User login
Should we still use electrocardiography to diagnose pericardial disease?
Yes. Acute pericarditis has a unique clinical presentation, physical findings, and electrocardiographic (ECG) changes. ECG is always ordered to look for ischemic changes in patients with chest pain. Acute pericarditis develops in stages, which makes it easy to differentiate from early repolarization and, more significantly, myocardial infarction. The ECG changes, along with the clinical presentation and physical findings, can make the diagnosis of pericarditis.
In atypical and complicated cases, advanced imaging studies (ie, echocardiography and cardiac magnetic resonance imaging) have been used to confirm the diagnosis and to follow the course of the disease. However, ECG remains a useful, cost-effective test.
PERICARDIAL DISEASE IS DIVERSE
The pericardium is a thin layer that covers the heart and separates it from other structures in the mediastinum.
Pericardial syndromes include acute, recurrent, constrictive, and effusive-constrictive pericarditis, as well as pericardial effusion with or without tamponade. Causes include viral or bacterial infection, postpericardiotomy syndrome (Dressler syndrome), postmyocardial infarction, primary and metastatic tumors, trauma, uremia, radiation, and autoimmune disease, but pericardial syndromes can also be idiopathic.1
Acute pericarditis is the most common pericardial syndrome and occurs in all age groups. Once diagnosed, it can easily be treated with antiinflammatory drugs. However, recurrent pericarditis, reported in 30% of patients experiencing a first attack of pericarditis, can be difficult to manage, can have a significant impact on the patient’s health, and can be life-threatening.2
CHANGES OF ACUTE PERICARDITIS DEVELOP IN STAGES
Pericarditis can be diagnosed on the basis of ECG changes, clinical signs and symptoms, and laboratory and imaging findings.3 ECG criteria of acute pericarditis have been published.4,5
The characteristic chest pain in acute pericarditis is usually sudden in onset and sharp and occurs over the anterior chest wall. The pain is exacerbated by inspiration and decreases when the patient sits up and leans forward.4
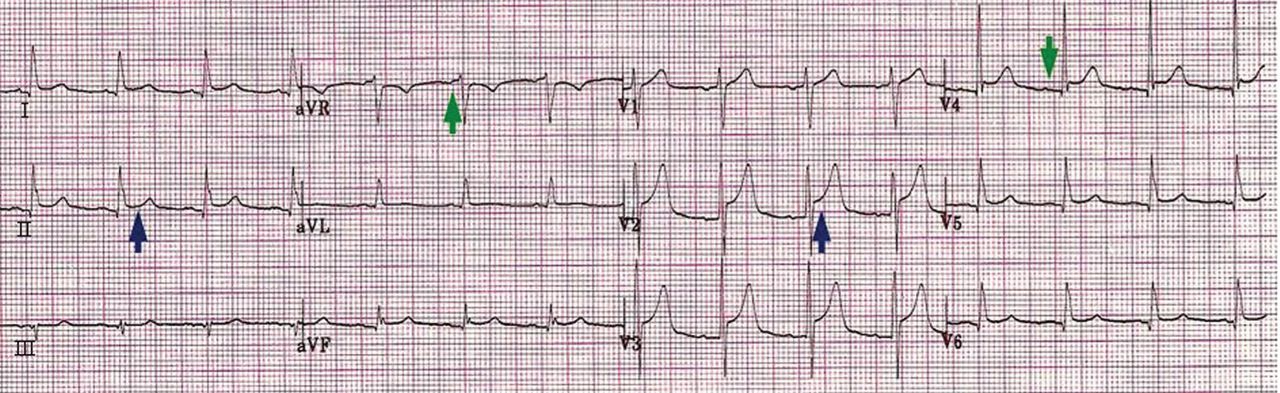
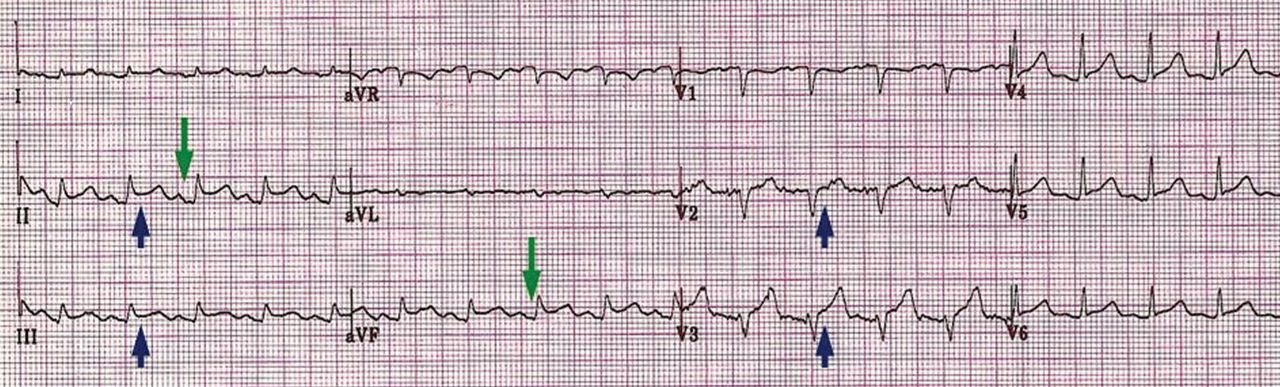
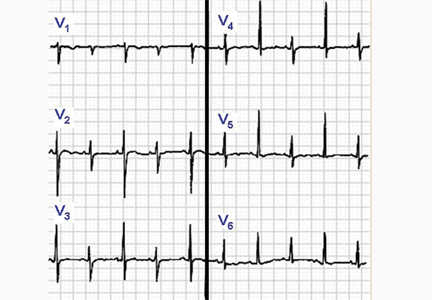
ECG classically shows a widespread saddle-shaped (upward concave) ST-segment elevation in the precordial and limb leads, reflecting subepicardial inflammation. PR-segment depression (with PR-segment elevation in lead aVR) can accompany or precede the ST changes and is known as the “discordant ST-PR segment sign” (Figures 1 and 2). These changes are seen in 60% of patients.
The ECG changes develop in stages, making them easy to differentiate from early repolarization and, more significantly, from myocardial infarction. Four stages are apparent1,4,6–9:
- Stage I occurs in a few hours to days, with diffuse, up-sloping ST-segment elevation and upright T waves, the result of an alteration in ventricular repolarization caused by pericardial inflammation. Because of alteration in repolarization of the atrium secondary to inflammation, the PR segment is elevated in aVR and depressed in the rest of the limb and chest leads.
- Stage II—the ST and PR segments normalize.
- Stage III—widespread T-wave inversion.
- Stage IV—normalization of the T waves.
There is no pathologic Q-wave formation or loss of R-wave progression in acute pericarditis.
The ECG changes of pericarditis vary widely from one patient to another, depending on the extent and severity of pericardial inflammation and the timing of the patient’s presentation. Changes vary in duration. In some cases, ST elevation returns to baseline within a few days without T-wave inversions; in other cases, T-wave inversions can persist for weeks to months. Sometimes the abnormalities resolve by the time symptoms develop.
ASSOCIATED CONDITIONS
Myocardial involvement
In acute myocarditis, findings on ECG can be normal unless the pericardium is involved. Changes that can be seen in myocarditis and that indicate a deeper involvement of inflammation include ST-segment abnormalities, arrhythmias (eg, premature ventricular or atrial contractions), pathologic Q waves, intraventricular conduction delay, and right or left bundle branch block.1,10–12
Elevated troponin and new focal or global left ventricular dysfunction on cardiac imaging indicates myocarditis, especially in a patient with a normal coronary angiogram.10–13
Pericardial effusion: Tachycardia and low QRS voltage
Pericardial effusion is often a complication of pericarditis, but it can also develop from other conditions, such as myxedema, uremia, malignancy, connective tissue disease, aortic dissection, and postpericardiotomy syndrome, and it can also be iatrogenic.
The most common ECG sign of pericardial effusion is tachycardia and low voltage of the QRS complexes. Low voltage is defined as a total amplitude of the QRS complexes in each of the six limb leads less than or equal to 5 mm, and less than or equal to 10 mm in V1 through V6. However, low voltage is not always present in the chest leads.
Mechanisms proposed to explain low QRS voltage associated with pericardial effusion include internal short-circuiting of the electrical currents by accumulated fluids within the pericardial sac, greater distance of the heart from body surface electrodes, reduced cardiac size caused by effusion, and change in the generation and propagation of electrical current in the myocardium.14,15
Cardiac tamponade: Tachycardia, electrical alternans, low QRS voltage
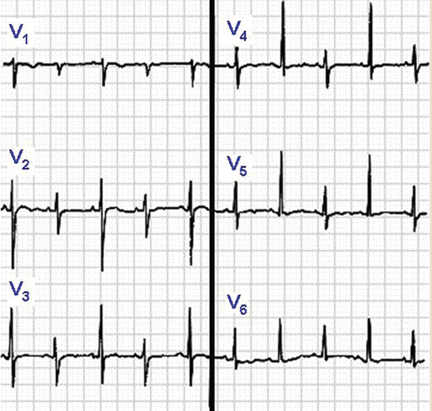
Sinus tachycardia and electrical alternans are specific but not sensitive signs of pericardial tamponade (Figure 3).16,17 Electrical alternans is characterized by beat-to-beat alterations in the axis of QRS complexes in the limb and precordial leads as a result of the mechanical swinging of the heart in a large pericardial effusion.17 There is evidence to suggest that low QRS voltage is more the result of the tamponade than the effusion.18
Treating tamponade with pericardiocentesis, surgical creation of a fistula (“window”) between the pericardial space and the pleural cavity, or anti-inflammatory drugs can resolve low QRS voltage within 1 week.
DIFFERENTIAL DIAGNOSIS OF ACUTE PERICARDITIS
Acute myocardial infarction
ECG changes in acute pericarditis differ from those in acute myocardial infarction in many ways.
ST-segment elevation in pericarditis rarely exceeds 5 mm, in contrast to acute myocardial infarction, in which ST elevation at the J point has to be more than 2 mm and in two anatomically contiguous leads.19
In pericarditis, the changes occur more slowly and in stages, reflecting the evolving inflammation of different areas of the pericardium.
The ST segment is elevated diffusely in the precordial and limb leads in pericarditis, indicating involvement of more than one coronary vascular territory, differentiating it from characteristic regional changes in myocardial infarction.19,20
If concomitant atrial injury is present with acute pericarditis, then PR elevation in aVR with PR depression in other leads may be seen.
Finally, pathologic Q waves or high-grade heart block reflects acute myocardial infarction.
Early repolarization: Elevation of the J point
Early repolarization is sometimes seen in healthy young people, especially in black men.
Early repolarization is characterized by elevation of the J point (ie, the junction between the end of the QRS complex and the beginning of the ST segment). Elevation of the J point causes elevation of the ST segment in the mid to lateral precordial leads (V3–V6) with an up-right T wave.21
Acute pericarditis tends to cause ST-segment elevation in both the limb and precordial leads, whereas ST elevation in early repolarization mainly involves the lateral chest leads.
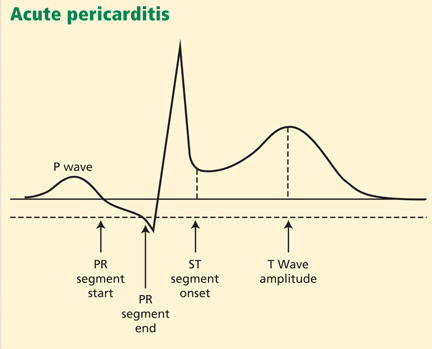
The PR segment is more prominent in acute pericarditis, especially in lead aVR.
Another finding that strongly favors acute pericarditis is the ratio of the height of the ST-segment junction to the height of the apex of the T wave of more than 0.25 in leads I, V4, V5, and V6 (Figure 4).5,8,22
- Imazio M, Trinchero R. Triage and management of acute pericarditis. Int J Cardiol 2007; 118:286–294.
- Little WC, Freeman GL. Pericardial disease. Circulation 2006; 113:1622–1632.
- Imazio M, Spodick DH, Brucato A, Trinchero R, Markel G, Adler Y. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract 2010; 64:1384–1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Shabetai R. Acute pericarditis. Cardiol Clin 1990; 8:639–644.
- Baljepally R, Spodick DH. PR-segment deviation as the initial electrocardiographic response in acute pericarditis. Am J Cardiol 1998; 81:1505–1506.
- Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
- Spodick D, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:46–64.
- Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation 1997; 95:163–168.
- Sarda L, Colin P, Boccara F, et al. Myocarditis in patients with clinical presentation of myocardial infarction and normal coronary angiograms. J Am Coll Cardiol 2001; 37:786–792.
- Spodick DH. Arrhythmias during acute pericarditis. A prospective study of 100 consecutive cases. JAMA 1976; 235:39–41.
- Imazio M, Trinchero R. Myopericarditis: etiology, management, and prognosis. Int J Cardiol 2008; 127:17–26.
- Toney JC, Kolmen SN. Cardiac tamponade: fluid and pressure effects on electrocardiographic changes. Proc Soc Exp Biol Med 1966; 121:642–648.
- Karatay CM, Fruehan CT, Lighty GW, Spear RM, Smulyan H. Acute pericardial distension in pigs: effect of fluid conductance on body surface electrocardiogram QRS size. Cardiovasc Res 1993; 27:1033–1038.
- Spodick DH. Acute cardiac tamponade. Pathologic physiology, diagnosis and management. Prog Cardiovasc Dis 1967; 10:64–96.
- Eisenberg MJ, de Romeral LM, Heidenreich PA, Schiller NB, Evans GT. The diagnosis of pericardial effusion and cardiac tamponade by 12-lead ECG. A technology assessment. Chest 1996; 110:318–324.
- Bruch C, Schmermund A, Dagres N, et al. Changes in QRS voltage in cardiac tamponade and pericardial effusion: reversibility after pericardiocentesis and after anti-inflammatory drug treatment. J Am Coll Cardiol 2001; 38:219–226.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brady WJ, Perron A, Ullman E. Errors in emergency physician interpretation of ST-segment elevation in emergency department chest pain patients. Acad Emerg Med 2000; 7:1256–1260.
- Kambara H, Phillips J. Long-term evaluation of early repolarization syndrome (normal variant RS-T segment elevation). Am J Cardiol 1976; 38:157–166.
- Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
Yes. Acute pericarditis has a unique clinical presentation, physical findings, and electrocardiographic (ECG) changes. ECG is always ordered to look for ischemic changes in patients with chest pain. Acute pericarditis develops in stages, which makes it easy to differentiate from early repolarization and, more significantly, myocardial infarction. The ECG changes, along with the clinical presentation and physical findings, can make the diagnosis of pericarditis.
In atypical and complicated cases, advanced imaging studies (ie, echocardiography and cardiac magnetic resonance imaging) have been used to confirm the diagnosis and to follow the course of the disease. However, ECG remains a useful, cost-effective test.
PERICARDIAL DISEASE IS DIVERSE
The pericardium is a thin layer that covers the heart and separates it from other structures in the mediastinum.
Pericardial syndromes include acute, recurrent, constrictive, and effusive-constrictive pericarditis, as well as pericardial effusion with or without tamponade. Causes include viral or bacterial infection, postpericardiotomy syndrome (Dressler syndrome), postmyocardial infarction, primary and metastatic tumors, trauma, uremia, radiation, and autoimmune disease, but pericardial syndromes can also be idiopathic.1
Acute pericarditis is the most common pericardial syndrome and occurs in all age groups. Once diagnosed, it can easily be treated with antiinflammatory drugs. However, recurrent pericarditis, reported in 30% of patients experiencing a first attack of pericarditis, can be difficult to manage, can have a significant impact on the patient’s health, and can be life-threatening.2
CHANGES OF ACUTE PERICARDITIS DEVELOP IN STAGES
Pericarditis can be diagnosed on the basis of ECG changes, clinical signs and symptoms, and laboratory and imaging findings.3 ECG criteria of acute pericarditis have been published.4,5
The characteristic chest pain in acute pericarditis is usually sudden in onset and sharp and occurs over the anterior chest wall. The pain is exacerbated by inspiration and decreases when the patient sits up and leans forward.4
ECG classically shows a widespread saddle-shaped (upward concave) ST-segment elevation in the precordial and limb leads, reflecting subepicardial inflammation. PR-segment depression (with PR-segment elevation in lead aVR) can accompany or precede the ST changes and is known as the “discordant ST-PR segment sign” (Figures 1 and 2). These changes are seen in 60% of patients.
The ECG changes develop in stages, making them easy to differentiate from early repolarization and, more significantly, from myocardial infarction. Four stages are apparent1,4,6–9:
- Stage I occurs in a few hours to days, with diffuse, up-sloping ST-segment elevation and upright T waves, the result of an alteration in ventricular repolarization caused by pericardial inflammation. Because of alteration in repolarization of the atrium secondary to inflammation, the PR segment is elevated in aVR and depressed in the rest of the limb and chest leads.
- Stage II—the ST and PR segments normalize.
- Stage III—widespread T-wave inversion.
- Stage IV—normalization of the T waves.
There is no pathologic Q-wave formation or loss of R-wave progression in acute pericarditis.
The ECG changes of pericarditis vary widely from one patient to another, depending on the extent and severity of pericardial inflammation and the timing of the patient’s presentation. Changes vary in duration. In some cases, ST elevation returns to baseline within a few days without T-wave inversions; in other cases, T-wave inversions can persist for weeks to months. Sometimes the abnormalities resolve by the time symptoms develop.
ASSOCIATED CONDITIONS
Myocardial involvement
In acute myocarditis, findings on ECG can be normal unless the pericardium is involved. Changes that can be seen in myocarditis and that indicate a deeper involvement of inflammation include ST-segment abnormalities, arrhythmias (eg, premature ventricular or atrial contractions), pathologic Q waves, intraventricular conduction delay, and right or left bundle branch block.1,10–12
Elevated troponin and new focal or global left ventricular dysfunction on cardiac imaging indicates myocarditis, especially in a patient with a normal coronary angiogram.10–13
Pericardial effusion: Tachycardia and low QRS voltage
Pericardial effusion is often a complication of pericarditis, but it can also develop from other conditions, such as myxedema, uremia, malignancy, connective tissue disease, aortic dissection, and postpericardiotomy syndrome, and it can also be iatrogenic.
The most common ECG sign of pericardial effusion is tachycardia and low voltage of the QRS complexes. Low voltage is defined as a total amplitude of the QRS complexes in each of the six limb leads less than or equal to 5 mm, and less than or equal to 10 mm in V1 through V6. However, low voltage is not always present in the chest leads.
Mechanisms proposed to explain low QRS voltage associated with pericardial effusion include internal short-circuiting of the electrical currents by accumulated fluids within the pericardial sac, greater distance of the heart from body surface electrodes, reduced cardiac size caused by effusion, and change in the generation and propagation of electrical current in the myocardium.14,15
Cardiac tamponade: Tachycardia, electrical alternans, low QRS voltage
Sinus tachycardia and electrical alternans are specific but not sensitive signs of pericardial tamponade (Figure 3).16,17 Electrical alternans is characterized by beat-to-beat alterations in the axis of QRS complexes in the limb and precordial leads as a result of the mechanical swinging of the heart in a large pericardial effusion.17 There is evidence to suggest that low QRS voltage is more the result of the tamponade than the effusion.18
Treating tamponade with pericardiocentesis, surgical creation of a fistula (“window”) between the pericardial space and the pleural cavity, or anti-inflammatory drugs can resolve low QRS voltage within 1 week.
DIFFERENTIAL DIAGNOSIS OF ACUTE PERICARDITIS
Acute myocardial infarction
ECG changes in acute pericarditis differ from those in acute myocardial infarction in many ways.
ST-segment elevation in pericarditis rarely exceeds 5 mm, in contrast to acute myocardial infarction, in which ST elevation at the J point has to be more than 2 mm and in two anatomically contiguous leads.19
In pericarditis, the changes occur more slowly and in stages, reflecting the evolving inflammation of different areas of the pericardium.
The ST segment is elevated diffusely in the precordial and limb leads in pericarditis, indicating involvement of more than one coronary vascular territory, differentiating it from characteristic regional changes in myocardial infarction.19,20
If concomitant atrial injury is present with acute pericarditis, then PR elevation in aVR with PR depression in other leads may be seen.
Finally, pathologic Q waves or high-grade heart block reflects acute myocardial infarction.
Early repolarization: Elevation of the J point
Early repolarization is sometimes seen in healthy young people, especially in black men.
Early repolarization is characterized by elevation of the J point (ie, the junction between the end of the QRS complex and the beginning of the ST segment). Elevation of the J point causes elevation of the ST segment in the mid to lateral precordial leads (V3–V6) with an up-right T wave.21
Acute pericarditis tends to cause ST-segment elevation in both the limb and precordial leads, whereas ST elevation in early repolarization mainly involves the lateral chest leads.
The PR segment is more prominent in acute pericarditis, especially in lead aVR.
Another finding that strongly favors acute pericarditis is the ratio of the height of the ST-segment junction to the height of the apex of the T wave of more than 0.25 in leads I, V4, V5, and V6 (Figure 4).5,8,22
Yes. Acute pericarditis has a unique clinical presentation, physical findings, and electrocardiographic (ECG) changes. ECG is always ordered to look for ischemic changes in patients with chest pain. Acute pericarditis develops in stages, which makes it easy to differentiate from early repolarization and, more significantly, myocardial infarction. The ECG changes, along with the clinical presentation and physical findings, can make the diagnosis of pericarditis.
In atypical and complicated cases, advanced imaging studies (ie, echocardiography and cardiac magnetic resonance imaging) have been used to confirm the diagnosis and to follow the course of the disease. However, ECG remains a useful, cost-effective test.
PERICARDIAL DISEASE IS DIVERSE
The pericardium is a thin layer that covers the heart and separates it from other structures in the mediastinum.
Pericardial syndromes include acute, recurrent, constrictive, and effusive-constrictive pericarditis, as well as pericardial effusion with or without tamponade. Causes include viral or bacterial infection, postpericardiotomy syndrome (Dressler syndrome), postmyocardial infarction, primary and metastatic tumors, trauma, uremia, radiation, and autoimmune disease, but pericardial syndromes can also be idiopathic.1
Acute pericarditis is the most common pericardial syndrome and occurs in all age groups. Once diagnosed, it can easily be treated with antiinflammatory drugs. However, recurrent pericarditis, reported in 30% of patients experiencing a first attack of pericarditis, can be difficult to manage, can have a significant impact on the patient’s health, and can be life-threatening.2
CHANGES OF ACUTE PERICARDITIS DEVELOP IN STAGES
Pericarditis can be diagnosed on the basis of ECG changes, clinical signs and symptoms, and laboratory and imaging findings.3 ECG criteria of acute pericarditis have been published.4,5
The characteristic chest pain in acute pericarditis is usually sudden in onset and sharp and occurs over the anterior chest wall. The pain is exacerbated by inspiration and decreases when the patient sits up and leans forward.4
ECG classically shows a widespread saddle-shaped (upward concave) ST-segment elevation in the precordial and limb leads, reflecting subepicardial inflammation. PR-segment depression (with PR-segment elevation in lead aVR) can accompany or precede the ST changes and is known as the “discordant ST-PR segment sign” (Figures 1 and 2). These changes are seen in 60% of patients.
The ECG changes develop in stages, making them easy to differentiate from early repolarization and, more significantly, from myocardial infarction. Four stages are apparent1,4,6–9:
- Stage I occurs in a few hours to days, with diffuse, up-sloping ST-segment elevation and upright T waves, the result of an alteration in ventricular repolarization caused by pericardial inflammation. Because of alteration in repolarization of the atrium secondary to inflammation, the PR segment is elevated in aVR and depressed in the rest of the limb and chest leads.
- Stage II—the ST and PR segments normalize.
- Stage III—widespread T-wave inversion.
- Stage IV—normalization of the T waves.
There is no pathologic Q-wave formation or loss of R-wave progression in acute pericarditis.
The ECG changes of pericarditis vary widely from one patient to another, depending on the extent and severity of pericardial inflammation and the timing of the patient’s presentation. Changes vary in duration. In some cases, ST elevation returns to baseline within a few days without T-wave inversions; in other cases, T-wave inversions can persist for weeks to months. Sometimes the abnormalities resolve by the time symptoms develop.
ASSOCIATED CONDITIONS
Myocardial involvement
In acute myocarditis, findings on ECG can be normal unless the pericardium is involved. Changes that can be seen in myocarditis and that indicate a deeper involvement of inflammation include ST-segment abnormalities, arrhythmias (eg, premature ventricular or atrial contractions), pathologic Q waves, intraventricular conduction delay, and right or left bundle branch block.1,10–12
Elevated troponin and new focal or global left ventricular dysfunction on cardiac imaging indicates myocarditis, especially in a patient with a normal coronary angiogram.10–13
Pericardial effusion: Tachycardia and low QRS voltage
Pericardial effusion is often a complication of pericarditis, but it can also develop from other conditions, such as myxedema, uremia, malignancy, connective tissue disease, aortic dissection, and postpericardiotomy syndrome, and it can also be iatrogenic.
The most common ECG sign of pericardial effusion is tachycardia and low voltage of the QRS complexes. Low voltage is defined as a total amplitude of the QRS complexes in each of the six limb leads less than or equal to 5 mm, and less than or equal to 10 mm in V1 through V6. However, low voltage is not always present in the chest leads.
Mechanisms proposed to explain low QRS voltage associated with pericardial effusion include internal short-circuiting of the electrical currents by accumulated fluids within the pericardial sac, greater distance of the heart from body surface electrodes, reduced cardiac size caused by effusion, and change in the generation and propagation of electrical current in the myocardium.14,15
Cardiac tamponade: Tachycardia, electrical alternans, low QRS voltage
Sinus tachycardia and electrical alternans are specific but not sensitive signs of pericardial tamponade (Figure 3).16,17 Electrical alternans is characterized by beat-to-beat alterations in the axis of QRS complexes in the limb and precordial leads as a result of the mechanical swinging of the heart in a large pericardial effusion.17 There is evidence to suggest that low QRS voltage is more the result of the tamponade than the effusion.18
Treating tamponade with pericardiocentesis, surgical creation of a fistula (“window”) between the pericardial space and the pleural cavity, or anti-inflammatory drugs can resolve low QRS voltage within 1 week.
DIFFERENTIAL DIAGNOSIS OF ACUTE PERICARDITIS
Acute myocardial infarction
ECG changes in acute pericarditis differ from those in acute myocardial infarction in many ways.
ST-segment elevation in pericarditis rarely exceeds 5 mm, in contrast to acute myocardial infarction, in which ST elevation at the J point has to be more than 2 mm and in two anatomically contiguous leads.19
In pericarditis, the changes occur more slowly and in stages, reflecting the evolving inflammation of different areas of the pericardium.
The ST segment is elevated diffusely in the precordial and limb leads in pericarditis, indicating involvement of more than one coronary vascular territory, differentiating it from characteristic regional changes in myocardial infarction.19,20
If concomitant atrial injury is present with acute pericarditis, then PR elevation in aVR with PR depression in other leads may be seen.
Finally, pathologic Q waves or high-grade heart block reflects acute myocardial infarction.
Early repolarization: Elevation of the J point
Early repolarization is sometimes seen in healthy young people, especially in black men.
Early repolarization is characterized by elevation of the J point (ie, the junction between the end of the QRS complex and the beginning of the ST segment). Elevation of the J point causes elevation of the ST segment in the mid to lateral precordial leads (V3–V6) with an up-right T wave.21
Acute pericarditis tends to cause ST-segment elevation in both the limb and precordial leads, whereas ST elevation in early repolarization mainly involves the lateral chest leads.
The PR segment is more prominent in acute pericarditis, especially in lead aVR.
Another finding that strongly favors acute pericarditis is the ratio of the height of the ST-segment junction to the height of the apex of the T wave of more than 0.25 in leads I, V4, V5, and V6 (Figure 4).5,8,22
- Imazio M, Trinchero R. Triage and management of acute pericarditis. Int J Cardiol 2007; 118:286–294.
- Little WC, Freeman GL. Pericardial disease. Circulation 2006; 113:1622–1632.
- Imazio M, Spodick DH, Brucato A, Trinchero R, Markel G, Adler Y. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract 2010; 64:1384–1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Shabetai R. Acute pericarditis. Cardiol Clin 1990; 8:639–644.
- Baljepally R, Spodick DH. PR-segment deviation as the initial electrocardiographic response in acute pericarditis. Am J Cardiol 1998; 81:1505–1506.
- Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
- Spodick D, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:46–64.
- Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation 1997; 95:163–168.
- Sarda L, Colin P, Boccara F, et al. Myocarditis in patients with clinical presentation of myocardial infarction and normal coronary angiograms. J Am Coll Cardiol 2001; 37:786–792.
- Spodick DH. Arrhythmias during acute pericarditis. A prospective study of 100 consecutive cases. JAMA 1976; 235:39–41.
- Imazio M, Trinchero R. Myopericarditis: etiology, management, and prognosis. Int J Cardiol 2008; 127:17–26.
- Toney JC, Kolmen SN. Cardiac tamponade: fluid and pressure effects on electrocardiographic changes. Proc Soc Exp Biol Med 1966; 121:642–648.
- Karatay CM, Fruehan CT, Lighty GW, Spear RM, Smulyan H. Acute pericardial distension in pigs: effect of fluid conductance on body surface electrocardiogram QRS size. Cardiovasc Res 1993; 27:1033–1038.
- Spodick DH. Acute cardiac tamponade. Pathologic physiology, diagnosis and management. Prog Cardiovasc Dis 1967; 10:64–96.
- Eisenberg MJ, de Romeral LM, Heidenreich PA, Schiller NB, Evans GT. The diagnosis of pericardial effusion and cardiac tamponade by 12-lead ECG. A technology assessment. Chest 1996; 110:318–324.
- Bruch C, Schmermund A, Dagres N, et al. Changes in QRS voltage in cardiac tamponade and pericardial effusion: reversibility after pericardiocentesis and after anti-inflammatory drug treatment. J Am Coll Cardiol 2001; 38:219–226.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brady WJ, Perron A, Ullman E. Errors in emergency physician interpretation of ST-segment elevation in emergency department chest pain patients. Acad Emerg Med 2000; 7:1256–1260.
- Kambara H, Phillips J. Long-term evaluation of early repolarization syndrome (normal variant RS-T segment elevation). Am J Cardiol 1976; 38:157–166.
- Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
- Imazio M, Trinchero R. Triage and management of acute pericarditis. Int J Cardiol 2007; 118:286–294.
- Little WC, Freeman GL. Pericardial disease. Circulation 2006; 113:1622–1632.
- Imazio M, Spodick DH, Brucato A, Trinchero R, Markel G, Adler Y. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract 2010; 64:1384–1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Shabetai R. Acute pericarditis. Cardiol Clin 1990; 8:639–644.
- Baljepally R, Spodick DH. PR-segment deviation as the initial electrocardiographic response in acute pericarditis. Am J Cardiol 1998; 81:1505–1506.
- Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
- Spodick D, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:46–64.
- Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation 1997; 95:163–168.
- Sarda L, Colin P, Boccara F, et al. Myocarditis in patients with clinical presentation of myocardial infarction and normal coronary angiograms. J Am Coll Cardiol 2001; 37:786–792.
- Spodick DH. Arrhythmias during acute pericarditis. A prospective study of 100 consecutive cases. JAMA 1976; 235:39–41.
- Imazio M, Trinchero R. Myopericarditis: etiology, management, and prognosis. Int J Cardiol 2008; 127:17–26.
- Toney JC, Kolmen SN. Cardiac tamponade: fluid and pressure effects on electrocardiographic changes. Proc Soc Exp Biol Med 1966; 121:642–648.
- Karatay CM, Fruehan CT, Lighty GW, Spear RM, Smulyan H. Acute pericardial distension in pigs: effect of fluid conductance on body surface electrocardiogram QRS size. Cardiovasc Res 1993; 27:1033–1038.
- Spodick DH. Acute cardiac tamponade. Pathologic physiology, diagnosis and management. Prog Cardiovasc Dis 1967; 10:64–96.
- Eisenberg MJ, de Romeral LM, Heidenreich PA, Schiller NB, Evans GT. The diagnosis of pericardial effusion and cardiac tamponade by 12-lead ECG. A technology assessment. Chest 1996; 110:318–324.
- Bruch C, Schmermund A, Dagres N, et al. Changes in QRS voltage in cardiac tamponade and pericardial effusion: reversibility after pericardiocentesis and after anti-inflammatory drug treatment. J Am Coll Cardiol 2001; 38:219–226.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brady WJ, Perron A, Ullman E. Errors in emergency physician interpretation of ST-segment elevation in emergency department chest pain patients. Acad Emerg Med 2000; 7:1256–1260.
- Kambara H, Phillips J. Long-term evaluation of early repolarization syndrome (normal variant RS-T segment elevation). Am J Cardiol 1976; 38:157–166.
- Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
When do Raynaud symptoms merit a workup for autoimmune rheumatic disease?
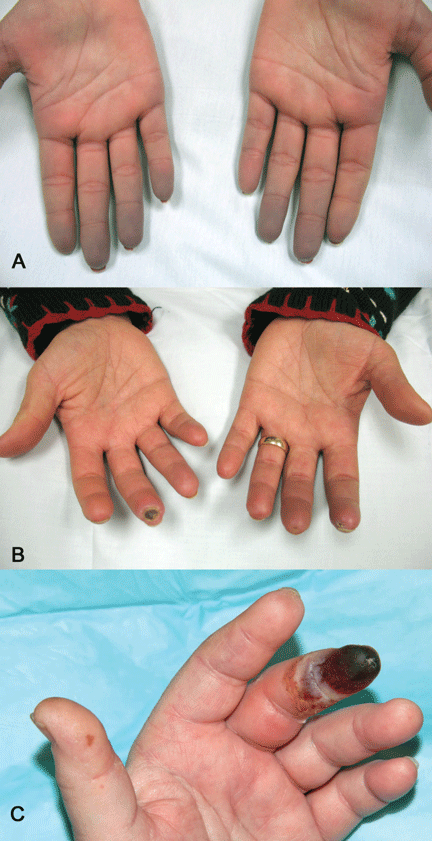
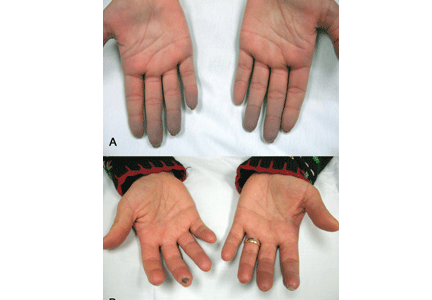
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
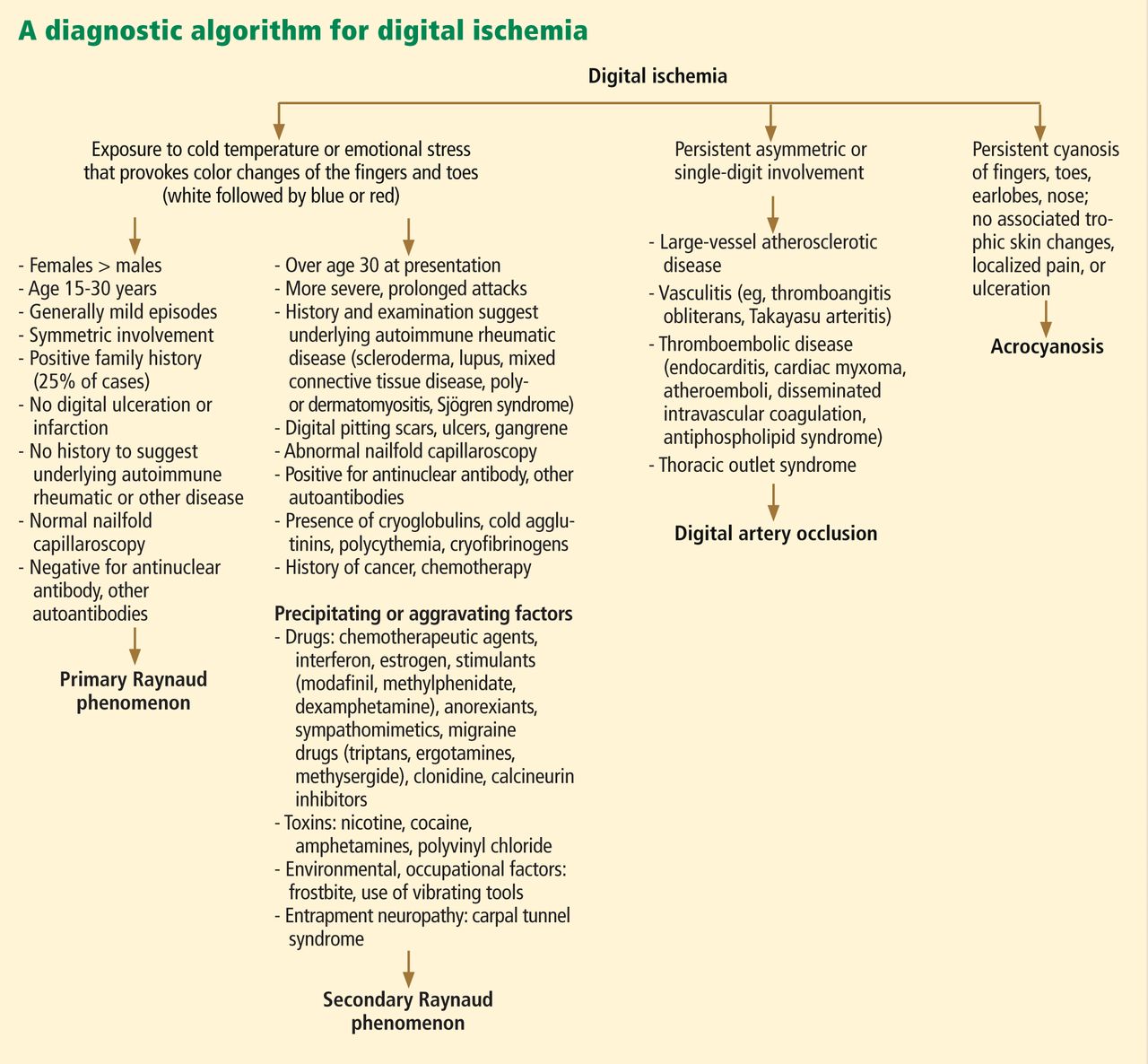
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
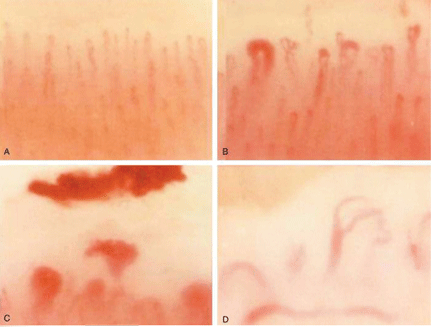
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
Should N-acetylcysteine be used routinely to prevent contrast-induced acute kidney injury?
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
Should I order an anti-CCP antibody test to diagnose rheumatoid arthritis?
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.

However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.
However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.
However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
Should target natriuretic peptide levels be used for outpatient management of chronic heart failure?
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
What is the best questionnaire to screen for alcohol use disorder in an office practice?
Popular questionnaires to screen for alcohol misuse include the CAGE, the TWEAK, and the short form of the Alcohol Use Disorder Identification Test (AUDIT-C). Any of these is recommended. The important thing is to be proactive about screening for this very common and underrecognized problem.
A COMMON PROBLEM, NOT OFTEN ADMITTED
Alcohol use disorder, which ranges from hazardous drinking to binge drinking and alcohol dependence, is more common than admitted and often goes undiagnosed. Its personal, societal, and economic consequences cannot be overemphasized. Alcohol use is responsible for 85,000 deaths each year in the United States, and it is linked to substantial medical and psychiatric consequences and injuries, especially motor vehicle accidents. The estimated annual cost of problems attributed to alcohol use is over $185 billion.1
About three in 10 US adults drink at levels that increase their risk for alcohol-related consequences, and about one in four adults currently abuses alcohol or is dependent on it.2 In 2009, 6.8% of the US population age 12 and above reported heavy drinking, with highest rates in those ages 21 to 29.3 The rate of alcohol use was higher in men than in women, but about 10% of pregnant women ages 15 to 44 reported current alcohol use.3
The prevalence of alcohol use disorder ranges from 2% to 29% in a typical ambulatory primary care medical practice.4 And only one-third of people with alcohol use disorder are diagnosed.
Studies and experience have shown that problem drinkers tend to not seek help until they have advanced dependence, often with associated medical and sociolegal complications. It is also well established that the earlier the diagnosis is made and appropriate intervention is offered, the better the prognosis.
WHAT IS THE GOAL OF SCREENING?
The goals of screening for alcohol use disorder are to estimate the patient’s risk level, to identify those at risk because they exceed defined limits, and to identify those with evidence of an active problem, ie, with adverse consequences related to their drinking. This screening paves the way for further assessment, definitive diagnosis, and a treatment plan.
The US Preventive Services Task Force recommends screening and behavioral counseling interventions (such as a brief intervention) in the primary care setting to reduce alcohol misuse by adults, including pregnant women.5 In addition, most primary care patients who screen positive for heavy drinking or alcohol use disorder show motivation and readiness to change, and those with the most severe symptoms tend to be the most ready.6
THE IDEAL QUESTIONNAIRE: SENSITIVE, SPECIFIC, AND SHORT
The ideal alcohol screening questionnaire for a busy practice should be brief and highly sensitive and specific for identifying the spectrum of alcohol misuse. Also, it should be easy to recall so it can be part of routine face-to-face discussion with the patient during an office visit.
Further, it should include questions that focus on the consequences of drinking as well as on quantity and frequency. It should also take into account factors such as the patient’s age, sex, race or ethnicity, and pregnancy status, as these can influence the effectiveness of the screening method.
Problems with focusing on quantity alone
“Risky use” is defined (in a non-alcohol-dependent person or one with no alcohol-related consequences) as more than seven standard drinks per week or more than three per occasion for women, and more than 14 standard drinks per week or more than four per occasion for men.2
A standard drink in the United States contains about 12 to 14 g of ethanol: a 12-oz can or bottle of beer, a 5-oz glass of wine, or about 1.5 oz of 80-proof liquor.2
The common single-item screening test asks, “How many times in the past year have you had more than four drinks (for women) or five drinks (for men) in a day?” This is recommended by the National Institute on Alcohol Abuse and Alcoholism for brief screening in primary care. However, a positive answer (ie, one or more times in the past year) has a sensitivity of only 82% and a specificity of only 79% for detecting unhealthy alcohol use, and an even lower specificity (67%) for detecting current alcohol use disorder.7
The CAGE questionnaire
The four-item CAGE questionnaire8 focuses on the consequences of drinking:
- C: Have you felt the need to cut down on your drinking?
- A: Have you ever felt annoyed by someone criticizing your drinking?
- G: Have you ever felt bad or guilty about your drinking?
- E: Have you ever had an eye-opener—a drink the first thing in the morning to steady your nerves?
A yes to one or more of the questions denotes a need for further assessment.
The CAGE questionnaire is simple, non-threatening, brief, and easy to remember. A yes answer to two or more items has a sensitivity of 75% to 95% and a specificity of 84% to 97% for alcohol dependence.9 However, CAGE is less sensitive for identifying nonalcohol-dependent at-risk drinkers. The patient’s sex and ethnicity have also been found to affect its performance somewhat, with some studies showing a sensitivity as low as 50% in adult white women and as low as 40% in at-risk groups ages 60 and over.
The TWEAK questionnaire
The TWEAK is a modification of the CAGE and includes a question about tolerance; it has a sensitivity of 87% for harmful drinking and 84% for dependence, especially in trauma-related cases.9 It has also been found to be better than the CAGE for screening pregnant patients.
- Tolerance: How many drinks can you hold without falling asleep or passing out? (2 points if six drinks or more)
- Worried: Have friends or relatives worried about your drinking? (2 points if yes)
- Eye-opener: Do you sometimes take a drink in the morning when you first get up? (1 point if yes)
- Amnesia: Have friends or relatives told you about things you said or did while drinking that you could not remember? (1 point if yes)
- Cut down: Do you sometimes feel the need to cut down on your drinking? (1 point if yes)
An answer of ≥ 6 to the first question or a total score of 3 or more denotes a problem with alcohol use and a need for further assessment.10
The AUDIT-C
The AUDIT-C, a shorter form of the 10-item AUDIT developed by the World Health Organization, uses only the first three questions of the full-length AUDIT. The three-item AUDIT-C has a sensitivity ranging from 85% in Hispanic women to 95% in white men.9,11 The questions center on the quantity and frequency of alcohol use:
- How often do you have a drink containing alcohol? Answer choices: never; monthly or less often; 2 to 4 times a month; 2 to 3 times a week; 4 or more times a week.
- How many standard drinks containing alcohol do you have on a typical day when you are drinking? Answer choices: one or two; three or four; five or six; seven to nine; 10 or more.
- How often do you have six or more drinks on one occasion? Answer choices: never, less than monthly; monthly; weekly; daily or almost.
Scoring is 0 for never, and 1, 2, 3, or 4 for the subsequent answer choices in each question.
The cut-off score for the AUDIT-C is usually a total of 3 points for women and 4 for men: ie, a score of 3 or higher for women and a score of 4 or higher for men indicate alcohol use disorder and the need for further assessment.
The AUDIT questionnaire has been found not only to have a high sensitivity (83%) and specificity (90%) for identifying alcohol dependence, but also to be more sensitive than the CAGE questionnaire (85% vs 75%) for identifying harmful drinking, hazardous drinking, and at-risk drinking. (Note: The full version of AUDIT performed similarly to the three-item AUDIT-C for detecting heavy drinking and active abuse or dependence.12) Furthermore, it has performed well as a screening test in many multinational trials of alcohol brief intervention. The questions about quantity of alcohol consumed may be even more suitable for adolescents and young adults, who tend to fall into the harmful-hazardous drinking category rather than the dependent category. In some studies, patients tended to reveal less with the CAGE questionnaire when it was preceded by direct and close-ended questions about the quantity and frequency of alcohol use, thus reducing its sensitivity.13
The AUDIT and TWEAK questionnaires showed greater sensitivity in both men and women than the CAGE questionnaire and were equally sensitive in African Americans.14
HOW TO FIT ALCOHOL SCREENING INTO AN OFFICE VISIT
A practical way to fit alcohol screening into an office visit is to include a questionnaire in the assessment papers completed by the patient while in the waiting room. In other settings, these questions may be asked by trained nursing staff as part of the initial assessment, ie, while obtaining the patient’s weight and vital statistics. This can be briefly reviewed by the physician during the face-to-face history and physical examination.
A concerted effort is needed to proactively screen for alcohol use. A combination of questions about the effect, the quantity, and the frequency of alcohol use is the best way to screen for the many different aspects of alcohol use disorder—many of which can be managed in the primary care setting through brief interventions without referral to a specialist.
When screening for alcohol misuse, it is also important to consider factors such as age, sex, race or ethnicity, pregnancy, and history of recent trauma or surgery.
- Saitz R. Clinical practice. Unhealthy alcohol use. N Engl J Med 2005; 352:596–607.
- National institute on Alcohol Abuse and Alcoholism (NIAAA). Helping patients who drink too much: A clinician’s guide and related professional support resources. http://pubs.niaaa.nih.gov/publications/practitioner/cliniciansguide2005/clinicians_guide.htm. Accessed July 29, 2011.
- Substance Abuse and Mental Health Services Administration (SAMHSA). Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings. http://www.oas.samhsa.gov/NSDUH/2k9NSDUH/2k9ResultsP.pdf. Accessed July 29, 2011.
- Fiellin DA, Reid MC, O’Connor PG. Screening for alcohol problems in primary care: a systematic review. Arch Intern Med 2000; 160:1977–1989.
- US Preventive Services Task Force (USPSTF). Screening and behavioral counseling interventions in primary care to reduce alcohol misuse. Release date: April 2004. http://www.uspreventiveservicestaskforce.org/uspstf/uspsdrin.htm. Accessed July 29, 2011.
- Williams EC, Kivlahan DR, Saitz R, et al. Readiness to change in primary care patients who screened positive for alcohol misuse. Ann Fam Med 2006; 4:213–220.
- Smith PC, Schmidt SM, Allensworth-Davies D, Saitz R. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009; 24:783–788.
- Ewing JA. Detecting alcoholism. The CAGE questionnaire. JAMA 1984; 252:1905–1907.
- Cherpitel CJ. Screening for alcohol problems in the emergency department. Ann Emerg Med 1995; 26:158–166.
- Russell M, Martier SS, Sokol RJ, et al. Screening for pregnancy risk-drinking. Alcohol Clin Exp Res 1994; 18:1156–1161.
- Frank D, DeBenedetti AF, Volk RJ, Williams EC, Kivlahan DR, Bradley KA. Effectiveness of the AUDIT-C as a screening test for alcohol misuse in three race/ethnic groups. J Gen Intern Med 2008; 23:781–787.
- Bush K, Kivlahan DR, McDonell MB, Fihn SD, Bradley KA. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med 1998; 158:1789–1795.
- Steinweg DL, Worth H. Alcoholism: the keys to the CAGE. Am J Med 1993; 94:520–523.
- Cherpitel CJ. Brief screening instruments for alcoholism. Alcohol Health Res World 1997; 21:348–351.
Popular questionnaires to screen for alcohol misuse include the CAGE, the TWEAK, and the short form of the Alcohol Use Disorder Identification Test (AUDIT-C). Any of these is recommended. The important thing is to be proactive about screening for this very common and underrecognized problem.
A COMMON PROBLEM, NOT OFTEN ADMITTED
Alcohol use disorder, which ranges from hazardous drinking to binge drinking and alcohol dependence, is more common than admitted and often goes undiagnosed. Its personal, societal, and economic consequences cannot be overemphasized. Alcohol use is responsible for 85,000 deaths each year in the United States, and it is linked to substantial medical and psychiatric consequences and injuries, especially motor vehicle accidents. The estimated annual cost of problems attributed to alcohol use is over $185 billion.1
About three in 10 US adults drink at levels that increase their risk for alcohol-related consequences, and about one in four adults currently abuses alcohol or is dependent on it.2 In 2009, 6.8% of the US population age 12 and above reported heavy drinking, with highest rates in those ages 21 to 29.3 The rate of alcohol use was higher in men than in women, but about 10% of pregnant women ages 15 to 44 reported current alcohol use.3
The prevalence of alcohol use disorder ranges from 2% to 29% in a typical ambulatory primary care medical practice.4 And only one-third of people with alcohol use disorder are diagnosed.
Studies and experience have shown that problem drinkers tend to not seek help until they have advanced dependence, often with associated medical and sociolegal complications. It is also well established that the earlier the diagnosis is made and appropriate intervention is offered, the better the prognosis.
WHAT IS THE GOAL OF SCREENING?
The goals of screening for alcohol use disorder are to estimate the patient’s risk level, to identify those at risk because they exceed defined limits, and to identify those with evidence of an active problem, ie, with adverse consequences related to their drinking. This screening paves the way for further assessment, definitive diagnosis, and a treatment plan.
The US Preventive Services Task Force recommends screening and behavioral counseling interventions (such as a brief intervention) in the primary care setting to reduce alcohol misuse by adults, including pregnant women.5 In addition, most primary care patients who screen positive for heavy drinking or alcohol use disorder show motivation and readiness to change, and those with the most severe symptoms tend to be the most ready.6
THE IDEAL QUESTIONNAIRE: SENSITIVE, SPECIFIC, AND SHORT
The ideal alcohol screening questionnaire for a busy practice should be brief and highly sensitive and specific for identifying the spectrum of alcohol misuse. Also, it should be easy to recall so it can be part of routine face-to-face discussion with the patient during an office visit.
Further, it should include questions that focus on the consequences of drinking as well as on quantity and frequency. It should also take into account factors such as the patient’s age, sex, race or ethnicity, and pregnancy status, as these can influence the effectiveness of the screening method.
Problems with focusing on quantity alone
“Risky use” is defined (in a non-alcohol-dependent person or one with no alcohol-related consequences) as more than seven standard drinks per week or more than three per occasion for women, and more than 14 standard drinks per week or more than four per occasion for men.2
A standard drink in the United States contains about 12 to 14 g of ethanol: a 12-oz can or bottle of beer, a 5-oz glass of wine, or about 1.5 oz of 80-proof liquor.2
The common single-item screening test asks, “How many times in the past year have you had more than four drinks (for women) or five drinks (for men) in a day?” This is recommended by the National Institute on Alcohol Abuse and Alcoholism for brief screening in primary care. However, a positive answer (ie, one or more times in the past year) has a sensitivity of only 82% and a specificity of only 79% for detecting unhealthy alcohol use, and an even lower specificity (67%) for detecting current alcohol use disorder.7
The CAGE questionnaire
The four-item CAGE questionnaire8 focuses on the consequences of drinking:
- C: Have you felt the need to cut down on your drinking?
- A: Have you ever felt annoyed by someone criticizing your drinking?
- G: Have you ever felt bad or guilty about your drinking?
- E: Have you ever had an eye-opener—a drink the first thing in the morning to steady your nerves?
A yes to one or more of the questions denotes a need for further assessment.
The CAGE questionnaire is simple, non-threatening, brief, and easy to remember. A yes answer to two or more items has a sensitivity of 75% to 95% and a specificity of 84% to 97% for alcohol dependence.9 However, CAGE is less sensitive for identifying nonalcohol-dependent at-risk drinkers. The patient’s sex and ethnicity have also been found to affect its performance somewhat, with some studies showing a sensitivity as low as 50% in adult white women and as low as 40% in at-risk groups ages 60 and over.
The TWEAK questionnaire
The TWEAK is a modification of the CAGE and includes a question about tolerance; it has a sensitivity of 87% for harmful drinking and 84% for dependence, especially in trauma-related cases.9 It has also been found to be better than the CAGE for screening pregnant patients.
- Tolerance: How many drinks can you hold without falling asleep or passing out? (2 points if six drinks or more)
- Worried: Have friends or relatives worried about your drinking? (2 points if yes)
- Eye-opener: Do you sometimes take a drink in the morning when you first get up? (1 point if yes)
- Amnesia: Have friends or relatives told you about things you said or did while drinking that you could not remember? (1 point if yes)
- Cut down: Do you sometimes feel the need to cut down on your drinking? (1 point if yes)
An answer of ≥ 6 to the first question or a total score of 3 or more denotes a problem with alcohol use and a need for further assessment.10
The AUDIT-C
The AUDIT-C, a shorter form of the 10-item AUDIT developed by the World Health Organization, uses only the first three questions of the full-length AUDIT. The three-item AUDIT-C has a sensitivity ranging from 85% in Hispanic women to 95% in white men.9,11 The questions center on the quantity and frequency of alcohol use:
- How often do you have a drink containing alcohol? Answer choices: never; monthly or less often; 2 to 4 times a month; 2 to 3 times a week; 4 or more times a week.
- How many standard drinks containing alcohol do you have on a typical day when you are drinking? Answer choices: one or two; three or four; five or six; seven to nine; 10 or more.
- How often do you have six or more drinks on one occasion? Answer choices: never, less than monthly; monthly; weekly; daily or almost.
Scoring is 0 for never, and 1, 2, 3, or 4 for the subsequent answer choices in each question.
The cut-off score for the AUDIT-C is usually a total of 3 points for women and 4 for men: ie, a score of 3 or higher for women and a score of 4 or higher for men indicate alcohol use disorder and the need for further assessment.
The AUDIT questionnaire has been found not only to have a high sensitivity (83%) and specificity (90%) for identifying alcohol dependence, but also to be more sensitive than the CAGE questionnaire (85% vs 75%) for identifying harmful drinking, hazardous drinking, and at-risk drinking. (Note: The full version of AUDIT performed similarly to the three-item AUDIT-C for detecting heavy drinking and active abuse or dependence.12) Furthermore, it has performed well as a screening test in many multinational trials of alcohol brief intervention. The questions about quantity of alcohol consumed may be even more suitable for adolescents and young adults, who tend to fall into the harmful-hazardous drinking category rather than the dependent category. In some studies, patients tended to reveal less with the CAGE questionnaire when it was preceded by direct and close-ended questions about the quantity and frequency of alcohol use, thus reducing its sensitivity.13
The AUDIT and TWEAK questionnaires showed greater sensitivity in both men and women than the CAGE questionnaire and were equally sensitive in African Americans.14
HOW TO FIT ALCOHOL SCREENING INTO AN OFFICE VISIT
A practical way to fit alcohol screening into an office visit is to include a questionnaire in the assessment papers completed by the patient while in the waiting room. In other settings, these questions may be asked by trained nursing staff as part of the initial assessment, ie, while obtaining the patient’s weight and vital statistics. This can be briefly reviewed by the physician during the face-to-face history and physical examination.
A concerted effort is needed to proactively screen for alcohol use. A combination of questions about the effect, the quantity, and the frequency of alcohol use is the best way to screen for the many different aspects of alcohol use disorder—many of which can be managed in the primary care setting through brief interventions without referral to a specialist.
When screening for alcohol misuse, it is also important to consider factors such as age, sex, race or ethnicity, pregnancy, and history of recent trauma or surgery.
Popular questionnaires to screen for alcohol misuse include the CAGE, the TWEAK, and the short form of the Alcohol Use Disorder Identification Test (AUDIT-C). Any of these is recommended. The important thing is to be proactive about screening for this very common and underrecognized problem.
A COMMON PROBLEM, NOT OFTEN ADMITTED
Alcohol use disorder, which ranges from hazardous drinking to binge drinking and alcohol dependence, is more common than admitted and often goes undiagnosed. Its personal, societal, and economic consequences cannot be overemphasized. Alcohol use is responsible for 85,000 deaths each year in the United States, and it is linked to substantial medical and psychiatric consequences and injuries, especially motor vehicle accidents. The estimated annual cost of problems attributed to alcohol use is over $185 billion.1
About three in 10 US adults drink at levels that increase their risk for alcohol-related consequences, and about one in four adults currently abuses alcohol or is dependent on it.2 In 2009, 6.8% of the US population age 12 and above reported heavy drinking, with highest rates in those ages 21 to 29.3 The rate of alcohol use was higher in men than in women, but about 10% of pregnant women ages 15 to 44 reported current alcohol use.3
The prevalence of alcohol use disorder ranges from 2% to 29% in a typical ambulatory primary care medical practice.4 And only one-third of people with alcohol use disorder are diagnosed.
Studies and experience have shown that problem drinkers tend to not seek help until they have advanced dependence, often with associated medical and sociolegal complications. It is also well established that the earlier the diagnosis is made and appropriate intervention is offered, the better the prognosis.
WHAT IS THE GOAL OF SCREENING?
The goals of screening for alcohol use disorder are to estimate the patient’s risk level, to identify those at risk because they exceed defined limits, and to identify those with evidence of an active problem, ie, with adverse consequences related to their drinking. This screening paves the way for further assessment, definitive diagnosis, and a treatment plan.
The US Preventive Services Task Force recommends screening and behavioral counseling interventions (such as a brief intervention) in the primary care setting to reduce alcohol misuse by adults, including pregnant women.5 In addition, most primary care patients who screen positive for heavy drinking or alcohol use disorder show motivation and readiness to change, and those with the most severe symptoms tend to be the most ready.6
THE IDEAL QUESTIONNAIRE: SENSITIVE, SPECIFIC, AND SHORT
The ideal alcohol screening questionnaire for a busy practice should be brief and highly sensitive and specific for identifying the spectrum of alcohol misuse. Also, it should be easy to recall so it can be part of routine face-to-face discussion with the patient during an office visit.
Further, it should include questions that focus on the consequences of drinking as well as on quantity and frequency. It should also take into account factors such as the patient’s age, sex, race or ethnicity, and pregnancy status, as these can influence the effectiveness of the screening method.
Problems with focusing on quantity alone
“Risky use” is defined (in a non-alcohol-dependent person or one with no alcohol-related consequences) as more than seven standard drinks per week or more than three per occasion for women, and more than 14 standard drinks per week or more than four per occasion for men.2
A standard drink in the United States contains about 12 to 14 g of ethanol: a 12-oz can or bottle of beer, a 5-oz glass of wine, or about 1.5 oz of 80-proof liquor.2
The common single-item screening test asks, “How many times in the past year have you had more than four drinks (for women) or five drinks (for men) in a day?” This is recommended by the National Institute on Alcohol Abuse and Alcoholism for brief screening in primary care. However, a positive answer (ie, one or more times in the past year) has a sensitivity of only 82% and a specificity of only 79% for detecting unhealthy alcohol use, and an even lower specificity (67%) for detecting current alcohol use disorder.7
The CAGE questionnaire
The four-item CAGE questionnaire8 focuses on the consequences of drinking:
- C: Have you felt the need to cut down on your drinking?
- A: Have you ever felt annoyed by someone criticizing your drinking?
- G: Have you ever felt bad or guilty about your drinking?
- E: Have you ever had an eye-opener—a drink the first thing in the morning to steady your nerves?
A yes to one or more of the questions denotes a need for further assessment.
The CAGE questionnaire is simple, non-threatening, brief, and easy to remember. A yes answer to two or more items has a sensitivity of 75% to 95% and a specificity of 84% to 97% for alcohol dependence.9 However, CAGE is less sensitive for identifying nonalcohol-dependent at-risk drinkers. The patient’s sex and ethnicity have also been found to affect its performance somewhat, with some studies showing a sensitivity as low as 50% in adult white women and as low as 40% in at-risk groups ages 60 and over.
The TWEAK questionnaire
The TWEAK is a modification of the CAGE and includes a question about tolerance; it has a sensitivity of 87% for harmful drinking and 84% for dependence, especially in trauma-related cases.9 It has also been found to be better than the CAGE for screening pregnant patients.
- Tolerance: How many drinks can you hold without falling asleep or passing out? (2 points if six drinks or more)
- Worried: Have friends or relatives worried about your drinking? (2 points if yes)
- Eye-opener: Do you sometimes take a drink in the morning when you first get up? (1 point if yes)
- Amnesia: Have friends or relatives told you about things you said or did while drinking that you could not remember? (1 point if yes)
- Cut down: Do you sometimes feel the need to cut down on your drinking? (1 point if yes)
An answer of ≥ 6 to the first question or a total score of 3 or more denotes a problem with alcohol use and a need for further assessment.10
The AUDIT-C
The AUDIT-C, a shorter form of the 10-item AUDIT developed by the World Health Organization, uses only the first three questions of the full-length AUDIT. The three-item AUDIT-C has a sensitivity ranging from 85% in Hispanic women to 95% in white men.9,11 The questions center on the quantity and frequency of alcohol use:
- How often do you have a drink containing alcohol? Answer choices: never; monthly or less often; 2 to 4 times a month; 2 to 3 times a week; 4 or more times a week.
- How many standard drinks containing alcohol do you have on a typical day when you are drinking? Answer choices: one or two; three or four; five or six; seven to nine; 10 or more.
- How often do you have six or more drinks on one occasion? Answer choices: never, less than monthly; monthly; weekly; daily or almost.
Scoring is 0 for never, and 1, 2, 3, or 4 for the subsequent answer choices in each question.
The cut-off score for the AUDIT-C is usually a total of 3 points for women and 4 for men: ie, a score of 3 or higher for women and a score of 4 or higher for men indicate alcohol use disorder and the need for further assessment.
The AUDIT questionnaire has been found not only to have a high sensitivity (83%) and specificity (90%) for identifying alcohol dependence, but also to be more sensitive than the CAGE questionnaire (85% vs 75%) for identifying harmful drinking, hazardous drinking, and at-risk drinking. (Note: The full version of AUDIT performed similarly to the three-item AUDIT-C for detecting heavy drinking and active abuse or dependence.12) Furthermore, it has performed well as a screening test in many multinational trials of alcohol brief intervention. The questions about quantity of alcohol consumed may be even more suitable for adolescents and young adults, who tend to fall into the harmful-hazardous drinking category rather than the dependent category. In some studies, patients tended to reveal less with the CAGE questionnaire when it was preceded by direct and close-ended questions about the quantity and frequency of alcohol use, thus reducing its sensitivity.13
The AUDIT and TWEAK questionnaires showed greater sensitivity in both men and women than the CAGE questionnaire and were equally sensitive in African Americans.14
HOW TO FIT ALCOHOL SCREENING INTO AN OFFICE VISIT
A practical way to fit alcohol screening into an office visit is to include a questionnaire in the assessment papers completed by the patient while in the waiting room. In other settings, these questions may be asked by trained nursing staff as part of the initial assessment, ie, while obtaining the patient’s weight and vital statistics. This can be briefly reviewed by the physician during the face-to-face history and physical examination.
A concerted effort is needed to proactively screen for alcohol use. A combination of questions about the effect, the quantity, and the frequency of alcohol use is the best way to screen for the many different aspects of alcohol use disorder—many of which can be managed in the primary care setting through brief interventions without referral to a specialist.
When screening for alcohol misuse, it is also important to consider factors such as age, sex, race or ethnicity, pregnancy, and history of recent trauma or surgery.
- Saitz R. Clinical practice. Unhealthy alcohol use. N Engl J Med 2005; 352:596–607.
- National institute on Alcohol Abuse and Alcoholism (NIAAA). Helping patients who drink too much: A clinician’s guide and related professional support resources. http://pubs.niaaa.nih.gov/publications/practitioner/cliniciansguide2005/clinicians_guide.htm. Accessed July 29, 2011.
- Substance Abuse and Mental Health Services Administration (SAMHSA). Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings. http://www.oas.samhsa.gov/NSDUH/2k9NSDUH/2k9ResultsP.pdf. Accessed July 29, 2011.
- Fiellin DA, Reid MC, O’Connor PG. Screening for alcohol problems in primary care: a systematic review. Arch Intern Med 2000; 160:1977–1989.
- US Preventive Services Task Force (USPSTF). Screening and behavioral counseling interventions in primary care to reduce alcohol misuse. Release date: April 2004. http://www.uspreventiveservicestaskforce.org/uspstf/uspsdrin.htm. Accessed July 29, 2011.
- Williams EC, Kivlahan DR, Saitz R, et al. Readiness to change in primary care patients who screened positive for alcohol misuse. Ann Fam Med 2006; 4:213–220.
- Smith PC, Schmidt SM, Allensworth-Davies D, Saitz R. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009; 24:783–788.
- Ewing JA. Detecting alcoholism. The CAGE questionnaire. JAMA 1984; 252:1905–1907.
- Cherpitel CJ. Screening for alcohol problems in the emergency department. Ann Emerg Med 1995; 26:158–166.
- Russell M, Martier SS, Sokol RJ, et al. Screening for pregnancy risk-drinking. Alcohol Clin Exp Res 1994; 18:1156–1161.
- Frank D, DeBenedetti AF, Volk RJ, Williams EC, Kivlahan DR, Bradley KA. Effectiveness of the AUDIT-C as a screening test for alcohol misuse in three race/ethnic groups. J Gen Intern Med 2008; 23:781–787.
- Bush K, Kivlahan DR, McDonell MB, Fihn SD, Bradley KA. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med 1998; 158:1789–1795.
- Steinweg DL, Worth H. Alcoholism: the keys to the CAGE. Am J Med 1993; 94:520–523.
- Cherpitel CJ. Brief screening instruments for alcoholism. Alcohol Health Res World 1997; 21:348–351.
- Saitz R. Clinical practice. Unhealthy alcohol use. N Engl J Med 2005; 352:596–607.
- National institute on Alcohol Abuse and Alcoholism (NIAAA). Helping patients who drink too much: A clinician’s guide and related professional support resources. http://pubs.niaaa.nih.gov/publications/practitioner/cliniciansguide2005/clinicians_guide.htm. Accessed July 29, 2011.
- Substance Abuse and Mental Health Services Administration (SAMHSA). Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings. http://www.oas.samhsa.gov/NSDUH/2k9NSDUH/2k9ResultsP.pdf. Accessed July 29, 2011.
- Fiellin DA, Reid MC, O’Connor PG. Screening for alcohol problems in primary care: a systematic review. Arch Intern Med 2000; 160:1977–1989.
- US Preventive Services Task Force (USPSTF). Screening and behavioral counseling interventions in primary care to reduce alcohol misuse. Release date: April 2004. http://www.uspreventiveservicestaskforce.org/uspstf/uspsdrin.htm. Accessed July 29, 2011.
- Williams EC, Kivlahan DR, Saitz R, et al. Readiness to change in primary care patients who screened positive for alcohol misuse. Ann Fam Med 2006; 4:213–220.
- Smith PC, Schmidt SM, Allensworth-Davies D, Saitz R. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009; 24:783–788.
- Ewing JA. Detecting alcoholism. The CAGE questionnaire. JAMA 1984; 252:1905–1907.
- Cherpitel CJ. Screening for alcohol problems in the emergency department. Ann Emerg Med 1995; 26:158–166.
- Russell M, Martier SS, Sokol RJ, et al. Screening for pregnancy risk-drinking. Alcohol Clin Exp Res 1994; 18:1156–1161.
- Frank D, DeBenedetti AF, Volk RJ, Williams EC, Kivlahan DR, Bradley KA. Effectiveness of the AUDIT-C as a screening test for alcohol misuse in three race/ethnic groups. J Gen Intern Med 2008; 23:781–787.
- Bush K, Kivlahan DR, McDonell MB, Fihn SD, Bradley KA. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med 1998; 158:1789–1795.
- Steinweg DL, Worth H. Alcoholism: the keys to the CAGE. Am J Med 1993; 94:520–523.
- Cherpitel CJ. Brief screening instruments for alcoholism. Alcohol Health Res World 1997; 21:348–351.
Is iron therapy for anemia harmful in the setting of infection?
The harmful effects of iron therapy in the setting of infection are more theoretical than observed, with no irrefutable data to support them. On the other hand, there are also no convincing data to support the benefit of this therapy. If iron is to be used, frequent monitoring of serum iron markers is prudent to avoid iron overload during treatment.
ANEMIA OF INFLAMMATION IS COMPLEX
Anemia that develops in the hospital, especially in the setting of infection or inflammation, is similar hematologically to anemia of chronic disease, except for its acute onset.1
The pathogenesis of anemia in such settings is complex, but the most important causes of this common syndrome include shortening of red cell survival, impaired erythropoietin production, blunted responsiveness of the bone marrow to endogenous erythropoietin, and impaired iron metabolism mediated through the action of inflammatory cytokines.2,3 Other important causes include nutritional deficiencies (iron, vitamin B12, and folic acid)4 and blood loss.5,6
Moreover, anemia of inflammation may be difficult to differentiate from iron-deficiency anemia because the serum iron markers are unreliable in inflammation.1
The reported prevalence of anemia during hospitalization has ranged from 55% on hospital wards7 to 95% in intensive care units.8
Transfusion of packed red blood cells is the fastest treatment for anemia in hospitalized patients and it is the one traditionally used, but many concerns have been raised about its efficacy and adverse effects.9 Erythropoietin, with or without iron therapy, has emerged as an alternative in treating anemia of inflammation.10,11
IRON THERAPY
Iron is widely used to treat anemia, especially in hospitalized patients and those with chronic kidney disease.2 The intravenous route is more commonly used than the oral route, since it has faster action, is better tolerated, and has better bioavailability.1,2
Controversy over benefit
Whether iron supplementation increases the red blood cell mass and reduces the need for blood transfusion is controversial.10,12 Pieracci et al13 documented these benefits in critically ill surgical patients, whereas van Iperen et al11 did not find such benefits in critically ill patients receiving intravenous iron and erythropoietin.
Harmful effects
Some authors1,14 object to giving iron to hospitalized patients (especially critically ill patients) who have infections on the grounds that it is risky, although definitive evidence is lacking.15
Most of the harmful effects of iron have been linked to elevated serum ferritin levels and to non–transferrin-bound iron, more than to iron per se.16 Ferritin is an acute-phase reactant; thus, ferritin levels may be elevated in inflammation and infection regardless of the body iron status.1
Anaphylactic reaction. This rare complication of iron dextran therapy is not much of a concern at present with the newer formulations of iron such as iron gluconate and iron sucrose.16
Oxidative stress. Iron-derived free radicals can cause a rise in inflammatory cytokine levels, especially if the ferritin level is elevated (> 500 μg/L). This cytokine rise is worrisome, as it may have acute detrimental effects on cellular homeostasis, leading to tissue injury,15 while chronically it might be related to enhanced atherosclerosis and cardiac disease.16
Iron overload. In vitro and animal studies have documented an association between elevated ferritin levels (500–650 μg/L) and decreases in T-cell function, polymorphonuclear neutrophil migration, phagocytosis, and bacterial eradication.15 Studies in hemodialysis patients have identified iron overload as an independent risk factor for bacterial infection, but the confounding role of the dialysis process cannot be disregarded.17,18
Bacterial growth. Many bacteria depend on iron for their growth; examples are Escherichia coli; Klebsiella, Pseudomonas, Salmonella, Yersinia, Listeria, and Staphylococcus species; and Haemophilus influenzae. In vitro studies have linked increased bacterial growth with increased transferrin saturation in plasma.15,19
Iron therapy and infection risk
The theory linking iron with risk of infection arose from the observation that patients with hemochromatosis are more susceptible to certain bacterial infections, especially Vibrio vulnificus.20 A few human studies, most of them in chronic hemodialysis patients, have examined the relation between iron therapy and infection risk, with conflicting results.21–26 Multiple studies13,19,21,22,25–27 found no relation between iron therapy and risk of infection or death.
Canziani et al23 found that the risk of infection was higher with higher intravenous doses of iron than with lower doses.
Collins et al24 found a higher risk of sepsis and hospitalization in patients who received iron for a prolonged duration (5–6 months) than in those who did not.
Feldman et al,27 in their report of a study of iron therapy in hemodialysis patients, suggested that previously observed associations between iron administration and higher death rates may have been confounded by other factors.
Iron therapy in concurrent infection
There are no data in humans on the effects of iron therapy on outcomes during concurrent infection or sepsis.15,28 However, mice with sepsis had worse outcomes when treated with intravenous iron.28
A CONUNDRUM IN CLINICAL PRACTICE
After reviewing the available literature, we concur with most of the authors1,15,16,18,19,29 that despite the worrisome theoretical adverse effects of iron therapy in patients with infections, there are no convincing data to support those fears. On the other hand, there are also no convincing data to favor its benefit.
More definitive studies are needed to answer this question, which has been a conundrum in clinical practice. Patients who might benefit from iron therapy should not be deprived of it on the basis of the available data. Frequent monitoring of serum iron markers during therapy to avoid iron overload seems prudent.
- Pieracci FM, Barie PS. Diagnosis and management of iron-related anemias in critical illness. Crit Care Med 2006; 34:1898–1905.
- Krantz SB. Pathogenesis and treatment of the anemia of chronic disease. Am J Med Sci 1994; 307:353–359.
- Price EA, Schrier SL. Unexplained aspects of anemia of inflammation. Review article. Adv Hematol 2010; 2010:508739.
- Rodriguez RM, Corwin HL, Gettinger A, Corwin MJ, Gubler D, Pearl RG. Nutritional deficiencies and blunted erythropoietin response as causes of the anemia of critical illness. J Crit Care 2001; 16:36–41.
- Wong P, Intragumtornchai T. Hospital-acquired anemia. J Med Assoc Thai 2006; 89:63–67.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Reade MC, Weissfeld L, Angus DC, Kellum JA, Milbrandt EB. The prevalence of anemia and its association with 90-day mortality in hospitalized community-acquired pneumonia. BMC Pulm Med 2010; 10:15.
- Debellis RJ. Anemia in critical care patients: incidence, etiology, impact, management, and use of treatment guidelines and protocols. Am J Health Syst Pharm 2007; 64:S14–S21.
- Marik PE. The hazards of blood transfusion. Br J Hosp Med (Lond) 2009; 70:12–15.
- Corwin HL, Gettinger A, Fabian TC, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med 2007; 357:965–976.
- van Iperen CE, Gaillard CA, Kraaijenhagen RJ, Braam BG, Marx JJ, van de Wiel A. Response of erythropoiesis and iron metabolism to recombinant human erythropoietin in intensive care unit patients. Crit Care Med 2000; 28:2773–2778.
- Muñoz M, Breymann C, García-Erce JA, Gómez-Ramirez S, Comin J, Bisbe E. Efficacy and safety of intravenous iron therapy as an alternative/adjunct to allogeneic blood transfusion. Vox Sang 2008; 94:172–183.
- Pieracci FM, Henderson P, Rodney JR, et al. Randomized, double-blind, placebo-controlled trial of effects of enteral iron supplementation on anemia and risk of infection during surgical critical illness. Surg Infect 2009; 10:9–19.
- Pieracci FM, Barie PS. Iron and the risk of infection. Surg Infect 2005; 6(suppl 1):S41–S46.
- Maynor L, Brophy DF. Risk of infections with intravenous iron therapy. Ann Pharmacother 2007; 41:1476–1480.
- Cavill I. Intravenous iron as adjuvant therapy: a two-edged sword? Nephrol Dial Transplant 2003; 18(suppl 8):viii24–viii28.
- Kessler M, Hoen B, Mayeux D, Hestin D, Fontenaille C. Bacteremia in patients on chronic hemodialysis. A multicenter prospective survey. Nephron 1993; 64:95–100.
- Hoen B, Kessler M, Hestin D, Mayeux D. Risk factors for bacterial infections in chronic haemodialysis adult patients: a multicentre prospective survey. Nephrol Dial Transplant 1995; 10:377–381.
- Cieri E. Does iron cause bacterial infections in patients with end stage renal disease? ANNA J 1999; 26:591–596.
- Jurado RL. Iron, infections, and anemia of inflammation. Clin Infect Dis 1997; 25:888–895.
- Brewster UC, Coca SG, Reilly RF, Perazella MA. Effect of intravenous iron on hemodialysis catheter microbial colonization and blood-borne infection. Nephrology 2005; 10:124–128.
- Aronoff GR, Bennett WM, Blumenthal S, et al; United States Iron Sucrose (Venofer) Clinical Trials Group. Iron sucrose in hemodialysis patients: safety of replacement and maintenance regimens. Kidney Int 2004; 66:1193–1198.
- Canziani ME, Yumiya ST, Rangel EB, Manfredi SR, Neto MC, Draibe SA. Risk of bacterial infection in patients under intravenous iron therapy: dose versus length of treatment. Artif Organs 2001; 25:866–869.
- Collins A, Ma J, Xia H, et al. I.V. iron dosing patterns and hospitalization. J Am Soc Nephrol 1998; 9:204A.
- Burns DL, Mascioli EA, Bistrian BR. Effect of iron-supplemented total parenteral nutrition in patients with iron deficiency anemia. Nutrition 1996; 12:411–415.
- Olijhoek G, Megens JG, Musto P, et al. Role of oral versus IV iron supplementation in the erythropoietic response to rHuEPO: a randomized, placebo-controlled trial. Transfusion 2001; 41:957–963.
- Feldman HI, Joffe M, Robinson B, et al. Administration of parenteral iron and mortality among hemodialysis patients. J Am Soc Nephrol 2004; 15:1623–1632.
- Javadi P, Buchman TG, Stromberg PE, et al. High-dose exogenous iron following cecal ligation and puncture increases mortality rate in mice and is associated with an increase in gut epithelial and splenic apoptosis. Crit Care Med 2004; 32:1178–1185.
- Lapointe M. Iron supplementation in the intensive care unit: when, how much, and by what route? Crit Care 2004; 8(suppl 2):S37–S41.
The harmful effects of iron therapy in the setting of infection are more theoretical than observed, with no irrefutable data to support them. On the other hand, there are also no convincing data to support the benefit of this therapy. If iron is to be used, frequent monitoring of serum iron markers is prudent to avoid iron overload during treatment.
ANEMIA OF INFLAMMATION IS COMPLEX
Anemia that develops in the hospital, especially in the setting of infection or inflammation, is similar hematologically to anemia of chronic disease, except for its acute onset.1
The pathogenesis of anemia in such settings is complex, but the most important causes of this common syndrome include shortening of red cell survival, impaired erythropoietin production, blunted responsiveness of the bone marrow to endogenous erythropoietin, and impaired iron metabolism mediated through the action of inflammatory cytokines.2,3 Other important causes include nutritional deficiencies (iron, vitamin B12, and folic acid)4 and blood loss.5,6
Moreover, anemia of inflammation may be difficult to differentiate from iron-deficiency anemia because the serum iron markers are unreliable in inflammation.1
The reported prevalence of anemia during hospitalization has ranged from 55% on hospital wards7 to 95% in intensive care units.8
Transfusion of packed red blood cells is the fastest treatment for anemia in hospitalized patients and it is the one traditionally used, but many concerns have been raised about its efficacy and adverse effects.9 Erythropoietin, with or without iron therapy, has emerged as an alternative in treating anemia of inflammation.10,11
IRON THERAPY
Iron is widely used to treat anemia, especially in hospitalized patients and those with chronic kidney disease.2 The intravenous route is more commonly used than the oral route, since it has faster action, is better tolerated, and has better bioavailability.1,2
Controversy over benefit
Whether iron supplementation increases the red blood cell mass and reduces the need for blood transfusion is controversial.10,12 Pieracci et al13 documented these benefits in critically ill surgical patients, whereas van Iperen et al11 did not find such benefits in critically ill patients receiving intravenous iron and erythropoietin.
Harmful effects
Some authors1,14 object to giving iron to hospitalized patients (especially critically ill patients) who have infections on the grounds that it is risky, although definitive evidence is lacking.15
Most of the harmful effects of iron have been linked to elevated serum ferritin levels and to non–transferrin-bound iron, more than to iron per se.16 Ferritin is an acute-phase reactant; thus, ferritin levels may be elevated in inflammation and infection regardless of the body iron status.1
Anaphylactic reaction. This rare complication of iron dextran therapy is not much of a concern at present with the newer formulations of iron such as iron gluconate and iron sucrose.16
Oxidative stress. Iron-derived free radicals can cause a rise in inflammatory cytokine levels, especially if the ferritin level is elevated (> 500 μg/L). This cytokine rise is worrisome, as it may have acute detrimental effects on cellular homeostasis, leading to tissue injury,15 while chronically it might be related to enhanced atherosclerosis and cardiac disease.16
Iron overload. In vitro and animal studies have documented an association between elevated ferritin levels (500–650 μg/L) and decreases in T-cell function, polymorphonuclear neutrophil migration, phagocytosis, and bacterial eradication.15 Studies in hemodialysis patients have identified iron overload as an independent risk factor for bacterial infection, but the confounding role of the dialysis process cannot be disregarded.17,18
Bacterial growth. Many bacteria depend on iron for their growth; examples are Escherichia coli; Klebsiella, Pseudomonas, Salmonella, Yersinia, Listeria, and Staphylococcus species; and Haemophilus influenzae. In vitro studies have linked increased bacterial growth with increased transferrin saturation in plasma.15,19
Iron therapy and infection risk
The theory linking iron with risk of infection arose from the observation that patients with hemochromatosis are more susceptible to certain bacterial infections, especially Vibrio vulnificus.20 A few human studies, most of them in chronic hemodialysis patients, have examined the relation between iron therapy and infection risk, with conflicting results.21–26 Multiple studies13,19,21,22,25–27 found no relation between iron therapy and risk of infection or death.
Canziani et al23 found that the risk of infection was higher with higher intravenous doses of iron than with lower doses.
Collins et al24 found a higher risk of sepsis and hospitalization in patients who received iron for a prolonged duration (5–6 months) than in those who did not.
Feldman et al,27 in their report of a study of iron therapy in hemodialysis patients, suggested that previously observed associations between iron administration and higher death rates may have been confounded by other factors.
Iron therapy in concurrent infection
There are no data in humans on the effects of iron therapy on outcomes during concurrent infection or sepsis.15,28 However, mice with sepsis had worse outcomes when treated with intravenous iron.28
A CONUNDRUM IN CLINICAL PRACTICE
After reviewing the available literature, we concur with most of the authors1,15,16,18,19,29 that despite the worrisome theoretical adverse effects of iron therapy in patients with infections, there are no convincing data to support those fears. On the other hand, there are also no convincing data to favor its benefit.
More definitive studies are needed to answer this question, which has been a conundrum in clinical practice. Patients who might benefit from iron therapy should not be deprived of it on the basis of the available data. Frequent monitoring of serum iron markers during therapy to avoid iron overload seems prudent.
The harmful effects of iron therapy in the setting of infection are more theoretical than observed, with no irrefutable data to support them. On the other hand, there are also no convincing data to support the benefit of this therapy. If iron is to be used, frequent monitoring of serum iron markers is prudent to avoid iron overload during treatment.
ANEMIA OF INFLAMMATION IS COMPLEX
Anemia that develops in the hospital, especially in the setting of infection or inflammation, is similar hematologically to anemia of chronic disease, except for its acute onset.1
The pathogenesis of anemia in such settings is complex, but the most important causes of this common syndrome include shortening of red cell survival, impaired erythropoietin production, blunted responsiveness of the bone marrow to endogenous erythropoietin, and impaired iron metabolism mediated through the action of inflammatory cytokines.2,3 Other important causes include nutritional deficiencies (iron, vitamin B12, and folic acid)4 and blood loss.5,6
Moreover, anemia of inflammation may be difficult to differentiate from iron-deficiency anemia because the serum iron markers are unreliable in inflammation.1
The reported prevalence of anemia during hospitalization has ranged from 55% on hospital wards7 to 95% in intensive care units.8
Transfusion of packed red blood cells is the fastest treatment for anemia in hospitalized patients and it is the one traditionally used, but many concerns have been raised about its efficacy and adverse effects.9 Erythropoietin, with or without iron therapy, has emerged as an alternative in treating anemia of inflammation.10,11
IRON THERAPY
Iron is widely used to treat anemia, especially in hospitalized patients and those with chronic kidney disease.2 The intravenous route is more commonly used than the oral route, since it has faster action, is better tolerated, and has better bioavailability.1,2
Controversy over benefit
Whether iron supplementation increases the red blood cell mass and reduces the need for blood transfusion is controversial.10,12 Pieracci et al13 documented these benefits in critically ill surgical patients, whereas van Iperen et al11 did not find such benefits in critically ill patients receiving intravenous iron and erythropoietin.
Harmful effects
Some authors1,14 object to giving iron to hospitalized patients (especially critically ill patients) who have infections on the grounds that it is risky, although definitive evidence is lacking.15
Most of the harmful effects of iron have been linked to elevated serum ferritin levels and to non–transferrin-bound iron, more than to iron per se.16 Ferritin is an acute-phase reactant; thus, ferritin levels may be elevated in inflammation and infection regardless of the body iron status.1
Anaphylactic reaction. This rare complication of iron dextran therapy is not much of a concern at present with the newer formulations of iron such as iron gluconate and iron sucrose.16
Oxidative stress. Iron-derived free radicals can cause a rise in inflammatory cytokine levels, especially if the ferritin level is elevated (> 500 μg/L). This cytokine rise is worrisome, as it may have acute detrimental effects on cellular homeostasis, leading to tissue injury,15 while chronically it might be related to enhanced atherosclerosis and cardiac disease.16
Iron overload. In vitro and animal studies have documented an association between elevated ferritin levels (500–650 μg/L) and decreases in T-cell function, polymorphonuclear neutrophil migration, phagocytosis, and bacterial eradication.15 Studies in hemodialysis patients have identified iron overload as an independent risk factor for bacterial infection, but the confounding role of the dialysis process cannot be disregarded.17,18
Bacterial growth. Many bacteria depend on iron for their growth; examples are Escherichia coli; Klebsiella, Pseudomonas, Salmonella, Yersinia, Listeria, and Staphylococcus species; and Haemophilus influenzae. In vitro studies have linked increased bacterial growth with increased transferrin saturation in plasma.15,19
Iron therapy and infection risk
The theory linking iron with risk of infection arose from the observation that patients with hemochromatosis are more susceptible to certain bacterial infections, especially Vibrio vulnificus.20 A few human studies, most of them in chronic hemodialysis patients, have examined the relation between iron therapy and infection risk, with conflicting results.21–26 Multiple studies13,19,21,22,25–27 found no relation between iron therapy and risk of infection or death.
Canziani et al23 found that the risk of infection was higher with higher intravenous doses of iron than with lower doses.
Collins et al24 found a higher risk of sepsis and hospitalization in patients who received iron for a prolonged duration (5–6 months) than in those who did not.
Feldman et al,27 in their report of a study of iron therapy in hemodialysis patients, suggested that previously observed associations between iron administration and higher death rates may have been confounded by other factors.
Iron therapy in concurrent infection
There are no data in humans on the effects of iron therapy on outcomes during concurrent infection or sepsis.15,28 However, mice with sepsis had worse outcomes when treated with intravenous iron.28
A CONUNDRUM IN CLINICAL PRACTICE
After reviewing the available literature, we concur with most of the authors1,15,16,18,19,29 that despite the worrisome theoretical adverse effects of iron therapy in patients with infections, there are no convincing data to support those fears. On the other hand, there are also no convincing data to favor its benefit.
More definitive studies are needed to answer this question, which has been a conundrum in clinical practice. Patients who might benefit from iron therapy should not be deprived of it on the basis of the available data. Frequent monitoring of serum iron markers during therapy to avoid iron overload seems prudent.
- Pieracci FM, Barie PS. Diagnosis and management of iron-related anemias in critical illness. Crit Care Med 2006; 34:1898–1905.
- Krantz SB. Pathogenesis and treatment of the anemia of chronic disease. Am J Med Sci 1994; 307:353–359.
- Price EA, Schrier SL. Unexplained aspects of anemia of inflammation. Review article. Adv Hematol 2010; 2010:508739.
- Rodriguez RM, Corwin HL, Gettinger A, Corwin MJ, Gubler D, Pearl RG. Nutritional deficiencies and blunted erythropoietin response as causes of the anemia of critical illness. J Crit Care 2001; 16:36–41.
- Wong P, Intragumtornchai T. Hospital-acquired anemia. J Med Assoc Thai 2006; 89:63–67.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Reade MC, Weissfeld L, Angus DC, Kellum JA, Milbrandt EB. The prevalence of anemia and its association with 90-day mortality in hospitalized community-acquired pneumonia. BMC Pulm Med 2010; 10:15.
- Debellis RJ. Anemia in critical care patients: incidence, etiology, impact, management, and use of treatment guidelines and protocols. Am J Health Syst Pharm 2007; 64:S14–S21.
- Marik PE. The hazards of blood transfusion. Br J Hosp Med (Lond) 2009; 70:12–15.
- Corwin HL, Gettinger A, Fabian TC, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med 2007; 357:965–976.
- van Iperen CE, Gaillard CA, Kraaijenhagen RJ, Braam BG, Marx JJ, van de Wiel A. Response of erythropoiesis and iron metabolism to recombinant human erythropoietin in intensive care unit patients. Crit Care Med 2000; 28:2773–2778.
- Muñoz M, Breymann C, García-Erce JA, Gómez-Ramirez S, Comin J, Bisbe E. Efficacy and safety of intravenous iron therapy as an alternative/adjunct to allogeneic blood transfusion. Vox Sang 2008; 94:172–183.
- Pieracci FM, Henderson P, Rodney JR, et al. Randomized, double-blind, placebo-controlled trial of effects of enteral iron supplementation on anemia and risk of infection during surgical critical illness. Surg Infect 2009; 10:9–19.
- Pieracci FM, Barie PS. Iron and the risk of infection. Surg Infect 2005; 6(suppl 1):S41–S46.
- Maynor L, Brophy DF. Risk of infections with intravenous iron therapy. Ann Pharmacother 2007; 41:1476–1480.
- Cavill I. Intravenous iron as adjuvant therapy: a two-edged sword? Nephrol Dial Transplant 2003; 18(suppl 8):viii24–viii28.
- Kessler M, Hoen B, Mayeux D, Hestin D, Fontenaille C. Bacteremia in patients on chronic hemodialysis. A multicenter prospective survey. Nephron 1993; 64:95–100.
- Hoen B, Kessler M, Hestin D, Mayeux D. Risk factors for bacterial infections in chronic haemodialysis adult patients: a multicentre prospective survey. Nephrol Dial Transplant 1995; 10:377–381.
- Cieri E. Does iron cause bacterial infections in patients with end stage renal disease? ANNA J 1999; 26:591–596.
- Jurado RL. Iron, infections, and anemia of inflammation. Clin Infect Dis 1997; 25:888–895.
- Brewster UC, Coca SG, Reilly RF, Perazella MA. Effect of intravenous iron on hemodialysis catheter microbial colonization and blood-borne infection. Nephrology 2005; 10:124–128.
- Aronoff GR, Bennett WM, Blumenthal S, et al; United States Iron Sucrose (Venofer) Clinical Trials Group. Iron sucrose in hemodialysis patients: safety of replacement and maintenance regimens. Kidney Int 2004; 66:1193–1198.
- Canziani ME, Yumiya ST, Rangel EB, Manfredi SR, Neto MC, Draibe SA. Risk of bacterial infection in patients under intravenous iron therapy: dose versus length of treatment. Artif Organs 2001; 25:866–869.
- Collins A, Ma J, Xia H, et al. I.V. iron dosing patterns and hospitalization. J Am Soc Nephrol 1998; 9:204A.
- Burns DL, Mascioli EA, Bistrian BR. Effect of iron-supplemented total parenteral nutrition in patients with iron deficiency anemia. Nutrition 1996; 12:411–415.
- Olijhoek G, Megens JG, Musto P, et al. Role of oral versus IV iron supplementation in the erythropoietic response to rHuEPO: a randomized, placebo-controlled trial. Transfusion 2001; 41:957–963.
- Feldman HI, Joffe M, Robinson B, et al. Administration of parenteral iron and mortality among hemodialysis patients. J Am Soc Nephrol 2004; 15:1623–1632.
- Javadi P, Buchman TG, Stromberg PE, et al. High-dose exogenous iron following cecal ligation and puncture increases mortality rate in mice and is associated with an increase in gut epithelial and splenic apoptosis. Crit Care Med 2004; 32:1178–1185.
- Lapointe M. Iron supplementation in the intensive care unit: when, how much, and by what route? Crit Care 2004; 8(suppl 2):S37–S41.
- Pieracci FM, Barie PS. Diagnosis and management of iron-related anemias in critical illness. Crit Care Med 2006; 34:1898–1905.
- Krantz SB. Pathogenesis and treatment of the anemia of chronic disease. Am J Med Sci 1994; 307:353–359.
- Price EA, Schrier SL. Unexplained aspects of anemia of inflammation. Review article. Adv Hematol 2010; 2010:508739.
- Rodriguez RM, Corwin HL, Gettinger A, Corwin MJ, Gubler D, Pearl RG. Nutritional deficiencies and blunted erythropoietin response as causes of the anemia of critical illness. J Crit Care 2001; 16:36–41.
- Wong P, Intragumtornchai T. Hospital-acquired anemia. J Med Assoc Thai 2006; 89:63–67.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Reade MC, Weissfeld L, Angus DC, Kellum JA, Milbrandt EB. The prevalence of anemia and its association with 90-day mortality in hospitalized community-acquired pneumonia. BMC Pulm Med 2010; 10:15.
- Debellis RJ. Anemia in critical care patients: incidence, etiology, impact, management, and use of treatment guidelines and protocols. Am J Health Syst Pharm 2007; 64:S14–S21.
- Marik PE. The hazards of blood transfusion. Br J Hosp Med (Lond) 2009; 70:12–15.
- Corwin HL, Gettinger A, Fabian TC, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med 2007; 357:965–976.
- van Iperen CE, Gaillard CA, Kraaijenhagen RJ, Braam BG, Marx JJ, van de Wiel A. Response of erythropoiesis and iron metabolism to recombinant human erythropoietin in intensive care unit patients. Crit Care Med 2000; 28:2773–2778.
- Muñoz M, Breymann C, García-Erce JA, Gómez-Ramirez S, Comin J, Bisbe E. Efficacy and safety of intravenous iron therapy as an alternative/adjunct to allogeneic blood transfusion. Vox Sang 2008; 94:172–183.
- Pieracci FM, Henderson P, Rodney JR, et al. Randomized, double-blind, placebo-controlled trial of effects of enteral iron supplementation on anemia and risk of infection during surgical critical illness. Surg Infect 2009; 10:9–19.
- Pieracci FM, Barie PS. Iron and the risk of infection. Surg Infect 2005; 6(suppl 1):S41–S46.
- Maynor L, Brophy DF. Risk of infections with intravenous iron therapy. Ann Pharmacother 2007; 41:1476–1480.
- Cavill I. Intravenous iron as adjuvant therapy: a two-edged sword? Nephrol Dial Transplant 2003; 18(suppl 8):viii24–viii28.
- Kessler M, Hoen B, Mayeux D, Hestin D, Fontenaille C. Bacteremia in patients on chronic hemodialysis. A multicenter prospective survey. Nephron 1993; 64:95–100.
- Hoen B, Kessler M, Hestin D, Mayeux D. Risk factors for bacterial infections in chronic haemodialysis adult patients: a multicentre prospective survey. Nephrol Dial Transplant 1995; 10:377–381.
- Cieri E. Does iron cause bacterial infections in patients with end stage renal disease? ANNA J 1999; 26:591–596.
- Jurado RL. Iron, infections, and anemia of inflammation. Clin Infect Dis 1997; 25:888–895.
- Brewster UC, Coca SG, Reilly RF, Perazella MA. Effect of intravenous iron on hemodialysis catheter microbial colonization and blood-borne infection. Nephrology 2005; 10:124–128.
- Aronoff GR, Bennett WM, Blumenthal S, et al; United States Iron Sucrose (Venofer) Clinical Trials Group. Iron sucrose in hemodialysis patients: safety of replacement and maintenance regimens. Kidney Int 2004; 66:1193–1198.
- Canziani ME, Yumiya ST, Rangel EB, Manfredi SR, Neto MC, Draibe SA. Risk of bacterial infection in patients under intravenous iron therapy: dose versus length of treatment. Artif Organs 2001; 25:866–869.
- Collins A, Ma J, Xia H, et al. I.V. iron dosing patterns and hospitalization. J Am Soc Nephrol 1998; 9:204A.
- Burns DL, Mascioli EA, Bistrian BR. Effect of iron-supplemented total parenteral nutrition in patients with iron deficiency anemia. Nutrition 1996; 12:411–415.
- Olijhoek G, Megens JG, Musto P, et al. Role of oral versus IV iron supplementation in the erythropoietic response to rHuEPO: a randomized, placebo-controlled trial. Transfusion 2001; 41:957–963.
- Feldman HI, Joffe M, Robinson B, et al. Administration of parenteral iron and mortality among hemodialysis patients. J Am Soc Nephrol 2004; 15:1623–1632.
- Javadi P, Buchman TG, Stromberg PE, et al. High-dose exogenous iron following cecal ligation and puncture increases mortality rate in mice and is associated with an increase in gut epithelial and splenic apoptosis. Crit Care Med 2004; 32:1178–1185.
- Lapointe M. Iron supplementation in the intensive care unit: when, how much, and by what route? Crit Care 2004; 8(suppl 2):S37–S41.
How should one investigate a chronic cough?
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH

Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
- Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
- Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
- Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
- Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
- Multiple causes of chronic cough can coexist.
- Therapeutic trials are part of the workup.
- Do not stop therapy if it is only partially successful: add to existing therapies
- Start the investigation with the most likely cause.
- Treatment is 84% to 98% successful.
- Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med 2000; 343:1715–1721.
- Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol 2010; 69:200–203.
- Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):1S–23S.
- Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):63S–71S.
- Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):80S–94S.
- Kahrilas PJ. Clinical practice. Gastroesophageal reflux disease. N Engl J Med 2008; 359:1700–1707.
- Dicpinigaitis PV. Chronic cough due to asthma: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):75S–79S.
- Brightling CE. Chronic cough due to nonasthmatic eosinophilic bronchitis: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):116S–121S.
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH
Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
- Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
- Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
- Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
- Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
- Multiple causes of chronic cough can coexist.
- Therapeutic trials are part of the workup.
- Do not stop therapy if it is only partially successful: add to existing therapies
- Start the investigation with the most likely cause.
- Treatment is 84% to 98% successful.
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH
Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
- Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
- Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
- Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
- Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
- Multiple causes of chronic cough can coexist.
- Therapeutic trials are part of the workup.
- Do not stop therapy if it is only partially successful: add to existing therapies
- Start the investigation with the most likely cause.
- Treatment is 84% to 98% successful.
- Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med 2000; 343:1715–1721.
- Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol 2010; 69:200–203.
- Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):1S–23S.
- Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):63S–71S.
- Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):80S–94S.
- Kahrilas PJ. Clinical practice. Gastroesophageal reflux disease. N Engl J Med 2008; 359:1700–1707.
- Dicpinigaitis PV. Chronic cough due to asthma: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):75S–79S.
- Brightling CE. Chronic cough due to nonasthmatic eosinophilic bronchitis: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):116S–121S.
- Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med 2000; 343:1715–1721.
- Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol 2010; 69:200–203.
- Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):1S–23S.
- Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):63S–71S.
- Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):80S–94S.
- Kahrilas PJ. Clinical practice. Gastroesophageal reflux disease. N Engl J Med 2008; 359:1700–1707.
- Dicpinigaitis PV. Chronic cough due to asthma: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):75S–79S.
- Brightling CE. Chronic cough due to nonasthmatic eosinophilic bronchitis: ACCP evidence-based clinical practice guidelines. Chest 2006; 129(suppl):116S–121S.
Do patients with prosthetic joints require dental antimicrobial prophylaxis?
We believe the available evidence does not support routine antimicrobial prophylaxis before dental procedures in patients who have undergone total joint replacement, even though the practice is very common1 and even though professional societies recommend it in patients at high risk,2 or even in all patients.3
On the other hand, good oral hygiene prevents dental disease and decreases the frequency of bacteremia from routine daily activities, and thus should be especially encouraged in patients with prosthetic joints or in those undergoing total joint arthroplasty.
AN UNCOMMON BUT SERIOUS PROBLEM
By 2030, an estimated 4 million total hip or total knee replacements per year will be performed in the United States.4 Most patients have a satisfactory outcome, but in a small percentage the prosthesis fails prematurely.
Prosthetic joint infection is the second most common cause of prosthetic failure leading to loss of joint function, after aseptic loosening.5 Its treatment often requires removal of the infected prosthesis and prolonged intravenous antimicrobial therapy. The cost incurred with each episode of prosthetic joint infection is estimated to exceed $50,000.1
Because of the morbidity and substantial cost associated with managing this condition, investigators have focused on identifying preventable risk factors for it.
RISK FACTORS FOR PROSTHETIC JOINT INFECTION
Factors associated with a higher risk of prosthetic joint infection include prior joint surgery, failure to give antimicrobial prophylaxis during surgery, immunosuppression, perioperative wound complications, a high American Society of Anesthesiologists (ASA) score, prolonged operative time, and a history of prosthetic joint infection.6,7 The primary predisposing factors are related to the foreign body itself and to the opportunities for and the degree of exposure of the prosthesis to microorganisms during surgery. Bacteremia, especially with Staphylococcus aureus, has been recognized as a risk factor for hematogenous prosthetic joint infection.6
Whether dental procedures pose a risk of prosthetic joint infection has been debated for decades. Common daily activities such as toothbrushing and chewing can cause transient bacteremia in up to 40% of episodes.8
Extrapolating from the guidelines for preventing endocarditis, the American Dental Association (ADA)2 and the American Academy of Orthopaedic Surgeons (AAOS)3 have issued guidelines favoring antimicrobial prophylaxis in patients with prosthetic joints. However, given the significant differences in the pathophysiology, microbiology, and anatomy of infection between infective endocarditis and prosthetic joint infection, extrapolating the recommendations may not be valid.
MICROBIOLOGY OF PROSTHETIC JOINT INFECTION AND DENTAL FLORA
Staphylococci, the most common cause of prosthetic joint infection, are uncommon commensals of the oral flora and have been rarely implicated in bacteremia occurring after dental procedures.9 In contrast, viridans-group streptococci constitute most of the facultative oral flora and are the most common cause of transient bacteremia after dental procedures that result in trauma to the gingival or oral mucosa.10 However, viridans-group streptococci account for only 2% of all hematogenous prosthetic joint infections.9
DO DENTAL PROCEDURES INCREASE THE RISK OF PROSTHETIC JOINT INFECTION?
Prolonged or high-grade bacteremia is associated with prosthetic joint infection. On the other hand, data are scant on the association between low-grade or transient bacteremia and prosthetic joint infection.
After dental procedures, bacteria can be found in the blood, but at much lower levels (< 104 cfu/mL) than that needed for hematogenous seeding of prostheses in animal studies (3–5 × 108 cfu/mL).11 Transient, low-grade bacteremia occurs not only after dental procedures but also, as mentioned, after common activities such as chewing, brushing, or flossing.1 The cumulative exposure to transient bacteremia through these daily activities is several times higher than the single exposure that a person is subjected to during dental procedures.12
WHAT IS THE EVIDENCE?
Most of the current evidence linking dental procedures or dental manipulation to prosthetic joint infection is based on reports of single cases of infections that occurred after dental procedures.
In two retrospective reviews, late hematogenous prosthetic joint infection associated with a dental source occurred after 0.2% of primary knee arthroplasties11 and 6% of primary hip arthroplasties.13
Ainscow and Denham14 followed 1,000 patients who underwent total joint replacement over 6 years. Of these, 226 subsequently underwent dental procedures without receiving antimicrobial prophylaxis, and none developed a prosthetic joint infection.
In a recently published case-control study,1 our group assessed 339 patients with prosthetic joint infection and 339 patients with prosthetic joints that did not become infected. In this study, neither low-risk nor high-risk dental procedures were associated with an increased risk of prosthetic knee or hip infections (odds ratio [OR] 0.8; 95% confidence interval [CI] 0.4–1.6). Moreover, prophylactic use of antimicrobials before dental procedures was not associated with a lower risk.
However, a factor that was associated with a lower risk of prosthetic joint infection was good oral hygiene (OR 0.7; 95% CI 0.5–1.03). Good oral hygiene and prevention of dental disease could potentially decrease the frequency of bacteremia from daily activities and may even protect against prosthetic joint infection. Further study of the association of poor dental health and the risk of prosthetic joint infection is warranted.
GUIDELINES AND RECOMMENDATIONS
Despite the lack of evidence suggesting an association between prosthetic joint infection and dental procedure, surveys of orthopedists, dentists, infectious disease specialists, and other health care professionals show that a significant number of them recommend antimicrobial prophylaxis for patients with a prosthetic joint prior to a dental procedure.1
In 2003, a consensus panel of the AAOS and the ADA recommended routine consideration of antimicrobial prophylaxis in patients at high risk due to both patient factors and the type of dental procedure.2 Patient factors thought to confer high risk are immunosuppression, diabetes, malnourishment, human immunodeficiency virus infection, prior prosthetic joint infection, hemophilia, malignancy, and a prosthesis less than 2 years old. High-risk dental procedures are tooth extractions, periodontal procedures, root canal surgery, and dental cleaning in which bleeding is anticipated.
In a recent statement, the AAOS recommended antimicrobial prophylaxis in all patients with prosthetic joints.3
Concerns about promoting antimicrobial resistance and about adverse reactions from antimicrobial use may outweigh any hypothetic benefit related to prevention of prosthetic joint infection. Analyses of cost, risks, and benefits argue against this practice.3
In summary, the current evidence does not support the use of antimicrobial therapy to prevent prosthetic joint infection in patients with total joint replacement undergoing dental procedures. However, good oral hygiene should be encouraged to prevent dental disease and to decrease the frequency of bacteremia from routine daily activities in patients who have undergone or will be undergoing total joint arthroplasty.
- Berbari EF, Osmon DR, Carr A, et al. Dental procedures as risk factors for prosthetic hip or knee infection: a hospital-based prospective case-control study. Clin Infect Dis 2010; 50:8–16.
- American Dental Association. Antibiotic prophylaxis for dental patients with total joint replacements. J Am Dent Assoc 2003; 134:895–899.
- American Academy of Orthopaedic Surgeons. Information statement: antibiotic prophylaxis for bacteremia in patients with joint replacements. http://www.aaos.org/about/papers/advistmt/1033.asp. Accessed October 28, 2010.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007; 89:780–785.
- Roberts VI, Esler CN, Harper WM. A 15-year follow-up study of 4606 primary total knee replacements. J Bone Joint Surg Br 2007; 89:1452–1456.
- Del Pozo JL, Patel R. Clinical practice. Infection associated with prosthetic joints. N Engl J Med 2009; 361:787–794.
- Berbari EF, Hanssen AD, Duffy MC, et al. Risk factors for prosthetic joint infection: case-control study. Clin Infect Dis 1998; 27:1247–1254.
- Durack DT. Prevention of infective endocarditis. N Engl J Med 1995; 332:38–44.
- Deacon JM, Pagliaro AJ, Zelicof SB, Horowitz HW. Prophylactic use of antibiotics for procedures after total joint replacement. Bone Joint Surg Am 1996; 78:1755–1770.
- Kaye D. Infective endocarditis. In:Rose LF, Kaye D, editors. Internal Medicine for Dentistry, 2nd ed. Mosby: St. Louis, MO; 1990:156–161.
- Waldman BJ, Mont MA, Hungerford DS. Total knee arthroplasty infections associated with dental procedures. Clin Orthop Relat Res 1997; 343:164–172.
- Guntheroth WG. How important are dental procedures as a cause of infective endocarditis? Am J Cardiol 1984; 54:797–801.
- LaPorte DM, Waldman BJ, Mont MA, Hungerford DS. Infections associated with dental procedures in total hip arthroplasty. J Bone Joint Surg Br 1999; 81:56–59.
- Ainscow DA, Denham RA. The risk of haematogenous infection in total joint replacements. J Bone Joint Surg Br 1984; 66:580–582.
We believe the available evidence does not support routine antimicrobial prophylaxis before dental procedures in patients who have undergone total joint replacement, even though the practice is very common1 and even though professional societies recommend it in patients at high risk,2 or even in all patients.3
On the other hand, good oral hygiene prevents dental disease and decreases the frequency of bacteremia from routine daily activities, and thus should be especially encouraged in patients with prosthetic joints or in those undergoing total joint arthroplasty.
AN UNCOMMON BUT SERIOUS PROBLEM
By 2030, an estimated 4 million total hip or total knee replacements per year will be performed in the United States.4 Most patients have a satisfactory outcome, but in a small percentage the prosthesis fails prematurely.
Prosthetic joint infection is the second most common cause of prosthetic failure leading to loss of joint function, after aseptic loosening.5 Its treatment often requires removal of the infected prosthesis and prolonged intravenous antimicrobial therapy. The cost incurred with each episode of prosthetic joint infection is estimated to exceed $50,000.1
Because of the morbidity and substantial cost associated with managing this condition, investigators have focused on identifying preventable risk factors for it.
RISK FACTORS FOR PROSTHETIC JOINT INFECTION
Factors associated with a higher risk of prosthetic joint infection include prior joint surgery, failure to give antimicrobial prophylaxis during surgery, immunosuppression, perioperative wound complications, a high American Society of Anesthesiologists (ASA) score, prolonged operative time, and a history of prosthetic joint infection.6,7 The primary predisposing factors are related to the foreign body itself and to the opportunities for and the degree of exposure of the prosthesis to microorganisms during surgery. Bacteremia, especially with Staphylococcus aureus, has been recognized as a risk factor for hematogenous prosthetic joint infection.6
Whether dental procedures pose a risk of prosthetic joint infection has been debated for decades. Common daily activities such as toothbrushing and chewing can cause transient bacteremia in up to 40% of episodes.8
Extrapolating from the guidelines for preventing endocarditis, the American Dental Association (ADA)2 and the American Academy of Orthopaedic Surgeons (AAOS)3 have issued guidelines favoring antimicrobial prophylaxis in patients with prosthetic joints. However, given the significant differences in the pathophysiology, microbiology, and anatomy of infection between infective endocarditis and prosthetic joint infection, extrapolating the recommendations may not be valid.
MICROBIOLOGY OF PROSTHETIC JOINT INFECTION AND DENTAL FLORA
Staphylococci, the most common cause of prosthetic joint infection, are uncommon commensals of the oral flora and have been rarely implicated in bacteremia occurring after dental procedures.9 In contrast, viridans-group streptococci constitute most of the facultative oral flora and are the most common cause of transient bacteremia after dental procedures that result in trauma to the gingival or oral mucosa.10 However, viridans-group streptococci account for only 2% of all hematogenous prosthetic joint infections.9
DO DENTAL PROCEDURES INCREASE THE RISK OF PROSTHETIC JOINT INFECTION?
Prolonged or high-grade bacteremia is associated with prosthetic joint infection. On the other hand, data are scant on the association between low-grade or transient bacteremia and prosthetic joint infection.
After dental procedures, bacteria can be found in the blood, but at much lower levels (< 104 cfu/mL) than that needed for hematogenous seeding of prostheses in animal studies (3–5 × 108 cfu/mL).11 Transient, low-grade bacteremia occurs not only after dental procedures but also, as mentioned, after common activities such as chewing, brushing, or flossing.1 The cumulative exposure to transient bacteremia through these daily activities is several times higher than the single exposure that a person is subjected to during dental procedures.12
WHAT IS THE EVIDENCE?
Most of the current evidence linking dental procedures or dental manipulation to prosthetic joint infection is based on reports of single cases of infections that occurred after dental procedures.
In two retrospective reviews, late hematogenous prosthetic joint infection associated with a dental source occurred after 0.2% of primary knee arthroplasties11 and 6% of primary hip arthroplasties.13
Ainscow and Denham14 followed 1,000 patients who underwent total joint replacement over 6 years. Of these, 226 subsequently underwent dental procedures without receiving antimicrobial prophylaxis, and none developed a prosthetic joint infection.
In a recently published case-control study,1 our group assessed 339 patients with prosthetic joint infection and 339 patients with prosthetic joints that did not become infected. In this study, neither low-risk nor high-risk dental procedures were associated with an increased risk of prosthetic knee or hip infections (odds ratio [OR] 0.8; 95% confidence interval [CI] 0.4–1.6). Moreover, prophylactic use of antimicrobials before dental procedures was not associated with a lower risk.
However, a factor that was associated with a lower risk of prosthetic joint infection was good oral hygiene (OR 0.7; 95% CI 0.5–1.03). Good oral hygiene and prevention of dental disease could potentially decrease the frequency of bacteremia from daily activities and may even protect against prosthetic joint infection. Further study of the association of poor dental health and the risk of prosthetic joint infection is warranted.
GUIDELINES AND RECOMMENDATIONS
Despite the lack of evidence suggesting an association between prosthetic joint infection and dental procedure, surveys of orthopedists, dentists, infectious disease specialists, and other health care professionals show that a significant number of them recommend antimicrobial prophylaxis for patients with a prosthetic joint prior to a dental procedure.1
In 2003, a consensus panel of the AAOS and the ADA recommended routine consideration of antimicrobial prophylaxis in patients at high risk due to both patient factors and the type of dental procedure.2 Patient factors thought to confer high risk are immunosuppression, diabetes, malnourishment, human immunodeficiency virus infection, prior prosthetic joint infection, hemophilia, malignancy, and a prosthesis less than 2 years old. High-risk dental procedures are tooth extractions, periodontal procedures, root canal surgery, and dental cleaning in which bleeding is anticipated.
In a recent statement, the AAOS recommended antimicrobial prophylaxis in all patients with prosthetic joints.3
Concerns about promoting antimicrobial resistance and about adverse reactions from antimicrobial use may outweigh any hypothetic benefit related to prevention of prosthetic joint infection. Analyses of cost, risks, and benefits argue against this practice.3
In summary, the current evidence does not support the use of antimicrobial therapy to prevent prosthetic joint infection in patients with total joint replacement undergoing dental procedures. However, good oral hygiene should be encouraged to prevent dental disease and to decrease the frequency of bacteremia from routine daily activities in patients who have undergone or will be undergoing total joint arthroplasty.
We believe the available evidence does not support routine antimicrobial prophylaxis before dental procedures in patients who have undergone total joint replacement, even though the practice is very common1 and even though professional societies recommend it in patients at high risk,2 or even in all patients.3
On the other hand, good oral hygiene prevents dental disease and decreases the frequency of bacteremia from routine daily activities, and thus should be especially encouraged in patients with prosthetic joints or in those undergoing total joint arthroplasty.
AN UNCOMMON BUT SERIOUS PROBLEM
By 2030, an estimated 4 million total hip or total knee replacements per year will be performed in the United States.4 Most patients have a satisfactory outcome, but in a small percentage the prosthesis fails prematurely.
Prosthetic joint infection is the second most common cause of prosthetic failure leading to loss of joint function, after aseptic loosening.5 Its treatment often requires removal of the infected prosthesis and prolonged intravenous antimicrobial therapy. The cost incurred with each episode of prosthetic joint infection is estimated to exceed $50,000.1
Because of the morbidity and substantial cost associated with managing this condition, investigators have focused on identifying preventable risk factors for it.
RISK FACTORS FOR PROSTHETIC JOINT INFECTION
Factors associated with a higher risk of prosthetic joint infection include prior joint surgery, failure to give antimicrobial prophylaxis during surgery, immunosuppression, perioperative wound complications, a high American Society of Anesthesiologists (ASA) score, prolonged operative time, and a history of prosthetic joint infection.6,7 The primary predisposing factors are related to the foreign body itself and to the opportunities for and the degree of exposure of the prosthesis to microorganisms during surgery. Bacteremia, especially with Staphylococcus aureus, has been recognized as a risk factor for hematogenous prosthetic joint infection.6
Whether dental procedures pose a risk of prosthetic joint infection has been debated for decades. Common daily activities such as toothbrushing and chewing can cause transient bacteremia in up to 40% of episodes.8
Extrapolating from the guidelines for preventing endocarditis, the American Dental Association (ADA)2 and the American Academy of Orthopaedic Surgeons (AAOS)3 have issued guidelines favoring antimicrobial prophylaxis in patients with prosthetic joints. However, given the significant differences in the pathophysiology, microbiology, and anatomy of infection between infective endocarditis and prosthetic joint infection, extrapolating the recommendations may not be valid.
MICROBIOLOGY OF PROSTHETIC JOINT INFECTION AND DENTAL FLORA
Staphylococci, the most common cause of prosthetic joint infection, are uncommon commensals of the oral flora and have been rarely implicated in bacteremia occurring after dental procedures.9 In contrast, viridans-group streptococci constitute most of the facultative oral flora and are the most common cause of transient bacteremia after dental procedures that result in trauma to the gingival or oral mucosa.10 However, viridans-group streptococci account for only 2% of all hematogenous prosthetic joint infections.9
DO DENTAL PROCEDURES INCREASE THE RISK OF PROSTHETIC JOINT INFECTION?
Prolonged or high-grade bacteremia is associated with prosthetic joint infection. On the other hand, data are scant on the association between low-grade or transient bacteremia and prosthetic joint infection.
After dental procedures, bacteria can be found in the blood, but at much lower levels (< 104 cfu/mL) than that needed for hematogenous seeding of prostheses in animal studies (3–5 × 108 cfu/mL).11 Transient, low-grade bacteremia occurs not only after dental procedures but also, as mentioned, after common activities such as chewing, brushing, or flossing.1 The cumulative exposure to transient bacteremia through these daily activities is several times higher than the single exposure that a person is subjected to during dental procedures.12
WHAT IS THE EVIDENCE?
Most of the current evidence linking dental procedures or dental manipulation to prosthetic joint infection is based on reports of single cases of infections that occurred after dental procedures.
In two retrospective reviews, late hematogenous prosthetic joint infection associated with a dental source occurred after 0.2% of primary knee arthroplasties11 and 6% of primary hip arthroplasties.13
Ainscow and Denham14 followed 1,000 patients who underwent total joint replacement over 6 years. Of these, 226 subsequently underwent dental procedures without receiving antimicrobial prophylaxis, and none developed a prosthetic joint infection.
In a recently published case-control study,1 our group assessed 339 patients with prosthetic joint infection and 339 patients with prosthetic joints that did not become infected. In this study, neither low-risk nor high-risk dental procedures were associated with an increased risk of prosthetic knee or hip infections (odds ratio [OR] 0.8; 95% confidence interval [CI] 0.4–1.6). Moreover, prophylactic use of antimicrobials before dental procedures was not associated with a lower risk.
However, a factor that was associated with a lower risk of prosthetic joint infection was good oral hygiene (OR 0.7; 95% CI 0.5–1.03). Good oral hygiene and prevention of dental disease could potentially decrease the frequency of bacteremia from daily activities and may even protect against prosthetic joint infection. Further study of the association of poor dental health and the risk of prosthetic joint infection is warranted.
GUIDELINES AND RECOMMENDATIONS
Despite the lack of evidence suggesting an association between prosthetic joint infection and dental procedure, surveys of orthopedists, dentists, infectious disease specialists, and other health care professionals show that a significant number of them recommend antimicrobial prophylaxis for patients with a prosthetic joint prior to a dental procedure.1
In 2003, a consensus panel of the AAOS and the ADA recommended routine consideration of antimicrobial prophylaxis in patients at high risk due to both patient factors and the type of dental procedure.2 Patient factors thought to confer high risk are immunosuppression, diabetes, malnourishment, human immunodeficiency virus infection, prior prosthetic joint infection, hemophilia, malignancy, and a prosthesis less than 2 years old. High-risk dental procedures are tooth extractions, periodontal procedures, root canal surgery, and dental cleaning in which bleeding is anticipated.
In a recent statement, the AAOS recommended antimicrobial prophylaxis in all patients with prosthetic joints.3
Concerns about promoting antimicrobial resistance and about adverse reactions from antimicrobial use may outweigh any hypothetic benefit related to prevention of prosthetic joint infection. Analyses of cost, risks, and benefits argue against this practice.3
In summary, the current evidence does not support the use of antimicrobial therapy to prevent prosthetic joint infection in patients with total joint replacement undergoing dental procedures. However, good oral hygiene should be encouraged to prevent dental disease and to decrease the frequency of bacteremia from routine daily activities in patients who have undergone or will be undergoing total joint arthroplasty.
- Berbari EF, Osmon DR, Carr A, et al. Dental procedures as risk factors for prosthetic hip or knee infection: a hospital-based prospective case-control study. Clin Infect Dis 2010; 50:8–16.
- American Dental Association. Antibiotic prophylaxis for dental patients with total joint replacements. J Am Dent Assoc 2003; 134:895–899.
- American Academy of Orthopaedic Surgeons. Information statement: antibiotic prophylaxis for bacteremia in patients with joint replacements. http://www.aaos.org/about/papers/advistmt/1033.asp. Accessed October 28, 2010.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007; 89:780–785.
- Roberts VI, Esler CN, Harper WM. A 15-year follow-up study of 4606 primary total knee replacements. J Bone Joint Surg Br 2007; 89:1452–1456.
- Del Pozo JL, Patel R. Clinical practice. Infection associated with prosthetic joints. N Engl J Med 2009; 361:787–794.
- Berbari EF, Hanssen AD, Duffy MC, et al. Risk factors for prosthetic joint infection: case-control study. Clin Infect Dis 1998; 27:1247–1254.
- Durack DT. Prevention of infective endocarditis. N Engl J Med 1995; 332:38–44.
- Deacon JM, Pagliaro AJ, Zelicof SB, Horowitz HW. Prophylactic use of antibiotics for procedures after total joint replacement. Bone Joint Surg Am 1996; 78:1755–1770.
- Kaye D. Infective endocarditis. In:Rose LF, Kaye D, editors. Internal Medicine for Dentistry, 2nd ed. Mosby: St. Louis, MO; 1990:156–161.
- Waldman BJ, Mont MA, Hungerford DS. Total knee arthroplasty infections associated with dental procedures. Clin Orthop Relat Res 1997; 343:164–172.
- Guntheroth WG. How important are dental procedures as a cause of infective endocarditis? Am J Cardiol 1984; 54:797–801.
- LaPorte DM, Waldman BJ, Mont MA, Hungerford DS. Infections associated with dental procedures in total hip arthroplasty. J Bone Joint Surg Br 1999; 81:56–59.
- Ainscow DA, Denham RA. The risk of haematogenous infection in total joint replacements. J Bone Joint Surg Br 1984; 66:580–582.
- Berbari EF, Osmon DR, Carr A, et al. Dental procedures as risk factors for prosthetic hip or knee infection: a hospital-based prospective case-control study. Clin Infect Dis 2010; 50:8–16.
- American Dental Association. Antibiotic prophylaxis for dental patients with total joint replacements. J Am Dent Assoc 2003; 134:895–899.
- American Academy of Orthopaedic Surgeons. Information statement: antibiotic prophylaxis for bacteremia in patients with joint replacements. http://www.aaos.org/about/papers/advistmt/1033.asp. Accessed October 28, 2010.
- Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 2007; 89:780–785.
- Roberts VI, Esler CN, Harper WM. A 15-year follow-up study of 4606 primary total knee replacements. J Bone Joint Surg Br 2007; 89:1452–1456.
- Del Pozo JL, Patel R. Clinical practice. Infection associated with prosthetic joints. N Engl J Med 2009; 361:787–794.
- Berbari EF, Hanssen AD, Duffy MC, et al. Risk factors for prosthetic joint infection: case-control study. Clin Infect Dis 1998; 27:1247–1254.
- Durack DT. Prevention of infective endocarditis. N Engl J Med 1995; 332:38–44.
- Deacon JM, Pagliaro AJ, Zelicof SB, Horowitz HW. Prophylactic use of antibiotics for procedures after total joint replacement. Bone Joint Surg Am 1996; 78:1755–1770.
- Kaye D. Infective endocarditis. In:Rose LF, Kaye D, editors. Internal Medicine for Dentistry, 2nd ed. Mosby: St. Louis, MO; 1990:156–161.
- Waldman BJ, Mont MA, Hungerford DS. Total knee arthroplasty infections associated with dental procedures. Clin Orthop Relat Res 1997; 343:164–172.
- Guntheroth WG. How important are dental procedures as a cause of infective endocarditis? Am J Cardiol 1984; 54:797–801.
- LaPorte DM, Waldman BJ, Mont MA, Hungerford DS. Infections associated with dental procedures in total hip arthroplasty. J Bone Joint Surg Br 1999; 81:56–59.
- Ainscow DA, Denham RA. The risk of haematogenous infection in total joint replacements. J Bone Joint Surg Br 1984; 66:580–582.
Should alpha-blockers ever be used as antihypertensive drugs?
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
- Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet 2000; 355:863–864.
- Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA 2000; 283:1967–1975.
- Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med 2002; 137:313–320.
- Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol 2001; 38:1295–1296.
- Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med 1986; 314:1547–1552.
- Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens 2007; 25:1751–1762.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA 2003; 289:2560–2572.
- Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation 2008; 118:42–48.
- de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol 2006; 47:271–276.
- Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol 2003; 41:866–869.
- Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension? J Hypertens 2010; 28:1796–1803.
- Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis 2005; 8:152–157.
- Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther 1999; 21:1732–1748.
- MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int 2004; 94:1263–1270.
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
Alpha-blockers should not be used as first-line therapy for hypertension. However, an alpha-blocker can be considered as a second-line or third-line add-on in a patient whose blood pressure is not under control despite treatment with other drugs.
In addition, alpha-blockers are useful in relieving lower urinary tract symptoms in patients with benign prostatic hypertrophy. However, even in a patient who has both hypertension and benign prostatic hypertrophy, we advise physicians to use alpha-blockers primarily to relieve the urinary symptoms, and we recommend lowering the blood pressure with a drug of a class shown to reduce rates of illness and death.
NOT FIRST-LINE THERAPY
All antihypertensive drugs, including alpha-blockers, lower blood pressure. Alpha-blockers have been approved by the US Food and Drug Administration for treating high blood pressure, and they are just as effective as other antihypertensive drugs—if efficacy is defined as a decrease in millimeters of mercury.
However, lowering the blood pressure is not the main goal of antihypertensive therapy. What we want to achieve when prescribing antihypertensive drugs is to reduce the rates of heart attacks, strokes, and other adverse cardiovascular adverse outcomes, including death.
Unfortunately, alpha-blockers fall short in this regard. In the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack (ALLHAT) trial,1,2 doxazosin (Cardura) was found to carry a higher risk of combined cardiovascular disease (relative risk 1.19, P = .04), mostly stroke. Alarmingly, the incidence of symptomatic heart failure in patients on doxazosin was twice that in patients on chlorthalidone (relative risk 2.04, P < .001). Doxazosin was minimally more effective in lowering blood pressure than chlorthalidone, but the small difference in blood pressure was unlikely to have accounted for the significant difference in the risk of heart failure.3
This experience with doxazosin illustrates a key drawback to surrogate end points: a treatment may produce a favorable outcome in the surrogate end point (blood pressure) but produce little or no benefit in terms of the real end point (stroke, myocardial infarction, and heart failure).4
Based on the ALLHAT data as well as on a Veterans Administration study in patients with chronic heart failure in which survival with prazosin (Minipress) was no better than with placebo,5 it seems reasonable to no longer use alpha-blockers as initial therapy for hypertension. This view is reflected by current European6 and American7 guidelines.
ALPHA-BLOCKERS AS PART OF COMBINATION THERAPY
In several clinical trials, alpha-blockers were allowed8 or were specified9,10 as add-on therapy if other drugs failed to control the blood pressure, but they were not used in a randomized fashion. Thus, we cannot judge their effect on cardiovascular outcomes such as heart attack and stroke.
The choice of drugs for combination therapy very often is still empirical and based on personal preference. Doxazosin as add-on therapy, in general, has been shown to be safe and well tolerated.11 But even if it is acceptable, it is not a preferred combination.
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),9 patients received extended-release doxazosin as a third drug if they did not reach their goal blood pressure with either the combination of amlodipine (Norvasc) plus perindopril (Aceon) or atenolol (Tenormin) plus bendroflumethiazide. Extended-release doxazosin was an effective add-on, and there was no apparent excess rate of heart failure in doxazosin users.
In other studies, in patients with uncontrolled hypertension, adding doxazosin as a second- or third-line agent to a gold-standard drug—calcium channel blocker, diuretic, beta-blocker, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker, or combinations of these—allowed significantly more participants to achieve their blood pressure goal.11
Personally, we consider doxazosin in patients whose blood pressure is not controlled with triple therapy with a renin-angiotensin system blocker, a diuretic, and a calcium channel antagonist in full doses. In patients with stage 3 or stage 4 kidney disease who can no longer tolerate renin-angiotensin system blockers, doxazosin may also be a useful adjunct. Whether the metabolic effects of alpha-blockers, such as a reduction in insulin resistance and a decrease in total and low-density lipoprotein cholesterol, will result in lower rates of morbidity and death has not been conclusively determined.
A point of view somewhat more favorable to the use of alpha-blockers has recently been put forward by Chapman et al.12
ALPHA-BLOCKERS ALLEVIATE SYMPTOMS OF BENIGN PROSTATIC HYPERTROPHY
Doxazosin and other alpha-blockers are commonly used to alleviate lower urinary tract symptoms in patients with benign prostatic hypertrophy.
Both high blood pressure and benign prostatic hypertrophy become more common with advancing age, and it has been estimated that both are present in more than 25% of men over age 60.13 Indeed, two trials documented that a significant reduction in symptoms of benign prostatic hypertrophy and in systolic and diastolic blood pressure can be achieved with an alpha-blocker.13,14
This raises the question whether such a “twofer” (treating two disease states with one drug) should be used in clinical practice. We have to consider that the principle of the twofer has never been tested and agree with Davis et al,3 who, in a further analysis of the ALLHAT data, stated that, “In older men with benign prostatic hypertrophy in whom an [alpha]-adrenergic blocker seems like the best treatment for the uropathy, coexisting hypertension should be treated with another antihypertensive drug as well.”3
Again, this would clearly relegate doxazosin to second-line or third-line status, even in patients with benign prostatic hypertrophy, in whom it has been shown to be indicated.
ADVERSE EFFECTS OF ALPHA-BLOCKERS
Dizziness, fatigue, and somnolence are occasionally reported but appear to be well tolerated. Postural hypotension is much less common with proper titration of standard doxazosin or with the use of controlled-release formulations.9–15 However, in patients with impaired autonomic function, even long-acting alpha-blockers can cause postural hypotension and syncope.
Patients using phosphodiesterase type 5 inhibitors—sildenafil (Viagra), vardenafil (Levitra), or tadalafil (Cialis)—for erectile dysfunction should avoid alpha-blockers because the blood-pressure-lowering effects of the two drug classes may be additive.
- Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet 2000; 355:863–864.
- Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA 2000; 283:1967–1975.
- Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med 2002; 137:313–320.
- Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol 2001; 38:1295–1296.
- Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med 1986; 314:1547–1552.
- Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens 2007; 25:1751–1762.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA 2003; 289:2560–2572.
- Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation 2008; 118:42–48.
- de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol 2006; 47:271–276.
- Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol 2003; 41:866–869.
- Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension? J Hypertens 2010; 28:1796–1803.
- Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis 2005; 8:152–157.
- Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther 1999; 21:1732–1748.
- MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int 2004; 94:1263–1270.
- Messerli FH. Implications of discontinuation of doxazosin arm of ALLHAT. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (commentary). Lancet 2000; 355:863–864.
- Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA 2000; 283:1967–1975.
- Davis BR, Cutler JA, Furberg CD, et al; ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the Antihypertensive and Lipid-Lowering treatment to prevent Heart Attack Trial. Ann Intern Med 2002; 137:313–320.
- Messerli FH. Doxazosin and congestive heart failure (viewpoint). J Am Coll Cardiol 2001; 38:1295–1296.
- Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med 1986; 314:1547–1552.
- Mancia G, De Backer G, Dominiczak A, et al; ESH-ESC Task Force on the Management of Arterial Hypertension. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens 2007; 25:1751–1762.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA 2003; 289:2560–2572.
- Jamerson K, Weber MA, Bakris GL, et al; for the ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Chapman N, Chang CL, Dahlöf B, Sever PS, Wedel H, Poulter NR; ASCOT Investigators. Effect of doxazosin gastrointestinal therapeutic system as third-line antihypertensive therapy on blood pressure and lipids in the Anglo-Scandinavian Cardiac Outcomes Trial. Circulation 2008; 118:42–48.
- de Alvaro F, Hernandez-Presa MAASOCIA Study. Effect of doxazosin gastrointestinal therapeutic system on patients with uncontrolled hypertension: the ASOCIA Study. J Cardiovasc Pharmacol 2006; 47:271–276.
- Black HR. Doxazosin as combination therapy for patients with stage 1 and stage 2 hypertension. J Cardiovasc Pharmacol 2003; 41:866–869.
- Chapman N, Chen C-Y, Fujita T, et al. Time to re-appraise the role of alpha-1 adrenoceptor antagonists in the management of hypertension? J Hypertens 2010; 28:1796–1803.
- Steers WD, Kirby RS. Clinical ease of using doxazosin in BPH patients with and without hypertension. Prostate Cancer Prostatic Dis 2005; 8:152–157.
- Guthrie RM, Siegel RL. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: the Hypertension and BPH Intervention Trial (HABIT). Clin Ther 1999; 21:1732–1748.
- MacDonald R, Wilt TJ, Howe RW. Doxazosin for treating lower urinary tract symptoms compatible with benign prostatic obstruction: a systematic review of efficacy and adverse effects. BJU Int 2004; 94:1263–1270.