died postoperative day 0 to 2 and those with no oral medications on postoperative day 0 to 14, those with low comorbidity burden, within subtypes of surgery, and by surgical episode. To evaluate confounding by indication, we examined subsets without major complications and after excluding patients who died postoperative day 0 to 14. We then developed a propensity score model using quintiles to estimate average treatment effects associated with no restart (014 days).[16] A propensity score reflecting the probability of ACE‐I administration at 14 days was developed using logistic regression accounting for all independent variables. For analyses, we considered a 2‐tailed P value of 0.05 as statistically significant. Stata 12.1 software (Stata Corp.) was used.
RESULTS
Table 1 describes the characteristics and 30‐day mortality rates for our cohort. By postoperative day 14, 75% of the study sample (n=220,317) had restarted an ACE‐I (Figure 1). Our sample consisted primarily of older men with a substantial comorbidity burden and multiple indications for an ACE‐I. Most patients had 1 surgical episode, with the largest fraction undergoing general surgery overall. A third of the cases had lengths of stay >1 week, and surgeries occurred throughout the study period. The largest number of surgeries was noted for centers in 75% to 100% surgical volume and 50% to 75% restart quartiles. Most surgeries had no or minor complications.
The no restart (014 days) group had a higher 30‐day mortality rate (7.3%) compared to those who restarted by postoperative day 14 (2.1%) or 30 (1.7%). The highest mortality rates were found in patients aged >90 years, with a >4 comorbidity index or hospital stays >3 weeks, and those experiencing major postoperative complications.
30‐Day Mortality
Table 2 indicates that nonresumption of an ACE‐I from postoperative day 0 to 14 was independently associated with an approximately 2.5‐fold increased risk of 30‐day mortality (hazard ratio [HR]: 3.44; 95% confidence interval [CI]: 3.30‐3.60; P<0.001). Lower hazard ratios were noted when patients who restarted postoperative days 15 to 30 were included in models (HR: 2.79; 95% CI: 2.67‐2.92; P<0.001).
The sensitivity analyses illustrate the durability of treatment effects (Table 3). After excluding patients who died during days 0 to 2 and without a record of receiving an oral medication by postoperative day 14, ACE‐I nonresumption was associated with an 88% increase in 30‐day mortality risk (HR: 1.88; 95% CI: 1.79‐1.98; P<0.001). Similar increased risks were seen in patients with less comorbidity for each specialty and for those who did not experience a major complication. In data not shown, adjusting by propensity score did not modulate treatment effects (HR for no restart [014 days]: 3.03; 95% CI: 2.78‐3.30; P<0.001).
| Population | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|
| ||
| Exclude patients who died day 02 or no record of oral medications days 014 | 2.29 (2.182.40) | 1.88 (1.791.98) |
| Cases with 02 comorbidity scoreb | 1.92 (1.742.12) | 1.72 (1.551.90) |
| Only cardiothoracic surgery casesb | 2.07 (1.832.35) | 1.94 (1.702.21) |
| Only neurosurgery casesb | 1.49 (1.102.02) | 1.46 (1.072.00) |
| Only orthopedic surgery casesb | 2.48 (2.122.91) | 2.17 (1.842.55) |
| Only urologic surgery casesb | 1.92 (1.582.34) | 1.37 (1.121.68) |
| Only first surgery casesb | 2.22 (2.092.35) | 1.86 (1.751.97) |
| Subsequent surgery casesb | 2.49 (2.272.73) | 1.96 (1.782.16) |
| Cases with no major complicationsb | 2.49 (2.362.64) | 2.25 (2.122.38) |
| Exclude patients who died within the first 14 days after surgeryc | 2.26 (2.112.41) | 1.66 (1.551.78) |
Other factors associated with increased 30‐day mortality are displayed in Table 4. The risk associated with not restarting an ACE‐I was similar to effect of age >90years and a >4 comorbidity index.
| Parameter | Reference Group | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|---|
| |||
| No restart (014 days)b | Restart (014 days)c | 2.92 (2.803.05) | 2.39 (2.292.50) |
| Age, y | |||
| 6170 | Age <60 years | 1.33 (1.241.43) | 1.36 (1.261.46) |
| 7190 | 2.72 (2.552.90) | 2.01 (1.892.30) | |
| >90 | 4.05 (3.454.76) | 2.70 (2.183.74) | |
| Male | Female | 2.11 (1.742.57) | 1.54 (1.271.88) |
| Comorbidity score | |||
| 24 | 1 | 2.19 (2.062.33) | 1.36 (1.271.45) |
| >4 | 4.57 (4.294.87) | 1.97 (1.822.13) | |
| Center surgical volume quartile | |||
| 025th percentile | 76th100th percentile | 1.35 (1.281.43) | 1.21 (1.141.29) |
| 26th50th percentile | 1.11 (1.041.18) | 1.05 (0.991.12) | |
| Indications | |||
| Heart failure | No heart failure | 2.23 (2.142.34) | 1.19 (1.121.26) |
| Year of surgery | |||
| 19992002 | 20062008 | 1.49 (1.411.58) | 1.07 (1.011.13) |
| 20032005 | 1.21 (1.451.29) | 1.13 (1.061.20) | |
DISCUSSION
The results from this national retrospective study confirm our hypothesis that nonresumption of an ACE‐I for 14 or more postoperative days occurs frequently for VA surgery patients. However, we found that nonresumption of an ACE‐I during the first 2 weeks after surgery is independently associated with increased 30‐day mortality. Our study is one of the first to examine the patterns and risks of postoperative ACE‐I management across a large and varied surgical population.[11, 17]
The lack of inpatient and outpatient ACE‐I prescription use by postoperative day 14 across multiple surgery classes suggests that surgical patients may be prone to short‐term nonresumption of an ACE‐I. Our intention in using a 14‐day window to evaluate restarting strategies was to account for immediate postoperative management. After surgery, careful appraisal of whether medications should be restarted is often necessary in the face of substantially deranged physiology, hypercoagulability, and blood loss.[18] After physiologic stabilization over several days, cardiovascular drugs are usually restarted thereafter to help manage chronic comorbidities.[19] One immediate conclusion from our findings is that ACE‐I are commonly discontinued perioperatively (potentially due to concerns for hypotension), and are often not restarted.[20, 21, 22, 23, 24, 25]
Our rates of ACE‐I nonresumption are comparable to rates of nonresumption reported postoperatively for other medications and raise concerns for inadequate medication reconciliation in surgical cohorts. Bell et al. conducted a population‐based cohort study of patients undergoing elective surgery and found that 11.4% of 45,220 patients chronically prescribed warfarin were not restarted by postoperative day 180.[22] A subsequent study showed intensive care unit (ICU) admission was associated with increased rates of not restarting 4 of 5 medication groups (range, 4.5%19.4%; statins, antiplatelet/anticoagulant agents, levothyroxine, respiratory inhalers, and gastric acid‐suppressing drugs).[21] One‐year follow‐up showed elevated odds for the secondary composite outcome of death in the statins group (odds ratio [OR]: 1.07; 95% CI: 1.03‐1.11) and antiplatelet/anticoagulant agents group (OR: 1.10; 95% CI: 1.03‐1.16). Drenger et al. noted a 50% rate for no restart of ACE‐I after CABG surgery; restarting was associated with a decreased composite outcome of cardiac, cerebral, and renal events and in‐hospital mortality (OR: 0.50; 95% CI: 0.38‐0.66).[26] Because medication management has been noted to be problematic at care transitions, the inpatient medication reconciliation recommendations articulated in recent Joint Commission National Patient Safety Goals may be particularly relevant for high‐risk surgical patients who experience multiple transitions of care (ie, operating room to ICU to surgical ward to rehabilitation unit to discharge).[19, 24, 27]
In examining the crucial interval for the surgical patientthe postoperative period when medication changes are commonwe found a nearly 2.5‐fold increase in risk for 30‐day mortality associated with nonresumption of an ACE‐I.[4, 19, 28] We also noted that those who were restarted later on day 15 to 30 fared better than those not restarted (Table 2). Similar effect sizes have been found with postoperative nonresumption of other cardiovascular medications. Not restarting chronic ‐blocker treatment after surgery is associated with a significant 1‐year mortality risk (HR: 2.7; 95% CI: 1.25.9).[29] Postoperative statin withdrawal (>4 days) is an independent predictor of postoperative myonecrosis (OR: 2.9; 95% CI: 1.6‐5.5).[30, 31] Biologic mechanisms contributing to mortality after a temporary failure to restart an ACE‐I are speculative and were not addressed in this study. Potential mechanisms may lie with hypertensive rebound and associated cardiac decompensation. Withdrawing an ACE‐I can cause rapid increases in blood pressure within 48 hours on home self‐measured blood pressure in hypertensive patients and in diabetic patients with chronic renal failure.[32, 33] Patients with heart failure or coronary artery disease may then experience myocardial ischemia in the context of elevated blood pressure. Not restarting an ACE‐I may also lead to compromised microcirculatory flow with renal complications and mortality.[34, 35]
Alternative explanations for the magnitude of our findings may lie with unmeasured confounders. Our analysis did not evaluate potential interactions arising from the failure to restart of all other medications (eg, ‐blockers) or evaluate changes to angiotensin receptor blockers (ARBs). In addition, our study lacked data on health system variations or emergent versus elective surgeries. However, a key starting point of our analysis was distinguishing between purposeful versus potentially unintentional nonresumption of an ACE‐I. To accomplish this, we included patients who had at least 3 prescription ACE‐I fills prior to surgery, evaluated the preoperative indications for an ACE‐I and the ability to take postoperative oral medications (eg, immortal time bias), and accounted for minor and major postoperative complications.
To address bias from unmeasured confounders, we conducted sensitivity analyses in more homogeneous subpopulations. With each sensitivity analysis, we found consistently strong associations between increased 30‐day mortality and nonresumption of an ACE‐I (Table 3). Strong effects were observed in patients without major complications and with low comorbidity burdens, patients in whom we would not expect an effect. Because deaths in postoperative day 0 to 2 could be attributed to surgical factors (ie, hemorrhage) or that patients who did not restart an ACE‐I in postoperative day 0 to 14 were too sick to tolerate oral medications, we excluded these patients along with patients who died before postoperative day 14. Both sensitivity analyses maintained our primary finding. Somewhat attenuated risks were found when we examined ACE‐I nonresumption by individual surgery types, perhaps reflective of differences in comorbidity burden.
Finally, although this study did not examine predictors of nonresumption, our models showed that in the context of postoperative ACE‐I management, factors including increasing age, being male, those with heart failure, and surgeries conducted in centers with low surgical volume were associated with increased 30‐day mortality (Table 4). Future research might consider how reinstitution of an ACE‐I occurs in these subpopulations to identify potential mechanisms for nonresumption.
Our study has several strengths. We examined patients over a decade, considered all major types of surgery, and studied patients across a healthcare system. Moreover, we used computerized prescription data and medical records (eg, discharge diagnosis, ICD‐9 codes) to derive risk factors. VA prescription data are standardized and accurate because of intensive efforts to contain costs.[36] Within VA data, the estimated sensitivity of computerized diagnoses exceeds 80% in the administrative files, with specificity of 91% to 100% for common diagnoses such as coronary artery disease.[37] These records also carefully and accurately identify death.[38]
We also identified potential limitations to our study. First, a retrospective, observational, cohort study may be prone to selection bias, and therefore we report associations that are not necessarily causal relationships. However, our methods are supported by the fact that we developed a large study sample consisting of consecutive surgical patients over a decade and noted large effect sizes across multiple subpopulations. Second, for group assignment, we used prescription records rather than medication administration data. Nevertheless, a cohort analysis focusing on exposure is standard for epidemiologic studies and shows outcomes of care resulting from daily clinical practice.[39] Third, we did not study the cause of death, data that may help to identify potential causal pathways between not restarting an ACE‐I and mortality. Fourth, our results come from VA medical centers and so may not be generalizable to non‐VA institutions. However, the length of observation under conditions of routine clinical practice at multiple medical centers and a diverse set of surgical procedures support the external validity of our study results. Fifth, we did not have clinical data accounting for surgeon‐level effects potentially affecting rates of nonresumption of an ACE‐I, American Society of Anesthesiology physical status, information on perioperative hypotension or vasopressors, or the presence of a postoperative primary care visit.
In conclusion, in the VA Healthcare System, temporary nonresumption of an ACE‐I is common. Postoperative nonresumption of an ACE‐I, although sometimes indicated and appropriate, is associated with increased risk of mortality. Careful attention to the issue of eventual reinstitution of medications for chronic conditions, such as an ACE‐I, is indicated to avoid unnecessary mortality. Because early experience showed that dose titration was a key for successful application of an ACE‐I, practitioners may also need to consider dose modification rather than simply continuation or not restarting.[40] Future research is needed to confirm our results in other healthcare systems and to define mechanisms that link postoperative nonresumption of an ACE‐I to mortality.
Acknowledgements
The authors acknowledge Dr. Edward R. Mariano, Chief Anesthesia Service, VA Palo Alto Health Care System, and Associate Professor, Stanford Department of Anesthesiology for general support of this research and critical review of the manuscript. We would also like to thank Dr. Ronald Pearl, Chair, Department of Anesthesiology, Perioperative, and Pain Medicine, Stanford University, for his support of our research. This material is the result of work supported with resources and the use of facilities at the Veterans Affairs Medical Center, San Francisco and Veterans Affairs Palo Alto Healthcare System.
Disclosure: The Northern California Institute for Research and Education and the Veterans Affairs Medical Center, San Francisco, California supported this work. This work was presented at the American Society of Anesthesiologists Annual Meeting, Chicago, Illinois, October 1519, 2011, and the Veterans Affairs National Health Services Research and Development National Conference, National Harbor, Maryland, July 1619, 2012.
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Perioperative medication management requires careful consideration, because surgical patients, especially older ones, may be receiving multiple medications for the treatment of acute or chronic comorbidities.[1] Because patients often present to surgery stabilized on their drug regimens, nonresumption of medications for chronic conditions may be problematic in controlling underlying diseases.[2] For example, nonresumption of cardiovascular medications such as ‐blockers postoperatively has been shown to lead to increased longer‐term mortality.[3] Little data, however, exist to guide practitioners on the postoperative management risks for another widely used class of cardiovascular medication: angiotensin‐converting enzyme inhibitors (ACE‐Is).[4]
About 170 million prescriptions for an ACE‐I are dispensed in the United States annually, which reflects a multiple criteria for their use including hypertension, heart failure, ischemic heart disease, coronary disease risk, diabetes mellitus, chronic kidney disease, recurrent stroke prevention, and vascular disease.[5, 6, 7] ACE‐Is have been shown to improve outcomes in patients with ischemic heart disease and heart failure.[8, 9] An observational study found that perioperative use of an ACE‐I in coronary artery bypass grafting (CABG) patients was associated with increased mortality, use of vasopressors, and postoperative acute renal failure.[10] Data also indicate that patients who continue the use of an ACE‐I perioperatively can experience severe hypotension.[11] As a result, some have recommended that consideration be given to not restarting the ACE‐I perioperatively, especially with hypertensive patients undergoing noncardiac surgery.[12] However, little evidence exists to document benefits and risks of not restarting an ACE‐I in surgical patients for various intervals. To evaluate these risks, we tested the hypothesis that postoperative nonresumption of an ACE‐I occurs frequently for broad cohorts of Veterans Affairs (VA) surgery patients within the first 14 days and is associated with increased 30‐day mortality.
MATERIALS AND METHODS
After institutional review board approval (University of California, San Francisco), we examined surgeries conducted at hospitals at 120 stations within the VA Health Care System (VAHCS). The VAHCS is the largest integrated healthcare system in the United States, with long‐standing electronic medical records capturing detailed demographic, pharmacy, and mortality information.[13] Data were extracted from Medical Statistical Analysis System (SAS) and Corporate Data Warehouse (CDW) files in the VA Informatics and Computing Infrastructure.[14]
Development of the Study Population
To identify surgery patients who were consistently prescribed an ACE‐I preoperatively (Figure 1), we first located 1,213,086 surgical admissions in 846,454 patients from 1999 to 2012 using Medical SAS files and classified them by specialty of the surgeon (eg, neurosurgery, orthopedic, urology, cardiothoracic, general, vascular, plastic, and other [such as gynecology]). We identified comorbidities and cardiovascular risk factors from inpatient/outpatient diagnosis files in the CDW using International Classification of Diseases (ICD‐9) diagnosis codes (see Supporting Information, Tables 1 and 2, in the online version of this article). To ensure chronic preoperative ACE‐I use, we included surgeries with 3 outpatient prescription fills of an ACE‐I and <180‐day gap. ACE‐Is included benazepril, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, and ramipril. We excluded cases with a surgery in the prior 90 days and missing diagnosis codes. Our final population was comprised of 294,505 surgical admissions in 240,978 patients.
| Parameter | Surgeries, No. (%), Total=294,505 | Died by 30‐Days, Total=9,227 | P Value |
|---|---|---|---|
| |||
| No restart, 014 daysa | 59,949 (20%) | 7.3% | <0.001 |
| Restart, 014 daysb | 220,317 (75%) | 2.1% | |
| Restart, 1530 daysc | 14,239 (5%) | 1.7% | |
| Age, y | |||
| <60 | 74,326 (14%) | 1.7% | <0.001 |
| 6170 | 97,731 (24%) | 2.3% | |
| 7190 | 119,775 (60%) | 4.6% | |
| >90 | 2,673 (1%) | 6.9% | |
| Gender | |||
| Female | 7,186 (2%) | 1.6% | <0.001 |
| Male | 287,319 (98%) | 3.2% | |
| Indications for use of ACE‐I | |||
| Hypertension | 270,486 (92%) | 2.8% | <0.001 |
| Ischemic heart disease | 129,212 (44%) | 3.8% | <0.001 |
| Vascular disease | 75,410 (26%) | 3.7% | <0.001 |
| Heart failure | 59,809 (20%) | 5.7% | <0.001 |
| Chronic kidney disease | 8,804 (3%) | 4.9% | <0.001 |
| Diabetes mellitus | 170,320 (58%) | 3.0% | <0.001 |
| Coronary disease riskd | 280,958 (95%) | 3.1% | <0.001 |
| Stroke | 22,285 (8%) | 5.2% | <0.001 |
| Comorbidity scoree | |||
| 0 | 72,126 (24%) | 1.4% | <0.001 |
| 1 | 59,609 (20%) | 1.5% | |
| 2 4 | 116,914 (40%) | 3.5% | |
| >4 | 45,856 (16%) | 7.0% | |
| Preoperative ACE‐I gap, daysf | |||
| 045 | 21,383 (7%) | 3.7% | <0.001 |
| 4690 | 30,237 (10%) | 3.8% | |
| 91180 | 242,885 (83%) | 3.0% | |
| Surgical specialty | |||
| General | 98,210 (33%) | 4.6% | <0.001 |
| Neurosurgery | 15,423 (5%) | 2.3% | |
| Orthopedic | 51,600 (18%) | 1.9% | |
| Plastic | 12,547 (4%) | 3.8% | |
| Thoracic | 44,728 (15%) | 3.2% | |
| Urology | 34,595 (12%) | 1.5% | |
| Vascular | 34,228 (12%) | 2.8% | |
| Other (gynecology) | 3,174 (1%) | 1.4% | |
| Year of surgery | |||
| 19992002 | 66,689 (23%) | 4.2% | <0.001 |
| 20032005 | 75,420 (26%) | 3.4% | |
| 20062008 | 76,563 (26%) | 2.8% | |
| 20092012 | 75,833 (26%) | 2.2% | |
| No. of prior surgeries | |||
| 0 | 215,443 (74%) | 3.2% | 0.413 |
| 1 | 56,419 (19%) | 3.1% | |
| 2 | 22,643 (7%) | 3.1% | |
| Length of stay, d | |||
| 1 | 40,538 (14%) | 1.4% | <0.001 |
| 23 | 59,817 (20%) | 1.4% | |
| 47 | 83,366 (28%) | 2.0% | |
| 821 | 83,379 (28%) | 4.7% | |
| >21 | 27,405 (9%) | 8.0% | |
| Center surgical volume quartileg | |||
| 0%25% | 74,846 (25%) | 3.7% | <0.001 |
| 25%50% | 74,569 (25%) | 3.1% | |
| 50%75% | 69,947 (24%) | 2.8% | |
| 75%100% | 75,143 (26%) | 2.8% | |
| Center restart quartileh | |||
| 0%25% | 73,750 (25%) | 3.1% | 0.014 |
| 25%50% | 81,071 (28%) | 3.0% | |
| 50%75% | 83,952 (29%) | 3.3% | |
| 75%100% | 55,732 (19%) | 3.2% | |
| No complication | 80,700 (27%) | 1.3% | <0.001 |
| Minor complicationi | 181,924 (62%) | 4.2% | <0.001 |
| Major complicationj | 46,977 (16%) | 8.3% | <0.001 |
| Complications | |||
| Arrhythmia | 3,037 (1%) | 2.0% | <0.001 |
| Bleeding | 12,887 (4%) | 4.8% | <0.001 |
| Deep venous thrombosis | 6,075 (2%) | 3.6% | <0.001 |
| Myocardial infarction | 9,114 (3%) | 7.7% | <0.001 |
| Pneumonia | 109,660 (37%) | 5.1% | <0.001 |
| Pulmonary embolism | 5,064 (2%) | 6.2% | <0.001 |
| Renal failure | 25,513 (9%) | 11.0% | <0.001 |
| Sepsis | 5,846 (2%) | 16.5% | <0.001 |
| Stroke | 19,546 (7%) | 5.0% | <0.001 |
| Urinary tract infection | 32,548 (11%) | 4.9% | <0.001 |
| Unadjusted Hazard for 30‐Day Mortality (OR [95% CI]) | Adjusted hazard for 30 day mortality (OR [95% CI]) | ||||
|---|---|---|---|---|---|
| Restart (014 Days) (Referent)a | No Restart, 014 Daysb | Restart, 1530 Daysc | Restart, 014 Days (Referent) | No Restart, 014 Days | Restart, 1530 Days |
| |||||
| 1 | 3.44 (3.303.60)d | 0.23 (0.200.26)d | 1 | 2.79 (2.672.92)d | 0.24 (0.210.28)d |
| Restart, 014 Days (Referent) | No Restart, 014 Days | NA | Restart, 014 Days (Referent) | No Restart, 014 Days | NA |
| 1 | 2.92 (2.803.05)d | NA27 | 1 | 2.39 (2.292.50)d | NA27 |
Postoperative Medication Use
We defined patients as postoperative restart (014 days) if an ACE‐I was administered in‐hospital (oral or intravenous) or a postdischarge outpatient ACE‐I prescription was filled in the 14 days following surgery. In absence of ACE‐I administration or prescription during postoperative days 0 to 14, patients were classified as no restart (014 days). Intraclass changes from one ACE‐I to another were considered a restart if they occurred within 0 to 14 days of surgery. We also tracked ACE‐I prescription fills through postoperative day 15 to 30 (ie, restart [1530 days]) and noted administration or filling of oral medications. Oral medications were classified as tablets or caplets in formularies.
Patient Characteristics
We categorized patients by age strata: <60, 61 to 70, 71 to 90, and >90 years old; gender; and epochs (every 34 years starting from calendar year 1999). We tracked prior surgery admissions and length of stay.
Hospital Factors
To account for clustering of surgeries and hospital‐related factors affecting ACE‐I use practices, we divided hospitals into quartiles of (1) total surgical volume based on total number of surgeries done at a hospital from 1999 to 2012 (0%25%, n<2378; 50%, n=3498; 75%, n=4531; highest surgical volume, 8162); and (2) percent of cases restarted on ACE‐I at 14 days (71%, 76%, 79%, and 100%).
Indications, Patient Illness Severity, and Complications
We determined probable indications for ACE‐I usage (ie, heart failure) and comorbidities using ICD‐9 codes in medical records prior to surgical admissions (see Supporting Information, Tables 1 and 2, in the online version of this article). Comorbidities were aggregated using algorithms developed by Gagne aggregating comorbidity conditions (defined by Elixhauser) into scores similar to Charlson scores.[15] The Gagne score has higher correlation with 30‐day, 90‐day, 180‐day, and 1‐year mortality than Charlson scores.[15]
After evaluating secondary diagnosis codes in the clinic or hospital visits prior to surgery date, complications were defined using codes newly incident after surgery and up to 90 days following discharge. We organized complications into major and minor. Major complications were myocardial infarction, renal failure, and stroke; minor complications included arrhythmia, postoperative bleeding, deep venous thrombosis, pneumonia, pulmonary embolism, sepsis, and urinary tract infection.
Mortality
Deaths were ascertained from VA Vital Status files.
Statistical Analysis
The unit of analysis was surgical episode; surgeries were stratified by 30‐day mortality. We evaluated differences between the 2 groups using 2 tests accounting for restarting of an ACE‐I through day 30, risk factors, patient, and hospital‐stay characteristics. We also compared those who did not restart from postoperative day 0 to 14 and 15 to 30 to all others who did not restart at any point up to 90 days. Independent variables included age, gender, indications for ACE‐I, comorbidity burden, type and year of surgery, previous hospitalizations, length of stay, and complications. To account for site‐related effects and clustering of observations (ie, surgeries within hospitals), we included quartiles of hospital volume and hospital rates of ACE‐I restart in models and used cluster command in Stata (StataCorp, College Station, TX).
Risk of Mortality
We developed Cox regression models to examine 30‐day mortality risks between restart (015 days) and restart (1530 days) groups to a reference group of patients who did not restart in the first 14 days after surgery (ie, no restart [014 days]). We considered those who had restarted their ACE‐I beyond day 14 and excluded these from comparisons to the no restart group. Independent variables included age, gender, indications for ACE‐I usage, comorbidity, type and year of surgery, previous hospitalizations, length of stay, quartiles of hospital surgical volume and rates of restarting an ACE‐I, and complications.
Sensitivity Analyses
Using Cox regression, we tested robustness of results regarding no restart (014 days) versus restart (014 days) in subsets after excluding patients who died postoperative day 0 to 2 and those with no oral medications on postoperative day 0 to 14, those with low comorbidity burden, within subtypes of surgery, and by surgical episode. To evaluate confounding by indication, we examined subsets without major complications and after excluding patients who died postoperative day 0 to 14. We then developed a propensity score model using quintiles to estimate average treatment effects associated with no restart (014 days).[16] A propensity score reflecting the probability of ACE‐I administration at 14 days was developed using logistic regression accounting for all independent variables. For analyses, we considered a 2‐tailed P value of 0.05 as statistically significant. Stata 12.1 software (Stata Corp.) was used.
RESULTS
Table 1 describes the characteristics and 30‐day mortality rates for our cohort. By postoperative day 14, 75% of the study sample (n=220,317) had restarted an ACE‐I (Figure 1). Our sample consisted primarily of older men with a substantial comorbidity burden and multiple indications for an ACE‐I. Most patients had 1 surgical episode, with the largest fraction undergoing general surgery overall. A third of the cases had lengths of stay >1 week, and surgeries occurred throughout the study period. The largest number of surgeries was noted for centers in 75% to 100% surgical volume and 50% to 75% restart quartiles. Most surgeries had no or minor complications.
The no restart (014 days) group had a higher 30‐day mortality rate (7.3%) compared to those who restarted by postoperative day 14 (2.1%) or 30 (1.7%). The highest mortality rates were found in patients aged >90 years, with a >4 comorbidity index or hospital stays >3 weeks, and those experiencing major postoperative complications.
30‐Day Mortality
Table 2 indicates that nonresumption of an ACE‐I from postoperative day 0 to 14 was independently associated with an approximately 2.5‐fold increased risk of 30‐day mortality (hazard ratio [HR]: 3.44; 95% confidence interval [CI]: 3.30‐3.60; P<0.001). Lower hazard ratios were noted when patients who restarted postoperative days 15 to 30 were included in models (HR: 2.79; 95% CI: 2.67‐2.92; P<0.001).
The sensitivity analyses illustrate the durability of treatment effects (Table 3). After excluding patients who died during days 0 to 2 and without a record of receiving an oral medication by postoperative day 14, ACE‐I nonresumption was associated with an 88% increase in 30‐day mortality risk (HR: 1.88; 95% CI: 1.79‐1.98; P<0.001). Similar increased risks were seen in patients with less comorbidity for each specialty and for those who did not experience a major complication. In data not shown, adjusting by propensity score did not modulate treatment effects (HR for no restart [014 days]: 3.03; 95% CI: 2.78‐3.30; P<0.001).
| Population | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|
| ||
| Exclude patients who died day 02 or no record of oral medications days 014 | 2.29 (2.182.40) | 1.88 (1.791.98) |
| Cases with 02 comorbidity scoreb | 1.92 (1.742.12) | 1.72 (1.551.90) |
| Only cardiothoracic surgery casesb | 2.07 (1.832.35) | 1.94 (1.702.21) |
| Only neurosurgery casesb | 1.49 (1.102.02) | 1.46 (1.072.00) |
| Only orthopedic surgery casesb | 2.48 (2.122.91) | 2.17 (1.842.55) |
| Only urologic surgery casesb | 1.92 (1.582.34) | 1.37 (1.121.68) |
| Only first surgery casesb | 2.22 (2.092.35) | 1.86 (1.751.97) |
| Subsequent surgery casesb | 2.49 (2.272.73) | 1.96 (1.782.16) |
| Cases with no major complicationsb | 2.49 (2.362.64) | 2.25 (2.122.38) |
| Exclude patients who died within the first 14 days after surgeryc | 2.26 (2.112.41) | 1.66 (1.551.78) |
Other factors associated with increased 30‐day mortality are displayed in Table 4. The risk associated with not restarting an ACE‐I was similar to effect of age >90years and a >4 comorbidity index.
| Parameter | Reference Group | Unadjusted Hazard Ratio (95% CI)a | Adjusted Hazard Ratio (95% CI)a |
|---|---|---|---|
| |||
| No restart (014 days)b | Restart (014 days)c | 2.92 (2.803.05) | 2.39 (2.292.50) |
| Age, y | |||
| 6170 | Age <60 years | 1.33 (1.241.43) | 1.36 (1.261.46) |
| 7190 | 2.72 (2.552.90) | 2.01 (1.892.30) | |
| >90 | 4.05 (3.454.76) | 2.70 (2.183.74) | |
| Male | Female | 2.11 (1.742.57) | 1.54 (1.271.88) |
| Comorbidity score | |||
| 24 | 1 | 2.19 (2.062.33) | 1.36 (1.271.45) |
| >4 | 4.57 (4.294.87) | 1.97 (1.822.13) | |
| Center surgical volume quartile | |||
| 025th percentile | 76th100th percentile | 1.35 (1.281.43) | 1.21 (1.141.29) |
| 26th50th percentile | 1.11 (1.041.18) | 1.05 (0.991.12) | |
| Indications | |||
| Heart failure | No heart failure | 2.23 (2.142.34) | 1.19 (1.121.26) |
| Year of surgery | |||
| 19992002 | 20062008 | 1.49 (1.411.58) | 1.07 (1.011.13) |
| 20032005 | 1.21 (1.451.29) | 1.13 (1.061.20) | |
DISCUSSION
The results from this national retrospective study confirm our hypothesis that nonresumption of an ACE‐I for 14 or more postoperative days occurs frequently for VA surgery patients. However, we found that nonresumption of an ACE‐I during the first 2 weeks after surgery is independently associated with increased 30‐day mortality. Our study is one of the first to examine the patterns and risks of postoperative ACE‐I management across a large and varied surgical population.[11, 17]
The lack of inpatient and outpatient ACE‐I prescription use by postoperative day 14 across multiple surgery classes suggests that surgical patients may be prone to short‐term nonresumption of an ACE‐I. Our intention in using a 14‐day window to evaluate restarting strategies was to account for immediate postoperative management. After surgery, careful appraisal of whether medications should be restarted is often necessary in the face of substantially deranged physiology, hypercoagulability, and blood loss.[18] After physiologic stabilization over several days, cardiovascular drugs are usually restarted thereafter to help manage chronic comorbidities.[19] One immediate conclusion from our findings is that ACE‐I are commonly discontinued perioperatively (potentially due to concerns for hypotension), and are often not restarted.[20, 21, 22, 23, 24, 25]
Our rates of ACE‐I nonresumption are comparable to rates of nonresumption reported postoperatively for other medications and raise concerns for inadequate medication reconciliation in surgical cohorts. Bell et al. conducted a population‐based cohort study of patients undergoing elective surgery and found that 11.4% of 45,220 patients chronically prescribed warfarin were not restarted by postoperative day 180.[22] A subsequent study showed intensive care unit (ICU) admission was associated with increased rates of not restarting 4 of 5 medication groups (range, 4.5%19.4%; statins, antiplatelet/anticoagulant agents, levothyroxine, respiratory inhalers, and gastric acid‐suppressing drugs).[21] One‐year follow‐up showed elevated odds for the secondary composite outcome of death in the statins group (odds ratio [OR]: 1.07; 95% CI: 1.03‐1.11) and antiplatelet/anticoagulant agents group (OR: 1.10; 95% CI: 1.03‐1.16). Drenger et al. noted a 50% rate for no restart of ACE‐I after CABG surgery; restarting was associated with a decreased composite outcome of cardiac, cerebral, and renal events and in‐hospital mortality (OR: 0.50; 95% CI: 0.38‐0.66).[26] Because medication management has been noted to be problematic at care transitions, the inpatient medication reconciliation recommendations articulated in recent Joint Commission National Patient Safety Goals may be particularly relevant for high‐risk surgical patients who experience multiple transitions of care (ie, operating room to ICU to surgical ward to rehabilitation unit to discharge).[19, 24, 27]
In examining the crucial interval for the surgical patientthe postoperative period when medication changes are commonwe found a nearly 2.5‐fold increase in risk for 30‐day mortality associated with nonresumption of an ACE‐I.[4, 19, 28] We also noted that those who were restarted later on day 15 to 30 fared better than those not restarted (Table 2). Similar effect sizes have been found with postoperative nonresumption of other cardiovascular medications. Not restarting chronic ‐blocker treatment after surgery is associated with a significant 1‐year mortality risk (HR: 2.7; 95% CI: 1.25.9).[29] Postoperative statin withdrawal (>4 days) is an independent predictor of postoperative myonecrosis (OR: 2.9; 95% CI: 1.6‐5.5).[30, 31] Biologic mechanisms contributing to mortality after a temporary failure to restart an ACE‐I are speculative and were not addressed in this study. Potential mechanisms may lie with hypertensive rebound and associated cardiac decompensation. Withdrawing an ACE‐I can cause rapid increases in blood pressure within 48 hours on home self‐measured blood pressure in hypertensive patients and in diabetic patients with chronic renal failure.[32, 33] Patients with heart failure or coronary artery disease may then experience myocardial ischemia in the context of elevated blood pressure. Not restarting an ACE‐I may also lead to compromised microcirculatory flow with renal complications and mortality.[34, 35]
Alternative explanations for the magnitude of our findings may lie with unmeasured confounders. Our analysis did not evaluate potential interactions arising from the failure to restart of all other medications (eg, ‐blockers) or evaluate changes to angiotensin receptor blockers (ARBs). In addition, our study lacked data on health system variations or emergent versus elective surgeries. However, a key starting point of our analysis was distinguishing between purposeful versus potentially unintentional nonresumption of an ACE‐I. To accomplish this, we included patients who had at least 3 prescription ACE‐I fills prior to surgery, evaluated the preoperative indications for an ACE‐I and the ability to take postoperative oral medications (eg, immortal time bias), and accounted for minor and major postoperative complications.
To address bias from unmeasured confounders, we conducted sensitivity analyses in more homogeneous subpopulations. With each sensitivity analysis, we found consistently strong associations between increased 30‐day mortality and nonresumption of an ACE‐I (Table 3). Strong effects were observed in patients without major complications and with low comorbidity burdens, patients in whom we would not expect an effect. Because deaths in postoperative day 0 to 2 could be attributed to surgical factors (ie, hemorrhage) or that patients who did not restart an ACE‐I in postoperative day 0 to 14 were too sick to tolerate oral medications, we excluded these patients along with patients who died before postoperative day 14. Both sensitivity analyses maintained our primary finding. Somewhat attenuated risks were found when we examined ACE‐I nonresumption by individual surgery types, perhaps reflective of differences in comorbidity burden.
Finally, although this study did not examine predictors of nonresumption, our models showed that in the context of postoperative ACE‐I management, factors including increasing age, being male, those with heart failure, and surgeries conducted in centers with low surgical volume were associated with increased 30‐day mortality (Table 4). Future research might consider how reinstitution of an ACE‐I occurs in these subpopulations to identify potential mechanisms for nonresumption.
Our study has several strengths. We examined patients over a decade, considered all major types of surgery, and studied patients across a healthcare system. Moreover, we used computerized prescription data and medical records (eg, discharge diagnosis, ICD‐9 codes) to derive risk factors. VA prescription data are standardized and accurate because of intensive efforts to contain costs.[36] Within VA data, the estimated sensitivity of computerized diagnoses exceeds 80% in the administrative files, with specificity of 91% to 100% for common diagnoses such as coronary artery disease.[37] These records also carefully and accurately identify death.[38]
We also identified potential limitations to our study. First, a retrospective, observational, cohort study may be prone to selection bias, and therefore we report associations that are not necessarily causal relationships. However, our methods are supported by the fact that we developed a large study sample consisting of consecutive surgical patients over a decade and noted large effect sizes across multiple subpopulations. Second, for group assignment, we used prescription records rather than medication administration data. Nevertheless, a cohort analysis focusing on exposure is standard for epidemiologic studies and shows outcomes of care resulting from daily clinical practice.[39] Third, we did not study the cause of death, data that may help to identify potential causal pathways between not restarting an ACE‐I and mortality. Fourth, our results come from VA medical centers and so may not be generalizable to non‐VA institutions. However, the length of observation under conditions of routine clinical practice at multiple medical centers and a diverse set of surgical procedures support the external validity of our study results. Fifth, we did not have clinical data accounting for surgeon‐level effects potentially affecting rates of nonresumption of an ACE‐I, American Society of Anesthesiology physical status, information on perioperative hypotension or vasopressors, or the presence of a postoperative primary care visit.
In conclusion, in the VA Healthcare System, temporary nonresumption of an ACE‐I is common. Postoperative nonresumption of an ACE‐I, although sometimes indicated and appropriate, is associated with increased risk of mortality. Careful attention to the issue of eventual reinstitution of medications for chronic conditions, such as an ACE‐I, is indicated to avoid unnecessary mortality. Because early experience showed that dose titration was a key for successful application of an ACE‐I, practitioners may also need to consider dose modification rather than simply continuation or not restarting.[40] Future research is needed to confirm our results in other healthcare systems and to define mechanisms that link postoperative nonresumption of an ACE‐I to mortality.
Acknowledgements
The authors acknowledge Dr. Edward R. Mariano, Chief Anesthesia Service, VA Palo Alto Health Care System, and Associate Professor, Stanford Department of Anesthesiology for general support of this research and critical review of the manuscript. We would also like to thank Dr. Ronald Pearl, Chair, Department of Anesthesiology, Perioperative, and Pain Medicine, Stanford University, for his support of our research. This material is the result of work supported with resources and the use of facilities at the Veterans Affairs Medical Center, San Francisco and Veterans Affairs Palo Alto Healthcare System.
Disclosure: The Northern California Institute for Research and Education and the Veterans Affairs Medical Center, San Francisco, California supported this work. This work was presented at the American Society of Anesthesiologists Annual Meeting, Chicago, Illinois, October 1519, 2011, and the Veterans Affairs National Health Services Research and Development National Conference, National Harbor, Maryland, July 1619, 2012.
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
- , , , . The development of polypharmacy. A longitudinal study. Fam Pract. 2000;17(3):261–267.
- , , , , . Polypharmacy in a general surgical unit and consequences of drug withdrawal. Br J Clin Pharmacol. 2000;49(4):353–362.
- , , , , , . Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients. Am Heart J. 2001;141(1):148–153.
- . Perioperative medication management: general principles and practical applications. Cleve Clin J Med. 2009;76(suppl 4):S126–S132.
- , , , et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572.
- IMS Health. Top therapeutic classes by U.S. dispensed prescriptions. April 7, 2011. Available at: http://www.imshealth.com/deployedfiles/imshealth/Global/Content/StaticFile/Top_Line_Data/2010_Top_Therap eutic_Classes_by_RX.pdf. Accessed September 5, 2011.
- , , , et al. The consistency of the treatment effect of an ACE‐inhibitor based treatment regimen in patients with vascular disease or high risk of vascular disease: a combined analysis of individual data of ADVANCE, EUROPA, and PROGRESS trials. Eur Heart J. 2009;30(11):1385–1394.
- , , , , , . Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153.
- , , , , , . Effects of the early administration of enalapril on mortality in patients with acute myocardial infarction. Results of the Cooperative New Scandinavian Enalapril Survival Study II (CONSENSUS II). N Engl J Med. 1992;327(10):678–684.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636–644.
- , , . Guidelines for pre‐operative cardiac risk assessment and perioperative cardiac management in non‐cardiac surgery: The Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non‐cardiac Surgery of the European Society of Cardiology (ESC) and endorsed by the European Society of Anaesthesiology (ESA). Eur Heart J. 2009;30(22):2769–2812.
- , , , . Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218–2227.
- VA Information Resource Center; VIReC Research User Guide: VHA Decision support system clinical national data extracts. 2nd ed. Hines, IL: U.S. Department of VA, Health Services Research and Development Service, VA Information Resource Center, 2009. Available at: http://www.virec.research.va.gov/RUGs/RUGs-Index.htm. Accessed February 27, 2013.
- , , , , . A combined comorbidity score predicted mortality in elderly patients better than existing scores. J Clin Epidemiol. 2011;64(7):749–759.
- . A tutorial and case study in propensity score analysis: an application to estimating the effect of in‐hospital smoking cessation counseling on mortality. Multi Behav Res. 2011;46(1):119–151.
- , . Stopping and restarting medications in the perioperative period. J Gen Intern Med. 1987;2(4):270–283.
- , , . Perioperative management of drug therapy, clinical considerations. Drugs. 1996;51(2):238–259.
- , , , et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation. 2007;116(17):1971–1996.
- , , , et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med. 2008;23(9):1414–1422.
- , , , et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA. 2011;306(8):840–847.
- , , , , , . Potentially unintended discontinuation of long‐term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525–2531.
- , , , . Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314–323.
- , . Discontinuation and reinstitution of medications during the perioperative period. Am J Health Syst Pharm. 2004;61(9):899–912.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med. 2010;5(8):477–485.
- , , . Guidelines for the management of chronic medication in the perioperative period: systematic review and formal consensus. J Clin Pharm Therap. 2011;36(4):446–467.
- , , , et al. Increase of 1‐year mortality after perioperative beta‐blocker withdrawal in endovascular and vascular surgery patients. Eur J Vasc Endovasc Surg. 2007;33(1):13–19.
- , , , et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg. 2007;104(6):1326–1333.
- , , , et al. Effect of statin withdrawal on frequency of cardiac events after vascular surgery. Am J Cardiol. 2007;100(2):316–320.
- , , , et al. Short‐term effects of withdrawing angiotensin converting enzyme inhibitor therapy on home self‐measured blood pressure in hypertensive patients. Am J Hypertens. 1998;11(2):165–173.
- , , , et al. Hypertensive rebound after angiotensin converting enzyme inhibitor withdrawal in diabetic patients with chronic renal failure. Nephrol Dial Trans. 2001;16(5):1084–1085.
- , , . Vascular protective effects of angiotensin converting enzyme inhibitors and their relation to clinical events. J Cardiovasc Pharmacol. 2001;37(suppl 1):S21–S30.
- , , , et al. Angiotensin‐converting enzyme inhibitor withdrawal and ACE gene polymorphism. Clin Nephrol. 2003;60(4):225–232.
- , . Pharmacy data in the VA health care system. Med Care Res Rev. 2003;60(3 suppl):92S–123S.
- , , , , . Accuracy of computerized outpatient diagnoses in a Veterans Affairs general medicine clinic. Am J Manag Care. 2002;8(1):37–43.
- , , , . Mortality ascertainment in the veteran population: alternatives to the National Death Index. Am J Epidemiol. 1995;141(3):242–250.
- . Statistical considerations in the intent‐to‐treat principle. Control Clin Trials. 2000;21(3):167–189.
- , , . ACE inhibitors in cardiac surgery: current studies and controversies. Hypertens Res. 2010;34(1):15–22.
- , , , . The development of polypharmacy. A longitudinal study. Fam Pract. 2000;17(3):261–267.
- , , , , . Polypharmacy in a general surgical unit and consequences of drug withdrawal. Br J Clin Pharmacol. 2000;49(4):353–362.
- , , , , , . Perioperative beta‐blocker withdrawal and mortality in vascular surgical patients. Am Heart J. 2001;141(1):148–153.
- . Perioperative medication management: general principles and practical applications. Cleve Clin J Med. 2009;76(suppl 4):S126–S132.
- , , , et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572.
- IMS Health. Top therapeutic classes by U.S. dispensed prescriptions. April 7, 2011. Available at: http://www.imshealth.com/deployedfiles/imshealth/Global/Content/StaticFile/Top_Line_Data/2010_Top_Therap eutic_Classes_by_RX.pdf. Accessed September 5, 2011.
- , , , et al. The consistency of the treatment effect of an ACE‐inhibitor based treatment regimen in patients with vascular disease or high risk of vascular disease: a combined analysis of individual data of ADVANCE, EUROPA, and PROGRESS trials. Eur Heart J. 2009;30(11):1385–1394.
- , , , , , . Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153.
- , , , , , . Effects of the early administration of enalapril on mortality in patients with acute myocardial infarction. Results of the Cooperative New Scandinavian Enalapril Survival Study II (CONSENSUS II). N Engl J Med. 1992;327(10):678–684.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54(19):1778–1784.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636–644.
- , , . Guidelines for pre‐operative cardiac risk assessment and perioperative cardiac management in non‐cardiac surgery: The Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non‐cardiac Surgery of the European Society of Cardiology (ESC) and endorsed by the European Society of Anaesthesiology (ESA). Eur Heart J. 2009;30(22):2769–2812.
- , , , . Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218–2227.
- VA Information Resource Center; VIReC Research User Guide: VHA Decision support system clinical national data extracts. 2nd ed. Hines, IL: U.S. Department of VA, Health Services Research and Development Service, VA Information Resource Center, 2009. Available at: http://www.virec.research.va.gov/RUGs/RUGs-Index.htm. Accessed February 27, 2013.
- , , , , . A combined comorbidity score predicted mortality in elderly patients better than existing scores. J Clin Epidemiol. 2011;64(7):749–759.
- . A tutorial and case study in propensity score analysis: an application to estimating the effect of in‐hospital smoking cessation counseling on mortality. Multi Behav Res. 2011;46(1):119–151.
- , . Stopping and restarting medications in the perioperative period. J Gen Intern Med. 1987;2(4):270–283.
- , , . Perioperative management of drug therapy, clinical considerations. Drugs. 1996;51(2):238–259.
- , , , et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation. 2007;116(17):1971–1996.
- , , , et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med. 2008;23(9):1414–1422.
- , , , et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA. 2011;306(8):840–847.
- , , , , , . Potentially unintended discontinuation of long‐term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525–2531.
- , , , . Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314–323.
- , . Discontinuation and reinstitution of medications during the perioperative period. Am J Health Syst Pharm. 2004;61(9):899–912.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319–325.
- , , , et al. Patterns of use of perioperative angiotensin‐converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in‐hospital morbidity and mortality. Circulation. 2012;126(3):261–269.
- , , , et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med. 2010;5(8):477–485.
- , , . Guidelines for the management of chronic medication in the perioperative period: systematic review and formal consensus. J Clin Pharm Therap. 2011;36(4):446–467.
- , , , et al. Increase of 1‐year mortality after perioperative beta‐blocker withdrawal in endovascular and vascular surgery patients. Eur J Vasc Endovasc Surg. 2007;33(1):13–19.
- , , , et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg. 2007;104(6):1326–1333.
- , , , et al. Effect of statin withdrawal on frequency of cardiac events after vascular surgery. Am J Cardiol. 2007;100(2):316–320.
- , , , et al. Short‐term effects of withdrawing angiotensin converting enzyme inhibitor therapy on home self‐measured blood pressure in hypertensive patients. Am J Hypertens. 1998;11(2):165–173.
- , , , et al. Hypertensive rebound after angiotensin converting enzyme inhibitor withdrawal in diabetic patients with chronic renal failure. Nephrol Dial Trans. 2001;16(5):1084–1085.
- , , . Vascular protective effects of angiotensin converting enzyme inhibitors and their relation to clinical events. J Cardiovasc Pharmacol. 2001;37(suppl 1):S21–S30.
- , , , et al. Angiotensin‐converting enzyme inhibitor withdrawal and ACE gene polymorphism. Clin Nephrol. 2003;60(4):225–232.
- , . Pharmacy data in the VA health care system. Med Care Res Rev. 2003;60(3 suppl):92S–123S.
- , , , , . Accuracy of computerized outpatient diagnoses in a Veterans Affairs general medicine clinic. Am J Manag Care. 2002;8(1):37–43.
- , , , . Mortality ascertainment in the veteran population: alternatives to the National Death Index. Am J Epidemiol. 1995;141(3):242–250.
- . Statistical considerations in the intent‐to‐treat principle. Control Clin Trials. 2000;21(3):167–189.
- , , . ACE inhibitors in cardiac surgery: current studies and controversies. Hypertens Res. 2010;34(1):15–22.
© 2014 Society of Hospital Medicine
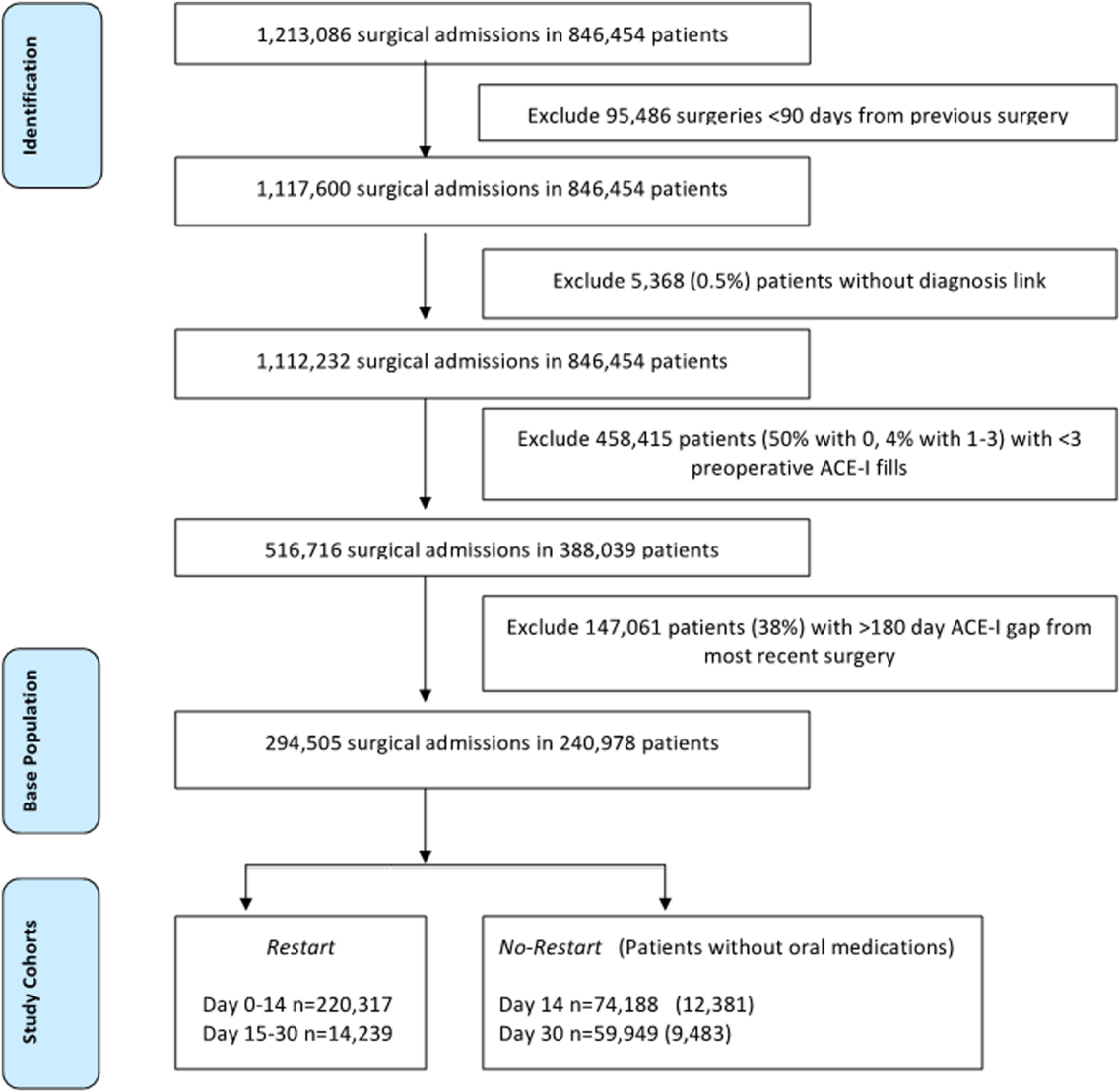
Hospira announces device correction for infusion pump docking station

Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing [email protected].
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program. ![]()
Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing [email protected].
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program. ![]()
Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing [email protected].
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program. ![]()
Baxter issues Class I recall of infusion pumps

of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program. ![]()
of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program. ![]()
of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program. ![]()
Pyoderma Gangrenosum Following Gastric Bypass Surgery
Chlorpromazine-Induced Skin Pigmentation With Corneal and Lens Opacities
Congenital Candidiasis: An Uncommon Skin Eruption Presenting at Birth
Making sure patients never walk alone
We are all painfully aware that falls are not uncommon in hospitalized patients, but I was shocked and appalled to learn that there are approximately 11,000 falls that are ultimately fatal in U.S. hospitals each year, according to the Joint Commission Center for Transforming Healthcare.
Fortunately, there appear to be some very viable solutions at hand.
Hospitals that use the center’s new measuring systems and solution have been able to slash the number of patients who fall by 35%, as well cutting the percentage of patients injured when they fall by 62%. Extrapolating these results to an average 200-bed hospital, an estimated $1 million could be saved each year through fall prevention efforts.

Hospitals participating in this study ranged from small community-based hospitals with fewer than 200 beds to large medical centers with more than 1,700 beds. All used a data-driven, Lean Six Sigma–inspired "Robust Process Improvement" methodology to determine the causes of falls and create solutions to prevent them. One solution was simply providing hourly rounding which included proactive toileting. When you think about it, this makes perfect sense. Patients with an immediate need may not be able to hold it until it is "their turn" for the nurse to assist them. In their haste to avoid soiling their clothes, a mechanical fall is very understandable, especially if they are impaired due to weakness or medication.
Other solutions included teaching patients how to actively participate in their own safety, engaging patients and family members in their fall safety program, using a validated fall risk assessment tool, and increasing awareness and participation among staff so that patients did not walk alone.
Most of us have received that dreaded call from the nurse about a patient who was injured from a fall. I once had a patient who slipped and fell, breaking a hip while in the hospital for a relatively minor issue. If she had been more stable on her feet, she probably could have caught herself prior to hitting the hard floor. Had a close relative not witnessed the incident, it would have been very difficult (and embarrassing) to explain to the family why their loved one experienced such a traumatic event while in a seemingly protected environment. Her son was very understanding, though the event was very disconcerting all the same.
Based on their staffing, resources, and creativity, different hospitals may develop different innovative solutions to prevent falls. There is room for a wide variety of options. Yes, early ambulation is crucial to help prevent unnecessary complications, such as pneumonia and blood clots, but we should be mindful of the individual patient’s circumstances. A simple order requesting the nurse to ambulate a patient in the room or down the hall two or three times a day may be adequate for some, while in other cases a formal physical therapy consultation may clearly be in order. If there are any concerns over the patient’s ability to ambulate safely and I am not sure if a physical therapy consult is really needed, I sometimes call the nurse into the room and the two of us walk with the patient. That way, I can get an immediate sense of the likelihood of falls, the need for dedicated strengthening exercises, and, on occasion, the impact that medications are having on gait. And for those difficult, unmotivated patients, family members can frequently provide invaluable encouragement, as well as the emotional, and sometimes physical safety net many patients desire.
With safety interventions and highly engaged care teams in place, even steady patients never walk truly alone.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected]
We are all painfully aware that falls are not uncommon in hospitalized patients, but I was shocked and appalled to learn that there are approximately 11,000 falls that are ultimately fatal in U.S. hospitals each year, according to the Joint Commission Center for Transforming Healthcare.
Fortunately, there appear to be some very viable solutions at hand.
Hospitals that use the center’s new measuring systems and solution have been able to slash the number of patients who fall by 35%, as well cutting the percentage of patients injured when they fall by 62%. Extrapolating these results to an average 200-bed hospital, an estimated $1 million could be saved each year through fall prevention efforts.
Hospitals participating in this study ranged from small community-based hospitals with fewer than 200 beds to large medical centers with more than 1,700 beds. All used a data-driven, Lean Six Sigma–inspired "Robust Process Improvement" methodology to determine the causes of falls and create solutions to prevent them. One solution was simply providing hourly rounding which included proactive toileting. When you think about it, this makes perfect sense. Patients with an immediate need may not be able to hold it until it is "their turn" for the nurse to assist them. In their haste to avoid soiling their clothes, a mechanical fall is very understandable, especially if they are impaired due to weakness or medication.
Other solutions included teaching patients how to actively participate in their own safety, engaging patients and family members in their fall safety program, using a validated fall risk assessment tool, and increasing awareness and participation among staff so that patients did not walk alone.
Most of us have received that dreaded call from the nurse about a patient who was injured from a fall. I once had a patient who slipped and fell, breaking a hip while in the hospital for a relatively minor issue. If she had been more stable on her feet, she probably could have caught herself prior to hitting the hard floor. Had a close relative not witnessed the incident, it would have been very difficult (and embarrassing) to explain to the family why their loved one experienced such a traumatic event while in a seemingly protected environment. Her son was very understanding, though the event was very disconcerting all the same.
Based on their staffing, resources, and creativity, different hospitals may develop different innovative solutions to prevent falls. There is room for a wide variety of options. Yes, early ambulation is crucial to help prevent unnecessary complications, such as pneumonia and blood clots, but we should be mindful of the individual patient’s circumstances. A simple order requesting the nurse to ambulate a patient in the room or down the hall two or three times a day may be adequate for some, while in other cases a formal physical therapy consultation may clearly be in order. If there are any concerns over the patient’s ability to ambulate safely and I am not sure if a physical therapy consult is really needed, I sometimes call the nurse into the room and the two of us walk with the patient. That way, I can get an immediate sense of the likelihood of falls, the need for dedicated strengthening exercises, and, on occasion, the impact that medications are having on gait. And for those difficult, unmotivated patients, family members can frequently provide invaluable encouragement, as well as the emotional, and sometimes physical safety net many patients desire.
With safety interventions and highly engaged care teams in place, even steady patients never walk truly alone.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected]
We are all painfully aware that falls are not uncommon in hospitalized patients, but I was shocked and appalled to learn that there are approximately 11,000 falls that are ultimately fatal in U.S. hospitals each year, according to the Joint Commission Center for Transforming Healthcare.
Fortunately, there appear to be some very viable solutions at hand.
Hospitals that use the center’s new measuring systems and solution have been able to slash the number of patients who fall by 35%, as well cutting the percentage of patients injured when they fall by 62%. Extrapolating these results to an average 200-bed hospital, an estimated $1 million could be saved each year through fall prevention efforts.
Hospitals participating in this study ranged from small community-based hospitals with fewer than 200 beds to large medical centers with more than 1,700 beds. All used a data-driven, Lean Six Sigma–inspired "Robust Process Improvement" methodology to determine the causes of falls and create solutions to prevent them. One solution was simply providing hourly rounding which included proactive toileting. When you think about it, this makes perfect sense. Patients with an immediate need may not be able to hold it until it is "their turn" for the nurse to assist them. In their haste to avoid soiling their clothes, a mechanical fall is very understandable, especially if they are impaired due to weakness or medication.
Other solutions included teaching patients how to actively participate in their own safety, engaging patients and family members in their fall safety program, using a validated fall risk assessment tool, and increasing awareness and participation among staff so that patients did not walk alone.
Most of us have received that dreaded call from the nurse about a patient who was injured from a fall. I once had a patient who slipped and fell, breaking a hip while in the hospital for a relatively minor issue. If she had been more stable on her feet, she probably could have caught herself prior to hitting the hard floor. Had a close relative not witnessed the incident, it would have been very difficult (and embarrassing) to explain to the family why their loved one experienced such a traumatic event while in a seemingly protected environment. Her son was very understanding, though the event was very disconcerting all the same.
Based on their staffing, resources, and creativity, different hospitals may develop different innovative solutions to prevent falls. There is room for a wide variety of options. Yes, early ambulation is crucial to help prevent unnecessary complications, such as pneumonia and blood clots, but we should be mindful of the individual patient’s circumstances. A simple order requesting the nurse to ambulate a patient in the room or down the hall two or three times a day may be adequate for some, while in other cases a formal physical therapy consultation may clearly be in order. If there are any concerns over the patient’s ability to ambulate safely and I am not sure if a physical therapy consult is really needed, I sometimes call the nurse into the room and the two of us walk with the patient. That way, I can get an immediate sense of the likelihood of falls, the need for dedicated strengthening exercises, and, on occasion, the impact that medications are having on gait. And for those difficult, unmotivated patients, family members can frequently provide invaluable encouragement, as well as the emotional, and sometimes physical safety net many patients desire.
With safety interventions and highly engaged care teams in place, even steady patients never walk truly alone.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected]

Solitary Adult Myofibroma
Using chromatin conformation data to classify leukemia

Chromatin conformation can guide the classification of leukemia, according to research published in Genome Biology.
Investigators mapped the conformation of the homeobox A (HOXA) gene cluster—11 genes encoding proteins that are highly relevant to many cancers—in a panel of leukemia cell lines.
And the team found they could use this information to distinguish subtypes of leukemia from one another.
“Previous studies have shown that looking at gene expression—the specific proteins produced by the genes—is a good predictor of whether patients have leukemia,” said study author Mathieu Blanchette, PhD, of McGill University in Montréal, Québec, Canada.
“We found that different types of leukemia cells also have a distinctive chromatin interaction—how the chromatin that makes up the genome is folded.”
The investigators used 5C chromosome conformation capture technology to analyze the HOXA gene cluster and then used the data to train and test a support vector machine classifier called 3D-SP.
They found 3D-SP could distinguish leukemias expressing MLL-fusion proteins from those expressing wild-type MLL. It could also classify leukemia subtypes according to MLL fusion partner.
The team noted that it is not clear whether the genome shape plays a role in causing leukemia or whether the leukemia causes the genome to change shape. And additional studies are needed to determine whether genome shape is as useful for classifying other types of cancer.
“Our study validates a new research avenue: the application of 3D genomics for developing medical diagnostics or treatments that could be explored for diseases where current technologies, including gene expression data, have failed to improve patient care,” said Josée Dostie, PhD, also of McGill University.
“While the use of 3D genomics in the clinic is still remote when considering the technical challenges required for translating the information to the bedside, we discovered a new approach for classifying human disease that must be explored further, if only for what it can reveal about how the human genome works.” ![]()
Chromatin conformation can guide the classification of leukemia, according to research published in Genome Biology.
Investigators mapped the conformation of the homeobox A (HOXA) gene cluster—11 genes encoding proteins that are highly relevant to many cancers—in a panel of leukemia cell lines.
And the team found they could use this information to distinguish subtypes of leukemia from one another.
“Previous studies have shown that looking at gene expression—the specific proteins produced by the genes—is a good predictor of whether patients have leukemia,” said study author Mathieu Blanchette, PhD, of McGill University in Montréal, Québec, Canada.
“We found that different types of leukemia cells also have a distinctive chromatin interaction—how the chromatin that makes up the genome is folded.”
The investigators used 5C chromosome conformation capture technology to analyze the HOXA gene cluster and then used the data to train and test a support vector machine classifier called 3D-SP.
They found 3D-SP could distinguish leukemias expressing MLL-fusion proteins from those expressing wild-type MLL. It could also classify leukemia subtypes according to MLL fusion partner.
The team noted that it is not clear whether the genome shape plays a role in causing leukemia or whether the leukemia causes the genome to change shape. And additional studies are needed to determine whether genome shape is as useful for classifying other types of cancer.
“Our study validates a new research avenue: the application of 3D genomics for developing medical diagnostics or treatments that could be explored for diseases where current technologies, including gene expression data, have failed to improve patient care,” said Josée Dostie, PhD, also of McGill University.
“While the use of 3D genomics in the clinic is still remote when considering the technical challenges required for translating the information to the bedside, we discovered a new approach for classifying human disease that must be explored further, if only for what it can reveal about how the human genome works.” ![]()
Chromatin conformation can guide the classification of leukemia, according to research published in Genome Biology.
Investigators mapped the conformation of the homeobox A (HOXA) gene cluster—11 genes encoding proteins that are highly relevant to many cancers—in a panel of leukemia cell lines.
And the team found they could use this information to distinguish subtypes of leukemia from one another.
“Previous studies have shown that looking at gene expression—the specific proteins produced by the genes—is a good predictor of whether patients have leukemia,” said study author Mathieu Blanchette, PhD, of McGill University in Montréal, Québec, Canada.
“We found that different types of leukemia cells also have a distinctive chromatin interaction—how the chromatin that makes up the genome is folded.”
The investigators used 5C chromosome conformation capture technology to analyze the HOXA gene cluster and then used the data to train and test a support vector machine classifier called 3D-SP.
They found 3D-SP could distinguish leukemias expressing MLL-fusion proteins from those expressing wild-type MLL. It could also classify leukemia subtypes according to MLL fusion partner.
The team noted that it is not clear whether the genome shape plays a role in causing leukemia or whether the leukemia causes the genome to change shape. And additional studies are needed to determine whether genome shape is as useful for classifying other types of cancer.
“Our study validates a new research avenue: the application of 3D genomics for developing medical diagnostics or treatments that could be explored for diseases where current technologies, including gene expression data, have failed to improve patient care,” said Josée Dostie, PhD, also of McGill University.
“While the use of 3D genomics in the clinic is still remote when considering the technical challenges required for translating the information to the bedside, we discovered a new approach for classifying human disease that must be explored further, if only for what it can reveal about how the human genome works.” ![]()