User login
The IOM’s report on calcium and vitamin D: Should it change the way you practice?
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
How to avoid this medical emergency
• Suspect malignant hyperthermia (MH) if a patient has night sweats, cramping, mottled skin, low-grade fever, and excessive sweating, or has elevated creatine kinase and rhabdomyolysis on lab studies. B
• Make sure patients and their family members know that MH is life threatening, familial, and can even occur in patients whose previous experiences with anesthesia have been uneventful. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
You are asked to perform a preoperative evaluation of a 6-year-old boy who is due to have a tonsillectomy. The family history reveals that his father had an episode that sounds like malignant hyperthermia (MH). Also, a paternal uncle experienced a high fever and almost died after undergoing anesthesia. The boy’s parents tell you they took the boy to a pediatrician, who did a “blood test” for MH. They hand you a written report of an enzyme-linked immunosorbent assay, which tested negative for an allergy to succinylcholine.
Has this child been adequately screened for MH?
If you answered No, you are correct. The review that follows explains why.
The “disease of anesthesia”
MH is a pharmacogenetic disease process that occurs when predisposed individuals are exposed to certain triggering agents—specifically, anesthetics. Succinylcholine and all potent inhalational anesthetic agents have been implicated (TABLE 1). Most episodes occur in the intraoperative period.
MH is a familial disease and follows an autosomal dominant pattern, but with incomplete penetrance. Surprisingly, the disease was not described until 1961, when Denborough et al reported a string of anesthetic-related deaths in a family.1,2 A similar condition was described in pigs in 1966.3 This condition, porcine stress syndrome, was noted during research in which pigs had received succinylcholine. This syndrome has become the animal model for the study of MH.4,5
Over time, this condition came to be known as malignant hyperthermia because a rapid rise in temperature was a common feature in all reported cases. Additional possible signs and symptoms include skin mottling, arrhythmias, elevated creatine kinase (CK), and rhabdomyolysis, among others (TABLE 2).
Associated conditions. MH may occur with any condition requiring intervention with anesthesia. It was once believed that strabismus and MH were linked, but this assumption was based on a statistical error related to an increased number of surgical procedures in children with strabismus. Currently, a propensity toward MH seems associated only with rare myopathic conditions such as central core disease, hypokalemic periodic paralysis, Evans myopathy, and King-Denborough syndrome.6 Precise genetic mapping will determine what, if any, relationship there is between these processes and MH.
TABLE 1
Triggering and nontriggering anesthetic agents
| Triggering agents | Nontriggering agents |
|---|---|
| Succinylcholine (most common) | Barbiturates |
| Desflurane | Benzodiazepines |
| Halothane | Ketamine |
| Isoflurane | Local anesthetics |
| Sevoflurane | Nitrous oxide |
| Nondepolarizing muscle relaxants | |
| Opioids | |
| Propofol | |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. | |
TABLE 2
Signs and symptoms of malignant hyperthermia
| Arrhythmias |
| Coagulopathy |
| Elevated creatine kinase |
| Elevated temperature |
| Hypercarbia |
| Hyperkalemia |
| Increased oxygen consumption |
| Masseter muscle spasm |
| Metabolic acidosis |
| Muscle rigidity |
| Rhabdomyolysis |
| Skin mottling |
| Tachycardia |
| Tachypnea |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. |
Awake triggering: Similar disorder without anesthesia
Since 1980 there have been several reports of “awake triggering” in genetically predisposed individuals, whereby stressful conditions alone unrelated to general anesthesia cause MH.7-10 Often, the presenting condition has been heat-stroke, but other symptoms are also common, such as rhabdomyolysis, increased CK, muscle pain, and cardiovascular collapse.11 Relatives of those with a history of MH have also exhibited chronic muscle pain or chronic CK elevation. All of these people, when tested, have had a positive reaction to the caffeine-halothane contracture test (CHCT), which is the gold standard for confirming MH.12
The Malignant Hyperthermia Association of the United States (MHAUS) lists signs and symptoms that accompany awake triggering on its Web site, www.mhaus.org. They include heat sensitivity, night sweats, cramping, mottled skin, low-grade fever, and excessive sweating.
Should such findings—especially elevated CK and rhabdomyolysis—come to your attention by a patient’s report or during physical examination, consider further workup for MH.
How MH develops
MH occurs because of a defect in the ryanodine receptor, RYR1. This receptor is responsible for intracellular calcium release by its mediation of the sarcoplasmic reticulum. RYR1 is found in all skeletal muscle.13 During an episode of MH, exposure to the triggering agent causes intracellular calcium release by the sarcoplasmic reticulum and sustained skeletal muscle contractions and rigidity. Increased oxygen consumption occurs, and this hypermetabolic state leads to hypercarbia, severe metabolic acidosis, tachycardia, arrhythmias, hyperkalemia, and elevated temperature. Rhabdomyolysis and elevations in CK also occur because of skeletal muscle breakdown.
How to treat this medical emergency
Only rapid, specific treatment can save a patient with MH from death. Before investigators discovered that dantrolene sodium is an effective treatment for MH, >70% of patients who developed the disease died.12 Mortality has since dropped to ≤15%.14 With a presumptive diagnosis of MH, dantrolene should be prepared and given immediately. The drug inhibits the release of calcium from the sarcoplasmic reticulum of skeletal muscle by limiting the activation of RYR1.
Successful resuscitation of patients with MH hinges on the following:
Making a prompt diagnosis. While most episodes of MH occur shortly after induction of anesthesia or during the intraoperative course, nearly 2% of MH cases occur postoperatively15—some as long as 12 hours after exposure to the presumed trigger.
Immediately discontinuing triggering anesthetics and starting dantrolene. The patient should be kept anesthetized using nontriggering agents (TABLE 1), and surgery should be concluded as quickly as possible.
Preparation of dantrolene is a tedious process because of its poor water solubility. One to 2 minutes are needed for the preparation to solubilize. Initial treatment with dantrolene is 2.5 mg/kg administered intravenously (via peripheral or central line). This dose (or up to 10 mg/kg) can be repeated every 5 to 10 minutes until major symptoms such as hypercarbia, arrhythmias, hyperpyrexia, and metabolic acidosis abate. Thereafter, dantrolene is continued at 2.5 mg/kg every 4 hours for 24 hours.
Cooling the patient. A patient should be cooled using ice packs, gastric lavage with ice water, cold intravenous fluid, or ice water lavage at the surgical site. Active cooling should stop when the patient’s temperature reaches 100°F (so as not to cause hypothermia). Cooling should not, however, prevent or slow the administration of dantrolene.
Treating acidosis, arrhythmias, electrolyte disturbances, coagulopathy, excess myoglobin, and acute renal failure. Arrhythmias should not be treated with calcium channel blockers.
Monitoring patients in the ICU for potential adverse effects of dantrolene that can include sedation and muscle weakness. Mechanical ventilation may be needed. (The malignant hyperthermia hotline [800-644-9737] at MHAUS is available 24 hours a day to assist clinicians with questions regarding diagnosis and treatment.)
Keeping patients safe by properly testing them
The CHCT is 97% sensitive and 78% specific for MH.16 It is an invasive procedure, requiring the patient to undergo anesthesia. The test costs more than $5,000, which most insurance companies will cover.17
The CHCT is performed only on fresh muscle, and only 5 centers in North America perform the test. The number of centers is kept to a minimum to maintain high procedural quality. The biopsy specimen is taken from the vastus lateralis. Because of the size of the specimen required to perform the test, a child weighing <20 kg or <5 years of age is usually not tested but simply presumed to be MH susceptible.18
The number of reported cases of malignant hyperthermia (MH) has increased over the last 20 years, but the exact incidence is unknown. It varies not only by country, but, within the United States, from state to state, and even within states, presumably due to variations in the genetic pool. Michigan, Nebraska, West Virginia, and Wisconsin report especially high incidences.19
MH is more common in men than women (58% vs 42%, respectively, of affected patients ). The last major survey of MH incidence was published in 2009.20 In the survey years (2000-2005), MH incidence increased from 10.2 to 13.3 patients per million hospital discharges. Mortality in the same period decreased from 16.1% to 6.5%.
Counseling patients and their families
Make sure affected individuals and family members know the life-threatening implications of this condition; that it is familial and that its onset does not always occur with a first exposure to anesthesia, but sometimes with a subsequent exposure. Explain that MH is not an “allergy to anesthesia” or an “allergy to succinylcholine.” Patients can undergo anesthesia safely in the future if performed with a nontriggering agent and associated technique. Anesthesiologists today are well trained to manage these patients with minimum risk.
Counsel first-degree relatives of MH patients about undergoing the CHCT. Molecular genetic testing, which requires a blood draw but is less sensitive than the CHCT, may be another option. If an MH patient was diagnosed using this blood test, relatives may opt to be tested this way, as well. If genetic test results for relatives are positive, these individuals are presumed to have MH and should be treated as such, saving them the expense and risk of undergoing muscle biopsy. If genetic test results are negative, the CHCT should be considered or patients should be presumed to be MH susceptible.
Why is testing of relatives needed if a nontriggering anesthetic can be administered? Nontriggering anesthetic agents are not routinely used in surgery, and problems can arise in, say, emergency situations when MH susceptibility in a patient is unknown to the surgical team. Testing enables patients to learn their status and to obtain a medical alert bracelet.
What about the child in the opening scenario?
It is evident that the child in the scenario at the beginning of this article was not adequately screened for MH. Given that the child is >5 years of age, he should undergo the CHCT. You would be wise to presume that his first-degree relatives are also susceptible until proven otherwise by the CHCT. With children not meeting the age or weight requirement for CHCT, make sure the family understands the potential severity of MH and immediately inform the surgeon and anesthesiologist of the family history so appropriate precautions, including arrangements for nontriggering anesthetics, can be put into place.
CORRESPONDENCE
Gregory L. Rose, MD, Department of Anesthesia, University of Kentucky College of Medicine, 800 Rose Street, Lexington, KY 40536; [email protected]
1. Denborough MA, Lovell RRH. Anaesthetic deaths in a family. Lancet. 1960;276:45.
2. Denborough MA, Forster JF, Lovell RRH, et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395-396.
3. Hall L, Woolf N, Bradley J, et al. Unusual reaction to suxamethonium chloride. BMJ. 1966;2:1305.
4. Stalder K, Conatser G. Porcine stress syndrome and its effects on maternal, feedlot and carcass quantitative and qualitative traits. Agricultural Extension Service, The University of Tennessee. Available at: http://www.utextension.utk.edu/publications/pbfiles/PB1606.pdf. Accessed December 7, 2010.
5. Harrison G. Control of the malignant hyperpyrexic syndrome in MHS swine using dantrolene sodium. Br. J. Anaesthesia. 1975;47:62-65.
6. Allen GC. Malignant hyperthermia and associated disorders. Curr Opin Rheumatol. 1993;5:719-724.
7. Kolb M, Horne M, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56:254-262.
8. Hopkins PM. Malignant hyperthermia: advances in clinical management and diagnosis. Br J Anaesth. 2000;85:118-128.
9. Tobin JR, Jason DR, Nelson TE, et al. Malignant hyperthermia and apparent heat stroke (Correspondence). JAMA. 2001;286:168.
10. Hopkins PM. Is there a link between malignant hyperthermia and exertional heat illness? Br J Sports Med. 2007;41:283-284.
11. Hines RL, Marschall KE. eds. Stoelting’s Anesthesia and Co-Existing Disease. 5th ed. Philadelphia, PA: Saunders; 2008.
12. Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131-1136.
13. Parness J, et al. eds. Ryanodine Receptors Structure, Function and Dysfunction in Clinical Disease (Developments in Cardiovascular Medicine). New York, NY: Springer Press; 2005.
14. Rosenberg H, Davis M, James D, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21.
15. Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1-51.
16. Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. Anesthesiology. 1998;88:579-588.
17. Malignant Hyperthermia Association of the United States. Available at: http://www.mhaus.org/index.cfm/fuseaction/OnlineBrochures.Display/BrochurePK/71A5AFFC-1BC7-4A36-970C6FDB27999FE5.cfm. Accessed: November 22, 2010.
18. Rosenberg H, et al. Malignant hyperthermia susceptibility. In: Pagon RA, et al, eds. Gene Reviews [Internet]. Last Update: January 19, 2010. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1146/#mhs. Accessed December 9, 2010.
19. Litman R, Flood C, Kaplan R, et al. Postoperative malignant hyperthermia: an analysis of cases from the North American Malignant Hyperthermia Registry. Anesthesiology. 2008;109:825-829.
20. Rosero E, Adesanya A, Timaran C, et al. Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology. 2009;110:89-94.
• Suspect malignant hyperthermia (MH) if a patient has night sweats, cramping, mottled skin, low-grade fever, and excessive sweating, or has elevated creatine kinase and rhabdomyolysis on lab studies. B
• Make sure patients and their family members know that MH is life threatening, familial, and can even occur in patients whose previous experiences with anesthesia have been uneventful. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
You are asked to perform a preoperative evaluation of a 6-year-old boy who is due to have a tonsillectomy. The family history reveals that his father had an episode that sounds like malignant hyperthermia (MH). Also, a paternal uncle experienced a high fever and almost died after undergoing anesthesia. The boy’s parents tell you they took the boy to a pediatrician, who did a “blood test” for MH. They hand you a written report of an enzyme-linked immunosorbent assay, which tested negative for an allergy to succinylcholine.
Has this child been adequately screened for MH?
If you answered No, you are correct. The review that follows explains why.
The “disease of anesthesia”
MH is a pharmacogenetic disease process that occurs when predisposed individuals are exposed to certain triggering agents—specifically, anesthetics. Succinylcholine and all potent inhalational anesthetic agents have been implicated (TABLE 1). Most episodes occur in the intraoperative period.
MH is a familial disease and follows an autosomal dominant pattern, but with incomplete penetrance. Surprisingly, the disease was not described until 1961, when Denborough et al reported a string of anesthetic-related deaths in a family.1,2 A similar condition was described in pigs in 1966.3 This condition, porcine stress syndrome, was noted during research in which pigs had received succinylcholine. This syndrome has become the animal model for the study of MH.4,5
Over time, this condition came to be known as malignant hyperthermia because a rapid rise in temperature was a common feature in all reported cases. Additional possible signs and symptoms include skin mottling, arrhythmias, elevated creatine kinase (CK), and rhabdomyolysis, among others (TABLE 2).
Associated conditions. MH may occur with any condition requiring intervention with anesthesia. It was once believed that strabismus and MH were linked, but this assumption was based on a statistical error related to an increased number of surgical procedures in children with strabismus. Currently, a propensity toward MH seems associated only with rare myopathic conditions such as central core disease, hypokalemic periodic paralysis, Evans myopathy, and King-Denborough syndrome.6 Precise genetic mapping will determine what, if any, relationship there is between these processes and MH.
TABLE 1
Triggering and nontriggering anesthetic agents
| Triggering agents | Nontriggering agents |
|---|---|
| Succinylcholine (most common) | Barbiturates |
| Desflurane | Benzodiazepines |
| Halothane | Ketamine |
| Isoflurane | Local anesthetics |
| Sevoflurane | Nitrous oxide |
| Nondepolarizing muscle relaxants | |
| Opioids | |
| Propofol | |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. | |
TABLE 2
Signs and symptoms of malignant hyperthermia
| Arrhythmias |
| Coagulopathy |
| Elevated creatine kinase |
| Elevated temperature |
| Hypercarbia |
| Hyperkalemia |
| Increased oxygen consumption |
| Masseter muscle spasm |
| Metabolic acidosis |
| Muscle rigidity |
| Rhabdomyolysis |
| Skin mottling |
| Tachycardia |
| Tachypnea |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. |
Awake triggering: Similar disorder without anesthesia
Since 1980 there have been several reports of “awake triggering” in genetically predisposed individuals, whereby stressful conditions alone unrelated to general anesthesia cause MH.7-10 Often, the presenting condition has been heat-stroke, but other symptoms are also common, such as rhabdomyolysis, increased CK, muscle pain, and cardiovascular collapse.11 Relatives of those with a history of MH have also exhibited chronic muscle pain or chronic CK elevation. All of these people, when tested, have had a positive reaction to the caffeine-halothane contracture test (CHCT), which is the gold standard for confirming MH.12
The Malignant Hyperthermia Association of the United States (MHAUS) lists signs and symptoms that accompany awake triggering on its Web site, www.mhaus.org. They include heat sensitivity, night sweats, cramping, mottled skin, low-grade fever, and excessive sweating.
Should such findings—especially elevated CK and rhabdomyolysis—come to your attention by a patient’s report or during physical examination, consider further workup for MH.
How MH develops
MH occurs because of a defect in the ryanodine receptor, RYR1. This receptor is responsible for intracellular calcium release by its mediation of the sarcoplasmic reticulum. RYR1 is found in all skeletal muscle.13 During an episode of MH, exposure to the triggering agent causes intracellular calcium release by the sarcoplasmic reticulum and sustained skeletal muscle contractions and rigidity. Increased oxygen consumption occurs, and this hypermetabolic state leads to hypercarbia, severe metabolic acidosis, tachycardia, arrhythmias, hyperkalemia, and elevated temperature. Rhabdomyolysis and elevations in CK also occur because of skeletal muscle breakdown.
How to treat this medical emergency
Only rapid, specific treatment can save a patient with MH from death. Before investigators discovered that dantrolene sodium is an effective treatment for MH, >70% of patients who developed the disease died.12 Mortality has since dropped to ≤15%.14 With a presumptive diagnosis of MH, dantrolene should be prepared and given immediately. The drug inhibits the release of calcium from the sarcoplasmic reticulum of skeletal muscle by limiting the activation of RYR1.
Successful resuscitation of patients with MH hinges on the following:
Making a prompt diagnosis. While most episodes of MH occur shortly after induction of anesthesia or during the intraoperative course, nearly 2% of MH cases occur postoperatively15—some as long as 12 hours after exposure to the presumed trigger.
Immediately discontinuing triggering anesthetics and starting dantrolene. The patient should be kept anesthetized using nontriggering agents (TABLE 1), and surgery should be concluded as quickly as possible.
Preparation of dantrolene is a tedious process because of its poor water solubility. One to 2 minutes are needed for the preparation to solubilize. Initial treatment with dantrolene is 2.5 mg/kg administered intravenously (via peripheral or central line). This dose (or up to 10 mg/kg) can be repeated every 5 to 10 minutes until major symptoms such as hypercarbia, arrhythmias, hyperpyrexia, and metabolic acidosis abate. Thereafter, dantrolene is continued at 2.5 mg/kg every 4 hours for 24 hours.
Cooling the patient. A patient should be cooled using ice packs, gastric lavage with ice water, cold intravenous fluid, or ice water lavage at the surgical site. Active cooling should stop when the patient’s temperature reaches 100°F (so as not to cause hypothermia). Cooling should not, however, prevent or slow the administration of dantrolene.
Treating acidosis, arrhythmias, electrolyte disturbances, coagulopathy, excess myoglobin, and acute renal failure. Arrhythmias should not be treated with calcium channel blockers.
Monitoring patients in the ICU for potential adverse effects of dantrolene that can include sedation and muscle weakness. Mechanical ventilation may be needed. (The malignant hyperthermia hotline [800-644-9737] at MHAUS is available 24 hours a day to assist clinicians with questions regarding diagnosis and treatment.)
Keeping patients safe by properly testing them
The CHCT is 97% sensitive and 78% specific for MH.16 It is an invasive procedure, requiring the patient to undergo anesthesia. The test costs more than $5,000, which most insurance companies will cover.17
The CHCT is performed only on fresh muscle, and only 5 centers in North America perform the test. The number of centers is kept to a minimum to maintain high procedural quality. The biopsy specimen is taken from the vastus lateralis. Because of the size of the specimen required to perform the test, a child weighing <20 kg or <5 years of age is usually not tested but simply presumed to be MH susceptible.18
The number of reported cases of malignant hyperthermia (MH) has increased over the last 20 years, but the exact incidence is unknown. It varies not only by country, but, within the United States, from state to state, and even within states, presumably due to variations in the genetic pool. Michigan, Nebraska, West Virginia, and Wisconsin report especially high incidences.19
MH is more common in men than women (58% vs 42%, respectively, of affected patients ). The last major survey of MH incidence was published in 2009.20 In the survey years (2000-2005), MH incidence increased from 10.2 to 13.3 patients per million hospital discharges. Mortality in the same period decreased from 16.1% to 6.5%.
Counseling patients and their families
Make sure affected individuals and family members know the life-threatening implications of this condition; that it is familial and that its onset does not always occur with a first exposure to anesthesia, but sometimes with a subsequent exposure. Explain that MH is not an “allergy to anesthesia” or an “allergy to succinylcholine.” Patients can undergo anesthesia safely in the future if performed with a nontriggering agent and associated technique. Anesthesiologists today are well trained to manage these patients with minimum risk.
Counsel first-degree relatives of MH patients about undergoing the CHCT. Molecular genetic testing, which requires a blood draw but is less sensitive than the CHCT, may be another option. If an MH patient was diagnosed using this blood test, relatives may opt to be tested this way, as well. If genetic test results for relatives are positive, these individuals are presumed to have MH and should be treated as such, saving them the expense and risk of undergoing muscle biopsy. If genetic test results are negative, the CHCT should be considered or patients should be presumed to be MH susceptible.
Why is testing of relatives needed if a nontriggering anesthetic can be administered? Nontriggering anesthetic agents are not routinely used in surgery, and problems can arise in, say, emergency situations when MH susceptibility in a patient is unknown to the surgical team. Testing enables patients to learn their status and to obtain a medical alert bracelet.
What about the child in the opening scenario?
It is evident that the child in the scenario at the beginning of this article was not adequately screened for MH. Given that the child is >5 years of age, he should undergo the CHCT. You would be wise to presume that his first-degree relatives are also susceptible until proven otherwise by the CHCT. With children not meeting the age or weight requirement for CHCT, make sure the family understands the potential severity of MH and immediately inform the surgeon and anesthesiologist of the family history so appropriate precautions, including arrangements for nontriggering anesthetics, can be put into place.
CORRESPONDENCE
Gregory L. Rose, MD, Department of Anesthesia, University of Kentucky College of Medicine, 800 Rose Street, Lexington, KY 40536; [email protected]
• Suspect malignant hyperthermia (MH) if a patient has night sweats, cramping, mottled skin, low-grade fever, and excessive sweating, or has elevated creatine kinase and rhabdomyolysis on lab studies. B
• Make sure patients and their family members know that MH is life threatening, familial, and can even occur in patients whose previous experiences with anesthesia have been uneventful. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
You are asked to perform a preoperative evaluation of a 6-year-old boy who is due to have a tonsillectomy. The family history reveals that his father had an episode that sounds like malignant hyperthermia (MH). Also, a paternal uncle experienced a high fever and almost died after undergoing anesthesia. The boy’s parents tell you they took the boy to a pediatrician, who did a “blood test” for MH. They hand you a written report of an enzyme-linked immunosorbent assay, which tested negative for an allergy to succinylcholine.
Has this child been adequately screened for MH?
If you answered No, you are correct. The review that follows explains why.
The “disease of anesthesia”
MH is a pharmacogenetic disease process that occurs when predisposed individuals are exposed to certain triggering agents—specifically, anesthetics. Succinylcholine and all potent inhalational anesthetic agents have been implicated (TABLE 1). Most episodes occur in the intraoperative period.
MH is a familial disease and follows an autosomal dominant pattern, but with incomplete penetrance. Surprisingly, the disease was not described until 1961, when Denborough et al reported a string of anesthetic-related deaths in a family.1,2 A similar condition was described in pigs in 1966.3 This condition, porcine stress syndrome, was noted during research in which pigs had received succinylcholine. This syndrome has become the animal model for the study of MH.4,5
Over time, this condition came to be known as malignant hyperthermia because a rapid rise in temperature was a common feature in all reported cases. Additional possible signs and symptoms include skin mottling, arrhythmias, elevated creatine kinase (CK), and rhabdomyolysis, among others (TABLE 2).
Associated conditions. MH may occur with any condition requiring intervention with anesthesia. It was once believed that strabismus and MH were linked, but this assumption was based on a statistical error related to an increased number of surgical procedures in children with strabismus. Currently, a propensity toward MH seems associated only with rare myopathic conditions such as central core disease, hypokalemic periodic paralysis, Evans myopathy, and King-Denborough syndrome.6 Precise genetic mapping will determine what, if any, relationship there is between these processes and MH.
TABLE 1
Triggering and nontriggering anesthetic agents
| Triggering agents | Nontriggering agents |
|---|---|
| Succinylcholine (most common) | Barbiturates |
| Desflurane | Benzodiazepines |
| Halothane | Ketamine |
| Isoflurane | Local anesthetics |
| Sevoflurane | Nitrous oxide |
| Nondepolarizing muscle relaxants | |
| Opioids | |
| Propofol | |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. | |
TABLE 2
Signs and symptoms of malignant hyperthermia
| Arrhythmias |
| Coagulopathy |
| Elevated creatine kinase |
| Elevated temperature |
| Hypercarbia |
| Hyperkalemia |
| Increased oxygen consumption |
| Masseter muscle spasm |
| Metabolic acidosis |
| Muscle rigidity |
| Rhabdomyolysis |
| Skin mottling |
| Tachycardia |
| Tachypnea |
| Adapted from: Barash PG, et al, eds. Clinical Anesthesia. 6th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2009. |
Awake triggering: Similar disorder without anesthesia
Since 1980 there have been several reports of “awake triggering” in genetically predisposed individuals, whereby stressful conditions alone unrelated to general anesthesia cause MH.7-10 Often, the presenting condition has been heat-stroke, but other symptoms are also common, such as rhabdomyolysis, increased CK, muscle pain, and cardiovascular collapse.11 Relatives of those with a history of MH have also exhibited chronic muscle pain or chronic CK elevation. All of these people, when tested, have had a positive reaction to the caffeine-halothane contracture test (CHCT), which is the gold standard for confirming MH.12
The Malignant Hyperthermia Association of the United States (MHAUS) lists signs and symptoms that accompany awake triggering on its Web site, www.mhaus.org. They include heat sensitivity, night sweats, cramping, mottled skin, low-grade fever, and excessive sweating.
Should such findings—especially elevated CK and rhabdomyolysis—come to your attention by a patient’s report or during physical examination, consider further workup for MH.
How MH develops
MH occurs because of a defect in the ryanodine receptor, RYR1. This receptor is responsible for intracellular calcium release by its mediation of the sarcoplasmic reticulum. RYR1 is found in all skeletal muscle.13 During an episode of MH, exposure to the triggering agent causes intracellular calcium release by the sarcoplasmic reticulum and sustained skeletal muscle contractions and rigidity. Increased oxygen consumption occurs, and this hypermetabolic state leads to hypercarbia, severe metabolic acidosis, tachycardia, arrhythmias, hyperkalemia, and elevated temperature. Rhabdomyolysis and elevations in CK also occur because of skeletal muscle breakdown.
How to treat this medical emergency
Only rapid, specific treatment can save a patient with MH from death. Before investigators discovered that dantrolene sodium is an effective treatment for MH, >70% of patients who developed the disease died.12 Mortality has since dropped to ≤15%.14 With a presumptive diagnosis of MH, dantrolene should be prepared and given immediately. The drug inhibits the release of calcium from the sarcoplasmic reticulum of skeletal muscle by limiting the activation of RYR1.
Successful resuscitation of patients with MH hinges on the following:
Making a prompt diagnosis. While most episodes of MH occur shortly after induction of anesthesia or during the intraoperative course, nearly 2% of MH cases occur postoperatively15—some as long as 12 hours after exposure to the presumed trigger.
Immediately discontinuing triggering anesthetics and starting dantrolene. The patient should be kept anesthetized using nontriggering agents (TABLE 1), and surgery should be concluded as quickly as possible.
Preparation of dantrolene is a tedious process because of its poor water solubility. One to 2 minutes are needed for the preparation to solubilize. Initial treatment with dantrolene is 2.5 mg/kg administered intravenously (via peripheral or central line). This dose (or up to 10 mg/kg) can be repeated every 5 to 10 minutes until major symptoms such as hypercarbia, arrhythmias, hyperpyrexia, and metabolic acidosis abate. Thereafter, dantrolene is continued at 2.5 mg/kg every 4 hours for 24 hours.
Cooling the patient. A patient should be cooled using ice packs, gastric lavage with ice water, cold intravenous fluid, or ice water lavage at the surgical site. Active cooling should stop when the patient’s temperature reaches 100°F (so as not to cause hypothermia). Cooling should not, however, prevent or slow the administration of dantrolene.
Treating acidosis, arrhythmias, electrolyte disturbances, coagulopathy, excess myoglobin, and acute renal failure. Arrhythmias should not be treated with calcium channel blockers.
Monitoring patients in the ICU for potential adverse effects of dantrolene that can include sedation and muscle weakness. Mechanical ventilation may be needed. (The malignant hyperthermia hotline [800-644-9737] at MHAUS is available 24 hours a day to assist clinicians with questions regarding diagnosis and treatment.)
Keeping patients safe by properly testing them
The CHCT is 97% sensitive and 78% specific for MH.16 It is an invasive procedure, requiring the patient to undergo anesthesia. The test costs more than $5,000, which most insurance companies will cover.17
The CHCT is performed only on fresh muscle, and only 5 centers in North America perform the test. The number of centers is kept to a minimum to maintain high procedural quality. The biopsy specimen is taken from the vastus lateralis. Because of the size of the specimen required to perform the test, a child weighing <20 kg or <5 years of age is usually not tested but simply presumed to be MH susceptible.18
The number of reported cases of malignant hyperthermia (MH) has increased over the last 20 years, but the exact incidence is unknown. It varies not only by country, but, within the United States, from state to state, and even within states, presumably due to variations in the genetic pool. Michigan, Nebraska, West Virginia, and Wisconsin report especially high incidences.19
MH is more common in men than women (58% vs 42%, respectively, of affected patients ). The last major survey of MH incidence was published in 2009.20 In the survey years (2000-2005), MH incidence increased from 10.2 to 13.3 patients per million hospital discharges. Mortality in the same period decreased from 16.1% to 6.5%.
Counseling patients and their families
Make sure affected individuals and family members know the life-threatening implications of this condition; that it is familial and that its onset does not always occur with a first exposure to anesthesia, but sometimes with a subsequent exposure. Explain that MH is not an “allergy to anesthesia” or an “allergy to succinylcholine.” Patients can undergo anesthesia safely in the future if performed with a nontriggering agent and associated technique. Anesthesiologists today are well trained to manage these patients with minimum risk.
Counsel first-degree relatives of MH patients about undergoing the CHCT. Molecular genetic testing, which requires a blood draw but is less sensitive than the CHCT, may be another option. If an MH patient was diagnosed using this blood test, relatives may opt to be tested this way, as well. If genetic test results for relatives are positive, these individuals are presumed to have MH and should be treated as such, saving them the expense and risk of undergoing muscle biopsy. If genetic test results are negative, the CHCT should be considered or patients should be presumed to be MH susceptible.
Why is testing of relatives needed if a nontriggering anesthetic can be administered? Nontriggering anesthetic agents are not routinely used in surgery, and problems can arise in, say, emergency situations when MH susceptibility in a patient is unknown to the surgical team. Testing enables patients to learn their status and to obtain a medical alert bracelet.
What about the child in the opening scenario?
It is evident that the child in the scenario at the beginning of this article was not adequately screened for MH. Given that the child is >5 years of age, he should undergo the CHCT. You would be wise to presume that his first-degree relatives are also susceptible until proven otherwise by the CHCT. With children not meeting the age or weight requirement for CHCT, make sure the family understands the potential severity of MH and immediately inform the surgeon and anesthesiologist of the family history so appropriate precautions, including arrangements for nontriggering anesthetics, can be put into place.
CORRESPONDENCE
Gregory L. Rose, MD, Department of Anesthesia, University of Kentucky College of Medicine, 800 Rose Street, Lexington, KY 40536; [email protected]
1. Denborough MA, Lovell RRH. Anaesthetic deaths in a family. Lancet. 1960;276:45.
2. Denborough MA, Forster JF, Lovell RRH, et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395-396.
3. Hall L, Woolf N, Bradley J, et al. Unusual reaction to suxamethonium chloride. BMJ. 1966;2:1305.
4. Stalder K, Conatser G. Porcine stress syndrome and its effects on maternal, feedlot and carcass quantitative and qualitative traits. Agricultural Extension Service, The University of Tennessee. Available at: http://www.utextension.utk.edu/publications/pbfiles/PB1606.pdf. Accessed December 7, 2010.
5. Harrison G. Control of the malignant hyperpyrexic syndrome in MHS swine using dantrolene sodium. Br. J. Anaesthesia. 1975;47:62-65.
6. Allen GC. Malignant hyperthermia and associated disorders. Curr Opin Rheumatol. 1993;5:719-724.
7. Kolb M, Horne M, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56:254-262.
8. Hopkins PM. Malignant hyperthermia: advances in clinical management and diagnosis. Br J Anaesth. 2000;85:118-128.
9. Tobin JR, Jason DR, Nelson TE, et al. Malignant hyperthermia and apparent heat stroke (Correspondence). JAMA. 2001;286:168.
10. Hopkins PM. Is there a link between malignant hyperthermia and exertional heat illness? Br J Sports Med. 2007;41:283-284.
11. Hines RL, Marschall KE. eds. Stoelting’s Anesthesia and Co-Existing Disease. 5th ed. Philadelphia, PA: Saunders; 2008.
12. Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131-1136.
13. Parness J, et al. eds. Ryanodine Receptors Structure, Function and Dysfunction in Clinical Disease (Developments in Cardiovascular Medicine). New York, NY: Springer Press; 2005.
14. Rosenberg H, Davis M, James D, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21.
15. Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1-51.
16. Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. Anesthesiology. 1998;88:579-588.
17. Malignant Hyperthermia Association of the United States. Available at: http://www.mhaus.org/index.cfm/fuseaction/OnlineBrochures.Display/BrochurePK/71A5AFFC-1BC7-4A36-970C6FDB27999FE5.cfm. Accessed: November 22, 2010.
18. Rosenberg H, et al. Malignant hyperthermia susceptibility. In: Pagon RA, et al, eds. Gene Reviews [Internet]. Last Update: January 19, 2010. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1146/#mhs. Accessed December 9, 2010.
19. Litman R, Flood C, Kaplan R, et al. Postoperative malignant hyperthermia: an analysis of cases from the North American Malignant Hyperthermia Registry. Anesthesiology. 2008;109:825-829.
20. Rosero E, Adesanya A, Timaran C, et al. Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology. 2009;110:89-94.
1. Denborough MA, Lovell RRH. Anaesthetic deaths in a family. Lancet. 1960;276:45.
2. Denborough MA, Forster JF, Lovell RRH, et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395-396.
3. Hall L, Woolf N, Bradley J, et al. Unusual reaction to suxamethonium chloride. BMJ. 1966;2:1305.
4. Stalder K, Conatser G. Porcine stress syndrome and its effects on maternal, feedlot and carcass quantitative and qualitative traits. Agricultural Extension Service, The University of Tennessee. Available at: http://www.utextension.utk.edu/publications/pbfiles/PB1606.pdf. Accessed December 7, 2010.
5. Harrison G. Control of the malignant hyperpyrexic syndrome in MHS swine using dantrolene sodium. Br. J. Anaesthesia. 1975;47:62-65.
6. Allen GC. Malignant hyperthermia and associated disorders. Curr Opin Rheumatol. 1993;5:719-724.
7. Kolb M, Horne M, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56:254-262.
8. Hopkins PM. Malignant hyperthermia: advances in clinical management and diagnosis. Br J Anaesth. 2000;85:118-128.
9. Tobin JR, Jason DR, Nelson TE, et al. Malignant hyperthermia and apparent heat stroke (Correspondence). JAMA. 2001;286:168.
10. Hopkins PM. Is there a link between malignant hyperthermia and exertional heat illness? Br J Sports Med. 2007;41:283-284.
11. Hines RL, Marschall KE. eds. Stoelting’s Anesthesia and Co-Existing Disease. 5th ed. Philadelphia, PA: Saunders; 2008.
12. Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131-1136.
13. Parness J, et al. eds. Ryanodine Receptors Structure, Function and Dysfunction in Clinical Disease (Developments in Cardiovascular Medicine). New York, NY: Springer Press; 2005.
14. Rosenberg H, Davis M, James D, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21.
15. Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1-51.
16. Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. Anesthesiology. 1998;88:579-588.
17. Malignant Hyperthermia Association of the United States. Available at: http://www.mhaus.org/index.cfm/fuseaction/OnlineBrochures.Display/BrochurePK/71A5AFFC-1BC7-4A36-970C6FDB27999FE5.cfm. Accessed: November 22, 2010.
18. Rosenberg H, et al. Malignant hyperthermia susceptibility. In: Pagon RA, et al, eds. Gene Reviews [Internet]. Last Update: January 19, 2010. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1146/#mhs. Accessed December 9, 2010.
19. Litman R, Flood C, Kaplan R, et al. Postoperative malignant hyperthermia: an analysis of cases from the North American Malignant Hyperthermia Registry. Anesthesiology. 2008;109:825-829.
20. Rosero E, Adesanya A, Timaran C, et al. Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology. 2009;110:89-94.
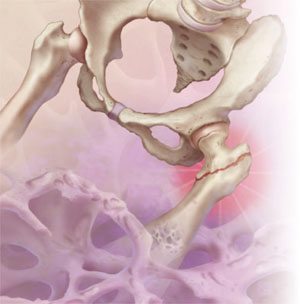
When do bisphosphonates make the most sense?
An estimated 10 million US residents, most of them women over the age of 50, suffer from osteoporosis, and another 33 million have low bone mass.1 Together, they incur more than 2 million osteoporotic fractures annually.1,2 In addition to the high cost of a single osteoporotic fracture in terms of morbidity, mortality, and health care spending, individuals who sustain one such fracture are at high risk for another. That risk can be greatly reduced with appropriate treatment.
Bisphosphonates, which act on osteoclasts to inhibit bone resorption, are first-line therapy for prevention of osteoporotic fractures. Four bisphosphonates—alendronate, ibandronate, risedronate, and zoledronic acid—are approved by the US Food and Drug Administration (FDA) for the treatment of postmenopausal osteoporosis.
While menopause itself increases a woman’s risk for osteoporotic fracture, questions remain about when to initiate preventive therapy, which patients are candidates for bisphosphonates, and whether bisphosphonates are effective for primary as well as secondary prevention. This overview from the Cochrane Musculoskeletal Group (CMSG) addresses those questions.
To help you provide optimal treatment for postmenopausal patients, we present the findings of recently conducted systematic reviews of 2 bisphosphonates—alendronate and risedronate—from the Cochrane Database of Systematic Reviews,3,4 in context with the available evidence on the efficacy of ibandronate and zoledronic acid. Cochrane reviews of ibandronate and zoledronic acid are underway, but not yet completed.5,6
This is the second in a series of articles based on the findings of the Cochrane Musculoskeletal Group (CMSG), one of the largest review groups in the Cochrane Collaboration. The CMSG synthesizes the results of high-quality clinical trials to determine whether interventions for the prevention, treatment, and rehabilitation of musculoskeletal disorders are safe and effective. In this article and those that follow, CMSG’s aim is to bring its findings to the attention of family physicians in a context that is relevant to clinical practice.
Alendronate reduces vertebral fracture risk across the board
Wells et al identified 11 RCTs for the alendronate review (3 primary and 8 secondary prevention trials), representing a total of 12,068 women.3 (For definitions of what constituted a primary vs a secondary prevention trial, see the box.)
Doses of alendronate ranged from 1 to 20 mg daily, with most studies using doses of 5 or 10 mg. Treatment duration ranged from 1 to 4 years.
A look at the relative risk (RR) for primary and secondary prevention at different fracture sites (TABLE 1) highlights similarities and differences. The risk reduction for vertebral fractures was statistically significant—and about the same—for women being treated with alendronate for primary and secondary prevention (RR=0.55; 95% confidence interval [CI], 0.38-0.80; RR=0.55; 95% CI, 0.43-0.69, respectively). For all other (nonvertebral) fractures in patients being treated with alendronate, only the outcomes for secondary prevention were statistically significant.
The Cochrane reviewers studied the effects of alendronate and risedronate3,4 for both primary and secondary prevention of osteoporotic fractures in postmenopausal women, using the following definitions (with slight variations in definitions between trials):
Primary prevention. Randomized controlled trials were classified as primary prevention trials if the participants had baseline T-scores >–2.0 or a baseline prevalence of vertebral fracture <20%.
Secondary prevention. Studies were classified as secondary prevention trials if the women had baseline T-scores ≤–2.0 (ie, bone mineral density [BMD] ≥2 standard deviations below peak bone mass) or previous vertebral compression fractures. (In the ibandronate individual patient meta-analysis,14 secondary prevention was defined as lumbar spine T-score <–2.5 or baseline vertebral fracture prevalence >20% or mean age of participants >60 years.)
Age-based criterion. When data on T-scores and/or vertebral compression fractures were unavailable, age was the determinant: Trials were considered secondary prevention if the average age of the participants was >62 years, and primary prevention if the average age was ≤62.
Risedronate is effective only for secondary prevention
Seven RCTs, including 2 primary and 5 secondary prevention trials, were included in the Cochrane review of risedronate, representing a total of 14,049 women.4 Doses ranged from 2.5 to 5 mg daily, but also included cyclical dosing—for example, taking 5 mg/d for the first 2 weeks of every month. Treatment duration ranged from 2 to 3 years.
At doses of 5 mg/d, there were no statistically significant decreases in fracture risk at any site in the primary prevention trials (TABLE 1), although the quality of evidence assessed was low. For secondary prevention, however, the risk reduction for vertebral fracture was significant (RR=0.61; 95% CI, 0.50-0.76), as were the reductions in risk for nonvertebral and hip fractures.
TABLE 1
Fracture risk reduction: How the bisphosphonates compare*
| Study | Vertebral fracture RR (95% CI) | Nonvertebral fracture (hip, wrist, others) RR (95% CI) | Hip fracture RR (95% CI) | Wrist fracture RR (95% CI) |
|---|---|---|---|---|
| Primary prevention | ||||
| Alendronate3 | 0.55 (0.38-0.80) | 0.89 (0.76-1.04) | 0.79 (0.44-1.44) | 1.19 (0.87-1.62) |
| Risedronate4 | 0.97 (0.42-2.25) | 0.81 (0.25-2.58) | N/A | N/A |
| Secondary prevention | ||||
| Alendronate3 | 0.55 (0.43-0.69) | 0.77 (0.64-0.92) | 0.47 (0.26-0.85) | 0.50 (0.34-0.73) |
| Risedronate4 | 0.61 (0.50-0.76) | 0.80 (0.72-0.90) | 0.74 (0.59-0.94) | 0.67 (0.42-1.07)† |
| Ibandronate15,16 Oral daily Oral intermittent | 0.62 (0.42-0.75) 0.50 (0.26-0.66) | No effect)‡ No effect | N/A N/A | N/A N/A |
| Zoledronic acid22 | 0.30 (0.2-0.38) | 0.75)§ (N/A) | 0.59¶ (0.42-0.83) | N/A |
| CI, confidence interval; N/A, not available; RR, relative risk. *Bold type indicates statistical significance (P<.05). †P=.10. ‡RR of nonvertebral fracture was 0.69 (P=.013) for daily oral ibandronate in the subgroup with femoral neck BMD T-score <–3.0. §P<.001. ¶Hazard ratio. | ||||
What are the absolute benefits? A look at number needed to treat
In addition to looking at the RR, the authors of both the alendronate and risedronate reviews calculated the number needed to treat (NNT) to prevent one fracture (TABLE 2) in the trial participants;3,4 they focused on the secondary prevention outcomes, as these were statistically significant. The reviewers also estimated what the NNT would be if the risk reductions achieved with alendronate and risedronate in the reviews occurred when treating community-based samples of women at moderate compared with high fracture risk.
The biggest differences involved hip fracture: For alendronate, if a community-based sample of women at moderate risk of fracture were treated with the drug and the reduction in RR seen in the secondary prevention trials applied, the NNT would be 100. Thus, for every 100 women treated for 5 years with alendronate, 1 hip fracture would be prevented. However, if this same RR reduction were applied to women at high risk of fracture, the NNT would be only 22.3 For risedronate, the estimated NNT to prevent one hip fracture in women at moderate risk was 203, compared with only 45 for women at high risk.4 These estimates indicate that the benefits of bisphosphonate therapy in preventing fractures are greatest in women with a high underlying fracture risk.
TABLE 2
NNT analysis: Women at higher risk are most likely to benefit3,4
| NNT | |||
|---|---|---|---|
| Observed in secondary prevention trials in reviews | Estimated for community-based sample of women with | ||
| High fracture risk* | Moderate fracture risk* | ||
| Alendronate (10 mg/d) | |||
| Vertebral fracture | 19 | 20 | 42 |
| Nonvertebral fracture | 47 | 16 | 27 |
| Hip fracture | 146 | 22 | 100 |
| Wrist fracture | 69 | N/A | N/A |
| Risedronate (5 mg/d) | |||
| Vertebral fracture | 19 | 23 | 49 |
| Nonvertebral fracture | 49 | 19 | 31 |
| Hip fracture | 138 | 45 | 203 |
| Wrist fracture | N/A | N/A | N/A |
| N/A, not available; NNT, number needed to treat. *NNT calculated by applying the relative risk reduction observed in the reviews to published estimates of 5-year fracture risk in a community-based sample of women >50 years of age at moderate and high risk. | |||
Adverse effects do not increase with longer-term treatment
In both the alendronate and risedronate reviews, adverse effects and the risk of discontinuing treatment due to adverse events were similar in the intervention and control groups.3,4 Postmarketing data suggest that there is potential for upper gastrointestinal (GI) problems, however;7 osteonecrosis of the jaw has also been reported infrequently.8,9 More recently, there have been reports of a possible link between bisphosphonates and atypical femoral fractures, which we’ll say more about in a bit.
Some potential adverse events—eg, osteonecrosis of the jaw and atypical femoral fractures—may be related to treatment duration. The maximum duration of the trials included in these meta-analyses was 4 years for alendronate and 3 years for risedronate. However, additional published data do not appear to support a relation between adverse events and treatment duration.
For alendronate, researchers extended the Fracture Incidence Trial (FIT) for a 10-year follow-up,10,11 comparing women who took the drug for the first 5 years with women who took it for 10 years. Adverse effects were similar in both groups.
For risedronate, researchers followed a small subsample (n=164) of the participants in the Vertical Efficacy with Risedronate Therapy (VERT) Study Group for up to 7 years.12,13 For the first 5 years, half of the participants took 5 mg/d risedronate, while the other half took a placebo. During the final 2 years, all participants received 5 mg/d risedronate. The incidence of adverse events among those who took the drug for 7 years was similar to that reported in the first 3 years of the original trial.13
Ibandronate studies focus on dose
Nonvertebral fracture. The Cochrane systematic review examining ibandronate for postmenopausal osteoporosis is not yet completed.5 However, Cranney et al performed a pooled analysis of individual patient data from 8 RCTs to examine the efficacy of different doses of the drug for the secondary prevention of nonvertebral fracture.14 (No studies of the drug for primary prevention have been done.) After 2 years of treatment at higher doses of ibandronate (annual cumulative exposure ≥10.8 mg, equivalent to 150 mg orally/month, 3 mg IV quarterly, or 2 mg IV every 2 months), the hazard ratio was 0.62 (95% CI, 0.396-0.974), compared with those taking lower doses (annual cumulative exposure of 5.5 mg). The individual results of the 2 largest trials did not demonstrate an effect on nonvertebral fracture, except in the subgroup of women with very low femoral neck bone mineral density (BMD) (T-scores <–3.0). 15-17
Vertebral fracture. There is no meta-analysis available with vertebral fracture outcomes for ibandronate, so we present the results of individual secondary prevention trials.
One was a double-blind RCT with 2496 participants, comparing women taking either 2.5 mg/d of ibandronate or 20 mg on alternate days with a group on placebo.15,16 The results? Those in both the daily and the intermittent treatment arms had significant risk reductions (RR=0.62; 95% CI, 0.42-0.75; RR=0.50; 95% CI, 0.26-0.66, respectively), after taking the drug for 3 years (TABLE 1), compared with those on placebo.15,16 The other RCT—a trial in which 2862 women received either quarterly intravenous (IV) injections of 1 or 0.5 mg ibandronate or placebo—did not demonstrate a significant reduction in vertebral fracture.17 This was attributed to an insufficient dose of the drug, a supposition supported by improvements in BMD in patients receiving higher doses of ibandronate.18,19
Oral ibandronate has been well tolerated in clinical trials in terms of GI side effects.20,21 Injection site reactions have been reported in those receiving IV infusions,17 and both IV and monthly oral ibandronate may be associated with mild, self-limiting flu-like symptoms.
Zoledronic acid RCTs show reduced fracture, mortality risk
Black et al studied the efficacy of zoledronic acid in a randomized, double-blind, placebo-controlled trial of 7736 postmenopausal women between the ages of 65 and 89 years.22 The women, all of whom had osteoporosis, received an IV infusion of either zoledronic acid (5 mg) or placebo at baseline, and again at 12 and 24 months. Vertebral and nonvertebral fractures, as well as hip fracture, were significantly reduced in the treatment group compared with placebo (TABLE 1).
In another RCT with 2127 participants, Lyles et al examined the effectiveness of 5 mg zoledronic acid IV given within 90 days of surgical repair of a hip fracture. In the intervention group, there was a 35% risk reduction in new clinical fractures (8.6% vs 13.9% for those on placebo; P=.001); mortality was also lower in the zoledronic acid group (9.6% vs 13.3%; P=.01).23
In both trials, the number of patients who had serious adverse events or dropped out because of an adverse event was similar in the treatment and placebo groups. In both studies, too, a sizeable number of patients treated with zoledronic acid reported flu-like symptoms up to 3 days after receiving an infusion, particularly after the first one. Cardiovascular events were similar across intervention groups in both studies, with one exception: In Black’s study,22 there was an increased incidence of serious atrial fibrillation in the zoledronic acid group (1.3% vs 0.5% for the placebo group).
Other issues to keep in mind
Atypical femoral fractures. Published data suggest an association between bisphosphonate use and atypical femoral fractures, particularly with longer-term use,24 although whether there is a causal link is unclear. Atypical femoral fractures occur with little or no trauma along the femur from just distal to the lesser trochanter to just proximal to the supracondylar flare.
In 2010, the FDA announced requirements for a black box warning about a possible link,25 highlighting the uncertainty about both the optimal duration of bisphosphonate therapy and the cause of these fractures.
While concerns about such a link remain, it is important to note that atypical femoral fractures are very uncommon: Current estimates are that they account for less than 1% of hip/femoral fractures. What’s more, far more fractures are prevented by the use of bisphosphonates than are associated with their use, with an estimated ratio of up to 29:1.24
Dosing schedules. Adherence to treatment is of key importance in maximizing outcomes from osteoporosis treatments, and is frequently low.26,27 One way of improving adherence is to reduce the frequency of dosing required.27 With that in mind, researchers have tested intermittent dosing regimens, using noninferiority or bridging trials.
Such studies have led to a number of approved dosing regimens—70 mg weekly for alendronate; 150 mg monthly and 35 mg weekly for risedronate; and 150 mg PO monthly and 3 mg IV quarterly for ibandronate among them. In making decisions about dosing, family physicians should consider patient preferences, but be aware that there are no direct efficacy data from RCTs to support these dosing regimens.
Calcium and vitamin D. The major fracture prevention trials of bisphosphonates have featured women who are calcium- and vitamin D-replete. In a recent study of 1515 women undergoing treatment with alendronate, risedronate, or raloxifene, however, that wasn’t always the case. 28 After 13 months, 115 participants suffered from a new clinical fracture. The adjusted odds ratio for fractures in women with vitamin D deficiency compared with those with normal levels of vitamin D was 1.77 (95% CI, 1.20-2.59; P=.004), an indication of the importance of maintaining adequate vitamin D levels in patients taking bisphosphonates.
In clinical practice, it is important to ensure that patients being treated with bisphosphonates are not deficient in vitamin D. While direct evidence of poorer outcomes associated with low calcium levels is lacking, it is reasonable to also assess calcium intake and to ensure that patients have adequate intake of both. (For more on calcium and vitamin D requirements, see the Institute of Medicine’s recent report at http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D/Report-Brief.aspx) and “The IOM’s report on calcium and vitamin D: Should it change the way you practice?”.
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
What’s best for your patients?
All these bisphosphonates have demonstrated efficacy for the secondary prevention of vertebral fracture. All except ibandronate have demonstrated efficacy for nonvertebral fracture, as well, and the evidence suggests that ibandronate will also be effective if adequate doses are given. Thus, for women at significant risk for fracture, it seems clear that the benefits of treatment outweigh the risks. The case is not so clearcut for women at lower risk. Evidence to support the use of bisphosphonates for primary prevention is limited, other than for alendronate—which has been shown to provide primary prevention of vertebral fracture.
Which bisphosphonate is best depends on patient preferences and individual profiles. (See “How would you treat these patients?”.) In the absence of head-to-head RCTs, it isn’t possible to comment on the relative efficacy of the various bisphosphonates or their adverse event profiles. Indeed, the authors of the 2 Cochrane reviews completed to date note that trial participants have been healthier, with fewer comorbidities, than many of the postmenopausal women seen by primary care physicians. Head-to-head studies conducted in family practice settings would be an important addition to the body of evidence for the prevention of osteoporotic fracture.
How would you treat these patients?
CASE 1 Mrs. A is an active 67-year-old in good health. On a recent hike, she lost her footing and sustained a Colles’ fracture when she fell, although her fall was only from standing height. Now, you are concerned that she might have osteoporosis.
A dual-energy x-ray absorptiometry (DXA) scan confirms this suspicion: Mrs. A’s lumbar spine T-score is –2.6. A dietary review reveals that she has a satisfactory calcium intake, and lab work shows that her serum vitamin D levels are normal. Mrs. A wants to discuss treatment options with you.
What immediate treatment do you consider?
Mrs. A has no contraindications to any FDA-approved treatment for osteoporosis; you suggest she begin taking bisphosphonates, explaining that they are first-line treatment to prevent subsequent osteoporotic fractures. You briefly discuss other options, but note that raloxifene only reduces the risk of vertebral fractures and parathyroid hormone is effective (but very expensive) and requires daily injections, and is therefore generally used for severe osteoporosis. Your patient asks about bisphosphonates’ side effects, particularly the serious jaw problems she’s heard about.
You explain that for the most part, oral bisphosphonates are well tolerated, but that there is a potential for upper gastrointestinal (GI) problems—which is why it’s important to remain upright for at least 30 minutes after taking the medication. You tell her that the risk of developing osteonecrosis of the jaw is very low when the medication is taken at the doses needed for osteoporosis treatment, but that the risk may increase after tooth extraction or dental surgery. Mrs. A has no current dental symptoms and at her usual yearly dental check-up 9 months ago, there were no problems noted, so dental review before starting treatment is not needed. Should she develop any jaw pain, however, she should see you or her dentist immediately.
You also advise her of the possible link between bisphosphonates and atypical femoral fracture, but point out that such fractures are extremely rare—and that the medication prevents far more fractures than it has the potential to cause. You tell her to contact you immediately if she develops pain in the groin or thigh or experiences GI distress.
Which bisphosphonate do you prescribe?
You inform Mrs. A that alendronate has the longest follow-up data of the oral bisphosphonates and has demonstrated efficacy for the secondary prevention of wrist fractures, that risedronate and ibandronate have the advantage of being able to be taken monthly rather than weekly, and that zoledronic acid can be administered in a yearly infusion. She opts for alendronate. You prescribe a weekly dose of 70 mg and ask her to return in 3 months, and to call before then if any problems arise.
CASE 2 Mrs. Y, age 82, recently sustained a fractured femoral neck, which was treated surgically at the local hospital. She was discharged with a prescription for alendronate to treat her osteoporosis and prevent further fractures; her husband has brought her in today to get a new prescription.
During the visit, he reminds you that Mrs. Y has problems with memory. He also says he’s finding it increasingly difficult to ensure that his wife remains upright for 30 minutes after taking alendronate, and that she has begun complaining of indigestion.
What do you decide to do?
An inability to stay upright for 30 minutes after drug administration is a contraindication to the use of oral bisphosphonates. The presence of upper GI symptoms is also a concern. You offer Mrs. Y the option of a once-yearly IV infusion of zoledronic acid instead, and she and her husband agree to this. Before scheduling a follow-up visit, you discuss the patient’s nutritional intake, and discover that she consumes only a moderate amount of calcium—at most 2 servings of dairy products per day. You also note that her serum vitamin D level was not checked in the hospital. You order lab work, with a view to correcting any deficiency before proceeding with a zoledronic infusion (due to the risk of tetany) and to maintaining her on an appropriate level of calcium and vitamin D intake, using supplements only if necessary.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
An estimated 10 million US residents, most of them women over the age of 50, suffer from osteoporosis, and another 33 million have low bone mass.1 Together, they incur more than 2 million osteoporotic fractures annually.1,2 In addition to the high cost of a single osteoporotic fracture in terms of morbidity, mortality, and health care spending, individuals who sustain one such fracture are at high risk for another. That risk can be greatly reduced with appropriate treatment.
Bisphosphonates, which act on osteoclasts to inhibit bone resorption, are first-line therapy for prevention of osteoporotic fractures. Four bisphosphonates—alendronate, ibandronate, risedronate, and zoledronic acid—are approved by the US Food and Drug Administration (FDA) for the treatment of postmenopausal osteoporosis.
While menopause itself increases a woman’s risk for osteoporotic fracture, questions remain about when to initiate preventive therapy, which patients are candidates for bisphosphonates, and whether bisphosphonates are effective for primary as well as secondary prevention. This overview from the Cochrane Musculoskeletal Group (CMSG) addresses those questions.
To help you provide optimal treatment for postmenopausal patients, we present the findings of recently conducted systematic reviews of 2 bisphosphonates—alendronate and risedronate—from the Cochrane Database of Systematic Reviews,3,4 in context with the available evidence on the efficacy of ibandronate and zoledronic acid. Cochrane reviews of ibandronate and zoledronic acid are underway, but not yet completed.5,6
This is the second in a series of articles based on the findings of the Cochrane Musculoskeletal Group (CMSG), one of the largest review groups in the Cochrane Collaboration. The CMSG synthesizes the results of high-quality clinical trials to determine whether interventions for the prevention, treatment, and rehabilitation of musculoskeletal disorders are safe and effective. In this article and those that follow, CMSG’s aim is to bring its findings to the attention of family physicians in a context that is relevant to clinical practice.
Alendronate reduces vertebral fracture risk across the board
Wells et al identified 11 RCTs for the alendronate review (3 primary and 8 secondary prevention trials), representing a total of 12,068 women.3 (For definitions of what constituted a primary vs a secondary prevention trial, see the box.)
Doses of alendronate ranged from 1 to 20 mg daily, with most studies using doses of 5 or 10 mg. Treatment duration ranged from 1 to 4 years.
A look at the relative risk (RR) for primary and secondary prevention at different fracture sites (TABLE 1) highlights similarities and differences. The risk reduction for vertebral fractures was statistically significant—and about the same—for women being treated with alendronate for primary and secondary prevention (RR=0.55; 95% confidence interval [CI], 0.38-0.80; RR=0.55; 95% CI, 0.43-0.69, respectively). For all other (nonvertebral) fractures in patients being treated with alendronate, only the outcomes for secondary prevention were statistically significant.
The Cochrane reviewers studied the effects of alendronate and risedronate3,4 for both primary and secondary prevention of osteoporotic fractures in postmenopausal women, using the following definitions (with slight variations in definitions between trials):
Primary prevention. Randomized controlled trials were classified as primary prevention trials if the participants had baseline T-scores >–2.0 or a baseline prevalence of vertebral fracture <20%.
Secondary prevention. Studies were classified as secondary prevention trials if the women had baseline T-scores ≤–2.0 (ie, bone mineral density [BMD] ≥2 standard deviations below peak bone mass) or previous vertebral compression fractures. (In the ibandronate individual patient meta-analysis,14 secondary prevention was defined as lumbar spine T-score <–2.5 or baseline vertebral fracture prevalence >20% or mean age of participants >60 years.)
Age-based criterion. When data on T-scores and/or vertebral compression fractures were unavailable, age was the determinant: Trials were considered secondary prevention if the average age of the participants was >62 years, and primary prevention if the average age was ≤62.
Risedronate is effective only for secondary prevention
Seven RCTs, including 2 primary and 5 secondary prevention trials, were included in the Cochrane review of risedronate, representing a total of 14,049 women.4 Doses ranged from 2.5 to 5 mg daily, but also included cyclical dosing—for example, taking 5 mg/d for the first 2 weeks of every month. Treatment duration ranged from 2 to 3 years.
At doses of 5 mg/d, there were no statistically significant decreases in fracture risk at any site in the primary prevention trials (TABLE 1), although the quality of evidence assessed was low. For secondary prevention, however, the risk reduction for vertebral fracture was significant (RR=0.61; 95% CI, 0.50-0.76), as were the reductions in risk for nonvertebral and hip fractures.
TABLE 1
Fracture risk reduction: How the bisphosphonates compare*
| Study | Vertebral fracture RR (95% CI) | Nonvertebral fracture (hip, wrist, others) RR (95% CI) | Hip fracture RR (95% CI) | Wrist fracture RR (95% CI) |
|---|---|---|---|---|
| Primary prevention | ||||
| Alendronate3 | 0.55 (0.38-0.80) | 0.89 (0.76-1.04) | 0.79 (0.44-1.44) | 1.19 (0.87-1.62) |
| Risedronate4 | 0.97 (0.42-2.25) | 0.81 (0.25-2.58) | N/A | N/A |
| Secondary prevention | ||||
| Alendronate3 | 0.55 (0.43-0.69) | 0.77 (0.64-0.92) | 0.47 (0.26-0.85) | 0.50 (0.34-0.73) |
| Risedronate4 | 0.61 (0.50-0.76) | 0.80 (0.72-0.90) | 0.74 (0.59-0.94) | 0.67 (0.42-1.07)† |
| Ibandronate15,16 Oral daily Oral intermittent | 0.62 (0.42-0.75) 0.50 (0.26-0.66) | No effect)‡ No effect | N/A N/A | N/A N/A |
| Zoledronic acid22 | 0.30 (0.2-0.38) | 0.75)§ (N/A) | 0.59¶ (0.42-0.83) | N/A |
| CI, confidence interval; N/A, not available; RR, relative risk. *Bold type indicates statistical significance (P<.05). †P=.10. ‡RR of nonvertebral fracture was 0.69 (P=.013) for daily oral ibandronate in the subgroup with femoral neck BMD T-score <–3.0. §P<.001. ¶Hazard ratio. | ||||
What are the absolute benefits? A look at number needed to treat
In addition to looking at the RR, the authors of both the alendronate and risedronate reviews calculated the number needed to treat (NNT) to prevent one fracture (TABLE 2) in the trial participants;3,4 they focused on the secondary prevention outcomes, as these were statistically significant. The reviewers also estimated what the NNT would be if the risk reductions achieved with alendronate and risedronate in the reviews occurred when treating community-based samples of women at moderate compared with high fracture risk.
The biggest differences involved hip fracture: For alendronate, if a community-based sample of women at moderate risk of fracture were treated with the drug and the reduction in RR seen in the secondary prevention trials applied, the NNT would be 100. Thus, for every 100 women treated for 5 years with alendronate, 1 hip fracture would be prevented. However, if this same RR reduction were applied to women at high risk of fracture, the NNT would be only 22.3 For risedronate, the estimated NNT to prevent one hip fracture in women at moderate risk was 203, compared with only 45 for women at high risk.4 These estimates indicate that the benefits of bisphosphonate therapy in preventing fractures are greatest in women with a high underlying fracture risk.
TABLE 2
NNT analysis: Women at higher risk are most likely to benefit3,4
| NNT | |||
|---|---|---|---|
| Observed in secondary prevention trials in reviews | Estimated for community-based sample of women with | ||
| High fracture risk* | Moderate fracture risk* | ||
| Alendronate (10 mg/d) | |||
| Vertebral fracture | 19 | 20 | 42 |
| Nonvertebral fracture | 47 | 16 | 27 |
| Hip fracture | 146 | 22 | 100 |
| Wrist fracture | 69 | N/A | N/A |
| Risedronate (5 mg/d) | |||
| Vertebral fracture | 19 | 23 | 49 |
| Nonvertebral fracture | 49 | 19 | 31 |
| Hip fracture | 138 | 45 | 203 |
| Wrist fracture | N/A | N/A | N/A |
| N/A, not available; NNT, number needed to treat. *NNT calculated by applying the relative risk reduction observed in the reviews to published estimates of 5-year fracture risk in a community-based sample of women >50 years of age at moderate and high risk. | |||
Adverse effects do not increase with longer-term treatment
In both the alendronate and risedronate reviews, adverse effects and the risk of discontinuing treatment due to adverse events were similar in the intervention and control groups.3,4 Postmarketing data suggest that there is potential for upper gastrointestinal (GI) problems, however;7 osteonecrosis of the jaw has also been reported infrequently.8,9 More recently, there have been reports of a possible link between bisphosphonates and atypical femoral fractures, which we’ll say more about in a bit.
Some potential adverse events—eg, osteonecrosis of the jaw and atypical femoral fractures—may be related to treatment duration. The maximum duration of the trials included in these meta-analyses was 4 years for alendronate and 3 years for risedronate. However, additional published data do not appear to support a relation between adverse events and treatment duration.
For alendronate, researchers extended the Fracture Incidence Trial (FIT) for a 10-year follow-up,10,11 comparing women who took the drug for the first 5 years with women who took it for 10 years. Adverse effects were similar in both groups.
For risedronate, researchers followed a small subsample (n=164) of the participants in the Vertical Efficacy with Risedronate Therapy (VERT) Study Group for up to 7 years.12,13 For the first 5 years, half of the participants took 5 mg/d risedronate, while the other half took a placebo. During the final 2 years, all participants received 5 mg/d risedronate. The incidence of adverse events among those who took the drug for 7 years was similar to that reported in the first 3 years of the original trial.13
Ibandronate studies focus on dose
Nonvertebral fracture. The Cochrane systematic review examining ibandronate for postmenopausal osteoporosis is not yet completed.5 However, Cranney et al performed a pooled analysis of individual patient data from 8 RCTs to examine the efficacy of different doses of the drug for the secondary prevention of nonvertebral fracture.14 (No studies of the drug for primary prevention have been done.) After 2 years of treatment at higher doses of ibandronate (annual cumulative exposure ≥10.8 mg, equivalent to 150 mg orally/month, 3 mg IV quarterly, or 2 mg IV every 2 months), the hazard ratio was 0.62 (95% CI, 0.396-0.974), compared with those taking lower doses (annual cumulative exposure of 5.5 mg). The individual results of the 2 largest trials did not demonstrate an effect on nonvertebral fracture, except in the subgroup of women with very low femoral neck bone mineral density (BMD) (T-scores <–3.0). 15-17
Vertebral fracture. There is no meta-analysis available with vertebral fracture outcomes for ibandronate, so we present the results of individual secondary prevention trials.
One was a double-blind RCT with 2496 participants, comparing women taking either 2.5 mg/d of ibandronate or 20 mg on alternate days with a group on placebo.15,16 The results? Those in both the daily and the intermittent treatment arms had significant risk reductions (RR=0.62; 95% CI, 0.42-0.75; RR=0.50; 95% CI, 0.26-0.66, respectively), after taking the drug for 3 years (TABLE 1), compared with those on placebo.15,16 The other RCT—a trial in which 2862 women received either quarterly intravenous (IV) injections of 1 or 0.5 mg ibandronate or placebo—did not demonstrate a significant reduction in vertebral fracture.17 This was attributed to an insufficient dose of the drug, a supposition supported by improvements in BMD in patients receiving higher doses of ibandronate.18,19
Oral ibandronate has been well tolerated in clinical trials in terms of GI side effects.20,21 Injection site reactions have been reported in those receiving IV infusions,17 and both IV and monthly oral ibandronate may be associated with mild, self-limiting flu-like symptoms.
Zoledronic acid RCTs show reduced fracture, mortality risk
Black et al studied the efficacy of zoledronic acid in a randomized, double-blind, placebo-controlled trial of 7736 postmenopausal women between the ages of 65 and 89 years.22 The women, all of whom had osteoporosis, received an IV infusion of either zoledronic acid (5 mg) or placebo at baseline, and again at 12 and 24 months. Vertebral and nonvertebral fractures, as well as hip fracture, were significantly reduced in the treatment group compared with placebo (TABLE 1).
In another RCT with 2127 participants, Lyles et al examined the effectiveness of 5 mg zoledronic acid IV given within 90 days of surgical repair of a hip fracture. In the intervention group, there was a 35% risk reduction in new clinical fractures (8.6% vs 13.9% for those on placebo; P=.001); mortality was also lower in the zoledronic acid group (9.6% vs 13.3%; P=.01).23
In both trials, the number of patients who had serious adverse events or dropped out because of an adverse event was similar in the treatment and placebo groups. In both studies, too, a sizeable number of patients treated with zoledronic acid reported flu-like symptoms up to 3 days after receiving an infusion, particularly after the first one. Cardiovascular events were similar across intervention groups in both studies, with one exception: In Black’s study,22 there was an increased incidence of serious atrial fibrillation in the zoledronic acid group (1.3% vs 0.5% for the placebo group).
Other issues to keep in mind
Atypical femoral fractures. Published data suggest an association between bisphosphonate use and atypical femoral fractures, particularly with longer-term use,24 although whether there is a causal link is unclear. Atypical femoral fractures occur with little or no trauma along the femur from just distal to the lesser trochanter to just proximal to the supracondylar flare.
In 2010, the FDA announced requirements for a black box warning about a possible link,25 highlighting the uncertainty about both the optimal duration of bisphosphonate therapy and the cause of these fractures.
While concerns about such a link remain, it is important to note that atypical femoral fractures are very uncommon: Current estimates are that they account for less than 1% of hip/femoral fractures. What’s more, far more fractures are prevented by the use of bisphosphonates than are associated with their use, with an estimated ratio of up to 29:1.24
Dosing schedules. Adherence to treatment is of key importance in maximizing outcomes from osteoporosis treatments, and is frequently low.26,27 One way of improving adherence is to reduce the frequency of dosing required.27 With that in mind, researchers have tested intermittent dosing regimens, using noninferiority or bridging trials.
Such studies have led to a number of approved dosing regimens—70 mg weekly for alendronate; 150 mg monthly and 35 mg weekly for risedronate; and 150 mg PO monthly and 3 mg IV quarterly for ibandronate among them. In making decisions about dosing, family physicians should consider patient preferences, but be aware that there are no direct efficacy data from RCTs to support these dosing regimens.
Calcium and vitamin D. The major fracture prevention trials of bisphosphonates have featured women who are calcium- and vitamin D-replete. In a recent study of 1515 women undergoing treatment with alendronate, risedronate, or raloxifene, however, that wasn’t always the case. 28 After 13 months, 115 participants suffered from a new clinical fracture. The adjusted odds ratio for fractures in women with vitamin D deficiency compared with those with normal levels of vitamin D was 1.77 (95% CI, 1.20-2.59; P=.004), an indication of the importance of maintaining adequate vitamin D levels in patients taking bisphosphonates.
In clinical practice, it is important to ensure that patients being treated with bisphosphonates are not deficient in vitamin D. While direct evidence of poorer outcomes associated with low calcium levels is lacking, it is reasonable to also assess calcium intake and to ensure that patients have adequate intake of both. (For more on calcium and vitamin D requirements, see the Institute of Medicine’s recent report at http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D/Report-Brief.aspx) and “The IOM’s report on calcium and vitamin D: Should it change the way you practice?”.
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
What’s best for your patients?
All these bisphosphonates have demonstrated efficacy for the secondary prevention of vertebral fracture. All except ibandronate have demonstrated efficacy for nonvertebral fracture, as well, and the evidence suggests that ibandronate will also be effective if adequate doses are given. Thus, for women at significant risk for fracture, it seems clear that the benefits of treatment outweigh the risks. The case is not so clearcut for women at lower risk. Evidence to support the use of bisphosphonates for primary prevention is limited, other than for alendronate—which has been shown to provide primary prevention of vertebral fracture.
Which bisphosphonate is best depends on patient preferences and individual profiles. (See “How would you treat these patients?”.) In the absence of head-to-head RCTs, it isn’t possible to comment on the relative efficacy of the various bisphosphonates or their adverse event profiles. Indeed, the authors of the 2 Cochrane reviews completed to date note that trial participants have been healthier, with fewer comorbidities, than many of the postmenopausal women seen by primary care physicians. Head-to-head studies conducted in family practice settings would be an important addition to the body of evidence for the prevention of osteoporotic fracture.
How would you treat these patients?
CASE 1 Mrs. A is an active 67-year-old in good health. On a recent hike, she lost her footing and sustained a Colles’ fracture when she fell, although her fall was only from standing height. Now, you are concerned that she might have osteoporosis.
A dual-energy x-ray absorptiometry (DXA) scan confirms this suspicion: Mrs. A’s lumbar spine T-score is –2.6. A dietary review reveals that she has a satisfactory calcium intake, and lab work shows that her serum vitamin D levels are normal. Mrs. A wants to discuss treatment options with you.
What immediate treatment do you consider?
Mrs. A has no contraindications to any FDA-approved treatment for osteoporosis; you suggest she begin taking bisphosphonates, explaining that they are first-line treatment to prevent subsequent osteoporotic fractures. You briefly discuss other options, but note that raloxifene only reduces the risk of vertebral fractures and parathyroid hormone is effective (but very expensive) and requires daily injections, and is therefore generally used for severe osteoporosis. Your patient asks about bisphosphonates’ side effects, particularly the serious jaw problems she’s heard about.
You explain that for the most part, oral bisphosphonates are well tolerated, but that there is a potential for upper gastrointestinal (GI) problems—which is why it’s important to remain upright for at least 30 minutes after taking the medication. You tell her that the risk of developing osteonecrosis of the jaw is very low when the medication is taken at the doses needed for osteoporosis treatment, but that the risk may increase after tooth extraction or dental surgery. Mrs. A has no current dental symptoms and at her usual yearly dental check-up 9 months ago, there were no problems noted, so dental review before starting treatment is not needed. Should she develop any jaw pain, however, she should see you or her dentist immediately.
You also advise her of the possible link between bisphosphonates and atypical femoral fracture, but point out that such fractures are extremely rare—and that the medication prevents far more fractures than it has the potential to cause. You tell her to contact you immediately if she develops pain in the groin or thigh or experiences GI distress.
Which bisphosphonate do you prescribe?
You inform Mrs. A that alendronate has the longest follow-up data of the oral bisphosphonates and has demonstrated efficacy for the secondary prevention of wrist fractures, that risedronate and ibandronate have the advantage of being able to be taken monthly rather than weekly, and that zoledronic acid can be administered in a yearly infusion. She opts for alendronate. You prescribe a weekly dose of 70 mg and ask her to return in 3 months, and to call before then if any problems arise.
CASE 2 Mrs. Y, age 82, recently sustained a fractured femoral neck, which was treated surgically at the local hospital. She was discharged with a prescription for alendronate to treat her osteoporosis and prevent further fractures; her husband has brought her in today to get a new prescription.
During the visit, he reminds you that Mrs. Y has problems with memory. He also says he’s finding it increasingly difficult to ensure that his wife remains upright for 30 minutes after taking alendronate, and that she has begun complaining of indigestion.
What do you decide to do?
An inability to stay upright for 30 minutes after drug administration is a contraindication to the use of oral bisphosphonates. The presence of upper GI symptoms is also a concern. You offer Mrs. Y the option of a once-yearly IV infusion of zoledronic acid instead, and she and her husband agree to this. Before scheduling a follow-up visit, you discuss the patient’s nutritional intake, and discover that she consumes only a moderate amount of calcium—at most 2 servings of dairy products per day. You also note that her serum vitamin D level was not checked in the hospital. You order lab work, with a view to correcting any deficiency before proceeding with a zoledronic infusion (due to the risk of tetany) and to maintaining her on an appropriate level of calcium and vitamin D intake, using supplements only if necessary.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
An estimated 10 million US residents, most of them women over the age of 50, suffer from osteoporosis, and another 33 million have low bone mass.1 Together, they incur more than 2 million osteoporotic fractures annually.1,2 In addition to the high cost of a single osteoporotic fracture in terms of morbidity, mortality, and health care spending, individuals who sustain one such fracture are at high risk for another. That risk can be greatly reduced with appropriate treatment.
Bisphosphonates, which act on osteoclasts to inhibit bone resorption, are first-line therapy for prevention of osteoporotic fractures. Four bisphosphonates—alendronate, ibandronate, risedronate, and zoledronic acid—are approved by the US Food and Drug Administration (FDA) for the treatment of postmenopausal osteoporosis.
While menopause itself increases a woman’s risk for osteoporotic fracture, questions remain about when to initiate preventive therapy, which patients are candidates for bisphosphonates, and whether bisphosphonates are effective for primary as well as secondary prevention. This overview from the Cochrane Musculoskeletal Group (CMSG) addresses those questions.
To help you provide optimal treatment for postmenopausal patients, we present the findings of recently conducted systematic reviews of 2 bisphosphonates—alendronate and risedronate—from the Cochrane Database of Systematic Reviews,3,4 in context with the available evidence on the efficacy of ibandronate and zoledronic acid. Cochrane reviews of ibandronate and zoledronic acid are underway, but not yet completed.5,6
This is the second in a series of articles based on the findings of the Cochrane Musculoskeletal Group (CMSG), one of the largest review groups in the Cochrane Collaboration. The CMSG synthesizes the results of high-quality clinical trials to determine whether interventions for the prevention, treatment, and rehabilitation of musculoskeletal disorders are safe and effective. In this article and those that follow, CMSG’s aim is to bring its findings to the attention of family physicians in a context that is relevant to clinical practice.
Alendronate reduces vertebral fracture risk across the board
Wells et al identified 11 RCTs for the alendronate review (3 primary and 8 secondary prevention trials), representing a total of 12,068 women.3 (For definitions of what constituted a primary vs a secondary prevention trial, see the box.)
Doses of alendronate ranged from 1 to 20 mg daily, with most studies using doses of 5 or 10 mg. Treatment duration ranged from 1 to 4 years.
A look at the relative risk (RR) for primary and secondary prevention at different fracture sites (TABLE 1) highlights similarities and differences. The risk reduction for vertebral fractures was statistically significant—and about the same—for women being treated with alendronate for primary and secondary prevention (RR=0.55; 95% confidence interval [CI], 0.38-0.80; RR=0.55; 95% CI, 0.43-0.69, respectively). For all other (nonvertebral) fractures in patients being treated with alendronate, only the outcomes for secondary prevention were statistically significant.
The Cochrane reviewers studied the effects of alendronate and risedronate3,4 for both primary and secondary prevention of osteoporotic fractures in postmenopausal women, using the following definitions (with slight variations in definitions between trials):
Primary prevention. Randomized controlled trials were classified as primary prevention trials if the participants had baseline T-scores >–2.0 or a baseline prevalence of vertebral fracture <20%.
Secondary prevention. Studies were classified as secondary prevention trials if the women had baseline T-scores ≤–2.0 (ie, bone mineral density [BMD] ≥2 standard deviations below peak bone mass) or previous vertebral compression fractures. (In the ibandronate individual patient meta-analysis,14 secondary prevention was defined as lumbar spine T-score <–2.5 or baseline vertebral fracture prevalence >20% or mean age of participants >60 years.)
Age-based criterion. When data on T-scores and/or vertebral compression fractures were unavailable, age was the determinant: Trials were considered secondary prevention if the average age of the participants was >62 years, and primary prevention if the average age was ≤62.
Risedronate is effective only for secondary prevention
Seven RCTs, including 2 primary and 5 secondary prevention trials, were included in the Cochrane review of risedronate, representing a total of 14,049 women.4 Doses ranged from 2.5 to 5 mg daily, but also included cyclical dosing—for example, taking 5 mg/d for the first 2 weeks of every month. Treatment duration ranged from 2 to 3 years.
At doses of 5 mg/d, there were no statistically significant decreases in fracture risk at any site in the primary prevention trials (TABLE 1), although the quality of evidence assessed was low. For secondary prevention, however, the risk reduction for vertebral fracture was significant (RR=0.61; 95% CI, 0.50-0.76), as were the reductions in risk for nonvertebral and hip fractures.
TABLE 1
Fracture risk reduction: How the bisphosphonates compare*
| Study | Vertebral fracture RR (95% CI) | Nonvertebral fracture (hip, wrist, others) RR (95% CI) | Hip fracture RR (95% CI) | Wrist fracture RR (95% CI) |
|---|---|---|---|---|
| Primary prevention | ||||
| Alendronate3 | 0.55 (0.38-0.80) | 0.89 (0.76-1.04) | 0.79 (0.44-1.44) | 1.19 (0.87-1.62) |
| Risedronate4 | 0.97 (0.42-2.25) | 0.81 (0.25-2.58) | N/A | N/A |
| Secondary prevention | ||||
| Alendronate3 | 0.55 (0.43-0.69) | 0.77 (0.64-0.92) | 0.47 (0.26-0.85) | 0.50 (0.34-0.73) |
| Risedronate4 | 0.61 (0.50-0.76) | 0.80 (0.72-0.90) | 0.74 (0.59-0.94) | 0.67 (0.42-1.07)† |
| Ibandronate15,16 Oral daily Oral intermittent | 0.62 (0.42-0.75) 0.50 (0.26-0.66) | No effect)‡ No effect | N/A N/A | N/A N/A |
| Zoledronic acid22 | 0.30 (0.2-0.38) | 0.75)§ (N/A) | 0.59¶ (0.42-0.83) | N/A |
| CI, confidence interval; N/A, not available; RR, relative risk. *Bold type indicates statistical significance (P<.05). †P=.10. ‡RR of nonvertebral fracture was 0.69 (P=.013) for daily oral ibandronate in the subgroup with femoral neck BMD T-score <–3.0. §P<.001. ¶Hazard ratio. | ||||
What are the absolute benefits? A look at number needed to treat
In addition to looking at the RR, the authors of both the alendronate and risedronate reviews calculated the number needed to treat (NNT) to prevent one fracture (TABLE 2) in the trial participants;3,4 they focused on the secondary prevention outcomes, as these were statistically significant. The reviewers also estimated what the NNT would be if the risk reductions achieved with alendronate and risedronate in the reviews occurred when treating community-based samples of women at moderate compared with high fracture risk.
The biggest differences involved hip fracture: For alendronate, if a community-based sample of women at moderate risk of fracture were treated with the drug and the reduction in RR seen in the secondary prevention trials applied, the NNT would be 100. Thus, for every 100 women treated for 5 years with alendronate, 1 hip fracture would be prevented. However, if this same RR reduction were applied to women at high risk of fracture, the NNT would be only 22.3 For risedronate, the estimated NNT to prevent one hip fracture in women at moderate risk was 203, compared with only 45 for women at high risk.4 These estimates indicate that the benefits of bisphosphonate therapy in preventing fractures are greatest in women with a high underlying fracture risk.
TABLE 2
NNT analysis: Women at higher risk are most likely to benefit3,4
| NNT | |||
|---|---|---|---|
| Observed in secondary prevention trials in reviews | Estimated for community-based sample of women with | ||
| High fracture risk* | Moderate fracture risk* | ||
| Alendronate (10 mg/d) | |||
| Vertebral fracture | 19 | 20 | 42 |
| Nonvertebral fracture | 47 | 16 | 27 |
| Hip fracture | 146 | 22 | 100 |
| Wrist fracture | 69 | N/A | N/A |
| Risedronate (5 mg/d) | |||
| Vertebral fracture | 19 | 23 | 49 |
| Nonvertebral fracture | 49 | 19 | 31 |
| Hip fracture | 138 | 45 | 203 |
| Wrist fracture | N/A | N/A | N/A |
| N/A, not available; NNT, number needed to treat. *NNT calculated by applying the relative risk reduction observed in the reviews to published estimates of 5-year fracture risk in a community-based sample of women >50 years of age at moderate and high risk. | |||
Adverse effects do not increase with longer-term treatment
In both the alendronate and risedronate reviews, adverse effects and the risk of discontinuing treatment due to adverse events were similar in the intervention and control groups.3,4 Postmarketing data suggest that there is potential for upper gastrointestinal (GI) problems, however;7 osteonecrosis of the jaw has also been reported infrequently.8,9 More recently, there have been reports of a possible link between bisphosphonates and atypical femoral fractures, which we’ll say more about in a bit.
Some potential adverse events—eg, osteonecrosis of the jaw and atypical femoral fractures—may be related to treatment duration. The maximum duration of the trials included in these meta-analyses was 4 years for alendronate and 3 years for risedronate. However, additional published data do not appear to support a relation between adverse events and treatment duration.
For alendronate, researchers extended the Fracture Incidence Trial (FIT) for a 10-year follow-up,10,11 comparing women who took the drug for the first 5 years with women who took it for 10 years. Adverse effects were similar in both groups.
For risedronate, researchers followed a small subsample (n=164) of the participants in the Vertical Efficacy with Risedronate Therapy (VERT) Study Group for up to 7 years.12,13 For the first 5 years, half of the participants took 5 mg/d risedronate, while the other half took a placebo. During the final 2 years, all participants received 5 mg/d risedronate. The incidence of adverse events among those who took the drug for 7 years was similar to that reported in the first 3 years of the original trial.13
Ibandronate studies focus on dose
Nonvertebral fracture. The Cochrane systematic review examining ibandronate for postmenopausal osteoporosis is not yet completed.5 However, Cranney et al performed a pooled analysis of individual patient data from 8 RCTs to examine the efficacy of different doses of the drug for the secondary prevention of nonvertebral fracture.14 (No studies of the drug for primary prevention have been done.) After 2 years of treatment at higher doses of ibandronate (annual cumulative exposure ≥10.8 mg, equivalent to 150 mg orally/month, 3 mg IV quarterly, or 2 mg IV every 2 months), the hazard ratio was 0.62 (95% CI, 0.396-0.974), compared with those taking lower doses (annual cumulative exposure of 5.5 mg). The individual results of the 2 largest trials did not demonstrate an effect on nonvertebral fracture, except in the subgroup of women with very low femoral neck bone mineral density (BMD) (T-scores <–3.0). 15-17
Vertebral fracture. There is no meta-analysis available with vertebral fracture outcomes for ibandronate, so we present the results of individual secondary prevention trials.
One was a double-blind RCT with 2496 participants, comparing women taking either 2.5 mg/d of ibandronate or 20 mg on alternate days with a group on placebo.15,16 The results? Those in both the daily and the intermittent treatment arms had significant risk reductions (RR=0.62; 95% CI, 0.42-0.75; RR=0.50; 95% CI, 0.26-0.66, respectively), after taking the drug for 3 years (TABLE 1), compared with those on placebo.15,16 The other RCT—a trial in which 2862 women received either quarterly intravenous (IV) injections of 1 or 0.5 mg ibandronate or placebo—did not demonstrate a significant reduction in vertebral fracture.17 This was attributed to an insufficient dose of the drug, a supposition supported by improvements in BMD in patients receiving higher doses of ibandronate.18,19
Oral ibandronate has been well tolerated in clinical trials in terms of GI side effects.20,21 Injection site reactions have been reported in those receiving IV infusions,17 and both IV and monthly oral ibandronate may be associated with mild, self-limiting flu-like symptoms.
Zoledronic acid RCTs show reduced fracture, mortality risk
Black et al studied the efficacy of zoledronic acid in a randomized, double-blind, placebo-controlled trial of 7736 postmenopausal women between the ages of 65 and 89 years.22 The women, all of whom had osteoporosis, received an IV infusion of either zoledronic acid (5 mg) or placebo at baseline, and again at 12 and 24 months. Vertebral and nonvertebral fractures, as well as hip fracture, were significantly reduced in the treatment group compared with placebo (TABLE 1).
In another RCT with 2127 participants, Lyles et al examined the effectiveness of 5 mg zoledronic acid IV given within 90 days of surgical repair of a hip fracture. In the intervention group, there was a 35% risk reduction in new clinical fractures (8.6% vs 13.9% for those on placebo; P=.001); mortality was also lower in the zoledronic acid group (9.6% vs 13.3%; P=.01).23
In both trials, the number of patients who had serious adverse events or dropped out because of an adverse event was similar in the treatment and placebo groups. In both studies, too, a sizeable number of patients treated with zoledronic acid reported flu-like symptoms up to 3 days after receiving an infusion, particularly after the first one. Cardiovascular events were similar across intervention groups in both studies, with one exception: In Black’s study,22 there was an increased incidence of serious atrial fibrillation in the zoledronic acid group (1.3% vs 0.5% for the placebo group).
Other issues to keep in mind
Atypical femoral fractures. Published data suggest an association between bisphosphonate use and atypical femoral fractures, particularly with longer-term use,24 although whether there is a causal link is unclear. Atypical femoral fractures occur with little or no trauma along the femur from just distal to the lesser trochanter to just proximal to the supracondylar flare.
In 2010, the FDA announced requirements for a black box warning about a possible link,25 highlighting the uncertainty about both the optimal duration of bisphosphonate therapy and the cause of these fractures.
While concerns about such a link remain, it is important to note that atypical femoral fractures are very uncommon: Current estimates are that they account for less than 1% of hip/femoral fractures. What’s more, far more fractures are prevented by the use of bisphosphonates than are associated with their use, with an estimated ratio of up to 29:1.24
Dosing schedules. Adherence to treatment is of key importance in maximizing outcomes from osteoporosis treatments, and is frequently low.26,27 One way of improving adherence is to reduce the frequency of dosing required.27 With that in mind, researchers have tested intermittent dosing regimens, using noninferiority or bridging trials.
Such studies have led to a number of approved dosing regimens—70 mg weekly for alendronate; 150 mg monthly and 35 mg weekly for risedronate; and 150 mg PO monthly and 3 mg IV quarterly for ibandronate among them. In making decisions about dosing, family physicians should consider patient preferences, but be aware that there are no direct efficacy data from RCTs to support these dosing regimens.
Calcium and vitamin D. The major fracture prevention trials of bisphosphonates have featured women who are calcium- and vitamin D-replete. In a recent study of 1515 women undergoing treatment with alendronate, risedronate, or raloxifene, however, that wasn’t always the case. 28 After 13 months, 115 participants suffered from a new clinical fracture. The adjusted odds ratio for fractures in women with vitamin D deficiency compared with those with normal levels of vitamin D was 1.77 (95% CI, 1.20-2.59; P=.004), an indication of the importance of maintaining adequate vitamin D levels in patients taking bisphosphonates.
In clinical practice, it is important to ensure that patients being treated with bisphosphonates are not deficient in vitamin D. While direct evidence of poorer outcomes associated with low calcium levels is lacking, it is reasonable to also assess calcium intake and to ensure that patients have adequate intake of both. (For more on calcium and vitamin D requirements, see the Institute of Medicine’s recent report at http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D/Report-Brief.aspx) and “The IOM’s report on calcium and vitamin D: Should it change the way you practice?”.
“Dietary Reference Intakes for Calcium and Vitamin D,” the consensus report released by the Institute of Medicine (IOM) late last year (http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) generated a great deal of attention because it concluded that postmenopausal women taking supplements may be getting too much calcium, and that few people need to take vitamin D. These findings, among others, left many physicians wondering how, or if, the IOM’s report should change the way they practice.
The Journal of Family Practice posed that question to Susan Williams, MD, MS, FACN, FACP, an internist at the Cleveland Clinic and a diplomate with the American Board of Physician Nutrition Specialists. Her response: The report probably shouldn’t change the way you practice.
Here, Dr. Williams explains why.
Recommended daily allowances are guidelines. The new dietary reference intakes (DRIs), like the recommended daily allowances (RDAs) they replace, are quantitative estimates of nutrient intakes intended for planning and assessing diets of healthy populations. They were never intended to be applied “across the board,” or used as a benchmark for the dietary adequacy of individual patients.
Testing is still advisable when there is clinical suspicion of a calcium or vitamin D deficiency. Because parathyroid hormone (PTH) compensates for calcium deficiency by drawing calcium from the bones, an adequate serum calcium level alone does not necessarily reflect an adequate calcium intake. In fact, a low serum calcium level is likely to be the result of abnormally low levels of vitamin D. Thus, the best way to get an accurate picture of a patient’s status is to simultaneously test serum calcium, vitamin D, and PTH levels.
Some patients require considerably larger doses of vitamin D than the recommended quantities.1,2 This is particularly true for obese individuals and patients who have undergone bariatric surgery, for example.3-5 The safety of daily dosing of vitamin D in far greater quantities has been established,6,7 and the risks of chronic undersupplementation8-10 outweigh the risks associated with hypervitaminosis D, particularly when D3 (cholecalciferol) supplements are recommended.
Calcium supplementation is safe for postmenopausal women. Many older women have poor dietary intake of calcium, and again, the consequences of a deficiency are far greater than those associated with an excess. The risk of kidney stones in women taking calcium supplements can be averted by advising patients to take calcium citrate, which tends to neutralize urine and has better fractional uptake into the bone than calcium carbonate.
The IOM report serves to remind us that getting adequate calcium and vitamin D is important for everyone. Age and gender-specific recommendations should be emphasized, remembering that in general, the IOM’s DRIs are likely to meet the actual needs of most healthy patients, but may well fall short in the presence of chronic illness and disease.
Remember, too, that while we should always emphasize the importance of eating foods that are rich in calcium and vitamin D, patients’ diets often fall short. In such cases—with the exception of patients with certain conditions (eg, renal failure or hyperparathyroidism)—supplements such as calcium citrate and vitamin D3 can be safely and confidently recommended.
Susan Williams, MD, MS, FACN, FACP, reported no potential conflict of interest relevant to this article.
References
1. Holick MF. The role of vitamin D for bone health and fracture prevention. Curr Osteoporos Rep. 2006;4:96-102.
2. Grant WB, Holick MF. Benefits and requirements of vitamin D for optimal health. Altern Med Rev. 2005;10:94-111.
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
4. Bischoff-Ferrari HA, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18-28.
5. Flores L, et al. Calcium and vitamin D supplementation after gastric bypass should be individualized to improve or avoid hyperparathyroidism. Obes Surg. 2010;20:738-743.
6. Vieth R, et al. Efficacy and safety of vitamin D intake exceeding the lowest observed adverse eff ect level. Am J Clin Nutr. 2001;73:288-294.
7. Barger-Lux MJ, et al. Vitamin D and its major metabolite: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8:222-230.
8. Sakuma M, et al. Vitamin D and intact PTH status in patients with hip fracture. Osteoporos Int. 2006;17:1608-1614.
9. Broe KE, et al. A higher dose of vitamin D reduces the risk of falls in nursing home residents. J Am Geriatr Soc. 2007;55:234-239.
10. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly. Endocr Rev. 2001;22:477-501.
What’s best for your patients?
All these bisphosphonates have demonstrated efficacy for the secondary prevention of vertebral fracture. All except ibandronate have demonstrated efficacy for nonvertebral fracture, as well, and the evidence suggests that ibandronate will also be effective if adequate doses are given. Thus, for women at significant risk for fracture, it seems clear that the benefits of treatment outweigh the risks. The case is not so clearcut for women at lower risk. Evidence to support the use of bisphosphonates for primary prevention is limited, other than for alendronate—which has been shown to provide primary prevention of vertebral fracture.
Which bisphosphonate is best depends on patient preferences and individual profiles. (See “How would you treat these patients?”.) In the absence of head-to-head RCTs, it isn’t possible to comment on the relative efficacy of the various bisphosphonates or their adverse event profiles. Indeed, the authors of the 2 Cochrane reviews completed to date note that trial participants have been healthier, with fewer comorbidities, than many of the postmenopausal women seen by primary care physicians. Head-to-head studies conducted in family practice settings would be an important addition to the body of evidence for the prevention of osteoporotic fracture.
How would you treat these patients?
CASE 1 Mrs. A is an active 67-year-old in good health. On a recent hike, she lost her footing and sustained a Colles’ fracture when she fell, although her fall was only from standing height. Now, you are concerned that she might have osteoporosis.
A dual-energy x-ray absorptiometry (DXA) scan confirms this suspicion: Mrs. A’s lumbar spine T-score is –2.6. A dietary review reveals that she has a satisfactory calcium intake, and lab work shows that her serum vitamin D levels are normal. Mrs. A wants to discuss treatment options with you.
What immediate treatment do you consider?
Mrs. A has no contraindications to any FDA-approved treatment for osteoporosis; you suggest she begin taking bisphosphonates, explaining that they are first-line treatment to prevent subsequent osteoporotic fractures. You briefly discuss other options, but note that raloxifene only reduces the risk of vertebral fractures and parathyroid hormone is effective (but very expensive) and requires daily injections, and is therefore generally used for severe osteoporosis. Your patient asks about bisphosphonates’ side effects, particularly the serious jaw problems she’s heard about.
You explain that for the most part, oral bisphosphonates are well tolerated, but that there is a potential for upper gastrointestinal (GI) problems—which is why it’s important to remain upright for at least 30 minutes after taking the medication. You tell her that the risk of developing osteonecrosis of the jaw is very low when the medication is taken at the doses needed for osteoporosis treatment, but that the risk may increase after tooth extraction or dental surgery. Mrs. A has no current dental symptoms and at her usual yearly dental check-up 9 months ago, there were no problems noted, so dental review before starting treatment is not needed. Should she develop any jaw pain, however, she should see you or her dentist immediately.
You also advise her of the possible link between bisphosphonates and atypical femoral fracture, but point out that such fractures are extremely rare—and that the medication prevents far more fractures than it has the potential to cause. You tell her to contact you immediately if she develops pain in the groin or thigh or experiences GI distress.
Which bisphosphonate do you prescribe?
You inform Mrs. A that alendronate has the longest follow-up data of the oral bisphosphonates and has demonstrated efficacy for the secondary prevention of wrist fractures, that risedronate and ibandronate have the advantage of being able to be taken monthly rather than weekly, and that zoledronic acid can be administered in a yearly infusion. She opts for alendronate. You prescribe a weekly dose of 70 mg and ask her to return in 3 months, and to call before then if any problems arise.
CASE 2 Mrs. Y, age 82, recently sustained a fractured femoral neck, which was treated surgically at the local hospital. She was discharged with a prescription for alendronate to treat her osteoporosis and prevent further fractures; her husband has brought her in today to get a new prescription.
During the visit, he reminds you that Mrs. Y has problems with memory. He also says he’s finding it increasingly difficult to ensure that his wife remains upright for 30 minutes after taking alendronate, and that she has begun complaining of indigestion.
What do you decide to do?
An inability to stay upright for 30 minutes after drug administration is a contraindication to the use of oral bisphosphonates. The presence of upper GI symptoms is also a concern. You offer Mrs. Y the option of a once-yearly IV infusion of zoledronic acid instead, and she and her husband agree to this. Before scheduling a follow-up visit, you discuss the patient’s nutritional intake, and discover that she consumes only a moderate amount of calcium—at most 2 servings of dairy products per day. You also note that her serum vitamin D level was not checked in the hospital. You order lab work, with a view to correcting any deficiency before proceeding with a zoledronic infusion (due to the risk of tetany) and to maintaining her on an appropriate level of calcium and vitamin D intake, using supplements only if necessary.
CORRESPONDENCE Tania Winzenberg, MBBS, Menzies Research Institute, Private Bag 23, Hobart, Tasmania, Australia 7001; [email protected]
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
1. National Osteoporosis Foundation. America’s bone health: the state of osteoporosis and low bone mass in our nation. Washington, DC: National Osteoporosis Foundation; 2002.
2. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
3. Wells GA, Cranney A, Peterson J, et al. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD001155.-
4. Wells G, Cranney A, Peterson J, et al. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst Rev. 2008;(1):CD004523.-
5. Wang Q, Decai C. Ibandronate sodium for osteoporosis in post-menopausal women (Protocol). Cochrane Database Syst Rev. 2007;CD006514.-DOI:10.1002/14651858.
6. Albergaria BH, Gomes Silva BN, Atallah AN, et al. Intravenous zoledronate for postmenopausal osteoporosis (Protocol). Cochrane Database Syst Rev. 2010;(1):CD008332.-DOI:10.1002/14651858.
7. Anonymous. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd; 2007.
8. Silverman SL, Landesberg R. Osteonecrosis of the jaw and the role of bisphosphonates: a critical review. Am J Med. 2009;122 (suppl 2):S33-S45.
9. Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44:4-10.
10. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA. 2006;296:2927-2938.
11. Cummings SR, Black DM, Thompson DE, et al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA. 1998;280:2077-2082.
12. Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83-91.
13. Mellstrom DD, Sorensen OH, Goemaere S, et al. Seven years of treatment with risedronate in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;75:462-468.
14. Cranney A, Wells GA, Yetisir E, et al. Ibandronate for the prevention of nonvertebral fractures: a pooled analysis of individual patient data. Osteoporos Int. 2009;20:291-297.
15. Chesnut IC, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241-1249.
16. Delmas PD, Recker RR, Chesnut CH, et al. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporosis Int. 2004;15:792-798.
17. Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently dosed intravenous ibandronate injections are associated with suboptimal antifracture efficacy in postmenopausal osteoporosis. Bone. 2004;34:890-899.
18. Adami S, Felsenberg D, Christiansen C, et al. Efficacy and safety of ibandronate given by intravenous injection once every 3 months. Bone. 2004;34:881-889.
19. Delmas PD, Adami S, Strugala C, et al. Intravenous ibandronate injections in postmenopausal women with osteoporosis. One-year results from the dosing intravenous administration study. Arthritis Rheum. 2006;54:1838-1846.
20. Epstein S, Delmas PD, Emkey R, et al. Oral ibandronate in the management of postmenopausal osteoporosis: review of upper gastrointestinal safety. Maturitas. 2006;54:1-10.
21. Ettinger MP, Felsenberg D, Harris ST, et al. Safety and tolerability of oral daily and intermittent ibandronate are not influenced by age. J Rheumatol. 2005;32:1968-1974.
22. Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809-1822.
23. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
24. Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267-2294.
25. US Food and Drug Administration. FDA Drug Safety Communication: Safety update for osteoporosis drugs, bisphosphonates, and atypical fractures. October 13, 2010. Available at: http://www.fda.gov/drugs/drugsafety/ucm229009.htm. Accessed December 7, 2010.
26. Seeman E, Compston J, Adachi J, et al. Non-compliance: the Achilles’ heel of anti-fracture efficacy. Osteoporos Int. 2007;18:711-719.
27. Cramer JA, Gold DT, Silverman SL, et al. A systematic review of persistence and compliance with bisphosphonates for osteoporosis. Osteoporos Int. 2007;18:1023-1031.
28. Adami S, Giannini S, Bianchi G, et al. Vitamin D status and response to treatment in post-menopausal osteoporosis. Osteoporos Int. 2009;20:239-244.
Battling shingles: Fine-tune your care
• Reserve laboratory testing for unclear or complicated cases of herpes zoster (HZ), as the condition can be diagnosed clinically in most cases. B
• Whenever possible, initiate oral antiviral therapy within 72 hours of the onset of the shingles rash to accelerate healing and reduce the duration and severity of pain. A
• Offer the HZ vaccine (Zostavax) to patients ages 60 and older to reduce the risk of shingles and postherpetic neuralgia. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane T, a 53-year-old patient, calls her family physician’s office for a same-day appointment; she has severe right upper quadrant pain that developed a few hours ago. When Jane comes in, she tells her physician that the pain feels like the gallbladder attacks she used to have—before her gallbladder was removed 2 years ago. She has nausea but no vomiting, is afebrile, and has no urinary symptoms.
A series of in-office tests—urinalysis, complete blood count, comprehensive metabolic profile, amylase, and lipase— are all normal. The physician gives Jane an intramuscular injection of ketorolac and a prescription for oral tramadol, and sends her for a right upper quadrant ultrasound, which is also normal. The next morning, the pain intensifies, and Jane goes to the emergency department, where she undergoes computed tomography of the abdomen and pelvis—also normal. After a night in the hospital for observation, she is released. She returns to the family physician 3 days later, this time with an erythematous papular rash in a dermatomal distribution over her back and right upper quadrant that her doctor immediately recognizes as shingles.
Jane is not alone. Each year, about 1 million US residents develop herpes zoster (HZ),1 and 70% to 80% of them experience prodromal pain in the affected dermatome.2 For some, the pain is significant enough to prompt a medical work-up to rule out other potential causes, such as myocardial infarction, nephrolithiasis, pancreatitis, cholecystitis, and appendicitis.
The time and resources spent on such a work-up may be unavoidable when a patient presents with severe pain. But alert primary care physicians can avoid unnecessary diagnostic tests by being aware of the prodromal symptoms of HZ and being on the lookout for the rash that follows.
Age alone is a key factor in the detection of HZ, of course. One in 3 people will develop shingles during their lifetime,1 with half of all cases occurring in people ages 60 and older.3 In addition to early detection and treatment, physicians can do their part to battle HZ by routinely recommending the shingles vaccine (Zostavax) to patients in this age group.
Diagnosing HZ: From prodrome to rash
For many patients, an abnormal skin sensation on one side of the body, such as itching, burning, or altered sensitivity to touch, is the first symptom of HZ. These sensations, including pain that can range from mild to severe, may precede the rash by days or even weeks.4 Systemic symptoms are far less common; less than 20% of patients develop fever, headache, malaise, or fatigue as part of the HZ prodrome.2
The shingles rash is usually unilateral and does not cross the midline. However, it may occur in up to 3 adjacent dermatomes. The trunk is the most common site for the rash, but it may also develop on the face, buttocks, or other parts of the body. The lesions start as erythematous papules and evolve into vesicles within 12 to 24 hours.5
Once the rash emerges, HZ can usually be diagnosed clinically in a primary care setting, and laboratory testing should be considered only when diagnosis is uncertain. In a cohort study of 260 patients older than 50 years with clinically diagnosed HZ, 236 (91%) cases were confirmed on serologic testing.6 In an Icelandic study, 93% of 505 cases were diagnosed correctly by general practitioners, using expert opinion and clinical course as the gold standard.7
If diagnostic testing is necessary, physicians have a number of choices:
Varicella zoster polymerase chain reaction (PCR) test. PCR testing for HZ can provide rapid and reliable diagnosis and is becoming more widely available, but its use should be limited because of the cost (approximately $300).
Tzanck smear. This test is quick and inexpensive, and can reliably diagnose a herpetic lesion based on the presence of acantholytic and multinucleated giant cells in a sample collected from the base of a vesicle.8 However, this technique cannot differentiate HZ from herpes simplex infection, and lack of experience with collection or interpretation of the Tzanck smear limits its usefulness.
Serologic testing. Serology has limited utility in the diagnosis of acute HZ, but may provide a retrospective diagnosis, if needed. In a study of 260 patients older than 50 years with clinically diagnosed acute HZ, varicella zoster IgA or IgM was positive in 61% of patients at the time of presentation, and in 91% of patients 5 to 10 days later.6
Viral culture. Obtaining a viral culture of varicella zoster is another option, but it is not recommended, as sensitivity is poor and incubation requires several days.
The course of illness, and risk of complications
For some patients, the acute pain and hypersensitivity at the site of the rash resolve in several days; for others, this may take several weeks or more. One recent study found a median of 32.5 days for pain duration in patients ages 50 and older.9 The suffering can last far longer, however, if complications arise.
Postherpetic neuralgia (PHN), a chronic and often debilitating condition with pain that can last months, or even indefinitely, affects about 10% to 15% of patients with HZ.10 Risk factors for the development of PHN include advanced age, female sex, presence of a prodrome, severe acute HZ pain, and a severe rash.11
Trigeminal nerve involvement. HZ affects the first branch of the trigeminal nerve in about 10% to 15% of patients.5 In such cases, the rash may erupt on the forehead, periocular area, and nose. Patients with trigeminal nerve involvement are at significant risk for ocular complications (HZ ophthalmicus), including keratitis, iritis, and possible vision loss, and should be treated and referred to an ophthalmologist without delay.
In addition to these complications, others reported in a review of 859 patients include bacterial skin infection, motor neuropathy, and, rarely, meningitis and HZ oticus.12 Advanced age markedly increased the likelihood of complications.
Initiate treatment without delay
Multiple randomized controlled clinical trials have demonstrated the efficacy of oral antiviral treatment in reducing the duration of viral shedding, accelerating rash healing, and reducing the severity and duration of acute HZ pain.2 In all the studies, however, antiviral therapy was started within 72 hours of the onset of the rash; the efficacy of initiating antiviral treatment after >72 hours has not been systematically studied.2 When it’s not possible to begin therapy within this time frame, however, many experts recommend initiating therapy as soon as possible thereafter.1
Three antiviral agents—acyclovir, famciclovir, and valacyclovir—have been approved by the US Food and Drug Administration to treat HZ. Evidence suggests that famciclovir and valacyclovir have comparable efficacy with regard to resolution of both the rash and the acute pain,13 and result in more rapid pain resolution compared with acyclovir.14,15 Famciclovir and valacyclovir also offer simpler dosing schedules—both are taken 3 times a day, while acyclovir requires 5 daily doses— but they are significantly more expensive (TABLE).
Oral antiviral therapy is strongly recommended for patients who are older than 50 and those who have moderate-to-severe pain or rash—or whose rash appears somewhere other than the trunk.2 But given the safety of oral antiviral agents, treatment can be considered for younger patients and those with milder cases of HZ, as well. (Routine use of antiviral agents is not indicated for unexplained unilateral pain, as varicella zoster infection without the classic rash [zoster sine herpete] is a rare cause.16)
Results regarding the efficacy of oral antiviral agents for reducing the duration of chronic pain and preventing PHN are mixed.2 A recent Cochrane review concluded that oral acyclovir does not significantly reduce the incidence of PHN and that there is insufficient evidence to determine if other antivirals do.17
TABLE
FDA-approved oral antiviral regimens
| Drug | Dosing regimen | Cost of regimen* |
|---|---|---|
| Acyclovir | 800 mg 5 times/d for 7-10 days | $55.97 |
| Famciclovir | 500 mg tid for 7 days | $319.99 |
| Valacyclovir | 1000 mg tid for 7 days | $315.99 |
| *Source: http://www.drugstore.com. Accessed December 6, 2010. | ||
Add a corticosteroid? The evidence is mixed
Oral corticosteroids have been studied as an adjunct to antiviral therapy for the treatment of HZ. One clinical trial of 208 immunocompetent adults older than 50 found that the addition of a 21-day prednisone taper to acyclovir led to accelerated healing of cutaneous lesions, cessation of analgesic use, and return to normal activities and uninterrupted sleep.18 However, this study and another randomized trial of oral corticosteroids did not show that steroids had any effect on longer-term pain relief or the development of PHN.18,19 A recent Cochrane review concluded that there is insufficient evidence that corticosteroids are safe or effective in the prevention of PHN.20 Given the potential adverse effects of oral corticosteroids, their use should be weighed carefully.
Give analgesics for shingles pain
Many clinical trials have looked at the efficacy of different analgesics in the treatment of PHN, but data regarding analgesics for the treatment of acute HZ pain are limited. One RCT of 87 patients older than 50 divided participants into 3 groups: One group took controlled-release oxycodone, another received a placebo, and the third group took gabapentin.21 (The study did not include patients with mild pain, for whom nonnarcotic analgesics would likely be more appropriate.)
The researchers found that the oxycodone significantly reduced acute HZ pain during the first 2 weeks of treatment as compared with placebo or gabapentin. There was no statistically significant reduction in pain for the gabapentin group, compared with those on placebo. The oxycodone group did have the highest dropout rate (27.6% vs 6.9% for the placebo group), however, primarily because of constipation. This study showed that narcotic analgesics are effective and relatively well tolerated for the treatment of acute HZ pain.21
Because severe pain with acute HZ is a well-established risk factor for the development of PHN, there is interest in determining whether effective pain control in the acute setting decreases the risk of chronic pain. One placebo-controlled trial of 72 patients older than 60 years found that 25 mg amitriptyline daily, started within 48 hours of rash onset and continued for 90 days, reduced pain prevalence by more than half at 6 months from diagnosis.22 This study did not control for the use of antiviral agents, however, so further investigation is needed.
CASE Jane T began a course of antiviral therapy with valacyclovir shortly after her rash appeared, and continued to take oral tramadol for the pain. Her rash and pain level improved over the next several days, and within 2 weeks she was fully recovered.
Prevention: Vaccination holds the key
HZ is contagious and can cause primary varicella in people who are susceptible. Indeed, one study found that 15.5% of susceptible household contacts developed varicella after exposure to HZ.1 Advise patients with HZ to avoid contact with those at high risk for severe varicella—including pregnant women, premature infants, and immunocompromised individuals of all ages—until their lesions are crusted. They can further avoid transmission by keeping the lesions covered.23
Increased use of the HZ vaccine, however, is the key to prevention of shingles. The Centers for Disease Control and Prevention (CDC) recommends Zostavax, a live attenuated varicella zoster vaccine given as a single subcutaneous injection, for people ages 60 and older. Compared with the varicella vaccines designed for children, Zostavax has a significantly higher potency in order to elicit a significant and durable response in older adults.24
The vaccine is generally safe, with a mild injection site reaction being the most common adverse event. No evidence exists of transmission of virus from vaccine recipients to contacts.1 Like other live virus vaccines, Zostavax is contraindicated in pregnant women and immunocompromised patients. It can be given to patients regardless of their history of chicken pox or previous episodes of HZ, as studies have shown that the recurrence rate for HZ is similar to the rate for initial episodes.
How well does it work? In a Shingles Prevention Study Group trial of 38,546 people 60 years of age or older, the vaccine reduced the incidence of HZ by 51% and the incidence of PHN by 67% over a 3-year follow-up period.25 The vaccine was most effective for the prevention of HZ in the 60- to 69-year age group, but there was no significant difference in its efficacy in preventing PHN or reducing the burden of illness (a measure based on the incidence, severity, and duration of pain and discomfort) in 60- to 69-year-olds vs those ages 70 and older.25 An analysis by the CDC suggests that approximately 17 people would need to be vaccinated with Zostavax to prevent one case of HZ, and approximately 31 people would need to be vaccinated to prevent one case of PHN.24
Ongoing studies are examining the safety and efficacy of Zostavax for patients ages 50 to 59.26 Although results thus far look promising, there is no recommendation for routine vaccination for this age group, and insurance companies do not routinely cover the cost of vaccinating them.
While primary care physicians generally favor the concept of HZ vaccination,27 a CDC survey conducted in 2007—a year after the vaccine received FDA approval—found that only 1.9% of eligible patients received the vaccine.28 Physicians cite concerns about reimbursement as a barrier to its use.28
Zostavax is not covered by Medicare Part B, in contrast to other adult vaccines, such as influenza and pneumococcal.29 However, the HZ vaccine is covered by many private insurers, as well as by Medicare Part D. Advise patients to check with their insurance carrier for specific details regarding their coverage, as some plans require the patient to purchase the vaccine from a pharmacy prior to administration in a physician’s office.
CORRESPONDENCE Sarah L. Cartwright, MD, Wake Forest University School of Medicine, Department of Family and Community Medicine, Medical Center Boulevard, Winston-Salem, NC 27157; [email protected]
1. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
2. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
3. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
4. Gilden DH, Dueland AN, Cohrs R, et al. Preherpetic neuralgia. Neurology. 1991;41:1215-1218.
5. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57(6 suppl):S130-S135.
6. Opstelten W, van Loon AM, Schuller M, et al. Clinical diagnosis of herpes zoster in family practice. Ann Fam Med. 2007;5:305-309.
7. Helgason S, Sigurdsson J, Gudmundsson S. The clinical course of herpes zoster: a prospective study in primary care. European J Gen Pract. 1996;2:12-16.
8. Durdu M, Baba M, Seckin D. The value of Tzanck smear test in diagnosis of erosive, vesicular, bullous, and pustular skin lesions. J Am Acad Dermatol. 2008;59:958-964.
9. Brisson DM, Levin MJ, Schmader KE, et al. A prospective study of the herpes zoster severity of illness. Clin J Pain. 2010;26:656-666.
10. Rowbotham M, Harden N, Stacey B, et al. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA. 1998;280:1837-1842.
11. Jung BF, Johnson RW, Griffin DR, et al. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545-1551.
12. Galil K, Choo PW, Donahue JG, et al. The sequelae of herpes zoster. Arch Intern Med. 1997;157:1209-1213.
13. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9:863-869.
14. Beutner KR, Friedman DJ, Forszpaniak C, et al. Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob Agents Chemother. 1995;39:1546-1553.
15. Degreef H. Famciclovir, a new oral antiherpes drug: results of the first controlled clinical study demonstrating its efficacy and safety in the treatment of uncomplicated herpes zoster in immunocompetent patients. Int J Antimicrob Agents. 1994;4:241-246.
16. McKendrick MW, Care CC, Kudesia G, et al. Is VZV reactivation a common cause of unexplained unilateral pain? Results of a prospective study of 57 patients. J Infect. 1999;39:209-212.
17. Li Q, Chen N, Yang J, et al. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2009;(2):CD006866.-
18. Whitley RJ, Weiss H, Gnann JW, Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster. A randomized, placebo-controlled trial. The National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Ann Intern Med. 1996;125:376-383.
19. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
20. He L, Zhang D, Zhou M, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2008;(1):CD005582.-
21. Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain. 2009;142:209-217.
22. Bowsher D. The effects of pre-emptive treatment of postherpetic neuralgia with amitriptyline: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage. 1997;13:327-331.
23. Bolyard EA, Tablan OC, Williams WW, et al. Guideline for infection control in healthcare personnel, 1998. Hospital Infection Control Practices Advisory Committee. Infect Control Hosp Epidemiol. 1998;19:407-463.
24. Kimberlin DW, Whitley RJ. Varicella-zoster vaccine for the prevention of herpes zoster. N Engl J Med. 2007;356:1338-1343.
25. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
26. Sutradhar SC, Wang WW, Schlienger K, et al. Comparison of the levels of immunogenicity and safety of Zostavax in adults 50 to 59 years old and in adults 60 years old or older. Clin Vaccine Immunol. 2009;16:646-652.
27. Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis. 2008;197(suppl 2):S216-S223.
28. Lu PJ, Euler GL, Jumaan AO, et al. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine. 2009;27:882-887.
29. Orenstein WA, Mootrey GT, Pazol K, et al. Financing immunization of adults in the United States. Clin Pharmacol Ther. 2007;82:764-768.
• Reserve laboratory testing for unclear or complicated cases of herpes zoster (HZ), as the condition can be diagnosed clinically in most cases. B
• Whenever possible, initiate oral antiviral therapy within 72 hours of the onset of the shingles rash to accelerate healing and reduce the duration and severity of pain. A
• Offer the HZ vaccine (Zostavax) to patients ages 60 and older to reduce the risk of shingles and postherpetic neuralgia. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane T, a 53-year-old patient, calls her family physician’s office for a same-day appointment; she has severe right upper quadrant pain that developed a few hours ago. When Jane comes in, she tells her physician that the pain feels like the gallbladder attacks she used to have—before her gallbladder was removed 2 years ago. She has nausea but no vomiting, is afebrile, and has no urinary symptoms.
A series of in-office tests—urinalysis, complete blood count, comprehensive metabolic profile, amylase, and lipase— are all normal. The physician gives Jane an intramuscular injection of ketorolac and a prescription for oral tramadol, and sends her for a right upper quadrant ultrasound, which is also normal. The next morning, the pain intensifies, and Jane goes to the emergency department, where she undergoes computed tomography of the abdomen and pelvis—also normal. After a night in the hospital for observation, she is released. She returns to the family physician 3 days later, this time with an erythematous papular rash in a dermatomal distribution over her back and right upper quadrant that her doctor immediately recognizes as shingles.
Jane is not alone. Each year, about 1 million US residents develop herpes zoster (HZ),1 and 70% to 80% of them experience prodromal pain in the affected dermatome.2 For some, the pain is significant enough to prompt a medical work-up to rule out other potential causes, such as myocardial infarction, nephrolithiasis, pancreatitis, cholecystitis, and appendicitis.
The time and resources spent on such a work-up may be unavoidable when a patient presents with severe pain. But alert primary care physicians can avoid unnecessary diagnostic tests by being aware of the prodromal symptoms of HZ and being on the lookout for the rash that follows.
Age alone is a key factor in the detection of HZ, of course. One in 3 people will develop shingles during their lifetime,1 with half of all cases occurring in people ages 60 and older.3 In addition to early detection and treatment, physicians can do their part to battle HZ by routinely recommending the shingles vaccine (Zostavax) to patients in this age group.
Diagnosing HZ: From prodrome to rash
For many patients, an abnormal skin sensation on one side of the body, such as itching, burning, or altered sensitivity to touch, is the first symptom of HZ. These sensations, including pain that can range from mild to severe, may precede the rash by days or even weeks.4 Systemic symptoms are far less common; less than 20% of patients develop fever, headache, malaise, or fatigue as part of the HZ prodrome.2
The shingles rash is usually unilateral and does not cross the midline. However, it may occur in up to 3 adjacent dermatomes. The trunk is the most common site for the rash, but it may also develop on the face, buttocks, or other parts of the body. The lesions start as erythematous papules and evolve into vesicles within 12 to 24 hours.5
Once the rash emerges, HZ can usually be diagnosed clinically in a primary care setting, and laboratory testing should be considered only when diagnosis is uncertain. In a cohort study of 260 patients older than 50 years with clinically diagnosed HZ, 236 (91%) cases were confirmed on serologic testing.6 In an Icelandic study, 93% of 505 cases were diagnosed correctly by general practitioners, using expert opinion and clinical course as the gold standard.7
If diagnostic testing is necessary, physicians have a number of choices:
Varicella zoster polymerase chain reaction (PCR) test. PCR testing for HZ can provide rapid and reliable diagnosis and is becoming more widely available, but its use should be limited because of the cost (approximately $300).
Tzanck smear. This test is quick and inexpensive, and can reliably diagnose a herpetic lesion based on the presence of acantholytic and multinucleated giant cells in a sample collected from the base of a vesicle.8 However, this technique cannot differentiate HZ from herpes simplex infection, and lack of experience with collection or interpretation of the Tzanck smear limits its usefulness.
Serologic testing. Serology has limited utility in the diagnosis of acute HZ, but may provide a retrospective diagnosis, if needed. In a study of 260 patients older than 50 years with clinically diagnosed acute HZ, varicella zoster IgA or IgM was positive in 61% of patients at the time of presentation, and in 91% of patients 5 to 10 days later.6
Viral culture. Obtaining a viral culture of varicella zoster is another option, but it is not recommended, as sensitivity is poor and incubation requires several days.
The course of illness, and risk of complications
For some patients, the acute pain and hypersensitivity at the site of the rash resolve in several days; for others, this may take several weeks or more. One recent study found a median of 32.5 days for pain duration in patients ages 50 and older.9 The suffering can last far longer, however, if complications arise.
Postherpetic neuralgia (PHN), a chronic and often debilitating condition with pain that can last months, or even indefinitely, affects about 10% to 15% of patients with HZ.10 Risk factors for the development of PHN include advanced age, female sex, presence of a prodrome, severe acute HZ pain, and a severe rash.11
Trigeminal nerve involvement. HZ affects the first branch of the trigeminal nerve in about 10% to 15% of patients.5 In such cases, the rash may erupt on the forehead, periocular area, and nose. Patients with trigeminal nerve involvement are at significant risk for ocular complications (HZ ophthalmicus), including keratitis, iritis, and possible vision loss, and should be treated and referred to an ophthalmologist without delay.
In addition to these complications, others reported in a review of 859 patients include bacterial skin infection, motor neuropathy, and, rarely, meningitis and HZ oticus.12 Advanced age markedly increased the likelihood of complications.
Initiate treatment without delay
Multiple randomized controlled clinical trials have demonstrated the efficacy of oral antiviral treatment in reducing the duration of viral shedding, accelerating rash healing, and reducing the severity and duration of acute HZ pain.2 In all the studies, however, antiviral therapy was started within 72 hours of the onset of the rash; the efficacy of initiating antiviral treatment after >72 hours has not been systematically studied.2 When it’s not possible to begin therapy within this time frame, however, many experts recommend initiating therapy as soon as possible thereafter.1
Three antiviral agents—acyclovir, famciclovir, and valacyclovir—have been approved by the US Food and Drug Administration to treat HZ. Evidence suggests that famciclovir and valacyclovir have comparable efficacy with regard to resolution of both the rash and the acute pain,13 and result in more rapid pain resolution compared with acyclovir.14,15 Famciclovir and valacyclovir also offer simpler dosing schedules—both are taken 3 times a day, while acyclovir requires 5 daily doses— but they are significantly more expensive (TABLE).
Oral antiviral therapy is strongly recommended for patients who are older than 50 and those who have moderate-to-severe pain or rash—or whose rash appears somewhere other than the trunk.2 But given the safety of oral antiviral agents, treatment can be considered for younger patients and those with milder cases of HZ, as well. (Routine use of antiviral agents is not indicated for unexplained unilateral pain, as varicella zoster infection without the classic rash [zoster sine herpete] is a rare cause.16)
Results regarding the efficacy of oral antiviral agents for reducing the duration of chronic pain and preventing PHN are mixed.2 A recent Cochrane review concluded that oral acyclovir does not significantly reduce the incidence of PHN and that there is insufficient evidence to determine if other antivirals do.17
TABLE
FDA-approved oral antiviral regimens
| Drug | Dosing regimen | Cost of regimen* |
|---|---|---|
| Acyclovir | 800 mg 5 times/d for 7-10 days | $55.97 |
| Famciclovir | 500 mg tid for 7 days | $319.99 |
| Valacyclovir | 1000 mg tid for 7 days | $315.99 |
| *Source: http://www.drugstore.com. Accessed December 6, 2010. | ||
Add a corticosteroid? The evidence is mixed
Oral corticosteroids have been studied as an adjunct to antiviral therapy for the treatment of HZ. One clinical trial of 208 immunocompetent adults older than 50 found that the addition of a 21-day prednisone taper to acyclovir led to accelerated healing of cutaneous lesions, cessation of analgesic use, and return to normal activities and uninterrupted sleep.18 However, this study and another randomized trial of oral corticosteroids did not show that steroids had any effect on longer-term pain relief or the development of PHN.18,19 A recent Cochrane review concluded that there is insufficient evidence that corticosteroids are safe or effective in the prevention of PHN.20 Given the potential adverse effects of oral corticosteroids, their use should be weighed carefully.
Give analgesics for shingles pain
Many clinical trials have looked at the efficacy of different analgesics in the treatment of PHN, but data regarding analgesics for the treatment of acute HZ pain are limited. One RCT of 87 patients older than 50 divided participants into 3 groups: One group took controlled-release oxycodone, another received a placebo, and the third group took gabapentin.21 (The study did not include patients with mild pain, for whom nonnarcotic analgesics would likely be more appropriate.)
The researchers found that the oxycodone significantly reduced acute HZ pain during the first 2 weeks of treatment as compared with placebo or gabapentin. There was no statistically significant reduction in pain for the gabapentin group, compared with those on placebo. The oxycodone group did have the highest dropout rate (27.6% vs 6.9% for the placebo group), however, primarily because of constipation. This study showed that narcotic analgesics are effective and relatively well tolerated for the treatment of acute HZ pain.21
Because severe pain with acute HZ is a well-established risk factor for the development of PHN, there is interest in determining whether effective pain control in the acute setting decreases the risk of chronic pain. One placebo-controlled trial of 72 patients older than 60 years found that 25 mg amitriptyline daily, started within 48 hours of rash onset and continued for 90 days, reduced pain prevalence by more than half at 6 months from diagnosis.22 This study did not control for the use of antiviral agents, however, so further investigation is needed.
CASE Jane T began a course of antiviral therapy with valacyclovir shortly after her rash appeared, and continued to take oral tramadol for the pain. Her rash and pain level improved over the next several days, and within 2 weeks she was fully recovered.
Prevention: Vaccination holds the key
HZ is contagious and can cause primary varicella in people who are susceptible. Indeed, one study found that 15.5% of susceptible household contacts developed varicella after exposure to HZ.1 Advise patients with HZ to avoid contact with those at high risk for severe varicella—including pregnant women, premature infants, and immunocompromised individuals of all ages—until their lesions are crusted. They can further avoid transmission by keeping the lesions covered.23
Increased use of the HZ vaccine, however, is the key to prevention of shingles. The Centers for Disease Control and Prevention (CDC) recommends Zostavax, a live attenuated varicella zoster vaccine given as a single subcutaneous injection, for people ages 60 and older. Compared with the varicella vaccines designed for children, Zostavax has a significantly higher potency in order to elicit a significant and durable response in older adults.24
The vaccine is generally safe, with a mild injection site reaction being the most common adverse event. No evidence exists of transmission of virus from vaccine recipients to contacts.1 Like other live virus vaccines, Zostavax is contraindicated in pregnant women and immunocompromised patients. It can be given to patients regardless of their history of chicken pox or previous episodes of HZ, as studies have shown that the recurrence rate for HZ is similar to the rate for initial episodes.
How well does it work? In a Shingles Prevention Study Group trial of 38,546 people 60 years of age or older, the vaccine reduced the incidence of HZ by 51% and the incidence of PHN by 67% over a 3-year follow-up period.25 The vaccine was most effective for the prevention of HZ in the 60- to 69-year age group, but there was no significant difference in its efficacy in preventing PHN or reducing the burden of illness (a measure based on the incidence, severity, and duration of pain and discomfort) in 60- to 69-year-olds vs those ages 70 and older.25 An analysis by the CDC suggests that approximately 17 people would need to be vaccinated with Zostavax to prevent one case of HZ, and approximately 31 people would need to be vaccinated to prevent one case of PHN.24
Ongoing studies are examining the safety and efficacy of Zostavax for patients ages 50 to 59.26 Although results thus far look promising, there is no recommendation for routine vaccination for this age group, and insurance companies do not routinely cover the cost of vaccinating them.
While primary care physicians generally favor the concept of HZ vaccination,27 a CDC survey conducted in 2007—a year after the vaccine received FDA approval—found that only 1.9% of eligible patients received the vaccine.28 Physicians cite concerns about reimbursement as a barrier to its use.28
Zostavax is not covered by Medicare Part B, in contrast to other adult vaccines, such as influenza and pneumococcal.29 However, the HZ vaccine is covered by many private insurers, as well as by Medicare Part D. Advise patients to check with their insurance carrier for specific details regarding their coverage, as some plans require the patient to purchase the vaccine from a pharmacy prior to administration in a physician’s office.
CORRESPONDENCE Sarah L. Cartwright, MD, Wake Forest University School of Medicine, Department of Family and Community Medicine, Medical Center Boulevard, Winston-Salem, NC 27157; [email protected]
• Reserve laboratory testing for unclear or complicated cases of herpes zoster (HZ), as the condition can be diagnosed clinically in most cases. B
• Whenever possible, initiate oral antiviral therapy within 72 hours of the onset of the shingles rash to accelerate healing and reduce the duration and severity of pain. A
• Offer the HZ vaccine (Zostavax) to patients ages 60 and older to reduce the risk of shingles and postherpetic neuralgia. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane T, a 53-year-old patient, calls her family physician’s office for a same-day appointment; she has severe right upper quadrant pain that developed a few hours ago. When Jane comes in, she tells her physician that the pain feels like the gallbladder attacks she used to have—before her gallbladder was removed 2 years ago. She has nausea but no vomiting, is afebrile, and has no urinary symptoms.
A series of in-office tests—urinalysis, complete blood count, comprehensive metabolic profile, amylase, and lipase— are all normal. The physician gives Jane an intramuscular injection of ketorolac and a prescription for oral tramadol, and sends her for a right upper quadrant ultrasound, which is also normal. The next morning, the pain intensifies, and Jane goes to the emergency department, where she undergoes computed tomography of the abdomen and pelvis—also normal. After a night in the hospital for observation, she is released. She returns to the family physician 3 days later, this time with an erythematous papular rash in a dermatomal distribution over her back and right upper quadrant that her doctor immediately recognizes as shingles.
Jane is not alone. Each year, about 1 million US residents develop herpes zoster (HZ),1 and 70% to 80% of them experience prodromal pain in the affected dermatome.2 For some, the pain is significant enough to prompt a medical work-up to rule out other potential causes, such as myocardial infarction, nephrolithiasis, pancreatitis, cholecystitis, and appendicitis.
The time and resources spent on such a work-up may be unavoidable when a patient presents with severe pain. But alert primary care physicians can avoid unnecessary diagnostic tests by being aware of the prodromal symptoms of HZ and being on the lookout for the rash that follows.
Age alone is a key factor in the detection of HZ, of course. One in 3 people will develop shingles during their lifetime,1 with half of all cases occurring in people ages 60 and older.3 In addition to early detection and treatment, physicians can do their part to battle HZ by routinely recommending the shingles vaccine (Zostavax) to patients in this age group.
Diagnosing HZ: From prodrome to rash
For many patients, an abnormal skin sensation on one side of the body, such as itching, burning, or altered sensitivity to touch, is the first symptom of HZ. These sensations, including pain that can range from mild to severe, may precede the rash by days or even weeks.4 Systemic symptoms are far less common; less than 20% of patients develop fever, headache, malaise, or fatigue as part of the HZ prodrome.2
The shingles rash is usually unilateral and does not cross the midline. However, it may occur in up to 3 adjacent dermatomes. The trunk is the most common site for the rash, but it may also develop on the face, buttocks, or other parts of the body. The lesions start as erythematous papules and evolve into vesicles within 12 to 24 hours.5
Once the rash emerges, HZ can usually be diagnosed clinically in a primary care setting, and laboratory testing should be considered only when diagnosis is uncertain. In a cohort study of 260 patients older than 50 years with clinically diagnosed HZ, 236 (91%) cases were confirmed on serologic testing.6 In an Icelandic study, 93% of 505 cases were diagnosed correctly by general practitioners, using expert opinion and clinical course as the gold standard.7
If diagnostic testing is necessary, physicians have a number of choices:
Varicella zoster polymerase chain reaction (PCR) test. PCR testing for HZ can provide rapid and reliable diagnosis and is becoming more widely available, but its use should be limited because of the cost (approximately $300).
Tzanck smear. This test is quick and inexpensive, and can reliably diagnose a herpetic lesion based on the presence of acantholytic and multinucleated giant cells in a sample collected from the base of a vesicle.8 However, this technique cannot differentiate HZ from herpes simplex infection, and lack of experience with collection or interpretation of the Tzanck smear limits its usefulness.
Serologic testing. Serology has limited utility in the diagnosis of acute HZ, but may provide a retrospective diagnosis, if needed. In a study of 260 patients older than 50 years with clinically diagnosed acute HZ, varicella zoster IgA or IgM was positive in 61% of patients at the time of presentation, and in 91% of patients 5 to 10 days later.6
Viral culture. Obtaining a viral culture of varicella zoster is another option, but it is not recommended, as sensitivity is poor and incubation requires several days.
The course of illness, and risk of complications
For some patients, the acute pain and hypersensitivity at the site of the rash resolve in several days; for others, this may take several weeks or more. One recent study found a median of 32.5 days for pain duration in patients ages 50 and older.9 The suffering can last far longer, however, if complications arise.
Postherpetic neuralgia (PHN), a chronic and often debilitating condition with pain that can last months, or even indefinitely, affects about 10% to 15% of patients with HZ.10 Risk factors for the development of PHN include advanced age, female sex, presence of a prodrome, severe acute HZ pain, and a severe rash.11
Trigeminal nerve involvement. HZ affects the first branch of the trigeminal nerve in about 10% to 15% of patients.5 In such cases, the rash may erupt on the forehead, periocular area, and nose. Patients with trigeminal nerve involvement are at significant risk for ocular complications (HZ ophthalmicus), including keratitis, iritis, and possible vision loss, and should be treated and referred to an ophthalmologist without delay.
In addition to these complications, others reported in a review of 859 patients include bacterial skin infection, motor neuropathy, and, rarely, meningitis and HZ oticus.12 Advanced age markedly increased the likelihood of complications.
Initiate treatment without delay
Multiple randomized controlled clinical trials have demonstrated the efficacy of oral antiviral treatment in reducing the duration of viral shedding, accelerating rash healing, and reducing the severity and duration of acute HZ pain.2 In all the studies, however, antiviral therapy was started within 72 hours of the onset of the rash; the efficacy of initiating antiviral treatment after >72 hours has not been systematically studied.2 When it’s not possible to begin therapy within this time frame, however, many experts recommend initiating therapy as soon as possible thereafter.1
Three antiviral agents—acyclovir, famciclovir, and valacyclovir—have been approved by the US Food and Drug Administration to treat HZ. Evidence suggests that famciclovir and valacyclovir have comparable efficacy with regard to resolution of both the rash and the acute pain,13 and result in more rapid pain resolution compared with acyclovir.14,15 Famciclovir and valacyclovir also offer simpler dosing schedules—both are taken 3 times a day, while acyclovir requires 5 daily doses— but they are significantly more expensive (TABLE).
Oral antiviral therapy is strongly recommended for patients who are older than 50 and those who have moderate-to-severe pain or rash—or whose rash appears somewhere other than the trunk.2 But given the safety of oral antiviral agents, treatment can be considered for younger patients and those with milder cases of HZ, as well. (Routine use of antiviral agents is not indicated for unexplained unilateral pain, as varicella zoster infection without the classic rash [zoster sine herpete] is a rare cause.16)
Results regarding the efficacy of oral antiviral agents for reducing the duration of chronic pain and preventing PHN are mixed.2 A recent Cochrane review concluded that oral acyclovir does not significantly reduce the incidence of PHN and that there is insufficient evidence to determine if other antivirals do.17
TABLE
FDA-approved oral antiviral regimens
| Drug | Dosing regimen | Cost of regimen* |
|---|---|---|
| Acyclovir | 800 mg 5 times/d for 7-10 days | $55.97 |
| Famciclovir | 500 mg tid for 7 days | $319.99 |
| Valacyclovir | 1000 mg tid for 7 days | $315.99 |
| *Source: http://www.drugstore.com. Accessed December 6, 2010. | ||
Add a corticosteroid? The evidence is mixed
Oral corticosteroids have been studied as an adjunct to antiviral therapy for the treatment of HZ. One clinical trial of 208 immunocompetent adults older than 50 found that the addition of a 21-day prednisone taper to acyclovir led to accelerated healing of cutaneous lesions, cessation of analgesic use, and return to normal activities and uninterrupted sleep.18 However, this study and another randomized trial of oral corticosteroids did not show that steroids had any effect on longer-term pain relief or the development of PHN.18,19 A recent Cochrane review concluded that there is insufficient evidence that corticosteroids are safe or effective in the prevention of PHN.20 Given the potential adverse effects of oral corticosteroids, their use should be weighed carefully.
Give analgesics for shingles pain
Many clinical trials have looked at the efficacy of different analgesics in the treatment of PHN, but data regarding analgesics for the treatment of acute HZ pain are limited. One RCT of 87 patients older than 50 divided participants into 3 groups: One group took controlled-release oxycodone, another received a placebo, and the third group took gabapentin.21 (The study did not include patients with mild pain, for whom nonnarcotic analgesics would likely be more appropriate.)
The researchers found that the oxycodone significantly reduced acute HZ pain during the first 2 weeks of treatment as compared with placebo or gabapentin. There was no statistically significant reduction in pain for the gabapentin group, compared with those on placebo. The oxycodone group did have the highest dropout rate (27.6% vs 6.9% for the placebo group), however, primarily because of constipation. This study showed that narcotic analgesics are effective and relatively well tolerated for the treatment of acute HZ pain.21
Because severe pain with acute HZ is a well-established risk factor for the development of PHN, there is interest in determining whether effective pain control in the acute setting decreases the risk of chronic pain. One placebo-controlled trial of 72 patients older than 60 years found that 25 mg amitriptyline daily, started within 48 hours of rash onset and continued for 90 days, reduced pain prevalence by more than half at 6 months from diagnosis.22 This study did not control for the use of antiviral agents, however, so further investigation is needed.
CASE Jane T began a course of antiviral therapy with valacyclovir shortly after her rash appeared, and continued to take oral tramadol for the pain. Her rash and pain level improved over the next several days, and within 2 weeks she was fully recovered.
Prevention: Vaccination holds the key
HZ is contagious and can cause primary varicella in people who are susceptible. Indeed, one study found that 15.5% of susceptible household contacts developed varicella after exposure to HZ.1 Advise patients with HZ to avoid contact with those at high risk for severe varicella—including pregnant women, premature infants, and immunocompromised individuals of all ages—until their lesions are crusted. They can further avoid transmission by keeping the lesions covered.23
Increased use of the HZ vaccine, however, is the key to prevention of shingles. The Centers for Disease Control and Prevention (CDC) recommends Zostavax, a live attenuated varicella zoster vaccine given as a single subcutaneous injection, for people ages 60 and older. Compared with the varicella vaccines designed for children, Zostavax has a significantly higher potency in order to elicit a significant and durable response in older adults.24
The vaccine is generally safe, with a mild injection site reaction being the most common adverse event. No evidence exists of transmission of virus from vaccine recipients to contacts.1 Like other live virus vaccines, Zostavax is contraindicated in pregnant women and immunocompromised patients. It can be given to patients regardless of their history of chicken pox or previous episodes of HZ, as studies have shown that the recurrence rate for HZ is similar to the rate for initial episodes.
How well does it work? In a Shingles Prevention Study Group trial of 38,546 people 60 years of age or older, the vaccine reduced the incidence of HZ by 51% and the incidence of PHN by 67% over a 3-year follow-up period.25 The vaccine was most effective for the prevention of HZ in the 60- to 69-year age group, but there was no significant difference in its efficacy in preventing PHN or reducing the burden of illness (a measure based on the incidence, severity, and duration of pain and discomfort) in 60- to 69-year-olds vs those ages 70 and older.25 An analysis by the CDC suggests that approximately 17 people would need to be vaccinated with Zostavax to prevent one case of HZ, and approximately 31 people would need to be vaccinated to prevent one case of PHN.24
Ongoing studies are examining the safety and efficacy of Zostavax for patients ages 50 to 59.26 Although results thus far look promising, there is no recommendation for routine vaccination for this age group, and insurance companies do not routinely cover the cost of vaccinating them.
While primary care physicians generally favor the concept of HZ vaccination,27 a CDC survey conducted in 2007—a year after the vaccine received FDA approval—found that only 1.9% of eligible patients received the vaccine.28 Physicians cite concerns about reimbursement as a barrier to its use.28
Zostavax is not covered by Medicare Part B, in contrast to other adult vaccines, such as influenza and pneumococcal.29 However, the HZ vaccine is covered by many private insurers, as well as by Medicare Part D. Advise patients to check with their insurance carrier for specific details regarding their coverage, as some plans require the patient to purchase the vaccine from a pharmacy prior to administration in a physician’s office.
CORRESPONDENCE Sarah L. Cartwright, MD, Wake Forest University School of Medicine, Department of Family and Community Medicine, Medical Center Boulevard, Winston-Salem, NC 27157; [email protected]
1. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
2. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
3. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
4. Gilden DH, Dueland AN, Cohrs R, et al. Preherpetic neuralgia. Neurology. 1991;41:1215-1218.
5. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57(6 suppl):S130-S135.
6. Opstelten W, van Loon AM, Schuller M, et al. Clinical diagnosis of herpes zoster in family practice. Ann Fam Med. 2007;5:305-309.
7. Helgason S, Sigurdsson J, Gudmundsson S. The clinical course of herpes zoster: a prospective study in primary care. European J Gen Pract. 1996;2:12-16.
8. Durdu M, Baba M, Seckin D. The value of Tzanck smear test in diagnosis of erosive, vesicular, bullous, and pustular skin lesions. J Am Acad Dermatol. 2008;59:958-964.
9. Brisson DM, Levin MJ, Schmader KE, et al. A prospective study of the herpes zoster severity of illness. Clin J Pain. 2010;26:656-666.
10. Rowbotham M, Harden N, Stacey B, et al. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA. 1998;280:1837-1842.
11. Jung BF, Johnson RW, Griffin DR, et al. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545-1551.
12. Galil K, Choo PW, Donahue JG, et al. The sequelae of herpes zoster. Arch Intern Med. 1997;157:1209-1213.
13. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9:863-869.
14. Beutner KR, Friedman DJ, Forszpaniak C, et al. Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob Agents Chemother. 1995;39:1546-1553.
15. Degreef H. Famciclovir, a new oral antiherpes drug: results of the first controlled clinical study demonstrating its efficacy and safety in the treatment of uncomplicated herpes zoster in immunocompetent patients. Int J Antimicrob Agents. 1994;4:241-246.
16. McKendrick MW, Care CC, Kudesia G, et al. Is VZV reactivation a common cause of unexplained unilateral pain? Results of a prospective study of 57 patients. J Infect. 1999;39:209-212.
17. Li Q, Chen N, Yang J, et al. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2009;(2):CD006866.-
18. Whitley RJ, Weiss H, Gnann JW, Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster. A randomized, placebo-controlled trial. The National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Ann Intern Med. 1996;125:376-383.
19. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
20. He L, Zhang D, Zhou M, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2008;(1):CD005582.-
21. Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain. 2009;142:209-217.
22. Bowsher D. The effects of pre-emptive treatment of postherpetic neuralgia with amitriptyline: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage. 1997;13:327-331.
23. Bolyard EA, Tablan OC, Williams WW, et al. Guideline for infection control in healthcare personnel, 1998. Hospital Infection Control Practices Advisory Committee. Infect Control Hosp Epidemiol. 1998;19:407-463.
24. Kimberlin DW, Whitley RJ. Varicella-zoster vaccine for the prevention of herpes zoster. N Engl J Med. 2007;356:1338-1343.
25. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
26. Sutradhar SC, Wang WW, Schlienger K, et al. Comparison of the levels of immunogenicity and safety of Zostavax in adults 50 to 59 years old and in adults 60 years old or older. Clin Vaccine Immunol. 2009;16:646-652.
27. Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis. 2008;197(suppl 2):S216-S223.
28. Lu PJ, Euler GL, Jumaan AO, et al. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine. 2009;27:882-887.
29. Orenstein WA, Mootrey GT, Pazol K, et al. Financing immunization of adults in the United States. Clin Pharmacol Ther. 2007;82:764-768.
1. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
2. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
3. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
4. Gilden DH, Dueland AN, Cohrs R, et al. Preherpetic neuralgia. Neurology. 1991;41:1215-1218.
5. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57(6 suppl):S130-S135.
6. Opstelten W, van Loon AM, Schuller M, et al. Clinical diagnosis of herpes zoster in family practice. Ann Fam Med. 2007;5:305-309.
7. Helgason S, Sigurdsson J, Gudmundsson S. The clinical course of herpes zoster: a prospective study in primary care. European J Gen Pract. 1996;2:12-16.
8. Durdu M, Baba M, Seckin D. The value of Tzanck smear test in diagnosis of erosive, vesicular, bullous, and pustular skin lesions. J Am Acad Dermatol. 2008;59:958-964.
9. Brisson DM, Levin MJ, Schmader KE, et al. A prospective study of the herpes zoster severity of illness. Clin J Pain. 2010;26:656-666.
10. Rowbotham M, Harden N, Stacey B, et al. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA. 1998;280:1837-1842.
11. Jung BF, Johnson RW, Griffin DR, et al. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545-1551.
12. Galil K, Choo PW, Donahue JG, et al. The sequelae of herpes zoster. Arch Intern Med. 1997;157:1209-1213.
13. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9:863-869.
14. Beutner KR, Friedman DJ, Forszpaniak C, et al. Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob Agents Chemother. 1995;39:1546-1553.
15. Degreef H. Famciclovir, a new oral antiherpes drug: results of the first controlled clinical study demonstrating its efficacy and safety in the treatment of uncomplicated herpes zoster in immunocompetent patients. Int J Antimicrob Agents. 1994;4:241-246.
16. McKendrick MW, Care CC, Kudesia G, et al. Is VZV reactivation a common cause of unexplained unilateral pain? Results of a prospective study of 57 patients. J Infect. 1999;39:209-212.
17. Li Q, Chen N, Yang J, et al. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2009;(2):CD006866.-
18. Whitley RJ, Weiss H, Gnann JW, Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster. A randomized, placebo-controlled trial. The National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Ann Intern Med. 1996;125:376-383.
19. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
20. He L, Zhang D, Zhou M, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2008;(1):CD005582.-
21. Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain. 2009;142:209-217.
22. Bowsher D. The effects of pre-emptive treatment of postherpetic neuralgia with amitriptyline: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage. 1997;13:327-331.
23. Bolyard EA, Tablan OC, Williams WW, et al. Guideline for infection control in healthcare personnel, 1998. Hospital Infection Control Practices Advisory Committee. Infect Control Hosp Epidemiol. 1998;19:407-463.
24. Kimberlin DW, Whitley RJ. Varicella-zoster vaccine for the prevention of herpes zoster. N Engl J Med. 2007;356:1338-1343.
25. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
26. Sutradhar SC, Wang WW, Schlienger K, et al. Comparison of the levels of immunogenicity and safety of Zostavax in adults 50 to 59 years old and in adults 60 years old or older. Clin Vaccine Immunol. 2009;16:646-652.
27. Hurley LP, Harpaz R, Daley MF, et al. National survey of primary care physicians regarding herpes zoster and the herpes zoster vaccine. J Infect Dis. 2008;197(suppl 2):S216-S223.
28. Lu PJ, Euler GL, Jumaan AO, et al. Herpes zoster vaccination among adults aged 60 years or older in the United States, 2007: uptake of the first new vaccine to target seniors. Vaccine. 2009;27:882-887.
29. Orenstein WA, Mootrey GT, Pazol K, et al. Financing immunization of adults in the United States. Clin Pharmacol Ther. 2007;82:764-768.
Hormone therapy for menopausal symptoms: Putting benefits and risks into perspective
• After assessing an individual’s benefit-risk profile, consider prescribing estrogen therapy (ET) or combined estrogen/progestin therapy (EPT) for management of vasomotor and vaginal symptoms of menopause (vaginal ET for local symptoms only). A
• Use the lowest effective doses of ET and EPT, as they may be better tolerated and have a more favorable benefit–risk ratio compared with standard doses. A
• Do not use hormone therapy for coronary protection A, although initiation by women ages 50 to 59 years or by those within 10 years of menopause may reduce cardiovascular risk. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Findings from the Women’s Health Initiative (WHI) and the Heart and Estrogen/progestin Replacement Study1,2 have left physicians and patients confused about the risks and benefits of hormone therapy (HT) and have dramatically affected prescription patterns.3 After the WHI trial findings were published in 2002,1 use of HT declined dramatically; many women discontinued therapy or switched to lower doses, while others turned to alternate therapies.4 This, despite long-standing evidence that HT administered as estrogen alone (ET; for hysterectomized women) or in combination with progestin (EPT; for nonhysterectomized women) effectively controls menopausal symptoms—hot flashes, vaginal atrophy, insomnia, and sexual problems.5
When interpreting results of recent clinical trials, it is important to consider how closely the trial subjects resemble patients in your practice. Patients in HT clinical studies may range from younger women who are newly menopausal to older women who experienced menopause decades ago. Women also have differing risk factors that determine whether HT is appropriate treatment.
Recent reanalyses of WHI data and other studies, as well as new guidelines from the North American Menopause Society (NAMS), have helped to clarify the benefit–risk profile of HT according to patient characteristics. This article places clinical trial evidence in perspective and explains how you can evaluate the benefit–risk profile of HT for individuals.
What are the benefits of HT?
The primary indication for HT is treatment of vasomotor symptoms, which are common at the time of menopause and can diminish quality of life.5 The efficacy of HT in alleviating these symptoms is well established.6 Hot flash rates are highest in women during the first 2 years postmenopause,7 and most women use HT for up to 2 years.8 A study of women who had recently become postmenopausal (45-58 years of age) showed a significant reduction in vasomotor symptoms over 5 years with ET/EPT.9
Both oral and vaginal ET effectively relieve vaginal dryness.5,10 A meta-analysis of 10 clinical trials showed that low-dose vaginal ET was as effective as systemic ET in providing relief of the signs and symptoms of urogenital atrophy.11
Nonhormonal treatments are also sometimes prescribed off label to treat vasomotor symptoms for women who cannot or choose not to use estrogens. Such agents include selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors, clonidine, and gabapentin.
A meta-analysis found that these treatments were more effective than placebo in reducing hot flashes in postmenopausal women, but the magnitude of symptom relief with these drugs has been less than that observed with estrogens.12 In another study, off-label use of antidepressants greatly attenuated hot flashes for some patients.13
NAMS recommends that women with moderate-to-severe menopause-related hot flashes who have concerns with, or contraindications to, estrogen-containing treatments, consider other treatments, such as SSRIs or gabapentin;7 however, high-quality studies evaluating these therapies in women with moderate-to-severe hot flashes are lacking.12
Phytoestrogens such as soy compounds and black cohosh may be helpful, although results have been variable in clinical trials.14 Common adverse events associated with black cohosh treatment include gastrointestinal complaints and rashes. There have been rare reports of liver toxicity, suggesting the need for further investigation.15
Protecting bone mass density and reducing risk of fractures
Estrogen therapy. In the WHI study, ET reduced the rates of hip fractures (P=.01), clinical vertebral fractures (P=.02), and total osteoporotic fractures (P<.001).16 The reduced risk was not affected by patient age.17 The randomized Women’s Health, Osteoporosis, Progestin, Estrogen (HOPE) study showed protection against early bone loss with ET vs placebo. After 2 years of follow-up, 55% of placebo-treated patients exhibited >2% loss of spine bone-mass density, compared with just 7% of women using ET (0.625 mg/d).18
Estrogen-progestin therapy. The WHI study1,19 confirmed the reduced risk of osteoporotic fractures with EPT seen in previous clinical studies, and the Women’s HOPE study18 confirmed EPT’s protective effect against bone loss. In a meta-analysis of HT studies (most of which used EPT), the benefits associated with HT in fracture prevention were particularly marked in women younger than 60 years,20 although no effect of age or time since menopause was observed in the WHI study.19 Initiation of EPT soon after menopause has been shown to improve postural balance to levels seen in premenopausal women, which may contribute to protection against fracture.21
Nonhormonal therapy for bone health. For women who are not candidates for HT, therapeutic options for maintaining bone health include bisphosphonates, raloxifene, teriparatide, and calcitonin.22 In addition to calcium and vitamin D supplementation, the NAMS guidelines recommend bisphosphonates as first-line treatment, followed by raloxifene, for postmenopausal women with low bone mass or younger postmenopausal women with osteoporosis who are at greater risk of spine fracture than hip fracture.23 Teriparatide is generally reserved for women at high risk of fracture.23 Calcitonin, typically administered as a nasal spray, is approved for osteoporosis treatment, but not prevention. It is generally considered an alternative for patients who cannot tolerate other therapies.23 Denosumab, a monoclonal antibody, is a new drug indicated in women at high risk for fracture or who cannot tolerate other therapies.
What are the potential risks of HT?
Risk factors associated with HT relate to a woman’s baseline disease risks: age; age at menopause; cause of menopause; time elapsed since menopause; prior use of any hormone; types, routes of administration, and doses of HT used; and medical conditions emerging during treatment.5 When assessing the benefit–risk profile of HT for any patient, take into account the woman’s health profile as well as the chance of harm associated with any particular therapy.
Cardiovascular disease: Timing of HT matters
Estrogen therapy. Although observational studies,24,25 such as the Nurses’ Health Study, suggest a reduced risk of cardiovascular events with ET, randomized clinical trials16,26 have shown either no effect or increased risk of cardiovascular disease among women using ET. In the ET arm of the WHI study,16,26,27 there was an increased risk of stroke and a trend toward increased risk of peripheral arterial disease, but no effect on the incidence of coronary heart disease (including myocardial infarction and coronary death).
The disparity between observational and randomized clinical trial results is now believed to be a result of differences in patient characteristics (particularly age) and timing of initiation of HT in both types of studies.5 Demographic or biologic differences influence the effects of HT on cardiovascular risk. This timing hypothesis is supported by data from an observational study,28 meta-analysis of clinical trials,29 secondary analyses from the WHI,30 and a substudy of the WHI (WHI-Coronary Artery Calcium Study).31
Estrogen-progestin therapy. As with ET, observational studies24,25 have indicated a reduced risk of cardiovascular events with EPT, whereas randomized clinical trials1,26,32 have shown either no effect or increased risk of cardiovascular disease in women using EPT. A recent observational study of women taking primarily EPT (87%; 13% on ET) for a mean duration of 8.3 years found no significant difference in the risk of cardiovascular disease between groups exposed to HT and those unexposed (relative risk, 0.84; 95% confidence interval [CI], 0.16-4.13).33
The WHI study demonstrated an increase in cardiovascular event risk with EPT, particularly during the first year of treatment.1 However, when results were adjusted for age and time since menopause, this risk was isolated to women ≥20 years past menopause, contrasting with a trend toward reduced risk of coronary heart disease in women who initiated HT within 10 years of menopause.30
In the Women’s International Study of Long Duration Oestrogen After Menopause (WISDOM), there was a significant increase in the number of major cardiovascular events with EPT vs placebo.32 However, as in the original WHI study, most women in the WISDOM study were age 65 or older and thus did not fall into the younger age category that experiences cardiovascular benefit from HT.
Influence of age on cardiovascular risk. In the WHI and WISDOM studies,1,16,32 women tended to be at least 10 years postmenopause, whereas the observational studies included younger women who started HT sooner after menopause. The WHI data have shown no increased risk of cardiovascular disease with ET overall and have shown lower coronary artery disease risk in women ages 50 to 59 years.26 There was also a trend for reduced cardiovascular risk with EPT among women who were up to 10 years postmenopause.30
In a meta-analysis34 of randomized studies, there was a reduction in the risk of cardiovascular events with HT in women younger than 60 years, but an increased risk of events during the first year of treatment in older women. HT has been associated with reduced blood pressure in women who are <5 years postmenopause but not in women ≥5 years postmenopause.35 Thus, the data appear to support the hypothesis of a “therapeutic window” during which ET or EPT may be cardioprotective in younger, newly menopausal women, and an increased risk for cardiovascular disease with EPT, principally confined to older women at an increased distance from menopause.
Thromboembolism: Patient age makes a difference
Estrogen therapy. Observational data from the UK General Practice Research Database, which included women ages 55 to 79 years, demonstrated a reduced risk of deep vein thrombosis (P=.008) and a trend toward reduced risk of venous thromboembolism (VTE; P=.057) among users of ET.36 However, the ET arm of the WHI showed an early increased risk of venous thrombosis, particularly within the first 2 years of use.37 The absolute incidence of VTE (including deep vein thrombosis and pulmonary embolism) was relatively low in the study, and risk of pulmonary embolism alone was not significantly different from that seen with placebo; however, the use of conjugated estrogens did increase the relative risk of VTE in postmenopausal women without a uterus. Risk also increased with obesity.37
Estrogen-progestin therapy. The WHI study demonstrated an increased risk of VTE with EPT compared with placebo, the risk increasing with advancing age and obesity.38 In addition, the risk of VTE was significantly greater with EPT than with ET in the same study.37 In women younger than 60 years, the projected 5-year risk associated with EPT was 1.4% in obese women, compared with less than 0.5% in women of normal weight. In the WISDOM study, which involved women older than 65 years, there was a significant increase in VTE incidence with EPT vs placebo (hazard ratio [HR], 7.36; 95% CI, 2.20-24.60).32
Thrombotic risk in perspective. The risk of VTE is an important determinant of the benefit–risk profile when prescribing HT. Data from observational and randomized trials have shown an increased risk of VTE with oral HT.5,39 In women with preexisting cardiovascular disease, the use of statins appeared to negate the increased risk of thromboembolism with EPT.40 In the WHI trials, the absolute VTE risk associated with either EPT (7 per 10,000 women per year of use) or ET (4 per 10,000 women per year of use) in women younger than 60 years was lower than in older women37—and considered rare by NAMS consensus. Thus, for otherwise healthy newly menopausal women younger than 60 years, carefully consider the benefits of ET or EPT against the negligible risk of thromboembolism.
Limited observational data suggest lower risks of VTE with transdermal ET compared with oral ET,41 but there is no conclusive evidence from randomized controlled trials on this subject.5 Low-dose oral and transdermal formulations may provide promising routes of administration, pending further studies. Evidence suggests that women with a history of VTE or women who have factor V Leiden are at increased risk for VTE with HT use.39 Use caution, therefore, when considering HT in women at higher risk of VTE, such as those with prior VTE or thrombogenic mutations, those undergoing surgery, or those who are immobilized.39
Estrogen therapy. Observational studies have suggested an increased incidence of breast cancer among women using ET for more than 1 year, with the risk increasing as use continues.36,42 In contrast, results of the WHI study showed that invasive breast cancer was diagnosed at a 23% lower rate in the ET group than in the placebo group, although this difference did not reach statistical significance (P=.06).16 The Women’s Health Study showed no association between current use of ET and the risk of total breast cancer or invasive breast cancer.43 The degree of breast cancer risk may depend on dose, as a meta-analysis of studies showed no increase in breast cancer risk with use of ET at ≤0.625 mg/d.44 In addition, the incidence of breast cancer has been shown to be lower in women who do not have benign breast disease or first-degree relatives with breast cancer.45
Estrogen-progestin therapy. Observational studies have shown an increased risk of breast cancer with EPT.42,46 In the WHI study, there was a significantly increased relative risk of invasive breast cancer in women receiving EPT over a follow-up of 5.6 years (HR, 1.24; 95% CI, 1.02-1.50).47 However, some have noted that the observed increase in the incidence of invasive breast cancer in the EPT arm vs placebo was not statistically significant and could have resulted from chance alone.48 A recent analysis of breast cancer incidence in the United States found a sharp decrease from 2002 to 2003,49 suggesting that breast cancer risk diminished soon after discontinuation of EPT for many women following the publication of the WHI results.
A newly published WHI follow-up study has yielded similar findings regarding the incidence of invasive breast cancer with EPT. The small increase in cancer incidence compared with placebo was associated with positive nodes and the death rate in this group was also higher (2.6 deaths vs 1.3 per 10,000 women). These findings do not apply to ET alone.50
Breast cancer risk in perspective. When interpreting increased risk, consider the absolute risk. In the WHI study, the absolute risk of invasive breast cancer increased by 4 to 6 cases per 10,000 women per year in the EPT group vs placebo.47 Similarly, a systematic review of clinical studies showed that EPT was associated with an increase of 4 breast cancer cases per 10,000 women per year.51 The increased risk of breast cancer with combined EPT is similar to that associated with early menarche or late menopause and is smaller than that associated with nulliparity or having children after 30 years of age.52
Assessing risks and benefits for the potential HT patient
The first step in treating patients who have hot flashes is to determine the extent of their symptoms and the effect on their quality of life (TABLE).5,10,23 Two hot flashes a day is considered mild and will usually respond to lifestyle measures such as exercising, avoiding alcohol and spicy foods, and staying in a cool environment. If the patient wakes in the night with hot flashes and night sweats that lead to insomnia, this may be more serious and require treatment. Consider HT for moderate-to-severe hot flashes—ie, 5 to 7 a day. HT is the only pharmacologic therapy indicated for the treatment of hot flashes.
Although professional guidelines recommend appropriate use of HT, publication of the WHI study caused many patients to mistrust and fear hormonal approaches to managing menopausal symptoms.3,4,53 Among those who discontinued HT, many have had vasomotor symptoms recur, and some patients remain untreated.53 A thorough discussion of individual needs and risk factors can help assess whether a patient is a suitable candidate for HT, and patient education and counseling may help alleviate concerns.54
When considering HT for a patient, take into account risk factors, such as baseline disease, age at menopause, cause of menopause, prior hormone use, variations in HT used, and age and time elapsed since menopause.5 An individual’s risks for cardiovascular disease, breast cancer, and osteoporotic fractures will help determine the most appropriate treatment.55 In the WHI, symptomatic women who were younger and closer to the menopausal transition experienced the greatest relief of vasomotor symptoms with EPT and were less likely to experience adverse effects compared with older women.56 The WHI data also showed that the prevalence of menopausal symptoms decreased with increasing age, occurring most commonly in women ages 50 to 54 years.56 The WHI findings have been shown to apply to HT regimens in general.57
Specific recommendations. Current prescribing guidelines5 recommend using ET/EPT to treat moderate-to-severe vasomotor symptoms associated with menopause when the benefits of short-term therapy outweigh the risks. For women who experience mainly vaginal symptoms rather than vasomotor symptoms, vaginal ET is recommended.
HT is not recommended for coronary protection in women of any age, as there is evidence that use in older women increases the risk of cardiovascular events.5 However, HT does not appear to increase the risk of CV events if initiated by women ages 50 to 59 years or by those within 10 years of menopause. There is evidence of an increased risk of VTE with oral HT, although absolute risk is low in women ages 50 to 59 years.5
To prevent further bone loss and reduce the risk of osteoporotic fracture in women with established reduction in bone mass, the guidelines recommend extended use of HT, regardless of menopausal symptoms, when alternate therapies are not appropriate or cause side effects or when the safety and hazards of extended use of alternate therapies are not well established.5
Breast cancer risk increases with EPT use beyond 3 to 5 years, although the absolute risk is still considered rare.5 Clinical evidence, including findings from the WHI study16 and the Women’s Health Study,43 shows no increase in the risk of breast cancer in women receiving ET. Further, the risk of breast cancer with ET may be lower in certain subgroups of women, such as those with lower Gail risk estimates based on age, history of benign breast disease, age of menarche, age of first birth, race/ ethnicity, and mothers and sisters with breast cancer;58 women with no first-degree relatives with breast cancer; women without benign breast disease; and women with no prior hormone use.45
Initiating HT for symptom control in newly menopausal women may provide additional benefits, such as reduced osteoporosis and cardiovascular risk, that outweigh the small risks associated with HT in this younger age group.
Evaluate the relative risks vs benefits, and use the lowest effective dose. Evaluate older women in a similar fashion. Those who continue to experience symptoms after discontinuing HT can be restarted on low-dose HT if symptoms do not abate.
TABLE
Select hormone therapy according to nature and severity of symptoms5,10,23
| Symptoms | Severity | Treatment |
|---|---|---|
| 2 hot flashes per day | Mild | Exercise Diet Environmental temperature regulation |
| 5-7 hot flashes per day Nighttime awakenings Night sweats/insomnia | Moderate-to-severe | HT for appropriate patients |
| Vaginal symptoms only (atrophic vaginitis) | Moderate-to-severe | Vaginal estrogen therapy |
| Osteoporosis | Established reduction in bone mass | Calcium + vitamin D plus bisphosphonate or raloxifene or extended HT for appropriate patients when preceding therapies are not tolerated or not appropriate |
Optimal candidates for HT:
| ||
| HT, hormone therapy. | ||
CORRESPONDENCE Michelle P. Warren, MD, Presbyterian Hospital, 622 West 168th Street, New York, NY 10032; [email protected]
1. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321-333.
2. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
3. Hoffmann M, Hammar M, Kjellgren KI, et al. Changes in women’s attitudes towards and use of hormone therapy after HERS and WHI. Maturitas. 2005;52:11-17.
4. Hersh AL, Stefanick ML, Stafford RS. National use of postmenopausal hormone therapy: annual trends and response to recent evidence. JAMA. 2004;291:47-53.
5. North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:242-255.
6. Maclennan AH, Broadbent JL, Lester S, et al. Oral oestrogen and combined oestrogen/progestogen therapy versus placebo for hot flushes. Cochrane Database Syst Rev. 2004;(4):CD002978.-
7. North American Menopause Society. Treatment of menopause-associated vasomotor symptoms: position statement of The North American Menopause Society. Menopause. 2004;11:11-33.
8. Grady D, Sawaya GF. Discontinuation of postmenopausal hormone therapy. Am J Med. 2005;118:163-165.
9. Vestergaard P, Hermann AP, Stilgren L, et al. Effects of 5 years of hormonal replacement therapy on menopausal symptoms and blood pressure—a randomised controlled study. Maturitas. 2003;46:123-132.
10. North American Menopause Society. The role of local vaginal estrogen for treatment of vaginal atrophy in postmenopausal women: 2007 position statement of The North American Menopause Society. Menopause. 2007;14:357-369.
11. Cardozo L, Bachmann G, McClish D, et al. Meta-analysis of estrogen therapy in the management of urogenital atrophy in postmenopausal women: second report of the Hormones and Urogenital Therapy Committee. Obstet Gynecol. 1998;92:722-727.
12. Nelson HD, Vesco KK, Haney E, et al. Nonhormonal therapies for menopausal hot flashes: systematic review and meta-analysis. JAMA. 2006;295:2057-2071.
13. Cheema D, Coomarasamy A, El-Toukhy T. Non-hormonal therapy of post-menopausal vasomotor symptoms: a structured evidence-based review. Arch Gynecol Obstet. 2007;276:463-469.
14. Whelan AM, Jurgens TM, Bowles SK. Natural health products in the prevention and treatment of osteoporosis: systematic review of randomized controlled trials. Ann Pharmacother. 2006;40:836-849.
15. Borrelli F, Ernst E. Black cohosh (Cimicifuga racemosa): a systematic review of adverse events. Am J Obstet Gynecol. 2008;199:455-466.
16. Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-1712.
17. Jackson RD, Wactawski-Wende J, LaCroix AZ, et al. Effects of conjugated equine estrogen on risk of fractures and BMD in postmenopausal women with hysterectomy: results from the women’s health initiative randomized trial. J Bone Miner Res. 2006;21:817-828.
18. Lindsay R, Gallagher JC, Kleerekoper M, et al. Bone response to treatment with lower doses of conjugated estrogens with and without medroxyprogesterone acetate in early postmenopausal women. Osteoporos Int. 2005;16:372-379.
19. Cauley JA, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA. 2003;290:1729-1738.
20. Torgerson DJ, Bell-Syer SE. Hormone replacement therapy and prevention of nonvertebral fractures: a meta-analysis of randomized trials. JAMA. 2001;285:2891-2897.
21. Naessen T, Lindmark B, Lagerstrom C, et al. Early postmenopausal hormone therapy improves postural balance. Menopause. 2007;14:14-19.
22. Jenkins MR, Sikon AL. Update on nonhormonal approaches to menopausal management. Cleve Clin J Med. 2008;75(suppl 4):S17-S24.
23. North American Menopause Society. Management of osteoporosis in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:25-54.
24. Ferrara A, Quesenberry CP, Karter AJ, et al. Current use of unopposed estrogen and estrogen plus progestin and the risk of acute myocardial infarction among women with diabetes: the Northern California Kaiser Permanente Diabetes Registry, 1995-1998. Circulation. 2003;107:43-48.
25. Grodstein F, Manson JE, Colditz GA, et al. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med. 2000;133:933-941.
26. Hsia J, Langer RD, Manson JE, et al. Conjugated equine estrogens and coronary heart disease: the Women’s Health Initiative. Arch Intern Med. 2006;166:357-365.
27. Hendrix SL, Wassertheil-Smoller S, Johnson KC, et al. Effects of conjugated equine estrogen on stroke in the Women’s Health Initiative. Circulation. 2006;113:2425-2434.
28. Grodstein F, Manson JE, Stampfer MJ. Hormone therapy and coronary heart disease: the role of time since menopause and age at hormone initiation. J Womens Health (Larchmt). 2006;15:35-44.
29. Hernán MA, Alonso A, Logan R, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008;19:766-779.
30. Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465-1477.
31. Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591-2602.
32. Vickers MR, Maclennan AH, Lawton B, et al. Main morbidities recorded in the women’s international study of long duration oestrogen after menopause (WISDOM): a randomised controlled trial of hormone replacement therapy in postmenopausal women. BMJ. 2007;335:239.-
33. Mares P, Chevallier T, Micheletti MC, et al. Coronary heart disease and HRT in France: MISSION study prospective phase results. Gynecol Endocrinol. 2008;24:696-700.
34. Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med. 2006;21:363-366.
35. Brownley KA, Hinderliter AL, West SG, et al. Cardiovascular effects of 6 months of hormone replacement therapy versus placebo: differences associated with years since menopause. Am J Obstet Gynecol. 2004;190:1052-1058.
36. Tannen RL, Weiner MG, Xie D, et al. Estrogen affects postmenopausal women differently than estrogen plus progestin replacement therapy. Hum Reprod. 2007;22:1769-1777.
37. Curb JD, Prentice RL, Bray PF, et al. Venous thrombosis and conjugated equine estrogen in women without a uterus. Arch Intern Med. 2006;166:772-780.
38. Cushman M, Kuller LH, Prentice R, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292:1573-1580.
39. McLaren J, Barnhart K. Hormone therapy and venous thromboembolism in menopausal women. Menopausal Med. 2008;16(4):S1-S7.
40. Grady D, Herrington D, Bittner V, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA. 2002;288:49-57.
41. Canonico M, Oger E, Plu-Bureau G, et al. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation. 2007;115:840-845.
42. Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
43. Zhang SM, Manson JE, Rexrode KM, et al. Use of oral conjugated estrogen alone and risk of breast cancer. Am J Epidemiol. 2007;165:524-529.
44. Dupont WD, Page DL. Menopausal estrogen replacement therapy and breast cancer. Arch Intern Med. 1991;151:67-72.
45. Stefanick ML, Anderson GL, Margolis KL, et al. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA. 2006;295:1647-1657.
46. Ross RK, Paganini-Hill A, Wan PC, et al. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Anderson GL, Chlebowski RT, Rossouw JE, et al. Prior hormone therapy and breast cancer risk in the Women’s Health Initiative randomized trial of estrogen plus progestin. Maturitas. 2006;55:103-115.
48. Goodman N, Goldzieher J, Ayala C. Critique of the report from the Writing Group of the WHI. Menopausal Med. 2003;10(4):1-4.
49. Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356:1670-1674.
50. Chlebowski RT, Anderson GL, Gass M, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. 2010;304:1684-1692.
51. Collins JA, Blake JM, Crosignani PG. Breast cancer risk with postmenopausal hormonal treatment. Hum Reprod Update. 2005;11:545-560.
52. Singletary SE. Rating the risk factors for breast cancer. Ann Surg. 2003;237:474-482.
53. Helenius IM, Korenstein D, Halm EA. Changing use of hormone therapy among minority women since the Women’s Health Initiative. Menopause. 2007;14:216-222.
54. Theroux R, Taylor K. Women’s decision making about the use of hormonal and nonhormonal remedies for the menopausal transition. J Obstet Gynecol Neonatal Nurs. 2003;32:712-723.
55. Col NF, Pauker SG, Goldberg RJ, et al. Individualizing therapy to prevent long-term consequences of estrogen deficiency in postmenopausal women. Arch Intern Med. 1999;159:1458-1466.
56. Barnabei VM, Cochrane BB, Aragaki AK, et al. Menopausal symptoms and treatment-related effects of estrogen and progestin in the Women’s Health Initiative. Obstet Gynecol. 2005;105:1063-1073.
57. Warren MP. A comparative review of the risks and benefits of hormone replacement therapy regimens. Am J Obstet Gynecol. 2004;190:1141-1167.
58. Gail MH, Costantino JP, Bryant J, et al. Weighing the risks and benefits of tamoxifen treatment for preventing breast cancer. J Natl Cancer Inst. 1999;91:1829-1846.
• After assessing an individual’s benefit-risk profile, consider prescribing estrogen therapy (ET) or combined estrogen/progestin therapy (EPT) for management of vasomotor and vaginal symptoms of menopause (vaginal ET for local symptoms only). A
• Use the lowest effective doses of ET and EPT, as they may be better tolerated and have a more favorable benefit–risk ratio compared with standard doses. A
• Do not use hormone therapy for coronary protection A, although initiation by women ages 50 to 59 years or by those within 10 years of menopause may reduce cardiovascular risk. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Findings from the Women’s Health Initiative (WHI) and the Heart and Estrogen/progestin Replacement Study1,2 have left physicians and patients confused about the risks and benefits of hormone therapy (HT) and have dramatically affected prescription patterns.3 After the WHI trial findings were published in 2002,1 use of HT declined dramatically; many women discontinued therapy or switched to lower doses, while others turned to alternate therapies.4 This, despite long-standing evidence that HT administered as estrogen alone (ET; for hysterectomized women) or in combination with progestin (EPT; for nonhysterectomized women) effectively controls menopausal symptoms—hot flashes, vaginal atrophy, insomnia, and sexual problems.5
When interpreting results of recent clinical trials, it is important to consider how closely the trial subjects resemble patients in your practice. Patients in HT clinical studies may range from younger women who are newly menopausal to older women who experienced menopause decades ago. Women also have differing risk factors that determine whether HT is appropriate treatment.
Recent reanalyses of WHI data and other studies, as well as new guidelines from the North American Menopause Society (NAMS), have helped to clarify the benefit–risk profile of HT according to patient characteristics. This article places clinical trial evidence in perspective and explains how you can evaluate the benefit–risk profile of HT for individuals.
What are the benefits of HT?
The primary indication for HT is treatment of vasomotor symptoms, which are common at the time of menopause and can diminish quality of life.5 The efficacy of HT in alleviating these symptoms is well established.6 Hot flash rates are highest in women during the first 2 years postmenopause,7 and most women use HT for up to 2 years.8 A study of women who had recently become postmenopausal (45-58 years of age) showed a significant reduction in vasomotor symptoms over 5 years with ET/EPT.9
Both oral and vaginal ET effectively relieve vaginal dryness.5,10 A meta-analysis of 10 clinical trials showed that low-dose vaginal ET was as effective as systemic ET in providing relief of the signs and symptoms of urogenital atrophy.11
Nonhormonal treatments are also sometimes prescribed off label to treat vasomotor symptoms for women who cannot or choose not to use estrogens. Such agents include selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors, clonidine, and gabapentin.
A meta-analysis found that these treatments were more effective than placebo in reducing hot flashes in postmenopausal women, but the magnitude of symptom relief with these drugs has been less than that observed with estrogens.12 In another study, off-label use of antidepressants greatly attenuated hot flashes for some patients.13
NAMS recommends that women with moderate-to-severe menopause-related hot flashes who have concerns with, or contraindications to, estrogen-containing treatments, consider other treatments, such as SSRIs or gabapentin;7 however, high-quality studies evaluating these therapies in women with moderate-to-severe hot flashes are lacking.12
Phytoestrogens such as soy compounds and black cohosh may be helpful, although results have been variable in clinical trials.14 Common adverse events associated with black cohosh treatment include gastrointestinal complaints and rashes. There have been rare reports of liver toxicity, suggesting the need for further investigation.15
Protecting bone mass density and reducing risk of fractures
Estrogen therapy. In the WHI study, ET reduced the rates of hip fractures (P=.01), clinical vertebral fractures (P=.02), and total osteoporotic fractures (P<.001).16 The reduced risk was not affected by patient age.17 The randomized Women’s Health, Osteoporosis, Progestin, Estrogen (HOPE) study showed protection against early bone loss with ET vs placebo. After 2 years of follow-up, 55% of placebo-treated patients exhibited >2% loss of spine bone-mass density, compared with just 7% of women using ET (0.625 mg/d).18
Estrogen-progestin therapy. The WHI study1,19 confirmed the reduced risk of osteoporotic fractures with EPT seen in previous clinical studies, and the Women’s HOPE study18 confirmed EPT’s protective effect against bone loss. In a meta-analysis of HT studies (most of which used EPT), the benefits associated with HT in fracture prevention were particularly marked in women younger than 60 years,20 although no effect of age or time since menopause was observed in the WHI study.19 Initiation of EPT soon after menopause has been shown to improve postural balance to levels seen in premenopausal women, which may contribute to protection against fracture.21
Nonhormonal therapy for bone health. For women who are not candidates for HT, therapeutic options for maintaining bone health include bisphosphonates, raloxifene, teriparatide, and calcitonin.22 In addition to calcium and vitamin D supplementation, the NAMS guidelines recommend bisphosphonates as first-line treatment, followed by raloxifene, for postmenopausal women with low bone mass or younger postmenopausal women with osteoporosis who are at greater risk of spine fracture than hip fracture.23 Teriparatide is generally reserved for women at high risk of fracture.23 Calcitonin, typically administered as a nasal spray, is approved for osteoporosis treatment, but not prevention. It is generally considered an alternative for patients who cannot tolerate other therapies.23 Denosumab, a monoclonal antibody, is a new drug indicated in women at high risk for fracture or who cannot tolerate other therapies.
What are the potential risks of HT?
Risk factors associated with HT relate to a woman’s baseline disease risks: age; age at menopause; cause of menopause; time elapsed since menopause; prior use of any hormone; types, routes of administration, and doses of HT used; and medical conditions emerging during treatment.5 When assessing the benefit–risk profile of HT for any patient, take into account the woman’s health profile as well as the chance of harm associated with any particular therapy.
Cardiovascular disease: Timing of HT matters
Estrogen therapy. Although observational studies,24,25 such as the Nurses’ Health Study, suggest a reduced risk of cardiovascular events with ET, randomized clinical trials16,26 have shown either no effect or increased risk of cardiovascular disease among women using ET. In the ET arm of the WHI study,16,26,27 there was an increased risk of stroke and a trend toward increased risk of peripheral arterial disease, but no effect on the incidence of coronary heart disease (including myocardial infarction and coronary death).
The disparity between observational and randomized clinical trial results is now believed to be a result of differences in patient characteristics (particularly age) and timing of initiation of HT in both types of studies.5 Demographic or biologic differences influence the effects of HT on cardiovascular risk. This timing hypothesis is supported by data from an observational study,28 meta-analysis of clinical trials,29 secondary analyses from the WHI,30 and a substudy of the WHI (WHI-Coronary Artery Calcium Study).31
Estrogen-progestin therapy. As with ET, observational studies24,25 have indicated a reduced risk of cardiovascular events with EPT, whereas randomized clinical trials1,26,32 have shown either no effect or increased risk of cardiovascular disease in women using EPT. A recent observational study of women taking primarily EPT (87%; 13% on ET) for a mean duration of 8.3 years found no significant difference in the risk of cardiovascular disease between groups exposed to HT and those unexposed (relative risk, 0.84; 95% confidence interval [CI], 0.16-4.13).33
The WHI study demonstrated an increase in cardiovascular event risk with EPT, particularly during the first year of treatment.1 However, when results were adjusted for age and time since menopause, this risk was isolated to women ≥20 years past menopause, contrasting with a trend toward reduced risk of coronary heart disease in women who initiated HT within 10 years of menopause.30
In the Women’s International Study of Long Duration Oestrogen After Menopause (WISDOM), there was a significant increase in the number of major cardiovascular events with EPT vs placebo.32 However, as in the original WHI study, most women in the WISDOM study were age 65 or older and thus did not fall into the younger age category that experiences cardiovascular benefit from HT.
Influence of age on cardiovascular risk. In the WHI and WISDOM studies,1,16,32 women tended to be at least 10 years postmenopause, whereas the observational studies included younger women who started HT sooner after menopause. The WHI data have shown no increased risk of cardiovascular disease with ET overall and have shown lower coronary artery disease risk in women ages 50 to 59 years.26 There was also a trend for reduced cardiovascular risk with EPT among women who were up to 10 years postmenopause.30
In a meta-analysis34 of randomized studies, there was a reduction in the risk of cardiovascular events with HT in women younger than 60 years, but an increased risk of events during the first year of treatment in older women. HT has been associated with reduced blood pressure in women who are <5 years postmenopause but not in women ≥5 years postmenopause.35 Thus, the data appear to support the hypothesis of a “therapeutic window” during which ET or EPT may be cardioprotective in younger, newly menopausal women, and an increased risk for cardiovascular disease with EPT, principally confined to older women at an increased distance from menopause.
Thromboembolism: Patient age makes a difference
Estrogen therapy. Observational data from the UK General Practice Research Database, which included women ages 55 to 79 years, demonstrated a reduced risk of deep vein thrombosis (P=.008) and a trend toward reduced risk of venous thromboembolism (VTE; P=.057) among users of ET.36 However, the ET arm of the WHI showed an early increased risk of venous thrombosis, particularly within the first 2 years of use.37 The absolute incidence of VTE (including deep vein thrombosis and pulmonary embolism) was relatively low in the study, and risk of pulmonary embolism alone was not significantly different from that seen with placebo; however, the use of conjugated estrogens did increase the relative risk of VTE in postmenopausal women without a uterus. Risk also increased with obesity.37
Estrogen-progestin therapy. The WHI study demonstrated an increased risk of VTE with EPT compared with placebo, the risk increasing with advancing age and obesity.38 In addition, the risk of VTE was significantly greater with EPT than with ET in the same study.37 In women younger than 60 years, the projected 5-year risk associated with EPT was 1.4% in obese women, compared with less than 0.5% in women of normal weight. In the WISDOM study, which involved women older than 65 years, there was a significant increase in VTE incidence with EPT vs placebo (hazard ratio [HR], 7.36; 95% CI, 2.20-24.60).32
Thrombotic risk in perspective. The risk of VTE is an important determinant of the benefit–risk profile when prescribing HT. Data from observational and randomized trials have shown an increased risk of VTE with oral HT.5,39 In women with preexisting cardiovascular disease, the use of statins appeared to negate the increased risk of thromboembolism with EPT.40 In the WHI trials, the absolute VTE risk associated with either EPT (7 per 10,000 women per year of use) or ET (4 per 10,000 women per year of use) in women younger than 60 years was lower than in older women37—and considered rare by NAMS consensus. Thus, for otherwise healthy newly menopausal women younger than 60 years, carefully consider the benefits of ET or EPT against the negligible risk of thromboembolism.
Limited observational data suggest lower risks of VTE with transdermal ET compared with oral ET,41 but there is no conclusive evidence from randomized controlled trials on this subject.5 Low-dose oral and transdermal formulations may provide promising routes of administration, pending further studies. Evidence suggests that women with a history of VTE or women who have factor V Leiden are at increased risk for VTE with HT use.39 Use caution, therefore, when considering HT in women at higher risk of VTE, such as those with prior VTE or thrombogenic mutations, those undergoing surgery, or those who are immobilized.39
Estrogen therapy. Observational studies have suggested an increased incidence of breast cancer among women using ET for more than 1 year, with the risk increasing as use continues.36,42 In contrast, results of the WHI study showed that invasive breast cancer was diagnosed at a 23% lower rate in the ET group than in the placebo group, although this difference did not reach statistical significance (P=.06).16 The Women’s Health Study showed no association between current use of ET and the risk of total breast cancer or invasive breast cancer.43 The degree of breast cancer risk may depend on dose, as a meta-analysis of studies showed no increase in breast cancer risk with use of ET at ≤0.625 mg/d.44 In addition, the incidence of breast cancer has been shown to be lower in women who do not have benign breast disease or first-degree relatives with breast cancer.45
Estrogen-progestin therapy. Observational studies have shown an increased risk of breast cancer with EPT.42,46 In the WHI study, there was a significantly increased relative risk of invasive breast cancer in women receiving EPT over a follow-up of 5.6 years (HR, 1.24; 95% CI, 1.02-1.50).47 However, some have noted that the observed increase in the incidence of invasive breast cancer in the EPT arm vs placebo was not statistically significant and could have resulted from chance alone.48 A recent analysis of breast cancer incidence in the United States found a sharp decrease from 2002 to 2003,49 suggesting that breast cancer risk diminished soon after discontinuation of EPT for many women following the publication of the WHI results.
A newly published WHI follow-up study has yielded similar findings regarding the incidence of invasive breast cancer with EPT. The small increase in cancer incidence compared with placebo was associated with positive nodes and the death rate in this group was also higher (2.6 deaths vs 1.3 per 10,000 women). These findings do not apply to ET alone.50
Breast cancer risk in perspective. When interpreting increased risk, consider the absolute risk. In the WHI study, the absolute risk of invasive breast cancer increased by 4 to 6 cases per 10,000 women per year in the EPT group vs placebo.47 Similarly, a systematic review of clinical studies showed that EPT was associated with an increase of 4 breast cancer cases per 10,000 women per year.51 The increased risk of breast cancer with combined EPT is similar to that associated with early menarche or late menopause and is smaller than that associated with nulliparity or having children after 30 years of age.52
Assessing risks and benefits for the potential HT patient
The first step in treating patients who have hot flashes is to determine the extent of their symptoms and the effect on their quality of life (TABLE).5,10,23 Two hot flashes a day is considered mild and will usually respond to lifestyle measures such as exercising, avoiding alcohol and spicy foods, and staying in a cool environment. If the patient wakes in the night with hot flashes and night sweats that lead to insomnia, this may be more serious and require treatment. Consider HT for moderate-to-severe hot flashes—ie, 5 to 7 a day. HT is the only pharmacologic therapy indicated for the treatment of hot flashes.
Although professional guidelines recommend appropriate use of HT, publication of the WHI study caused many patients to mistrust and fear hormonal approaches to managing menopausal symptoms.3,4,53 Among those who discontinued HT, many have had vasomotor symptoms recur, and some patients remain untreated.53 A thorough discussion of individual needs and risk factors can help assess whether a patient is a suitable candidate for HT, and patient education and counseling may help alleviate concerns.54
When considering HT for a patient, take into account risk factors, such as baseline disease, age at menopause, cause of menopause, prior hormone use, variations in HT used, and age and time elapsed since menopause.5 An individual’s risks for cardiovascular disease, breast cancer, and osteoporotic fractures will help determine the most appropriate treatment.55 In the WHI, symptomatic women who were younger and closer to the menopausal transition experienced the greatest relief of vasomotor symptoms with EPT and were less likely to experience adverse effects compared with older women.56 The WHI data also showed that the prevalence of menopausal symptoms decreased with increasing age, occurring most commonly in women ages 50 to 54 years.56 The WHI findings have been shown to apply to HT regimens in general.57
Specific recommendations. Current prescribing guidelines5 recommend using ET/EPT to treat moderate-to-severe vasomotor symptoms associated with menopause when the benefits of short-term therapy outweigh the risks. For women who experience mainly vaginal symptoms rather than vasomotor symptoms, vaginal ET is recommended.
HT is not recommended for coronary protection in women of any age, as there is evidence that use in older women increases the risk of cardiovascular events.5 However, HT does not appear to increase the risk of CV events if initiated by women ages 50 to 59 years or by those within 10 years of menopause. There is evidence of an increased risk of VTE with oral HT, although absolute risk is low in women ages 50 to 59 years.5
To prevent further bone loss and reduce the risk of osteoporotic fracture in women with established reduction in bone mass, the guidelines recommend extended use of HT, regardless of menopausal symptoms, when alternate therapies are not appropriate or cause side effects or when the safety and hazards of extended use of alternate therapies are not well established.5
Breast cancer risk increases with EPT use beyond 3 to 5 years, although the absolute risk is still considered rare.5 Clinical evidence, including findings from the WHI study16 and the Women’s Health Study,43 shows no increase in the risk of breast cancer in women receiving ET. Further, the risk of breast cancer with ET may be lower in certain subgroups of women, such as those with lower Gail risk estimates based on age, history of benign breast disease, age of menarche, age of first birth, race/ ethnicity, and mothers and sisters with breast cancer;58 women with no first-degree relatives with breast cancer; women without benign breast disease; and women with no prior hormone use.45
Initiating HT for symptom control in newly menopausal women may provide additional benefits, such as reduced osteoporosis and cardiovascular risk, that outweigh the small risks associated with HT in this younger age group.
Evaluate the relative risks vs benefits, and use the lowest effective dose. Evaluate older women in a similar fashion. Those who continue to experience symptoms after discontinuing HT can be restarted on low-dose HT if symptoms do not abate.
TABLE
Select hormone therapy according to nature and severity of symptoms5,10,23
| Symptoms | Severity | Treatment |
|---|---|---|
| 2 hot flashes per day | Mild | Exercise Diet Environmental temperature regulation |
| 5-7 hot flashes per day Nighttime awakenings Night sweats/insomnia | Moderate-to-severe | HT for appropriate patients |
| Vaginal symptoms only (atrophic vaginitis) | Moderate-to-severe | Vaginal estrogen therapy |
| Osteoporosis | Established reduction in bone mass | Calcium + vitamin D plus bisphosphonate or raloxifene or extended HT for appropriate patients when preceding therapies are not tolerated or not appropriate |
Optimal candidates for HT:
| ||
| HT, hormone therapy. | ||
CORRESPONDENCE Michelle P. Warren, MD, Presbyterian Hospital, 622 West 168th Street, New York, NY 10032; [email protected]
• After assessing an individual’s benefit-risk profile, consider prescribing estrogen therapy (ET) or combined estrogen/progestin therapy (EPT) for management of vasomotor and vaginal symptoms of menopause (vaginal ET for local symptoms only). A
• Use the lowest effective doses of ET and EPT, as they may be better tolerated and have a more favorable benefit–risk ratio compared with standard doses. A
• Do not use hormone therapy for coronary protection A, although initiation by women ages 50 to 59 years or by those within 10 years of menopause may reduce cardiovascular risk. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Findings from the Women’s Health Initiative (WHI) and the Heart and Estrogen/progestin Replacement Study1,2 have left physicians and patients confused about the risks and benefits of hormone therapy (HT) and have dramatically affected prescription patterns.3 After the WHI trial findings were published in 2002,1 use of HT declined dramatically; many women discontinued therapy or switched to lower doses, while others turned to alternate therapies.4 This, despite long-standing evidence that HT administered as estrogen alone (ET; for hysterectomized women) or in combination with progestin (EPT; for nonhysterectomized women) effectively controls menopausal symptoms—hot flashes, vaginal atrophy, insomnia, and sexual problems.5
When interpreting results of recent clinical trials, it is important to consider how closely the trial subjects resemble patients in your practice. Patients in HT clinical studies may range from younger women who are newly menopausal to older women who experienced menopause decades ago. Women also have differing risk factors that determine whether HT is appropriate treatment.
Recent reanalyses of WHI data and other studies, as well as new guidelines from the North American Menopause Society (NAMS), have helped to clarify the benefit–risk profile of HT according to patient characteristics. This article places clinical trial evidence in perspective and explains how you can evaluate the benefit–risk profile of HT for individuals.
What are the benefits of HT?
The primary indication for HT is treatment of vasomotor symptoms, which are common at the time of menopause and can diminish quality of life.5 The efficacy of HT in alleviating these symptoms is well established.6 Hot flash rates are highest in women during the first 2 years postmenopause,7 and most women use HT for up to 2 years.8 A study of women who had recently become postmenopausal (45-58 years of age) showed a significant reduction in vasomotor symptoms over 5 years with ET/EPT.9
Both oral and vaginal ET effectively relieve vaginal dryness.5,10 A meta-analysis of 10 clinical trials showed that low-dose vaginal ET was as effective as systemic ET in providing relief of the signs and symptoms of urogenital atrophy.11
Nonhormonal treatments are also sometimes prescribed off label to treat vasomotor symptoms for women who cannot or choose not to use estrogens. Such agents include selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors, clonidine, and gabapentin.
A meta-analysis found that these treatments were more effective than placebo in reducing hot flashes in postmenopausal women, but the magnitude of symptom relief with these drugs has been less than that observed with estrogens.12 In another study, off-label use of antidepressants greatly attenuated hot flashes for some patients.13
NAMS recommends that women with moderate-to-severe menopause-related hot flashes who have concerns with, or contraindications to, estrogen-containing treatments, consider other treatments, such as SSRIs or gabapentin;7 however, high-quality studies evaluating these therapies in women with moderate-to-severe hot flashes are lacking.12
Phytoestrogens such as soy compounds and black cohosh may be helpful, although results have been variable in clinical trials.14 Common adverse events associated with black cohosh treatment include gastrointestinal complaints and rashes. There have been rare reports of liver toxicity, suggesting the need for further investigation.15
Protecting bone mass density and reducing risk of fractures
Estrogen therapy. In the WHI study, ET reduced the rates of hip fractures (P=.01), clinical vertebral fractures (P=.02), and total osteoporotic fractures (P<.001).16 The reduced risk was not affected by patient age.17 The randomized Women’s Health, Osteoporosis, Progestin, Estrogen (HOPE) study showed protection against early bone loss with ET vs placebo. After 2 years of follow-up, 55% of placebo-treated patients exhibited >2% loss of spine bone-mass density, compared with just 7% of women using ET (0.625 mg/d).18
Estrogen-progestin therapy. The WHI study1,19 confirmed the reduced risk of osteoporotic fractures with EPT seen in previous clinical studies, and the Women’s HOPE study18 confirmed EPT’s protective effect against bone loss. In a meta-analysis of HT studies (most of which used EPT), the benefits associated with HT in fracture prevention were particularly marked in women younger than 60 years,20 although no effect of age or time since menopause was observed in the WHI study.19 Initiation of EPT soon after menopause has been shown to improve postural balance to levels seen in premenopausal women, which may contribute to protection against fracture.21
Nonhormonal therapy for bone health. For women who are not candidates for HT, therapeutic options for maintaining bone health include bisphosphonates, raloxifene, teriparatide, and calcitonin.22 In addition to calcium and vitamin D supplementation, the NAMS guidelines recommend bisphosphonates as first-line treatment, followed by raloxifene, for postmenopausal women with low bone mass or younger postmenopausal women with osteoporosis who are at greater risk of spine fracture than hip fracture.23 Teriparatide is generally reserved for women at high risk of fracture.23 Calcitonin, typically administered as a nasal spray, is approved for osteoporosis treatment, but not prevention. It is generally considered an alternative for patients who cannot tolerate other therapies.23 Denosumab, a monoclonal antibody, is a new drug indicated in women at high risk for fracture or who cannot tolerate other therapies.
What are the potential risks of HT?
Risk factors associated with HT relate to a woman’s baseline disease risks: age; age at menopause; cause of menopause; time elapsed since menopause; prior use of any hormone; types, routes of administration, and doses of HT used; and medical conditions emerging during treatment.5 When assessing the benefit–risk profile of HT for any patient, take into account the woman’s health profile as well as the chance of harm associated with any particular therapy.
Cardiovascular disease: Timing of HT matters
Estrogen therapy. Although observational studies,24,25 such as the Nurses’ Health Study, suggest a reduced risk of cardiovascular events with ET, randomized clinical trials16,26 have shown either no effect or increased risk of cardiovascular disease among women using ET. In the ET arm of the WHI study,16,26,27 there was an increased risk of stroke and a trend toward increased risk of peripheral arterial disease, but no effect on the incidence of coronary heart disease (including myocardial infarction and coronary death).
The disparity between observational and randomized clinical trial results is now believed to be a result of differences in patient characteristics (particularly age) and timing of initiation of HT in both types of studies.5 Demographic or biologic differences influence the effects of HT on cardiovascular risk. This timing hypothesis is supported by data from an observational study,28 meta-analysis of clinical trials,29 secondary analyses from the WHI,30 and a substudy of the WHI (WHI-Coronary Artery Calcium Study).31
Estrogen-progestin therapy. As with ET, observational studies24,25 have indicated a reduced risk of cardiovascular events with EPT, whereas randomized clinical trials1,26,32 have shown either no effect or increased risk of cardiovascular disease in women using EPT. A recent observational study of women taking primarily EPT (87%; 13% on ET) for a mean duration of 8.3 years found no significant difference in the risk of cardiovascular disease between groups exposed to HT and those unexposed (relative risk, 0.84; 95% confidence interval [CI], 0.16-4.13).33
The WHI study demonstrated an increase in cardiovascular event risk with EPT, particularly during the first year of treatment.1 However, when results were adjusted for age and time since menopause, this risk was isolated to women ≥20 years past menopause, contrasting with a trend toward reduced risk of coronary heart disease in women who initiated HT within 10 years of menopause.30
In the Women’s International Study of Long Duration Oestrogen After Menopause (WISDOM), there was a significant increase in the number of major cardiovascular events with EPT vs placebo.32 However, as in the original WHI study, most women in the WISDOM study were age 65 or older and thus did not fall into the younger age category that experiences cardiovascular benefit from HT.
Influence of age on cardiovascular risk. In the WHI and WISDOM studies,1,16,32 women tended to be at least 10 years postmenopause, whereas the observational studies included younger women who started HT sooner after menopause. The WHI data have shown no increased risk of cardiovascular disease with ET overall and have shown lower coronary artery disease risk in women ages 50 to 59 years.26 There was also a trend for reduced cardiovascular risk with EPT among women who were up to 10 years postmenopause.30
In a meta-analysis34 of randomized studies, there was a reduction in the risk of cardiovascular events with HT in women younger than 60 years, but an increased risk of events during the first year of treatment in older women. HT has been associated with reduced blood pressure in women who are <5 years postmenopause but not in women ≥5 years postmenopause.35 Thus, the data appear to support the hypothesis of a “therapeutic window” during which ET or EPT may be cardioprotective in younger, newly menopausal women, and an increased risk for cardiovascular disease with EPT, principally confined to older women at an increased distance from menopause.
Thromboembolism: Patient age makes a difference
Estrogen therapy. Observational data from the UK General Practice Research Database, which included women ages 55 to 79 years, demonstrated a reduced risk of deep vein thrombosis (P=.008) and a trend toward reduced risk of venous thromboembolism (VTE; P=.057) among users of ET.36 However, the ET arm of the WHI showed an early increased risk of venous thrombosis, particularly within the first 2 years of use.37 The absolute incidence of VTE (including deep vein thrombosis and pulmonary embolism) was relatively low in the study, and risk of pulmonary embolism alone was not significantly different from that seen with placebo; however, the use of conjugated estrogens did increase the relative risk of VTE in postmenopausal women without a uterus. Risk also increased with obesity.37
Estrogen-progestin therapy. The WHI study demonstrated an increased risk of VTE with EPT compared with placebo, the risk increasing with advancing age and obesity.38 In addition, the risk of VTE was significantly greater with EPT than with ET in the same study.37 In women younger than 60 years, the projected 5-year risk associated with EPT was 1.4% in obese women, compared with less than 0.5% in women of normal weight. In the WISDOM study, which involved women older than 65 years, there was a significant increase in VTE incidence with EPT vs placebo (hazard ratio [HR], 7.36; 95% CI, 2.20-24.60).32
Thrombotic risk in perspective. The risk of VTE is an important determinant of the benefit–risk profile when prescribing HT. Data from observational and randomized trials have shown an increased risk of VTE with oral HT.5,39 In women with preexisting cardiovascular disease, the use of statins appeared to negate the increased risk of thromboembolism with EPT.40 In the WHI trials, the absolute VTE risk associated with either EPT (7 per 10,000 women per year of use) or ET (4 per 10,000 women per year of use) in women younger than 60 years was lower than in older women37—and considered rare by NAMS consensus. Thus, for otherwise healthy newly menopausal women younger than 60 years, carefully consider the benefits of ET or EPT against the negligible risk of thromboembolism.
Limited observational data suggest lower risks of VTE with transdermal ET compared with oral ET,41 but there is no conclusive evidence from randomized controlled trials on this subject.5 Low-dose oral and transdermal formulations may provide promising routes of administration, pending further studies. Evidence suggests that women with a history of VTE or women who have factor V Leiden are at increased risk for VTE with HT use.39 Use caution, therefore, when considering HT in women at higher risk of VTE, such as those with prior VTE or thrombogenic mutations, those undergoing surgery, or those who are immobilized.39
Estrogen therapy. Observational studies have suggested an increased incidence of breast cancer among women using ET for more than 1 year, with the risk increasing as use continues.36,42 In contrast, results of the WHI study showed that invasive breast cancer was diagnosed at a 23% lower rate in the ET group than in the placebo group, although this difference did not reach statistical significance (P=.06).16 The Women’s Health Study showed no association between current use of ET and the risk of total breast cancer or invasive breast cancer.43 The degree of breast cancer risk may depend on dose, as a meta-analysis of studies showed no increase in breast cancer risk with use of ET at ≤0.625 mg/d.44 In addition, the incidence of breast cancer has been shown to be lower in women who do not have benign breast disease or first-degree relatives with breast cancer.45
Estrogen-progestin therapy. Observational studies have shown an increased risk of breast cancer with EPT.42,46 In the WHI study, there was a significantly increased relative risk of invasive breast cancer in women receiving EPT over a follow-up of 5.6 years (HR, 1.24; 95% CI, 1.02-1.50).47 However, some have noted that the observed increase in the incidence of invasive breast cancer in the EPT arm vs placebo was not statistically significant and could have resulted from chance alone.48 A recent analysis of breast cancer incidence in the United States found a sharp decrease from 2002 to 2003,49 suggesting that breast cancer risk diminished soon after discontinuation of EPT for many women following the publication of the WHI results.
A newly published WHI follow-up study has yielded similar findings regarding the incidence of invasive breast cancer with EPT. The small increase in cancer incidence compared with placebo was associated with positive nodes and the death rate in this group was also higher (2.6 deaths vs 1.3 per 10,000 women). These findings do not apply to ET alone.50
Breast cancer risk in perspective. When interpreting increased risk, consider the absolute risk. In the WHI study, the absolute risk of invasive breast cancer increased by 4 to 6 cases per 10,000 women per year in the EPT group vs placebo.47 Similarly, a systematic review of clinical studies showed that EPT was associated with an increase of 4 breast cancer cases per 10,000 women per year.51 The increased risk of breast cancer with combined EPT is similar to that associated with early menarche or late menopause and is smaller than that associated with nulliparity or having children after 30 years of age.52
Assessing risks and benefits for the potential HT patient
The first step in treating patients who have hot flashes is to determine the extent of their symptoms and the effect on their quality of life (TABLE).5,10,23 Two hot flashes a day is considered mild and will usually respond to lifestyle measures such as exercising, avoiding alcohol and spicy foods, and staying in a cool environment. If the patient wakes in the night with hot flashes and night sweats that lead to insomnia, this may be more serious and require treatment. Consider HT for moderate-to-severe hot flashes—ie, 5 to 7 a day. HT is the only pharmacologic therapy indicated for the treatment of hot flashes.
Although professional guidelines recommend appropriate use of HT, publication of the WHI study caused many patients to mistrust and fear hormonal approaches to managing menopausal symptoms.3,4,53 Among those who discontinued HT, many have had vasomotor symptoms recur, and some patients remain untreated.53 A thorough discussion of individual needs and risk factors can help assess whether a patient is a suitable candidate for HT, and patient education and counseling may help alleviate concerns.54
When considering HT for a patient, take into account risk factors, such as baseline disease, age at menopause, cause of menopause, prior hormone use, variations in HT used, and age and time elapsed since menopause.5 An individual’s risks for cardiovascular disease, breast cancer, and osteoporotic fractures will help determine the most appropriate treatment.55 In the WHI, symptomatic women who were younger and closer to the menopausal transition experienced the greatest relief of vasomotor symptoms with EPT and were less likely to experience adverse effects compared with older women.56 The WHI data also showed that the prevalence of menopausal symptoms decreased with increasing age, occurring most commonly in women ages 50 to 54 years.56 The WHI findings have been shown to apply to HT regimens in general.57
Specific recommendations. Current prescribing guidelines5 recommend using ET/EPT to treat moderate-to-severe vasomotor symptoms associated with menopause when the benefits of short-term therapy outweigh the risks. For women who experience mainly vaginal symptoms rather than vasomotor symptoms, vaginal ET is recommended.
HT is not recommended for coronary protection in women of any age, as there is evidence that use in older women increases the risk of cardiovascular events.5 However, HT does not appear to increase the risk of CV events if initiated by women ages 50 to 59 years or by those within 10 years of menopause. There is evidence of an increased risk of VTE with oral HT, although absolute risk is low in women ages 50 to 59 years.5
To prevent further bone loss and reduce the risk of osteoporotic fracture in women with established reduction in bone mass, the guidelines recommend extended use of HT, regardless of menopausal symptoms, when alternate therapies are not appropriate or cause side effects or when the safety and hazards of extended use of alternate therapies are not well established.5
Breast cancer risk increases with EPT use beyond 3 to 5 years, although the absolute risk is still considered rare.5 Clinical evidence, including findings from the WHI study16 and the Women’s Health Study,43 shows no increase in the risk of breast cancer in women receiving ET. Further, the risk of breast cancer with ET may be lower in certain subgroups of women, such as those with lower Gail risk estimates based on age, history of benign breast disease, age of menarche, age of first birth, race/ ethnicity, and mothers and sisters with breast cancer;58 women with no first-degree relatives with breast cancer; women without benign breast disease; and women with no prior hormone use.45
Initiating HT for symptom control in newly menopausal women may provide additional benefits, such as reduced osteoporosis and cardiovascular risk, that outweigh the small risks associated with HT in this younger age group.
Evaluate the relative risks vs benefits, and use the lowest effective dose. Evaluate older women in a similar fashion. Those who continue to experience symptoms after discontinuing HT can be restarted on low-dose HT if symptoms do not abate.
TABLE
Select hormone therapy according to nature and severity of symptoms5,10,23
| Symptoms | Severity | Treatment |
|---|---|---|
| 2 hot flashes per day | Mild | Exercise Diet Environmental temperature regulation |
| 5-7 hot flashes per day Nighttime awakenings Night sweats/insomnia | Moderate-to-severe | HT for appropriate patients |
| Vaginal symptoms only (atrophic vaginitis) | Moderate-to-severe | Vaginal estrogen therapy |
| Osteoporosis | Established reduction in bone mass | Calcium + vitamin D plus bisphosphonate or raloxifene or extended HT for appropriate patients when preceding therapies are not tolerated or not appropriate |
Optimal candidates for HT:
| ||
| HT, hormone therapy. | ||
CORRESPONDENCE Michelle P. Warren, MD, Presbyterian Hospital, 622 West 168th Street, New York, NY 10032; [email protected]
1. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321-333.
2. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
3. Hoffmann M, Hammar M, Kjellgren KI, et al. Changes in women’s attitudes towards and use of hormone therapy after HERS and WHI. Maturitas. 2005;52:11-17.
4. Hersh AL, Stefanick ML, Stafford RS. National use of postmenopausal hormone therapy: annual trends and response to recent evidence. JAMA. 2004;291:47-53.
5. North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:242-255.
6. Maclennan AH, Broadbent JL, Lester S, et al. Oral oestrogen and combined oestrogen/progestogen therapy versus placebo for hot flushes. Cochrane Database Syst Rev. 2004;(4):CD002978.-
7. North American Menopause Society. Treatment of menopause-associated vasomotor symptoms: position statement of The North American Menopause Society. Menopause. 2004;11:11-33.
8. Grady D, Sawaya GF. Discontinuation of postmenopausal hormone therapy. Am J Med. 2005;118:163-165.
9. Vestergaard P, Hermann AP, Stilgren L, et al. Effects of 5 years of hormonal replacement therapy on menopausal symptoms and blood pressure—a randomised controlled study. Maturitas. 2003;46:123-132.
10. North American Menopause Society. The role of local vaginal estrogen for treatment of vaginal atrophy in postmenopausal women: 2007 position statement of The North American Menopause Society. Menopause. 2007;14:357-369.
11. Cardozo L, Bachmann G, McClish D, et al. Meta-analysis of estrogen therapy in the management of urogenital atrophy in postmenopausal women: second report of the Hormones and Urogenital Therapy Committee. Obstet Gynecol. 1998;92:722-727.
12. Nelson HD, Vesco KK, Haney E, et al. Nonhormonal therapies for menopausal hot flashes: systematic review and meta-analysis. JAMA. 2006;295:2057-2071.
13. Cheema D, Coomarasamy A, El-Toukhy T. Non-hormonal therapy of post-menopausal vasomotor symptoms: a structured evidence-based review. Arch Gynecol Obstet. 2007;276:463-469.
14. Whelan AM, Jurgens TM, Bowles SK. Natural health products in the prevention and treatment of osteoporosis: systematic review of randomized controlled trials. Ann Pharmacother. 2006;40:836-849.
15. Borrelli F, Ernst E. Black cohosh (Cimicifuga racemosa): a systematic review of adverse events. Am J Obstet Gynecol. 2008;199:455-466.
16. Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-1712.
17. Jackson RD, Wactawski-Wende J, LaCroix AZ, et al. Effects of conjugated equine estrogen on risk of fractures and BMD in postmenopausal women with hysterectomy: results from the women’s health initiative randomized trial. J Bone Miner Res. 2006;21:817-828.
18. Lindsay R, Gallagher JC, Kleerekoper M, et al. Bone response to treatment with lower doses of conjugated estrogens with and without medroxyprogesterone acetate in early postmenopausal women. Osteoporos Int. 2005;16:372-379.
19. Cauley JA, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA. 2003;290:1729-1738.
20. Torgerson DJ, Bell-Syer SE. Hormone replacement therapy and prevention of nonvertebral fractures: a meta-analysis of randomized trials. JAMA. 2001;285:2891-2897.
21. Naessen T, Lindmark B, Lagerstrom C, et al. Early postmenopausal hormone therapy improves postural balance. Menopause. 2007;14:14-19.
22. Jenkins MR, Sikon AL. Update on nonhormonal approaches to menopausal management. Cleve Clin J Med. 2008;75(suppl 4):S17-S24.
23. North American Menopause Society. Management of osteoporosis in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:25-54.
24. Ferrara A, Quesenberry CP, Karter AJ, et al. Current use of unopposed estrogen and estrogen plus progestin and the risk of acute myocardial infarction among women with diabetes: the Northern California Kaiser Permanente Diabetes Registry, 1995-1998. Circulation. 2003;107:43-48.
25. Grodstein F, Manson JE, Colditz GA, et al. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med. 2000;133:933-941.
26. Hsia J, Langer RD, Manson JE, et al. Conjugated equine estrogens and coronary heart disease: the Women’s Health Initiative. Arch Intern Med. 2006;166:357-365.
27. Hendrix SL, Wassertheil-Smoller S, Johnson KC, et al. Effects of conjugated equine estrogen on stroke in the Women’s Health Initiative. Circulation. 2006;113:2425-2434.
28. Grodstein F, Manson JE, Stampfer MJ. Hormone therapy and coronary heart disease: the role of time since menopause and age at hormone initiation. J Womens Health (Larchmt). 2006;15:35-44.
29. Hernán MA, Alonso A, Logan R, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008;19:766-779.
30. Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465-1477.
31. Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591-2602.
32. Vickers MR, Maclennan AH, Lawton B, et al. Main morbidities recorded in the women’s international study of long duration oestrogen after menopause (WISDOM): a randomised controlled trial of hormone replacement therapy in postmenopausal women. BMJ. 2007;335:239.-
33. Mares P, Chevallier T, Micheletti MC, et al. Coronary heart disease and HRT in France: MISSION study prospective phase results. Gynecol Endocrinol. 2008;24:696-700.
34. Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med. 2006;21:363-366.
35. Brownley KA, Hinderliter AL, West SG, et al. Cardiovascular effects of 6 months of hormone replacement therapy versus placebo: differences associated with years since menopause. Am J Obstet Gynecol. 2004;190:1052-1058.
36. Tannen RL, Weiner MG, Xie D, et al. Estrogen affects postmenopausal women differently than estrogen plus progestin replacement therapy. Hum Reprod. 2007;22:1769-1777.
37. Curb JD, Prentice RL, Bray PF, et al. Venous thrombosis and conjugated equine estrogen in women without a uterus. Arch Intern Med. 2006;166:772-780.
38. Cushman M, Kuller LH, Prentice R, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292:1573-1580.
39. McLaren J, Barnhart K. Hormone therapy and venous thromboembolism in menopausal women. Menopausal Med. 2008;16(4):S1-S7.
40. Grady D, Herrington D, Bittner V, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA. 2002;288:49-57.
41. Canonico M, Oger E, Plu-Bureau G, et al. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation. 2007;115:840-845.
42. Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
43. Zhang SM, Manson JE, Rexrode KM, et al. Use of oral conjugated estrogen alone and risk of breast cancer. Am J Epidemiol. 2007;165:524-529.
44. Dupont WD, Page DL. Menopausal estrogen replacement therapy and breast cancer. Arch Intern Med. 1991;151:67-72.
45. Stefanick ML, Anderson GL, Margolis KL, et al. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA. 2006;295:1647-1657.
46. Ross RK, Paganini-Hill A, Wan PC, et al. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Anderson GL, Chlebowski RT, Rossouw JE, et al. Prior hormone therapy and breast cancer risk in the Women’s Health Initiative randomized trial of estrogen plus progestin. Maturitas. 2006;55:103-115.
48. Goodman N, Goldzieher J, Ayala C. Critique of the report from the Writing Group of the WHI. Menopausal Med. 2003;10(4):1-4.
49. Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356:1670-1674.
50. Chlebowski RT, Anderson GL, Gass M, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. 2010;304:1684-1692.
51. Collins JA, Blake JM, Crosignani PG. Breast cancer risk with postmenopausal hormonal treatment. Hum Reprod Update. 2005;11:545-560.
52. Singletary SE. Rating the risk factors for breast cancer. Ann Surg. 2003;237:474-482.
53. Helenius IM, Korenstein D, Halm EA. Changing use of hormone therapy among minority women since the Women’s Health Initiative. Menopause. 2007;14:216-222.
54. Theroux R, Taylor K. Women’s decision making about the use of hormonal and nonhormonal remedies for the menopausal transition. J Obstet Gynecol Neonatal Nurs. 2003;32:712-723.
55. Col NF, Pauker SG, Goldberg RJ, et al. Individualizing therapy to prevent long-term consequences of estrogen deficiency in postmenopausal women. Arch Intern Med. 1999;159:1458-1466.
56. Barnabei VM, Cochrane BB, Aragaki AK, et al. Menopausal symptoms and treatment-related effects of estrogen and progestin in the Women’s Health Initiative. Obstet Gynecol. 2005;105:1063-1073.
57. Warren MP. A comparative review of the risks and benefits of hormone replacement therapy regimens. Am J Obstet Gynecol. 2004;190:1141-1167.
58. Gail MH, Costantino JP, Bryant J, et al. Weighing the risks and benefits of tamoxifen treatment for preventing breast cancer. J Natl Cancer Inst. 1999;91:1829-1846.
1. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321-333.
2. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
3. Hoffmann M, Hammar M, Kjellgren KI, et al. Changes in women’s attitudes towards and use of hormone therapy after HERS and WHI. Maturitas. 2005;52:11-17.
4. Hersh AL, Stefanick ML, Stafford RS. National use of postmenopausal hormone therapy: annual trends and response to recent evidence. JAMA. 2004;291:47-53.
5. North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:242-255.
6. Maclennan AH, Broadbent JL, Lester S, et al. Oral oestrogen and combined oestrogen/progestogen therapy versus placebo for hot flushes. Cochrane Database Syst Rev. 2004;(4):CD002978.-
7. North American Menopause Society. Treatment of menopause-associated vasomotor symptoms: position statement of The North American Menopause Society. Menopause. 2004;11:11-33.
8. Grady D, Sawaya GF. Discontinuation of postmenopausal hormone therapy. Am J Med. 2005;118:163-165.
9. Vestergaard P, Hermann AP, Stilgren L, et al. Effects of 5 years of hormonal replacement therapy on menopausal symptoms and blood pressure—a randomised controlled study. Maturitas. 2003;46:123-132.
10. North American Menopause Society. The role of local vaginal estrogen for treatment of vaginal atrophy in postmenopausal women: 2007 position statement of The North American Menopause Society. Menopause. 2007;14:357-369.
11. Cardozo L, Bachmann G, McClish D, et al. Meta-analysis of estrogen therapy in the management of urogenital atrophy in postmenopausal women: second report of the Hormones and Urogenital Therapy Committee. Obstet Gynecol. 1998;92:722-727.
12. Nelson HD, Vesco KK, Haney E, et al. Nonhormonal therapies for menopausal hot flashes: systematic review and meta-analysis. JAMA. 2006;295:2057-2071.
13. Cheema D, Coomarasamy A, El-Toukhy T. Non-hormonal therapy of post-menopausal vasomotor symptoms: a structured evidence-based review. Arch Gynecol Obstet. 2007;276:463-469.
14. Whelan AM, Jurgens TM, Bowles SK. Natural health products in the prevention and treatment of osteoporosis: systematic review of randomized controlled trials. Ann Pharmacother. 2006;40:836-849.
15. Borrelli F, Ernst E. Black cohosh (Cimicifuga racemosa): a systematic review of adverse events. Am J Obstet Gynecol. 2008;199:455-466.
16. Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-1712.
17. Jackson RD, Wactawski-Wende J, LaCroix AZ, et al. Effects of conjugated equine estrogen on risk of fractures and BMD in postmenopausal women with hysterectomy: results from the women’s health initiative randomized trial. J Bone Miner Res. 2006;21:817-828.
18. Lindsay R, Gallagher JC, Kleerekoper M, et al. Bone response to treatment with lower doses of conjugated estrogens with and without medroxyprogesterone acetate in early postmenopausal women. Osteoporos Int. 2005;16:372-379.
19. Cauley JA, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA. 2003;290:1729-1738.
20. Torgerson DJ, Bell-Syer SE. Hormone replacement therapy and prevention of nonvertebral fractures: a meta-analysis of randomized trials. JAMA. 2001;285:2891-2897.
21. Naessen T, Lindmark B, Lagerstrom C, et al. Early postmenopausal hormone therapy improves postural balance. Menopause. 2007;14:14-19.
22. Jenkins MR, Sikon AL. Update on nonhormonal approaches to menopausal management. Cleve Clin J Med. 2008;75(suppl 4):S17-S24.
23. North American Menopause Society. Management of osteoporosis in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:25-54.
24. Ferrara A, Quesenberry CP, Karter AJ, et al. Current use of unopposed estrogen and estrogen plus progestin and the risk of acute myocardial infarction among women with diabetes: the Northern California Kaiser Permanente Diabetes Registry, 1995-1998. Circulation. 2003;107:43-48.
25. Grodstein F, Manson JE, Colditz GA, et al. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med. 2000;133:933-941.
26. Hsia J, Langer RD, Manson JE, et al. Conjugated equine estrogens and coronary heart disease: the Women’s Health Initiative. Arch Intern Med. 2006;166:357-365.
27. Hendrix SL, Wassertheil-Smoller S, Johnson KC, et al. Effects of conjugated equine estrogen on stroke in the Women’s Health Initiative. Circulation. 2006;113:2425-2434.
28. Grodstein F, Manson JE, Stampfer MJ. Hormone therapy and coronary heart disease: the role of time since menopause and age at hormone initiation. J Womens Health (Larchmt). 2006;15:35-44.
29. Hernán MA, Alonso A, Logan R, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008;19:766-779.
30. Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465-1477.
31. Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591-2602.
32. Vickers MR, Maclennan AH, Lawton B, et al. Main morbidities recorded in the women’s international study of long duration oestrogen after menopause (WISDOM): a randomised controlled trial of hormone replacement therapy in postmenopausal women. BMJ. 2007;335:239.-
33. Mares P, Chevallier T, Micheletti MC, et al. Coronary heart disease and HRT in France: MISSION study prospective phase results. Gynecol Endocrinol. 2008;24:696-700.
34. Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med. 2006;21:363-366.
35. Brownley KA, Hinderliter AL, West SG, et al. Cardiovascular effects of 6 months of hormone replacement therapy versus placebo: differences associated with years since menopause. Am J Obstet Gynecol. 2004;190:1052-1058.
36. Tannen RL, Weiner MG, Xie D, et al. Estrogen affects postmenopausal women differently than estrogen plus progestin replacement therapy. Hum Reprod. 2007;22:1769-1777.
37. Curb JD, Prentice RL, Bray PF, et al. Venous thrombosis and conjugated equine estrogen in women without a uterus. Arch Intern Med. 2006;166:772-780.
38. Cushman M, Kuller LH, Prentice R, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292:1573-1580.
39. McLaren J, Barnhart K. Hormone therapy and venous thromboembolism in menopausal women. Menopausal Med. 2008;16(4):S1-S7.
40. Grady D, Herrington D, Bittner V, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA. 2002;288:49-57.
41. Canonico M, Oger E, Plu-Bureau G, et al. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation. 2007;115:840-845.
42. Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
43. Zhang SM, Manson JE, Rexrode KM, et al. Use of oral conjugated estrogen alone and risk of breast cancer. Am J Epidemiol. 2007;165:524-529.
44. Dupont WD, Page DL. Menopausal estrogen replacement therapy and breast cancer. Arch Intern Med. 1991;151:67-72.
45. Stefanick ML, Anderson GL, Margolis KL, et al. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA. 2006;295:1647-1657.
46. Ross RK, Paganini-Hill A, Wan PC, et al. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Anderson GL, Chlebowski RT, Rossouw JE, et al. Prior hormone therapy and breast cancer risk in the Women’s Health Initiative randomized trial of estrogen plus progestin. Maturitas. 2006;55:103-115.
48. Goodman N, Goldzieher J, Ayala C. Critique of the report from the Writing Group of the WHI. Menopausal Med. 2003;10(4):1-4.
49. Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356:1670-1674.
50. Chlebowski RT, Anderson GL, Gass M, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. 2010;304:1684-1692.
51. Collins JA, Blake JM, Crosignani PG. Breast cancer risk with postmenopausal hormonal treatment. Hum Reprod Update. 2005;11:545-560.
52. Singletary SE. Rating the risk factors for breast cancer. Ann Surg. 2003;237:474-482.
53. Helenius IM, Korenstein D, Halm EA. Changing use of hormone therapy among minority women since the Women’s Health Initiative. Menopause. 2007;14:216-222.
54. Theroux R, Taylor K. Women’s decision making about the use of hormonal and nonhormonal remedies for the menopausal transition. J Obstet Gynecol Neonatal Nurs. 2003;32:712-723.
55. Col NF, Pauker SG, Goldberg RJ, et al. Individualizing therapy to prevent long-term consequences of estrogen deficiency in postmenopausal women. Arch Intern Med. 1999;159:1458-1466.
56. Barnabei VM, Cochrane BB, Aragaki AK, et al. Menopausal symptoms and treatment-related effects of estrogen and progestin in the Women’s Health Initiative. Obstet Gynecol. 2005;105:1063-1073.
57. Warren MP. A comparative review of the risks and benefits of hormone replacement therapy regimens. Am J Obstet Gynecol. 2004;190:1141-1167.
58. Gail MH, Costantino JP, Bryant J, et al. Weighing the risks and benefits of tamoxifen treatment for preventing breast cancer. J Natl Cancer Inst. 1999;91:1829-1846.
A view from retirement
Being in practice for 42 years was like running a marathon. Things seem easy and pleasant at first, but then as time goes by, you hit the “wall” and you feel like you can’t go on. “It’s just too hard,” you think. And you wonder: “What am I doing here?”
In an actual marathon, you hit that wall somewhere around the 20-mile mark. (At least that’s what my son tells me.) But in my family medicine practice, I hit the wall at the 10-year mark.
If, like me, you decide not to quit, the endorphins kick in. You feel a high and know you could go on like this forever. You wonder to yourself: “Can life really be this good?”
And then, as the years pass by, you and your patients change and you know the race is coming to an end. It’s time to stop running. Yet, there are many losses in giving up practice. After spending nearly a lifetime as a doctor, it’s hard to give up that identity. That’s who you are, and who you have been.
In my case, I saw the doctor-patient relationship as a “covenant, not a contract,” as Gayle Stephens, MD, described it, and my role as a physician was to prescribe myself as my most potent therapy, as taught by Michael Balint.1
David Loxterkamp has written about “being there” as the prime service of the family doctor.2 But in retiring you are not there—at least not the way you once were.
How about lunch, doc?
When I retired 5 years ago, many patients wanted to “go out to lunch” or in some way maintain our relationship. I avoided this, saying that I thought it was important for them to develop a relationship with their new doctor. This was (and is) true, but I’ve come to realize that it is not the most important reason to pass on such invitations.
Lovers breaking up say they can “still be friends,” even though they know that is impossible. They can neither give up the special feelings they have had, nor the memories of those feelings that will always be a relevant part of their lives. Similarly, I have too much invested in these relationships to “just be friends.”
Moving on
I have moved on. My wife of 52 years and I travel and visit our children and grandchildren. I take and teach classes at a program for retired people. I have more free time than I have ever had, and I don’t miss the constant sense of responsibility for others, or the time spent agonizing over mistakes. But it was the right time to leave practice when technological advancements were accelerating at lightning speed, and my energy level was no longer keeping pace.
Mixed emotions when I talk to patients
Despite not wanting to have lunch with my former patients, I must confess that I periodically call some of them to see how they are doing. I realize that it is really more for me than for them—but I try not to make that obvious. Our conversations leave me with such mixed emotions.
Feeling guilty
Bob and his family were patients of mine almost from the day I started. I attended their daughters’ weddings, shared in their tragedies, cared for multiple illnesses, and counseled the children. When Bob was diagnosed with Alzheimer’s disease, I told him it was very early and we would go through it together and learn from each other. Then I retired.
I know through my conversations with him and his family that he has gone on with good care. But he has gone on without me.
I feel guilty.
I realize that some of this is ego—a loss of importance. But mainly I feel badly that I am not fulfilling that promise I made to him. And I have “cheated” myself out of the pleasure of learning and giving.
Feeling incomplete
I was particularly close with Marylou and her family. I attended birthday parties, cared for her and her husband’s chronic illnesses, supported them through the illness and death of their daughter, and listened when that’s all I could do. Last year, Marylou called me when she was diagnosed with breast cancer. I stayed in touch and expressed my pleasure when she did well. But, I wasn’t involved in the therapy decisions and I wasn’t there when it was time to cry or talk to the family.
It made me feel incomplete.
Feeling humbled
Recently I got a letter from a urologist regarding a former patient of mine, Robin.
Robin was diagnosed with prostate cancer about 10 years ago, when I was still his physician. Obviously, the new urologist didn’t know that I had retired. So I forwarded the note to Robin’s new family physician and called Robin to see how he was.
I still felt a tremendous sense of responsibility for Robin’s diagnosis. I had never screened him for prostate cancer. But as he reminded me at the time of his diagnosis, he and I had discussed screening. It’s just that Robin, who knows much about medicine and was always involved in his own decisions, had chosen not to pursue it.
Now 10 years later, Robin and I were catching up. As we talked, Robin revealed that he had multiple complications requiring permanent catheters and that he’d had to give up work.
“I wish you were still in practice,” he said to me. “I miss our talks.”
With that, I felt humbled.
Talking to Robin got me thinking. As doctors, we spend so much time worrying about doing the right thing and giving the right advice that we sometimes forget that we need to have confidence in our patients and their ability to make their own decisions. We need to know when to let go.
“Being there”
Jane was another person who emerged from my professional past. I had known her for years. Not only was she my patient, but I saw her when she came in with her father, sister, and mother for their appointments. Together, we had cared for her family members through their illnesses and deaths.
One day after my retirement, she called to get some advice for a problem she was having with her stepson. I listened, gave some suggestions about whom to see, and offered to stay in touch. She thanked me, saying she didn’t know who else to call.
I hung up thinking how hard it is to “be there” when you are not there.
Jane’s call reminded me of a lesson I’d given years ago to a class of first-year medical students. I had brought in a patient of mine and together, in front of the class, we discussed the doctor-patient relationship.
I asked my patient what was most important about our relationship. She said that when she was diagnosed with diabetes, I gave her my private home phone number.
I responded, “Mrs. E, in our 15 years together, how many times have you used that number?”
“None,” was her reply.
Med students, take note
I really don’t know if my retirement has been easier for my patients than for me. I certainly hope so. Part of my job was to encourage their independence and self-sufficiency. My emotional dependence on them is my problem and I suspect one that is not that uncommon among doctors. I am still teaching and doing some research. Some of my retired friends still go to grand rounds and travel to medical meetings, even though they don’t see any patients.
I have few regrets in retiring from my practice. It was the right thing to do at the right time. Do I miss it every day? Yes, but I also feel so lucky to have worked as a family physician for 42 years.
I once heard a British family physician define the family doctor as someone you can go to and talk to about anything you want. To me, the family doctor is someone who knows you—really knows you—in a way that no one else does. A family doctor is someone who can cry with a patient about a loss, not because the physician can appreciate the loss, but because the patient’s loss is the physician’s loss, too.
I wish more young medical students understood the depth of the connections we make as family physicians, and just how rewarding the work can be. If they did, there would certainly be more students choosing a career in family medicine.
Acknowledgement
I thank Nancy W. Merenstein, the first reader of everything I write and my constant supporter; Paula Preisach, manager and organizer of my academic career; and Jonathan Han, MD, David Loxterkamp, MD, Jennifer Middleton, MD, MPH, and Allen Shaughnessy, PharmD, for helpful comments and suggestions on earlier drafts of this paper.
CORRESPONDENCE
Joel H. Merenstein, MD, UPMC St. Margaret, 3937 Butler Street, Pittsburgh, PA 15201; [email protected]

Joel H. Merenstein, MD
Department of Family Medicine, University of Pittsburgh, Pittsburgh, Pa
[email protected]
Joel H. Merenstein, MD
Department of Family Medicine, University of Pittsburgh, Pittsburgh, Pa
[email protected]
Joel H. Merenstein, MD
Department of Family Medicine, University of Pittsburgh, Pittsburgh, Pa
[email protected]
Being in practice for 42 years was like running a marathon. Things seem easy and pleasant at first, but then as time goes by, you hit the “wall” and you feel like you can’t go on. “It’s just too hard,” you think. And you wonder: “What am I doing here?”
In an actual marathon, you hit that wall somewhere around the 20-mile mark. (At least that’s what my son tells me.) But in my family medicine practice, I hit the wall at the 10-year mark.
If, like me, you decide not to quit, the endorphins kick in. You feel a high and know you could go on like this forever. You wonder to yourself: “Can life really be this good?”
And then, as the years pass by, you and your patients change and you know the race is coming to an end. It’s time to stop running. Yet, there are many losses in giving up practice. After spending nearly a lifetime as a doctor, it’s hard to give up that identity. That’s who you are, and who you have been.
In my case, I saw the doctor-patient relationship as a “covenant, not a contract,” as Gayle Stephens, MD, described it, and my role as a physician was to prescribe myself as my most potent therapy, as taught by Michael Balint.1
David Loxterkamp has written about “being there” as the prime service of the family doctor.2 But in retiring you are not there—at least not the way you once were.
How about lunch, doc?
When I retired 5 years ago, many patients wanted to “go out to lunch” or in some way maintain our relationship. I avoided this, saying that I thought it was important for them to develop a relationship with their new doctor. This was (and is) true, but I’ve come to realize that it is not the most important reason to pass on such invitations.
Lovers breaking up say they can “still be friends,” even though they know that is impossible. They can neither give up the special feelings they have had, nor the memories of those feelings that will always be a relevant part of their lives. Similarly, I have too much invested in these relationships to “just be friends.”
Moving on
I have moved on. My wife of 52 years and I travel and visit our children and grandchildren. I take and teach classes at a program for retired people. I have more free time than I have ever had, and I don’t miss the constant sense of responsibility for others, or the time spent agonizing over mistakes. But it was the right time to leave practice when technological advancements were accelerating at lightning speed, and my energy level was no longer keeping pace.
Mixed emotions when I talk to patients
Despite not wanting to have lunch with my former patients, I must confess that I periodically call some of them to see how they are doing. I realize that it is really more for me than for them—but I try not to make that obvious. Our conversations leave me with such mixed emotions.
Feeling guilty
Bob and his family were patients of mine almost from the day I started. I attended their daughters’ weddings, shared in their tragedies, cared for multiple illnesses, and counseled the children. When Bob was diagnosed with Alzheimer’s disease, I told him it was very early and we would go through it together and learn from each other. Then I retired.
I know through my conversations with him and his family that he has gone on with good care. But he has gone on without me.
I feel guilty.
I realize that some of this is ego—a loss of importance. But mainly I feel badly that I am not fulfilling that promise I made to him. And I have “cheated” myself out of the pleasure of learning and giving.
Feeling incomplete
I was particularly close with Marylou and her family. I attended birthday parties, cared for her and her husband’s chronic illnesses, supported them through the illness and death of their daughter, and listened when that’s all I could do. Last year, Marylou called me when she was diagnosed with breast cancer. I stayed in touch and expressed my pleasure when she did well. But, I wasn’t involved in the therapy decisions and I wasn’t there when it was time to cry or talk to the family.
It made me feel incomplete.
Feeling humbled
Recently I got a letter from a urologist regarding a former patient of mine, Robin.
Robin was diagnosed with prostate cancer about 10 years ago, when I was still his physician. Obviously, the new urologist didn’t know that I had retired. So I forwarded the note to Robin’s new family physician and called Robin to see how he was.
I still felt a tremendous sense of responsibility for Robin’s diagnosis. I had never screened him for prostate cancer. But as he reminded me at the time of his diagnosis, he and I had discussed screening. It’s just that Robin, who knows much about medicine and was always involved in his own decisions, had chosen not to pursue it.
Now 10 years later, Robin and I were catching up. As we talked, Robin revealed that he had multiple complications requiring permanent catheters and that he’d had to give up work.
“I wish you were still in practice,” he said to me. “I miss our talks.”
With that, I felt humbled.
Talking to Robin got me thinking. As doctors, we spend so much time worrying about doing the right thing and giving the right advice that we sometimes forget that we need to have confidence in our patients and their ability to make their own decisions. We need to know when to let go.
“Being there”
Jane was another person who emerged from my professional past. I had known her for years. Not only was she my patient, but I saw her when she came in with her father, sister, and mother for their appointments. Together, we had cared for her family members through their illnesses and deaths.
One day after my retirement, she called to get some advice for a problem she was having with her stepson. I listened, gave some suggestions about whom to see, and offered to stay in touch. She thanked me, saying she didn’t know who else to call.
I hung up thinking how hard it is to “be there” when you are not there.
Jane’s call reminded me of a lesson I’d given years ago to a class of first-year medical students. I had brought in a patient of mine and together, in front of the class, we discussed the doctor-patient relationship.
I asked my patient what was most important about our relationship. She said that when she was diagnosed with diabetes, I gave her my private home phone number.
I responded, “Mrs. E, in our 15 years together, how many times have you used that number?”
“None,” was her reply.
Med students, take note
I really don’t know if my retirement has been easier for my patients than for me. I certainly hope so. Part of my job was to encourage their independence and self-sufficiency. My emotional dependence on them is my problem and I suspect one that is not that uncommon among doctors. I am still teaching and doing some research. Some of my retired friends still go to grand rounds and travel to medical meetings, even though they don’t see any patients.
I have few regrets in retiring from my practice. It was the right thing to do at the right time. Do I miss it every day? Yes, but I also feel so lucky to have worked as a family physician for 42 years.
I once heard a British family physician define the family doctor as someone you can go to and talk to about anything you want. To me, the family doctor is someone who knows you—really knows you—in a way that no one else does. A family doctor is someone who can cry with a patient about a loss, not because the physician can appreciate the loss, but because the patient’s loss is the physician’s loss, too.
I wish more young medical students understood the depth of the connections we make as family physicians, and just how rewarding the work can be. If they did, there would certainly be more students choosing a career in family medicine.
Acknowledgement
I thank Nancy W. Merenstein, the first reader of everything I write and my constant supporter; Paula Preisach, manager and organizer of my academic career; and Jonathan Han, MD, David Loxterkamp, MD, Jennifer Middleton, MD, MPH, and Allen Shaughnessy, PharmD, for helpful comments and suggestions on earlier drafts of this paper.
CORRESPONDENCE
Joel H. Merenstein, MD, UPMC St. Margaret, 3937 Butler Street, Pittsburgh, PA 15201; [email protected]
Being in practice for 42 years was like running a marathon. Things seem easy and pleasant at first, but then as time goes by, you hit the “wall” and you feel like you can’t go on. “It’s just too hard,” you think. And you wonder: “What am I doing here?”
In an actual marathon, you hit that wall somewhere around the 20-mile mark. (At least that’s what my son tells me.) But in my family medicine practice, I hit the wall at the 10-year mark.
If, like me, you decide not to quit, the endorphins kick in. You feel a high and know you could go on like this forever. You wonder to yourself: “Can life really be this good?”
And then, as the years pass by, you and your patients change and you know the race is coming to an end. It’s time to stop running. Yet, there are many losses in giving up practice. After spending nearly a lifetime as a doctor, it’s hard to give up that identity. That’s who you are, and who you have been.
In my case, I saw the doctor-patient relationship as a “covenant, not a contract,” as Gayle Stephens, MD, described it, and my role as a physician was to prescribe myself as my most potent therapy, as taught by Michael Balint.1
David Loxterkamp has written about “being there” as the prime service of the family doctor.2 But in retiring you are not there—at least not the way you once were.
How about lunch, doc?
When I retired 5 years ago, many patients wanted to “go out to lunch” or in some way maintain our relationship. I avoided this, saying that I thought it was important for them to develop a relationship with their new doctor. This was (and is) true, but I’ve come to realize that it is not the most important reason to pass on such invitations.
Lovers breaking up say they can “still be friends,” even though they know that is impossible. They can neither give up the special feelings they have had, nor the memories of those feelings that will always be a relevant part of their lives. Similarly, I have too much invested in these relationships to “just be friends.”
Moving on
I have moved on. My wife of 52 years and I travel and visit our children and grandchildren. I take and teach classes at a program for retired people. I have more free time than I have ever had, and I don’t miss the constant sense of responsibility for others, or the time spent agonizing over mistakes. But it was the right time to leave practice when technological advancements were accelerating at lightning speed, and my energy level was no longer keeping pace.
Mixed emotions when I talk to patients
Despite not wanting to have lunch with my former patients, I must confess that I periodically call some of them to see how they are doing. I realize that it is really more for me than for them—but I try not to make that obvious. Our conversations leave me with such mixed emotions.
Feeling guilty
Bob and his family were patients of mine almost from the day I started. I attended their daughters’ weddings, shared in their tragedies, cared for multiple illnesses, and counseled the children. When Bob was diagnosed with Alzheimer’s disease, I told him it was very early and we would go through it together and learn from each other. Then I retired.
I know through my conversations with him and his family that he has gone on with good care. But he has gone on without me.
I feel guilty.
I realize that some of this is ego—a loss of importance. But mainly I feel badly that I am not fulfilling that promise I made to him. And I have “cheated” myself out of the pleasure of learning and giving.
Feeling incomplete
I was particularly close with Marylou and her family. I attended birthday parties, cared for her and her husband’s chronic illnesses, supported them through the illness and death of their daughter, and listened when that’s all I could do. Last year, Marylou called me when she was diagnosed with breast cancer. I stayed in touch and expressed my pleasure when she did well. But, I wasn’t involved in the therapy decisions and I wasn’t there when it was time to cry or talk to the family.
It made me feel incomplete.
Feeling humbled
Recently I got a letter from a urologist regarding a former patient of mine, Robin.
Robin was diagnosed with prostate cancer about 10 years ago, when I was still his physician. Obviously, the new urologist didn’t know that I had retired. So I forwarded the note to Robin’s new family physician and called Robin to see how he was.
I still felt a tremendous sense of responsibility for Robin’s diagnosis. I had never screened him for prostate cancer. But as he reminded me at the time of his diagnosis, he and I had discussed screening. It’s just that Robin, who knows much about medicine and was always involved in his own decisions, had chosen not to pursue it.
Now 10 years later, Robin and I were catching up. As we talked, Robin revealed that he had multiple complications requiring permanent catheters and that he’d had to give up work.
“I wish you were still in practice,” he said to me. “I miss our talks.”
With that, I felt humbled.
Talking to Robin got me thinking. As doctors, we spend so much time worrying about doing the right thing and giving the right advice that we sometimes forget that we need to have confidence in our patients and their ability to make their own decisions. We need to know when to let go.
“Being there”
Jane was another person who emerged from my professional past. I had known her for years. Not only was she my patient, but I saw her when she came in with her father, sister, and mother for their appointments. Together, we had cared for her family members through their illnesses and deaths.
One day after my retirement, she called to get some advice for a problem she was having with her stepson. I listened, gave some suggestions about whom to see, and offered to stay in touch. She thanked me, saying she didn’t know who else to call.
I hung up thinking how hard it is to “be there” when you are not there.
Jane’s call reminded me of a lesson I’d given years ago to a class of first-year medical students. I had brought in a patient of mine and together, in front of the class, we discussed the doctor-patient relationship.
I asked my patient what was most important about our relationship. She said that when she was diagnosed with diabetes, I gave her my private home phone number.
I responded, “Mrs. E, in our 15 years together, how many times have you used that number?”
“None,” was her reply.
Med students, take note
I really don’t know if my retirement has been easier for my patients than for me. I certainly hope so. Part of my job was to encourage their independence and self-sufficiency. My emotional dependence on them is my problem and I suspect one that is not that uncommon among doctors. I am still teaching and doing some research. Some of my retired friends still go to grand rounds and travel to medical meetings, even though they don’t see any patients.
I have few regrets in retiring from my practice. It was the right thing to do at the right time. Do I miss it every day? Yes, but I also feel so lucky to have worked as a family physician for 42 years.
I once heard a British family physician define the family doctor as someone you can go to and talk to about anything you want. To me, the family doctor is someone who knows you—really knows you—in a way that no one else does. A family doctor is someone who can cry with a patient about a loss, not because the physician can appreciate the loss, but because the patient’s loss is the physician’s loss, too.
I wish more young medical students understood the depth of the connections we make as family physicians, and just how rewarding the work can be. If they did, there would certainly be more students choosing a career in family medicine.
Acknowledgement
I thank Nancy W. Merenstein, the first reader of everything I write and my constant supporter; Paula Preisach, manager and organizer of my academic career; and Jonathan Han, MD, David Loxterkamp, MD, Jennifer Middleton, MD, MPH, and Allen Shaughnessy, PharmD, for helpful comments and suggestions on earlier drafts of this paper.
CORRESPONDENCE
Joel H. Merenstein, MD, UPMC St. Margaret, 3937 Butler Street, Pittsburgh, PA 15201; [email protected]
When to suspect bipolar disorder
• Before prescribing antidepressant therapy to depressed patients, screen for bipolar illness, either by taking a detailed medical and family history or by administering the Mood Disorder Questionnaire. A
• Be alert to medical and psychiatric comorbidities in patients with bipolar illness, particularly anxiety disorders and substance abuse. A
• Prescribe a mood stabilizer for acutely depressed patients with bipolar disorder; if the depression does not resolve, add an agent with relapse prevention properties. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
As a family physician, you are better positioned than you might think to make a difference in the lives of patients with bipolar illness. Not only are you likely to be involved in monitoring such patients, but you may frequently be the first clinician patients with bipolar symptoms seek help from.
All it takes to provide that help is a heightened awareness of bipolar disorder, the ways in which bipolar patients present, and the signs and symptoms to look for. Yet evidence suggests that many physicians do not have adequate knowledge of this chronic and debilitating condition. While close to one-third of patients with bipolar disorder seek medical care within a year of the onset of bipolar symptoms, nearly 70% do not receive an accurate diagnosis until they have seen an average of 4 physicians.1 Misdiagnosis—both underdetection1 and over-inclusion2—often results in improper treatment. And even when the diagnosis is correct, patients with bipolar disease often receive inadequate or harmful treatment.3
Ongoing care for bipolar illness is best provided in collaboration with a psychiatrist. With the disorder affecting about 3% to 5% of the US population,4 family physicians will inevitably play a key role in diagnosis and treatment. The text, tables, and screening tool that follow will help with both.
Bipolar diagnosis hinges on this characteristic
The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) defines 4 types of bipolar illness: bipolar I, bipolar II, cyclothymia (the most mild form), and not otherwise specified (TABLE 1).5 The key feature of all 4 types—and the distinguishing characteristic that diagnosis typically hinges on—is a manic or hypomanic episode (TABLE 2).5
Although a full-blown manic episode may not be hard to identify, hypomania is easily missed. By definition, hypomania—with its heightened sense of well-being and productivity—is not problematic and is rarely a patient’s primary complaint.
Mixed mania, a feature of bipolar I, is the worst of both worlds: It is a state in which a full manic episode is superimposed on a full depressive episode—a depression with all the energy and force of a mania. Patients who have experienced one episode of mixed mania have a 12-fold increase in mixed states, 6.5 times more depression, and 1.7 times more dysthymia than those who experience manic episodes without the overlay of depression.6
TABLE 1
Types of bipolar disease: DSM-IV diagnoses5
| Bipolar I | Bipolar II | Cyclothymia | Bipolar disorder not otherwise specified |
|---|---|---|---|
| ≥1 manic or mixed lifetime episode, frequently accompanied by major depressive episodes | ≥1 major depressive episode, accompanied by ≥1 lifetime hypomanic episode | ≥2 years of numerous periods of hypomanic and subsyndromal depressive symptoms | Symptoms resemble, but do not meet, criteria for any specific bipolar disorder |
TABLE 2
Mania and hypomania: DSM-IV criteria5
To identify an episode of mania or hypomania, all of the following criteria must be met. Of note: Hypomania has the same criteria as mania, with 2 notable exceptions: (1) the minimum duration of hypomania is 4 days, rather than 7;* and (2) hypomania is not significantly problematic.
|
| *There is no maximal duration, but the average manic episode lasts 1 to 2 months. |
Complicating matters: Numerous comorbidities
Bipolar illness predisposes patients to multiple medical and psychiatric comorbidities. Cardiovascular, cerebrovascular, and metabolic disorders and sleep disturbances are common in those with bipolar disorder.7 So is obesity, which affects about 50% of patients with bipolar disease.8
Bipolar patients also suffer from an extremely high rate of comorbid psychiatric conditions. Overall, 93% of those with bipolar I also have an anxiety disorder, 71% suffer from drug or alcohol dependence, and 50% suffer from dysthymia, according to the National Comorbidity Survey.9 In addition, about two-thirds of bipolar patients suffer from various personality disorders10—a comorbidity that is particularly disturbing because it is associated with a chronically dysfunctional pattern of problem-solving.
Even without comorbidities, the impact of bipolar disorder is significant: In a study of 1468 patients with bipolar disorder, complaints of difficulties with work, leisure activities, and family and social interactions were common.11 Women were more likely to cite disruption of social and family life, while men often reported having been convicted of crimes. Younger patients reported a greater number of symptomatic days compared with their older counterparts.11
Suicide risk. Patients with bipolar disorder also face an increased risk of suicide, particularly in the depressive phase of the illness. Among 12,662 Oregon Medicaid patients diagnosed with, and treated for, bipolar disorder between 1998 and 2003, there were 11 deaths by suicide and 79 significant suicide attempts.12
Suspect bipolar disorder?
One of the best ways by which family physicians can speed up and improve the accuracy of bipolar diagnosis is to utilize the Mood Disorder Questionnaire (MDQ) (TABLE 3).13 Patients with any mood complaint are its target population. Within that group, the MDQ has been found to have excellent specificity (0.90) and acceptable sensitivity (0.73).13 (For more on identifying patients with bipolar disease, see “A blood test for bipolar disorder?”.)
TABLE 3
The Mood Disorder Questionnaire bipolar screening tool
| Please answer each question to the best of your ability. | ||
| 1. Has there ever been a period of time when you were not your usual self and … | ||
| YES | NO | |
| you felt so good or so hyper that other people thought you were not your normal self or you were so hyper that you got into trouble? | ||
| you were so irritable that you shouted at people or started fights or arguments? | ||
| you felt much more self-confident than usual? | ||
| you got much less sleep than usual and found you didn’t really miss it? | ||
| you were much more talkative or spoke much faster than usual? | ||
| thoughts raced through your head or you couldn’t slow your mind down? | ||
| you were so easily distracted by things around you that you had trouble concentrating or staying on track? | ||
| you had much more energy than usual? | ||
| you were much more active or did many more things than usual? | ||
| you were much more social or outgoing than usual; for example, you telephoned friends in the middle of the night? | ||
| you were much more interested in sex than usual? | ||
| you did things that were unusual for you or that other people might have thought were excessive, foolish, or risky? | ||
| spending money got you or your family into trouble? | ||
| 2. If you checked YES to more than one of the above, have several of these ever happened during the same period of time? | ||
3. How much of a problem did any of these cause you—like being unable to work; having family, money, or legal troubles; getting into arguments or fights? Please circle one response only.
| ||
| Have any of your blood relatives (ie, children, siblings, parents, grandparents, aunts, uncles) had manic-depressive illness or bipolar disorder? | ||
| For a positive screen, 7 of the 13 items in No. 1 must be Yes, No. 2 must be Yes, and No. 3 must be moderate or serious. Source: Hirschfeld RM, et al. Am J Psychiatry. 2000.13 Reprinted with permission. | ||
When to administer the MDQ
Because patients with bipolar disease are more likely to seek help when they are suffering from a depressive episode, it is important to maintain a high index of suspicion. Before ruling out bipolar disease, take a complete medical history, inquiring about comorbidities, family history, and whether the patient can recall any episodes of agitation, intense irritation, or other manifestations of mania or hypomania (TABLE 2).5 If, based on the history, you continue to suspect bipolar disorder, administer the MDQ.
If the patient has a positive screen, your next step would be to initiate treatment for bipolar disorder, even if depression is the presenting symptom. A referral to a psychiatrist would be indicated, as well.
Complexities of bipolar treatment
In recent years, numerous agents have been approved by the US Food and Drug Administration (FDA) for the treatment of bipolar illness in general, and for acute mania in particular. Nearly all of the second-generation, or atypical, antipsychotics have been approved for use in acute mania.14 (Mixed states should be treated the same as mania.) Most of these agents have also been found to be useful as maintenance medications, to prevent relapse (TABLE 4).
Considerable evidence suggests that lithium significantly reduces the risk of relapse, particularly in classic euphoric mania. Other agents that are approved for maintenance therapy include aripiprazole, lamotrigine, and olanzapine as monotherapy, and olanzapine, quetiapine, and ziprasidone as addon agents to lithium or divalproex. Combining a mood stabilizer and an antipsychotic agent generally leads to better outcomes, both in acute mania15 and relapse prevention,16 compared with a mood stabilizer alone. However, bipolar depression, which is more common than either mania17 or hypomania,18 is the major clinical challenge.
TABLE 4
FDA-approved treatments for bipolar disorder32,33
| Treatment | Mania | Bipolar depression | Maintenance |
|---|---|---|---|
| Aripiprazole | √ | √ | |
| Asenapine | √ | ||
| Carbamazepine ER | √ | ||
| Chlorpromazine | √ | ||
| Divalproex; divalproex ER | √ | ||
| Lamotrigine | √ | ||
| Lithium | √* | √ | |
| Olanzapine | à | à | à |
| Quetiapine; quetiapine XR | √ | √ | √ |
| Risperidone | √ | ||
| Ziprasidone | √ | √ | |
| ER, extended release; FDA, US Food and Drug Administration; XR, extended release. *Not approved for mixed mania. † Approved for bipolar depression in combination with fluoxetine. | |||
What’s best for bipolar depression?
For the acutely depressed bipolar patient, optimizing mood stabilization therapy is typically the first step. If the depression doesn’t resolve in 4 or 5 weeks, adding an agent with relapse prevention properties is a preferred approach. Antidepressants may be harmful to patients with bipolar disorder (possibly triggering manic episodes, rapid cycling, or a chronic dysphoric state),3,19 and are usually tried only after other options have been exhausted.
Quetiapine, which is effective for the treatment of mania at doses around 600 mg daily, appears to also be effective for the treatment of acute bipolar depression at doses around 300 mg/d.20 In 2-year studies in which quetiapine was added to either lithium or divalproex, the 2-drug combination was found to reduce the risk for relapse approximately 3-fold compared with the mood stabilizer alone.16
Olanzapine/fluoxetine. Besides quetiapine, this drug combination is the only other agent with FDA approval for the treatment of bipolar depression. A 24-week open extension trial found that the risk for a manic episode due to the coadministration of fluoxetine was low, but 27% of those studied relapsed into depression.21
Drugs that do not have FDA approval specifically for bipolar depression may also be used to treat it.
Lithium, which has antidepressant activity, particularly at levels exceeding 0.8 mEq/L, is one such drug. In addition to its effectiveness in treating bipolar depression, lithium appears to have an antisuicide effect.12
In a recent study of patients with bipolar disorder, lithium was found to be more protective than other mood stabilizers. The hazard ratio (HR) for suicide attempts was significantly greater for patients taking divalproex (HR=2.7; P<.001) or carbamazepine (HR=2.8; P<.001) compared with patients taking lithium.12
Modafinil, a nonstimulant used to increase alertness in patients with daytime sleepiness due to a variety of conditions, has been tested as an adjunctive agent in depressed bipolar patients. In a blinded study, patients were randomly assigned to have modafinil (n=41) or placebo (n=44) added to their existing treatment regimen.22 Response, defined as ≥50% improvement in mood, occurred at twice the rate in those treated with modafinil (44%) compared with those on placebo (23%; P<.05).22 In the brief (6 week) study, modafinil did not appear to cause an increase in manic or hypomanic episodes.22
Pramipexole, a dopamine agonist used for early-stage Parkinson’s disease, has been tested in patients with bipolar depression in 2 small, short-term placebo-controlled trials. A total of 15 patients with type I disease and 28 patients with type II disease were studied for a 6-week period. The results: 60% to 67% of patients taking pramipexole responded, vs 9% to 20% of those on placebo.23,24
Electroconvulsive therapy (ECT) is an underutilized treatment that is effective for depressed patients who are resistant to pharmacological treatment. In fact, bipolar depression may improve more rapidly than unipolar depression with ultra-brief pulse treatment—a therapy in which the pulse width of the electrical stimulus is much briefer (<0.5 msec) than that of standard ECT.25 ECT has also been shown to be effective for mixed states, in which depression and mania coexist.26 The cognitive adverse effects associated with ECT can be reduced while maintaining the same efficacy by using bifrontal (instead of the typical bitemporal) electrode replacement.27
Although the DSM-IV identifies bipolar disorder on the basis of symptoms, there have been increasing attempts to diagnose the disease biologically. Most have been unsuccessful. However, an initial study found that a recently developed blood test (PsychNostics LLC, Baltimore, Md; http://psychnostics.com) that uses the membrane potential as a biological marker had a specificity of 0.88 and a sensitivity of 0.78. The blood test is a promising approach, but is still not ready for prime time.31
Predicting the course of disease, preventing relapse
Generally, the polarity of the current episode predicts that of future episodes. Studies have found that, independent of whether patients were on effective mood-stabilizing agents or placebo, those who relapsed had an episode like their most recent one by a ratio of more than 2 to 1.28
Research suggests a link between age at onset of illness and cycling time and response to particular agents. In 1 trial, those with early-onset bipolar disorder (in adolescence) had briefer euthymic periods and responded better to carbamazepine compared with those who developed symptoms of bipolar disorder at a later age.29 Late-onset bipolar illness (in the 30s or older) was characterized by longer euthymic periods and manic episodes that responded well to lithium.29 The average age of onset is about 25 years.30
Regardless of patient age or age of onset of symptoms, however, prevention of relapse is the goal of ongoing treatment. You can help by assessing the patient’s mood, reviewing the medication regimen and level of compliance, and offering support at every visit—and by consulting with the patient’s psychiatrist, as needed.
CORRESPONDENCE
Rif S. El-Mallakh, MD, Mood Disorders Research Program, Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, MedCenter One, 501 East Broadway, Suite 340, Louisville, KY 40202; [email protected]
1. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64:161-174.
2. Stewart C, El-Mallakh RS. Is bipolar disorder over diagnosed among patients with substance abuse? Bipolar Disord. 2007;9:646-648.
3. El-Mallakh RS, Karippot A. Chronic depression in bipolar disorder. Am J Psychiatry. 2006;163:1137-1341.
4. Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression, 2nd ed. Oxford University Press: New York; 2007.
5. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed, text rev (DSM-IV-TR). American Psychiatric Association: Arlington, VA; 2000.
6. Baldessarini RJ, Salvatore P, Khalsa HM, et al. Dissimilar morbidity following initial mania versus mixed-states in type-I bipolar disorder. J Affect Disord. 2010;126:299-302.
7. Kilbourne AM, Cornelius JR, Hans X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord. 2004;6:368-373.
8. Shah A, Shen N, El-Mallakh RS. Weight gain occurs after onset of bipolar illness in overweight bipolar patients. Ann Clin Psychiatry. 2006;18:239-241.
9. Kessler RC, Nelson CB, McGonagle KA, et al. The epidemiology of co-occurring addictive and mental disorders: implications for prevention and service utilization. Am J Orthopsychiatry. 1996;66:17-31.
10. Mantere O, Melartin TK, Suominen K, et al. Differences in Axis I and II comorbidity between bipolar I and II disorders and major depressive disorder. J Clin Psychiatry. 2006;67:584-593.
11. Calabrese JR, Hirschfeld RM, Reed M, et al. Impact of bipolar disorder on a US community sample. J Clin Psychiatry. 2003;64:425-432.
12. Collins JC, McFarland BH. Divalproex, lithium and suicide among Medicaid patients with bipolar disorder. J Affect Disord. 2008;107:23-28.
13. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157:1873-1875.
14. Surja AAS, Tamas RL, El-Mallakh RS. Antipsychotic medications in the treatment of bipolar disorder. Curr Drug Targets. 2006;7:1217-1224.
15. Scherk H, Pajonk FG, Leucht S. Second-generation antipsychotic agents in the treatment of acute mania: a systematic review and meta-analysis of randomized controlled trials. Arch Gen Psychiatry. 2007;64:442-455.
16. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166:476-488.
17. Judd LL, Akiskal HS, Schettler PJ. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59:530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60:261-269.
19. El-Mallakh RS, Karippot A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
20. Suppes T, Datto C, Minkwitz M, et al. Effectiveness of the extended release formulation of quetiapine as monotherapy for the treatment of acute bipolar depression. J Affect Disord. 2010;121:106-115.
21. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67:798-806.
22. Frey MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164:1242-1249. .
23. Goldberg JF, Burdick KE, Endick CJ. Preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
24. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
25. Sienaert P, Vansteelandt K, Demyttenaere K, et al. Ultra-brief pulse ECT in bipolar and unipolar depressive disorder: differences in speed of response. Bipolar Disord. 2009;11:418-424.
26. Valenti M, Benabarre A, Garcia-Amador M, et al. Electroconvulsive therapy in the treatment of mixed states in bipolar disorder. Eur Psychiatry. 2008;23:53-56.
27. Barekatain M, Jahangard L, Haghighi M, et al. Bifrontal versus bitemporal electroconvulsive therapy in severe manic patients. J ECT. 2008;24:199-202.
28. Calabrese JR, Vieta E, El-Mallakh R, et al. Mood state at study entry as predictor of the polarity of relapse in bipolar disorder. Biol Psychiatry. 2004;56:957-963.
29. Fujiwara Y, Honda T, Tanaka Y, et al. Comparison of early- and late-onset rapid cycling affective disorders: clinical course and response to pharmacotherapy. J Clin Psychopharmacol. 1998;18:282-288.
30. Bellivier F, Golmard J, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160:999-1001.
31. Thiruvengadam AP, Chandrasekaran K. Evaluating the validity of blood-based membrane potential changes for the identification of bipolar disorder I. J Affect Disord. 2007;100:75-82.
32. National Institute of Mental Health. How is bipolar disorder treated? Available at: http://www.nimh.nih.gov/health/publications/bipolar-disorder/how-is-bipolar-disorder-treated.shtml. Accessed November 9, 2010.
33. US Food and Drug Administration. FDA approves Saphris to treat schizophrenia and bipolar disorder. Aug. 14, 2009. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm177401.htm. Accessed November 17, 2010.
• Before prescribing antidepressant therapy to depressed patients, screen for bipolar illness, either by taking a detailed medical and family history or by administering the Mood Disorder Questionnaire. A
• Be alert to medical and psychiatric comorbidities in patients with bipolar illness, particularly anxiety disorders and substance abuse. A
• Prescribe a mood stabilizer for acutely depressed patients with bipolar disorder; if the depression does not resolve, add an agent with relapse prevention properties. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
As a family physician, you are better positioned than you might think to make a difference in the lives of patients with bipolar illness. Not only are you likely to be involved in monitoring such patients, but you may frequently be the first clinician patients with bipolar symptoms seek help from.
All it takes to provide that help is a heightened awareness of bipolar disorder, the ways in which bipolar patients present, and the signs and symptoms to look for. Yet evidence suggests that many physicians do not have adequate knowledge of this chronic and debilitating condition. While close to one-third of patients with bipolar disorder seek medical care within a year of the onset of bipolar symptoms, nearly 70% do not receive an accurate diagnosis until they have seen an average of 4 physicians.1 Misdiagnosis—both underdetection1 and over-inclusion2—often results in improper treatment. And even when the diagnosis is correct, patients with bipolar disease often receive inadequate or harmful treatment.3
Ongoing care for bipolar illness is best provided in collaboration with a psychiatrist. With the disorder affecting about 3% to 5% of the US population,4 family physicians will inevitably play a key role in diagnosis and treatment. The text, tables, and screening tool that follow will help with both.
Bipolar diagnosis hinges on this characteristic
The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) defines 4 types of bipolar illness: bipolar I, bipolar II, cyclothymia (the most mild form), and not otherwise specified (TABLE 1).5 The key feature of all 4 types—and the distinguishing characteristic that diagnosis typically hinges on—is a manic or hypomanic episode (TABLE 2).5
Although a full-blown manic episode may not be hard to identify, hypomania is easily missed. By definition, hypomania—with its heightened sense of well-being and productivity—is not problematic and is rarely a patient’s primary complaint.
Mixed mania, a feature of bipolar I, is the worst of both worlds: It is a state in which a full manic episode is superimposed on a full depressive episode—a depression with all the energy and force of a mania. Patients who have experienced one episode of mixed mania have a 12-fold increase in mixed states, 6.5 times more depression, and 1.7 times more dysthymia than those who experience manic episodes without the overlay of depression.6
TABLE 1
Types of bipolar disease: DSM-IV diagnoses5
| Bipolar I | Bipolar II | Cyclothymia | Bipolar disorder not otherwise specified |
|---|---|---|---|
| ≥1 manic or mixed lifetime episode, frequently accompanied by major depressive episodes | ≥1 major depressive episode, accompanied by ≥1 lifetime hypomanic episode | ≥2 years of numerous periods of hypomanic and subsyndromal depressive symptoms | Symptoms resemble, but do not meet, criteria for any specific bipolar disorder |
TABLE 2
Mania and hypomania: DSM-IV criteria5
To identify an episode of mania or hypomania, all of the following criteria must be met. Of note: Hypomania has the same criteria as mania, with 2 notable exceptions: (1) the minimum duration of hypomania is 4 days, rather than 7;* and (2) hypomania is not significantly problematic.
|
| *There is no maximal duration, but the average manic episode lasts 1 to 2 months. |
Complicating matters: Numerous comorbidities
Bipolar illness predisposes patients to multiple medical and psychiatric comorbidities. Cardiovascular, cerebrovascular, and metabolic disorders and sleep disturbances are common in those with bipolar disorder.7 So is obesity, which affects about 50% of patients with bipolar disease.8
Bipolar patients also suffer from an extremely high rate of comorbid psychiatric conditions. Overall, 93% of those with bipolar I also have an anxiety disorder, 71% suffer from drug or alcohol dependence, and 50% suffer from dysthymia, according to the National Comorbidity Survey.9 In addition, about two-thirds of bipolar patients suffer from various personality disorders10—a comorbidity that is particularly disturbing because it is associated with a chronically dysfunctional pattern of problem-solving.
Even without comorbidities, the impact of bipolar disorder is significant: In a study of 1468 patients with bipolar disorder, complaints of difficulties with work, leisure activities, and family and social interactions were common.11 Women were more likely to cite disruption of social and family life, while men often reported having been convicted of crimes. Younger patients reported a greater number of symptomatic days compared with their older counterparts.11
Suicide risk. Patients with bipolar disorder also face an increased risk of suicide, particularly in the depressive phase of the illness. Among 12,662 Oregon Medicaid patients diagnosed with, and treated for, bipolar disorder between 1998 and 2003, there were 11 deaths by suicide and 79 significant suicide attempts.12
Suspect bipolar disorder?
One of the best ways by which family physicians can speed up and improve the accuracy of bipolar diagnosis is to utilize the Mood Disorder Questionnaire (MDQ) (TABLE 3).13 Patients with any mood complaint are its target population. Within that group, the MDQ has been found to have excellent specificity (0.90) and acceptable sensitivity (0.73).13 (For more on identifying patients with bipolar disease, see “A blood test for bipolar disorder?”.)
TABLE 3
The Mood Disorder Questionnaire bipolar screening tool
| Please answer each question to the best of your ability. | ||
| 1. Has there ever been a period of time when you were not your usual self and … | ||
| YES | NO | |
| you felt so good or so hyper that other people thought you were not your normal self or you were so hyper that you got into trouble? | ||
| you were so irritable that you shouted at people or started fights or arguments? | ||
| you felt much more self-confident than usual? | ||
| you got much less sleep than usual and found you didn’t really miss it? | ||
| you were much more talkative or spoke much faster than usual? | ||
| thoughts raced through your head or you couldn’t slow your mind down? | ||
| you were so easily distracted by things around you that you had trouble concentrating or staying on track? | ||
| you had much more energy than usual? | ||
| you were much more active or did many more things than usual? | ||
| you were much more social or outgoing than usual; for example, you telephoned friends in the middle of the night? | ||
| you were much more interested in sex than usual? | ||
| you did things that were unusual for you or that other people might have thought were excessive, foolish, or risky? | ||
| spending money got you or your family into trouble? | ||
| 2. If you checked YES to more than one of the above, have several of these ever happened during the same period of time? | ||
3. How much of a problem did any of these cause you—like being unable to work; having family, money, or legal troubles; getting into arguments or fights? Please circle one response only.
| ||
| Have any of your blood relatives (ie, children, siblings, parents, grandparents, aunts, uncles) had manic-depressive illness or bipolar disorder? | ||
| For a positive screen, 7 of the 13 items in No. 1 must be Yes, No. 2 must be Yes, and No. 3 must be moderate or serious. Source: Hirschfeld RM, et al. Am J Psychiatry. 2000.13 Reprinted with permission. | ||
When to administer the MDQ
Because patients with bipolar disease are more likely to seek help when they are suffering from a depressive episode, it is important to maintain a high index of suspicion. Before ruling out bipolar disease, take a complete medical history, inquiring about comorbidities, family history, and whether the patient can recall any episodes of agitation, intense irritation, or other manifestations of mania or hypomania (TABLE 2).5 If, based on the history, you continue to suspect bipolar disorder, administer the MDQ.
If the patient has a positive screen, your next step would be to initiate treatment for bipolar disorder, even if depression is the presenting symptom. A referral to a psychiatrist would be indicated, as well.
Complexities of bipolar treatment
In recent years, numerous agents have been approved by the US Food and Drug Administration (FDA) for the treatment of bipolar illness in general, and for acute mania in particular. Nearly all of the second-generation, or atypical, antipsychotics have been approved for use in acute mania.14 (Mixed states should be treated the same as mania.) Most of these agents have also been found to be useful as maintenance medications, to prevent relapse (TABLE 4).
Considerable evidence suggests that lithium significantly reduces the risk of relapse, particularly in classic euphoric mania. Other agents that are approved for maintenance therapy include aripiprazole, lamotrigine, and olanzapine as monotherapy, and olanzapine, quetiapine, and ziprasidone as addon agents to lithium or divalproex. Combining a mood stabilizer and an antipsychotic agent generally leads to better outcomes, both in acute mania15 and relapse prevention,16 compared with a mood stabilizer alone. However, bipolar depression, which is more common than either mania17 or hypomania,18 is the major clinical challenge.
TABLE 4
FDA-approved treatments for bipolar disorder32,33
| Treatment | Mania | Bipolar depression | Maintenance |
|---|---|---|---|
| Aripiprazole | √ | √ | |
| Asenapine | √ | ||
| Carbamazepine ER | √ | ||
| Chlorpromazine | √ | ||
| Divalproex; divalproex ER | √ | ||
| Lamotrigine | √ | ||
| Lithium | √* | √ | |
| Olanzapine | à | à | à |
| Quetiapine; quetiapine XR | √ | √ | √ |
| Risperidone | √ | ||
| Ziprasidone | √ | √ | |
| ER, extended release; FDA, US Food and Drug Administration; XR, extended release. *Not approved for mixed mania. † Approved for bipolar depression in combination with fluoxetine. | |||
What’s best for bipolar depression?
For the acutely depressed bipolar patient, optimizing mood stabilization therapy is typically the first step. If the depression doesn’t resolve in 4 or 5 weeks, adding an agent with relapse prevention properties is a preferred approach. Antidepressants may be harmful to patients with bipolar disorder (possibly triggering manic episodes, rapid cycling, or a chronic dysphoric state),3,19 and are usually tried only after other options have been exhausted.
Quetiapine, which is effective for the treatment of mania at doses around 600 mg daily, appears to also be effective for the treatment of acute bipolar depression at doses around 300 mg/d.20 In 2-year studies in which quetiapine was added to either lithium or divalproex, the 2-drug combination was found to reduce the risk for relapse approximately 3-fold compared with the mood stabilizer alone.16
Olanzapine/fluoxetine. Besides quetiapine, this drug combination is the only other agent with FDA approval for the treatment of bipolar depression. A 24-week open extension trial found that the risk for a manic episode due to the coadministration of fluoxetine was low, but 27% of those studied relapsed into depression.21
Drugs that do not have FDA approval specifically for bipolar depression may also be used to treat it.
Lithium, which has antidepressant activity, particularly at levels exceeding 0.8 mEq/L, is one such drug. In addition to its effectiveness in treating bipolar depression, lithium appears to have an antisuicide effect.12
In a recent study of patients with bipolar disorder, lithium was found to be more protective than other mood stabilizers. The hazard ratio (HR) for suicide attempts was significantly greater for patients taking divalproex (HR=2.7; P<.001) or carbamazepine (HR=2.8; P<.001) compared with patients taking lithium.12
Modafinil, a nonstimulant used to increase alertness in patients with daytime sleepiness due to a variety of conditions, has been tested as an adjunctive agent in depressed bipolar patients. In a blinded study, patients were randomly assigned to have modafinil (n=41) or placebo (n=44) added to their existing treatment regimen.22 Response, defined as ≥50% improvement in mood, occurred at twice the rate in those treated with modafinil (44%) compared with those on placebo (23%; P<.05).22 In the brief (6 week) study, modafinil did not appear to cause an increase in manic or hypomanic episodes.22
Pramipexole, a dopamine agonist used for early-stage Parkinson’s disease, has been tested in patients with bipolar depression in 2 small, short-term placebo-controlled trials. A total of 15 patients with type I disease and 28 patients with type II disease were studied for a 6-week period. The results: 60% to 67% of patients taking pramipexole responded, vs 9% to 20% of those on placebo.23,24
Electroconvulsive therapy (ECT) is an underutilized treatment that is effective for depressed patients who are resistant to pharmacological treatment. In fact, bipolar depression may improve more rapidly than unipolar depression with ultra-brief pulse treatment—a therapy in which the pulse width of the electrical stimulus is much briefer (<0.5 msec) than that of standard ECT.25 ECT has also been shown to be effective for mixed states, in which depression and mania coexist.26 The cognitive adverse effects associated with ECT can be reduced while maintaining the same efficacy by using bifrontal (instead of the typical bitemporal) electrode replacement.27
Although the DSM-IV identifies bipolar disorder on the basis of symptoms, there have been increasing attempts to diagnose the disease biologically. Most have been unsuccessful. However, an initial study found that a recently developed blood test (PsychNostics LLC, Baltimore, Md; http://psychnostics.com) that uses the membrane potential as a biological marker had a specificity of 0.88 and a sensitivity of 0.78. The blood test is a promising approach, but is still not ready for prime time.31
Predicting the course of disease, preventing relapse
Generally, the polarity of the current episode predicts that of future episodes. Studies have found that, independent of whether patients were on effective mood-stabilizing agents or placebo, those who relapsed had an episode like their most recent one by a ratio of more than 2 to 1.28
Research suggests a link between age at onset of illness and cycling time and response to particular agents. In 1 trial, those with early-onset bipolar disorder (in adolescence) had briefer euthymic periods and responded better to carbamazepine compared with those who developed symptoms of bipolar disorder at a later age.29 Late-onset bipolar illness (in the 30s or older) was characterized by longer euthymic periods and manic episodes that responded well to lithium.29 The average age of onset is about 25 years.30
Regardless of patient age or age of onset of symptoms, however, prevention of relapse is the goal of ongoing treatment. You can help by assessing the patient’s mood, reviewing the medication regimen and level of compliance, and offering support at every visit—and by consulting with the patient’s psychiatrist, as needed.
CORRESPONDENCE
Rif S. El-Mallakh, MD, Mood Disorders Research Program, Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, MedCenter One, 501 East Broadway, Suite 340, Louisville, KY 40202; [email protected]
• Before prescribing antidepressant therapy to depressed patients, screen for bipolar illness, either by taking a detailed medical and family history or by administering the Mood Disorder Questionnaire. A
• Be alert to medical and psychiatric comorbidities in patients with bipolar illness, particularly anxiety disorders and substance abuse. A
• Prescribe a mood stabilizer for acutely depressed patients with bipolar disorder; if the depression does not resolve, add an agent with relapse prevention properties. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
As a family physician, you are better positioned than you might think to make a difference in the lives of patients with bipolar illness. Not only are you likely to be involved in monitoring such patients, but you may frequently be the first clinician patients with bipolar symptoms seek help from.
All it takes to provide that help is a heightened awareness of bipolar disorder, the ways in which bipolar patients present, and the signs and symptoms to look for. Yet evidence suggests that many physicians do not have adequate knowledge of this chronic and debilitating condition. While close to one-third of patients with bipolar disorder seek medical care within a year of the onset of bipolar symptoms, nearly 70% do not receive an accurate diagnosis until they have seen an average of 4 physicians.1 Misdiagnosis—both underdetection1 and over-inclusion2—often results in improper treatment. And even when the diagnosis is correct, patients with bipolar disease often receive inadequate or harmful treatment.3
Ongoing care for bipolar illness is best provided in collaboration with a psychiatrist. With the disorder affecting about 3% to 5% of the US population,4 family physicians will inevitably play a key role in diagnosis and treatment. The text, tables, and screening tool that follow will help with both.
Bipolar diagnosis hinges on this characteristic
The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) defines 4 types of bipolar illness: bipolar I, bipolar II, cyclothymia (the most mild form), and not otherwise specified (TABLE 1).5 The key feature of all 4 types—and the distinguishing characteristic that diagnosis typically hinges on—is a manic or hypomanic episode (TABLE 2).5
Although a full-blown manic episode may not be hard to identify, hypomania is easily missed. By definition, hypomania—with its heightened sense of well-being and productivity—is not problematic and is rarely a patient’s primary complaint.
Mixed mania, a feature of bipolar I, is the worst of both worlds: It is a state in which a full manic episode is superimposed on a full depressive episode—a depression with all the energy and force of a mania. Patients who have experienced one episode of mixed mania have a 12-fold increase in mixed states, 6.5 times more depression, and 1.7 times more dysthymia than those who experience manic episodes without the overlay of depression.6
TABLE 1
Types of bipolar disease: DSM-IV diagnoses5
| Bipolar I | Bipolar II | Cyclothymia | Bipolar disorder not otherwise specified |
|---|---|---|---|
| ≥1 manic or mixed lifetime episode, frequently accompanied by major depressive episodes | ≥1 major depressive episode, accompanied by ≥1 lifetime hypomanic episode | ≥2 years of numerous periods of hypomanic and subsyndromal depressive symptoms | Symptoms resemble, but do not meet, criteria for any specific bipolar disorder |
TABLE 2
Mania and hypomania: DSM-IV criteria5
To identify an episode of mania or hypomania, all of the following criteria must be met. Of note: Hypomania has the same criteria as mania, with 2 notable exceptions: (1) the minimum duration of hypomania is 4 days, rather than 7;* and (2) hypomania is not significantly problematic.
|
| *There is no maximal duration, but the average manic episode lasts 1 to 2 months. |
Complicating matters: Numerous comorbidities
Bipolar illness predisposes patients to multiple medical and psychiatric comorbidities. Cardiovascular, cerebrovascular, and metabolic disorders and sleep disturbances are common in those with bipolar disorder.7 So is obesity, which affects about 50% of patients with bipolar disease.8
Bipolar patients also suffer from an extremely high rate of comorbid psychiatric conditions. Overall, 93% of those with bipolar I also have an anxiety disorder, 71% suffer from drug or alcohol dependence, and 50% suffer from dysthymia, according to the National Comorbidity Survey.9 In addition, about two-thirds of bipolar patients suffer from various personality disorders10—a comorbidity that is particularly disturbing because it is associated with a chronically dysfunctional pattern of problem-solving.
Even without comorbidities, the impact of bipolar disorder is significant: In a study of 1468 patients with bipolar disorder, complaints of difficulties with work, leisure activities, and family and social interactions were common.11 Women were more likely to cite disruption of social and family life, while men often reported having been convicted of crimes. Younger patients reported a greater number of symptomatic days compared with their older counterparts.11
Suicide risk. Patients with bipolar disorder also face an increased risk of suicide, particularly in the depressive phase of the illness. Among 12,662 Oregon Medicaid patients diagnosed with, and treated for, bipolar disorder between 1998 and 2003, there were 11 deaths by suicide and 79 significant suicide attempts.12
Suspect bipolar disorder?
One of the best ways by which family physicians can speed up and improve the accuracy of bipolar diagnosis is to utilize the Mood Disorder Questionnaire (MDQ) (TABLE 3).13 Patients with any mood complaint are its target population. Within that group, the MDQ has been found to have excellent specificity (0.90) and acceptable sensitivity (0.73).13 (For more on identifying patients with bipolar disease, see “A blood test for bipolar disorder?”.)
TABLE 3
The Mood Disorder Questionnaire bipolar screening tool
| Please answer each question to the best of your ability. | ||
| 1. Has there ever been a period of time when you were not your usual self and … | ||
| YES | NO | |
| you felt so good or so hyper that other people thought you were not your normal self or you were so hyper that you got into trouble? | ||
| you were so irritable that you shouted at people or started fights or arguments? | ||
| you felt much more self-confident than usual? | ||
| you got much less sleep than usual and found you didn’t really miss it? | ||
| you were much more talkative or spoke much faster than usual? | ||
| thoughts raced through your head or you couldn’t slow your mind down? | ||
| you were so easily distracted by things around you that you had trouble concentrating or staying on track? | ||
| you had much more energy than usual? | ||
| you were much more active or did many more things than usual? | ||
| you were much more social or outgoing than usual; for example, you telephoned friends in the middle of the night? | ||
| you were much more interested in sex than usual? | ||
| you did things that were unusual for you or that other people might have thought were excessive, foolish, or risky? | ||
| spending money got you or your family into trouble? | ||
| 2. If you checked YES to more than one of the above, have several of these ever happened during the same period of time? | ||
3. How much of a problem did any of these cause you—like being unable to work; having family, money, or legal troubles; getting into arguments or fights? Please circle one response only.
| ||
| Have any of your blood relatives (ie, children, siblings, parents, grandparents, aunts, uncles) had manic-depressive illness or bipolar disorder? | ||
| For a positive screen, 7 of the 13 items in No. 1 must be Yes, No. 2 must be Yes, and No. 3 must be moderate or serious. Source: Hirschfeld RM, et al. Am J Psychiatry. 2000.13 Reprinted with permission. | ||
When to administer the MDQ
Because patients with bipolar disease are more likely to seek help when they are suffering from a depressive episode, it is important to maintain a high index of suspicion. Before ruling out bipolar disease, take a complete medical history, inquiring about comorbidities, family history, and whether the patient can recall any episodes of agitation, intense irritation, or other manifestations of mania or hypomania (TABLE 2).5 If, based on the history, you continue to suspect bipolar disorder, administer the MDQ.
If the patient has a positive screen, your next step would be to initiate treatment for bipolar disorder, even if depression is the presenting symptom. A referral to a psychiatrist would be indicated, as well.
Complexities of bipolar treatment
In recent years, numerous agents have been approved by the US Food and Drug Administration (FDA) for the treatment of bipolar illness in general, and for acute mania in particular. Nearly all of the second-generation, or atypical, antipsychotics have been approved for use in acute mania.14 (Mixed states should be treated the same as mania.) Most of these agents have also been found to be useful as maintenance medications, to prevent relapse (TABLE 4).
Considerable evidence suggests that lithium significantly reduces the risk of relapse, particularly in classic euphoric mania. Other agents that are approved for maintenance therapy include aripiprazole, lamotrigine, and olanzapine as monotherapy, and olanzapine, quetiapine, and ziprasidone as addon agents to lithium or divalproex. Combining a mood stabilizer and an antipsychotic agent generally leads to better outcomes, both in acute mania15 and relapse prevention,16 compared with a mood stabilizer alone. However, bipolar depression, which is more common than either mania17 or hypomania,18 is the major clinical challenge.
TABLE 4
FDA-approved treatments for bipolar disorder32,33
| Treatment | Mania | Bipolar depression | Maintenance |
|---|---|---|---|
| Aripiprazole | √ | √ | |
| Asenapine | √ | ||
| Carbamazepine ER | √ | ||
| Chlorpromazine | √ | ||
| Divalproex; divalproex ER | √ | ||
| Lamotrigine | √ | ||
| Lithium | √* | √ | |
| Olanzapine | à | à | à |
| Quetiapine; quetiapine XR | √ | √ | √ |
| Risperidone | √ | ||
| Ziprasidone | √ | √ | |
| ER, extended release; FDA, US Food and Drug Administration; XR, extended release. *Not approved for mixed mania. † Approved for bipolar depression in combination with fluoxetine. | |||
What’s best for bipolar depression?
For the acutely depressed bipolar patient, optimizing mood stabilization therapy is typically the first step. If the depression doesn’t resolve in 4 or 5 weeks, adding an agent with relapse prevention properties is a preferred approach. Antidepressants may be harmful to patients with bipolar disorder (possibly triggering manic episodes, rapid cycling, or a chronic dysphoric state),3,19 and are usually tried only after other options have been exhausted.
Quetiapine, which is effective for the treatment of mania at doses around 600 mg daily, appears to also be effective for the treatment of acute bipolar depression at doses around 300 mg/d.20 In 2-year studies in which quetiapine was added to either lithium or divalproex, the 2-drug combination was found to reduce the risk for relapse approximately 3-fold compared with the mood stabilizer alone.16
Olanzapine/fluoxetine. Besides quetiapine, this drug combination is the only other agent with FDA approval for the treatment of bipolar depression. A 24-week open extension trial found that the risk for a manic episode due to the coadministration of fluoxetine was low, but 27% of those studied relapsed into depression.21
Drugs that do not have FDA approval specifically for bipolar depression may also be used to treat it.
Lithium, which has antidepressant activity, particularly at levels exceeding 0.8 mEq/L, is one such drug. In addition to its effectiveness in treating bipolar depression, lithium appears to have an antisuicide effect.12
In a recent study of patients with bipolar disorder, lithium was found to be more protective than other mood stabilizers. The hazard ratio (HR) for suicide attempts was significantly greater for patients taking divalproex (HR=2.7; P<.001) or carbamazepine (HR=2.8; P<.001) compared with patients taking lithium.12
Modafinil, a nonstimulant used to increase alertness in patients with daytime sleepiness due to a variety of conditions, has been tested as an adjunctive agent in depressed bipolar patients. In a blinded study, patients were randomly assigned to have modafinil (n=41) or placebo (n=44) added to their existing treatment regimen.22 Response, defined as ≥50% improvement in mood, occurred at twice the rate in those treated with modafinil (44%) compared with those on placebo (23%; P<.05).22 In the brief (6 week) study, modafinil did not appear to cause an increase in manic or hypomanic episodes.22
Pramipexole, a dopamine agonist used for early-stage Parkinson’s disease, has been tested in patients with bipolar depression in 2 small, short-term placebo-controlled trials. A total of 15 patients with type I disease and 28 patients with type II disease were studied for a 6-week period. The results: 60% to 67% of patients taking pramipexole responded, vs 9% to 20% of those on placebo.23,24
Electroconvulsive therapy (ECT) is an underutilized treatment that is effective for depressed patients who are resistant to pharmacological treatment. In fact, bipolar depression may improve more rapidly than unipolar depression with ultra-brief pulse treatment—a therapy in which the pulse width of the electrical stimulus is much briefer (<0.5 msec) than that of standard ECT.25 ECT has also been shown to be effective for mixed states, in which depression and mania coexist.26 The cognitive adverse effects associated with ECT can be reduced while maintaining the same efficacy by using bifrontal (instead of the typical bitemporal) electrode replacement.27
Although the DSM-IV identifies bipolar disorder on the basis of symptoms, there have been increasing attempts to diagnose the disease biologically. Most have been unsuccessful. However, an initial study found that a recently developed blood test (PsychNostics LLC, Baltimore, Md; http://psychnostics.com) that uses the membrane potential as a biological marker had a specificity of 0.88 and a sensitivity of 0.78. The blood test is a promising approach, but is still not ready for prime time.31
Predicting the course of disease, preventing relapse
Generally, the polarity of the current episode predicts that of future episodes. Studies have found that, independent of whether patients were on effective mood-stabilizing agents or placebo, those who relapsed had an episode like their most recent one by a ratio of more than 2 to 1.28
Research suggests a link between age at onset of illness and cycling time and response to particular agents. In 1 trial, those with early-onset bipolar disorder (in adolescence) had briefer euthymic periods and responded better to carbamazepine compared with those who developed symptoms of bipolar disorder at a later age.29 Late-onset bipolar illness (in the 30s or older) was characterized by longer euthymic periods and manic episodes that responded well to lithium.29 The average age of onset is about 25 years.30
Regardless of patient age or age of onset of symptoms, however, prevention of relapse is the goal of ongoing treatment. You can help by assessing the patient’s mood, reviewing the medication regimen and level of compliance, and offering support at every visit—and by consulting with the patient’s psychiatrist, as needed.
CORRESPONDENCE
Rif S. El-Mallakh, MD, Mood Disorders Research Program, Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, MedCenter One, 501 East Broadway, Suite 340, Louisville, KY 40202; [email protected]
1. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64:161-174.
2. Stewart C, El-Mallakh RS. Is bipolar disorder over diagnosed among patients with substance abuse? Bipolar Disord. 2007;9:646-648.
3. El-Mallakh RS, Karippot A. Chronic depression in bipolar disorder. Am J Psychiatry. 2006;163:1137-1341.
4. Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression, 2nd ed. Oxford University Press: New York; 2007.
5. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed, text rev (DSM-IV-TR). American Psychiatric Association: Arlington, VA; 2000.
6. Baldessarini RJ, Salvatore P, Khalsa HM, et al. Dissimilar morbidity following initial mania versus mixed-states in type-I bipolar disorder. J Affect Disord. 2010;126:299-302.
7. Kilbourne AM, Cornelius JR, Hans X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord. 2004;6:368-373.
8. Shah A, Shen N, El-Mallakh RS. Weight gain occurs after onset of bipolar illness in overweight bipolar patients. Ann Clin Psychiatry. 2006;18:239-241.
9. Kessler RC, Nelson CB, McGonagle KA, et al. The epidemiology of co-occurring addictive and mental disorders: implications for prevention and service utilization. Am J Orthopsychiatry. 1996;66:17-31.
10. Mantere O, Melartin TK, Suominen K, et al. Differences in Axis I and II comorbidity between bipolar I and II disorders and major depressive disorder. J Clin Psychiatry. 2006;67:584-593.
11. Calabrese JR, Hirschfeld RM, Reed M, et al. Impact of bipolar disorder on a US community sample. J Clin Psychiatry. 2003;64:425-432.
12. Collins JC, McFarland BH. Divalproex, lithium and suicide among Medicaid patients with bipolar disorder. J Affect Disord. 2008;107:23-28.
13. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157:1873-1875.
14. Surja AAS, Tamas RL, El-Mallakh RS. Antipsychotic medications in the treatment of bipolar disorder. Curr Drug Targets. 2006;7:1217-1224.
15. Scherk H, Pajonk FG, Leucht S. Second-generation antipsychotic agents in the treatment of acute mania: a systematic review and meta-analysis of randomized controlled trials. Arch Gen Psychiatry. 2007;64:442-455.
16. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166:476-488.
17. Judd LL, Akiskal HS, Schettler PJ. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59:530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60:261-269.
19. El-Mallakh RS, Karippot A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
20. Suppes T, Datto C, Minkwitz M, et al. Effectiveness of the extended release formulation of quetiapine as monotherapy for the treatment of acute bipolar depression. J Affect Disord. 2010;121:106-115.
21. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67:798-806.
22. Frey MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164:1242-1249. .
23. Goldberg JF, Burdick KE, Endick CJ. Preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
24. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
25. Sienaert P, Vansteelandt K, Demyttenaere K, et al. Ultra-brief pulse ECT in bipolar and unipolar depressive disorder: differences in speed of response. Bipolar Disord. 2009;11:418-424.
26. Valenti M, Benabarre A, Garcia-Amador M, et al. Electroconvulsive therapy in the treatment of mixed states in bipolar disorder. Eur Psychiatry. 2008;23:53-56.
27. Barekatain M, Jahangard L, Haghighi M, et al. Bifrontal versus bitemporal electroconvulsive therapy in severe manic patients. J ECT. 2008;24:199-202.
28. Calabrese JR, Vieta E, El-Mallakh R, et al. Mood state at study entry as predictor of the polarity of relapse in bipolar disorder. Biol Psychiatry. 2004;56:957-963.
29. Fujiwara Y, Honda T, Tanaka Y, et al. Comparison of early- and late-onset rapid cycling affective disorders: clinical course and response to pharmacotherapy. J Clin Psychopharmacol. 1998;18:282-288.
30. Bellivier F, Golmard J, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160:999-1001.
31. Thiruvengadam AP, Chandrasekaran K. Evaluating the validity of blood-based membrane potential changes for the identification of bipolar disorder I. J Affect Disord. 2007;100:75-82.
32. National Institute of Mental Health. How is bipolar disorder treated? Available at: http://www.nimh.nih.gov/health/publications/bipolar-disorder/how-is-bipolar-disorder-treated.shtml. Accessed November 9, 2010.
33. US Food and Drug Administration. FDA approves Saphris to treat schizophrenia and bipolar disorder. Aug. 14, 2009. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm177401.htm. Accessed November 17, 2010.
1. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64:161-174.
2. Stewart C, El-Mallakh RS. Is bipolar disorder over diagnosed among patients with substance abuse? Bipolar Disord. 2007;9:646-648.
3. El-Mallakh RS, Karippot A. Chronic depression in bipolar disorder. Am J Psychiatry. 2006;163:1137-1341.
4. Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression, 2nd ed. Oxford University Press: New York; 2007.
5. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed, text rev (DSM-IV-TR). American Psychiatric Association: Arlington, VA; 2000.
6. Baldessarini RJ, Salvatore P, Khalsa HM, et al. Dissimilar morbidity following initial mania versus mixed-states in type-I bipolar disorder. J Affect Disord. 2010;126:299-302.
7. Kilbourne AM, Cornelius JR, Hans X, et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord. 2004;6:368-373.
8. Shah A, Shen N, El-Mallakh RS. Weight gain occurs after onset of bipolar illness in overweight bipolar patients. Ann Clin Psychiatry. 2006;18:239-241.
9. Kessler RC, Nelson CB, McGonagle KA, et al. The epidemiology of co-occurring addictive and mental disorders: implications for prevention and service utilization. Am J Orthopsychiatry. 1996;66:17-31.
10. Mantere O, Melartin TK, Suominen K, et al. Differences in Axis I and II comorbidity between bipolar I and II disorders and major depressive disorder. J Clin Psychiatry. 2006;67:584-593.
11. Calabrese JR, Hirschfeld RM, Reed M, et al. Impact of bipolar disorder on a US community sample. J Clin Psychiatry. 2003;64:425-432.
12. Collins JC, McFarland BH. Divalproex, lithium and suicide among Medicaid patients with bipolar disorder. J Affect Disord. 2008;107:23-28.
13. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157:1873-1875.
14. Surja AAS, Tamas RL, El-Mallakh RS. Antipsychotic medications in the treatment of bipolar disorder. Curr Drug Targets. 2006;7:1217-1224.
15. Scherk H, Pajonk FG, Leucht S. Second-generation antipsychotic agents in the treatment of acute mania: a systematic review and meta-analysis of randomized controlled trials. Arch Gen Psychiatry. 2007;64:442-455.
16. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166:476-488.
17. Judd LL, Akiskal HS, Schettler PJ. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59:530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60:261-269.
19. El-Mallakh RS, Karippot A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
20. Suppes T, Datto C, Minkwitz M, et al. Effectiveness of the extended release formulation of quetiapine as monotherapy for the treatment of acute bipolar depression. J Affect Disord. 2010;121:106-115.
21. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67:798-806.
22. Frey MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164:1242-1249. .
23. Goldberg JF, Burdick KE, Endick CJ. Preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
24. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
25. Sienaert P, Vansteelandt K, Demyttenaere K, et al. Ultra-brief pulse ECT in bipolar and unipolar depressive disorder: differences in speed of response. Bipolar Disord. 2009;11:418-424.
26. Valenti M, Benabarre A, Garcia-Amador M, et al. Electroconvulsive therapy in the treatment of mixed states in bipolar disorder. Eur Psychiatry. 2008;23:53-56.
27. Barekatain M, Jahangard L, Haghighi M, et al. Bifrontal versus bitemporal electroconvulsive therapy in severe manic patients. J ECT. 2008;24:199-202.
28. Calabrese JR, Vieta E, El-Mallakh R, et al. Mood state at study entry as predictor of the polarity of relapse in bipolar disorder. Biol Psychiatry. 2004;56:957-963.
29. Fujiwara Y, Honda T, Tanaka Y, et al. Comparison of early- and late-onset rapid cycling affective disorders: clinical course and response to pharmacotherapy. J Clin Psychopharmacol. 1998;18:282-288.
30. Bellivier F, Golmard J, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160:999-1001.
31. Thiruvengadam AP, Chandrasekaran K. Evaluating the validity of blood-based membrane potential changes for the identification of bipolar disorder I. J Affect Disord. 2007;100:75-82.
32. National Institute of Mental Health. How is bipolar disorder treated? Available at: http://www.nimh.nih.gov/health/publications/bipolar-disorder/how-is-bipolar-disorder-treated.shtml. Accessed November 9, 2010.
33. US Food and Drug Administration. FDA approves Saphris to treat schizophrenia and bipolar disorder. Aug. 14, 2009. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm177401.htm. Accessed November 17, 2010.
Pregnancy and epilepsy—when you’re managing both
• Use the dose of antiepileptic drug (AED) at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy. C
• Avoid switching a pregnant patient to an AED that she has not taken before. C
• Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 500,000 women in the United States suffer from epilepsy and are of childbearing age.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma: Infants born to women with epilepsy have a 2- to 3-fold risk of congenital malformations compared with those whose mothers do not have epilepsy, mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neuro-cognitive deficits, even when there are no major congenital malformations.1,3,4
Yet discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In 1 recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is not known.5
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancies and a neurologist or an epileptologist. In reality, those who live in areas with limited access to such specialized care or have limited health coverage may be cared for throughout their pregnancy by a family physician. This evidence-based review was developed with this family physician in mind.
Safeguarding mom and fetus starts before pregnancy
Mechanisms by which AEDs cause fetal and embryonic harm remain unclear. Possible causes include drug toxicity, drug-drug interactions, folic acid deficiency, the effect of suboptimally controlled convulsions, genetic predisposition, enhanced apoptotic neurodegeneration, and alterations in thyroid hormone status, among others.6-9 Major congenital malformations may occur in a dose-dependent manner, and physicians should aim to use the most effective AED at the lowest effective dose for women of childbearing age.2
In reviewing antiseizure therapy for such patients, here are some considerations to keep in mind:
Avoid polytherapy, if possible. Taking multiple AEDs may increase the risk of major congenital malformations, especially when valproate is one of the drugs.1 Hence, an attempt should be made to switch women with epilepsy who are of childbearing age to monotherapy. Ideally, this should be done a year before planned conception so that good seizure control can be achieved and documented prior to pregnancy.
Avoid high-risk AEDs. Overall, an increased risk of major congenital malformations has been most convincingly found with valproate and phenobarbital.1 Specific types of malformations have also been linked to certain drugs (TABLE). Cardiac malformations are associated with carbamazepine, phenobarbital, and valproate; spina bifida, hypospadias, porencephaly, and other brain anomalies, as well as limb reduction defects, are associated with valproate, particularly at doses >1100 mg/d.10
Choose newer agents, whenever possible. The risk of malformations with newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, and topiramate—remains unclear, but preliminary data for monotherapy with these agents suggest a lower teratogenic risk compared with traditional AEDs, such as phenobarbital and valproate.10
Initiate folic acid supplementation. Drug-induced folate deficiency has been proposed as a contributing factor in the teratogenicity of AEDs, so diligence is essential in ensuring that patients who have epilepsy and are of childbearing age take folic acid.11 Studies have demonstrated a significant reduction in spontaneous abortion in women who are receiving AED therapy and taking folic acid supplements, and the benefits of folic acid have been found to be especially notable for women taking valproate.12
Folic acid supplementation, of course, is important for all women of childbearing age. At a dose of 0.8 mg/d, folate has been shown to reduce the risk of neural tube and ventricular septal defects in the general population. The American Academy of Neurology/American Epilepsy Society (AAN/AES) Practice Parameters recommend that all women of child-bearing age taking AEDs also take folate supplements (0.4-4 mg/d).13 An optimal dose has not been determined for this patient population, but we routinely recommend 1 mg/d for women with epilepsy of childbearing age and increase the dose to 4 mg/d after conception.
TABLE
Commonly used antiepileptic drugs: Major teratogenic risks1,10,19
| AED | FDA pregnancy category* | Associated risks | Recommendations for use during pregnancy |
| Carbamazepine | C | Cardiac malformations | Lower teratogenic potential compared with phenobarbital and valproate |
| Gabapentin | C | No MCMs associated with monotherapy | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Lamotrigine | C | No distinctive pattern of MCMs | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Levetiracetam | C | Pyloric stenosis (in polytherapy with lamotrigine); spina bifida (in polytherapy with carbamazepine and valproate) | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Oxcarbazepine | C | Urogenital malformations | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Phenobarbital | D | Cardiac malformations | Best avoided in women of childbearing age |
| Phenytoin | D | Bradycardia and hypotension; fetal hydantoin syndrome | Best avoided in women of childbearing age |
| Topiramate | C | Hypospadias; oral clefts | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Valproate | D | Cardiac malformations; hypospadias; limb reduction defects; neural tube defects; porencephaly; spina bifida | Best avoided in women of childbearing age |
| AED, antiepileptic drug; FDA, US Food and Drug Administration; MCMs, major congenital malformations. | |||
| *Category C indicates that human data are lacking and animal studies were positive OR not done; Category D indicates that human data have shown a teratogenic risk but the benefits may outweigh the risk. | |||
| †Traditional AEDs include carbamazepine, phenobarbital, phenytoin, and valproate. | |||
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that patients may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year prior to planned pregnancy so good seizure control can be achieved before then.
In attempting a change in medication, start by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That allows you to determine the baseline therapeutic drug level and dose at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dose, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
What about stopping AED therapy entirely if your patient wants to get pregnant? Stopping AEDs is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis, and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests she may have recurrent seizures, explain that the risk of discontinuing the medication is greater than the risk of fetal exposure to AEDs. It is also important to point out that more than 90% of women with epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider in the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry, available at http://aedpregnancyregistry.org. We recommend that physicians caring for pregnant women with epilepsy encourage them to enroll early on, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women taking AEDs—particularly those on higher-risk drugs such as valproate—undergo a detailed first trimester ultrasound study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible; if needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk for major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 while a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in the pregnancy. In some cases, dramatic increases—several multiples of the preconception dose—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman with epilepsy includes the following:
- Check levels at baseline—prior to conception, whenever possible—and monthly throughout the pregnancy, with more frequent checks for women with recurrent seizures and those taking lamotrigine.
- Use the dose at which the patient was seizure-free prior to conception as a target level during pregnancy.
- Adjust the dose as needed to maintain the preconception serum drug level.
Drug-specific considerations. As phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the free level of the drug with the following formula:
Free phenytoin = measured level/ [(0.2 × albumin level) + 0.1].
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/d folic acid for pregnant women with epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women with epilepsy (phytonadione 10 mg/d) starting at 36 weeks’ gestation and continuing until delivery despite the lack of a proven benefit because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes AEDs—to prevent hemorrhagic disease.13
Should women taking AEDs breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with AEDs are generally concerned about the possibility of contaminated breast milk. While antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with AEDs.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients with epilepsy who are being treated with AEDs, and is not contra-indicated by the AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women with epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the factors to consider in managing epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
CORRESPONDENCE Nitin K. Sethi, MD, Comprehensive Epilepsy Center, New York-Presbyterian Hospital, Weill Cornell Medical Center, 525 East 68th Street, Room K-615, New York, NY 10021; [email protected]
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69 (suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11:270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22:162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71 (suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of anti-epileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38:981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci. 2002;99:15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322:494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67:407-412.
10. Kaaja R, Hiilesmaa V. Major malformations in off spring of women with epilepsy. Neurology. 2003;60:575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57:A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255:1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. Available at: www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm. Accessed November 4, 2010.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K, folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:142-149.
15. Tettenborn B. Management of epilepsy in women of child-bearing age: practice recommendations. CNS Drugs. 2006;20:373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49:1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, et al. Antiepileptic drug use of women with epilepsy and congenital malformations in off spring. Neurology. 2005;64:1874-1878.
• Use the dose of antiepileptic drug (AED) at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy. C
• Avoid switching a pregnant patient to an AED that she has not taken before. C
• Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 500,000 women in the United States suffer from epilepsy and are of childbearing age.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma: Infants born to women with epilepsy have a 2- to 3-fold risk of congenital malformations compared with those whose mothers do not have epilepsy, mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neuro-cognitive deficits, even when there are no major congenital malformations.1,3,4
Yet discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In 1 recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is not known.5
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancies and a neurologist or an epileptologist. In reality, those who live in areas with limited access to such specialized care or have limited health coverage may be cared for throughout their pregnancy by a family physician. This evidence-based review was developed with this family physician in mind.
Safeguarding mom and fetus starts before pregnancy
Mechanisms by which AEDs cause fetal and embryonic harm remain unclear. Possible causes include drug toxicity, drug-drug interactions, folic acid deficiency, the effect of suboptimally controlled convulsions, genetic predisposition, enhanced apoptotic neurodegeneration, and alterations in thyroid hormone status, among others.6-9 Major congenital malformations may occur in a dose-dependent manner, and physicians should aim to use the most effective AED at the lowest effective dose for women of childbearing age.2
In reviewing antiseizure therapy for such patients, here are some considerations to keep in mind:
Avoid polytherapy, if possible. Taking multiple AEDs may increase the risk of major congenital malformations, especially when valproate is one of the drugs.1 Hence, an attempt should be made to switch women with epilepsy who are of childbearing age to monotherapy. Ideally, this should be done a year before planned conception so that good seizure control can be achieved and documented prior to pregnancy.
Avoid high-risk AEDs. Overall, an increased risk of major congenital malformations has been most convincingly found with valproate and phenobarbital.1 Specific types of malformations have also been linked to certain drugs (TABLE). Cardiac malformations are associated with carbamazepine, phenobarbital, and valproate; spina bifida, hypospadias, porencephaly, and other brain anomalies, as well as limb reduction defects, are associated with valproate, particularly at doses >1100 mg/d.10
Choose newer agents, whenever possible. The risk of malformations with newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, and topiramate—remains unclear, but preliminary data for monotherapy with these agents suggest a lower teratogenic risk compared with traditional AEDs, such as phenobarbital and valproate.10
Initiate folic acid supplementation. Drug-induced folate deficiency has been proposed as a contributing factor in the teratogenicity of AEDs, so diligence is essential in ensuring that patients who have epilepsy and are of childbearing age take folic acid.11 Studies have demonstrated a significant reduction in spontaneous abortion in women who are receiving AED therapy and taking folic acid supplements, and the benefits of folic acid have been found to be especially notable for women taking valproate.12
Folic acid supplementation, of course, is important for all women of childbearing age. At a dose of 0.8 mg/d, folate has been shown to reduce the risk of neural tube and ventricular septal defects in the general population. The American Academy of Neurology/American Epilepsy Society (AAN/AES) Practice Parameters recommend that all women of child-bearing age taking AEDs also take folate supplements (0.4-4 mg/d).13 An optimal dose has not been determined for this patient population, but we routinely recommend 1 mg/d for women with epilepsy of childbearing age and increase the dose to 4 mg/d after conception.
TABLE
Commonly used antiepileptic drugs: Major teratogenic risks1,10,19
| AED | FDA pregnancy category* | Associated risks | Recommendations for use during pregnancy |
| Carbamazepine | C | Cardiac malformations | Lower teratogenic potential compared with phenobarbital and valproate |
| Gabapentin | C | No MCMs associated with monotherapy | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Lamotrigine | C | No distinctive pattern of MCMs | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Levetiracetam | C | Pyloric stenosis (in polytherapy with lamotrigine); spina bifida (in polytherapy with carbamazepine and valproate) | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Oxcarbazepine | C | Urogenital malformations | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Phenobarbital | D | Cardiac malformations | Best avoided in women of childbearing age |
| Phenytoin | D | Bradycardia and hypotension; fetal hydantoin syndrome | Best avoided in women of childbearing age |
| Topiramate | C | Hypospadias; oral clefts | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Valproate | D | Cardiac malformations; hypospadias; limb reduction defects; neural tube defects; porencephaly; spina bifida | Best avoided in women of childbearing age |
| AED, antiepileptic drug; FDA, US Food and Drug Administration; MCMs, major congenital malformations. | |||
| *Category C indicates that human data are lacking and animal studies were positive OR not done; Category D indicates that human data have shown a teratogenic risk but the benefits may outweigh the risk. | |||
| †Traditional AEDs include carbamazepine, phenobarbital, phenytoin, and valproate. | |||
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that patients may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year prior to planned pregnancy so good seizure control can be achieved before then.
In attempting a change in medication, start by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That allows you to determine the baseline therapeutic drug level and dose at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dose, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
What about stopping AED therapy entirely if your patient wants to get pregnant? Stopping AEDs is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis, and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests she may have recurrent seizures, explain that the risk of discontinuing the medication is greater than the risk of fetal exposure to AEDs. It is also important to point out that more than 90% of women with epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider in the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry, available at http://aedpregnancyregistry.org. We recommend that physicians caring for pregnant women with epilepsy encourage them to enroll early on, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women taking AEDs—particularly those on higher-risk drugs such as valproate—undergo a detailed first trimester ultrasound study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible; if needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk for major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 while a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in the pregnancy. In some cases, dramatic increases—several multiples of the preconception dose—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman with epilepsy includes the following:
- Check levels at baseline—prior to conception, whenever possible—and monthly throughout the pregnancy, with more frequent checks for women with recurrent seizures and those taking lamotrigine.
- Use the dose at which the patient was seizure-free prior to conception as a target level during pregnancy.
- Adjust the dose as needed to maintain the preconception serum drug level.
Drug-specific considerations. As phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the free level of the drug with the following formula:
Free phenytoin = measured level/ [(0.2 × albumin level) + 0.1].
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/d folic acid for pregnant women with epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women with epilepsy (phytonadione 10 mg/d) starting at 36 weeks’ gestation and continuing until delivery despite the lack of a proven benefit because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes AEDs—to prevent hemorrhagic disease.13
Should women taking AEDs breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with AEDs are generally concerned about the possibility of contaminated breast milk. While antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with AEDs.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients with epilepsy who are being treated with AEDs, and is not contra-indicated by the AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women with epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the factors to consider in managing epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
CORRESPONDENCE Nitin K. Sethi, MD, Comprehensive Epilepsy Center, New York-Presbyterian Hospital, Weill Cornell Medical Center, 525 East 68th Street, Room K-615, New York, NY 10021; [email protected]
• Use the dose of antiepileptic drug (AED) at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy. C
• Avoid switching a pregnant patient to an AED that she has not taken before. C
• Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
About 500,000 women in the United States suffer from epilepsy and are of childbearing age.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma: Infants born to women with epilepsy have a 2- to 3-fold risk of congenital malformations compared with those whose mothers do not have epilepsy, mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neuro-cognitive deficits, even when there are no major congenital malformations.1,3,4
Yet discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In 1 recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is not known.5
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancies and a neurologist or an epileptologist. In reality, those who live in areas with limited access to such specialized care or have limited health coverage may be cared for throughout their pregnancy by a family physician. This evidence-based review was developed with this family physician in mind.
Safeguarding mom and fetus starts before pregnancy
Mechanisms by which AEDs cause fetal and embryonic harm remain unclear. Possible causes include drug toxicity, drug-drug interactions, folic acid deficiency, the effect of suboptimally controlled convulsions, genetic predisposition, enhanced apoptotic neurodegeneration, and alterations in thyroid hormone status, among others.6-9 Major congenital malformations may occur in a dose-dependent manner, and physicians should aim to use the most effective AED at the lowest effective dose for women of childbearing age.2
In reviewing antiseizure therapy for such patients, here are some considerations to keep in mind:
Avoid polytherapy, if possible. Taking multiple AEDs may increase the risk of major congenital malformations, especially when valproate is one of the drugs.1 Hence, an attempt should be made to switch women with epilepsy who are of childbearing age to monotherapy. Ideally, this should be done a year before planned conception so that good seizure control can be achieved and documented prior to pregnancy.
Avoid high-risk AEDs. Overall, an increased risk of major congenital malformations has been most convincingly found with valproate and phenobarbital.1 Specific types of malformations have also been linked to certain drugs (TABLE). Cardiac malformations are associated with carbamazepine, phenobarbital, and valproate; spina bifida, hypospadias, porencephaly, and other brain anomalies, as well as limb reduction defects, are associated with valproate, particularly at doses >1100 mg/d.10
Choose newer agents, whenever possible. The risk of malformations with newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, and topiramate—remains unclear, but preliminary data for monotherapy with these agents suggest a lower teratogenic risk compared with traditional AEDs, such as phenobarbital and valproate.10
Initiate folic acid supplementation. Drug-induced folate deficiency has been proposed as a contributing factor in the teratogenicity of AEDs, so diligence is essential in ensuring that patients who have epilepsy and are of childbearing age take folic acid.11 Studies have demonstrated a significant reduction in spontaneous abortion in women who are receiving AED therapy and taking folic acid supplements, and the benefits of folic acid have been found to be especially notable for women taking valproate.12
Folic acid supplementation, of course, is important for all women of childbearing age. At a dose of 0.8 mg/d, folate has been shown to reduce the risk of neural tube and ventricular septal defects in the general population. The American Academy of Neurology/American Epilepsy Society (AAN/AES) Practice Parameters recommend that all women of child-bearing age taking AEDs also take folate supplements (0.4-4 mg/d).13 An optimal dose has not been determined for this patient population, but we routinely recommend 1 mg/d for women with epilepsy of childbearing age and increase the dose to 4 mg/d after conception.
TABLE
Commonly used antiepileptic drugs: Major teratogenic risks1,10,19
| AED | FDA pregnancy category* | Associated risks | Recommendations for use during pregnancy |
| Carbamazepine | C | Cardiac malformations | Lower teratogenic potential compared with phenobarbital and valproate |
| Gabapentin | C | No MCMs associated with monotherapy | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Lamotrigine | C | No distinctive pattern of MCMs | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Levetiracetam | C | Pyloric stenosis (in polytherapy with lamotrigine); spina bifida (in polytherapy with carbamazepine and valproate) | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Oxcarbazepine | C | Urogenital malformations | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Phenobarbital | D | Cardiac malformations | Best avoided in women of childbearing age |
| Phenytoin | D | Bradycardia and hypotension; fetal hydantoin syndrome | Best avoided in women of childbearing age |
| Topiramate | C | Hypospadias; oral clefts | Limited data suggest lower teratogenic risk compared with traditional AEDs† |
| Valproate | D | Cardiac malformations; hypospadias; limb reduction defects; neural tube defects; porencephaly; spina bifida | Best avoided in women of childbearing age |
| AED, antiepileptic drug; FDA, US Food and Drug Administration; MCMs, major congenital malformations. | |||
| *Category C indicates that human data are lacking and animal studies were positive OR not done; Category D indicates that human data have shown a teratogenic risk but the benefits may outweigh the risk. | |||
| †Traditional AEDs include carbamazepine, phenobarbital, phenytoin, and valproate. | |||
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that patients may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year prior to planned pregnancy so good seizure control can be achieved before then.
In attempting a change in medication, start by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That allows you to determine the baseline therapeutic drug level and dose at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dose, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
What about stopping AED therapy entirely if your patient wants to get pregnant? Stopping AEDs is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis, and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests she may have recurrent seizures, explain that the risk of discontinuing the medication is greater than the risk of fetal exposure to AEDs. It is also important to point out that more than 90% of women with epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider in the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry, available at http://aedpregnancyregistry.org. We recommend that physicians caring for pregnant women with epilepsy encourage them to enroll early on, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women taking AEDs—particularly those on higher-risk drugs such as valproate—undergo a detailed first trimester ultrasound study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible; if needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk for major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 while a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in the pregnancy. In some cases, dramatic increases—several multiples of the preconception dose—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman with epilepsy includes the following:
- Check levels at baseline—prior to conception, whenever possible—and monthly throughout the pregnancy, with more frequent checks for women with recurrent seizures and those taking lamotrigine.
- Use the dose at which the patient was seizure-free prior to conception as a target level during pregnancy.
- Adjust the dose as needed to maintain the preconception serum drug level.
Drug-specific considerations. As phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the free level of the drug with the following formula:
Free phenytoin = measured level/ [(0.2 × albumin level) + 0.1].
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/d folic acid for pregnant women with epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women with epilepsy (phytonadione 10 mg/d) starting at 36 weeks’ gestation and continuing until delivery despite the lack of a proven benefit because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes AEDs—to prevent hemorrhagic disease.13
Should women taking AEDs breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with AEDs are generally concerned about the possibility of contaminated breast milk. While antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with AEDs.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients with epilepsy who are being treated with AEDs, and is not contra-indicated by the AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women with epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the factors to consider in managing epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
CORRESPONDENCE Nitin K. Sethi, MD, Comprehensive Epilepsy Center, New York-Presbyterian Hospital, Weill Cornell Medical Center, 525 East 68th Street, Room K-615, New York, NY 10021; [email protected]
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69 (suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11:270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22:162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71 (suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of anti-epileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38:981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci. 2002;99:15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322:494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67:407-412.
10. Kaaja R, Hiilesmaa V. Major malformations in off spring of women with epilepsy. Neurology. 2003;60:575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57:A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255:1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. Available at: www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm. Accessed November 4, 2010.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K, folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:142-149.
15. Tettenborn B. Management of epilepsy in women of child-bearing age: practice recommendations. CNS Drugs. 2006;20:373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49:1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, et al. Antiepileptic drug use of women with epilepsy and congenital malformations in off spring. Neurology. 2005;64:1874-1878.
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69 (suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11:270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22:162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71 (suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of anti-epileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38:981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci. 2002;99:15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322:494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67:407-412.
10. Kaaja R, Hiilesmaa V. Major malformations in off spring of women with epilepsy. Neurology. 2003;60:575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57:A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255:1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. Available at: www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm. Accessed November 4, 2010.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K, folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:142-149.
15. Tettenborn B. Management of epilepsy in women of child-bearing age: practice recommendations. CNS Drugs. 2006;20:373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49:1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, et al. Antiepileptic drug use of women with epilepsy and congenital malformations in off spring. Neurology. 2005;64:1874-1878.
Treating DVT: Answers to 7 key questions
• Start patients with a new-onset venous thrombosis on a low-molecular-weight heparin (LMWH), unfractionated heparin (UFH), or fondaparinux as well as warfarin therapy. A
• Continue LMWH,UFH, or fondaparinux with warfarin for a minimum of 5 days until the international normalized ratio (INR) is ≥2 for 24 hours. A
• Educate patients about anticoagulant therapy, dietary and medication interactions with warfarin, and signs and symptoms of bleeding. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Arterial and venous thromboses are major causes of morbidity and mortality in the United States. Each year, about 100 out of 100,000 Americans (0.1%) experience a venous thromboembolism (VTE), and the incidence is considerably higher among hospitalized patients.1 Incidence and early mortality after a first-time event increase with age. Mortality and the potential for a pulmonary embolism (PE) to occur after a deep vein thrombosis (DVT) depend on the location of the DVT and how well the DVT is managed. Proximal DVTs are more likely to develop into a PE. Mortality rates for patients with PE are as high as 17% 3 months after diagnosis.2
Anticoagulant therapy is the foundation for prevention and treatment of thromboembolic disease, and family physicians are on the front line of management when patients with DVT are discharged from the hospital. There are many therapeutic options to choose from: unfractionated heparin (UFH), low-molecular-weight heparins (LMWHs), the factor Xa inhibitor fondaparinux, direct thrombin inhibitors, or vitamin K antagonists (VKAs). All of these agents are effective, but you’ll need to keep clinical considerations and drug limitations in mind to use them properly.
The salient details for optimal use of these agents are set out in the 8th edition of the American College of Chest Physicians (ACCP) Evidence-based Clinical Practice Guidelines, released in 2008.3 But following these complex guidelines to maximize patient safety and minimize both cost and inconvenience raises many questions for the busy family physician. This article provides the answers you need to maximize your care.
1. What therapies can be used in the outpatient setting to treat acute DVT or PE?
You can manage DVT with LMWH—dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep)—or the factor Xa inhibitor fondaparinux (Arixtra) overlapped with warfarin (Coumadin). UFH is generally not recommended in the outpatient setting. Patients who are obese or have a creatinine clearance <30 mL/min will need inpatient treatment with UFH in most cases.
Outpatient management of PE based on clinical prediction rules that stratify patients by risk factors has been attempted, although the safety and efficacy of this practice have not been conclusively demonstrated. Prediction rules are available at http://www.medicalcriteria.com/criteria/car_thrombosis.htm. Note that LMWH and fondaparinux are not approved by the US Food and Drug Administration (FDA) for the outpatient treatment of PE.
Dosing guidelines for LMWH agents and fondaparinux are given in TABLE 1, and recommendations for treatment of DVT are summarized in TABLE 2.
TABLE 1
Low-molecular-weight heparins and fondaparinux dosing for DVT
| Agent | Dose |
|---|---|
| Dalteparin (Fragmin)21 | 100 units/kg SQ every 12 h or 200 units/kg SQ every 24 h |
| Enoxaparin (Lovenox)22 | 1 mg/kg SQ every 12 h or 1.5 mg/kg every 24 h |
| Tinzaparin (Innohep)23 | 175 anti-Xa IU/kg SQ every 24 h |
| Fondaparinux (Arixtra)24 | Weight <50 kg: 5 mg SQ every 24 h Weight 50-100 kg: 7.5 mg SQ every 24 h Weight >100 kg: 10 mg SQ every 24 h |
| DVT, deep vein thrombosis; SQ, subcutaneously. | |
TABLE 2
Treating DVT: Recommended options3,4,21-24
| Warfarin | UFH | LMWH |
|---|---|---|
| Starting dose 5-10 mg/d for first 1-2 days. Lower starting dose for patients with liver impairment, malnourishment, heart failure, or recent major surgery; for debilitated and elderly patients; and for patients on medications known to inhibit CYP-450 enzyme. Initial monitoring after the first 2-3 doses. Maintenance monitoring at least every 4 weeks. For acute DVT, overlap with LMWH, UFH, or fondaparinux for at least 5 days and until INR is ≥2 for 24 hours. Continue therapy for ≥3 months for patients with upper extremity DVT. | UFH is recommended for patients who are obese or have a creatinine clearance <30 mL/min; UFH is generally an inpatient treatment option, and patients may need to be admitted for therapy. | For acute DVT, LMWH daily or twice daily is recommended over UFH. Exceptions include patients who are obese or have a creatinine clearance <30 mL/min. Anti-factor Xa levels should be monitored in pregnant patients on therapeutic doses of LMWH. |
| DVT, deep vein thrombosis; INR, international normalized ratio; LMWH, low-molecular-weight heparin; UFH, unfractionated heparin. | ||
2. When, and at what dosage, should i initiate warfarin?
With a medically stable patient, you can start warfarin shortly after the first dose of LMWH or fondaparinux, and overlap both therapies for at least 5 days until the patient’s international normalized ratio (INR) is ≥2 for 24 hours. If the INR does not reach 2 within 5 days, LMWH or fondaparinux should be continued. The target INR for DVT is 2.5.
The initial dose of warfarin for most patients should be between 5 and 10 mg per day for the first 2 doses, with 10-mg doses reserved for younger patients without significant drug interactions or comorbidities.4 Consider a starting dose ≤5 mg in elderly patients, those with certain medical conditions (eg, liver disease or heart failure), and patients taking medications known to significantly inhibit warfarin metabolism.3,4TABLE 3 provides a suggested method for initiation of warfarin in ambulatory patients.
Continue warfarin for at least 3 months, and possibly longer, depending on the cause of DVT/PE and underlying or ongoing risk factors. Evaluate the risk vs benefit of continued therapy 3 months after the initial thromboembolic event. Patients with cancer, whose risk for VTE is greater, should receive LMWH for the first 3 to 6 months, followed by long-term therapy with warfarin or LMWH until the cancer is resolved.3,4
TABLE 3
Average warfarin daily dosing for INR goal 2-3
| Dosage change | Patients nonsensitive to warfarin | Patients sensitive to warfarin* | |
|---|---|---|---|
| Initial dose | 5 mg/d | 2.5 mg/d | |
| First INR | 3 days after initial dose | 3 days after initial dose | |
| <1.5 | Increase dose by 50% | 7.5 mg/d | 5 mg/d |
| 1.5-1.9 | Maintain current dose | 5 mg/d | 2.5 mg/d |
| 2-3 | Decrease dose by 50% | 2.5 mg/d | 1.25 mg/d |
| 3.1-4 | Decrease dose by ~75% | 1.25 mg/d | 0.5 mg/d |
| >4 | Hold dose | Hold | Hold |
| Next INR | 2-3 days | 2-3 days | |
| INR, international normalized ratio. | |||
| *Factors that influence sensitivity to warfarin include age >75 years, clinical congestive heart failure, diarrhea, drug interactions, elevated baseline INR, hyperthyroidism, malignancy, malnutrition, or nothing by mouth for >3 days. | |||
| Source: University of Washington Medical Center. Average daily dosing method. Available at: http://vte.son.washington.edu/docs/VTE_flexible_initiation.pdf. Accessed September 26, 2010. | |||
3. Is it time to customize anticoagulant therapy based on genetic testing?
No. Currently, FDA and ACCP guidelines do not recommend genetic testing before initiating warfarin.5,6 Theoretically, genetic testing should be helpful in predicting an individual’s optimal starting warfarin dose. At present, however, no good clinical data support this practice.5 If randomized trials show improved clinical outcomes with pharmacogenetic dosing of warfarin, genotyping may become part of clinical practice in the future.
An estimated one-third of patients on warfarin therapy may be at higher risk for adverse outcomes because they carry genes that make them more or less sensitive to warfarin.5 Variants of 2 genes—cytochrome P450 2C9 (CYP2C9) and the vitamin K oxide reductase complex 1 (VKORC1)—are thought to be responsible for this variance in warfarin response.5
Patients with variations of CYP2C9 may need lower starting doses of warfarin. Mutations in the VKORC1 gene affect the enzymes that activate vitamin K, which are the target for warfarin’s inhibitory effect on clotting. Mutations in this gene therefore result in varying sensitivities to warfarin and may be the cause of hereditary warfarin resistance in some individuals. Genetic variations in VKORC1 are estimated to occur in 14% to 37% of Caucasians and African Americans and may exist in as many as 89% of Asians.5 Several tests to detect some variants in these genes have been approved by the FDA.
In August 2007, a labeling change for Coumadin and its generics detailed the influence of gene variations on warfarin sensitivity.7 A report from the American Enterprise Institute-Brookings Joint Center for Regulatory Studies estimated that genetic testing could prevent 85,000 serious bleeding events and 17,000 strokes per year, resulting in a $1.1 billion reduction in warfarin-related health care spending. Costs of genetic testing for the 2 million Americans who begin warfarin therapy each year would be approximately $1 billion.6
4. Which warfarin–drug interactions are clinically important?
Drugs, supplements, and foods that potentiate or inhibit warfarin’s anticoagulant effect or increase the risk of bleeding are clinically important. The list of such interactions has been referred to as the 8 “As”: antibiotics, antifungals, antidepressants, antiplatelets, amiodarone, anti-inflammatories, high-dose acetaminophen, and alternative remedies.8 (For details on common warfarin interactions, see TABLE W1.)
These and other medications can affect how warfarin is absorbed, distributed, and metabolized. For example, sucralfate and bile-acid sequestrants such as cholestyramine can inhibit absorption. You can minimize this interaction by staggering the time each medication is ingested. Drugs that induce cytochrome P450 enzymes (eg, rifampin, carbamazepine) enhance warfarin clearance, while drugs that inhibit CYP enzymes (amiodarone or itraconazole) decrease warfarin clearance.2 Most clinically relevant interactions affect warfarin metabolism.
5. How should i proceed when a patient taking warfarin also needs antiplatelet medications?
Monitor warfarin more frequently in such patients and target the lower end of the INR therapeutic range (2-2.5).9 Keep an eye on your patient’s overall medication regimen and avoid medications like nonsteroidal anti-inflammatory drugs (NSAIDs) that increase bleeding risk. If NSAIDs must be used, avoid chronic use, high doses, and NSAIDs with a long half-life. You may also want to consider referral to an anticoagulation clinic.
Many of these patients have cardiac conditions for which dual antiplatelet therapy is recommended. For example, patients with coronary stents may need aspirin and clopidogrel for a specified period of time. They may have underlying atrial fibrillation or valve replacement requiring warfarin therapy. Data examining triple therapy (aspirin, clopidogrel, and warfarin) are primarily limited to patients with acute coronary syndrome or those who have had percutaneous coronary intervention. Unfortunately, the data are also retrospective, based on a small sample, and inconsistent.10 In these patients, you need to weigh the increased risk of bleeding against the proven preventive value of each of these modalities.
For patients with stents, current guidelines recommend a lower dose of aspirin and discontinuation of clopidogrel after a certain length of time, depending on the type of stent.10,11 However, 1 study showed that aspirin dose and INR values did not influence bleeding risk in patients on triple therapy.12
It is imperative that you counsel patients on triple therapy to report the first sign of bleeding.
6. What is the best approach when a patient’s INR is elevated?
You’ll need to minimize the risk of bleeding while at the same time ensuring adequate levels of anticoagulation. You can use oral vitamin K (phytonadione [Mephyton]) to reverse the effects of warfarin without inducing warfarin resistance. Avoid subcutaneous administration; the effects are unpredictable and response is delayed.4
Send patients with active or life-threatening bleeding to the emergency department. Reserve intravenous vitamin K administration for patients who are bleeding or have an INR >20. The ACCP guidelines provide recommendations on managing elevated INRs in patients receiving warfarin (TABLE 4).4
TABLE 4
Managing elevated INR
| For any INR above therapeutic range | Monitor more frequently and resume anticoagulation at an appropriately adjusted dose when the INR is at a therapeutic level. |
| INR above therapeutic range, but ≤5.0 and no significant bleeding | Lower the dose or omit a dose; INR only minimally above therapeutic range or associated with a transient causative factor may not require dose reduction. |
| INR >5.0 but <9.0, and no significant bleeding | Omit 1 to 2 doses. Alternatively, if the patient is at increased risk of bleeding, omit a dose and administer vitamin K (1-2.5 mg) orally. If more rapid reversal is required because the patient requires urgent surgery, vitamin K (<5 mg orally) will reduce INR within 24 hours. If INR remains high, give additional vitamin K (1-2 mg) orally. |
| INR ≥9.0 | Hold warfarin therapy and administer vitamin K (2.5-5 mg orally); INR will be reduced substantially in 24-48 hours. Administer additional vitamin K if necessary. |
| Serious bleeding regardless of INR | Hold warfarin and give vitamin K (10 mg by slow IV infusion). may repeat in 12 hours if necessary. Administer FFP, PCC, or rVIIa if necessary. |
| Life-threatening bleeding | Hold warfarin. Administer vitamin K (10 mg by slow IV infusion). May repeat if necessary. Administer FFP, PCC, or rVIIa along with vitamin K. |
| FFP, fresh frozen plasma; INR, international normalized ratio; PCC, prothrombin complex concentrate; rVIIa, recombinant factor Viia. | |
| Adapted from: Ansell J, et al. Chest. 2008.4 | |
7. What new anticoagulants are on the horizon?
Several alternative treatments for DVT are currently in clinical trials, and 1 recently received FDA approval.
Ximelagatran, a direct thrombin inhibitor, appeared to hold promise as an oral anticoagulant, but was denied FDA approval and eventually withdrawn by its manufacturer when reports of hepatotoxicity and possible myocardial ischemia surfaced.13,14 Other oral treatment options to be aware of include another direct thrombin inhibitor, dabigatran, and the factor Xa inhibitors apixaban and rivaroxaban.
Dabigatran (Pradaxa), an oral direct thrombin inhibitor similar to ximelagatran, received FDA approval last month for stroke prevention in atrial fibrillation. One study, RE-COVER, studied dabigatran vs warfarin for the treatment of acute VTE—both lower extremity DVT and PE. This noninferiority trial compared dabigatran 150 mg twice daily with daily warfarin adjusted to achieve an INR of 2.0 to 3.0, with the 6-month recurrence of VTE as the primary outcome. Dabigatran was found to be as effective as warfarin at preventing recurrent or fatal VTE. There was no difference in major bleeding between the dabigatran and warfarin groups, although the dabigatran group did show more major or clinically relevant nonmajor bleeding. No differences in other adverse events were observed between the 2 groups.15
Three studies, RE-MOBILIZE, RENOVATE, and RE-MODEL, compared dabigatran’s efficacy and safety with enoxaparin for the prevention of VTE after knee and hip replacement surgery. In the RE-MOBILIZE trial, dabigatran was effective compared with enoxaparin once daily, but not effective compared with twice-daily enoxaparin.16 The RE-NOVATE and RE-MODEL studies also showed dabigatran’s efficacy compared with once-daily enoxaparin.17-19 Major bleeding occurred in approximately 1% of patients in both the dabigatran and enoxaparin treatment groups, and the incidence of hepatotoxicity was similar.17-19
The RE-LY trial studied warfarin vs 2 different doses of dabigatran in atrial fibrillation for the prevention of stroke or systemic embolism. Both doses of dabigatran (110 mg twice daily or 150 mg once daily) were similar to warfarin for the study’s primary outcome, and dabigatran at the 110-mg dose had a significantly lower incidence of hemorrhagic stroke.19 Studies on the use of dabigatran in acute coronary syndrome are ongoing.
Apixaban and rivaroxaban are oral inhibitors of both free and fibrin-bound factor Xa. They are similar in activity to the currently available, injectable fondaparinux. In the RECORD 1, 2, 3, and 4 trials, rivaroxaban was compared with once- or twice-daily enoxaparin in patients undergoing hip and knee replacement surgery.20 Rivaroxaban was significantly better in preventing VTE and it had a comparable rate of major bleeding (approximately 0.2%). Rivaroxaban has been approved in Canada and Europe for thromboprophylaxis after major orthopedic surgery. Rivaroxaban was recommended for approval by an FDA advisory panel, but the FDA has not issued an approval as yet.
Phase III trials for other indications of rivaroxaban and apixaban are currently underway. The long-term safety and adverse event profiles are as yet unclear. If and when these new medications are approved, they should be used judiciously while issues related to reversibility, long-term adverse events, and monitoring are still unresolved. For some patients, warfarin may continue to be the most appropriate oral anticoagulant medication.
Your patient, a 64-year-old man, has a 4-day history of warmth and tenderness in his right calf. Two weeks earlier, he had knee replacement surgery. he left the hospital with a prescription for enoxaparin (Lovenox) 30 mg every 12 hours for 5 days (for a total of 10 doses), but he tells you that he did not get the prescription filled because of the cost. (Even with the copay, it was more than he thought the medication was worth.)
Your clinical diagnosis is a deep vein thrombosis (DVT), and this is confirmed by a venous Doppler ultrasound study showing a clot extending from the popliteal to the femoral vein. He has no signs of a pulmonary embolism (PE), no shortness of breath or chest pain, and according to your office PE prediction calculator, his probability of PE is low. He doesn’t want to go back to the hospital for treatment, and you agree that he is capable of managing his condition at home. At a weight of 98 kg, he isn’t obese and his serum creatinine and complete blood count are within normal limits.
Q How would you treat this patient?
You decide to start the patient on enoxaparin 100 mg subcutaneously every 12 hours. You teach him proper injection technique and write a prescription for 10 syringes with 1 refill. He now understands that the medication is essential and is ready to cover the copay. You also start him on warfarin 5 mg daily. You explain that when he is taking warfarin, he needs to have his blood clotting time tested frequently. He’ll need a lab test of his international normalized ratio (INR) on Day 3 and Day 5 of warfarin. If the INR is ≥2 after 5 days, he can stop enoxaparin therapy. If the INR is <2 on Day 5, he will need to continue enoxaparin until the INR is ≥2.
On Day 5, your patient’s INR is 2.5, so you tell him to stop taking enoxaparin and continue regular INR testing, getting his next test within 1 week of this office visit. His INR remains stable for 3 months on 5 mg warfarin daily. Then you get a call from the lab, telling you the patient’s INR is elevated at 4.2.
Q What could be causing your patient’s INR to be elevated?
You call the patient and ask if he has been taking his medication faithfully and whether he has been eating normally. You also ask whether he has started any new medications.
He tells you he has been taking his warfarin and hasn’t made any changes in his diet, but he is on the last day of a 7-day treatment with metronidazole for pseudomembranous colitis. He says he has had no bleeding and has not noticed any large bruises or dark stools. The elevated INR is probably a drug interaction with the metronidazole. You tell him to skip his warfarin for 1 night and then have his INR rechecked. The next day, the INR is back in the normal range. He continues on warfarin therapy. His INR remains stable and his leg pain does not recur.
TABLE W1
Important warfarin interactions*
| Anti-infectives | Cardiovascular drugs | Analgesics | Agents that affect the central nervous system | Agents that affect the GI tract | Herbal supplements | Other | |
|---|---|---|---|---|---|---|---|
| Potentiation | Ciprofloxacin Clarithromycin Cotrimoxazole Erythromycin Fluconazole Gatifloxacin Itraconazole Levofloxacin Metronidazole Tetracycline Voriconazole | Amiodarone Atorvastatin Fenofibrate Fluvastatin Gemfibrozil Lovastatin Propafenone Ropinirole Simvastatin | Acetaminophen Celecoxib Interferon Piroxicam Propoxyphene Tramadol | Alcohol (binge) Citalopram Entacapone Phenytoin Sertraline | Cimetidine Fish oil Mango Omeprazole | Boldo-fenugreek Danshen Dong quai Lyceum barbarum L Quilinggao | Anabolic steroids Fluorouracil Gemcitabine Levamisole/fluorouracil Levothyroxine Tamoxifen Tolterodine Zileuton |
| Inhibition | Dicloxacillin Griseofulvin Nafcillin Rifampin | Bosentan Cholestyramine | Azathioprine Mesalamine | Alcohol Barbiturates Carbamazepine | Avocado (large amounts) Foods and enteral nutrition high in vitamin K Soy milk Sucralfate | Ginseng Green tea | Chelation therapy Mercaptopurine Methimazole Multivitamins Propylthiouracil Raloxifene |
| Increase bleeding risk | Anticoagulants Antiplatelets | NSAIDs | Alcohol | Garlic Ginkgo Ginseng | |||
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. | |||||||
| *Not a complete list. | |||||||
| Adapted from: Ansell J, et al. Chest. 2008.4 | |||||||
CORRESPONDENCE Anne H. Metzger, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Avenue, Cincinnati, OH 45267; [email protected]
1. Minichiello T, Fogarty PF. Diagnosis and management of venous thromboembolism. Med Clin North Am. 2008;92:443-465.
2. Deitcher SR, Carman TL. Deep venous thrombosis and pulmonary embolism. Curr Treat Options Cardiovasc Med. 2002;4:223-238.
3. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):454S-545S.
4. Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):160S-198S.
5. Bussey HL, Wittkowsky AK, Hylek EM, et al. Genetic testing for warfarin dosing? Not yet ready for prime time. Pharmacotherapy. 2008;28:141-143.
6. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalizing medicine using genetic testing: the case of warfarin. American Enterprise Institute-Brookings Joint Center for Regulatory Studies. November 2006. Available at: www.reg-markets.org/publications/abstract.php?pid=1127&printversion=1. Accessed October 2, 2010.
7. Coumadin [package insert]. Princeton, NJ. Bristol-Myers Squibb; 2010.
8. Juurlink DN. Drug interactions with warfarin: what clinicians need to know. CMAJ. 2007;177:369-371.
9. Holmes DR, Kereiakes DJ, Kleiman NS, et al. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol. 2009;54:95-109.
10. Hermosillo AJ, Spinler SA. Aspirin, clopidogrel and warfarin: Is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother. 2008;42:790-805.
11. Becker RC, Meade TW, Berger PB, et al. The primary and secondary prevention of coronary artery disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):776S-814S.
12. Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol. 2006;18:162-164.
13. Bauer KA. New anticoagulants. Hematology Am Soc Hematol Educ Program. 2006;450-456.
14. Fiessinger JN, Huisman MV, Davidson BL, et al. Ximelagatran vs low-molecular weight heparin and warfarin for the treatment of deep vein thrombosis. JAMA. 2005;293:681-689.
15. Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342-2352.
16. The RE-MOBILIZE Writing Committee. Oral thrombin inhibitor dabigatran etexilate versus the North American enoxaparin regimen for the prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty. 2009;24:1-9.
17. Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost. 2007;5:2178-2185.
18. Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement. Lancet. 2007;370:949-956.
19. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Re-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
20. Hughes S. RECORD 1, 2 and 3 rivaroxaban trials published. Heartwire July 1, 2008. Available at: http://www.theheart.org/article/878097.do. Accessed November 24, 2008.
21. Fragmin [package insert]. Woodcliff Lake, NJ: Eisai; 2010.
22. Lovenox [package insert]. Greenville, NC: Sanofi-Aventis; 2009.
23. Innohep [package insert] Parsippany, NJ: LEO Pharmaceuticals Products; 2010.
24. Arixtra [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
• Start patients with a new-onset venous thrombosis on a low-molecular-weight heparin (LMWH), unfractionated heparin (UFH), or fondaparinux as well as warfarin therapy. A
• Continue LMWH,UFH, or fondaparinux with warfarin for a minimum of 5 days until the international normalized ratio (INR) is ≥2 for 24 hours. A
• Educate patients about anticoagulant therapy, dietary and medication interactions with warfarin, and signs and symptoms of bleeding. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Arterial and venous thromboses are major causes of morbidity and mortality in the United States. Each year, about 100 out of 100,000 Americans (0.1%) experience a venous thromboembolism (VTE), and the incidence is considerably higher among hospitalized patients.1 Incidence and early mortality after a first-time event increase with age. Mortality and the potential for a pulmonary embolism (PE) to occur after a deep vein thrombosis (DVT) depend on the location of the DVT and how well the DVT is managed. Proximal DVTs are more likely to develop into a PE. Mortality rates for patients with PE are as high as 17% 3 months after diagnosis.2
Anticoagulant therapy is the foundation for prevention and treatment of thromboembolic disease, and family physicians are on the front line of management when patients with DVT are discharged from the hospital. There are many therapeutic options to choose from: unfractionated heparin (UFH), low-molecular-weight heparins (LMWHs), the factor Xa inhibitor fondaparinux, direct thrombin inhibitors, or vitamin K antagonists (VKAs). All of these agents are effective, but you’ll need to keep clinical considerations and drug limitations in mind to use them properly.
The salient details for optimal use of these agents are set out in the 8th edition of the American College of Chest Physicians (ACCP) Evidence-based Clinical Practice Guidelines, released in 2008.3 But following these complex guidelines to maximize patient safety and minimize both cost and inconvenience raises many questions for the busy family physician. This article provides the answers you need to maximize your care.
1. What therapies can be used in the outpatient setting to treat acute DVT or PE?
You can manage DVT with LMWH—dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep)—or the factor Xa inhibitor fondaparinux (Arixtra) overlapped with warfarin (Coumadin). UFH is generally not recommended in the outpatient setting. Patients who are obese or have a creatinine clearance <30 mL/min will need inpatient treatment with UFH in most cases.
Outpatient management of PE based on clinical prediction rules that stratify patients by risk factors has been attempted, although the safety and efficacy of this practice have not been conclusively demonstrated. Prediction rules are available at http://www.medicalcriteria.com/criteria/car_thrombosis.htm. Note that LMWH and fondaparinux are not approved by the US Food and Drug Administration (FDA) for the outpatient treatment of PE.
Dosing guidelines for LMWH agents and fondaparinux are given in TABLE 1, and recommendations for treatment of DVT are summarized in TABLE 2.
TABLE 1
Low-molecular-weight heparins and fondaparinux dosing for DVT
| Agent | Dose |
|---|---|
| Dalteparin (Fragmin)21 | 100 units/kg SQ every 12 h or 200 units/kg SQ every 24 h |
| Enoxaparin (Lovenox)22 | 1 mg/kg SQ every 12 h or 1.5 mg/kg every 24 h |
| Tinzaparin (Innohep)23 | 175 anti-Xa IU/kg SQ every 24 h |
| Fondaparinux (Arixtra)24 | Weight <50 kg: 5 mg SQ every 24 h Weight 50-100 kg: 7.5 mg SQ every 24 h Weight >100 kg: 10 mg SQ every 24 h |
| DVT, deep vein thrombosis; SQ, subcutaneously. | |
TABLE 2
Treating DVT: Recommended options3,4,21-24
| Warfarin | UFH | LMWH |
|---|---|---|
| Starting dose 5-10 mg/d for first 1-2 days. Lower starting dose for patients with liver impairment, malnourishment, heart failure, or recent major surgery; for debilitated and elderly patients; and for patients on medications known to inhibit CYP-450 enzyme. Initial monitoring after the first 2-3 doses. Maintenance monitoring at least every 4 weeks. For acute DVT, overlap with LMWH, UFH, or fondaparinux for at least 5 days and until INR is ≥2 for 24 hours. Continue therapy for ≥3 months for patients with upper extremity DVT. | UFH is recommended for patients who are obese or have a creatinine clearance <30 mL/min; UFH is generally an inpatient treatment option, and patients may need to be admitted for therapy. | For acute DVT, LMWH daily or twice daily is recommended over UFH. Exceptions include patients who are obese or have a creatinine clearance <30 mL/min. Anti-factor Xa levels should be monitored in pregnant patients on therapeutic doses of LMWH. |
| DVT, deep vein thrombosis; INR, international normalized ratio; LMWH, low-molecular-weight heparin; UFH, unfractionated heparin. | ||
2. When, and at what dosage, should i initiate warfarin?
With a medically stable patient, you can start warfarin shortly after the first dose of LMWH or fondaparinux, and overlap both therapies for at least 5 days until the patient’s international normalized ratio (INR) is ≥2 for 24 hours. If the INR does not reach 2 within 5 days, LMWH or fondaparinux should be continued. The target INR for DVT is 2.5.
The initial dose of warfarin for most patients should be between 5 and 10 mg per day for the first 2 doses, with 10-mg doses reserved for younger patients without significant drug interactions or comorbidities.4 Consider a starting dose ≤5 mg in elderly patients, those with certain medical conditions (eg, liver disease or heart failure), and patients taking medications known to significantly inhibit warfarin metabolism.3,4TABLE 3 provides a suggested method for initiation of warfarin in ambulatory patients.
Continue warfarin for at least 3 months, and possibly longer, depending on the cause of DVT/PE and underlying or ongoing risk factors. Evaluate the risk vs benefit of continued therapy 3 months after the initial thromboembolic event. Patients with cancer, whose risk for VTE is greater, should receive LMWH for the first 3 to 6 months, followed by long-term therapy with warfarin or LMWH until the cancer is resolved.3,4
TABLE 3
Average warfarin daily dosing for INR goal 2-3
| Dosage change | Patients nonsensitive to warfarin | Patients sensitive to warfarin* | |
|---|---|---|---|
| Initial dose | 5 mg/d | 2.5 mg/d | |
| First INR | 3 days after initial dose | 3 days after initial dose | |
| <1.5 | Increase dose by 50% | 7.5 mg/d | 5 mg/d |
| 1.5-1.9 | Maintain current dose | 5 mg/d | 2.5 mg/d |
| 2-3 | Decrease dose by 50% | 2.5 mg/d | 1.25 mg/d |
| 3.1-4 | Decrease dose by ~75% | 1.25 mg/d | 0.5 mg/d |
| >4 | Hold dose | Hold | Hold |
| Next INR | 2-3 days | 2-3 days | |
| INR, international normalized ratio. | |||
| *Factors that influence sensitivity to warfarin include age >75 years, clinical congestive heart failure, diarrhea, drug interactions, elevated baseline INR, hyperthyroidism, malignancy, malnutrition, or nothing by mouth for >3 days. | |||
| Source: University of Washington Medical Center. Average daily dosing method. Available at: http://vte.son.washington.edu/docs/VTE_flexible_initiation.pdf. Accessed September 26, 2010. | |||
3. Is it time to customize anticoagulant therapy based on genetic testing?
No. Currently, FDA and ACCP guidelines do not recommend genetic testing before initiating warfarin.5,6 Theoretically, genetic testing should be helpful in predicting an individual’s optimal starting warfarin dose. At present, however, no good clinical data support this practice.5 If randomized trials show improved clinical outcomes with pharmacogenetic dosing of warfarin, genotyping may become part of clinical practice in the future.
An estimated one-third of patients on warfarin therapy may be at higher risk for adverse outcomes because they carry genes that make them more or less sensitive to warfarin.5 Variants of 2 genes—cytochrome P450 2C9 (CYP2C9) and the vitamin K oxide reductase complex 1 (VKORC1)—are thought to be responsible for this variance in warfarin response.5
Patients with variations of CYP2C9 may need lower starting doses of warfarin. Mutations in the VKORC1 gene affect the enzymes that activate vitamin K, which are the target for warfarin’s inhibitory effect on clotting. Mutations in this gene therefore result in varying sensitivities to warfarin and may be the cause of hereditary warfarin resistance in some individuals. Genetic variations in VKORC1 are estimated to occur in 14% to 37% of Caucasians and African Americans and may exist in as many as 89% of Asians.5 Several tests to detect some variants in these genes have been approved by the FDA.
In August 2007, a labeling change for Coumadin and its generics detailed the influence of gene variations on warfarin sensitivity.7 A report from the American Enterprise Institute-Brookings Joint Center for Regulatory Studies estimated that genetic testing could prevent 85,000 serious bleeding events and 17,000 strokes per year, resulting in a $1.1 billion reduction in warfarin-related health care spending. Costs of genetic testing for the 2 million Americans who begin warfarin therapy each year would be approximately $1 billion.6
4. Which warfarin–drug interactions are clinically important?
Drugs, supplements, and foods that potentiate or inhibit warfarin’s anticoagulant effect or increase the risk of bleeding are clinically important. The list of such interactions has been referred to as the 8 “As”: antibiotics, antifungals, antidepressants, antiplatelets, amiodarone, anti-inflammatories, high-dose acetaminophen, and alternative remedies.8 (For details on common warfarin interactions, see TABLE W1.)
These and other medications can affect how warfarin is absorbed, distributed, and metabolized. For example, sucralfate and bile-acid sequestrants such as cholestyramine can inhibit absorption. You can minimize this interaction by staggering the time each medication is ingested. Drugs that induce cytochrome P450 enzymes (eg, rifampin, carbamazepine) enhance warfarin clearance, while drugs that inhibit CYP enzymes (amiodarone or itraconazole) decrease warfarin clearance.2 Most clinically relevant interactions affect warfarin metabolism.
5. How should i proceed when a patient taking warfarin also needs antiplatelet medications?
Monitor warfarin more frequently in such patients and target the lower end of the INR therapeutic range (2-2.5).9 Keep an eye on your patient’s overall medication regimen and avoid medications like nonsteroidal anti-inflammatory drugs (NSAIDs) that increase bleeding risk. If NSAIDs must be used, avoid chronic use, high doses, and NSAIDs with a long half-life. You may also want to consider referral to an anticoagulation clinic.
Many of these patients have cardiac conditions for which dual antiplatelet therapy is recommended. For example, patients with coronary stents may need aspirin and clopidogrel for a specified period of time. They may have underlying atrial fibrillation or valve replacement requiring warfarin therapy. Data examining triple therapy (aspirin, clopidogrel, and warfarin) are primarily limited to patients with acute coronary syndrome or those who have had percutaneous coronary intervention. Unfortunately, the data are also retrospective, based on a small sample, and inconsistent.10 In these patients, you need to weigh the increased risk of bleeding against the proven preventive value of each of these modalities.
For patients with stents, current guidelines recommend a lower dose of aspirin and discontinuation of clopidogrel after a certain length of time, depending on the type of stent.10,11 However, 1 study showed that aspirin dose and INR values did not influence bleeding risk in patients on triple therapy.12
It is imperative that you counsel patients on triple therapy to report the first sign of bleeding.
6. What is the best approach when a patient’s INR is elevated?
You’ll need to minimize the risk of bleeding while at the same time ensuring adequate levels of anticoagulation. You can use oral vitamin K (phytonadione [Mephyton]) to reverse the effects of warfarin without inducing warfarin resistance. Avoid subcutaneous administration; the effects are unpredictable and response is delayed.4
Send patients with active or life-threatening bleeding to the emergency department. Reserve intravenous vitamin K administration for patients who are bleeding or have an INR >20. The ACCP guidelines provide recommendations on managing elevated INRs in patients receiving warfarin (TABLE 4).4
TABLE 4
Managing elevated INR
| For any INR above therapeutic range | Monitor more frequently and resume anticoagulation at an appropriately adjusted dose when the INR is at a therapeutic level. |
| INR above therapeutic range, but ≤5.0 and no significant bleeding | Lower the dose or omit a dose; INR only minimally above therapeutic range or associated with a transient causative factor may not require dose reduction. |
| INR >5.0 but <9.0, and no significant bleeding | Omit 1 to 2 doses. Alternatively, if the patient is at increased risk of bleeding, omit a dose and administer vitamin K (1-2.5 mg) orally. If more rapid reversal is required because the patient requires urgent surgery, vitamin K (<5 mg orally) will reduce INR within 24 hours. If INR remains high, give additional vitamin K (1-2 mg) orally. |
| INR ≥9.0 | Hold warfarin therapy and administer vitamin K (2.5-5 mg orally); INR will be reduced substantially in 24-48 hours. Administer additional vitamin K if necessary. |
| Serious bleeding regardless of INR | Hold warfarin and give vitamin K (10 mg by slow IV infusion). may repeat in 12 hours if necessary. Administer FFP, PCC, or rVIIa if necessary. |
| Life-threatening bleeding | Hold warfarin. Administer vitamin K (10 mg by slow IV infusion). May repeat if necessary. Administer FFP, PCC, or rVIIa along with vitamin K. |
| FFP, fresh frozen plasma; INR, international normalized ratio; PCC, prothrombin complex concentrate; rVIIa, recombinant factor Viia. | |
| Adapted from: Ansell J, et al. Chest. 2008.4 | |
7. What new anticoagulants are on the horizon?
Several alternative treatments for DVT are currently in clinical trials, and 1 recently received FDA approval.
Ximelagatran, a direct thrombin inhibitor, appeared to hold promise as an oral anticoagulant, but was denied FDA approval and eventually withdrawn by its manufacturer when reports of hepatotoxicity and possible myocardial ischemia surfaced.13,14 Other oral treatment options to be aware of include another direct thrombin inhibitor, dabigatran, and the factor Xa inhibitors apixaban and rivaroxaban.
Dabigatran (Pradaxa), an oral direct thrombin inhibitor similar to ximelagatran, received FDA approval last month for stroke prevention in atrial fibrillation. One study, RE-COVER, studied dabigatran vs warfarin for the treatment of acute VTE—both lower extremity DVT and PE. This noninferiority trial compared dabigatran 150 mg twice daily with daily warfarin adjusted to achieve an INR of 2.0 to 3.0, with the 6-month recurrence of VTE as the primary outcome. Dabigatran was found to be as effective as warfarin at preventing recurrent or fatal VTE. There was no difference in major bleeding between the dabigatran and warfarin groups, although the dabigatran group did show more major or clinically relevant nonmajor bleeding. No differences in other adverse events were observed between the 2 groups.15
Three studies, RE-MOBILIZE, RENOVATE, and RE-MODEL, compared dabigatran’s efficacy and safety with enoxaparin for the prevention of VTE after knee and hip replacement surgery. In the RE-MOBILIZE trial, dabigatran was effective compared with enoxaparin once daily, but not effective compared with twice-daily enoxaparin.16 The RE-NOVATE and RE-MODEL studies also showed dabigatran’s efficacy compared with once-daily enoxaparin.17-19 Major bleeding occurred in approximately 1% of patients in both the dabigatran and enoxaparin treatment groups, and the incidence of hepatotoxicity was similar.17-19
The RE-LY trial studied warfarin vs 2 different doses of dabigatran in atrial fibrillation for the prevention of stroke or systemic embolism. Both doses of dabigatran (110 mg twice daily or 150 mg once daily) were similar to warfarin for the study’s primary outcome, and dabigatran at the 110-mg dose had a significantly lower incidence of hemorrhagic stroke.19 Studies on the use of dabigatran in acute coronary syndrome are ongoing.
Apixaban and rivaroxaban are oral inhibitors of both free and fibrin-bound factor Xa. They are similar in activity to the currently available, injectable fondaparinux. In the RECORD 1, 2, 3, and 4 trials, rivaroxaban was compared with once- or twice-daily enoxaparin in patients undergoing hip and knee replacement surgery.20 Rivaroxaban was significantly better in preventing VTE and it had a comparable rate of major bleeding (approximately 0.2%). Rivaroxaban has been approved in Canada and Europe for thromboprophylaxis after major orthopedic surgery. Rivaroxaban was recommended for approval by an FDA advisory panel, but the FDA has not issued an approval as yet.
Phase III trials for other indications of rivaroxaban and apixaban are currently underway. The long-term safety and adverse event profiles are as yet unclear. If and when these new medications are approved, they should be used judiciously while issues related to reversibility, long-term adverse events, and monitoring are still unresolved. For some patients, warfarin may continue to be the most appropriate oral anticoagulant medication.
Your patient, a 64-year-old man, has a 4-day history of warmth and tenderness in his right calf. Two weeks earlier, he had knee replacement surgery. he left the hospital with a prescription for enoxaparin (Lovenox) 30 mg every 12 hours for 5 days (for a total of 10 doses), but he tells you that he did not get the prescription filled because of the cost. (Even with the copay, it was more than he thought the medication was worth.)
Your clinical diagnosis is a deep vein thrombosis (DVT), and this is confirmed by a venous Doppler ultrasound study showing a clot extending from the popliteal to the femoral vein. He has no signs of a pulmonary embolism (PE), no shortness of breath or chest pain, and according to your office PE prediction calculator, his probability of PE is low. He doesn’t want to go back to the hospital for treatment, and you agree that he is capable of managing his condition at home. At a weight of 98 kg, he isn’t obese and his serum creatinine and complete blood count are within normal limits.
Q How would you treat this patient?
You decide to start the patient on enoxaparin 100 mg subcutaneously every 12 hours. You teach him proper injection technique and write a prescription for 10 syringes with 1 refill. He now understands that the medication is essential and is ready to cover the copay. You also start him on warfarin 5 mg daily. You explain that when he is taking warfarin, he needs to have his blood clotting time tested frequently. He’ll need a lab test of his international normalized ratio (INR) on Day 3 and Day 5 of warfarin. If the INR is ≥2 after 5 days, he can stop enoxaparin therapy. If the INR is <2 on Day 5, he will need to continue enoxaparin until the INR is ≥2.
On Day 5, your patient’s INR is 2.5, so you tell him to stop taking enoxaparin and continue regular INR testing, getting his next test within 1 week of this office visit. His INR remains stable for 3 months on 5 mg warfarin daily. Then you get a call from the lab, telling you the patient’s INR is elevated at 4.2.
Q What could be causing your patient’s INR to be elevated?
You call the patient and ask if he has been taking his medication faithfully and whether he has been eating normally. You also ask whether he has started any new medications.
He tells you he has been taking his warfarin and hasn’t made any changes in his diet, but he is on the last day of a 7-day treatment with metronidazole for pseudomembranous colitis. He says he has had no bleeding and has not noticed any large bruises or dark stools. The elevated INR is probably a drug interaction with the metronidazole. You tell him to skip his warfarin for 1 night and then have his INR rechecked. The next day, the INR is back in the normal range. He continues on warfarin therapy. His INR remains stable and his leg pain does not recur.
TABLE W1
Important warfarin interactions*
| Anti-infectives | Cardiovascular drugs | Analgesics | Agents that affect the central nervous system | Agents that affect the GI tract | Herbal supplements | Other | |
|---|---|---|---|---|---|---|---|
| Potentiation | Ciprofloxacin Clarithromycin Cotrimoxazole Erythromycin Fluconazole Gatifloxacin Itraconazole Levofloxacin Metronidazole Tetracycline Voriconazole | Amiodarone Atorvastatin Fenofibrate Fluvastatin Gemfibrozil Lovastatin Propafenone Ropinirole Simvastatin | Acetaminophen Celecoxib Interferon Piroxicam Propoxyphene Tramadol | Alcohol (binge) Citalopram Entacapone Phenytoin Sertraline | Cimetidine Fish oil Mango Omeprazole | Boldo-fenugreek Danshen Dong quai Lyceum barbarum L Quilinggao | Anabolic steroids Fluorouracil Gemcitabine Levamisole/fluorouracil Levothyroxine Tamoxifen Tolterodine Zileuton |
| Inhibition | Dicloxacillin Griseofulvin Nafcillin Rifampin | Bosentan Cholestyramine | Azathioprine Mesalamine | Alcohol Barbiturates Carbamazepine | Avocado (large amounts) Foods and enteral nutrition high in vitamin K Soy milk Sucralfate | Ginseng Green tea | Chelation therapy Mercaptopurine Methimazole Multivitamins Propylthiouracil Raloxifene |
| Increase bleeding risk | Anticoagulants Antiplatelets | NSAIDs | Alcohol | Garlic Ginkgo Ginseng | |||
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. | |||||||
| *Not a complete list. | |||||||
| Adapted from: Ansell J, et al. Chest. 2008.4 | |||||||
CORRESPONDENCE Anne H. Metzger, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Avenue, Cincinnati, OH 45267; [email protected]
• Start patients with a new-onset venous thrombosis on a low-molecular-weight heparin (LMWH), unfractionated heparin (UFH), or fondaparinux as well as warfarin therapy. A
• Continue LMWH,UFH, or fondaparinux with warfarin for a minimum of 5 days until the international normalized ratio (INR) is ≥2 for 24 hours. A
• Educate patients about anticoagulant therapy, dietary and medication interactions with warfarin, and signs and symptoms of bleeding. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Arterial and venous thromboses are major causes of morbidity and mortality in the United States. Each year, about 100 out of 100,000 Americans (0.1%) experience a venous thromboembolism (VTE), and the incidence is considerably higher among hospitalized patients.1 Incidence and early mortality after a first-time event increase with age. Mortality and the potential for a pulmonary embolism (PE) to occur after a deep vein thrombosis (DVT) depend on the location of the DVT and how well the DVT is managed. Proximal DVTs are more likely to develop into a PE. Mortality rates for patients with PE are as high as 17% 3 months after diagnosis.2
Anticoagulant therapy is the foundation for prevention and treatment of thromboembolic disease, and family physicians are on the front line of management when patients with DVT are discharged from the hospital. There are many therapeutic options to choose from: unfractionated heparin (UFH), low-molecular-weight heparins (LMWHs), the factor Xa inhibitor fondaparinux, direct thrombin inhibitors, or vitamin K antagonists (VKAs). All of these agents are effective, but you’ll need to keep clinical considerations and drug limitations in mind to use them properly.
The salient details for optimal use of these agents are set out in the 8th edition of the American College of Chest Physicians (ACCP) Evidence-based Clinical Practice Guidelines, released in 2008.3 But following these complex guidelines to maximize patient safety and minimize both cost and inconvenience raises many questions for the busy family physician. This article provides the answers you need to maximize your care.
1. What therapies can be used in the outpatient setting to treat acute DVT or PE?
You can manage DVT with LMWH—dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep)—or the factor Xa inhibitor fondaparinux (Arixtra) overlapped with warfarin (Coumadin). UFH is generally not recommended in the outpatient setting. Patients who are obese or have a creatinine clearance <30 mL/min will need inpatient treatment with UFH in most cases.
Outpatient management of PE based on clinical prediction rules that stratify patients by risk factors has been attempted, although the safety and efficacy of this practice have not been conclusively demonstrated. Prediction rules are available at http://www.medicalcriteria.com/criteria/car_thrombosis.htm. Note that LMWH and fondaparinux are not approved by the US Food and Drug Administration (FDA) for the outpatient treatment of PE.
Dosing guidelines for LMWH agents and fondaparinux are given in TABLE 1, and recommendations for treatment of DVT are summarized in TABLE 2.
TABLE 1
Low-molecular-weight heparins and fondaparinux dosing for DVT
| Agent | Dose |
|---|---|
| Dalteparin (Fragmin)21 | 100 units/kg SQ every 12 h or 200 units/kg SQ every 24 h |
| Enoxaparin (Lovenox)22 | 1 mg/kg SQ every 12 h or 1.5 mg/kg every 24 h |
| Tinzaparin (Innohep)23 | 175 anti-Xa IU/kg SQ every 24 h |
| Fondaparinux (Arixtra)24 | Weight <50 kg: 5 mg SQ every 24 h Weight 50-100 kg: 7.5 mg SQ every 24 h Weight >100 kg: 10 mg SQ every 24 h |
| DVT, deep vein thrombosis; SQ, subcutaneously. | |
TABLE 2
Treating DVT: Recommended options3,4,21-24
| Warfarin | UFH | LMWH |
|---|---|---|
| Starting dose 5-10 mg/d for first 1-2 days. Lower starting dose for patients with liver impairment, malnourishment, heart failure, or recent major surgery; for debilitated and elderly patients; and for patients on medications known to inhibit CYP-450 enzyme. Initial monitoring after the first 2-3 doses. Maintenance monitoring at least every 4 weeks. For acute DVT, overlap with LMWH, UFH, or fondaparinux for at least 5 days and until INR is ≥2 for 24 hours. Continue therapy for ≥3 months for patients with upper extremity DVT. | UFH is recommended for patients who are obese or have a creatinine clearance <30 mL/min; UFH is generally an inpatient treatment option, and patients may need to be admitted for therapy. | For acute DVT, LMWH daily or twice daily is recommended over UFH. Exceptions include patients who are obese or have a creatinine clearance <30 mL/min. Anti-factor Xa levels should be monitored in pregnant patients on therapeutic doses of LMWH. |
| DVT, deep vein thrombosis; INR, international normalized ratio; LMWH, low-molecular-weight heparin; UFH, unfractionated heparin. | ||
2. When, and at what dosage, should i initiate warfarin?
With a medically stable patient, you can start warfarin shortly after the first dose of LMWH or fondaparinux, and overlap both therapies for at least 5 days until the patient’s international normalized ratio (INR) is ≥2 for 24 hours. If the INR does not reach 2 within 5 days, LMWH or fondaparinux should be continued. The target INR for DVT is 2.5.
The initial dose of warfarin for most patients should be between 5 and 10 mg per day for the first 2 doses, with 10-mg doses reserved for younger patients without significant drug interactions or comorbidities.4 Consider a starting dose ≤5 mg in elderly patients, those with certain medical conditions (eg, liver disease or heart failure), and patients taking medications known to significantly inhibit warfarin metabolism.3,4TABLE 3 provides a suggested method for initiation of warfarin in ambulatory patients.
Continue warfarin for at least 3 months, and possibly longer, depending on the cause of DVT/PE and underlying or ongoing risk factors. Evaluate the risk vs benefit of continued therapy 3 months after the initial thromboembolic event. Patients with cancer, whose risk for VTE is greater, should receive LMWH for the first 3 to 6 months, followed by long-term therapy with warfarin or LMWH until the cancer is resolved.3,4
TABLE 3
Average warfarin daily dosing for INR goal 2-3
| Dosage change | Patients nonsensitive to warfarin | Patients sensitive to warfarin* | |
|---|---|---|---|
| Initial dose | 5 mg/d | 2.5 mg/d | |
| First INR | 3 days after initial dose | 3 days after initial dose | |
| <1.5 | Increase dose by 50% | 7.5 mg/d | 5 mg/d |
| 1.5-1.9 | Maintain current dose | 5 mg/d | 2.5 mg/d |
| 2-3 | Decrease dose by 50% | 2.5 mg/d | 1.25 mg/d |
| 3.1-4 | Decrease dose by ~75% | 1.25 mg/d | 0.5 mg/d |
| >4 | Hold dose | Hold | Hold |
| Next INR | 2-3 days | 2-3 days | |
| INR, international normalized ratio. | |||
| *Factors that influence sensitivity to warfarin include age >75 years, clinical congestive heart failure, diarrhea, drug interactions, elevated baseline INR, hyperthyroidism, malignancy, malnutrition, or nothing by mouth for >3 days. | |||
| Source: University of Washington Medical Center. Average daily dosing method. Available at: http://vte.son.washington.edu/docs/VTE_flexible_initiation.pdf. Accessed September 26, 2010. | |||
3. Is it time to customize anticoagulant therapy based on genetic testing?
No. Currently, FDA and ACCP guidelines do not recommend genetic testing before initiating warfarin.5,6 Theoretically, genetic testing should be helpful in predicting an individual’s optimal starting warfarin dose. At present, however, no good clinical data support this practice.5 If randomized trials show improved clinical outcomes with pharmacogenetic dosing of warfarin, genotyping may become part of clinical practice in the future.
An estimated one-third of patients on warfarin therapy may be at higher risk for adverse outcomes because they carry genes that make them more or less sensitive to warfarin.5 Variants of 2 genes—cytochrome P450 2C9 (CYP2C9) and the vitamin K oxide reductase complex 1 (VKORC1)—are thought to be responsible for this variance in warfarin response.5
Patients with variations of CYP2C9 may need lower starting doses of warfarin. Mutations in the VKORC1 gene affect the enzymes that activate vitamin K, which are the target for warfarin’s inhibitory effect on clotting. Mutations in this gene therefore result in varying sensitivities to warfarin and may be the cause of hereditary warfarin resistance in some individuals. Genetic variations in VKORC1 are estimated to occur in 14% to 37% of Caucasians and African Americans and may exist in as many as 89% of Asians.5 Several tests to detect some variants in these genes have been approved by the FDA.
In August 2007, a labeling change for Coumadin and its generics detailed the influence of gene variations on warfarin sensitivity.7 A report from the American Enterprise Institute-Brookings Joint Center for Regulatory Studies estimated that genetic testing could prevent 85,000 serious bleeding events and 17,000 strokes per year, resulting in a $1.1 billion reduction in warfarin-related health care spending. Costs of genetic testing for the 2 million Americans who begin warfarin therapy each year would be approximately $1 billion.6
4. Which warfarin–drug interactions are clinically important?
Drugs, supplements, and foods that potentiate or inhibit warfarin’s anticoagulant effect or increase the risk of bleeding are clinically important. The list of such interactions has been referred to as the 8 “As”: antibiotics, antifungals, antidepressants, antiplatelets, amiodarone, anti-inflammatories, high-dose acetaminophen, and alternative remedies.8 (For details on common warfarin interactions, see TABLE W1.)
These and other medications can affect how warfarin is absorbed, distributed, and metabolized. For example, sucralfate and bile-acid sequestrants such as cholestyramine can inhibit absorption. You can minimize this interaction by staggering the time each medication is ingested. Drugs that induce cytochrome P450 enzymes (eg, rifampin, carbamazepine) enhance warfarin clearance, while drugs that inhibit CYP enzymes (amiodarone or itraconazole) decrease warfarin clearance.2 Most clinically relevant interactions affect warfarin metabolism.
5. How should i proceed when a patient taking warfarin also needs antiplatelet medications?
Monitor warfarin more frequently in such patients and target the lower end of the INR therapeutic range (2-2.5).9 Keep an eye on your patient’s overall medication regimen and avoid medications like nonsteroidal anti-inflammatory drugs (NSAIDs) that increase bleeding risk. If NSAIDs must be used, avoid chronic use, high doses, and NSAIDs with a long half-life. You may also want to consider referral to an anticoagulation clinic.
Many of these patients have cardiac conditions for which dual antiplatelet therapy is recommended. For example, patients with coronary stents may need aspirin and clopidogrel for a specified period of time. They may have underlying atrial fibrillation or valve replacement requiring warfarin therapy. Data examining triple therapy (aspirin, clopidogrel, and warfarin) are primarily limited to patients with acute coronary syndrome or those who have had percutaneous coronary intervention. Unfortunately, the data are also retrospective, based on a small sample, and inconsistent.10 In these patients, you need to weigh the increased risk of bleeding against the proven preventive value of each of these modalities.
For patients with stents, current guidelines recommend a lower dose of aspirin and discontinuation of clopidogrel after a certain length of time, depending on the type of stent.10,11 However, 1 study showed that aspirin dose and INR values did not influence bleeding risk in patients on triple therapy.12
It is imperative that you counsel patients on triple therapy to report the first sign of bleeding.
6. What is the best approach when a patient’s INR is elevated?
You’ll need to minimize the risk of bleeding while at the same time ensuring adequate levels of anticoagulation. You can use oral vitamin K (phytonadione [Mephyton]) to reverse the effects of warfarin without inducing warfarin resistance. Avoid subcutaneous administration; the effects are unpredictable and response is delayed.4
Send patients with active or life-threatening bleeding to the emergency department. Reserve intravenous vitamin K administration for patients who are bleeding or have an INR >20. The ACCP guidelines provide recommendations on managing elevated INRs in patients receiving warfarin (TABLE 4).4
TABLE 4
Managing elevated INR
| For any INR above therapeutic range | Monitor more frequently and resume anticoagulation at an appropriately adjusted dose when the INR is at a therapeutic level. |
| INR above therapeutic range, but ≤5.0 and no significant bleeding | Lower the dose or omit a dose; INR only minimally above therapeutic range or associated with a transient causative factor may not require dose reduction. |
| INR >5.0 but <9.0, and no significant bleeding | Omit 1 to 2 doses. Alternatively, if the patient is at increased risk of bleeding, omit a dose and administer vitamin K (1-2.5 mg) orally. If more rapid reversal is required because the patient requires urgent surgery, vitamin K (<5 mg orally) will reduce INR within 24 hours. If INR remains high, give additional vitamin K (1-2 mg) orally. |
| INR ≥9.0 | Hold warfarin therapy and administer vitamin K (2.5-5 mg orally); INR will be reduced substantially in 24-48 hours. Administer additional vitamin K if necessary. |
| Serious bleeding regardless of INR | Hold warfarin and give vitamin K (10 mg by slow IV infusion). may repeat in 12 hours if necessary. Administer FFP, PCC, or rVIIa if necessary. |
| Life-threatening bleeding | Hold warfarin. Administer vitamin K (10 mg by slow IV infusion). May repeat if necessary. Administer FFP, PCC, or rVIIa along with vitamin K. |
| FFP, fresh frozen plasma; INR, international normalized ratio; PCC, prothrombin complex concentrate; rVIIa, recombinant factor Viia. | |
| Adapted from: Ansell J, et al. Chest. 2008.4 | |
7. What new anticoagulants are on the horizon?
Several alternative treatments for DVT are currently in clinical trials, and 1 recently received FDA approval.
Ximelagatran, a direct thrombin inhibitor, appeared to hold promise as an oral anticoagulant, but was denied FDA approval and eventually withdrawn by its manufacturer when reports of hepatotoxicity and possible myocardial ischemia surfaced.13,14 Other oral treatment options to be aware of include another direct thrombin inhibitor, dabigatran, and the factor Xa inhibitors apixaban and rivaroxaban.
Dabigatran (Pradaxa), an oral direct thrombin inhibitor similar to ximelagatran, received FDA approval last month for stroke prevention in atrial fibrillation. One study, RE-COVER, studied dabigatran vs warfarin for the treatment of acute VTE—both lower extremity DVT and PE. This noninferiority trial compared dabigatran 150 mg twice daily with daily warfarin adjusted to achieve an INR of 2.0 to 3.0, with the 6-month recurrence of VTE as the primary outcome. Dabigatran was found to be as effective as warfarin at preventing recurrent or fatal VTE. There was no difference in major bleeding between the dabigatran and warfarin groups, although the dabigatran group did show more major or clinically relevant nonmajor bleeding. No differences in other adverse events were observed between the 2 groups.15
Three studies, RE-MOBILIZE, RENOVATE, and RE-MODEL, compared dabigatran’s efficacy and safety with enoxaparin for the prevention of VTE after knee and hip replacement surgery. In the RE-MOBILIZE trial, dabigatran was effective compared with enoxaparin once daily, but not effective compared with twice-daily enoxaparin.16 The RE-NOVATE and RE-MODEL studies also showed dabigatran’s efficacy compared with once-daily enoxaparin.17-19 Major bleeding occurred in approximately 1% of patients in both the dabigatran and enoxaparin treatment groups, and the incidence of hepatotoxicity was similar.17-19
The RE-LY trial studied warfarin vs 2 different doses of dabigatran in atrial fibrillation for the prevention of stroke or systemic embolism. Both doses of dabigatran (110 mg twice daily or 150 mg once daily) were similar to warfarin for the study’s primary outcome, and dabigatran at the 110-mg dose had a significantly lower incidence of hemorrhagic stroke.19 Studies on the use of dabigatran in acute coronary syndrome are ongoing.
Apixaban and rivaroxaban are oral inhibitors of both free and fibrin-bound factor Xa. They are similar in activity to the currently available, injectable fondaparinux. In the RECORD 1, 2, 3, and 4 trials, rivaroxaban was compared with once- or twice-daily enoxaparin in patients undergoing hip and knee replacement surgery.20 Rivaroxaban was significantly better in preventing VTE and it had a comparable rate of major bleeding (approximately 0.2%). Rivaroxaban has been approved in Canada and Europe for thromboprophylaxis after major orthopedic surgery. Rivaroxaban was recommended for approval by an FDA advisory panel, but the FDA has not issued an approval as yet.
Phase III trials for other indications of rivaroxaban and apixaban are currently underway. The long-term safety and adverse event profiles are as yet unclear. If and when these new medications are approved, they should be used judiciously while issues related to reversibility, long-term adverse events, and monitoring are still unresolved. For some patients, warfarin may continue to be the most appropriate oral anticoagulant medication.
Your patient, a 64-year-old man, has a 4-day history of warmth and tenderness in his right calf. Two weeks earlier, he had knee replacement surgery. he left the hospital with a prescription for enoxaparin (Lovenox) 30 mg every 12 hours for 5 days (for a total of 10 doses), but he tells you that he did not get the prescription filled because of the cost. (Even with the copay, it was more than he thought the medication was worth.)
Your clinical diagnosis is a deep vein thrombosis (DVT), and this is confirmed by a venous Doppler ultrasound study showing a clot extending from the popliteal to the femoral vein. He has no signs of a pulmonary embolism (PE), no shortness of breath or chest pain, and according to your office PE prediction calculator, his probability of PE is low. He doesn’t want to go back to the hospital for treatment, and you agree that he is capable of managing his condition at home. At a weight of 98 kg, he isn’t obese and his serum creatinine and complete blood count are within normal limits.
Q How would you treat this patient?
You decide to start the patient on enoxaparin 100 mg subcutaneously every 12 hours. You teach him proper injection technique and write a prescription for 10 syringes with 1 refill. He now understands that the medication is essential and is ready to cover the copay. You also start him on warfarin 5 mg daily. You explain that when he is taking warfarin, he needs to have his blood clotting time tested frequently. He’ll need a lab test of his international normalized ratio (INR) on Day 3 and Day 5 of warfarin. If the INR is ≥2 after 5 days, he can stop enoxaparin therapy. If the INR is <2 on Day 5, he will need to continue enoxaparin until the INR is ≥2.
On Day 5, your patient’s INR is 2.5, so you tell him to stop taking enoxaparin and continue regular INR testing, getting his next test within 1 week of this office visit. His INR remains stable for 3 months on 5 mg warfarin daily. Then you get a call from the lab, telling you the patient’s INR is elevated at 4.2.
Q What could be causing your patient’s INR to be elevated?
You call the patient and ask if he has been taking his medication faithfully and whether he has been eating normally. You also ask whether he has started any new medications.
He tells you he has been taking his warfarin and hasn’t made any changes in his diet, but he is on the last day of a 7-day treatment with metronidazole for pseudomembranous colitis. He says he has had no bleeding and has not noticed any large bruises or dark stools. The elevated INR is probably a drug interaction with the metronidazole. You tell him to skip his warfarin for 1 night and then have his INR rechecked. The next day, the INR is back in the normal range. He continues on warfarin therapy. His INR remains stable and his leg pain does not recur.
TABLE W1
Important warfarin interactions*
| Anti-infectives | Cardiovascular drugs | Analgesics | Agents that affect the central nervous system | Agents that affect the GI tract | Herbal supplements | Other | |
|---|---|---|---|---|---|---|---|
| Potentiation | Ciprofloxacin Clarithromycin Cotrimoxazole Erythromycin Fluconazole Gatifloxacin Itraconazole Levofloxacin Metronidazole Tetracycline Voriconazole | Amiodarone Atorvastatin Fenofibrate Fluvastatin Gemfibrozil Lovastatin Propafenone Ropinirole Simvastatin | Acetaminophen Celecoxib Interferon Piroxicam Propoxyphene Tramadol | Alcohol (binge) Citalopram Entacapone Phenytoin Sertraline | Cimetidine Fish oil Mango Omeprazole | Boldo-fenugreek Danshen Dong quai Lyceum barbarum L Quilinggao | Anabolic steroids Fluorouracil Gemcitabine Levamisole/fluorouracil Levothyroxine Tamoxifen Tolterodine Zileuton |
| Inhibition | Dicloxacillin Griseofulvin Nafcillin Rifampin | Bosentan Cholestyramine | Azathioprine Mesalamine | Alcohol Barbiturates Carbamazepine | Avocado (large amounts) Foods and enteral nutrition high in vitamin K Soy milk Sucralfate | Ginseng Green tea | Chelation therapy Mercaptopurine Methimazole Multivitamins Propylthiouracil Raloxifene |
| Increase bleeding risk | Anticoagulants Antiplatelets | NSAIDs | Alcohol | Garlic Ginkgo Ginseng | |||
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. | |||||||
| *Not a complete list. | |||||||
| Adapted from: Ansell J, et al. Chest. 2008.4 | |||||||
CORRESPONDENCE Anne H. Metzger, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Avenue, Cincinnati, OH 45267; [email protected]
1. Minichiello T, Fogarty PF. Diagnosis and management of venous thromboembolism. Med Clin North Am. 2008;92:443-465.
2. Deitcher SR, Carman TL. Deep venous thrombosis and pulmonary embolism. Curr Treat Options Cardiovasc Med. 2002;4:223-238.
3. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):454S-545S.
4. Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):160S-198S.
5. Bussey HL, Wittkowsky AK, Hylek EM, et al. Genetic testing for warfarin dosing? Not yet ready for prime time. Pharmacotherapy. 2008;28:141-143.
6. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalizing medicine using genetic testing: the case of warfarin. American Enterprise Institute-Brookings Joint Center for Regulatory Studies. November 2006. Available at: www.reg-markets.org/publications/abstract.php?pid=1127&printversion=1. Accessed October 2, 2010.
7. Coumadin [package insert]. Princeton, NJ. Bristol-Myers Squibb; 2010.
8. Juurlink DN. Drug interactions with warfarin: what clinicians need to know. CMAJ. 2007;177:369-371.
9. Holmes DR, Kereiakes DJ, Kleiman NS, et al. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol. 2009;54:95-109.
10. Hermosillo AJ, Spinler SA. Aspirin, clopidogrel and warfarin: Is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother. 2008;42:790-805.
11. Becker RC, Meade TW, Berger PB, et al. The primary and secondary prevention of coronary artery disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):776S-814S.
12. Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol. 2006;18:162-164.
13. Bauer KA. New anticoagulants. Hematology Am Soc Hematol Educ Program. 2006;450-456.
14. Fiessinger JN, Huisman MV, Davidson BL, et al. Ximelagatran vs low-molecular weight heparin and warfarin for the treatment of deep vein thrombosis. JAMA. 2005;293:681-689.
15. Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342-2352.
16. The RE-MOBILIZE Writing Committee. Oral thrombin inhibitor dabigatran etexilate versus the North American enoxaparin regimen for the prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty. 2009;24:1-9.
17. Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost. 2007;5:2178-2185.
18. Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement. Lancet. 2007;370:949-956.
19. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Re-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
20. Hughes S. RECORD 1, 2 and 3 rivaroxaban trials published. Heartwire July 1, 2008. Available at: http://www.theheart.org/article/878097.do. Accessed November 24, 2008.
21. Fragmin [package insert]. Woodcliff Lake, NJ: Eisai; 2010.
22. Lovenox [package insert]. Greenville, NC: Sanofi-Aventis; 2009.
23. Innohep [package insert] Parsippany, NJ: LEO Pharmaceuticals Products; 2010.
24. Arixtra [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
1. Minichiello T, Fogarty PF. Diagnosis and management of venous thromboembolism. Med Clin North Am. 2008;92:443-465.
2. Deitcher SR, Carman TL. Deep venous thrombosis and pulmonary embolism. Curr Treat Options Cardiovasc Med. 2002;4:223-238.
3. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):454S-545S.
4. Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):160S-198S.
5. Bussey HL, Wittkowsky AK, Hylek EM, et al. Genetic testing for warfarin dosing? Not yet ready for prime time. Pharmacotherapy. 2008;28:141-143.
6. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalizing medicine using genetic testing: the case of warfarin. American Enterprise Institute-Brookings Joint Center for Regulatory Studies. November 2006. Available at: www.reg-markets.org/publications/abstract.php?pid=1127&printversion=1. Accessed October 2, 2010.
7. Coumadin [package insert]. Princeton, NJ. Bristol-Myers Squibb; 2010.
8. Juurlink DN. Drug interactions with warfarin: what clinicians need to know. CMAJ. 2007;177:369-371.
9. Holmes DR, Kereiakes DJ, Kleiman NS, et al. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol. 2009;54:95-109.
10. Hermosillo AJ, Spinler SA. Aspirin, clopidogrel and warfarin: Is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother. 2008;42:790-805.
11. Becker RC, Meade TW, Berger PB, et al. The primary and secondary prevention of coronary artery disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):776S-814S.
12. Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol. 2006;18:162-164.
13. Bauer KA. New anticoagulants. Hematology Am Soc Hematol Educ Program. 2006;450-456.
14. Fiessinger JN, Huisman MV, Davidson BL, et al. Ximelagatran vs low-molecular weight heparin and warfarin for the treatment of deep vein thrombosis. JAMA. 2005;293:681-689.
15. Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342-2352.
16. The RE-MOBILIZE Writing Committee. Oral thrombin inhibitor dabigatran etexilate versus the North American enoxaparin regimen for the prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty. 2009;24:1-9.
17. Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost. 2007;5:2178-2185.
18. Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement. Lancet. 2007;370:949-956.
19. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Re-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
20. Hughes S. RECORD 1, 2 and 3 rivaroxaban trials published. Heartwire July 1, 2008. Available at: http://www.theheart.org/article/878097.do. Accessed November 24, 2008.
21. Fragmin [package insert]. Woodcliff Lake, NJ: Eisai; 2010.
22. Lovenox [package insert]. Greenville, NC: Sanofi-Aventis; 2009.
23. Innohep [package insert] Parsippany, NJ: LEO Pharmaceuticals Products; 2010.
24. Arixtra [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
Which NSAID for your patient with osteoarthritis?
• In selecting an NSAID, assess a patient’s baseline cardiovascular (CV) and gastrointestinal (GI) risks and the potential for medication-related incremental CV and GI toxicity. C
• For patients with increased CV risk (taking aspirin for established CV risk) and low GI risk, the preferred agent is naproxen. Consider adding a proton pump inhibitor (PPI) or misoprostol, as dual therapy with aspirin and naproxen may warrant gastroprotection. A
• For patients with moderate GI risk and low CV risk, use a nonselective NSAID with a PPI or misoprostol; if GI risk is high, use a cyclooxygenase (COX)-2–selective NSAID and gastroprotection. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although clinicians have considerable experience in using analgesics and nonsteroidal anti-inflammatory drugs (NSAIDs) to relieve the pain of osteoarthritis (OA), emerging data have made the task of weighing benefits and risks of each agent more complex.1 In this article, we review the latest evidence for NSAIDs and provide a foundation on which you can make more informed decisions for controlling OA pain and—in conjunction with education, physical therapy, exercise, and cognitive and behavioral approaches2,3—improve patients’ daily function and quality of life.
Agents for OA pain relief: Benefits and trade-offs
Treatment options for OA pain are the analgesic acetaminophen and the NSAIDs, comprising both nonselective agents and the cyclooxygenase (COX)-2–selective inhibitors.
NSAIDs inhibit COX, a key enzyme in the biosynthesis of prostanoids, including the prostaglandins and leukotrienes, which are important mediators of pain. The COX-1 isoform is constantly expressed in tissue. It regulates protection of the gastric mucosa, platelet activation, and renal function. In contrast, COX-2 is induced primarily in response to inflammatory stimuli.
The nonselective NSAIDs inhibit both isoforms of COX. The anti-inflammatory and analgesic effects of the NSAIDs result primarily from COX-2 inhibition. Inhibition of COX-1 is largely responsible for the gastrointestinal (GI) ulceration and anti-platelet-promoted bleeding that can occur with these drugs.4
The COX-2–selective inhibitors were developed to spare the normal “housekeeping” functions of COX-1. This benefit, however, has been diminished by the adverse cardiovascular (CV) events occurring with selective inhibition of COX-2, owing to the expression of this isoform in vasculature and the kidneys.4 Increased risk of CV events may also occur with nonselective NSAIDs.
Acetaminophen’s mechanism of action is poorly understood. It is a weak inhibitor of COX-1 and COX-2, but it most likely acts centrally in the hypothalamus and spinal cord, rather than peripherally in joint cartilage where inflammation and damage occur.5
Revised treatment guidelines in brief
The American College of Rheumatology (ACR) and the Osteoarthritis Research Society International (OARSI) have published treatment recommendations for OA.1-3
The recent ACR publication noted that nonselective NSAIDs are more effective than acetaminophen for treating OA pain, but that the differences are small.1 Because of costs and the risk of adverse events associated with NSAID use, the ACR guidelines recommend that patients with mild to moderate OA pain receive a trial of acetaminophen initially; patients who do not respond could then receive NSAIDs. With moderate to severe OA pain, initial treatment with nonselective NSAIDs is appropriate.1,3
The OARSI guidelines2 state that “acetaminophen (up to 4 g/d) can be an effective initial oral analgesic for treatment of mild to moderate pain in patients with knee or hip OA.” The guidelines warn, however, that recent evidence has questioned both the efficacy and safety of long-term acetaminophen use in doses up to 4 g/d. The OARSI guidelines, like the ACR guidelines, recommend alternative pharmacotherapy when patients do not respond to acetaminophen for mild to moderate OA pain, or when OA pain is more severe. NSAIDs are most appropriately prescribed at the lowest effective dose for the shortest possible time.2
Accounting for risk factors. Current guidelines emphasize the importance of selecting treatments based on a patient’s CV and GI risk profiles. For patients with CV risk factors, use nonselective NSAIDs and COX-2–selective inhibitors with caution. For patients with increased GI risk, use either a COX-2–selective inhibitor or a nonselective NSAID with a proton pump inhibitor (PPI) or misoprostol.2
The evidence underlying guideline revisions
Selecting an agent that optimally balances efficacy and safety requires that we consider the complexities of 3 competing clinical concerns—relief of arthritis pain, CV toxicity, and GI toxicity.6 We review here the evidence supporting the revised recommendations.
Acetaminophen: A good option, but there are better ones
Acetaminophen relieves OA pain, but not as effectively as nonselective NSAIDs.1,3,7 A Cochrane meta-analysis showed that although acetaminophen was superior to placebo for reducing OA pain, it was less effective than either nonselective NSAIDs or COX-2–selective NSAIDs for reducing pain and improving functional status, especially in patients with moderate pain.7
Acetaminophen at higher doses has been associated with GI toxicity.8 In a case-control study, acetaminophen at doses ≥2 g/d increased the risk of upper GI bleeding or perforation.9 A cohort study showed that doses of acetaminophen >3 g/d led to higher rates of upper GI events (GI hospitalization, ulcer, and dyspepsia) comparable to those seen with NSAIDs.10 It remains unclear if the acetaminophen in this trial caused GI adverse events among all patients due to the higher doses alone, or if the rates reflected increases in adverse events expected among high-risk GI patients or concomitant NSAID users.10 Furthermore, healthy adults who ingested 4 g acetaminophen each day for 2 weeks exhibited significant elevations of serum alanine aminotransferase levels, suggestive of liver injury.11
Caution is justified with prolonged use of acetaminophen at high doses, particularly in alcohol users. In cohort studies with women and men, acetaminophen has been associated with an increased risk of incident hypertension.12,13 In case-control studies, long-term use has also been dose-dependently associated with an increased risk of chronic renal failure.14,15
Nonselective NSAIDs: Keep GI risks in mind
All nonselective NSAIDs, when administered at equivalent therapeutic doses (same degree of COX inhibition), appear to have comparable efficacy in relieving OA pain. Analgesia is dose dependent, which enables patients to start therapy at lower over-the-counter (OTC) doses and escalate to higher prescription doses as needed.3 The OTC dose range of ibuprofen is 200 to 400 mg 3 times a day, to a maximum of 1200 mg/d;16 the maximum prescription dose is 3200 mg/d.17 Similarly, the maximum dose of OTC naproxen is 660 mg/d,18 although by prescription it can be given up to 1500 mg/d.19
NSAIDs confer a dose-related risk for GI adverse events, including ulcers and bleeding. Patients with a history of ulcers and those at advanced age are at greater risk;20 those with a history of an ulcer bleed are at the greatest risk for an adverse event. Also at increased risk are those taking high doses of an NSAID, multiple NSAIDs (eg, concomitant low-dose aspirin), or anticoagulant or antiplatelet agents.21
Recent data suggest that nonselective NSAIDs, with the exception of naproxen, may increase CV risk on a level seen with COX-2–selective inhibitors.22,23 In a meta-analysis of 91 randomized active-controlled trials, a comparison of COX-2–selective inhibitors and non-naproxen nonselective NSAIDs showed no significant difference in the risk of myocardial infarction (MI) (relative risk [RR]=1.20; 95% confidence interval [CI], 0.85–1.68); however, COX-2–selective inhibitors had an increased risk compared with naproxen (RR=2.04; 95% CI, 1.41–2.96).23 In another meta-analysis of 11 observational studies, naproxen reduced the risk of MI compared with COX-2–selective inhibitors and other nonselective NSAIDs (RR=0.86; 95% CI, 0.75–0.99).22 An increased risk of incident hypertension has been associated with frequent NSAID use in cohort studies in women and men.12,13
COX-2–selective NSAIDs: Good on gut, but increase MI risk
COX-2–selective NSAIDs lower the incidence of upper GI tract complications compared with nonselective agents, while maintaining comparable efficacy in pain relief, both when used alone (without concomitant aspirin therapy)20,24,25 and in combination with PPIs.26,27
But despite their GI safety profile, the COX-2–selective NSAIDs increased the risk of MI and ischemic cerebrovascular events in trials where they were being studied for arthritis pain and for GI polyp prevention.22,28,29 Among the proposed mechanisms for this effect is that selective COX-2 inhibition reduces the level of the antithrombotic prostanoid, prostacyclin, relative to the level of the prothrombotic prostanoid, thromboxane, thereby leading to a prothrombotic tendency.30
Rofecoxib and valdecoxib were withdrawn from the market in the United States by the manufacturers after the drugs were linked to serious CV adverse effects—and in the case of valdecoxib, to a serious skin reaction.30 Celecoxib remains commercially available in the United States.30 The CV risks associated with celecoxib are dose related, with once-daily dosing (400 mg/d) associated with a much lower risk than twice-daily dosing (200 or 400 mg twice a day).31 The recommended dose is 200 mg/d.32
The deleterious impact of combining low-dose aspirin with NSAIDs
Many patients who take NSAIDs also require aspirin for cardioprotection. Catella-Lawson and colleagues33 investigated the potential interactions between aspirin and several NSAIDs used in managing OA. They found that ibuprofen, when taken before aspirin, reduced aspirin’s inhibition of platelet aggregation, demonstrating potential impairment of aspirin’s cardioprotective effect.33 Subsequent observational studies have supported these in vitro findings.34
The US Food and Drug Administration (FDA) states that “healthcare professionals should be aware of an interaction between low-dose aspirin (81 mg/d) and ibuprofen, which might render aspirin less effective when used for its antiplatelet cardioprotective effect.” To minimize the interaction, the FDA recommends taking ibuprofen 8 hours before or 30 minutes after the ingestion of immediate-release (not enteric-coated) aspirin.35 It is not clear if this strategy can circumvent the interaction. For those who depend on aspirin’s lifesaving antiplatelet activity, it would seem more prudent to avoid medications known to interact with it.
This interaction, thought to be due to the competitive binding of ibuprofen and aspirin to the COX-1 molecule, has not been clinically demonstrated with other NSAIDs, such as diclofenac33 and naproxen, or with acetaminophen.36,37
A small, open-label, crossover study in healthy volunteers showed that both low-dose aspirin and naproxen (500 mg, twice daily) produced persistent and nearly complete suppression of platelet thromboxane production when naproxen was given 2 hours before aspirin or 2 hours after aspirin, suggesting no interference with aspirin’s effect.
An additional analysis in the same study examined thromboxane production in ex vivo platelets and showed that naproxen, like aspirin, inhibited thromboxane production in a concentration-dependent fashion, but reversibly, whereas aspirin’s effect was irreversible.38 Lower, nonprescription doses of naproxen 220 mg 2 and 3 times a day resulted in antiplatelet effects similar to the 550 mg twice-daily prescription dose used in a study of healthy volunteers whose blood was tested for inhibition of serum thromboxane as a measure of platelet COX-1 activity and inhibition of platelet aggregation.39
The propensity of aspirin cotherapy to increase the risk of NSAID-related GI adverse events is an underappreciated concern. A recent review of low-dose aspirin use emphasizes that concomitant NSAID use exacerbates GI bleeding, and low-dose aspirin may significantly offset the reduced GI toxicity of COX-2–selective NSAIDs.40
New recommendations in detail
In choosing an NSAID for a patient with OA, consider the patient’s baseline health risks, the potential for incremental medication-related GI and CV risks, and known hyper-sensitivity reactions or drug intolerance.41 The following recommendations also take into account the impact of aspirin cotherapy.
The presence of GI risk may necessitate using a PPI or misoprostol with the selected NSAID. Both PPIs and misoprostol decrease the rate of gastroduodenal ulceration in NSAID users. Additionally, misoprostol reduces ulcer complications, and PPIs reduce recurrent ulcer bleeding.42-44 One drawback with misoprostol is that it is not well tolerated. Thus PPIs, given their once-daily administration and superiority to histamine-2 (H2) blockers, are the preferred gastroprotective agent.42 The TABLE summarizes the following recommendations.7,41
Patients with no CV risk (not receiving aspirin) and little or no GI risk. Any non-selective NSAID would be reasonable initial therapy for patients with uncomplicated, mild to moderate OA pain.7,41,45 Acetaminophen at doses of up to 4 g/d is an acceptable alternative, but does not relieve pain as effectively as a nonselective NSAID.1,3,8 The risk of GI adverse events is very low with short-term use of OTC NSAID doses.46
Patients with no CV risk (not receiving aspirin) but moderate to high GI risk. For patients with moderate GI risk (eg, age ≥70 years, receiving concomitant corticosteroids or anticoagulants), a COX-2–selective NSAID or any nonselective NSAID with a gastroprotective agent (PPI) is appropriate. If all else is equal in your clinical assessment, cost favors low OTC dosing with nonselective NSAIDs over more costly COX-2–selective agents.7,41,45,47 However, for patients with very high GI risk (eg, prior complicated upper GI event or multiple GI risk factors), choose a COX-2–selective NSAID in combination with a PPI for gastroprotection.7,41,45
Patients with no GI risk and increased CV risk (receiving aspirin). For patients who have an increased CV risk (10-year risk ≥10% according to the Framingham equation for primary prevention; or a history of ischemic heart disease, or cerebrovascular or peripheral vascular disease [secondary prevention]), avoid NSAIDs, with the exception of naproxen.7,45 Large meta-analyses have shown that naproxen is associated with a lower risk of adverse CV events compared with other nonselective NSAIDs and COX-2–selective agents.22,23 Concomitant treatment with a PPI may be appropriate for patients taking naproxen and aspirin, because the risk of gastric ulcers may be increased with cotherapy.7,41,45
Patients with both CV risk (receiving aspirin) and GI risk. Gastroprotection is essential for the aspirin-related risk of bleeding, and PPIs reduce this risk.7,21,41 If an NSAID is required, naproxen in combination with a PPI may be the best choice.45 If naproxen is ineffective, you may consider another NSAID, but limit your selection to those agents without proven aspirin antagonism, such as the nonselective agents diclofenac and sulindac or low-dose celecoxib.33,48 Patients with elevated CV risk commonly take aspirin, potentially reducing the gastroprotective benefits of COX-2–selective NSAIDs; prescribe a concomitant PPI.20
A low-dose COX-2–selective NSAID with a PPIis an evidence-based recommendation for patients who have both CV and GI risks and who have had a previous GI ulcer bleed. Use the lowest possible dose of a COX-2–selective agent, because lower doses are associated with fewer CV adverse events.30,31
TABLE
Choose NSAID options according to CV and GI risks
| None or low risk | Moderate to high NSAID GI risk* | |
|---|---|---|
| No CV risk (without aspirin) | Any nonselective NSAID (cost consideration) | COX-2–selective inhibitor or any nonselective NSAID + PPI† COX-2–selective inhibitor + PPI for patients with prior ulcer GI bleeding |
| CV risk (with aspirin) | Naproxen‡ Add PPI if GI risk of aspirin/NSAID combination warrants gastroprotection | Add PPI regardless of NSAID COX-2–selective inhibitor + PPI for patients with previous ulcer GI bleeding |
| COX, cyclooxygenase; CV, cardiovascular; GI, gastrointestinal; NSAID, nonsteroidal anti-inflammatory drug; PPI, proton-pump inhibitor. | ||
| *Age ≥70 years or receiving concomitant corticosteroids or anticoagulants; highest GI risk is a prior ulcer bleed. | ||
| †Misoprostol at full dose (200 mcg, 4 times a day) may be substituted for a PPI. | ||
| ‡If naproxen is ineffective, use a nonselective or COX-2–selective (low-dose) inhibitor without established aspirin interaction—eg, diclofenac or sulindac. | ||
| Adapted from: Scheiman JM, et al. Lancet. 2007.41 | ||
CORRESPONDENCE James M. Scheiman, MD, University of Michigan Medical Center, 3912 Taubman Center, Box 0362, Ann Arbor, MI 48109; [email protected]
1. American College of Rheumatology ad hoc group on use of selective and nonselective nonsteroidal antiinflammatory drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum. 2008;59:1058-1073.
2. Zhang W, Muskowitz RW, Niki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage. 2008;16:137-162.
3. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum. 2000;43:1905-1915.
4. Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4-15.
5. Shamoon M, Hochberg MC. Treatment of osteoarthritis with acetaminophen: efficacy, safety, and comparison with nonsteroidal anti-inflammatory drugs. Curr Rheumatol Rep. 2000;2:454-458.
6. Jones R, Rubin G, Berenbaum F, et al. Gastrointestinal and cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Med. 2008;121:464-474.
7. Towheed TE, Judd MJ, Hochberg MC, et al. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;(1):CD004257.-
8. Bonnet CS, Walsh DA. Osteoarthritis, angiogenesis and inflammation. Rheumatology (Oxford). 2005;44:7-16.
9. Garcia Rodriguez LA, Hernandez-Diaz S. The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents. Arthritis Res. 2001;3:98-101.
10. Rahme E, Pettitt D, LeLorier J. Determinants and sequelae associated with utilization of acetaminophen versus traditional nonsteroidal antiinflammatory drugs in an elderly population. Arthritis Rheum. 2002;46:3046-3054.
11. Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296:87-93.
12. Curhan GC, Willett WC, Rosner B, et al. Frequency of analgesic use and risk of hypertension in younger women. Arch Intern Med. 2002;162:2204-2208.
13. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167:394-399.
14. Fored CM, Ejerblad E, Lindblad P, et al. Acetaminophen, aspirin, and chronic renal failure. N Engl J Med. 2001;345:1801-1808.
15. Perneger TV, Whelton PK, Klag MJ. Risk of kidney failure associated with the use of acetaminophen, aspirin, and nonsteroidal antiinflammatory drugs. N Engl J Med. 1994;331:1675-1679.
16. Pfizer Consumer Healthcare. Advil. 2010. Available at: http://www.advil.com/OurProducts/Advil.aspx. Accessed October 21, 2010.
17. Motrin [package insert]. New York, NY: Pharmacia & Upjohn Company, a division of Pfizer Inc; 2007.
18. US Food and Drug Administration. Naproxen - patient information sheet. 2004. Available at: http://www.fda.gov/downloads/Drugs/DrugsSafety/…/UCM164733.pdf. Accessed October 21, 2010.
19. Naprosyn [package insert]. Nutley, NJ: Roche Laboratories Inc.; 2008.
20. Wilcox CM, Allison J, Benzuly K, et al. Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-1089.
21. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation. 2008;118:1894-1909.
22. Juni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021-2029.
23. Kearney PM, Baigent C, Godwin J, et al. Do selective cyclooxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302-1308.
24. Schnitzer TJ, Burmester GR, Mysler E, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665-674.
25. Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247-1255.
26. Chan FK, Wong VW, Suen BY, et al. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
27. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:1-10.
28. Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-1102.
29. Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071-1080.
30. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634-1642.
31. Solomon SD, Wittes J, Finn PV, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104-2113.
32. Celebrex [package insert]. New York, NY: Pfizer Inc; 2008.
33. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
34. MacDonald TM, Wei L. Is there an interaction between the cardiovascular protective effects of low-dose aspirin and ibuprofen? Basic Clin Pharmacol Toxicol. 2006;98:275-280.
35. US Food and Drug Administration. Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. 2006. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM161282.pdf. Accessed October 19, 2010.
36. Brune K, Hochberg MC, Schiff M, et al. The platelet inhibitory effects of the combination of naproxen sodium or acetaminophen with low dose aspirin [abstract]. Arthritis Rheum. 2007;56:S359.-
37. Farkouh ME, Greenberg JD, Jeger RV, et al. Cardiovascular outcomes in high risk patients with osteoarthritis treated with ibuprofen, naproxen or lumiracoxib. Ann Rheum Dis. 2007;66:764-770.
38. Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295-1301.
39. Zlotnick S, Oldenhof J, Schuller R, et al. Effect of over-the-counter doses of naproxen sodium on inhibition of platelet cyclooxygenase-1 in healthy volunteers. Poster presented at: the American College of Rheumatology; November 13, 2006; Washington, DC. Poster L33.
40. Lanas A, Scheiman J. Low-dose aspirin and upper gastrointestinal damage: epidemiology, prevention and treatment. Curr Med Res Opin. 2007;23:163-173.
41. Scheiman JM, Fendrick AM. Summing the risk of NSAID therapy. Lancet. 2007;369:1580-1581.
42. Rostom A, Dube C, Wells G, et al. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev. 2002;(4):CD002296.-
43. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241-249.
44. Chan FK, Wong VW, Suen BY. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
45. Chan FK, Abraham NS, Scheiman JM, et al. Management of patients on nonsteroidal anti-inflammatory drugs: a clinical practice recommendation from the First International Working Party on Gastrointestinal and Cardiovascular Effects of Nonsteroidal Anti-inflammatory Drugs and Anti-platelet Agents. Am J Gastroenterol. 2008;103:2908-2918.
46. Lewis SC, Langman MJ, Laporte JR, et al. Dose-response relationships between individual nonaspirin nonsteroidal anti-inflammatory drugs (NANSAIDs) and serious upper gastrointestinal bleeding: a meta-analysis based on individual patient data. Br J Clin Pharmacol. 2002;54:320-326.
47. Spiegel BM, Chiou CF, Ofman JJ. Minimizing complications from nonsteroidal antiinflammatory drugs: cost-effectiveness of competing strategies in varying risk groups. Arthritis Rheum. 2005;53:185-197.
48. Gladding PA, Webster MW, Farrell HB, et al. The antiplatelet effect of six non-steroidal anti-inflammatory drugs and their pharmacodynamic interaction with aspirin in healthy volunteers. Am J Cardiol. 2008;101:1060-1063.
• In selecting an NSAID, assess a patient’s baseline cardiovascular (CV) and gastrointestinal (GI) risks and the potential for medication-related incremental CV and GI toxicity. C
• For patients with increased CV risk (taking aspirin for established CV risk) and low GI risk, the preferred agent is naproxen. Consider adding a proton pump inhibitor (PPI) or misoprostol, as dual therapy with aspirin and naproxen may warrant gastroprotection. A
• For patients with moderate GI risk and low CV risk, use a nonselective NSAID with a PPI or misoprostol; if GI risk is high, use a cyclooxygenase (COX)-2–selective NSAID and gastroprotection. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although clinicians have considerable experience in using analgesics and nonsteroidal anti-inflammatory drugs (NSAIDs) to relieve the pain of osteoarthritis (OA), emerging data have made the task of weighing benefits and risks of each agent more complex.1 In this article, we review the latest evidence for NSAIDs and provide a foundation on which you can make more informed decisions for controlling OA pain and—in conjunction with education, physical therapy, exercise, and cognitive and behavioral approaches2,3—improve patients’ daily function and quality of life.
Agents for OA pain relief: Benefits and trade-offs
Treatment options for OA pain are the analgesic acetaminophen and the NSAIDs, comprising both nonselective agents and the cyclooxygenase (COX)-2–selective inhibitors.
NSAIDs inhibit COX, a key enzyme in the biosynthesis of prostanoids, including the prostaglandins and leukotrienes, which are important mediators of pain. The COX-1 isoform is constantly expressed in tissue. It regulates protection of the gastric mucosa, platelet activation, and renal function. In contrast, COX-2 is induced primarily in response to inflammatory stimuli.
The nonselective NSAIDs inhibit both isoforms of COX. The anti-inflammatory and analgesic effects of the NSAIDs result primarily from COX-2 inhibition. Inhibition of COX-1 is largely responsible for the gastrointestinal (GI) ulceration and anti-platelet-promoted bleeding that can occur with these drugs.4
The COX-2–selective inhibitors were developed to spare the normal “housekeeping” functions of COX-1. This benefit, however, has been diminished by the adverse cardiovascular (CV) events occurring with selective inhibition of COX-2, owing to the expression of this isoform in vasculature and the kidneys.4 Increased risk of CV events may also occur with nonselective NSAIDs.
Acetaminophen’s mechanism of action is poorly understood. It is a weak inhibitor of COX-1 and COX-2, but it most likely acts centrally in the hypothalamus and spinal cord, rather than peripherally in joint cartilage where inflammation and damage occur.5
Revised treatment guidelines in brief
The American College of Rheumatology (ACR) and the Osteoarthritis Research Society International (OARSI) have published treatment recommendations for OA.1-3
The recent ACR publication noted that nonselective NSAIDs are more effective than acetaminophen for treating OA pain, but that the differences are small.1 Because of costs and the risk of adverse events associated with NSAID use, the ACR guidelines recommend that patients with mild to moderate OA pain receive a trial of acetaminophen initially; patients who do not respond could then receive NSAIDs. With moderate to severe OA pain, initial treatment with nonselective NSAIDs is appropriate.1,3
The OARSI guidelines2 state that “acetaminophen (up to 4 g/d) can be an effective initial oral analgesic for treatment of mild to moderate pain in patients with knee or hip OA.” The guidelines warn, however, that recent evidence has questioned both the efficacy and safety of long-term acetaminophen use in doses up to 4 g/d. The OARSI guidelines, like the ACR guidelines, recommend alternative pharmacotherapy when patients do not respond to acetaminophen for mild to moderate OA pain, or when OA pain is more severe. NSAIDs are most appropriately prescribed at the lowest effective dose for the shortest possible time.2
Accounting for risk factors. Current guidelines emphasize the importance of selecting treatments based on a patient’s CV and GI risk profiles. For patients with CV risk factors, use nonselective NSAIDs and COX-2–selective inhibitors with caution. For patients with increased GI risk, use either a COX-2–selective inhibitor or a nonselective NSAID with a proton pump inhibitor (PPI) or misoprostol.2
The evidence underlying guideline revisions
Selecting an agent that optimally balances efficacy and safety requires that we consider the complexities of 3 competing clinical concerns—relief of arthritis pain, CV toxicity, and GI toxicity.6 We review here the evidence supporting the revised recommendations.
Acetaminophen: A good option, but there are better ones
Acetaminophen relieves OA pain, but not as effectively as nonselective NSAIDs.1,3,7 A Cochrane meta-analysis showed that although acetaminophen was superior to placebo for reducing OA pain, it was less effective than either nonselective NSAIDs or COX-2–selective NSAIDs for reducing pain and improving functional status, especially in patients with moderate pain.7
Acetaminophen at higher doses has been associated with GI toxicity.8 In a case-control study, acetaminophen at doses ≥2 g/d increased the risk of upper GI bleeding or perforation.9 A cohort study showed that doses of acetaminophen >3 g/d led to higher rates of upper GI events (GI hospitalization, ulcer, and dyspepsia) comparable to those seen with NSAIDs.10 It remains unclear if the acetaminophen in this trial caused GI adverse events among all patients due to the higher doses alone, or if the rates reflected increases in adverse events expected among high-risk GI patients or concomitant NSAID users.10 Furthermore, healthy adults who ingested 4 g acetaminophen each day for 2 weeks exhibited significant elevations of serum alanine aminotransferase levels, suggestive of liver injury.11
Caution is justified with prolonged use of acetaminophen at high doses, particularly in alcohol users. In cohort studies with women and men, acetaminophen has been associated with an increased risk of incident hypertension.12,13 In case-control studies, long-term use has also been dose-dependently associated with an increased risk of chronic renal failure.14,15
Nonselective NSAIDs: Keep GI risks in mind
All nonselective NSAIDs, when administered at equivalent therapeutic doses (same degree of COX inhibition), appear to have comparable efficacy in relieving OA pain. Analgesia is dose dependent, which enables patients to start therapy at lower over-the-counter (OTC) doses and escalate to higher prescription doses as needed.3 The OTC dose range of ibuprofen is 200 to 400 mg 3 times a day, to a maximum of 1200 mg/d;16 the maximum prescription dose is 3200 mg/d.17 Similarly, the maximum dose of OTC naproxen is 660 mg/d,18 although by prescription it can be given up to 1500 mg/d.19
NSAIDs confer a dose-related risk for GI adverse events, including ulcers and bleeding. Patients with a history of ulcers and those at advanced age are at greater risk;20 those with a history of an ulcer bleed are at the greatest risk for an adverse event. Also at increased risk are those taking high doses of an NSAID, multiple NSAIDs (eg, concomitant low-dose aspirin), or anticoagulant or antiplatelet agents.21
Recent data suggest that nonselective NSAIDs, with the exception of naproxen, may increase CV risk on a level seen with COX-2–selective inhibitors.22,23 In a meta-analysis of 91 randomized active-controlled trials, a comparison of COX-2–selective inhibitors and non-naproxen nonselective NSAIDs showed no significant difference in the risk of myocardial infarction (MI) (relative risk [RR]=1.20; 95% confidence interval [CI], 0.85–1.68); however, COX-2–selective inhibitors had an increased risk compared with naproxen (RR=2.04; 95% CI, 1.41–2.96).23 In another meta-analysis of 11 observational studies, naproxen reduced the risk of MI compared with COX-2–selective inhibitors and other nonselective NSAIDs (RR=0.86; 95% CI, 0.75–0.99).22 An increased risk of incident hypertension has been associated with frequent NSAID use in cohort studies in women and men.12,13
COX-2–selective NSAIDs: Good on gut, but increase MI risk
COX-2–selective NSAIDs lower the incidence of upper GI tract complications compared with nonselective agents, while maintaining comparable efficacy in pain relief, both when used alone (without concomitant aspirin therapy)20,24,25 and in combination with PPIs.26,27
But despite their GI safety profile, the COX-2–selective NSAIDs increased the risk of MI and ischemic cerebrovascular events in trials where they were being studied for arthritis pain and for GI polyp prevention.22,28,29 Among the proposed mechanisms for this effect is that selective COX-2 inhibition reduces the level of the antithrombotic prostanoid, prostacyclin, relative to the level of the prothrombotic prostanoid, thromboxane, thereby leading to a prothrombotic tendency.30
Rofecoxib and valdecoxib were withdrawn from the market in the United States by the manufacturers after the drugs were linked to serious CV adverse effects—and in the case of valdecoxib, to a serious skin reaction.30 Celecoxib remains commercially available in the United States.30 The CV risks associated with celecoxib are dose related, with once-daily dosing (400 mg/d) associated with a much lower risk than twice-daily dosing (200 or 400 mg twice a day).31 The recommended dose is 200 mg/d.32
The deleterious impact of combining low-dose aspirin with NSAIDs
Many patients who take NSAIDs also require aspirin for cardioprotection. Catella-Lawson and colleagues33 investigated the potential interactions between aspirin and several NSAIDs used in managing OA. They found that ibuprofen, when taken before aspirin, reduced aspirin’s inhibition of platelet aggregation, demonstrating potential impairment of aspirin’s cardioprotective effect.33 Subsequent observational studies have supported these in vitro findings.34
The US Food and Drug Administration (FDA) states that “healthcare professionals should be aware of an interaction between low-dose aspirin (81 mg/d) and ibuprofen, which might render aspirin less effective when used for its antiplatelet cardioprotective effect.” To minimize the interaction, the FDA recommends taking ibuprofen 8 hours before or 30 minutes after the ingestion of immediate-release (not enteric-coated) aspirin.35 It is not clear if this strategy can circumvent the interaction. For those who depend on aspirin’s lifesaving antiplatelet activity, it would seem more prudent to avoid medications known to interact with it.
This interaction, thought to be due to the competitive binding of ibuprofen and aspirin to the COX-1 molecule, has not been clinically demonstrated with other NSAIDs, such as diclofenac33 and naproxen, or with acetaminophen.36,37
A small, open-label, crossover study in healthy volunteers showed that both low-dose aspirin and naproxen (500 mg, twice daily) produced persistent and nearly complete suppression of platelet thromboxane production when naproxen was given 2 hours before aspirin or 2 hours after aspirin, suggesting no interference with aspirin’s effect.
An additional analysis in the same study examined thromboxane production in ex vivo platelets and showed that naproxen, like aspirin, inhibited thromboxane production in a concentration-dependent fashion, but reversibly, whereas aspirin’s effect was irreversible.38 Lower, nonprescription doses of naproxen 220 mg 2 and 3 times a day resulted in antiplatelet effects similar to the 550 mg twice-daily prescription dose used in a study of healthy volunteers whose blood was tested for inhibition of serum thromboxane as a measure of platelet COX-1 activity and inhibition of platelet aggregation.39
The propensity of aspirin cotherapy to increase the risk of NSAID-related GI adverse events is an underappreciated concern. A recent review of low-dose aspirin use emphasizes that concomitant NSAID use exacerbates GI bleeding, and low-dose aspirin may significantly offset the reduced GI toxicity of COX-2–selective NSAIDs.40
New recommendations in detail
In choosing an NSAID for a patient with OA, consider the patient’s baseline health risks, the potential for incremental medication-related GI and CV risks, and known hyper-sensitivity reactions or drug intolerance.41 The following recommendations also take into account the impact of aspirin cotherapy.
The presence of GI risk may necessitate using a PPI or misoprostol with the selected NSAID. Both PPIs and misoprostol decrease the rate of gastroduodenal ulceration in NSAID users. Additionally, misoprostol reduces ulcer complications, and PPIs reduce recurrent ulcer bleeding.42-44 One drawback with misoprostol is that it is not well tolerated. Thus PPIs, given their once-daily administration and superiority to histamine-2 (H2) blockers, are the preferred gastroprotective agent.42 The TABLE summarizes the following recommendations.7,41
Patients with no CV risk (not receiving aspirin) and little or no GI risk. Any non-selective NSAID would be reasonable initial therapy for patients with uncomplicated, mild to moderate OA pain.7,41,45 Acetaminophen at doses of up to 4 g/d is an acceptable alternative, but does not relieve pain as effectively as a nonselective NSAID.1,3,8 The risk of GI adverse events is very low with short-term use of OTC NSAID doses.46
Patients with no CV risk (not receiving aspirin) but moderate to high GI risk. For patients with moderate GI risk (eg, age ≥70 years, receiving concomitant corticosteroids or anticoagulants), a COX-2–selective NSAID or any nonselective NSAID with a gastroprotective agent (PPI) is appropriate. If all else is equal in your clinical assessment, cost favors low OTC dosing with nonselective NSAIDs over more costly COX-2–selective agents.7,41,45,47 However, for patients with very high GI risk (eg, prior complicated upper GI event or multiple GI risk factors), choose a COX-2–selective NSAID in combination with a PPI for gastroprotection.7,41,45
Patients with no GI risk and increased CV risk (receiving aspirin). For patients who have an increased CV risk (10-year risk ≥10% according to the Framingham equation for primary prevention; or a history of ischemic heart disease, or cerebrovascular or peripheral vascular disease [secondary prevention]), avoid NSAIDs, with the exception of naproxen.7,45 Large meta-analyses have shown that naproxen is associated with a lower risk of adverse CV events compared with other nonselective NSAIDs and COX-2–selective agents.22,23 Concomitant treatment with a PPI may be appropriate for patients taking naproxen and aspirin, because the risk of gastric ulcers may be increased with cotherapy.7,41,45
Patients with both CV risk (receiving aspirin) and GI risk. Gastroprotection is essential for the aspirin-related risk of bleeding, and PPIs reduce this risk.7,21,41 If an NSAID is required, naproxen in combination with a PPI may be the best choice.45 If naproxen is ineffective, you may consider another NSAID, but limit your selection to those agents without proven aspirin antagonism, such as the nonselective agents diclofenac and sulindac or low-dose celecoxib.33,48 Patients with elevated CV risk commonly take aspirin, potentially reducing the gastroprotective benefits of COX-2–selective NSAIDs; prescribe a concomitant PPI.20
A low-dose COX-2–selective NSAID with a PPIis an evidence-based recommendation for patients who have both CV and GI risks and who have had a previous GI ulcer bleed. Use the lowest possible dose of a COX-2–selective agent, because lower doses are associated with fewer CV adverse events.30,31
TABLE
Choose NSAID options according to CV and GI risks
| None or low risk | Moderate to high NSAID GI risk* | |
|---|---|---|
| No CV risk (without aspirin) | Any nonselective NSAID (cost consideration) | COX-2–selective inhibitor or any nonselective NSAID + PPI† COX-2–selective inhibitor + PPI for patients with prior ulcer GI bleeding |
| CV risk (with aspirin) | Naproxen‡ Add PPI if GI risk of aspirin/NSAID combination warrants gastroprotection | Add PPI regardless of NSAID COX-2–selective inhibitor + PPI for patients with previous ulcer GI bleeding |
| COX, cyclooxygenase; CV, cardiovascular; GI, gastrointestinal; NSAID, nonsteroidal anti-inflammatory drug; PPI, proton-pump inhibitor. | ||
| *Age ≥70 years or receiving concomitant corticosteroids or anticoagulants; highest GI risk is a prior ulcer bleed. | ||
| †Misoprostol at full dose (200 mcg, 4 times a day) may be substituted for a PPI. | ||
| ‡If naproxen is ineffective, use a nonselective or COX-2–selective (low-dose) inhibitor without established aspirin interaction—eg, diclofenac or sulindac. | ||
| Adapted from: Scheiman JM, et al. Lancet. 2007.41 | ||
CORRESPONDENCE James M. Scheiman, MD, University of Michigan Medical Center, 3912 Taubman Center, Box 0362, Ann Arbor, MI 48109; [email protected]
• In selecting an NSAID, assess a patient’s baseline cardiovascular (CV) and gastrointestinal (GI) risks and the potential for medication-related incremental CV and GI toxicity. C
• For patients with increased CV risk (taking aspirin for established CV risk) and low GI risk, the preferred agent is naproxen. Consider adding a proton pump inhibitor (PPI) or misoprostol, as dual therapy with aspirin and naproxen may warrant gastroprotection. A
• For patients with moderate GI risk and low CV risk, use a nonselective NSAID with a PPI or misoprostol; if GI risk is high, use a cyclooxygenase (COX)-2–selective NSAID and gastroprotection. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although clinicians have considerable experience in using analgesics and nonsteroidal anti-inflammatory drugs (NSAIDs) to relieve the pain of osteoarthritis (OA), emerging data have made the task of weighing benefits and risks of each agent more complex.1 In this article, we review the latest evidence for NSAIDs and provide a foundation on which you can make more informed decisions for controlling OA pain and—in conjunction with education, physical therapy, exercise, and cognitive and behavioral approaches2,3—improve patients’ daily function and quality of life.
Agents for OA pain relief: Benefits and trade-offs
Treatment options for OA pain are the analgesic acetaminophen and the NSAIDs, comprising both nonselective agents and the cyclooxygenase (COX)-2–selective inhibitors.
NSAIDs inhibit COX, a key enzyme in the biosynthesis of prostanoids, including the prostaglandins and leukotrienes, which are important mediators of pain. The COX-1 isoform is constantly expressed in tissue. It regulates protection of the gastric mucosa, platelet activation, and renal function. In contrast, COX-2 is induced primarily in response to inflammatory stimuli.
The nonselective NSAIDs inhibit both isoforms of COX. The anti-inflammatory and analgesic effects of the NSAIDs result primarily from COX-2 inhibition. Inhibition of COX-1 is largely responsible for the gastrointestinal (GI) ulceration and anti-platelet-promoted bleeding that can occur with these drugs.4
The COX-2–selective inhibitors were developed to spare the normal “housekeeping” functions of COX-1. This benefit, however, has been diminished by the adverse cardiovascular (CV) events occurring with selective inhibition of COX-2, owing to the expression of this isoform in vasculature and the kidneys.4 Increased risk of CV events may also occur with nonselective NSAIDs.
Acetaminophen’s mechanism of action is poorly understood. It is a weak inhibitor of COX-1 and COX-2, but it most likely acts centrally in the hypothalamus and spinal cord, rather than peripherally in joint cartilage where inflammation and damage occur.5
Revised treatment guidelines in brief
The American College of Rheumatology (ACR) and the Osteoarthritis Research Society International (OARSI) have published treatment recommendations for OA.1-3
The recent ACR publication noted that nonselective NSAIDs are more effective than acetaminophen for treating OA pain, but that the differences are small.1 Because of costs and the risk of adverse events associated with NSAID use, the ACR guidelines recommend that patients with mild to moderate OA pain receive a trial of acetaminophen initially; patients who do not respond could then receive NSAIDs. With moderate to severe OA pain, initial treatment with nonselective NSAIDs is appropriate.1,3
The OARSI guidelines2 state that “acetaminophen (up to 4 g/d) can be an effective initial oral analgesic for treatment of mild to moderate pain in patients with knee or hip OA.” The guidelines warn, however, that recent evidence has questioned both the efficacy and safety of long-term acetaminophen use in doses up to 4 g/d. The OARSI guidelines, like the ACR guidelines, recommend alternative pharmacotherapy when patients do not respond to acetaminophen for mild to moderate OA pain, or when OA pain is more severe. NSAIDs are most appropriately prescribed at the lowest effective dose for the shortest possible time.2
Accounting for risk factors. Current guidelines emphasize the importance of selecting treatments based on a patient’s CV and GI risk profiles. For patients with CV risk factors, use nonselective NSAIDs and COX-2–selective inhibitors with caution. For patients with increased GI risk, use either a COX-2–selective inhibitor or a nonselective NSAID with a proton pump inhibitor (PPI) or misoprostol.2
The evidence underlying guideline revisions
Selecting an agent that optimally balances efficacy and safety requires that we consider the complexities of 3 competing clinical concerns—relief of arthritis pain, CV toxicity, and GI toxicity.6 We review here the evidence supporting the revised recommendations.
Acetaminophen: A good option, but there are better ones
Acetaminophen relieves OA pain, but not as effectively as nonselective NSAIDs.1,3,7 A Cochrane meta-analysis showed that although acetaminophen was superior to placebo for reducing OA pain, it was less effective than either nonselective NSAIDs or COX-2–selective NSAIDs for reducing pain and improving functional status, especially in patients with moderate pain.7
Acetaminophen at higher doses has been associated with GI toxicity.8 In a case-control study, acetaminophen at doses ≥2 g/d increased the risk of upper GI bleeding or perforation.9 A cohort study showed that doses of acetaminophen >3 g/d led to higher rates of upper GI events (GI hospitalization, ulcer, and dyspepsia) comparable to those seen with NSAIDs.10 It remains unclear if the acetaminophen in this trial caused GI adverse events among all patients due to the higher doses alone, or if the rates reflected increases in adverse events expected among high-risk GI patients or concomitant NSAID users.10 Furthermore, healthy adults who ingested 4 g acetaminophen each day for 2 weeks exhibited significant elevations of serum alanine aminotransferase levels, suggestive of liver injury.11
Caution is justified with prolonged use of acetaminophen at high doses, particularly in alcohol users. In cohort studies with women and men, acetaminophen has been associated with an increased risk of incident hypertension.12,13 In case-control studies, long-term use has also been dose-dependently associated with an increased risk of chronic renal failure.14,15
Nonselective NSAIDs: Keep GI risks in mind
All nonselective NSAIDs, when administered at equivalent therapeutic doses (same degree of COX inhibition), appear to have comparable efficacy in relieving OA pain. Analgesia is dose dependent, which enables patients to start therapy at lower over-the-counter (OTC) doses and escalate to higher prescription doses as needed.3 The OTC dose range of ibuprofen is 200 to 400 mg 3 times a day, to a maximum of 1200 mg/d;16 the maximum prescription dose is 3200 mg/d.17 Similarly, the maximum dose of OTC naproxen is 660 mg/d,18 although by prescription it can be given up to 1500 mg/d.19
NSAIDs confer a dose-related risk for GI adverse events, including ulcers and bleeding. Patients with a history of ulcers and those at advanced age are at greater risk;20 those with a history of an ulcer bleed are at the greatest risk for an adverse event. Also at increased risk are those taking high doses of an NSAID, multiple NSAIDs (eg, concomitant low-dose aspirin), or anticoagulant or antiplatelet agents.21
Recent data suggest that nonselective NSAIDs, with the exception of naproxen, may increase CV risk on a level seen with COX-2–selective inhibitors.22,23 In a meta-analysis of 91 randomized active-controlled trials, a comparison of COX-2–selective inhibitors and non-naproxen nonselective NSAIDs showed no significant difference in the risk of myocardial infarction (MI) (relative risk [RR]=1.20; 95% confidence interval [CI], 0.85–1.68); however, COX-2–selective inhibitors had an increased risk compared with naproxen (RR=2.04; 95% CI, 1.41–2.96).23 In another meta-analysis of 11 observational studies, naproxen reduced the risk of MI compared with COX-2–selective inhibitors and other nonselective NSAIDs (RR=0.86; 95% CI, 0.75–0.99).22 An increased risk of incident hypertension has been associated with frequent NSAID use in cohort studies in women and men.12,13
COX-2–selective NSAIDs: Good on gut, but increase MI risk
COX-2–selective NSAIDs lower the incidence of upper GI tract complications compared with nonselective agents, while maintaining comparable efficacy in pain relief, both when used alone (without concomitant aspirin therapy)20,24,25 and in combination with PPIs.26,27
But despite their GI safety profile, the COX-2–selective NSAIDs increased the risk of MI and ischemic cerebrovascular events in trials where they were being studied for arthritis pain and for GI polyp prevention.22,28,29 Among the proposed mechanisms for this effect is that selective COX-2 inhibition reduces the level of the antithrombotic prostanoid, prostacyclin, relative to the level of the prothrombotic prostanoid, thromboxane, thereby leading to a prothrombotic tendency.30
Rofecoxib and valdecoxib were withdrawn from the market in the United States by the manufacturers after the drugs were linked to serious CV adverse effects—and in the case of valdecoxib, to a serious skin reaction.30 Celecoxib remains commercially available in the United States.30 The CV risks associated with celecoxib are dose related, with once-daily dosing (400 mg/d) associated with a much lower risk than twice-daily dosing (200 or 400 mg twice a day).31 The recommended dose is 200 mg/d.32
The deleterious impact of combining low-dose aspirin with NSAIDs
Many patients who take NSAIDs also require aspirin for cardioprotection. Catella-Lawson and colleagues33 investigated the potential interactions between aspirin and several NSAIDs used in managing OA. They found that ibuprofen, when taken before aspirin, reduced aspirin’s inhibition of platelet aggregation, demonstrating potential impairment of aspirin’s cardioprotective effect.33 Subsequent observational studies have supported these in vitro findings.34
The US Food and Drug Administration (FDA) states that “healthcare professionals should be aware of an interaction between low-dose aspirin (81 mg/d) and ibuprofen, which might render aspirin less effective when used for its antiplatelet cardioprotective effect.” To minimize the interaction, the FDA recommends taking ibuprofen 8 hours before or 30 minutes after the ingestion of immediate-release (not enteric-coated) aspirin.35 It is not clear if this strategy can circumvent the interaction. For those who depend on aspirin’s lifesaving antiplatelet activity, it would seem more prudent to avoid medications known to interact with it.
This interaction, thought to be due to the competitive binding of ibuprofen and aspirin to the COX-1 molecule, has not been clinically demonstrated with other NSAIDs, such as diclofenac33 and naproxen, or with acetaminophen.36,37
A small, open-label, crossover study in healthy volunteers showed that both low-dose aspirin and naproxen (500 mg, twice daily) produced persistent and nearly complete suppression of platelet thromboxane production when naproxen was given 2 hours before aspirin or 2 hours after aspirin, suggesting no interference with aspirin’s effect.
An additional analysis in the same study examined thromboxane production in ex vivo platelets and showed that naproxen, like aspirin, inhibited thromboxane production in a concentration-dependent fashion, but reversibly, whereas aspirin’s effect was irreversible.38 Lower, nonprescription doses of naproxen 220 mg 2 and 3 times a day resulted in antiplatelet effects similar to the 550 mg twice-daily prescription dose used in a study of healthy volunteers whose blood was tested for inhibition of serum thromboxane as a measure of platelet COX-1 activity and inhibition of platelet aggregation.39
The propensity of aspirin cotherapy to increase the risk of NSAID-related GI adverse events is an underappreciated concern. A recent review of low-dose aspirin use emphasizes that concomitant NSAID use exacerbates GI bleeding, and low-dose aspirin may significantly offset the reduced GI toxicity of COX-2–selective NSAIDs.40
New recommendations in detail
In choosing an NSAID for a patient with OA, consider the patient’s baseline health risks, the potential for incremental medication-related GI and CV risks, and known hyper-sensitivity reactions or drug intolerance.41 The following recommendations also take into account the impact of aspirin cotherapy.
The presence of GI risk may necessitate using a PPI or misoprostol with the selected NSAID. Both PPIs and misoprostol decrease the rate of gastroduodenal ulceration in NSAID users. Additionally, misoprostol reduces ulcer complications, and PPIs reduce recurrent ulcer bleeding.42-44 One drawback with misoprostol is that it is not well tolerated. Thus PPIs, given their once-daily administration and superiority to histamine-2 (H2) blockers, are the preferred gastroprotective agent.42 The TABLE summarizes the following recommendations.7,41
Patients with no CV risk (not receiving aspirin) and little or no GI risk. Any non-selective NSAID would be reasonable initial therapy for patients with uncomplicated, mild to moderate OA pain.7,41,45 Acetaminophen at doses of up to 4 g/d is an acceptable alternative, but does not relieve pain as effectively as a nonselective NSAID.1,3,8 The risk of GI adverse events is very low with short-term use of OTC NSAID doses.46
Patients with no CV risk (not receiving aspirin) but moderate to high GI risk. For patients with moderate GI risk (eg, age ≥70 years, receiving concomitant corticosteroids or anticoagulants), a COX-2–selective NSAID or any nonselective NSAID with a gastroprotective agent (PPI) is appropriate. If all else is equal in your clinical assessment, cost favors low OTC dosing with nonselective NSAIDs over more costly COX-2–selective agents.7,41,45,47 However, for patients with very high GI risk (eg, prior complicated upper GI event or multiple GI risk factors), choose a COX-2–selective NSAID in combination with a PPI for gastroprotection.7,41,45
Patients with no GI risk and increased CV risk (receiving aspirin). For patients who have an increased CV risk (10-year risk ≥10% according to the Framingham equation for primary prevention; or a history of ischemic heart disease, or cerebrovascular or peripheral vascular disease [secondary prevention]), avoid NSAIDs, with the exception of naproxen.7,45 Large meta-analyses have shown that naproxen is associated with a lower risk of adverse CV events compared with other nonselective NSAIDs and COX-2–selective agents.22,23 Concomitant treatment with a PPI may be appropriate for patients taking naproxen and aspirin, because the risk of gastric ulcers may be increased with cotherapy.7,41,45
Patients with both CV risk (receiving aspirin) and GI risk. Gastroprotection is essential for the aspirin-related risk of bleeding, and PPIs reduce this risk.7,21,41 If an NSAID is required, naproxen in combination with a PPI may be the best choice.45 If naproxen is ineffective, you may consider another NSAID, but limit your selection to those agents without proven aspirin antagonism, such as the nonselective agents diclofenac and sulindac or low-dose celecoxib.33,48 Patients with elevated CV risk commonly take aspirin, potentially reducing the gastroprotective benefits of COX-2–selective NSAIDs; prescribe a concomitant PPI.20
A low-dose COX-2–selective NSAID with a PPIis an evidence-based recommendation for patients who have both CV and GI risks and who have had a previous GI ulcer bleed. Use the lowest possible dose of a COX-2–selective agent, because lower doses are associated with fewer CV adverse events.30,31
TABLE
Choose NSAID options according to CV and GI risks
| None or low risk | Moderate to high NSAID GI risk* | |
|---|---|---|
| No CV risk (without aspirin) | Any nonselective NSAID (cost consideration) | COX-2–selective inhibitor or any nonselective NSAID + PPI† COX-2–selective inhibitor + PPI for patients with prior ulcer GI bleeding |
| CV risk (with aspirin) | Naproxen‡ Add PPI if GI risk of aspirin/NSAID combination warrants gastroprotection | Add PPI regardless of NSAID COX-2–selective inhibitor + PPI for patients with previous ulcer GI bleeding |
| COX, cyclooxygenase; CV, cardiovascular; GI, gastrointestinal; NSAID, nonsteroidal anti-inflammatory drug; PPI, proton-pump inhibitor. | ||
| *Age ≥70 years or receiving concomitant corticosteroids or anticoagulants; highest GI risk is a prior ulcer bleed. | ||
| †Misoprostol at full dose (200 mcg, 4 times a day) may be substituted for a PPI. | ||
| ‡If naproxen is ineffective, use a nonselective or COX-2–selective (low-dose) inhibitor without established aspirin interaction—eg, diclofenac or sulindac. | ||
| Adapted from: Scheiman JM, et al. Lancet. 2007.41 | ||
CORRESPONDENCE James M. Scheiman, MD, University of Michigan Medical Center, 3912 Taubman Center, Box 0362, Ann Arbor, MI 48109; [email protected]
1. American College of Rheumatology ad hoc group on use of selective and nonselective nonsteroidal antiinflammatory drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum. 2008;59:1058-1073.
2. Zhang W, Muskowitz RW, Niki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage. 2008;16:137-162.
3. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum. 2000;43:1905-1915.
4. Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4-15.
5. Shamoon M, Hochberg MC. Treatment of osteoarthritis with acetaminophen: efficacy, safety, and comparison with nonsteroidal anti-inflammatory drugs. Curr Rheumatol Rep. 2000;2:454-458.
6. Jones R, Rubin G, Berenbaum F, et al. Gastrointestinal and cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Med. 2008;121:464-474.
7. Towheed TE, Judd MJ, Hochberg MC, et al. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;(1):CD004257.-
8. Bonnet CS, Walsh DA. Osteoarthritis, angiogenesis and inflammation. Rheumatology (Oxford). 2005;44:7-16.
9. Garcia Rodriguez LA, Hernandez-Diaz S. The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents. Arthritis Res. 2001;3:98-101.
10. Rahme E, Pettitt D, LeLorier J. Determinants and sequelae associated with utilization of acetaminophen versus traditional nonsteroidal antiinflammatory drugs in an elderly population. Arthritis Rheum. 2002;46:3046-3054.
11. Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296:87-93.
12. Curhan GC, Willett WC, Rosner B, et al. Frequency of analgesic use and risk of hypertension in younger women. Arch Intern Med. 2002;162:2204-2208.
13. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167:394-399.
14. Fored CM, Ejerblad E, Lindblad P, et al. Acetaminophen, aspirin, and chronic renal failure. N Engl J Med. 2001;345:1801-1808.
15. Perneger TV, Whelton PK, Klag MJ. Risk of kidney failure associated with the use of acetaminophen, aspirin, and nonsteroidal antiinflammatory drugs. N Engl J Med. 1994;331:1675-1679.
16. Pfizer Consumer Healthcare. Advil. 2010. Available at: http://www.advil.com/OurProducts/Advil.aspx. Accessed October 21, 2010.
17. Motrin [package insert]. New York, NY: Pharmacia & Upjohn Company, a division of Pfizer Inc; 2007.
18. US Food and Drug Administration. Naproxen - patient information sheet. 2004. Available at: http://www.fda.gov/downloads/Drugs/DrugsSafety/…/UCM164733.pdf. Accessed October 21, 2010.
19. Naprosyn [package insert]. Nutley, NJ: Roche Laboratories Inc.; 2008.
20. Wilcox CM, Allison J, Benzuly K, et al. Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-1089.
21. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation. 2008;118:1894-1909.
22. Juni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021-2029.
23. Kearney PM, Baigent C, Godwin J, et al. Do selective cyclooxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302-1308.
24. Schnitzer TJ, Burmester GR, Mysler E, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665-674.
25. Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247-1255.
26. Chan FK, Wong VW, Suen BY, et al. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
27. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:1-10.
28. Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-1102.
29. Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071-1080.
30. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634-1642.
31. Solomon SD, Wittes J, Finn PV, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104-2113.
32. Celebrex [package insert]. New York, NY: Pfizer Inc; 2008.
33. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
34. MacDonald TM, Wei L. Is there an interaction between the cardiovascular protective effects of low-dose aspirin and ibuprofen? Basic Clin Pharmacol Toxicol. 2006;98:275-280.
35. US Food and Drug Administration. Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. 2006. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM161282.pdf. Accessed October 19, 2010.
36. Brune K, Hochberg MC, Schiff M, et al. The platelet inhibitory effects of the combination of naproxen sodium or acetaminophen with low dose aspirin [abstract]. Arthritis Rheum. 2007;56:S359.-
37. Farkouh ME, Greenberg JD, Jeger RV, et al. Cardiovascular outcomes in high risk patients with osteoarthritis treated with ibuprofen, naproxen or lumiracoxib. Ann Rheum Dis. 2007;66:764-770.
38. Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295-1301.
39. Zlotnick S, Oldenhof J, Schuller R, et al. Effect of over-the-counter doses of naproxen sodium on inhibition of platelet cyclooxygenase-1 in healthy volunteers. Poster presented at: the American College of Rheumatology; November 13, 2006; Washington, DC. Poster L33.
40. Lanas A, Scheiman J. Low-dose aspirin and upper gastrointestinal damage: epidemiology, prevention and treatment. Curr Med Res Opin. 2007;23:163-173.
41. Scheiman JM, Fendrick AM. Summing the risk of NSAID therapy. Lancet. 2007;369:1580-1581.
42. Rostom A, Dube C, Wells G, et al. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev. 2002;(4):CD002296.-
43. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241-249.
44. Chan FK, Wong VW, Suen BY. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
45. Chan FK, Abraham NS, Scheiman JM, et al. Management of patients on nonsteroidal anti-inflammatory drugs: a clinical practice recommendation from the First International Working Party on Gastrointestinal and Cardiovascular Effects of Nonsteroidal Anti-inflammatory Drugs and Anti-platelet Agents. Am J Gastroenterol. 2008;103:2908-2918.
46. Lewis SC, Langman MJ, Laporte JR, et al. Dose-response relationships between individual nonaspirin nonsteroidal anti-inflammatory drugs (NANSAIDs) and serious upper gastrointestinal bleeding: a meta-analysis based on individual patient data. Br J Clin Pharmacol. 2002;54:320-326.
47. Spiegel BM, Chiou CF, Ofman JJ. Minimizing complications from nonsteroidal antiinflammatory drugs: cost-effectiveness of competing strategies in varying risk groups. Arthritis Rheum. 2005;53:185-197.
48. Gladding PA, Webster MW, Farrell HB, et al. The antiplatelet effect of six non-steroidal anti-inflammatory drugs and their pharmacodynamic interaction with aspirin in healthy volunteers. Am J Cardiol. 2008;101:1060-1063.
1. American College of Rheumatology ad hoc group on use of selective and nonselective nonsteroidal antiinflammatory drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum. 2008;59:1058-1073.
2. Zhang W, Muskowitz RW, Niki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage. 2008;16:137-162.
3. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum. 2000;43:1905-1915.
4. Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4-15.
5. Shamoon M, Hochberg MC. Treatment of osteoarthritis with acetaminophen: efficacy, safety, and comparison with nonsteroidal anti-inflammatory drugs. Curr Rheumatol Rep. 2000;2:454-458.
6. Jones R, Rubin G, Berenbaum F, et al. Gastrointestinal and cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Med. 2008;121:464-474.
7. Towheed TE, Judd MJ, Hochberg MC, et al. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;(1):CD004257.-
8. Bonnet CS, Walsh DA. Osteoarthritis, angiogenesis and inflammation. Rheumatology (Oxford). 2005;44:7-16.
9. Garcia Rodriguez LA, Hernandez-Diaz S. The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents. Arthritis Res. 2001;3:98-101.
10. Rahme E, Pettitt D, LeLorier J. Determinants and sequelae associated with utilization of acetaminophen versus traditional nonsteroidal antiinflammatory drugs in an elderly population. Arthritis Rheum. 2002;46:3046-3054.
11. Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296:87-93.
12. Curhan GC, Willett WC, Rosner B, et al. Frequency of analgesic use and risk of hypertension in younger women. Arch Intern Med. 2002;162:2204-2208.
13. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167:394-399.
14. Fored CM, Ejerblad E, Lindblad P, et al. Acetaminophen, aspirin, and chronic renal failure. N Engl J Med. 2001;345:1801-1808.
15. Perneger TV, Whelton PK, Klag MJ. Risk of kidney failure associated with the use of acetaminophen, aspirin, and nonsteroidal antiinflammatory drugs. N Engl J Med. 1994;331:1675-1679.
16. Pfizer Consumer Healthcare. Advil. 2010. Available at: http://www.advil.com/OurProducts/Advil.aspx. Accessed October 21, 2010.
17. Motrin [package insert]. New York, NY: Pharmacia & Upjohn Company, a division of Pfizer Inc; 2007.
18. US Food and Drug Administration. Naproxen - patient information sheet. 2004. Available at: http://www.fda.gov/downloads/Drugs/DrugsSafety/…/UCM164733.pdf. Accessed October 21, 2010.
19. Naprosyn [package insert]. Nutley, NJ: Roche Laboratories Inc.; 2008.
20. Wilcox CM, Allison J, Benzuly K, et al. Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-1089.
21. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation. 2008;118:1894-1909.
22. Juni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021-2029.
23. Kearney PM, Baigent C, Godwin J, et al. Do selective cyclooxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302-1308.
24. Schnitzer TJ, Burmester GR, Mysler E, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665-674.
25. Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247-1255.
26. Chan FK, Wong VW, Suen BY, et al. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
27. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:1-10.
28. Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-1102.
29. Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071-1080.
30. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634-1642.
31. Solomon SD, Wittes J, Finn PV, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104-2113.
32. Celebrex [package insert]. New York, NY: Pfizer Inc; 2008.
33. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
34. MacDonald TM, Wei L. Is there an interaction between the cardiovascular protective effects of low-dose aspirin and ibuprofen? Basic Clin Pharmacol Toxicol. 2006;98:275-280.
35. US Food and Drug Administration. Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. 2006. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM161282.pdf. Accessed October 19, 2010.
36. Brune K, Hochberg MC, Schiff M, et al. The platelet inhibitory effects of the combination of naproxen sodium or acetaminophen with low dose aspirin [abstract]. Arthritis Rheum. 2007;56:S359.-
37. Farkouh ME, Greenberg JD, Jeger RV, et al. Cardiovascular outcomes in high risk patients with osteoarthritis treated with ibuprofen, naproxen or lumiracoxib. Ann Rheum Dis. 2007;66:764-770.
38. Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295-1301.
39. Zlotnick S, Oldenhof J, Schuller R, et al. Effect of over-the-counter doses of naproxen sodium on inhibition of platelet cyclooxygenase-1 in healthy volunteers. Poster presented at: the American College of Rheumatology; November 13, 2006; Washington, DC. Poster L33.
40. Lanas A, Scheiman J. Low-dose aspirin and upper gastrointestinal damage: epidemiology, prevention and treatment. Curr Med Res Opin. 2007;23:163-173.
41. Scheiman JM, Fendrick AM. Summing the risk of NSAID therapy. Lancet. 2007;369:1580-1581.
42. Rostom A, Dube C, Wells G, et al. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev. 2002;(4):CD002296.-
43. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241-249.
44. Chan FK, Wong VW, Suen BY. Combination of a cyclooxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369:1621-1626.
45. Chan FK, Abraham NS, Scheiman JM, et al. Management of patients on nonsteroidal anti-inflammatory drugs: a clinical practice recommendation from the First International Working Party on Gastrointestinal and Cardiovascular Effects of Nonsteroidal Anti-inflammatory Drugs and Anti-platelet Agents. Am J Gastroenterol. 2008;103:2908-2918.
46. Lewis SC, Langman MJ, Laporte JR, et al. Dose-response relationships between individual nonaspirin nonsteroidal anti-inflammatory drugs (NANSAIDs) and serious upper gastrointestinal bleeding: a meta-analysis based on individual patient data. Br J Clin Pharmacol. 2002;54:320-326.
47. Spiegel BM, Chiou CF, Ofman JJ. Minimizing complications from nonsteroidal antiinflammatory drugs: cost-effectiveness of competing strategies in varying risk groups. Arthritis Rheum. 2005;53:185-197.
48. Gladding PA, Webster MW, Farrell HB, et al. The antiplatelet effect of six non-steroidal anti-inflammatory drugs and their pharmacodynamic interaction with aspirin in healthy volunteers. Am J Cardiol. 2008;101:1060-1063.