User login
How soon after hip fracture surgery should a patient start bisphosphonates?
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
- In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
- The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
- The type of intervention to stabilize the fracture site (eg, surgical repair)
- The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
- Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res 2009; 24:1308–1313.
- Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res 2010; 25:91–97.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab 2010; 95:1174–1181.
- Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol 2008; 19:459–466.
- Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res 2007; 22:867–876.
- Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res 1999; 14:969–979.
- Fleisch H. Can bisphosphonates be given to patients with fractures? J Bone Miner Res 2001; 16:437–440.
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
- In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
- The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
- The type of intervention to stabilize the fracture site (eg, surgical repair)
- The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
Patients with an osteoporotic hip fracture suffer from profound morbidity and are at a heightened risk of death. It is therefore essential that they receive treatment with a bisphosphonate known to modify the subsequent risk of fracture at any site—eg, alendronate (Fosamax), risedronate (Actonel), or zoledronic acid (Reclast).
However, there is concern that starting a bisphosphonate too soon after surgery could disrupt bone remodeling and delay fracture repair.
Only one clinical study addressed the timing of bisphosphonate therapy after hip fracture repair. In this study, Eriksen et al1 performed a post hoc analysis of data from the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Recurrent Fracture Trial (HORIZON-RFT)2 and concluded that the optimal time to give intravenous zoledronic acid is 2 to 12 weeks after surgical repair of the fracture.
In a frail, elderly patient with comorbidities, a single intravenous 5-mg dose of zoledronic acid guarantees adequate treatment, obviating issues of poor compliance and oral absorption and loss to follow-up. Sufficient levels of vitamin D and calcium should be ensured.
THE EVIDENCE
The original HORIZON-RFT study,2 published in 2007, compared intravenous zoledronic acid against placebo in elderly patients with osteoporotic hip fracture. Most of the patients were white women; their mean age was 74; 1,065 received intravenous zoledronic acid, and 1,062 received placebo. All received vitamin D and calcium.
The trial showed a clear reduction in the rate of recurrent fractures at other sites (a primary end point) and a reduction in the rate of all-cause mortality in patients treated within 90 days of fracture. A total of 424 fractures occurred in 231 patients. The risk of any new clinical fracture was 35% lower with treatment than with placebo (occurring in 8.6% vs 13.9% of patients, P = .001), and the number of deaths due to any cause was 28% lower with treatment than with placebo (occurring in 101 vs 141, P = .01).2
The mean time to fracture was 39.8 months in the treated group vs 36.4 in the placebo group. The fracture risk reduction began to be apparent by 12 months, and the reduction in mortality rate by 16 months.2
In a post hoc analysis of the trial, Eriksen et al1 attempted to ascertain the optimal time for therapy in terms of fracture risk and mortality reduction. Analyzing the data by 2-week intervals beginning after the surgical repair of the fracture, the authors found that only 56 patients (5.3%) had received zoledronic acid within 2 weeks of surgery and only 47 had received placebo, and they saw no advantage to intravenous zoledronic acid compared with placebo in these first 2 weeks with respect to bone mineral density, fracture risk, or risk of death. However, excluding this small subset, antifracture efficacy and reduction in mortality rate were present when patients were treated with zoledronic acid in the 2 to 12 weeks after hip fracture repair, and improvement in bone mineral density at the hip was noted at 12 months in all cohorts.
Colón-Emeric et al3 performed another post hoc analysis, attempting to explain the lower mortality rate seen in patients treated with zoledronic acid. It had been an unexpected finding, and determinants of mortality rate reduction were hampered by a limited knowledge of the true cause of death or the circumstances of care after fracture. The authors concluded that only 8% of the reduction in mortality rate evident early in the second year of treatment with zoledronic acid could be attributed to a reduction in fractures.3 Other mechanisms by which the mortality rate reduction occurred remained unclear.
Curiously, in another large randomized controlled trial of zoledronic acid, in women with postmenopausal osteoporosis, Black et al4 reported that more patients died in the treated group (130 of 3,862) than in the placebo group (112 of 3,852). This difference was not statistically significant, but neither was it explained.
A meta-analysis by Bolland et al5 examined the effect of other osteoporosis treatments on mortality rate, using randomized controlled trials that lasted more than 12 months and that reported more than 10 deaths. The authors concluded the following:
- In the trials in which bisphosphonates reduced the mortality rate, the mortality rate in the placebo group was higher than 10 per 1,000 patient-years
- The effect of osteoporosis treatment on the mortality rate in a frail, elderly population is evident using agents with proven efficacy in reducing vertebral and nonvertebral fractures, eg, alendronate, risedronate, and zoledronic acid.5
THE SCIENCE
Osteoporotic fractures occur with minimal trauma, with the failure of bone attributed to impaired integrity of bone microarchitecture. The ultimate goal of fracture repair is to restore bone size, shape, and tissue properties. The issue of when to treat with a bisphosphonate after hip fracture arises because bisphosphonates are known to disrupt bone remodeling and so delay fracture repair.
After fracture, both anabolic and catabolic phases occur.6 The final outcome depends on the following:
- The type of intervention to stabilize the fracture site (eg, surgical repair)
- The inflammatory cytokines and growth factors released by the cellular elements in bloody and disrupted tissue.
Oxygen tension, angiogenesis, and osteoblasts are critical to primary bone formation, and osteoclasts are essential in remodeling this initial bone deposition. These late phases of fracture repair are most vulnerable to the bisphosphonates, through suppression of osteoclast resorption and possibly through decreased angiogenesis.6 Callus formation is sustained, but bone remodeling is delayed.
Amanat et al7 examined the timing of a single dose of zoledronic acid after fracture repair in a rat model of diaphyseal fracture and found that the callus was larger and stronger if the bisphosphonate dose had been delayed 1 or 2 weeks. The animals treated with zoledronic acid showed a remarkable trabecular network of bone between the original femoral cortex and the new cortical bone that was not present in the control group, perhaps contributing to the enhanced mechanical properties of the callus. Other studies suggest single dosing rather than continuous dosing may be advantageous in fracture healing.8
THE REALITY
Healthy dogs or growing rats with linear diaphyseal fractures are imperfect models for elderly osteoporotic patients with hip fracture, as Dr. Herbert Fleisch noted in his editorial, “Can bisphosphonates be given to patients with fractures?”9 Still, if retained primary bone can be used in the process of fracture repair to gain an early mechanical advantage, then perhaps delayed remodeling will permit early mobilization and further fracture prevention in humans.
How soon after hip fracture surgery should a patient start a bisphosphonate? The only data we have are from a single randomized controlled trial designed to measure fracture risk reduction in osteoporotic patients with hip fracture using intravenous zoledronic acid 5 mg compared with placebo.2 A post hoc analysis of this study1 generated the limited clinical data we have on the optimal timing of the treatment. Linking these study data with the laboratory data, one would intuit that delaying the infusion of zoledronic acid for at least 2 weeks after hip fracture repair would offer a clinical reduction in fracture risk and improvement (or stabilization) in bone mineral density by 12 months, and a reduction in the rate of all-cause mortality beginning at 16 months.
- Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res 2009; 24:1308–1313.
- Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res 2010; 25:91–97.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab 2010; 95:1174–1181.
- Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol 2008; 19:459–466.
- Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res 2007; 22:867–876.
- Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res 1999; 14:969–979.
- Fleisch H. Can bisphosphonates be given to patients with fractures? J Bone Miner Res 2001; 16:437–440.
- Eriksen EF, Lyles KW, Colón-Emeric CS, et al. Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res 2009; 24:1308–1313.
- Lyles KW, Colón-Emeric CS, Magaziner JS, et al; for the HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 2007; 357:1799–1809.
- Colón-Emeric CS, Mesenbrink P, Lyles KW, et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res 2010; 25:91–97.
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. J Clin Endocrinol Metab 2010; 95:1174–1181.
- Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol 2008; 19:459–466.
- Amanat N, McDonald M, Godfrey C, Bilston L, Little D. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res 2007; 22:867–876.
- Li J, Mori S, Kaji Y, Mashiba T, Kawanishi J, Norimatsu H. Effect of bisphosphonate (incadronate) on fracture healing of long bones in rats. J Bone Miner Res 1999; 14:969–979.
- Fleisch H. Can bisphosphonates be given to patients with fractures? J Bone Miner Res 2001; 16:437–440.
Should healthy people take a multivitamin?
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
No. There is no scientific basis for recommending vitamin-mineral supplements to the healthy population.
(This commentary deals only with healthy people in the general US population. There are well-established guidelines for the use of supplements in pregnant and lactating women, infants, and individuals with a wide variety of health conditions.)
TAKE A PILL, OR EAT A HEALTHIER DIET?
The Dietary Guidelines for Americans1 reported that the US population consumes insufficient amounts of green leafy vegetables, fresh fruits, whole grains, and fiber and excessive amounts of refined carbohydrates, saturated fat, and sodium. This may result in inadequate intake of some nutrients. (The term “inadequate” intake is being used to differentiate this situation from “deficiency,” which is rare in the general population.)
So the real question is, Should we pop a vitamin pill every day and forget about it, or try to eat a healthier diet?
To answer that question, consider this: no supplement trial has ever been able to reproduce the health benefits of eating adequate amounts of fresh fruits and vegetables.
One reason is that natural foods contain far more compounds than the few we know about and can put in a supplement pill. For example, vegetables contain hundreds of antioxidant compounds, many perhaps acting synergistically, while so far we have been able to identify and isolate only a handful.
Second, nutrients have different health effects depending on the host’s conditions. A calcium supplement will not increase bone mineral density unless accompanied by regular, weight-bearing exercise that stimulates bone accretion.
This is why most supplement trials have shown disappointing results. A recent National Institutes of Health state-of-the-science conference on multivitamin-mineral supplements2 concluded that there is no consistent evidence that single-vitamin or multivitamin supplements help in preventing a wide range of diseases studied.
‘AT LEAST IT WON’T HURT’ MAY NOT BE TRUE
In spite of the lack of evidence, many will go on taking supplements, with the argument that “at least it won’t hurt.” They should be reminded that several supplement trials had to be stopped prematurely due to unexpected adverse effects.
In the Selenium and Vitamin E Cancer Prevention Trial (SELECT),3 which evaluated supplementation to prevent prostate cancer, the group receiving vitamin E had more cases of prostate cancer than controls, and the group taking selenium had more diabetes cases. While these differences were not statistically significant, they were of enough concern to stop the trial.
A meta-analysis of vitamin E trials4 showed a slight increase in the rate of all-cause mortality in those receiving the active supplement.
The bottom line: the evidence that supplements “won’t hurt” is even more limited than the evidence for their efficacy, because trials are usually not designed to address safety outcomes.
TELL YOUR PATIENTS THE THINGS THEY DO NOT WANT TO HEAR
Unfortunately, this means you have to tell your patients all the things they do not want to hear: cut the ice cream, eat more broccoli, exercise regularly. But because of their position of authority and credibility, physicians can play a crucial role in helping the US population improve its dietary and lifestyle habits.
The key is to introduce and support minor but sustained changes in the diet and physical activity. For example, we have shown that simply reducing consumption of caloric beverages (soft drinks) can result in significant weight loss in overweight adults, without any other dietary intervention.5
The other key is of course to modify the obesogenic environment we live in. Only by creating conditions that facilitate healthy eating and regular activity will we have a significant impact on public health.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
- United States Department of Agriculture Center for Nutrition Policy and Promotion. Report of the Dietary Guidelines Advisory Committee on the Dietary Guidelines for Americans, 2010. www.cnpp.usda.gov/DGAs2010-DGACReport.htm. Accessed 8/25/2010.
- Huang HY, Caballero B, Chang S, et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a National Institutes of Health state-of-the-science conference. Ann Intern Med 2006; 145:372–385.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005; 142:37–46.
- Chen L, Appel LJ, Loria C, et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: the PREMIER trial. Am J Clin Nutr 2009; 89:1299–1306.
Are antibiotics indicated for the treatment of aspiration pneumonia?
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
Do incretin drugs for type 2 diabetes increase the risk of acute pancreatitis?
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
Can patients with COPD or asthma take a beta-blocker?
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS

- Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol 2001; 37:1950–1956.
- The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med 1997; 157:2413–2446.
- Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol 1978; 5:415–419.
- Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med 1984; 311:1441.
- Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med 1998; 339:489–497.
- Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med 2003; 97:1094–1101.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med 2002; 137:715–725.
- van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest 2005; 127:818–824.
- Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol 2004; 44:497–502.
- Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant 2002; 21:1290–1295.
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS
- Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol 2001; 37:1950–1956.
- The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med 1997; 157:2413–2446.
- Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol 1978; 5:415–419.
- Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med 1984; 311:1441.
- Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med 1998; 339:489–497.
- Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med 2003; 97:1094–1101.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med 2002; 137:715–725.
- van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest 2005; 127:818–824.
- Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol 2004; 44:497–502.
- Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant 2002; 21:1290–1295.
- Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol 2001; 37:1950–1956.
- The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med 1997; 157:2413–2446.
- Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol 1978; 5:415–419.
- Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med 1984; 311:1441.
- Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med 1998; 339:489–497.
- Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med 2003; 97:1094–1101.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med 2002; 137:715–725.
- van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest 2005; 127:818–824.
- Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol 2004; 44:497–502.
- Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant 2002; 21:1290–1295.
Should patients with mild asthma use inhaled steroids?
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
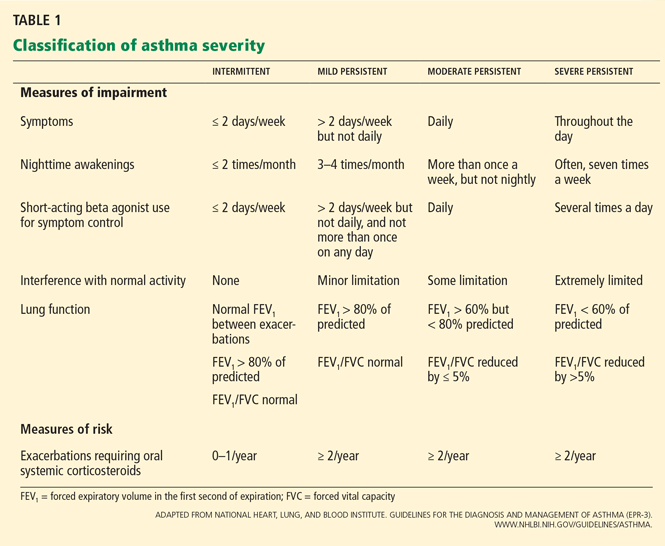
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT

The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
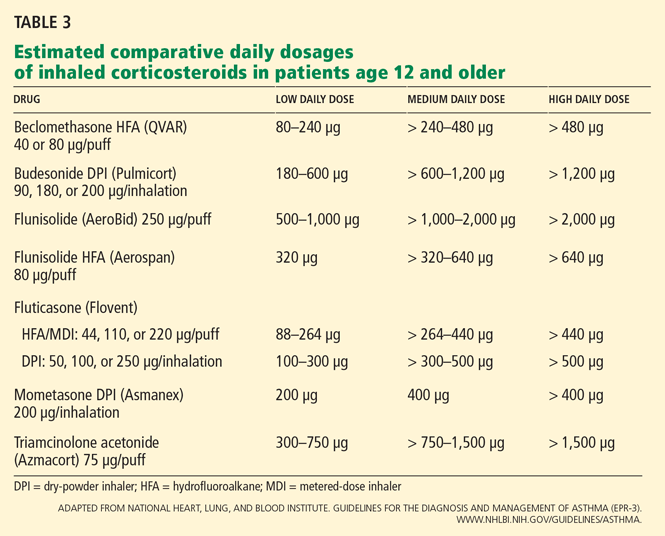
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival?
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
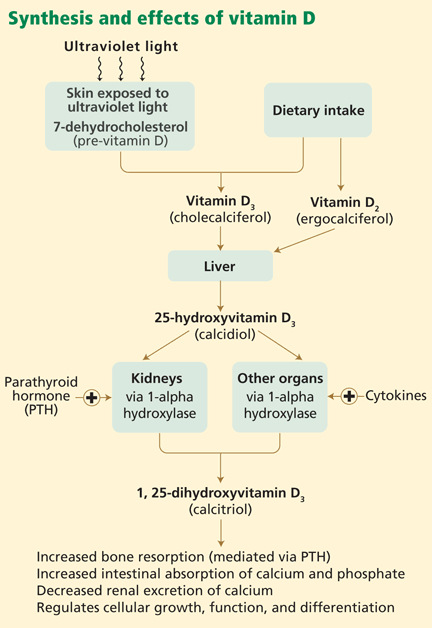
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE

The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
When should serum amylase and lipase levels be repeated in a patient with acute pancreatitis?
In general, repeating serum amylase and lipase levels has no value once the diagnosis of acute pancreatitis has been made. In gallstone-related acute pancreatitis (ie, in most cases), delaying surgery for several days for the pancreas to “cool down” is common practice, but repeating serum pancreatic enzyme levels daily during this period is of no prognostic value, as the levels do not correlate with the severity, course, or outcome of the acute pancreatitis.1–3 Rather, the decision to proceed with treatment should be based on clinical measures, such as improvement of pain or increasing appetite.
Repeated pancreatic enzyme tests have diagnostic value, though. For example, in mild acute pancreatitis, symptoms tend to resolve in less than 1 week, whereas in severe cases, not only do symptoms persist beyond 1 week, but complications (new symptoms) also develop after the first week. In such cases, serum amylase and lipase levels may be repeated when the patient has signs and symptoms of persisting pancreatic or peripancreatic inflammation, blockage of the pancreatic duct, or development of a pseudocyst, 3 but the purpose of retesting the levels is to diagnose complications, not to monitor the status of the pancreas. However, imaging tests generally have a higher sensitivity than serum amylase and lipase levels for diagnosing complications of acute pancreatitis.
MAKING BEST USE OF SERUM PANCREATIC ENZYME LEVELS
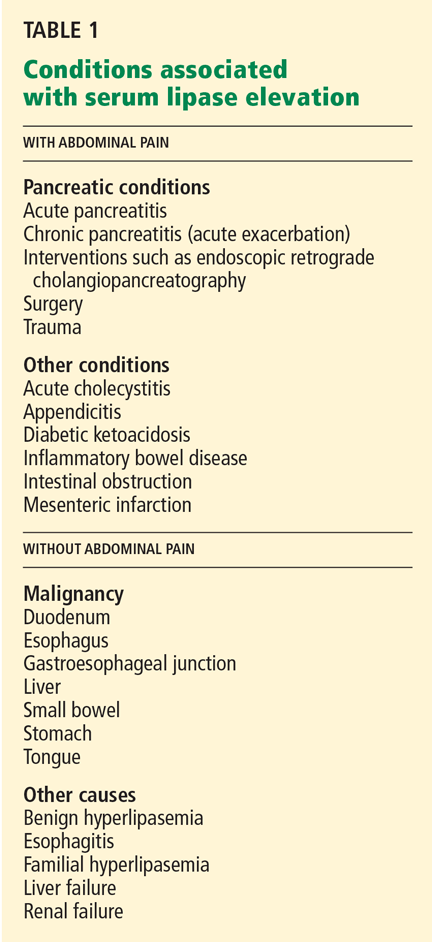

Amylase is also noted in salivary glands, fallopian tubes and cyst fluid, testes, lungs, thyroid, tonsils, breast milk, sweat, tears, and some malignant neoplasms. Serum lipase is often considered a more specific marker of acute pancreatitis than serum amylase, but recent data cast doubt on this.5
- Kim YS, Lee BS, Kim SH, Seong JK, Jeong HY, Lee HY. Is there correlation between pancreatic enzyme and radiological severity in acute pancreatitis? World J Gastroenterol 2008; 14:2401–2405.
- Lankisch PG, Burchard-Reckert S, Lehnick D. Underestimation of acute pancreatitis: patients with only a small increase in amylase/lipase levels can also have or develop severe acute pancreatitis. Gut 1999; 44:542–544.
- Banks PA, Freeman MLPractice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Gumaste VV, Roditis N, Mehta D, Dave PB. Serum lipase levels in nonpancreatic abdominal pain versus acute pancreatitis. Am J Gastroenterol 1993; 88:2051–2055.
- Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol 2002; 97:1309–1318.
In general, repeating serum amylase and lipase levels has no value once the diagnosis of acute pancreatitis has been made. In gallstone-related acute pancreatitis (ie, in most cases), delaying surgery for several days for the pancreas to “cool down” is common practice, but repeating serum pancreatic enzyme levels daily during this period is of no prognostic value, as the levels do not correlate with the severity, course, or outcome of the acute pancreatitis.1–3 Rather, the decision to proceed with treatment should be based on clinical measures, such as improvement of pain or increasing appetite.
Repeated pancreatic enzyme tests have diagnostic value, though. For example, in mild acute pancreatitis, symptoms tend to resolve in less than 1 week, whereas in severe cases, not only do symptoms persist beyond 1 week, but complications (new symptoms) also develop after the first week. In such cases, serum amylase and lipase levels may be repeated when the patient has signs and symptoms of persisting pancreatic or peripancreatic inflammation, blockage of the pancreatic duct, or development of a pseudocyst, 3 but the purpose of retesting the levels is to diagnose complications, not to monitor the status of the pancreas. However, imaging tests generally have a higher sensitivity than serum amylase and lipase levels for diagnosing complications of acute pancreatitis.
MAKING BEST USE OF SERUM PANCREATIC ENZYME LEVELS
Amylase is also noted in salivary glands, fallopian tubes and cyst fluid, testes, lungs, thyroid, tonsils, breast milk, sweat, tears, and some malignant neoplasms. Serum lipase is often considered a more specific marker of acute pancreatitis than serum amylase, but recent data cast doubt on this.5
In general, repeating serum amylase and lipase levels has no value once the diagnosis of acute pancreatitis has been made. In gallstone-related acute pancreatitis (ie, in most cases), delaying surgery for several days for the pancreas to “cool down” is common practice, but repeating serum pancreatic enzyme levels daily during this period is of no prognostic value, as the levels do not correlate with the severity, course, or outcome of the acute pancreatitis.1–3 Rather, the decision to proceed with treatment should be based on clinical measures, such as improvement of pain or increasing appetite.
Repeated pancreatic enzyme tests have diagnostic value, though. For example, in mild acute pancreatitis, symptoms tend to resolve in less than 1 week, whereas in severe cases, not only do symptoms persist beyond 1 week, but complications (new symptoms) also develop after the first week. In such cases, serum amylase and lipase levels may be repeated when the patient has signs and symptoms of persisting pancreatic or peripancreatic inflammation, blockage of the pancreatic duct, or development of a pseudocyst, 3 but the purpose of retesting the levels is to diagnose complications, not to monitor the status of the pancreas. However, imaging tests generally have a higher sensitivity than serum amylase and lipase levels for diagnosing complications of acute pancreatitis.
MAKING BEST USE OF SERUM PANCREATIC ENZYME LEVELS
Amylase is also noted in salivary glands, fallopian tubes and cyst fluid, testes, lungs, thyroid, tonsils, breast milk, sweat, tears, and some malignant neoplasms. Serum lipase is often considered a more specific marker of acute pancreatitis than serum amylase, but recent data cast doubt on this.5
- Kim YS, Lee BS, Kim SH, Seong JK, Jeong HY, Lee HY. Is there correlation between pancreatic enzyme and radiological severity in acute pancreatitis? World J Gastroenterol 2008; 14:2401–2405.
- Lankisch PG, Burchard-Reckert S, Lehnick D. Underestimation of acute pancreatitis: patients with only a small increase in amylase/lipase levels can also have or develop severe acute pancreatitis. Gut 1999; 44:542–544.
- Banks PA, Freeman MLPractice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Gumaste VV, Roditis N, Mehta D, Dave PB. Serum lipase levels in nonpancreatic abdominal pain versus acute pancreatitis. Am J Gastroenterol 1993; 88:2051–2055.
- Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol 2002; 97:1309–1318.
- Kim YS, Lee BS, Kim SH, Seong JK, Jeong HY, Lee HY. Is there correlation between pancreatic enzyme and radiological severity in acute pancreatitis? World J Gastroenterol 2008; 14:2401–2405.
- Lankisch PG, Burchard-Reckert S, Lehnick D. Underestimation of acute pancreatitis: patients with only a small increase in amylase/lipase levels can also have or develop severe acute pancreatitis. Gut 1999; 44:542–544.
- Banks PA, Freeman MLPractice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Gumaste VV, Roditis N, Mehta D, Dave PB. Serum lipase levels in nonpancreatic abdominal pain versus acute pancreatitis. Am J Gastroenterol 1993; 88:2051–2055.
- Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol 2002; 97:1309–1318.
Should we routinely screen for hypercapnia in sleep apnea patients before elective noncardiac surgery?
Yes. Obesity hypoventilation syndrome (OHS) is often undiagnosed and greatly increases perioperative risk. Therefore, we recommend trying to detect OHS in a timely manner. Treatment should begin without delay to avoid adverse perioperative outcomes, which can include acute-on-chronic respiratory failure requiring intensive-care monitoring and invasive mechanical ventilation, or death.
ALSO CALLED PICKWICKIAN SYNDROME
OHS is also known as Pickwickian syndrome, named for a character—a “fat boy” who is constantly falling asleep—in The Posthumous Papers of the Pickwick Club by Charles Dickens.
Salient features of OHS are:
- Obesity (body mass index ≥ 30 kg/m2)
- Sleep-disordered breathing (most patients with OHS are morbidly obese and have severe obstructive sleep apnea1)
- Chronic daytime alveolar hypoventilation: ie, Paco2 ≥ 45 mm Hg (normal range 35–45 mm Hg) and Pao2 < 70 mm Hg1 (normal range 85–95 mm Hg)
- No other identifiable cause of hypoventilation such as pulmonary disease (severe obstructive or restrictive), chest wall deformities, severe hypothyroidism, or neuromuscular disease.
WHY SCREEN FOR OHS?
Both obstructive sleep apnea and OHS worsen quality of life and increase the risk of serious disease and death.2–3 Patients with severe sleep apnea, particularly those with hypercapnia (ie, OHS) are at higher risk of cardiopulmonary complications in the perioperative period.
Compared with eucapnic patients with obstructive sleep apnea, patients with OHS have higher health care expenses, are at higher risk of developing serious cardiovascular diseases such as pulmonary hypertension and congestive heart failure, and are more likely to die sooner.4,5
Nowbar et al5 prospectively followed a group of severely obese patients after hospital discharge. At 18 months, 23% of those with OHS had died, compared with 9% of those without OHS. The groups were well matched for body mass index, age, and a number of comorbid conditions. Most of the deaths occurred in the first 3 months after hospital discharge. During the hospital stay, more patients with OHS were admitted to the intensive care unit and needed endotracheal intubation and mechanical ventilation, and more were discharged to a long-term facility.
A high level of suspicion can lead to early recognition and treatment, which may reduce the rate of adverse outcomes associated with undiagnosed and untreated OHS. Routine screening for hypercapnia in patients with sleep apnea might help to identify patients with OHS and allow for modifications in surgical approach, anesthetic technique, and postoperative monitoring, increasing patient safety.
HOW PREVALENT IS OHS?
Obstructive sleep apnea affects up to 20% of US adults and is undiagnosed and untreated in up to 90% of cases.6 Simple screening questionnaires have been shown to reliably identify patients at risk.7,8
To date, no population-based prevalence studies of OHS have been done.
The overall prevalence of OHS in patients with obstructive sleep apnea is better studied: multiple prospective and retrospective studies across various geographic regions with a variety of racial or ethnic populations have shown it to be between 10% and 20%.1,9 This range is very consistent among studies performed in Europe, the United States, and Japan, whether retrospective or prospective, and whether large or small.
The prevalence of OHS in the general adult population in the United States can, however, be estimated. If approximately 5% of the general US population has severe obesity (body mass index ≤ 40 kg/m2), if half of patients with severe obesity have obstructive sleep apnea,10 and if 15% of severely obese patients with sleep apnea have OHS, then a conservative estimated prevalence of OHS in the general adult US population is 0.37% (1 in 270 adults).
WHAT CAN BE DONE BEFORE ELECTIVE SURGERY?
Patients with OHS have an elevated serum bicarbonate level due to metabolic compensation for chronic respiratory acidosis. Moreover, they may have mild hypoxemia during wakefulness as measured by finger pulse oximetry.
The serum venous bicarbonate level is an easy and reasonable test to screen for hypercapnia in obese patients with obstructive sleep apnea because it is readily available, physiologically sensible, and less invasive than arterial puncture to measure blood gases.9
Arterial blood gas measurements, however, should be obtained to confirm the presence and severity of daytime hypercapnia in obese patients with hypoxemia during wakefulness or an elevated serum bicarbonate level.
Pulmonary function testing and chest imaging can exclude other causes of hypercapnia if hypercapnia is confirmed.
An overnight, attended polysomnographic study in a sleep laboratory is ultimately needed to establish the diagnosis and severity of obstructive sleep apnea and to titrate continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) therapy. Since most patients with OHS have severe obstructive sleep apnea, in-laboratory attended polysomnography allows the clinician to both diagnose and intervene with PAP therapy (a “split-night” study). Home titration with an auto-CPAP device is not recommended because it does not have the ability to titrate PAP pressures in response to hypoxemia or hypoventilation. Patients with OHS require attended, laboratory-based PAP titration with or without supplemental oxygen.
CPAP or BPAP therapy should be started during the few days or weeks before surgery, and adherence should be emphasized. Anesthesiologists might reconsider the choice of anesthetic technique in favor of regional anesthesia and modify postoperative pain management to reduce opioid requirements. Reinstituting CPAP or BPAP therapy upon extubation or arrival in the postoperative recovery unit can further reduce the risk of respiratory complications. Additional monitoring such as continuous pulse oximetry when the patient is on the general ward should be considered.
- Mokhlesi B, Kryger MH, Grunstein RR. Assessment and management of patients with obesity hypoventilation syndrome. Proc Am Thorac Soc 2008; 5:218–225.
- Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA 2005; 293:1861–1867.
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31:1071–1078.
- Berg G, Delaive K, Manfreda J, Walld R, Kryger MH. The use of health-care resources in obesity-hypoventilation syndrome. Chest 2001; 120:377–383.
- Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. Am J Med 2004; 116:1–7.
- Kapur V, Strohl KP, Redline S, Iber C, O'Connor G, Nieto J. Underdiagnosis of sleep apnea syndrome in U.S. communities. Sleep Breath 2002; 6:49–54.
- Finkel KJ, Searleman AC, Tymkew H, et al. Prevalence of undiagnosed obstructive sleep apnea among adult surgical patients in an academic medical center. Sleep Med 2009; 10:753–758.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breath 2007; 11:117–124.
- Lee W, Nagubadi S, Kryger MH, Mokhlesi B. Epidemiology of obstructive sleep apnea: a population-based perspective. Expert Rev Respir Med 2008; 2:349–364.
Yes. Obesity hypoventilation syndrome (OHS) is often undiagnosed and greatly increases perioperative risk. Therefore, we recommend trying to detect OHS in a timely manner. Treatment should begin without delay to avoid adverse perioperative outcomes, which can include acute-on-chronic respiratory failure requiring intensive-care monitoring and invasive mechanical ventilation, or death.
ALSO CALLED PICKWICKIAN SYNDROME
OHS is also known as Pickwickian syndrome, named for a character—a “fat boy” who is constantly falling asleep—in The Posthumous Papers of the Pickwick Club by Charles Dickens.
Salient features of OHS are:
- Obesity (body mass index ≥ 30 kg/m2)
- Sleep-disordered breathing (most patients with OHS are morbidly obese and have severe obstructive sleep apnea1)
- Chronic daytime alveolar hypoventilation: ie, Paco2 ≥ 45 mm Hg (normal range 35–45 mm Hg) and Pao2 < 70 mm Hg1 (normal range 85–95 mm Hg)
- No other identifiable cause of hypoventilation such as pulmonary disease (severe obstructive or restrictive), chest wall deformities, severe hypothyroidism, or neuromuscular disease.
WHY SCREEN FOR OHS?
Both obstructive sleep apnea and OHS worsen quality of life and increase the risk of serious disease and death.2–3 Patients with severe sleep apnea, particularly those with hypercapnia (ie, OHS) are at higher risk of cardiopulmonary complications in the perioperative period.
Compared with eucapnic patients with obstructive sleep apnea, patients with OHS have higher health care expenses, are at higher risk of developing serious cardiovascular diseases such as pulmonary hypertension and congestive heart failure, and are more likely to die sooner.4,5
Nowbar et al5 prospectively followed a group of severely obese patients after hospital discharge. At 18 months, 23% of those with OHS had died, compared with 9% of those without OHS. The groups were well matched for body mass index, age, and a number of comorbid conditions. Most of the deaths occurred in the first 3 months after hospital discharge. During the hospital stay, more patients with OHS were admitted to the intensive care unit and needed endotracheal intubation and mechanical ventilation, and more were discharged to a long-term facility.
A high level of suspicion can lead to early recognition and treatment, which may reduce the rate of adverse outcomes associated with undiagnosed and untreated OHS. Routine screening for hypercapnia in patients with sleep apnea might help to identify patients with OHS and allow for modifications in surgical approach, anesthetic technique, and postoperative monitoring, increasing patient safety.
HOW PREVALENT IS OHS?
Obstructive sleep apnea affects up to 20% of US adults and is undiagnosed and untreated in up to 90% of cases.6 Simple screening questionnaires have been shown to reliably identify patients at risk.7,8
To date, no population-based prevalence studies of OHS have been done.
The overall prevalence of OHS in patients with obstructive sleep apnea is better studied: multiple prospective and retrospective studies across various geographic regions with a variety of racial or ethnic populations have shown it to be between 10% and 20%.1,9 This range is very consistent among studies performed in Europe, the United States, and Japan, whether retrospective or prospective, and whether large or small.
The prevalence of OHS in the general adult population in the United States can, however, be estimated. If approximately 5% of the general US population has severe obesity (body mass index ≤ 40 kg/m2), if half of patients with severe obesity have obstructive sleep apnea,10 and if 15% of severely obese patients with sleep apnea have OHS, then a conservative estimated prevalence of OHS in the general adult US population is 0.37% (1 in 270 adults).
WHAT CAN BE DONE BEFORE ELECTIVE SURGERY?
Patients with OHS have an elevated serum bicarbonate level due to metabolic compensation for chronic respiratory acidosis. Moreover, they may have mild hypoxemia during wakefulness as measured by finger pulse oximetry.
The serum venous bicarbonate level is an easy and reasonable test to screen for hypercapnia in obese patients with obstructive sleep apnea because it is readily available, physiologically sensible, and less invasive than arterial puncture to measure blood gases.9
Arterial blood gas measurements, however, should be obtained to confirm the presence and severity of daytime hypercapnia in obese patients with hypoxemia during wakefulness or an elevated serum bicarbonate level.
Pulmonary function testing and chest imaging can exclude other causes of hypercapnia if hypercapnia is confirmed.
An overnight, attended polysomnographic study in a sleep laboratory is ultimately needed to establish the diagnosis and severity of obstructive sleep apnea and to titrate continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) therapy. Since most patients with OHS have severe obstructive sleep apnea, in-laboratory attended polysomnography allows the clinician to both diagnose and intervene with PAP therapy (a “split-night” study). Home titration with an auto-CPAP device is not recommended because it does not have the ability to titrate PAP pressures in response to hypoxemia or hypoventilation. Patients with OHS require attended, laboratory-based PAP titration with or without supplemental oxygen.
CPAP or BPAP therapy should be started during the few days or weeks before surgery, and adherence should be emphasized. Anesthesiologists might reconsider the choice of anesthetic technique in favor of regional anesthesia and modify postoperative pain management to reduce opioid requirements. Reinstituting CPAP or BPAP therapy upon extubation or arrival in the postoperative recovery unit can further reduce the risk of respiratory complications. Additional monitoring such as continuous pulse oximetry when the patient is on the general ward should be considered.
Yes. Obesity hypoventilation syndrome (OHS) is often undiagnosed and greatly increases perioperative risk. Therefore, we recommend trying to detect OHS in a timely manner. Treatment should begin without delay to avoid adverse perioperative outcomes, which can include acute-on-chronic respiratory failure requiring intensive-care monitoring and invasive mechanical ventilation, or death.
ALSO CALLED PICKWICKIAN SYNDROME
OHS is also known as Pickwickian syndrome, named for a character—a “fat boy” who is constantly falling asleep—in The Posthumous Papers of the Pickwick Club by Charles Dickens.
Salient features of OHS are:
- Obesity (body mass index ≥ 30 kg/m2)
- Sleep-disordered breathing (most patients with OHS are morbidly obese and have severe obstructive sleep apnea1)
- Chronic daytime alveolar hypoventilation: ie, Paco2 ≥ 45 mm Hg (normal range 35–45 mm Hg) and Pao2 < 70 mm Hg1 (normal range 85–95 mm Hg)
- No other identifiable cause of hypoventilation such as pulmonary disease (severe obstructive or restrictive), chest wall deformities, severe hypothyroidism, or neuromuscular disease.
WHY SCREEN FOR OHS?
Both obstructive sleep apnea and OHS worsen quality of life and increase the risk of serious disease and death.2–3 Patients with severe sleep apnea, particularly those with hypercapnia (ie, OHS) are at higher risk of cardiopulmonary complications in the perioperative period.
Compared with eucapnic patients with obstructive sleep apnea, patients with OHS have higher health care expenses, are at higher risk of developing serious cardiovascular diseases such as pulmonary hypertension and congestive heart failure, and are more likely to die sooner.4,5
Nowbar et al5 prospectively followed a group of severely obese patients after hospital discharge. At 18 months, 23% of those with OHS had died, compared with 9% of those without OHS. The groups were well matched for body mass index, age, and a number of comorbid conditions. Most of the deaths occurred in the first 3 months after hospital discharge. During the hospital stay, more patients with OHS were admitted to the intensive care unit and needed endotracheal intubation and mechanical ventilation, and more were discharged to a long-term facility.
A high level of suspicion can lead to early recognition and treatment, which may reduce the rate of adverse outcomes associated with undiagnosed and untreated OHS. Routine screening for hypercapnia in patients with sleep apnea might help to identify patients with OHS and allow for modifications in surgical approach, anesthetic technique, and postoperative monitoring, increasing patient safety.
HOW PREVALENT IS OHS?
Obstructive sleep apnea affects up to 20% of US adults and is undiagnosed and untreated in up to 90% of cases.6 Simple screening questionnaires have been shown to reliably identify patients at risk.7,8
To date, no population-based prevalence studies of OHS have been done.
The overall prevalence of OHS in patients with obstructive sleep apnea is better studied: multiple prospective and retrospective studies across various geographic regions with a variety of racial or ethnic populations have shown it to be between 10% and 20%.1,9 This range is very consistent among studies performed in Europe, the United States, and Japan, whether retrospective or prospective, and whether large or small.
The prevalence of OHS in the general adult population in the United States can, however, be estimated. If approximately 5% of the general US population has severe obesity (body mass index ≤ 40 kg/m2), if half of patients with severe obesity have obstructive sleep apnea,10 and if 15% of severely obese patients with sleep apnea have OHS, then a conservative estimated prevalence of OHS in the general adult US population is 0.37% (1 in 270 adults).
WHAT CAN BE DONE BEFORE ELECTIVE SURGERY?
Patients with OHS have an elevated serum bicarbonate level due to metabolic compensation for chronic respiratory acidosis. Moreover, they may have mild hypoxemia during wakefulness as measured by finger pulse oximetry.
The serum venous bicarbonate level is an easy and reasonable test to screen for hypercapnia in obese patients with obstructive sleep apnea because it is readily available, physiologically sensible, and less invasive than arterial puncture to measure blood gases.9
Arterial blood gas measurements, however, should be obtained to confirm the presence and severity of daytime hypercapnia in obese patients with hypoxemia during wakefulness or an elevated serum bicarbonate level.
Pulmonary function testing and chest imaging can exclude other causes of hypercapnia if hypercapnia is confirmed.
An overnight, attended polysomnographic study in a sleep laboratory is ultimately needed to establish the diagnosis and severity of obstructive sleep apnea and to titrate continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) therapy. Since most patients with OHS have severe obstructive sleep apnea, in-laboratory attended polysomnography allows the clinician to both diagnose and intervene with PAP therapy (a “split-night” study). Home titration with an auto-CPAP device is not recommended because it does not have the ability to titrate PAP pressures in response to hypoxemia or hypoventilation. Patients with OHS require attended, laboratory-based PAP titration with or without supplemental oxygen.
CPAP or BPAP therapy should be started during the few days or weeks before surgery, and adherence should be emphasized. Anesthesiologists might reconsider the choice of anesthetic technique in favor of regional anesthesia and modify postoperative pain management to reduce opioid requirements. Reinstituting CPAP or BPAP therapy upon extubation or arrival in the postoperative recovery unit can further reduce the risk of respiratory complications. Additional monitoring such as continuous pulse oximetry when the patient is on the general ward should be considered.
- Mokhlesi B, Kryger MH, Grunstein RR. Assessment and management of patients with obesity hypoventilation syndrome. Proc Am Thorac Soc 2008; 5:218–225.
- Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA 2005; 293:1861–1867.
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31:1071–1078.
- Berg G, Delaive K, Manfreda J, Walld R, Kryger MH. The use of health-care resources in obesity-hypoventilation syndrome. Chest 2001; 120:377–383.
- Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. Am J Med 2004; 116:1–7.
- Kapur V, Strohl KP, Redline S, Iber C, O'Connor G, Nieto J. Underdiagnosis of sleep apnea syndrome in U.S. communities. Sleep Breath 2002; 6:49–54.
- Finkel KJ, Searleman AC, Tymkew H, et al. Prevalence of undiagnosed obstructive sleep apnea among adult surgical patients in an academic medical center. Sleep Med 2009; 10:753–758.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breath 2007; 11:117–124.
- Lee W, Nagubadi S, Kryger MH, Mokhlesi B. Epidemiology of obstructive sleep apnea: a population-based perspective. Expert Rev Respir Med 2008; 2:349–364.
- Mokhlesi B, Kryger MH, Grunstein RR. Assessment and management of patients with obesity hypoventilation syndrome. Proc Am Thorac Soc 2008; 5:218–225.
- Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA 2005; 293:1861–1867.
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31:1071–1078.
- Berg G, Delaive K, Manfreda J, Walld R, Kryger MH. The use of health-care resources in obesity-hypoventilation syndrome. Chest 2001; 120:377–383.
- Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. Am J Med 2004; 116:1–7.
- Kapur V, Strohl KP, Redline S, Iber C, O'Connor G, Nieto J. Underdiagnosis of sleep apnea syndrome in U.S. communities. Sleep Breath 2002; 6:49–54.
- Finkel KJ, Searleman AC, Tymkew H, et al. Prevalence of undiagnosed obstructive sleep apnea among adult surgical patients in an academic medical center. Sleep Med 2009; 10:753–758.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breath 2007; 11:117–124.
- Lee W, Nagubadi S, Kryger MH, Mokhlesi B. Epidemiology of obstructive sleep apnea: a population-based perspective. Expert Rev Respir Med 2008; 2:349–364.
Is an ACE inhibitor plus an ARB more effective than either drug alone?
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
No. Although randomized, controlled trials have shown convincingly that angiotensin-converting enzyme (ACE) inhibitors reduce the rates of death, myocardial infarction, stroke, and heart failure in patients with known coronary artery disease or left ventricular dysfunction,1 and that angiotensin receptor blockers (ARBs) are “noninferior” to and better tolerated than ACE inhibitors, causing less angioedema and cough but costing more,2 dual renin-angiotensin system (RAS) blockade—an ACE inhibitor plus an ARB—has never been shown to reduce the rates of morbidity or death from any cause.
In fact, the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)3,4 found that dual RAS blockade was no more beneficial than monotherapy with an ACE inhibitor or an ARB in preventing serious outcomes in patients with known vascular disease or diabetes with end-organ damage. Furthermore, patients on dual RAS blockade had higher rates of renal insufficiency, hyperkalemia, and hypotension.
THE RATIONALE FOR DUAL RAS BLOCKADE
Dual RAS blockade was first proposed in the early 1990s as a way to avoid the “escape phenomenon” (incomplete suppression of angiotensin II) with ACE inhibitor monotherapy.5 Indeed, studies in rats showed a synergistic effect on blood pressure with an ACE inhibitor combined with an ARB,6 and these results were encouraging enough for the medical community to make a remarkably quick transition to adopting dual RAS blockade in clinical practice.
The concept of dual RAS blockade was so appealing that effects on surrogate end points—lower blood pressure, less protein in the urine, and improved endothelial function—were accepted as free passes, obviating the need for evidence of an effect on hard end points such as lower rates of cardiovascular death or kidney failure. Currently, in the United States, about 1.5% of all patients on RAS blockers are currently receiving both an ACE inhibitor and an ARB.
CONDITIONS IN WHICH DUAL RAS BLOCKADE WAS THOUGHT BENEFICIAL
Hypertension
The European Society of Cardiology’s 2007 clinical practice guidelines7 say that treatment with an ACE inhibitor plus an ARB is preferred for hypertensive patients with metabolic syndrome and its major components (eg, abdominal obesity, insulin resistance, frank diabetes).
Dulton et al, in a meta-analysis,8 calculated that the combination of an ACE inhibitor and an ARB lowered 24-hour blood pressure by 4/3 mm Hg more than monotherapy did. However, most of the studies were of short duration (6 to 8 weeks) and used submaximal doses or once-daily doses of a short-acting ACE inhibitor. Interestingly, studies that used a long-acting ACE inhibitor or a larger dose of a short-acting ACE inhibitor generally showed no additive effect on blood pressure when an ARB was added.
Hence, more evidence from larger randomized and appropriately designed studies is needed before we can conclude that dual RAS blockade is safe and significantly superior to monotherapy in blood pressure control.
Proteinuria
Proteinuria is a surrogate end point for cardiovascular death and is a marker as well as a cause of progressive renal insufficiency. It therefore seemed rational that modifying the degree of proteinuria would translate into robust clinical benefits. Several studies9 showed better renal outcomes, such as fewer patients needing dialysis with combination therapy than with an ACE inhibitor or ARB alone. However, this has never been proven in an adequately powered trial.
ONTARGET was a perfect opportunity to convert what seemed like reliable mechanistic information into solid outcome data.3 The trial enrolled 25,620 patients with established atherosclerotic disease or with diabetes and evidence of end-organ damage. At baseline, 13.1% had microalbuminuria and 4.0% had macroalbuminuria.3 The amount of protein in the urine increased by a significantly lesser amount in the ARB group and in the dualtherapy group than in the group taking only an ACE inhibitor, but in the dual-therapy group this apparent advantage came at the expense of hard end points: more patients reached the primary composite end point of needing dialysis, doubling of their serum creatinine level, or death.
Reducing proteinuria could be an important benefit, but it certainly does not outweigh the risk of increased rates of renal failure and death.
Atherosclerosis and acute coronary syndrome
The road to myocardial infarction begins with inflammation in the “shoulders” of atherosclerotic plaques, which subsequently rupture. Tissue ACE activity and expression of the angiotensin II type 1 receptor are significantly increased in patients with acute coronary syndrome and primarily co-localized to the shoulder regions of the plaque.10 Giving an ACE inhibitor or an ARB to patients who have unstable angina or who have had a myocardial infarction may decrease the rate of reinfarction and lessens the inflammatory process in the atherosclerotic plaque.
Large randomized clinical trials such as HOPE (Heart Outcomes Prevention Evaluation)11 and EUROPA (European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease)12 showed a lower rate of cardiovascular death in patients with established coronary artery disease and normal left ventricular function if they received an ACE inhibitor. In the HOPE trial, the rate of cardiovascular death was 25% lower in patients treated with ramipril (Altace) vs placebo.11 (The year after HOPE was published, the number of prescriptions for ramipril went up 400%). Interestingly, studies of ARBs for secondary prevention failed to show any lowering of the rate of cardiovascular death or myocardial infarction.13
In ONTARGET,4 although the combination of telmisartan (Micardis) and ramipril had a greater effect on blood pressure, it was not significantly better than ramipril alone in terms of the primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure (relative risk 0.99).
Heart failure
The bulk of data on dual RAS blockade in heart failure patients comes from three large randomized trials: CHARM-Added (Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity),14 VALIANT (Valsartan in Acute Myocardial Infarction Trial),15 and VAL-HeFT (Valsartan Heart Failure Trial).16
CHARM-Added14 was the only trial that showed a reduction in cardiovascular deaths with dual RAS therapy (absolute risk reduction 3.6%). It also showed a lower rate of hospitalization for heart failure (absolute risk reduction 4%). However, the rate of allcause mortality was not different between the groups. Of note, more patients receiving dual RAS blockade had to stop taking the study drug because of adverse effects.
Val-HeFT16 showed, in a post hoc analysis, higher rates of morbidity (cardiac arrest, hospitalization for heart failure, or receipt of intravenous inotropic or vasodilator therapy for at least 4 hours) and death when the ARB valsartan (Diovan) was added to the combination of an ACE inhibitor plus a beta-blocker.
A recent meta-analysis17 of safety and tolerability of dual RAS blockade compared with an ACE inhibitor alone found a higher risk of discontinuation because of adverse effects such as hyperkalemia, renal dysfunction, and hypotension in patients on dual RAS blockade. The authors concluded that, given the adverse effects and the lack of consistent survival benefit, the available data do not support the routine addition of an ARB to ACE inhibitor therapy in heart failure patients.
WHAT ABOUT DIRECT RENIN INHIBITORS?
Another class of RAS blockers is available: direct renin inhibitors. Therefore, dual RAS blockade can be achieved by combining an ACE inhibitor with an ARB, an ACE inhibitor with a direct renin inhibitor, or an ARB with a direct renin inhibitor.
We have some outcome data on the combination of an ACE inhibitor plus an ARB,3,4,17 but none for the other two possible dual RAS combinations. Thus far, we know that dual RAS blockade with an ARB and an ACE inhibitor is not beneficial in patients like those in ONTARGET, and that it has questionable benefit in heart failure. However, little is known about combining a direct renin inhibitor with either an ACE inhibitor or an ARB.
Since ARBs and ACE inhibitors both increase plasma renin activity and only partially block the RAS, the argument has been put forward that the addition of a drug such as a direct renin inhibitor, which really decreases plasma renin activity, has the potential to be more beneficial than blockade with either an ACE inhibitor or an ARB. In theory, this is an attractive concept and certainly deserves scrutiny in outcome studies such as ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints).18
SURROGATE END POINTS: A CAVEAT
As defined by Temple,19 a surrogate end point of a clinical trial is a laboratory measurement or a physical sign used as a substitute for a clinically meaningful end point that measures directly how patients feel or function, or if they survive. Effects on surrogate end points often fail to predict the true clinical effects of an intervention, as the ONTARGET data demonstrated. Among several explanations for this failure is that interventions may affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms that operate independently of the disease process.20 Nonetheless, surrogate end point cosmetics remains attractive for many clinicians.
The ONTARGET findings indicate that there is no clinically important benefit in adding an ARB for patients with hypertension, proteinuria, heart failure, or coronary artery disease if they are already being treated with an ACE inhibitor. This would indicate that dual RAS blockade should be avoided in clinical practice until we are provided with better evidence.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.
- Father MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left ventricular dysfunction: a systemic overview of data from individual patients. ACEInhibitor Myocardial Infarction Collaborative Group. Lancet 2000; 355:1575–1581.
- Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 2000; 355:1582–1587.
- Mann JF, Schmiede RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomized, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Yusuf S, Teo KK, Pogue J, et al; ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events, N Engl J Med 2008; 358:1547–1559.
- van den Meiracker AH, Man in ‘t Veld AJ, Admiraal PJ, et al. Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: dose it exist and does it affect the antihypertensive response? J Hypertens 1992; 10:803–812.
- Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation 1997; 96:3072–3078.
- Mancia G, De Backer G, Dominiczak A, et al; Task Force for the Management of Arterial Hypertension of the European Society of Hypertension. 2007 guidelines for the management of arterial hypertension. J Hypertens 2007; 25:1105–1187.
- Dulton TW, He FJ, MacGregor GA. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45:880–886.
- Kunz R, Fredrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000; 101:1372–1378.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342:145–153.
- Fox KM; European trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomized, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003; 362:782–788.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002; 359:995–1003.
- McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced leftventricular systolic function taking angiotensin converting enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362:767–771.
- Pfeffer MA, McMurray JJ, Velzquez E, et al; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- Cohn JN, Tognoni GValsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med 2001: 345:1667–1675.
- Lakhdar R, Al-Mallah MH, Lanfear DE. Safety and tolerability of angiotensin-converting enzyme inhibitor versus the combination of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker in patients with left ventricular dysfunction: a systematic review and meta-analysis of randomized controlled trials. J Card Fail 2008; 14:181–188.
- Parving H-H, Brenner BM, McMurray JJV, et al. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design [published online ahead of print January 14, 2009]. Nephrol Dial Transplant. doi:10.1093/ndt/gfn721.
- Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In:Nimmo WS, Tucker GT, eds. Clinical Measurement in Drug Evaluation. J Wiley: New York, 1995.
- Messerli FH. The sudden demise of dual renin-angiotensin system blockade or the soft science of the surrogate end point. J Am Coll Cardiol 2009; 53:468–470.