User login
HIV update: Which single-tablet regimens, and when
› Offer all patients with human immunodeficiency virus (HIV) disease antiretroviral therapy (ART) regardless of disease state or CD4 cell lymphocyte count. A
› Consider one of 6 recommended ART regimens for ART-naive patients. A
› Offer one of 6 alternative antiretroviral regimens to patients unable to tolerate one of the recommended regimens for reasons of toxicity, a pre-existing medical condition, or baseline viral resistance. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.
A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 years have changed everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the US Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer half-life than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of single-tablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of antiretroviral therapy
The US Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see TABLE 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.

The DHHS guidelines divide treatment options into 3 categories:2
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently recommended first-line therapies
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (TABLE 2).2
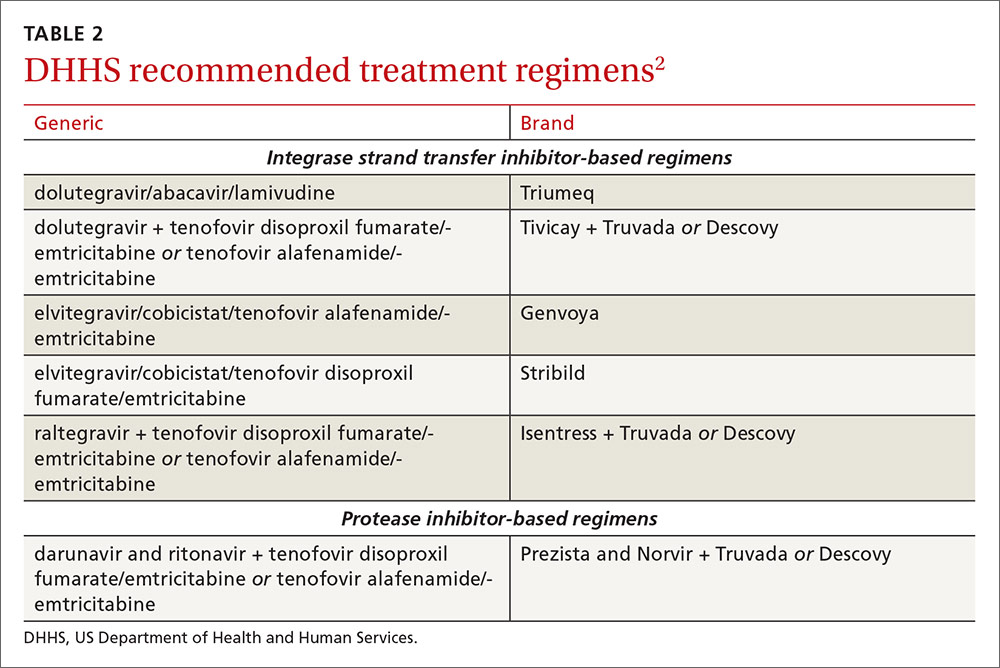
INSTI-based regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
Dolutegravir plus tenofovir disoproxil fumarate/emtricitabine (Tivicay plus Truvada). The combination of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12
Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine that was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15
Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. There is a once-daily formulation of raltegravir that's expected to be available late in 2017.20
Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-based regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and self-limiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in late 2017, it will provide yet another daily STR option.23
The addition of fixed-dose tenofovir alafenamide/emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS “alternative” and“other” regimens
The DHHS guidelines also include “alternative” (TABLE 32) and “other” regimens (available at: http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not. These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice.
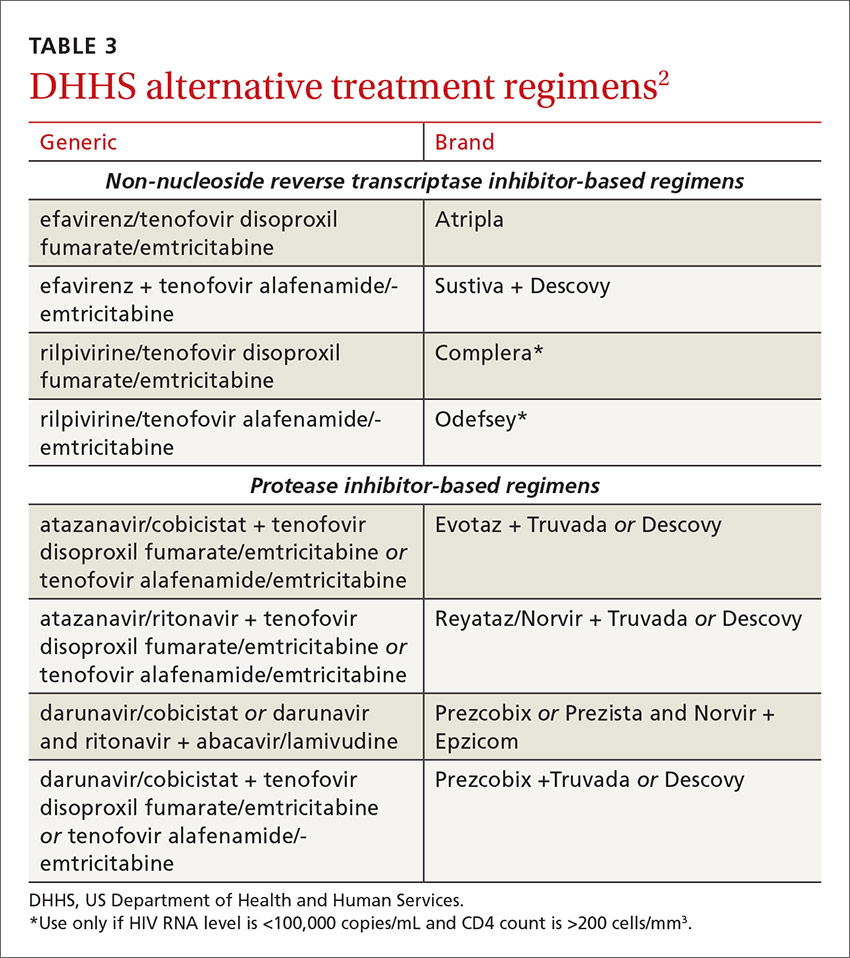
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir- or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The future of ART
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE › At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
CORRESPONDENCE
Jeffrey T. Kirchner, DO, FAAFP, AAHIVS, Medical Director, LGHP Comprehensive Care, 554 North Duke St., 3rd Floor, Lancaster, PA 1760; [email protected].
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J,et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drugs/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://prescribersletter.therapeuticresearch.com/ce/cecourse.aspx?pc=15-215. Accessed October 6
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
› Offer all patients with human immunodeficiency virus (HIV) disease antiretroviral therapy (ART) regardless of disease state or CD4 cell lymphocyte count. A
› Consider one of 6 recommended ART regimens for ART-naive patients. A
› Offer one of 6 alternative antiretroviral regimens to patients unable to tolerate one of the recommended regimens for reasons of toxicity, a pre-existing medical condition, or baseline viral resistance. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.
A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 years have changed everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the US Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer half-life than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of single-tablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of antiretroviral therapy
The US Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see TABLE 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.
The DHHS guidelines divide treatment options into 3 categories:2
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently recommended first-line therapies
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (TABLE 2).2
INSTI-based regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
Dolutegravir plus tenofovir disoproxil fumarate/emtricitabine (Tivicay plus Truvada). The combination of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12
Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine that was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15
Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. There is a once-daily formulation of raltegravir that's expected to be available late in 2017.20
Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-based regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and self-limiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in late 2017, it will provide yet another daily STR option.23
The addition of fixed-dose tenofovir alafenamide/emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS “alternative” and“other” regimens
The DHHS guidelines also include “alternative” (TABLE 32) and “other” regimens (available at: http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not. These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice.
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir- or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The future of ART
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE › At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
CORRESPONDENCE
Jeffrey T. Kirchner, DO, FAAFP, AAHIVS, Medical Director, LGHP Comprehensive Care, 554 North Duke St., 3rd Floor, Lancaster, PA 1760; [email protected].
› Offer all patients with human immunodeficiency virus (HIV) disease antiretroviral therapy (ART) regardless of disease state or CD4 cell lymphocyte count. A
› Consider one of 6 recommended ART regimens for ART-naive patients. A
› Offer one of 6 alternative antiretroviral regimens to patients unable to tolerate one of the recommended regimens for reasons of toxicity, a pre-existing medical condition, or baseline viral resistance. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.
A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 years have changed everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the US Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer half-life than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of single-tablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of antiretroviral therapy
The US Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see TABLE 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.
The DHHS guidelines divide treatment options into 3 categories:2
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently recommended first-line therapies
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (TABLE 2).2
INSTI-based regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
Dolutegravir plus tenofovir disoproxil fumarate/emtricitabine (Tivicay plus Truvada). The combination of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12
Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine that was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15
Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. There is a once-daily formulation of raltegravir that's expected to be available late in 2017.20
Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-based regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and self-limiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in late 2017, it will provide yet another daily STR option.23
The addition of fixed-dose tenofovir alafenamide/emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS “alternative” and“other” regimens
The DHHS guidelines also include “alternative” (TABLE 32) and “other” regimens (available at: http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not. These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice.
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir- or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The future of ART
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE › At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
CORRESPONDENCE
Jeffrey T. Kirchner, DO, FAAFP, AAHIVS, Medical Director, LGHP Comprehensive Care, 554 North Duke St., 3rd Floor, Lancaster, PA 1760; [email protected].
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J,et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drugs/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://prescribersletter.therapeuticresearch.com/ce/cecourse.aspx?pc=15-215. Accessed October 6
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J,et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drugs/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://prescribersletter.therapeuticresearch.com/ce/cecourse.aspx?pc=15-215. Accessed October 6
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.

A step-wise approach to exertional leg pain
PRACTICE RECOMMENDATIONS
› Consider the possibility of vascular and neurologic problems as the source of exertional leg pain (ELP). C
› Order magnetic resonance imaging to evaluate patients with ELP and negative x-rays for stress fractures. C
› Measure lower extremity intracompartmental pressures both before and after exercise when you suspect chronic exertional compartmental syndrome. Doing so is the gold standard for the diagnosis of this condition. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Most family physicians are accustomed to treating active patients with shin splints and stress fractures. But many are less familiar with, and slower to recognize, other sources of exertional leg pain (ELP), defined as exercise-related pain that localizes in the lower extremity distal to the knee and proximal to the talocrural joint.1
ELP has a broad differential diagnosis that includes other musculoskeletal conditions—most notably chronic exertional compartment syndrome (CECS), which has been found to affect 33% of athletes with chronic ELP1—as well as a number of vascular and neurologic causes.2-4 In addition, etiologies may overlap. Greater awareness of the many causes of ELP can help you to avoid the unnecessary use of expensive diagnostic tests as well as delayed diagnosis and treatment.
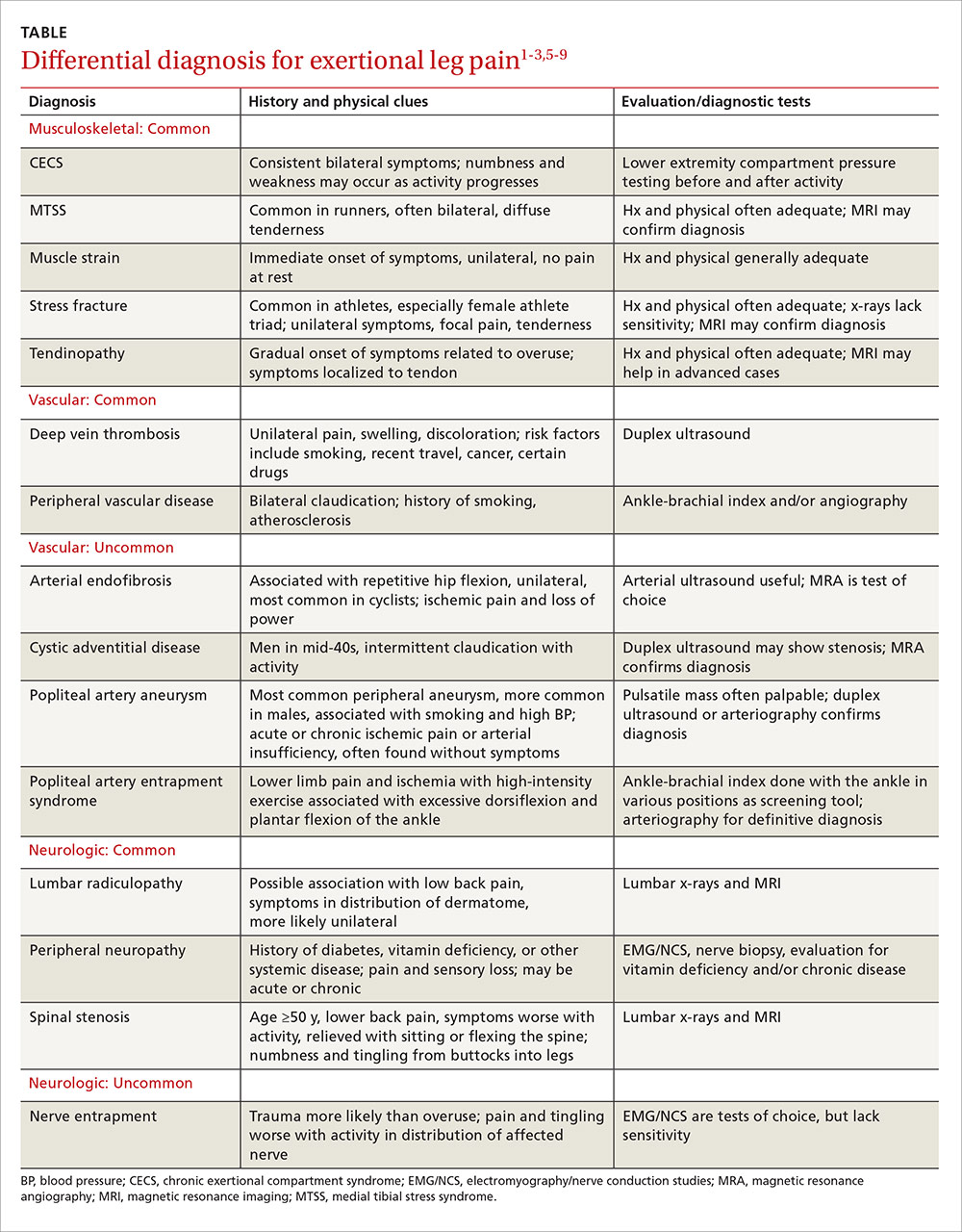
A thorough medical and activity history, symptom review, and physical examination are your most important tools when patients present with ELP. When the cause is not obvious or the patient fails to respond to conservative measures, x-rays, magnetic resonance imaging (MRI), vascular studies, electromyography and nerve conduction studies, and/or intracompartmental pressure testing may be needed to find the source of the symptoms. In the text that follows, we review both common and relatively uncommon sources of ELP, using a stepwise diagnostic approach. You’ll find a diagnostic challenge, in which you can test your skills and a more comprehensive differential diagnosis in the TABLE.1-3,5-9
Musculoskeletal injuries: Shin splints and beyond
Medial tibial stress syndrome (MTSS), commonly known as shin splints, is characterized by pain and tenderness over the posteromedial aspect of the distal tibia.3 It typically results in diffuse pain that occurs with exercise, but may persist at rest in severe cases.3-6 Less often, localized swelling may also be present.2
MTSS accounts for between 6% and 16% of all running injuries.2,10 It is associated with a spectrum of tibial stress injuries, including periostitis, tendinopathy, and stress reaction, with dysfunction of the tibialis posterior, tibialis anterior, and soleus muscles thought to be contributing factors.2,11 Intrinsic factors include high body mass index (BMI), female sex, excessive internal and external hip rotation, hyperpronation, and hyper plantar flexion.2,10,12
X-rays of the leg are typically normal in patients with MTSS and should be considered only if the clinical presentation suggests the possibility of an alternative diagnosis, such as a stress fracture or tumor.2-4,13 Advanced imaging such as MRI or triple phase bone scans (TPBS) are useful when the diagnosis is in question and will reveal an abnormally high signal along the posterior medial tibial surface or the classic train-track appearance of nucleotide uptake in patients with MTSS.2 MRI readily shows periosteal reaction and bony edema and has a sensitivity of 78% to 89% and a specificity of 33% to 100% for the diagnosis of MTSS.14,15
Initial management of MTSS is conservative, with the mainstay of treatment consisting of rest, ice, and nonsteroidal anti-inflammatory drugs (NSAIDs).3,13,16 While ice, NSAIDs, proper conditioning, physical therapy to stretch and strengthen the calf musculature, rigid orthotics to correct foot hyperpronation, and activity modification are all appropriate treatments, randomized controlled trials have shown none of these interventions to be more effective than rest alone.2 Non-operative treatment is usually successful, but surgery may be required for severe or refractory cases. Procedures include posteromedial fasciotomy, release of the medial soleus fascial bridge, deep compartment fasciotomy, or removal of a section of the distal tibia periosteum.3,4
Lower extremity stress fracture. Stress fractures are caused by repetitive loading that results in microtrauma, including bony microfractures. The vast majority of cases—80% to 95% of stress fractures—affect the lower extremities, and most involve the tibia.2-4,6,13,17 The most common presentation is an insidious onset of pain over a specific bony area with a normal appearance, although localized swelling or erythema may occasionally be present.3,14,17,18 The pain may be reproduced or worsened by weight-bearing activities and relieved by rest.14,18
Consider the female athlete triad. In evaluating a patient with a stress fracture, pay close attention to dietary history, BMI, and, in female athletes, take a detailed menstrual history. Such patients are at risk for amenorrhea, low bone mineral density, and nutritional deficits—the “female athlete triad,” which carries an increased risk of stress fractures.3,14,17-19
Stress fractures can often be diagnosed with a thorough medical history and physical, with imaging used for confirmation.6,14,17,18 Historical features of a stress fracture that may differentiate it from MTSS include pain that is unilateral and absent at rest and occurs with more prolonged activity, as well as post-exercise and/or nocturnal pain. Notable physical exam features include pain that is reproduced in a focal area with a single leg hop or percussion with a tuning fork or ultrasound.5,11,17
Initially, sensitivity for a plain radiograph is as low as 10%.2,11 Abnormalities on x-ray are usually seen after 2 to 8 weeks of symptoms2,7,11 and may include a faint periosteal reaction, a fluffy area of callus, or a cortical lucency sometimes referred to as the “dreaded black line.”3,6,17 If a radiographic exam shows evidence of a stress fracture, further imaging is typically unnecessary. MRI or TPBS is suggested, however, when x-rays appear normal but suspicion of a stress fracture remains.3,17,18 MRI may show edema within 3 days of symptom onset and is more sensitive and specific than computed tomography (CT) or TPBS for diagnosing stress fractures of the tibia.2,16
Treatment of tibial stress fractures is typically non-operative and consists of alterations in activity (eg, non weight-bearing), correction of nutritional deficits, such as inadequate caloric intake or too little calcium or iron, and addressing problems with footwear, training regimen, and/or running surface.3,14,18 Fibular and posteromedial tibial stress fractures are considered low risk and heal with weight-bearing restrictions and rest, initially for a minimum of 2 to 4 weeks.3
Posteromedial tibia injuries tend to heal well because they are on the compression side of the bone. Anterior tibia stress fractures, which are located on the tension side of the bone2,7 and account for approximately 5% of all tibia stress fractures, are more prone to non-union or progression to a complete fracture.7,20 Thus, anterior tibia stress injuries warrant a more aggressive approach, with treatment options including non-weight bearing status that may last longer than 8 weeks, pneumatic brace casting, and/or orthopedic referral to evaluate for surgical intervention.7,20-22 Time for radiographic evidence of healing may exceed 8 months, so early surgical intervention should be considered, especially for high-level athletes.7,20,21
Test your skills: A diagnostic challenge
Janine T, a 24-year-old long-distance runner, presents with left lower leg pain that occurs with activity. There was no injury, Ms. T reports; the pain began about 6 weeks ago, shortly after she began training for a marathon and running more than 30 miles per week. The pain is not relieved with intermittent rest or over-the-counter analgesics, she says. But it usually abates within 15 to 30 minutes after she completes her run.
Ms. T is underweight, with a body mass index <17 kg/m2. She denies any dietary restrictions and has normal menstrual cycles. The patient reports taking oral contraceptives, but no other medications. An initial x-ray is normal, as is magnetic resonance imaging to evaluate for a stress fracture.
You suspect Ms. T has shin splints, advise her to rest for a few weeks and to consider getting orthotics for her running shoes, and schedule a follow-up visit.
When she comes in 6 weeks later, the patient reports that she resumed running after a 3-week rest; shortly after, she noticed pain in both legs. What’s more, she now experiences tingling in her feet after running a few miles.
What’s wrong with this patient?
Ms. T’s symptoms—bilateral persistent leg pain, with tingling in both feet, and little improvement with rest—strongly suggest that she has chronic exertional compartment syndrome. Intracompartmental pressure testing, which reveals pre-exercise values ≥15 mm Hg and post-exercise values of ≥30 mm Hg at one minute, confirms the diagnosis.
Activity avoidance or modification will allow Ms. T’s symptoms to subside, but they’re highly likely to recur when she resumes running. The definitive treatment is intracompartmental fasciotomy, which has a success rate of approximately 80%.1,28
When to suspect chronic exertional compartment syndrome
Leg pain in CECS results from increased pressure within the lower extremity fascial compartments temporally related to exercise.2,23,24 Its incidence in the general population is unknown, but CECS has been found to range from 14% to 27% in patients with previously undiagnosed leg pain1,14,25 and to affect about a third of athletes with chronic ELP. In addition, CECS has been found in 90% of patients who have both diabetes and ELP with normal findings on vascular studies.1,3,4,26
The anterior compartment is most commonly affected, followed by the lateral, deep posterior, and superficial posterior compartments.3,13,23,27 Symptoms are bilateral 60% to 95% of the time.2,13,14,25 Factors contributing to CECS include fixed muscular compartment constraints, muscle swelling, thickened fascia, muscle hypertrophy related to resistance training, dynamic muscular contraction patterns, and low muscle capillary supply. Stretching of fascial pain receptors and pressure fibers and inadequate myocyte response to increased metabolism may play a role, as well.14,28
The initial clinical presentation is usually predictable leg pain—ie, pain that begins at about the same time, distance, or intensity of a workout and resolves with rest; numbness and weakness may occur as the workout progresses. In time, leg pain associated with CECS may be present with everyday activity or at rest. The physical exam may be normal or reveal swelling, tenderness over the involved compartments, pain with passive digit or ankle motion, and palpable muscle herniation.14
Measurement of intracompartmental pressure before and after exercise is the gold standard for diagnosis of CECS.2,14,27 Pre-exercise values ≥15 mm Hg and post-exercise values ≥30 mm Hg at one minute or ≥20 mm Hg at 5 minutes are all considered diagnostic of CECS,11 although these widely accepted criteria for bilateral testing of all compartments yields a false-positive rate of 5%.27 CECS is almost always bilateral,29 and some clinicians advocate limiting the number of needle insertions by taking only post-exercise measurements and testing only symptomatic compartments in one limb.
Imaging has limited value, as both x-rays and MRIs are usually normal.14 However, post-exertional T2-weighted MRI findings of muscular edema correspond to increased intracompartmental pressures, with a sensitivity of 87% and a specificity of 62%.14,24,30,31 Infrared spectroscopy, which measures levels of oxygenated and deoxygenated blood, is sensitive for CECS when the post-exercise ratio of deoxygenated to oxygenated blood remains elevated.14,24 Neither of these screening modalities is routinely obtained or considered diagnostic, however. Their chief role is to exclude an alternative diagnosis.14
Treatment and symptom relief. Discontinuing or modifying the aggravating activity typically brings relief of CECS. But this is not a long-term solution, as symptoms are likely to recur when the patient returns to the activity in question.1 The definitive treatment is compartment release via fasciotomy. Success rates for anterior and lateral compartment releases are >80%.1,28 The success of fasciotomy of posterior compartments, however, is <50%—a finding attributed to more complex anatomy, difficult visualization, and the presence of additional compartments.1,32
When the cause is vascular
Arterial endofibrosis—the fibrotic thickening of the intima of an artery—is thought to be caused by repetitive hip flexion.8 This results in hyperplasia, wall thickening, and eventual stenosis of the vessel, with 90% of cases affecting the external iliac artery.8,33 The condition is most common in activities such as cycling, but is also seen in such activities as running, skiing, soccer, and rugby. Symptoms are typically unilateral, but an estimated 15% of patients experience bilateral symptoms.8,33
Loss of power in the affected leg, with intermittent claudication and pain due to presumed ischemia from the vascular defect, is the usual presentation, although some patients develop cramping of the buttocks and/or paresthesia of the affected leg and foot during uphill running or cycling.8,33 The physical exam is often normal, but there may be a post-exercise arterial bruit over the femoral artery when the hip is flexed.8,34
Pre-exercise ankle-brachial index (ABI) <0.5 and post-exercise ABI <0.66 at one minute is suggestive of moderate arterial endofibrosis, with 90% sensitivity and 87% specificity.8,33,34 Arterial ultrasound and color Doppler may also be used for diagnosis, but are often operator dependent. Magnetic resonance angiography (MRA), while more expensive, can detect excessive kinking or compression of the vessel and is not operator dependent.8,33 Angioplastic balloon catheter dilation and stenting, bypass surgery, vascular reconstruction and endarterectomy with vein patch are the options for treatment. The success rates of the various interventions are unknown due to a lack of head-to-head studies and long-term follow-up.8,33
Popliteal artery entrapment syndrome (PAES) is a constellation of symptoms caused by vascular impingement in the popliteal fossa of the knee.8,34 The typical presenting symptoms are lower limb ischemia and pain caused by intense exercise that resolves quickly afterwards. Symptoms correlate more with the intensity than the duration of exercise.3,8
PAES is usually caused by a variant of the gastrocnemius muscle in which a medial head passes behind the popliteal artery in males younger than 30 years.8,33-35 Less commonly, it is the result of an overuse or acute orthopedic injury that irritates structures surrounding the popliteal fossa.8,34 PAES affects football, basketball, and soccer players, as well as runners because of excessive dorsiflexion and plantar flexion of the ankle.3,4
The physical exam for a patient with PAES is typically normal, but a post-exercise popliteal bruit with weak peripheral pulses may be elicited.8,33 An ABI in the neutral, forced dorsiflexion and forced plantar flexion positions can serve as a useful screening tool. An ABI <0.9 is abnormal, with a sensitivity and specificity of 90% and 98%, respectively, for stenosis >50%.2,36
Arteriography is the gold standard for diagnosis of PAES. Contrast arteriography is most commonly used because of its availability and cost. But MRA better differentiates functional from anatomic entrapment—a differentiation that less invasive tests, such as duplex ultrasound studies, lack the specificity to reveal.8,34 Treatment requires either surgical removal of the offending musculotendinous structures or arterial bypass and grafting of the chronically impinged area, as conservative therapies lack efficacy.2,8,34
Cystic adventitial disease (CAD) is the narrowing of an artery by mucoid cysts in the arterial wall or adventitia.8,9 It is a rare condition, accounting for just 0.1% of all vascular diseases, most commonly occurring in men in their mid-40s.8,33 CAD is thought to be the result of mucin-producing cells being haphazardly incorporated into the adventitia during arterial development. About 85% of patients whose popliteal artery is affected in this way will experience intermittent claudication with activity.8,9
On exam, such patients often have diminished ankle-brachial pressure indices, and duplex ultrasound often reveals stenosis in the affected artery, as well as a collection of mucoid cysts in the adventitia.8,9
Diagnosis can be confirmed by MRA.8,37 Evidence for the treatment of CAD is largely anecdotal.9 Cysts may be aspirated but tend to recur, and stenting does not correct the cystic-induced narrowing of the vessel. Surgical removal of the cysts is the only successful treatment.8,9
Neurologic causes to consider
Spinal stenosis is caused by central canal narrowing secondary to congenital abnormalities, trauma, or, most commonly, degenerative changes in the lumbar spine. Spinal stenosis is generally seen in men or women ages 50 to 70 years.38 Patients experience unilateral or bilateral claudication that improves with sitting or flexion of the spine5 and may develop bilateral lower extremity numbness and tingling from the buttocks that radiates down the legs. Diagnosis is typically made with a combination of a lumbar x-ray and an MRI, which will show nerve compression and bony overgrowth.38 CT myelogram, another imaging option, isless sensitive in the acute phase, but can be used to monitor the disease course.
Initial treatment includes physical therapy and NSAIDs.5 If conservative therapy fails, epidural or nerve root corticosteroid injections and surgical decompression or laminectomy are options.38
Nerve entrapment is a less common source of lower extremity pain in which the superficial peroneal nerve is most often affected.4,12,17,39 Trauma is the usual cause of nerve entrapment, but it may also be associated with overuse, most notably related to dance, soccer, or tennis.2,14,40,41 The most likely anatomic site is where the nerve exits the deep fascia within the lateral compartment in the lower third of the leg.39,40 Less frequently, the common peroneal nerve at the fibular neck, the saphenous nerve as it passes through Hunter’s canal, the posterior tibial nerve at the tarsal tunnel, and the sural nerve in the posterior calf may be affected.3,4,12,17,20,40,41 Entrapment of the peroneal nerve may be associated with activities involving repetitive inversion and eversion, such as running and cycling. Injury of the saphenous nerve is seen in sports involving repetitive knee flexion like rowing and cycling. Sural nerve entrapment is a result of crural fascia compression of the nerve during activities like running and track.3,14,40,42,43
Patients typically experience burning, tingling, and radiation of pain with activity. Symptoms worsen with continued exercise. The physical exam is often normal, especially early in the disease process, but may reveal sensory loss, motor weakness, and a loss of reflexes.2,40 Patients with superficial peroneal nerve involvement may have distal lateral leg pain that radiates into the dorsum of the foot, often exacerbated by lower leg percussion and resulting in diminished sensation.1 Common peroneal nerve involvement may alter sensation of the lateral leg, as well, but may also cause foot drop.2 The saphenous nerve can cause medial knee or leg symptoms, while the sural nerve can yield pain in the lateral ankle and foot.2
To diagnose nerve entrapment, electromyography and nerve conduction velocities at the level of the lesion may yield positive results 3 to 4 weeks after symptom onset.2,13,40 There are wide ranges of sensitivity and specificity for these studies, but they are nonetheless considered the tests of choice for nerve entrapment.1,44 Conservative treatment with activity modification, physical therapy, massage, and NSAIDs is often sufficient,2 with surgical management warranted only for refractory cases.2,14,40,41
CORRESPONDENCE
Jonathan A. Becker, MD, CAQSM, University of Louisville Department of Family and Geriatric Medicine, 201 Abraham Flexner Way, Suite 690, Louisville, KY 40202; jon.becker@louisville@edu.
1. Rajasekaran S, Kvinlaug K, Finnoff JT. Exertional leg pain in the athlete. PM R. 2012;4:985-1000.
2. Brewer RB, Gregory AJ. Chronic lower leg pain in athletes: a guide for the differential diagnosis, evaluation, and treatment. Sports Health. 2012;4:121-127.
3. Edwards PH Jr, Wright ML, Hartman JF. A practical approach for the differential diagnosis of chronic leg pain in the athlete. Am J Sports Med. 2005;33:1241-1249.
4. Clanton TO, Solcher BW. Chronic leg pain in the athlete. Clin Sports Med. 1994;13:743-759.
5. Fredericson M, Wun C. Differential diagnosis of leg pain in the athlete. J Am Podiatr Med Assoc. 2003;93:321-324.
6. Pell RF 4th, Khanuja HS, Cooley GR. Leg pain in the running athlete. J Am Acad Orthop Surg. 2004;12:396-404.
7. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8:344-353.
8. Pham TT, Kapur R, Harwood MI. Exertional leg pain: teasing out arterial entrapments. Curr Sports Med Rep. 2007;6:371-375.
9. Wright LB, Matchett WJ, Cruz CP, et al. Popliteal artery disease: diagnosis and treatment. Radiographics. 2004;24:467-479.
10. Yates B, White S. The incidence and risk factors in the development of medial tibial stress syndrome among naval recruits. Am J Sports Med. 2004;32:772-780.
11. Fredericson M, Jennings F, Beaulieu C, et al. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17:309-325.
12. Plisky MS, Rauh MJ, Heiderscheit B, et al. Medial tibial stress syndrome in high school cross-country runners: incidence and risk factors. J Orthop Sports Phys Ther. 2007;37:40-47.
13. Touliopolous S, Hershman EB. Lower leg pain. Diagnosis and treatment of compartment syndromes and other pain syndromes of the leg. Sports Med. 1999;27:193-204.
14. Burrus MT, Werner BC, Starman JS, et al. Chronic leg pain in athletes. Am J Sports Med. 2015;43:1538-1547.
15. Batt ME, Ugalde V, Anderson MW, et al. A prospective controlled study of diagnostic imaging for acute shin splints. Med Sci Sports Exerc. 1998;30:1564-1571.
16. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235:553-561.
17. Sterling JC, Edelstein DW, Calvo RD, et al. Stress fractures in the athlete. Diagnosis and management. Sports Med. 1992;14:336-346.
18. Brukner P. Exercise-related lower leg pain: bone. Med Sci Sports Exerc. 2000;32:S15-S26.
19. Tuan K, Wu S, Sennett B. Stress fractures in athletes: risk factors, diagnosis, and management. Orthopedics. 2004;27:583-591.
20. Harrast MA, Colonno D. Stress fractures in runners. Clin Sports Med. 2010;29:399-416.
21. Varner KE, Younas SA, Lintner DM, et al. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33:1071-1076.
22. Kaeding CC, Yu JR, Wright R, et al. Management and return to play of stress fractures. Clin J Sport Med. 2005;15:442-447.
23. Blackman PG. A review of chronic exertional compartment syndrome in the lower leg. Med Sci Sports Exerc. 2000;32:S4-S10.
24. Brennan FH Jr, Kane SF. Diagnosis, treatment options, and rehabilitation of chronic lower leg exertional compartment syndrome. Curr Sports Med Rep. 2003;2:247-250.
25. Davis DE, Raikin S, Garras DN, et al. Characteristics of patients with chronic exertional compartment syndrome. Foot Ankle Int. 2013;34:1349-1354.
26. Edmundsson D, Toolanen G. Chronic exertional compartment syndrome in diabetes mellitus. Diabet Med. 2011;28:81-85.
27. Pedowitz RA, Hargens AR, Mubarak SJ, et al. Modified criteria for the objective diagnosis of chronic compartment syndrome of the leg. Am J Sports Med. 1990;18:35-40.
28. Bong MR, Polatsch DB, Jazrawi LM, et al. Chronic exertional compartment syndrome: diagnosis and management. Bull Hosp Jt Dis. 2005;62:77-84.
29. Hislop M, Batt ME. Chronic exertional compartment syndrome testing: a minimalist approach. Br J Sports Med. 2011;45:954-955.
30. Brown RR, Rosenberg ZS. MR imaging of exercise-induced lower leg pain. Magn Reson Imaging Clin N Am. 2001;9:533-552.
31. Ringler MD, Litwiller DV, Felmlee JP, et al. MRI accurately detects chronic exertional compartment syndrome: a validation study. Skeletal Radiol. 2013;42:385-392.
32. Schepsis AA, Martini D, Corbett M. Surgical management of exertional compartment syndrome of the lower leg. Long-term followup. Am J Sports Med. 1993;21:811-817.
33. Ehsan O, Darwish A, Edmundson C, et al. Non-traumatic lower limb vascular complications in endurance athletes. Review of literature. Eur J Vasc Endovasc Surg. 2004;28:1-8.
34. Turnipseed WD. Popliteal entrapment syndrome. J Vasc Surg. 2002;35:910-915.
35. Baltopoulos P, Filippou DK, Sigala F. Popliteal artery entrapment syndrome: anatomic or functional syndrome? Clin J Sport Med. 2004;14:8-12.
36. McDermott MM, Criqui MH, Liu K, et al. Lower ankle/brachial index, as calculated by averaging the dorsalis pedis and posterior tibial arterial pressures, and association with leg functioning in peripheral arterial disease. J Vasc Surg. 2000;32:1164-1171.
37. Elias DA, White LM, Rubenstein JD, et al. Clinical evaluation and MR imaging features of popliteal artery entrapment and cystic adventitial disease. AJR Am J Roentgenol. 2003;180:627-632.
38. Genevay S, Atlas SJ. Lumbar spinal stenosis. Best Pract Res Clin Rheumatol. 2010;24:253-265.
39. Korkola M, Amendola A. Exercise-induced leg pain: sifting through a broad differential. Phys Sportsmed. 2001;29:35-50.
40. McCrory P, Bell S, Bradshaw C. Nerve entrapments of the lower leg, ankle and foot in sport. Sports Med. 2002;32:371-391.
41. Schon LC. Nerve entrapment, neuropathy, and nerve dysfunction in athletes. Orthop Clin North Am. 1994;25:47-59.
42. Anselmi SJ. Common peroneal nerve compression. J Am Podiatr Med Assoc. 2006;96:413-417.
43. Maalla R, Youssef M, Ben Lassoued N, et al. Peroneal nerve entrapment at the fibular head: outcomes of neurolysis. Orthop Traumatol Surg Res. 2013;99:719-722.
44. Marciniak C, Armon C, Wilson J, et al. Practice parameter: utility of electrodiagnostic techniques in evaluating patients with suspected peroneal neuropathy: an evidence-based review. Muscle Nerve. 2005;31:520-527.
PRACTICE RECOMMENDATIONS
› Consider the possibility of vascular and neurologic problems as the source of exertional leg pain (ELP). C
› Order magnetic resonance imaging to evaluate patients with ELP and negative x-rays for stress fractures. C
› Measure lower extremity intracompartmental pressures both before and after exercise when you suspect chronic exertional compartmental syndrome. Doing so is the gold standard for the diagnosis of this condition. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Most family physicians are accustomed to treating active patients with shin splints and stress fractures. But many are less familiar with, and slower to recognize, other sources of exertional leg pain (ELP), defined as exercise-related pain that localizes in the lower extremity distal to the knee and proximal to the talocrural joint.1
ELP has a broad differential diagnosis that includes other musculoskeletal conditions—most notably chronic exertional compartment syndrome (CECS), which has been found to affect 33% of athletes with chronic ELP1—as well as a number of vascular and neurologic causes.2-4 In addition, etiologies may overlap. Greater awareness of the many causes of ELP can help you to avoid the unnecessary use of expensive diagnostic tests as well as delayed diagnosis and treatment.
A thorough medical and activity history, symptom review, and physical examination are your most important tools when patients present with ELP. When the cause is not obvious or the patient fails to respond to conservative measures, x-rays, magnetic resonance imaging (MRI), vascular studies, electromyography and nerve conduction studies, and/or intracompartmental pressure testing may be needed to find the source of the symptoms. In the text that follows, we review both common and relatively uncommon sources of ELP, using a stepwise diagnostic approach. You’ll find a diagnostic challenge, in which you can test your skills and a more comprehensive differential diagnosis in the TABLE.1-3,5-9
Musculoskeletal injuries: Shin splints and beyond
Medial tibial stress syndrome (MTSS), commonly known as shin splints, is characterized by pain and tenderness over the posteromedial aspect of the distal tibia.3 It typically results in diffuse pain that occurs with exercise, but may persist at rest in severe cases.3-6 Less often, localized swelling may also be present.2
MTSS accounts for between 6% and 16% of all running injuries.2,10 It is associated with a spectrum of tibial stress injuries, including periostitis, tendinopathy, and stress reaction, with dysfunction of the tibialis posterior, tibialis anterior, and soleus muscles thought to be contributing factors.2,11 Intrinsic factors include high body mass index (BMI), female sex, excessive internal and external hip rotation, hyperpronation, and hyper plantar flexion.2,10,12
X-rays of the leg are typically normal in patients with MTSS and should be considered only if the clinical presentation suggests the possibility of an alternative diagnosis, such as a stress fracture or tumor.2-4,13 Advanced imaging such as MRI or triple phase bone scans (TPBS) are useful when the diagnosis is in question and will reveal an abnormally high signal along the posterior medial tibial surface or the classic train-track appearance of nucleotide uptake in patients with MTSS.2 MRI readily shows periosteal reaction and bony edema and has a sensitivity of 78% to 89% and a specificity of 33% to 100% for the diagnosis of MTSS.14,15
Initial management of MTSS is conservative, with the mainstay of treatment consisting of rest, ice, and nonsteroidal anti-inflammatory drugs (NSAIDs).3,13,16 While ice, NSAIDs, proper conditioning, physical therapy to stretch and strengthen the calf musculature, rigid orthotics to correct foot hyperpronation, and activity modification are all appropriate treatments, randomized controlled trials have shown none of these interventions to be more effective than rest alone.2 Non-operative treatment is usually successful, but surgery may be required for severe or refractory cases. Procedures include posteromedial fasciotomy, release of the medial soleus fascial bridge, deep compartment fasciotomy, or removal of a section of the distal tibia periosteum.3,4
Lower extremity stress fracture. Stress fractures are caused by repetitive loading that results in microtrauma, including bony microfractures. The vast majority of cases—80% to 95% of stress fractures—affect the lower extremities, and most involve the tibia.2-4,6,13,17 The most common presentation is an insidious onset of pain over a specific bony area with a normal appearance, although localized swelling or erythema may occasionally be present.3,14,17,18 The pain may be reproduced or worsened by weight-bearing activities and relieved by rest.14,18
Consider the female athlete triad. In evaluating a patient with a stress fracture, pay close attention to dietary history, BMI, and, in female athletes, take a detailed menstrual history. Such patients are at risk for amenorrhea, low bone mineral density, and nutritional deficits—the “female athlete triad,” which carries an increased risk of stress fractures.3,14,17-19
Stress fractures can often be diagnosed with a thorough medical history and physical, with imaging used for confirmation.6,14,17,18 Historical features of a stress fracture that may differentiate it from MTSS include pain that is unilateral and absent at rest and occurs with more prolonged activity, as well as post-exercise and/or nocturnal pain. Notable physical exam features include pain that is reproduced in a focal area with a single leg hop or percussion with a tuning fork or ultrasound.5,11,17
Initially, sensitivity for a plain radiograph is as low as 10%.2,11 Abnormalities on x-ray are usually seen after 2 to 8 weeks of symptoms2,7,11 and may include a faint periosteal reaction, a fluffy area of callus, or a cortical lucency sometimes referred to as the “dreaded black line.”3,6,17 If a radiographic exam shows evidence of a stress fracture, further imaging is typically unnecessary. MRI or TPBS is suggested, however, when x-rays appear normal but suspicion of a stress fracture remains.3,17,18 MRI may show edema within 3 days of symptom onset and is more sensitive and specific than computed tomography (CT) or TPBS for diagnosing stress fractures of the tibia.2,16
Treatment of tibial stress fractures is typically non-operative and consists of alterations in activity (eg, non weight-bearing), correction of nutritional deficits, such as inadequate caloric intake or too little calcium or iron, and addressing problems with footwear, training regimen, and/or running surface.3,14,18 Fibular and posteromedial tibial stress fractures are considered low risk and heal with weight-bearing restrictions and rest, initially for a minimum of 2 to 4 weeks.3
Posteromedial tibia injuries tend to heal well because they are on the compression side of the bone. Anterior tibia stress fractures, which are located on the tension side of the bone2,7 and account for approximately 5% of all tibia stress fractures, are more prone to non-union or progression to a complete fracture.7,20 Thus, anterior tibia stress injuries warrant a more aggressive approach, with treatment options including non-weight bearing status that may last longer than 8 weeks, pneumatic brace casting, and/or orthopedic referral to evaluate for surgical intervention.7,20-22 Time for radiographic evidence of healing may exceed 8 months, so early surgical intervention should be considered, especially for high-level athletes.7,20,21
Test your skills: A diagnostic challenge
Janine T, a 24-year-old long-distance runner, presents with left lower leg pain that occurs with activity. There was no injury, Ms. T reports; the pain began about 6 weeks ago, shortly after she began training for a marathon and running more than 30 miles per week. The pain is not relieved with intermittent rest or over-the-counter analgesics, she says. But it usually abates within 15 to 30 minutes after she completes her run.
Ms. T is underweight, with a body mass index <17 kg/m2. She denies any dietary restrictions and has normal menstrual cycles. The patient reports taking oral contraceptives, but no other medications. An initial x-ray is normal, as is magnetic resonance imaging to evaluate for a stress fracture.
You suspect Ms. T has shin splints, advise her to rest for a few weeks and to consider getting orthotics for her running shoes, and schedule a follow-up visit.
When she comes in 6 weeks later, the patient reports that she resumed running after a 3-week rest; shortly after, she noticed pain in both legs. What’s more, she now experiences tingling in her feet after running a few miles.
What’s wrong with this patient?
Ms. T’s symptoms—bilateral persistent leg pain, with tingling in both feet, and little improvement with rest—strongly suggest that she has chronic exertional compartment syndrome. Intracompartmental pressure testing, which reveals pre-exercise values ≥15 mm Hg and post-exercise values of ≥30 mm Hg at one minute, confirms the diagnosis.
Activity avoidance or modification will allow Ms. T’s symptoms to subside, but they’re highly likely to recur when she resumes running. The definitive treatment is intracompartmental fasciotomy, which has a success rate of approximately 80%.1,28
When to suspect chronic exertional compartment syndrome
Leg pain in CECS results from increased pressure within the lower extremity fascial compartments temporally related to exercise.2,23,24 Its incidence in the general population is unknown, but CECS has been found to range from 14% to 27% in patients with previously undiagnosed leg pain1,14,25 and to affect about a third of athletes with chronic ELP. In addition, CECS has been found in 90% of patients who have both diabetes and ELP with normal findings on vascular studies.1,3,4,26
The anterior compartment is most commonly affected, followed by the lateral, deep posterior, and superficial posterior compartments.3,13,23,27 Symptoms are bilateral 60% to 95% of the time.2,13,14,25 Factors contributing to CECS include fixed muscular compartment constraints, muscle swelling, thickened fascia, muscle hypertrophy related to resistance training, dynamic muscular contraction patterns, and low muscle capillary supply. Stretching of fascial pain receptors and pressure fibers and inadequate myocyte response to increased metabolism may play a role, as well.14,28
The initial clinical presentation is usually predictable leg pain—ie, pain that begins at about the same time, distance, or intensity of a workout and resolves with rest; numbness and weakness may occur as the workout progresses. In time, leg pain associated with CECS may be present with everyday activity or at rest. The physical exam may be normal or reveal swelling, tenderness over the involved compartments, pain with passive digit or ankle motion, and palpable muscle herniation.14
Measurement of intracompartmental pressure before and after exercise is the gold standard for diagnosis of CECS.2,14,27 Pre-exercise values ≥15 mm Hg and post-exercise values ≥30 mm Hg at one minute or ≥20 mm Hg at 5 minutes are all considered diagnostic of CECS,11 although these widely accepted criteria for bilateral testing of all compartments yields a false-positive rate of 5%.27 CECS is almost always bilateral,29 and some clinicians advocate limiting the number of needle insertions by taking only post-exercise measurements and testing only symptomatic compartments in one limb.
Imaging has limited value, as both x-rays and MRIs are usually normal.14 However, post-exertional T2-weighted MRI findings of muscular edema correspond to increased intracompartmental pressures, with a sensitivity of 87% and a specificity of 62%.14,24,30,31 Infrared spectroscopy, which measures levels of oxygenated and deoxygenated blood, is sensitive for CECS when the post-exercise ratio of deoxygenated to oxygenated blood remains elevated.14,24 Neither of these screening modalities is routinely obtained or considered diagnostic, however. Their chief role is to exclude an alternative diagnosis.14
Treatment and symptom relief. Discontinuing or modifying the aggravating activity typically brings relief of CECS. But this is not a long-term solution, as symptoms are likely to recur when the patient returns to the activity in question.1 The definitive treatment is compartment release via fasciotomy. Success rates for anterior and lateral compartment releases are >80%.1,28 The success of fasciotomy of posterior compartments, however, is <50%—a finding attributed to more complex anatomy, difficult visualization, and the presence of additional compartments.1,32
When the cause is vascular
Arterial endofibrosis—the fibrotic thickening of the intima of an artery—is thought to be caused by repetitive hip flexion.8 This results in hyperplasia, wall thickening, and eventual stenosis of the vessel, with 90% of cases affecting the external iliac artery.8,33 The condition is most common in activities such as cycling, but is also seen in such activities as running, skiing, soccer, and rugby. Symptoms are typically unilateral, but an estimated 15% of patients experience bilateral symptoms.8,33
Loss of power in the affected leg, with intermittent claudication and pain due to presumed ischemia from the vascular defect, is the usual presentation, although some patients develop cramping of the buttocks and/or paresthesia of the affected leg and foot during uphill running or cycling.8,33 The physical exam is often normal, but there may be a post-exercise arterial bruit over the femoral artery when the hip is flexed.8,34
Pre-exercise ankle-brachial index (ABI) <0.5 and post-exercise ABI <0.66 at one minute is suggestive of moderate arterial endofibrosis, with 90% sensitivity and 87% specificity.8,33,34 Arterial ultrasound and color Doppler may also be used for diagnosis, but are often operator dependent. Magnetic resonance angiography (MRA), while more expensive, can detect excessive kinking or compression of the vessel and is not operator dependent.8,33 Angioplastic balloon catheter dilation and stenting, bypass surgery, vascular reconstruction and endarterectomy with vein patch are the options for treatment. The success rates of the various interventions are unknown due to a lack of head-to-head studies and long-term follow-up.8,33
Popliteal artery entrapment syndrome (PAES) is a constellation of symptoms caused by vascular impingement in the popliteal fossa of the knee.8,34 The typical presenting symptoms are lower limb ischemia and pain caused by intense exercise that resolves quickly afterwards. Symptoms correlate more with the intensity than the duration of exercise.3,8
PAES is usually caused by a variant of the gastrocnemius muscle in which a medial head passes behind the popliteal artery in males younger than 30 years.8,33-35 Less commonly, it is the result of an overuse or acute orthopedic injury that irritates structures surrounding the popliteal fossa.8,34 PAES affects football, basketball, and soccer players, as well as runners because of excessive dorsiflexion and plantar flexion of the ankle.3,4
The physical exam for a patient with PAES is typically normal, but a post-exercise popliteal bruit with weak peripheral pulses may be elicited.8,33 An ABI in the neutral, forced dorsiflexion and forced plantar flexion positions can serve as a useful screening tool. An ABI <0.9 is abnormal, with a sensitivity and specificity of 90% and 98%, respectively, for stenosis >50%.2,36
Arteriography is the gold standard for diagnosis of PAES. Contrast arteriography is most commonly used because of its availability and cost. But MRA better differentiates functional from anatomic entrapment—a differentiation that less invasive tests, such as duplex ultrasound studies, lack the specificity to reveal.8,34 Treatment requires either surgical removal of the offending musculotendinous structures or arterial bypass and grafting of the chronically impinged area, as conservative therapies lack efficacy.2,8,34
Cystic adventitial disease (CAD) is the narrowing of an artery by mucoid cysts in the arterial wall or adventitia.8,9 It is a rare condition, accounting for just 0.1% of all vascular diseases, most commonly occurring in men in their mid-40s.8,33 CAD is thought to be the result of mucin-producing cells being haphazardly incorporated into the adventitia during arterial development. About 85% of patients whose popliteal artery is affected in this way will experience intermittent claudication with activity.8,9
On exam, such patients often have diminished ankle-brachial pressure indices, and duplex ultrasound often reveals stenosis in the affected artery, as well as a collection of mucoid cysts in the adventitia.8,9
Diagnosis can be confirmed by MRA.8,37 Evidence for the treatment of CAD is largely anecdotal.9 Cysts may be aspirated but tend to recur, and stenting does not correct the cystic-induced narrowing of the vessel. Surgical removal of the cysts is the only successful treatment.8,9
Neurologic causes to consider
Spinal stenosis is caused by central canal narrowing secondary to congenital abnormalities, trauma, or, most commonly, degenerative changes in the lumbar spine. Spinal stenosis is generally seen in men or women ages 50 to 70 years.38 Patients experience unilateral or bilateral claudication that improves with sitting or flexion of the spine5 and may develop bilateral lower extremity numbness and tingling from the buttocks that radiates down the legs. Diagnosis is typically made with a combination of a lumbar x-ray and an MRI, which will show nerve compression and bony overgrowth.38 CT myelogram, another imaging option, isless sensitive in the acute phase, but can be used to monitor the disease course.
Initial treatment includes physical therapy and NSAIDs.5 If conservative therapy fails, epidural or nerve root corticosteroid injections and surgical decompression or laminectomy are options.38
Nerve entrapment is a less common source of lower extremity pain in which the superficial peroneal nerve is most often affected.4,12,17,39 Trauma is the usual cause of nerve entrapment, but it may also be associated with overuse, most notably related to dance, soccer, or tennis.2,14,40,41 The most likely anatomic site is where the nerve exits the deep fascia within the lateral compartment in the lower third of the leg.39,40 Less frequently, the common peroneal nerve at the fibular neck, the saphenous nerve as it passes through Hunter’s canal, the posterior tibial nerve at the tarsal tunnel, and the sural nerve in the posterior calf may be affected.3,4,12,17,20,40,41 Entrapment of the peroneal nerve may be associated with activities involving repetitive inversion and eversion, such as running and cycling. Injury of the saphenous nerve is seen in sports involving repetitive knee flexion like rowing and cycling. Sural nerve entrapment is a result of crural fascia compression of the nerve during activities like running and track.3,14,40,42,43
Patients typically experience burning, tingling, and radiation of pain with activity. Symptoms worsen with continued exercise. The physical exam is often normal, especially early in the disease process, but may reveal sensory loss, motor weakness, and a loss of reflexes.2,40 Patients with superficial peroneal nerve involvement may have distal lateral leg pain that radiates into the dorsum of the foot, often exacerbated by lower leg percussion and resulting in diminished sensation.1 Common peroneal nerve involvement may alter sensation of the lateral leg, as well, but may also cause foot drop.2 The saphenous nerve can cause medial knee or leg symptoms, while the sural nerve can yield pain in the lateral ankle and foot.2
To diagnose nerve entrapment, electromyography and nerve conduction velocities at the level of the lesion may yield positive results 3 to 4 weeks after symptom onset.2,13,40 There are wide ranges of sensitivity and specificity for these studies, but they are nonetheless considered the tests of choice for nerve entrapment.1,44 Conservative treatment with activity modification, physical therapy, massage, and NSAIDs is often sufficient,2 with surgical management warranted only for refractory cases.2,14,40,41
CORRESPONDENCE
Jonathan A. Becker, MD, CAQSM, University of Louisville Department of Family and Geriatric Medicine, 201 Abraham Flexner Way, Suite 690, Louisville, KY 40202; jon.becker@louisville@edu.
PRACTICE RECOMMENDATIONS
› Consider the possibility of vascular and neurologic problems as the source of exertional leg pain (ELP). C
› Order magnetic resonance imaging to evaluate patients with ELP and negative x-rays for stress fractures. C
› Measure lower extremity intracompartmental pressures both before and after exercise when you suspect chronic exertional compartmental syndrome. Doing so is the gold standard for the diagnosis of this condition. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Most family physicians are accustomed to treating active patients with shin splints and stress fractures. But many are less familiar with, and slower to recognize, other sources of exertional leg pain (ELP), defined as exercise-related pain that localizes in the lower extremity distal to the knee and proximal to the talocrural joint.1
ELP has a broad differential diagnosis that includes other musculoskeletal conditions—most notably chronic exertional compartment syndrome (CECS), which has been found to affect 33% of athletes with chronic ELP1—as well as a number of vascular and neurologic causes.2-4 In addition, etiologies may overlap. Greater awareness of the many causes of ELP can help you to avoid the unnecessary use of expensive diagnostic tests as well as delayed diagnosis and treatment.
A thorough medical and activity history, symptom review, and physical examination are your most important tools when patients present with ELP. When the cause is not obvious or the patient fails to respond to conservative measures, x-rays, magnetic resonance imaging (MRI), vascular studies, electromyography and nerve conduction studies, and/or intracompartmental pressure testing may be needed to find the source of the symptoms. In the text that follows, we review both common and relatively uncommon sources of ELP, using a stepwise diagnostic approach. You’ll find a diagnostic challenge, in which you can test your skills and a more comprehensive differential diagnosis in the TABLE.1-3,5-9
Musculoskeletal injuries: Shin splints and beyond
Medial tibial stress syndrome (MTSS), commonly known as shin splints, is characterized by pain and tenderness over the posteromedial aspect of the distal tibia.3 It typically results in diffuse pain that occurs with exercise, but may persist at rest in severe cases.3-6 Less often, localized swelling may also be present.2
MTSS accounts for between 6% and 16% of all running injuries.2,10 It is associated with a spectrum of tibial stress injuries, including periostitis, tendinopathy, and stress reaction, with dysfunction of the tibialis posterior, tibialis anterior, and soleus muscles thought to be contributing factors.2,11 Intrinsic factors include high body mass index (BMI), female sex, excessive internal and external hip rotation, hyperpronation, and hyper plantar flexion.2,10,12
X-rays of the leg are typically normal in patients with MTSS and should be considered only if the clinical presentation suggests the possibility of an alternative diagnosis, such as a stress fracture or tumor.2-4,13 Advanced imaging such as MRI or triple phase bone scans (TPBS) are useful when the diagnosis is in question and will reveal an abnormally high signal along the posterior medial tibial surface or the classic train-track appearance of nucleotide uptake in patients with MTSS.2 MRI readily shows periosteal reaction and bony edema and has a sensitivity of 78% to 89% and a specificity of 33% to 100% for the diagnosis of MTSS.14,15
Initial management of MTSS is conservative, with the mainstay of treatment consisting of rest, ice, and nonsteroidal anti-inflammatory drugs (NSAIDs).3,13,16 While ice, NSAIDs, proper conditioning, physical therapy to stretch and strengthen the calf musculature, rigid orthotics to correct foot hyperpronation, and activity modification are all appropriate treatments, randomized controlled trials have shown none of these interventions to be more effective than rest alone.2 Non-operative treatment is usually successful, but surgery may be required for severe or refractory cases. Procedures include posteromedial fasciotomy, release of the medial soleus fascial bridge, deep compartment fasciotomy, or removal of a section of the distal tibia periosteum.3,4
Lower extremity stress fracture. Stress fractures are caused by repetitive loading that results in microtrauma, including bony microfractures. The vast majority of cases—80% to 95% of stress fractures—affect the lower extremities, and most involve the tibia.2-4,6,13,17 The most common presentation is an insidious onset of pain over a specific bony area with a normal appearance, although localized swelling or erythema may occasionally be present.3,14,17,18 The pain may be reproduced or worsened by weight-bearing activities and relieved by rest.14,18
Consider the female athlete triad. In evaluating a patient with a stress fracture, pay close attention to dietary history, BMI, and, in female athletes, take a detailed menstrual history. Such patients are at risk for amenorrhea, low bone mineral density, and nutritional deficits—the “female athlete triad,” which carries an increased risk of stress fractures.3,14,17-19
Stress fractures can often be diagnosed with a thorough medical history and physical, with imaging used for confirmation.6,14,17,18 Historical features of a stress fracture that may differentiate it from MTSS include pain that is unilateral and absent at rest and occurs with more prolonged activity, as well as post-exercise and/or nocturnal pain. Notable physical exam features include pain that is reproduced in a focal area with a single leg hop or percussion with a tuning fork or ultrasound.5,11,17
Initially, sensitivity for a plain radiograph is as low as 10%.2,11 Abnormalities on x-ray are usually seen after 2 to 8 weeks of symptoms2,7,11 and may include a faint periosteal reaction, a fluffy area of callus, or a cortical lucency sometimes referred to as the “dreaded black line.”3,6,17 If a radiographic exam shows evidence of a stress fracture, further imaging is typically unnecessary. MRI or TPBS is suggested, however, when x-rays appear normal but suspicion of a stress fracture remains.3,17,18 MRI may show edema within 3 days of symptom onset and is more sensitive and specific than computed tomography (CT) or TPBS for diagnosing stress fractures of the tibia.2,16
Treatment of tibial stress fractures is typically non-operative and consists of alterations in activity (eg, non weight-bearing), correction of nutritional deficits, such as inadequate caloric intake or too little calcium or iron, and addressing problems with footwear, training regimen, and/or running surface.3,14,18 Fibular and posteromedial tibial stress fractures are considered low risk and heal with weight-bearing restrictions and rest, initially for a minimum of 2 to 4 weeks.3
Posteromedial tibia injuries tend to heal well because they are on the compression side of the bone. Anterior tibia stress fractures, which are located on the tension side of the bone2,7 and account for approximately 5% of all tibia stress fractures, are more prone to non-union or progression to a complete fracture.7,20 Thus, anterior tibia stress injuries warrant a more aggressive approach, with treatment options including non-weight bearing status that may last longer than 8 weeks, pneumatic brace casting, and/or orthopedic referral to evaluate for surgical intervention.7,20-22 Time for radiographic evidence of healing may exceed 8 months, so early surgical intervention should be considered, especially for high-level athletes.7,20,21
Test your skills: A diagnostic challenge
Janine T, a 24-year-old long-distance runner, presents with left lower leg pain that occurs with activity. There was no injury, Ms. T reports; the pain began about 6 weeks ago, shortly after she began training for a marathon and running more than 30 miles per week. The pain is not relieved with intermittent rest or over-the-counter analgesics, she says. But it usually abates within 15 to 30 minutes after she completes her run.
Ms. T is underweight, with a body mass index <17 kg/m2. She denies any dietary restrictions and has normal menstrual cycles. The patient reports taking oral contraceptives, but no other medications. An initial x-ray is normal, as is magnetic resonance imaging to evaluate for a stress fracture.
You suspect Ms. T has shin splints, advise her to rest for a few weeks and to consider getting orthotics for her running shoes, and schedule a follow-up visit.
When she comes in 6 weeks later, the patient reports that she resumed running after a 3-week rest; shortly after, she noticed pain in both legs. What’s more, she now experiences tingling in her feet after running a few miles.
What’s wrong with this patient?
Ms. T’s symptoms—bilateral persistent leg pain, with tingling in both feet, and little improvement with rest—strongly suggest that she has chronic exertional compartment syndrome. Intracompartmental pressure testing, which reveals pre-exercise values ≥15 mm Hg and post-exercise values of ≥30 mm Hg at one minute, confirms the diagnosis.
Activity avoidance or modification will allow Ms. T’s symptoms to subside, but they’re highly likely to recur when she resumes running. The definitive treatment is intracompartmental fasciotomy, which has a success rate of approximately 80%.1,28
When to suspect chronic exertional compartment syndrome
Leg pain in CECS results from increased pressure within the lower extremity fascial compartments temporally related to exercise.2,23,24 Its incidence in the general population is unknown, but CECS has been found to range from 14% to 27% in patients with previously undiagnosed leg pain1,14,25 and to affect about a third of athletes with chronic ELP. In addition, CECS has been found in 90% of patients who have both diabetes and ELP with normal findings on vascular studies.1,3,4,26
The anterior compartment is most commonly affected, followed by the lateral, deep posterior, and superficial posterior compartments.3,13,23,27 Symptoms are bilateral 60% to 95% of the time.2,13,14,25 Factors contributing to CECS include fixed muscular compartment constraints, muscle swelling, thickened fascia, muscle hypertrophy related to resistance training, dynamic muscular contraction patterns, and low muscle capillary supply. Stretching of fascial pain receptors and pressure fibers and inadequate myocyte response to increased metabolism may play a role, as well.14,28
The initial clinical presentation is usually predictable leg pain—ie, pain that begins at about the same time, distance, or intensity of a workout and resolves with rest; numbness and weakness may occur as the workout progresses. In time, leg pain associated with CECS may be present with everyday activity or at rest. The physical exam may be normal or reveal swelling, tenderness over the involved compartments, pain with passive digit or ankle motion, and palpable muscle herniation.14
Measurement of intracompartmental pressure before and after exercise is the gold standard for diagnosis of CECS.2,14,27 Pre-exercise values ≥15 mm Hg and post-exercise values ≥30 mm Hg at one minute or ≥20 mm Hg at 5 minutes are all considered diagnostic of CECS,11 although these widely accepted criteria for bilateral testing of all compartments yields a false-positive rate of 5%.27 CECS is almost always bilateral,29 and some clinicians advocate limiting the number of needle insertions by taking only post-exercise measurements and testing only symptomatic compartments in one limb.
Imaging has limited value, as both x-rays and MRIs are usually normal.14 However, post-exertional T2-weighted MRI findings of muscular edema correspond to increased intracompartmental pressures, with a sensitivity of 87% and a specificity of 62%.14,24,30,31 Infrared spectroscopy, which measures levels of oxygenated and deoxygenated blood, is sensitive for CECS when the post-exercise ratio of deoxygenated to oxygenated blood remains elevated.14,24 Neither of these screening modalities is routinely obtained or considered diagnostic, however. Their chief role is to exclude an alternative diagnosis.14
Treatment and symptom relief. Discontinuing or modifying the aggravating activity typically brings relief of CECS. But this is not a long-term solution, as symptoms are likely to recur when the patient returns to the activity in question.1 The definitive treatment is compartment release via fasciotomy. Success rates for anterior and lateral compartment releases are >80%.1,28 The success of fasciotomy of posterior compartments, however, is <50%—a finding attributed to more complex anatomy, difficult visualization, and the presence of additional compartments.1,32
When the cause is vascular
Arterial endofibrosis—the fibrotic thickening of the intima of an artery—is thought to be caused by repetitive hip flexion.8 This results in hyperplasia, wall thickening, and eventual stenosis of the vessel, with 90% of cases affecting the external iliac artery.8,33 The condition is most common in activities such as cycling, but is also seen in such activities as running, skiing, soccer, and rugby. Symptoms are typically unilateral, but an estimated 15% of patients experience bilateral symptoms.8,33
Loss of power in the affected leg, with intermittent claudication and pain due to presumed ischemia from the vascular defect, is the usual presentation, although some patients develop cramping of the buttocks and/or paresthesia of the affected leg and foot during uphill running or cycling.8,33 The physical exam is often normal, but there may be a post-exercise arterial bruit over the femoral artery when the hip is flexed.8,34
Pre-exercise ankle-brachial index (ABI) <0.5 and post-exercise ABI <0.66 at one minute is suggestive of moderate arterial endofibrosis, with 90% sensitivity and 87% specificity.8,33,34 Arterial ultrasound and color Doppler may also be used for diagnosis, but are often operator dependent. Magnetic resonance angiography (MRA), while more expensive, can detect excessive kinking or compression of the vessel and is not operator dependent.8,33 Angioplastic balloon catheter dilation and stenting, bypass surgery, vascular reconstruction and endarterectomy with vein patch are the options for treatment. The success rates of the various interventions are unknown due to a lack of head-to-head studies and long-term follow-up.8,33
Popliteal artery entrapment syndrome (PAES) is a constellation of symptoms caused by vascular impingement in the popliteal fossa of the knee.8,34 The typical presenting symptoms are lower limb ischemia and pain caused by intense exercise that resolves quickly afterwards. Symptoms correlate more with the intensity than the duration of exercise.3,8
PAES is usually caused by a variant of the gastrocnemius muscle in which a medial head passes behind the popliteal artery in males younger than 30 years.8,33-35 Less commonly, it is the result of an overuse or acute orthopedic injury that irritates structures surrounding the popliteal fossa.8,34 PAES affects football, basketball, and soccer players, as well as runners because of excessive dorsiflexion and plantar flexion of the ankle.3,4
The physical exam for a patient with PAES is typically normal, but a post-exercise popliteal bruit with weak peripheral pulses may be elicited.8,33 An ABI in the neutral, forced dorsiflexion and forced plantar flexion positions can serve as a useful screening tool. An ABI <0.9 is abnormal, with a sensitivity and specificity of 90% and 98%, respectively, for stenosis >50%.2,36
Arteriography is the gold standard for diagnosis of PAES. Contrast arteriography is most commonly used because of its availability and cost. But MRA better differentiates functional from anatomic entrapment—a differentiation that less invasive tests, such as duplex ultrasound studies, lack the specificity to reveal.8,34 Treatment requires either surgical removal of the offending musculotendinous structures or arterial bypass and grafting of the chronically impinged area, as conservative therapies lack efficacy.2,8,34
Cystic adventitial disease (CAD) is the narrowing of an artery by mucoid cysts in the arterial wall or adventitia.8,9 It is a rare condition, accounting for just 0.1% of all vascular diseases, most commonly occurring in men in their mid-40s.8,33 CAD is thought to be the result of mucin-producing cells being haphazardly incorporated into the adventitia during arterial development. About 85% of patients whose popliteal artery is affected in this way will experience intermittent claudication with activity.8,9
On exam, such patients often have diminished ankle-brachial pressure indices, and duplex ultrasound often reveals stenosis in the affected artery, as well as a collection of mucoid cysts in the adventitia.8,9
Diagnosis can be confirmed by MRA.8,37 Evidence for the treatment of CAD is largely anecdotal.9 Cysts may be aspirated but tend to recur, and stenting does not correct the cystic-induced narrowing of the vessel. Surgical removal of the cysts is the only successful treatment.8,9
Neurologic causes to consider
Spinal stenosis is caused by central canal narrowing secondary to congenital abnormalities, trauma, or, most commonly, degenerative changes in the lumbar spine. Spinal stenosis is generally seen in men or women ages 50 to 70 years.38 Patients experience unilateral or bilateral claudication that improves with sitting or flexion of the spine5 and may develop bilateral lower extremity numbness and tingling from the buttocks that radiates down the legs. Diagnosis is typically made with a combination of a lumbar x-ray and an MRI, which will show nerve compression and bony overgrowth.38 CT myelogram, another imaging option, isless sensitive in the acute phase, but can be used to monitor the disease course.
Initial treatment includes physical therapy and NSAIDs.5 If conservative therapy fails, epidural or nerve root corticosteroid injections and surgical decompression or laminectomy are options.38
Nerve entrapment is a less common source of lower extremity pain in which the superficial peroneal nerve is most often affected.4,12,17,39 Trauma is the usual cause of nerve entrapment, but it may also be associated with overuse, most notably related to dance, soccer, or tennis.2,14,40,41 The most likely anatomic site is where the nerve exits the deep fascia within the lateral compartment in the lower third of the leg.39,40 Less frequently, the common peroneal nerve at the fibular neck, the saphenous nerve as it passes through Hunter’s canal, the posterior tibial nerve at the tarsal tunnel, and the sural nerve in the posterior calf may be affected.3,4,12,17,20,40,41 Entrapment of the peroneal nerve may be associated with activities involving repetitive inversion and eversion, such as running and cycling. Injury of the saphenous nerve is seen in sports involving repetitive knee flexion like rowing and cycling. Sural nerve entrapment is a result of crural fascia compression of the nerve during activities like running and track.3,14,40,42,43
Patients typically experience burning, tingling, and radiation of pain with activity. Symptoms worsen with continued exercise. The physical exam is often normal, especially early in the disease process, but may reveal sensory loss, motor weakness, and a loss of reflexes.2,40 Patients with superficial peroneal nerve involvement may have distal lateral leg pain that radiates into the dorsum of the foot, often exacerbated by lower leg percussion and resulting in diminished sensation.1 Common peroneal nerve involvement may alter sensation of the lateral leg, as well, but may also cause foot drop.2 The saphenous nerve can cause medial knee or leg symptoms, while the sural nerve can yield pain in the lateral ankle and foot.2
To diagnose nerve entrapment, electromyography and nerve conduction velocities at the level of the lesion may yield positive results 3 to 4 weeks after symptom onset.2,13,40 There are wide ranges of sensitivity and specificity for these studies, but they are nonetheless considered the tests of choice for nerve entrapment.1,44 Conservative treatment with activity modification, physical therapy, massage, and NSAIDs is often sufficient,2 with surgical management warranted only for refractory cases.2,14,40,41
CORRESPONDENCE
Jonathan A. Becker, MD, CAQSM, University of Louisville Department of Family and Geriatric Medicine, 201 Abraham Flexner Way, Suite 690, Louisville, KY 40202; jon.becker@louisville@edu.
1. Rajasekaran S, Kvinlaug K, Finnoff JT. Exertional leg pain in the athlete. PM R. 2012;4:985-1000.
2. Brewer RB, Gregory AJ. Chronic lower leg pain in athletes: a guide for the differential diagnosis, evaluation, and treatment. Sports Health. 2012;4:121-127.
3. Edwards PH Jr, Wright ML, Hartman JF. A practical approach for the differential diagnosis of chronic leg pain in the athlete. Am J Sports Med. 2005;33:1241-1249.
4. Clanton TO, Solcher BW. Chronic leg pain in the athlete. Clin Sports Med. 1994;13:743-759.
5. Fredericson M, Wun C. Differential diagnosis of leg pain in the athlete. J Am Podiatr Med Assoc. 2003;93:321-324.
6. Pell RF 4th, Khanuja HS, Cooley GR. Leg pain in the running athlete. J Am Acad Orthop Surg. 2004;12:396-404.
7. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8:344-353.
8. Pham TT, Kapur R, Harwood MI. Exertional leg pain: teasing out arterial entrapments. Curr Sports Med Rep. 2007;6:371-375.
9. Wright LB, Matchett WJ, Cruz CP, et al. Popliteal artery disease: diagnosis and treatment. Radiographics. 2004;24:467-479.
10. Yates B, White S. The incidence and risk factors in the development of medial tibial stress syndrome among naval recruits. Am J Sports Med. 2004;32:772-780.
11. Fredericson M, Jennings F, Beaulieu C, et al. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17:309-325.
12. Plisky MS, Rauh MJ, Heiderscheit B, et al. Medial tibial stress syndrome in high school cross-country runners: incidence and risk factors. J Orthop Sports Phys Ther. 2007;37:40-47.
13. Touliopolous S, Hershman EB. Lower leg pain. Diagnosis and treatment of compartment syndromes and other pain syndromes of the leg. Sports Med. 1999;27:193-204.
14. Burrus MT, Werner BC, Starman JS, et al. Chronic leg pain in athletes. Am J Sports Med. 2015;43:1538-1547.
15. Batt ME, Ugalde V, Anderson MW, et al. A prospective controlled study of diagnostic imaging for acute shin splints. Med Sci Sports Exerc. 1998;30:1564-1571.
16. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235:553-561.
17. Sterling JC, Edelstein DW, Calvo RD, et al. Stress fractures in the athlete. Diagnosis and management. Sports Med. 1992;14:336-346.
18. Brukner P. Exercise-related lower leg pain: bone. Med Sci Sports Exerc. 2000;32:S15-S26.
19. Tuan K, Wu S, Sennett B. Stress fractures in athletes: risk factors, diagnosis, and management. Orthopedics. 2004;27:583-591.
20. Harrast MA, Colonno D. Stress fractures in runners. Clin Sports Med. 2010;29:399-416.
21. Varner KE, Younas SA, Lintner DM, et al. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33:1071-1076.
22. Kaeding CC, Yu JR, Wright R, et al. Management and return to play of stress fractures. Clin J Sport Med. 2005;15:442-447.
23. Blackman PG. A review of chronic exertional compartment syndrome in the lower leg. Med Sci Sports Exerc. 2000;32:S4-S10.
24. Brennan FH Jr, Kane SF. Diagnosis, treatment options, and rehabilitation of chronic lower leg exertional compartment syndrome. Curr Sports Med Rep. 2003;2:247-250.
25. Davis DE, Raikin S, Garras DN, et al. Characteristics of patients with chronic exertional compartment syndrome. Foot Ankle Int. 2013;34:1349-1354.
26. Edmundsson D, Toolanen G. Chronic exertional compartment syndrome in diabetes mellitus. Diabet Med. 2011;28:81-85.
27. Pedowitz RA, Hargens AR, Mubarak SJ, et al. Modified criteria for the objective diagnosis of chronic compartment syndrome of the leg. Am J Sports Med. 1990;18:35-40.
28. Bong MR, Polatsch DB, Jazrawi LM, et al. Chronic exertional compartment syndrome: diagnosis and management. Bull Hosp Jt Dis. 2005;62:77-84.
29. Hislop M, Batt ME. Chronic exertional compartment syndrome testing: a minimalist approach. Br J Sports Med. 2011;45:954-955.
30. Brown RR, Rosenberg ZS. MR imaging of exercise-induced lower leg pain. Magn Reson Imaging Clin N Am. 2001;9:533-552.
31. Ringler MD, Litwiller DV, Felmlee JP, et al. MRI accurately detects chronic exertional compartment syndrome: a validation study. Skeletal Radiol. 2013;42:385-392.
32. Schepsis AA, Martini D, Corbett M. Surgical management of exertional compartment syndrome of the lower leg. Long-term followup. Am J Sports Med. 1993;21:811-817.
33. Ehsan O, Darwish A, Edmundson C, et al. Non-traumatic lower limb vascular complications in endurance athletes. Review of literature. Eur J Vasc Endovasc Surg. 2004;28:1-8.
34. Turnipseed WD. Popliteal entrapment syndrome. J Vasc Surg. 2002;35:910-915.
35. Baltopoulos P, Filippou DK, Sigala F. Popliteal artery entrapment syndrome: anatomic or functional syndrome? Clin J Sport Med. 2004;14:8-12.
36. McDermott MM, Criqui MH, Liu K, et al. Lower ankle/brachial index, as calculated by averaging the dorsalis pedis and posterior tibial arterial pressures, and association with leg functioning in peripheral arterial disease. J Vasc Surg. 2000;32:1164-1171.
37. Elias DA, White LM, Rubenstein JD, et al. Clinical evaluation and MR imaging features of popliteal artery entrapment and cystic adventitial disease. AJR Am J Roentgenol. 2003;180:627-632.
38. Genevay S, Atlas SJ. Lumbar spinal stenosis. Best Pract Res Clin Rheumatol. 2010;24:253-265.
39. Korkola M, Amendola A. Exercise-induced leg pain: sifting through a broad differential. Phys Sportsmed. 2001;29:35-50.
40. McCrory P, Bell S, Bradshaw C. Nerve entrapments of the lower leg, ankle and foot in sport. Sports Med. 2002;32:371-391.
41. Schon LC. Nerve entrapment, neuropathy, and nerve dysfunction in athletes. Orthop Clin North Am. 1994;25:47-59.
42. Anselmi SJ. Common peroneal nerve compression. J Am Podiatr Med Assoc. 2006;96:413-417.
43. Maalla R, Youssef M, Ben Lassoued N, et al. Peroneal nerve entrapment at the fibular head: outcomes of neurolysis. Orthop Traumatol Surg Res. 2013;99:719-722.
44. Marciniak C, Armon C, Wilson J, et al. Practice parameter: utility of electrodiagnostic techniques in evaluating patients with suspected peroneal neuropathy: an evidence-based review. Muscle Nerve. 2005;31:520-527.
1. Rajasekaran S, Kvinlaug K, Finnoff JT. Exertional leg pain in the athlete. PM R. 2012;4:985-1000.
2. Brewer RB, Gregory AJ. Chronic lower leg pain in athletes: a guide for the differential diagnosis, evaluation, and treatment. Sports Health. 2012;4:121-127.
3. Edwards PH Jr, Wright ML, Hartman JF. A practical approach for the differential diagnosis of chronic leg pain in the athlete. Am J Sports Med. 2005;33:1241-1249.
4. Clanton TO, Solcher BW. Chronic leg pain in the athlete. Clin Sports Med. 1994;13:743-759.
5. Fredericson M, Wun C. Differential diagnosis of leg pain in the athlete. J Am Podiatr Med Assoc. 2003;93:321-324.
6. Pell RF 4th, Khanuja HS, Cooley GR. Leg pain in the running athlete. J Am Acad Orthop Surg. 2004;12:396-404.
7. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8:344-353.
8. Pham TT, Kapur R, Harwood MI. Exertional leg pain: teasing out arterial entrapments. Curr Sports Med Rep. 2007;6:371-375.
9. Wright LB, Matchett WJ, Cruz CP, et al. Popliteal artery disease: diagnosis and treatment. Radiographics. 2004;24:467-479.
10. Yates B, White S. The incidence and risk factors in the development of medial tibial stress syndrome among naval recruits. Am J Sports Med. 2004;32:772-780.
11. Fredericson M, Jennings F, Beaulieu C, et al. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17:309-325.
12. Plisky MS, Rauh MJ, Heiderscheit B, et al. Medial tibial stress syndrome in high school cross-country runners: incidence and risk factors. J Orthop Sports Phys Ther. 2007;37:40-47.
13. Touliopolous S, Hershman EB. Lower leg pain. Diagnosis and treatment of compartment syndromes and other pain syndromes of the leg. Sports Med. 1999;27:193-204.
14. Burrus MT, Werner BC, Starman JS, et al. Chronic leg pain in athletes. Am J Sports Med. 2015;43:1538-1547.
15. Batt ME, Ugalde V, Anderson MW, et al. A prospective controlled study of diagnostic imaging for acute shin splints. Med Sci Sports Exerc. 1998;30:1564-1571.
16. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235:553-561.
17. Sterling JC, Edelstein DW, Calvo RD, et al. Stress fractures in the athlete. Diagnosis and management. Sports Med. 1992;14:336-346.
18. Brukner P. Exercise-related lower leg pain: bone. Med Sci Sports Exerc. 2000;32:S15-S26.
19. Tuan K, Wu S, Sennett B. Stress fractures in athletes: risk factors, diagnosis, and management. Orthopedics. 2004;27:583-591.
20. Harrast MA, Colonno D. Stress fractures in runners. Clin Sports Med. 2010;29:399-416.
21. Varner KE, Younas SA, Lintner DM, et al. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33:1071-1076.
22. Kaeding CC, Yu JR, Wright R, et al. Management and return to play of stress fractures. Clin J Sport Med. 2005;15:442-447.
23. Blackman PG. A review of chronic exertional compartment syndrome in the lower leg. Med Sci Sports Exerc. 2000;32:S4-S10.
24. Brennan FH Jr, Kane SF. Diagnosis, treatment options, and rehabilitation of chronic lower leg exertional compartment syndrome. Curr Sports Med Rep. 2003;2:247-250.
25. Davis DE, Raikin S, Garras DN, et al. Characteristics of patients with chronic exertional compartment syndrome. Foot Ankle Int. 2013;34:1349-1354.
26. Edmundsson D, Toolanen G. Chronic exertional compartment syndrome in diabetes mellitus. Diabet Med. 2011;28:81-85.
27. Pedowitz RA, Hargens AR, Mubarak SJ, et al. Modified criteria for the objective diagnosis of chronic compartment syndrome of the leg. Am J Sports Med. 1990;18:35-40.
28. Bong MR, Polatsch DB, Jazrawi LM, et al. Chronic exertional compartment syndrome: diagnosis and management. Bull Hosp Jt Dis. 2005;62:77-84.
29. Hislop M, Batt ME. Chronic exertional compartment syndrome testing: a minimalist approach. Br J Sports Med. 2011;45:954-955.
30. Brown RR, Rosenberg ZS. MR imaging of exercise-induced lower leg pain. Magn Reson Imaging Clin N Am. 2001;9:533-552.
31. Ringler MD, Litwiller DV, Felmlee JP, et al. MRI accurately detects chronic exertional compartment syndrome: a validation study. Skeletal Radiol. 2013;42:385-392.
32. Schepsis AA, Martini D, Corbett M. Surgical management of exertional compartment syndrome of the lower leg. Long-term followup. Am J Sports Med. 1993;21:811-817.
33. Ehsan O, Darwish A, Edmundson C, et al. Non-traumatic lower limb vascular complications in endurance athletes. Review of literature. Eur J Vasc Endovasc Surg. 2004;28:1-8.
34. Turnipseed WD. Popliteal entrapment syndrome. J Vasc Surg. 2002;35:910-915.
35. Baltopoulos P, Filippou DK, Sigala F. Popliteal artery entrapment syndrome: anatomic or functional syndrome? Clin J Sport Med. 2004;14:8-12.
36. McDermott MM, Criqui MH, Liu K, et al. Lower ankle/brachial index, as calculated by averaging the dorsalis pedis and posterior tibial arterial pressures, and association with leg functioning in peripheral arterial disease. J Vasc Surg. 2000;32:1164-1171.
37. Elias DA, White LM, Rubenstein JD, et al. Clinical evaluation and MR imaging features of popliteal artery entrapment and cystic adventitial disease. AJR Am J Roentgenol. 2003;180:627-632.
38. Genevay S, Atlas SJ. Lumbar spinal stenosis. Best Pract Res Clin Rheumatol. 2010;24:253-265.
39. Korkola M, Amendola A. Exercise-induced leg pain: sifting through a broad differential. Phys Sportsmed. 2001;29:35-50.
40. McCrory P, Bell S, Bradshaw C. Nerve entrapments of the lower leg, ankle and foot in sport. Sports Med. 2002;32:371-391.
41. Schon LC. Nerve entrapment, neuropathy, and nerve dysfunction in athletes. Orthop Clin North Am. 1994;25:47-59.
42. Anselmi SJ. Common peroneal nerve compression. J Am Podiatr Med Assoc. 2006;96:413-417.
43. Maalla R, Youssef M, Ben Lassoued N, et al. Peroneal nerve entrapment at the fibular head: outcomes of neurolysis. Orthop Traumatol Surg Res. 2013;99:719-722.
44. Marciniak C, Armon C, Wilson J, et al. Practice parameter: utility of electrodiagnostic techniques in evaluating patients with suspected peroneal neuropathy: an evidence-based review. Muscle Nerve. 2005;31:520-527.

Preoperative evaluation: A time-saving algorithm
PRACTICE RECOMMENDATIONS
› Recommend that patients quit smoking 8 weeks before surgery; keep in mind, though, that quitting closer to the date of surgery does not increase the risk of complications. A
› Use the online American College of Surgeons/National Surgical Quality Improvement Program surgical risk calculator to estimate a patient’s surgical risk. C
› Send a patient directly to surgery if he or she has an estimated cardiac risk <1% or <2 risk factors of the Revised Cardiac Risk Index. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 27 million Americans undergo surgery every year1 and before doing so, they turn to you—their primary care physician—or their cardiologist for a preoperative evaluation. Of course, the goal of this evaluation is to determine an individual patient’s risk and compare it to procedural averages in an effort to identify opportunities for risk mitigation. But the preoperative evaluation is also an opportunity to make recommendations regarding perioperative management of medications. And certainly we want to conduct these evaluations in a way that is both expeditious and in keeping with the latest guidelines.
Current guidelines for preoperative evaluations are less complicated than they used to be and focus on cardiac and pulmonary risk stratification. While a risk calculator remains your primary tool, elements such as smoking cessation and identifying sleep apnea are important parts of the preop equation. In the review that follows, we present a simple algorithm (FIGURE 12-6) that we developed that can be completed in a single visit.

Cardiac assessment: A risk calculator is the primary tool
Cardiac risk estimation is perhaps the most important element in determining a patient’s overall surgical risk. But before you begin, you'll need to determine whether the preoperative evaluation of the patient is best handled by you or a specialist. Current guidelines recommend preoperative evaluation by a specialist when a patient has certain conditions, such as moderate or greater valvular stenosis/regurgitation, a cardiac implantable electronic device, pulmonary hypertension, congenital heart disease, or severe systemic disease.2
If these conditions are not present (and an immediate referral is not required), you can turn your attention to the cardiac assessment. The first step is to determine which cardiac risk calculator you’d like to use. In its comprehensive guideline on perioperative cardiovascular evaluation, the American College of Cardiology/American Heart Association (ACC/AHA) recommends the use of one of the 2 calculators described below.2
Revised Cardiac Risk Index. The most well-known cardiac risk calculator is the 6-element Revised Cardiac Risk Index (RCRI) (TABLE 1).7 Published in 1999, the RCRI was derived from a cohort of 2800 patients, verified in 1400 patients, and has been validated in numerous studies.2 Each element increases the odds of a cardiac complication by a factor of 2 to 3, and more than one positive response indicates the patient is at high risk for complications.7
The ACS NSQIP risk calculator has been criticized because it has not been validated in a group separate from the initial patient population used in its development.2 Another criticism is the inclusion of the American Society of Anesthesiologists' (ASA) classification of the overall health of the patient, a simple yet subjective and unreliable method of patient characterization.2
Choosing a calculator. The ACS NSQIP calculator may be more useful for primary care physicians because it provides individualized risks for numerous complications and is easy to use. The output page can be printed as documentation of the preoperative evaluation, and is useful for counseling patients about reconsideration of surgery or risk-reduction strategies. The RCRI is also simple to use, but considers only cardiac risk. Although the RCRI has been validated in numerous studies, the ACS NSQIP was derived from a more substantial 1.4 million patients.
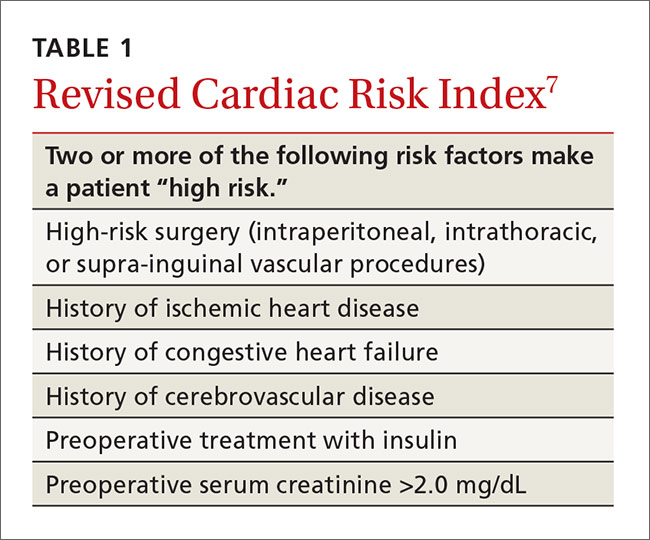
Mapping out next steps based on risk score
The next step in the preoperative evaluation process is to calculate your patient’s risk score and determine whether it is low or high. If the risk is determined to be low—either an RCRI score <2 or an ACS NSQIP cardiac complication risk <1%—the patient can be referred to surgery without further evaluation.2
If the calculator suggests higher risk, the patient’s functional status should be assessed. If the patient has a functional status of >4 metabolic equivalents (METs), then the patient can be recommended for surgery without further evaluation.2 Examples of activities that are greater than 4 METs are yard work such as raking leaves, weeding, or pushing a power mower; sexual relations; climbing a flight of stairs; walking up a hill; and participating in moderate recreational activities like golf, bowling, dancing, doubles tennis, or throwing a baseball or football.9
Patient can’t perform >4 METs? If the patient does not have a functional capacity of >4 METs, further risk stratification should be considered if the results would change management.2 Prior guidelines recommended either perioperative beta-blockers to mitigate risk or coronary interventions, but both are controversial due to lack of proven benefit.
Perioperative beta-blocker use. A recommendation to consider starting beta-blockers at least one day prior to surgery remains in the 2014 ACC/AHA guidelines for patients with 3 or more RCRI risk factors.2 But a group of studies supporting beta-blocker use has been discredited due to serious flaws and fabricated data. At the same time, a large study arguing against perioperative beta-blockers has been criticized for starting high doses of beta-blockers on the day of surgery.2,10,11 In the end, mortality benefit from perioperative beta-blockers is uncertain, and the suggested reduction in cardiac events is partially offset by an increased risk of stroke.2
Stress testing is of questionable value. A patient with high cardiac risk (as evaluated with a calculator) may need to forego the surgical procedure or undergo a modified procedure. Alternatively, he or she may need to be referred to a cardiologist for consultation and possible pharmacologic nuclear stress testing. Although a normal stress test has a high negative predictive value, an abnormal test often leads to percutaneous coronary intervention or bypass surgery, and neither has been shown to reduce cardiac surgical risk.2 Percutaneous coronary interventions require a period of dual antiplatelet therapy, delaying surgery for unproven benefit.2
EKGs and echocardiograms are of limited use. An anesthesia group or surgical center will often require an electrocardiogram (EKG) as part of a preoperative evaluation, but preoperative evaluation by EKG or echocardiogram is controversial due to unproven benefits and potential risks. The 2014 ACC/AHA guidelines recommend against a 12-lead EKG for patients with low cardiac risk using the RCRI or ACS NSQIP or those who are having a low-risk procedure, such as endoscopy or cataract surgery.2 The United States Preventive Health Services Task Force also recommends against screening low-risk patients and says that screening EKGs and stress testing in asymptomatic medium- to high-risk patients is of undetermined value.12 They noted no evidence of benefit from resting or exercise EKG, with harm from a 1.7% complication rate of angiography, which is performed after up to 2.9% of exercise EKG testing.12
There are no recommendations for preoperative echocardiogram in the asymptomatic patient. Only unexplained dyspnea or other clinical signs of heart failure require an echocardiogram. For patients with known heart failure that is clinically stable, the ACC/AHA guidelines suggest that an echocardiogram should be performed within the year prior to surgery, although this is based on expert opinion.2
Because of the controversy over both coronary interventions and perioperative beta-blocker therapy, consider cardiology referral for a patient with poor functional activity level who does not meet low-risk criteria. While stress testing is acceptable, it may not lead to improved patient outcomes.
Optimize preventive care
Begin by ensuring that blood pressure (BP) and cholesterol are managed according to ACC/AHA guidelines. Then consider whether to start preoperative medications. You'll also want to screen for sleep apnea and discuss smoking status and cessation, if appropriate.
Initiate medications preoperatively?
In addition to having value as long-term primary prevention, there is some evidence that statins help prevent cardiac events during surgery. A randomized trial of over 200 vascular surgery patients showed that starting statins an average of 30 days prior to surgery significantly reduced cardiac complications.13 Another systematic review demonstrated that preoperative statins significantly reduce acute kidney injury from surgery.14
Unlike statins, aspirin started prior to surgery does not confer benefit. Aspirin was shown to significantly increase bleeding risk without improving cardiac outcomes in a large trial.15
Screen for sleep apnea
Sleep apnea increases the rate of respiratory failure between 2 and 3 times and the rate of cardiac complications about 1.5 times.16 This is a potentially correctable risk factor. The European Society of Anaesthesiology recommends clinical screening for obstructive sleep apnea using the STOP-Bang screening tool5 (TABLE 24). For its part, the ASA combines screening questions from the STOP-Bang screening questionnaire with a review of the medical record and physical exam, because the STOP-Bang questionnaire alone has an insufficient negative predictive value, ranging from 30% and 82%.6 Obstructive sleep apnea is not addressed on published pulmonary and cardiac risk tools.2,3,7,8
Is the patient a smoker?
Smoking is a reversible risk factor for pulmonary, cardiac, and infectious complications, as well as overall mortality. Perioperative smoking cessation counseling has been complicated by concerns that stopping smoking within 8 weeks of surgery might worsen postoperative outcomes. A study that looked at intraoperative sputum retrieved via tracheal suction showed that patients who had stopped smoking for 2 months or more prior to surgery had the same amount of sputum as non-smokers, while those that quit smoking <2 months before surgery were more likely to have increased intraoperative sputum volume.18 This study did not demonstrate a difference in postoperative pulmonary complications, likely because it included patients receiving minor surgeries only. But based on this study, a cessation period of 2 months was often recommended.
A recent systematic review showed that smoking cessation shortly before surgery does not increase risk.19 In fact, although the review did not show a statistically significant reduction in postoperative complications among recent quitters as compared with continued smokers, there was a trend toward a reduction in overall complications with only a slight increase in pulmonary complications.19
Address potential pulmonary complications
In addition to screening for issues that could lead to cardiac complications, it’s important to address the potential for pulmonary complications. Postoperative pulmonary complications are at least as common as cardiac complications, and include all possible respiratory related outcomes of surgery, from pneumonia to respiratory failure.
The seminal study on postoperative pulmonary complications is a systematic review published in 2006, which showed that the most important risk factors were surgery type, advanced age, ASA classification of overall health ≥II, and congestive heart failure.3 Chronic lung diseases and cigarette use were less predictive of pulmonary issues. All of these factors are included in the ACS NSQIP risk calculator.
Perioperative medication management
One aspect of the preoperative evaluation that should not be overlooked is a thorough medication reconciliation. Primary care providers can support the operative team by recommending medication adjustments prior to surgery. Several classes of medications have specific perioperative recommendations, which are summarized here.
Hypertension medications
- Beta-blockers. A patient who regularly takes a beta-blocker should continue the medication on the day of surgery and restart after surgery.2,20
- Calcium channel blockers. Calcium channel blockers can be continued through the day of surgery.2,20
- Renin-angiotensin system antagonists. Given the increased risk of hypotension following anesthesia induction, have patients refrain from taking angiotensin-converting enzyme inhibitors and angiotensin receptor blocker medications for at least 10 hours prior to surgery.20
- Diuretics. Diuretics can be given on the day of surgery, although they increase the risk of hypovolemia and electrolyte disturbances.20
Diabetes medications
- Insulin. For patients with type I diabetes, recommend basal insulin of 0.2 to 0.3 units/kg/day of long-acting insulin.21 If the patient is using an insulin pump, basal rate should be continued. For patients with type 2 diabetes, the simplest method is to use one-half the normal long-acting insulin dose on the morning of surgery.22
- Metformin. Discontinue metformin 24 hours prior to surgery because of the risk for lactic acidosis.21,22 While the risk of lactic acidosis from metformin is low, mortality rates as high as 50% have been documented after lactic acidosis occurred with similar medications.22
- Sulfonylureas. Sulfonylureas should be held on the day of surgery due to the risk of hypoglycemia and a possible increased risk of ischemia.21,22
- Thiazolidinediones, dipeptidyl peptidase-4 inhibitors, and glucagon-like peptide-1 agonists. All should be held on the day of surgery.21
Anticoagulant medications
- Vitamin K antagonists (warfarin). Discontinue warfarin 5 days prior to the procedure. The half-life is approximately 40 hours, requiring at least 5 days for the anticoagulant effect to be eliminated from the body.23 Use of bridging therapy with regular- or low-molecular weight heparin remains controversial due to increased surgical bleeding risk without evidence of a decrease in cardiovascular events.24 The patient’s risks of stroke and venous thromboembolism should be taken into account when deciding whether to use bridging therapy or not.
- Factor Xa inhibitor. Management of factor Xa inhibitors (rivaroxaban, apixaban) depends on the bleeding risk of the surgery and the patient’s renal function.24,25 For instance, a patient undergoing cataract surgery (low risk) needs a shorter cessation time than a patient undergoing hip arthroplasty (high risk). Discontinuation times are listed in TABLE 3.24
- Direct thrombin inhibitor. Management of direct thrombin inhibitors (dabigatran) is also dependent on surgical bleeding risk and renal function (TABLE 3).23,24
- Aspirin, clopidogrel, ticlopidine, prasugrel. All should be stopped 7 to 10 days prior to surgery to allow new platelet growth. Low-dose aspirin for secondary prevention of cardiovascular disease or primary prevention in a high-risk patient can be continued through surgery.23
Other
- Corticosteroids. Recent evidence suggests that stress-dose steroids are not needed to prevent adrenal insufficiency in patients taking corticosteroids chronically.26 These patients should continue maintenance therapy at regular dosing.26,27 Stress dosing of corticosteroids is only required when a patient has signs of adrenal insufficiency.26
- Statins. Statin medications should be continued on the day of surgery.2
- Nonsteroidal anti-inflammatory drugs. NSAIDs should be stopped 5 days prior to surgery to reverse antiplatelet effects.23
CORRESPONDENCE
CDR Michael J. Arnold, Naval Hospital, 2080 Child Street, Jacksonville, FL 32214; [email protected].
ACKNOWLEDGEMENT
The authors thank CDR Kristian Sanchack and LCDR Dustin Smith for their assistance with this manuscript.
1. Wier LM, Steiner CA, Owens PL. Surgeries in hospital-owned outpatient facilities, 2012. HCUP Statistical Brief #188. February 2015. Available at: https://www.hcup-us.ahrq.gov/reports/statbriefs/sb188-Surgeries-Hospital-Outpatient-Facilities-2012.jsp. Accessed September 15, 2016.
2. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. J Am Coll Cardiol. 2014;64:e77-e137.
3. Smetana GW, Lawrence VA, Cornell JE. Preoperative pulmonary risk stratification for noncardiothoracic surgery: systematic review for the American College of Physicians. Ann Intern Med. 2006;144:581-595.
4. Chung F, Abdullah HR, Liao P. STOP-Bang questionnaire: a prac tical approach to screen for obstructive sleep apnea. Chest. 2016;149:631-638.
5. De Hert S, Imberger G, Carlisle J, et al. Preoperative evaluation of the adult patient undergoing non-cardiac surgery: guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol. 2011;28:684-722.
6. American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Practice guidelines for the perioperative management of patients with obstructive sleep apnea; an updated report by the American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Anesthesiology. 2014;120:268-286.
7. Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation. 1999;100:1043-1049.
8. Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aide and informed consent tool for patients and surgeons. J Am Coll Surg. 2013;217:833-842.
9. Hlatky MA, Boineau RE, Higginbotham MB, et al. A brief self-administered questionnaire to determine functional capacity (the Duke Activity Status Index). Am J Cardiol. 1989;64:651-654.
10. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomized controlled trials of B-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
11. Mounsey A, Roque JM, Egan M. Why you shouldn’t start beta-blockers before surgery. J Fam Pract. 2014;63:E15-E16.
12. Chou R, Arora B, Dana T, et al. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155:375-385.
13. Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg. 2004;39:967-975.
14. Pan SY, Wu VC, Huang TM, et al. Effect of preoperative statin therapy on postoperative acute kidney injury in patients undergoing major surgery: systemic review and meta-analysis. Nephrology. 2014;19:750-763.
15. Devereux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med. 2014;370:1494-1503.
16. Adesanya AO, Lee W, Greilich NB, et al. Perioperative management of obstructive sleep apnea. Chest. 2010;138:1489-1498.
17. Chung F, Nagappa M, Singh M, et al. CPAP in the perioperative setting: evidence of support. Chest. 2016;149:586-597.
18. Yamashita S, Yamaguchi H, Sakaguchi M, et al. Effect of smoking on intraoperative sputum and postoperative pulmonary complication in minor surgical patients. Respir Med. 2004;98:760-766.
19. Myers K, Hajek P, Hinds
20. Lonjaret L, Lairez O, Minville V, et al. Optimal perioperative management of arterial blood pressure. Integr Blood Press Control. 2014;7:49-59.
21. Sudhakaran S, Surani SR. Guidelines for perioperative management of the diabetic patient. Surg Res Pract. 2015;2015:284063.
22. Duncan AE. Hyperglycemia and perioperative glucose management. Curr Pharm Des. 2012;18:6195-6203.
23. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e326S-e350S.
24. Faraoni D, Levy JH, Albaladejo P, et al. Updates in the perioperative and emergency management of non-vitamin K antagonist oral anticoagulants. Crit Care. 2015;19:203.
25. Shamoun F, Obeid H, Ramakrishna H. Novel anticoagulants in atrial fibrillation: monitoring, reversal and perioperative management. Biomed Res Int. 2015;2015:424031.
26. Kelly KN, Domajnko B. Perioperative stress-dose steroids. Clin Colon Rectal Surg. 2013;26:163-167.
27. Scanzello CR, Nestor BJ. Perioperative management of medications used in the treatment of rheumatoid arthritis. HSSJ. 2006;2:141-147.
PRACTICE RECOMMENDATIONS
› Recommend that patients quit smoking 8 weeks before surgery; keep in mind, though, that quitting closer to the date of surgery does not increase the risk of complications. A
› Use the online American College of Surgeons/National Surgical Quality Improvement Program surgical risk calculator to estimate a patient’s surgical risk. C
› Send a patient directly to surgery if he or she has an estimated cardiac risk <1% or <2 risk factors of the Revised Cardiac Risk Index. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 27 million Americans undergo surgery every year1 and before doing so, they turn to you—their primary care physician—or their cardiologist for a preoperative evaluation. Of course, the goal of this evaluation is to determine an individual patient’s risk and compare it to procedural averages in an effort to identify opportunities for risk mitigation. But the preoperative evaluation is also an opportunity to make recommendations regarding perioperative management of medications. And certainly we want to conduct these evaluations in a way that is both expeditious and in keeping with the latest guidelines.
Current guidelines for preoperative evaluations are less complicated than they used to be and focus on cardiac and pulmonary risk stratification. While a risk calculator remains your primary tool, elements such as smoking cessation and identifying sleep apnea are important parts of the preop equation. In the review that follows, we present a simple algorithm (FIGURE 12-6) that we developed that can be completed in a single visit.
Cardiac assessment: A risk calculator is the primary tool
Cardiac risk estimation is perhaps the most important element in determining a patient’s overall surgical risk. But before you begin, you'll need to determine whether the preoperative evaluation of the patient is best handled by you or a specialist. Current guidelines recommend preoperative evaluation by a specialist when a patient has certain conditions, such as moderate or greater valvular stenosis/regurgitation, a cardiac implantable electronic device, pulmonary hypertension, congenital heart disease, or severe systemic disease.2
If these conditions are not present (and an immediate referral is not required), you can turn your attention to the cardiac assessment. The first step is to determine which cardiac risk calculator you’d like to use. In its comprehensive guideline on perioperative cardiovascular evaluation, the American College of Cardiology/American Heart Association (ACC/AHA) recommends the use of one of the 2 calculators described below.2
Revised Cardiac Risk Index. The most well-known cardiac risk calculator is the 6-element Revised Cardiac Risk Index (RCRI) (TABLE 1).7 Published in 1999, the RCRI was derived from a cohort of 2800 patients, verified in 1400 patients, and has been validated in numerous studies.2 Each element increases the odds of a cardiac complication by a factor of 2 to 3, and more than one positive response indicates the patient is at high risk for complications.7
The ACS NSQIP risk calculator has been criticized because it has not been validated in a group separate from the initial patient population used in its development.2 Another criticism is the inclusion of the American Society of Anesthesiologists' (ASA) classification of the overall health of the patient, a simple yet subjective and unreliable method of patient characterization.2
Choosing a calculator. The ACS NSQIP calculator may be more useful for primary care physicians because it provides individualized risks for numerous complications and is easy to use. The output page can be printed as documentation of the preoperative evaluation, and is useful for counseling patients about reconsideration of surgery or risk-reduction strategies. The RCRI is also simple to use, but considers only cardiac risk. Although the RCRI has been validated in numerous studies, the ACS NSQIP was derived from a more substantial 1.4 million patients.
Mapping out next steps based on risk score
The next step in the preoperative evaluation process is to calculate your patient’s risk score and determine whether it is low or high. If the risk is determined to be low—either an RCRI score <2 or an ACS NSQIP cardiac complication risk <1%—the patient can be referred to surgery without further evaluation.2
If the calculator suggests higher risk, the patient’s functional status should be assessed. If the patient has a functional status of >4 metabolic equivalents (METs), then the patient can be recommended for surgery without further evaluation.2 Examples of activities that are greater than 4 METs are yard work such as raking leaves, weeding, or pushing a power mower; sexual relations; climbing a flight of stairs; walking up a hill; and participating in moderate recreational activities like golf, bowling, dancing, doubles tennis, or throwing a baseball or football.9
Patient can’t perform >4 METs? If the patient does not have a functional capacity of >4 METs, further risk stratification should be considered if the results would change management.2 Prior guidelines recommended either perioperative beta-blockers to mitigate risk or coronary interventions, but both are controversial due to lack of proven benefit.
Perioperative beta-blocker use. A recommendation to consider starting beta-blockers at least one day prior to surgery remains in the 2014 ACC/AHA guidelines for patients with 3 or more RCRI risk factors.2 But a group of studies supporting beta-blocker use has been discredited due to serious flaws and fabricated data. At the same time, a large study arguing against perioperative beta-blockers has been criticized for starting high doses of beta-blockers on the day of surgery.2,10,11 In the end, mortality benefit from perioperative beta-blockers is uncertain, and the suggested reduction in cardiac events is partially offset by an increased risk of stroke.2
Stress testing is of questionable value. A patient with high cardiac risk (as evaluated with a calculator) may need to forego the surgical procedure or undergo a modified procedure. Alternatively, he or she may need to be referred to a cardiologist for consultation and possible pharmacologic nuclear stress testing. Although a normal stress test has a high negative predictive value, an abnormal test often leads to percutaneous coronary intervention or bypass surgery, and neither has been shown to reduce cardiac surgical risk.2 Percutaneous coronary interventions require a period of dual antiplatelet therapy, delaying surgery for unproven benefit.2
EKGs and echocardiograms are of limited use. An anesthesia group or surgical center will often require an electrocardiogram (EKG) as part of a preoperative evaluation, but preoperative evaluation by EKG or echocardiogram is controversial due to unproven benefits and potential risks. The 2014 ACC/AHA guidelines recommend against a 12-lead EKG for patients with low cardiac risk using the RCRI or ACS NSQIP or those who are having a low-risk procedure, such as endoscopy or cataract surgery.2 The United States Preventive Health Services Task Force also recommends against screening low-risk patients and says that screening EKGs and stress testing in asymptomatic medium- to high-risk patients is of undetermined value.12 They noted no evidence of benefit from resting or exercise EKG, with harm from a 1.7% complication rate of angiography, which is performed after up to 2.9% of exercise EKG testing.12
There are no recommendations for preoperative echocardiogram in the asymptomatic patient. Only unexplained dyspnea or other clinical signs of heart failure require an echocardiogram. For patients with known heart failure that is clinically stable, the ACC/AHA guidelines suggest that an echocardiogram should be performed within the year prior to surgery, although this is based on expert opinion.2
Because of the controversy over both coronary interventions and perioperative beta-blocker therapy, consider cardiology referral for a patient with poor functional activity level who does not meet low-risk criteria. While stress testing is acceptable, it may not lead to improved patient outcomes.
Optimize preventive care
Begin by ensuring that blood pressure (BP) and cholesterol are managed according to ACC/AHA guidelines. Then consider whether to start preoperative medications. You'll also want to screen for sleep apnea and discuss smoking status and cessation, if appropriate.
Initiate medications preoperatively?
In addition to having value as long-term primary prevention, there is some evidence that statins help prevent cardiac events during surgery. A randomized trial of over 200 vascular surgery patients showed that starting statins an average of 30 days prior to surgery significantly reduced cardiac complications.13 Another systematic review demonstrated that preoperative statins significantly reduce acute kidney injury from surgery.14
Unlike statins, aspirin started prior to surgery does not confer benefit. Aspirin was shown to significantly increase bleeding risk without improving cardiac outcomes in a large trial.15
Screen for sleep apnea
Sleep apnea increases the rate of respiratory failure between 2 and 3 times and the rate of cardiac complications about 1.5 times.16 This is a potentially correctable risk factor. The European Society of Anaesthesiology recommends clinical screening for obstructive sleep apnea using the STOP-Bang screening tool5 (TABLE 24). For its part, the ASA combines screening questions from the STOP-Bang screening questionnaire with a review of the medical record and physical exam, because the STOP-Bang questionnaire alone has an insufficient negative predictive value, ranging from 30% and 82%.6 Obstructive sleep apnea is not addressed on published pulmonary and cardiac risk tools.2,3,7,8
Is the patient a smoker?
Smoking is a reversible risk factor for pulmonary, cardiac, and infectious complications, as well as overall mortality. Perioperative smoking cessation counseling has been complicated by concerns that stopping smoking within 8 weeks of surgery might worsen postoperative outcomes. A study that looked at intraoperative sputum retrieved via tracheal suction showed that patients who had stopped smoking for 2 months or more prior to surgery had the same amount of sputum as non-smokers, while those that quit smoking <2 months before surgery were more likely to have increased intraoperative sputum volume.18 This study did not demonstrate a difference in postoperative pulmonary complications, likely because it included patients receiving minor surgeries only. But based on this study, a cessation period of 2 months was often recommended.
A recent systematic review showed that smoking cessation shortly before surgery does not increase risk.19 In fact, although the review did not show a statistically significant reduction in postoperative complications among recent quitters as compared with continued smokers, there was a trend toward a reduction in overall complications with only a slight increase in pulmonary complications.19
Address potential pulmonary complications
In addition to screening for issues that could lead to cardiac complications, it’s important to address the potential for pulmonary complications. Postoperative pulmonary complications are at least as common as cardiac complications, and include all possible respiratory related outcomes of surgery, from pneumonia to respiratory failure.
The seminal study on postoperative pulmonary complications is a systematic review published in 2006, which showed that the most important risk factors were surgery type, advanced age, ASA classification of overall health ≥II, and congestive heart failure.3 Chronic lung diseases and cigarette use were less predictive of pulmonary issues. All of these factors are included in the ACS NSQIP risk calculator.
Perioperative medication management
One aspect of the preoperative evaluation that should not be overlooked is a thorough medication reconciliation. Primary care providers can support the operative team by recommending medication adjustments prior to surgery. Several classes of medications have specific perioperative recommendations, which are summarized here.
Hypertension medications
- Beta-blockers. A patient who regularly takes a beta-blocker should continue the medication on the day of surgery and restart after surgery.2,20
- Calcium channel blockers. Calcium channel blockers can be continued through the day of surgery.2,20
- Renin-angiotensin system antagonists. Given the increased risk of hypotension following anesthesia induction, have patients refrain from taking angiotensin-converting enzyme inhibitors and angiotensin receptor blocker medications for at least 10 hours prior to surgery.20
- Diuretics. Diuretics can be given on the day of surgery, although they increase the risk of hypovolemia and electrolyte disturbances.20
Diabetes medications
- Insulin. For patients with type I diabetes, recommend basal insulin of 0.2 to 0.3 units/kg/day of long-acting insulin.21 If the patient is using an insulin pump, basal rate should be continued. For patients with type 2 diabetes, the simplest method is to use one-half the normal long-acting insulin dose on the morning of surgery.22
- Metformin. Discontinue metformin 24 hours prior to surgery because of the risk for lactic acidosis.21,22 While the risk of lactic acidosis from metformin is low, mortality rates as high as 50% have been documented after lactic acidosis occurred with similar medications.22
- Sulfonylureas. Sulfonylureas should be held on the day of surgery due to the risk of hypoglycemia and a possible increased risk of ischemia.21,22
- Thiazolidinediones, dipeptidyl peptidase-4 inhibitors, and glucagon-like peptide-1 agonists. All should be held on the day of surgery.21
Anticoagulant medications
- Vitamin K antagonists (warfarin). Discontinue warfarin 5 days prior to the procedure. The half-life is approximately 40 hours, requiring at least 5 days for the anticoagulant effect to be eliminated from the body.23 Use of bridging therapy with regular- or low-molecular weight heparin remains controversial due to increased surgical bleeding risk without evidence of a decrease in cardiovascular events.24 The patient’s risks of stroke and venous thromboembolism should be taken into account when deciding whether to use bridging therapy or not.
- Factor Xa inhibitor. Management of factor Xa inhibitors (rivaroxaban, apixaban) depends on the bleeding risk of the surgery and the patient’s renal function.24,25 For instance, a patient undergoing cataract surgery (low risk) needs a shorter cessation time than a patient undergoing hip arthroplasty (high risk). Discontinuation times are listed in TABLE 3.24
- Direct thrombin inhibitor. Management of direct thrombin inhibitors (dabigatran) is also dependent on surgical bleeding risk and renal function (TABLE 3).23,24
- Aspirin, clopidogrel, ticlopidine, prasugrel. All should be stopped 7 to 10 days prior to surgery to allow new platelet growth. Low-dose aspirin for secondary prevention of cardiovascular disease or primary prevention in a high-risk patient can be continued through surgery.23
Other
- Corticosteroids. Recent evidence suggests that stress-dose steroids are not needed to prevent adrenal insufficiency in patients taking corticosteroids chronically.26 These patients should continue maintenance therapy at regular dosing.26,27 Stress dosing of corticosteroids is only required when a patient has signs of adrenal insufficiency.26
- Statins. Statin medications should be continued on the day of surgery.2
- Nonsteroidal anti-inflammatory drugs. NSAIDs should be stopped 5 days prior to surgery to reverse antiplatelet effects.23
CORRESPONDENCE
CDR Michael J. Arnold, Naval Hospital, 2080 Child Street, Jacksonville, FL 32214; [email protected].
ACKNOWLEDGEMENT
The authors thank CDR Kristian Sanchack and LCDR Dustin Smith for their assistance with this manuscript.
PRACTICE RECOMMENDATIONS
› Recommend that patients quit smoking 8 weeks before surgery; keep in mind, though, that quitting closer to the date of surgery does not increase the risk of complications. A
› Use the online American College of Surgeons/National Surgical Quality Improvement Program surgical risk calculator to estimate a patient’s surgical risk. C
› Send a patient directly to surgery if he or she has an estimated cardiac risk <1% or <2 risk factors of the Revised Cardiac Risk Index. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 27 million Americans undergo surgery every year1 and before doing so, they turn to you—their primary care physician—or their cardiologist for a preoperative evaluation. Of course, the goal of this evaluation is to determine an individual patient’s risk and compare it to procedural averages in an effort to identify opportunities for risk mitigation. But the preoperative evaluation is also an opportunity to make recommendations regarding perioperative management of medications. And certainly we want to conduct these evaluations in a way that is both expeditious and in keeping with the latest guidelines.
Current guidelines for preoperative evaluations are less complicated than they used to be and focus on cardiac and pulmonary risk stratification. While a risk calculator remains your primary tool, elements such as smoking cessation and identifying sleep apnea are important parts of the preop equation. In the review that follows, we present a simple algorithm (FIGURE 12-6) that we developed that can be completed in a single visit.
Cardiac assessment: A risk calculator is the primary tool
Cardiac risk estimation is perhaps the most important element in determining a patient’s overall surgical risk. But before you begin, you'll need to determine whether the preoperative evaluation of the patient is best handled by you or a specialist. Current guidelines recommend preoperative evaluation by a specialist when a patient has certain conditions, such as moderate or greater valvular stenosis/regurgitation, a cardiac implantable electronic device, pulmonary hypertension, congenital heart disease, or severe systemic disease.2
If these conditions are not present (and an immediate referral is not required), you can turn your attention to the cardiac assessment. The first step is to determine which cardiac risk calculator you’d like to use. In its comprehensive guideline on perioperative cardiovascular evaluation, the American College of Cardiology/American Heart Association (ACC/AHA) recommends the use of one of the 2 calculators described below.2
Revised Cardiac Risk Index. The most well-known cardiac risk calculator is the 6-element Revised Cardiac Risk Index (RCRI) (TABLE 1).7 Published in 1999, the RCRI was derived from a cohort of 2800 patients, verified in 1400 patients, and has been validated in numerous studies.2 Each element increases the odds of a cardiac complication by a factor of 2 to 3, and more than one positive response indicates the patient is at high risk for complications.7
The ACS NSQIP risk calculator has been criticized because it has not been validated in a group separate from the initial patient population used in its development.2 Another criticism is the inclusion of the American Society of Anesthesiologists' (ASA) classification of the overall health of the patient, a simple yet subjective and unreliable method of patient characterization.2
Choosing a calculator. The ACS NSQIP calculator may be more useful for primary care physicians because it provides individualized risks for numerous complications and is easy to use. The output page can be printed as documentation of the preoperative evaluation, and is useful for counseling patients about reconsideration of surgery or risk-reduction strategies. The RCRI is also simple to use, but considers only cardiac risk. Although the RCRI has been validated in numerous studies, the ACS NSQIP was derived from a more substantial 1.4 million patients.
Mapping out next steps based on risk score
The next step in the preoperative evaluation process is to calculate your patient’s risk score and determine whether it is low or high. If the risk is determined to be low—either an RCRI score <2 or an ACS NSQIP cardiac complication risk <1%—the patient can be referred to surgery without further evaluation.2
If the calculator suggests higher risk, the patient’s functional status should be assessed. If the patient has a functional status of >4 metabolic equivalents (METs), then the patient can be recommended for surgery without further evaluation.2 Examples of activities that are greater than 4 METs are yard work such as raking leaves, weeding, or pushing a power mower; sexual relations; climbing a flight of stairs; walking up a hill; and participating in moderate recreational activities like golf, bowling, dancing, doubles tennis, or throwing a baseball or football.9
Patient can’t perform >4 METs? If the patient does not have a functional capacity of >4 METs, further risk stratification should be considered if the results would change management.2 Prior guidelines recommended either perioperative beta-blockers to mitigate risk or coronary interventions, but both are controversial due to lack of proven benefit.
Perioperative beta-blocker use. A recommendation to consider starting beta-blockers at least one day prior to surgery remains in the 2014 ACC/AHA guidelines for patients with 3 or more RCRI risk factors.2 But a group of studies supporting beta-blocker use has been discredited due to serious flaws and fabricated data. At the same time, a large study arguing against perioperative beta-blockers has been criticized for starting high doses of beta-blockers on the day of surgery.2,10,11 In the end, mortality benefit from perioperative beta-blockers is uncertain, and the suggested reduction in cardiac events is partially offset by an increased risk of stroke.2
Stress testing is of questionable value. A patient with high cardiac risk (as evaluated with a calculator) may need to forego the surgical procedure or undergo a modified procedure. Alternatively, he or she may need to be referred to a cardiologist for consultation and possible pharmacologic nuclear stress testing. Although a normal stress test has a high negative predictive value, an abnormal test often leads to percutaneous coronary intervention or bypass surgery, and neither has been shown to reduce cardiac surgical risk.2 Percutaneous coronary interventions require a period of dual antiplatelet therapy, delaying surgery for unproven benefit.2
EKGs and echocardiograms are of limited use. An anesthesia group or surgical center will often require an electrocardiogram (EKG) as part of a preoperative evaluation, but preoperative evaluation by EKG or echocardiogram is controversial due to unproven benefits and potential risks. The 2014 ACC/AHA guidelines recommend against a 12-lead EKG for patients with low cardiac risk using the RCRI or ACS NSQIP or those who are having a low-risk procedure, such as endoscopy or cataract surgery.2 The United States Preventive Health Services Task Force also recommends against screening low-risk patients and says that screening EKGs and stress testing in asymptomatic medium- to high-risk patients is of undetermined value.12 They noted no evidence of benefit from resting or exercise EKG, with harm from a 1.7% complication rate of angiography, which is performed after up to 2.9% of exercise EKG testing.12
There are no recommendations for preoperative echocardiogram in the asymptomatic patient. Only unexplained dyspnea or other clinical signs of heart failure require an echocardiogram. For patients with known heart failure that is clinically stable, the ACC/AHA guidelines suggest that an echocardiogram should be performed within the year prior to surgery, although this is based on expert opinion.2
Because of the controversy over both coronary interventions and perioperative beta-blocker therapy, consider cardiology referral for a patient with poor functional activity level who does not meet low-risk criteria. While stress testing is acceptable, it may not lead to improved patient outcomes.
Optimize preventive care
Begin by ensuring that blood pressure (BP) and cholesterol are managed according to ACC/AHA guidelines. Then consider whether to start preoperative medications. You'll also want to screen for sleep apnea and discuss smoking status and cessation, if appropriate.
Initiate medications preoperatively?
In addition to having value as long-term primary prevention, there is some evidence that statins help prevent cardiac events during surgery. A randomized trial of over 200 vascular surgery patients showed that starting statins an average of 30 days prior to surgery significantly reduced cardiac complications.13 Another systematic review demonstrated that preoperative statins significantly reduce acute kidney injury from surgery.14
Unlike statins, aspirin started prior to surgery does not confer benefit. Aspirin was shown to significantly increase bleeding risk without improving cardiac outcomes in a large trial.15
Screen for sleep apnea
Sleep apnea increases the rate of respiratory failure between 2 and 3 times and the rate of cardiac complications about 1.5 times.16 This is a potentially correctable risk factor. The European Society of Anaesthesiology recommends clinical screening for obstructive sleep apnea using the STOP-Bang screening tool5 (TABLE 24). For its part, the ASA combines screening questions from the STOP-Bang screening questionnaire with a review of the medical record and physical exam, because the STOP-Bang questionnaire alone has an insufficient negative predictive value, ranging from 30% and 82%.6 Obstructive sleep apnea is not addressed on published pulmonary and cardiac risk tools.2,3,7,8
Is the patient a smoker?
Smoking is a reversible risk factor for pulmonary, cardiac, and infectious complications, as well as overall mortality. Perioperative smoking cessation counseling has been complicated by concerns that stopping smoking within 8 weeks of surgery might worsen postoperative outcomes. A study that looked at intraoperative sputum retrieved via tracheal suction showed that patients who had stopped smoking for 2 months or more prior to surgery had the same amount of sputum as non-smokers, while those that quit smoking <2 months before surgery were more likely to have increased intraoperative sputum volume.18 This study did not demonstrate a difference in postoperative pulmonary complications, likely because it included patients receiving minor surgeries only. But based on this study, a cessation period of 2 months was often recommended.
A recent systematic review showed that smoking cessation shortly before surgery does not increase risk.19 In fact, although the review did not show a statistically significant reduction in postoperative complications among recent quitters as compared with continued smokers, there was a trend toward a reduction in overall complications with only a slight increase in pulmonary complications.19
Address potential pulmonary complications
In addition to screening for issues that could lead to cardiac complications, it’s important to address the potential for pulmonary complications. Postoperative pulmonary complications are at least as common as cardiac complications, and include all possible respiratory related outcomes of surgery, from pneumonia to respiratory failure.
The seminal study on postoperative pulmonary complications is a systematic review published in 2006, which showed that the most important risk factors were surgery type, advanced age, ASA classification of overall health ≥II, and congestive heart failure.3 Chronic lung diseases and cigarette use were less predictive of pulmonary issues. All of these factors are included in the ACS NSQIP risk calculator.
Perioperative medication management
One aspect of the preoperative evaluation that should not be overlooked is a thorough medication reconciliation. Primary care providers can support the operative team by recommending medication adjustments prior to surgery. Several classes of medications have specific perioperative recommendations, which are summarized here.
Hypertension medications
- Beta-blockers. A patient who regularly takes a beta-blocker should continue the medication on the day of surgery and restart after surgery.2,20
- Calcium channel blockers. Calcium channel blockers can be continued through the day of surgery.2,20
- Renin-angiotensin system antagonists. Given the increased risk of hypotension following anesthesia induction, have patients refrain from taking angiotensin-converting enzyme inhibitors and angiotensin receptor blocker medications for at least 10 hours prior to surgery.20
- Diuretics. Diuretics can be given on the day of surgery, although they increase the risk of hypovolemia and electrolyte disturbances.20
Diabetes medications
- Insulin. For patients with type I diabetes, recommend basal insulin of 0.2 to 0.3 units/kg/day of long-acting insulin.21 If the patient is using an insulin pump, basal rate should be continued. For patients with type 2 diabetes, the simplest method is to use one-half the normal long-acting insulin dose on the morning of surgery.22
- Metformin. Discontinue metformin 24 hours prior to surgery because of the risk for lactic acidosis.21,22 While the risk of lactic acidosis from metformin is low, mortality rates as high as 50% have been documented after lactic acidosis occurred with similar medications.22
- Sulfonylureas. Sulfonylureas should be held on the day of surgery due to the risk of hypoglycemia and a possible increased risk of ischemia.21,22
- Thiazolidinediones, dipeptidyl peptidase-4 inhibitors, and glucagon-like peptide-1 agonists. All should be held on the day of surgery.21
Anticoagulant medications
- Vitamin K antagonists (warfarin). Discontinue warfarin 5 days prior to the procedure. The half-life is approximately 40 hours, requiring at least 5 days for the anticoagulant effect to be eliminated from the body.23 Use of bridging therapy with regular- or low-molecular weight heparin remains controversial due to increased surgical bleeding risk without evidence of a decrease in cardiovascular events.24 The patient’s risks of stroke and venous thromboembolism should be taken into account when deciding whether to use bridging therapy or not.
- Factor Xa inhibitor. Management of factor Xa inhibitors (rivaroxaban, apixaban) depends on the bleeding risk of the surgery and the patient’s renal function.24,25 For instance, a patient undergoing cataract surgery (low risk) needs a shorter cessation time than a patient undergoing hip arthroplasty (high risk). Discontinuation times are listed in TABLE 3.24
- Direct thrombin inhibitor. Management of direct thrombin inhibitors (dabigatran) is also dependent on surgical bleeding risk and renal function (TABLE 3).23,24
- Aspirin, clopidogrel, ticlopidine, prasugrel. All should be stopped 7 to 10 days prior to surgery to allow new platelet growth. Low-dose aspirin for secondary prevention of cardiovascular disease or primary prevention in a high-risk patient can be continued through surgery.23
Other
- Corticosteroids. Recent evidence suggests that stress-dose steroids are not needed to prevent adrenal insufficiency in patients taking corticosteroids chronically.26 These patients should continue maintenance therapy at regular dosing.26,27 Stress dosing of corticosteroids is only required when a patient has signs of adrenal insufficiency.26
- Statins. Statin medications should be continued on the day of surgery.2
- Nonsteroidal anti-inflammatory drugs. NSAIDs should be stopped 5 days prior to surgery to reverse antiplatelet effects.23
CORRESPONDENCE
CDR Michael J. Arnold, Naval Hospital, 2080 Child Street, Jacksonville, FL 32214; [email protected].
ACKNOWLEDGEMENT
The authors thank CDR Kristian Sanchack and LCDR Dustin Smith for their assistance with this manuscript.
1. Wier LM, Steiner CA, Owens PL. Surgeries in hospital-owned outpatient facilities, 2012. HCUP Statistical Brief #188. February 2015. Available at: https://www.hcup-us.ahrq.gov/reports/statbriefs/sb188-Surgeries-Hospital-Outpatient-Facilities-2012.jsp. Accessed September 15, 2016.
2. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. J Am Coll Cardiol. 2014;64:e77-e137.
3. Smetana GW, Lawrence VA, Cornell JE. Preoperative pulmonary risk stratification for noncardiothoracic surgery: systematic review for the American College of Physicians. Ann Intern Med. 2006;144:581-595.
4. Chung F, Abdullah HR, Liao P. STOP-Bang questionnaire: a prac tical approach to screen for obstructive sleep apnea. Chest. 2016;149:631-638.
5. De Hert S, Imberger G, Carlisle J, et al. Preoperative evaluation of the adult patient undergoing non-cardiac surgery: guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol. 2011;28:684-722.
6. American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Practice guidelines for the perioperative management of patients with obstructive sleep apnea; an updated report by the American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Anesthesiology. 2014;120:268-286.
7. Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation. 1999;100:1043-1049.
8. Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aide and informed consent tool for patients and surgeons. J Am Coll Surg. 2013;217:833-842.
9. Hlatky MA, Boineau RE, Higginbotham MB, et al. A brief self-administered questionnaire to determine functional capacity (the Duke Activity Status Index). Am J Cardiol. 1989;64:651-654.
10. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomized controlled trials of B-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
11. Mounsey A, Roque JM, Egan M. Why you shouldn’t start beta-blockers before surgery. J Fam Pract. 2014;63:E15-E16.
12. Chou R, Arora B, Dana T, et al. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155:375-385.
13. Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg. 2004;39:967-975.
14. Pan SY, Wu VC, Huang TM, et al. Effect of preoperative statin therapy on postoperative acute kidney injury in patients undergoing major surgery: systemic review and meta-analysis. Nephrology. 2014;19:750-763.
15. Devereux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med. 2014;370:1494-1503.
16. Adesanya AO, Lee W, Greilich NB, et al. Perioperative management of obstructive sleep apnea. Chest. 2010;138:1489-1498.
17. Chung F, Nagappa M, Singh M, et al. CPAP in the perioperative setting: evidence of support. Chest. 2016;149:586-597.
18. Yamashita S, Yamaguchi H, Sakaguchi M, et al. Effect of smoking on intraoperative sputum and postoperative pulmonary complication in minor surgical patients. Respir Med. 2004;98:760-766.
19. Myers K, Hajek P, Hinds
20. Lonjaret L, Lairez O, Minville V, et al. Optimal perioperative management of arterial blood pressure. Integr Blood Press Control. 2014;7:49-59.
21. Sudhakaran S, Surani SR. Guidelines for perioperative management of the diabetic patient. Surg Res Pract. 2015;2015:284063.
22. Duncan AE. Hyperglycemia and perioperative glucose management. Curr Pharm Des. 2012;18:6195-6203.
23. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e326S-e350S.
24. Faraoni D, Levy JH, Albaladejo P, et al. Updates in the perioperative and emergency management of non-vitamin K antagonist oral anticoagulants. Crit Care. 2015;19:203.
25. Shamoun F, Obeid H, Ramakrishna H. Novel anticoagulants in atrial fibrillation: monitoring, reversal and perioperative management. Biomed Res Int. 2015;2015:424031.
26. Kelly KN, Domajnko B. Perioperative stress-dose steroids. Clin Colon Rectal Surg. 2013;26:163-167.
27. Scanzello CR, Nestor BJ. Perioperative management of medications used in the treatment of rheumatoid arthritis. HSSJ. 2006;2:141-147.
1. Wier LM, Steiner CA, Owens PL. Surgeries in hospital-owned outpatient facilities, 2012. HCUP Statistical Brief #188. February 2015. Available at: https://www.hcup-us.ahrq.gov/reports/statbriefs/sb188-Surgeries-Hospital-Outpatient-Facilities-2012.jsp. Accessed September 15, 2016.
2. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. J Am Coll Cardiol. 2014;64:e77-e137.
3. Smetana GW, Lawrence VA, Cornell JE. Preoperative pulmonary risk stratification for noncardiothoracic surgery: systematic review for the American College of Physicians. Ann Intern Med. 2006;144:581-595.
4. Chung F, Abdullah HR, Liao P. STOP-Bang questionnaire: a prac tical approach to screen for obstructive sleep apnea. Chest. 2016;149:631-638.
5. De Hert S, Imberger G, Carlisle J, et al. Preoperative evaluation of the adult patient undergoing non-cardiac surgery: guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol. 2011;28:684-722.
6. American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Practice guidelines for the perioperative management of patients with obstructive sleep apnea; an updated report by the American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Obstructive Sleep Apnea. Anesthesiology. 2014;120:268-286.
7. Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation. 1999;100:1043-1049.
8. Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aide and informed consent tool for patients and surgeons. J Am Coll Surg. 2013;217:833-842.
9. Hlatky MA, Boineau RE, Higginbotham MB, et al. A brief self-administered questionnaire to determine functional capacity (the Duke Activity Status Index). Am J Cardiol. 1989;64:651-654.
10. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomized controlled trials of B-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
11. Mounsey A, Roque JM, Egan M. Why you shouldn’t start beta-blockers before surgery. J Fam Pract. 2014;63:E15-E16.
12. Chou R, Arora B, Dana T, et al. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155:375-385.
13. Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg. 2004;39:967-975.
14. Pan SY, Wu VC, Huang TM, et al. Effect of preoperative statin therapy on postoperative acute kidney injury in patients undergoing major surgery: systemic review and meta-analysis. Nephrology. 2014;19:750-763.
15. Devereux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med. 2014;370:1494-1503.
16. Adesanya AO, Lee W, Greilich NB, et al. Perioperative management of obstructive sleep apnea. Chest. 2010;138:1489-1498.
17. Chung F, Nagappa M, Singh M, et al. CPAP in the perioperative setting: evidence of support. Chest. 2016;149:586-597.
18. Yamashita S, Yamaguchi H, Sakaguchi M, et al. Effect of smoking on intraoperative sputum and postoperative pulmonary complication in minor surgical patients. Respir Med. 2004;98:760-766.
19. Myers K, Hajek P, Hinds
20. Lonjaret L, Lairez O, Minville V, et al. Optimal perioperative management of arterial blood pressure. Integr Blood Press Control. 2014;7:49-59.
21. Sudhakaran S, Surani SR. Guidelines for perioperative management of the diabetic patient. Surg Res Pract. 2015;2015:284063.
22. Duncan AE. Hyperglycemia and perioperative glucose management. Curr Pharm Des. 2012;18:6195-6203.
23. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e326S-e350S.
24. Faraoni D, Levy JH, Albaladejo P, et al. Updates in the perioperative and emergency management of non-vitamin K antagonist oral anticoagulants. Crit Care. 2015;19:203.
25. Shamoun F, Obeid H, Ramakrishna H. Novel anticoagulants in atrial fibrillation: monitoring, reversal and perioperative management. Biomed Res Int. 2015;2015:424031.
26. Kelly KN, Domajnko B. Perioperative stress-dose steroids. Clin Colon Rectal Surg. 2013;26:163-167.
27. Scanzello CR, Nestor BJ. Perioperative management of medications used in the treatment of rheumatoid arthritis. HSSJ. 2006;2:141-147.

Menstrual migraines: Which options and when?
› Consider recommending that patients with menstrual migraines try using prophylactic triptans 2 days before the onset of menses. B
› Advise against estrogen-containing contraception for women who have menstrual migraines with aura, who smoke, or are over 35, due to the increased risk of stroke (absolute contraindication). A
› Consider estrogen-containing contraception if the benefits outweigh the risks for women with migraines who are under 35 and do not have aura (relative contraindication). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mary, a 34-year-old woman, is a new patient to your practice after moving to the area for a job. She has a history of migraine headaches triggered by her menstrual periods. She has been taking combined oral contraceptives (COCs) since she was 17, with a few years off when she had 2 children. Her migraines improved when she was pregnant, but worsened postpartum with each of her daughters to a point where she had to stop breastfeeding at 4 months to go back on the pills.
On the COCs, she gets one or 2 mild-to-moderate headaches a month. She uses sumatriptan for abortive treatment with good relief. She has not missed work in the past 4 years because of her migraines. During the 6 months she was off COCs when trying to get pregnant, she routinely missed 2 to 3 workdays per month due to migraines. She knows when she is going to get a headache because she sees flashing lights in her left visual field. She has no other neurologic symptoms with the headaches, and the character of the headaches has not changed. She is a non-smoker, has normal blood pressure and lipid levels, and no other vascular risk factors.
You review her history and talk to her about the risk of stroke with migraines and with COCs. She is almost 35 years of age and you recommend stopping the COCs due to the risk. She feels strongly that she wants to continue taking the COCs, saying her quality of life is poor when she is off the pills. What should you do?
Migraine headaches are 2 to 3 times more prevalent in women than in men,1 with a lifetime risk of 43% vs 18%, respectively.2 Women account for about 80% of the $1 billion spent each year in the United States in medical expenses and lost work productivity related to migraines.1,2
Clinical patterns suggestive of menstrual migraine. About half of women affected by migraine have menstrually-related migraines (MRM); 3% to 12% have pure menstrual migraines (PMM).3 MRM and PMM are both characterized by the presence of symptoms in at least 2 to 3 consecutive cycles, with symptoms occurring from between 2 days before to 3 days after the onset of menstruation. However, in PMM, symptoms do not occur at any other time of the menstrual cycle; in MRM, symptoms can occur at other times of the cycle. PMM is more likely to respond to hormone therapy than is MRM.
Multiple studies in the United States, Europe, and Asia have noted that migraines related to menses typically last longer, are more severe, less likely to be associated with aura, and more likely to be recurrent and recalcitrant to treatment than non-menstrual migraines.1 TABLE 13 describes diagnostic criteria for migraine without aura.
Possible mechanisms of MRM and PMM. The etiology of migraine is not well understood and is likely multifactorial.4 Incidence of menstrual migraines is related to cyclic changes in female hormones—specifically, the decreasing levels of estrogen that typically happen the week before onset of menses.1 The mechanism is not yet clear, though it is thought that a decline in estrogen levels triggers a decline in serotonin levels, which may lead to cranial vasodilation and sensitization of the trigeminal nerve.5,6 Estrogen decline has also been linked to increased cranial nociception as well as decreased endogenous opioid activity. A study using positron emission tomography found increased activity of serotonergic neurons in migraineurs.7 The evidence that triptans and serotonin receptor agonists are effective in the treatment of migraine also supports the theory that serotonin neurohormonal signaling pathways play a critical role in the pathogenesis of migraines.7
Prevalence patterns point to the role of estrogen. The prevalence of migraines in women increases around puberty, peaks between ages 30 and 40, and decreases after natural menopause.6 Migraine prevalence increases during the first week postpartum, when levels of estrogen and progesterone decrease suddenly and significantly.1 Migraine frequency and intensity decrease in the second and third trimesters of pregnancy and after menopause, when estrogen levels fluctuate significantly less.1 In the Women’s Health Initiative study, women who used hormone replacement therapy (HRT) had a 42% increased risk of migraines compared with women in the study who had never used HRT.8
The association of migraine with female hormones was further supported by a Dutch study of male-to-female transgender patients on estrogen therapy, who had a 26% incidence of migraine, equivalent to the 25% prevalence in natal female controls in this study, compared with just 7.5% in male controls.9 The association between migraine and estrogen withdrawal was investigated in studies performed more than 40 years ago, when women experiencing migraines around the time of menses were given intramuscular estradiol and experienced a delay in symptom onset.10
Abortive and prophylactic treatments: Factors that guide selection
In considering probable menstrual migraine, take a detailed history, review headache diaries if available to determine association of headaches with menses, and perform a thorough neurologic examination. If a diagnosis of menstrual migraine is established, discuss the benefits of different treatment options, both abortive and prophylactic.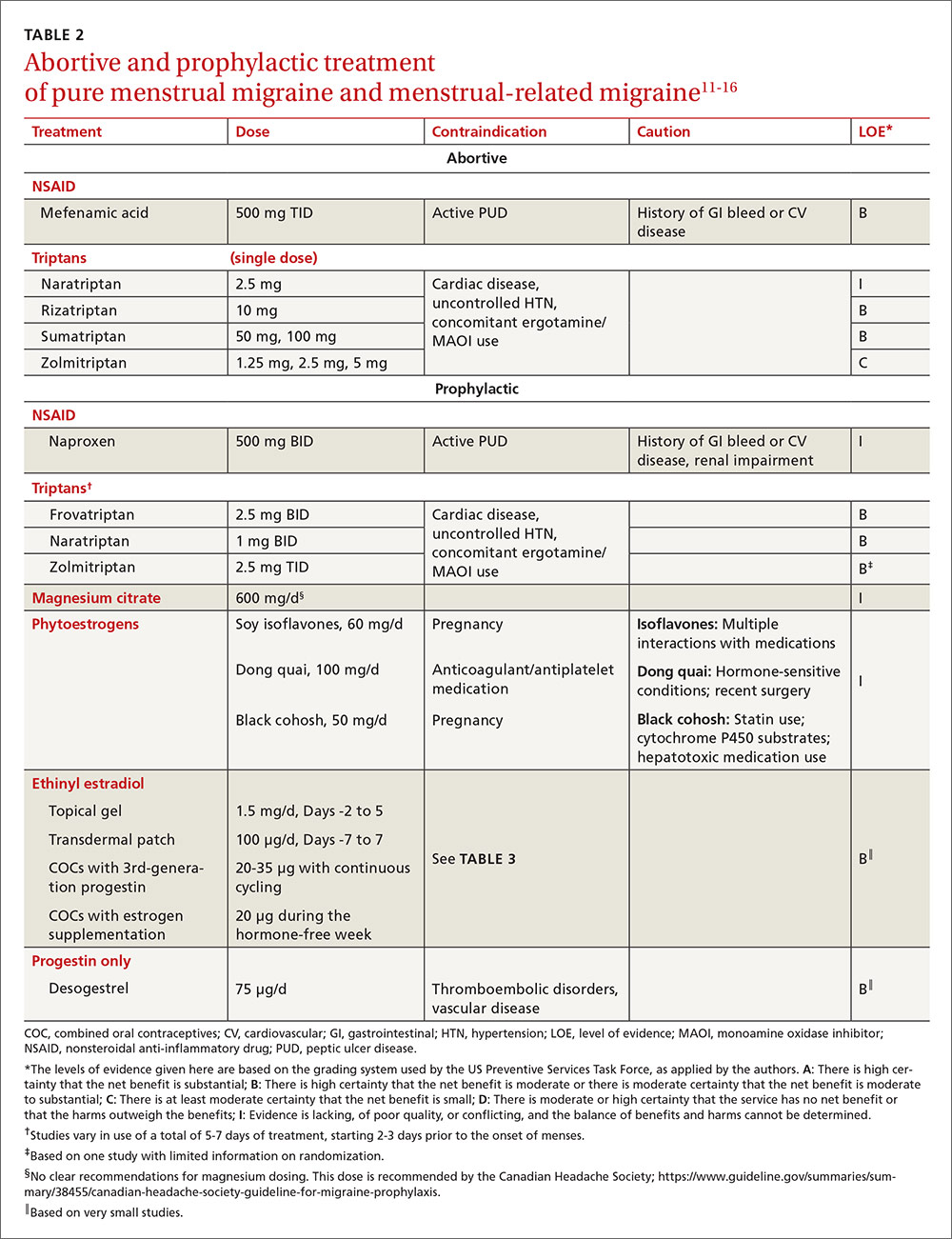
For the patient with MRM, take into account frequency of symptoms, predictability of menstruation, medication costs, and comorbidities. Both triptans and nonsteroidal anti-inflammatory drugs (NSAIDs) can be effective treatments for MRM.11 Abortive therapy may be appropriate if a patient prefers to take medication intermittently, if her menses are unpredictable, or if she does not get migraine headaches with every menses. Mefenamic acid, sumatriptan, and rizatriptan have category B recommendations for abortive treatment for menstrual migraines (TABLE 211-16). (For the patient who has regular MRM but unpredictable menses, ovulation predictor kits can be used to help predict the onset of menses, although this would involve additional cost.)
For the patient who has predictable menses and regularly occurring menstrual migraine, some data show that a short-term prophylactic regimen with triptans started 2 to 3 days before the onset of menses and continued for 5 to 7 days total can reduce the incidence of menstrual migraine (TABLE 211-16). At least one high-quality randomized controlled trial (RCT) showed a significant reduction in the incidence of MRM when women were treated prophylactically with frovatriptan, a long-acting triptan with a half-life of approximately 26 hours. Participants received frovatriptan 2.5 mg once a day or twice a day or placebo in the perimenstrual period (day -2 to +3). The incidence of MRM was 52%, 41%, and 67%, respectively (P<.0001).11,17
Another RCT of fair quality examined the effect of naratriptan (half-life 6-8 hours) on the median number of menstrual migraines over 4 menstrual cycles. Women who received 1 mg of naratriptan BID for 2 to 3 days before menses had 2 MRM episodes over the 4 cycles compared with 4 MRM episodes in women who received placebo over the same time period (P<.05).11,18 A third RCT, also of fair quality, compared 2 different regimens of zolmitriptan (half-life 3 hours) with placebo and found that women who received 2.5 mg of zolmitriptan either BID or TID 2 to 3 days prior to menses had a reduction both in frequency of menstrual migraines and in the mean number of breakthrough headaches per menstrual cycle, as well as a reduction in the need for rescue medications.12,19 Triptans are contraindicated in women with a history of cardiac disease or uncontrolled hypertension. Also, triptans can be expensive, precluding their use for some patients.
Evidence is insufficient to recommend for or against the use of NSAIDs as prophylaxis for MRM.11 NSAIDs may be contraindicated in women with a history of peptic ulcer disease or gastrointestinal bleeding. That said, if NSAIDs are not contraindicated, a trial may be reasonable given their low cost.
Data are sparse on the use of vitamins and supplements in treating and preventing PMM or MRM. In one very small double-blind, placebo-controlled study in 1991 (N=24, with efficacy data for 20), participants received a 2-week course of oral magnesium premenstrually. There was a statistically significant reduction in the number of days with headache per month (from 4.7±3.1 days to 2.4±2.2 days; P<.01) and in the total pain index (P<.03).20 A number of studies have demonstrated a correlation between hypomagnesemia and migraine headaches.5,21 The exact mechanism for this relationship is unclear.
Some recent evidence-based reviews have examined the efficacy of nutraceuticals such as magnesium, feverfew, butterbur, coenzyme Q10, and riboflavin on typical migraine, but it is not clear if these results are translatable to the treatment and prophylaxis of menstrual migraine.11,22 A multicenter, single-blind, RCT is underway to examine the efficacy of acupuncture as prophylaxis for MRM.23
Estrogen: Prescribing criteria are strict
Most studies examining the role of exogenous estrogen in reducing menstrual migraines have used topical estrogen (either in patch or gel formulations) in the perimenstrual window (TABLE 211-16). The topical estrogen route has been examined, in particular, as it is presumed to confer less risk of hypercoagulability by avoiding first-pass metabolism. However, there is conflicting evidence on this issue, in particular regarding premenopausal women.13,25 Additionally, many of the studies of estrogen supplementation show a trend toward increased headache once estrogen is discontinued, presumably due to estrogen withdrawal.10,24
That said, one study by MacGregor, et al demonstrates that the use of estradiol gel in the perimenstrual window leads to a 22% reduction in migraine days as well as less severe migrainous symptoms.26 This trend has been demonstrated in other studies examining estrogen supplementation. Of note, the estrogen studies generally are small, older, and of fair to poor quality.11 These studies have used higher doses of estrogen than are commonly used for contraception today because lower doses of estrogen seem not to have the same impact on migraine.5,24
As for COCs, with either normal or extended cycling, data are more mixed than for estrogen supplementation alone; equivalent numbers of women experience improvement, no change, or worsening of their headache pattern. Many women have continuing or worsening migraines in the hormone-free week, and thus most studies have examined the use of extended cycling COCs.5 Sulak, et al demonstrated a statistically significant reduction in headache frequency using extended-cycling COCs, though they did not examine MRM in particular.27 The efficacy of extended-cycling COCs for reduction of MRM was confirmed by Coffee and colleagues with a small but statistically significant decrease in daily headache scores.28
Adverse effects. All estrogen therapies pose the risk of adverse effects (deep vein thrombosis, hypertension, breast tenderness, nausea, etc). Additionally, estrogen supplementation may actually trigger migraines in some women if, when it is discontinued, the blood estrogen level does not remain above a threshold concentration.5,10,24 Estrogen may also trigger migraine in previously headache-free women and may convert migraine without aura into migraine with aura. In either case, therapy should be stopped.5,24
There is promising evidence from 2 small RCTs and one observational trial that progestin-only contraceptive pills (POP) may reduce the frequency and severity of menstrual migraines (TABLE 211-16). More prospective data are needed to confirm this reduction, as there have not been specific studies examining other progesterone-only preparations to prevent menstrual migraines.
Risk of ischemic stroke. Unfortunately, there are population data showing that second-generation and, to a smaller degree, third-generation progestins, which include the desogestrel used in the above studies, may increase the risk of ischemic stroke. This is a particular concern in women who experience migraine.29 Second-generation progestins include levonorgestrel, which is in the levonorgestrel IUD; however, there is no direct evidence for increased ischemic stroke in this particular preparation, and the circulating plasma levels are low. Etonorgestrel, the active ingredient in the contraceptive implant, is a third-generation progestin, though there is no direct evidence of increased ischemic stroke with use of the etonorgestrel implant.
There is a 2- to 4-fold increased risk of ischemic stroke in women who experience migraine.1,5,30 As stated above, this risk may be further increased by some progesterone formulations. But there is also a demonstrable increase in ischemic stroke risk with the use of estrogen, particularly at the higher concentrations that have been shown to prevent MRM.31,32 The overall incidence of ischemic stroke in menstrual-age women is low, which has limited the number of studies with enough power to quantify the absolute increased risk of stroke in conjunction with estrogen use. Nevertheless, exogenous estrogen is thought to increase the risk of ischemic stroke an additional 2- to 4-fold.1,5,29,30,32-34
Women who experience aura. MRM, as it is defined, typically excludes women who experience aura; however, the number of women who experience aura with migraine either in proximity to their menses or throughout the month has not been well documented. The risk of ischemic stroke is higher for women who experience migraine with aura than those with migraine alone, possibly because aura is associated with reduced regional vascular flow leading to hypoperfusion, which sets the stage for a possible ischemic event.4,5,35 The risk of ischemic stroke is amplified further for women who are over 35, who smoke, or who have additional vascular risk factors (eg, uncontrolled hypertension, diabetes, or known vascular or cardiac disease).1,5,34 This array of evidence serves as the basis for the US Medical Eligibility Criteria (USMEC) recommendations36 for hormonal contraceptive use, in particular the absolute contraindication for estrogen use in women who experience migraine with aura (TABLE 336-38).
CASE › Given Mary’s experience of aura with migraine, you talk with her at length about the risk of ischemic stroke and the USMEC recommendation that she absolutely should not be taking COCs. You suggest a progestin-only method of contraception such as depot medroxyprogesterone acetate, a progestin intrauterine device, or a hormonal implant, which may suppress ovulation and decrease her headaches. You discuss that while some women may have headaches with these progestin-only methods, stroke risk is significantly reduced. You also suggest a trial of prophylactic triptans as another possible option.
She says she understands the increased risk of stroke but is still unwilling to try anything else right now due to worries about her quality of life. You decide jointly to refill COCs for 3 months, and you document the shared decision process in the chart. After advising the patient that you will not continue to prescribe COCs for an extended period of time, you also schedule a follow-up appointment to further discuss risks and benefits of migraine treatment and means of reducing other risk factors for stroke.
CORRESPONDENCE
Sarina Schrager, MD, MS, University of Wisconsin, Department of Family Medicine, 1100 Delaplaine Ct, Madison, WI 53715; [email protected].
1. MacGregor EA, Rosenberg JD, Kurth T. Sex-related differences in epidemiological and clinic-based headache studies. Headache. 2011;51:843-859.
2. Stewart WF, Wood C, Reed ML, et al. Cumulative lifetime migraine incidence in women and men. Cephalalgia. 2008;28:1170-1178.
3. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd ed. Cephalalgia. 2013;33:629-808.
4. Garza I, Swanson JW, Cheshire WP Jr, et al. Headache and other craniofacial pain. In: Daroff RB, Fenichel GM, Jankovic J, et al, eds. Bradley’s Neurology in Clinical Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2012:1703-1744.
5. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis – part 2. Headache. 2006;46:365-386.
6. Brandes JL. The influence of estrogen on migraine: a systematic review. JAMA. 2006; 295:1824-1830.
7. Loder EW. Menstrual migraine: pathophysiology, diagnosis and impact. Headache. 2006;46 (Suppl 2):S55-S60.
8. Misakian AL, Langer RD, Bensenor IM, et al. Postmenopausal hormone therapy and migraine headache. J Women’s Health (Larchmt). 2003;12:1027-1036.
9. Pringsheim T, Gooren L. Migraine prevalence in male to female transsexuals on hormone therapy. Neurology. 2004;63:593-594.
10. Somerville BW. The role of estradiol withdrawal in the etiology of menstrual migraine. Neurology. 1972;22:355-365.
11. Pringsheim T, Davenport WJ, Dodick D. Acute treatment and prevention of menstrually related migraine headache: evidence-based review. Neurology. 2008;70:1555-1563.
12. Hu Y, Guan X, Fan L, et al. Triptans in prevention of menstrual migraine: a systematic review with meta-analysis. J Headache Pain. 2013;14:7.
13. Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ. 2008;336:1227-1231.
14. Merki-Feld GS, Imthurn B, Langner R, et al. Headache frequency and intensity in female migraineurs using desogestrel-only contraception: a retrospective pilot diary study. Cephalalgia. 2013;33:340-346.
15.
16. Morotti M, Remorgida V, Venturini PL, et al. Progestin-only contraception compared with extended combined oral contraceptive in women with migraine without aura: a retrospective pilot study. Eur J Obstet Gynecol Reprod Biol. 2014;183:178-182.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Newman L, Mannix LK, Landy S, et al. Naratriptan as short-term prophylaxis of menstrually associated migraine: a randomized double-blind, placebo-controlled study. Headache. 2001;41:248-256.
19. Tuchman MM, Hee A, Emeribe U, et al. Oral zolmitriptan in the short-term prevention of menstrual migraine: a randomized, placebo-controlled study. CNS Drugs. 2008;22:877-886.
20. Facchinetti F, Sances G, Borella P, et al. Magnesium prophylaxis of menstrual migraine: effects on intracellular magnesium. Headache. 1991;31:298-301.
21. Teigen L, Boes CJ. An evidence-based review of oral magnesium supplementation in the preventive treatment of migraine. Cephalalgia. 2014;35:912-922.
22. Taylor FR. Nutraceuticals and headache: the biological basis. Headache. 2011;51:484-501.
23. Zhang XZ, Zhang L, Guo J, et al. Acupuncture as prophylaxis for menstrual-related migraine: study protocol for a multicenter randomized controlled trial. Trials. 2013;14:374.
24. MacGregor EA. Oestrogen and attacks of migraine with and without aura. Lancet Neurol. 2004;3:354-361.
25. Cole JA, Norman H, Doherty M, et al. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol. 2007;109:339-346.
26. MacGregor EA, Frith A, Ellis J, et al. Prevention of menstrual attacks of migraine: a double blind placebo-controlled crossover study. Neurology. 2006;67:2159-2163.
27. Sulak P, Willis S, Kuehl T, et al. Headaches and oral contraceptives: impact of eliminating the standard 7-day placebo interval. Headache. 2007;47:27-37.
28. Coffee AL, Sulak PJ, Hill AJ, et al. Extended cycle combined oral contraceptives and prophylactic frovatriptan during the hormone-free interval in women with menstrual-related migraines. J Womens Health. 2014;23:310-317.
29. Lidegaard Ø, Kreiner S. Contraceptives and cerebral thrombosis: a five-year national case-control study. Contraception. 2002;65:197-205.
30. Bousser MG. Estrogen, migraine, and stroke. Stroke. 2004;35(Suppl 1):2652-2656.
31. Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta-analysis. JAMA. 2000;284:72-78.
32. Donaghy M, Chang CL, Poulter N. Duration, frequency, recency, and type of migraine and the risk of ischaemic stroke in women of childbearing age. J Neurol Neurosurg Psychiatry. 2002;73:747-750.
33. Sacco S, Ricci S, Degan D. Migraine in women: the role of hormones and their impact on vascular diseases. J Headache Pain. 2012;12:177-189.
34. Merikangas KR, Fenton BT, Cheng SH, et al. Association between migraine and stroke in a large-scale epidemiological study of the United States. Arch Neurol. 1997;54:362-368.
35. MacClellan LR, Giles W, Cole J, et al. Probable migraine with visual aura and risk of ischemic stroke: the stroke prevention in young women study. Stroke. 2007;38:2438-2445.
36. Centers for Disease Control and Prevention. U.S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep
37. Chang CL, Donaghy M, Poulter N. Migraine and stroke in young women: case-control study. The World Health Organisation Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. BMJ. 1999;318:13-18.
38. Tzourio C, Tehindrazanarivelo A, Iglésias S, et al. Case-control study of migraine and risk of ischaemic stroke in young women. BMJ. 1995;310:830-833.
› Consider recommending that patients with menstrual migraines try using prophylactic triptans 2 days before the onset of menses. B
› Advise against estrogen-containing contraception for women who have menstrual migraines with aura, who smoke, or are over 35, due to the increased risk of stroke (absolute contraindication). A
› Consider estrogen-containing contraception if the benefits outweigh the risks for women with migraines who are under 35 and do not have aura (relative contraindication). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mary, a 34-year-old woman, is a new patient to your practice after moving to the area for a job. She has a history of migraine headaches triggered by her menstrual periods. She has been taking combined oral contraceptives (COCs) since she was 17, with a few years off when she had 2 children. Her migraines improved when she was pregnant, but worsened postpartum with each of her daughters to a point where she had to stop breastfeeding at 4 months to go back on the pills.
On the COCs, she gets one or 2 mild-to-moderate headaches a month. She uses sumatriptan for abortive treatment with good relief. She has not missed work in the past 4 years because of her migraines. During the 6 months she was off COCs when trying to get pregnant, she routinely missed 2 to 3 workdays per month due to migraines. She knows when she is going to get a headache because she sees flashing lights in her left visual field. She has no other neurologic symptoms with the headaches, and the character of the headaches has not changed. She is a non-smoker, has normal blood pressure and lipid levels, and no other vascular risk factors.
You review her history and talk to her about the risk of stroke with migraines and with COCs. She is almost 35 years of age and you recommend stopping the COCs due to the risk. She feels strongly that she wants to continue taking the COCs, saying her quality of life is poor when she is off the pills. What should you do?
Migraine headaches are 2 to 3 times more prevalent in women than in men,1 with a lifetime risk of 43% vs 18%, respectively.2 Women account for about 80% of the $1 billion spent each year in the United States in medical expenses and lost work productivity related to migraines.1,2
Clinical patterns suggestive of menstrual migraine. About half of women affected by migraine have menstrually-related migraines (MRM); 3% to 12% have pure menstrual migraines (PMM).3 MRM and PMM are both characterized by the presence of symptoms in at least 2 to 3 consecutive cycles, with symptoms occurring from between 2 days before to 3 days after the onset of menstruation. However, in PMM, symptoms do not occur at any other time of the menstrual cycle; in MRM, symptoms can occur at other times of the cycle. PMM is more likely to respond to hormone therapy than is MRM.
Multiple studies in the United States, Europe, and Asia have noted that migraines related to menses typically last longer, are more severe, less likely to be associated with aura, and more likely to be recurrent and recalcitrant to treatment than non-menstrual migraines.1 TABLE 13 describes diagnostic criteria for migraine without aura.
Possible mechanisms of MRM and PMM. The etiology of migraine is not well understood and is likely multifactorial.4 Incidence of menstrual migraines is related to cyclic changes in female hormones—specifically, the decreasing levels of estrogen that typically happen the week before onset of menses.1 The mechanism is not yet clear, though it is thought that a decline in estrogen levels triggers a decline in serotonin levels, which may lead to cranial vasodilation and sensitization of the trigeminal nerve.5,6 Estrogen decline has also been linked to increased cranial nociception as well as decreased endogenous opioid activity. A study using positron emission tomography found increased activity of serotonergic neurons in migraineurs.7 The evidence that triptans and serotonin receptor agonists are effective in the treatment of migraine also supports the theory that serotonin neurohormonal signaling pathways play a critical role in the pathogenesis of migraines.7
Prevalence patterns point to the role of estrogen. The prevalence of migraines in women increases around puberty, peaks between ages 30 and 40, and decreases after natural menopause.6 Migraine prevalence increases during the first week postpartum, when levels of estrogen and progesterone decrease suddenly and significantly.1 Migraine frequency and intensity decrease in the second and third trimesters of pregnancy and after menopause, when estrogen levels fluctuate significantly less.1 In the Women’s Health Initiative study, women who used hormone replacement therapy (HRT) had a 42% increased risk of migraines compared with women in the study who had never used HRT.8
The association of migraine with female hormones was further supported by a Dutch study of male-to-female transgender patients on estrogen therapy, who had a 26% incidence of migraine, equivalent to the 25% prevalence in natal female controls in this study, compared with just 7.5% in male controls.9 The association between migraine and estrogen withdrawal was investigated in studies performed more than 40 years ago, when women experiencing migraines around the time of menses were given intramuscular estradiol and experienced a delay in symptom onset.10
Abortive and prophylactic treatments: Factors that guide selection
In considering probable menstrual migraine, take a detailed history, review headache diaries if available to determine association of headaches with menses, and perform a thorough neurologic examination. If a diagnosis of menstrual migraine is established, discuss the benefits of different treatment options, both abortive and prophylactic.
For the patient with MRM, take into account frequency of symptoms, predictability of menstruation, medication costs, and comorbidities. Both triptans and nonsteroidal anti-inflammatory drugs (NSAIDs) can be effective treatments for MRM.11 Abortive therapy may be appropriate if a patient prefers to take medication intermittently, if her menses are unpredictable, or if she does not get migraine headaches with every menses. Mefenamic acid, sumatriptan, and rizatriptan have category B recommendations for abortive treatment for menstrual migraines (TABLE 211-16). (For the patient who has regular MRM but unpredictable menses, ovulation predictor kits can be used to help predict the onset of menses, although this would involve additional cost.)
For the patient who has predictable menses and regularly occurring menstrual migraine, some data show that a short-term prophylactic regimen with triptans started 2 to 3 days before the onset of menses and continued for 5 to 7 days total can reduce the incidence of menstrual migraine (TABLE 211-16). At least one high-quality randomized controlled trial (RCT) showed a significant reduction in the incidence of MRM when women were treated prophylactically with frovatriptan, a long-acting triptan with a half-life of approximately 26 hours. Participants received frovatriptan 2.5 mg once a day or twice a day or placebo in the perimenstrual period (day -2 to +3). The incidence of MRM was 52%, 41%, and 67%, respectively (P<.0001).11,17
Another RCT of fair quality examined the effect of naratriptan (half-life 6-8 hours) on the median number of menstrual migraines over 4 menstrual cycles. Women who received 1 mg of naratriptan BID for 2 to 3 days before menses had 2 MRM episodes over the 4 cycles compared with 4 MRM episodes in women who received placebo over the same time period (P<.05).11,18 A third RCT, also of fair quality, compared 2 different regimens of zolmitriptan (half-life 3 hours) with placebo and found that women who received 2.5 mg of zolmitriptan either BID or TID 2 to 3 days prior to menses had a reduction both in frequency of menstrual migraines and in the mean number of breakthrough headaches per menstrual cycle, as well as a reduction in the need for rescue medications.12,19 Triptans are contraindicated in women with a history of cardiac disease or uncontrolled hypertension. Also, triptans can be expensive, precluding their use for some patients.
Evidence is insufficient to recommend for or against the use of NSAIDs as prophylaxis for MRM.11 NSAIDs may be contraindicated in women with a history of peptic ulcer disease or gastrointestinal bleeding. That said, if NSAIDs are not contraindicated, a trial may be reasonable given their low cost.
Data are sparse on the use of vitamins and supplements in treating and preventing PMM or MRM. In one very small double-blind, placebo-controlled study in 1991 (N=24, with efficacy data for 20), participants received a 2-week course of oral magnesium premenstrually. There was a statistically significant reduction in the number of days with headache per month (from 4.7±3.1 days to 2.4±2.2 days; P<.01) and in the total pain index (P<.03).20 A number of studies have demonstrated a correlation between hypomagnesemia and migraine headaches.5,21 The exact mechanism for this relationship is unclear.
Some recent evidence-based reviews have examined the efficacy of nutraceuticals such as magnesium, feverfew, butterbur, coenzyme Q10, and riboflavin on typical migraine, but it is not clear if these results are translatable to the treatment and prophylaxis of menstrual migraine.11,22 A multicenter, single-blind, RCT is underway to examine the efficacy of acupuncture as prophylaxis for MRM.23
Estrogen: Prescribing criteria are strict
Most studies examining the role of exogenous estrogen in reducing menstrual migraines have used topical estrogen (either in patch or gel formulations) in the perimenstrual window (TABLE 211-16). The topical estrogen route has been examined, in particular, as it is presumed to confer less risk of hypercoagulability by avoiding first-pass metabolism. However, there is conflicting evidence on this issue, in particular regarding premenopausal women.13,25 Additionally, many of the studies of estrogen supplementation show a trend toward increased headache once estrogen is discontinued, presumably due to estrogen withdrawal.10,24
That said, one study by MacGregor, et al demonstrates that the use of estradiol gel in the perimenstrual window leads to a 22% reduction in migraine days as well as less severe migrainous symptoms.26 This trend has been demonstrated in other studies examining estrogen supplementation. Of note, the estrogen studies generally are small, older, and of fair to poor quality.11 These studies have used higher doses of estrogen than are commonly used for contraception today because lower doses of estrogen seem not to have the same impact on migraine.5,24
As for COCs, with either normal or extended cycling, data are more mixed than for estrogen supplementation alone; equivalent numbers of women experience improvement, no change, or worsening of their headache pattern. Many women have continuing or worsening migraines in the hormone-free week, and thus most studies have examined the use of extended cycling COCs.5 Sulak, et al demonstrated a statistically significant reduction in headache frequency using extended-cycling COCs, though they did not examine MRM in particular.27 The efficacy of extended-cycling COCs for reduction of MRM was confirmed by Coffee and colleagues with a small but statistically significant decrease in daily headache scores.28
Adverse effects. All estrogen therapies pose the risk of adverse effects (deep vein thrombosis, hypertension, breast tenderness, nausea, etc). Additionally, estrogen supplementation may actually trigger migraines in some women if, when it is discontinued, the blood estrogen level does not remain above a threshold concentration.5,10,24 Estrogen may also trigger migraine in previously headache-free women and may convert migraine without aura into migraine with aura. In either case, therapy should be stopped.5,24
There is promising evidence from 2 small RCTs and one observational trial that progestin-only contraceptive pills (POP) may reduce the frequency and severity of menstrual migraines (TABLE 211-16). More prospective data are needed to confirm this reduction, as there have not been specific studies examining other progesterone-only preparations to prevent menstrual migraines.
Risk of ischemic stroke. Unfortunately, there are population data showing that second-generation and, to a smaller degree, third-generation progestins, which include the desogestrel used in the above studies, may increase the risk of ischemic stroke. This is a particular concern in women who experience migraine.29 Second-generation progestins include levonorgestrel, which is in the levonorgestrel IUD; however, there is no direct evidence for increased ischemic stroke in this particular preparation, and the circulating plasma levels are low. Etonorgestrel, the active ingredient in the contraceptive implant, is a third-generation progestin, though there is no direct evidence of increased ischemic stroke with use of the etonorgestrel implant.
There is a 2- to 4-fold increased risk of ischemic stroke in women who experience migraine.1,5,30 As stated above, this risk may be further increased by some progesterone formulations. But there is also a demonstrable increase in ischemic stroke risk with the use of estrogen, particularly at the higher concentrations that have been shown to prevent MRM.31,32 The overall incidence of ischemic stroke in menstrual-age women is low, which has limited the number of studies with enough power to quantify the absolute increased risk of stroke in conjunction with estrogen use. Nevertheless, exogenous estrogen is thought to increase the risk of ischemic stroke an additional 2- to 4-fold.1,5,29,30,32-34
Women who experience aura. MRM, as it is defined, typically excludes women who experience aura; however, the number of women who experience aura with migraine either in proximity to their menses or throughout the month has not been well documented. The risk of ischemic stroke is higher for women who experience migraine with aura than those with migraine alone, possibly because aura is associated with reduced regional vascular flow leading to hypoperfusion, which sets the stage for a possible ischemic event.4,5,35 The risk of ischemic stroke is amplified further for women who are over 35, who smoke, or who have additional vascular risk factors (eg, uncontrolled hypertension, diabetes, or known vascular or cardiac disease).1,5,34 This array of evidence serves as the basis for the US Medical Eligibility Criteria (USMEC) recommendations36 for hormonal contraceptive use, in particular the absolute contraindication for estrogen use in women who experience migraine with aura (TABLE 336-38).
CASE › Given Mary’s experience of aura with migraine, you talk with her at length about the risk of ischemic stroke and the USMEC recommendation that she absolutely should not be taking COCs. You suggest a progestin-only method of contraception such as depot medroxyprogesterone acetate, a progestin intrauterine device, or a hormonal implant, which may suppress ovulation and decrease her headaches. You discuss that while some women may have headaches with these progestin-only methods, stroke risk is significantly reduced. You also suggest a trial of prophylactic triptans as another possible option.
She says she understands the increased risk of stroke but is still unwilling to try anything else right now due to worries about her quality of life. You decide jointly to refill COCs for 3 months, and you document the shared decision process in the chart. After advising the patient that you will not continue to prescribe COCs for an extended period of time, you also schedule a follow-up appointment to further discuss risks and benefits of migraine treatment and means of reducing other risk factors for stroke.
CORRESPONDENCE
Sarina Schrager, MD, MS, University of Wisconsin, Department of Family Medicine, 1100 Delaplaine Ct, Madison, WI 53715; [email protected].
› Consider recommending that patients with menstrual migraines try using prophylactic triptans 2 days before the onset of menses. B
› Advise against estrogen-containing contraception for women who have menstrual migraines with aura, who smoke, or are over 35, due to the increased risk of stroke (absolute contraindication). A
› Consider estrogen-containing contraception if the benefits outweigh the risks for women with migraines who are under 35 and do not have aura (relative contraindication). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mary, a 34-year-old woman, is a new patient to your practice after moving to the area for a job. She has a history of migraine headaches triggered by her menstrual periods. She has been taking combined oral contraceptives (COCs) since she was 17, with a few years off when she had 2 children. Her migraines improved when she was pregnant, but worsened postpartum with each of her daughters to a point where she had to stop breastfeeding at 4 months to go back on the pills.
On the COCs, she gets one or 2 mild-to-moderate headaches a month. She uses sumatriptan for abortive treatment with good relief. She has not missed work in the past 4 years because of her migraines. During the 6 months she was off COCs when trying to get pregnant, she routinely missed 2 to 3 workdays per month due to migraines. She knows when she is going to get a headache because she sees flashing lights in her left visual field. She has no other neurologic symptoms with the headaches, and the character of the headaches has not changed. She is a non-smoker, has normal blood pressure and lipid levels, and no other vascular risk factors.
You review her history and talk to her about the risk of stroke with migraines and with COCs. She is almost 35 years of age and you recommend stopping the COCs due to the risk. She feels strongly that she wants to continue taking the COCs, saying her quality of life is poor when she is off the pills. What should you do?
Migraine headaches are 2 to 3 times more prevalent in women than in men,1 with a lifetime risk of 43% vs 18%, respectively.2 Women account for about 80% of the $1 billion spent each year in the United States in medical expenses and lost work productivity related to migraines.1,2
Clinical patterns suggestive of menstrual migraine. About half of women affected by migraine have menstrually-related migraines (MRM); 3% to 12% have pure menstrual migraines (PMM).3 MRM and PMM are both characterized by the presence of symptoms in at least 2 to 3 consecutive cycles, with symptoms occurring from between 2 days before to 3 days after the onset of menstruation. However, in PMM, symptoms do not occur at any other time of the menstrual cycle; in MRM, symptoms can occur at other times of the cycle. PMM is more likely to respond to hormone therapy than is MRM.
Multiple studies in the United States, Europe, and Asia have noted that migraines related to menses typically last longer, are more severe, less likely to be associated with aura, and more likely to be recurrent and recalcitrant to treatment than non-menstrual migraines.1 TABLE 13 describes diagnostic criteria for migraine without aura.
Possible mechanisms of MRM and PMM. The etiology of migraine is not well understood and is likely multifactorial.4 Incidence of menstrual migraines is related to cyclic changes in female hormones—specifically, the decreasing levels of estrogen that typically happen the week before onset of menses.1 The mechanism is not yet clear, though it is thought that a decline in estrogen levels triggers a decline in serotonin levels, which may lead to cranial vasodilation and sensitization of the trigeminal nerve.5,6 Estrogen decline has also been linked to increased cranial nociception as well as decreased endogenous opioid activity. A study using positron emission tomography found increased activity of serotonergic neurons in migraineurs.7 The evidence that triptans and serotonin receptor agonists are effective in the treatment of migraine also supports the theory that serotonin neurohormonal signaling pathways play a critical role in the pathogenesis of migraines.7
Prevalence patterns point to the role of estrogen. The prevalence of migraines in women increases around puberty, peaks between ages 30 and 40, and decreases after natural menopause.6 Migraine prevalence increases during the first week postpartum, when levels of estrogen and progesterone decrease suddenly and significantly.1 Migraine frequency and intensity decrease in the second and third trimesters of pregnancy and after menopause, when estrogen levels fluctuate significantly less.1 In the Women’s Health Initiative study, women who used hormone replacement therapy (HRT) had a 42% increased risk of migraines compared with women in the study who had never used HRT.8
The association of migraine with female hormones was further supported by a Dutch study of male-to-female transgender patients on estrogen therapy, who had a 26% incidence of migraine, equivalent to the 25% prevalence in natal female controls in this study, compared with just 7.5% in male controls.9 The association between migraine and estrogen withdrawal was investigated in studies performed more than 40 years ago, when women experiencing migraines around the time of menses were given intramuscular estradiol and experienced a delay in symptom onset.10
Abortive and prophylactic treatments: Factors that guide selection
In considering probable menstrual migraine, take a detailed history, review headache diaries if available to determine association of headaches with menses, and perform a thorough neurologic examination. If a diagnosis of menstrual migraine is established, discuss the benefits of different treatment options, both abortive and prophylactic.
For the patient with MRM, take into account frequency of symptoms, predictability of menstruation, medication costs, and comorbidities. Both triptans and nonsteroidal anti-inflammatory drugs (NSAIDs) can be effective treatments for MRM.11 Abortive therapy may be appropriate if a patient prefers to take medication intermittently, if her menses are unpredictable, or if she does not get migraine headaches with every menses. Mefenamic acid, sumatriptan, and rizatriptan have category B recommendations for abortive treatment for menstrual migraines (TABLE 211-16). (For the patient who has regular MRM but unpredictable menses, ovulation predictor kits can be used to help predict the onset of menses, although this would involve additional cost.)
For the patient who has predictable menses and regularly occurring menstrual migraine, some data show that a short-term prophylactic regimen with triptans started 2 to 3 days before the onset of menses and continued for 5 to 7 days total can reduce the incidence of menstrual migraine (TABLE 211-16). At least one high-quality randomized controlled trial (RCT) showed a significant reduction in the incidence of MRM when women were treated prophylactically with frovatriptan, a long-acting triptan with a half-life of approximately 26 hours. Participants received frovatriptan 2.5 mg once a day or twice a day or placebo in the perimenstrual period (day -2 to +3). The incidence of MRM was 52%, 41%, and 67%, respectively (P<.0001).11,17
Another RCT of fair quality examined the effect of naratriptan (half-life 6-8 hours) on the median number of menstrual migraines over 4 menstrual cycles. Women who received 1 mg of naratriptan BID for 2 to 3 days before menses had 2 MRM episodes over the 4 cycles compared with 4 MRM episodes in women who received placebo over the same time period (P<.05).11,18 A third RCT, also of fair quality, compared 2 different regimens of zolmitriptan (half-life 3 hours) with placebo and found that women who received 2.5 mg of zolmitriptan either BID or TID 2 to 3 days prior to menses had a reduction both in frequency of menstrual migraines and in the mean number of breakthrough headaches per menstrual cycle, as well as a reduction in the need for rescue medications.12,19 Triptans are contraindicated in women with a history of cardiac disease or uncontrolled hypertension. Also, triptans can be expensive, precluding their use for some patients.
Evidence is insufficient to recommend for or against the use of NSAIDs as prophylaxis for MRM.11 NSAIDs may be contraindicated in women with a history of peptic ulcer disease or gastrointestinal bleeding. That said, if NSAIDs are not contraindicated, a trial may be reasonable given their low cost.
Data are sparse on the use of vitamins and supplements in treating and preventing PMM or MRM. In one very small double-blind, placebo-controlled study in 1991 (N=24, with efficacy data for 20), participants received a 2-week course of oral magnesium premenstrually. There was a statistically significant reduction in the number of days with headache per month (from 4.7±3.1 days to 2.4±2.2 days; P<.01) and in the total pain index (P<.03).20 A number of studies have demonstrated a correlation between hypomagnesemia and migraine headaches.5,21 The exact mechanism for this relationship is unclear.
Some recent evidence-based reviews have examined the efficacy of nutraceuticals such as magnesium, feverfew, butterbur, coenzyme Q10, and riboflavin on typical migraine, but it is not clear if these results are translatable to the treatment and prophylaxis of menstrual migraine.11,22 A multicenter, single-blind, RCT is underway to examine the efficacy of acupuncture as prophylaxis for MRM.23
Estrogen: Prescribing criteria are strict
Most studies examining the role of exogenous estrogen in reducing menstrual migraines have used topical estrogen (either in patch or gel formulations) in the perimenstrual window (TABLE 211-16). The topical estrogen route has been examined, in particular, as it is presumed to confer less risk of hypercoagulability by avoiding first-pass metabolism. However, there is conflicting evidence on this issue, in particular regarding premenopausal women.13,25 Additionally, many of the studies of estrogen supplementation show a trend toward increased headache once estrogen is discontinued, presumably due to estrogen withdrawal.10,24
That said, one study by MacGregor, et al demonstrates that the use of estradiol gel in the perimenstrual window leads to a 22% reduction in migraine days as well as less severe migrainous symptoms.26 This trend has been demonstrated in other studies examining estrogen supplementation. Of note, the estrogen studies generally are small, older, and of fair to poor quality.11 These studies have used higher doses of estrogen than are commonly used for contraception today because lower doses of estrogen seem not to have the same impact on migraine.5,24
As for COCs, with either normal or extended cycling, data are more mixed than for estrogen supplementation alone; equivalent numbers of women experience improvement, no change, or worsening of their headache pattern. Many women have continuing or worsening migraines in the hormone-free week, and thus most studies have examined the use of extended cycling COCs.5 Sulak, et al demonstrated a statistically significant reduction in headache frequency using extended-cycling COCs, though they did not examine MRM in particular.27 The efficacy of extended-cycling COCs for reduction of MRM was confirmed by Coffee and colleagues with a small but statistically significant decrease in daily headache scores.28
Adverse effects. All estrogen therapies pose the risk of adverse effects (deep vein thrombosis, hypertension, breast tenderness, nausea, etc). Additionally, estrogen supplementation may actually trigger migraines in some women if, when it is discontinued, the blood estrogen level does not remain above a threshold concentration.5,10,24 Estrogen may also trigger migraine in previously headache-free women and may convert migraine without aura into migraine with aura. In either case, therapy should be stopped.5,24
There is promising evidence from 2 small RCTs and one observational trial that progestin-only contraceptive pills (POP) may reduce the frequency and severity of menstrual migraines (TABLE 211-16). More prospective data are needed to confirm this reduction, as there have not been specific studies examining other progesterone-only preparations to prevent menstrual migraines.
Risk of ischemic stroke. Unfortunately, there are population data showing that second-generation and, to a smaller degree, third-generation progestins, which include the desogestrel used in the above studies, may increase the risk of ischemic stroke. This is a particular concern in women who experience migraine.29 Second-generation progestins include levonorgestrel, which is in the levonorgestrel IUD; however, there is no direct evidence for increased ischemic stroke in this particular preparation, and the circulating plasma levels are low. Etonorgestrel, the active ingredient in the contraceptive implant, is a third-generation progestin, though there is no direct evidence of increased ischemic stroke with use of the etonorgestrel implant.
There is a 2- to 4-fold increased risk of ischemic stroke in women who experience migraine.1,5,30 As stated above, this risk may be further increased by some progesterone formulations. But there is also a demonstrable increase in ischemic stroke risk with the use of estrogen, particularly at the higher concentrations that have been shown to prevent MRM.31,32 The overall incidence of ischemic stroke in menstrual-age women is low, which has limited the number of studies with enough power to quantify the absolute increased risk of stroke in conjunction with estrogen use. Nevertheless, exogenous estrogen is thought to increase the risk of ischemic stroke an additional 2- to 4-fold.1,5,29,30,32-34
Women who experience aura. MRM, as it is defined, typically excludes women who experience aura; however, the number of women who experience aura with migraine either in proximity to their menses or throughout the month has not been well documented. The risk of ischemic stroke is higher for women who experience migraine with aura than those with migraine alone, possibly because aura is associated with reduced regional vascular flow leading to hypoperfusion, which sets the stage for a possible ischemic event.4,5,35 The risk of ischemic stroke is amplified further for women who are over 35, who smoke, or who have additional vascular risk factors (eg, uncontrolled hypertension, diabetes, or known vascular or cardiac disease).1,5,34 This array of evidence serves as the basis for the US Medical Eligibility Criteria (USMEC) recommendations36 for hormonal contraceptive use, in particular the absolute contraindication for estrogen use in women who experience migraine with aura (TABLE 336-38).
CASE › Given Mary’s experience of aura with migraine, you talk with her at length about the risk of ischemic stroke and the USMEC recommendation that she absolutely should not be taking COCs. You suggest a progestin-only method of contraception such as depot medroxyprogesterone acetate, a progestin intrauterine device, or a hormonal implant, which may suppress ovulation and decrease her headaches. You discuss that while some women may have headaches with these progestin-only methods, stroke risk is significantly reduced. You also suggest a trial of prophylactic triptans as another possible option.
She says she understands the increased risk of stroke but is still unwilling to try anything else right now due to worries about her quality of life. You decide jointly to refill COCs for 3 months, and you document the shared decision process in the chart. After advising the patient that you will not continue to prescribe COCs for an extended period of time, you also schedule a follow-up appointment to further discuss risks and benefits of migraine treatment and means of reducing other risk factors for stroke.
CORRESPONDENCE
Sarina Schrager, MD, MS, University of Wisconsin, Department of Family Medicine, 1100 Delaplaine Ct, Madison, WI 53715; [email protected].
1. MacGregor EA, Rosenberg JD, Kurth T. Sex-related differences in epidemiological and clinic-based headache studies. Headache. 2011;51:843-859.
2. Stewart WF, Wood C, Reed ML, et al. Cumulative lifetime migraine incidence in women and men. Cephalalgia. 2008;28:1170-1178.
3. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd ed. Cephalalgia. 2013;33:629-808.
4. Garza I, Swanson JW, Cheshire WP Jr, et al. Headache and other craniofacial pain. In: Daroff RB, Fenichel GM, Jankovic J, et al, eds. Bradley’s Neurology in Clinical Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2012:1703-1744.
5. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis – part 2. Headache. 2006;46:365-386.
6. Brandes JL. The influence of estrogen on migraine: a systematic review. JAMA. 2006; 295:1824-1830.
7. Loder EW. Menstrual migraine: pathophysiology, diagnosis and impact. Headache. 2006;46 (Suppl 2):S55-S60.
8. Misakian AL, Langer RD, Bensenor IM, et al. Postmenopausal hormone therapy and migraine headache. J Women’s Health (Larchmt). 2003;12:1027-1036.
9. Pringsheim T, Gooren L. Migraine prevalence in male to female transsexuals on hormone therapy. Neurology. 2004;63:593-594.
10. Somerville BW. The role of estradiol withdrawal in the etiology of menstrual migraine. Neurology. 1972;22:355-365.
11. Pringsheim T, Davenport WJ, Dodick D. Acute treatment and prevention of menstrually related migraine headache: evidence-based review. Neurology. 2008;70:1555-1563.
12. Hu Y, Guan X, Fan L, et al. Triptans in prevention of menstrual migraine: a systematic review with meta-analysis. J Headache Pain. 2013;14:7.
13. Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ. 2008;336:1227-1231.
14. Merki-Feld GS, Imthurn B, Langner R, et al. Headache frequency and intensity in female migraineurs using desogestrel-only contraception: a retrospective pilot diary study. Cephalalgia. 2013;33:340-346.
15.
16. Morotti M, Remorgida V, Venturini PL, et al. Progestin-only contraception compared with extended combined oral contraceptive in women with migraine without aura: a retrospective pilot study. Eur J Obstet Gynecol Reprod Biol. 2014;183:178-182.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Newman L, Mannix LK, Landy S, et al. Naratriptan as short-term prophylaxis of menstrually associated migraine: a randomized double-blind, placebo-controlled study. Headache. 2001;41:248-256.
19. Tuchman MM, Hee A, Emeribe U, et al. Oral zolmitriptan in the short-term prevention of menstrual migraine: a randomized, placebo-controlled study. CNS Drugs. 2008;22:877-886.
20. Facchinetti F, Sances G, Borella P, et al. Magnesium prophylaxis of menstrual migraine: effects on intracellular magnesium. Headache. 1991;31:298-301.
21. Teigen L, Boes CJ. An evidence-based review of oral magnesium supplementation in the preventive treatment of migraine. Cephalalgia. 2014;35:912-922.
22. Taylor FR. Nutraceuticals and headache: the biological basis. Headache. 2011;51:484-501.
23. Zhang XZ, Zhang L, Guo J, et al. Acupuncture as prophylaxis for menstrual-related migraine: study protocol for a multicenter randomized controlled trial. Trials. 2013;14:374.
24. MacGregor EA. Oestrogen and attacks of migraine with and without aura. Lancet Neurol. 2004;3:354-361.
25. Cole JA, Norman H, Doherty M, et al. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol. 2007;109:339-346.
26. MacGregor EA, Frith A, Ellis J, et al. Prevention of menstrual attacks of migraine: a double blind placebo-controlled crossover study. Neurology. 2006;67:2159-2163.
27. Sulak P, Willis S, Kuehl T, et al. Headaches and oral contraceptives: impact of eliminating the standard 7-day placebo interval. Headache. 2007;47:27-37.
28. Coffee AL, Sulak PJ, Hill AJ, et al. Extended cycle combined oral contraceptives and prophylactic frovatriptan during the hormone-free interval in women with menstrual-related migraines. J Womens Health. 2014;23:310-317.
29. Lidegaard Ø, Kreiner S. Contraceptives and cerebral thrombosis: a five-year national case-control study. Contraception. 2002;65:197-205.
30. Bousser MG. Estrogen, migraine, and stroke. Stroke. 2004;35(Suppl 1):2652-2656.
31. Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta-analysis. JAMA. 2000;284:72-78.
32. Donaghy M, Chang CL, Poulter N. Duration, frequency, recency, and type of migraine and the risk of ischaemic stroke in women of childbearing age. J Neurol Neurosurg Psychiatry. 2002;73:747-750.
33. Sacco S, Ricci S, Degan D. Migraine in women: the role of hormones and their impact on vascular diseases. J Headache Pain. 2012;12:177-189.
34. Merikangas KR, Fenton BT, Cheng SH, et al. Association between migraine and stroke in a large-scale epidemiological study of the United States. Arch Neurol. 1997;54:362-368.
35. MacClellan LR, Giles W, Cole J, et al. Probable migraine with visual aura and risk of ischemic stroke: the stroke prevention in young women study. Stroke. 2007;38:2438-2445.
36. Centers for Disease Control and Prevention. U.S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep
37. Chang CL, Donaghy M, Poulter N. Migraine and stroke in young women: case-control study. The World Health Organisation Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. BMJ. 1999;318:13-18.
38. Tzourio C, Tehindrazanarivelo A, Iglésias S, et al. Case-control study of migraine and risk of ischaemic stroke in young women. BMJ. 1995;310:830-833.
1. MacGregor EA, Rosenberg JD, Kurth T. Sex-related differences in epidemiological and clinic-based headache studies. Headache. 2011;51:843-859.
2. Stewart WF, Wood C, Reed ML, et al. Cumulative lifetime migraine incidence in women and men. Cephalalgia. 2008;28:1170-1178.
3. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd ed. Cephalalgia. 2013;33:629-808.
4. Garza I, Swanson JW, Cheshire WP Jr, et al. Headache and other craniofacial pain. In: Daroff RB, Fenichel GM, Jankovic J, et al, eds. Bradley’s Neurology in Clinical Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2012:1703-1744.
5. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis – part 2. Headache. 2006;46:365-386.
6. Brandes JL. The influence of estrogen on migraine: a systematic review. JAMA. 2006; 295:1824-1830.
7. Loder EW. Menstrual migraine: pathophysiology, diagnosis and impact. Headache. 2006;46 (Suppl 2):S55-S60.
8. Misakian AL, Langer RD, Bensenor IM, et al. Postmenopausal hormone therapy and migraine headache. J Women’s Health (Larchmt). 2003;12:1027-1036.
9. Pringsheim T, Gooren L. Migraine prevalence in male to female transsexuals on hormone therapy. Neurology. 2004;63:593-594.
10. Somerville BW. The role of estradiol withdrawal in the etiology of menstrual migraine. Neurology. 1972;22:355-365.
11. Pringsheim T, Davenport WJ, Dodick D. Acute treatment and prevention of menstrually related migraine headache: evidence-based review. Neurology. 2008;70:1555-1563.
12. Hu Y, Guan X, Fan L, et al. Triptans in prevention of menstrual migraine: a systematic review with meta-analysis. J Headache Pain. 2013;14:7.
13. Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ. 2008;336:1227-1231.
14. Merki-Feld GS, Imthurn B, Langner R, et al. Headache frequency and intensity in female migraineurs using desogestrel-only contraception: a retrospective pilot diary study. Cephalalgia. 2013;33:340-346.
15.
16. Morotti M, Remorgida V, Venturini PL, et al. Progestin-only contraception compared with extended combined oral contraceptive in women with migraine without aura: a retrospective pilot study. Eur J Obstet Gynecol Reprod Biol. 2014;183:178-182.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Newman L, Mannix LK, Landy S, et al. Naratriptan as short-term prophylaxis of menstrually associated migraine: a randomized double-blind, placebo-controlled study. Headache. 2001;41:248-256.
19. Tuchman MM, Hee A, Emeribe U, et al. Oral zolmitriptan in the short-term prevention of menstrual migraine: a randomized, placebo-controlled study. CNS Drugs. 2008;22:877-886.
20. Facchinetti F, Sances G, Borella P, et al. Magnesium prophylaxis of menstrual migraine: effects on intracellular magnesium. Headache. 1991;31:298-301.
21. Teigen L, Boes CJ. An evidence-based review of oral magnesium supplementation in the preventive treatment of migraine. Cephalalgia. 2014;35:912-922.
22. Taylor FR. Nutraceuticals and headache: the biological basis. Headache. 2011;51:484-501.
23. Zhang XZ, Zhang L, Guo J, et al. Acupuncture as prophylaxis for menstrual-related migraine: study protocol for a multicenter randomized controlled trial. Trials. 2013;14:374.
24. MacGregor EA. Oestrogen and attacks of migraine with and without aura. Lancet Neurol. 2004;3:354-361.
25. Cole JA, Norman H, Doherty M, et al. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol. 2007;109:339-346.
26. MacGregor EA, Frith A, Ellis J, et al. Prevention of menstrual attacks of migraine: a double blind placebo-controlled crossover study. Neurology. 2006;67:2159-2163.
27. Sulak P, Willis S, Kuehl T, et al. Headaches and oral contraceptives: impact of eliminating the standard 7-day placebo interval. Headache. 2007;47:27-37.
28. Coffee AL, Sulak PJ, Hill AJ, et al. Extended cycle combined oral contraceptives and prophylactic frovatriptan during the hormone-free interval in women with menstrual-related migraines. J Womens Health. 2014;23:310-317.
29. Lidegaard Ø, Kreiner S. Contraceptives and cerebral thrombosis: a five-year national case-control study. Contraception. 2002;65:197-205.
30. Bousser MG. Estrogen, migraine, and stroke. Stroke. 2004;35(Suppl 1):2652-2656.
31. Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta-analysis. JAMA. 2000;284:72-78.
32. Donaghy M, Chang CL, Poulter N. Duration, frequency, recency, and type of migraine and the risk of ischaemic stroke in women of childbearing age. J Neurol Neurosurg Psychiatry. 2002;73:747-750.
33. Sacco S, Ricci S, Degan D. Migraine in women: the role of hormones and their impact on vascular diseases. J Headache Pain. 2012;12:177-189.
34. Merikangas KR, Fenton BT, Cheng SH, et al. Association between migraine and stroke in a large-scale epidemiological study of the United States. Arch Neurol. 1997;54:362-368.
35. MacClellan LR, Giles W, Cole J, et al. Probable migraine with visual aura and risk of ischemic stroke: the stroke prevention in young women study. Stroke. 2007;38:2438-2445.
36. Centers for Disease Control and Prevention. U.S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep
37. Chang CL, Donaghy M, Poulter N. Migraine and stroke in young women: case-control study. The World Health Organisation Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. BMJ. 1999;318:13-18.
38. Tzourio C, Tehindrazanarivelo A, Iglésias S, et al. Case-control study of migraine and risk of ischaemic stroke in young women. BMJ. 1995;310:830-833.

Bone disease in patients with kidney disease: A tricky interplay
PRACTICE RECOMMENDATIONS
› Perform laboratory testing for chronic kidney disease (CKD)-induced bone disease at CKD stage 3. B
› Avoid calcium-based phosphate binders in patients with known vascular calcifications. B
› Consider the use of phosphate binders in non-dialysis patients on a case-by-case basis, particularly in those with hyperphosphatemia not controlled by dietary measures. B
› Prescribe native vitamin D (ergocalciferol or cholecalciferol) to patients with CKD stages 3 to 4 who have secondary hyperparathyroidism and vitamin D deficiency. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 14% of the US general population has chronic kidney disease (CKD).1 Limited data exist regarding the exact prevalence of CKD-mineral and bone disorder (MBD), but abnormal mineral metabolism is believed to start in stage 3 CKD, implying that 8% of the adult US population could be at risk for, or already have established, CKD-MBD.2 Although the disorder has traditionally been managed by nephrologists, this earlier onset suggests that many patients should be screened and treated by their primary care physicians.
Because CKD-MBD can lead to significant morbidity (ie, increased fracture risk) and mortality, identification and treatment are of utmost importance.3 This review provides information from the current literature and the KDIGO (Kidney Disease: Improving Global Outcomes) guidelines, and focuses primarily on the non-dialysis CKD population.
CKD-MBD: A broad spectrum of disorders
CKD-MBD is defined as a systemic disorder of mineral and bone metabolism due to CKD. Traditionally referred to as renal osteodystrophy, the term CKD-MBD is meant to indicate and describe a broad clinical spectrum of CKD-associated bone mineral metabolism disorders that manifest from one or a combination of the following:
- Abnormalities of calcium, phosphorus, parathyroid hormone (PTH), or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.4
Renal bone disease can be divided into low bone turnover (adynamic bone disease) and high bone turnover states. Both can lead to a decrease in bone strength and an increase in pathological fractures.5
Pathophysiology: Difficult to know where the cascade begins
Understanding the pathophysiology and treatment of bone disease in patients with CKD can be challenging. Because of abnormalities of mineral metabolism and changes in hormones and cytokines, bone remodeling is severely disrupted in patients with CKD, and it remains unclear where this cascade begins.
As an adaptive response to decreased kidney function, PTH levels increase. Elevations of both fibroblast growth factor 23 (FGF23) lower blood phosphate levels by inhibiting phosphate reabsorption in the kidneys, thus increasing urinary excretion of phosphorus. Secondary hyperparathyroidism (SHPT), driven by hypocalcemia, responds to normalize serum calcium levels by increasing the number and size of osteoclasts actively breaking down bone matrix. This escalates fracture risk. In addition, the inability of damaged kidneys to convert vitamin D to an active form further deranges calcium and phosphate homeostasis.
Successful management of serum levels begins with monitoring
KDIGO, an independent nonprofit foundation that seeks to improve the care and outcomes of kidney disease patients worldwide, developed guidelines for the diagnosis, evaluation, prevention, and treatment of CKD-MBD in 2009.6 These guidelines recommend that treatment of CKD-MBD be aimed at managing serum phosphate, PTH, and calcium levels. The recommended frequency for laboratory monitoring of these levels varies by stage of CKD and is described in TABLE 1.6 (For more on chronic kidney disease staging, see KDIGO’s 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease, available at: http://www.kdigo.org/clinical_practice_guidelines/pdf/CKD/KDIGO_2012_CKD_GL.pdf.)
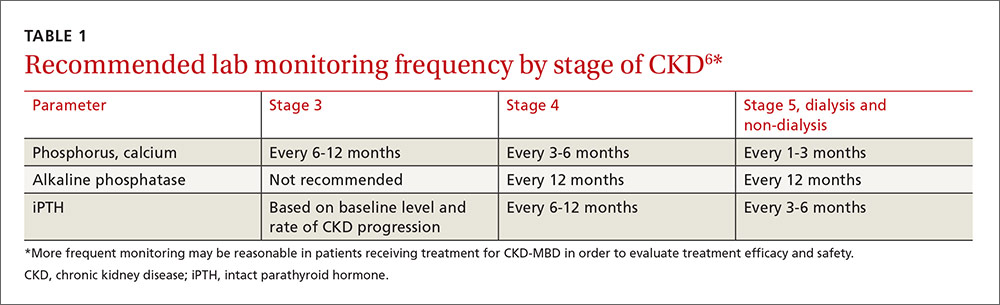
Because of the interrelated nature of these minerals and hormones, drug therapy aimed at treating one may impact the others. This must be considered when designing treatment regimens.
Hyperphosphatemia: Manage with diet, drugs, dialysis
Observational studies have shown an association between higher serum phosphate levels and mortality.6-8 KDIGO recommends maintaining serum phosphorus levels within the normal range of the assay in patients with CKD who are not receiving dialysis.6 For dialyzed patients, the recommendation is to lower the phosphorus level toward the normal range as much as possible.6 Maintaining an appropriate phosphorus level is accomplished through dietary phosphate restriction, the use of phosphate binders, and, in dialyzed patients, dialytic removal of phosphate.6
Dietary phosphate restriction is often challenging for patients, in part, because phosphorous content is not always included on food labels in the United States.9 Phosphorus is highly absorbed from additives in processed food (approaching 100% absorption), less absorbed from animal sources such as meat and dairy products (40%-60%), and is the least absorbed from plant sources such as beans and nuts (20%-40%).10 Advise patients to avoid fast food, processed foods, cheese, frozen meals, colas, and certain ready-to-eat cereals and prepared meats, as these products may have additives from which phosphorus is readily absorbed.11 A patient-friendly list of high-phosphorus foods, as well as other dietary advice and recipes, can be found on the National Kidney Foundation Web site at https://www.kidney.org/atoz/content/phosphorus. Tables listing the phosphorus content of common foods are also available in the literature and online.11,12 Keep in mind that not all resources take into account phosphate bioavailability. Dietician referral may be helpful to assure that patients maintain adequate protein intake while restricting dietary phosphate.
Phosphate binders are recommended by the KDIGO guidelines for use in patients with kidney disease and hyperphosphatemia.6 Most of the data to support the use of phosphate binders was gleaned from the dialysis population. The use of phosphate binders in non-dialyzed patients with CKD has both proponents and opponents, with literature supporting both positions.13,14 A recent KDIGO conference on controversies in CKD-MBD identified this as an area that should be evaluated further for the next guideline update.15
Phosphate binders—which bind the phosphorus in food to prevent absorption—should be taken with meals or high-phosphorus snacks. Products and formulations of commonly used phosphate binders are shown in TABLE 2.16,17 Taste, formulation, adverse effects, pill burden, and cost are issues to discuss with patients when initiating or adjusting phosphate binder therapy. It’s estimated that more than half of all patients receiving dialysis do not adhere to their prescribed phosphate binder regimen, highlighting the need to assess adherence before adjusting dose and to involve the patient in the decision-making process to select a phosphate binder product.18
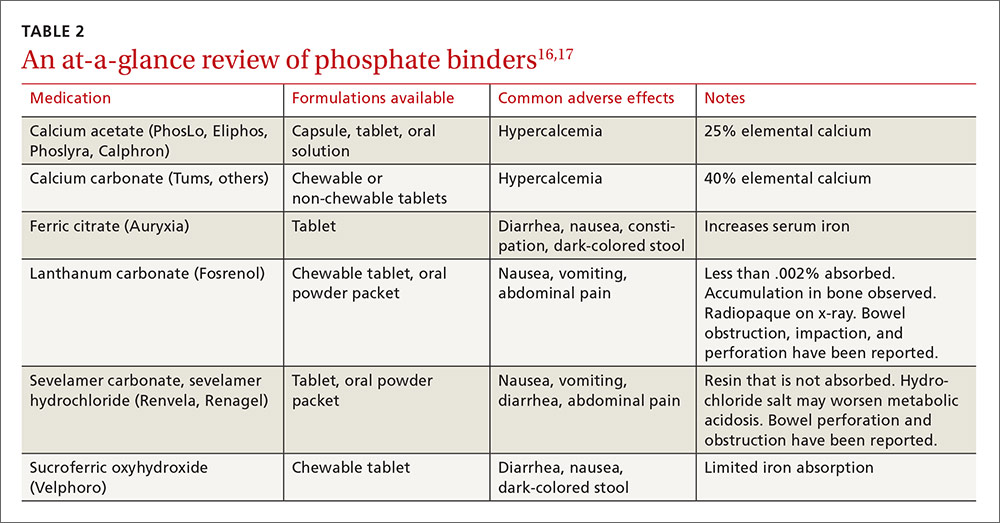
Avoid calcium-based binders? The risk of hypercalcemia and the potentially increased risk of vascular calcifications with calcium-based binders have led some nephrologists to favor non-calcium-based products. Two recent meta-analyses found a reduced risk of all-cause mortality with the non-calcium-based binders sevelamer or lanthanum as compared to calcium-based binders.19,20 Current KDIGO guidelines were published prior to these meta-analyses and do not recommend one phosphate binder over another. They do, however, recommend restricting the dose of calcium-based binders in the setting of persistent or recurrent hypercalcemia, known vascular calcification, or low PTH levels.6
Secondary hyperparathyroidism
Due to a lack of data, the goal PTH level in patients not receiving dialysis is unknown.6 A reasonable approach in non-dialyzed patients, however, is to correct 25-OH vitamin D (25[OH]D) deficiency, elevations in serum phosphate, and hypocalcemia when the level of intact PTH (iPTH) exceeds the normal range for the assay because correcting these derangements may result in a decline in iPTH.6,21 If this approach fails and PTH levels continue to rise, use of calcitriol or vitamin D analogues is recommended.6 Characteristics of medications used to treat SHPT are presented in TABLE 3.16,17
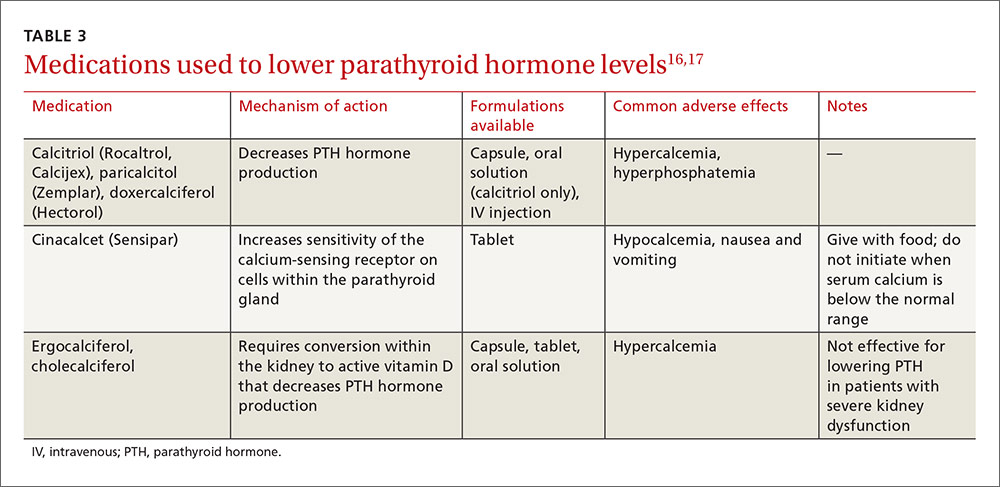
In dialysis patients, the target iPTH range suggested by KDIGO is 2 to 9 times the upper limit of normal for the assay.6 Elevated PTH levels in the dialysis population may be managed with activated vitamin D and/or cinacalcet.
Native vitamin D (ergocalciferol, cholecalciferol) and activated vitamin D analogs (calcitriol, doxercalciferol, paricalcitol). Native vitamin D products are recommended for non-dialyzed patients with CKD to correct vitamin D deficiencies. Although many approaches may be used clinically to replenish low vitamin D stores, one reasonable recommendation in patients with a 25(OH)D level <30 ng/mL is to prescribe ergocalciferol 50,000 units/week for 8 weeks and then to repeat the serum 25-OH vitamin D test. If the level is still <30 ng/mL, a second 8-week course of weekly ergocalciferol 50,000 IU may be administered.21
Following repletion with ergocalciferol, maintenance doses of cholecalciferol (1000-2000 IU/d) or ergocalciferol (50,000 IU/-month) may be initiated.21 Discontinue native vitamin D in patients who develop hypercalcemia.
Native vitamin D becomes less effective at reducing PTH levels as kidney disease advances. This is likely due to a decline in renal conversion of 25(OH)D to 1,25-(OH)2vitamin D (1,25[OH]2D), the most active form of vitamin D and the form of vitamin D that decreases PTH production. By stage 5 CKD, it is unlikely that native vitamin D will significantly decrease PTH levels; treatment with activated vitamin D products or cinacalcet is generally required.
Because the enzyme responsible for converting 25(OH)D into the most active form can be found in multiple tissues outside of the kidney, and the 1,25(OH)2D converted for use by these organs may help prevent such conditions/events as hypertension, type 2 diabetes, myocardial infarction, and stroke (in patients with and without kidney disease), some specialists prescribe native vitamin D to patients with CKD for reasons unrelated to PTH suppression. There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.21
Don’t forget calcium
All of the active vitamin D products can increase serum calcium and phosphate levels. Calcitriol, however, may cause more hypercalcemia than paricalcitol.22 If hypercalcemia develops, you may need to stop, or reduce the dose of, vitamin D analogues. Or you may need to switch patients from calcium-based to non-calcium-based phosphate binders. If hyperphosphatemia develops, intensify phosphate binder therapy or reduce the dose of, or stop, vitamin D analogues. If iPTH levels go below the target range, reduce the dose of the vitamin D analogue to avoid iatrogenic adynamic bone disease.
Avoid this agent in the non-dialyzed patient. Cinacalcet effectively treats SHPT in patients receiving dialysis, but is not recommended for use in undialyzed patients.23 That’s because unacceptably high rates of hypocalcemia have been observed in non-dialyzed patients who were taking the drug.23,24 In addition, while cinacalcet neutrally affects, or causes a slight decrease in, serum phosphate in patients receiving dialysis, it increases serum phosphate in patients who are not.24,25
Drug therapy for osteoporosis
Therapy to prevent and treat fractures in patients with CKD is controversial because patients with CKD stage 3 to 5 with and without MBD were excluded from clinical trials of commercially available treatments. Furthermore, in adynamic bone disease, bones are capable of neither breaking down nor building (ie, reduced resorption). Bisphosphonates and other antiresorptive therapies are more effective at decreasing fractures in patients who are in a state of increased bone resorption, such as menopausal women, so the benefits of these medications in terms of their ability to reduce fractures in CKD patients are questionable, as is their safety.26,27
In addition, while dual-energy x-ray absorptiometry (DXA) is typically used to identify patients who would benefit from these agents, studies have recently demonstrated that femoral neck bone density measured via DXA may underestimate fracture risk in patients with CKD-MBD (ie, bone density may actually be lower than measured).26,28
Antiresorptive agents and teriparatide
Osteoporosis treatments include antiresorptive agents (ie, the bisphosphonates, raloxifene, denosumab), and the anabolic bone agent teriparatide.
Evidence supports treating patients with stage 1 to 3 CKD the same as patients without CKD.15 Bisphosphonates are labeled as contraindicated in patients with a glomerular filtration rate (GFR) <30 mL/min/1.73m2, due to concerns arising from animal trials and subsequent human case reports (both with intravenous formulations only) regarding acute kidney injury.27
While raloxifene lacks a warning regarding use in patients with stage 3 to 5 CKD, it has not been shown to prevent hip fractures in any population.29
Denosumab is not contraindicated for use in patients with CKD stage 3 to 5 without MBD, but it can worsen hypocalcemia, particularly in patients receiving dialysis.30
Teriparatide is contraindicated in patients with CKD and SHPT,31 and there are no studies of its use in patients with CKD-MBD.
What the guidelines say about antiresorptive treatment
For patients with stage 3 to 5 CKD with manifestations of MBD, 2009 KDIGO guidelines recommend a bone biopsy to evaluate for adynamic bone disease before initiating antiresorptive treatment.6 Because few physicians in most communities are trained to conduct and evaluate bone biopsies, this recommendation is infrequently followed. Without a bone biopsy to rule out adynamic bone disease, options to prevent or treat fractures in the setting of CKD-MBD are limited.
CORRESPONDENCE
Karly Pippitt, MD, Department of Family and Preventive Medicine, University of Utah School of Medicine, 375 Chipeta Way, Suite A, Salt Lake City, UT 84108; [email protected].
1. United States Renal Data System. Chapter 1: CKD in the general population. Available at: https://www.usrds.org/2015/download/vol1_01_General_Pop_15.pdf. Accessed July 27, 2016.
2. Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038-2047.
3. Uhlig K, Berns JS, Kestenbaum B, et al. KDOQI US commentary on the 2009 KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, and Treatment of CKD-Mineral and Bone Disorder (CKD-MBD). Am J Kidney Dis. 2010;55:773-799.
4. Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol. 2007;18:875-885.
5. Roberts DM, Singer RF. Management of renal bone disease. Aust Prescr. 2010;33:34-37.
6. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009:S1-130.
7. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305:1119-1127.
8. Cannata-Andia JB, Martin KJ. The challenge of controlling phosphorus in chronic kidney disease. Nephrol Dial Transplant. 2016;31:541-547.
9. US Food and Drug Administration. Food Labeling Guide. Available at: http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/LabelingNutrition/ucm2006828.htm. Accessed July 25, 2016.
10. Kalantar-Zadeh K. Patient education for phosphorus management in chronic kidney disease. Patient Prefer Adherence. 2013;7:379-390.
11. Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:519-530.
12. USDA National Nutrient Database for Standard Reference. 2015; Available at: https://ndb.nal.usda.gov. Accessed April 25, 2016.
13. Bellasi A. Pro: Should phosphate binders be used in chronic kidney disease stage 3-4? Nephrol Dial Transplant. 2016;31:184-188.
14. Kestenbaum B. Con: Phosphate binders in chronic kidney disease. Nephrol Dial Transplant. 2016;31:189-194.
15. Ketteler M, Elder GJ, Evenepoel P, et al. Revisiting KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: a commentary from a Kidney Disease: Improving Global Outcomes controversies conference. Kidney Int. 2015;87:502-528.
16. Wolters Kluwer. Lexicomp. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed April 26, 2016.
17. Truven Health Analytics. Micromedex Solutions. Available at: http://micromedex.com/. Accessed April 26, 2016.
18. Wang S, Anum EA, Ramakrishnan K, et al. Reasons for phosphate binder discontinuation vary by binder type. J Ren Nutr. 2014;24:105-109.
19. Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016;11:232-244.
20. Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382:1268-1277.
21. Nigwekar SU, Bhan I, Thadhani R. Ergocalciferol and cholecalciferol in CKD. Am J Kidney Dis. 2012;60:139-156.
22. Teng M, Wolf M, Lowrie E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446-456.
23. Sensipar package insert. Thousand Oaks, California: Amgen Pharmaceuticals; 2014. Available at: http://pi.amgen.com/united_states/sensipar/sensipar_pi_hcp_english.pdf. Accessed April 25, 2016.
24. Chonchol M, Locatelli F, Abboud HE, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53:197-207.
25. Ballinger AE, Palmer SC, Nistor I,et al. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst Rev. 2014;12:CD006254.
26. Miller PD. Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis. 2014;64:290-304.
27. Ott SM. Bisphosphonate safety and efficacy in chronic kidney disease. Kidney Int. 2012;82:833-835.
28. Yencheck RH, Ix JH, Shlipak MG, et al. Bone mineral density and fracture risk in older individuals with CKD. Clin J Am Soc Nephrol. 2012;7:1130-1136.
29. Crandall CJ, Newberry SJ, Diamant A, et al. Treatments to prevent fractures in men and women with low bone density or osteoporosis: update of a 2007 report. Comparative Effectiveness Reviews, No. 53. Rockville, MD: Agency for Healthcare Research and Quality; March 2012. Available at: www.effectivehealthcare.ahrq.gov/lbd.cfm. Accessed August 14, 2016.
30. Amgen. Prolia package insert. Available at: http://pi.amgen.com/united_states/prolia/prolia_pi.pdf. Accessed April 26, 2016.
31. Eli Lilly and Company. Fortio package insert. Available at: https://pi.lilly.com/us/forteo-pi.pdf. Accessed April 26, 2016.
PRACTICE RECOMMENDATIONS
› Perform laboratory testing for chronic kidney disease (CKD)-induced bone disease at CKD stage 3. B
› Avoid calcium-based phosphate binders in patients with known vascular calcifications. B
› Consider the use of phosphate binders in non-dialysis patients on a case-by-case basis, particularly in those with hyperphosphatemia not controlled by dietary measures. B
› Prescribe native vitamin D (ergocalciferol or cholecalciferol) to patients with CKD stages 3 to 4 who have secondary hyperparathyroidism and vitamin D deficiency. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 14% of the US general population has chronic kidney disease (CKD).1 Limited data exist regarding the exact prevalence of CKD-mineral and bone disorder (MBD), but abnormal mineral metabolism is believed to start in stage 3 CKD, implying that 8% of the adult US population could be at risk for, or already have established, CKD-MBD.2 Although the disorder has traditionally been managed by nephrologists, this earlier onset suggests that many patients should be screened and treated by their primary care physicians.
Because CKD-MBD can lead to significant morbidity (ie, increased fracture risk) and mortality, identification and treatment are of utmost importance.3 This review provides information from the current literature and the KDIGO (Kidney Disease: Improving Global Outcomes) guidelines, and focuses primarily on the non-dialysis CKD population.
CKD-MBD: A broad spectrum of disorders
CKD-MBD is defined as a systemic disorder of mineral and bone metabolism due to CKD. Traditionally referred to as renal osteodystrophy, the term CKD-MBD is meant to indicate and describe a broad clinical spectrum of CKD-associated bone mineral metabolism disorders that manifest from one or a combination of the following:
- Abnormalities of calcium, phosphorus, parathyroid hormone (PTH), or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.4
Renal bone disease can be divided into low bone turnover (adynamic bone disease) and high bone turnover states. Both can lead to a decrease in bone strength and an increase in pathological fractures.5
Pathophysiology: Difficult to know where the cascade begins
Understanding the pathophysiology and treatment of bone disease in patients with CKD can be challenging. Because of abnormalities of mineral metabolism and changes in hormones and cytokines, bone remodeling is severely disrupted in patients with CKD, and it remains unclear where this cascade begins.
As an adaptive response to decreased kidney function, PTH levels increase. Elevations of both fibroblast growth factor 23 (FGF23) lower blood phosphate levels by inhibiting phosphate reabsorption in the kidneys, thus increasing urinary excretion of phosphorus. Secondary hyperparathyroidism (SHPT), driven by hypocalcemia, responds to normalize serum calcium levels by increasing the number and size of osteoclasts actively breaking down bone matrix. This escalates fracture risk. In addition, the inability of damaged kidneys to convert vitamin D to an active form further deranges calcium and phosphate homeostasis.
Successful management of serum levels begins with monitoring
KDIGO, an independent nonprofit foundation that seeks to improve the care and outcomes of kidney disease patients worldwide, developed guidelines for the diagnosis, evaluation, prevention, and treatment of CKD-MBD in 2009.6 These guidelines recommend that treatment of CKD-MBD be aimed at managing serum phosphate, PTH, and calcium levels. The recommended frequency for laboratory monitoring of these levels varies by stage of CKD and is described in TABLE 1.6 (For more on chronic kidney disease staging, see KDIGO’s 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease, available at: http://www.kdigo.org/clinical_practice_guidelines/pdf/CKD/KDIGO_2012_CKD_GL.pdf.)
Because of the interrelated nature of these minerals and hormones, drug therapy aimed at treating one may impact the others. This must be considered when designing treatment regimens.
Hyperphosphatemia: Manage with diet, drugs, dialysis
Observational studies have shown an association between higher serum phosphate levels and mortality.6-8 KDIGO recommends maintaining serum phosphorus levels within the normal range of the assay in patients with CKD who are not receiving dialysis.6 For dialyzed patients, the recommendation is to lower the phosphorus level toward the normal range as much as possible.6 Maintaining an appropriate phosphorus level is accomplished through dietary phosphate restriction, the use of phosphate binders, and, in dialyzed patients, dialytic removal of phosphate.6
Dietary phosphate restriction is often challenging for patients, in part, because phosphorous content is not always included on food labels in the United States.9 Phosphorus is highly absorbed from additives in processed food (approaching 100% absorption), less absorbed from animal sources such as meat and dairy products (40%-60%), and is the least absorbed from plant sources such as beans and nuts (20%-40%).10 Advise patients to avoid fast food, processed foods, cheese, frozen meals, colas, and certain ready-to-eat cereals and prepared meats, as these products may have additives from which phosphorus is readily absorbed.11 A patient-friendly list of high-phosphorus foods, as well as other dietary advice and recipes, can be found on the National Kidney Foundation Web site at https://www.kidney.org/atoz/content/phosphorus. Tables listing the phosphorus content of common foods are also available in the literature and online.11,12 Keep in mind that not all resources take into account phosphate bioavailability. Dietician referral may be helpful to assure that patients maintain adequate protein intake while restricting dietary phosphate.
Phosphate binders are recommended by the KDIGO guidelines for use in patients with kidney disease and hyperphosphatemia.6 Most of the data to support the use of phosphate binders was gleaned from the dialysis population. The use of phosphate binders in non-dialyzed patients with CKD has both proponents and opponents, with literature supporting both positions.13,14 A recent KDIGO conference on controversies in CKD-MBD identified this as an area that should be evaluated further for the next guideline update.15
Phosphate binders—which bind the phosphorus in food to prevent absorption—should be taken with meals or high-phosphorus snacks. Products and formulations of commonly used phosphate binders are shown in TABLE 2.16,17 Taste, formulation, adverse effects, pill burden, and cost are issues to discuss with patients when initiating or adjusting phosphate binder therapy. It’s estimated that more than half of all patients receiving dialysis do not adhere to their prescribed phosphate binder regimen, highlighting the need to assess adherence before adjusting dose and to involve the patient in the decision-making process to select a phosphate binder product.18
Avoid calcium-based binders? The risk of hypercalcemia and the potentially increased risk of vascular calcifications with calcium-based binders have led some nephrologists to favor non-calcium-based products. Two recent meta-analyses found a reduced risk of all-cause mortality with the non-calcium-based binders sevelamer or lanthanum as compared to calcium-based binders.19,20 Current KDIGO guidelines were published prior to these meta-analyses and do not recommend one phosphate binder over another. They do, however, recommend restricting the dose of calcium-based binders in the setting of persistent or recurrent hypercalcemia, known vascular calcification, or low PTH levels.6
Secondary hyperparathyroidism
Due to a lack of data, the goal PTH level in patients not receiving dialysis is unknown.6 A reasonable approach in non-dialyzed patients, however, is to correct 25-OH vitamin D (25[OH]D) deficiency, elevations in serum phosphate, and hypocalcemia when the level of intact PTH (iPTH) exceeds the normal range for the assay because correcting these derangements may result in a decline in iPTH.6,21 If this approach fails and PTH levels continue to rise, use of calcitriol or vitamin D analogues is recommended.6 Characteristics of medications used to treat SHPT are presented in TABLE 3.16,17
In dialysis patients, the target iPTH range suggested by KDIGO is 2 to 9 times the upper limit of normal for the assay.6 Elevated PTH levels in the dialysis population may be managed with activated vitamin D and/or cinacalcet.
Native vitamin D (ergocalciferol, cholecalciferol) and activated vitamin D analogs (calcitriol, doxercalciferol, paricalcitol). Native vitamin D products are recommended for non-dialyzed patients with CKD to correct vitamin D deficiencies. Although many approaches may be used clinically to replenish low vitamin D stores, one reasonable recommendation in patients with a 25(OH)D level <30 ng/mL is to prescribe ergocalciferol 50,000 units/week for 8 weeks and then to repeat the serum 25-OH vitamin D test. If the level is still <30 ng/mL, a second 8-week course of weekly ergocalciferol 50,000 IU may be administered.21
Following repletion with ergocalciferol, maintenance doses of cholecalciferol (1000-2000 IU/d) or ergocalciferol (50,000 IU/-month) may be initiated.21 Discontinue native vitamin D in patients who develop hypercalcemia.
Native vitamin D becomes less effective at reducing PTH levels as kidney disease advances. This is likely due to a decline in renal conversion of 25(OH)D to 1,25-(OH)2vitamin D (1,25[OH]2D), the most active form of vitamin D and the form of vitamin D that decreases PTH production. By stage 5 CKD, it is unlikely that native vitamin D will significantly decrease PTH levels; treatment with activated vitamin D products or cinacalcet is generally required.
Because the enzyme responsible for converting 25(OH)D into the most active form can be found in multiple tissues outside of the kidney, and the 1,25(OH)2D converted for use by these organs may help prevent such conditions/events as hypertension, type 2 diabetes, myocardial infarction, and stroke (in patients with and without kidney disease), some specialists prescribe native vitamin D to patients with CKD for reasons unrelated to PTH suppression. There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.21
Don’t forget calcium
All of the active vitamin D products can increase serum calcium and phosphate levels. Calcitriol, however, may cause more hypercalcemia than paricalcitol.22 If hypercalcemia develops, you may need to stop, or reduce the dose of, vitamin D analogues. Or you may need to switch patients from calcium-based to non-calcium-based phosphate binders. If hyperphosphatemia develops, intensify phosphate binder therapy or reduce the dose of, or stop, vitamin D analogues. If iPTH levels go below the target range, reduce the dose of the vitamin D analogue to avoid iatrogenic adynamic bone disease.
Avoid this agent in the non-dialyzed patient. Cinacalcet effectively treats SHPT in patients receiving dialysis, but is not recommended for use in undialyzed patients.23 That’s because unacceptably high rates of hypocalcemia have been observed in non-dialyzed patients who were taking the drug.23,24 In addition, while cinacalcet neutrally affects, or causes a slight decrease in, serum phosphate in patients receiving dialysis, it increases serum phosphate in patients who are not.24,25
Drug therapy for osteoporosis
Therapy to prevent and treat fractures in patients with CKD is controversial because patients with CKD stage 3 to 5 with and without MBD were excluded from clinical trials of commercially available treatments. Furthermore, in adynamic bone disease, bones are capable of neither breaking down nor building (ie, reduced resorption). Bisphosphonates and other antiresorptive therapies are more effective at decreasing fractures in patients who are in a state of increased bone resorption, such as menopausal women, so the benefits of these medications in terms of their ability to reduce fractures in CKD patients are questionable, as is their safety.26,27
In addition, while dual-energy x-ray absorptiometry (DXA) is typically used to identify patients who would benefit from these agents, studies have recently demonstrated that femoral neck bone density measured via DXA may underestimate fracture risk in patients with CKD-MBD (ie, bone density may actually be lower than measured).26,28
Antiresorptive agents and teriparatide
Osteoporosis treatments include antiresorptive agents (ie, the bisphosphonates, raloxifene, denosumab), and the anabolic bone agent teriparatide.
Evidence supports treating patients with stage 1 to 3 CKD the same as patients without CKD.15 Bisphosphonates are labeled as contraindicated in patients with a glomerular filtration rate (GFR) <30 mL/min/1.73m2, due to concerns arising from animal trials and subsequent human case reports (both with intravenous formulations only) regarding acute kidney injury.27
While raloxifene lacks a warning regarding use in patients with stage 3 to 5 CKD, it has not been shown to prevent hip fractures in any population.29
Denosumab is not contraindicated for use in patients with CKD stage 3 to 5 without MBD, but it can worsen hypocalcemia, particularly in patients receiving dialysis.30
Teriparatide is contraindicated in patients with CKD and SHPT,31 and there are no studies of its use in patients with CKD-MBD.
What the guidelines say about antiresorptive treatment
For patients with stage 3 to 5 CKD with manifestations of MBD, 2009 KDIGO guidelines recommend a bone biopsy to evaluate for adynamic bone disease before initiating antiresorptive treatment.6 Because few physicians in most communities are trained to conduct and evaluate bone biopsies, this recommendation is infrequently followed. Without a bone biopsy to rule out adynamic bone disease, options to prevent or treat fractures in the setting of CKD-MBD are limited.
CORRESPONDENCE
Karly Pippitt, MD, Department of Family and Preventive Medicine, University of Utah School of Medicine, 375 Chipeta Way, Suite A, Salt Lake City, UT 84108; [email protected].
PRACTICE RECOMMENDATIONS
› Perform laboratory testing for chronic kidney disease (CKD)-induced bone disease at CKD stage 3. B
› Avoid calcium-based phosphate binders in patients with known vascular calcifications. B
› Consider the use of phosphate binders in non-dialysis patients on a case-by-case basis, particularly in those with hyperphosphatemia not controlled by dietary measures. B
› Prescribe native vitamin D (ergocalciferol or cholecalciferol) to patients with CKD stages 3 to 4 who have secondary hyperparathyroidism and vitamin D deficiency. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 14% of the US general population has chronic kidney disease (CKD).1 Limited data exist regarding the exact prevalence of CKD-mineral and bone disorder (MBD), but abnormal mineral metabolism is believed to start in stage 3 CKD, implying that 8% of the adult US population could be at risk for, or already have established, CKD-MBD.2 Although the disorder has traditionally been managed by nephrologists, this earlier onset suggests that many patients should be screened and treated by their primary care physicians.
Because CKD-MBD can lead to significant morbidity (ie, increased fracture risk) and mortality, identification and treatment are of utmost importance.3 This review provides information from the current literature and the KDIGO (Kidney Disease: Improving Global Outcomes) guidelines, and focuses primarily on the non-dialysis CKD population.
CKD-MBD: A broad spectrum of disorders
CKD-MBD is defined as a systemic disorder of mineral and bone metabolism due to CKD. Traditionally referred to as renal osteodystrophy, the term CKD-MBD is meant to indicate and describe a broad clinical spectrum of CKD-associated bone mineral metabolism disorders that manifest from one or a combination of the following:
- Abnormalities of calcium, phosphorus, parathyroid hormone (PTH), or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.4
Renal bone disease can be divided into low bone turnover (adynamic bone disease) and high bone turnover states. Both can lead to a decrease in bone strength and an increase in pathological fractures.5
Pathophysiology: Difficult to know where the cascade begins
Understanding the pathophysiology and treatment of bone disease in patients with CKD can be challenging. Because of abnormalities of mineral metabolism and changes in hormones and cytokines, bone remodeling is severely disrupted in patients with CKD, and it remains unclear where this cascade begins.
As an adaptive response to decreased kidney function, PTH levels increase. Elevations of both fibroblast growth factor 23 (FGF23) lower blood phosphate levels by inhibiting phosphate reabsorption in the kidneys, thus increasing urinary excretion of phosphorus. Secondary hyperparathyroidism (SHPT), driven by hypocalcemia, responds to normalize serum calcium levels by increasing the number and size of osteoclasts actively breaking down bone matrix. This escalates fracture risk. In addition, the inability of damaged kidneys to convert vitamin D to an active form further deranges calcium and phosphate homeostasis.
Successful management of serum levels begins with monitoring
KDIGO, an independent nonprofit foundation that seeks to improve the care and outcomes of kidney disease patients worldwide, developed guidelines for the diagnosis, evaluation, prevention, and treatment of CKD-MBD in 2009.6 These guidelines recommend that treatment of CKD-MBD be aimed at managing serum phosphate, PTH, and calcium levels. The recommended frequency for laboratory monitoring of these levels varies by stage of CKD and is described in TABLE 1.6 (For more on chronic kidney disease staging, see KDIGO’s 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease, available at: http://www.kdigo.org/clinical_practice_guidelines/pdf/CKD/KDIGO_2012_CKD_GL.pdf.)
Because of the interrelated nature of these minerals and hormones, drug therapy aimed at treating one may impact the others. This must be considered when designing treatment regimens.
Hyperphosphatemia: Manage with diet, drugs, dialysis
Observational studies have shown an association between higher serum phosphate levels and mortality.6-8 KDIGO recommends maintaining serum phosphorus levels within the normal range of the assay in patients with CKD who are not receiving dialysis.6 For dialyzed patients, the recommendation is to lower the phosphorus level toward the normal range as much as possible.6 Maintaining an appropriate phosphorus level is accomplished through dietary phosphate restriction, the use of phosphate binders, and, in dialyzed patients, dialytic removal of phosphate.6
Dietary phosphate restriction is often challenging for patients, in part, because phosphorous content is not always included on food labels in the United States.9 Phosphorus is highly absorbed from additives in processed food (approaching 100% absorption), less absorbed from animal sources such as meat and dairy products (40%-60%), and is the least absorbed from plant sources such as beans and nuts (20%-40%).10 Advise patients to avoid fast food, processed foods, cheese, frozen meals, colas, and certain ready-to-eat cereals and prepared meats, as these products may have additives from which phosphorus is readily absorbed.11 A patient-friendly list of high-phosphorus foods, as well as other dietary advice and recipes, can be found on the National Kidney Foundation Web site at https://www.kidney.org/atoz/content/phosphorus. Tables listing the phosphorus content of common foods are also available in the literature and online.11,12 Keep in mind that not all resources take into account phosphate bioavailability. Dietician referral may be helpful to assure that patients maintain adequate protein intake while restricting dietary phosphate.
Phosphate binders are recommended by the KDIGO guidelines for use in patients with kidney disease and hyperphosphatemia.6 Most of the data to support the use of phosphate binders was gleaned from the dialysis population. The use of phosphate binders in non-dialyzed patients with CKD has both proponents and opponents, with literature supporting both positions.13,14 A recent KDIGO conference on controversies in CKD-MBD identified this as an area that should be evaluated further for the next guideline update.15
Phosphate binders—which bind the phosphorus in food to prevent absorption—should be taken with meals or high-phosphorus snacks. Products and formulations of commonly used phosphate binders are shown in TABLE 2.16,17 Taste, formulation, adverse effects, pill burden, and cost are issues to discuss with patients when initiating or adjusting phosphate binder therapy. It’s estimated that more than half of all patients receiving dialysis do not adhere to their prescribed phosphate binder regimen, highlighting the need to assess adherence before adjusting dose and to involve the patient in the decision-making process to select a phosphate binder product.18
Avoid calcium-based binders? The risk of hypercalcemia and the potentially increased risk of vascular calcifications with calcium-based binders have led some nephrologists to favor non-calcium-based products. Two recent meta-analyses found a reduced risk of all-cause mortality with the non-calcium-based binders sevelamer or lanthanum as compared to calcium-based binders.19,20 Current KDIGO guidelines were published prior to these meta-analyses and do not recommend one phosphate binder over another. They do, however, recommend restricting the dose of calcium-based binders in the setting of persistent or recurrent hypercalcemia, known vascular calcification, or low PTH levels.6
Secondary hyperparathyroidism
Due to a lack of data, the goal PTH level in patients not receiving dialysis is unknown.6 A reasonable approach in non-dialyzed patients, however, is to correct 25-OH vitamin D (25[OH]D) deficiency, elevations in serum phosphate, and hypocalcemia when the level of intact PTH (iPTH) exceeds the normal range for the assay because correcting these derangements may result in a decline in iPTH.6,21 If this approach fails and PTH levels continue to rise, use of calcitriol or vitamin D analogues is recommended.6 Characteristics of medications used to treat SHPT are presented in TABLE 3.16,17
In dialysis patients, the target iPTH range suggested by KDIGO is 2 to 9 times the upper limit of normal for the assay.6 Elevated PTH levels in the dialysis population may be managed with activated vitamin D and/or cinacalcet.
Native vitamin D (ergocalciferol, cholecalciferol) and activated vitamin D analogs (calcitriol, doxercalciferol, paricalcitol). Native vitamin D products are recommended for non-dialyzed patients with CKD to correct vitamin D deficiencies. Although many approaches may be used clinically to replenish low vitamin D stores, one reasonable recommendation in patients with a 25(OH)D level <30 ng/mL is to prescribe ergocalciferol 50,000 units/week for 8 weeks and then to repeat the serum 25-OH vitamin D test. If the level is still <30 ng/mL, a second 8-week course of weekly ergocalciferol 50,000 IU may be administered.21
Following repletion with ergocalciferol, maintenance doses of cholecalciferol (1000-2000 IU/d) or ergocalciferol (50,000 IU/-month) may be initiated.21 Discontinue native vitamin D in patients who develop hypercalcemia.
Native vitamin D becomes less effective at reducing PTH levels as kidney disease advances. This is likely due to a decline in renal conversion of 25(OH)D to 1,25-(OH)2vitamin D (1,25[OH]2D), the most active form of vitamin D and the form of vitamin D that decreases PTH production. By stage 5 CKD, it is unlikely that native vitamin D will significantly decrease PTH levels; treatment with activated vitamin D products or cinacalcet is generally required.
Because the enzyme responsible for converting 25(OH)D into the most active form can be found in multiple tissues outside of the kidney, and the 1,25(OH)2D converted for use by these organs may help prevent such conditions/events as hypertension, type 2 diabetes, myocardial infarction, and stroke (in patients with and without kidney disease), some specialists prescribe native vitamin D to patients with CKD for reasons unrelated to PTH suppression. There are no data, however, confirming that 25(OH)D supplementation mitigates these outcomes.21
Don’t forget calcium
All of the active vitamin D products can increase serum calcium and phosphate levels. Calcitriol, however, may cause more hypercalcemia than paricalcitol.22 If hypercalcemia develops, you may need to stop, or reduce the dose of, vitamin D analogues. Or you may need to switch patients from calcium-based to non-calcium-based phosphate binders. If hyperphosphatemia develops, intensify phosphate binder therapy or reduce the dose of, or stop, vitamin D analogues. If iPTH levels go below the target range, reduce the dose of the vitamin D analogue to avoid iatrogenic adynamic bone disease.
Avoid this agent in the non-dialyzed patient. Cinacalcet effectively treats SHPT in patients receiving dialysis, but is not recommended for use in undialyzed patients.23 That’s because unacceptably high rates of hypocalcemia have been observed in non-dialyzed patients who were taking the drug.23,24 In addition, while cinacalcet neutrally affects, or causes a slight decrease in, serum phosphate in patients receiving dialysis, it increases serum phosphate in patients who are not.24,25
Drug therapy for osteoporosis
Therapy to prevent and treat fractures in patients with CKD is controversial because patients with CKD stage 3 to 5 with and without MBD were excluded from clinical trials of commercially available treatments. Furthermore, in adynamic bone disease, bones are capable of neither breaking down nor building (ie, reduced resorption). Bisphosphonates and other antiresorptive therapies are more effective at decreasing fractures in patients who are in a state of increased bone resorption, such as menopausal women, so the benefits of these medications in terms of their ability to reduce fractures in CKD patients are questionable, as is their safety.26,27
In addition, while dual-energy x-ray absorptiometry (DXA) is typically used to identify patients who would benefit from these agents, studies have recently demonstrated that femoral neck bone density measured via DXA may underestimate fracture risk in patients with CKD-MBD (ie, bone density may actually be lower than measured).26,28
Antiresorptive agents and teriparatide
Osteoporosis treatments include antiresorptive agents (ie, the bisphosphonates, raloxifene, denosumab), and the anabolic bone agent teriparatide.
Evidence supports treating patients with stage 1 to 3 CKD the same as patients without CKD.15 Bisphosphonates are labeled as contraindicated in patients with a glomerular filtration rate (GFR) <30 mL/min/1.73m2, due to concerns arising from animal trials and subsequent human case reports (both with intravenous formulations only) regarding acute kidney injury.27
While raloxifene lacks a warning regarding use in patients with stage 3 to 5 CKD, it has not been shown to prevent hip fractures in any population.29
Denosumab is not contraindicated for use in patients with CKD stage 3 to 5 without MBD, but it can worsen hypocalcemia, particularly in patients receiving dialysis.30
Teriparatide is contraindicated in patients with CKD and SHPT,31 and there are no studies of its use in patients with CKD-MBD.
What the guidelines say about antiresorptive treatment
For patients with stage 3 to 5 CKD with manifestations of MBD, 2009 KDIGO guidelines recommend a bone biopsy to evaluate for adynamic bone disease before initiating antiresorptive treatment.6 Because few physicians in most communities are trained to conduct and evaluate bone biopsies, this recommendation is infrequently followed. Without a bone biopsy to rule out adynamic bone disease, options to prevent or treat fractures in the setting of CKD-MBD are limited.
CORRESPONDENCE
Karly Pippitt, MD, Department of Family and Preventive Medicine, University of Utah School of Medicine, 375 Chipeta Way, Suite A, Salt Lake City, UT 84108; [email protected].
1. United States Renal Data System. Chapter 1: CKD in the general population. Available at: https://www.usrds.org/2015/download/vol1_01_General_Pop_15.pdf. Accessed July 27, 2016.
2. Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038-2047.
3. Uhlig K, Berns JS, Kestenbaum B, et al. KDOQI US commentary on the 2009 KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, and Treatment of CKD-Mineral and Bone Disorder (CKD-MBD). Am J Kidney Dis. 2010;55:773-799.
4. Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol. 2007;18:875-885.
5. Roberts DM, Singer RF. Management of renal bone disease. Aust Prescr. 2010;33:34-37.
6. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009:S1-130.
7. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305:1119-1127.
8. Cannata-Andia JB, Martin KJ. The challenge of controlling phosphorus in chronic kidney disease. Nephrol Dial Transplant. 2016;31:541-547.
9. US Food and Drug Administration. Food Labeling Guide. Available at: http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/LabelingNutrition/ucm2006828.htm. Accessed July 25, 2016.
10. Kalantar-Zadeh K. Patient education for phosphorus management in chronic kidney disease. Patient Prefer Adherence. 2013;7:379-390.
11. Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:519-530.
12. USDA National Nutrient Database for Standard Reference. 2015; Available at: https://ndb.nal.usda.gov. Accessed April 25, 2016.
13. Bellasi A. Pro: Should phosphate binders be used in chronic kidney disease stage 3-4? Nephrol Dial Transplant. 2016;31:184-188.
14. Kestenbaum B. Con: Phosphate binders in chronic kidney disease. Nephrol Dial Transplant. 2016;31:189-194.
15. Ketteler M, Elder GJ, Evenepoel P, et al. Revisiting KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: a commentary from a Kidney Disease: Improving Global Outcomes controversies conference. Kidney Int. 2015;87:502-528.
16. Wolters Kluwer. Lexicomp. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed April 26, 2016.
17. Truven Health Analytics. Micromedex Solutions. Available at: http://micromedex.com/. Accessed April 26, 2016.
18. Wang S, Anum EA, Ramakrishnan K, et al. Reasons for phosphate binder discontinuation vary by binder type. J Ren Nutr. 2014;24:105-109.
19. Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016;11:232-244.
20. Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382:1268-1277.
21. Nigwekar SU, Bhan I, Thadhani R. Ergocalciferol and cholecalciferol in CKD. Am J Kidney Dis. 2012;60:139-156.
22. Teng M, Wolf M, Lowrie E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446-456.
23. Sensipar package insert. Thousand Oaks, California: Amgen Pharmaceuticals; 2014. Available at: http://pi.amgen.com/united_states/sensipar/sensipar_pi_hcp_english.pdf. Accessed April 25, 2016.
24. Chonchol M, Locatelli F, Abboud HE, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53:197-207.
25. Ballinger AE, Palmer SC, Nistor I,et al. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst Rev. 2014;12:CD006254.
26. Miller PD. Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis. 2014;64:290-304.
27. Ott SM. Bisphosphonate safety and efficacy in chronic kidney disease. Kidney Int. 2012;82:833-835.
28. Yencheck RH, Ix JH, Shlipak MG, et al. Bone mineral density and fracture risk in older individuals with CKD. Clin J Am Soc Nephrol. 2012;7:1130-1136.
29. Crandall CJ, Newberry SJ, Diamant A, et al. Treatments to prevent fractures in men and women with low bone density or osteoporosis: update of a 2007 report. Comparative Effectiveness Reviews, No. 53. Rockville, MD: Agency for Healthcare Research and Quality; March 2012. Available at: www.effectivehealthcare.ahrq.gov/lbd.cfm. Accessed August 14, 2016.
30. Amgen. Prolia package insert. Available at: http://pi.amgen.com/united_states/prolia/prolia_pi.pdf. Accessed April 26, 2016.
31. Eli Lilly and Company. Fortio package insert. Available at: https://pi.lilly.com/us/forteo-pi.pdf. Accessed April 26, 2016.
1. United States Renal Data System. Chapter 1: CKD in the general population. Available at: https://www.usrds.org/2015/download/vol1_01_General_Pop_15.pdf. Accessed July 27, 2016.
2. Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038-2047.
3. Uhlig K, Berns JS, Kestenbaum B, et al. KDOQI US commentary on the 2009 KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, and Treatment of CKD-Mineral and Bone Disorder (CKD-MBD). Am J Kidney Dis. 2010;55:773-799.
4. Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol. 2007;18:875-885.
5. Roberts DM, Singer RF. Management of renal bone disease. Aust Prescr. 2010;33:34-37.
6. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009:S1-130.
7. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305:1119-1127.
8. Cannata-Andia JB, Martin KJ. The challenge of controlling phosphorus in chronic kidney disease. Nephrol Dial Transplant. 2016;31:541-547.
9. US Food and Drug Administration. Food Labeling Guide. Available at: http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/LabelingNutrition/ucm2006828.htm. Accessed July 25, 2016.
10. Kalantar-Zadeh K. Patient education for phosphorus management in chronic kidney disease. Patient Prefer Adherence. 2013;7:379-390.
11. Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:519-530.
12. USDA National Nutrient Database for Standard Reference. 2015; Available at: https://ndb.nal.usda.gov. Accessed April 25, 2016.
13. Bellasi A. Pro: Should phosphate binders be used in chronic kidney disease stage 3-4? Nephrol Dial Transplant. 2016;31:184-188.
14. Kestenbaum B. Con: Phosphate binders in chronic kidney disease. Nephrol Dial Transplant. 2016;31:189-194.
15. Ketteler M, Elder GJ, Evenepoel P, et al. Revisiting KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: a commentary from a Kidney Disease: Improving Global Outcomes controversies conference. Kidney Int. 2015;87:502-528.
16. Wolters Kluwer. Lexicomp. Clinical Drug Information. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed April 26, 2016.
17. Truven Health Analytics. Micromedex Solutions. Available at: http://micromedex.com/. Accessed April 26, 2016.
18. Wang S, Anum EA, Ramakrishnan K, et al. Reasons for phosphate binder discontinuation vary by binder type. J Ren Nutr. 2014;24:105-109.
19. Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016;11:232-244.
20. Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382:1268-1277.
21. Nigwekar SU, Bhan I, Thadhani R. Ergocalciferol and cholecalciferol in CKD. Am J Kidney Dis. 2012;60:139-156.
22. Teng M, Wolf M, Lowrie E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446-456.
23. Sensipar package insert. Thousand Oaks, California: Amgen Pharmaceuticals; 2014. Available at: http://pi.amgen.com/united_states/sensipar/sensipar_pi_hcp_english.pdf. Accessed April 25, 2016.
24. Chonchol M, Locatelli F, Abboud HE, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53:197-207.
25. Ballinger AE, Palmer SC, Nistor I,et al. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst Rev. 2014;12:CD006254.
26. Miller PD. Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis. 2014;64:290-304.
27. Ott SM. Bisphosphonate safety and efficacy in chronic kidney disease. Kidney Int. 2012;82:833-835.
28. Yencheck RH, Ix JH, Shlipak MG, et al. Bone mineral density and fracture risk in older individuals with CKD. Clin J Am Soc Nephrol. 2012;7:1130-1136.
29. Crandall CJ, Newberry SJ, Diamant A, et al. Treatments to prevent fractures in men and women with low bone density or osteoporosis: update of a 2007 report. Comparative Effectiveness Reviews, No. 53. Rockville, MD: Agency for Healthcare Research and Quality; March 2012. Available at: www.effectivehealthcare.ahrq.gov/lbd.cfm. Accessed August 14, 2016.
30. Amgen. Prolia package insert. Available at: http://pi.amgen.com/united_states/prolia/prolia_pi.pdf. Accessed April 26, 2016.
31. Eli Lilly and Company. Fortio package insert. Available at: https://pi.lilly.com/us/forteo-pi.pdf. Accessed April 26, 2016.
From The Journal of Family Practice | 2016;65(9):606-608,610-612

Is an SGLT2 inhibitor right for your patient with type 2 diabetes?
› Consider sodium-glucose cotransporter 2 (SGLT2) inhibitors as second-line agents in patients with type 2 diabetes mellitus who need mild hemoglobin A1c reductions (≤1%) and who would benefit from mild to modest weight and blood pressure reductions. A
› Avoid using SGLT2 inhibitors in patients with a history of recurrent genital mycotic or urinary tract infections. B
› Use SGLT2 inhibitors with caution in patients at risk for volume-related adverse effects (dizziness and hypotension), such as the elderly, those with moderate renal dysfunction, and those taking concomitant diuretic therapy. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Joe S is a 41-year-old African-American man who comes to your clinic after his employee health screening revealed elevated triglycerides. The patient has a 3-year history of type 2 diabetes mellitus (T2DM); he also has a history of hypertension, gastroesophageal reflux disease, and obstructive sleep apnea. Mr. S tells you he takes metformin 1000 mg twice daily, but stopped taking his glipizide because he didn’t think it was helping his blood sugar. His last hemoglobin (Hb) A1c result was 8.8%, and he is very resistant to starting insulin therapy.
The patient’s other medications include enalapril 10 mg/d, atorvastatin 10 mg/d, and omeprazole 20 mg/d. Mr. S weighs 255.6 lbs (body mass index=34.7), his BP is 140/88 mm Hg, and his heart rate is 82 beats per minute. Laboratory values include: serum creatinine, 1.01 mg/dL; estimated glomerular filtration rate (eGFR) >100 mL/min/1.73 m2; potassium (K), 4.3 mmol/L; serum phosphorous (Phos), 2.8 mg/dL; magnesium (Mg), 1.9 mg/dL; total cholesterol, 167 mg/dL; low-density lipoprotein (LDL), 78 mg/dL; high-density lipoprotein (HDL), 38 mg/dL; and triglycerides, 256 mg/dL.
CASE 2 › Susan R, a 68-year-old Caucasian woman, returns to your clinic for a follow-up visit 3 months after you prescribed dapagliflozin 10 mg/d for her T2DM. Her glucose levels have improved, but she complains of vaginal pruritus and is worried that she has a yeast infection.
You diagnose vulvovaginal candidiasis in this patient and prescribe a single dose of fluconazole 150 mg. After reviewing her laboratory test results, you notice that since starting the dapagliflozin, her HbA1c level has improved slightly from 9.8% to 9.3%, but is still not where it needs to be. Her eGFR is 49 mL/min/1.73 m2.
What would you recommend to improve control of these patients’ blood glucose levels?
When to consider an SGLT2 inhibitor
Consider therapy with SGLT2 inhibitors in adult patients with T2DM who:3-9,13-15,17-24
- have an HbA1c between 7% and 9%
- would benefit from weight and/or blood pressure reductions
- have metabolic syndrome
- have adequate means to pay for the medication (ie, prescription coverage or the ability to afford it).
In addition, consider an SGLT2 inhibitor as initial monotherapy if metformin is contraindicated or not tolerated, or as add-on therapy to metformin, sulfonylureas, thiazolidinediones, dipeptidyl peptidase IV inhibitors, or insulin.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are the newest class of agents to enter the T2DM management arena. They act in the proximal renal tubules to decrease the reabsorption of glucose by targeting the SGLT2 transmembrane protein, which reabsorbs about 90% of the body’s glucose.1,2 The class is currently made up of 3 agents—canagliflozin, dapagliflozin, and empagliflozin—all of which are approved by the US Food and Drug Administration (FDA) for the treatment of T2DM (TABLE 1).2
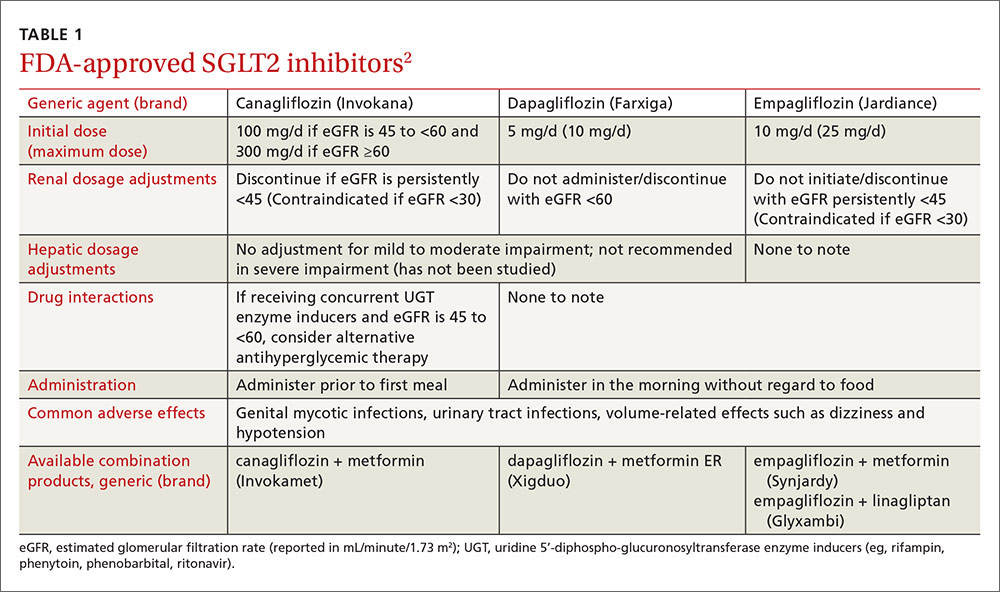
The American Diabetes Association and the European Association for the Study of Diabetes published updated guidelines for T2DM management in 2015.1 In addition to lifestyle modifications, the guidelines recommend the use of metformin as first-line therapy unless it is contraindicated or patients are unable to tolerate it (eg, because of gastrointestinal adverse effects). They recommend other pharmacologic therapies as second-line options based on specific patient characteristics. Thus, SGLT2 inhibitors may be used as add-on therapy after metformin, or as a first-line option if metformin is contraindicated or not tolerated. Because the mechanism of action of SGLT2 inhibitors is independent of insulin secretion, these agents may be used at any stage of the diabetes continuum.
SGLT2 agents as monotherapy, or as add-on therapy
All SGLT2 agents have been studied as monotherapy accompanied by diet and exercise and shown to produce HbA1c reductions of 0.34% to 1.11%.3-6 In trials, the effect was similar regardless of study duration (18-104 weeks); generally, higher doses corresponded with larger HbA1c reductions.3-6
SGLT2 inhibitors have also been studied as add-on therapy to several oral agents including metformin, sulfonylureas, thiazolidinediones (TZDs), and the combination of metformin plus sulfonylureas or TZDs or dipeptidyl peptidase IV (DPP-IV) inhibitors.1 When used in any of these combinations, each SGLT2 agent demonstrated a consistent HbA1c lowering effect of 0.62% to 1.19%.7-14
Additionally, SGLT2 inhibitors have been studied in combination with insulin therapy (median or mean daily doses >60 units), which yielded further reductions in HbA1c of 0.58% to 1.02% without significant insulin adjustments or an increase in major hypoglycemia events.15-17 Patients receiving insulin and an SGLT2 inhibitor had lower insulin doses and more weight loss compared to placebo groups.
SGLT2 inhibitors offer additional benefits
Secondary analyses of most studies of SGLT2 inhibitors include changes in BP and weight from baseline as well as minor changes (some positive, some not) in several lipid parameters.3-5,7-9,13-15,17-24 In general, these effects do not appear to be dose-dependent (with the exception of canagliflozin and its associated lipid effects25) and are similar among the 3 medications.3-5,7-9,13-15,17-24 (For more on who would benefit from these agents, see “When to consider an SGLT2 inhibitor” above.)
BP reduction. Although the mean baseline BP was controlled in most studies, SGLT2 inhibitors have been shown to significantly reduce BP. Reductions in BP with all 3 SGLT2 medications range from approximately 2 to 5 mm Hg systolic and 0.5 to 2.5 mm Hg diastolic, which may be due to weight loss and diuresis.4-8,10-16,20-23 While the reductions were modest at best, one study involving empagliflozin reported that more than one-quarter of patients with uncontrolled BP at baseline achieved a BP <130/80 mm Hg 24 weeks later.5 While these agents should not be used solely for their BP lowering effects, they may help a small number of patients with mildly elevated BP achieve their goal without an additional antihypertensive agent.
Weight reduction. Modest weight loss, likely due to the loss of calories through urine, was seen with SGLT2 inhibitors in most studies, with reductions persisting beyond one year of use. In most studies, including those involving obese patients on insulin therapy,15,17,21 patients’ body weights were reduced by approximately 2 to 4 kg from baseline.3-16,18,21-23,26
Lipid effects. Although the mechanism is unclear, use of SGLT2 inhibitors can have varying effects on lipid panels. In most studies, total and LDL cholesterol levels were increased with elevations ranging from 0.7 to 10 mg/dL.3,7,8,18,19,22,23 Conversely, at least one study demonstrated mild reductions in total and LDL cholesterol levels with higher doses of empagliflozin.13 Additionally, modest reductions in triglycerides and increases in HDL across all doses of canagliflozin, dapagliflozin, and empagliflozin have been seen.8,9,13,15,19 While the clinical relevance of these lipid changes is unknown, monitoring is recommended.2
These agents are well tolerated
SGLT2 inhibitors were generally well tolerated in studies. The most common adverse effects include mycotic infections (2.4%-21.6%) and urinary tract infections (UTIs) (4.0%-19.6%) (both with higher incidences in females); volume-related effects such as dizziness and hypotension (0.3%-8.3%); and nasopharyngitis (5.4%-18.3%).4-14,16-23,26-28 Hypoglycemia was observed more often when an SGLT2 inhibitor was used in combination with a sulfonylurea or insulin therapy.4-14,16-23,26-28 The number of times adverse events led to discontinuation was low and similar to that in control groups.4-14,16-23,26-28
Mycotic and urinary infections should be diagnosed and treated according to current standards of care and do not require discontinuation of the SGLT2 inhibitor. Canagli-flozin therapy was associated with electrolyte abnormalities including hyperkalemia, hypermagnesemia, and hyperphosphatemia.25 Thus, levels should be monitored periodically, especially in patients predisposed to elevations due to other conditions or medications.25
Two additional warnings are worth noting
Diabetic ketoacidosis (DKA) has been reported with all 3 agents, and bone fractures have been reported with canagliflozin.
The FDA issued a warning in May 2015 regarding the increased risk of DKA with the use of SGLT2 inhibitor single and combination products.29 This warning was prompted by several case reports of DKA with uncharacteristically mild to moderate glucose elevations in patients with type 1 diabetes mellitus (T1DM) and T2DM who were taking an SGLT2 inhibitor. The absence of significant hyperglycemia delayed diagnosis in many cases. Therefore, patients should be counseled on the signs and symptoms of DKA, as well as when to seek medical attention.
Patients with diabetes and symptoms of ketoacidosis (eg, difficulty breathing, nausea, vomiting, abdominal pain, confusion, and fatigue) should be evaluated regardless of current blood glucose levels, and SGLT2 inhibitors should be discontinued if acidosis is confirmed. Identified potential triggers include illness, reduced food and fluid intake, reduced insulin dose, and history of alcohol intake. Use of SGLT2 inhibitors should be avoided in patients with T1DM until safety and efficacy are established in large randomized controlled trials. The European Medicines Agency announced that a thorough review of all currently approved SGLT2 agents is underway to evaluate the risk for DKA.30
In addition, the FDA called for a revision of the label of canagliflozin to reflect a strengthened warning about an increased risk of bone fractures and decreased bone mineral density (BMD).31 Fractures can occur as early as 12 weeks after initiating treatment and with only minor trauma.31
Over a 2-year period, canagliflozin also significantly decreased BMD in the hip and lower spine compared to placebo.31 Patients should be evaluated for additional risk factors for fracture before taking canagliflozin.31 The FDA is continuing to evaluate whether the other approved SGLT2 inhibitors are associated with an increased risk for fractures.
Drug interactions: Proceed carefully with diuretics
The number of drugs that interact with SGLT2 inhibitors is minimal. Because these agents can cause volume-related effects such as hypotension, dizziness, and osmotic diuresis, patients—particularly the elderly and those with renal impairment—taking concomitant diuretics, especially loop diuretics, may be at increased risk for these effects and should be monitored accordingly.2,25
Canagliflozin is primarily metabolized via glucuronidation by the uridine 5'-diphospho-glucuronosyltransferase (UGT) enzymes. Therefore, UGT enzyme inducers (eg, rifampin, phenytoin, phenobarbital, ritonavir) decrease canagliflozin’s serum concentration. If a patient has an eGFR >60 mL/min/1.73 m2 and is tolerating a dose of 100 mg/d, consider increasing the dose to 300 mg/d during concomitant treatment.
In addition, researchers have found that canagliflozin increases serum levels of digoxin by between 20% and 36%.25 Experts suspect this occurs because canagliflozin inhibits P-glycoprotein efflux of digoxin. Although monitoring of digoxin levels is recommended, this interaction is considered to be minor.25
Cost consideration: SGLT2 inhibitors are more expensive
The SGLT2 inhibitors are available only as brand name products and are more expensive than agents that have generic options (eg, metformin, sulfonylureas, TZDs). The average wholesale cost is approximately $400 for a 30-day supply of all SGLT2 agents.32 When considering an SGLT2 inhibitor, the patient should ideally have medication prescription coverage. Depending on the specific insurance plan, these agents are classified as tier 2 to 4, which is comparable to other oral brand name options.
Research looks at CV outcomes and cancer risk
Cardiovascular (CV) risk reduction. To date, only one study evaluating the effect of SGLT2 inhibitors on CV outcomes is complete.33 Two large randomized controlled trials involving canagliflozin and dapagliflozin designed to evaluate treatment effects on major CV endpoints are ongoing.34,35
In the EMPA-REG OUTCOME trial,33 researchers found that empagliflozin had beneficial effects on CV outcomes, making it one of the only antidiabetic agents on the market to have such benefits. The study, which involved more than 7000 patients with a history of T2DM and existing cardiovascular disease (CVD), found that 10.5% of patients in the empagliflozin group vs 12.1% in the placebo group died from a CV cause or experienced a nonfatal myocardial infarction or stroke over a median of 3.1 years. Results were similar with both doses (10 mg vs 25 mg) of empagliflozin. The mechanisms behind the CV benefits are likely multifactorial and may be related to reductions in weight and BP,33 but additional research is needed to fully elucidate the role of empagliflozin in this population.
Canagliflozin is being evaluated in the Canagliflozin Cardiovascular Assessment Study (CANVAS) for its effect on major CV events—CV death, nonfatal myocardial infarction, and nonfatal stroke—in patients with either a history of CVD or who are at increased risk of CVD and have uncontrolled diabetes.34 The trial is expected to wrap up in June 2017.
And dapagliflozin is being studied in the DECLARE-TIMI 58 trial (the Effect of Dapagliflozin on the Incidence of Cardiovascular Events) in patients with T2DM and either known CVD or at least 2 risk factors for CVD.35 The study is designed to assess dapagliflozin’s effect on the incidence of CV death, myocardial infarction, and ischemic stroke and has an estimated completion date of April 2019, which will provide a median follow-up of 4.5 years.
Cancer. All 3 agents have been examined for any possible carcinogenic links. In 2011, the FDA issued a request for further investigation surrounding the risk of cancer associated with dapagliflozin.36 As of November 2013, 10 of 6045 patients treated with dapagliflozin developed bladder cancer compared to 1 of 3512 controls.36 Furthermore, 9 of 2223 patients treated with dapagliflozin developed breast cancer compared to 1 of 1053 controls.36
Although the trials were not designed to detect an increase in risk, the number of observed cases warranted further investigation. No official warning for breast cancer exists since the characteristics of the malignancies led the FDA to believe dapagliflozin was unlikely the cause.36
Given what we know to date, it appears to be prudent to avoid prescribing SGLT2 inhibitors in patients with active bladder cancer, and to use them with caution in those with a history of the disease.2
Other studies. Initially, animal studies suggested an increased risk of various malignancies associated with canagliflozin use in rats,37 but consistent results were not seen in human studies. Similarly, at least one study found that empagliflozin was associated with lung cancer and melanoma, but closer examination found that most patients who developed these cancers had risk factors.38 Large, long-term studies of these agents in various populations are needed to thoroughly investigate possible carcinogenicity.
Additional considerations: Kidney function, age, and pregnancy
Consider avoiding SGLT2 inhibitors in patients with moderate kidney dysfunction (eGFR 30-59 mL/min/1.73 m2). Studies have shown that SGLT2 inhibitors are not as effective at lowering blood glucose in those with reduced eGFR, although adverse events were similar to those in placebo groups.24,39,40 Dapagliflozin is not recommended in patients with an eGFR <60 mL/min/1.73 m2 due to lack of efficacy.2,24 Empagliflozin does not require dose adjustments if eGFR is ≥45 mL/min/1.73 m2. A lower dose of canagliflozin (ie, 100 mg/d) is recommended in those with an eGFR of 45 to 59 mL/min/1.73 m2.2 All agents are contraindicated in patients with severe renal impairment (eGFR <30 mL/min/1.73 m2).
Older patients are at higher risk for dehydration, hypotension, and falls; therefore, SGLT2 inhibitors should be used with caution in this population. Similarly, they should not be used in patients with T1DM and should be avoided in those with active, or a history of, DKA.
There are no data on the use of SGLT2 inhibitors in pregnancy; thus, these agents should be avoided unless the potential benefits outweigh the potential risks to the unborn fetus.2
CASE 1 › An SGLT2 inhibitor is an acceptable option for Mr. S. Because he is resistant to starting insulin therapy and his HbA1c is <9%, an additional oral medication is reasonable. Adding an SGLT2 inhibitor may reduce his HbA1c up to ~1%, and education on lifestyle modifications may help bring him to goal. An SGLT2 inhibitor may also benefit his BP and weight, both of which could be improved.
Given the drugs he’s taking, drug interactions should not be an issue, and his renal function and pertinent labs (K, Phos, Mg) are within normal limits. Nevertheless, monitor these labs periodically and monitor Mr. S for adverse effects, such as UTIs, although these are more common in women. Canagliflozin is the preferred SGLT2 inhibitor on his insurance formulary, so you could initiate therapy at 100 mg/d, administered prior to the first meal, and increase to 300 mg/d if needed. As an alternative, consider prescribing the metformin/canagliflozin combination agent.
CASE 2 › Ms. R is likely experiencing a yeast infection as an adverse effect of the dapagliflozin. Although one yeast infection is insufficient grounds for discontinuation of the drug, recurrent infections should prompt a risk-to-benefit analysis to determine whether it’s worth continuing the medication. Her recent eGFR (<60 mL/min/1.73 m2) is, however, a contraindication to dapagliflozin, and therapy should be discontinued. Canagliflozin and empagliflozin may be considered since her eGFR is >45 mL/min/1.73 m2, but given her current HbA1c and recent adverse drug event, alternative therapies, such as basal insulin, are more appropriate treatment choices.
CORRESPONDENCE
Katelin M. Lisenby, PharmD, BCPS, University of Alabama College of Community Health Sciences, University Medical Center, Box 870374, Tuscaloosa, AL 35487; [email protected].
1. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2015;38:140-149.
2. Canagliflozin, dapagliflozin, empagliflozin. Lexicomp, Inc. (Lexi-Drugs®). Accessed October 12, 2015.
3. Stenlöf K, Cefalu WT, Kim KA, et al. Long-term efficacy and safety of canagliflozin monotherapy in patients with type 2 diabetes mellitus inadequately controlled with diet and exercise: findings from the 52-week CANTATA-M study. Curr Med Res Opin. 2014;30:163-175.
4. Ferrannini E, Ramos SJ, Salsali A, et al. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010:33:2217-2224.
5. Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1:208-219.
6. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36:4015-4021.
7. Wilding JPH, Charpentier G, Hollander P, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: a randomised trial. Int J Clin Pract. 2013;67:1267-1282.
8. Forst T, Guthrie R, Goldenberg R, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes on background metformin and pioglitazone. Diabetes Obes Metab. 2014;16:467-477.
9. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36:2508-2515.
10. Bristol-Myers Squibb [press release]. New phase III data showed dapagliflozin significantly reduced HbA1c compared to placebo at 24 weeks in patients with type 2 diabetes inadequately controlled with the combination of metformin plus sulfonylurea. Available at: http://news.bms.com/press-release/rd-news/new-phase-iii-data-showed-dapagliflozin-significantly-reduced-hba1c-compared-p&t=635156160653787526. Accessed September 17, 2015.
11. Jabbour SA, Hardy E, Sugg J, et al. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24- week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care. 2014;37:740-750.
12. DeFronzo RA, Lewin A, Patel S, et al. Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care. 2015;38:384-393.
13. Kovacs CS, Seshiah V, Merker L, et al. Empagliflozin as add-on therapy to pioglitazone with or without metformin in patients with type 2 diabetes mellitus. Clin Ther. 2015;37:1773-1788.
14. Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized double-blind, placebo-controlled trial. Diabetes Care. 2013;36:3396-3404.
15. Neal B, Percovik V, de Zeeuw D, et al. Efficacy and safety of canagliflozin, an inhibitor of sodium–glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38:403-411.
16. Wilding JPH, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156:405-415.
17. Rosenstock J, Jelaska A, Frappin G, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37:1815-1823.
18. Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941-950.
19. Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010:375:2223-2233.
20. Bailey CJ, Gross JL, Hennicken D, et al. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
21. Rosenstock J, Vico M, Wei L, et al. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473-1478.
22. Merker L, Häring HU, Christiansen AV, et al. Empagliflozin as add-on to metformin in people with type 2 diabetes. Diabet Med. 2015;32:1555-1567.
23. Ridderstråle M, Anderson KR, Zeller C, et al. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2:691-700.
24. Kohan DE, Fioretto P, Tang W, et al. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85:962-971.
25. Invokana (canagliflozin) tablets [product information]. Titusville, NJ: Janssen Pharmaceuticals Inc. Available at: https://www.invokana.com. Accessed March 15, 2013.
26. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56:2582-2592.
27. Strojek K, Yoon KH, Hruba V, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928-938.
28. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38:355-364.
29. US Food and Drug Administration. FDA drug safety communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Accessed July 11, 2016.
30. Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38:1638-1642.
31. US Food and Drug Administration. FDA drug safety communication: FDA revises label of diabetes drug canagliflozin (Invokana, Invokamet) to include updates on bone fracture risk and new information on decreased bone mineral density. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm461449.htm. Acces-sed July 11, 2016.
32. Canagliflozin, dapagliflozin, empagliflozin. In: RED BOOK [AUHSOP intranet database]. Greenwood Village, CO: Truven Health Analytics; [updated daily]. Available at: http://www.micromedexsolutions.com/micromedex2/librarian/ND_T/evidencexpert/ND_PR/evidencexpert/CS/BB1644/ND_AppProduct/evidencexpert/DUPLICATIONSHIELDSYNC/FAF693/ND_PG/evidencexpert/ND_B/evidencexpert/ND_P/evidencexpert/PFActionId/redbook.ShowProductSearchResults?SearchTerm=JARDIANCE&searchType=redbookProductName&searchTermId=42798&searchContent=%24searchContent&searchFilterAD=filterADActive&searchFilterRepackager=filterExcludeRepackager&searchPattern=%5Ejard. Accessed March 15, 2016.
33. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
34. CANagliflozin cardioVascular Assessment Study (CANVAS). Available at: http://clinicaltrials.gov/show/NCT01032629. Accessed October 12, 2015.
35. Multicenter trial to evaluate the effect of dapagliflozin on the incidence of cardiovascular events (DECLARE-TIMI 58). Available at: http://clinicaltrials.gov/show/NCT01730534. Accessed October 12, 2015.
36. FDA background document. BMS-512148 NDA 202293. In: Proceedings of the US Food and Drug Administration Endocrinologic & Metabolic Drug Advisory Committee Meeting, 2013. Available at: http://www.fda.gov/downloads/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm378079.pdf. Accessed October 12, 2015.
37. Lin HW, Tseng CH. A review of the relationship between SGLT2 inhibitors and cancer. Int J Endocrinol. 2014;2014:719578.
38. Center for Drug Evaluation and Research. Risk assessment and risk mitigation review(s). July 28, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/ 204629Orig1s000RiskR.pdf. Accessed September 21, 2015.
39. Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes Metab. 2014;16:1016-1027.
40. Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2: 369-384.
› Consider sodium-glucose cotransporter 2 (SGLT2) inhibitors as second-line agents in patients with type 2 diabetes mellitus who need mild hemoglobin A1c reductions (≤1%) and who would benefit from mild to modest weight and blood pressure reductions. A
› Avoid using SGLT2 inhibitors in patients with a history of recurrent genital mycotic or urinary tract infections. B
› Use SGLT2 inhibitors with caution in patients at risk for volume-related adverse effects (dizziness and hypotension), such as the elderly, those with moderate renal dysfunction, and those taking concomitant diuretic therapy. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Joe S is a 41-year-old African-American man who comes to your clinic after his employee health screening revealed elevated triglycerides. The patient has a 3-year history of type 2 diabetes mellitus (T2DM); he also has a history of hypertension, gastroesophageal reflux disease, and obstructive sleep apnea. Mr. S tells you he takes metformin 1000 mg twice daily, but stopped taking his glipizide because he didn’t think it was helping his blood sugar. His last hemoglobin (Hb) A1c result was 8.8%, and he is very resistant to starting insulin therapy.
The patient’s other medications include enalapril 10 mg/d, atorvastatin 10 mg/d, and omeprazole 20 mg/d. Mr. S weighs 255.6 lbs (body mass index=34.7), his BP is 140/88 mm Hg, and his heart rate is 82 beats per minute. Laboratory values include: serum creatinine, 1.01 mg/dL; estimated glomerular filtration rate (eGFR) >100 mL/min/1.73 m2; potassium (K), 4.3 mmol/L; serum phosphorous (Phos), 2.8 mg/dL; magnesium (Mg), 1.9 mg/dL; total cholesterol, 167 mg/dL; low-density lipoprotein (LDL), 78 mg/dL; high-density lipoprotein (HDL), 38 mg/dL; and triglycerides, 256 mg/dL.
CASE 2 › Susan R, a 68-year-old Caucasian woman, returns to your clinic for a follow-up visit 3 months after you prescribed dapagliflozin 10 mg/d for her T2DM. Her glucose levels have improved, but she complains of vaginal pruritus and is worried that she has a yeast infection.
You diagnose vulvovaginal candidiasis in this patient and prescribe a single dose of fluconazole 150 mg. After reviewing her laboratory test results, you notice that since starting the dapagliflozin, her HbA1c level has improved slightly from 9.8% to 9.3%, but is still not where it needs to be. Her eGFR is 49 mL/min/1.73 m2.
What would you recommend to improve control of these patients’ blood glucose levels?
When to consider an SGLT2 inhibitor
Consider therapy with SGLT2 inhibitors in adult patients with T2DM who:3-9,13-15,17-24
- have an HbA1c between 7% and 9%
- would benefit from weight and/or blood pressure reductions
- have metabolic syndrome
- have adequate means to pay for the medication (ie, prescription coverage or the ability to afford it).
In addition, consider an SGLT2 inhibitor as initial monotherapy if metformin is contraindicated or not tolerated, or as add-on therapy to metformin, sulfonylureas, thiazolidinediones, dipeptidyl peptidase IV inhibitors, or insulin.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are the newest class of agents to enter the T2DM management arena. They act in the proximal renal tubules to decrease the reabsorption of glucose by targeting the SGLT2 transmembrane protein, which reabsorbs about 90% of the body’s glucose.1,2 The class is currently made up of 3 agents—canagliflozin, dapagliflozin, and empagliflozin—all of which are approved by the US Food and Drug Administration (FDA) for the treatment of T2DM (TABLE 1).2
The American Diabetes Association and the European Association for the Study of Diabetes published updated guidelines for T2DM management in 2015.1 In addition to lifestyle modifications, the guidelines recommend the use of metformin as first-line therapy unless it is contraindicated or patients are unable to tolerate it (eg, because of gastrointestinal adverse effects). They recommend other pharmacologic therapies as second-line options based on specific patient characteristics. Thus, SGLT2 inhibitors may be used as add-on therapy after metformin, or as a first-line option if metformin is contraindicated or not tolerated. Because the mechanism of action of SGLT2 inhibitors is independent of insulin secretion, these agents may be used at any stage of the diabetes continuum.
SGLT2 agents as monotherapy, or as add-on therapy
All SGLT2 agents have been studied as monotherapy accompanied by diet and exercise and shown to produce HbA1c reductions of 0.34% to 1.11%.3-6 In trials, the effect was similar regardless of study duration (18-104 weeks); generally, higher doses corresponded with larger HbA1c reductions.3-6
SGLT2 inhibitors have also been studied as add-on therapy to several oral agents including metformin, sulfonylureas, thiazolidinediones (TZDs), and the combination of metformin plus sulfonylureas or TZDs or dipeptidyl peptidase IV (DPP-IV) inhibitors.1 When used in any of these combinations, each SGLT2 agent demonstrated a consistent HbA1c lowering effect of 0.62% to 1.19%.7-14
Additionally, SGLT2 inhibitors have been studied in combination with insulin therapy (median or mean daily doses >60 units), which yielded further reductions in HbA1c of 0.58% to 1.02% without significant insulin adjustments or an increase in major hypoglycemia events.15-17 Patients receiving insulin and an SGLT2 inhibitor had lower insulin doses and more weight loss compared to placebo groups.
SGLT2 inhibitors offer additional benefits
Secondary analyses of most studies of SGLT2 inhibitors include changes in BP and weight from baseline as well as minor changes (some positive, some not) in several lipid parameters.3-5,7-9,13-15,17-24 In general, these effects do not appear to be dose-dependent (with the exception of canagliflozin and its associated lipid effects25) and are similar among the 3 medications.3-5,7-9,13-15,17-24 (For more on who would benefit from these agents, see “When to consider an SGLT2 inhibitor” above.)
BP reduction. Although the mean baseline BP was controlled in most studies, SGLT2 inhibitors have been shown to significantly reduce BP. Reductions in BP with all 3 SGLT2 medications range from approximately 2 to 5 mm Hg systolic and 0.5 to 2.5 mm Hg diastolic, which may be due to weight loss and diuresis.4-8,10-16,20-23 While the reductions were modest at best, one study involving empagliflozin reported that more than one-quarter of patients with uncontrolled BP at baseline achieved a BP <130/80 mm Hg 24 weeks later.5 While these agents should not be used solely for their BP lowering effects, they may help a small number of patients with mildly elevated BP achieve their goal without an additional antihypertensive agent.
Weight reduction. Modest weight loss, likely due to the loss of calories through urine, was seen with SGLT2 inhibitors in most studies, with reductions persisting beyond one year of use. In most studies, including those involving obese patients on insulin therapy,15,17,21 patients’ body weights were reduced by approximately 2 to 4 kg from baseline.3-16,18,21-23,26
Lipid effects. Although the mechanism is unclear, use of SGLT2 inhibitors can have varying effects on lipid panels. In most studies, total and LDL cholesterol levels were increased with elevations ranging from 0.7 to 10 mg/dL.3,7,8,18,19,22,23 Conversely, at least one study demonstrated mild reductions in total and LDL cholesterol levels with higher doses of empagliflozin.13 Additionally, modest reductions in triglycerides and increases in HDL across all doses of canagliflozin, dapagliflozin, and empagliflozin have been seen.8,9,13,15,19 While the clinical relevance of these lipid changes is unknown, monitoring is recommended.2
These agents are well tolerated
SGLT2 inhibitors were generally well tolerated in studies. The most common adverse effects include mycotic infections (2.4%-21.6%) and urinary tract infections (UTIs) (4.0%-19.6%) (both with higher incidences in females); volume-related effects such as dizziness and hypotension (0.3%-8.3%); and nasopharyngitis (5.4%-18.3%).4-14,16-23,26-28 Hypoglycemia was observed more often when an SGLT2 inhibitor was used in combination with a sulfonylurea or insulin therapy.4-14,16-23,26-28 The number of times adverse events led to discontinuation was low and similar to that in control groups.4-14,16-23,26-28
Mycotic and urinary infections should be diagnosed and treated according to current standards of care and do not require discontinuation of the SGLT2 inhibitor. Canagli-flozin therapy was associated with electrolyte abnormalities including hyperkalemia, hypermagnesemia, and hyperphosphatemia.25 Thus, levels should be monitored periodically, especially in patients predisposed to elevations due to other conditions or medications.25
Two additional warnings are worth noting
Diabetic ketoacidosis (DKA) has been reported with all 3 agents, and bone fractures have been reported with canagliflozin.
The FDA issued a warning in May 2015 regarding the increased risk of DKA with the use of SGLT2 inhibitor single and combination products.29 This warning was prompted by several case reports of DKA with uncharacteristically mild to moderate glucose elevations in patients with type 1 diabetes mellitus (T1DM) and T2DM who were taking an SGLT2 inhibitor. The absence of significant hyperglycemia delayed diagnosis in many cases. Therefore, patients should be counseled on the signs and symptoms of DKA, as well as when to seek medical attention.
Patients with diabetes and symptoms of ketoacidosis (eg, difficulty breathing, nausea, vomiting, abdominal pain, confusion, and fatigue) should be evaluated regardless of current blood glucose levels, and SGLT2 inhibitors should be discontinued if acidosis is confirmed. Identified potential triggers include illness, reduced food and fluid intake, reduced insulin dose, and history of alcohol intake. Use of SGLT2 inhibitors should be avoided in patients with T1DM until safety and efficacy are established in large randomized controlled trials. The European Medicines Agency announced that a thorough review of all currently approved SGLT2 agents is underway to evaluate the risk for DKA.30
In addition, the FDA called for a revision of the label of canagliflozin to reflect a strengthened warning about an increased risk of bone fractures and decreased bone mineral density (BMD).31 Fractures can occur as early as 12 weeks after initiating treatment and with only minor trauma.31
Over a 2-year period, canagliflozin also significantly decreased BMD in the hip and lower spine compared to placebo.31 Patients should be evaluated for additional risk factors for fracture before taking canagliflozin.31 The FDA is continuing to evaluate whether the other approved SGLT2 inhibitors are associated with an increased risk for fractures.
Drug interactions: Proceed carefully with diuretics
The number of drugs that interact with SGLT2 inhibitors is minimal. Because these agents can cause volume-related effects such as hypotension, dizziness, and osmotic diuresis, patients—particularly the elderly and those with renal impairment—taking concomitant diuretics, especially loop diuretics, may be at increased risk for these effects and should be monitored accordingly.2,25
Canagliflozin is primarily metabolized via glucuronidation by the uridine 5'-diphospho-glucuronosyltransferase (UGT) enzymes. Therefore, UGT enzyme inducers (eg, rifampin, phenytoin, phenobarbital, ritonavir) decrease canagliflozin’s serum concentration. If a patient has an eGFR >60 mL/min/1.73 m2 and is tolerating a dose of 100 mg/d, consider increasing the dose to 300 mg/d during concomitant treatment.
In addition, researchers have found that canagliflozin increases serum levels of digoxin by between 20% and 36%.25 Experts suspect this occurs because canagliflozin inhibits P-glycoprotein efflux of digoxin. Although monitoring of digoxin levels is recommended, this interaction is considered to be minor.25
Cost consideration: SGLT2 inhibitors are more expensive
The SGLT2 inhibitors are available only as brand name products and are more expensive than agents that have generic options (eg, metformin, sulfonylureas, TZDs). The average wholesale cost is approximately $400 for a 30-day supply of all SGLT2 agents.32 When considering an SGLT2 inhibitor, the patient should ideally have medication prescription coverage. Depending on the specific insurance plan, these agents are classified as tier 2 to 4, which is comparable to other oral brand name options.
Research looks at CV outcomes and cancer risk
Cardiovascular (CV) risk reduction. To date, only one study evaluating the effect of SGLT2 inhibitors on CV outcomes is complete.33 Two large randomized controlled trials involving canagliflozin and dapagliflozin designed to evaluate treatment effects on major CV endpoints are ongoing.34,35
In the EMPA-REG OUTCOME trial,33 researchers found that empagliflozin had beneficial effects on CV outcomes, making it one of the only antidiabetic agents on the market to have such benefits. The study, which involved more than 7000 patients with a history of T2DM and existing cardiovascular disease (CVD), found that 10.5% of patients in the empagliflozin group vs 12.1% in the placebo group died from a CV cause or experienced a nonfatal myocardial infarction or stroke over a median of 3.1 years. Results were similar with both doses (10 mg vs 25 mg) of empagliflozin. The mechanisms behind the CV benefits are likely multifactorial and may be related to reductions in weight and BP,33 but additional research is needed to fully elucidate the role of empagliflozin in this population.
Canagliflozin is being evaluated in the Canagliflozin Cardiovascular Assessment Study (CANVAS) for its effect on major CV events—CV death, nonfatal myocardial infarction, and nonfatal stroke—in patients with either a history of CVD or who are at increased risk of CVD and have uncontrolled diabetes.34 The trial is expected to wrap up in June 2017.
And dapagliflozin is being studied in the DECLARE-TIMI 58 trial (the Effect of Dapagliflozin on the Incidence of Cardiovascular Events) in patients with T2DM and either known CVD or at least 2 risk factors for CVD.35 The study is designed to assess dapagliflozin’s effect on the incidence of CV death, myocardial infarction, and ischemic stroke and has an estimated completion date of April 2019, which will provide a median follow-up of 4.5 years.
Cancer. All 3 agents have been examined for any possible carcinogenic links. In 2011, the FDA issued a request for further investigation surrounding the risk of cancer associated with dapagliflozin.36 As of November 2013, 10 of 6045 patients treated with dapagliflozin developed bladder cancer compared to 1 of 3512 controls.36 Furthermore, 9 of 2223 patients treated with dapagliflozin developed breast cancer compared to 1 of 1053 controls.36
Although the trials were not designed to detect an increase in risk, the number of observed cases warranted further investigation. No official warning for breast cancer exists since the characteristics of the malignancies led the FDA to believe dapagliflozin was unlikely the cause.36
Given what we know to date, it appears to be prudent to avoid prescribing SGLT2 inhibitors in patients with active bladder cancer, and to use them with caution in those with a history of the disease.2
Other studies. Initially, animal studies suggested an increased risk of various malignancies associated with canagliflozin use in rats,37 but consistent results were not seen in human studies. Similarly, at least one study found that empagliflozin was associated with lung cancer and melanoma, but closer examination found that most patients who developed these cancers had risk factors.38 Large, long-term studies of these agents in various populations are needed to thoroughly investigate possible carcinogenicity.
Additional considerations: Kidney function, age, and pregnancy
Consider avoiding SGLT2 inhibitors in patients with moderate kidney dysfunction (eGFR 30-59 mL/min/1.73 m2). Studies have shown that SGLT2 inhibitors are not as effective at lowering blood glucose in those with reduced eGFR, although adverse events were similar to those in placebo groups.24,39,40 Dapagliflozin is not recommended in patients with an eGFR <60 mL/min/1.73 m2 due to lack of efficacy.2,24 Empagliflozin does not require dose adjustments if eGFR is ≥45 mL/min/1.73 m2. A lower dose of canagliflozin (ie, 100 mg/d) is recommended in those with an eGFR of 45 to 59 mL/min/1.73 m2.2 All agents are contraindicated in patients with severe renal impairment (eGFR <30 mL/min/1.73 m2).
Older patients are at higher risk for dehydration, hypotension, and falls; therefore, SGLT2 inhibitors should be used with caution in this population. Similarly, they should not be used in patients with T1DM and should be avoided in those with active, or a history of, DKA.
There are no data on the use of SGLT2 inhibitors in pregnancy; thus, these agents should be avoided unless the potential benefits outweigh the potential risks to the unborn fetus.2
CASE 1 › An SGLT2 inhibitor is an acceptable option for Mr. S. Because he is resistant to starting insulin therapy and his HbA1c is <9%, an additional oral medication is reasonable. Adding an SGLT2 inhibitor may reduce his HbA1c up to ~1%, and education on lifestyle modifications may help bring him to goal. An SGLT2 inhibitor may also benefit his BP and weight, both of which could be improved.
Given the drugs he’s taking, drug interactions should not be an issue, and his renal function and pertinent labs (K, Phos, Mg) are within normal limits. Nevertheless, monitor these labs periodically and monitor Mr. S for adverse effects, such as UTIs, although these are more common in women. Canagliflozin is the preferred SGLT2 inhibitor on his insurance formulary, so you could initiate therapy at 100 mg/d, administered prior to the first meal, and increase to 300 mg/d if needed. As an alternative, consider prescribing the metformin/canagliflozin combination agent.
CASE 2 › Ms. R is likely experiencing a yeast infection as an adverse effect of the dapagliflozin. Although one yeast infection is insufficient grounds for discontinuation of the drug, recurrent infections should prompt a risk-to-benefit analysis to determine whether it’s worth continuing the medication. Her recent eGFR (<60 mL/min/1.73 m2) is, however, a contraindication to dapagliflozin, and therapy should be discontinued. Canagliflozin and empagliflozin may be considered since her eGFR is >45 mL/min/1.73 m2, but given her current HbA1c and recent adverse drug event, alternative therapies, such as basal insulin, are more appropriate treatment choices.
CORRESPONDENCE
Katelin M. Lisenby, PharmD, BCPS, University of Alabama College of Community Health Sciences, University Medical Center, Box 870374, Tuscaloosa, AL 35487; [email protected].
› Consider sodium-glucose cotransporter 2 (SGLT2) inhibitors as second-line agents in patients with type 2 diabetes mellitus who need mild hemoglobin A1c reductions (≤1%) and who would benefit from mild to modest weight and blood pressure reductions. A
› Avoid using SGLT2 inhibitors in patients with a history of recurrent genital mycotic or urinary tract infections. B
› Use SGLT2 inhibitors with caution in patients at risk for volume-related adverse effects (dizziness and hypotension), such as the elderly, those with moderate renal dysfunction, and those taking concomitant diuretic therapy. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Joe S is a 41-year-old African-American man who comes to your clinic after his employee health screening revealed elevated triglycerides. The patient has a 3-year history of type 2 diabetes mellitus (T2DM); he also has a history of hypertension, gastroesophageal reflux disease, and obstructive sleep apnea. Mr. S tells you he takes metformin 1000 mg twice daily, but stopped taking his glipizide because he didn’t think it was helping his blood sugar. His last hemoglobin (Hb) A1c result was 8.8%, and he is very resistant to starting insulin therapy.
The patient’s other medications include enalapril 10 mg/d, atorvastatin 10 mg/d, and omeprazole 20 mg/d. Mr. S weighs 255.6 lbs (body mass index=34.7), his BP is 140/88 mm Hg, and his heart rate is 82 beats per minute. Laboratory values include: serum creatinine, 1.01 mg/dL; estimated glomerular filtration rate (eGFR) >100 mL/min/1.73 m2; potassium (K), 4.3 mmol/L; serum phosphorous (Phos), 2.8 mg/dL; magnesium (Mg), 1.9 mg/dL; total cholesterol, 167 mg/dL; low-density lipoprotein (LDL), 78 mg/dL; high-density lipoprotein (HDL), 38 mg/dL; and triglycerides, 256 mg/dL.
CASE 2 › Susan R, a 68-year-old Caucasian woman, returns to your clinic for a follow-up visit 3 months after you prescribed dapagliflozin 10 mg/d for her T2DM. Her glucose levels have improved, but she complains of vaginal pruritus and is worried that she has a yeast infection.
You diagnose vulvovaginal candidiasis in this patient and prescribe a single dose of fluconazole 150 mg. After reviewing her laboratory test results, you notice that since starting the dapagliflozin, her HbA1c level has improved slightly from 9.8% to 9.3%, but is still not where it needs to be. Her eGFR is 49 mL/min/1.73 m2.
What would you recommend to improve control of these patients’ blood glucose levels?
When to consider an SGLT2 inhibitor
Consider therapy with SGLT2 inhibitors in adult patients with T2DM who:3-9,13-15,17-24
- have an HbA1c between 7% and 9%
- would benefit from weight and/or blood pressure reductions
- have metabolic syndrome
- have adequate means to pay for the medication (ie, prescription coverage or the ability to afford it).
In addition, consider an SGLT2 inhibitor as initial monotherapy if metformin is contraindicated or not tolerated, or as add-on therapy to metformin, sulfonylureas, thiazolidinediones, dipeptidyl peptidase IV inhibitors, or insulin.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are the newest class of agents to enter the T2DM management arena. They act in the proximal renal tubules to decrease the reabsorption of glucose by targeting the SGLT2 transmembrane protein, which reabsorbs about 90% of the body’s glucose.1,2 The class is currently made up of 3 agents—canagliflozin, dapagliflozin, and empagliflozin—all of which are approved by the US Food and Drug Administration (FDA) for the treatment of T2DM (TABLE 1).2
The American Diabetes Association and the European Association for the Study of Diabetes published updated guidelines for T2DM management in 2015.1 In addition to lifestyle modifications, the guidelines recommend the use of metformin as first-line therapy unless it is contraindicated or patients are unable to tolerate it (eg, because of gastrointestinal adverse effects). They recommend other pharmacologic therapies as second-line options based on specific patient characteristics. Thus, SGLT2 inhibitors may be used as add-on therapy after metformin, or as a first-line option if metformin is contraindicated or not tolerated. Because the mechanism of action of SGLT2 inhibitors is independent of insulin secretion, these agents may be used at any stage of the diabetes continuum.
SGLT2 agents as monotherapy, or as add-on therapy
All SGLT2 agents have been studied as monotherapy accompanied by diet and exercise and shown to produce HbA1c reductions of 0.34% to 1.11%.3-6 In trials, the effect was similar regardless of study duration (18-104 weeks); generally, higher doses corresponded with larger HbA1c reductions.3-6
SGLT2 inhibitors have also been studied as add-on therapy to several oral agents including metformin, sulfonylureas, thiazolidinediones (TZDs), and the combination of metformin plus sulfonylureas or TZDs or dipeptidyl peptidase IV (DPP-IV) inhibitors.1 When used in any of these combinations, each SGLT2 agent demonstrated a consistent HbA1c lowering effect of 0.62% to 1.19%.7-14
Additionally, SGLT2 inhibitors have been studied in combination with insulin therapy (median or mean daily doses >60 units), which yielded further reductions in HbA1c of 0.58% to 1.02% without significant insulin adjustments or an increase in major hypoglycemia events.15-17 Patients receiving insulin and an SGLT2 inhibitor had lower insulin doses and more weight loss compared to placebo groups.
SGLT2 inhibitors offer additional benefits
Secondary analyses of most studies of SGLT2 inhibitors include changes in BP and weight from baseline as well as minor changes (some positive, some not) in several lipid parameters.3-5,7-9,13-15,17-24 In general, these effects do not appear to be dose-dependent (with the exception of canagliflozin and its associated lipid effects25) and are similar among the 3 medications.3-5,7-9,13-15,17-24 (For more on who would benefit from these agents, see “When to consider an SGLT2 inhibitor” above.)
BP reduction. Although the mean baseline BP was controlled in most studies, SGLT2 inhibitors have been shown to significantly reduce BP. Reductions in BP with all 3 SGLT2 medications range from approximately 2 to 5 mm Hg systolic and 0.5 to 2.5 mm Hg diastolic, which may be due to weight loss and diuresis.4-8,10-16,20-23 While the reductions were modest at best, one study involving empagliflozin reported that more than one-quarter of patients with uncontrolled BP at baseline achieved a BP <130/80 mm Hg 24 weeks later.5 While these agents should not be used solely for their BP lowering effects, they may help a small number of patients with mildly elevated BP achieve their goal without an additional antihypertensive agent.
Weight reduction. Modest weight loss, likely due to the loss of calories through urine, was seen with SGLT2 inhibitors in most studies, with reductions persisting beyond one year of use. In most studies, including those involving obese patients on insulin therapy,15,17,21 patients’ body weights were reduced by approximately 2 to 4 kg from baseline.3-16,18,21-23,26
Lipid effects. Although the mechanism is unclear, use of SGLT2 inhibitors can have varying effects on lipid panels. In most studies, total and LDL cholesterol levels were increased with elevations ranging from 0.7 to 10 mg/dL.3,7,8,18,19,22,23 Conversely, at least one study demonstrated mild reductions in total and LDL cholesterol levels with higher doses of empagliflozin.13 Additionally, modest reductions in triglycerides and increases in HDL across all doses of canagliflozin, dapagliflozin, and empagliflozin have been seen.8,9,13,15,19 While the clinical relevance of these lipid changes is unknown, monitoring is recommended.2
These agents are well tolerated
SGLT2 inhibitors were generally well tolerated in studies. The most common adverse effects include mycotic infections (2.4%-21.6%) and urinary tract infections (UTIs) (4.0%-19.6%) (both with higher incidences in females); volume-related effects such as dizziness and hypotension (0.3%-8.3%); and nasopharyngitis (5.4%-18.3%).4-14,16-23,26-28 Hypoglycemia was observed more often when an SGLT2 inhibitor was used in combination with a sulfonylurea or insulin therapy.4-14,16-23,26-28 The number of times adverse events led to discontinuation was low and similar to that in control groups.4-14,16-23,26-28
Mycotic and urinary infections should be diagnosed and treated according to current standards of care and do not require discontinuation of the SGLT2 inhibitor. Canagli-flozin therapy was associated with electrolyte abnormalities including hyperkalemia, hypermagnesemia, and hyperphosphatemia.25 Thus, levels should be monitored periodically, especially in patients predisposed to elevations due to other conditions or medications.25
Two additional warnings are worth noting
Diabetic ketoacidosis (DKA) has been reported with all 3 agents, and bone fractures have been reported with canagliflozin.
The FDA issued a warning in May 2015 regarding the increased risk of DKA with the use of SGLT2 inhibitor single and combination products.29 This warning was prompted by several case reports of DKA with uncharacteristically mild to moderate glucose elevations in patients with type 1 diabetes mellitus (T1DM) and T2DM who were taking an SGLT2 inhibitor. The absence of significant hyperglycemia delayed diagnosis in many cases. Therefore, patients should be counseled on the signs and symptoms of DKA, as well as when to seek medical attention.
Patients with diabetes and symptoms of ketoacidosis (eg, difficulty breathing, nausea, vomiting, abdominal pain, confusion, and fatigue) should be evaluated regardless of current blood glucose levels, and SGLT2 inhibitors should be discontinued if acidosis is confirmed. Identified potential triggers include illness, reduced food and fluid intake, reduced insulin dose, and history of alcohol intake. Use of SGLT2 inhibitors should be avoided in patients with T1DM until safety and efficacy are established in large randomized controlled trials. The European Medicines Agency announced that a thorough review of all currently approved SGLT2 agents is underway to evaluate the risk for DKA.30
In addition, the FDA called for a revision of the label of canagliflozin to reflect a strengthened warning about an increased risk of bone fractures and decreased bone mineral density (BMD).31 Fractures can occur as early as 12 weeks after initiating treatment and with only minor trauma.31
Over a 2-year period, canagliflozin also significantly decreased BMD in the hip and lower spine compared to placebo.31 Patients should be evaluated for additional risk factors for fracture before taking canagliflozin.31 The FDA is continuing to evaluate whether the other approved SGLT2 inhibitors are associated with an increased risk for fractures.
Drug interactions: Proceed carefully with diuretics
The number of drugs that interact with SGLT2 inhibitors is minimal. Because these agents can cause volume-related effects such as hypotension, dizziness, and osmotic diuresis, patients—particularly the elderly and those with renal impairment—taking concomitant diuretics, especially loop diuretics, may be at increased risk for these effects and should be monitored accordingly.2,25
Canagliflozin is primarily metabolized via glucuronidation by the uridine 5'-diphospho-glucuronosyltransferase (UGT) enzymes. Therefore, UGT enzyme inducers (eg, rifampin, phenytoin, phenobarbital, ritonavir) decrease canagliflozin’s serum concentration. If a patient has an eGFR >60 mL/min/1.73 m2 and is tolerating a dose of 100 mg/d, consider increasing the dose to 300 mg/d during concomitant treatment.
In addition, researchers have found that canagliflozin increases serum levels of digoxin by between 20% and 36%.25 Experts suspect this occurs because canagliflozin inhibits P-glycoprotein efflux of digoxin. Although monitoring of digoxin levels is recommended, this interaction is considered to be minor.25
Cost consideration: SGLT2 inhibitors are more expensive
The SGLT2 inhibitors are available only as brand name products and are more expensive than agents that have generic options (eg, metformin, sulfonylureas, TZDs). The average wholesale cost is approximately $400 for a 30-day supply of all SGLT2 agents.32 When considering an SGLT2 inhibitor, the patient should ideally have medication prescription coverage. Depending on the specific insurance plan, these agents are classified as tier 2 to 4, which is comparable to other oral brand name options.
Research looks at CV outcomes and cancer risk
Cardiovascular (CV) risk reduction. To date, only one study evaluating the effect of SGLT2 inhibitors on CV outcomes is complete.33 Two large randomized controlled trials involving canagliflozin and dapagliflozin designed to evaluate treatment effects on major CV endpoints are ongoing.34,35
In the EMPA-REG OUTCOME trial,33 researchers found that empagliflozin had beneficial effects on CV outcomes, making it one of the only antidiabetic agents on the market to have such benefits. The study, which involved more than 7000 patients with a history of T2DM and existing cardiovascular disease (CVD), found that 10.5% of patients in the empagliflozin group vs 12.1% in the placebo group died from a CV cause or experienced a nonfatal myocardial infarction or stroke over a median of 3.1 years. Results were similar with both doses (10 mg vs 25 mg) of empagliflozin. The mechanisms behind the CV benefits are likely multifactorial and may be related to reductions in weight and BP,33 but additional research is needed to fully elucidate the role of empagliflozin in this population.
Canagliflozin is being evaluated in the Canagliflozin Cardiovascular Assessment Study (CANVAS) for its effect on major CV events—CV death, nonfatal myocardial infarction, and nonfatal stroke—in patients with either a history of CVD or who are at increased risk of CVD and have uncontrolled diabetes.34 The trial is expected to wrap up in June 2017.
And dapagliflozin is being studied in the DECLARE-TIMI 58 trial (the Effect of Dapagliflozin on the Incidence of Cardiovascular Events) in patients with T2DM and either known CVD or at least 2 risk factors for CVD.35 The study is designed to assess dapagliflozin’s effect on the incidence of CV death, myocardial infarction, and ischemic stroke and has an estimated completion date of April 2019, which will provide a median follow-up of 4.5 years.
Cancer. All 3 agents have been examined for any possible carcinogenic links. In 2011, the FDA issued a request for further investigation surrounding the risk of cancer associated with dapagliflozin.36 As of November 2013, 10 of 6045 patients treated with dapagliflozin developed bladder cancer compared to 1 of 3512 controls.36 Furthermore, 9 of 2223 patients treated with dapagliflozin developed breast cancer compared to 1 of 1053 controls.36
Although the trials were not designed to detect an increase in risk, the number of observed cases warranted further investigation. No official warning for breast cancer exists since the characteristics of the malignancies led the FDA to believe dapagliflozin was unlikely the cause.36
Given what we know to date, it appears to be prudent to avoid prescribing SGLT2 inhibitors in patients with active bladder cancer, and to use them with caution in those with a history of the disease.2
Other studies. Initially, animal studies suggested an increased risk of various malignancies associated with canagliflozin use in rats,37 but consistent results were not seen in human studies. Similarly, at least one study found that empagliflozin was associated with lung cancer and melanoma, but closer examination found that most patients who developed these cancers had risk factors.38 Large, long-term studies of these agents in various populations are needed to thoroughly investigate possible carcinogenicity.
Additional considerations: Kidney function, age, and pregnancy
Consider avoiding SGLT2 inhibitors in patients with moderate kidney dysfunction (eGFR 30-59 mL/min/1.73 m2). Studies have shown that SGLT2 inhibitors are not as effective at lowering blood glucose in those with reduced eGFR, although adverse events were similar to those in placebo groups.24,39,40 Dapagliflozin is not recommended in patients with an eGFR <60 mL/min/1.73 m2 due to lack of efficacy.2,24 Empagliflozin does not require dose adjustments if eGFR is ≥45 mL/min/1.73 m2. A lower dose of canagliflozin (ie, 100 mg/d) is recommended in those with an eGFR of 45 to 59 mL/min/1.73 m2.2 All agents are contraindicated in patients with severe renal impairment (eGFR <30 mL/min/1.73 m2).
Older patients are at higher risk for dehydration, hypotension, and falls; therefore, SGLT2 inhibitors should be used with caution in this population. Similarly, they should not be used in patients with T1DM and should be avoided in those with active, or a history of, DKA.
There are no data on the use of SGLT2 inhibitors in pregnancy; thus, these agents should be avoided unless the potential benefits outweigh the potential risks to the unborn fetus.2
CASE 1 › An SGLT2 inhibitor is an acceptable option for Mr. S. Because he is resistant to starting insulin therapy and his HbA1c is <9%, an additional oral medication is reasonable. Adding an SGLT2 inhibitor may reduce his HbA1c up to ~1%, and education on lifestyle modifications may help bring him to goal. An SGLT2 inhibitor may also benefit his BP and weight, both of which could be improved.
Given the drugs he’s taking, drug interactions should not be an issue, and his renal function and pertinent labs (K, Phos, Mg) are within normal limits. Nevertheless, monitor these labs periodically and monitor Mr. S for adverse effects, such as UTIs, although these are more common in women. Canagliflozin is the preferred SGLT2 inhibitor on his insurance formulary, so you could initiate therapy at 100 mg/d, administered prior to the first meal, and increase to 300 mg/d if needed. As an alternative, consider prescribing the metformin/canagliflozin combination agent.
CASE 2 › Ms. R is likely experiencing a yeast infection as an adverse effect of the dapagliflozin. Although one yeast infection is insufficient grounds for discontinuation of the drug, recurrent infections should prompt a risk-to-benefit analysis to determine whether it’s worth continuing the medication. Her recent eGFR (<60 mL/min/1.73 m2) is, however, a contraindication to dapagliflozin, and therapy should be discontinued. Canagliflozin and empagliflozin may be considered since her eGFR is >45 mL/min/1.73 m2, but given her current HbA1c and recent adverse drug event, alternative therapies, such as basal insulin, are more appropriate treatment choices.
CORRESPONDENCE
Katelin M. Lisenby, PharmD, BCPS, University of Alabama College of Community Health Sciences, University Medical Center, Box 870374, Tuscaloosa, AL 35487; [email protected].
1. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2015;38:140-149.
2. Canagliflozin, dapagliflozin, empagliflozin. Lexicomp, Inc. (Lexi-Drugs®). Accessed October 12, 2015.
3. Stenlöf K, Cefalu WT, Kim KA, et al. Long-term efficacy and safety of canagliflozin monotherapy in patients with type 2 diabetes mellitus inadequately controlled with diet and exercise: findings from the 52-week CANTATA-M study. Curr Med Res Opin. 2014;30:163-175.
4. Ferrannini E, Ramos SJ, Salsali A, et al. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010:33:2217-2224.
5. Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1:208-219.
6. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36:4015-4021.
7. Wilding JPH, Charpentier G, Hollander P, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: a randomised trial. Int J Clin Pract. 2013;67:1267-1282.
8. Forst T, Guthrie R, Goldenberg R, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes on background metformin and pioglitazone. Diabetes Obes Metab. 2014;16:467-477.
9. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36:2508-2515.
10. Bristol-Myers Squibb [press release]. New phase III data showed dapagliflozin significantly reduced HbA1c compared to placebo at 24 weeks in patients with type 2 diabetes inadequately controlled with the combination of metformin plus sulfonylurea. Available at: http://news.bms.com/press-release/rd-news/new-phase-iii-data-showed-dapagliflozin-significantly-reduced-hba1c-compared-p&t=635156160653787526. Accessed September 17, 2015.
11. Jabbour SA, Hardy E, Sugg J, et al. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24- week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care. 2014;37:740-750.
12. DeFronzo RA, Lewin A, Patel S, et al. Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care. 2015;38:384-393.
13. Kovacs CS, Seshiah V, Merker L, et al. Empagliflozin as add-on therapy to pioglitazone with or without metformin in patients with type 2 diabetes mellitus. Clin Ther. 2015;37:1773-1788.
14. Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized double-blind, placebo-controlled trial. Diabetes Care. 2013;36:3396-3404.
15. Neal B, Percovik V, de Zeeuw D, et al. Efficacy and safety of canagliflozin, an inhibitor of sodium–glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38:403-411.
16. Wilding JPH, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156:405-415.
17. Rosenstock J, Jelaska A, Frappin G, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37:1815-1823.
18. Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941-950.
19. Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010:375:2223-2233.
20. Bailey CJ, Gross JL, Hennicken D, et al. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
21. Rosenstock J, Vico M, Wei L, et al. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473-1478.
22. Merker L, Häring HU, Christiansen AV, et al. Empagliflozin as add-on to metformin in people with type 2 diabetes. Diabet Med. 2015;32:1555-1567.
23. Ridderstråle M, Anderson KR, Zeller C, et al. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2:691-700.
24. Kohan DE, Fioretto P, Tang W, et al. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85:962-971.
25. Invokana (canagliflozin) tablets [product information]. Titusville, NJ: Janssen Pharmaceuticals Inc. Available at: https://www.invokana.com. Accessed March 15, 2013.
26. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56:2582-2592.
27. Strojek K, Yoon KH, Hruba V, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928-938.
28. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38:355-364.
29. US Food and Drug Administration. FDA drug safety communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Accessed July 11, 2016.
30. Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38:1638-1642.
31. US Food and Drug Administration. FDA drug safety communication: FDA revises label of diabetes drug canagliflozin (Invokana, Invokamet) to include updates on bone fracture risk and new information on decreased bone mineral density. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm461449.htm. Acces-sed July 11, 2016.
32. Canagliflozin, dapagliflozin, empagliflozin. In: RED BOOK [AUHSOP intranet database]. Greenwood Village, CO: Truven Health Analytics; [updated daily]. Available at: http://www.micromedexsolutions.com/micromedex2/librarian/ND_T/evidencexpert/ND_PR/evidencexpert/CS/BB1644/ND_AppProduct/evidencexpert/DUPLICATIONSHIELDSYNC/FAF693/ND_PG/evidencexpert/ND_B/evidencexpert/ND_P/evidencexpert/PFActionId/redbook.ShowProductSearchResults?SearchTerm=JARDIANCE&searchType=redbookProductName&searchTermId=42798&searchContent=%24searchContent&searchFilterAD=filterADActive&searchFilterRepackager=filterExcludeRepackager&searchPattern=%5Ejard. Accessed March 15, 2016.
33. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
34. CANagliflozin cardioVascular Assessment Study (CANVAS). Available at: http://clinicaltrials.gov/show/NCT01032629. Accessed October 12, 2015.
35. Multicenter trial to evaluate the effect of dapagliflozin on the incidence of cardiovascular events (DECLARE-TIMI 58). Available at: http://clinicaltrials.gov/show/NCT01730534. Accessed October 12, 2015.
36. FDA background document. BMS-512148 NDA 202293. In: Proceedings of the US Food and Drug Administration Endocrinologic & Metabolic Drug Advisory Committee Meeting, 2013. Available at: http://www.fda.gov/downloads/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm378079.pdf. Accessed October 12, 2015.
37. Lin HW, Tseng CH. A review of the relationship between SGLT2 inhibitors and cancer. Int J Endocrinol. 2014;2014:719578.
38. Center for Drug Evaluation and Research. Risk assessment and risk mitigation review(s). July 28, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/ 204629Orig1s000RiskR.pdf. Accessed September 21, 2015.
39. Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes Metab. 2014;16:1016-1027.
40. Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2: 369-384.
1. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2015;38:140-149.
2. Canagliflozin, dapagliflozin, empagliflozin. Lexicomp, Inc. (Lexi-Drugs®). Accessed October 12, 2015.
3. Stenlöf K, Cefalu WT, Kim KA, et al. Long-term efficacy and safety of canagliflozin monotherapy in patients with type 2 diabetes mellitus inadequately controlled with diet and exercise: findings from the 52-week CANTATA-M study. Curr Med Res Opin. 2014;30:163-175.
4. Ferrannini E, Ramos SJ, Salsali A, et al. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010:33:2217-2224.
5. Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1:208-219.
6. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36:4015-4021.
7. Wilding JPH, Charpentier G, Hollander P, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: a randomised trial. Int J Clin Pract. 2013;67:1267-1282.
8. Forst T, Guthrie R, Goldenberg R, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes on background metformin and pioglitazone. Diabetes Obes Metab. 2014;16:467-477.
9. Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36:2508-2515.
10. Bristol-Myers Squibb [press release]. New phase III data showed dapagliflozin significantly reduced HbA1c compared to placebo at 24 weeks in patients with type 2 diabetes inadequately controlled with the combination of metformin plus sulfonylurea. Available at: http://news.bms.com/press-release/rd-news/new-phase-iii-data-showed-dapagliflozin-significantly-reduced-hba1c-compared-p&t=635156160653787526. Accessed September 17, 2015.
11. Jabbour SA, Hardy E, Sugg J, et al. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24- week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care. 2014;37:740-750.
12. DeFronzo RA, Lewin A, Patel S, et al. Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care. 2015;38:384-393.
13. Kovacs CS, Seshiah V, Merker L, et al. Empagliflozin as add-on therapy to pioglitazone with or without metformin in patients with type 2 diabetes mellitus. Clin Ther. 2015;37:1773-1788.
14. Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized double-blind, placebo-controlled trial. Diabetes Care. 2013;36:3396-3404.
15. Neal B, Percovik V, de Zeeuw D, et al. Efficacy and safety of canagliflozin, an inhibitor of sodium–glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38:403-411.
16. Wilding JPH, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156:405-415.
17. Rosenstock J, Jelaska A, Frappin G, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37:1815-1823.
18. Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941-950.
19. Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010:375:2223-2233.
20. Bailey CJ, Gross JL, Hennicken D, et al. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
21. Rosenstock J, Vico M, Wei L, et al. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473-1478.
22. Merker L, Häring HU, Christiansen AV, et al. Empagliflozin as add-on to metformin in people with type 2 diabetes. Diabet Med. 2015;32:1555-1567.
23. Ridderstråle M, Anderson KR, Zeller C, et al. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2:691-700.
24. Kohan DE, Fioretto P, Tang W, et al. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85:962-971.
25. Invokana (canagliflozin) tablets [product information]. Titusville, NJ: Janssen Pharmaceuticals Inc. Available at: https://www.invokana.com. Accessed March 15, 2013.
26. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56:2582-2592.
27. Strojek K, Yoon KH, Hruba V, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928-938.
28. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38:355-364.
29. US Food and Drug Administration. FDA drug safety communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Accessed July 11, 2016.
30. Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38:1638-1642.
31. US Food and Drug Administration. FDA drug safety communication: FDA revises label of diabetes drug canagliflozin (Invokana, Invokamet) to include updates on bone fracture risk and new information on decreased bone mineral density. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm461449.htm. Acces-sed July 11, 2016.
32. Canagliflozin, dapagliflozin, empagliflozin. In: RED BOOK [AUHSOP intranet database]. Greenwood Village, CO: Truven Health Analytics; [updated daily]. Available at: http://www.micromedexsolutions.com/micromedex2/librarian/ND_T/evidencexpert/ND_PR/evidencexpert/CS/BB1644/ND_AppProduct/evidencexpert/DUPLICATIONSHIELDSYNC/FAF693/ND_PG/evidencexpert/ND_B/evidencexpert/ND_P/evidencexpert/PFActionId/redbook.ShowProductSearchResults?SearchTerm=JARDIANCE&searchType=redbookProductName&searchTermId=42798&searchContent=%24searchContent&searchFilterAD=filterADActive&searchFilterRepackager=filterExcludeRepackager&searchPattern=%5Ejard. Accessed March 15, 2016.
33. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
34. CANagliflozin cardioVascular Assessment Study (CANVAS). Available at: http://clinicaltrials.gov/show/NCT01032629. Accessed October 12, 2015.
35. Multicenter trial to evaluate the effect of dapagliflozin on the incidence of cardiovascular events (DECLARE-TIMI 58). Available at: http://clinicaltrials.gov/show/NCT01730534. Accessed October 12, 2015.
36. FDA background document. BMS-512148 NDA 202293. In: Proceedings of the US Food and Drug Administration Endocrinologic & Metabolic Drug Advisory Committee Meeting, 2013. Available at: http://www.fda.gov/downloads/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm378079.pdf. Accessed October 12, 2015.
37. Lin HW, Tseng CH. A review of the relationship between SGLT2 inhibitors and cancer. Int J Endocrinol. 2014;2014:719578.
38. Center for Drug Evaluation and Research. Risk assessment and risk mitigation review(s). July 28, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/ 204629Orig1s000RiskR.pdf. Accessed September 21, 2015.
39. Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes Metab. 2014;16:1016-1027.
40. Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2: 369-384.
A new paradigm for pain?
The care of people with pain has been wrought with ineffective and unnecessary treatment, including the misuse of opioids, largely because we do not have an accurate conceptualization of pain. The absence of animal and human models of central nervous system (CNS) pain processing ensures that our understanding of pain will remain incomplete for the foreseeable future, but enough evidence exists to help family physicians develop an understanding of pain that goes beyond what we learned in medical school and that can help us more effectively treat patients with pain.
In this review, we will briefly discuss the established concepts of nociceptive and neuropathic pain. And then, with those concepts in mind, we will explore a third type of pain that for lack of a better term, we will call “pain for psychological reasons.” We hypothesize that this pain may be the consequence of changes in nervous system function that arise from developmental trauma, other traumatic experiences in a patient’s life, or mental health disorders. It is this third type of pain that may offer us insights into conditions such as fibromyalgia.
While we do not yet have validated diagnostic criteria for this third type of pain, we believe that there is enough information to present initial criteria so that one may distinguish it from nociceptive and neuropathic pain.
Nociceptive and neuropathic pain: The current paradigm
Nociceptive pain. The sensory pain experience, or nociceptive pain, is produced by noxious stimuli that either damage, or are capable of damaging, tissues (eg, burns, cuts, fractures, inflammation, and increased pressure in a hollow viscus). Noxious stimuli are detected at the molecular level by specific pain sensory receptors embedded in our tissues called nociceptors.
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps—transduction, transmission, modulation, and perception—which are described in “From periphery to brain: The process of nociceptive pain.”1-4
Neuropathic pain. While nociceptive pain can be easily traced from a peripheral nociceptive fiber to the brain and typically resolves when the nociceptive stimulus stops, neuropathic pain (NPP) results from changes to the function of the nervous system and is typically caused by injury to the nerves. Such changes, referred to as neuronal sensitization, may not quickly resolve, as is the case with postherpetic neuralgia. In fact, the changes can become permanent. NPP fundamentally differs from nociceptive pain because it results from changes in the central processing of pain that can lead a person to perceive pain sensations even in the absence of tissue pathology.
Common causes of NPP that persists even after tissue damage has healed include trauma (eg, amputation of a limb), ischemia (eg, pressure palsy), disease (eg, the metabolic injury of diabetes or the injury caused by a shingles infection), and drug treatment (eg, chemotherapy). The underlying mechanisms of NPP and the neuronal plasticity (the ability of the nervous system to rewire itself) that initiate and then maintain NPP are important areas of active research that may eventually lead to the development of more effective treatments.
Timing is critical. Neuroplastic changes in the nervous system following nerve injury are time-dependent. Synaptic plasticity can occur within seconds to minutes, while cellular plasticity occurs within hours to days. Synaptic and cellular plasticity happen relatively fast and may be reversible.
In contrast, systems plasticity (when new CNS neuronal connections are formed in response to nerve injury) takes place over the months and years following nerve injury and is often irreversible. When we recognize NPP and intervene before system neuroplastic changes occur, it may be possible to prevent pain from becoming chronic (TABLE 15). In cases of nerve injury, researchers have long suspected that early and aggressive pain treatment within the first few months that may include sympathetic and peripheral neural blockade reduces the likelihood that the patient will have chronic pain.6,7
It’s time to update our understanding of pain
The International Association for the Study of Pain (IASP)—a group of health care providers, scientists, and policymakers seeking to improve pain relief worldwide—notes in its definition of pain that the complaint, “I hurt” does not necessarily imply that there is a painful stimulus in the form of tissue injury.8 Yet most of us have been taught to think of pain solely as the result of tissue pathology, and we assume that emotional factors merely modify how the physical damage is perceived. This traditional concept of pain is incomplete. It leads clinicians to misdiagnose the cause of pain, initiate expensive and unnecessary treatment, engage in well-meaning but misguided prescribing behavior, and miss opportunities to help patients.
SIDEBAR
From periphery to brain: The process of nociceptive pain1-4
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps:
In transduction, nociceptors containing special molecular proteins respond to noxious modalities, such as thermal, mechanical, or chemical stimuli, and trigger nerve impulses in the nociceptive nerve fibers (nerves dedicated to pain sensation).
During transmission—the second stage of the process—information from the nociceptors in the periphery (skin, muscle, viscera) is relayed to the spinal cord mainly by 2 types of nociceptive neurons: C-fibers and A delta (Aδ) fibers. Both approach the spinal cord in a peripheral nerve and then enter the spinal cord in the dorsal root entry zone. Because Aδ fibers are thinly myelinated, they send impulses faster than unmyelinated C fibers. This is why when injury occurs, we first feel sharp, acute pain that then slowly diffuses into a duller ache.
Once the incoming signal is transmitted to the CNS at the spinal cord, primary afferent neurons synapse on second order neurons. From there, information travels on to the thalamus via multiple neurons that have the capacity to change their response patterns when activity of nociceptive fibers is sustained (as occurs in the setting of a tissue or nerve injury and perhaps in the setting of psychological trauma). This is known as modulation of the incoming nociceptive stimulus. During this step of the process, stimuli can be amplified, suppressed, or even transformed from one type to another (eg, a light touch can be modulated in such a way that it will be perceived as a burning sensation). Also, it is this step that is affected by many medications, by intrathecal drug infusions, and by spinal neurostimulators.
In perception, the thalamus then directs the pain sensation to multiple brain centers. At this step, the stimulus is finally consciously perceived as pain by the individual.
Cortical pain circuits can be activated without physical input (ie, no tissue damage, noxious stimuli, or nerve injury). This becomes important in understanding pain syndromes, such as fibromyalgia.
Pain in the absence of any pathophysiologic cause or injury
The clinician’s search for a pain diagnosis is typically predicated on the notion that there must be an underlying tissue injury of severity equal to the severity of the patient’s pain complaints. This approach to a pain evaluation rests on 2 assumptions that are not true for all patients:
- Pain is simply a sensory experience that is always caused by tissue damage of some type.
- The severity of the pain experienced by a patient should be tightly bound to the severity of the pain stimulus (ie, tissue damage).
These assumptions are true of acute nociceptive pain, they may or may not be true for NPP, but they do not apply to the third type of pain—pain for psychological reasons. While tissue pathology in humans and animals with nociceptive pain is usually visible, measurable, and correlates with observed pain behaviors, the damage to nerve tissue and the ensuing changes in nervous system function with NPP are not always visible or able to be imaged. These changes produce pain that can appear more severe than expected based on a brief exam. Some of the time, however, characteristic symptoms and physical signs of NPP will be present, and perhaps electrodiagnostic or other tests will be abnormal, thus providing some objective sense of changes in nervous system function.
In contrast, pain behavior due to the third type of pain usually appears very much out of proportion, and unbound to, tissue pathology. Furthermore, the patient’s pain behaviors often reflect heightened emotional pain processing (TABLE 29). The resulting emotionally charged presentation can be alarming and suggestive of extreme tissue injury, but there may be absolutely no evidence of tissue injury or pathology.

Functional change in the CNS
There is evidence from experimental studies that psychologic factors change nervous system function. In one review, the authors concluded, “Pain…can vary widely between people and even within an individual depending on…the psychological state of the person.”10 In a second review, the authors concluded that our emotional state has an enormous influence on pain; a negative emotional state increases pain, whereas a positive state lowers pain.11
But can psychological factors induce long-term changes in nervous system function analogous to the systems neuroplasticity responsible for irreversible changes in NPP? And can psychologically induced changes in nervous system sensory processing lead to pain without any tissue or nerve damage?
We theorize that a functional change in the CNS can occur in response to certain emotional states or traumatic experiences (eg, child abuse, assault, accidents). (More on this in a bit.) When such changes occur, mildly painful stimuli are amplified and processed through overly sensitized, dysregulated, ramped-up emotional and somatosensory pain circuits in the brain. This is analogous to the functional changes in the nervous system that occur with NPP; however, when the nervous system changes are due to psychological factors, there may be no tissue or nerve injury.
Childhood trauma influences adult pain. One of the more compelling narratives emerging in health care has to do with the influence that childhood developmental trauma can have on health, including pain. In his chapter on the impact of early life trauma on health and disease, Lanius states:12
“Women were 50% more likely than men to have experienced 5 or more categories of adverse childhood experiences. We believe that here is a key to what in mainstream epidemiology appears as women’s natural proneness to ill-defined health problems like fibromyalgia, chronic fatigue syndrome, obesity, irritable bowel syndrome, and chronic non-malignant pain syndromes. In light of our findings, we now see these as medical constructs, artifacts resulting from medical blindness to social realities and ignorance of the impact of gender.”
Lanius12 suggests that adverse childhood experiences13 (trauma such as abuse and sexual assault) can lead to long-term changes within the nervous system, including areas of pain processing. My coauthor and I describe these changes here in terms of nervous system sensitization or dysregulation, and we believe that these changes lead to a bias toward hyperactivation of emotional pain circuits, which leads to the emotionally laden pain behaviors that often seem out of proportion to tissue pathology.
1. Dubner R, Gold M. The neurobiology of pain. Proc Natl Acad Sci U S A. 1999;96:7627-7630.
2. Markenson JA. Mechanisms of chronic pain. Am J Med. 1996;101:S6-S18.
3. Rainville P, Duncan GH, Price DD, et al. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968-971.
4. Bushnell MC, Duncan GH. Sensory and affective aspects of pain perception: is medial thalamus restricted to emotional issues? Exp Brain Res. 1989;78:415-418.
5. Galer BS, Jensen MP. Development and preliminary validation of a pain measure specific to neuropathic pain: the Neuropathic Pain Scale. Neurology. 1997;48:332-338.
6. Fassoulaki A, Triga A, Melemeni A, et al. Multimodal analgesia with gabapentin and local anesthetics prevents acute and chronic pain after breast surgery for cancer. Anesth Analg. 2005;101:1427-1432.
7. Woolf CJ, Chong MS. Preemptive analgesia—treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg. 1993;77:362-379.
8. International Association for the Study of Pain Web site. IASP Taxonomy. Available at: http://www.iasp-pain.org/Taxonomy. Accessed January 10, 2016.
9. Waddell G, McCulloch JA, Kummel E, et al. Nonorganic physical signs in low-back pain. Spine. 1980;5:117-125.
10. Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14:502-511.
11. Villemure C, Bushnell MC. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95:195-199.
12. Felitti VJ, Anda RF. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders, and sexual behavior: implications for healthcare. In: Lanius R, Vermetten E, eds. The Hidden Epidemic: The Impact of Early Life Trauma on Health and Disease. Cambridge University Press; 2010. Available at: http://www.unnaturalcauses.org/assets/uploads/file/ACE%20Study-Lanius.pdf. Accessed January 11, 2016.
13. Centers for Disease Control and Prevention. Injury prevention and control: Division of violence prevention. Adverse childhood experiences. Available at: http://www.cdc.gov/violenceprevention/acestudy/. Accessed January 11, 2016.
14. Kross E, Berman MG, Mischel W, et al. Social rejection shares somatosensory representations with physical pain. Proc Natl Acad Sci. 2011;108:6270-6275.
15. Geuze E, Westenberg HGM, Jochims A, et al. Altered pain processing in veterans with posttraumatic stress disorder. Arch Gen Psychiatry. 2007;64:76-85.
16. Mickleborough MJ, Daniels JK, Coupland NJ. Effects of trauma-related cues on pain processing in posttraumatic stress disorder: an fMRI investigation. J Psychiatry Neurosci. 2011;36: 6-14.
17. Noll-Hussong M, Otti A, Laeer L, et al. Aftermath of sexual abuse history on adult patients suffering from chronic functional pain syndromes: an fMRI pilot study. J Psychoso Res. 2010; 68:483-487.
The care of people with pain has been wrought with ineffective and unnecessary treatment, including the misuse of opioids, largely because we do not have an accurate conceptualization of pain. The absence of animal and human models of central nervous system (CNS) pain processing ensures that our understanding of pain will remain incomplete for the foreseeable future, but enough evidence exists to help family physicians develop an understanding of pain that goes beyond what we learned in medical school and that can help us more effectively treat patients with pain.
In this review, we will briefly discuss the established concepts of nociceptive and neuropathic pain. And then, with those concepts in mind, we will explore a third type of pain that for lack of a better term, we will call “pain for psychological reasons.” We hypothesize that this pain may be the consequence of changes in nervous system function that arise from developmental trauma, other traumatic experiences in a patient’s life, or mental health disorders. It is this third type of pain that may offer us insights into conditions such as fibromyalgia.
While we do not yet have validated diagnostic criteria for this third type of pain, we believe that there is enough information to present initial criteria so that one may distinguish it from nociceptive and neuropathic pain.
Nociceptive and neuropathic pain: The current paradigm
Nociceptive pain. The sensory pain experience, or nociceptive pain, is produced by noxious stimuli that either damage, or are capable of damaging, tissues (eg, burns, cuts, fractures, inflammation, and increased pressure in a hollow viscus). Noxious stimuli are detected at the molecular level by specific pain sensory receptors embedded in our tissues called nociceptors.
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps—transduction, transmission, modulation, and perception—which are described in “From periphery to brain: The process of nociceptive pain.”1-4
Neuropathic pain. While nociceptive pain can be easily traced from a peripheral nociceptive fiber to the brain and typically resolves when the nociceptive stimulus stops, neuropathic pain (NPP) results from changes to the function of the nervous system and is typically caused by injury to the nerves. Such changes, referred to as neuronal sensitization, may not quickly resolve, as is the case with postherpetic neuralgia. In fact, the changes can become permanent. NPP fundamentally differs from nociceptive pain because it results from changes in the central processing of pain that can lead a person to perceive pain sensations even in the absence of tissue pathology.
Common causes of NPP that persists even after tissue damage has healed include trauma (eg, amputation of a limb), ischemia (eg, pressure palsy), disease (eg, the metabolic injury of diabetes or the injury caused by a shingles infection), and drug treatment (eg, chemotherapy). The underlying mechanisms of NPP and the neuronal plasticity (the ability of the nervous system to rewire itself) that initiate and then maintain NPP are important areas of active research that may eventually lead to the development of more effective treatments.
Timing is critical. Neuroplastic changes in the nervous system following nerve injury are time-dependent. Synaptic plasticity can occur within seconds to minutes, while cellular plasticity occurs within hours to days. Synaptic and cellular plasticity happen relatively fast and may be reversible.
In contrast, systems plasticity (when new CNS neuronal connections are formed in response to nerve injury) takes place over the months and years following nerve injury and is often irreversible. When we recognize NPP and intervene before system neuroplastic changes occur, it may be possible to prevent pain from becoming chronic (TABLE 15). In cases of nerve injury, researchers have long suspected that early and aggressive pain treatment within the first few months that may include sympathetic and peripheral neural blockade reduces the likelihood that the patient will have chronic pain.6,7
It’s time to update our understanding of pain
The International Association for the Study of Pain (IASP)—a group of health care providers, scientists, and policymakers seeking to improve pain relief worldwide—notes in its definition of pain that the complaint, “I hurt” does not necessarily imply that there is a painful stimulus in the form of tissue injury.8 Yet most of us have been taught to think of pain solely as the result of tissue pathology, and we assume that emotional factors merely modify how the physical damage is perceived. This traditional concept of pain is incomplete. It leads clinicians to misdiagnose the cause of pain, initiate expensive and unnecessary treatment, engage in well-meaning but misguided prescribing behavior, and miss opportunities to help patients.
SIDEBAR
From periphery to brain: The process of nociceptive pain1-4
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps:
In transduction, nociceptors containing special molecular proteins respond to noxious modalities, such as thermal, mechanical, or chemical stimuli, and trigger nerve impulses in the nociceptive nerve fibers (nerves dedicated to pain sensation).
During transmission—the second stage of the process—information from the nociceptors in the periphery (skin, muscle, viscera) is relayed to the spinal cord mainly by 2 types of nociceptive neurons: C-fibers and A delta (Aδ) fibers. Both approach the spinal cord in a peripheral nerve and then enter the spinal cord in the dorsal root entry zone. Because Aδ fibers are thinly myelinated, they send impulses faster than unmyelinated C fibers. This is why when injury occurs, we first feel sharp, acute pain that then slowly diffuses into a duller ache.
Once the incoming signal is transmitted to the CNS at the spinal cord, primary afferent neurons synapse on second order neurons. From there, information travels on to the thalamus via multiple neurons that have the capacity to change their response patterns when activity of nociceptive fibers is sustained (as occurs in the setting of a tissue or nerve injury and perhaps in the setting of psychological trauma). This is known as modulation of the incoming nociceptive stimulus. During this step of the process, stimuli can be amplified, suppressed, or even transformed from one type to another (eg, a light touch can be modulated in such a way that it will be perceived as a burning sensation). Also, it is this step that is affected by many medications, by intrathecal drug infusions, and by spinal neurostimulators.
In perception, the thalamus then directs the pain sensation to multiple brain centers. At this step, the stimulus is finally consciously perceived as pain by the individual.
Cortical pain circuits can be activated without physical input (ie, no tissue damage, noxious stimuli, or nerve injury). This becomes important in understanding pain syndromes, such as fibromyalgia.
Pain in the absence of any pathophysiologic cause or injury
The clinician’s search for a pain diagnosis is typically predicated on the notion that there must be an underlying tissue injury of severity equal to the severity of the patient’s pain complaints. This approach to a pain evaluation rests on 2 assumptions that are not true for all patients:
- Pain is simply a sensory experience that is always caused by tissue damage of some type.
- The severity of the pain experienced by a patient should be tightly bound to the severity of the pain stimulus (ie, tissue damage).
These assumptions are true of acute nociceptive pain, they may or may not be true for NPP, but they do not apply to the third type of pain—pain for psychological reasons. While tissue pathology in humans and animals with nociceptive pain is usually visible, measurable, and correlates with observed pain behaviors, the damage to nerve tissue and the ensuing changes in nervous system function with NPP are not always visible or able to be imaged. These changes produce pain that can appear more severe than expected based on a brief exam. Some of the time, however, characteristic symptoms and physical signs of NPP will be present, and perhaps electrodiagnostic or other tests will be abnormal, thus providing some objective sense of changes in nervous system function.
In contrast, pain behavior due to the third type of pain usually appears very much out of proportion, and unbound to, tissue pathology. Furthermore, the patient’s pain behaviors often reflect heightened emotional pain processing (TABLE 29). The resulting emotionally charged presentation can be alarming and suggestive of extreme tissue injury, but there may be absolutely no evidence of tissue injury or pathology.
Functional change in the CNS
There is evidence from experimental studies that psychologic factors change nervous system function. In one review, the authors concluded, “Pain…can vary widely between people and even within an individual depending on…the psychological state of the person.”10 In a second review, the authors concluded that our emotional state has an enormous influence on pain; a negative emotional state increases pain, whereas a positive state lowers pain.11
But can psychological factors induce long-term changes in nervous system function analogous to the systems neuroplasticity responsible for irreversible changes in NPP? And can psychologically induced changes in nervous system sensory processing lead to pain without any tissue or nerve damage?
We theorize that a functional change in the CNS can occur in response to certain emotional states or traumatic experiences (eg, child abuse, assault, accidents). (More on this in a bit.) When such changes occur, mildly painful stimuli are amplified and processed through overly sensitized, dysregulated, ramped-up emotional and somatosensory pain circuits in the brain. This is analogous to the functional changes in the nervous system that occur with NPP; however, when the nervous system changes are due to psychological factors, there may be no tissue or nerve injury.
Childhood trauma influences adult pain. One of the more compelling narratives emerging in health care has to do with the influence that childhood developmental trauma can have on health, including pain. In his chapter on the impact of early life trauma on health and disease, Lanius states:12
“Women were 50% more likely than men to have experienced 5 or more categories of adverse childhood experiences. We believe that here is a key to what in mainstream epidemiology appears as women’s natural proneness to ill-defined health problems like fibromyalgia, chronic fatigue syndrome, obesity, irritable bowel syndrome, and chronic non-malignant pain syndromes. In light of our findings, we now see these as medical constructs, artifacts resulting from medical blindness to social realities and ignorance of the impact of gender.”
Lanius12 suggests that adverse childhood experiences13 (trauma such as abuse and sexual assault) can lead to long-term changes within the nervous system, including areas of pain processing. My coauthor and I describe these changes here in terms of nervous system sensitization or dysregulation, and we believe that these changes lead to a bias toward hyperactivation of emotional pain circuits, which leads to the emotionally laden pain behaviors that often seem out of proportion to tissue pathology.
The care of people with pain has been wrought with ineffective and unnecessary treatment, including the misuse of opioids, largely because we do not have an accurate conceptualization of pain. The absence of animal and human models of central nervous system (CNS) pain processing ensures that our understanding of pain will remain incomplete for the foreseeable future, but enough evidence exists to help family physicians develop an understanding of pain that goes beyond what we learned in medical school and that can help us more effectively treat patients with pain.
In this review, we will briefly discuss the established concepts of nociceptive and neuropathic pain. And then, with those concepts in mind, we will explore a third type of pain that for lack of a better term, we will call “pain for psychological reasons.” We hypothesize that this pain may be the consequence of changes in nervous system function that arise from developmental trauma, other traumatic experiences in a patient’s life, or mental health disorders. It is this third type of pain that may offer us insights into conditions such as fibromyalgia.
While we do not yet have validated diagnostic criteria for this third type of pain, we believe that there is enough information to present initial criteria so that one may distinguish it from nociceptive and neuropathic pain.
Nociceptive and neuropathic pain: The current paradigm
Nociceptive pain. The sensory pain experience, or nociceptive pain, is produced by noxious stimuli that either damage, or are capable of damaging, tissues (eg, burns, cuts, fractures, inflammation, and increased pressure in a hollow viscus). Noxious stimuli are detected at the molecular level by specific pain sensory receptors embedded in our tissues called nociceptors.
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps—transduction, transmission, modulation, and perception—which are described in “From periphery to brain: The process of nociceptive pain.”1-4
Neuropathic pain. While nociceptive pain can be easily traced from a peripheral nociceptive fiber to the brain and typically resolves when the nociceptive stimulus stops, neuropathic pain (NPP) results from changes to the function of the nervous system and is typically caused by injury to the nerves. Such changes, referred to as neuronal sensitization, may not quickly resolve, as is the case with postherpetic neuralgia. In fact, the changes can become permanent. NPP fundamentally differs from nociceptive pain because it results from changes in the central processing of pain that can lead a person to perceive pain sensations even in the absence of tissue pathology.
Common causes of NPP that persists even after tissue damage has healed include trauma (eg, amputation of a limb), ischemia (eg, pressure palsy), disease (eg, the metabolic injury of diabetes or the injury caused by a shingles infection), and drug treatment (eg, chemotherapy). The underlying mechanisms of NPP and the neuronal plasticity (the ability of the nervous system to rewire itself) that initiate and then maintain NPP are important areas of active research that may eventually lead to the development of more effective treatments.
Timing is critical. Neuroplastic changes in the nervous system following nerve injury are time-dependent. Synaptic plasticity can occur within seconds to minutes, while cellular plasticity occurs within hours to days. Synaptic and cellular plasticity happen relatively fast and may be reversible.
In contrast, systems plasticity (when new CNS neuronal connections are formed in response to nerve injury) takes place over the months and years following nerve injury and is often irreversible. When we recognize NPP and intervene before system neuroplastic changes occur, it may be possible to prevent pain from becoming chronic (TABLE 15). In cases of nerve injury, researchers have long suspected that early and aggressive pain treatment within the first few months that may include sympathetic and peripheral neural blockade reduces the likelihood that the patient will have chronic pain.6,7
It’s time to update our understanding of pain
The International Association for the Study of Pain (IASP)—a group of health care providers, scientists, and policymakers seeking to improve pain relief worldwide—notes in its definition of pain that the complaint, “I hurt” does not necessarily imply that there is a painful stimulus in the form of tissue injury.8 Yet most of us have been taught to think of pain solely as the result of tissue pathology, and we assume that emotional factors merely modify how the physical damage is perceived. This traditional concept of pain is incomplete. It leads clinicians to misdiagnose the cause of pain, initiate expensive and unnecessary treatment, engage in well-meaning but misguided prescribing behavior, and miss opportunities to help patients.
SIDEBAR
From periphery to brain: The process of nociceptive pain1-4
The process by which noxious stimuli lead to the experience of sensory pain consists of 4 steps:
In transduction, nociceptors containing special molecular proteins respond to noxious modalities, such as thermal, mechanical, or chemical stimuli, and trigger nerve impulses in the nociceptive nerve fibers (nerves dedicated to pain sensation).
During transmission—the second stage of the process—information from the nociceptors in the periphery (skin, muscle, viscera) is relayed to the spinal cord mainly by 2 types of nociceptive neurons: C-fibers and A delta (Aδ) fibers. Both approach the spinal cord in a peripheral nerve and then enter the spinal cord in the dorsal root entry zone. Because Aδ fibers are thinly myelinated, they send impulses faster than unmyelinated C fibers. This is why when injury occurs, we first feel sharp, acute pain that then slowly diffuses into a duller ache.
Once the incoming signal is transmitted to the CNS at the spinal cord, primary afferent neurons synapse on second order neurons. From there, information travels on to the thalamus via multiple neurons that have the capacity to change their response patterns when activity of nociceptive fibers is sustained (as occurs in the setting of a tissue or nerve injury and perhaps in the setting of psychological trauma). This is known as modulation of the incoming nociceptive stimulus. During this step of the process, stimuli can be amplified, suppressed, or even transformed from one type to another (eg, a light touch can be modulated in such a way that it will be perceived as a burning sensation). Also, it is this step that is affected by many medications, by intrathecal drug infusions, and by spinal neurostimulators.
In perception, the thalamus then directs the pain sensation to multiple brain centers. At this step, the stimulus is finally consciously perceived as pain by the individual.
Cortical pain circuits can be activated without physical input (ie, no tissue damage, noxious stimuli, or nerve injury). This becomes important in understanding pain syndromes, such as fibromyalgia.
Pain in the absence of any pathophysiologic cause or injury
The clinician’s search for a pain diagnosis is typically predicated on the notion that there must be an underlying tissue injury of severity equal to the severity of the patient’s pain complaints. This approach to a pain evaluation rests on 2 assumptions that are not true for all patients:
- Pain is simply a sensory experience that is always caused by tissue damage of some type.
- The severity of the pain experienced by a patient should be tightly bound to the severity of the pain stimulus (ie, tissue damage).
These assumptions are true of acute nociceptive pain, they may or may not be true for NPP, but they do not apply to the third type of pain—pain for psychological reasons. While tissue pathology in humans and animals with nociceptive pain is usually visible, measurable, and correlates with observed pain behaviors, the damage to nerve tissue and the ensuing changes in nervous system function with NPP are not always visible or able to be imaged. These changes produce pain that can appear more severe than expected based on a brief exam. Some of the time, however, characteristic symptoms and physical signs of NPP will be present, and perhaps electrodiagnostic or other tests will be abnormal, thus providing some objective sense of changes in nervous system function.
In contrast, pain behavior due to the third type of pain usually appears very much out of proportion, and unbound to, tissue pathology. Furthermore, the patient’s pain behaviors often reflect heightened emotional pain processing (TABLE 29). The resulting emotionally charged presentation can be alarming and suggestive of extreme tissue injury, but there may be absolutely no evidence of tissue injury or pathology.
Functional change in the CNS
There is evidence from experimental studies that psychologic factors change nervous system function. In one review, the authors concluded, “Pain…can vary widely between people and even within an individual depending on…the psychological state of the person.”10 In a second review, the authors concluded that our emotional state has an enormous influence on pain; a negative emotional state increases pain, whereas a positive state lowers pain.11
But can psychological factors induce long-term changes in nervous system function analogous to the systems neuroplasticity responsible for irreversible changes in NPP? And can psychologically induced changes in nervous system sensory processing lead to pain without any tissue or nerve damage?
We theorize that a functional change in the CNS can occur in response to certain emotional states or traumatic experiences (eg, child abuse, assault, accidents). (More on this in a bit.) When such changes occur, mildly painful stimuli are amplified and processed through overly sensitized, dysregulated, ramped-up emotional and somatosensory pain circuits in the brain. This is analogous to the functional changes in the nervous system that occur with NPP; however, when the nervous system changes are due to psychological factors, there may be no tissue or nerve injury.
Childhood trauma influences adult pain. One of the more compelling narratives emerging in health care has to do with the influence that childhood developmental trauma can have on health, including pain. In his chapter on the impact of early life trauma on health and disease, Lanius states:12
“Women were 50% more likely than men to have experienced 5 or more categories of adverse childhood experiences. We believe that here is a key to what in mainstream epidemiology appears as women’s natural proneness to ill-defined health problems like fibromyalgia, chronic fatigue syndrome, obesity, irritable bowel syndrome, and chronic non-malignant pain syndromes. In light of our findings, we now see these as medical constructs, artifacts resulting from medical blindness to social realities and ignorance of the impact of gender.”
Lanius12 suggests that adverse childhood experiences13 (trauma such as abuse and sexual assault) can lead to long-term changes within the nervous system, including areas of pain processing. My coauthor and I describe these changes here in terms of nervous system sensitization or dysregulation, and we believe that these changes lead to a bias toward hyperactivation of emotional pain circuits, which leads to the emotionally laden pain behaviors that often seem out of proportion to tissue pathology.
1. Dubner R, Gold M. The neurobiology of pain. Proc Natl Acad Sci U S A. 1999;96:7627-7630.
2. Markenson JA. Mechanisms of chronic pain. Am J Med. 1996;101:S6-S18.
3. Rainville P, Duncan GH, Price DD, et al. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968-971.
4. Bushnell MC, Duncan GH. Sensory and affective aspects of pain perception: is medial thalamus restricted to emotional issues? Exp Brain Res. 1989;78:415-418.
5. Galer BS, Jensen MP. Development and preliminary validation of a pain measure specific to neuropathic pain: the Neuropathic Pain Scale. Neurology. 1997;48:332-338.
6. Fassoulaki A, Triga A, Melemeni A, et al. Multimodal analgesia with gabapentin and local anesthetics prevents acute and chronic pain after breast surgery for cancer. Anesth Analg. 2005;101:1427-1432.
7. Woolf CJ, Chong MS. Preemptive analgesia—treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg. 1993;77:362-379.
8. International Association for the Study of Pain Web site. IASP Taxonomy. Available at: http://www.iasp-pain.org/Taxonomy. Accessed January 10, 2016.
9. Waddell G, McCulloch JA, Kummel E, et al. Nonorganic physical signs in low-back pain. Spine. 1980;5:117-125.
10. Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14:502-511.
11. Villemure C, Bushnell MC. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95:195-199.
12. Felitti VJ, Anda RF. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders, and sexual behavior: implications for healthcare. In: Lanius R, Vermetten E, eds. The Hidden Epidemic: The Impact of Early Life Trauma on Health and Disease. Cambridge University Press; 2010. Available at: http://www.unnaturalcauses.org/assets/uploads/file/ACE%20Study-Lanius.pdf. Accessed January 11, 2016.
13. Centers for Disease Control and Prevention. Injury prevention and control: Division of violence prevention. Adverse childhood experiences. Available at: http://www.cdc.gov/violenceprevention/acestudy/. Accessed January 11, 2016.
14. Kross E, Berman MG, Mischel W, et al. Social rejection shares somatosensory representations with physical pain. Proc Natl Acad Sci. 2011;108:6270-6275.
15. Geuze E, Westenberg HGM, Jochims A, et al. Altered pain processing in veterans with posttraumatic stress disorder. Arch Gen Psychiatry. 2007;64:76-85.
16. Mickleborough MJ, Daniels JK, Coupland NJ. Effects of trauma-related cues on pain processing in posttraumatic stress disorder: an fMRI investigation. J Psychiatry Neurosci. 2011;36: 6-14.
17. Noll-Hussong M, Otti A, Laeer L, et al. Aftermath of sexual abuse history on adult patients suffering from chronic functional pain syndromes: an fMRI pilot study. J Psychoso Res. 2010; 68:483-487.
1. Dubner R, Gold M. The neurobiology of pain. Proc Natl Acad Sci U S A. 1999;96:7627-7630.
2. Markenson JA. Mechanisms of chronic pain. Am J Med. 1996;101:S6-S18.
3. Rainville P, Duncan GH, Price DD, et al. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968-971.
4. Bushnell MC, Duncan GH. Sensory and affective aspects of pain perception: is medial thalamus restricted to emotional issues? Exp Brain Res. 1989;78:415-418.
5. Galer BS, Jensen MP. Development and preliminary validation of a pain measure specific to neuropathic pain: the Neuropathic Pain Scale. Neurology. 1997;48:332-338.
6. Fassoulaki A, Triga A, Melemeni A, et al. Multimodal analgesia with gabapentin and local anesthetics prevents acute and chronic pain after breast surgery for cancer. Anesth Analg. 2005;101:1427-1432.
7. Woolf CJ, Chong MS. Preemptive analgesia—treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg. 1993;77:362-379.
8. International Association for the Study of Pain Web site. IASP Taxonomy. Available at: http://www.iasp-pain.org/Taxonomy. Accessed January 10, 2016.
9. Waddell G, McCulloch JA, Kummel E, et al. Nonorganic physical signs in low-back pain. Spine. 1980;5:117-125.
10. Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14:502-511.
11. Villemure C, Bushnell MC. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95:195-199.
12. Felitti VJ, Anda RF. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders, and sexual behavior: implications for healthcare. In: Lanius R, Vermetten E, eds. The Hidden Epidemic: The Impact of Early Life Trauma on Health and Disease. Cambridge University Press; 2010. Available at: http://www.unnaturalcauses.org/assets/uploads/file/ACE%20Study-Lanius.pdf. Accessed January 11, 2016.
13. Centers for Disease Control and Prevention. Injury prevention and control: Division of violence prevention. Adverse childhood experiences. Available at: http://www.cdc.gov/violenceprevention/acestudy/. Accessed January 11, 2016.
14. Kross E, Berman MG, Mischel W, et al. Social rejection shares somatosensory representations with physical pain. Proc Natl Acad Sci. 2011;108:6270-6275.
15. Geuze E, Westenberg HGM, Jochims A, et al. Altered pain processing in veterans with posttraumatic stress disorder. Arch Gen Psychiatry. 2007;64:76-85.
16. Mickleborough MJ, Daniels JK, Coupland NJ. Effects of trauma-related cues on pain processing in posttraumatic stress disorder: an fMRI investigation. J Psychiatry Neurosci. 2011;36: 6-14.
17. Noll-Hussong M, Otti A, Laeer L, et al. Aftermath of sexual abuse history on adult patients suffering from chronic functional pain syndromes: an fMRI pilot study. J Psychoso Res. 2010; 68:483-487.
From The Journal of Family Practice | 2016;65(9):598-600,602-605.