User login
Immune Response Modifier Therapy in Anogenital Warts and Actinic Keratoses
A supplement to Family Practice News and Internal Medicine News.
This Journal Scan is supported by 3M Pharmaceuticals.
•Introduction
•Topic Highlights

To view the supplement, click the image above.
Introduction
Introduction
Thomas J. Zuber, MD, MPH, MBA
Adjunct Associate Professor
Brody School of Medicine, East Carolina University
Private Practice, Mountain Family Medicine
Boone, North Carolina
Nothing to disclose.
Topic Highlights
• Condyloma Recurrence Rates Improve With Immune Response Modifier Treatment
• 3-Year Experience With Imiquimod in Vulvar Warts
• Placebo-Controlled Trial in External Anogenital Warts
• Phase III Trials of Imiquimod in Actinic Keratosis
• Observational Long-Term Study of Imiquimod in Actinic Keratosis
• Immune Response Modifier Mechanism of Action
A supplement to Family Practice News and Internal Medicine News.
This Journal Scan is supported by 3M Pharmaceuticals.
•Introduction
•Topic Highlights
To view the supplement, click the image above.
Introduction
Introduction
Thomas J. Zuber, MD, MPH, MBA
Adjunct Associate Professor
Brody School of Medicine, East Carolina University
Private Practice, Mountain Family Medicine
Boone, North Carolina
Nothing to disclose.
Topic Highlights
• Condyloma Recurrence Rates Improve With Immune Response Modifier Treatment
• 3-Year Experience With Imiquimod in Vulvar Warts
• Placebo-Controlled Trial in External Anogenital Warts
• Phase III Trials of Imiquimod in Actinic Keratosis
• Observational Long-Term Study of Imiquimod in Actinic Keratosis
• Immune Response Modifier Mechanism of Action
A supplement to Family Practice News and Internal Medicine News.
This Journal Scan is supported by 3M Pharmaceuticals.
•Introduction
•Topic Highlights
To view the supplement, click the image above.
Introduction
Introduction
Thomas J. Zuber, MD, MPH, MBA
Adjunct Associate Professor
Brody School of Medicine, East Carolina University
Private Practice, Mountain Family Medicine
Boone, North Carolina
Nothing to disclose.
Topic Highlights
• Condyloma Recurrence Rates Improve With Immune Response Modifier Treatment
• 3-Year Experience With Imiquimod in Vulvar Warts
• Placebo-Controlled Trial in External Anogenital Warts
• Phase III Trials of Imiquimod in Actinic Keratosis
• Observational Long-Term Study of Imiquimod in Actinic Keratosis
• Immune Response Modifier Mechanism of Action

Effective Communication Ensures Patient Safety

Effective Communication Ensures Patient Safety
Can you explain to me what is meant by SBAR? I heard this acronym mentioned during a session at HM09, but I did not understand the term.
S. East, MD
Pullman, Wash.
Dr. Hospitalist responds: SBAR (pronounced “ess-bar”) is a standardized method of communication that originated in the Navy’s nuclear submarine program. It stands for:
- Situation: What is happening presently?
- Background: What circumstances led to this situation?
- Assessment: What do I think is the problem?
- Recommendation: What should we do to correct the problem?
The SBAR system was developed to prevent simple communication errors that could lead to global disaster.
Kaiser Permanente of Colorado was among the first to adopt this model of communication among its staff and has since popularized its use in healthcare. Numerous hospitals and healthcare organizations have implemented SBAR as an approach to minimize communication errors between healthcare providers. The idea is that eliminating communication errors between healthcare providers improves patient safety. SBAR encourages all providers (doctors, nurses, pharmacists, etc.) to communicate with a shared mental model for information transfer.
SBAR requires providers to organize their thoughts, understand what it is they want to convey, and make requests in an organized fashion. Adherence to SBAR allows providers to transmit factual information in a concise manner.
Highly effective communication is essential to any hospitalist program. The SBAR approach should not be limited to nurse-doctor communication. I encourage you to implement this tool at your institution.
For more information, an SBAR toolkit is available at www.azhha.org/patient_safety/sbar.aspx. TH
Effective Communication Ensures Patient Safety
Can you explain to me what is meant by SBAR? I heard this acronym mentioned during a session at HM09, but I did not understand the term.
S. East, MD
Pullman, Wash.
Dr. Hospitalist responds: SBAR (pronounced “ess-bar”) is a standardized method of communication that originated in the Navy’s nuclear submarine program. It stands for:
- Situation: What is happening presently?
- Background: What circumstances led to this situation?
- Assessment: What do I think is the problem?
- Recommendation: What should we do to correct the problem?
The SBAR system was developed to prevent simple communication errors that could lead to global disaster.
Kaiser Permanente of Colorado was among the first to adopt this model of communication among its staff and has since popularized its use in healthcare. Numerous hospitals and healthcare organizations have implemented SBAR as an approach to minimize communication errors between healthcare providers. The idea is that eliminating communication errors between healthcare providers improves patient safety. SBAR encourages all providers (doctors, nurses, pharmacists, etc.) to communicate with a shared mental model for information transfer.
SBAR requires providers to organize their thoughts, understand what it is they want to convey, and make requests in an organized fashion. Adherence to SBAR allows providers to transmit factual information in a concise manner.
Highly effective communication is essential to any hospitalist program. The SBAR approach should not be limited to nurse-doctor communication. I encourage you to implement this tool at your institution.
For more information, an SBAR toolkit is available at www.azhha.org/patient_safety/sbar.aspx. TH
Effective Communication Ensures Patient Safety
Can you explain to me what is meant by SBAR? I heard this acronym mentioned during a session at HM09, but I did not understand the term.
S. East, MD
Pullman, Wash.
Dr. Hospitalist responds: SBAR (pronounced “ess-bar”) is a standardized method of communication that originated in the Navy’s nuclear submarine program. It stands for:
- Situation: What is happening presently?
- Background: What circumstances led to this situation?
- Assessment: What do I think is the problem?
- Recommendation: What should we do to correct the problem?
The SBAR system was developed to prevent simple communication errors that could lead to global disaster.
Kaiser Permanente of Colorado was among the first to adopt this model of communication among its staff and has since popularized its use in healthcare. Numerous hospitals and healthcare organizations have implemented SBAR as an approach to minimize communication errors between healthcare providers. The idea is that eliminating communication errors between healthcare providers improves patient safety. SBAR encourages all providers (doctors, nurses, pharmacists, etc.) to communicate with a shared mental model for information transfer.
SBAR requires providers to organize their thoughts, understand what it is they want to convey, and make requests in an organized fashion. Adherence to SBAR allows providers to transmit factual information in a concise manner.
Highly effective communication is essential to any hospitalist program. The SBAR approach should not be limited to nurse-doctor communication. I encourage you to implement this tool at your institution.
For more information, an SBAR toolkit is available at www.azhha.org/patient_safety/sbar.aspx. TH
Necessary Evil: Change

The amount and complexity of medical knowledge we need to keep up with is changing and growing at a remarkable rate. I was trained in an era in which it was taken as a given that congestive heart failure patients should not receive beta-blockers; now it is a big mistake if we don’t prescribe them in most cases. But even before starting medical school, most of us realize that things will change a lot, and many of us see that as a good thing. It keeps our work interesting. Just recently, our hospital had a guest speaker who talked about potential medical applications of nanotechnology. It was way over my head, but it sounded pretty cool.
While I was prepared for ongoing changes in medical knowledge, I failed to anticipate how quickly the business of medicine would change during my career. I think the need to keep up with ever-increasing financial and regulatory issues siphons a lot of time and energy that could be used to keep up with the medical knowledge base. I wasn’t prepared for this when I started my career.
Because it is the start of a new year, I thought I would highlight one issue related to CPT coding: Medicare stopped recognizing consult codes as of Jan. 1 (see “Consultation Elimination,” p. 31).
What It Means for Hospitalists
The good news is that we can just use initial hospital visit codes, inpatient or observation, for all new visits. For example, it won’t matter anymore whether I’m admitting and serving as attending for a patient, or whether a surgeon admitted the patient and asked me to consult for preoperative medical evaluation (“clearance”). I should use the same CPT code in either situation, simply appending a modifier if I’m the admitting physician. And for billing purposes, we won’t have to worry about documenting which doctor requested that we see the patient, though it is a good idea to document it as part of the clinical record anyway.
But it gets a little more complicated. The codes aren’t going away or being removed from the CPT “bible” published by the American Medical Association (AMA). Instead, Medicare simply won’t recognize them anymore. Other payors probably will follow suit within a few months, but that isn’t certain. So it is possible that when asked by a surgeon to provide a preoperative evaluation, you will need to bill an initial hospital (or office or nursing facility) care visit if the patient is on Medicare but bill a consult code if the patient has other insurance. You should check with your billers to ensure you’re doing this correctly.
Medicare-paid consults are at a slightly higher rate than the equivalent service billed as initial hospital care (e.g., when the hospitalist is attending). So a higher reimbursing code has been replaced with one that pays a little less. For example, a 99253 consultation code requires a detailed history, detailed examination, and medical decision-making of low complexity; last year, 99253 was reimbursed by Medicare at an average rate of $114.69. The equivalent admission code for a detailed history, detailed examination, and low-complexity medical decision-making is a 99221 code, for which Medicare pays about $99.90. This represents a difference of about 14%.
However, the net financial impact of this change probably will be positive for most HM groups because you probably bill very few initial consult codes, and instead were stuck billing a follow-up visit code when seeing co-management “consults” (i.e., a patient admitted by a surgeon who asks you to follow and manage diabetes and other medical issues). Now, at least in the case of Medicare, it is appropriate for us to bill an initial hospital visit code, which provides significantly higher reimbursement than follow-up codes.
In addition, there is a modest (about 0.3%) proposed increase in work relative value units attached to the initial hospital visit codes, which will benefit us not only when we’re consulting, but also when we admit and serve as a patient’s attending.
Some specialists may be less interested in consulting on our patients because the initial visit codes will reimburse a little less than similar consultation codes. I don’t anticipate this will be a significant problem for most of us, particularly since many specialists bill the highest level of consultation code (99255), which pays about the same as the equivalent admission code (99223).
Although I think elimination of the use of consultation codes seems like a reasonable step toward simplifying how hospitalists bill for our services, keeping up with these frequent coding changes requires a high level of diligence on our part, and on the part of our administrative and clerical staffs. And it consumes time and resources that I—and my team—could better spend keeping up with changes in clinical practice.
Perhaps when all the dust settles around the healthcare reform debate, we will begin to move toward new, more creative payment models that will allow us to focus on what we do best. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
The amount and complexity of medical knowledge we need to keep up with is changing and growing at a remarkable rate. I was trained in an era in which it was taken as a given that congestive heart failure patients should not receive beta-blockers; now it is a big mistake if we don’t prescribe them in most cases. But even before starting medical school, most of us realize that things will change a lot, and many of us see that as a good thing. It keeps our work interesting. Just recently, our hospital had a guest speaker who talked about potential medical applications of nanotechnology. It was way over my head, but it sounded pretty cool.
While I was prepared for ongoing changes in medical knowledge, I failed to anticipate how quickly the business of medicine would change during my career. I think the need to keep up with ever-increasing financial and regulatory issues siphons a lot of time and energy that could be used to keep up with the medical knowledge base. I wasn’t prepared for this when I started my career.
Because it is the start of a new year, I thought I would highlight one issue related to CPT coding: Medicare stopped recognizing consult codes as of Jan. 1 (see “Consultation Elimination,” p. 31).
What It Means for Hospitalists
The good news is that we can just use initial hospital visit codes, inpatient or observation, for all new visits. For example, it won’t matter anymore whether I’m admitting and serving as attending for a patient, or whether a surgeon admitted the patient and asked me to consult for preoperative medical evaluation (“clearance”). I should use the same CPT code in either situation, simply appending a modifier if I’m the admitting physician. And for billing purposes, we won’t have to worry about documenting which doctor requested that we see the patient, though it is a good idea to document it as part of the clinical record anyway.
But it gets a little more complicated. The codes aren’t going away or being removed from the CPT “bible” published by the American Medical Association (AMA). Instead, Medicare simply won’t recognize them anymore. Other payors probably will follow suit within a few months, but that isn’t certain. So it is possible that when asked by a surgeon to provide a preoperative evaluation, you will need to bill an initial hospital (or office or nursing facility) care visit if the patient is on Medicare but bill a consult code if the patient has other insurance. You should check with your billers to ensure you’re doing this correctly.
Medicare-paid consults are at a slightly higher rate than the equivalent service billed as initial hospital care (e.g., when the hospitalist is attending). So a higher reimbursing code has been replaced with one that pays a little less. For example, a 99253 consultation code requires a detailed history, detailed examination, and medical decision-making of low complexity; last year, 99253 was reimbursed by Medicare at an average rate of $114.69. The equivalent admission code for a detailed history, detailed examination, and low-complexity medical decision-making is a 99221 code, for which Medicare pays about $99.90. This represents a difference of about 14%.
However, the net financial impact of this change probably will be positive for most HM groups because you probably bill very few initial consult codes, and instead were stuck billing a follow-up visit code when seeing co-management “consults” (i.e., a patient admitted by a surgeon who asks you to follow and manage diabetes and other medical issues). Now, at least in the case of Medicare, it is appropriate for us to bill an initial hospital visit code, which provides significantly higher reimbursement than follow-up codes.
In addition, there is a modest (about 0.3%) proposed increase in work relative value units attached to the initial hospital visit codes, which will benefit us not only when we’re consulting, but also when we admit and serve as a patient’s attending.
Some specialists may be less interested in consulting on our patients because the initial visit codes will reimburse a little less than similar consultation codes. I don’t anticipate this will be a significant problem for most of us, particularly since many specialists bill the highest level of consultation code (99255), which pays about the same as the equivalent admission code (99223).
Although I think elimination of the use of consultation codes seems like a reasonable step toward simplifying how hospitalists bill for our services, keeping up with these frequent coding changes requires a high level of diligence on our part, and on the part of our administrative and clerical staffs. And it consumes time and resources that I—and my team—could better spend keeping up with changes in clinical practice.
Perhaps when all the dust settles around the healthcare reform debate, we will begin to move toward new, more creative payment models that will allow us to focus on what we do best. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
The amount and complexity of medical knowledge we need to keep up with is changing and growing at a remarkable rate. I was trained in an era in which it was taken as a given that congestive heart failure patients should not receive beta-blockers; now it is a big mistake if we don’t prescribe them in most cases. But even before starting medical school, most of us realize that things will change a lot, and many of us see that as a good thing. It keeps our work interesting. Just recently, our hospital had a guest speaker who talked about potential medical applications of nanotechnology. It was way over my head, but it sounded pretty cool.
While I was prepared for ongoing changes in medical knowledge, I failed to anticipate how quickly the business of medicine would change during my career. I think the need to keep up with ever-increasing financial and regulatory issues siphons a lot of time and energy that could be used to keep up with the medical knowledge base. I wasn’t prepared for this when I started my career.
Because it is the start of a new year, I thought I would highlight one issue related to CPT coding: Medicare stopped recognizing consult codes as of Jan. 1 (see “Consultation Elimination,” p. 31).
What It Means for Hospitalists
The good news is that we can just use initial hospital visit codes, inpatient or observation, for all new visits. For example, it won’t matter anymore whether I’m admitting and serving as attending for a patient, or whether a surgeon admitted the patient and asked me to consult for preoperative medical evaluation (“clearance”). I should use the same CPT code in either situation, simply appending a modifier if I’m the admitting physician. And for billing purposes, we won’t have to worry about documenting which doctor requested that we see the patient, though it is a good idea to document it as part of the clinical record anyway.
But it gets a little more complicated. The codes aren’t going away or being removed from the CPT “bible” published by the American Medical Association (AMA). Instead, Medicare simply won’t recognize them anymore. Other payors probably will follow suit within a few months, but that isn’t certain. So it is possible that when asked by a surgeon to provide a preoperative evaluation, you will need to bill an initial hospital (or office or nursing facility) care visit if the patient is on Medicare but bill a consult code if the patient has other insurance. You should check with your billers to ensure you’re doing this correctly.
Medicare-paid consults are at a slightly higher rate than the equivalent service billed as initial hospital care (e.g., when the hospitalist is attending). So a higher reimbursing code has been replaced with one that pays a little less. For example, a 99253 consultation code requires a detailed history, detailed examination, and medical decision-making of low complexity; last year, 99253 was reimbursed by Medicare at an average rate of $114.69. The equivalent admission code for a detailed history, detailed examination, and low-complexity medical decision-making is a 99221 code, for which Medicare pays about $99.90. This represents a difference of about 14%.
However, the net financial impact of this change probably will be positive for most HM groups because you probably bill very few initial consult codes, and instead were stuck billing a follow-up visit code when seeing co-management “consults” (i.e., a patient admitted by a surgeon who asks you to follow and manage diabetes and other medical issues). Now, at least in the case of Medicare, it is appropriate for us to bill an initial hospital visit code, which provides significantly higher reimbursement than follow-up codes.
In addition, there is a modest (about 0.3%) proposed increase in work relative value units attached to the initial hospital visit codes, which will benefit us not only when we’re consulting, but also when we admit and serve as a patient’s attending.
Some specialists may be less interested in consulting on our patients because the initial visit codes will reimburse a little less than similar consultation codes. I don’t anticipate this will be a significant problem for most of us, particularly since many specialists bill the highest level of consultation code (99255), which pays about the same as the equivalent admission code (99223).
Although I think elimination of the use of consultation codes seems like a reasonable step toward simplifying how hospitalists bill for our services, keeping up with these frequent coding changes requires a high level of diligence on our part, and on the part of our administrative and clerical staffs. And it consumes time and resources that I—and my team—could better spend keeping up with changes in clinical practice.
Perhaps when all the dust settles around the healthcare reform debate, we will begin to move toward new, more creative payment models that will allow us to focus on what we do best. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Standing Ovation

Is this really happening? That’s what I was thinking as my mind quickly ran a differential of the possible explanations for the 80 people before me, positioned erectly, hands audibly moving together and apart. This had never happened to me before. Was this some sort of group yoga stretch aimed at quelling DVT formation, perhaps a pre-determined signal alerting security to have me removed for hitting an unconscionable level of boredom, or, most likely, a synchronized form of mass exit (my talks are accustomed to a certain level of attrition)? Beyond these possibilities lies just one alternative: I actually was receiving a standing ovation.
First, let me dispense with one ever-important technicality. The ovation, coming at the close of the recent four-day Academic Hospitalist Academy held outside Atlanta, was more rightly intended for the efforts of the entire eight-member faculty than me alone. I just happened to be giving the closing session.
The fact of the matter is that as a faculty member for this program, which aimed to provide early-career development for junior academic hospitalists, I, too, was in awe at the tremendous, unparalleled work of the faculty: Drs. Brad Sharpe, Vikas Parekh, Andy Auerbach, Jeff Wiese, Shobi Chheda, Bob Centor, and Jen Myers.
Yet there I was, just moments after uttering a few closing comments, being showered with praise, each clap further pumping my chest fuller with pride. It was then, with a deflating wheeze, that I realized what was happening—they weren’t clapping for me, or even the rest of the faculty. It turns out the more obvious cause for their enthusiasm had been staring me in the face the whole time. Or, more to the point, the faces staring back at me were reflecting my numerous missteps that this course had ensured they’d never make. And that was very ovation-worthy.
Take-Home Points
Clapping excitedly at table No. 4 was a young first-year hospitalist from a major academic medical center. Looking at her, I could tell she would not, like I had, make the mistake of waiting too long to find a mentor. It wasn’t until my fourth year in academics that I found a mentor. That was four years of unproductive wandering, chasing dead ends, grabbing at wrong straws. So after multiple sessions covering the importance and means of finding mentors, as well as the role of mentees, it was clear that this young hospitalist would build her career foundation on more firm footing.
Applauding from table No. 7 was a second-year hospitalist from a community teaching hospital with aspirations of making a splash on the national hospitalist scene. Unlike my early fruitless attempts to get involved outside of my institution, the session on the importance of peer and national networking provided his quiver with several time-honored arrows that took me years to acquire.
The eyes of a hospitalist at table No. 8 foretold the story of a young faculty member who wouldn’t struggle with the process of promotion, as I had. After sitting through talks that lifted the veil on both the inner workings of an academic medical center and the mysteries by which said centers promote their members, she had a head start on ensuring her academic success.
Table No. 3 offered several hospitalists who wouldn’t make the errors I’ve made—repeatedly—with e-mail, phone conversations and running a meeting. A presentation on the basics of communications ensured that they wouldn’t send those irretrievable e-missives carrying unintentional messages or waste hours trying to cover in e-mail what would be better solved over the phone or in a face-to-face meeting.
A person at table one patted me on the shoulder, the glint in his eye signaling that the sessions on how to run a teaching team and be a more effective teacher at both the bedside and at the chalkboard would help him avoid the lower teaching scores that plagued my early academic years.
Focus on Fundamentals
I could go on, but the point is that while my fellow faculty and I reveled in the pleasure of a standing ovation, the truth is that the clapping had less to do with us or how we imparted information and more to do with the fact that we had shared with them the ingredients of their future success, something they actively longed for—a means to enhance their career success and satisfaction.
Mind you, none of these revelations were shocking; indeed, most are mundane and straightforward. However, the reality is that more often than not we just need help getting started—a little enzymatic push in the right direction. The fact that these needs were finally being met was evident in every heartfelt clap of the hands.
Lest you think these lessons are only important for us academic eggheads, I’d submit to you that the same, or at least similarly important, points are just as critical for young community hospitalists.
In fact, hospitalists and the field of HM are all very young; most of us are in desperate need of career guidance. And I’d go so far as to say the success of our field depends on meeting these needs as much or more than our ability to improve the quality of healthcare. The reality is that without sated, successful, career-oriented hospitalists, there can be no HM movement to improve the quality of care.
It’s tempting to feel that our residency training should prepare us for our jobs. However, the reality is that while residency prepares us reasonably well to practice clinic medicine, it does very little to prepare us for the wide-ranging rigors of medical practice. And while it’s easy to dismiss early-career development as touchy-feely nonsense, we do so at our own peril.
That message is written in the conflicts that abound within our HM groups and our hospitals, the burnout and low satisfaction that fuel our high turnover rate, and the unfulfilled careers that litter the HM landscape. It’s a message that threatens our beloved specialty—a message that all the clapping couldn’t drown out. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Is this really happening? That’s what I was thinking as my mind quickly ran a differential of the possible explanations for the 80 people before me, positioned erectly, hands audibly moving together and apart. This had never happened to me before. Was this some sort of group yoga stretch aimed at quelling DVT formation, perhaps a pre-determined signal alerting security to have me removed for hitting an unconscionable level of boredom, or, most likely, a synchronized form of mass exit (my talks are accustomed to a certain level of attrition)? Beyond these possibilities lies just one alternative: I actually was receiving a standing ovation.
First, let me dispense with one ever-important technicality. The ovation, coming at the close of the recent four-day Academic Hospitalist Academy held outside Atlanta, was more rightly intended for the efforts of the entire eight-member faculty than me alone. I just happened to be giving the closing session.
The fact of the matter is that as a faculty member for this program, which aimed to provide early-career development for junior academic hospitalists, I, too, was in awe at the tremendous, unparalleled work of the faculty: Drs. Brad Sharpe, Vikas Parekh, Andy Auerbach, Jeff Wiese, Shobi Chheda, Bob Centor, and Jen Myers.
Yet there I was, just moments after uttering a few closing comments, being showered with praise, each clap further pumping my chest fuller with pride. It was then, with a deflating wheeze, that I realized what was happening—they weren’t clapping for me, or even the rest of the faculty. It turns out the more obvious cause for their enthusiasm had been staring me in the face the whole time. Or, more to the point, the faces staring back at me were reflecting my numerous missteps that this course had ensured they’d never make. And that was very ovation-worthy.
Take-Home Points
Clapping excitedly at table No. 4 was a young first-year hospitalist from a major academic medical center. Looking at her, I could tell she would not, like I had, make the mistake of waiting too long to find a mentor. It wasn’t until my fourth year in academics that I found a mentor. That was four years of unproductive wandering, chasing dead ends, grabbing at wrong straws. So after multiple sessions covering the importance and means of finding mentors, as well as the role of mentees, it was clear that this young hospitalist would build her career foundation on more firm footing.
Applauding from table No. 7 was a second-year hospitalist from a community teaching hospital with aspirations of making a splash on the national hospitalist scene. Unlike my early fruitless attempts to get involved outside of my institution, the session on the importance of peer and national networking provided his quiver with several time-honored arrows that took me years to acquire.
The eyes of a hospitalist at table No. 8 foretold the story of a young faculty member who wouldn’t struggle with the process of promotion, as I had. After sitting through talks that lifted the veil on both the inner workings of an academic medical center and the mysteries by which said centers promote their members, she had a head start on ensuring her academic success.
Table No. 3 offered several hospitalists who wouldn’t make the errors I’ve made—repeatedly—with e-mail, phone conversations and running a meeting. A presentation on the basics of communications ensured that they wouldn’t send those irretrievable e-missives carrying unintentional messages or waste hours trying to cover in e-mail what would be better solved over the phone or in a face-to-face meeting.
A person at table one patted me on the shoulder, the glint in his eye signaling that the sessions on how to run a teaching team and be a more effective teacher at both the bedside and at the chalkboard would help him avoid the lower teaching scores that plagued my early academic years.
Focus on Fundamentals
I could go on, but the point is that while my fellow faculty and I reveled in the pleasure of a standing ovation, the truth is that the clapping had less to do with us or how we imparted information and more to do with the fact that we had shared with them the ingredients of their future success, something they actively longed for—a means to enhance their career success and satisfaction.
Mind you, none of these revelations were shocking; indeed, most are mundane and straightforward. However, the reality is that more often than not we just need help getting started—a little enzymatic push in the right direction. The fact that these needs were finally being met was evident in every heartfelt clap of the hands.
Lest you think these lessons are only important for us academic eggheads, I’d submit to you that the same, or at least similarly important, points are just as critical for young community hospitalists.
In fact, hospitalists and the field of HM are all very young; most of us are in desperate need of career guidance. And I’d go so far as to say the success of our field depends on meeting these needs as much or more than our ability to improve the quality of healthcare. The reality is that without sated, successful, career-oriented hospitalists, there can be no HM movement to improve the quality of care.
It’s tempting to feel that our residency training should prepare us for our jobs. However, the reality is that while residency prepares us reasonably well to practice clinic medicine, it does very little to prepare us for the wide-ranging rigors of medical practice. And while it’s easy to dismiss early-career development as touchy-feely nonsense, we do so at our own peril.
That message is written in the conflicts that abound within our HM groups and our hospitals, the burnout and low satisfaction that fuel our high turnover rate, and the unfulfilled careers that litter the HM landscape. It’s a message that threatens our beloved specialty—a message that all the clapping couldn’t drown out. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Is this really happening? That’s what I was thinking as my mind quickly ran a differential of the possible explanations for the 80 people before me, positioned erectly, hands audibly moving together and apart. This had never happened to me before. Was this some sort of group yoga stretch aimed at quelling DVT formation, perhaps a pre-determined signal alerting security to have me removed for hitting an unconscionable level of boredom, or, most likely, a synchronized form of mass exit (my talks are accustomed to a certain level of attrition)? Beyond these possibilities lies just one alternative: I actually was receiving a standing ovation.
First, let me dispense with one ever-important technicality. The ovation, coming at the close of the recent four-day Academic Hospitalist Academy held outside Atlanta, was more rightly intended for the efforts of the entire eight-member faculty than me alone. I just happened to be giving the closing session.
The fact of the matter is that as a faculty member for this program, which aimed to provide early-career development for junior academic hospitalists, I, too, was in awe at the tremendous, unparalleled work of the faculty: Drs. Brad Sharpe, Vikas Parekh, Andy Auerbach, Jeff Wiese, Shobi Chheda, Bob Centor, and Jen Myers.
Yet there I was, just moments after uttering a few closing comments, being showered with praise, each clap further pumping my chest fuller with pride. It was then, with a deflating wheeze, that I realized what was happening—they weren’t clapping for me, or even the rest of the faculty. It turns out the more obvious cause for their enthusiasm had been staring me in the face the whole time. Or, more to the point, the faces staring back at me were reflecting my numerous missteps that this course had ensured they’d never make. And that was very ovation-worthy.
Take-Home Points
Clapping excitedly at table No. 4 was a young first-year hospitalist from a major academic medical center. Looking at her, I could tell she would not, like I had, make the mistake of waiting too long to find a mentor. It wasn’t until my fourth year in academics that I found a mentor. That was four years of unproductive wandering, chasing dead ends, grabbing at wrong straws. So after multiple sessions covering the importance and means of finding mentors, as well as the role of mentees, it was clear that this young hospitalist would build her career foundation on more firm footing.
Applauding from table No. 7 was a second-year hospitalist from a community teaching hospital with aspirations of making a splash on the national hospitalist scene. Unlike my early fruitless attempts to get involved outside of my institution, the session on the importance of peer and national networking provided his quiver with several time-honored arrows that took me years to acquire.
The eyes of a hospitalist at table No. 8 foretold the story of a young faculty member who wouldn’t struggle with the process of promotion, as I had. After sitting through talks that lifted the veil on both the inner workings of an academic medical center and the mysteries by which said centers promote their members, she had a head start on ensuring her academic success.
Table No. 3 offered several hospitalists who wouldn’t make the errors I’ve made—repeatedly—with e-mail, phone conversations and running a meeting. A presentation on the basics of communications ensured that they wouldn’t send those irretrievable e-missives carrying unintentional messages or waste hours trying to cover in e-mail what would be better solved over the phone or in a face-to-face meeting.
A person at table one patted me on the shoulder, the glint in his eye signaling that the sessions on how to run a teaching team and be a more effective teacher at both the bedside and at the chalkboard would help him avoid the lower teaching scores that plagued my early academic years.
Focus on Fundamentals
I could go on, but the point is that while my fellow faculty and I reveled in the pleasure of a standing ovation, the truth is that the clapping had less to do with us or how we imparted information and more to do with the fact that we had shared with them the ingredients of their future success, something they actively longed for—a means to enhance their career success and satisfaction.
Mind you, none of these revelations were shocking; indeed, most are mundane and straightforward. However, the reality is that more often than not we just need help getting started—a little enzymatic push in the right direction. The fact that these needs were finally being met was evident in every heartfelt clap of the hands.
Lest you think these lessons are only important for us academic eggheads, I’d submit to you that the same, or at least similarly important, points are just as critical for young community hospitalists.
In fact, hospitalists and the field of HM are all very young; most of us are in desperate need of career guidance. And I’d go so far as to say the success of our field depends on meeting these needs as much or more than our ability to improve the quality of healthcare. The reality is that without sated, successful, career-oriented hospitalists, there can be no HM movement to improve the quality of care.
It’s tempting to feel that our residency training should prepare us for our jobs. However, the reality is that while residency prepares us reasonably well to practice clinic medicine, it does very little to prepare us for the wide-ranging rigors of medical practice. And while it’s easy to dismiss early-career development as touchy-feely nonsense, we do so at our own peril.
That message is written in the conflicts that abound within our HM groups and our hospitals, the burnout and low satisfaction that fuel our high turnover rate, and the unfulfilled careers that litter the HM landscape. It’s a message that threatens our beloved specialty—a message that all the clapping couldn’t drown out. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
An Imperfect Solution

There is no doubt we are getting healthcare reform, and in the end, Democrats will declare victory for the first meaningful progress since the 1960s, when Medicare and Medicaid were passed. Of course, in the interim, we have had legislation facilitating the development of HMOs under President Nixon and a senior pharmacy benefit under President George W. Bush, but many presidents have flailed at taking a crack at making major changes.
Republicans will declare victory, too, for stopping many bad ideas and trying to hold the line on costs. And everyone will complain about all the things that are not in the bill President Obama will sign this year.
And everyone will be right.
One Out of Three
To oversimplify things, all of the talk about healthcare reform has focused on three main areas:
- Increasing access for the uninsured and underinsured;
- Reigning in healthcare costs; and
- Designing a new system that rewards performance and safety.
At best, all we are getting is a down payment on access—and it will come with a substantial cost.
But what we are more likely beginning is an unraveling of business as usual and a reshuffling of the deck—and some key stakeholders won’t like the cards they will be dealt. The best way to think of what is happening in 2010 is that this is the first step toward having the healthcare system we will have in 2020.
Civic Obligation
It is a national embarrassment for the U.S. to be the only developed country that has not come up with a solution that offers most of its citizens access to healthcare. As a culture, we have decided that every child deserves a free education, that all families should have access to fire and police protection, and that we all should have access to due process and “an attorney who will be appointed to you if you cannot afford one.”
But right now in our country, about 47 million people live sicker and die quicker because of a healthcare system that doesn’t include them. A more sorry aspect is the “underinsured,” the constantly employed person with “good” insurance who is unfortunate enough to be diagnosed with cancer only to find out that their $1 million lifetime benefit runs out in year two or three. Those families face the tough choices between bankruptcy and foreclosure, or allowing Mom or Dad to give up another year or two or three of life. Is this the America we are living in?
Reform, Part I
To get this partial loaf of healthcare reform, Obama and Congressional leaders had to be creative. What has torpedoed previous efforts has been the vast power and reach of large, well-funded stakeholders who see any change as a threat and take a “what’s in it for me” approach. These industries have not been shy about using power and money to influence Congress and the White House, and even more insidiously have gone “direct” with advertisements and commentators who use “Harry and Louise” tactics to frighten an underinformed public about this complex process.
But this time, Obama promised the doctors, the insurance industry, the pharmaceutical companies, the hospitals, the device-makers, and just about anybody who would listen that “they” would not be hurt by these reforms. In fact, in the access discussion for many of these stakeholders, the initial result would be 47 million more customers paying for healthcare products and services. Is it any wonder that the price tag must go up, and by trillions of dollars?
It is the price of admission, at least to get the ball rolling. Now we all are in the box. With a price tag approaching $3 trillion a year, and an aging population and a taxpaying workforce shrinking relative to those they must support with entitlements (think Medicare and Social Security), the die is cast for “Healthcare Reform: The Sequel.”
Trust me—the next round of change will be more cataclysmic. In the aggregate, physicians will make less than the nearly $500 billion we make now. Sure, the primary-care physicians (PCPs) and lower-paid specialties might not be hit (and could even move up), but some physicians will see a marked change in their compensation.
Hospitals will need to adapt as well. They must become more efficient. We saw this in California, Washington, Oregon, Massachusetts, and elsewhere, as capitation and managed care ratcheted down on the old “cost-plus” payment method and moved the industry to reward value and efficiency. Those who are efficient and effective will do very well. Those who have lived by just doing more and more without demonstrating their performance or achieving standards will suffer and be dissatisfied.
More Reforms Possible
The future of the insurance industry will be very different as well, maybe because of government’s more intrusive role (think Medicare for most people) or by evolving to a model like Germany’s, where 200 nonprofit insurance companies compete for business. We will demand that insurance companies return $0.95 on the dollar for patient care, not $0.75 or less, as is common practice today.
Device-makers and Big Pharma might start to see a glimpse into the future as comparative-effectiveness research looks at the value of new, expensive technology and advances in treatments. As medications become “included” in the standard benefits bundle, just like physician fees and hospitalizations, we will see a relentless push downward on pricing. Drugs will become just one more line item to be budgeted for, especially if MedPAC and Congress are involved. We will get what we can afford, not everything that is possible or available.
Because this is 21st-century America, under the cacophony of Glenn Beck and Keith Olbermann and Rush Limbaugh and Rachel Maddow, the potential losers will be loud. They will trumpet any fact or pseudo-fact to alarm the populace. Phrases like “government takeover” and “you will lose the great healthcare you have,” and “death squads” and “illegal immigrants” and “back to 19th-century healthcare,” will bounce around the 24-hour news cycle. They will make real, positive change difficult.
But the beauty of what we are passing now, in 2010, is that the train is leaving the station. We are burning the boats. The healthcare system shakeup officially is under way. There is no turning back.
HM was not borne of a new law or mandate. We are an innovation of a system that must change and evolve. And while HM is not all it eventually will be, there are hints of what we can become. For a new healthcare system that offers greater access and is grounded in documented performance and efficiency, HM will be a solution for hospitals with hospitalist groups.
A lot of uncertainty remains out there, and the next decade promises to be even more turbulent, but hospitalists are as well positioned as any stakeholder in healthcare.
We are ready to be an active, contributing, and solution-oriented profession that will add value to our patients and our healthcare communities.
Stay tuned. TH
Dr. Wellikson is CEO of SHM.
There is no doubt we are getting healthcare reform, and in the end, Democrats will declare victory for the first meaningful progress since the 1960s, when Medicare and Medicaid were passed. Of course, in the interim, we have had legislation facilitating the development of HMOs under President Nixon and a senior pharmacy benefit under President George W. Bush, but many presidents have flailed at taking a crack at making major changes.
Republicans will declare victory, too, for stopping many bad ideas and trying to hold the line on costs. And everyone will complain about all the things that are not in the bill President Obama will sign this year.
And everyone will be right.
One Out of Three
To oversimplify things, all of the talk about healthcare reform has focused on three main areas:
- Increasing access for the uninsured and underinsured;
- Reigning in healthcare costs; and
- Designing a new system that rewards performance and safety.
At best, all we are getting is a down payment on access—and it will come with a substantial cost.
But what we are more likely beginning is an unraveling of business as usual and a reshuffling of the deck—and some key stakeholders won’t like the cards they will be dealt. The best way to think of what is happening in 2010 is that this is the first step toward having the healthcare system we will have in 2020.
Civic Obligation
It is a national embarrassment for the U.S. to be the only developed country that has not come up with a solution that offers most of its citizens access to healthcare. As a culture, we have decided that every child deserves a free education, that all families should have access to fire and police protection, and that we all should have access to due process and “an attorney who will be appointed to you if you cannot afford one.”
But right now in our country, about 47 million people live sicker and die quicker because of a healthcare system that doesn’t include them. A more sorry aspect is the “underinsured,” the constantly employed person with “good” insurance who is unfortunate enough to be diagnosed with cancer only to find out that their $1 million lifetime benefit runs out in year two or three. Those families face the tough choices between bankruptcy and foreclosure, or allowing Mom or Dad to give up another year or two or three of life. Is this the America we are living in?
Reform, Part I
To get this partial loaf of healthcare reform, Obama and Congressional leaders had to be creative. What has torpedoed previous efforts has been the vast power and reach of large, well-funded stakeholders who see any change as a threat and take a “what’s in it for me” approach. These industries have not been shy about using power and money to influence Congress and the White House, and even more insidiously have gone “direct” with advertisements and commentators who use “Harry and Louise” tactics to frighten an underinformed public about this complex process.
But this time, Obama promised the doctors, the insurance industry, the pharmaceutical companies, the hospitals, the device-makers, and just about anybody who would listen that “they” would not be hurt by these reforms. In fact, in the access discussion for many of these stakeholders, the initial result would be 47 million more customers paying for healthcare products and services. Is it any wonder that the price tag must go up, and by trillions of dollars?
It is the price of admission, at least to get the ball rolling. Now we all are in the box. With a price tag approaching $3 trillion a year, and an aging population and a taxpaying workforce shrinking relative to those they must support with entitlements (think Medicare and Social Security), the die is cast for “Healthcare Reform: The Sequel.”
Trust me—the next round of change will be more cataclysmic. In the aggregate, physicians will make less than the nearly $500 billion we make now. Sure, the primary-care physicians (PCPs) and lower-paid specialties might not be hit (and could even move up), but some physicians will see a marked change in their compensation.
Hospitals will need to adapt as well. They must become more efficient. We saw this in California, Washington, Oregon, Massachusetts, and elsewhere, as capitation and managed care ratcheted down on the old “cost-plus” payment method and moved the industry to reward value and efficiency. Those who are efficient and effective will do very well. Those who have lived by just doing more and more without demonstrating their performance or achieving standards will suffer and be dissatisfied.
More Reforms Possible
The future of the insurance industry will be very different as well, maybe because of government’s more intrusive role (think Medicare for most people) or by evolving to a model like Germany’s, where 200 nonprofit insurance companies compete for business. We will demand that insurance companies return $0.95 on the dollar for patient care, not $0.75 or less, as is common practice today.
Device-makers and Big Pharma might start to see a glimpse into the future as comparative-effectiveness research looks at the value of new, expensive technology and advances in treatments. As medications become “included” in the standard benefits bundle, just like physician fees and hospitalizations, we will see a relentless push downward on pricing. Drugs will become just one more line item to be budgeted for, especially if MedPAC and Congress are involved. We will get what we can afford, not everything that is possible or available.
Because this is 21st-century America, under the cacophony of Glenn Beck and Keith Olbermann and Rush Limbaugh and Rachel Maddow, the potential losers will be loud. They will trumpet any fact or pseudo-fact to alarm the populace. Phrases like “government takeover” and “you will lose the great healthcare you have,” and “death squads” and “illegal immigrants” and “back to 19th-century healthcare,” will bounce around the 24-hour news cycle. They will make real, positive change difficult.
But the beauty of what we are passing now, in 2010, is that the train is leaving the station. We are burning the boats. The healthcare system shakeup officially is under way. There is no turning back.
HM was not borne of a new law or mandate. We are an innovation of a system that must change and evolve. And while HM is not all it eventually will be, there are hints of what we can become. For a new healthcare system that offers greater access and is grounded in documented performance and efficiency, HM will be a solution for hospitals with hospitalist groups.
A lot of uncertainty remains out there, and the next decade promises to be even more turbulent, but hospitalists are as well positioned as any stakeholder in healthcare.
We are ready to be an active, contributing, and solution-oriented profession that will add value to our patients and our healthcare communities.
Stay tuned. TH
Dr. Wellikson is CEO of SHM.
There is no doubt we are getting healthcare reform, and in the end, Democrats will declare victory for the first meaningful progress since the 1960s, when Medicare and Medicaid were passed. Of course, in the interim, we have had legislation facilitating the development of HMOs under President Nixon and a senior pharmacy benefit under President George W. Bush, but many presidents have flailed at taking a crack at making major changes.
Republicans will declare victory, too, for stopping many bad ideas and trying to hold the line on costs. And everyone will complain about all the things that are not in the bill President Obama will sign this year.
And everyone will be right.
One Out of Three
To oversimplify things, all of the talk about healthcare reform has focused on three main areas:
- Increasing access for the uninsured and underinsured;
- Reigning in healthcare costs; and
- Designing a new system that rewards performance and safety.
At best, all we are getting is a down payment on access—and it will come with a substantial cost.
But what we are more likely beginning is an unraveling of business as usual and a reshuffling of the deck—and some key stakeholders won’t like the cards they will be dealt. The best way to think of what is happening in 2010 is that this is the first step toward having the healthcare system we will have in 2020.
Civic Obligation
It is a national embarrassment for the U.S. to be the only developed country that has not come up with a solution that offers most of its citizens access to healthcare. As a culture, we have decided that every child deserves a free education, that all families should have access to fire and police protection, and that we all should have access to due process and “an attorney who will be appointed to you if you cannot afford one.”
But right now in our country, about 47 million people live sicker and die quicker because of a healthcare system that doesn’t include them. A more sorry aspect is the “underinsured,” the constantly employed person with “good” insurance who is unfortunate enough to be diagnosed with cancer only to find out that their $1 million lifetime benefit runs out in year two or three. Those families face the tough choices between bankruptcy and foreclosure, or allowing Mom or Dad to give up another year or two or three of life. Is this the America we are living in?
Reform, Part I
To get this partial loaf of healthcare reform, Obama and Congressional leaders had to be creative. What has torpedoed previous efforts has been the vast power and reach of large, well-funded stakeholders who see any change as a threat and take a “what’s in it for me” approach. These industries have not been shy about using power and money to influence Congress and the White House, and even more insidiously have gone “direct” with advertisements and commentators who use “Harry and Louise” tactics to frighten an underinformed public about this complex process.
But this time, Obama promised the doctors, the insurance industry, the pharmaceutical companies, the hospitals, the device-makers, and just about anybody who would listen that “they” would not be hurt by these reforms. In fact, in the access discussion for many of these stakeholders, the initial result would be 47 million more customers paying for healthcare products and services. Is it any wonder that the price tag must go up, and by trillions of dollars?
It is the price of admission, at least to get the ball rolling. Now we all are in the box. With a price tag approaching $3 trillion a year, and an aging population and a taxpaying workforce shrinking relative to those they must support with entitlements (think Medicare and Social Security), the die is cast for “Healthcare Reform: The Sequel.”
Trust me—the next round of change will be more cataclysmic. In the aggregate, physicians will make less than the nearly $500 billion we make now. Sure, the primary-care physicians (PCPs) and lower-paid specialties might not be hit (and could even move up), but some physicians will see a marked change in their compensation.
Hospitals will need to adapt as well. They must become more efficient. We saw this in California, Washington, Oregon, Massachusetts, and elsewhere, as capitation and managed care ratcheted down on the old “cost-plus” payment method and moved the industry to reward value and efficiency. Those who are efficient and effective will do very well. Those who have lived by just doing more and more without demonstrating their performance or achieving standards will suffer and be dissatisfied.
More Reforms Possible
The future of the insurance industry will be very different as well, maybe because of government’s more intrusive role (think Medicare for most people) or by evolving to a model like Germany’s, where 200 nonprofit insurance companies compete for business. We will demand that insurance companies return $0.95 on the dollar for patient care, not $0.75 or less, as is common practice today.
Device-makers and Big Pharma might start to see a glimpse into the future as comparative-effectiveness research looks at the value of new, expensive technology and advances in treatments. As medications become “included” in the standard benefits bundle, just like physician fees and hospitalizations, we will see a relentless push downward on pricing. Drugs will become just one more line item to be budgeted for, especially if MedPAC and Congress are involved. We will get what we can afford, not everything that is possible or available.
Because this is 21st-century America, under the cacophony of Glenn Beck and Keith Olbermann and Rush Limbaugh and Rachel Maddow, the potential losers will be loud. They will trumpet any fact or pseudo-fact to alarm the populace. Phrases like “government takeover” and “you will lose the great healthcare you have,” and “death squads” and “illegal immigrants” and “back to 19th-century healthcare,” will bounce around the 24-hour news cycle. They will make real, positive change difficult.
But the beauty of what we are passing now, in 2010, is that the train is leaving the station. We are burning the boats. The healthcare system shakeup officially is under way. There is no turning back.
HM was not borne of a new law or mandate. We are an innovation of a system that must change and evolve. And while HM is not all it eventually will be, there are hints of what we can become. For a new healthcare system that offers greater access and is grounded in documented performance and efficiency, HM will be a solution for hospitals with hospitalist groups.
A lot of uncertainty remains out there, and the next decade promises to be even more turbulent, but hospitalists are as well positioned as any stakeholder in healthcare.
We are ready to be an active, contributing, and solution-oriented profession that will add value to our patients and our healthcare communities.
Stay tuned. TH
Dr. Wellikson is CEO of SHM.
Medicare Fee Inspection
No one can call 2009 a dull year for healthcare policy. And 2010 already is shaping up as another humdinger, with several issues bubbling to the surface. One of the biggest comes courtesy of the Dartmouth Atlas of Health Care (www.dartmouthatlas.org), as politicians, analysts, researchers, and physicians grapple over how to resolve the contentious issue of geographical disparities in healthcare spending.
One of the main bodies of evidence driving the debate, the interactive Dartmouth map, depicts a color-coded nation in which wide swaths of the Midwest and West are colored with a pale green hue, which represents a significantly reduced amount of Medicare reimbursements. Meanwhile, states such as New York, New Jersey, Massachusetts, Florida, Texas, and Louisiana are marked by a darker shade of green—representing the nation’s most expensive per capita reimbursement rates.
Tucked within 2009’s massive Affordable Health Care for America Act passed by the House is a provision calling for a study of “geographic variation in healthcare spending and promoting high-value healthcare,” which is aiming for a more evenly colored landscape.
More than 50 legislators, hailing primarily from the Midwest and Pacific Northwest and calling themselves the Quality Care Coalition, pushed through the wording as a condition for supporting the larger healthcare reform bill. One measure would direct the nonpartisan Institute of Medicine (IOM) to check the accuracy of the geographic adjustment factors that underlie existing Medicare reimbursements and suggest necessary revisions. The second would call upon the IOM “to conduct a study on geographic variation and growth in volume and intensity of services in per capita healthcare spending among the Medicare, Medicaid, privately insured, and uninsured populations.”
Recommendations to Secretary of Health and Human Services Kathleen Sebelius as a result of that study would go into effect unless the House and Senate passed a joint resolution of disapproval with a two-thirds vote.
Reimbursement Battles
The implicit message is that some states, cities, and health providers have been shortchanged in their reimbursements—a complaint that flows into the larger meme that the country’s dysfunctional payment system rewards quantity, not quality. Officials at the Mayo Clinic in Rochester, Minn., have suggested in media accounts that the current Medicare formula cost the clinic $840 million in lost reimbursements in 2008 alone.
Rep. Jay Inslee (D-Washington), whose district lies northwest of Seattle, served as one of the lead negotiators on the issue. According to Inslee spokesman Robert Kellar, the geographical disparity in healthcare spending has been a perennial concern for the Washington delegation due to reimbursement rates that lag by as much as 50%, depending on the procedure. “Hospitals haven’t been able to keep or attract the personnel that they could have because of this issue,” Kellar says. In Washington state, per capita Medicare reimbursements in 2006 hovered about $1,200 below the national average, though 15 other states, led by Hawaii, received even less.
Despite the specter of a skirmish between urban and rural states and hospitals, however, the Dartmouth Atlas suggests that many disparities are more geographically nuanced. In 2006, for example, the Miami hospital referral region received more than $16,300 in Medicare reimbursements per enrollee, while nearby Fort Lauderdale received $9,800 and Atlanta less than $7,400. By comparison, New York netted $12,100, Seattle received $7,200, Rochester, Minn., received $6,700, and Honolulu was reimbursed only $5,300.
Representatives of higher-spending areas have complained that the atlas doesn’t tell the whole story—that steep living costs, poorer populations seeking medical care, and infrastructure necessary for teaching institutions can drive up Medicare expenses. As part of a compromise negotiated with the Quality Care Coalition, the examination of per capita spending will not include expenses related to graduate medical education, disproportionate share hospital (DSH) payments, and health information technology.
In attempting to get at the source of remaining cost disparities, however, the IOM has been charged with considering such factors as a local population’s relative health and socioeconomic status (race, ethnicity, gender, age, income, and education). The study will scrutinize healthcare providers’ organizational models, practice patterns, healthcare outcomes, quality benchmarks, and doctors’ discretion in making treatment decisions, among other criteria.
Differences of Opinion
Dylan Roby, an assistant professor at the UCLA Center for Health Policy Research, says the general expectation among healthcare analysts is that significant differences will remain even with additional sophisticated modeling techniques. “The main hypothesis by most people in the field is that it’s differences in practice patterns that are really driving this, not differences in need or differences in disease burden,” he says.
But what about outcomes? A recent study of heart failure patients at six California hospitals seemed to throw cold water on the notion that higher resource use doesn’t equate with better results with patients.1 The study found more treatment did lead to higher odds of survival.
Roby thinks the study’s results lay the framework for looking at hospital-to-hospital differences in how providers deliver care and allocate resources, but he cautions that they shouldn’t be overanalyzed. All six of the California hospitals in the study are linked to universities and have ample access to resources, he points out.
HM at the Forefront
As for hospitalists, Roby hopes they will be increasingly called upon as focal points for improving efficiencies within provider networks. He concedes that plenty of challenges remain: An institution’s internal politics, for instance, could stymie even the most efficient and proactive physician. Even so, Roby is hopeful that an independent study could at least spur a dialogue about best practices. “I think what the study could potentially do, rather than just act as a way to penalize hospitals that might not be efficient with care, is really offer the ability for us to look at the characteristics of hospitals, in terms of how the care is delivered,” he says.
Ideally, the ability to learn would be followed by the impetus to change. But as analysts have noted, a panel’s recommendations on how to improve healthcare delivery don’t always neatly translate into federal policy.
Consider November’s uproar over mammogram recommendations. When the 16-member U.S. Preventive Services Task Force recommended that women wait until age 50 for routine mammograms instead of starting the screening process at 40, in large part to prevent overtreatment, the fallout was fast and furious. Sebelius quickly signaled in a strongly worded statement that federal policy wasn’t about to change, despite the evidence-based conclusions of a panel convened by her department’s Agency for Healthcare Research and Quality. A group of Republican legislators decried the recommendation as evidence of bureaucrats intruding on healthcare decisions, and even Rep. Debbie Wasserman Schulz (D-Florida), herself a breast-cancer survivor, called the panel’s recommendations “disturbing” and considered Congressional hearings.
The take-home message is readily transferrable to hospitalists: The perception that patients might receive less care can spark public upheaval and force policy makers to beat a hasty retreat away from evidence-based medicine.
Despite the best intentions, a federal panel’s recommendations over resolving geographical disparities in spending could unleash far more drama. Inevitably, such a study will identify both winners and losers, the latter of whom might not accept reduced payments willingly or quietly. TH
Bryn Nelson is a freelance writer based in Seattle.
Reference
- Ellis SG, Miller D, Keys TF. Comparing physician-specific two-year patient outcomes after coronary angiography. J Am Coll Cardiol. 1999;33:1278-1285.
No one can call 2009 a dull year for healthcare policy. And 2010 already is shaping up as another humdinger, with several issues bubbling to the surface. One of the biggest comes courtesy of the Dartmouth Atlas of Health Care (www.dartmouthatlas.org), as politicians, analysts, researchers, and physicians grapple over how to resolve the contentious issue of geographical disparities in healthcare spending.
One of the main bodies of evidence driving the debate, the interactive Dartmouth map, depicts a color-coded nation in which wide swaths of the Midwest and West are colored with a pale green hue, which represents a significantly reduced amount of Medicare reimbursements. Meanwhile, states such as New York, New Jersey, Massachusetts, Florida, Texas, and Louisiana are marked by a darker shade of green—representing the nation’s most expensive per capita reimbursement rates.
Tucked within 2009’s massive Affordable Health Care for America Act passed by the House is a provision calling for a study of “geographic variation in healthcare spending and promoting high-value healthcare,” which is aiming for a more evenly colored landscape.
More than 50 legislators, hailing primarily from the Midwest and Pacific Northwest and calling themselves the Quality Care Coalition, pushed through the wording as a condition for supporting the larger healthcare reform bill. One measure would direct the nonpartisan Institute of Medicine (IOM) to check the accuracy of the geographic adjustment factors that underlie existing Medicare reimbursements and suggest necessary revisions. The second would call upon the IOM “to conduct a study on geographic variation and growth in volume and intensity of services in per capita healthcare spending among the Medicare, Medicaid, privately insured, and uninsured populations.”
Recommendations to Secretary of Health and Human Services Kathleen Sebelius as a result of that study would go into effect unless the House and Senate passed a joint resolution of disapproval with a two-thirds vote.
Reimbursement Battles
The implicit message is that some states, cities, and health providers have been shortchanged in their reimbursements—a complaint that flows into the larger meme that the country’s dysfunctional payment system rewards quantity, not quality. Officials at the Mayo Clinic in Rochester, Minn., have suggested in media accounts that the current Medicare formula cost the clinic $840 million in lost reimbursements in 2008 alone.
Rep. Jay Inslee (D-Washington), whose district lies northwest of Seattle, served as one of the lead negotiators on the issue. According to Inslee spokesman Robert Kellar, the geographical disparity in healthcare spending has been a perennial concern for the Washington delegation due to reimbursement rates that lag by as much as 50%, depending on the procedure. “Hospitals haven’t been able to keep or attract the personnel that they could have because of this issue,” Kellar says. In Washington state, per capita Medicare reimbursements in 2006 hovered about $1,200 below the national average, though 15 other states, led by Hawaii, received even less.
Despite the specter of a skirmish between urban and rural states and hospitals, however, the Dartmouth Atlas suggests that many disparities are more geographically nuanced. In 2006, for example, the Miami hospital referral region received more than $16,300 in Medicare reimbursements per enrollee, while nearby Fort Lauderdale received $9,800 and Atlanta less than $7,400. By comparison, New York netted $12,100, Seattle received $7,200, Rochester, Minn., received $6,700, and Honolulu was reimbursed only $5,300.
Representatives of higher-spending areas have complained that the atlas doesn’t tell the whole story—that steep living costs, poorer populations seeking medical care, and infrastructure necessary for teaching institutions can drive up Medicare expenses. As part of a compromise negotiated with the Quality Care Coalition, the examination of per capita spending will not include expenses related to graduate medical education, disproportionate share hospital (DSH) payments, and health information technology.
In attempting to get at the source of remaining cost disparities, however, the IOM has been charged with considering such factors as a local population’s relative health and socioeconomic status (race, ethnicity, gender, age, income, and education). The study will scrutinize healthcare providers’ organizational models, practice patterns, healthcare outcomes, quality benchmarks, and doctors’ discretion in making treatment decisions, among other criteria.
Differences of Opinion
Dylan Roby, an assistant professor at the UCLA Center for Health Policy Research, says the general expectation among healthcare analysts is that significant differences will remain even with additional sophisticated modeling techniques. “The main hypothesis by most people in the field is that it’s differences in practice patterns that are really driving this, not differences in need or differences in disease burden,” he says.
But what about outcomes? A recent study of heart failure patients at six California hospitals seemed to throw cold water on the notion that higher resource use doesn’t equate with better results with patients.1 The study found more treatment did lead to higher odds of survival.
Roby thinks the study’s results lay the framework for looking at hospital-to-hospital differences in how providers deliver care and allocate resources, but he cautions that they shouldn’t be overanalyzed. All six of the California hospitals in the study are linked to universities and have ample access to resources, he points out.
HM at the Forefront
As for hospitalists, Roby hopes they will be increasingly called upon as focal points for improving efficiencies within provider networks. He concedes that plenty of challenges remain: An institution’s internal politics, for instance, could stymie even the most efficient and proactive physician. Even so, Roby is hopeful that an independent study could at least spur a dialogue about best practices. “I think what the study could potentially do, rather than just act as a way to penalize hospitals that might not be efficient with care, is really offer the ability for us to look at the characteristics of hospitals, in terms of how the care is delivered,” he says.
Ideally, the ability to learn would be followed by the impetus to change. But as analysts have noted, a panel’s recommendations on how to improve healthcare delivery don’t always neatly translate into federal policy.
Consider November’s uproar over mammogram recommendations. When the 16-member U.S. Preventive Services Task Force recommended that women wait until age 50 for routine mammograms instead of starting the screening process at 40, in large part to prevent overtreatment, the fallout was fast and furious. Sebelius quickly signaled in a strongly worded statement that federal policy wasn’t about to change, despite the evidence-based conclusions of a panel convened by her department’s Agency for Healthcare Research and Quality. A group of Republican legislators decried the recommendation as evidence of bureaucrats intruding on healthcare decisions, and even Rep. Debbie Wasserman Schulz (D-Florida), herself a breast-cancer survivor, called the panel’s recommendations “disturbing” and considered Congressional hearings.
The take-home message is readily transferrable to hospitalists: The perception that patients might receive less care can spark public upheaval and force policy makers to beat a hasty retreat away from evidence-based medicine.
Despite the best intentions, a federal panel’s recommendations over resolving geographical disparities in spending could unleash far more drama. Inevitably, such a study will identify both winners and losers, the latter of whom might not accept reduced payments willingly or quietly. TH
Bryn Nelson is a freelance writer based in Seattle.
Reference
- Ellis SG, Miller D, Keys TF. Comparing physician-specific two-year patient outcomes after coronary angiography. J Am Coll Cardiol. 1999;33:1278-1285.
No one can call 2009 a dull year for healthcare policy. And 2010 already is shaping up as another humdinger, with several issues bubbling to the surface. One of the biggest comes courtesy of the Dartmouth Atlas of Health Care (www.dartmouthatlas.org), as politicians, analysts, researchers, and physicians grapple over how to resolve the contentious issue of geographical disparities in healthcare spending.
One of the main bodies of evidence driving the debate, the interactive Dartmouth map, depicts a color-coded nation in which wide swaths of the Midwest and West are colored with a pale green hue, which represents a significantly reduced amount of Medicare reimbursements. Meanwhile, states such as New York, New Jersey, Massachusetts, Florida, Texas, and Louisiana are marked by a darker shade of green—representing the nation’s most expensive per capita reimbursement rates.
Tucked within 2009’s massive Affordable Health Care for America Act passed by the House is a provision calling for a study of “geographic variation in healthcare spending and promoting high-value healthcare,” which is aiming for a more evenly colored landscape.
More than 50 legislators, hailing primarily from the Midwest and Pacific Northwest and calling themselves the Quality Care Coalition, pushed through the wording as a condition for supporting the larger healthcare reform bill. One measure would direct the nonpartisan Institute of Medicine (IOM) to check the accuracy of the geographic adjustment factors that underlie existing Medicare reimbursements and suggest necessary revisions. The second would call upon the IOM “to conduct a study on geographic variation and growth in volume and intensity of services in per capita healthcare spending among the Medicare, Medicaid, privately insured, and uninsured populations.”
Recommendations to Secretary of Health and Human Services Kathleen Sebelius as a result of that study would go into effect unless the House and Senate passed a joint resolution of disapproval with a two-thirds vote.
Reimbursement Battles
The implicit message is that some states, cities, and health providers have been shortchanged in their reimbursements—a complaint that flows into the larger meme that the country’s dysfunctional payment system rewards quantity, not quality. Officials at the Mayo Clinic in Rochester, Minn., have suggested in media accounts that the current Medicare formula cost the clinic $840 million in lost reimbursements in 2008 alone.
Rep. Jay Inslee (D-Washington), whose district lies northwest of Seattle, served as one of the lead negotiators on the issue. According to Inslee spokesman Robert Kellar, the geographical disparity in healthcare spending has been a perennial concern for the Washington delegation due to reimbursement rates that lag by as much as 50%, depending on the procedure. “Hospitals haven’t been able to keep or attract the personnel that they could have because of this issue,” Kellar says. In Washington state, per capita Medicare reimbursements in 2006 hovered about $1,200 below the national average, though 15 other states, led by Hawaii, received even less.
Despite the specter of a skirmish between urban and rural states and hospitals, however, the Dartmouth Atlas suggests that many disparities are more geographically nuanced. In 2006, for example, the Miami hospital referral region received more than $16,300 in Medicare reimbursements per enrollee, while nearby Fort Lauderdale received $9,800 and Atlanta less than $7,400. By comparison, New York netted $12,100, Seattle received $7,200, Rochester, Minn., received $6,700, and Honolulu was reimbursed only $5,300.
Representatives of higher-spending areas have complained that the atlas doesn’t tell the whole story—that steep living costs, poorer populations seeking medical care, and infrastructure necessary for teaching institutions can drive up Medicare expenses. As part of a compromise negotiated with the Quality Care Coalition, the examination of per capita spending will not include expenses related to graduate medical education, disproportionate share hospital (DSH) payments, and health information technology.
In attempting to get at the source of remaining cost disparities, however, the IOM has been charged with considering such factors as a local population’s relative health and socioeconomic status (race, ethnicity, gender, age, income, and education). The study will scrutinize healthcare providers’ organizational models, practice patterns, healthcare outcomes, quality benchmarks, and doctors’ discretion in making treatment decisions, among other criteria.
Differences of Opinion
Dylan Roby, an assistant professor at the UCLA Center for Health Policy Research, says the general expectation among healthcare analysts is that significant differences will remain even with additional sophisticated modeling techniques. “The main hypothesis by most people in the field is that it’s differences in practice patterns that are really driving this, not differences in need or differences in disease burden,” he says.
But what about outcomes? A recent study of heart failure patients at six California hospitals seemed to throw cold water on the notion that higher resource use doesn’t equate with better results with patients.1 The study found more treatment did lead to higher odds of survival.
Roby thinks the study’s results lay the framework for looking at hospital-to-hospital differences in how providers deliver care and allocate resources, but he cautions that they shouldn’t be overanalyzed. All six of the California hospitals in the study are linked to universities and have ample access to resources, he points out.
HM at the Forefront
As for hospitalists, Roby hopes they will be increasingly called upon as focal points for improving efficiencies within provider networks. He concedes that plenty of challenges remain: An institution’s internal politics, for instance, could stymie even the most efficient and proactive physician. Even so, Roby is hopeful that an independent study could at least spur a dialogue about best practices. “I think what the study could potentially do, rather than just act as a way to penalize hospitals that might not be efficient with care, is really offer the ability for us to look at the characteristics of hospitals, in terms of how the care is delivered,” he says.
Ideally, the ability to learn would be followed by the impetus to change. But as analysts have noted, a panel’s recommendations on how to improve healthcare delivery don’t always neatly translate into federal policy.
Consider November’s uproar over mammogram recommendations. When the 16-member U.S. Preventive Services Task Force recommended that women wait until age 50 for routine mammograms instead of starting the screening process at 40, in large part to prevent overtreatment, the fallout was fast and furious. Sebelius quickly signaled in a strongly worded statement that federal policy wasn’t about to change, despite the evidence-based conclusions of a panel convened by her department’s Agency for Healthcare Research and Quality. A group of Republican legislators decried the recommendation as evidence of bureaucrats intruding on healthcare decisions, and even Rep. Debbie Wasserman Schulz (D-Florida), herself a breast-cancer survivor, called the panel’s recommendations “disturbing” and considered Congressional hearings.
The take-home message is readily transferrable to hospitalists: The perception that patients might receive less care can spark public upheaval and force policy makers to beat a hasty retreat away from evidence-based medicine.
Despite the best intentions, a federal panel’s recommendations over resolving geographical disparities in spending could unleash far more drama. Inevitably, such a study will identify both winners and losers, the latter of whom might not accept reduced payments willingly or quietly. TH
Bryn Nelson is a freelance writer based in Seattle.
Reference
- Ellis SG, Miller D, Keys TF. Comparing physician-specific two-year patient outcomes after coronary angiography. J Am Coll Cardiol. 1999;33:1278-1285.
Consultation Elimination
As of Jan. 1, the Centers for Medicare and Medicaid Services (CMS) ceased physician payment for consultations. The elimination of consult codes will affect physician group payments as well as relative-value-unit (RVU)-based incentive payments to individual physicians.
The Medicare-designated status of outpatient consultation (99241-99245) and inpatient consultation (99251-99255) codes has changed from “A” (separately payable under the physician fee schedule, when covered) to “I” (not valid for Medicare purposes; Medicare uses another code for the reporting of and the payment for these services). So if you submit consultation codes for Medicare beneficiaries, the result will be nonpayment.
While many physicians fear the negative impact of this ruling, hospitalists should consider its potential. Let’s take a look at a scenario hospitalists encounter on a routine basis.
Typical HM Scenario
A surgeon admits a 76-year-old man for aortic valve replacement. The patient’s history also includes well-controlled hypertension and chronic obstructive pulmonary disease (COPD). Postoperatively, the patient experiences an exacerbation of COPD related to anesthesia, elevated blood pressure, and hyperglycemia. The surgeon requests the hospitalist’s advice on appropriate medical interventions of these conditions. How should the hospitalist report the initial encounter with this Medicare beneficiary?
The hospitalist should select the CPT code that best fits the service and the payor. While most physicians regard this requested service as an inpatient consultation (99251-99255), Medicare no longer recognizes those codes. Instead, the hospitalist should report this encounter as an initial hospital care service (99221-99223).
Comanagement Issues
CMS and Medicare administrative contractors regularly uncover reporting errors for co-management requests. CMS decided the nature of these services were not consultative because the surgeon is not asking the physician or qualified nonphysician provider’s (NPP’s) opinion or advice for the surgeon’s use in treating the patient. Instead, these services constituted concurrent care and should have been billed using subsequent hospital care codes (99231-99233) in the hospital inpatient setting, subsequent NF care codes (99307-99310) in the SNF/NF setting, or office or other outpatient visit codes (99201-99215) in the office or outpatient settings.1
The new ruling simplifies coding and reduces reporting errors. The initial encounter with the patient is reported as such. Regardless of who is the attending of record or the consultant, the first physician from a particular provider group reports initial hospital care codes (i.e., 99221-99223) to represent the first patient encounter, even when this encounter does not occur on the admission date. Other physicians of the same specialty within the same provider group will not be permitted to report initial hospital care codes for their own initial encounter if someone from the group and specialty has already seen the patient during that hospitalization. In other words, the first hospitalist in the provider group reports 9922x, while the remaining hospitalists use subsequent hospital care codes (9923x).
In order to differentiate “consultant” services from “attending” services, CMS will be creating a modifier. The anticipated “AI” modifier must be appended to the attending physician’s initial encounter. Other initial hospital care codes reported throughout the hospital stay, as appropriate, are presumed to be that of “consultants” (i.e., physicians with a different specialty designation than the attending physician) participating in the case. Therefore, the hospitalist now can rightfully recover the increased work effort of the initial patient encounter (99223: 3.79 relative value units, ~$147 vs. 99233: 2.0 relative value units, ~$78, based on 2010 Medicare rates). Physicians will be required to meet the minimum documentation required for the selected visit code.
Other and Undefined Service Locations
Consultations in nursing facilities are handled much like inpatient hospital care. Physicians should report initial nursing facility services (99203-99306) for the first patient encounter, and subsequent nursing facility care codes (99307-99310) for each encounter thereafter. The attending physician of record appends the assigned modifier (presumed to be “AI”) when submitting their initial care service. All other initial care codes are presumed to be those of “consulting” physicians.
Initial information from CMS does not address observation services. Logically, these hospital-based services would follow the same methodology as inpatient care: report initial observation care (99218-99220) for the first “consulting” encounter. However, this might not be appropriate given Medicare’s existing rules for observation services, which guide physicians other than the admitting physician/group to “bill the office and other outpatient service codes or outpatient consultation codes as appropriate when they provide services to the patient.”2 With Medicare’s elimination of consultation codes, the consultant reports “office and other outpatient service codes” (i.e., new patient, 99201-99205, or established patient codes, 99212-99215) by default.
Without further clarification on observation services, hospitalists should report new or established patient service codes, depending on whether the patient has been seen by a group member within the last three years.
Medicare also has existing guidelines for the ED, which suggest that any physician not meeting the consultation criteria report ED service codes (99281-99285). Without further clarification, hospitalists should continue to follow this instruction for Medicare beneficiaries.
Nonphysician Providers
Medicare’s split/shared billing guidelines apply to most hospital inpatient, hospital outpatient, and ED evaluation and management (E/M) services, with consultations as one exception. Now, in accordance with the new ruling, hospitalists should select the appropriate initial service codes that correspond to patient’s location (e.g., 99223 for inpatients). NPPs can participate in the initial service provided to patients in these locations without the hospitalist having to replicate the entire service. The hospitalist can submit the claim in their name after selecting the visit level based upon the cumulative service personally provided on the same calendar day by both the NPP and the physician. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She also is faculty for SHM’s inpatient coding course.
References
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.10I. Centers for Medicare and Medicaid Services Web site. Available at: www.cms.hhs.gov/ manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.8A. CMS Web site. Available at: www. cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- PFS Federal Regulation Notices: Proposed Revisions to Payment Policies Under the Physician Fee Schedule and Part B for CY 2010. CMS Web site. Available at: www.cms.hhs.gov/PhysicianFeeSched/PFSFRN/itemdetail.asp?filterType=none&filterByDID=99&sortByDID=4&sortOrder=descending&itemID=CMS1223902&intNumPerPage=10. Accessed Nov. 12, 2009.
- Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B (for CY 2010). CMS Web site. Available at: www.federalregister.gov/OFR Upload/OFRData/2009-26502_PI.pdf. Accessed Nov. 10, 2009.
The Bottom Line
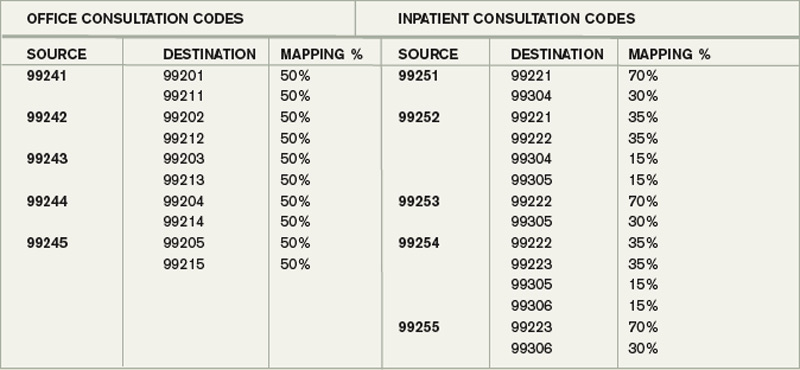
The elimination of consults will affect physician group payments as well as RVU-based incentive payments to individual physicians. HM group leaders will want to refer to the CMS formula, also known as a crosswalk. The crosswalk outlines the utilization of consult codes and E/M codes for the sole purpose of establishing aggregate budget neutrality.3
The payment differential for consultations has been redistributed to increase payments for existing E/M services.
Here’s how it works: Use the “mapping percentage” column to project the distribution of consultative services between the “replacement” (i.e., “destination”) codes. For example, presumably 70% of 99251 will now be reported as initial hospital care (99221), whereas 30% will be reported as initial nursing facility care (99304). Please note that this table does not represent billing guidance or documentation equivalencies, but it does attempt to quantify sites of consultative service and their corresponding code categories for the basis of fiscal-year projections.
Physicians should select the visit code in the appropriate code category for the service location and in accordance with CMS documentation guidelines, which are available at www.cms.hhs.gov/MLNEdWebGuide/25_EMDOC.asp.
As of Jan. 1, the Centers for Medicare and Medicaid Services (CMS) ceased physician payment for consultations. The elimination of consult codes will affect physician group payments as well as relative-value-unit (RVU)-based incentive payments to individual physicians.
The Medicare-designated status of outpatient consultation (99241-99245) and inpatient consultation (99251-99255) codes has changed from “A” (separately payable under the physician fee schedule, when covered) to “I” (not valid for Medicare purposes; Medicare uses another code for the reporting of and the payment for these services). So if you submit consultation codes for Medicare beneficiaries, the result will be nonpayment.
While many physicians fear the negative impact of this ruling, hospitalists should consider its potential. Let’s take a look at a scenario hospitalists encounter on a routine basis.
Typical HM Scenario
A surgeon admits a 76-year-old man for aortic valve replacement. The patient’s history also includes well-controlled hypertension and chronic obstructive pulmonary disease (COPD). Postoperatively, the patient experiences an exacerbation of COPD related to anesthesia, elevated blood pressure, and hyperglycemia. The surgeon requests the hospitalist’s advice on appropriate medical interventions of these conditions. How should the hospitalist report the initial encounter with this Medicare beneficiary?
The hospitalist should select the CPT code that best fits the service and the payor. While most physicians regard this requested service as an inpatient consultation (99251-99255), Medicare no longer recognizes those codes. Instead, the hospitalist should report this encounter as an initial hospital care service (99221-99223).
Comanagement Issues
CMS and Medicare administrative contractors regularly uncover reporting errors for co-management requests. CMS decided the nature of these services were not consultative because the surgeon is not asking the physician or qualified nonphysician provider’s (NPP’s) opinion or advice for the surgeon’s use in treating the patient. Instead, these services constituted concurrent care and should have been billed using subsequent hospital care codes (99231-99233) in the hospital inpatient setting, subsequent NF care codes (99307-99310) in the SNF/NF setting, or office or other outpatient visit codes (99201-99215) in the office or outpatient settings.1
The new ruling simplifies coding and reduces reporting errors. The initial encounter with the patient is reported as such. Regardless of who is the attending of record or the consultant, the first physician from a particular provider group reports initial hospital care codes (i.e., 99221-99223) to represent the first patient encounter, even when this encounter does not occur on the admission date. Other physicians of the same specialty within the same provider group will not be permitted to report initial hospital care codes for their own initial encounter if someone from the group and specialty has already seen the patient during that hospitalization. In other words, the first hospitalist in the provider group reports 9922x, while the remaining hospitalists use subsequent hospital care codes (9923x).
In order to differentiate “consultant” services from “attending” services, CMS will be creating a modifier. The anticipated “AI” modifier must be appended to the attending physician’s initial encounter. Other initial hospital care codes reported throughout the hospital stay, as appropriate, are presumed to be that of “consultants” (i.e., physicians with a different specialty designation than the attending physician) participating in the case. Therefore, the hospitalist now can rightfully recover the increased work effort of the initial patient encounter (99223: 3.79 relative value units, ~$147 vs. 99233: 2.0 relative value units, ~$78, based on 2010 Medicare rates). Physicians will be required to meet the minimum documentation required for the selected visit code.
Other and Undefined Service Locations
Consultations in nursing facilities are handled much like inpatient hospital care. Physicians should report initial nursing facility services (99203-99306) for the first patient encounter, and subsequent nursing facility care codes (99307-99310) for each encounter thereafter. The attending physician of record appends the assigned modifier (presumed to be “AI”) when submitting their initial care service. All other initial care codes are presumed to be those of “consulting” physicians.
Initial information from CMS does not address observation services. Logically, these hospital-based services would follow the same methodology as inpatient care: report initial observation care (99218-99220) for the first “consulting” encounter. However, this might not be appropriate given Medicare’s existing rules for observation services, which guide physicians other than the admitting physician/group to “bill the office and other outpatient service codes or outpatient consultation codes as appropriate when they provide services to the patient.”2 With Medicare’s elimination of consultation codes, the consultant reports “office and other outpatient service codes” (i.e., new patient, 99201-99205, or established patient codes, 99212-99215) by default.
Without further clarification on observation services, hospitalists should report new or established patient service codes, depending on whether the patient has been seen by a group member within the last three years.
Medicare also has existing guidelines for the ED, which suggest that any physician not meeting the consultation criteria report ED service codes (99281-99285). Without further clarification, hospitalists should continue to follow this instruction for Medicare beneficiaries.
Nonphysician Providers
Medicare’s split/shared billing guidelines apply to most hospital inpatient, hospital outpatient, and ED evaluation and management (E/M) services, with consultations as one exception. Now, in accordance with the new ruling, hospitalists should select the appropriate initial service codes that correspond to patient’s location (e.g., 99223 for inpatients). NPPs can participate in the initial service provided to patients in these locations without the hospitalist having to replicate the entire service. The hospitalist can submit the claim in their name after selecting the visit level based upon the cumulative service personally provided on the same calendar day by both the NPP and the physician. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She also is faculty for SHM’s inpatient coding course.
References
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.10I. Centers for Medicare and Medicaid Services Web site. Available at: www.cms.hhs.gov/ manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.8A. CMS Web site. Available at: www. cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- PFS Federal Regulation Notices: Proposed Revisions to Payment Policies Under the Physician Fee Schedule and Part B for CY 2010. CMS Web site. Available at: www.cms.hhs.gov/PhysicianFeeSched/PFSFRN/itemdetail.asp?filterType=none&filterByDID=99&sortByDID=4&sortOrder=descending&itemID=CMS1223902&intNumPerPage=10. Accessed Nov. 12, 2009.
- Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B (for CY 2010). CMS Web site. Available at: www.federalregister.gov/OFR Upload/OFRData/2009-26502_PI.pdf. Accessed Nov. 10, 2009.
The Bottom Line
The elimination of consults will affect physician group payments as well as RVU-based incentive payments to individual physicians. HM group leaders will want to refer to the CMS formula, also known as a crosswalk. The crosswalk outlines the utilization of consult codes and E/M codes for the sole purpose of establishing aggregate budget neutrality.3
The payment differential for consultations has been redistributed to increase payments for existing E/M services.
Here’s how it works: Use the “mapping percentage” column to project the distribution of consultative services between the “replacement” (i.e., “destination”) codes. For example, presumably 70% of 99251 will now be reported as initial hospital care (99221), whereas 30% will be reported as initial nursing facility care (99304). Please note that this table does not represent billing guidance or documentation equivalencies, but it does attempt to quantify sites of consultative service and their corresponding code categories for the basis of fiscal-year projections.
Physicians should select the visit code in the appropriate code category for the service location and in accordance with CMS documentation guidelines, which are available at www.cms.hhs.gov/MLNEdWebGuide/25_EMDOC.asp.
As of Jan. 1, the Centers for Medicare and Medicaid Services (CMS) ceased physician payment for consultations. The elimination of consult codes will affect physician group payments as well as relative-value-unit (RVU)-based incentive payments to individual physicians.
The Medicare-designated status of outpatient consultation (99241-99245) and inpatient consultation (99251-99255) codes has changed from “A” (separately payable under the physician fee schedule, when covered) to “I” (not valid for Medicare purposes; Medicare uses another code for the reporting of and the payment for these services). So if you submit consultation codes for Medicare beneficiaries, the result will be nonpayment.
While many physicians fear the negative impact of this ruling, hospitalists should consider its potential. Let’s take a look at a scenario hospitalists encounter on a routine basis.
Typical HM Scenario
A surgeon admits a 76-year-old man for aortic valve replacement. The patient’s history also includes well-controlled hypertension and chronic obstructive pulmonary disease (COPD). Postoperatively, the patient experiences an exacerbation of COPD related to anesthesia, elevated blood pressure, and hyperglycemia. The surgeon requests the hospitalist’s advice on appropriate medical interventions of these conditions. How should the hospitalist report the initial encounter with this Medicare beneficiary?
The hospitalist should select the CPT code that best fits the service and the payor. While most physicians regard this requested service as an inpatient consultation (99251-99255), Medicare no longer recognizes those codes. Instead, the hospitalist should report this encounter as an initial hospital care service (99221-99223).
Comanagement Issues
CMS and Medicare administrative contractors regularly uncover reporting errors for co-management requests. CMS decided the nature of these services were not consultative because the surgeon is not asking the physician or qualified nonphysician provider’s (NPP’s) opinion or advice for the surgeon’s use in treating the patient. Instead, these services constituted concurrent care and should have been billed using subsequent hospital care codes (99231-99233) in the hospital inpatient setting, subsequent NF care codes (99307-99310) in the SNF/NF setting, or office or other outpatient visit codes (99201-99215) in the office or outpatient settings.1
The new ruling simplifies coding and reduces reporting errors. The initial encounter with the patient is reported as such. Regardless of who is the attending of record or the consultant, the first physician from a particular provider group reports initial hospital care codes (i.e., 99221-99223) to represent the first patient encounter, even when this encounter does not occur on the admission date. Other physicians of the same specialty within the same provider group will not be permitted to report initial hospital care codes for their own initial encounter if someone from the group and specialty has already seen the patient during that hospitalization. In other words, the first hospitalist in the provider group reports 9922x, while the remaining hospitalists use subsequent hospital care codes (9923x).
In order to differentiate “consultant” services from “attending” services, CMS will be creating a modifier. The anticipated “AI” modifier must be appended to the attending physician’s initial encounter. Other initial hospital care codes reported throughout the hospital stay, as appropriate, are presumed to be that of “consultants” (i.e., physicians with a different specialty designation than the attending physician) participating in the case. Therefore, the hospitalist now can rightfully recover the increased work effort of the initial patient encounter (99223: 3.79 relative value units, ~$147 vs. 99233: 2.0 relative value units, ~$78, based on 2010 Medicare rates). Physicians will be required to meet the minimum documentation required for the selected visit code.
Other and Undefined Service Locations
Consultations in nursing facilities are handled much like inpatient hospital care. Physicians should report initial nursing facility services (99203-99306) for the first patient encounter, and subsequent nursing facility care codes (99307-99310) for each encounter thereafter. The attending physician of record appends the assigned modifier (presumed to be “AI”) when submitting their initial care service. All other initial care codes are presumed to be those of “consulting” physicians.
Initial information from CMS does not address observation services. Logically, these hospital-based services would follow the same methodology as inpatient care: report initial observation care (99218-99220) for the first “consulting” encounter. However, this might not be appropriate given Medicare’s existing rules for observation services, which guide physicians other than the admitting physician/group to “bill the office and other outpatient service codes or outpatient consultation codes as appropriate when they provide services to the patient.”2 With Medicare’s elimination of consultation codes, the consultant reports “office and other outpatient service codes” (i.e., new patient, 99201-99205, or established patient codes, 99212-99215) by default.
Without further clarification on observation services, hospitalists should report new or established patient service codes, depending on whether the patient has been seen by a group member within the last three years.
Medicare also has existing guidelines for the ED, which suggest that any physician not meeting the consultation criteria report ED service codes (99281-99285). Without further clarification, hospitalists should continue to follow this instruction for Medicare beneficiaries.
Nonphysician Providers
Medicare’s split/shared billing guidelines apply to most hospital inpatient, hospital outpatient, and ED evaluation and management (E/M) services, with consultations as one exception. Now, in accordance with the new ruling, hospitalists should select the appropriate initial service codes that correspond to patient’s location (e.g., 99223 for inpatients). NPPs can participate in the initial service provided to patients in these locations without the hospitalist having to replicate the entire service. The hospitalist can submit the claim in their name after selecting the visit level based upon the cumulative service personally provided on the same calendar day by both the NPP and the physician. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She also is faculty for SHM’s inpatient coding course.
References
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.10I. Centers for Medicare and Medicaid Services Web site. Available at: www.cms.hhs.gov/ manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- Medicare Claims Processing Manual: Chapter 12, Section 30.6.8A. CMS Web site. Available at: www. cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed Nov. 14, 2009.
- PFS Federal Regulation Notices: Proposed Revisions to Payment Policies Under the Physician Fee Schedule and Part B for CY 2010. CMS Web site. Available at: www.cms.hhs.gov/PhysicianFeeSched/PFSFRN/itemdetail.asp?filterType=none&filterByDID=99&sortByDID=4&sortOrder=descending&itemID=CMS1223902&intNumPerPage=10. Accessed Nov. 12, 2009.
- Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B (for CY 2010). CMS Web site. Available at: www.federalregister.gov/OFR Upload/OFRData/2009-26502_PI.pdf. Accessed Nov. 10, 2009.
The Bottom Line
The elimination of consults will affect physician group payments as well as RVU-based incentive payments to individual physicians. HM group leaders will want to refer to the CMS formula, also known as a crosswalk. The crosswalk outlines the utilization of consult codes and E/M codes for the sole purpose of establishing aggregate budget neutrality.3
The payment differential for consultations has been redistributed to increase payments for existing E/M services.
Here’s how it works: Use the “mapping percentage” column to project the distribution of consultative services between the “replacement” (i.e., “destination”) codes. For example, presumably 70% of 99251 will now be reported as initial hospital care (99221), whereas 30% will be reported as initial nursing facility care (99304). Please note that this table does not represent billing guidance or documentation equivalencies, but it does attempt to quantify sites of consultative service and their corresponding code categories for the basis of fiscal-year projections.
Physicians should select the visit code in the appropriate code category for the service location and in accordance with CMS documentation guidelines, which are available at www.cms.hhs.gov/MLNEdWebGuide/25_EMDOC.asp.
What Is the Appropriate Evaluation and Treatment of Funguria?
Case
A previously healthy 74-year-old female was admitted to the ICU nine days ago for treatment of severe streptococcal pneumonia. Her initial urine culture, which was collected on hospital day one, showed no growth; however, Candida albicans was isolated from a urine culture collected seven days later. Her second urinalysis revealed mild pyuria. What is the appropriate evaluation and treatment of funguria?
Background
Funguria is a relatively common clinical finding, and it is considerably more prevalent in patients with severe illnesses compared with healthy individuals. One study found that 2.2% of healthy, community-dwelling patients have Candida species (spp) in their urine.1 Candida spp are opportunistic organisms, which is implied by the fact that they can be isolated from 22% of patients admitted to an ICU.2
Despite the frequent isolation of Candida spp from urine cultures, the clinical significance is often unclear. It is difficult to determine if the funguria is caused by contamination, colonization, or a true urinary tract infection (UTI)—there is no test to reliably differentiate between these three possibilities. This is in contrast to bacterial UTIs, in which the findings of pyuria, bacteriuria, and a defined number of colony-forming units strongly support this diagnosis.3
Since it often is difficult to determine the true importance of funguria, its treatment has been controversial.3 The presence of a chronically indwelling urinary catheter often results in the funguria development, and, in many instances, simply removing the catheter will lead to its resolution.4 Furthermore, it has been demonstrated that for most patients with asymptomatic funguria, treatment with antifungal therapy has no effect on morbidity or mortality.5,6 Also, the propensity for funguria recurrence after completion of a course of antifungal therapy often discourages clinicians from ordering pharmacologic therapy.7,8
Review of the Data
The prevalence of funguria is increasing worldwide, primarily due to the increased use of antibiotics and immunosuppressive therapy, as well as the more frequent utilization of invasive procedures.1,7 Candida spp cause as many as 30% of all nosocomial UTIs, and they are most commonly isolated from patients who require ICU treatment.9 In fact, in one large study, only 10.9% of 861 patients with funguria had no underlying illnesses.10
Common risk factors for funguria development include the use of urinary tract drainage devices, hyperalimentation, steroids, recent antibiotic therapy, diabetes mellitus, increased age, urinary tract abnormalities, female sex, malignancy, and a previous surgical procedure.1,2,3,7,10,11,12,13,14
By far, the most common cause of funguria is Candida spp. C. albicans is responsible for at least 50% of all cases of funguria.1,10 Other yeasts that cause funguria include C. glabrata (15.6%), C. tropicalis (7.9%), C. parapsilosis (4.1%), and C. krusei (1%).10
C. glabrata most often is isolated from individuals who have been treated with fluconazole, while C. parapsilos is seen most frequently in neonates. It is noteworthy that for approximately 10% of patients with candiduria, at least two types of Candida spp are isolated from the same urine culture.6,7,14 Other types of fungi that are infrequently isolated from the urine include Aspergillus, Cryptococcus, Fusarium, Trichosporon, and such dimorphic fungi as Histoplasma capsulatum and Coccidioides immitis.6 The latter organisms tend to cause funguria in individuals who have a disseminated fungal infection.
Although funguria is a relatively common finding in some patient populations, there is uncertainty about its importance. This is because funguria might be present for one of several reasons, and differentiating colonization from true infection is difficult. Unfortunately, there are no established criteria that reliably differentiate the two entities.3 Most patients with funguria have no symptoms suggestive of a UTI (e.g., dysuria, suprapubic tenderness, or hematuria).1,6,7,10
For bacterial UTIs, the presence of pyuria, and a defined minimum number of colony-forming units (CFUs), is helpful in establishing this diagnosis.1,2,3,6 However, with funguria, neither of these parameters is helpful in distinguishing between colonization, contamination, or a true UTI. The reason is that pyuria commonly develops as a result of either coexistent bacteriuria or local irritation caused by the presence of an indwelling urinary catheter.1,7 Large numbers of fungal CFUs might indicate colonization only, which has no clinical significance.6,14
Candida spp often live as saprophytes on the skin in the genital and perineal areas.13 Women have a 10% to 65% rate of colonization of the vulvovestibular area with Candida spp.7 This readily allows for contamination of urine specimens during the collection process, and facilitates the introduction of organisms into the urinary bladder, particularly through the use of indwelling urinary catheters.6
Colonization of indwelling urinary devices universally occurs, as long as they remain inserted for substantial periods of time.6 Funguria is commonly observed with the use of either urethral or suprapubic catheters.4,7 Fortunately, intermittent urinary catheterization rarely is associated with the development of funguria.15 Additional substrates for the development of colonization include ureteral stents and nephrostomy tubes.7,14
UTIs due to fungi can present in several ways, including asymptomatic funguria, lower-tract UTIs, upper-tract UTIs, and renal candidiasis.7 Asymptomatic funguria is most commonly found in hospitalized patients who have indwelling urinary catheterization devices. Fungal lower-tract UTIs (i.e., cystitis) resulting in symptoms are uncommon in both catheterized and noncatheterized patients.15 Fungal upper-tract UTIs, which usually manifest as pyelonephritis or sepsis due to a UTI, cannot be distinguished from those with a bacterial etiology because their clinical presentations are similar.7 Upper-tract UTIs tend to occur in patients who have either urinary obstruction or a disorder that results in urinary stasis.7 Fungal balls (bezoars) might develop as a serious complication of an upper UTI, and can result in obstruction. Renal candidiasis usually develops as a result of hematogenous dissemination of a fungal infection.7,14
Funguria generally does not predispose to the development of fungemia, but when it does occur, it usually is due to the presence of an upper-urinary-tract obstruction.3,4,6,8,12,14
It has been estimated that disseminated infection can be expected to occur in 1.3% to 10.5% of immunosuppressed patients with funguria.1,3,6,10,15
Until fairly recently, it was thought that renal transplant patients with funguria were at increased risk for developing fungemia, but this assertion is now known to be false.5
In deciding whether to treat funguria, it is important to consider the clinical setting in which it occurs. For example, when funguria occurs in asymptomatic patients with an indwelling urinary catheter, it often is due to colonization of the catheter. In such instances, simply removing the catheter will result in the resolution of between 33% and 40% of funguria cases.1,12 For most patients with asymptomatic funguria, it has been shown that the administration of antifungal therapy has no significant effect on morbidity or mortality.6,16
However, for certain patient populations who are at increased risk for developing a disseminated fungal infection, the treatment of asymptomatic funguria is indicated. This includes neutropenic patients, those with a known urologic obstruction, and those who will undergo a urologic procedure.7 It also is important to recognize that for oncology patients, or those with sepsis, funguria might be the only manifestation of a disseminated fungal infection.6
All patients with symptomatic funguria require treatment with antifungal therapy.6 Septic patients who have funguria require blood cultures and radiologic imaging studies; the latter are obtained in order to localize the anatomic source of infection, and also to evaluate for urinary obstruction.6 Such patients require the prompt administration of appropriate systemic antifungal therapy; failure to do so doubles the risk of in-hospital mortality.2
Fluconazole is the most utilized medication for funguria treatment. Unlike itraconazole, ketoconazole, and voriconazole, it achieves high concentrations in the urine.7,12 The efficacy of Capsofungin for the treatment of funguria has not been established firmly.14,17 Flucytosine has a limited role in the treatment of funguria, but it is very useful in the treatment of non-C. albicans species, which are increasing in frequency and often are resistant to fluconazole.6,14
Amphotericin B bladder irrigation no longer is recognized as a first-line treatment for candiduria, although some investigators still support its use, particularly in special circumstances.15 With the ready availability of an oral agent, IV amphotericin B is not commonly utilized for the treatment of asymptomatic funguria. However, either IV amphotericin B or IV fluconazole are options for the treatment of renal candidiasis.3,7 Unfortunately, the recurrence of funguria after the completion of an appropriate course of antifungal therapy commonly occurs.3,6,8,14,15
Back to the Case
The patient’s initial blood and sputum cultures grew Streptococcus pneumoniae, which was adequately covered by the initial treatments of piperacillin/tazobactam and levofloxacin at the time of admission. Although the patient’s clinical condition improved gradually, she required ICU management throughout her hospital stay. Due to her poor mobility, an indwelling urinary catheter was inserted. The placement of this catheter, along with her age, sex, current antibiotic therapy, and debility, all increased her likelihood of developing funguria.
It is noteworthy that the patient had no suprapubic tenderness, and she had been afebrile for the preceeding 48 hours.
The finding of funguria in this patient should not create undue concern. The true source of her acute illness (Streptococcus pneumoniae) was identified. In this instance, there would be no expected benefit from the initiation of antifungal therapy. Instead, removal of the indwelling urinary drainage device would be advisable.
Bottom Line
Asymptomatic funguria is a common clinical finding, one in which further workups or the administration of antifungal therapy is not necessary in most cases. Symptomatic funguria always requires treatment. TH
Dr. Clarke is a clinical instructor in the section of hospital medicine at Emory University Medical Center in Atlanta. Dr. Razavi is an assistant professor in the section of hospital medicine at Emory.
References
- Colodner R, Nuri Y, Chazan B, Raz R. Community-acquired and hospital-acquired candiduria: comparison of prevalence and clinical characteristics. Eur J Clin Microbiol Infect Dis. 2008;27:301-305.
- Blot S, Dimopoulos G, Rello J, Vogelaers D. Is Candida really a threat in the ICU? Curr Opin Crit Care. 2008;14:600-604.
- Kauffman, CA. Candiduria. Clin Infect Dis. 2005;41:S371-376.
- Goetz LL, Howard M, Cipher D, Revankar SG. Occurrence of candiduria in a population of chronically catheterized patients with spinal cord injury. Spinal Cord. 2009;doi:10.1038/SC.2009.81.
- Safdar N, Slattery WR, Knasinski V, et al. Predictors and outcomes of candiduria in renal transplant recipients. Clin Infect Dis. 2005;40:1413-1421.
- Hollenbach E. To treat or not to treat—critically ill patients with candiduria. Mycoses. 2008;51(Suppl2):12-24.
- Lundstrom T, Sobel J. Nosocomial candiduria: a review. Clin Infect Dis. 2001;32:1602-1607.
- Sobel JD, Kauffman CA, McKinsey D, et al. Candiduria: A randomized, double-blind study of treatment with Fluconazole and placebo. Clin Infect Dis. 2000;30:19-24.
- Chen SC, Tong ZS, Lee OC, et al. Clinician response to Candida organisms in the urine of patients attending hospital. Eur J Clin Microbiol Infect Dis. 2008;27:201-208.
- Kauffman CA, Vazquez JA, Sobel JD, et al. Prospective multicenter surveillance study of funguria in hospitalized patients. Clin Infect Dis. 2000;30:14-18.
- Gubbins PO, McConnell SA, Penzak SR. Current management of funguria. Am J Health Syst Pharm. 1999;56(19):1929-1935.
- Drew RH, Arthur RR, Perfect JR. Is it time to abandon the use of amphotericin B bladder irrigation? Clin Infect Dis. 2005;40:1465-1470.
- Bromberg WD. How do UTIs due to Candida differ from other infections? Cortlandt Forum. 1998;11(2):210.
- Bukhary ZA. Candiduria: a review of clinical significance and management. Saudi J Kidney Dis Transplant. 2008;19(3):350-360.
- Tuon FF, Amato VS, Filho SR. Bladder irrigation with amphotericin B and fungal urinary tract infection—systematic review with meta-analysis. Int J infect Dis. 2009;13(6):701-706.
- Simpson C, Blitz S, Shafran SD. The effect of current management on morbidity and mortality in hospitalized adults with funguria. J Infect. 2004;49(3):248-252.
- JD, Bradshaw SK, Lipka CJ, Kartsonis NA. Capsofungin in the treatment of symptomatic candiduria. Clin Infect Dis. 2007;44:e46.
Case
A previously healthy 74-year-old female was admitted to the ICU nine days ago for treatment of severe streptococcal pneumonia. Her initial urine culture, which was collected on hospital day one, showed no growth; however, Candida albicans was isolated from a urine culture collected seven days later. Her second urinalysis revealed mild pyuria. What is the appropriate evaluation and treatment of funguria?
Background
Funguria is a relatively common clinical finding, and it is considerably more prevalent in patients with severe illnesses compared with healthy individuals. One study found that 2.2% of healthy, community-dwelling patients have Candida species (spp) in their urine.1 Candida spp are opportunistic organisms, which is implied by the fact that they can be isolated from 22% of patients admitted to an ICU.2
Despite the frequent isolation of Candida spp from urine cultures, the clinical significance is often unclear. It is difficult to determine if the funguria is caused by contamination, colonization, or a true urinary tract infection (UTI)—there is no test to reliably differentiate between these three possibilities. This is in contrast to bacterial UTIs, in which the findings of pyuria, bacteriuria, and a defined number of colony-forming units strongly support this diagnosis.3
Since it often is difficult to determine the true importance of funguria, its treatment has been controversial.3 The presence of a chronically indwelling urinary catheter often results in the funguria development, and, in many instances, simply removing the catheter will lead to its resolution.4 Furthermore, it has been demonstrated that for most patients with asymptomatic funguria, treatment with antifungal therapy has no effect on morbidity or mortality.5,6 Also, the propensity for funguria recurrence after completion of a course of antifungal therapy often discourages clinicians from ordering pharmacologic therapy.7,8
Review of the Data
The prevalence of funguria is increasing worldwide, primarily due to the increased use of antibiotics and immunosuppressive therapy, as well as the more frequent utilization of invasive procedures.1,7 Candida spp cause as many as 30% of all nosocomial UTIs, and they are most commonly isolated from patients who require ICU treatment.9 In fact, in one large study, only 10.9% of 861 patients with funguria had no underlying illnesses.10
Common risk factors for funguria development include the use of urinary tract drainage devices, hyperalimentation, steroids, recent antibiotic therapy, diabetes mellitus, increased age, urinary tract abnormalities, female sex, malignancy, and a previous surgical procedure.1,2,3,7,10,11,12,13,14
By far, the most common cause of funguria is Candida spp. C. albicans is responsible for at least 50% of all cases of funguria.1,10 Other yeasts that cause funguria include C. glabrata (15.6%), C. tropicalis (7.9%), C. parapsilosis (4.1%), and C. krusei (1%).10
C. glabrata most often is isolated from individuals who have been treated with fluconazole, while C. parapsilos is seen most frequently in neonates. It is noteworthy that for approximately 10% of patients with candiduria, at least two types of Candida spp are isolated from the same urine culture.6,7,14 Other types of fungi that are infrequently isolated from the urine include Aspergillus, Cryptococcus, Fusarium, Trichosporon, and such dimorphic fungi as Histoplasma capsulatum and Coccidioides immitis.6 The latter organisms tend to cause funguria in individuals who have a disseminated fungal infection.
Although funguria is a relatively common finding in some patient populations, there is uncertainty about its importance. This is because funguria might be present for one of several reasons, and differentiating colonization from true infection is difficult. Unfortunately, there are no established criteria that reliably differentiate the two entities.3 Most patients with funguria have no symptoms suggestive of a UTI (e.g., dysuria, suprapubic tenderness, or hematuria).1,6,7,10
For bacterial UTIs, the presence of pyuria, and a defined minimum number of colony-forming units (CFUs), is helpful in establishing this diagnosis.1,2,3,6 However, with funguria, neither of these parameters is helpful in distinguishing between colonization, contamination, or a true UTI. The reason is that pyuria commonly develops as a result of either coexistent bacteriuria or local irritation caused by the presence of an indwelling urinary catheter.1,7 Large numbers of fungal CFUs might indicate colonization only, which has no clinical significance.6,14
Candida spp often live as saprophytes on the skin in the genital and perineal areas.13 Women have a 10% to 65% rate of colonization of the vulvovestibular area with Candida spp.7 This readily allows for contamination of urine specimens during the collection process, and facilitates the introduction of organisms into the urinary bladder, particularly through the use of indwelling urinary catheters.6
Colonization of indwelling urinary devices universally occurs, as long as they remain inserted for substantial periods of time.6 Funguria is commonly observed with the use of either urethral or suprapubic catheters.4,7 Fortunately, intermittent urinary catheterization rarely is associated with the development of funguria.15 Additional substrates for the development of colonization include ureteral stents and nephrostomy tubes.7,14
UTIs due to fungi can present in several ways, including asymptomatic funguria, lower-tract UTIs, upper-tract UTIs, and renal candidiasis.7 Asymptomatic funguria is most commonly found in hospitalized patients who have indwelling urinary catheterization devices. Fungal lower-tract UTIs (i.e., cystitis) resulting in symptoms are uncommon in both catheterized and noncatheterized patients.15 Fungal upper-tract UTIs, which usually manifest as pyelonephritis or sepsis due to a UTI, cannot be distinguished from those with a bacterial etiology because their clinical presentations are similar.7 Upper-tract UTIs tend to occur in patients who have either urinary obstruction or a disorder that results in urinary stasis.7 Fungal balls (bezoars) might develop as a serious complication of an upper UTI, and can result in obstruction. Renal candidiasis usually develops as a result of hematogenous dissemination of a fungal infection.7,14
Funguria generally does not predispose to the development of fungemia, but when it does occur, it usually is due to the presence of an upper-urinary-tract obstruction.3,4,6,8,12,14
It has been estimated that disseminated infection can be expected to occur in 1.3% to 10.5% of immunosuppressed patients with funguria.1,3,6,10,15
Until fairly recently, it was thought that renal transplant patients with funguria were at increased risk for developing fungemia, but this assertion is now known to be false.5
In deciding whether to treat funguria, it is important to consider the clinical setting in which it occurs. For example, when funguria occurs in asymptomatic patients with an indwelling urinary catheter, it often is due to colonization of the catheter. In such instances, simply removing the catheter will result in the resolution of between 33% and 40% of funguria cases.1,12 For most patients with asymptomatic funguria, it has been shown that the administration of antifungal therapy has no significant effect on morbidity or mortality.6,16
However, for certain patient populations who are at increased risk for developing a disseminated fungal infection, the treatment of asymptomatic funguria is indicated. This includes neutropenic patients, those with a known urologic obstruction, and those who will undergo a urologic procedure.7 It also is important to recognize that for oncology patients, or those with sepsis, funguria might be the only manifestation of a disseminated fungal infection.6
All patients with symptomatic funguria require treatment with antifungal therapy.6 Septic patients who have funguria require blood cultures and radiologic imaging studies; the latter are obtained in order to localize the anatomic source of infection, and also to evaluate for urinary obstruction.6 Such patients require the prompt administration of appropriate systemic antifungal therapy; failure to do so doubles the risk of in-hospital mortality.2
Fluconazole is the most utilized medication for funguria treatment. Unlike itraconazole, ketoconazole, and voriconazole, it achieves high concentrations in the urine.7,12 The efficacy of Capsofungin for the treatment of funguria has not been established firmly.14,17 Flucytosine has a limited role in the treatment of funguria, but it is very useful in the treatment of non-C. albicans species, which are increasing in frequency and often are resistant to fluconazole.6,14
Amphotericin B bladder irrigation no longer is recognized as a first-line treatment for candiduria, although some investigators still support its use, particularly in special circumstances.15 With the ready availability of an oral agent, IV amphotericin B is not commonly utilized for the treatment of asymptomatic funguria. However, either IV amphotericin B or IV fluconazole are options for the treatment of renal candidiasis.3,7 Unfortunately, the recurrence of funguria after the completion of an appropriate course of antifungal therapy commonly occurs.3,6,8,14,15
Back to the Case
The patient’s initial blood and sputum cultures grew Streptococcus pneumoniae, which was adequately covered by the initial treatments of piperacillin/tazobactam and levofloxacin at the time of admission. Although the patient’s clinical condition improved gradually, she required ICU management throughout her hospital stay. Due to her poor mobility, an indwelling urinary catheter was inserted. The placement of this catheter, along with her age, sex, current antibiotic therapy, and debility, all increased her likelihood of developing funguria.
It is noteworthy that the patient had no suprapubic tenderness, and she had been afebrile for the preceeding 48 hours.
The finding of funguria in this patient should not create undue concern. The true source of her acute illness (Streptococcus pneumoniae) was identified. In this instance, there would be no expected benefit from the initiation of antifungal therapy. Instead, removal of the indwelling urinary drainage device would be advisable.
Bottom Line
Asymptomatic funguria is a common clinical finding, one in which further workups or the administration of antifungal therapy is not necessary in most cases. Symptomatic funguria always requires treatment. TH
Dr. Clarke is a clinical instructor in the section of hospital medicine at Emory University Medical Center in Atlanta. Dr. Razavi is an assistant professor in the section of hospital medicine at Emory.
References
- Colodner R, Nuri Y, Chazan B, Raz R. Community-acquired and hospital-acquired candiduria: comparison of prevalence and clinical characteristics. Eur J Clin Microbiol Infect Dis. 2008;27:301-305.
- Blot S, Dimopoulos G, Rello J, Vogelaers D. Is Candida really a threat in the ICU? Curr Opin Crit Care. 2008;14:600-604.
- Kauffman, CA. Candiduria. Clin Infect Dis. 2005;41:S371-376.
- Goetz LL, Howard M, Cipher D, Revankar SG. Occurrence of candiduria in a population of chronically catheterized patients with spinal cord injury. Spinal Cord. 2009;doi:10.1038/SC.2009.81.
- Safdar N, Slattery WR, Knasinski V, et al. Predictors and outcomes of candiduria in renal transplant recipients. Clin Infect Dis. 2005;40:1413-1421.
- Hollenbach E. To treat or not to treat—critically ill patients with candiduria. Mycoses. 2008;51(Suppl2):12-24.
- Lundstrom T, Sobel J. Nosocomial candiduria: a review. Clin Infect Dis. 2001;32:1602-1607.
- Sobel JD, Kauffman CA, McKinsey D, et al. Candiduria: A randomized, double-blind study of treatment with Fluconazole and placebo. Clin Infect Dis. 2000;30:19-24.
- Chen SC, Tong ZS, Lee OC, et al. Clinician response to Candida organisms in the urine of patients attending hospital. Eur J Clin Microbiol Infect Dis. 2008;27:201-208.
- Kauffman CA, Vazquez JA, Sobel JD, et al. Prospective multicenter surveillance study of funguria in hospitalized patients. Clin Infect Dis. 2000;30:14-18.
- Gubbins PO, McConnell SA, Penzak SR. Current management of funguria. Am J Health Syst Pharm. 1999;56(19):1929-1935.
- Drew RH, Arthur RR, Perfect JR. Is it time to abandon the use of amphotericin B bladder irrigation? Clin Infect Dis. 2005;40:1465-1470.
- Bromberg WD. How do UTIs due to Candida differ from other infections? Cortlandt Forum. 1998;11(2):210.
- Bukhary ZA. Candiduria: a review of clinical significance and management. Saudi J Kidney Dis Transplant. 2008;19(3):350-360.
- Tuon FF, Amato VS, Filho SR. Bladder irrigation with amphotericin B and fungal urinary tract infection—systematic review with meta-analysis. Int J infect Dis. 2009;13(6):701-706.
- Simpson C, Blitz S, Shafran SD. The effect of current management on morbidity and mortality in hospitalized adults with funguria. J Infect. 2004;49(3):248-252.
- JD, Bradshaw SK, Lipka CJ, Kartsonis NA. Capsofungin in the treatment of symptomatic candiduria. Clin Infect Dis. 2007;44:e46.
Case
A previously healthy 74-year-old female was admitted to the ICU nine days ago for treatment of severe streptococcal pneumonia. Her initial urine culture, which was collected on hospital day one, showed no growth; however, Candida albicans was isolated from a urine culture collected seven days later. Her second urinalysis revealed mild pyuria. What is the appropriate evaluation and treatment of funguria?
Background
Funguria is a relatively common clinical finding, and it is considerably more prevalent in patients with severe illnesses compared with healthy individuals. One study found that 2.2% of healthy, community-dwelling patients have Candida species (spp) in their urine.1 Candida spp are opportunistic organisms, which is implied by the fact that they can be isolated from 22% of patients admitted to an ICU.2
Despite the frequent isolation of Candida spp from urine cultures, the clinical significance is often unclear. It is difficult to determine if the funguria is caused by contamination, colonization, or a true urinary tract infection (UTI)—there is no test to reliably differentiate between these three possibilities. This is in contrast to bacterial UTIs, in which the findings of pyuria, bacteriuria, and a defined number of colony-forming units strongly support this diagnosis.3
Since it often is difficult to determine the true importance of funguria, its treatment has been controversial.3 The presence of a chronically indwelling urinary catheter often results in the funguria development, and, in many instances, simply removing the catheter will lead to its resolution.4 Furthermore, it has been demonstrated that for most patients with asymptomatic funguria, treatment with antifungal therapy has no effect on morbidity or mortality.5,6 Also, the propensity for funguria recurrence after completion of a course of antifungal therapy often discourages clinicians from ordering pharmacologic therapy.7,8
Review of the Data
The prevalence of funguria is increasing worldwide, primarily due to the increased use of antibiotics and immunosuppressive therapy, as well as the more frequent utilization of invasive procedures.1,7 Candida spp cause as many as 30% of all nosocomial UTIs, and they are most commonly isolated from patients who require ICU treatment.9 In fact, in one large study, only 10.9% of 861 patients with funguria had no underlying illnesses.10
Common risk factors for funguria development include the use of urinary tract drainage devices, hyperalimentation, steroids, recent antibiotic therapy, diabetes mellitus, increased age, urinary tract abnormalities, female sex, malignancy, and a previous surgical procedure.1,2,3,7,10,11,12,13,14
By far, the most common cause of funguria is Candida spp. C. albicans is responsible for at least 50% of all cases of funguria.1,10 Other yeasts that cause funguria include C. glabrata (15.6%), C. tropicalis (7.9%), C. parapsilosis (4.1%), and C. krusei (1%).10
C. glabrata most often is isolated from individuals who have been treated with fluconazole, while C. parapsilos is seen most frequently in neonates. It is noteworthy that for approximately 10% of patients with candiduria, at least two types of Candida spp are isolated from the same urine culture.6,7,14 Other types of fungi that are infrequently isolated from the urine include Aspergillus, Cryptococcus, Fusarium, Trichosporon, and such dimorphic fungi as Histoplasma capsulatum and Coccidioides immitis.6 The latter organisms tend to cause funguria in individuals who have a disseminated fungal infection.
Although funguria is a relatively common finding in some patient populations, there is uncertainty about its importance. This is because funguria might be present for one of several reasons, and differentiating colonization from true infection is difficult. Unfortunately, there are no established criteria that reliably differentiate the two entities.3 Most patients with funguria have no symptoms suggestive of a UTI (e.g., dysuria, suprapubic tenderness, or hematuria).1,6,7,10
For bacterial UTIs, the presence of pyuria, and a defined minimum number of colony-forming units (CFUs), is helpful in establishing this diagnosis.1,2,3,6 However, with funguria, neither of these parameters is helpful in distinguishing between colonization, contamination, or a true UTI. The reason is that pyuria commonly develops as a result of either coexistent bacteriuria or local irritation caused by the presence of an indwelling urinary catheter.1,7 Large numbers of fungal CFUs might indicate colonization only, which has no clinical significance.6,14
Candida spp often live as saprophytes on the skin in the genital and perineal areas.13 Women have a 10% to 65% rate of colonization of the vulvovestibular area with Candida spp.7 This readily allows for contamination of urine specimens during the collection process, and facilitates the introduction of organisms into the urinary bladder, particularly through the use of indwelling urinary catheters.6
Colonization of indwelling urinary devices universally occurs, as long as they remain inserted for substantial periods of time.6 Funguria is commonly observed with the use of either urethral or suprapubic catheters.4,7 Fortunately, intermittent urinary catheterization rarely is associated with the development of funguria.15 Additional substrates for the development of colonization include ureteral stents and nephrostomy tubes.7,14
UTIs due to fungi can present in several ways, including asymptomatic funguria, lower-tract UTIs, upper-tract UTIs, and renal candidiasis.7 Asymptomatic funguria is most commonly found in hospitalized patients who have indwelling urinary catheterization devices. Fungal lower-tract UTIs (i.e., cystitis) resulting in symptoms are uncommon in both catheterized and noncatheterized patients.15 Fungal upper-tract UTIs, which usually manifest as pyelonephritis or sepsis due to a UTI, cannot be distinguished from those with a bacterial etiology because their clinical presentations are similar.7 Upper-tract UTIs tend to occur in patients who have either urinary obstruction or a disorder that results in urinary stasis.7 Fungal balls (bezoars) might develop as a serious complication of an upper UTI, and can result in obstruction. Renal candidiasis usually develops as a result of hematogenous dissemination of a fungal infection.7,14
Funguria generally does not predispose to the development of fungemia, but when it does occur, it usually is due to the presence of an upper-urinary-tract obstruction.3,4,6,8,12,14
It has been estimated that disseminated infection can be expected to occur in 1.3% to 10.5% of immunosuppressed patients with funguria.1,3,6,10,15
Until fairly recently, it was thought that renal transplant patients with funguria were at increased risk for developing fungemia, but this assertion is now known to be false.5
In deciding whether to treat funguria, it is important to consider the clinical setting in which it occurs. For example, when funguria occurs in asymptomatic patients with an indwelling urinary catheter, it often is due to colonization of the catheter. In such instances, simply removing the catheter will result in the resolution of between 33% and 40% of funguria cases.1,12 For most patients with asymptomatic funguria, it has been shown that the administration of antifungal therapy has no significant effect on morbidity or mortality.6,16
However, for certain patient populations who are at increased risk for developing a disseminated fungal infection, the treatment of asymptomatic funguria is indicated. This includes neutropenic patients, those with a known urologic obstruction, and those who will undergo a urologic procedure.7 It also is important to recognize that for oncology patients, or those with sepsis, funguria might be the only manifestation of a disseminated fungal infection.6
All patients with symptomatic funguria require treatment with antifungal therapy.6 Septic patients who have funguria require blood cultures and radiologic imaging studies; the latter are obtained in order to localize the anatomic source of infection, and also to evaluate for urinary obstruction.6 Such patients require the prompt administration of appropriate systemic antifungal therapy; failure to do so doubles the risk of in-hospital mortality.2
Fluconazole is the most utilized medication for funguria treatment. Unlike itraconazole, ketoconazole, and voriconazole, it achieves high concentrations in the urine.7,12 The efficacy of Capsofungin for the treatment of funguria has not been established firmly.14,17 Flucytosine has a limited role in the treatment of funguria, but it is very useful in the treatment of non-C. albicans species, which are increasing in frequency and often are resistant to fluconazole.6,14
Amphotericin B bladder irrigation no longer is recognized as a first-line treatment for candiduria, although some investigators still support its use, particularly in special circumstances.15 With the ready availability of an oral agent, IV amphotericin B is not commonly utilized for the treatment of asymptomatic funguria. However, either IV amphotericin B or IV fluconazole are options for the treatment of renal candidiasis.3,7 Unfortunately, the recurrence of funguria after the completion of an appropriate course of antifungal therapy commonly occurs.3,6,8,14,15
Back to the Case
The patient’s initial blood and sputum cultures grew Streptococcus pneumoniae, which was adequately covered by the initial treatments of piperacillin/tazobactam and levofloxacin at the time of admission. Although the patient’s clinical condition improved gradually, she required ICU management throughout her hospital stay. Due to her poor mobility, an indwelling urinary catheter was inserted. The placement of this catheter, along with her age, sex, current antibiotic therapy, and debility, all increased her likelihood of developing funguria.
It is noteworthy that the patient had no suprapubic tenderness, and she had been afebrile for the preceeding 48 hours.
The finding of funguria in this patient should not create undue concern. The true source of her acute illness (Streptococcus pneumoniae) was identified. In this instance, there would be no expected benefit from the initiation of antifungal therapy. Instead, removal of the indwelling urinary drainage device would be advisable.
Bottom Line
Asymptomatic funguria is a common clinical finding, one in which further workups or the administration of antifungal therapy is not necessary in most cases. Symptomatic funguria always requires treatment. TH
Dr. Clarke is a clinical instructor in the section of hospital medicine at Emory University Medical Center in Atlanta. Dr. Razavi is an assistant professor in the section of hospital medicine at Emory.
References
- Colodner R, Nuri Y, Chazan B, Raz R. Community-acquired and hospital-acquired candiduria: comparison of prevalence and clinical characteristics. Eur J Clin Microbiol Infect Dis. 2008;27:301-305.
- Blot S, Dimopoulos G, Rello J, Vogelaers D. Is Candida really a threat in the ICU? Curr Opin Crit Care. 2008;14:600-604.
- Kauffman, CA. Candiduria. Clin Infect Dis. 2005;41:S371-376.
- Goetz LL, Howard M, Cipher D, Revankar SG. Occurrence of candiduria in a population of chronically catheterized patients with spinal cord injury. Spinal Cord. 2009;doi:10.1038/SC.2009.81.
- Safdar N, Slattery WR, Knasinski V, et al. Predictors and outcomes of candiduria in renal transplant recipients. Clin Infect Dis. 2005;40:1413-1421.
- Hollenbach E. To treat or not to treat—critically ill patients with candiduria. Mycoses. 2008;51(Suppl2):12-24.
- Lundstrom T, Sobel J. Nosocomial candiduria: a review. Clin Infect Dis. 2001;32:1602-1607.
- Sobel JD, Kauffman CA, McKinsey D, et al. Candiduria: A randomized, double-blind study of treatment with Fluconazole and placebo. Clin Infect Dis. 2000;30:19-24.
- Chen SC, Tong ZS, Lee OC, et al. Clinician response to Candida organisms in the urine of patients attending hospital. Eur J Clin Microbiol Infect Dis. 2008;27:201-208.
- Kauffman CA, Vazquez JA, Sobel JD, et al. Prospective multicenter surveillance study of funguria in hospitalized patients. Clin Infect Dis. 2000;30:14-18.
- Gubbins PO, McConnell SA, Penzak SR. Current management of funguria. Am J Health Syst Pharm. 1999;56(19):1929-1935.
- Drew RH, Arthur RR, Perfect JR. Is it time to abandon the use of amphotericin B bladder irrigation? Clin Infect Dis. 2005;40:1465-1470.
- Bromberg WD. How do UTIs due to Candida differ from other infections? Cortlandt Forum. 1998;11(2):210.
- Bukhary ZA. Candiduria: a review of clinical significance and management. Saudi J Kidney Dis Transplant. 2008;19(3):350-360.
- Tuon FF, Amato VS, Filho SR. Bladder irrigation with amphotericin B and fungal urinary tract infection—systematic review with meta-analysis. Int J infect Dis. 2009;13(6):701-706.
- Simpson C, Blitz S, Shafran SD. The effect of current management on morbidity and mortality in hospitalized adults with funguria. J Infect. 2004;49(3):248-252.
- JD, Bradshaw SK, Lipka CJ, Kartsonis NA. Capsofungin in the treatment of symptomatic candiduria. Clin Infect Dis. 2007;44:e46.
Market Watch
New Generics
- Tamsulosin (generic Flomax) capsules should be available March 2, 20101
New Drugs, Indications, Dosage Forms, and Approvals
- C1 Esterase inhibitor [human] (Berinert) has been approved by the Food and Drug Administration (FDA) to treat acute abdominal attacks and facial edema associated with hereditary angioedema (HAE) in adolescents and adults. It is derived from human plasma and regulates clotting and inflammatory reactions. HAE, a genetic disorder affecting 6,000 to 10,000 Americans, is caused by a deficit of C1-INH.2
- Colesevelam HCl tablets (Welchol) have been approved by the FDA as an adjunct to diet and exercise for reducing LDL-C levels in boys and postmenarchal girls ages 10 to 17, with heterozygous familial hypercholesterolemia as monotherapy, or in combination with a statin after failing an adequate trial of diet therapy.3
- Colesevelam HCl (Welchol) has been approved by the FDA as an oral solution providing an alternate dosage form to the large oral tablets currently available.3
- Oxycodone HCl has been recommended for FDA approval. When this new formulation is dissolved in water, it forms a gel, which makes it difficult to abuse. If approved, the new formulation will keep the OxyContin name and will be available in seven dosages. The older product will be phased out and only the newer product will be available.4
- Peginterferon alpha-2b injection (PegIntron) has been recommended for FDA approval for the treatment of patients with stage-III malignant melanoma. Peginterferon alpha-2b currently is approved for treating hepatitis C in combination with ribavirin. It is a once-weekly, subcutaneous injection.5
- Ustekinumab (Stelara) has been approved by the FDA for treating moderate to severe plaque psoriasis by disabling two interleukin (IL) cytokines, IL-12 and IL-23.6 It is a monoclonal antibody administered via subcutaneous injection. Recommended dosing is a baseline injection followed by another injection at week four, followed by subsequent injections every 12 weeks.7 Serious infections have been reported in clinical trials. Therefore, the company has developed a Risk Evaluation and Mitigation Strategy (REMS), as well as targeted healthcare provider education and a patient guide. The product label also contains cautions related to potential immunosuppression, as well as information on avoiding live vaccines while being treated with the agent.
Pipeline
- Cladribine, originally approved by the FDA in an intravenous formulation in the 1990s to treat hairy cell leukemia, has been reformulated as an oral product to manage patients with multiple sclerosis.8 Merck has submitted oral cladribine as a disease-modifying therapy for multiple sclerosis. If approved, it will be the first oral disease modifying agent for treating multiple sclerosis patients.9
- Dapagliflozin, a new mechanism renal sodium-glucose co-transporter 2 (SGLT2) inhibitor, has been shown to reduce fasting plasma glucose and significantly reduce HbA1c levels in patients with Type 2 diabetes mellitus, compared with patients treated with a metformin and placebo combination.10,11 Additionally, about 25% of patients treated with dapagliflozin (vs. 6% of the placebo-metformin-treated patients) had at least a 5% decrease in body weight. Diastolic blood pressure and uric acid level also decreased but not significantly. Serious adverse events were similar between the two treated groups.
- Fingolimod, an oral, disease-modifying agent to treat multiple sclerosis, is the first in a new class of agents known as sphingosine 1-phosphate receptor modulators (S1P-R). A recent two-year study showed it significantly reduced both relapses and disability progression (compared with placebo) in patients with relapsing remitting multiple sclerosis.12,13 More information will be available on this agent.
- Naproxcinod is a cyclo-oxygenase-inhibiting nitric oxide donator (CINOD) anti-inflammatory agent.14 The proposed indication is for the relief of the signs and symptoms of osteoarthritis, predominantly for pain management.
Safety Information
- Promethazine injection has undergone a label change to include a boxed warning. The warning is to emphasize the risk of serious tissue injury when promethazine is incorrectly administered.15 The preferred route is deep intramuscular injection; if administered in or near a vein, severe tissue injury might occur. The FDA previously informed healthcare professionals about the risks of incorrectly administered promethazine in December 2006 and again in February 2008. Post-marketing adverse events reported from 1969 to 2009 have identified cases of gangrene requiring amputation associated with administration of injectable promethazine.
- Since its original FDA approval in October 2006, sitagliptin, the first oral dipeptidyl peptidase-4 (DPP-4) inhibitor, is undergoing a safety label change.16 There have been 88 post-marketing cases reported of acute pancreatitis, including two cases of necrotizing or hemorrhagic pancreatitis reported between Oct. 16, 2006, and Feb. 9, 2009.17 The updated labeling discusses more information on the pancreatitis cases reported, and recommends that healthcare professionals carefully monitor patients for the development of pancreatitis, either upon beginning therapy or around dose increases. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Japan’s Astellas settles with Impax on prostate drug. Reuters Web site. Available at: http://www.reuters.com/article/rbssPharmaceuticals%20-%20Generic%20&%20Specialty/idUST28251320091007. Accessed Oct. 13, 2009.
- FDA approves Berinert. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/csl-behring-announces-fda-approval-berinert-first-only-therapy-approved-acute-abdominal-facial-1681.html. Accessed Oct. 13, 2009.
- Welchol package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022362lbl.pdf. Accessed Oct. 13, 2009.
- Walker E. FDA Panel recommends approval of new oxycodone formulation. Medpage Today Web site. Available at: http://www.medpagetoday.com/Neurology/PainManagement/16132?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Todoruk M. FDA panel supports approval of Schering-Plough’s PegIntron for patients with melanoma. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=8E68078692384C8A8249EA4A4C036635&logRowId=330581. Accessed Oct. 13, 2009.
- Gever J, Agus ZS. FDA approves biologic drug for psoriasis. Medpage Today Web site. Available at: http://www.medpagetoday.com/InfectiousDisease/PublicHealth/16147?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Stelara package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125261lbl.pdf. Accessed Oct. 13, 2009.
- Leustatin package label. FDA Web site. Available at: http://www.cancerconsultants.com/druginserts/Cladribine.pdf. Accessed Oct. 13, 2009.
- Todoruk M. Merck KGaA files cladribine for FDA approval. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=08CF20114B2A4F1DA8FD33AAC7240AB8&logRowId=329839. Accessed Oct. 2, 2009.
- Dennis M. AstraZeneca, Bristol-Myers Squibb’s dapagliflozin meets endpoints in Phase III study. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=3B158484CA7A47B99CEA373CB2ABFFA2&logRowId=330336. Accessed Oct. 13, 2009.
- Gever J, Zaleznik DF, Caputo D. EASD: Phase III data look good for novel diabetes drug. Medpage Today Web site. Available at: http://www.medpagetoday.com/MeetingCoverage/EASD/16270. Accessed Oct. 11, 2009.
- Dennis M. Novartis: Phase III study shows oral MS drug fingolimod reduces relapses, disability progression. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=EDBA0E3366E2477DB714674E9C19821D&logRowId=329838. Accessed Oct. 13, 2009.
- Multiple sclerosis therapy FTY720 reduces relapses and disability progression. The Multiple Sclerosis Resource Centre Web site. Available at: http://www.msrc.co.uk/printable.cfm?pageid=1309. Accessed Oct. 13, 2009.
- NicOx submits New Drug Application (NDA) for naproxcinod to the US FDA. NicOx Web site. Available at: http://www.nicox.com/upload/PR_NDA_submission-250909__EN.pdf. Accessed Oct. 13, 2009.
- Riley K. FDA requires boxed warning for promethazine hydrochloride injection. FDA Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm182498.htm. Accessed Oct. 13, 2009.
- Januvia package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021995lbl.pdf. Accessed Oct. 13, 2009.
- Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). FDA Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed Sept. 26, 2009.
New Generics
- Tamsulosin (generic Flomax) capsules should be available March 2, 20101
New Drugs, Indications, Dosage Forms, and Approvals
- C1 Esterase inhibitor [human] (Berinert) has been approved by the Food and Drug Administration (FDA) to treat acute abdominal attacks and facial edema associated with hereditary angioedema (HAE) in adolescents and adults. It is derived from human plasma and regulates clotting and inflammatory reactions. HAE, a genetic disorder affecting 6,000 to 10,000 Americans, is caused by a deficit of C1-INH.2
- Colesevelam HCl tablets (Welchol) have been approved by the FDA as an adjunct to diet and exercise for reducing LDL-C levels in boys and postmenarchal girls ages 10 to 17, with heterozygous familial hypercholesterolemia as monotherapy, or in combination with a statin after failing an adequate trial of diet therapy.3
- Colesevelam HCl (Welchol) has been approved by the FDA as an oral solution providing an alternate dosage form to the large oral tablets currently available.3
- Oxycodone HCl has been recommended for FDA approval. When this new formulation is dissolved in water, it forms a gel, which makes it difficult to abuse. If approved, the new formulation will keep the OxyContin name and will be available in seven dosages. The older product will be phased out and only the newer product will be available.4
- Peginterferon alpha-2b injection (PegIntron) has been recommended for FDA approval for the treatment of patients with stage-III malignant melanoma. Peginterferon alpha-2b currently is approved for treating hepatitis C in combination with ribavirin. It is a once-weekly, subcutaneous injection.5
- Ustekinumab (Stelara) has been approved by the FDA for treating moderate to severe plaque psoriasis by disabling two interleukin (IL) cytokines, IL-12 and IL-23.6 It is a monoclonal antibody administered via subcutaneous injection. Recommended dosing is a baseline injection followed by another injection at week four, followed by subsequent injections every 12 weeks.7 Serious infections have been reported in clinical trials. Therefore, the company has developed a Risk Evaluation and Mitigation Strategy (REMS), as well as targeted healthcare provider education and a patient guide. The product label also contains cautions related to potential immunosuppression, as well as information on avoiding live vaccines while being treated with the agent.
Pipeline
- Cladribine, originally approved by the FDA in an intravenous formulation in the 1990s to treat hairy cell leukemia, has been reformulated as an oral product to manage patients with multiple sclerosis.8 Merck has submitted oral cladribine as a disease-modifying therapy for multiple sclerosis. If approved, it will be the first oral disease modifying agent for treating multiple sclerosis patients.9
- Dapagliflozin, a new mechanism renal sodium-glucose co-transporter 2 (SGLT2) inhibitor, has been shown to reduce fasting plasma glucose and significantly reduce HbA1c levels in patients with Type 2 diabetes mellitus, compared with patients treated with a metformin and placebo combination.10,11 Additionally, about 25% of patients treated with dapagliflozin (vs. 6% of the placebo-metformin-treated patients) had at least a 5% decrease in body weight. Diastolic blood pressure and uric acid level also decreased but not significantly. Serious adverse events were similar between the two treated groups.
- Fingolimod, an oral, disease-modifying agent to treat multiple sclerosis, is the first in a new class of agents known as sphingosine 1-phosphate receptor modulators (S1P-R). A recent two-year study showed it significantly reduced both relapses and disability progression (compared with placebo) in patients with relapsing remitting multiple sclerosis.12,13 More information will be available on this agent.
- Naproxcinod is a cyclo-oxygenase-inhibiting nitric oxide donator (CINOD) anti-inflammatory agent.14 The proposed indication is for the relief of the signs and symptoms of osteoarthritis, predominantly for pain management.
Safety Information
- Promethazine injection has undergone a label change to include a boxed warning. The warning is to emphasize the risk of serious tissue injury when promethazine is incorrectly administered.15 The preferred route is deep intramuscular injection; if administered in or near a vein, severe tissue injury might occur. The FDA previously informed healthcare professionals about the risks of incorrectly administered promethazine in December 2006 and again in February 2008. Post-marketing adverse events reported from 1969 to 2009 have identified cases of gangrene requiring amputation associated with administration of injectable promethazine.
- Since its original FDA approval in October 2006, sitagliptin, the first oral dipeptidyl peptidase-4 (DPP-4) inhibitor, is undergoing a safety label change.16 There have been 88 post-marketing cases reported of acute pancreatitis, including two cases of necrotizing or hemorrhagic pancreatitis reported between Oct. 16, 2006, and Feb. 9, 2009.17 The updated labeling discusses more information on the pancreatitis cases reported, and recommends that healthcare professionals carefully monitor patients for the development of pancreatitis, either upon beginning therapy or around dose increases. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Japan’s Astellas settles with Impax on prostate drug. Reuters Web site. Available at: http://www.reuters.com/article/rbssPharmaceuticals%20-%20Generic%20&%20Specialty/idUST28251320091007. Accessed Oct. 13, 2009.
- FDA approves Berinert. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/csl-behring-announces-fda-approval-berinert-first-only-therapy-approved-acute-abdominal-facial-1681.html. Accessed Oct. 13, 2009.
- Welchol package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022362lbl.pdf. Accessed Oct. 13, 2009.
- Walker E. FDA Panel recommends approval of new oxycodone formulation. Medpage Today Web site. Available at: http://www.medpagetoday.com/Neurology/PainManagement/16132?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Todoruk M. FDA panel supports approval of Schering-Plough’s PegIntron for patients with melanoma. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=8E68078692384C8A8249EA4A4C036635&logRowId=330581. Accessed Oct. 13, 2009.
- Gever J, Agus ZS. FDA approves biologic drug for psoriasis. Medpage Today Web site. Available at: http://www.medpagetoday.com/InfectiousDisease/PublicHealth/16147?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Stelara package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125261lbl.pdf. Accessed Oct. 13, 2009.
- Leustatin package label. FDA Web site. Available at: http://www.cancerconsultants.com/druginserts/Cladribine.pdf. Accessed Oct. 13, 2009.
- Todoruk M. Merck KGaA files cladribine for FDA approval. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=08CF20114B2A4F1DA8FD33AAC7240AB8&logRowId=329839. Accessed Oct. 2, 2009.
- Dennis M. AstraZeneca, Bristol-Myers Squibb’s dapagliflozin meets endpoints in Phase III study. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=3B158484CA7A47B99CEA373CB2ABFFA2&logRowId=330336. Accessed Oct. 13, 2009.
- Gever J, Zaleznik DF, Caputo D. EASD: Phase III data look good for novel diabetes drug. Medpage Today Web site. Available at: http://www.medpagetoday.com/MeetingCoverage/EASD/16270. Accessed Oct. 11, 2009.
- Dennis M. Novartis: Phase III study shows oral MS drug fingolimod reduces relapses, disability progression. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=EDBA0E3366E2477DB714674E9C19821D&logRowId=329838. Accessed Oct. 13, 2009.
- Multiple sclerosis therapy FTY720 reduces relapses and disability progression. The Multiple Sclerosis Resource Centre Web site. Available at: http://www.msrc.co.uk/printable.cfm?pageid=1309. Accessed Oct. 13, 2009.
- NicOx submits New Drug Application (NDA) for naproxcinod to the US FDA. NicOx Web site. Available at: http://www.nicox.com/upload/PR_NDA_submission-250909__EN.pdf. Accessed Oct. 13, 2009.
- Riley K. FDA requires boxed warning for promethazine hydrochloride injection. FDA Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm182498.htm. Accessed Oct. 13, 2009.
- Januvia package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021995lbl.pdf. Accessed Oct. 13, 2009.
- Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). FDA Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed Sept. 26, 2009.
New Generics
- Tamsulosin (generic Flomax) capsules should be available March 2, 20101
New Drugs, Indications, Dosage Forms, and Approvals
- C1 Esterase inhibitor [human] (Berinert) has been approved by the Food and Drug Administration (FDA) to treat acute abdominal attacks and facial edema associated with hereditary angioedema (HAE) in adolescents and adults. It is derived from human plasma and regulates clotting and inflammatory reactions. HAE, a genetic disorder affecting 6,000 to 10,000 Americans, is caused by a deficit of C1-INH.2
- Colesevelam HCl tablets (Welchol) have been approved by the FDA as an adjunct to diet and exercise for reducing LDL-C levels in boys and postmenarchal girls ages 10 to 17, with heterozygous familial hypercholesterolemia as monotherapy, or in combination with a statin after failing an adequate trial of diet therapy.3
- Colesevelam HCl (Welchol) has been approved by the FDA as an oral solution providing an alternate dosage form to the large oral tablets currently available.3
- Oxycodone HCl has been recommended for FDA approval. When this new formulation is dissolved in water, it forms a gel, which makes it difficult to abuse. If approved, the new formulation will keep the OxyContin name and will be available in seven dosages. The older product will be phased out and only the newer product will be available.4
- Peginterferon alpha-2b injection (PegIntron) has been recommended for FDA approval for the treatment of patients with stage-III malignant melanoma. Peginterferon alpha-2b currently is approved for treating hepatitis C in combination with ribavirin. It is a once-weekly, subcutaneous injection.5
- Ustekinumab (Stelara) has been approved by the FDA for treating moderate to severe plaque psoriasis by disabling two interleukin (IL) cytokines, IL-12 and IL-23.6 It is a monoclonal antibody administered via subcutaneous injection. Recommended dosing is a baseline injection followed by another injection at week four, followed by subsequent injections every 12 weeks.7 Serious infections have been reported in clinical trials. Therefore, the company has developed a Risk Evaluation and Mitigation Strategy (REMS), as well as targeted healthcare provider education and a patient guide. The product label also contains cautions related to potential immunosuppression, as well as information on avoiding live vaccines while being treated with the agent.
Pipeline
- Cladribine, originally approved by the FDA in an intravenous formulation in the 1990s to treat hairy cell leukemia, has been reformulated as an oral product to manage patients with multiple sclerosis.8 Merck has submitted oral cladribine as a disease-modifying therapy for multiple sclerosis. If approved, it will be the first oral disease modifying agent for treating multiple sclerosis patients.9
- Dapagliflozin, a new mechanism renal sodium-glucose co-transporter 2 (SGLT2) inhibitor, has been shown to reduce fasting plasma glucose and significantly reduce HbA1c levels in patients with Type 2 diabetes mellitus, compared with patients treated with a metformin and placebo combination.10,11 Additionally, about 25% of patients treated with dapagliflozin (vs. 6% of the placebo-metformin-treated patients) had at least a 5% decrease in body weight. Diastolic blood pressure and uric acid level also decreased but not significantly. Serious adverse events were similar between the two treated groups.
- Fingolimod, an oral, disease-modifying agent to treat multiple sclerosis, is the first in a new class of agents known as sphingosine 1-phosphate receptor modulators (S1P-R). A recent two-year study showed it significantly reduced both relapses and disability progression (compared with placebo) in patients with relapsing remitting multiple sclerosis.12,13 More information will be available on this agent.
- Naproxcinod is a cyclo-oxygenase-inhibiting nitric oxide donator (CINOD) anti-inflammatory agent.14 The proposed indication is for the relief of the signs and symptoms of osteoarthritis, predominantly for pain management.
Safety Information
- Promethazine injection has undergone a label change to include a boxed warning. The warning is to emphasize the risk of serious tissue injury when promethazine is incorrectly administered.15 The preferred route is deep intramuscular injection; if administered in or near a vein, severe tissue injury might occur. The FDA previously informed healthcare professionals about the risks of incorrectly administered promethazine in December 2006 and again in February 2008. Post-marketing adverse events reported from 1969 to 2009 have identified cases of gangrene requiring amputation associated with administration of injectable promethazine.
- Since its original FDA approval in October 2006, sitagliptin, the first oral dipeptidyl peptidase-4 (DPP-4) inhibitor, is undergoing a safety label change.16 There have been 88 post-marketing cases reported of acute pancreatitis, including two cases of necrotizing or hemorrhagic pancreatitis reported between Oct. 16, 2006, and Feb. 9, 2009.17 The updated labeling discusses more information on the pancreatitis cases reported, and recommends that healthcare professionals carefully monitor patients for the development of pancreatitis, either upon beginning therapy or around dose increases. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Japan’s Astellas settles with Impax on prostate drug. Reuters Web site. Available at: http://www.reuters.com/article/rbssPharmaceuticals%20-%20Generic%20&%20Specialty/idUST28251320091007. Accessed Oct. 13, 2009.
- FDA approves Berinert. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/csl-behring-announces-fda-approval-berinert-first-only-therapy-approved-acute-abdominal-facial-1681.html. Accessed Oct. 13, 2009.
- Welchol package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022362lbl.pdf. Accessed Oct. 13, 2009.
- Walker E. FDA Panel recommends approval of new oxycodone formulation. Medpage Today Web site. Available at: http://www.medpagetoday.com/Neurology/PainManagement/16132?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Todoruk M. FDA panel supports approval of Schering-Plough’s PegIntron for patients with melanoma. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=8E68078692384C8A8249EA4A4C036635&logRowId=330581. Accessed Oct. 13, 2009.
- Gever J, Agus ZS. FDA approves biologic drug for psoriasis. Medpage Today Web site. Available at: http://www.medpagetoday.com/InfectiousDisease/PublicHealth/16147?utm_source=breaking-news&utm_medium=email&utm_campaign=breaking-news. Accessed Oct. 13, 2009.
- Stelara package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125261lbl.pdf. Accessed Oct. 13, 2009.
- Leustatin package label. FDA Web site. Available at: http://www.cancerconsultants.com/druginserts/Cladribine.pdf. Accessed Oct. 13, 2009.
- Todoruk M. Merck KGaA files cladribine for FDA approval. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=08CF20114B2A4F1DA8FD33AAC7240AB8&logRowId=329839. Accessed Oct. 2, 2009.
- Dennis M. AstraZeneca, Bristol-Myers Squibb’s dapagliflozin meets endpoints in Phase III study. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=3B158484CA7A47B99CEA373CB2ABFFA2&logRowId=330336. Accessed Oct. 13, 2009.
- Gever J, Zaleznik DF, Caputo D. EASD: Phase III data look good for novel diabetes drug. Medpage Today Web site. Available at: http://www.medpagetoday.com/MeetingCoverage/EASD/16270. Accessed Oct. 11, 2009.
- Dennis M. Novartis: Phase III study shows oral MS drug fingolimod reduces relapses, disability progression. FirstWord Web site. Available at: http://www.firstwordplus.com/Fws.do?articleid=EDBA0E3366E2477DB714674E9C19821D&logRowId=329838. Accessed Oct. 13, 2009.
- Multiple sclerosis therapy FTY720 reduces relapses and disability progression. The Multiple Sclerosis Resource Centre Web site. Available at: http://www.msrc.co.uk/printable.cfm?pageid=1309. Accessed Oct. 13, 2009.
- NicOx submits New Drug Application (NDA) for naproxcinod to the US FDA. NicOx Web site. Available at: http://www.nicox.com/upload/PR_NDA_submission-250909__EN.pdf. Accessed Oct. 13, 2009.
- Riley K. FDA requires boxed warning for promethazine hydrochloride injection. FDA Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm182498.htm. Accessed Oct. 13, 2009.
- Januvia package label. FDA Web site. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021995lbl.pdf. Accessed Oct. 13, 2009.
- Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). FDA Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed Sept. 26, 2009.
In the Literature: January 2010
In This Edition
Literature at a Glance
A guide to this month’s studies
- Resident duty hours and ICU patient outcomes
- Effect of ER boarding on patient outcomes
- ED voicemail signout of patients
- Adequacy of patient signouts
- D-dimer use in patients with low suspicion of pulmonary embolism
- Patient involvement in medication reconciliation
- Effects of on-screen reminders on outcomes
Decreased ICU Duty Hours Does Not Affect Patient Mortality
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Emergency Department “Boarding” Results in Undesirable Events
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Emergency Department Signout via Voicemail Yields Mixed Reviews
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Patient Signout Is Not Uniformly Comprehensive and Often Lacks Critical Information
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Negative D-Dimer Test Can Safely Exclude Pulmonary Embolism in Patients at Low To Intermediate Clinical Risk
Clinical question: In patients with symptoms consistent with pulmonary embolism (PE), can evaluation with a clinical risk assessment tool and D-dimer assay identify patients who do not require CT angiography to exclude PE?
Background: D-dimer is a highly sensitive but nonspecific marker of VTE, and studies suggest that VTE can be ruled out without further imaging in patients with low clinical probability of disease and a negative D-dimer test. Nevertheless, this practice has not been adopted uniformly, and CT angiography (CTA) overuse continues.
Study design: Prospective registry cohort.
Setting: A 550-bed community teaching hospital in Chicago.
Synopsis: Consecutive patients presenting to the ED with symptoms suggestive of PE were evaluated with 1) revised Geneva score; 2) D-dimer assay; and 3) CTA. Among the 627 patients who underwent all three components of the evaluation, 44.8% were identified as low probability for PE by revised Geneva score, 52.6% as intermediate probability, and 2.6% as high probability. The overall prevalence of PE (using CTA as the gold standard) was very low (4.5%); just 2.1% of low-risk, 5.2% of intermediate-risk, and 31.2% of high-risk patients were ultimately found to have PE on CTA.
Using a cutoff of 1.2 mg/L, the D-dimer assay accurately detected all low- to intermediate-probability patients with PE (sensitivity and negative predictive value of 100%). One patient in the high probability group did have a PE, even though the patient had a D-dimer value <1.2 mg/L (sensitivity and NPV both 80%). Had diagnostic testing stopped after a negative D-dimer result in the low- to intermediate-probability patients, 172 CTAs (27%) would have been avoided.
Bottom line: In a low-prevalence cohort, no pulmonary emboli were identified by CTA in any patient with a low to intermediate clinical risk assessment and a negative quantitative D-dimer assay result.
Citation: Gupta RT, Kakarla RK, Kirshenbaum KJ, Tapson VF. D-dimers and efficacy of clinical risk estimation algorithms: sensitivity in evaluation of acute pulmonary embolism. AJR Am J Roentgenol. 2009;193(2):425-430.
Patient Participation in Medication Reconciliation at Discharge Helps Detect Prescribing Discrepancies
Clinical question: Does the inclusion of a medication adherence counseling session during a hospital discharge reconciliation process reduce discrepancies in the final medication regimen?
Background: Inadvertent medication prescribing errors are an important cause of preventable adverse drug events and commonly occur at transitions of care. Although medication reconciliation processes can identify errors, the best strategies for implementation remain unclear.
Study design: Prospective, observational cohort.
Setting: A 550-bed teaching hospital in the Netherlands.
Synopsis: Of 437 patients admitted to a pulmonary ward and screened for eligibility, 267 were included in the analysis. A pharmacy specialist reviewed all available community prescription records, inpatient documentation, and discharge medication lists in an effort to identify discrepancies. Potential errors were discussed with the prescriber. Then, the pharmacy specialist interviewed the patient and provided additional counseling. Any new discrepancies were discussed with the prescriber. All questions raised by the pharmacist were recorded, as were all subsequent prescriber interventions.
The primary outcome measure was the number of interventions made as a result of pharmacy review. A total of 940 questions were asked. At least one intervention was recorded for 87% of patients before counseling (mean 2.7 interventions/patient) and for 97% of patients after (mean 5.3 interventions/patient). Discrepancies were addressed for 63.7% of patients before counseling and 72.5% after. Pharmacotherapy was optimized for 67.2% of patients before counseling and 76.3% after.
Bottom line: Patient engagement in the medication reconciliation process incrementally improves the quality of the history and helps identify clinically meaningful discrepancies at the time of hospital discharge.
Citation: Karapinar-Carkit F, Borgsteede S, Zoer J, Smit HJ, Egberts AC, van den Bemt P. Effect of medication reconciliation with and without patient counseling on the number of pharmaceutical interventions among patients discharged from the hospital. Ann Pharmacother. 2009;43(6):1001-1010.
Computer-Based Reminders Have Small to Modest Effect on Care Processes
Clinical question: Do on-screen, computer-based clinical reminders improve adherence to target processes of care or clinical outcomes?
Background: Gaps between practice guidelines and routine care are caused, in part, by the inability of clinicians to access or recall information at the point of care. Although automated reminder systems offer the promise of “just in time” recommendations, studies of electronic reminders have demonstrated mixed results.
Study design: Literature review and meta-analysis.
Setting: Multiple databases and information repositories, including MEDLINE, EMBASE, and CINAHL.
Synopsis: The authors conducted a literature search to identify randomized and quasi-randomized controlled trials measuring the effect of computer-based reminders on process measures or clinical outcomes. To avoid statistical challenges inherent in unit-of-analysis errors, the authors reported median improvement in process adherence or median change in clinical endpoints.
Out of a pool of 2,036 citations, 28 studies detailing 32 comparative analyses were included. Across the 28 studies, reminders resulted in a median improvement in target process adherence of 4.2% (3.3% for prescribing behavior, 2.8% for test ordering). Eight comparisons reported dichotomous clinical endpoints and collectively showed a median absolute improvement of 2.5%.
The greatest contribution to measured treatment effects came from large academic centers with well-established electronic health records and robust informatics departments. No characteristics of the reminder system or the clinical context were associated with the magnitude of impact. A potential limitation in reporting median effects across studies is that all studies were given equal weight.
Bottom line: Electronic reminders appear to have a small, positive effect on clinician adherence to recommended processes, although it is uncertain what contextual or design features are responsible for the greatest treatment effect.
Citation: Shojania K, Jennings A, Mayhew A, Ramsay CR, Eccles MP, Grimshaw J. The effects of on-screen, point of care computer reminders on processes and outcomes of care. Cochrane Database Syst Rev. 2009(3):CD001096. TH
Pediatric HM Literature
Short Course of Oral Antibiotics Effective for Acute Osteomyelitis and Septic Arthritis in Children
By Mark Shen, MD

Clinical question: Is a short course (less than four weeks) of antibiotics effective for the treatment of acute osteomyelitis and septic arthritis?
Background: The optimal duration of treatment for acute bone and joint infections in children has not been assessed adequately in prospectively designed trials. Historically, intravenous (IV) antibiotics in four- to six-week durations have been recommended, although the evidence for this practice is limited. There is widespread variation in both the route of administration (oral vs. IV) and duration of this treatment.
Study design: Prospective cohort study.
Setting: Two children’s hospitals in Australia.
Synopsis: Seventy children ages 17 and under who presented to two tertiary-care children’s hospitals with osteomyelitis or septic arthritis were enrolled. Primary surgical drainage was performed for patients with septic arthritis. Intravenous antibiotics were administered for at least three days, and until clinical symptoms improved and the C-reactive protein levels had stabilized. Patients then were transitioned to oral antibiotics and discharged to complete a minimum of three weeks of therapy.
Fifty-nine percent of patients were converted to oral antibiotics by day three, 86% by day five of therapy. Based on clinical and hematologic assessment, 83% of patients had oral antibiotics stopped at the three-week followup and remained well through the 12-month follow-up period.
This study essentially involved prospective data collection for a cohort of children receiving standardized care. Although the results suggest that a majority of children can be treated with a three-week course of oral antibiotics, the results would have been further strengthened by an explicit protocol with well-defined criteria for the oral to IV transition and cessation of antibiotic therapy. Additional limitations include pathogens and antibiotic choices that might not be applicable to North American populations.
Bottom line: After initial intravenous therapy, a three-week course of oral antibiotics can be effective for acute osteomyelitis and septic arthritis in children.
Citation: Jagodzinski NA, Kanwar R, Graham K, Bache CE. Prospective evaluation of a shortened regimen of treatment for acute osteomyelitis and septic arthritis in children. J Pediatr Orthop. 2009;29(5):518-525.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Resident duty hours and ICU patient outcomes
- Effect of ER boarding on patient outcomes
- ED voicemail signout of patients
- Adequacy of patient signouts
- D-dimer use in patients with low suspicion of pulmonary embolism
- Patient involvement in medication reconciliation
- Effects of on-screen reminders on outcomes
Decreased ICU Duty Hours Does Not Affect Patient Mortality
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Emergency Department “Boarding” Results in Undesirable Events
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Emergency Department Signout via Voicemail Yields Mixed Reviews
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Patient Signout Is Not Uniformly Comprehensive and Often Lacks Critical Information
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Negative D-Dimer Test Can Safely Exclude Pulmonary Embolism in Patients at Low To Intermediate Clinical Risk
Clinical question: In patients with symptoms consistent with pulmonary embolism (PE), can evaluation with a clinical risk assessment tool and D-dimer assay identify patients who do not require CT angiography to exclude PE?
Background: D-dimer is a highly sensitive but nonspecific marker of VTE, and studies suggest that VTE can be ruled out without further imaging in patients with low clinical probability of disease and a negative D-dimer test. Nevertheless, this practice has not been adopted uniformly, and CT angiography (CTA) overuse continues.
Study design: Prospective registry cohort.
Setting: A 550-bed community teaching hospital in Chicago.
Synopsis: Consecutive patients presenting to the ED with symptoms suggestive of PE were evaluated with 1) revised Geneva score; 2) D-dimer assay; and 3) CTA. Among the 627 patients who underwent all three components of the evaluation, 44.8% were identified as low probability for PE by revised Geneva score, 52.6% as intermediate probability, and 2.6% as high probability. The overall prevalence of PE (using CTA as the gold standard) was very low (4.5%); just 2.1% of low-risk, 5.2% of intermediate-risk, and 31.2% of high-risk patients were ultimately found to have PE on CTA.
Using a cutoff of 1.2 mg/L, the D-dimer assay accurately detected all low- to intermediate-probability patients with PE (sensitivity and negative predictive value of 100%). One patient in the high probability group did have a PE, even though the patient had a D-dimer value <1.2 mg/L (sensitivity and NPV both 80%). Had diagnostic testing stopped after a negative D-dimer result in the low- to intermediate-probability patients, 172 CTAs (27%) would have been avoided.
Bottom line: In a low-prevalence cohort, no pulmonary emboli were identified by CTA in any patient with a low to intermediate clinical risk assessment and a negative quantitative D-dimer assay result.
Citation: Gupta RT, Kakarla RK, Kirshenbaum KJ, Tapson VF. D-dimers and efficacy of clinical risk estimation algorithms: sensitivity in evaluation of acute pulmonary embolism. AJR Am J Roentgenol. 2009;193(2):425-430.
Patient Participation in Medication Reconciliation at Discharge Helps Detect Prescribing Discrepancies
Clinical question: Does the inclusion of a medication adherence counseling session during a hospital discharge reconciliation process reduce discrepancies in the final medication regimen?
Background: Inadvertent medication prescribing errors are an important cause of preventable adverse drug events and commonly occur at transitions of care. Although medication reconciliation processes can identify errors, the best strategies for implementation remain unclear.
Study design: Prospective, observational cohort.
Setting: A 550-bed teaching hospital in the Netherlands.
Synopsis: Of 437 patients admitted to a pulmonary ward and screened for eligibility, 267 were included in the analysis. A pharmacy specialist reviewed all available community prescription records, inpatient documentation, and discharge medication lists in an effort to identify discrepancies. Potential errors were discussed with the prescriber. Then, the pharmacy specialist interviewed the patient and provided additional counseling. Any new discrepancies were discussed with the prescriber. All questions raised by the pharmacist were recorded, as were all subsequent prescriber interventions.
The primary outcome measure was the number of interventions made as a result of pharmacy review. A total of 940 questions were asked. At least one intervention was recorded for 87% of patients before counseling (mean 2.7 interventions/patient) and for 97% of patients after (mean 5.3 interventions/patient). Discrepancies were addressed for 63.7% of patients before counseling and 72.5% after. Pharmacotherapy was optimized for 67.2% of patients before counseling and 76.3% after.
Bottom line: Patient engagement in the medication reconciliation process incrementally improves the quality of the history and helps identify clinically meaningful discrepancies at the time of hospital discharge.
Citation: Karapinar-Carkit F, Borgsteede S, Zoer J, Smit HJ, Egberts AC, van den Bemt P. Effect of medication reconciliation with and without patient counseling on the number of pharmaceutical interventions among patients discharged from the hospital. Ann Pharmacother. 2009;43(6):1001-1010.
Computer-Based Reminders Have Small to Modest Effect on Care Processes
Clinical question: Do on-screen, computer-based clinical reminders improve adherence to target processes of care or clinical outcomes?
Background: Gaps between practice guidelines and routine care are caused, in part, by the inability of clinicians to access or recall information at the point of care. Although automated reminder systems offer the promise of “just in time” recommendations, studies of electronic reminders have demonstrated mixed results.
Study design: Literature review and meta-analysis.
Setting: Multiple databases and information repositories, including MEDLINE, EMBASE, and CINAHL.
Synopsis: The authors conducted a literature search to identify randomized and quasi-randomized controlled trials measuring the effect of computer-based reminders on process measures or clinical outcomes. To avoid statistical challenges inherent in unit-of-analysis errors, the authors reported median improvement in process adherence or median change in clinical endpoints.
Out of a pool of 2,036 citations, 28 studies detailing 32 comparative analyses were included. Across the 28 studies, reminders resulted in a median improvement in target process adherence of 4.2% (3.3% for prescribing behavior, 2.8% for test ordering). Eight comparisons reported dichotomous clinical endpoints and collectively showed a median absolute improvement of 2.5%.
The greatest contribution to measured treatment effects came from large academic centers with well-established electronic health records and robust informatics departments. No characteristics of the reminder system or the clinical context were associated with the magnitude of impact. A potential limitation in reporting median effects across studies is that all studies were given equal weight.
Bottom line: Electronic reminders appear to have a small, positive effect on clinician adherence to recommended processes, although it is uncertain what contextual or design features are responsible for the greatest treatment effect.
Citation: Shojania K, Jennings A, Mayhew A, Ramsay CR, Eccles MP, Grimshaw J. The effects of on-screen, point of care computer reminders on processes and outcomes of care. Cochrane Database Syst Rev. 2009(3):CD001096. TH
Pediatric HM Literature
Short Course of Oral Antibiotics Effective for Acute Osteomyelitis and Septic Arthritis in Children
By Mark Shen, MD
Clinical question: Is a short course (less than four weeks) of antibiotics effective for the treatment of acute osteomyelitis and septic arthritis?
Background: The optimal duration of treatment for acute bone and joint infections in children has not been assessed adequately in prospectively designed trials. Historically, intravenous (IV) antibiotics in four- to six-week durations have been recommended, although the evidence for this practice is limited. There is widespread variation in both the route of administration (oral vs. IV) and duration of this treatment.
Study design: Prospective cohort study.
Setting: Two children’s hospitals in Australia.
Synopsis: Seventy children ages 17 and under who presented to two tertiary-care children’s hospitals with osteomyelitis or septic arthritis were enrolled. Primary surgical drainage was performed for patients with septic arthritis. Intravenous antibiotics were administered for at least three days, and until clinical symptoms improved and the C-reactive protein levels had stabilized. Patients then were transitioned to oral antibiotics and discharged to complete a minimum of three weeks of therapy.
Fifty-nine percent of patients were converted to oral antibiotics by day three, 86% by day five of therapy. Based on clinical and hematologic assessment, 83% of patients had oral antibiotics stopped at the three-week followup and remained well through the 12-month follow-up period.
This study essentially involved prospective data collection for a cohort of children receiving standardized care. Although the results suggest that a majority of children can be treated with a three-week course of oral antibiotics, the results would have been further strengthened by an explicit protocol with well-defined criteria for the oral to IV transition and cessation of antibiotic therapy. Additional limitations include pathogens and antibiotic choices that might not be applicable to North American populations.
Bottom line: After initial intravenous therapy, a three-week course of oral antibiotics can be effective for acute osteomyelitis and septic arthritis in children.
Citation: Jagodzinski NA, Kanwar R, Graham K, Bache CE. Prospective evaluation of a shortened regimen of treatment for acute osteomyelitis and septic arthritis in children. J Pediatr Orthop. 2009;29(5):518-525.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Resident duty hours and ICU patient outcomes
- Effect of ER boarding on patient outcomes
- ED voicemail signout of patients
- Adequacy of patient signouts
- D-dimer use in patients with low suspicion of pulmonary embolism
- Patient involvement in medication reconciliation
- Effects of on-screen reminders on outcomes
Decreased ICU Duty Hours Does Not Affect Patient Mortality
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Emergency Department “Boarding” Results in Undesirable Events
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Emergency Department Signout via Voicemail Yields Mixed Reviews
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Patient Signout Is Not Uniformly Comprehensive and Often Lacks Critical Information
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Negative D-Dimer Test Can Safely Exclude Pulmonary Embolism in Patients at Low To Intermediate Clinical Risk
Clinical question: In patients with symptoms consistent with pulmonary embolism (PE), can evaluation with a clinical risk assessment tool and D-dimer assay identify patients who do not require CT angiography to exclude PE?
Background: D-dimer is a highly sensitive but nonspecific marker of VTE, and studies suggest that VTE can be ruled out without further imaging in patients with low clinical probability of disease and a negative D-dimer test. Nevertheless, this practice has not been adopted uniformly, and CT angiography (CTA) overuse continues.
Study design: Prospective registry cohort.
Setting: A 550-bed community teaching hospital in Chicago.
Synopsis: Consecutive patients presenting to the ED with symptoms suggestive of PE were evaluated with 1) revised Geneva score; 2) D-dimer assay; and 3) CTA. Among the 627 patients who underwent all three components of the evaluation, 44.8% were identified as low probability for PE by revised Geneva score, 52.6% as intermediate probability, and 2.6% as high probability. The overall prevalence of PE (using CTA as the gold standard) was very low (4.5%); just 2.1% of low-risk, 5.2% of intermediate-risk, and 31.2% of high-risk patients were ultimately found to have PE on CTA.
Using a cutoff of 1.2 mg/L, the D-dimer assay accurately detected all low- to intermediate-probability patients with PE (sensitivity and negative predictive value of 100%). One patient in the high probability group did have a PE, even though the patient had a D-dimer value <1.2 mg/L (sensitivity and NPV both 80%). Had diagnostic testing stopped after a negative D-dimer result in the low- to intermediate-probability patients, 172 CTAs (27%) would have been avoided.
Bottom line: In a low-prevalence cohort, no pulmonary emboli were identified by CTA in any patient with a low to intermediate clinical risk assessment and a negative quantitative D-dimer assay result.
Citation: Gupta RT, Kakarla RK, Kirshenbaum KJ, Tapson VF. D-dimers and efficacy of clinical risk estimation algorithms: sensitivity in evaluation of acute pulmonary embolism. AJR Am J Roentgenol. 2009;193(2):425-430.
Patient Participation in Medication Reconciliation at Discharge Helps Detect Prescribing Discrepancies
Clinical question: Does the inclusion of a medication adherence counseling session during a hospital discharge reconciliation process reduce discrepancies in the final medication regimen?
Background: Inadvertent medication prescribing errors are an important cause of preventable adverse drug events and commonly occur at transitions of care. Although medication reconciliation processes can identify errors, the best strategies for implementation remain unclear.
Study design: Prospective, observational cohort.
Setting: A 550-bed teaching hospital in the Netherlands.
Synopsis: Of 437 patients admitted to a pulmonary ward and screened for eligibility, 267 were included in the analysis. A pharmacy specialist reviewed all available community prescription records, inpatient documentation, and discharge medication lists in an effort to identify discrepancies. Potential errors were discussed with the prescriber. Then, the pharmacy specialist interviewed the patient and provided additional counseling. Any new discrepancies were discussed with the prescriber. All questions raised by the pharmacist were recorded, as were all subsequent prescriber interventions.
The primary outcome measure was the number of interventions made as a result of pharmacy review. A total of 940 questions were asked. At least one intervention was recorded for 87% of patients before counseling (mean 2.7 interventions/patient) and for 97% of patients after (mean 5.3 interventions/patient). Discrepancies were addressed for 63.7% of patients before counseling and 72.5% after. Pharmacotherapy was optimized for 67.2% of patients before counseling and 76.3% after.
Bottom line: Patient engagement in the medication reconciliation process incrementally improves the quality of the history and helps identify clinically meaningful discrepancies at the time of hospital discharge.
Citation: Karapinar-Carkit F, Borgsteede S, Zoer J, Smit HJ, Egberts AC, van den Bemt P. Effect of medication reconciliation with and without patient counseling on the number of pharmaceutical interventions among patients discharged from the hospital. Ann Pharmacother. 2009;43(6):1001-1010.
Computer-Based Reminders Have Small to Modest Effect on Care Processes
Clinical question: Do on-screen, computer-based clinical reminders improve adherence to target processes of care or clinical outcomes?
Background: Gaps between practice guidelines and routine care are caused, in part, by the inability of clinicians to access or recall information at the point of care. Although automated reminder systems offer the promise of “just in time” recommendations, studies of electronic reminders have demonstrated mixed results.
Study design: Literature review and meta-analysis.
Setting: Multiple databases and information repositories, including MEDLINE, EMBASE, and CINAHL.
Synopsis: The authors conducted a literature search to identify randomized and quasi-randomized controlled trials measuring the effect of computer-based reminders on process measures or clinical outcomes. To avoid statistical challenges inherent in unit-of-analysis errors, the authors reported median improvement in process adherence or median change in clinical endpoints.
Out of a pool of 2,036 citations, 28 studies detailing 32 comparative analyses were included. Across the 28 studies, reminders resulted in a median improvement in target process adherence of 4.2% (3.3% for prescribing behavior, 2.8% for test ordering). Eight comparisons reported dichotomous clinical endpoints and collectively showed a median absolute improvement of 2.5%.
The greatest contribution to measured treatment effects came from large academic centers with well-established electronic health records and robust informatics departments. No characteristics of the reminder system or the clinical context were associated with the magnitude of impact. A potential limitation in reporting median effects across studies is that all studies were given equal weight.
Bottom line: Electronic reminders appear to have a small, positive effect on clinician adherence to recommended processes, although it is uncertain what contextual or design features are responsible for the greatest treatment effect.
Citation: Shojania K, Jennings A, Mayhew A, Ramsay CR, Eccles MP, Grimshaw J. The effects of on-screen, point of care computer reminders on processes and outcomes of care. Cochrane Database Syst Rev. 2009(3):CD001096. TH
Pediatric HM Literature
Short Course of Oral Antibiotics Effective for Acute Osteomyelitis and Septic Arthritis in Children
By Mark Shen, MD
Clinical question: Is a short course (less than four weeks) of antibiotics effective for the treatment of acute osteomyelitis and septic arthritis?
Background: The optimal duration of treatment for acute bone and joint infections in children has not been assessed adequately in prospectively designed trials. Historically, intravenous (IV) antibiotics in four- to six-week durations have been recommended, although the evidence for this practice is limited. There is widespread variation in both the route of administration (oral vs. IV) and duration of this treatment.
Study design: Prospective cohort study.
Setting: Two children’s hospitals in Australia.
Synopsis: Seventy children ages 17 and under who presented to two tertiary-care children’s hospitals with osteomyelitis or septic arthritis were enrolled. Primary surgical drainage was performed for patients with septic arthritis. Intravenous antibiotics were administered for at least three days, and until clinical symptoms improved and the C-reactive protein levels had stabilized. Patients then were transitioned to oral antibiotics and discharged to complete a minimum of three weeks of therapy.
Fifty-nine percent of patients were converted to oral antibiotics by day three, 86% by day five of therapy. Based on clinical and hematologic assessment, 83% of patients had oral antibiotics stopped at the three-week followup and remained well through the 12-month follow-up period.
This study essentially involved prospective data collection for a cohort of children receiving standardized care. Although the results suggest that a majority of children can be treated with a three-week course of oral antibiotics, the results would have been further strengthened by an explicit protocol with well-defined criteria for the oral to IV transition and cessation of antibiotic therapy. Additional limitations include pathogens and antibiotic choices that might not be applicable to North American populations.
Bottom line: After initial intravenous therapy, a three-week course of oral antibiotics can be effective for acute osteomyelitis and septic arthritis in children.
Citation: Jagodzinski NA, Kanwar R, Graham K, Bache CE. Prospective evaluation of a shortened regimen of treatment for acute osteomyelitis and septic arthritis in children. J Pediatr Orthop. 2009;29(5):518-525.