User login
When a child won’t speak
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
- Assess children with selective mutism for severe social anxiety (C).
- Drug therapy for anxiety and depressive conditions related to selective mutism and collaboration with school personnel and parents can help a child increase her frequency and audibility of speech in public settings (C).
- Exposure-based practices can help children with selective mutism to speak in public places (C).
Lucy’s parents were having difficulty reconciling the child they knew at home with the one they sent to school. At home, the 7-year-old spoke easily with her parents and siblings. At school and in other public places such as the supermarket, mall, or park, she would not speak at all. This had been going on for several years.
While Lucy was attending school without difficulty, she seemed sullen and withdrawn to her teachers. Lucy had friends at school, but would communicate with them by writing messages on paper or drawing letters in the air. Although Lucy had passed first grade on the basis of her written work, her parents worried about her ability to succeed in second grade, where her teachers expected her to participate in class and she would need to take standardized tests that required audible verbal responses. At the urging of a school counselor, Lucy’s parents took her to their family physician. They needed help in drawing her out so that the rest of the world could get to know the Lucy that they knew and loved.
Lucy’s case is typical of selective mutism
Lucy was suffering from selective mutism, the persistent failure to speak in specific social situations where speaking is expected, such as at school and with playmates.1 Lucy’s case is typical in that children with selective mutism speak well in other situations, typically at home. Thus, the disorder is not due to a communication disorder such as stuttering and it is not due to a lack of knowledge or comfort with language.
To meet diagnostic criteria, the disorder must last at least 1 month, though it can last for several years, and must interfere with a child’s education or ability to communicate socially.1
A little known disorder makes national headlines
Selective mutism occurs in 0.2%–2.0% of children, affects boys and girls equally, and often begins at 3 to 6 years of age.2-9 The disorder gained national attention this past spring when it was revealed that the shooter in the Virginia Tech massacre—Seung-Hui Cho—had, as an adolescent, been diagnosed with selective mutism.10 Though his diagnosis made plenty of headlines, the disorder itself occupies little space in the pediatric literature.11
What we do know is that selective mutism can have a chronic course that affects a child’s ability to form friendships, complete academic tasks, develop appropriate language and social skills, and participate in standardized testing.2-9 While parents often attribute their child’s behavior to shyness, this disorder goes beyond that. While shy children function, those with selective mutism struggle socially, emotionally, and academically. Children with selective mutism say the words won’t come out and their body won’t let them speak. One father of a 7-year-old girl with selective mutism said that his daughter “describes it as the words get stuck in her toes.”12
Many researchers theorize that children with selective mutism have severe social anxiety, and assessment and treatment strategies are typically based on this notion. Reports in the literature have also suggested that selective mutism is related to a developmental disorder or delay, as seen in autistic spectrum disorders, as well as anxiety disorders and depression.13 In addition, selective mutism has been linked to oppositional defiant disorder and subtle language impairments.14,15
Begin by excluding competing explanations
If a family is referred to you for possible selective mutism, you’ll first need to exclude competing explanations for the problem, such as hearing difficulties, speech and language disorders, school-based threats, and medical problems, such as asthma, which could prevent a child from speaking comfortably in a public setting. Assuming you are able to exclude these, and other competing explanations, you’ll need to ask 8 questions of parents and school officials. The answers you get will help you to screen for selective mutism and identify key antecedents and consequences to the behavior that are important in addressing the problem.
8 questions you’ll need to ask
1. What specific settings involve failure to speak? Children with selective mutism typically have difficulties in school, on the playground, in malls, and at restaurants.
2. Has the mutism lasted at least one month? Brief refusal to speak is not uncommon in children.
3. Does the child speak well at home with people she knows well? Most children with selective mutism speak well at home with family members, which belies a communication disorder.
4. Is failure to speak significantly interfering with the child’s academic or social development? Selective mutism must involve a significant interference in daily functioning.
5. What circumstances surround each episode of mutism? In particular, is the mutism associated with the desire to increase social attention or stimulation from others, decrease anxiety, or avoid or withdraw from adult commands or requests? A full understanding of selective mutism must include a review of triggers like these.
6. Can the child be encouraged to speak audibly in any way in certain public settings? Children with selective mutism who can speak to some extent in public situations may have a better prognosis than those who do not.9
7. How do others respond to, or compensate for, the child’s mutism? Do they complete tasks for the child? Order food for her? Allow whispers or communication via writing? Excessive accommodations enable a child with selective mutism to maintain the behavior.
8. Does the child appear anxious or depressed in situations involving mutism? Mutism is often linked to poor affect and social anxiety.
Still suspect selective mutism? Make a referral
If after asking these 8 questions you still suspect selective mutism, you will need to make a referral to a child psychologist or other mental health professional who specializes in behavioral strategies to treat selective mutism. The psychologist will meet with the child and utilize more formal methods of assessment.
The psychologist may use the Anxiety Disorders Interview Schedule for DSM-IV-TR (child and parent versions), a structured interview that emphasizes anxiety-based disorders and includes a section for selective mutism based on DSM-IV-TR criteria.16 Another helpful tool is the Functional Diagnostic Profile for Selective Mutism, which helps to assess contextual factors that surround refusal to speak.17 The challenge, of course, in conducting these interviews is that many children will not answer the psychologist’s questions verbally, but may communicate via nonverbal gestures, writing, or other creative ways such as drawing letters in the air.
Parent and teacher logs provide valuable insight
Ongoing behavioral observations or daily logbooks completed by parents and teachers are also important in treating a child with selective mutism. If parents aren’t doing so already at the suggestion of the therapist, ask them to keep a logbook and encourage them to ask the child’s teachers to keep one, as well. This log should include the number and volume of words spoken, to whom the child has spoken, where the child spoke, how others reacted (ie, Did they pay extra attention to the child?), and the child’s compensatory behaviors such as whispering, pointing, crying, nodding, or frowning.18 Parents and teachers should also note the antecedents to mutism, including demands (or requests) from others, or the presence of unfamiliar people.
A focus on systematic desensitization
Much of the literature regarding the treatment of selective mutism consists of case reports and small-scale studies.19 The paucity of data is, in part, a reflection of the rarity of the problem as well as a historical lack of consensus among researchers about the nature and diagnostic criteria for selective mutism.
The studies we do have on selective mutism indicate that behavior modification can be effective for these patients. Behavioral treatment primarily involves child- and parent-based practices.20
Child-based practices include management of physical sensations of anxiety often via relaxation training and breathing retraining. Neither technique requires verbal input from a child. In addition, the therapist will have the child practice audible words in progressively more difficult settings via systematic desensitization. These settings often include the following, in order: the child’s home (with the therapist), the therapist’s office, community settings such as a mall or a restaurant, and finally, school.
In these settings, the therapist might, for example, encourage the patient to:
- speak on the telephone or answer the door at home.
- stay at the therapist’s office until at least one word is audibly spoken. (One approach that the therapist may use to increase rapport and provide opportunities to prompt speech is to play games with the child.)
- order her own food at a local restaurant.
- read a story aloud to classmates at school.
In addition to systematic desensitization, the therapist may use audio or videotapes to help the youngster. In one scenario, the therapist may ask the parents to bring in an audiotape or videotape of the child at home when she is speaking. The therapist then provides ample reinforcement for the child’s speech by praising the child for having a beautiful voice and for making the tape. Audio and videotapes may also be used to help the child speak louder or work on her articulation.
Reinforce the positive, avoid “shaming”
Parent-based practices include reinforcing the child’s speech in different settings, developing new and independent exposures to generalize speech, and consulting with school officials on an ongoing basis to address social and academic problems from poor use of speech.
The therapist is likely to remind the parent that positive reinforcement is key, while punishing or “shaming” the child is ineffective.11 Some therapists will make use of a technique called “token economy,” where the child’s progress earns her colored chips that can be redeemed for a trip to a video game center, or a special treat, such as a sleep-over with a trusted friend.11
With the therapist’s help, parents can also model appropriate social interactions for a child with selective mutism because social skills may not be sufficiently developed. For example, the parent might encourage the child to answer the door or telephone. The parent might also monitor the child’s interactions with friends and provide feedback. In addition, the therapist may encourage the parents to ignore inappropriate compensatory behaviors as treatment progresses. Instead, a child with selective mutism may be increasingly challenged to provide audible speech to make requests or otherwise communicate.21
Drug therapy may include MAOIs and SSRIs
Medical treatment for selective mutism may include monoamine oxidase inhibitors (MAOIs) such as phenelzine (30–60 mg/day) or selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (10–60 mg/day), sertraline (100 mg/day), fluvoxamine (50–100 mg/day), citalopram (20–40 mg/day), or paroxetine (5 mg/day).22 However, no large-scale studies of pharmacotherapy for selective mutism have been conducted and medication effects are quite variable. The literature in this area has consisted largely of case studies.
After 9 months of treatment, Lucy speaks up at school
Lucy’s therapist (JLV) utilized many of the behavioral procedures described in this article. Treatment included exposure-based practices at home, in a clinic, in various community settings, and at school. By the end of her 9 months of treatment, Lucy was speaking independently to her classmates and teachers, though ongoing praise by her teachers was needed to encourage her to maintain an appropriate speech volume.
The literature on selective mutism suggests Lucy’s case was not unusual; it typically takes several months for these young patients to improve. Ongoing communication issues, though, often linger.9
- What specific settings involve failure to speak?
- Has the mutism lasted at least one month?
- Does the child speak well at home with people she knows well?
- Is failure to speak significantly interfering with the child’s academic or social development?
- What circumstances surround each episode of mutism?
- Can the child be encouraged to speak audibly in any way in certain public settings?
- How do others respond to, or compensate for, the child’s mutism?
- Does the child appear anxious or depressed in situations involving mutism?
Correspondence
Christopher A. Kearney, PhD, Department of Psychology, University of Nevada, Las Vegas, 4505 maryland Parkway, Las Vegas, Nv 89154-5030; [email protected]
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text revision). Washington, DC: Author.
2. Andersson CB, Thomsen PH. Electively mute children: an analysis of 37 Danish cases. Nordic J Psychiatry 1998;52:231-238.
3. Bergman RL, Piacentini J, McCracken JT. Prevalence and description of selective mutism in a school-based study. J Am Acad Child Adolesc Psychiatry 2002;41:938-946.
4. Black B, Uhde TW. Elective mutism as a variant of social phobia. J Am Acad Child Adolesc Psychiatry 1992;1090-1094.
5. Ford MA, Sladeczeck IE, Carlson J. Selective mutism: phenomenological characteristics. School Psychol Quarterly 1998;13:192-227.
6. Kopp S, Gillberg C. Selective mutism: a population-based study: a research note. J Child Psychol Psychiatry 1997;38:257-262.
7. Krysanski V. A brief review of selective mutism literature. J Psychol 2003;137:29-40.
8. Kumpulainen K, Rasanen E, Raaska H, Somppi V. Selective mutism among second-graders in elementary school. Euro Child Adolesc Psychiatry 1998;7:24-29.
9. Remschmidt H, Poller M, Herpertz-Dahlmann B, Hennighausen K, Gutenbrenner C. A follow-up study of 45 patients with elective mutism. European Archives of Psychiatry and Clinical Neuroscience 2001;251:284-296.
10. Urbina I. Virginia Tech criticized for actions in shooting. The New York Times. August 30, 2007. Available at: www.nytimes.com/2007/08/30/us/30school.html. Accessed on October 10, 2007.
11. Schwartz RH, Shipon-Blum E. “Shy”child? Don’t overlook selective mutism. Contemp Peds 2005;22(7):30-39.
12. ABC News. Selective mutism: When your child can’t speak. Available at: abcnews.go.com/GmA/AmericanFamily/story?id=1770308&page=1. Accessed on September 26, 2007.
13. Vecchio J, Kearney CA. Selective mutism in children: comparison to youths with and without anxiety disorders. J Psychopath Beh Assess 2005;28:139-144.
14. Yeganeh R, Beidel DC, Turner SM. Selective mutism: more than social anxiety? Depress Anx 2006;23:117-123.
15. Manassis K, Fung D, Tannock R, Sloman L, Fiksenbaum L, McInnes A. Characterizing selective mutism: is it more than social anxiety? Depress Anx 2003;18:153-161.
16. Silverman WK, Albano AM. The Anxiety Disorders Interview Schedule for DSM-IV, child and parent versions. New York: oxford University Press;1996.
17. Schill MT, Kratochwill TR, Gardner WI. An assessment protocol for selective mutism: analogue assessment using parents as facilitators. J Sch Psychol 1996;34:1-21.
18. Jackson MF, Allen RS, Boothe AB, Nava ML, Coates A. Innovative analyses and interventions in the treatment of selective mutism. Clin Case Studies 2005;4:81-112.
19. Cohan SL, Chavira DA, Stein MB. Practitioner review: psychosocial interventions for children with selective mutism: a critical evaluation of the literature from 1990-2005. J Child Psychol Psychiatry 2006;47:1085-1097.
20. Rye MS, Ullman D. The successful treatment of long-term selective mutism: a case study. J Beh Ther Exp Psychiatry 1999;30:313-323.
21. Fisak BJ, Oliveros A, Ehrenreich JT. Assessment and behavioral treatment of selective mutism. Clin Case Studies 2006;5:382-402.
22. Kumpulainen K. Phenomenology and treatment of selective mutism. CNS Drugs 2002;16:175-180.
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
- Assess children with selective mutism for severe social anxiety (C).
- Drug therapy for anxiety and depressive conditions related to selective mutism and collaboration with school personnel and parents can help a child increase her frequency and audibility of speech in public settings (C).
- Exposure-based practices can help children with selective mutism to speak in public places (C).
Lucy’s parents were having difficulty reconciling the child they knew at home with the one they sent to school. At home, the 7-year-old spoke easily with her parents and siblings. At school and in other public places such as the supermarket, mall, or park, she would not speak at all. This had been going on for several years.
While Lucy was attending school without difficulty, she seemed sullen and withdrawn to her teachers. Lucy had friends at school, but would communicate with them by writing messages on paper or drawing letters in the air. Although Lucy had passed first grade on the basis of her written work, her parents worried about her ability to succeed in second grade, where her teachers expected her to participate in class and she would need to take standardized tests that required audible verbal responses. At the urging of a school counselor, Lucy’s parents took her to their family physician. They needed help in drawing her out so that the rest of the world could get to know the Lucy that they knew and loved.
Lucy’s case is typical of selective mutism
Lucy was suffering from selective mutism, the persistent failure to speak in specific social situations where speaking is expected, such as at school and with playmates.1 Lucy’s case is typical in that children with selective mutism speak well in other situations, typically at home. Thus, the disorder is not due to a communication disorder such as stuttering and it is not due to a lack of knowledge or comfort with language.
To meet diagnostic criteria, the disorder must last at least 1 month, though it can last for several years, and must interfere with a child’s education or ability to communicate socially.1
A little known disorder makes national headlines
Selective mutism occurs in 0.2%–2.0% of children, affects boys and girls equally, and often begins at 3 to 6 years of age.2-9 The disorder gained national attention this past spring when it was revealed that the shooter in the Virginia Tech massacre—Seung-Hui Cho—had, as an adolescent, been diagnosed with selective mutism.10 Though his diagnosis made plenty of headlines, the disorder itself occupies little space in the pediatric literature.11
What we do know is that selective mutism can have a chronic course that affects a child’s ability to form friendships, complete academic tasks, develop appropriate language and social skills, and participate in standardized testing.2-9 While parents often attribute their child’s behavior to shyness, this disorder goes beyond that. While shy children function, those with selective mutism struggle socially, emotionally, and academically. Children with selective mutism say the words won’t come out and their body won’t let them speak. One father of a 7-year-old girl with selective mutism said that his daughter “describes it as the words get stuck in her toes.”12
Many researchers theorize that children with selective mutism have severe social anxiety, and assessment and treatment strategies are typically based on this notion. Reports in the literature have also suggested that selective mutism is related to a developmental disorder or delay, as seen in autistic spectrum disorders, as well as anxiety disorders and depression.13 In addition, selective mutism has been linked to oppositional defiant disorder and subtle language impairments.14,15
Begin by excluding competing explanations
If a family is referred to you for possible selective mutism, you’ll first need to exclude competing explanations for the problem, such as hearing difficulties, speech and language disorders, school-based threats, and medical problems, such as asthma, which could prevent a child from speaking comfortably in a public setting. Assuming you are able to exclude these, and other competing explanations, you’ll need to ask 8 questions of parents and school officials. The answers you get will help you to screen for selective mutism and identify key antecedents and consequences to the behavior that are important in addressing the problem.
8 questions you’ll need to ask
1. What specific settings involve failure to speak? Children with selective mutism typically have difficulties in school, on the playground, in malls, and at restaurants.
2. Has the mutism lasted at least one month? Brief refusal to speak is not uncommon in children.
3. Does the child speak well at home with people she knows well? Most children with selective mutism speak well at home with family members, which belies a communication disorder.
4. Is failure to speak significantly interfering with the child’s academic or social development? Selective mutism must involve a significant interference in daily functioning.
5. What circumstances surround each episode of mutism? In particular, is the mutism associated with the desire to increase social attention or stimulation from others, decrease anxiety, or avoid or withdraw from adult commands or requests? A full understanding of selective mutism must include a review of triggers like these.
6. Can the child be encouraged to speak audibly in any way in certain public settings? Children with selective mutism who can speak to some extent in public situations may have a better prognosis than those who do not.9
7. How do others respond to, or compensate for, the child’s mutism? Do they complete tasks for the child? Order food for her? Allow whispers or communication via writing? Excessive accommodations enable a child with selective mutism to maintain the behavior.
8. Does the child appear anxious or depressed in situations involving mutism? Mutism is often linked to poor affect and social anxiety.
Still suspect selective mutism? Make a referral
If after asking these 8 questions you still suspect selective mutism, you will need to make a referral to a child psychologist or other mental health professional who specializes in behavioral strategies to treat selective mutism. The psychologist will meet with the child and utilize more formal methods of assessment.
The psychologist may use the Anxiety Disorders Interview Schedule for DSM-IV-TR (child and parent versions), a structured interview that emphasizes anxiety-based disorders and includes a section for selective mutism based on DSM-IV-TR criteria.16 Another helpful tool is the Functional Diagnostic Profile for Selective Mutism, which helps to assess contextual factors that surround refusal to speak.17 The challenge, of course, in conducting these interviews is that many children will not answer the psychologist’s questions verbally, but may communicate via nonverbal gestures, writing, or other creative ways such as drawing letters in the air.
Parent and teacher logs provide valuable insight
Ongoing behavioral observations or daily logbooks completed by parents and teachers are also important in treating a child with selective mutism. If parents aren’t doing so already at the suggestion of the therapist, ask them to keep a logbook and encourage them to ask the child’s teachers to keep one, as well. This log should include the number and volume of words spoken, to whom the child has spoken, where the child spoke, how others reacted (ie, Did they pay extra attention to the child?), and the child’s compensatory behaviors such as whispering, pointing, crying, nodding, or frowning.18 Parents and teachers should also note the antecedents to mutism, including demands (or requests) from others, or the presence of unfamiliar people.
A focus on systematic desensitization
Much of the literature regarding the treatment of selective mutism consists of case reports and small-scale studies.19 The paucity of data is, in part, a reflection of the rarity of the problem as well as a historical lack of consensus among researchers about the nature and diagnostic criteria for selective mutism.
The studies we do have on selective mutism indicate that behavior modification can be effective for these patients. Behavioral treatment primarily involves child- and parent-based practices.20
Child-based practices include management of physical sensations of anxiety often via relaxation training and breathing retraining. Neither technique requires verbal input from a child. In addition, the therapist will have the child practice audible words in progressively more difficult settings via systematic desensitization. These settings often include the following, in order: the child’s home (with the therapist), the therapist’s office, community settings such as a mall or a restaurant, and finally, school.
In these settings, the therapist might, for example, encourage the patient to:
- speak on the telephone or answer the door at home.
- stay at the therapist’s office until at least one word is audibly spoken. (One approach that the therapist may use to increase rapport and provide opportunities to prompt speech is to play games with the child.)
- order her own food at a local restaurant.
- read a story aloud to classmates at school.
In addition to systematic desensitization, the therapist may use audio or videotapes to help the youngster. In one scenario, the therapist may ask the parents to bring in an audiotape or videotape of the child at home when she is speaking. The therapist then provides ample reinforcement for the child’s speech by praising the child for having a beautiful voice and for making the tape. Audio and videotapes may also be used to help the child speak louder or work on her articulation.
Reinforce the positive, avoid “shaming”
Parent-based practices include reinforcing the child’s speech in different settings, developing new and independent exposures to generalize speech, and consulting with school officials on an ongoing basis to address social and academic problems from poor use of speech.
The therapist is likely to remind the parent that positive reinforcement is key, while punishing or “shaming” the child is ineffective.11 Some therapists will make use of a technique called “token economy,” where the child’s progress earns her colored chips that can be redeemed for a trip to a video game center, or a special treat, such as a sleep-over with a trusted friend.11
With the therapist’s help, parents can also model appropriate social interactions for a child with selective mutism because social skills may not be sufficiently developed. For example, the parent might encourage the child to answer the door or telephone. The parent might also monitor the child’s interactions with friends and provide feedback. In addition, the therapist may encourage the parents to ignore inappropriate compensatory behaviors as treatment progresses. Instead, a child with selective mutism may be increasingly challenged to provide audible speech to make requests or otherwise communicate.21
Drug therapy may include MAOIs and SSRIs
Medical treatment for selective mutism may include monoamine oxidase inhibitors (MAOIs) such as phenelzine (30–60 mg/day) or selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (10–60 mg/day), sertraline (100 mg/day), fluvoxamine (50–100 mg/day), citalopram (20–40 mg/day), or paroxetine (5 mg/day).22 However, no large-scale studies of pharmacotherapy for selective mutism have been conducted and medication effects are quite variable. The literature in this area has consisted largely of case studies.
After 9 months of treatment, Lucy speaks up at school
Lucy’s therapist (JLV) utilized many of the behavioral procedures described in this article. Treatment included exposure-based practices at home, in a clinic, in various community settings, and at school. By the end of her 9 months of treatment, Lucy was speaking independently to her classmates and teachers, though ongoing praise by her teachers was needed to encourage her to maintain an appropriate speech volume.
The literature on selective mutism suggests Lucy’s case was not unusual; it typically takes several months for these young patients to improve. Ongoing communication issues, though, often linger.9
- What specific settings involve failure to speak?
- Has the mutism lasted at least one month?
- Does the child speak well at home with people she knows well?
- Is failure to speak significantly interfering with the child’s academic or social development?
- What circumstances surround each episode of mutism?
- Can the child be encouraged to speak audibly in any way in certain public settings?
- How do others respond to, or compensate for, the child’s mutism?
- Does the child appear anxious or depressed in situations involving mutism?
Correspondence
Christopher A. Kearney, PhD, Department of Psychology, University of Nevada, Las Vegas, 4505 maryland Parkway, Las Vegas, Nv 89154-5030; [email protected]
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
- Assess children with selective mutism for severe social anxiety (C).
- Drug therapy for anxiety and depressive conditions related to selective mutism and collaboration with school personnel and parents can help a child increase her frequency and audibility of speech in public settings (C).
- Exposure-based practices can help children with selective mutism to speak in public places (C).
Lucy’s parents were having difficulty reconciling the child they knew at home with the one they sent to school. At home, the 7-year-old spoke easily with her parents and siblings. At school and in other public places such as the supermarket, mall, or park, she would not speak at all. This had been going on for several years.
While Lucy was attending school without difficulty, she seemed sullen and withdrawn to her teachers. Lucy had friends at school, but would communicate with them by writing messages on paper or drawing letters in the air. Although Lucy had passed first grade on the basis of her written work, her parents worried about her ability to succeed in second grade, where her teachers expected her to participate in class and she would need to take standardized tests that required audible verbal responses. At the urging of a school counselor, Lucy’s parents took her to their family physician. They needed help in drawing her out so that the rest of the world could get to know the Lucy that they knew and loved.
Lucy’s case is typical of selective mutism
Lucy was suffering from selective mutism, the persistent failure to speak in specific social situations where speaking is expected, such as at school and with playmates.1 Lucy’s case is typical in that children with selective mutism speak well in other situations, typically at home. Thus, the disorder is not due to a communication disorder such as stuttering and it is not due to a lack of knowledge or comfort with language.
To meet diagnostic criteria, the disorder must last at least 1 month, though it can last for several years, and must interfere with a child’s education or ability to communicate socially.1
A little known disorder makes national headlines
Selective mutism occurs in 0.2%–2.0% of children, affects boys and girls equally, and often begins at 3 to 6 years of age.2-9 The disorder gained national attention this past spring when it was revealed that the shooter in the Virginia Tech massacre—Seung-Hui Cho—had, as an adolescent, been diagnosed with selective mutism.10 Though his diagnosis made plenty of headlines, the disorder itself occupies little space in the pediatric literature.11
What we do know is that selective mutism can have a chronic course that affects a child’s ability to form friendships, complete academic tasks, develop appropriate language and social skills, and participate in standardized testing.2-9 While parents often attribute their child’s behavior to shyness, this disorder goes beyond that. While shy children function, those with selective mutism struggle socially, emotionally, and academically. Children with selective mutism say the words won’t come out and their body won’t let them speak. One father of a 7-year-old girl with selective mutism said that his daughter “describes it as the words get stuck in her toes.”12
Many researchers theorize that children with selective mutism have severe social anxiety, and assessment and treatment strategies are typically based on this notion. Reports in the literature have also suggested that selective mutism is related to a developmental disorder or delay, as seen in autistic spectrum disorders, as well as anxiety disorders and depression.13 In addition, selective mutism has been linked to oppositional defiant disorder and subtle language impairments.14,15
Begin by excluding competing explanations
If a family is referred to you for possible selective mutism, you’ll first need to exclude competing explanations for the problem, such as hearing difficulties, speech and language disorders, school-based threats, and medical problems, such as asthma, which could prevent a child from speaking comfortably in a public setting. Assuming you are able to exclude these, and other competing explanations, you’ll need to ask 8 questions of parents and school officials. The answers you get will help you to screen for selective mutism and identify key antecedents and consequences to the behavior that are important in addressing the problem.
8 questions you’ll need to ask
1. What specific settings involve failure to speak? Children with selective mutism typically have difficulties in school, on the playground, in malls, and at restaurants.
2. Has the mutism lasted at least one month? Brief refusal to speak is not uncommon in children.
3. Does the child speak well at home with people she knows well? Most children with selective mutism speak well at home with family members, which belies a communication disorder.
4. Is failure to speak significantly interfering with the child’s academic or social development? Selective mutism must involve a significant interference in daily functioning.
5. What circumstances surround each episode of mutism? In particular, is the mutism associated with the desire to increase social attention or stimulation from others, decrease anxiety, or avoid or withdraw from adult commands or requests? A full understanding of selective mutism must include a review of triggers like these.
6. Can the child be encouraged to speak audibly in any way in certain public settings? Children with selective mutism who can speak to some extent in public situations may have a better prognosis than those who do not.9
7. How do others respond to, or compensate for, the child’s mutism? Do they complete tasks for the child? Order food for her? Allow whispers or communication via writing? Excessive accommodations enable a child with selective mutism to maintain the behavior.
8. Does the child appear anxious or depressed in situations involving mutism? Mutism is often linked to poor affect and social anxiety.
Still suspect selective mutism? Make a referral
If after asking these 8 questions you still suspect selective mutism, you will need to make a referral to a child psychologist or other mental health professional who specializes in behavioral strategies to treat selective mutism. The psychologist will meet with the child and utilize more formal methods of assessment.
The psychologist may use the Anxiety Disorders Interview Schedule for DSM-IV-TR (child and parent versions), a structured interview that emphasizes anxiety-based disorders and includes a section for selective mutism based on DSM-IV-TR criteria.16 Another helpful tool is the Functional Diagnostic Profile for Selective Mutism, which helps to assess contextual factors that surround refusal to speak.17 The challenge, of course, in conducting these interviews is that many children will not answer the psychologist’s questions verbally, but may communicate via nonverbal gestures, writing, or other creative ways such as drawing letters in the air.
Parent and teacher logs provide valuable insight
Ongoing behavioral observations or daily logbooks completed by parents and teachers are also important in treating a child with selective mutism. If parents aren’t doing so already at the suggestion of the therapist, ask them to keep a logbook and encourage them to ask the child’s teachers to keep one, as well. This log should include the number and volume of words spoken, to whom the child has spoken, where the child spoke, how others reacted (ie, Did they pay extra attention to the child?), and the child’s compensatory behaviors such as whispering, pointing, crying, nodding, or frowning.18 Parents and teachers should also note the antecedents to mutism, including demands (or requests) from others, or the presence of unfamiliar people.
A focus on systematic desensitization
Much of the literature regarding the treatment of selective mutism consists of case reports and small-scale studies.19 The paucity of data is, in part, a reflection of the rarity of the problem as well as a historical lack of consensus among researchers about the nature and diagnostic criteria for selective mutism.
The studies we do have on selective mutism indicate that behavior modification can be effective for these patients. Behavioral treatment primarily involves child- and parent-based practices.20
Child-based practices include management of physical sensations of anxiety often via relaxation training and breathing retraining. Neither technique requires verbal input from a child. In addition, the therapist will have the child practice audible words in progressively more difficult settings via systematic desensitization. These settings often include the following, in order: the child’s home (with the therapist), the therapist’s office, community settings such as a mall or a restaurant, and finally, school.
In these settings, the therapist might, for example, encourage the patient to:
- speak on the telephone or answer the door at home.
- stay at the therapist’s office until at least one word is audibly spoken. (One approach that the therapist may use to increase rapport and provide opportunities to prompt speech is to play games with the child.)
- order her own food at a local restaurant.
- read a story aloud to classmates at school.
In addition to systematic desensitization, the therapist may use audio or videotapes to help the youngster. In one scenario, the therapist may ask the parents to bring in an audiotape or videotape of the child at home when she is speaking. The therapist then provides ample reinforcement for the child’s speech by praising the child for having a beautiful voice and for making the tape. Audio and videotapes may also be used to help the child speak louder or work on her articulation.
Reinforce the positive, avoid “shaming”
Parent-based practices include reinforcing the child’s speech in different settings, developing new and independent exposures to generalize speech, and consulting with school officials on an ongoing basis to address social and academic problems from poor use of speech.
The therapist is likely to remind the parent that positive reinforcement is key, while punishing or “shaming” the child is ineffective.11 Some therapists will make use of a technique called “token economy,” where the child’s progress earns her colored chips that can be redeemed for a trip to a video game center, or a special treat, such as a sleep-over with a trusted friend.11
With the therapist’s help, parents can also model appropriate social interactions for a child with selective mutism because social skills may not be sufficiently developed. For example, the parent might encourage the child to answer the door or telephone. The parent might also monitor the child’s interactions with friends and provide feedback. In addition, the therapist may encourage the parents to ignore inappropriate compensatory behaviors as treatment progresses. Instead, a child with selective mutism may be increasingly challenged to provide audible speech to make requests or otherwise communicate.21
Drug therapy may include MAOIs and SSRIs
Medical treatment for selective mutism may include monoamine oxidase inhibitors (MAOIs) such as phenelzine (30–60 mg/day) or selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (10–60 mg/day), sertraline (100 mg/day), fluvoxamine (50–100 mg/day), citalopram (20–40 mg/day), or paroxetine (5 mg/day).22 However, no large-scale studies of pharmacotherapy for selective mutism have been conducted and medication effects are quite variable. The literature in this area has consisted largely of case studies.
After 9 months of treatment, Lucy speaks up at school
Lucy’s therapist (JLV) utilized many of the behavioral procedures described in this article. Treatment included exposure-based practices at home, in a clinic, in various community settings, and at school. By the end of her 9 months of treatment, Lucy was speaking independently to her classmates and teachers, though ongoing praise by her teachers was needed to encourage her to maintain an appropriate speech volume.
The literature on selective mutism suggests Lucy’s case was not unusual; it typically takes several months for these young patients to improve. Ongoing communication issues, though, often linger.9
- What specific settings involve failure to speak?
- Has the mutism lasted at least one month?
- Does the child speak well at home with people she knows well?
- Is failure to speak significantly interfering with the child’s academic or social development?
- What circumstances surround each episode of mutism?
- Can the child be encouraged to speak audibly in any way in certain public settings?
- How do others respond to, or compensate for, the child’s mutism?
- Does the child appear anxious or depressed in situations involving mutism?
Correspondence
Christopher A. Kearney, PhD, Department of Psychology, University of Nevada, Las Vegas, 4505 maryland Parkway, Las Vegas, Nv 89154-5030; [email protected]
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text revision). Washington, DC: Author.
2. Andersson CB, Thomsen PH. Electively mute children: an analysis of 37 Danish cases. Nordic J Psychiatry 1998;52:231-238.
3. Bergman RL, Piacentini J, McCracken JT. Prevalence and description of selective mutism in a school-based study. J Am Acad Child Adolesc Psychiatry 2002;41:938-946.
4. Black B, Uhde TW. Elective mutism as a variant of social phobia. J Am Acad Child Adolesc Psychiatry 1992;1090-1094.
5. Ford MA, Sladeczeck IE, Carlson J. Selective mutism: phenomenological characteristics. School Psychol Quarterly 1998;13:192-227.
6. Kopp S, Gillberg C. Selective mutism: a population-based study: a research note. J Child Psychol Psychiatry 1997;38:257-262.
7. Krysanski V. A brief review of selective mutism literature. J Psychol 2003;137:29-40.
8. Kumpulainen K, Rasanen E, Raaska H, Somppi V. Selective mutism among second-graders in elementary school. Euro Child Adolesc Psychiatry 1998;7:24-29.
9. Remschmidt H, Poller M, Herpertz-Dahlmann B, Hennighausen K, Gutenbrenner C. A follow-up study of 45 patients with elective mutism. European Archives of Psychiatry and Clinical Neuroscience 2001;251:284-296.
10. Urbina I. Virginia Tech criticized for actions in shooting. The New York Times. August 30, 2007. Available at: www.nytimes.com/2007/08/30/us/30school.html. Accessed on October 10, 2007.
11. Schwartz RH, Shipon-Blum E. “Shy”child? Don’t overlook selective mutism. Contemp Peds 2005;22(7):30-39.
12. ABC News. Selective mutism: When your child can’t speak. Available at: abcnews.go.com/GmA/AmericanFamily/story?id=1770308&page=1. Accessed on September 26, 2007.
13. Vecchio J, Kearney CA. Selective mutism in children: comparison to youths with and without anxiety disorders. J Psychopath Beh Assess 2005;28:139-144.
14. Yeganeh R, Beidel DC, Turner SM. Selective mutism: more than social anxiety? Depress Anx 2006;23:117-123.
15. Manassis K, Fung D, Tannock R, Sloman L, Fiksenbaum L, McInnes A. Characterizing selective mutism: is it more than social anxiety? Depress Anx 2003;18:153-161.
16. Silverman WK, Albano AM. The Anxiety Disorders Interview Schedule for DSM-IV, child and parent versions. New York: oxford University Press;1996.
17. Schill MT, Kratochwill TR, Gardner WI. An assessment protocol for selective mutism: analogue assessment using parents as facilitators. J Sch Psychol 1996;34:1-21.
18. Jackson MF, Allen RS, Boothe AB, Nava ML, Coates A. Innovative analyses and interventions in the treatment of selective mutism. Clin Case Studies 2005;4:81-112.
19. Cohan SL, Chavira DA, Stein MB. Practitioner review: psychosocial interventions for children with selective mutism: a critical evaluation of the literature from 1990-2005. J Child Psychol Psychiatry 2006;47:1085-1097.
20. Rye MS, Ullman D. The successful treatment of long-term selective mutism: a case study. J Beh Ther Exp Psychiatry 1999;30:313-323.
21. Fisak BJ, Oliveros A, Ehrenreich JT. Assessment and behavioral treatment of selective mutism. Clin Case Studies 2006;5:382-402.
22. Kumpulainen K. Phenomenology and treatment of selective mutism. CNS Drugs 2002;16:175-180.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text revision). Washington, DC: Author.
2. Andersson CB, Thomsen PH. Electively mute children: an analysis of 37 Danish cases. Nordic J Psychiatry 1998;52:231-238.
3. Bergman RL, Piacentini J, McCracken JT. Prevalence and description of selective mutism in a school-based study. J Am Acad Child Adolesc Psychiatry 2002;41:938-946.
4. Black B, Uhde TW. Elective mutism as a variant of social phobia. J Am Acad Child Adolesc Psychiatry 1992;1090-1094.
5. Ford MA, Sladeczeck IE, Carlson J. Selective mutism: phenomenological characteristics. School Psychol Quarterly 1998;13:192-227.
6. Kopp S, Gillberg C. Selective mutism: a population-based study: a research note. J Child Psychol Psychiatry 1997;38:257-262.
7. Krysanski V. A brief review of selective mutism literature. J Psychol 2003;137:29-40.
8. Kumpulainen K, Rasanen E, Raaska H, Somppi V. Selective mutism among second-graders in elementary school. Euro Child Adolesc Psychiatry 1998;7:24-29.
9. Remschmidt H, Poller M, Herpertz-Dahlmann B, Hennighausen K, Gutenbrenner C. A follow-up study of 45 patients with elective mutism. European Archives of Psychiatry and Clinical Neuroscience 2001;251:284-296.
10. Urbina I. Virginia Tech criticized for actions in shooting. The New York Times. August 30, 2007. Available at: www.nytimes.com/2007/08/30/us/30school.html. Accessed on October 10, 2007.
11. Schwartz RH, Shipon-Blum E. “Shy”child? Don’t overlook selective mutism. Contemp Peds 2005;22(7):30-39.
12. ABC News. Selective mutism: When your child can’t speak. Available at: abcnews.go.com/GmA/AmericanFamily/story?id=1770308&page=1. Accessed on September 26, 2007.
13. Vecchio J, Kearney CA. Selective mutism in children: comparison to youths with and without anxiety disorders. J Psychopath Beh Assess 2005;28:139-144.
14. Yeganeh R, Beidel DC, Turner SM. Selective mutism: more than social anxiety? Depress Anx 2006;23:117-123.
15. Manassis K, Fung D, Tannock R, Sloman L, Fiksenbaum L, McInnes A. Characterizing selective mutism: is it more than social anxiety? Depress Anx 2003;18:153-161.
16. Silverman WK, Albano AM. The Anxiety Disorders Interview Schedule for DSM-IV, child and parent versions. New York: oxford University Press;1996.
17. Schill MT, Kratochwill TR, Gardner WI. An assessment protocol for selective mutism: analogue assessment using parents as facilitators. J Sch Psychol 1996;34:1-21.
18. Jackson MF, Allen RS, Boothe AB, Nava ML, Coates A. Innovative analyses and interventions in the treatment of selective mutism. Clin Case Studies 2005;4:81-112.
19. Cohan SL, Chavira DA, Stein MB. Practitioner review: psychosocial interventions for children with selective mutism: a critical evaluation of the literature from 1990-2005. J Child Psychol Psychiatry 2006;47:1085-1097.
20. Rye MS, Ullman D. The successful treatment of long-term selective mutism: a case study. J Beh Ther Exp Psychiatry 1999;30:313-323.
21. Fisak BJ, Oliveros A, Ehrenreich JT. Assessment and behavioral treatment of selective mutism. Clin Case Studies 2006;5:382-402.
22. Kumpulainen K. Phenomenology and treatment of selective mutism. CNS Drugs 2002;16:175-180.
Smoking cessation: Tactics that make a big difference
- Recommend that your patients take advantage of telephone counseling— it improves both quit rates and long-term abstinence rates (A). Web-based cessation programs also help to support smokers in all stages of quitting (B).
- Encourage patients to use both pharmacotherapy and counseling to improve abstinence (A). Several medications—including bupropion and varenicline—achieve comparable rates of both quitting and long-term abstinence (A).
- Train your office staff to assist in the identification and counseling of smokers (A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Ann G. is a 34-year-old mother of 2 who had been coming to the office for her annual Pap smear for several years. Her medical history is significant only for her vaginal deliveries and mild GERD. Her medications include oral contraceptive pills (OCPs) and over-the-counter Zantac as needed. on her most recent annual visit, my medical assistant, Tammy, took Ann’s vital signs. The chart had a section about smoking status, and Tammy noted that Ann smoked.
During the office visit, I explained to Ann that her smoking was a serious health risk, and that she needed to quit. She would also need to find a new form of birth control next year, as smoking increases the risks of using OCPs. She nervously laughed off the warning.
The following year, Anne confessed to Tammy that she was still a smoker. Tammy asked her again about quitting. Ann was still adamant: “No way—I can’t do it.” Nonetheless, during the office visit, I brought up the subject of her smoking. She admitted that she was afraid that quitting smoking would cause her to gain weight. I attempted to address her fears, and then talked about other birth control methods to consider. I gave her a 3-month prescription of OCPs, and told her in 3 months we would discuss what she wanted to do about birth control.
Ann faces an uphill battle. The amount of nicotine in cigarettes is increasing,1 making it harder for her to quit. The good news is that the treatment of tobacco addiction is constantly improving and the number of tools in our arsenal is growing. In fact, there are many resources that we can try before turning to the prescription pad. However, when needed, pharmacotherapy is an important adjunct for achieving abstinence.
“5-A” strategy sets stage for success
The Agency for Healthcare Research and Quality (AHRQ) has published Treating Tobacco Use and Dependence, a useful guide for helping patients quit.2,3 These guidelines discuss many aspects of tobacco cessation, from counseling to pharmacotherapy to reimbursement issues. The guidelines break down the smoking cessation process into the 5 A’s:
- Ask each patient about her smoking status.
- Advise each patient who smokes that she needs to stop smoking.
- Assess your patient’s willingness to make a quit attempt in the next 30 days.
- Assist your patient either in making this quit attempt or in motivating her to consider a quit attempt later.
- Arrange close follow-up of any quit attempts to help prevent relapse.
The Ask and Act program from the American Academy of Family Physicians (AAFP) outlines a similar strategy.4 The program instructs physicians to Ask every patient about her tobacco use and to Act to help her quit, via on-or off-site counseling, quitlines, patient education materials, self-help guides or Web sites, cessation classes, and pharmacotherapy.
Take advantage of every opportunity you have to discuss the issue with patients; short conversations can make a difference. A Cochrane Review of 39 trials including 31,000 smokers5 revealed that even brief advice—simply encouraging patients to quit—was statistically significant (odds ratio [OR]=1.74; 95% confidence interval [CI], 1.48–2.05). The pooled data generated a quit rate difference of 2.5%: for every 40 people who were told to quit, 1 more smoker would.
Empower the office staff
Enlisting the help of the office staff can have a significant impact on the health of the patients. A proactive approach was studied by Fiore et al.6 Medical assistants, while assessing smoking status, invited all smokers to participate in a cessation study. (The assistants received periodic thank-you gifts for their efforts.)
The participants were randomized to either self-selected treatment or nicotine replacement therapy (NRT) patches, with or without a support program. Some who received the patches and support program also received individual counseling. Fiore et al showed that the majority of smokers were open to attempts to quit smoking. The 13% point-prevalence abstinence rate 1 year out is comparable with the rate obtained (14%) with smokers volunteering for NRT studies in the Cochrane review of 39 trials, noted earlier.
Likewise, in a randomized controlled trial (RCT) involving community-based primary care clinics, Katz7 demonstrated that intake clinicians could also play an important role in smoking cessation (SOR: A). In the study, researchers trained intake clinicians (including registered nurses, license practical nurses, and medical assistants) to identify smokers, provide brief counseling, and assist in their preparation to quit. Patients were offered vouchers for patches and a counselor’s business card. Intake clinicians received periodic feedback on their performance based on exit interviews of the patients. The researchers found that these interventions had a statistically significant effect in moderate-to-heavy smokers in quit attempts, quit rates, and continuous abstinence.
Our patient has a change of heart
At the 3-month follow up, Tammy learned that Ann was still smoking—but she now wanted to quit. Ann said that she’d found a pack of cigarettes in her 14-year-old daughter’s backpack, and felt that the only way to prevent her from getting hooked was to set a good example.
Tammy gave her the state’s quitline number and suggested some online quitting programs. Tammy worked with Ann to choose her target quit date and to pick the Web-based program she was going to use. Ann said that she liked the idea that she could go online whenever she needed support. She also liked the fact that she could put her quit date into the system, so it would give her timely reminders of all her reasons to quit when she logged on.
I wrote Ann a prescription for varenicline and her OCPs, and told her I wanted to see her in 4 weeks. For her part, Tammy added Ann to her list of patients to call the day after her quit date. Tammy makes this her practice with patients because she knows that one well-timed phone call can be the key to a successful quitting attempt.
Outside support improves abstinence rates
Improving your patients’ chances of long-term abstinence hinges, in part, on making the most of outside support. In many cases, your patients can take advantage of them without leaving their homes.
Outside support improves abstinence rates
Quitlines increase quit rates, decrease relapse Telephone counseling is an effective support system.8 Smokers who call to a single number (800-QUITNOW)—a service provided by the National Cancer Institute—are directed to the quitline for their state. Also, smokers can call the National Cancer Institute directly at their quitline (877-44U-QUIT). Calling a quitline provides smokers with real-time counseling and information about how to quit smoking. Quitlines can be appealing to those patients who are uncomfortable discussing their smoking in a group—and it’s free to the patient.
The research supports the use of such help lines. Zhu’s study9 of the California Smokers’ Helpline (SOR: A) was a proactive protocol where smokers were funneled into a research trial when the help line was overwhelmed. The smokers in the treatment arm of this RCT were assigned a counselor who called the smokers as many as 6 times, following a relapse-sensitive schedule. The 12-month abstinence rate increased from 4.1% to 7.5% (P<.001) in the group that had close telephone contact. This improved quit rate reflected both an increase of percentage of smokers who quit and, more importantly, a decrease in quitters who relapsed.
Another prospective RCT10 (SOR: A) enrolled patients from Veterans Affairs (VA) medical centers and involved the same proactive telephone protocol as Zhu used. The treatment group was offered telephone counseling as well as pharmacotherapy; the control group had access to the regular smoking cessation program of the VA system. Regardless of which group an individual was assigned, if that participant used both the counseling and the pharmacotherapy, the quit rate was similar: control (12.7%) and treatment (11.9%). However, only 18% of the controls used both services. The treatment group accessed the combined programs of counseling and medications at a rate of 88%. This led to the difference in 6-month abstinence rates of 13.0% in the treatment group and 4.1% in the control group (OR=3.50; 95% CI, 1.99–6.15). Patients who were directed to and enrolled in treatment programs were therefore more likely to attempt to quit and remain abstinent for up to 6 months.
Web-based programs offer reminders
Like quitlines, Web-based programs offer smokers immediate feedback to help them quit. Many of the programs include links to quitting resources, stories from former smokers and cancer patients, live advice from counselors, and message boards (TABLE 1). Web-based programs have been shown to help improve quit rates.
One study11 compared 2 Web programs involving 11,969 smokers. This RCT (SOR: B) looked at an interactive program based, in part, on the AHRQ treatment guidelines. This program generates personalized letters for the participants along with monthly e-mail reminders. A modified program was used as the control. The control program was developed by a maker of NRT products, and contained more information about nicotine than about tobacco dependence and cessation. This program was also shorter than the interactive program, which was designed to assist smoking cessation.
Both programs improved quit rates: 10.9% for the interactive program and 8% for the modified/control program, compared with 3.3% for no treatment at all. Although this study was based on participant reports of abstinence over the previous 7 days, and had low follow-up rates (which Internet studies tend to have), the interactive program produced 1 more quitter for every 26 participants than the modified (control) program, using an intent-to-treat analysis (14.6% vs. 10.7%, P<.001, OR=1.43, 95% CI, 1.28–1.59).
Another study12 looked at the use of a more extensive Web site, combining video, audio, and text. This RCT (SOR: B) was fully automated and delivered entirely by computer. Again, using the AHRQ guidelines and other sources, researchers designed a series of 5 modules to simulate working with a live counselor. There were 13 different versions, to match the demographics of the participant. The modules ended with a “quit calendar” to pick a date within the next 30 days. The program had 20 hours of video, although no participant saw every section. The intent-to-treat analysis showed a significant difference from the treatment group at 12.3% vs the controls at 5.0% (OR=2.66, 95% CI, 1.18–5.99).
TABLE 1
Web-based support helps smokers quit
| www.quitnet.com Boston University School of Public Health | Personalized quit plans |
| www.ffsonline.org American Lung Association | “Freedom from smoking” modules to guide smokers through quit process |
| www.whyquit.com Privately supported | Support for “cold turkey” quitting |
| www.trytostop.org Massachusetts Department of Public Health | Personalized “Quit Wizard” program |
Text messages work
Text messaging may also have a place in supporting smoking cessation efforts. An interesting, although short, study13 looked at using text messaging to target younger smokers in New Zealand. This RCT (SOR: B) involved 1705 smokers who had cell phones with text messaging. Researchers sent participants up to 5 messages daily around their quit date, drawing from over 100 messages that could be personalized with individual names/nicknames. The quit rate was doubled 6 weeks out (28% vs 13%; relative risk=2.20; 95% CI, 1.79–2.70).
Rx in hand, support in place
When Ann left my office, she took with her a prescription for varenicline, the state’s quitline number, and the URL for an online support program. Ann was eager to try varenicline: a coworker of hers was using it and doing well. Ann had tried the nicotine patch in the past, but reported that it gave her nightmares. She’d also kept smoking while wearing it. This time, she hoped she’d finally be able to quit for good.
Weighing the drug treatment options
The AHRQ guidelines recommend several types of pharmacotherapy. First-line therapies include different forms of NRT and sustained-release bupropion (Zyban).2,3
Nicotine replacement therapy doubles the chances of quitting
With NRT, the nicotine in cigarettes is replaced with nicotine from another source. The thought is that by reducing the withdrawal symptoms, the patient is less likely to relapse and resume smoking. Nicotine replacement is available in several forms: gum, transdermal patches, intranasal spray, inhaler, and lozenges.
A Cochrane meta-analysis of NRT14 (SOR: A) analyzed 123 studies that followed patients for at least 6 months from their quit date. The authors concluded that NRT could almost double a patient’s chances of quitting smoking. The data from various types of NRT revealed the types to be similarly efficacious (TABLE 2). In the treated groups, 17% were abstinent and only 10% were abstinent in the control groups at the various endpoints of the trials. Smokers with higher levels of nicotine dependence as indicated by smoking 10 or more cigarettes daily have higher quit rates using replacement nicotine. Generally, treatments of 8 weeks are as effective as longer courses.
The Cochrane meta-analysis also revealed that:
- Duration of therapy ranges from 3 weeks to 12 months with the various forms of NRT.
- There was no benefit to tapering off the NRT as compared to an abrupt withdrawal.
- Patients are much more likely to relapse after NRT in the first 3 months.
- Combining several forms of NRT may aid a relapsed smoker in another quit attempt. However, the re-attempt should be delayed by a few months, as back-to-back courses are unlikely to improve quit rates.
TABLE 2
Nicotine replacement therapy: Methods are similarly efficacious11
| THERAPY | OR (95% CI) | N (PARTICIPANTS/ TRIALS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)‡ |
|---|---|---|---|---|---|
| Nasal spray | 2.35 (1.63–3.38) | 887/4 | 8.3 | 3–6 months | $560/NA |
| Inhaler | 2.14 (1.44–3.18) | 976/4 | 12.5 | 3 months, then 3-month taper | $504/NA |
| Lozenges | 2.05 (1.62–2.59) | 2739/5 | 14.3 | Up to 12 weeks | $300/$240 |
| Patch | 1.84 (1.65–2.06) | 16,228/37 | 16.7 | 8–12 weeks | $110/$92 |
| NR T (all) | 1.77 (1.66–1.88) | 39,503/105 | * | ||
| Gum | 1.66 (1.51–1.81) | 17,819/52 | 12.5 | Up to 12 weeks | 4 mg: $234/$180 2 mg: $204/$150 |
| * Numbers not available. | |||||
| † Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; NA, product not available. | |||||
Sustained-release bupropion: Similar results to NRT
The other first-line therapy suggested by the AHRQ guidelines is sustained-release bupropion.2,3 A separate Cochrane Review15 analyzed the data from 36 studies using antidepressants and revealed that two thirds of the studies in this meta-analysis used bupropion. The odds of quitting smoking essentially doubled in the placebo-controlled studies. This is a similar effect as NRT. Neither the AHRQ guidelines nor the Cochrane Review recommend bupropion over NRT or vice-versa.
According to the Cochrane Review, there was no benefit to increasing the dose of bupropion from 150 mg to 300 mg daily. Although the initial multi-dose study of bupropion16 showed a difference, it was not clinically significant by the end of the study. A larger, open-label randomized trial of 1524 smokers17 followed for 1 year also showed similar results. At the 3-month evaluation, the higher dose had superior efficacy, but that effect was not statistically significant by the end of the study. Lastly, there is no benefit to continuing the bupropion beyond 7 weeks after the target quit date.
With other antidepressants, results vary
The Cochrane Review also looked at other antidepressants. There were 4 RCTs of nortriptyline (Aventyl/Pamelor) without NRT, totaling 777 smokers followed for at least 6 months.18-21 The pooled data essentially doubled the odds of quitting smoking from 7.0% for the controls to 17.2% in the treated groups (OR=2.79; 95% CI, 1.70–4.59). Adding nortriptyline to NRT did increase the quit rates, but not significantly. The dose used in these studies, at 75 to 150 mg is much lower than that used for depression, where significant side effects often interfere with treatment. Generally the starting dose is 25 mg at bedtime. After 1 week, the dose is increased to 50 mg and the following week, it is increased again to 75 mg. Once on the 75 mg dose for a week, the dose is titrated up only if needed. The titration continues at an additional 25 mg weekly.
One of the 4 placebo-controlled studies20 included an arm of bupropion, producing a head-to-head assessment with nortriptyline (SOR: A). The abstinence rates as indicated by no smoking during the final week of treatment were comparable for the 2 groups receiving active medication. Treatment with bupropion or nortriptyline was significantly more efficacious than placebo. However, the effect was lost at the 1 year continuous abstinence mark; the 2 drugs did not differ from each other or placebo (TABLE 3).
Other antidepressants were evaluated in the Cochrane study.15 The tricyclic antidepressants doxepin and imipramine (Tofranil) had no long-term studies and neither showed statistically significant differences in smaller trials. Of the selective serotonin reuptake inhibitors (SSRIs), only fluoxetine (Prozac) had any long-term studies, and none noted statistically significant differences. Likewise, venlafaxine (Effexor) had only 1 trial in which the confidence interval did allow for a potentially useful clinical effect, but failed to show a statistically significant increase in 12-month quit rates.
Clonidine is an option, but side effects are an issue
Another Cochrane Review22 looked at the effectiveness of clonidine (Catapres) on smoking cessation. Most of the clonidine studies assessed withdrawal symptoms rather than abstinence. Of those that did assess quit rates, the pooled OR for clonidine compares favorably at 1.89 (95% CI, 1.30–2.74). Unfortunately, clonidine has significant side effects: sedation and postural hypotension. The starting dose is 0.1 mg twice daily, and it may be titrated up to a maximum dose of 0.4 mg daily. It should be used for 3 to 4 weeks only to decrease the symptoms of withdrawal. The smoker should then be weaned off the clonidine.
The anxiolytics were the subject of another Cochrane Review.23 This review, however, did not recommend any anxiolytics, including diazepam and buspirone, for smoking cessation.
A new category of therapy: Nicotinic receptor agonists
With the US Food and Drug Administration’s approval of varenicline (Chantix) in May 2006, a new class of drugs became available for treatment of tobacco dependence. This α4β2 nicotinic acetylcholine receptor partial agonist was designed as a smoking cessation drug. By releasing dopamine in the brain like nicotine, it prevents craving. However, it also blocks nicotine from binding, thereby preventing the reinforcing effect of continued smoking.
Two RCTs have assessed varenicline against both bupropion and placebo (TABLE 3). Jorenby24 (SOR: A) showed the varenicline-treated participants were significantly more likely to be continuously abstinent at 52 weeks than the placebo-or bupropion-treated groups (23% vs 10.3% placebo [OR=2.66; 95% CI, 1.72–4.11; P<.001] and 14.6% bupropion [OR=1.77; 95% CI, 1.19–2.63; P=.004]). Gonzales25 (SOR: A) likewise showed the varenicline treated smokers were more likely to be continuously abstinent at 52 weeks than the placebo group (21.9% vs 8.4% [OR=3.09; 95% CI, 1.95–4.91; P<.001]). However, the difference between varenicline and bupropion did not reach statistical significance (21.9% vs 16.1% [OR=1.46; 95% CI, 0.99–2.17; P=.057]).
As with other medications, varenicline should be started at a low dose. The patient begins with 0.5 mg nightly for the first 3 nights, then increases to 0.5 mg twice a day for 4 days. The second week, the patient begins the 1 mg twice-daily dosing that is continued through treatment.
TABLE 3
Varenicline, nortriptyline, bupropion—strong allies in patients’ efforts to quit
| THERAPY | OR (95% CI) | N (PARTICIPA NTS/TRAILS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)* |
|---|---|---|---|---|---|
| Varenicline24,25 | 2.80 (2.03–3.88) | 1161/2 | 7.6 | 12 weeks | $120/NA |
| Nortriptyline15 | 2.79 (1.70–4.59) | 703/4 | 9.8 | 12 weeks | $814/$8 |
| Sustained-release bupropion15 | 2.06 (1.77–2.40) | 6443/19 | 10.2 | 7–12 weeks | $210/$100 |
| Clonidine23 | 1.89 (1.30–2.74) | 776/6 | 9.4 | 3–4 weeks | $74/$4 |
| Venlafaxine15 | 1.33 (0.59–3.00) | 136/1 | 20.4 | $145/NA | |
| Diazepam23 | 1.00 (0.39–2.54) | 76/1 | No difference | $209/$27 | |
| SSRI15 | 0.90 (0.68–1.18) | 1768/6 | 20.7 | $170/$4 | |
| Buspirone23 | 0.71 (0.34–1.48) | 201/3 | 22.1 | $280/$84 | |
| *Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; SSRI, selective serotonin reuptake inhibitors; NA, not available. | |||||
Vaccines hold the promise of continued abstinence
Several promising ideas for the treatment of tobacco dependence are in development. There are several vaccines being studied.26 When the immune system produces antibodies to nicotine in response to the vaccine, and when these antibodies bind to the nicotine, the resultant compound is too large to cross the blood-brain barrier. This prevents the reinforcing effect of nicotine. Initial studies of vaccines show that smokers do decrease the amount they smoke, but more importantly, abstinence is easier to maintain. However, the vaccine requires frequent boosters to maintain antibody titers that are effective.
NicVAX from Nabi Biopharmaceuticals was placed on a fast track for approval by the Food and Drug Administration. It is, however, still at least a year away from approval. The other 2 nicotine vaccines are probably several years beyond that for approval.27
Researchers are also studying other compounds that block the euphoria associated with smoking.28 The initial studies of rimonabant (Acomplia), a cannabinoid blocker, have shown it is no better than other treatments already available. With its indication in some European countries for weight loss, it offered promise as an important option for patients who are concerned about the weight gain associated with smoking cessation. However, the FDA did not approve rimonabant for tobacco cessation when issuing its initial approval letter for weight loss in 2006. Because of safety concerns, the manufacturer subsequently withdrew the new drug application for rimonabant in 2007.
With much work, our patient kicks the habit
Ann began taking varenicline the day she left the office, and reached her quit date a week later.
At her 1-month follow-up, Ann reported that it was actually easy for her to stay off the cigarettes. With the varenicline, she had lost the desire to smoke. I reminded her to work on the triggers for her smoking: I urged her to make sure that she did not light up when she made her morning coffee or got in the car. I also suggested she put $4 each morning into a jar on her dresser; so she would see how much she saved now that she wasn’t buying cigarettes.
At Ann’s next annual exam, we marked her in the computer system as a reformed smoker. She was very proud of that label. I asked her what she was doing with all that extra cash. She laughed: “My daughter spends it all! but not on cigarettes!”
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Drug Abuse.
Correspondence
Agnes O. Coffay, MD, NIDA/IRP, 5500 Nathan Shock Drive, B altimore, MD 21224-6823. [email protected]
1. Brown D. Nicotine up sharply in many cigarettes. Washington Post, August 31, 2006. Available at: www.washingtonpost.com/wp-dyn/content/article/2006/08/30/Ar2006083001418.html. Accessed on September 4, 2007.
2. Agency for Healthcare Research and Quality. Clinical Practice Guideline. Treating Tobacco Use and Dependence. Rockville, Md: US Department of Health and Human Services Public Health Service;2000. Available at: www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat2. chapter.7644. Accessed on September 4, 2007.
3. Tobacco Use and Dependence clinical Practice Guideline Panel, Staff, and Consortium Representatives. A clinical practice guideline for treating tobacco use and dependence: A US Public Health Service report. JAMA. 2000;283:3244-3254.
4. Ask and Act: A Tobacco Cessation Program. Available at: www.aafp.org/online/en/home/clinical/publichealth/tobacco/askandact.html. Accessed on September 4, 2007.
5. Lancaster T, Stead LF. Physician advice for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000165.-
6. Fiore MC, McCarthy DE, Jackson TC, et al. Integrating smoking cessation treatment into primary care: an effectiveness study. Prev Med 2004;38:412-420.
7. Katz DA, Muehlenbruch DR, Brown RL. Effectiveness of implementing the Agency for Healthcare Research and Quality Smoking cessation clinical Practice Guidelines: a randomized, control trial. J Natl Cancer Inst 2004;96:594-603.
8. Stead LF, Lancaster T, Perera R. Telephone counseling for smoking cessation. Cochrane Database Syst Rev 2003;(1):CD002850.-
9. Zhu SH, Anderson CM, Tedeschi GJ, et al. Evidence of real-world effectiveness of a telephone quitline for smokers. N Engl J Med 2002;347:1087-1093.
10. An LC, Zhu SH, Nelson DB, et al. Benefits of telephone care over primary care for smoking cessation. Arch Intern Med 2006;166:536-542.
11. Etter JF. Comparing the efficacy of two internet-based, computer-tailored smoking cessation programs: a randomized trial. J Med Internet Res 2005;7(1):e2.-
12. Swartz LH, Noell JW, Schroeder SW, Ary DV. A randomized control study of a fully automated internet based smoking cessation programme. Tobacco Control 2006;15:7-12.
13. Rodgers A, Corbett T, Bramley D, et al. Do u smoke after txt? Results of a randomized trial of smoking cessation using mobile phone text messaging. Tobacco Control 2005;14:255-261.
14. Silagy C, Lancaster T, Stead I, Mant D, Fowler G. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000146.-
15. Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev 2004;(2):cD000031.-
16. Hurt RD, Sachs DP, Glover ED, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
17. Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting. Arch Intern Med 2003;163:2337-2344.
18. da Costa CL, Younes RN, Lourenco MT. A prospective, randomized, double-blind study comparing nortriptyline to placebo. Chest 2002;122:403-408.
19. Hall SM, Reus VI, Munoz RF, et al. Nortriptyline and cognitive-behavioral therapy in the treatment of cigarette smoking. Arch Gen Psychiatry 1998;55:683-690.
20. Hall SM, Humfleet GL, Reus VI, et al. Psychological intervention and antidepressant treatment in smoking cessation. Arch Gen Psychiatry 2002;59:930-936.
21. Prochazka AV, Weaver MJ, Keller RT, et al. A randomized trial of nortriptyline for smoking cessation. Arch Intern Med 1998;158:2035-2039.
22. Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database Syst Rev 2004;(3):CD000058.-
23. Hughes JR, Stead LF, Lancaster T. Anxiolytics for smoking cessation. Cochrane Database Syst Rev 2000;(4):CD002849.-
24. Jorenby De, Hays JT, Rigotti NA. Efficacy of varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation. JAMA 2006;296:56-63.
25. Gonzales D, Rennard SI, Nides M. Varenicline, an a4b2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation. JAMA 2006;296:47-55.
26. LeHouezec J. Why a nicotine vaccine? Clin Pharmacol Ther 2005;78:453-455.
27. Tuller D. Scientists testing vaccines to help smokers quit. New York Times,July 4, 2006. Available at: www. nytimes.com/2006/07/040health/04vacc.html?ex=13 09665600&en=d978add467c2b80a&ei=5088&partne r=rssnyt&emc=rss. Accessed on September 6, 2007.
28. Fagerström K, Balfour DJ. Neuropharmacology and potential efficacy of new treatments for tobacco dependence. Expert Opin Investig Drugs 2005;15:107-116.
- Recommend that your patients take advantage of telephone counseling— it improves both quit rates and long-term abstinence rates (A). Web-based cessation programs also help to support smokers in all stages of quitting (B).
- Encourage patients to use both pharmacotherapy and counseling to improve abstinence (A). Several medications—including bupropion and varenicline—achieve comparable rates of both quitting and long-term abstinence (A).
- Train your office staff to assist in the identification and counseling of smokers (A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Ann G. is a 34-year-old mother of 2 who had been coming to the office for her annual Pap smear for several years. Her medical history is significant only for her vaginal deliveries and mild GERD. Her medications include oral contraceptive pills (OCPs) and over-the-counter Zantac as needed. on her most recent annual visit, my medical assistant, Tammy, took Ann’s vital signs. The chart had a section about smoking status, and Tammy noted that Ann smoked.
During the office visit, I explained to Ann that her smoking was a serious health risk, and that she needed to quit. She would also need to find a new form of birth control next year, as smoking increases the risks of using OCPs. She nervously laughed off the warning.
The following year, Anne confessed to Tammy that she was still a smoker. Tammy asked her again about quitting. Ann was still adamant: “No way—I can’t do it.” Nonetheless, during the office visit, I brought up the subject of her smoking. She admitted that she was afraid that quitting smoking would cause her to gain weight. I attempted to address her fears, and then talked about other birth control methods to consider. I gave her a 3-month prescription of OCPs, and told her in 3 months we would discuss what she wanted to do about birth control.
Ann faces an uphill battle. The amount of nicotine in cigarettes is increasing,1 making it harder for her to quit. The good news is that the treatment of tobacco addiction is constantly improving and the number of tools in our arsenal is growing. In fact, there are many resources that we can try before turning to the prescription pad. However, when needed, pharmacotherapy is an important adjunct for achieving abstinence.
“5-A” strategy sets stage for success
The Agency for Healthcare Research and Quality (AHRQ) has published Treating Tobacco Use and Dependence, a useful guide for helping patients quit.2,3 These guidelines discuss many aspects of tobacco cessation, from counseling to pharmacotherapy to reimbursement issues. The guidelines break down the smoking cessation process into the 5 A’s:
- Ask each patient about her smoking status.
- Advise each patient who smokes that she needs to stop smoking.
- Assess your patient’s willingness to make a quit attempt in the next 30 days.
- Assist your patient either in making this quit attempt or in motivating her to consider a quit attempt later.
- Arrange close follow-up of any quit attempts to help prevent relapse.
The Ask and Act program from the American Academy of Family Physicians (AAFP) outlines a similar strategy.4 The program instructs physicians to Ask every patient about her tobacco use and to Act to help her quit, via on-or off-site counseling, quitlines, patient education materials, self-help guides or Web sites, cessation classes, and pharmacotherapy.
Take advantage of every opportunity you have to discuss the issue with patients; short conversations can make a difference. A Cochrane Review of 39 trials including 31,000 smokers5 revealed that even brief advice—simply encouraging patients to quit—was statistically significant (odds ratio [OR]=1.74; 95% confidence interval [CI], 1.48–2.05). The pooled data generated a quit rate difference of 2.5%: for every 40 people who were told to quit, 1 more smoker would.
Empower the office staff
Enlisting the help of the office staff can have a significant impact on the health of the patients. A proactive approach was studied by Fiore et al.6 Medical assistants, while assessing smoking status, invited all smokers to participate in a cessation study. (The assistants received periodic thank-you gifts for their efforts.)
The participants were randomized to either self-selected treatment or nicotine replacement therapy (NRT) patches, with or without a support program. Some who received the patches and support program also received individual counseling. Fiore et al showed that the majority of smokers were open to attempts to quit smoking. The 13% point-prevalence abstinence rate 1 year out is comparable with the rate obtained (14%) with smokers volunteering for NRT studies in the Cochrane review of 39 trials, noted earlier.
Likewise, in a randomized controlled trial (RCT) involving community-based primary care clinics, Katz7 demonstrated that intake clinicians could also play an important role in smoking cessation (SOR: A). In the study, researchers trained intake clinicians (including registered nurses, license practical nurses, and medical assistants) to identify smokers, provide brief counseling, and assist in their preparation to quit. Patients were offered vouchers for patches and a counselor’s business card. Intake clinicians received periodic feedback on their performance based on exit interviews of the patients. The researchers found that these interventions had a statistically significant effect in moderate-to-heavy smokers in quit attempts, quit rates, and continuous abstinence.
Our patient has a change of heart
At the 3-month follow up, Tammy learned that Ann was still smoking—but she now wanted to quit. Ann said that she’d found a pack of cigarettes in her 14-year-old daughter’s backpack, and felt that the only way to prevent her from getting hooked was to set a good example.
Tammy gave her the state’s quitline number and suggested some online quitting programs. Tammy worked with Ann to choose her target quit date and to pick the Web-based program she was going to use. Ann said that she liked the idea that she could go online whenever she needed support. She also liked the fact that she could put her quit date into the system, so it would give her timely reminders of all her reasons to quit when she logged on.
I wrote Ann a prescription for varenicline and her OCPs, and told her I wanted to see her in 4 weeks. For her part, Tammy added Ann to her list of patients to call the day after her quit date. Tammy makes this her practice with patients because she knows that one well-timed phone call can be the key to a successful quitting attempt.
Outside support improves abstinence rates
Improving your patients’ chances of long-term abstinence hinges, in part, on making the most of outside support. In many cases, your patients can take advantage of them without leaving their homes.
Outside support improves abstinence rates
Quitlines increase quit rates, decrease relapse Telephone counseling is an effective support system.8 Smokers who call to a single number (800-QUITNOW)—a service provided by the National Cancer Institute—are directed to the quitline for their state. Also, smokers can call the National Cancer Institute directly at their quitline (877-44U-QUIT). Calling a quitline provides smokers with real-time counseling and information about how to quit smoking. Quitlines can be appealing to those patients who are uncomfortable discussing their smoking in a group—and it’s free to the patient.
The research supports the use of such help lines. Zhu’s study9 of the California Smokers’ Helpline (SOR: A) was a proactive protocol where smokers were funneled into a research trial when the help line was overwhelmed. The smokers in the treatment arm of this RCT were assigned a counselor who called the smokers as many as 6 times, following a relapse-sensitive schedule. The 12-month abstinence rate increased from 4.1% to 7.5% (P<.001) in the group that had close telephone contact. This improved quit rate reflected both an increase of percentage of smokers who quit and, more importantly, a decrease in quitters who relapsed.
Another prospective RCT10 (SOR: A) enrolled patients from Veterans Affairs (VA) medical centers and involved the same proactive telephone protocol as Zhu used. The treatment group was offered telephone counseling as well as pharmacotherapy; the control group had access to the regular smoking cessation program of the VA system. Regardless of which group an individual was assigned, if that participant used both the counseling and the pharmacotherapy, the quit rate was similar: control (12.7%) and treatment (11.9%). However, only 18% of the controls used both services. The treatment group accessed the combined programs of counseling and medications at a rate of 88%. This led to the difference in 6-month abstinence rates of 13.0% in the treatment group and 4.1% in the control group (OR=3.50; 95% CI, 1.99–6.15). Patients who were directed to and enrolled in treatment programs were therefore more likely to attempt to quit and remain abstinent for up to 6 months.
Web-based programs offer reminders
Like quitlines, Web-based programs offer smokers immediate feedback to help them quit. Many of the programs include links to quitting resources, stories from former smokers and cancer patients, live advice from counselors, and message boards (TABLE 1). Web-based programs have been shown to help improve quit rates.
One study11 compared 2 Web programs involving 11,969 smokers. This RCT (SOR: B) looked at an interactive program based, in part, on the AHRQ treatment guidelines. This program generates personalized letters for the participants along with monthly e-mail reminders. A modified program was used as the control. The control program was developed by a maker of NRT products, and contained more information about nicotine than about tobacco dependence and cessation. This program was also shorter than the interactive program, which was designed to assist smoking cessation.
Both programs improved quit rates: 10.9% for the interactive program and 8% for the modified/control program, compared with 3.3% for no treatment at all. Although this study was based on participant reports of abstinence over the previous 7 days, and had low follow-up rates (which Internet studies tend to have), the interactive program produced 1 more quitter for every 26 participants than the modified (control) program, using an intent-to-treat analysis (14.6% vs. 10.7%, P<.001, OR=1.43, 95% CI, 1.28–1.59).
Another study12 looked at the use of a more extensive Web site, combining video, audio, and text. This RCT (SOR: B) was fully automated and delivered entirely by computer. Again, using the AHRQ guidelines and other sources, researchers designed a series of 5 modules to simulate working with a live counselor. There were 13 different versions, to match the demographics of the participant. The modules ended with a “quit calendar” to pick a date within the next 30 days. The program had 20 hours of video, although no participant saw every section. The intent-to-treat analysis showed a significant difference from the treatment group at 12.3% vs the controls at 5.0% (OR=2.66, 95% CI, 1.18–5.99).
TABLE 1
Web-based support helps smokers quit
| www.quitnet.com Boston University School of Public Health | Personalized quit plans |
| www.ffsonline.org American Lung Association | “Freedom from smoking” modules to guide smokers through quit process |
| www.whyquit.com Privately supported | Support for “cold turkey” quitting |
| www.trytostop.org Massachusetts Department of Public Health | Personalized “Quit Wizard” program |
Text messages work
Text messaging may also have a place in supporting smoking cessation efforts. An interesting, although short, study13 looked at using text messaging to target younger smokers in New Zealand. This RCT (SOR: B) involved 1705 smokers who had cell phones with text messaging. Researchers sent participants up to 5 messages daily around their quit date, drawing from over 100 messages that could be personalized with individual names/nicknames. The quit rate was doubled 6 weeks out (28% vs 13%; relative risk=2.20; 95% CI, 1.79–2.70).
Rx in hand, support in place
When Ann left my office, she took with her a prescription for varenicline, the state’s quitline number, and the URL for an online support program. Ann was eager to try varenicline: a coworker of hers was using it and doing well. Ann had tried the nicotine patch in the past, but reported that it gave her nightmares. She’d also kept smoking while wearing it. This time, she hoped she’d finally be able to quit for good.
Weighing the drug treatment options
The AHRQ guidelines recommend several types of pharmacotherapy. First-line therapies include different forms of NRT and sustained-release bupropion (Zyban).2,3
Nicotine replacement therapy doubles the chances of quitting
With NRT, the nicotine in cigarettes is replaced with nicotine from another source. The thought is that by reducing the withdrawal symptoms, the patient is less likely to relapse and resume smoking. Nicotine replacement is available in several forms: gum, transdermal patches, intranasal spray, inhaler, and lozenges.
A Cochrane meta-analysis of NRT14 (SOR: A) analyzed 123 studies that followed patients for at least 6 months from their quit date. The authors concluded that NRT could almost double a patient’s chances of quitting smoking. The data from various types of NRT revealed the types to be similarly efficacious (TABLE 2). In the treated groups, 17% were abstinent and only 10% were abstinent in the control groups at the various endpoints of the trials. Smokers with higher levels of nicotine dependence as indicated by smoking 10 or more cigarettes daily have higher quit rates using replacement nicotine. Generally, treatments of 8 weeks are as effective as longer courses.
The Cochrane meta-analysis also revealed that:
- Duration of therapy ranges from 3 weeks to 12 months with the various forms of NRT.
- There was no benefit to tapering off the NRT as compared to an abrupt withdrawal.
- Patients are much more likely to relapse after NRT in the first 3 months.
- Combining several forms of NRT may aid a relapsed smoker in another quit attempt. However, the re-attempt should be delayed by a few months, as back-to-back courses are unlikely to improve quit rates.
TABLE 2
Nicotine replacement therapy: Methods are similarly efficacious11
| THERAPY | OR (95% CI) | N (PARTICIPANTS/ TRIALS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)‡ |
|---|---|---|---|---|---|
| Nasal spray | 2.35 (1.63–3.38) | 887/4 | 8.3 | 3–6 months | $560/NA |
| Inhaler | 2.14 (1.44–3.18) | 976/4 | 12.5 | 3 months, then 3-month taper | $504/NA |
| Lozenges | 2.05 (1.62–2.59) | 2739/5 | 14.3 | Up to 12 weeks | $300/$240 |
| Patch | 1.84 (1.65–2.06) | 16,228/37 | 16.7 | 8–12 weeks | $110/$92 |
| NR T (all) | 1.77 (1.66–1.88) | 39,503/105 | * | ||
| Gum | 1.66 (1.51–1.81) | 17,819/52 | 12.5 | Up to 12 weeks | 4 mg: $234/$180 2 mg: $204/$150 |
| * Numbers not available. | |||||
| † Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; NA, product not available. | |||||
Sustained-release bupropion: Similar results to NRT
The other first-line therapy suggested by the AHRQ guidelines is sustained-release bupropion.2,3 A separate Cochrane Review15 analyzed the data from 36 studies using antidepressants and revealed that two thirds of the studies in this meta-analysis used bupropion. The odds of quitting smoking essentially doubled in the placebo-controlled studies. This is a similar effect as NRT. Neither the AHRQ guidelines nor the Cochrane Review recommend bupropion over NRT or vice-versa.
According to the Cochrane Review, there was no benefit to increasing the dose of bupropion from 150 mg to 300 mg daily. Although the initial multi-dose study of bupropion16 showed a difference, it was not clinically significant by the end of the study. A larger, open-label randomized trial of 1524 smokers17 followed for 1 year also showed similar results. At the 3-month evaluation, the higher dose had superior efficacy, but that effect was not statistically significant by the end of the study. Lastly, there is no benefit to continuing the bupropion beyond 7 weeks after the target quit date.
With other antidepressants, results vary
The Cochrane Review also looked at other antidepressants. There were 4 RCTs of nortriptyline (Aventyl/Pamelor) without NRT, totaling 777 smokers followed for at least 6 months.18-21 The pooled data essentially doubled the odds of quitting smoking from 7.0% for the controls to 17.2% in the treated groups (OR=2.79; 95% CI, 1.70–4.59). Adding nortriptyline to NRT did increase the quit rates, but not significantly. The dose used in these studies, at 75 to 150 mg is much lower than that used for depression, where significant side effects often interfere with treatment. Generally the starting dose is 25 mg at bedtime. After 1 week, the dose is increased to 50 mg and the following week, it is increased again to 75 mg. Once on the 75 mg dose for a week, the dose is titrated up only if needed. The titration continues at an additional 25 mg weekly.
One of the 4 placebo-controlled studies20 included an arm of bupropion, producing a head-to-head assessment with nortriptyline (SOR: A). The abstinence rates as indicated by no smoking during the final week of treatment were comparable for the 2 groups receiving active medication. Treatment with bupropion or nortriptyline was significantly more efficacious than placebo. However, the effect was lost at the 1 year continuous abstinence mark; the 2 drugs did not differ from each other or placebo (TABLE 3).
Other antidepressants were evaluated in the Cochrane study.15 The tricyclic antidepressants doxepin and imipramine (Tofranil) had no long-term studies and neither showed statistically significant differences in smaller trials. Of the selective serotonin reuptake inhibitors (SSRIs), only fluoxetine (Prozac) had any long-term studies, and none noted statistically significant differences. Likewise, venlafaxine (Effexor) had only 1 trial in which the confidence interval did allow for a potentially useful clinical effect, but failed to show a statistically significant increase in 12-month quit rates.
Clonidine is an option, but side effects are an issue
Another Cochrane Review22 looked at the effectiveness of clonidine (Catapres) on smoking cessation. Most of the clonidine studies assessed withdrawal symptoms rather than abstinence. Of those that did assess quit rates, the pooled OR for clonidine compares favorably at 1.89 (95% CI, 1.30–2.74). Unfortunately, clonidine has significant side effects: sedation and postural hypotension. The starting dose is 0.1 mg twice daily, and it may be titrated up to a maximum dose of 0.4 mg daily. It should be used for 3 to 4 weeks only to decrease the symptoms of withdrawal. The smoker should then be weaned off the clonidine.
The anxiolytics were the subject of another Cochrane Review.23 This review, however, did not recommend any anxiolytics, including diazepam and buspirone, for smoking cessation.
A new category of therapy: Nicotinic receptor agonists
With the US Food and Drug Administration’s approval of varenicline (Chantix) in May 2006, a new class of drugs became available for treatment of tobacco dependence. This α4β2 nicotinic acetylcholine receptor partial agonist was designed as a smoking cessation drug. By releasing dopamine in the brain like nicotine, it prevents craving. However, it also blocks nicotine from binding, thereby preventing the reinforcing effect of continued smoking.
Two RCTs have assessed varenicline against both bupropion and placebo (TABLE 3). Jorenby24 (SOR: A) showed the varenicline-treated participants were significantly more likely to be continuously abstinent at 52 weeks than the placebo-or bupropion-treated groups (23% vs 10.3% placebo [OR=2.66; 95% CI, 1.72–4.11; P<.001] and 14.6% bupropion [OR=1.77; 95% CI, 1.19–2.63; P=.004]). Gonzales25 (SOR: A) likewise showed the varenicline treated smokers were more likely to be continuously abstinent at 52 weeks than the placebo group (21.9% vs 8.4% [OR=3.09; 95% CI, 1.95–4.91; P<.001]). However, the difference between varenicline and bupropion did not reach statistical significance (21.9% vs 16.1% [OR=1.46; 95% CI, 0.99–2.17; P=.057]).
As with other medications, varenicline should be started at a low dose. The patient begins with 0.5 mg nightly for the first 3 nights, then increases to 0.5 mg twice a day for 4 days. The second week, the patient begins the 1 mg twice-daily dosing that is continued through treatment.
TABLE 3
Varenicline, nortriptyline, bupropion—strong allies in patients’ efforts to quit
| THERAPY | OR (95% CI) | N (PARTICIPA NTS/TRAILS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)* |
|---|---|---|---|---|---|
| Varenicline24,25 | 2.80 (2.03–3.88) | 1161/2 | 7.6 | 12 weeks | $120/NA |
| Nortriptyline15 | 2.79 (1.70–4.59) | 703/4 | 9.8 | 12 weeks | $814/$8 |
| Sustained-release bupropion15 | 2.06 (1.77–2.40) | 6443/19 | 10.2 | 7–12 weeks | $210/$100 |
| Clonidine23 | 1.89 (1.30–2.74) | 776/6 | 9.4 | 3–4 weeks | $74/$4 |
| Venlafaxine15 | 1.33 (0.59–3.00) | 136/1 | 20.4 | $145/NA | |
| Diazepam23 | 1.00 (0.39–2.54) | 76/1 | No difference | $209/$27 | |
| SSRI15 | 0.90 (0.68–1.18) | 1768/6 | 20.7 | $170/$4 | |
| Buspirone23 | 0.71 (0.34–1.48) | 201/3 | 22.1 | $280/$84 | |
| *Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; SSRI, selective serotonin reuptake inhibitors; NA, not available. | |||||
Vaccines hold the promise of continued abstinence
Several promising ideas for the treatment of tobacco dependence are in development. There are several vaccines being studied.26 When the immune system produces antibodies to nicotine in response to the vaccine, and when these antibodies bind to the nicotine, the resultant compound is too large to cross the blood-brain barrier. This prevents the reinforcing effect of nicotine. Initial studies of vaccines show that smokers do decrease the amount they smoke, but more importantly, abstinence is easier to maintain. However, the vaccine requires frequent boosters to maintain antibody titers that are effective.
NicVAX from Nabi Biopharmaceuticals was placed on a fast track for approval by the Food and Drug Administration. It is, however, still at least a year away from approval. The other 2 nicotine vaccines are probably several years beyond that for approval.27
Researchers are also studying other compounds that block the euphoria associated with smoking.28 The initial studies of rimonabant (Acomplia), a cannabinoid blocker, have shown it is no better than other treatments already available. With its indication in some European countries for weight loss, it offered promise as an important option for patients who are concerned about the weight gain associated with smoking cessation. However, the FDA did not approve rimonabant for tobacco cessation when issuing its initial approval letter for weight loss in 2006. Because of safety concerns, the manufacturer subsequently withdrew the new drug application for rimonabant in 2007.
With much work, our patient kicks the habit
Ann began taking varenicline the day she left the office, and reached her quit date a week later.
At her 1-month follow-up, Ann reported that it was actually easy for her to stay off the cigarettes. With the varenicline, she had lost the desire to smoke. I reminded her to work on the triggers for her smoking: I urged her to make sure that she did not light up when she made her morning coffee or got in the car. I also suggested she put $4 each morning into a jar on her dresser; so she would see how much she saved now that she wasn’t buying cigarettes.
At Ann’s next annual exam, we marked her in the computer system as a reformed smoker. She was very proud of that label. I asked her what she was doing with all that extra cash. She laughed: “My daughter spends it all! but not on cigarettes!”
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Drug Abuse.
Correspondence
Agnes O. Coffay, MD, NIDA/IRP, 5500 Nathan Shock Drive, B altimore, MD 21224-6823. [email protected]
- Recommend that your patients take advantage of telephone counseling— it improves both quit rates and long-term abstinence rates (A). Web-based cessation programs also help to support smokers in all stages of quitting (B).
- Encourage patients to use both pharmacotherapy and counseling to improve abstinence (A). Several medications—including bupropion and varenicline—achieve comparable rates of both quitting and long-term abstinence (A).
- Train your office staff to assist in the identification and counseling of smokers (A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Ann G. is a 34-year-old mother of 2 who had been coming to the office for her annual Pap smear for several years. Her medical history is significant only for her vaginal deliveries and mild GERD. Her medications include oral contraceptive pills (OCPs) and over-the-counter Zantac as needed. on her most recent annual visit, my medical assistant, Tammy, took Ann’s vital signs. The chart had a section about smoking status, and Tammy noted that Ann smoked.
During the office visit, I explained to Ann that her smoking was a serious health risk, and that she needed to quit. She would also need to find a new form of birth control next year, as smoking increases the risks of using OCPs. She nervously laughed off the warning.
The following year, Anne confessed to Tammy that she was still a smoker. Tammy asked her again about quitting. Ann was still adamant: “No way—I can’t do it.” Nonetheless, during the office visit, I brought up the subject of her smoking. She admitted that she was afraid that quitting smoking would cause her to gain weight. I attempted to address her fears, and then talked about other birth control methods to consider. I gave her a 3-month prescription of OCPs, and told her in 3 months we would discuss what she wanted to do about birth control.
Ann faces an uphill battle. The amount of nicotine in cigarettes is increasing,1 making it harder for her to quit. The good news is that the treatment of tobacco addiction is constantly improving and the number of tools in our arsenal is growing. In fact, there are many resources that we can try before turning to the prescription pad. However, when needed, pharmacotherapy is an important adjunct for achieving abstinence.
“5-A” strategy sets stage for success
The Agency for Healthcare Research and Quality (AHRQ) has published Treating Tobacco Use and Dependence, a useful guide for helping patients quit.2,3 These guidelines discuss many aspects of tobacco cessation, from counseling to pharmacotherapy to reimbursement issues. The guidelines break down the smoking cessation process into the 5 A’s:
- Ask each patient about her smoking status.
- Advise each patient who smokes that she needs to stop smoking.
- Assess your patient’s willingness to make a quit attempt in the next 30 days.
- Assist your patient either in making this quit attempt or in motivating her to consider a quit attempt later.
- Arrange close follow-up of any quit attempts to help prevent relapse.
The Ask and Act program from the American Academy of Family Physicians (AAFP) outlines a similar strategy.4 The program instructs physicians to Ask every patient about her tobacco use and to Act to help her quit, via on-or off-site counseling, quitlines, patient education materials, self-help guides or Web sites, cessation classes, and pharmacotherapy.
Take advantage of every opportunity you have to discuss the issue with patients; short conversations can make a difference. A Cochrane Review of 39 trials including 31,000 smokers5 revealed that even brief advice—simply encouraging patients to quit—was statistically significant (odds ratio [OR]=1.74; 95% confidence interval [CI], 1.48–2.05). The pooled data generated a quit rate difference of 2.5%: for every 40 people who were told to quit, 1 more smoker would.
Empower the office staff
Enlisting the help of the office staff can have a significant impact on the health of the patients. A proactive approach was studied by Fiore et al.6 Medical assistants, while assessing smoking status, invited all smokers to participate in a cessation study. (The assistants received periodic thank-you gifts for their efforts.)
The participants were randomized to either self-selected treatment or nicotine replacement therapy (NRT) patches, with or without a support program. Some who received the patches and support program also received individual counseling. Fiore et al showed that the majority of smokers were open to attempts to quit smoking. The 13% point-prevalence abstinence rate 1 year out is comparable with the rate obtained (14%) with smokers volunteering for NRT studies in the Cochrane review of 39 trials, noted earlier.
Likewise, in a randomized controlled trial (RCT) involving community-based primary care clinics, Katz7 demonstrated that intake clinicians could also play an important role in smoking cessation (SOR: A). In the study, researchers trained intake clinicians (including registered nurses, license practical nurses, and medical assistants) to identify smokers, provide brief counseling, and assist in their preparation to quit. Patients were offered vouchers for patches and a counselor’s business card. Intake clinicians received periodic feedback on their performance based on exit interviews of the patients. The researchers found that these interventions had a statistically significant effect in moderate-to-heavy smokers in quit attempts, quit rates, and continuous abstinence.
Our patient has a change of heart
At the 3-month follow up, Tammy learned that Ann was still smoking—but she now wanted to quit. Ann said that she’d found a pack of cigarettes in her 14-year-old daughter’s backpack, and felt that the only way to prevent her from getting hooked was to set a good example.
Tammy gave her the state’s quitline number and suggested some online quitting programs. Tammy worked with Ann to choose her target quit date and to pick the Web-based program she was going to use. Ann said that she liked the idea that she could go online whenever she needed support. She also liked the fact that she could put her quit date into the system, so it would give her timely reminders of all her reasons to quit when she logged on.
I wrote Ann a prescription for varenicline and her OCPs, and told her I wanted to see her in 4 weeks. For her part, Tammy added Ann to her list of patients to call the day after her quit date. Tammy makes this her practice with patients because she knows that one well-timed phone call can be the key to a successful quitting attempt.
Outside support improves abstinence rates
Improving your patients’ chances of long-term abstinence hinges, in part, on making the most of outside support. In many cases, your patients can take advantage of them without leaving their homes.
Outside support improves abstinence rates
Quitlines increase quit rates, decrease relapse Telephone counseling is an effective support system.8 Smokers who call to a single number (800-QUITNOW)—a service provided by the National Cancer Institute—are directed to the quitline for their state. Also, smokers can call the National Cancer Institute directly at their quitline (877-44U-QUIT). Calling a quitline provides smokers with real-time counseling and information about how to quit smoking. Quitlines can be appealing to those patients who are uncomfortable discussing their smoking in a group—and it’s free to the patient.
The research supports the use of such help lines. Zhu’s study9 of the California Smokers’ Helpline (SOR: A) was a proactive protocol where smokers were funneled into a research trial when the help line was overwhelmed. The smokers in the treatment arm of this RCT were assigned a counselor who called the smokers as many as 6 times, following a relapse-sensitive schedule. The 12-month abstinence rate increased from 4.1% to 7.5% (P<.001) in the group that had close telephone contact. This improved quit rate reflected both an increase of percentage of smokers who quit and, more importantly, a decrease in quitters who relapsed.
Another prospective RCT10 (SOR: A) enrolled patients from Veterans Affairs (VA) medical centers and involved the same proactive telephone protocol as Zhu used. The treatment group was offered telephone counseling as well as pharmacotherapy; the control group had access to the regular smoking cessation program of the VA system. Regardless of which group an individual was assigned, if that participant used both the counseling and the pharmacotherapy, the quit rate was similar: control (12.7%) and treatment (11.9%). However, only 18% of the controls used both services. The treatment group accessed the combined programs of counseling and medications at a rate of 88%. This led to the difference in 6-month abstinence rates of 13.0% in the treatment group and 4.1% in the control group (OR=3.50; 95% CI, 1.99–6.15). Patients who were directed to and enrolled in treatment programs were therefore more likely to attempt to quit and remain abstinent for up to 6 months.
Web-based programs offer reminders
Like quitlines, Web-based programs offer smokers immediate feedback to help them quit. Many of the programs include links to quitting resources, stories from former smokers and cancer patients, live advice from counselors, and message boards (TABLE 1). Web-based programs have been shown to help improve quit rates.
One study11 compared 2 Web programs involving 11,969 smokers. This RCT (SOR: B) looked at an interactive program based, in part, on the AHRQ treatment guidelines. This program generates personalized letters for the participants along with monthly e-mail reminders. A modified program was used as the control. The control program was developed by a maker of NRT products, and contained more information about nicotine than about tobacco dependence and cessation. This program was also shorter than the interactive program, which was designed to assist smoking cessation.
Both programs improved quit rates: 10.9% for the interactive program and 8% for the modified/control program, compared with 3.3% for no treatment at all. Although this study was based on participant reports of abstinence over the previous 7 days, and had low follow-up rates (which Internet studies tend to have), the interactive program produced 1 more quitter for every 26 participants than the modified (control) program, using an intent-to-treat analysis (14.6% vs. 10.7%, P<.001, OR=1.43, 95% CI, 1.28–1.59).
Another study12 looked at the use of a more extensive Web site, combining video, audio, and text. This RCT (SOR: B) was fully automated and delivered entirely by computer. Again, using the AHRQ guidelines and other sources, researchers designed a series of 5 modules to simulate working with a live counselor. There were 13 different versions, to match the demographics of the participant. The modules ended with a “quit calendar” to pick a date within the next 30 days. The program had 20 hours of video, although no participant saw every section. The intent-to-treat analysis showed a significant difference from the treatment group at 12.3% vs the controls at 5.0% (OR=2.66, 95% CI, 1.18–5.99).
TABLE 1
Web-based support helps smokers quit
| www.quitnet.com Boston University School of Public Health | Personalized quit plans |
| www.ffsonline.org American Lung Association | “Freedom from smoking” modules to guide smokers through quit process |
| www.whyquit.com Privately supported | Support for “cold turkey” quitting |
| www.trytostop.org Massachusetts Department of Public Health | Personalized “Quit Wizard” program |
Text messages work
Text messaging may also have a place in supporting smoking cessation efforts. An interesting, although short, study13 looked at using text messaging to target younger smokers in New Zealand. This RCT (SOR: B) involved 1705 smokers who had cell phones with text messaging. Researchers sent participants up to 5 messages daily around their quit date, drawing from over 100 messages that could be personalized with individual names/nicknames. The quit rate was doubled 6 weeks out (28% vs 13%; relative risk=2.20; 95% CI, 1.79–2.70).
Rx in hand, support in place
When Ann left my office, she took with her a prescription for varenicline, the state’s quitline number, and the URL for an online support program. Ann was eager to try varenicline: a coworker of hers was using it and doing well. Ann had tried the nicotine patch in the past, but reported that it gave her nightmares. She’d also kept smoking while wearing it. This time, she hoped she’d finally be able to quit for good.
Weighing the drug treatment options
The AHRQ guidelines recommend several types of pharmacotherapy. First-line therapies include different forms of NRT and sustained-release bupropion (Zyban).2,3
Nicotine replacement therapy doubles the chances of quitting
With NRT, the nicotine in cigarettes is replaced with nicotine from another source. The thought is that by reducing the withdrawal symptoms, the patient is less likely to relapse and resume smoking. Nicotine replacement is available in several forms: gum, transdermal patches, intranasal spray, inhaler, and lozenges.
A Cochrane meta-analysis of NRT14 (SOR: A) analyzed 123 studies that followed patients for at least 6 months from their quit date. The authors concluded that NRT could almost double a patient’s chances of quitting smoking. The data from various types of NRT revealed the types to be similarly efficacious (TABLE 2). In the treated groups, 17% were abstinent and only 10% were abstinent in the control groups at the various endpoints of the trials. Smokers with higher levels of nicotine dependence as indicated by smoking 10 or more cigarettes daily have higher quit rates using replacement nicotine. Generally, treatments of 8 weeks are as effective as longer courses.
The Cochrane meta-analysis also revealed that:
- Duration of therapy ranges from 3 weeks to 12 months with the various forms of NRT.
- There was no benefit to tapering off the NRT as compared to an abrupt withdrawal.
- Patients are much more likely to relapse after NRT in the first 3 months.
- Combining several forms of NRT may aid a relapsed smoker in another quit attempt. However, the re-attempt should be delayed by a few months, as back-to-back courses are unlikely to improve quit rates.
TABLE 2
Nicotine replacement therapy: Methods are similarly efficacious11
| THERAPY | OR (95% CI) | N (PARTICIPANTS/ TRIALS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)‡ |
|---|---|---|---|---|---|
| Nasal spray | 2.35 (1.63–3.38) | 887/4 | 8.3 | 3–6 months | $560/NA |
| Inhaler | 2.14 (1.44–3.18) | 976/4 | 12.5 | 3 months, then 3-month taper | $504/NA |
| Lozenges | 2.05 (1.62–2.59) | 2739/5 | 14.3 | Up to 12 weeks | $300/$240 |
| Patch | 1.84 (1.65–2.06) | 16,228/37 | 16.7 | 8–12 weeks | $110/$92 |
| NR T (all) | 1.77 (1.66–1.88) | 39,503/105 | * | ||
| Gum | 1.66 (1.51–1.81) | 17,819/52 | 12.5 | Up to 12 weeks | 4 mg: $234/$180 2 mg: $204/$150 |
| * Numbers not available. | |||||
| † Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; NA, product not available. | |||||
Sustained-release bupropion: Similar results to NRT
The other first-line therapy suggested by the AHRQ guidelines is sustained-release bupropion.2,3 A separate Cochrane Review15 analyzed the data from 36 studies using antidepressants and revealed that two thirds of the studies in this meta-analysis used bupropion. The odds of quitting smoking essentially doubled in the placebo-controlled studies. This is a similar effect as NRT. Neither the AHRQ guidelines nor the Cochrane Review recommend bupropion over NRT or vice-versa.
According to the Cochrane Review, there was no benefit to increasing the dose of bupropion from 150 mg to 300 mg daily. Although the initial multi-dose study of bupropion16 showed a difference, it was not clinically significant by the end of the study. A larger, open-label randomized trial of 1524 smokers17 followed for 1 year also showed similar results. At the 3-month evaluation, the higher dose had superior efficacy, but that effect was not statistically significant by the end of the study. Lastly, there is no benefit to continuing the bupropion beyond 7 weeks after the target quit date.
With other antidepressants, results vary
The Cochrane Review also looked at other antidepressants. There were 4 RCTs of nortriptyline (Aventyl/Pamelor) without NRT, totaling 777 smokers followed for at least 6 months.18-21 The pooled data essentially doubled the odds of quitting smoking from 7.0% for the controls to 17.2% in the treated groups (OR=2.79; 95% CI, 1.70–4.59). Adding nortriptyline to NRT did increase the quit rates, but not significantly. The dose used in these studies, at 75 to 150 mg is much lower than that used for depression, where significant side effects often interfere with treatment. Generally the starting dose is 25 mg at bedtime. After 1 week, the dose is increased to 50 mg and the following week, it is increased again to 75 mg. Once on the 75 mg dose for a week, the dose is titrated up only if needed. The titration continues at an additional 25 mg weekly.
One of the 4 placebo-controlled studies20 included an arm of bupropion, producing a head-to-head assessment with nortriptyline (SOR: A). The abstinence rates as indicated by no smoking during the final week of treatment were comparable for the 2 groups receiving active medication. Treatment with bupropion or nortriptyline was significantly more efficacious than placebo. However, the effect was lost at the 1 year continuous abstinence mark; the 2 drugs did not differ from each other or placebo (TABLE 3).
Other antidepressants were evaluated in the Cochrane study.15 The tricyclic antidepressants doxepin and imipramine (Tofranil) had no long-term studies and neither showed statistically significant differences in smaller trials. Of the selective serotonin reuptake inhibitors (SSRIs), only fluoxetine (Prozac) had any long-term studies, and none noted statistically significant differences. Likewise, venlafaxine (Effexor) had only 1 trial in which the confidence interval did allow for a potentially useful clinical effect, but failed to show a statistically significant increase in 12-month quit rates.
Clonidine is an option, but side effects are an issue
Another Cochrane Review22 looked at the effectiveness of clonidine (Catapres) on smoking cessation. Most of the clonidine studies assessed withdrawal symptoms rather than abstinence. Of those that did assess quit rates, the pooled OR for clonidine compares favorably at 1.89 (95% CI, 1.30–2.74). Unfortunately, clonidine has significant side effects: sedation and postural hypotension. The starting dose is 0.1 mg twice daily, and it may be titrated up to a maximum dose of 0.4 mg daily. It should be used for 3 to 4 weeks only to decrease the symptoms of withdrawal. The smoker should then be weaned off the clonidine.
The anxiolytics were the subject of another Cochrane Review.23 This review, however, did not recommend any anxiolytics, including diazepam and buspirone, for smoking cessation.
A new category of therapy: Nicotinic receptor agonists
With the US Food and Drug Administration’s approval of varenicline (Chantix) in May 2006, a new class of drugs became available for treatment of tobacco dependence. This α4β2 nicotinic acetylcholine receptor partial agonist was designed as a smoking cessation drug. By releasing dopamine in the brain like nicotine, it prevents craving. However, it also blocks nicotine from binding, thereby preventing the reinforcing effect of continued smoking.
Two RCTs have assessed varenicline against both bupropion and placebo (TABLE 3). Jorenby24 (SOR: A) showed the varenicline-treated participants were significantly more likely to be continuously abstinent at 52 weeks than the placebo-or bupropion-treated groups (23% vs 10.3% placebo [OR=2.66; 95% CI, 1.72–4.11; P<.001] and 14.6% bupropion [OR=1.77; 95% CI, 1.19–2.63; P=.004]). Gonzales25 (SOR: A) likewise showed the varenicline treated smokers were more likely to be continuously abstinent at 52 weeks than the placebo group (21.9% vs 8.4% [OR=3.09; 95% CI, 1.95–4.91; P<.001]). However, the difference between varenicline and bupropion did not reach statistical significance (21.9% vs 16.1% [OR=1.46; 95% CI, 0.99–2.17; P=.057]).
As with other medications, varenicline should be started at a low dose. The patient begins with 0.5 mg nightly for the first 3 nights, then increases to 0.5 mg twice a day for 4 days. The second week, the patient begins the 1 mg twice-daily dosing that is continued through treatment.
TABLE 3
Varenicline, nortriptyline, bupropion—strong allies in patients’ efforts to quit
| THERAPY | OR (95% CI) | N (PARTICIPA NTS/TRAILS) | NNT | DURATION OF THERAPY | COST OF 4 WEEKS (BRAND/GENERIC)* |
|---|---|---|---|---|---|
| Varenicline24,25 | 2.80 (2.03–3.88) | 1161/2 | 7.6 | 12 weeks | $120/NA |
| Nortriptyline15 | 2.79 (1.70–4.59) | 703/4 | 9.8 | 12 weeks | $814/$8 |
| Sustained-release bupropion15 | 2.06 (1.77–2.40) | 6443/19 | 10.2 | 7–12 weeks | $210/$100 |
| Clonidine23 | 1.89 (1.30–2.74) | 776/6 | 9.4 | 3–4 weeks | $74/$4 |
| Venlafaxine15 | 1.33 (0.59–3.00) | 136/1 | 20.4 | $145/NA | |
| Diazepam23 | 1.00 (0.39–2.54) | 76/1 | No difference | $209/$27 | |
| SSRI15 | 0.90 (0.68–1.18) | 1768/6 | 20.7 | $170/$4 | |
| Buspirone23 | 0.71 (0.34–1.48) | 201/3 | 22.1 | $280/$84 | |
| *Cost based on prices from Walgreen’s and Target Pharmacies, May and September 2007. | |||||
| OR, odds ratio; NNT, number needed to treat; SSRI, selective serotonin reuptake inhibitors; NA, not available. | |||||
Vaccines hold the promise of continued abstinence
Several promising ideas for the treatment of tobacco dependence are in development. There are several vaccines being studied.26 When the immune system produces antibodies to nicotine in response to the vaccine, and when these antibodies bind to the nicotine, the resultant compound is too large to cross the blood-brain barrier. This prevents the reinforcing effect of nicotine. Initial studies of vaccines show that smokers do decrease the amount they smoke, but more importantly, abstinence is easier to maintain. However, the vaccine requires frequent boosters to maintain antibody titers that are effective.
NicVAX from Nabi Biopharmaceuticals was placed on a fast track for approval by the Food and Drug Administration. It is, however, still at least a year away from approval. The other 2 nicotine vaccines are probably several years beyond that for approval.27
Researchers are also studying other compounds that block the euphoria associated with smoking.28 The initial studies of rimonabant (Acomplia), a cannabinoid blocker, have shown it is no better than other treatments already available. With its indication in some European countries for weight loss, it offered promise as an important option for patients who are concerned about the weight gain associated with smoking cessation. However, the FDA did not approve rimonabant for tobacco cessation when issuing its initial approval letter for weight loss in 2006. Because of safety concerns, the manufacturer subsequently withdrew the new drug application for rimonabant in 2007.
With much work, our patient kicks the habit
Ann began taking varenicline the day she left the office, and reached her quit date a week later.
At her 1-month follow-up, Ann reported that it was actually easy for her to stay off the cigarettes. With the varenicline, she had lost the desire to smoke. I reminded her to work on the triggers for her smoking: I urged her to make sure that she did not light up when she made her morning coffee or got in the car. I also suggested she put $4 each morning into a jar on her dresser; so she would see how much she saved now that she wasn’t buying cigarettes.
At Ann’s next annual exam, we marked her in the computer system as a reformed smoker. She was very proud of that label. I asked her what she was doing with all that extra cash. She laughed: “My daughter spends it all! but not on cigarettes!”
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Drug Abuse.
Correspondence
Agnes O. Coffay, MD, NIDA/IRP, 5500 Nathan Shock Drive, B altimore, MD 21224-6823. [email protected]
1. Brown D. Nicotine up sharply in many cigarettes. Washington Post, August 31, 2006. Available at: www.washingtonpost.com/wp-dyn/content/article/2006/08/30/Ar2006083001418.html. Accessed on September 4, 2007.
2. Agency for Healthcare Research and Quality. Clinical Practice Guideline. Treating Tobacco Use and Dependence. Rockville, Md: US Department of Health and Human Services Public Health Service;2000. Available at: www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat2. chapter.7644. Accessed on September 4, 2007.
3. Tobacco Use and Dependence clinical Practice Guideline Panel, Staff, and Consortium Representatives. A clinical practice guideline for treating tobacco use and dependence: A US Public Health Service report. JAMA. 2000;283:3244-3254.
4. Ask and Act: A Tobacco Cessation Program. Available at: www.aafp.org/online/en/home/clinical/publichealth/tobacco/askandact.html. Accessed on September 4, 2007.
5. Lancaster T, Stead LF. Physician advice for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000165.-
6. Fiore MC, McCarthy DE, Jackson TC, et al. Integrating smoking cessation treatment into primary care: an effectiveness study. Prev Med 2004;38:412-420.
7. Katz DA, Muehlenbruch DR, Brown RL. Effectiveness of implementing the Agency for Healthcare Research and Quality Smoking cessation clinical Practice Guidelines: a randomized, control trial. J Natl Cancer Inst 2004;96:594-603.
8. Stead LF, Lancaster T, Perera R. Telephone counseling for smoking cessation. Cochrane Database Syst Rev 2003;(1):CD002850.-
9. Zhu SH, Anderson CM, Tedeschi GJ, et al. Evidence of real-world effectiveness of a telephone quitline for smokers. N Engl J Med 2002;347:1087-1093.
10. An LC, Zhu SH, Nelson DB, et al. Benefits of telephone care over primary care for smoking cessation. Arch Intern Med 2006;166:536-542.
11. Etter JF. Comparing the efficacy of two internet-based, computer-tailored smoking cessation programs: a randomized trial. J Med Internet Res 2005;7(1):e2.-
12. Swartz LH, Noell JW, Schroeder SW, Ary DV. A randomized control study of a fully automated internet based smoking cessation programme. Tobacco Control 2006;15:7-12.
13. Rodgers A, Corbett T, Bramley D, et al. Do u smoke after txt? Results of a randomized trial of smoking cessation using mobile phone text messaging. Tobacco Control 2005;14:255-261.
14. Silagy C, Lancaster T, Stead I, Mant D, Fowler G. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000146.-
15. Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev 2004;(2):cD000031.-
16. Hurt RD, Sachs DP, Glover ED, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
17. Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting. Arch Intern Med 2003;163:2337-2344.
18. da Costa CL, Younes RN, Lourenco MT. A prospective, randomized, double-blind study comparing nortriptyline to placebo. Chest 2002;122:403-408.
19. Hall SM, Reus VI, Munoz RF, et al. Nortriptyline and cognitive-behavioral therapy in the treatment of cigarette smoking. Arch Gen Psychiatry 1998;55:683-690.
20. Hall SM, Humfleet GL, Reus VI, et al. Psychological intervention and antidepressant treatment in smoking cessation. Arch Gen Psychiatry 2002;59:930-936.
21. Prochazka AV, Weaver MJ, Keller RT, et al. A randomized trial of nortriptyline for smoking cessation. Arch Intern Med 1998;158:2035-2039.
22. Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database Syst Rev 2004;(3):CD000058.-
23. Hughes JR, Stead LF, Lancaster T. Anxiolytics for smoking cessation. Cochrane Database Syst Rev 2000;(4):CD002849.-
24. Jorenby De, Hays JT, Rigotti NA. Efficacy of varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation. JAMA 2006;296:56-63.
25. Gonzales D, Rennard SI, Nides M. Varenicline, an a4b2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation. JAMA 2006;296:47-55.
26. LeHouezec J. Why a nicotine vaccine? Clin Pharmacol Ther 2005;78:453-455.
27. Tuller D. Scientists testing vaccines to help smokers quit. New York Times,July 4, 2006. Available at: www. nytimes.com/2006/07/040health/04vacc.html?ex=13 09665600&en=d978add467c2b80a&ei=5088&partne r=rssnyt&emc=rss. Accessed on September 6, 2007.
28. Fagerström K, Balfour DJ. Neuropharmacology and potential efficacy of new treatments for tobacco dependence. Expert Opin Investig Drugs 2005;15:107-116.
1. Brown D. Nicotine up sharply in many cigarettes. Washington Post, August 31, 2006. Available at: www.washingtonpost.com/wp-dyn/content/article/2006/08/30/Ar2006083001418.html. Accessed on September 4, 2007.
2. Agency for Healthcare Research and Quality. Clinical Practice Guideline. Treating Tobacco Use and Dependence. Rockville, Md: US Department of Health and Human Services Public Health Service;2000. Available at: www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat2. chapter.7644. Accessed on September 4, 2007.
3. Tobacco Use and Dependence clinical Practice Guideline Panel, Staff, and Consortium Representatives. A clinical practice guideline for treating tobacco use and dependence: A US Public Health Service report. JAMA. 2000;283:3244-3254.
4. Ask and Act: A Tobacco Cessation Program. Available at: www.aafp.org/online/en/home/clinical/publichealth/tobacco/askandact.html. Accessed on September 4, 2007.
5. Lancaster T, Stead LF. Physician advice for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000165.-
6. Fiore MC, McCarthy DE, Jackson TC, et al. Integrating smoking cessation treatment into primary care: an effectiveness study. Prev Med 2004;38:412-420.
7. Katz DA, Muehlenbruch DR, Brown RL. Effectiveness of implementing the Agency for Healthcare Research and Quality Smoking cessation clinical Practice Guidelines: a randomized, control trial. J Natl Cancer Inst 2004;96:594-603.
8. Stead LF, Lancaster T, Perera R. Telephone counseling for smoking cessation. Cochrane Database Syst Rev 2003;(1):CD002850.-
9. Zhu SH, Anderson CM, Tedeschi GJ, et al. Evidence of real-world effectiveness of a telephone quitline for smokers. N Engl J Med 2002;347:1087-1093.
10. An LC, Zhu SH, Nelson DB, et al. Benefits of telephone care over primary care for smoking cessation. Arch Intern Med 2006;166:536-542.
11. Etter JF. Comparing the efficacy of two internet-based, computer-tailored smoking cessation programs: a randomized trial. J Med Internet Res 2005;7(1):e2.-
12. Swartz LH, Noell JW, Schroeder SW, Ary DV. A randomized control study of a fully automated internet based smoking cessation programme. Tobacco Control 2006;15:7-12.
13. Rodgers A, Corbett T, Bramley D, et al. Do u smoke after txt? Results of a randomized trial of smoking cessation using mobile phone text messaging. Tobacco Control 2005;14:255-261.
14. Silagy C, Lancaster T, Stead I, Mant D, Fowler G. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev 2004;(4):CD000146.-
15. Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev 2004;(2):cD000031.-
16. Hurt RD, Sachs DP, Glover ED, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
17. Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting. Arch Intern Med 2003;163:2337-2344.
18. da Costa CL, Younes RN, Lourenco MT. A prospective, randomized, double-blind study comparing nortriptyline to placebo. Chest 2002;122:403-408.
19. Hall SM, Reus VI, Munoz RF, et al. Nortriptyline and cognitive-behavioral therapy in the treatment of cigarette smoking. Arch Gen Psychiatry 1998;55:683-690.
20. Hall SM, Humfleet GL, Reus VI, et al. Psychological intervention and antidepressant treatment in smoking cessation. Arch Gen Psychiatry 2002;59:930-936.
21. Prochazka AV, Weaver MJ, Keller RT, et al. A randomized trial of nortriptyline for smoking cessation. Arch Intern Med 1998;158:2035-2039.
22. Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database Syst Rev 2004;(3):CD000058.-
23. Hughes JR, Stead LF, Lancaster T. Anxiolytics for smoking cessation. Cochrane Database Syst Rev 2000;(4):CD002849.-
24. Jorenby De, Hays JT, Rigotti NA. Efficacy of varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation. JAMA 2006;296:56-63.
25. Gonzales D, Rennard SI, Nides M. Varenicline, an a4b2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation. JAMA 2006;296:47-55.
26. LeHouezec J. Why a nicotine vaccine? Clin Pharmacol Ther 2005;78:453-455.
27. Tuller D. Scientists testing vaccines to help smokers quit. New York Times,July 4, 2006. Available at: www. nytimes.com/2006/07/040health/04vacc.html?ex=13 09665600&en=d978add467c2b80a&ei=5088&partne r=rssnyt&emc=rss. Accessed on September 6, 2007.
28. Fagerström K, Balfour DJ. Neuropharmacology and potential efficacy of new treatments for tobacco dependence. Expert Opin Investig Drugs 2005;15:107-116.
Newborn care: 12 beliefs that shape practice (But should they?)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Baby M is a 12-hour-old 40-week gestation boy, without any risk factors, whom you delivered vaginally to a first-time mother last evening. The following morning, his mother tells you that while she intends to breastfeed exclusively, she was told by a staff member in the nursery to give the baby some formula “until your milk comes in, so he won’t be so cranky.” The child appears entirely well.
The next morning, at 36-hours-of-age, Baby M still appears well, except that he is jaundiced. His bilirubin level is 15—just above the level at which phototherapy should be started, but well below the level at which an exchange transfusion is required. You order a transfer to the nursery for phototherapy.
You order IV fluids to prevent dehydration and to enhance the excretion of bilirubin. To bring his bilirubin level down more quickly, you advise the mother to temporarily stop breastfeeding and feed him a standard infant formula. The nursing staff gives the mother a breast pump to use until breastfeeding is re-started.
Forty-eight hours later Baby M is ready for discharge, and the mother has questions about infant feeding practices. You and the nurse explain to the mother that “breast milk provides complete nutrition” for her baby and that the baby will need no other nutrition until he starts solid foods at around 4 to 6 months of age.
Routine care, but was it appropriate?
The care you provided to Baby M and the advice you offered his mother might seem pretty routine, but look again. Was it appropriate?
In fact, your care—and that of the nursing staff—diverged from the evidence in 4 ways:
- There was no need to give Baby M formula until his mother’s milk came in (Strength of Recommendation [SOR]: C).
- It was not necessary to use IV fluids to treat jaundice in this otherwise healthy term newborn (SOR: C). IV fluids do not bring down bilirubin levels, and even with mild dehydration, the best fluid therapy is breast milk or formula.
- There was no need to temporarily discontinue breastfeeding during phototherapy to bring Baby M’s bilirubin level down more quickly (SOR: C). While doing so may bring levels down a bit faster, it is not worth the risk that the mother may opt to discontinue breastfeeding entirely.
- You should not have told Baby M’s mother that “breast milk provides complete nutrition.” In fact, the American Academy of Pediatrics (AAP) recommends that all breast-fed newborns receive supplemental vitamin D (SOR: B).
12 common beliefs that require a second look
Medicine has a long history of promoting practices like the ones described here, which are either not based upon scientific evidence or are in direct contradiction to existing evidence. Often, we do not realize how much of our advice is based on misunderstandings passed down from our teachers and absorbed from the cultural milieu in which we were raised.
In this article, we will review the most current evidence regarding 12 commonly held beliefs regarding newborn care. These beliefs relate to breast milk, breastfeeding, pacifier use, emesis, umbilical cord care, and jaundice. We’ve included them because these beliefs give rise to many of the common practices we’ve seen in our combined 60 years in pediatrics.
In our effort to look at the evidence behind these beliefs, we encountered some limitations. For most of the recommendations we’ve put forth here, the best evidence that exists is category C—that is, recommendations based on consensus, usual practice, opinion, disease-oriented evidence, or case series for studies of diagnosis, treatment, prevention, or screening.
Though no randomized, controlled trials exist for most of the common practices reviewed here, we have included consensus statements from the best available sources, such as the Centers for Disease Control and Prevention and the AAP. Where appropriate, we also included clear, pathophysiologic reasoning to support each recommendation.
BELIEF 1: Breast milk is a complete nutritional source for a healthy term newborn
THE EVIDENCE: Breast milk is not a complete nutritional source for healthy term newborns. In fact, breast milk provides the ideal source of nutrition, and it is almost a complete and perfect source of nutrition—with one important exception. The AAP recommends that all breast-fed newborns receive 200 IU/day of vitamin D until they are getting at least 500 ml/day of Vitamin-D formula or milk (SOR: B).1,2
The purpose of the supplementation is to prevent vitamin D deficiency and subsequent rickets. (Rickets does continue to occur in the United States.3,4) The AAP makes no mention in its recommendation of infant pigmentation or the expected amount of exposure to sunshine. The AAP recommends that vitamin D supplementation begin by the time the infant is 2 months old.
BELIEF 2: Supplementing with formula because the mother’s milk hasn’t come in yet is a reasonable, routine practice
THE EVIDENCE: Formula supplements are not necessary as routine practice (SOR: C).
Formula supplements are counterproductive,5 taking away the primary stimulus for breast milk production—nursing at the breast. Infant dissatisfaction with the initial volume of breast milk produced actually works to the infant’s advantage,5 driving the child to the breast more often, and thus increasing the likelihood of successful breastfeeding.
In certain circumstances, formula supplements can be reasonable, such as when an infant is hypoglycemic or when the baby is receiving phototherapy5 and experiences excessive weight loss and becomes severely dehydrated. However, formula supplementation is not reasonable or necessary as routine practice.
BELIEF 3: Mothers on magnesium therapy should not breastfeed their infants
THE EVIDENCE: Mothers on magnesium therapy may continue to breastfeed their babies (SOR: B).
The misguided recommendation that mothers who are being treated with magnesium therapy should not breastfeed6,7 is based on an unreasonable fear that magnesium therapy can cause hypermagnesemia in breastfed newborns due to excessive magnesium levels in the breast milk. Supplemental magnesium, usually given intravenously to mothers with severe preeclampsia, does not cross over into breast milk in any significant amount, even when the mother continues to need intravenous magnesium after the birth of her baby.8,9
BELIEF 4: Mothers who are positive for hepatitis B surface antigen or who are carriers for hepatitis C should not breastfeed
THE EVIDENCE: Mothers who are hepatitis B surface antigen positive or carriers for hepatitis C can safely breastfeed their newborns10 (SOR: C).
The idea that mothers who are infected with hepatitis B or C should not breast-feed their babies at first seems obvious to many who care for newborns, as the diseases are transmitted through blood exposure, and nipple cracks with associated blood loss are common in mothers when they begin to breastfeed.
The hepatitis B immunization protocol for infants born to hepatitis B surface antigen positive mothers takes care of the first infectious concern.11 In addition, no case of hepatitis C transmission from breast milk has ever been reported.11 The Centers for Disease Control and Prevention confirms that the transmission rate of hepatitis C from infected mothers is the same whether the babies are breast- or bottle-fed.12
BELIEF 5: Mothers who are febrile should not breastfeed
THE EVIDENCE: In most cases, febrile mothers may safely breastfeed their infants (SOR: C).
The advice for mothers not to breast-feed while febrile seems intuitively true because of concern that the infection might pass over into the breast milk to the baby. This rarely happens. There are only 4 contraindications to breastfeeding during maternal fever:10
- Active, untreated maternal tuberculosis
- Mothers who are human T-cell lymphotropic virus type I or II positive
- Mothers who are HIV-positive
- Mothers with a herpes simplex lesion on the breast.
BELIEF 6: Mothers who smoke or drink alcohol should not breastfeed
THE EVIDENCE: While this recommendation seems self-evident, the research proving harmful effects to the infant is lacking13 (SOR: C).
In fact, in its most recent statement on “The Transfer of Drugs and Other Chemicals Into Human Milk,” the AAP removed nicotine from a table of drugs for which adverse effects have been reported in infants during breastfeeding.13
While it would be ideal if no breastfeeding mother smoked or drank alcohol, the fact of the matter is that some do. In light of this, it’s wise to encourage the mother to smoke outside the home, and to change her clothes before holding her baby. In so doing, she will avoid exposing her baby to most of the effects of secondhand smoke. In addition, while mothers who breastfeed their infants should, of course, avoid alcohol abuse, a single, occasional, small alcoholic drink is acceptable.14
BELIEF 7: Pacifiers are bad for newborns
THE EVIDENCE: It is not clear whether pacifiers are “bad” for newborns (SOR: C).
The belief that newborns should not have pacifiers came into being for a well-intended reason: Breastfeeding advocates were concerned that newborns would spend too much time sucking on the pacifier and too little time sucking at the breast, undermining the mother’s ability to breastfeed successfully.15
Consensus on the matter, though, is lacking. The UNICEF-World Health Organization Baby-Friendly Initiative, for instance, recommends that pacifiers not be used.16 The AAP, however, advises that pacifiers can be used once breastfeeding is well established.17
The research is also mixed. On the one hand, new evidence indicates that pacifier use may decrease the incidence of sudden infant death syndrome.18 On the other hand, pacifier use for longer than 48 months has been linked to orthodontic problems and dental caries.19,20
Thus, while prolonged pacifier use may be harmful to dental hygiene, newer evidence allows that pacifiers may be acceptable in the first few years during breastfeeding.
BELIEF 8: Newborn emesis is an indication for a formula change
THE EVIDENCE: No literature supports the belief that it is appropriate to change an infant’s formula in response to emesis in the first 2 weeks of life (SOR: C).
The overwhelming majority of vomiting episodes in newborns have no accompanying medical problem.21 A 2002 study by Miyazawa et al that looked at more than 900 infants showed more than 47% of Japanese infants ≤1 month of age had daily occurrences of regurgitation or emesis without having an underlying medical disorder.22
Newborns vomit for any number of benign reasons, including swallowed maternal blood or overfeeding.21 Gastroesophageal reflux can be a manifestation of milk allergy. However, a newborn infant is too young to manifest an antibody response to the protein in the formula. Therefore, switching formula because of vomiting due to milk allergy is not prudent in the first 10 to 14 days of life, the length of time needed to mount an antibody response to an antigen23 and thus, the length of time needed to become allergic to an infant formula.
BELIEF 9: Umbilical cord care can prevent umbilical cord infections
THE EVIDENCE: There is no definitive evidence regarding the best method for preventing umbilical cord infections among babies living in developed countries (SOR: C).
In fact, there is no evidence that any topical preparation, be it a dye, an antiseptic, or an antibiotic, is any better at preventing umbilical cord infections than keeping the area clean and dry.24 Umbilical cord infections such as omphalitis or tetanus neonatorum are more common in developing countries than high-income countries.16 In developed countries, cord care with topical antimicrobial agents is frequently unnecessary.24
Infants who were delivered at home and those who room in with their mothers have no need of a topical antimicrobial therapy. If an infant is kept in a hospital nursery or intensive care unit, topical antimicrobial therapy to the cord may have some benefit in keeping down cord colonization with pathological bacteria such as methicillin-resistant Staphylococcus aureus.24
Umbilical cord infections sometimes occur even when the cord area is kept clean and dry,25 so healthcare providers must be attentive to signs of possible infection.16
BELIEF 10: It’s easy to spot when a newborn is jaundiced
THE EVIDENCE: Jaundice is actually difficult to detect in darkly pigmented babies,5 and in babies sent home within 24 hours of birth, because bilirubin levels reach maximum levels between the third and fourth days of life26 (SOR: C).
Years ago, when infants stayed longer in the nursery, doctors had the chance to see them when their bilirubin level was highest and when the babies were most jaundiced. The current emphasis on early discharge does not allow this practice.
The AAP recommends clinical assessment of a newborn’s state of jaundice and that a bilirubin level be obtained whenever a physician is in doubt about the degree of clinical jaundice. The AAP also recommends that physicians consider obtaining a routine screening bilirubin in all newborns at the time of hospital discharge even if, by clinical assessment, the child is not jaundiced.5 (The AAP stopped short, though, of saying that such a screening test is required.) The AAP made these recommendations because of an increasing concern that the incidence of kernicterus in America is rising.27
BELIEF 11: All infants who require phototherapy need IV fluids to prevent dehydration and enhance excretion of bilirubin
THE EVIDENCE: Unless the baby is clinically dehydrated, IV fluid therapy for infants under phototherapy is not needed28 (SOR: c).
Though IV fluid therapy is commonly used to increase the excretion of bilirubin and combat dehydration, the research tells us that IV fluids do not bring down bilirubin levels and that even with mild dehydration, the best fluid therapy is breast milk or formula because it inhibits the enterohepatic circulation of bilirubin.5 Intravenous fluid therapy should be reserved for jaundiced newborns with moderate to severe dehydration, or those with mild dehydration5 who are not able to take fluid by mouth.
BELIEF 12: Breast-milk jaundice is best treated by stopping breastfeeding for 24 to 48 hours
THE EVIDENCE: breastfeeding should not be discontinued as a way to treat breast-milk jaundice (SOR: C).
In fact, breastfeeding should not be discontinued for jaundice due to any cause, as demonstrated in the opening scenario, unless you believe a baby is at risk of requiring an exchange transfusion. The need for phototherapy alone is not a sufficient reason to discontinue breastfeeding.5
Breast-milk jaundice is a common problem facing parents and physicians, but it is not a disease and does not represent an abnormality in and of itself. Rather, this normal physiologic condition gains its importance only in that it must be distinguished from pathological causes of newborn jaundice.29 Breast-milk jaundice is believed to affect 1%21to 33%30 of breastfed infants.
One treatment measure—to stop breastfeeding—began, in part, as a cost-effective way to diagnose breast-milk jaundice.31 Rechecking the bilirubin 24 to 48 hours after breastfeeding is discontinued would reveal a significant drop in the bilirubin level, confirming the diagnosis of breast-milk jaundice32 and obviating the need for testing for more serious medical illness.
The consequence of this misguided treatment approach, ie, discontinuing breastfeeding, is that some mothers are more likely to stop breastfeeding altogether.33
Acknowledgments
The authors thank the following individuals for providing assistance with this manuscript: Mariateresa Esquivel, MD; Charlotte Hobbs, MD, PhD; Christopher Monnikendam, MD; and Clare Campbell Nesmith, MD.
Funding/Support
Dr Hall receives funding through DHS 1 P20 RR020146-01.
Correspondence
Bryan L. Burke, Jr., MD, FAAP, Associate Professor, Pediatrics and Neonatology, University of Arkansas for Medical Sciences, Arkansas Children’s Hospital, 4301 West Markham, Slot # 512-5B, Little Rock, AR 72205; [email protected]
1. Gartner LM, Greer FR. Section on Breastfeeding and Committee on Nutrition. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics 2003;111:908-910.
2. Greer FR. Issues in establishing vitamin D recommendations for infants and children. Am J Clin Nutr 2004;80(Suppl):1759S-1762S.
3. Kreiter SR, Schwartz RP, Kirkman HN, Jr, Charlton PA, Calikogl AS, Davenport ML. Nutritional rickets in African American breast-fed infants. J Pediatr 2000;137:153-157.
4. Pugliese MT, Blumberg DL, Hludzinski K, Kay S. Nutritional rickets in suburbia. J Am Coll Nutr 1998;17:637-641.
5. American Academy of Pediatrics Subcommittee on Hyperbilirubinemia, Maisels MJ, Baltz RD, Bhutani VK, Newman TB, Palmer H, et al. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004;114:297—316.
6. Lew M, Klonis E. Emergency management of eclampsia and severe pre-eclampsia. Emerg Med 2003;15:361-368.
7. Touyz RM. Role of magnesium in the pathogenesis of hypertension. Mol Aspects Med 2003;24:107-136.
8. Cruikshank DP, Varner MW, Pitkin RM. Breast milk magnesium and calcium concentrations following magnesium sulfate treatment. Am J Obstet Gynecol 1982;143:685-688.
9. Hale T. Medications and Mother’s Milk: A Manual of Lactational Pharmacology. 11th ed. Amarillo, TX: Pharmasoft Medical Publishing; 2004.
10. Lawrence RA, Lawrence RM. Breastfeeding: A Guide for the Medical Profession. 6th ed. Philadelphia: Mosby, Inc; 2005.
11. Hepatitis B. In: Pickering LK, Baker CJ, Overturf GD, Prober CG, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2003:118-122;318-334.
12. Centers for Disease Control and Prevention. Recommendations for Prevention and Control of Hepatitis C Virus (HCV) Infection and HCV-Related Chronic Disease. Atlanta: US Department of Health and Human Services; 1998.
13. American Academy of Pediatrics committee on Drugs. The transfer of drugs and other chemicals into human milk. Pediatrics 2001;108:776-789.
14. Anderson PO. Alcohol and breastfeeding. J Hum Lact 1995;11:321-324.
15. Howard CR, Howard FM, Lanphear B, deblieck EA, Eberly S, Lawrence RA. The effects of early pacifier use on breastfeeding duration. Pediatrics 1999;103:e33-e38.
16. World Health Organization, Family and Reproductive Health, Division, of Child Health and Development. Evidence for the ten steps to successful breastfeeding. 1998 [cited May 22, 2006]; Available at: www.who.int/child-adolescent-health/New_Publications/NUTRITION/WHO_CHD_98.9.pdf
17. American Academy of Pediatrics Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2005;115:496-506.
18. Li D-K, Willinger M, Petitti DB, Odouli R, Liu L, Hoffman HJ. Use of a dummy (pacifier) during sleep and risk of sudden infant death syndrome (SIDS): population base case-control study. BMJ 2005;332:18-22.
19. Ollila P, Niemela M, Uhari M, Larmas M. Prolonged pacifier-sucking and use of a nursing bottle at night: possible risk factors for dental caries in children. Acta Odontol Scan 1998;56:233-237.
20. Warren JJ, Bishara SE, Steinbock KL, Yonezu T, Nowak AJ. Effects of oral habits’ duration on dental characteristics in the primary dentition. J Am Dent Assoc 2001;132:1685-1693.
21. Berkowitz CD. Jaundice. In: Berkowitz CD, ed. Pediatrics: A Primary Care Approach. Philadelphia, PA: WB Saunders; 1996:357-363.
22. Miyazawa R, Tomomasa T, Kaneko H, Tachibana A, Ogawa T, Morikawa A. Prevalence of gastro-esophageal reflux-related symptoms in Japanese infants. Pediatr Int 2002;44:513-516.
23. Virella G. The humoral response and its induction by active immunization. In. Virella G, ed. Introduction to Medical Immunology. 3rd ed. New York, NY: Marcel Dekker; 1993:213-232.
24. Zupan J, Garner P, Omari AAA. Topical umbilical cord care at birth. Cochrane Database Syst Rev 2004;(3):CD001057.-
25. Janssen PA, Selwood BL, Dobson SR, Peacock D, Thiessen PN. To dye or not to dye: a randomized, clinical trial of a triple dye/alcohol regime versus dry cord care. Pediatrics 2003;111:15-20.
26. Bhutani VK, Johnson L, Sivieri EM. Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics 1999;103:6-14.
27. Bhutani VK, Johnson LH, Maisels MJ, Newman TB, Phibbs C, Stark AR, et al. Kernicterus: edpidemiological strategies for its prevention through system-based approaches. J Perinatol 2004;24:650-652.
28. Boo N-Y, Lee H-T. Randomized controlled trial of oral versus intravenous fluid supplementation on serum bilirubin level during phototherapy of term infants with severe hyperbilirubinaemia. J Paediatr Child Health 2002;38:151-155.
29. Rubatelli FF. Current drug treatment options in neonatal hyperbilirubinaemia and the prevention of kernicterus. Drugs 1998;56:23-30.
30. Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-399.
31. Amato M, Howald H, Von Muralt G. Interruption of breastfeeding versus phototherapy as treatment of hyperbilirubinemia in full-term infants. Helv Paediat Acta 1985;40:127-131.
32. Stoll BJ, Kliegman RM. Digestive system disorders. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics. 17th ed. Philadelphia, PA: WB Saunders; 2004:595-596.
33. DiGirolamo A, Thompson N, Martorell R, Fein S, Grummer-Strawm L. Intention or experience? Predictors of continued breastfeeding. Health Educ Behav 2005;32:208-226.
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Baby M is a 12-hour-old 40-week gestation boy, without any risk factors, whom you delivered vaginally to a first-time mother last evening. The following morning, his mother tells you that while she intends to breastfeed exclusively, she was told by a staff member in the nursery to give the baby some formula “until your milk comes in, so he won’t be so cranky.” The child appears entirely well.
The next morning, at 36-hours-of-age, Baby M still appears well, except that he is jaundiced. His bilirubin level is 15—just above the level at which phototherapy should be started, but well below the level at which an exchange transfusion is required. You order a transfer to the nursery for phototherapy.
You order IV fluids to prevent dehydration and to enhance the excretion of bilirubin. To bring his bilirubin level down more quickly, you advise the mother to temporarily stop breastfeeding and feed him a standard infant formula. The nursing staff gives the mother a breast pump to use until breastfeeding is re-started.
Forty-eight hours later Baby M is ready for discharge, and the mother has questions about infant feeding practices. You and the nurse explain to the mother that “breast milk provides complete nutrition” for her baby and that the baby will need no other nutrition until he starts solid foods at around 4 to 6 months of age.
Routine care, but was it appropriate?
The care you provided to Baby M and the advice you offered his mother might seem pretty routine, but look again. Was it appropriate?
In fact, your care—and that of the nursing staff—diverged from the evidence in 4 ways:
- There was no need to give Baby M formula until his mother’s milk came in (Strength of Recommendation [SOR]: C).
- It was not necessary to use IV fluids to treat jaundice in this otherwise healthy term newborn (SOR: C). IV fluids do not bring down bilirubin levels, and even with mild dehydration, the best fluid therapy is breast milk or formula.
- There was no need to temporarily discontinue breastfeeding during phototherapy to bring Baby M’s bilirubin level down more quickly (SOR: C). While doing so may bring levels down a bit faster, it is not worth the risk that the mother may opt to discontinue breastfeeding entirely.
- You should not have told Baby M’s mother that “breast milk provides complete nutrition.” In fact, the American Academy of Pediatrics (AAP) recommends that all breast-fed newborns receive supplemental vitamin D (SOR: B).
12 common beliefs that require a second look
Medicine has a long history of promoting practices like the ones described here, which are either not based upon scientific evidence or are in direct contradiction to existing evidence. Often, we do not realize how much of our advice is based on misunderstandings passed down from our teachers and absorbed from the cultural milieu in which we were raised.
In this article, we will review the most current evidence regarding 12 commonly held beliefs regarding newborn care. These beliefs relate to breast milk, breastfeeding, pacifier use, emesis, umbilical cord care, and jaundice. We’ve included them because these beliefs give rise to many of the common practices we’ve seen in our combined 60 years in pediatrics.
In our effort to look at the evidence behind these beliefs, we encountered some limitations. For most of the recommendations we’ve put forth here, the best evidence that exists is category C—that is, recommendations based on consensus, usual practice, opinion, disease-oriented evidence, or case series for studies of diagnosis, treatment, prevention, or screening.
Though no randomized, controlled trials exist for most of the common practices reviewed here, we have included consensus statements from the best available sources, such as the Centers for Disease Control and Prevention and the AAP. Where appropriate, we also included clear, pathophysiologic reasoning to support each recommendation.
BELIEF 1: Breast milk is a complete nutritional source for a healthy term newborn
THE EVIDENCE: Breast milk is not a complete nutritional source for healthy term newborns. In fact, breast milk provides the ideal source of nutrition, and it is almost a complete and perfect source of nutrition—with one important exception. The AAP recommends that all breast-fed newborns receive 200 IU/day of vitamin D until they are getting at least 500 ml/day of Vitamin-D formula or milk (SOR: B).1,2
The purpose of the supplementation is to prevent vitamin D deficiency and subsequent rickets. (Rickets does continue to occur in the United States.3,4) The AAP makes no mention in its recommendation of infant pigmentation or the expected amount of exposure to sunshine. The AAP recommends that vitamin D supplementation begin by the time the infant is 2 months old.
BELIEF 2: Supplementing with formula because the mother’s milk hasn’t come in yet is a reasonable, routine practice
THE EVIDENCE: Formula supplements are not necessary as routine practice (SOR: C).
Formula supplements are counterproductive,5 taking away the primary stimulus for breast milk production—nursing at the breast. Infant dissatisfaction with the initial volume of breast milk produced actually works to the infant’s advantage,5 driving the child to the breast more often, and thus increasing the likelihood of successful breastfeeding.
In certain circumstances, formula supplements can be reasonable, such as when an infant is hypoglycemic or when the baby is receiving phototherapy5 and experiences excessive weight loss and becomes severely dehydrated. However, formula supplementation is not reasonable or necessary as routine practice.
BELIEF 3: Mothers on magnesium therapy should not breastfeed their infants
THE EVIDENCE: Mothers on magnesium therapy may continue to breastfeed their babies (SOR: B).
The misguided recommendation that mothers who are being treated with magnesium therapy should not breastfeed6,7 is based on an unreasonable fear that magnesium therapy can cause hypermagnesemia in breastfed newborns due to excessive magnesium levels in the breast milk. Supplemental magnesium, usually given intravenously to mothers with severe preeclampsia, does not cross over into breast milk in any significant amount, even when the mother continues to need intravenous magnesium after the birth of her baby.8,9
BELIEF 4: Mothers who are positive for hepatitis B surface antigen or who are carriers for hepatitis C should not breastfeed
THE EVIDENCE: Mothers who are hepatitis B surface antigen positive or carriers for hepatitis C can safely breastfeed their newborns10 (SOR: C).
The idea that mothers who are infected with hepatitis B or C should not breast-feed their babies at first seems obvious to many who care for newborns, as the diseases are transmitted through blood exposure, and nipple cracks with associated blood loss are common in mothers when they begin to breastfeed.
The hepatitis B immunization protocol for infants born to hepatitis B surface antigen positive mothers takes care of the first infectious concern.11 In addition, no case of hepatitis C transmission from breast milk has ever been reported.11 The Centers for Disease Control and Prevention confirms that the transmission rate of hepatitis C from infected mothers is the same whether the babies are breast- or bottle-fed.12
BELIEF 5: Mothers who are febrile should not breastfeed
THE EVIDENCE: In most cases, febrile mothers may safely breastfeed their infants (SOR: C).
The advice for mothers not to breast-feed while febrile seems intuitively true because of concern that the infection might pass over into the breast milk to the baby. This rarely happens. There are only 4 contraindications to breastfeeding during maternal fever:10
- Active, untreated maternal tuberculosis
- Mothers who are human T-cell lymphotropic virus type I or II positive
- Mothers who are HIV-positive
- Mothers with a herpes simplex lesion on the breast.
BELIEF 6: Mothers who smoke or drink alcohol should not breastfeed
THE EVIDENCE: While this recommendation seems self-evident, the research proving harmful effects to the infant is lacking13 (SOR: C).
In fact, in its most recent statement on “The Transfer of Drugs and Other Chemicals Into Human Milk,” the AAP removed nicotine from a table of drugs for which adverse effects have been reported in infants during breastfeeding.13
While it would be ideal if no breastfeeding mother smoked or drank alcohol, the fact of the matter is that some do. In light of this, it’s wise to encourage the mother to smoke outside the home, and to change her clothes before holding her baby. In so doing, she will avoid exposing her baby to most of the effects of secondhand smoke. In addition, while mothers who breastfeed their infants should, of course, avoid alcohol abuse, a single, occasional, small alcoholic drink is acceptable.14
BELIEF 7: Pacifiers are bad for newborns
THE EVIDENCE: It is not clear whether pacifiers are “bad” for newborns (SOR: C).
The belief that newborns should not have pacifiers came into being for a well-intended reason: Breastfeeding advocates were concerned that newborns would spend too much time sucking on the pacifier and too little time sucking at the breast, undermining the mother’s ability to breastfeed successfully.15
Consensus on the matter, though, is lacking. The UNICEF-World Health Organization Baby-Friendly Initiative, for instance, recommends that pacifiers not be used.16 The AAP, however, advises that pacifiers can be used once breastfeeding is well established.17
The research is also mixed. On the one hand, new evidence indicates that pacifier use may decrease the incidence of sudden infant death syndrome.18 On the other hand, pacifier use for longer than 48 months has been linked to orthodontic problems and dental caries.19,20
Thus, while prolonged pacifier use may be harmful to dental hygiene, newer evidence allows that pacifiers may be acceptable in the first few years during breastfeeding.
BELIEF 8: Newborn emesis is an indication for a formula change
THE EVIDENCE: No literature supports the belief that it is appropriate to change an infant’s formula in response to emesis in the first 2 weeks of life (SOR: C).
The overwhelming majority of vomiting episodes in newborns have no accompanying medical problem.21 A 2002 study by Miyazawa et al that looked at more than 900 infants showed more than 47% of Japanese infants ≤1 month of age had daily occurrences of regurgitation or emesis without having an underlying medical disorder.22
Newborns vomit for any number of benign reasons, including swallowed maternal blood or overfeeding.21 Gastroesophageal reflux can be a manifestation of milk allergy. However, a newborn infant is too young to manifest an antibody response to the protein in the formula. Therefore, switching formula because of vomiting due to milk allergy is not prudent in the first 10 to 14 days of life, the length of time needed to mount an antibody response to an antigen23 and thus, the length of time needed to become allergic to an infant formula.
BELIEF 9: Umbilical cord care can prevent umbilical cord infections
THE EVIDENCE: There is no definitive evidence regarding the best method for preventing umbilical cord infections among babies living in developed countries (SOR: C).
In fact, there is no evidence that any topical preparation, be it a dye, an antiseptic, or an antibiotic, is any better at preventing umbilical cord infections than keeping the area clean and dry.24 Umbilical cord infections such as omphalitis or tetanus neonatorum are more common in developing countries than high-income countries.16 In developed countries, cord care with topical antimicrobial agents is frequently unnecessary.24
Infants who were delivered at home and those who room in with their mothers have no need of a topical antimicrobial therapy. If an infant is kept in a hospital nursery or intensive care unit, topical antimicrobial therapy to the cord may have some benefit in keeping down cord colonization with pathological bacteria such as methicillin-resistant Staphylococcus aureus.24
Umbilical cord infections sometimes occur even when the cord area is kept clean and dry,25 so healthcare providers must be attentive to signs of possible infection.16
BELIEF 10: It’s easy to spot when a newborn is jaundiced
THE EVIDENCE: Jaundice is actually difficult to detect in darkly pigmented babies,5 and in babies sent home within 24 hours of birth, because bilirubin levels reach maximum levels between the third and fourth days of life26 (SOR: C).
Years ago, when infants stayed longer in the nursery, doctors had the chance to see them when their bilirubin level was highest and when the babies were most jaundiced. The current emphasis on early discharge does not allow this practice.
The AAP recommends clinical assessment of a newborn’s state of jaundice and that a bilirubin level be obtained whenever a physician is in doubt about the degree of clinical jaundice. The AAP also recommends that physicians consider obtaining a routine screening bilirubin in all newborns at the time of hospital discharge even if, by clinical assessment, the child is not jaundiced.5 (The AAP stopped short, though, of saying that such a screening test is required.) The AAP made these recommendations because of an increasing concern that the incidence of kernicterus in America is rising.27
BELIEF 11: All infants who require phototherapy need IV fluids to prevent dehydration and enhance excretion of bilirubin
THE EVIDENCE: Unless the baby is clinically dehydrated, IV fluid therapy for infants under phototherapy is not needed28 (SOR: c).
Though IV fluid therapy is commonly used to increase the excretion of bilirubin and combat dehydration, the research tells us that IV fluids do not bring down bilirubin levels and that even with mild dehydration, the best fluid therapy is breast milk or formula because it inhibits the enterohepatic circulation of bilirubin.5 Intravenous fluid therapy should be reserved for jaundiced newborns with moderate to severe dehydration, or those with mild dehydration5 who are not able to take fluid by mouth.
BELIEF 12: Breast-milk jaundice is best treated by stopping breastfeeding for 24 to 48 hours
THE EVIDENCE: breastfeeding should not be discontinued as a way to treat breast-milk jaundice (SOR: C).
In fact, breastfeeding should not be discontinued for jaundice due to any cause, as demonstrated in the opening scenario, unless you believe a baby is at risk of requiring an exchange transfusion. The need for phototherapy alone is not a sufficient reason to discontinue breastfeeding.5
Breast-milk jaundice is a common problem facing parents and physicians, but it is not a disease and does not represent an abnormality in and of itself. Rather, this normal physiologic condition gains its importance only in that it must be distinguished from pathological causes of newborn jaundice.29 Breast-milk jaundice is believed to affect 1%21to 33%30 of breastfed infants.
One treatment measure—to stop breastfeeding—began, in part, as a cost-effective way to diagnose breast-milk jaundice.31 Rechecking the bilirubin 24 to 48 hours after breastfeeding is discontinued would reveal a significant drop in the bilirubin level, confirming the diagnosis of breast-milk jaundice32 and obviating the need for testing for more serious medical illness.
The consequence of this misguided treatment approach, ie, discontinuing breastfeeding, is that some mothers are more likely to stop breastfeeding altogether.33
Acknowledgments
The authors thank the following individuals for providing assistance with this manuscript: Mariateresa Esquivel, MD; Charlotte Hobbs, MD, PhD; Christopher Monnikendam, MD; and Clare Campbell Nesmith, MD.
Funding/Support
Dr Hall receives funding through DHS 1 P20 RR020146-01.
Correspondence
Bryan L. Burke, Jr., MD, FAAP, Associate Professor, Pediatrics and Neonatology, University of Arkansas for Medical Sciences, Arkansas Children’s Hospital, 4301 West Markham, Slot # 512-5B, Little Rock, AR 72205; [email protected]
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Baby M is a 12-hour-old 40-week gestation boy, without any risk factors, whom you delivered vaginally to a first-time mother last evening. The following morning, his mother tells you that while she intends to breastfeed exclusively, she was told by a staff member in the nursery to give the baby some formula “until your milk comes in, so he won’t be so cranky.” The child appears entirely well.
The next morning, at 36-hours-of-age, Baby M still appears well, except that he is jaundiced. His bilirubin level is 15—just above the level at which phototherapy should be started, but well below the level at which an exchange transfusion is required. You order a transfer to the nursery for phototherapy.
You order IV fluids to prevent dehydration and to enhance the excretion of bilirubin. To bring his bilirubin level down more quickly, you advise the mother to temporarily stop breastfeeding and feed him a standard infant formula. The nursing staff gives the mother a breast pump to use until breastfeeding is re-started.
Forty-eight hours later Baby M is ready for discharge, and the mother has questions about infant feeding practices. You and the nurse explain to the mother that “breast milk provides complete nutrition” for her baby and that the baby will need no other nutrition until he starts solid foods at around 4 to 6 months of age.
Routine care, but was it appropriate?
The care you provided to Baby M and the advice you offered his mother might seem pretty routine, but look again. Was it appropriate?
In fact, your care—and that of the nursing staff—diverged from the evidence in 4 ways:
- There was no need to give Baby M formula until his mother’s milk came in (Strength of Recommendation [SOR]: C).
- It was not necessary to use IV fluids to treat jaundice in this otherwise healthy term newborn (SOR: C). IV fluids do not bring down bilirubin levels, and even with mild dehydration, the best fluid therapy is breast milk or formula.
- There was no need to temporarily discontinue breastfeeding during phototherapy to bring Baby M’s bilirubin level down more quickly (SOR: C). While doing so may bring levels down a bit faster, it is not worth the risk that the mother may opt to discontinue breastfeeding entirely.
- You should not have told Baby M’s mother that “breast milk provides complete nutrition.” In fact, the American Academy of Pediatrics (AAP) recommends that all breast-fed newborns receive supplemental vitamin D (SOR: B).
12 common beliefs that require a second look
Medicine has a long history of promoting practices like the ones described here, which are either not based upon scientific evidence or are in direct contradiction to existing evidence. Often, we do not realize how much of our advice is based on misunderstandings passed down from our teachers and absorbed from the cultural milieu in which we were raised.
In this article, we will review the most current evidence regarding 12 commonly held beliefs regarding newborn care. These beliefs relate to breast milk, breastfeeding, pacifier use, emesis, umbilical cord care, and jaundice. We’ve included them because these beliefs give rise to many of the common practices we’ve seen in our combined 60 years in pediatrics.
In our effort to look at the evidence behind these beliefs, we encountered some limitations. For most of the recommendations we’ve put forth here, the best evidence that exists is category C—that is, recommendations based on consensus, usual practice, opinion, disease-oriented evidence, or case series for studies of diagnosis, treatment, prevention, or screening.
Though no randomized, controlled trials exist for most of the common practices reviewed here, we have included consensus statements from the best available sources, such as the Centers for Disease Control and Prevention and the AAP. Where appropriate, we also included clear, pathophysiologic reasoning to support each recommendation.
BELIEF 1: Breast milk is a complete nutritional source for a healthy term newborn
THE EVIDENCE: Breast milk is not a complete nutritional source for healthy term newborns. In fact, breast milk provides the ideal source of nutrition, and it is almost a complete and perfect source of nutrition—with one important exception. The AAP recommends that all breast-fed newborns receive 200 IU/day of vitamin D until they are getting at least 500 ml/day of Vitamin-D formula or milk (SOR: B).1,2
The purpose of the supplementation is to prevent vitamin D deficiency and subsequent rickets. (Rickets does continue to occur in the United States.3,4) The AAP makes no mention in its recommendation of infant pigmentation or the expected amount of exposure to sunshine. The AAP recommends that vitamin D supplementation begin by the time the infant is 2 months old.
BELIEF 2: Supplementing with formula because the mother’s milk hasn’t come in yet is a reasonable, routine practice
THE EVIDENCE: Formula supplements are not necessary as routine practice (SOR: C).
Formula supplements are counterproductive,5 taking away the primary stimulus for breast milk production—nursing at the breast. Infant dissatisfaction with the initial volume of breast milk produced actually works to the infant’s advantage,5 driving the child to the breast more often, and thus increasing the likelihood of successful breastfeeding.
In certain circumstances, formula supplements can be reasonable, such as when an infant is hypoglycemic or when the baby is receiving phototherapy5 and experiences excessive weight loss and becomes severely dehydrated. However, formula supplementation is not reasonable or necessary as routine practice.
BELIEF 3: Mothers on magnesium therapy should not breastfeed their infants
THE EVIDENCE: Mothers on magnesium therapy may continue to breastfeed their babies (SOR: B).
The misguided recommendation that mothers who are being treated with magnesium therapy should not breastfeed6,7 is based on an unreasonable fear that magnesium therapy can cause hypermagnesemia in breastfed newborns due to excessive magnesium levels in the breast milk. Supplemental magnesium, usually given intravenously to mothers with severe preeclampsia, does not cross over into breast milk in any significant amount, even when the mother continues to need intravenous magnesium after the birth of her baby.8,9
BELIEF 4: Mothers who are positive for hepatitis B surface antigen or who are carriers for hepatitis C should not breastfeed
THE EVIDENCE: Mothers who are hepatitis B surface antigen positive or carriers for hepatitis C can safely breastfeed their newborns10 (SOR: C).
The idea that mothers who are infected with hepatitis B or C should not breast-feed their babies at first seems obvious to many who care for newborns, as the diseases are transmitted through blood exposure, and nipple cracks with associated blood loss are common in mothers when they begin to breastfeed.
The hepatitis B immunization protocol for infants born to hepatitis B surface antigen positive mothers takes care of the first infectious concern.11 In addition, no case of hepatitis C transmission from breast milk has ever been reported.11 The Centers for Disease Control and Prevention confirms that the transmission rate of hepatitis C from infected mothers is the same whether the babies are breast- or bottle-fed.12
BELIEF 5: Mothers who are febrile should not breastfeed
THE EVIDENCE: In most cases, febrile mothers may safely breastfeed their infants (SOR: C).
The advice for mothers not to breast-feed while febrile seems intuitively true because of concern that the infection might pass over into the breast milk to the baby. This rarely happens. There are only 4 contraindications to breastfeeding during maternal fever:10
- Active, untreated maternal tuberculosis
- Mothers who are human T-cell lymphotropic virus type I or II positive
- Mothers who are HIV-positive
- Mothers with a herpes simplex lesion on the breast.
BELIEF 6: Mothers who smoke or drink alcohol should not breastfeed
THE EVIDENCE: While this recommendation seems self-evident, the research proving harmful effects to the infant is lacking13 (SOR: C).
In fact, in its most recent statement on “The Transfer of Drugs and Other Chemicals Into Human Milk,” the AAP removed nicotine from a table of drugs for which adverse effects have been reported in infants during breastfeeding.13
While it would be ideal if no breastfeeding mother smoked or drank alcohol, the fact of the matter is that some do. In light of this, it’s wise to encourage the mother to smoke outside the home, and to change her clothes before holding her baby. In so doing, she will avoid exposing her baby to most of the effects of secondhand smoke. In addition, while mothers who breastfeed their infants should, of course, avoid alcohol abuse, a single, occasional, small alcoholic drink is acceptable.14
BELIEF 7: Pacifiers are bad for newborns
THE EVIDENCE: It is not clear whether pacifiers are “bad” for newborns (SOR: C).
The belief that newborns should not have pacifiers came into being for a well-intended reason: Breastfeeding advocates were concerned that newborns would spend too much time sucking on the pacifier and too little time sucking at the breast, undermining the mother’s ability to breastfeed successfully.15
Consensus on the matter, though, is lacking. The UNICEF-World Health Organization Baby-Friendly Initiative, for instance, recommends that pacifiers not be used.16 The AAP, however, advises that pacifiers can be used once breastfeeding is well established.17
The research is also mixed. On the one hand, new evidence indicates that pacifier use may decrease the incidence of sudden infant death syndrome.18 On the other hand, pacifier use for longer than 48 months has been linked to orthodontic problems and dental caries.19,20
Thus, while prolonged pacifier use may be harmful to dental hygiene, newer evidence allows that pacifiers may be acceptable in the first few years during breastfeeding.
BELIEF 8: Newborn emesis is an indication for a formula change
THE EVIDENCE: No literature supports the belief that it is appropriate to change an infant’s formula in response to emesis in the first 2 weeks of life (SOR: C).
The overwhelming majority of vomiting episodes in newborns have no accompanying medical problem.21 A 2002 study by Miyazawa et al that looked at more than 900 infants showed more than 47% of Japanese infants ≤1 month of age had daily occurrences of regurgitation or emesis without having an underlying medical disorder.22
Newborns vomit for any number of benign reasons, including swallowed maternal blood or overfeeding.21 Gastroesophageal reflux can be a manifestation of milk allergy. However, a newborn infant is too young to manifest an antibody response to the protein in the formula. Therefore, switching formula because of vomiting due to milk allergy is not prudent in the first 10 to 14 days of life, the length of time needed to mount an antibody response to an antigen23 and thus, the length of time needed to become allergic to an infant formula.
BELIEF 9: Umbilical cord care can prevent umbilical cord infections
THE EVIDENCE: There is no definitive evidence regarding the best method for preventing umbilical cord infections among babies living in developed countries (SOR: C).
In fact, there is no evidence that any topical preparation, be it a dye, an antiseptic, or an antibiotic, is any better at preventing umbilical cord infections than keeping the area clean and dry.24 Umbilical cord infections such as omphalitis or tetanus neonatorum are more common in developing countries than high-income countries.16 In developed countries, cord care with topical antimicrobial agents is frequently unnecessary.24
Infants who were delivered at home and those who room in with their mothers have no need of a topical antimicrobial therapy. If an infant is kept in a hospital nursery or intensive care unit, topical antimicrobial therapy to the cord may have some benefit in keeping down cord colonization with pathological bacteria such as methicillin-resistant Staphylococcus aureus.24
Umbilical cord infections sometimes occur even when the cord area is kept clean and dry,25 so healthcare providers must be attentive to signs of possible infection.16
BELIEF 10: It’s easy to spot when a newborn is jaundiced
THE EVIDENCE: Jaundice is actually difficult to detect in darkly pigmented babies,5 and in babies sent home within 24 hours of birth, because bilirubin levels reach maximum levels between the third and fourth days of life26 (SOR: C).
Years ago, when infants stayed longer in the nursery, doctors had the chance to see them when their bilirubin level was highest and when the babies were most jaundiced. The current emphasis on early discharge does not allow this practice.
The AAP recommends clinical assessment of a newborn’s state of jaundice and that a bilirubin level be obtained whenever a physician is in doubt about the degree of clinical jaundice. The AAP also recommends that physicians consider obtaining a routine screening bilirubin in all newborns at the time of hospital discharge even if, by clinical assessment, the child is not jaundiced.5 (The AAP stopped short, though, of saying that such a screening test is required.) The AAP made these recommendations because of an increasing concern that the incidence of kernicterus in America is rising.27
BELIEF 11: All infants who require phototherapy need IV fluids to prevent dehydration and enhance excretion of bilirubin
THE EVIDENCE: Unless the baby is clinically dehydrated, IV fluid therapy for infants under phototherapy is not needed28 (SOR: c).
Though IV fluid therapy is commonly used to increase the excretion of bilirubin and combat dehydration, the research tells us that IV fluids do not bring down bilirubin levels and that even with mild dehydration, the best fluid therapy is breast milk or formula because it inhibits the enterohepatic circulation of bilirubin.5 Intravenous fluid therapy should be reserved for jaundiced newborns with moderate to severe dehydration, or those with mild dehydration5 who are not able to take fluid by mouth.
BELIEF 12: Breast-milk jaundice is best treated by stopping breastfeeding for 24 to 48 hours
THE EVIDENCE: breastfeeding should not be discontinued as a way to treat breast-milk jaundice (SOR: C).
In fact, breastfeeding should not be discontinued for jaundice due to any cause, as demonstrated in the opening scenario, unless you believe a baby is at risk of requiring an exchange transfusion. The need for phototherapy alone is not a sufficient reason to discontinue breastfeeding.5
Breast-milk jaundice is a common problem facing parents and physicians, but it is not a disease and does not represent an abnormality in and of itself. Rather, this normal physiologic condition gains its importance only in that it must be distinguished from pathological causes of newborn jaundice.29 Breast-milk jaundice is believed to affect 1%21to 33%30 of breastfed infants.
One treatment measure—to stop breastfeeding—began, in part, as a cost-effective way to diagnose breast-milk jaundice.31 Rechecking the bilirubin 24 to 48 hours after breastfeeding is discontinued would reveal a significant drop in the bilirubin level, confirming the diagnosis of breast-milk jaundice32 and obviating the need for testing for more serious medical illness.
The consequence of this misguided treatment approach, ie, discontinuing breastfeeding, is that some mothers are more likely to stop breastfeeding altogether.33
Acknowledgments
The authors thank the following individuals for providing assistance with this manuscript: Mariateresa Esquivel, MD; Charlotte Hobbs, MD, PhD; Christopher Monnikendam, MD; and Clare Campbell Nesmith, MD.
Funding/Support
Dr Hall receives funding through DHS 1 P20 RR020146-01.
Correspondence
Bryan L. Burke, Jr., MD, FAAP, Associate Professor, Pediatrics and Neonatology, University of Arkansas for Medical Sciences, Arkansas Children’s Hospital, 4301 West Markham, Slot # 512-5B, Little Rock, AR 72205; [email protected]
1. Gartner LM, Greer FR. Section on Breastfeeding and Committee on Nutrition. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics 2003;111:908-910.
2. Greer FR. Issues in establishing vitamin D recommendations for infants and children. Am J Clin Nutr 2004;80(Suppl):1759S-1762S.
3. Kreiter SR, Schwartz RP, Kirkman HN, Jr, Charlton PA, Calikogl AS, Davenport ML. Nutritional rickets in African American breast-fed infants. J Pediatr 2000;137:153-157.
4. Pugliese MT, Blumberg DL, Hludzinski K, Kay S. Nutritional rickets in suburbia. J Am Coll Nutr 1998;17:637-641.
5. American Academy of Pediatrics Subcommittee on Hyperbilirubinemia, Maisels MJ, Baltz RD, Bhutani VK, Newman TB, Palmer H, et al. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004;114:297—316.
6. Lew M, Klonis E. Emergency management of eclampsia and severe pre-eclampsia. Emerg Med 2003;15:361-368.
7. Touyz RM. Role of magnesium in the pathogenesis of hypertension. Mol Aspects Med 2003;24:107-136.
8. Cruikshank DP, Varner MW, Pitkin RM. Breast milk magnesium and calcium concentrations following magnesium sulfate treatment. Am J Obstet Gynecol 1982;143:685-688.
9. Hale T. Medications and Mother’s Milk: A Manual of Lactational Pharmacology. 11th ed. Amarillo, TX: Pharmasoft Medical Publishing; 2004.
10. Lawrence RA, Lawrence RM. Breastfeeding: A Guide for the Medical Profession. 6th ed. Philadelphia: Mosby, Inc; 2005.
11. Hepatitis B. In: Pickering LK, Baker CJ, Overturf GD, Prober CG, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2003:118-122;318-334.
12. Centers for Disease Control and Prevention. Recommendations for Prevention and Control of Hepatitis C Virus (HCV) Infection and HCV-Related Chronic Disease. Atlanta: US Department of Health and Human Services; 1998.
13. American Academy of Pediatrics committee on Drugs. The transfer of drugs and other chemicals into human milk. Pediatrics 2001;108:776-789.
14. Anderson PO. Alcohol and breastfeeding. J Hum Lact 1995;11:321-324.
15. Howard CR, Howard FM, Lanphear B, deblieck EA, Eberly S, Lawrence RA. The effects of early pacifier use on breastfeeding duration. Pediatrics 1999;103:e33-e38.
16. World Health Organization, Family and Reproductive Health, Division, of Child Health and Development. Evidence for the ten steps to successful breastfeeding. 1998 [cited May 22, 2006]; Available at: www.who.int/child-adolescent-health/New_Publications/NUTRITION/WHO_CHD_98.9.pdf
17. American Academy of Pediatrics Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2005;115:496-506.
18. Li D-K, Willinger M, Petitti DB, Odouli R, Liu L, Hoffman HJ. Use of a dummy (pacifier) during sleep and risk of sudden infant death syndrome (SIDS): population base case-control study. BMJ 2005;332:18-22.
19. Ollila P, Niemela M, Uhari M, Larmas M. Prolonged pacifier-sucking and use of a nursing bottle at night: possible risk factors for dental caries in children. Acta Odontol Scan 1998;56:233-237.
20. Warren JJ, Bishara SE, Steinbock KL, Yonezu T, Nowak AJ. Effects of oral habits’ duration on dental characteristics in the primary dentition. J Am Dent Assoc 2001;132:1685-1693.
21. Berkowitz CD. Jaundice. In: Berkowitz CD, ed. Pediatrics: A Primary Care Approach. Philadelphia, PA: WB Saunders; 1996:357-363.
22. Miyazawa R, Tomomasa T, Kaneko H, Tachibana A, Ogawa T, Morikawa A. Prevalence of gastro-esophageal reflux-related symptoms in Japanese infants. Pediatr Int 2002;44:513-516.
23. Virella G. The humoral response and its induction by active immunization. In. Virella G, ed. Introduction to Medical Immunology. 3rd ed. New York, NY: Marcel Dekker; 1993:213-232.
24. Zupan J, Garner P, Omari AAA. Topical umbilical cord care at birth. Cochrane Database Syst Rev 2004;(3):CD001057.-
25. Janssen PA, Selwood BL, Dobson SR, Peacock D, Thiessen PN. To dye or not to dye: a randomized, clinical trial of a triple dye/alcohol regime versus dry cord care. Pediatrics 2003;111:15-20.
26. Bhutani VK, Johnson L, Sivieri EM. Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics 1999;103:6-14.
27. Bhutani VK, Johnson LH, Maisels MJ, Newman TB, Phibbs C, Stark AR, et al. Kernicterus: edpidemiological strategies for its prevention through system-based approaches. J Perinatol 2004;24:650-652.
28. Boo N-Y, Lee H-T. Randomized controlled trial of oral versus intravenous fluid supplementation on serum bilirubin level during phototherapy of term infants with severe hyperbilirubinaemia. J Paediatr Child Health 2002;38:151-155.
29. Rubatelli FF. Current drug treatment options in neonatal hyperbilirubinaemia and the prevention of kernicterus. Drugs 1998;56:23-30.
30. Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-399.
31. Amato M, Howald H, Von Muralt G. Interruption of breastfeeding versus phototherapy as treatment of hyperbilirubinemia in full-term infants. Helv Paediat Acta 1985;40:127-131.
32. Stoll BJ, Kliegman RM. Digestive system disorders. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics. 17th ed. Philadelphia, PA: WB Saunders; 2004:595-596.
33. DiGirolamo A, Thompson N, Martorell R, Fein S, Grummer-Strawm L. Intention or experience? Predictors of continued breastfeeding. Health Educ Behav 2005;32:208-226.
1. Gartner LM, Greer FR. Section on Breastfeeding and Committee on Nutrition. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics 2003;111:908-910.
2. Greer FR. Issues in establishing vitamin D recommendations for infants and children. Am J Clin Nutr 2004;80(Suppl):1759S-1762S.
3. Kreiter SR, Schwartz RP, Kirkman HN, Jr, Charlton PA, Calikogl AS, Davenport ML. Nutritional rickets in African American breast-fed infants. J Pediatr 2000;137:153-157.
4. Pugliese MT, Blumberg DL, Hludzinski K, Kay S. Nutritional rickets in suburbia. J Am Coll Nutr 1998;17:637-641.
5. American Academy of Pediatrics Subcommittee on Hyperbilirubinemia, Maisels MJ, Baltz RD, Bhutani VK, Newman TB, Palmer H, et al. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 2004;114:297—316.
6. Lew M, Klonis E. Emergency management of eclampsia and severe pre-eclampsia. Emerg Med 2003;15:361-368.
7. Touyz RM. Role of magnesium in the pathogenesis of hypertension. Mol Aspects Med 2003;24:107-136.
8. Cruikshank DP, Varner MW, Pitkin RM. Breast milk magnesium and calcium concentrations following magnesium sulfate treatment. Am J Obstet Gynecol 1982;143:685-688.
9. Hale T. Medications and Mother’s Milk: A Manual of Lactational Pharmacology. 11th ed. Amarillo, TX: Pharmasoft Medical Publishing; 2004.
10. Lawrence RA, Lawrence RM. Breastfeeding: A Guide for the Medical Profession. 6th ed. Philadelphia: Mosby, Inc; 2005.
11. Hepatitis B. In: Pickering LK, Baker CJ, Overturf GD, Prober CG, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2003:118-122;318-334.
12. Centers for Disease Control and Prevention. Recommendations for Prevention and Control of Hepatitis C Virus (HCV) Infection and HCV-Related Chronic Disease. Atlanta: US Department of Health and Human Services; 1998.
13. American Academy of Pediatrics committee on Drugs. The transfer of drugs and other chemicals into human milk. Pediatrics 2001;108:776-789.
14. Anderson PO. Alcohol and breastfeeding. J Hum Lact 1995;11:321-324.
15. Howard CR, Howard FM, Lanphear B, deblieck EA, Eberly S, Lawrence RA. The effects of early pacifier use on breastfeeding duration. Pediatrics 1999;103:e33-e38.
16. World Health Organization, Family and Reproductive Health, Division, of Child Health and Development. Evidence for the ten steps to successful breastfeeding. 1998 [cited May 22, 2006]; Available at: www.who.int/child-adolescent-health/New_Publications/NUTRITION/WHO_CHD_98.9.pdf
17. American Academy of Pediatrics Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2005;115:496-506.
18. Li D-K, Willinger M, Petitti DB, Odouli R, Liu L, Hoffman HJ. Use of a dummy (pacifier) during sleep and risk of sudden infant death syndrome (SIDS): population base case-control study. BMJ 2005;332:18-22.
19. Ollila P, Niemela M, Uhari M, Larmas M. Prolonged pacifier-sucking and use of a nursing bottle at night: possible risk factors for dental caries in children. Acta Odontol Scan 1998;56:233-237.
20. Warren JJ, Bishara SE, Steinbock KL, Yonezu T, Nowak AJ. Effects of oral habits’ duration on dental characteristics in the primary dentition. J Am Dent Assoc 2001;132:1685-1693.
21. Berkowitz CD. Jaundice. In: Berkowitz CD, ed. Pediatrics: A Primary Care Approach. Philadelphia, PA: WB Saunders; 1996:357-363.
22. Miyazawa R, Tomomasa T, Kaneko H, Tachibana A, Ogawa T, Morikawa A. Prevalence of gastro-esophageal reflux-related symptoms in Japanese infants. Pediatr Int 2002;44:513-516.
23. Virella G. The humoral response and its induction by active immunization. In. Virella G, ed. Introduction to Medical Immunology. 3rd ed. New York, NY: Marcel Dekker; 1993:213-232.
24. Zupan J, Garner P, Omari AAA. Topical umbilical cord care at birth. Cochrane Database Syst Rev 2004;(3):CD001057.-
25. Janssen PA, Selwood BL, Dobson SR, Peacock D, Thiessen PN. To dye or not to dye: a randomized, clinical trial of a triple dye/alcohol regime versus dry cord care. Pediatrics 2003;111:15-20.
26. Bhutani VK, Johnson L, Sivieri EM. Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics 1999;103:6-14.
27. Bhutani VK, Johnson LH, Maisels MJ, Newman TB, Phibbs C, Stark AR, et al. Kernicterus: edpidemiological strategies for its prevention through system-based approaches. J Perinatol 2004;24:650-652.
28. Boo N-Y, Lee H-T. Randomized controlled trial of oral versus intravenous fluid supplementation on serum bilirubin level during phototherapy of term infants with severe hyperbilirubinaemia. J Paediatr Child Health 2002;38:151-155.
29. Rubatelli FF. Current drug treatment options in neonatal hyperbilirubinaemia and the prevention of kernicterus. Drugs 1998;56:23-30.
30. Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-399.
31. Amato M, Howald H, Von Muralt G. Interruption of breastfeeding versus phototherapy as treatment of hyperbilirubinemia in full-term infants. Helv Paediat Acta 1985;40:127-131.
32. Stoll BJ, Kliegman RM. Digestive system disorders. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics. 17th ed. Philadelphia, PA: WB Saunders; 2004:595-596.
33. DiGirolamo A, Thompson N, Martorell R, Fein S, Grummer-Strawm L. Intention or experience? Predictors of continued breastfeeding. Health Educ Behav 2005;32:208-226.
Using prandial insulin to achieve glycemic control in type 2 diabetes
- A stepwise approach to antidiabetic therapy allows for the treatment to change in response to disease progression. This usually means beginning with oral agents and adding insulin as required (B).
- Treatment strategies must address both fasting and prandial hyperglycemia because prandial hyperglycemia has been shown to be an independent risk factor for cardiovascular events and mortality (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
More than 80% of patients with type 2 diabetes—including more than a third of patients with good metabolic control—have excessive postprandial hyperglycemia.1 That’s unwelcome news for the 20 million Americans with type 2 diabetes, especially when you consider that post-prandial hyperglycemia is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
To help our type 2 diabetes patients gain ideal control, we need to do at least 2 things better:
- Measure and act on glycosylated hemoglobin (A1c) levels.
- Take a stepped approach to glycemic control, making full use of prandial insulin.
A1clevels and the important role they play
Analysis of A1c is the “gold standard” for monitoring glycemic control in patients with diabetes because it provides an indication of mean plasma glucose levels during the preceding 120 days.6 The relative contribution of fasting plasma glucose (FPG) and postprandial plasma glucose (PPG) to A1c levels is a dynamic function of the extent of day-long hyperglycemia; FPG has a greater influence at higher A1c levels and PPG has a predominant role at lower A1c levels.7
The relationship between hyperglycemia, as measured by A1c, and increased morbidity and mortality (including cardiovascular events) was demonstrated several years ago in the United Kingdom Prospective Diabetes Study (UKPDS).8 Interestingly, several studies have also found that fasting glucose levels alone are not a reliable predictor for hyperglycemia-related morbidity or mortality, whereas postprandial hyperglycemia, as noted in the introduction, is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
Continued management of A1c through tight control of both FPG and PPG may therefore improve patient long-term health outcomes. A1c should be evaluated every 3 to 6 months, and appropriate changes to the patients’ treatment regimens should be made accordingly.
An algorithm for the stepwise approach
We typically use oral antidiabetic drugs typically are used as initial therapy for patients with newly diagnosed type 2 diabetes, especially those with initial A1c levels of 6.0% to 8.0%.9 Three recent publications10-12 provide an excellent analysis of the rationale for combination therapy to address multiple physiologic defects, as well as the relative efficacy of agents.
In 2006 the American Diabetes Association (ADA) and the European Association for the Study of Diabetes published a consensus statement that presented an algorithm for the initiation and adjustment of type 2 diabetes therapy (Figure).13 In this evidence-and experience-based treatment algorithm, the authors emphasize the achievement and maintenance of normal glycemic goals, initiating therapy with lifestyle intervention and metformin (Glucophage), not delaying therapy and transitioning to new regimens when glycemic targets are not achieved, and adding insulin therapy early to the regimens of patients who are not meeting glycemic targets.9,13
You may also consider newer therapeutic options not included in the ADA’s 2007 treatment guidelines.14 Incretin mimetics and dipeptidyl-peptidase IV (DPP-IV) inhibitors are 2 new classes of antidiabetic agents that are effective for patients with type 2 diabetes. (See “2 new classes of antidiabetic agents: Incretin mimetics and DPP-IV inhibitors” 15-22 ) Most patients with type 2 diabetes will, however, eventually require insulin therapy to maintain optimal glycemic control.23
Advancement to insulin therapy
Basal insulin replacement achieves glycemic control
The addition of once-daily basal insulin to oral antidiabetic drug regimens is a simple way to introduce insulin therapy and achieve glycemic control. (See “A guide to basal insulin dosing and titration”14,24-28 )
- In a randomized, parallel, multi-center study, treatment with once-daily insulin glargine (Lantus) or neutral protamine Hagedorn (NPH) insulin (Humulin, Novolin) added to preexisting oral antidiabetic drug regimens for 756 patients with inadequately controlled type 2 diabetes (A1c >7.5%) effectively achieved the goal A1c of ≤7.0% for most patients. More patients in the insulin glargine group were able to reach goal without experiencing any nocturnal hypoglycemia, compared with those in the NPH group (33.2% vs 26.7%, P<.05).24
- A similar study used a forced-titration algorithm to compare NPH insulin with insulin detemir (Levemir).26 This study demonstrated that a similar reduction in A1c could be achieved with insulin detemir and NPH insulin over 24 weeks (1.8% and 1.9%, P=ns), but weight gain and nocturnal hypoglycemia incidence were significantly lower with insulin detemir compared with NPH insulin (1.2 kg vs 2.8 kg, and 160 vs 349 events, respectively, P<.001 for both).
Prandial insulin is as effective as carbohydrate counting
If a patient doesn’t reach his or her A1c targets despite appropriate titration of the basal insulin dose, injections of a rapid-acting insulin analog at mealtime may be necessary. You can add rapid-acting insulin to the regimen, starting with 1 injection at the largest meal of the day and then adding an injection at additional meals as needed.
- A typical starting dose of rapid-acting prandial insulin (insulin aspart [NovoLog], insulin glulisine [Apidra], or insulin lispro [Humalog]) would be 5 to 10 U per meal.29
- The range of daily dose, when used at 3 meals per day, would be 0.2 to 0.5 U/kg per day (ie, 0.1 to 0.15 U/kg per meal).29 For a patient who weighs 100 kg, that would mean 20 U (6–7 units before each meal) per day.
FIGURE
An evidence-based algorithm for achieving normal glycemic goals in patients with type 2 diabetes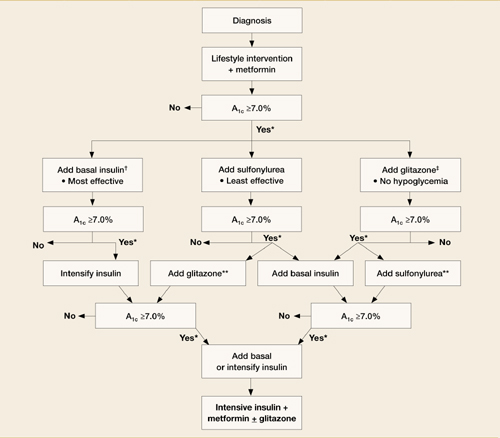
Note: Reinforce lifestyle intervention at every visit.
*Check A1c every 3 months until <7% and then at least every 6 months.
†At A1C >9%.
‡At A1C ≤8%.
**Although 3 oral agents can be used, initiation and intensification of insulin therapy is preferred based on effectiveness and expense.
Reprinted with permission from the American Diabetes Association.13
Incretin mimetics
Incretins, such as glucagonlike peptide-1 (GLP-1), enhance glucose-dependent insulin secretion by the pancreatic beta cells and exhibit other antihyperglycemic actions after release into circulation by the gut.
Exenatide (Byetta) is the first agent in this class to be approved by the Food and Drug Administration for use in type 2 diabetes. Exenatide may be used in combination with oral therapy (a sulfonylurea and/or metformin) by patients who have not achieved adequate glycemic control on oral therapy alone. Exenatide mimics the antihyperglycemic actions of GLP-1 but maintains a prolonged duration of action compared with endogenous GLP-1.15 This agent effectively addresses postprandial hyperglycemia by restoring a rapid postprandial insulin response; consider it when reduction of postprandial glycemic excursions is required.
When injected twice daily (before morning and evening meals), exenatide reduces hyperglycemia and promotes satiety, which in turn reduces caloric intake and body weight. In a recent study, exenatide achieved reductions in A1c (1.11%) similar to those observed with insulin glargine when it was added to sulfonylurea/metformin combination therapy for patients with type 2 diabetes.16 Exenatide reduced fasting plasma glucose (FPG) to a greater degree, whereas insulin glargine had a greater effect on FPG. This suggests that a basal insulin may be more appropriate when FPG levels are elevated (ie, patients with A1c levels >8.0%) and exenatide may be more useful for patients with A1c levels <8.0%, when elevated PPG levels are predominant.17 However, some patients with A1c levels >8.0% also may benefit from such intervention.
Liraglutide, another incretin mimetic, is in development for the treatment of type 2 diabetes. Liraglutide is a long-acting GLP-1 analog in phase 3 development for once-daily treatment of type 2 diabetes. The mechanism of action is similar to that of exenatide, but with a longer duration of action. Liraglutide may be suitable for once-daily administration. Initial data indicate that liraglutide improved glycemic control while providing modest weight loss for patients with type 2 diabetes.18 (Available online at: www.novonordisk.com/science/pipeline/rd_pipeline.asp. Accessed August 2, 2007.)
DPP-IV inhibitors
A new class of oral antidiabetic agents, DPP-IV inhibitors slow the degradation of incretin hormones, allowing these hormones to stimulate insulin secretion and decrease glucagon levels in the circulation in a glucose-dependent manner.19,20
Sitagliptin (Januvia) has been approved for use as monotherapy or in combination with metformin, pioglitazone, or rosiglitazone for the treatment of type 2 diabetes. Once-daily sitagliptin improves glycemic control, reducing A1c from 0.4% to 1.1% and decreasing fasting plasma glucose from 12 to 17 mg/dL and 2-hour postprandial glucose from 49 to 62 mg/dL in clinical trials. It is well-tolerated,21,22 but its long-term efficacy and safety are unknown.
Vildagliptin (Galvus), another DPP-IV inhibitor has completed phase 3 testing and is pending FDA approval at this time. (Available online at: www.diabeteshealth.com/read/2007/03/16/5039.html. Accessed August 2, 2007.)
Recent data indicate that a simple treatment algorithm based on preprandial glucose patterns can be as effective as carbohydrate counting for the dose titration of prandial insulin.30 In this 24-week study, insulin glulisine was added to basal-prandial insulin therapy, with insulin glargine as the basal insulin component. Glulisine was adjusted to target using either a simple algorithm of adding 1, 2, or 3 U based on premeal glucose patterns or standard carbohydrate counting. The carbohydrate counting–based dose adjustment and the algorithm-based titration treatment arms achieved similar A1c reductions. However, patients using the simple algorithm experienced significantly less symptomatic hypoglycemia (P=.02).
- Initiate basal insulin with a 10 U once-daily dose of insulin glargine, insulin detemir, or NPH insulin.24 (Note: NPH insulin and insulin detemir may require twice-daily dosing.)25
- Titrate weekly to a target fasting plasma glucose [FPG] of ≤100 mg/dL based on the average self-monitored FPG values from the preceding 2 days as follows:24
- If FPG is ≥180 mg/dL, increase insulin dosage by 8 U/d.
- If FPG is 140–180 mg/dL, increase insulin dosage by 6 U/d.
- If FPG is 120–140 mg/dL, increase insulin dosage by 4 U/d.
- If FPG is 100–120 mg/dL, increase insulin dosage by 2 U/d.
- If FPG is <72 mg/dL at any time during the week, do not increase insulin dosage.
- If FPG is <56 mg/dL, decrease insulin dosage by 2–4 U/d.
Keep in mind…
- A similar titration schedule to the one described here was effective in a study with insulin detemir and NPH insulin.26
- An alternative titration strategy to the one here would be to increase basal insulin dose by 2 U every 3 days to reach an FPG level of ≤100 mg/dL.27,28
- Less stringent A1c goals may be appropriate for patients with limited life expectancies, very young children, the elderly, and individuals with comorbid conditions.14
Rapid-acting analogs allow more flexible administration
Prior to the development of rapid-acting insulin analogs, regular human insulin (RHI) was the only available insulin suitable for prandial glycemic control. However, it had significant limitations, including the need for it to be injected 30 to 45 minutes before eating (and the poor compliance with this requirement), variability in peak levels (between patients and with the same patient), variability in absorption based on injection site, and frequent episodes of hypoglycemia.31,32
Newer rapid-acting insulin analogs such as insulin aspart, insulin glulisine, and insulin lispro demonstrate improved pharmacokinetic profiles with more rapid onset, faster time to peak activity, and shorter duration of action than RHI.32,33 These rapid-acting analogs allow administration right before or right after a meal, resulting in improved glycemic control without increased hypoglycemia or weight gain.34,35 Whereas the rapid onset of action of these analogs allows for administration 5 to 15 minutes before a meal, the patient can administer insulin glulisine within 20 minutes of the start of the meal.36 The addition of just 1 dose of prandial insulin to existing basal insulin plus oral antidiabetic drug therapy offers patients a substantial benefit.37
A new option: inhaled insulin
The US Food and Drug Administration recently approved an inhaled prandial insulin. Research has shown that it effectively addresses postprandial glucose excursions for patients with type 2 diabetes.38,39 A 12-week trial comparing A1c levels among patients switched to inhaled insulin (Exubera) before meals (n=76) or rosiglitazone (Avandia) 4 mg twice daily (n=69) found that inhaled insulin reduced A1c to a greater degree than rosiglitazone (–2.3% vs –1.4%); however, patients receiving inhaled insulin experienced a greater incidence of hypoglycemia (0.7 vs 0.05 episodes per subject-month).39
Inhaled insulin can be used as monotherapy or in conjunction with oral agents or a long-acting basal insulin. Inhaled insulin has a rapid onset of action (within 10–20 minutes, comparable with rapid-acting insulin analogs) and a duration of glucose-lowering activity of approximately 6 hours (comparable with RHI).40 This is useful for patients reluctant to begin insulin therapy because of injections; however, you will need to closely monitor hypoglycemia.
TABLE
How to use sensitivity factors to calculate 24-hour insulin need
| Characteristic | Dosage (U/kg) |
|---|---|
| Phenotype | |
| Normal weight | |
| Extremely physically active | 0.3 baseline |
| Moderately physically active | 0.4 baseline |
| Minimally active | 0.5 baseline |
| Obese | |
| Extremely physically active | 0.5 baseline |
| Moderately physically active | 0.6 baseline |
| Minimally active | 0.8 baseline |
| Renal failure | Subtract 0.2 |
| Coexisting illness raising risk of hypoglycemia | Subtract 0.2 |
| Eating habits (“big eater”) | Add 0.1 |
| New-onset type 1 diabetes, <30 years of age | 0.3 baseline |
| Reprinted with permission from Leahy, Insulin Therapy 2002.29 | |
Basal-prandial insulin in new type 2 diabetes
In certain cases, it may be more appropriate to initiate insulin therapy using a basal-prandial regimen that includes injections of prandial insulin with each meal of the day. Such cases include patients with newly diagnosed type 2 diabetes who have A1c levels >10.0%, or insulin-naive patients on oral antidiabetic drug regimens who have A1c levels >8.5%.25
You can calculate the starting total 24-hour insulin dosage for both the basal and prandial insulin components by multiplying body weight in kg by a factor based on the patient’s estimated insulin sensitivity ( TABLE ).29
Once you have this 24-hour insulin dose, you’ll then need to calculate the dose of basal insulin, which is 50% of the 24-hour total insulin dose, administered once daily. The remaining 50% of the total 24-hour dose provides prandial insulin coverage and is usually administered as follows:
- 30% to 40% at breakfast
- 30% at lunch
- 30% to 40% at dinner
Patients will need to adjust prandial insulin doses based on self-monitored blood glucose values.
Premixed insulin formulations
You should have your patients administer basal-prandial insulin as separate injections (eg, insulin glargine and insulin glulisine,30 or insulin detemir and insulin aspart25 ). The premixed (NPH based) formulations provide fixed doses of an intermediate-or long-acting insulin combined with a short-acting insulin. Although this method may be convenient to administer, it is more rigid and may not account for mealtimes and exercise. As a result, insulin levels will not match physiological insulin and thus, the risk for hypoglycemia increases. Another disadvantage is that adjustments to the dose based on self-monitored glucose levels are not possible with pre-mixed formulations.41
Separating the basal and prandial insulin components allows the insulin regimen to be adapted to an individual’s needs, thereby providing glycemic control with less propensity for hypoglycemia.
Acknowledgments
This article was supported by Sanofi-Aventis US. While the author is responsible for all content, he gratefully acknowledges the embryon scientific staff, who assisted in the preparation of a first draft of this article based on an author-approved outline, and also assisted in implementing author revisions.
Correspondence
George E. Dailey, MD, Senior Consultant, Division of Diabetes and Endocrinology, Head, Diabetes Research, Scripps Clinic, 10666 N. Torrey Pines Road, La Jolla, CA 92037; [email protected]
1. Bonora E, Corrao G, Bagnardi V, et al. Prevalence and correlates of post-prandial hyperglycaemia in a large sample of patients with type 2 diabetes mellitus. Diabetologia 2006;49:846-854.
2. Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care 1999;22:920-924.
3. The DECODE study group on behalf of the European Diabetes Epidemiology Group. Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. Lancet 1999;354:617-621.
4. Decode Study Group. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 2003;26:688-696.
5. Bonora E, Muggeo M. Postprandial blood glucose as a risk factor for cardiovascular disease in type II diabetes: the epidemiological evidence. Diabetologia 2001;44:2107-2114.
6. Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA1c: analysis of glucose profiles and HbA1c in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275-278.
7. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA1c. Diabetes Care 2003;26:881-885.
8. Stratton IM, Adler AI, Neil HAW, et al. on behalf of the UK Prospective Diabetes Study Group. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321:405-412.
9. ACE/AACE Diabetes Road Map Task Force. American Association of Clinical Endocrinologists Website. Road map for the prevention and treatment of type 2 diabetes. Available at: www.aace.com/meetings/consensus/odimplementation/roadmap.pdf. Accessed August 6, 2007.
10. Riddle MC, Rosenstock J. Oral monotherapy and combination therapy. In: Cefalu WT, Gerich JE, Le-Roith D, eds. The CADRE Handbook of Diabetes Management. New York, NY: Medical Information Press;2004:127-144.
11. Sheehan MT. Current therapeutic options in type 2 diabetes mellitus: a practical approach. Clin Med Res 2003;1:189-200.
12. Giorgino F, Laviola L, Leonardini A. Pathophysiology of type 2 diabetes: rationale for different oral antidiabetic treatment strategies. Diabetes Res Clin Pract 2005;68(suppl 1):S22-S29.
13. Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2006;29:1963-1972.
14. American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007;30(suppl 1):S4-S41.
15. Lam S, See S. Exenatide: a novel incretin mimetic agent for treating type 2 diabetes mellitus. Cardiol Rev 2006;14:205-211.
16. Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: A randomized trial. Ann Intern Med 2005;143:559-569.
17. Kennedy L. Exenatide versus glargine—complementary therapies rather than competing [electronic letter]. Ann Intern Med 2005; 143. Available at: www.annals.org/cgi/eletters/143/8/559#2404. Accessed August 3, 2007.
18. Feinglos MN, Saad MF, Pi-Sunyer FX, An B, Santiago O. on behalf of the Liraglutide Dose-Response Study Group. Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycaemic control and bodyweight in subjects with Type 2 diabetes. Diabetes Med 2005;22:1016-1023.
19. Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. for the Sitagliptin Study 021 Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006;29:2632-2637.
20. Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Sitagliptin Study 023 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006;49:2564-2571.
21. Miller SA, St Onge EL. Sitagliptin: a dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Ann Pharmacother 2006;40:1336-1343.
22. Herman GA, Bergman A, Liu F, et al. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol 2006;46:876-886.
23. Wright A, Burden ACF, Paisey RB, Cull CA, Holman RR. for the UK Prospective Diabetes Study Group. Sulfonylurea inadequacy: efficacy of addition of insulin over 6 years in patients with type 2 diabetes in the UK Prospective Diabetes Study (UKPDS 57). Diabetes Care 2002;25:330-336.
24. Riddle MC, Rosenstock J, Gerich J. on behalf of the Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080-3086.
25. Raslová K, Bogoev M, Raz I, Leth G, Gall MA, Hâncu N. Insulin detemir and insulin aspart: a promising basal-bolus regimen for type 2 diabetes. Diabetes Res Clin Pract 2004;66:193-201.
26. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. on behalf of the Levemir Treat-to-Target Study. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naïve people with type 2 diabetes. Diabetes Care 2006;29:1269-1274.
27. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R, Lantus Study Group. AT.LANTUS trial investigating treatment algorithms for insulin glargine (LANTUS): results of the Type 2 Study [abstract]. Diabetes 2004;53(suppl 2):A473.-Abstract 1980 PO.
28. Ryysy L, Yki-Jarvinen H, Hänninen J. Simplifying treat to target—the LANMET study. In: Program and abstracts of the 40th European Association for the Study of Diabetes Annual Meeting. 2004;No. 749. PS 064.
29. Leahy JL. Intensive insulin therapy in type 1 diabetes mellitus. In: Leahy, JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:87-112.
30. Bergenstal R, Johnson ML, Powers MA, Wynne AG, Vlajnic A, Hollander PA. Using a simple algorithm (ALG) to adjust mealtime glulisine (GLU) based on pre-prandial glucose patterns is a safe and effective alternative to carbohydrate counting (Carb Count). Diabetes 2006;55(suppl 1):A105.-
31. Overmann H, Heinemann L. Injection-meal interval: recommendations of diabetologists and how patients handle it. Diabetes Res Clin Pract 1999;43:137-142.
32. Wittlin SD, Woehrle HJ, Gerich JE. Insulin pharmacokinetics. In: Leahy JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:73-85.
33. Becker RHA, Frick AD, Burger F, Potgieter JH, Scholtz H. Insulin glulisine, a new rapid-acting insulin analogue, displays a rapid time-action profile in obese non-diabetic subjects. Exp Clin Endocrinol Diabetes 2005;13:435-443.
34. Chase HP, Lockspeiser T, Peery B, et al. The impact of the diabetes control and complications trial and humalog insulin on glycohemoglobin levels and severe hypoglycemia in type 1 diabetes. Diabetes Care 2001;24:430-434.
35. Dailey G, Rosenstock J, Moses RG, Ways K. Insulin glulisine provides improved glycemic control in patients with type 2 diabetes. Diabetes Care 2004;27:2363-2368.
36. Apidra [package insert] Kansas City, Mo: Aventis Pharmaceuticals Inc;2004.
37. Lankisch M, Stahr B, Alawi H, Ferlinz K, Scherbaum W. Basal insulin and oral antihyperglycemic therapy (BOT) plus a single dose of insulin glusine at breakfast or at the predominant meal lower HbA1c in patients with type 2 diabetes. Diabetes 2006;55(suppl 1).:Abstract 514-P.
38. Rosenstock J, Zinman B, Murphy LJ, et al. Inhaled insulin improves glycemic control when substituted for or added to oral combination therapy in type 2 diabetes: a randomized, controlled trial. Ann Intern Med 2005;143:549-558.
39. DeFronzo RA, Bergenstal RM, Cefalu WT, et al. Efficacy of inhaled insulin in patients with type 2 diabetes not controlled with diet and exercise: a 12-week, randomized, comparative trial. Diabetes Care 2005;28:1922-1928.
40. Exubera [package insert] New York, NY: Pfizer Inc;2006.
41. Hirsch IB. Insulin analogues. N Engl J Med 2005;352:174-183.
- A stepwise approach to antidiabetic therapy allows for the treatment to change in response to disease progression. This usually means beginning with oral agents and adding insulin as required (B).
- Treatment strategies must address both fasting and prandial hyperglycemia because prandial hyperglycemia has been shown to be an independent risk factor for cardiovascular events and mortality (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
More than 80% of patients with type 2 diabetes—including more than a third of patients with good metabolic control—have excessive postprandial hyperglycemia.1 That’s unwelcome news for the 20 million Americans with type 2 diabetes, especially when you consider that post-prandial hyperglycemia is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
To help our type 2 diabetes patients gain ideal control, we need to do at least 2 things better:
- Measure and act on glycosylated hemoglobin (A1c) levels.
- Take a stepped approach to glycemic control, making full use of prandial insulin.
A1clevels and the important role they play
Analysis of A1c is the “gold standard” for monitoring glycemic control in patients with diabetes because it provides an indication of mean plasma glucose levels during the preceding 120 days.6 The relative contribution of fasting plasma glucose (FPG) and postprandial plasma glucose (PPG) to A1c levels is a dynamic function of the extent of day-long hyperglycemia; FPG has a greater influence at higher A1c levels and PPG has a predominant role at lower A1c levels.7
The relationship between hyperglycemia, as measured by A1c, and increased morbidity and mortality (including cardiovascular events) was demonstrated several years ago in the United Kingdom Prospective Diabetes Study (UKPDS).8 Interestingly, several studies have also found that fasting glucose levels alone are not a reliable predictor for hyperglycemia-related morbidity or mortality, whereas postprandial hyperglycemia, as noted in the introduction, is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
Continued management of A1c through tight control of both FPG and PPG may therefore improve patient long-term health outcomes. A1c should be evaluated every 3 to 6 months, and appropriate changes to the patients’ treatment regimens should be made accordingly.
An algorithm for the stepwise approach
We typically use oral antidiabetic drugs typically are used as initial therapy for patients with newly diagnosed type 2 diabetes, especially those with initial A1c levels of 6.0% to 8.0%.9 Three recent publications10-12 provide an excellent analysis of the rationale for combination therapy to address multiple physiologic defects, as well as the relative efficacy of agents.
In 2006 the American Diabetes Association (ADA) and the European Association for the Study of Diabetes published a consensus statement that presented an algorithm for the initiation and adjustment of type 2 diabetes therapy (Figure).13 In this evidence-and experience-based treatment algorithm, the authors emphasize the achievement and maintenance of normal glycemic goals, initiating therapy with lifestyle intervention and metformin (Glucophage), not delaying therapy and transitioning to new regimens when glycemic targets are not achieved, and adding insulin therapy early to the regimens of patients who are not meeting glycemic targets.9,13
You may also consider newer therapeutic options not included in the ADA’s 2007 treatment guidelines.14 Incretin mimetics and dipeptidyl-peptidase IV (DPP-IV) inhibitors are 2 new classes of antidiabetic agents that are effective for patients with type 2 diabetes. (See “2 new classes of antidiabetic agents: Incretin mimetics and DPP-IV inhibitors” 15-22 ) Most patients with type 2 diabetes will, however, eventually require insulin therapy to maintain optimal glycemic control.23
Advancement to insulin therapy
Basal insulin replacement achieves glycemic control
The addition of once-daily basal insulin to oral antidiabetic drug regimens is a simple way to introduce insulin therapy and achieve glycemic control. (See “A guide to basal insulin dosing and titration”14,24-28 )
- In a randomized, parallel, multi-center study, treatment with once-daily insulin glargine (Lantus) or neutral protamine Hagedorn (NPH) insulin (Humulin, Novolin) added to preexisting oral antidiabetic drug regimens for 756 patients with inadequately controlled type 2 diabetes (A1c >7.5%) effectively achieved the goal A1c of ≤7.0% for most patients. More patients in the insulin glargine group were able to reach goal without experiencing any nocturnal hypoglycemia, compared with those in the NPH group (33.2% vs 26.7%, P<.05).24
- A similar study used a forced-titration algorithm to compare NPH insulin with insulin detemir (Levemir).26 This study demonstrated that a similar reduction in A1c could be achieved with insulin detemir and NPH insulin over 24 weeks (1.8% and 1.9%, P=ns), but weight gain and nocturnal hypoglycemia incidence were significantly lower with insulin detemir compared with NPH insulin (1.2 kg vs 2.8 kg, and 160 vs 349 events, respectively, P<.001 for both).
Prandial insulin is as effective as carbohydrate counting
If a patient doesn’t reach his or her A1c targets despite appropriate titration of the basal insulin dose, injections of a rapid-acting insulin analog at mealtime may be necessary. You can add rapid-acting insulin to the regimen, starting with 1 injection at the largest meal of the day and then adding an injection at additional meals as needed.
- A typical starting dose of rapid-acting prandial insulin (insulin aspart [NovoLog], insulin glulisine [Apidra], or insulin lispro [Humalog]) would be 5 to 10 U per meal.29
- The range of daily dose, when used at 3 meals per day, would be 0.2 to 0.5 U/kg per day (ie, 0.1 to 0.15 U/kg per meal).29 For a patient who weighs 100 kg, that would mean 20 U (6–7 units before each meal) per day.
FIGURE
An evidence-based algorithm for achieving normal glycemic goals in patients with type 2 diabetes
Note: Reinforce lifestyle intervention at every visit.
*Check A1c every 3 months until <7% and then at least every 6 months.
†At A1C >9%.
‡At A1C ≤8%.
**Although 3 oral agents can be used, initiation and intensification of insulin therapy is preferred based on effectiveness and expense.
Reprinted with permission from the American Diabetes Association.13
Incretin mimetics
Incretins, such as glucagonlike peptide-1 (GLP-1), enhance glucose-dependent insulin secretion by the pancreatic beta cells and exhibit other antihyperglycemic actions after release into circulation by the gut.
Exenatide (Byetta) is the first agent in this class to be approved by the Food and Drug Administration for use in type 2 diabetes. Exenatide may be used in combination with oral therapy (a sulfonylurea and/or metformin) by patients who have not achieved adequate glycemic control on oral therapy alone. Exenatide mimics the antihyperglycemic actions of GLP-1 but maintains a prolonged duration of action compared with endogenous GLP-1.15 This agent effectively addresses postprandial hyperglycemia by restoring a rapid postprandial insulin response; consider it when reduction of postprandial glycemic excursions is required.
When injected twice daily (before morning and evening meals), exenatide reduces hyperglycemia and promotes satiety, which in turn reduces caloric intake and body weight. In a recent study, exenatide achieved reductions in A1c (1.11%) similar to those observed with insulin glargine when it was added to sulfonylurea/metformin combination therapy for patients with type 2 diabetes.16 Exenatide reduced fasting plasma glucose (FPG) to a greater degree, whereas insulin glargine had a greater effect on FPG. This suggests that a basal insulin may be more appropriate when FPG levels are elevated (ie, patients with A1c levels >8.0%) and exenatide may be more useful for patients with A1c levels <8.0%, when elevated PPG levels are predominant.17 However, some patients with A1c levels >8.0% also may benefit from such intervention.
Liraglutide, another incretin mimetic, is in development for the treatment of type 2 diabetes. Liraglutide is a long-acting GLP-1 analog in phase 3 development for once-daily treatment of type 2 diabetes. The mechanism of action is similar to that of exenatide, but with a longer duration of action. Liraglutide may be suitable for once-daily administration. Initial data indicate that liraglutide improved glycemic control while providing modest weight loss for patients with type 2 diabetes.18 (Available online at: www.novonordisk.com/science/pipeline/rd_pipeline.asp. Accessed August 2, 2007.)
DPP-IV inhibitors
A new class of oral antidiabetic agents, DPP-IV inhibitors slow the degradation of incretin hormones, allowing these hormones to stimulate insulin secretion and decrease glucagon levels in the circulation in a glucose-dependent manner.19,20
Sitagliptin (Januvia) has been approved for use as monotherapy or in combination with metformin, pioglitazone, or rosiglitazone for the treatment of type 2 diabetes. Once-daily sitagliptin improves glycemic control, reducing A1c from 0.4% to 1.1% and decreasing fasting plasma glucose from 12 to 17 mg/dL and 2-hour postprandial glucose from 49 to 62 mg/dL in clinical trials. It is well-tolerated,21,22 but its long-term efficacy and safety are unknown.
Vildagliptin (Galvus), another DPP-IV inhibitor has completed phase 3 testing and is pending FDA approval at this time. (Available online at: www.diabeteshealth.com/read/2007/03/16/5039.html. Accessed August 2, 2007.)
Recent data indicate that a simple treatment algorithm based on preprandial glucose patterns can be as effective as carbohydrate counting for the dose titration of prandial insulin.30 In this 24-week study, insulin glulisine was added to basal-prandial insulin therapy, with insulin glargine as the basal insulin component. Glulisine was adjusted to target using either a simple algorithm of adding 1, 2, or 3 U based on premeal glucose patterns or standard carbohydrate counting. The carbohydrate counting–based dose adjustment and the algorithm-based titration treatment arms achieved similar A1c reductions. However, patients using the simple algorithm experienced significantly less symptomatic hypoglycemia (P=.02).
- Initiate basal insulin with a 10 U once-daily dose of insulin glargine, insulin detemir, or NPH insulin.24 (Note: NPH insulin and insulin detemir may require twice-daily dosing.)25
- Titrate weekly to a target fasting plasma glucose [FPG] of ≤100 mg/dL based on the average self-monitored FPG values from the preceding 2 days as follows:24
- If FPG is ≥180 mg/dL, increase insulin dosage by 8 U/d.
- If FPG is 140–180 mg/dL, increase insulin dosage by 6 U/d.
- If FPG is 120–140 mg/dL, increase insulin dosage by 4 U/d.
- If FPG is 100–120 mg/dL, increase insulin dosage by 2 U/d.
- If FPG is <72 mg/dL at any time during the week, do not increase insulin dosage.
- If FPG is <56 mg/dL, decrease insulin dosage by 2–4 U/d.
Keep in mind…
- A similar titration schedule to the one described here was effective in a study with insulin detemir and NPH insulin.26
- An alternative titration strategy to the one here would be to increase basal insulin dose by 2 U every 3 days to reach an FPG level of ≤100 mg/dL.27,28
- Less stringent A1c goals may be appropriate for patients with limited life expectancies, very young children, the elderly, and individuals with comorbid conditions.14
Rapid-acting analogs allow more flexible administration
Prior to the development of rapid-acting insulin analogs, regular human insulin (RHI) was the only available insulin suitable for prandial glycemic control. However, it had significant limitations, including the need for it to be injected 30 to 45 minutes before eating (and the poor compliance with this requirement), variability in peak levels (between patients and with the same patient), variability in absorption based on injection site, and frequent episodes of hypoglycemia.31,32
Newer rapid-acting insulin analogs such as insulin aspart, insulin glulisine, and insulin lispro demonstrate improved pharmacokinetic profiles with more rapid onset, faster time to peak activity, and shorter duration of action than RHI.32,33 These rapid-acting analogs allow administration right before or right after a meal, resulting in improved glycemic control without increased hypoglycemia or weight gain.34,35 Whereas the rapid onset of action of these analogs allows for administration 5 to 15 minutes before a meal, the patient can administer insulin glulisine within 20 minutes of the start of the meal.36 The addition of just 1 dose of prandial insulin to existing basal insulin plus oral antidiabetic drug therapy offers patients a substantial benefit.37
A new option: inhaled insulin
The US Food and Drug Administration recently approved an inhaled prandial insulin. Research has shown that it effectively addresses postprandial glucose excursions for patients with type 2 diabetes.38,39 A 12-week trial comparing A1c levels among patients switched to inhaled insulin (Exubera) before meals (n=76) or rosiglitazone (Avandia) 4 mg twice daily (n=69) found that inhaled insulin reduced A1c to a greater degree than rosiglitazone (–2.3% vs –1.4%); however, patients receiving inhaled insulin experienced a greater incidence of hypoglycemia (0.7 vs 0.05 episodes per subject-month).39
Inhaled insulin can be used as monotherapy or in conjunction with oral agents or a long-acting basal insulin. Inhaled insulin has a rapid onset of action (within 10–20 minutes, comparable with rapid-acting insulin analogs) and a duration of glucose-lowering activity of approximately 6 hours (comparable with RHI).40 This is useful for patients reluctant to begin insulin therapy because of injections; however, you will need to closely monitor hypoglycemia.
TABLE
How to use sensitivity factors to calculate 24-hour insulin need
| Characteristic | Dosage (U/kg) |
|---|---|
| Phenotype | |
| Normal weight | |
| Extremely physically active | 0.3 baseline |
| Moderately physically active | 0.4 baseline |
| Minimally active | 0.5 baseline |
| Obese | |
| Extremely physically active | 0.5 baseline |
| Moderately physically active | 0.6 baseline |
| Minimally active | 0.8 baseline |
| Renal failure | Subtract 0.2 |
| Coexisting illness raising risk of hypoglycemia | Subtract 0.2 |
| Eating habits (“big eater”) | Add 0.1 |
| New-onset type 1 diabetes, <30 years of age | 0.3 baseline |
| Reprinted with permission from Leahy, Insulin Therapy 2002.29 | |
Basal-prandial insulin in new type 2 diabetes
In certain cases, it may be more appropriate to initiate insulin therapy using a basal-prandial regimen that includes injections of prandial insulin with each meal of the day. Such cases include patients with newly diagnosed type 2 diabetes who have A1c levels >10.0%, or insulin-naive patients on oral antidiabetic drug regimens who have A1c levels >8.5%.25
You can calculate the starting total 24-hour insulin dosage for both the basal and prandial insulin components by multiplying body weight in kg by a factor based on the patient’s estimated insulin sensitivity ( TABLE ).29
Once you have this 24-hour insulin dose, you’ll then need to calculate the dose of basal insulin, which is 50% of the 24-hour total insulin dose, administered once daily. The remaining 50% of the total 24-hour dose provides prandial insulin coverage and is usually administered as follows:
- 30% to 40% at breakfast
- 30% at lunch
- 30% to 40% at dinner
Patients will need to adjust prandial insulin doses based on self-monitored blood glucose values.
Premixed insulin formulations
You should have your patients administer basal-prandial insulin as separate injections (eg, insulin glargine and insulin glulisine,30 or insulin detemir and insulin aspart25 ). The premixed (NPH based) formulations provide fixed doses of an intermediate-or long-acting insulin combined with a short-acting insulin. Although this method may be convenient to administer, it is more rigid and may not account for mealtimes and exercise. As a result, insulin levels will not match physiological insulin and thus, the risk for hypoglycemia increases. Another disadvantage is that adjustments to the dose based on self-monitored glucose levels are not possible with pre-mixed formulations.41
Separating the basal and prandial insulin components allows the insulin regimen to be adapted to an individual’s needs, thereby providing glycemic control with less propensity for hypoglycemia.
Acknowledgments
This article was supported by Sanofi-Aventis US. While the author is responsible for all content, he gratefully acknowledges the embryon scientific staff, who assisted in the preparation of a first draft of this article based on an author-approved outline, and also assisted in implementing author revisions.
Correspondence
George E. Dailey, MD, Senior Consultant, Division of Diabetes and Endocrinology, Head, Diabetes Research, Scripps Clinic, 10666 N. Torrey Pines Road, La Jolla, CA 92037; [email protected]
- A stepwise approach to antidiabetic therapy allows for the treatment to change in response to disease progression. This usually means beginning with oral agents and adding insulin as required (B).
- Treatment strategies must address both fasting and prandial hyperglycemia because prandial hyperglycemia has been shown to be an independent risk factor for cardiovascular events and mortality (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
More than 80% of patients with type 2 diabetes—including more than a third of patients with good metabolic control—have excessive postprandial hyperglycemia.1 That’s unwelcome news for the 20 million Americans with type 2 diabetes, especially when you consider that post-prandial hyperglycemia is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
To help our type 2 diabetes patients gain ideal control, we need to do at least 2 things better:
- Measure and act on glycosylated hemoglobin (A1c) levels.
- Take a stepped approach to glycemic control, making full use of prandial insulin.
A1clevels and the important role they play
Analysis of A1c is the “gold standard” for monitoring glycemic control in patients with diabetes because it provides an indication of mean plasma glucose levels during the preceding 120 days.6 The relative contribution of fasting plasma glucose (FPG) and postprandial plasma glucose (PPG) to A1c levels is a dynamic function of the extent of day-long hyperglycemia; FPG has a greater influence at higher A1c levels and PPG has a predominant role at lower A1c levels.7
The relationship between hyperglycemia, as measured by A1c, and increased morbidity and mortality (including cardiovascular events) was demonstrated several years ago in the United Kingdom Prospective Diabetes Study (UKPDS).8 Interestingly, several studies have also found that fasting glucose levels alone are not a reliable predictor for hyperglycemia-related morbidity or mortality, whereas postprandial hyperglycemia, as noted in the introduction, is a strong independent risk factor for all-cause mortality and cardiovascular events.2-5
Continued management of A1c through tight control of both FPG and PPG may therefore improve patient long-term health outcomes. A1c should be evaluated every 3 to 6 months, and appropriate changes to the patients’ treatment regimens should be made accordingly.
An algorithm for the stepwise approach
We typically use oral antidiabetic drugs typically are used as initial therapy for patients with newly diagnosed type 2 diabetes, especially those with initial A1c levels of 6.0% to 8.0%.9 Three recent publications10-12 provide an excellent analysis of the rationale for combination therapy to address multiple physiologic defects, as well as the relative efficacy of agents.
In 2006 the American Diabetes Association (ADA) and the European Association for the Study of Diabetes published a consensus statement that presented an algorithm for the initiation and adjustment of type 2 diabetes therapy (Figure).13 In this evidence-and experience-based treatment algorithm, the authors emphasize the achievement and maintenance of normal glycemic goals, initiating therapy with lifestyle intervention and metformin (Glucophage), not delaying therapy and transitioning to new regimens when glycemic targets are not achieved, and adding insulin therapy early to the regimens of patients who are not meeting glycemic targets.9,13
You may also consider newer therapeutic options not included in the ADA’s 2007 treatment guidelines.14 Incretin mimetics and dipeptidyl-peptidase IV (DPP-IV) inhibitors are 2 new classes of antidiabetic agents that are effective for patients with type 2 diabetes. (See “2 new classes of antidiabetic agents: Incretin mimetics and DPP-IV inhibitors” 15-22 ) Most patients with type 2 diabetes will, however, eventually require insulin therapy to maintain optimal glycemic control.23
Advancement to insulin therapy
Basal insulin replacement achieves glycemic control
The addition of once-daily basal insulin to oral antidiabetic drug regimens is a simple way to introduce insulin therapy and achieve glycemic control. (See “A guide to basal insulin dosing and titration”14,24-28 )
- In a randomized, parallel, multi-center study, treatment with once-daily insulin glargine (Lantus) or neutral protamine Hagedorn (NPH) insulin (Humulin, Novolin) added to preexisting oral antidiabetic drug regimens for 756 patients with inadequately controlled type 2 diabetes (A1c >7.5%) effectively achieved the goal A1c of ≤7.0% for most patients. More patients in the insulin glargine group were able to reach goal without experiencing any nocturnal hypoglycemia, compared with those in the NPH group (33.2% vs 26.7%, P<.05).24
- A similar study used a forced-titration algorithm to compare NPH insulin with insulin detemir (Levemir).26 This study demonstrated that a similar reduction in A1c could be achieved with insulin detemir and NPH insulin over 24 weeks (1.8% and 1.9%, P=ns), but weight gain and nocturnal hypoglycemia incidence were significantly lower with insulin detemir compared with NPH insulin (1.2 kg vs 2.8 kg, and 160 vs 349 events, respectively, P<.001 for both).
Prandial insulin is as effective as carbohydrate counting
If a patient doesn’t reach his or her A1c targets despite appropriate titration of the basal insulin dose, injections of a rapid-acting insulin analog at mealtime may be necessary. You can add rapid-acting insulin to the regimen, starting with 1 injection at the largest meal of the day and then adding an injection at additional meals as needed.
- A typical starting dose of rapid-acting prandial insulin (insulin aspart [NovoLog], insulin glulisine [Apidra], or insulin lispro [Humalog]) would be 5 to 10 U per meal.29
- The range of daily dose, when used at 3 meals per day, would be 0.2 to 0.5 U/kg per day (ie, 0.1 to 0.15 U/kg per meal).29 For a patient who weighs 100 kg, that would mean 20 U (6–7 units before each meal) per day.
FIGURE
An evidence-based algorithm for achieving normal glycemic goals in patients with type 2 diabetes
Note: Reinforce lifestyle intervention at every visit.
*Check A1c every 3 months until <7% and then at least every 6 months.
†At A1C >9%.
‡At A1C ≤8%.
**Although 3 oral agents can be used, initiation and intensification of insulin therapy is preferred based on effectiveness and expense.
Reprinted with permission from the American Diabetes Association.13
Incretin mimetics
Incretins, such as glucagonlike peptide-1 (GLP-1), enhance glucose-dependent insulin secretion by the pancreatic beta cells and exhibit other antihyperglycemic actions after release into circulation by the gut.
Exenatide (Byetta) is the first agent in this class to be approved by the Food and Drug Administration for use in type 2 diabetes. Exenatide may be used in combination with oral therapy (a sulfonylurea and/or metformin) by patients who have not achieved adequate glycemic control on oral therapy alone. Exenatide mimics the antihyperglycemic actions of GLP-1 but maintains a prolonged duration of action compared with endogenous GLP-1.15 This agent effectively addresses postprandial hyperglycemia by restoring a rapid postprandial insulin response; consider it when reduction of postprandial glycemic excursions is required.
When injected twice daily (before morning and evening meals), exenatide reduces hyperglycemia and promotes satiety, which in turn reduces caloric intake and body weight. In a recent study, exenatide achieved reductions in A1c (1.11%) similar to those observed with insulin glargine when it was added to sulfonylurea/metformin combination therapy for patients with type 2 diabetes.16 Exenatide reduced fasting plasma glucose (FPG) to a greater degree, whereas insulin glargine had a greater effect on FPG. This suggests that a basal insulin may be more appropriate when FPG levels are elevated (ie, patients with A1c levels >8.0%) and exenatide may be more useful for patients with A1c levels <8.0%, when elevated PPG levels are predominant.17 However, some patients with A1c levels >8.0% also may benefit from such intervention.
Liraglutide, another incretin mimetic, is in development for the treatment of type 2 diabetes. Liraglutide is a long-acting GLP-1 analog in phase 3 development for once-daily treatment of type 2 diabetes. The mechanism of action is similar to that of exenatide, but with a longer duration of action. Liraglutide may be suitable for once-daily administration. Initial data indicate that liraglutide improved glycemic control while providing modest weight loss for patients with type 2 diabetes.18 (Available online at: www.novonordisk.com/science/pipeline/rd_pipeline.asp. Accessed August 2, 2007.)
DPP-IV inhibitors
A new class of oral antidiabetic agents, DPP-IV inhibitors slow the degradation of incretin hormones, allowing these hormones to stimulate insulin secretion and decrease glucagon levels in the circulation in a glucose-dependent manner.19,20
Sitagliptin (Januvia) has been approved for use as monotherapy or in combination with metformin, pioglitazone, or rosiglitazone for the treatment of type 2 diabetes. Once-daily sitagliptin improves glycemic control, reducing A1c from 0.4% to 1.1% and decreasing fasting plasma glucose from 12 to 17 mg/dL and 2-hour postprandial glucose from 49 to 62 mg/dL in clinical trials. It is well-tolerated,21,22 but its long-term efficacy and safety are unknown.
Vildagliptin (Galvus), another DPP-IV inhibitor has completed phase 3 testing and is pending FDA approval at this time. (Available online at: www.diabeteshealth.com/read/2007/03/16/5039.html. Accessed August 2, 2007.)
Recent data indicate that a simple treatment algorithm based on preprandial glucose patterns can be as effective as carbohydrate counting for the dose titration of prandial insulin.30 In this 24-week study, insulin glulisine was added to basal-prandial insulin therapy, with insulin glargine as the basal insulin component. Glulisine was adjusted to target using either a simple algorithm of adding 1, 2, or 3 U based on premeal glucose patterns or standard carbohydrate counting. The carbohydrate counting–based dose adjustment and the algorithm-based titration treatment arms achieved similar A1c reductions. However, patients using the simple algorithm experienced significantly less symptomatic hypoglycemia (P=.02).
- Initiate basal insulin with a 10 U once-daily dose of insulin glargine, insulin detemir, or NPH insulin.24 (Note: NPH insulin and insulin detemir may require twice-daily dosing.)25
- Titrate weekly to a target fasting plasma glucose [FPG] of ≤100 mg/dL based on the average self-monitored FPG values from the preceding 2 days as follows:24
- If FPG is ≥180 mg/dL, increase insulin dosage by 8 U/d.
- If FPG is 140–180 mg/dL, increase insulin dosage by 6 U/d.
- If FPG is 120–140 mg/dL, increase insulin dosage by 4 U/d.
- If FPG is 100–120 mg/dL, increase insulin dosage by 2 U/d.
- If FPG is <72 mg/dL at any time during the week, do not increase insulin dosage.
- If FPG is <56 mg/dL, decrease insulin dosage by 2–4 U/d.
Keep in mind…
- A similar titration schedule to the one described here was effective in a study with insulin detemir and NPH insulin.26
- An alternative titration strategy to the one here would be to increase basal insulin dose by 2 U every 3 days to reach an FPG level of ≤100 mg/dL.27,28
- Less stringent A1c goals may be appropriate for patients with limited life expectancies, very young children, the elderly, and individuals with comorbid conditions.14
Rapid-acting analogs allow more flexible administration
Prior to the development of rapid-acting insulin analogs, regular human insulin (RHI) was the only available insulin suitable for prandial glycemic control. However, it had significant limitations, including the need for it to be injected 30 to 45 minutes before eating (and the poor compliance with this requirement), variability in peak levels (between patients and with the same patient), variability in absorption based on injection site, and frequent episodes of hypoglycemia.31,32
Newer rapid-acting insulin analogs such as insulin aspart, insulin glulisine, and insulin lispro demonstrate improved pharmacokinetic profiles with more rapid onset, faster time to peak activity, and shorter duration of action than RHI.32,33 These rapid-acting analogs allow administration right before or right after a meal, resulting in improved glycemic control without increased hypoglycemia or weight gain.34,35 Whereas the rapid onset of action of these analogs allows for administration 5 to 15 minutes before a meal, the patient can administer insulin glulisine within 20 minutes of the start of the meal.36 The addition of just 1 dose of prandial insulin to existing basal insulin plus oral antidiabetic drug therapy offers patients a substantial benefit.37
A new option: inhaled insulin
The US Food and Drug Administration recently approved an inhaled prandial insulin. Research has shown that it effectively addresses postprandial glucose excursions for patients with type 2 diabetes.38,39 A 12-week trial comparing A1c levels among patients switched to inhaled insulin (Exubera) before meals (n=76) or rosiglitazone (Avandia) 4 mg twice daily (n=69) found that inhaled insulin reduced A1c to a greater degree than rosiglitazone (–2.3% vs –1.4%); however, patients receiving inhaled insulin experienced a greater incidence of hypoglycemia (0.7 vs 0.05 episodes per subject-month).39
Inhaled insulin can be used as monotherapy or in conjunction with oral agents or a long-acting basal insulin. Inhaled insulin has a rapid onset of action (within 10–20 minutes, comparable with rapid-acting insulin analogs) and a duration of glucose-lowering activity of approximately 6 hours (comparable with RHI).40 This is useful for patients reluctant to begin insulin therapy because of injections; however, you will need to closely monitor hypoglycemia.
TABLE
How to use sensitivity factors to calculate 24-hour insulin need
| Characteristic | Dosage (U/kg) |
|---|---|
| Phenotype | |
| Normal weight | |
| Extremely physically active | 0.3 baseline |
| Moderately physically active | 0.4 baseline |
| Minimally active | 0.5 baseline |
| Obese | |
| Extremely physically active | 0.5 baseline |
| Moderately physically active | 0.6 baseline |
| Minimally active | 0.8 baseline |
| Renal failure | Subtract 0.2 |
| Coexisting illness raising risk of hypoglycemia | Subtract 0.2 |
| Eating habits (“big eater”) | Add 0.1 |
| New-onset type 1 diabetes, <30 years of age | 0.3 baseline |
| Reprinted with permission from Leahy, Insulin Therapy 2002.29 | |
Basal-prandial insulin in new type 2 diabetes
In certain cases, it may be more appropriate to initiate insulin therapy using a basal-prandial regimen that includes injections of prandial insulin with each meal of the day. Such cases include patients with newly diagnosed type 2 diabetes who have A1c levels >10.0%, or insulin-naive patients on oral antidiabetic drug regimens who have A1c levels >8.5%.25
You can calculate the starting total 24-hour insulin dosage for both the basal and prandial insulin components by multiplying body weight in kg by a factor based on the patient’s estimated insulin sensitivity ( TABLE ).29
Once you have this 24-hour insulin dose, you’ll then need to calculate the dose of basal insulin, which is 50% of the 24-hour total insulin dose, administered once daily. The remaining 50% of the total 24-hour dose provides prandial insulin coverage and is usually administered as follows:
- 30% to 40% at breakfast
- 30% at lunch
- 30% to 40% at dinner
Patients will need to adjust prandial insulin doses based on self-monitored blood glucose values.
Premixed insulin formulations
You should have your patients administer basal-prandial insulin as separate injections (eg, insulin glargine and insulin glulisine,30 or insulin detemir and insulin aspart25 ). The premixed (NPH based) formulations provide fixed doses of an intermediate-or long-acting insulin combined with a short-acting insulin. Although this method may be convenient to administer, it is more rigid and may not account for mealtimes and exercise. As a result, insulin levels will not match physiological insulin and thus, the risk for hypoglycemia increases. Another disadvantage is that adjustments to the dose based on self-monitored glucose levels are not possible with pre-mixed formulations.41
Separating the basal and prandial insulin components allows the insulin regimen to be adapted to an individual’s needs, thereby providing glycemic control with less propensity for hypoglycemia.
Acknowledgments
This article was supported by Sanofi-Aventis US. While the author is responsible for all content, he gratefully acknowledges the embryon scientific staff, who assisted in the preparation of a first draft of this article based on an author-approved outline, and also assisted in implementing author revisions.
Correspondence
George E. Dailey, MD, Senior Consultant, Division of Diabetes and Endocrinology, Head, Diabetes Research, Scripps Clinic, 10666 N. Torrey Pines Road, La Jolla, CA 92037; [email protected]
1. Bonora E, Corrao G, Bagnardi V, et al. Prevalence and correlates of post-prandial hyperglycaemia in a large sample of patients with type 2 diabetes mellitus. Diabetologia 2006;49:846-854.
2. Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care 1999;22:920-924.
3. The DECODE study group on behalf of the European Diabetes Epidemiology Group. Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. Lancet 1999;354:617-621.
4. Decode Study Group. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 2003;26:688-696.
5. Bonora E, Muggeo M. Postprandial blood glucose as a risk factor for cardiovascular disease in type II diabetes: the epidemiological evidence. Diabetologia 2001;44:2107-2114.
6. Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA1c: analysis of glucose profiles and HbA1c in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275-278.
7. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA1c. Diabetes Care 2003;26:881-885.
8. Stratton IM, Adler AI, Neil HAW, et al. on behalf of the UK Prospective Diabetes Study Group. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321:405-412.
9. ACE/AACE Diabetes Road Map Task Force. American Association of Clinical Endocrinologists Website. Road map for the prevention and treatment of type 2 diabetes. Available at: www.aace.com/meetings/consensus/odimplementation/roadmap.pdf. Accessed August 6, 2007.
10. Riddle MC, Rosenstock J. Oral monotherapy and combination therapy. In: Cefalu WT, Gerich JE, Le-Roith D, eds. The CADRE Handbook of Diabetes Management. New York, NY: Medical Information Press;2004:127-144.
11. Sheehan MT. Current therapeutic options in type 2 diabetes mellitus: a practical approach. Clin Med Res 2003;1:189-200.
12. Giorgino F, Laviola L, Leonardini A. Pathophysiology of type 2 diabetes: rationale for different oral antidiabetic treatment strategies. Diabetes Res Clin Pract 2005;68(suppl 1):S22-S29.
13. Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2006;29:1963-1972.
14. American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007;30(suppl 1):S4-S41.
15. Lam S, See S. Exenatide: a novel incretin mimetic agent for treating type 2 diabetes mellitus. Cardiol Rev 2006;14:205-211.
16. Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: A randomized trial. Ann Intern Med 2005;143:559-569.
17. Kennedy L. Exenatide versus glargine—complementary therapies rather than competing [electronic letter]. Ann Intern Med 2005; 143. Available at: www.annals.org/cgi/eletters/143/8/559#2404. Accessed August 3, 2007.
18. Feinglos MN, Saad MF, Pi-Sunyer FX, An B, Santiago O. on behalf of the Liraglutide Dose-Response Study Group. Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycaemic control and bodyweight in subjects with Type 2 diabetes. Diabetes Med 2005;22:1016-1023.
19. Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. for the Sitagliptin Study 021 Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006;29:2632-2637.
20. Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Sitagliptin Study 023 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006;49:2564-2571.
21. Miller SA, St Onge EL. Sitagliptin: a dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Ann Pharmacother 2006;40:1336-1343.
22. Herman GA, Bergman A, Liu F, et al. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol 2006;46:876-886.
23. Wright A, Burden ACF, Paisey RB, Cull CA, Holman RR. for the UK Prospective Diabetes Study Group. Sulfonylurea inadequacy: efficacy of addition of insulin over 6 years in patients with type 2 diabetes in the UK Prospective Diabetes Study (UKPDS 57). Diabetes Care 2002;25:330-336.
24. Riddle MC, Rosenstock J, Gerich J. on behalf of the Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080-3086.
25. Raslová K, Bogoev M, Raz I, Leth G, Gall MA, Hâncu N. Insulin detemir and insulin aspart: a promising basal-bolus regimen for type 2 diabetes. Diabetes Res Clin Pract 2004;66:193-201.
26. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. on behalf of the Levemir Treat-to-Target Study. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naïve people with type 2 diabetes. Diabetes Care 2006;29:1269-1274.
27. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R, Lantus Study Group. AT.LANTUS trial investigating treatment algorithms for insulin glargine (LANTUS): results of the Type 2 Study [abstract]. Diabetes 2004;53(suppl 2):A473.-Abstract 1980 PO.
28. Ryysy L, Yki-Jarvinen H, Hänninen J. Simplifying treat to target—the LANMET study. In: Program and abstracts of the 40th European Association for the Study of Diabetes Annual Meeting. 2004;No. 749. PS 064.
29. Leahy JL. Intensive insulin therapy in type 1 diabetes mellitus. In: Leahy, JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:87-112.
30. Bergenstal R, Johnson ML, Powers MA, Wynne AG, Vlajnic A, Hollander PA. Using a simple algorithm (ALG) to adjust mealtime glulisine (GLU) based on pre-prandial glucose patterns is a safe and effective alternative to carbohydrate counting (Carb Count). Diabetes 2006;55(suppl 1):A105.-
31. Overmann H, Heinemann L. Injection-meal interval: recommendations of diabetologists and how patients handle it. Diabetes Res Clin Pract 1999;43:137-142.
32. Wittlin SD, Woehrle HJ, Gerich JE. Insulin pharmacokinetics. In: Leahy JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:73-85.
33. Becker RHA, Frick AD, Burger F, Potgieter JH, Scholtz H. Insulin glulisine, a new rapid-acting insulin analogue, displays a rapid time-action profile in obese non-diabetic subjects. Exp Clin Endocrinol Diabetes 2005;13:435-443.
34. Chase HP, Lockspeiser T, Peery B, et al. The impact of the diabetes control and complications trial and humalog insulin on glycohemoglobin levels and severe hypoglycemia in type 1 diabetes. Diabetes Care 2001;24:430-434.
35. Dailey G, Rosenstock J, Moses RG, Ways K. Insulin glulisine provides improved glycemic control in patients with type 2 diabetes. Diabetes Care 2004;27:2363-2368.
36. Apidra [package insert] Kansas City, Mo: Aventis Pharmaceuticals Inc;2004.
37. Lankisch M, Stahr B, Alawi H, Ferlinz K, Scherbaum W. Basal insulin and oral antihyperglycemic therapy (BOT) plus a single dose of insulin glusine at breakfast or at the predominant meal lower HbA1c in patients with type 2 diabetes. Diabetes 2006;55(suppl 1).:Abstract 514-P.
38. Rosenstock J, Zinman B, Murphy LJ, et al. Inhaled insulin improves glycemic control when substituted for or added to oral combination therapy in type 2 diabetes: a randomized, controlled trial. Ann Intern Med 2005;143:549-558.
39. DeFronzo RA, Bergenstal RM, Cefalu WT, et al. Efficacy of inhaled insulin in patients with type 2 diabetes not controlled with diet and exercise: a 12-week, randomized, comparative trial. Diabetes Care 2005;28:1922-1928.
40. Exubera [package insert] New York, NY: Pfizer Inc;2006.
41. Hirsch IB. Insulin analogues. N Engl J Med 2005;352:174-183.
1. Bonora E, Corrao G, Bagnardi V, et al. Prevalence and correlates of post-prandial hyperglycaemia in a large sample of patients with type 2 diabetes mellitus. Diabetologia 2006;49:846-854.
2. Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care 1999;22:920-924.
3. The DECODE study group on behalf of the European Diabetes Epidemiology Group. Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. Lancet 1999;354:617-621.
4. Decode Study Group. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 2003;26:688-696.
5. Bonora E, Muggeo M. Postprandial blood glucose as a risk factor for cardiovascular disease in type II diabetes: the epidemiological evidence. Diabetologia 2001;44:2107-2114.
6. Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA1c: analysis of glucose profiles and HbA1c in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275-278.
7. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA1c. Diabetes Care 2003;26:881-885.
8. Stratton IM, Adler AI, Neil HAW, et al. on behalf of the UK Prospective Diabetes Study Group. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321:405-412.
9. ACE/AACE Diabetes Road Map Task Force. American Association of Clinical Endocrinologists Website. Road map for the prevention and treatment of type 2 diabetes. Available at: www.aace.com/meetings/consensus/odimplementation/roadmap.pdf. Accessed August 6, 2007.
10. Riddle MC, Rosenstock J. Oral monotherapy and combination therapy. In: Cefalu WT, Gerich JE, Le-Roith D, eds. The CADRE Handbook of Diabetes Management. New York, NY: Medical Information Press;2004:127-144.
11. Sheehan MT. Current therapeutic options in type 2 diabetes mellitus: a practical approach. Clin Med Res 2003;1:189-200.
12. Giorgino F, Laviola L, Leonardini A. Pathophysiology of type 2 diabetes: rationale for different oral antidiabetic treatment strategies. Diabetes Res Clin Pract 2005;68(suppl 1):S22-S29.
13. Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2006;29:1963-1972.
14. American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007;30(suppl 1):S4-S41.
15. Lam S, See S. Exenatide: a novel incretin mimetic agent for treating type 2 diabetes mellitus. Cardiol Rev 2006;14:205-211.
16. Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: A randomized trial. Ann Intern Med 2005;143:559-569.
17. Kennedy L. Exenatide versus glargine—complementary therapies rather than competing [electronic letter]. Ann Intern Med 2005; 143. Available at: www.annals.org/cgi/eletters/143/8/559#2404. Accessed August 3, 2007.
18. Feinglos MN, Saad MF, Pi-Sunyer FX, An B, Santiago O. on behalf of the Liraglutide Dose-Response Study Group. Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycaemic control and bodyweight in subjects with Type 2 diabetes. Diabetes Med 2005;22:1016-1023.
19. Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. for the Sitagliptin Study 021 Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006;29:2632-2637.
20. Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Sitagliptin Study 023 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006;49:2564-2571.
21. Miller SA, St Onge EL. Sitagliptin: a dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Ann Pharmacother 2006;40:1336-1343.
22. Herman GA, Bergman A, Liu F, et al. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol 2006;46:876-886.
23. Wright A, Burden ACF, Paisey RB, Cull CA, Holman RR. for the UK Prospective Diabetes Study Group. Sulfonylurea inadequacy: efficacy of addition of insulin over 6 years in patients with type 2 diabetes in the UK Prospective Diabetes Study (UKPDS 57). Diabetes Care 2002;25:330-336.
24. Riddle MC, Rosenstock J, Gerich J. on behalf of the Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003;26:3080-3086.
25. Raslová K, Bogoev M, Raz I, Leth G, Gall MA, Hâncu N. Insulin detemir and insulin aspart: a promising basal-bolus regimen for type 2 diabetes. Diabetes Res Clin Pract 2004;66:193-201.
26. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. on behalf of the Levemir Treat-to-Target Study. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naïve people with type 2 diabetes. Diabetes Care 2006;29:1269-1274.
27. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R, Lantus Study Group. AT.LANTUS trial investigating treatment algorithms for insulin glargine (LANTUS): results of the Type 2 Study [abstract]. Diabetes 2004;53(suppl 2):A473.-Abstract 1980 PO.
28. Ryysy L, Yki-Jarvinen H, Hänninen J. Simplifying treat to target—the LANMET study. In: Program and abstracts of the 40th European Association for the Study of Diabetes Annual Meeting. 2004;No. 749. PS 064.
29. Leahy JL. Intensive insulin therapy in type 1 diabetes mellitus. In: Leahy, JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:87-112.
30. Bergenstal R, Johnson ML, Powers MA, Wynne AG, Vlajnic A, Hollander PA. Using a simple algorithm (ALG) to adjust mealtime glulisine (GLU) based on pre-prandial glucose patterns is a safe and effective alternative to carbohydrate counting (Carb Count). Diabetes 2006;55(suppl 1):A105.-
31. Overmann H, Heinemann L. Injection-meal interval: recommendations of diabetologists and how patients handle it. Diabetes Res Clin Pract 1999;43:137-142.
32. Wittlin SD, Woehrle HJ, Gerich JE. Insulin pharmacokinetics. In: Leahy JL, Cefalu WT, eds. Insulin Therapy New York, NY: Marcel Dekker;2002:73-85.
33. Becker RHA, Frick AD, Burger F, Potgieter JH, Scholtz H. Insulin glulisine, a new rapid-acting insulin analogue, displays a rapid time-action profile in obese non-diabetic subjects. Exp Clin Endocrinol Diabetes 2005;13:435-443.
34. Chase HP, Lockspeiser T, Peery B, et al. The impact of the diabetes control and complications trial and humalog insulin on glycohemoglobin levels and severe hypoglycemia in type 1 diabetes. Diabetes Care 2001;24:430-434.
35. Dailey G, Rosenstock J, Moses RG, Ways K. Insulin glulisine provides improved glycemic control in patients with type 2 diabetes. Diabetes Care 2004;27:2363-2368.
36. Apidra [package insert] Kansas City, Mo: Aventis Pharmaceuticals Inc;2004.
37. Lankisch M, Stahr B, Alawi H, Ferlinz K, Scherbaum W. Basal insulin and oral antihyperglycemic therapy (BOT) plus a single dose of insulin glusine at breakfast or at the predominant meal lower HbA1c in patients with type 2 diabetes. Diabetes 2006;55(suppl 1).:Abstract 514-P.
38. Rosenstock J, Zinman B, Murphy LJ, et al. Inhaled insulin improves glycemic control when substituted for or added to oral combination therapy in type 2 diabetes: a randomized, controlled trial. Ann Intern Med 2005;143:549-558.
39. DeFronzo RA, Bergenstal RM, Cefalu WT, et al. Efficacy of inhaled insulin in patients with type 2 diabetes not controlled with diet and exercise: a 12-week, randomized, comparative trial. Diabetes Care 2005;28:1922-1928.
40. Exubera [package insert] New York, NY: Pfizer Inc;2006.
41. Hirsch IB. Insulin analogues. N Engl J Med 2005;352:174-183.
MANAGING CAP: An evidence-based algorithm
The 2007 guidelines from the Infectious Diseases Society of America (IDSA)/American Thoracic Society (ATS)1 are a blend of level-of-evidence strength and consensus opinion—a unified, evidence-based document. these new recommendations address prior discrepancies between the 2 specialties. We developed a CAP treatment algorithm based on the new advisory. (The following text includes levels of evidence.)
Site-of-care decisions
1. Let severity score be your guide
Based on evidence that physicians often hospitalize patients for CAP who could be managed as outpatients, the new guidelines recommend that we use an illness severity score (strong recommendation, level I evidence).
Previous guidelines advised only that we consider using a severity score.
- Use the validated Pneumonia Severity Index or the easier-to-use CURB-65. Patients with a CURB-65 score of 2 or more generally require hospitalization (moderate recommendation, level III evidence).
- Ability to reliably and safely take medications at home must also be taken into account (strong recommendation, level II evidence.)
2. Admit to ICU promptly if needed
The criteria for admission to the ICU is similar to the previous ATS guidelines, but the list of minor criteria is more extensive. This change reflects evidence demonstrating worse outcomes in patients whose transfer to the ICU was delayed. This new criteria has not been validated.
- Patients requiring vasopressors for blood pressure support or with hypoxemic respiratory failure should be admitted to the ICU—these are major criteria (strong recommendation, level II evidence).
- Patients with 3 or more minor criteria should also be directly admitted to the ICU (moderate recommendation, level II evidence).
3. Identify who needs more tests
In the wake of controversy about diagnostic testing recommendations, the new guidelines attempt to better identify patients who would benefit from further testing (TABLE).
- 12 indications. Prior ATS guidelines lacked specifics on required additional testing, but the new guidelines give 12 clinical indications for more extensive evaluation, and identify which tests are recommended for each indication (strong recommendation, level II evidence).
- Routine testing to identify the cause of CAP in outpatients is optional (moderate recommendation, level III evidence).
TABLE
Clinical indications for more extensive diagnostic testing
| CLINICAL INDICATION | RECOMMENDED DIAGNOSTIC TESTS | |||
|---|---|---|---|---|
| BLOOD CULTURE | SPUTUM CULTURE | LEGIONELLA URINARY ANTIGEN TEST | PNEUMOCOCCAL URINARY ANTIGEN TEST | |
| ICU admission* | ||||
| Failed outpatient therapy | ||||
| Cavitary infiltrates† | ||||
| Leukopenia | ||||
| Active alcohol abuse | ||||
| Chronic severe liver disease | ||||
| Severe lung disease | ||||
| Asplenia | ||||
| Anatomic or functional | ||||
| Recent travel‡ | ||||
| Within past 2 weeks | ||||
| Positive Legionella urinary antigen test | N/A | |||
| Positive Pneumococcal urinary antigen test | N/A | |||
| Pleural effusion** | ||||
| Additional tests: | ||||
| * Endotracheal aspirate if intubated, possibly bronchoscopy or nonbronchoscopic bronchoalveolar lavage. | ||||
| † Fungal and tuberculosis cultures. | ||||
| ‡ Region/type of travel related to Legionella, Coccidioides, Hantavirus, B pseudomallei, avian influenza, SARS. | ||||
| ** Thoracentesis and pleural fluid cultures. | ||||
| Adapted from Mandell et al.1 | ||||
Empiric antibiotics
The recommendations of IDSA/ATS are generally for a class of antibiotics rather than a specific drug, unless noted.
4. Assess DRSP risk factors
Growth of drug-resistant Streptococcus pneumoniae (DRSP) necessitated a more extensive list of risk factors for DRSP. Other recommendations did not change.
Outpatient treatment
- Adults who were previously healthy and who do not have risk factors for DRSP CAP should be treated with either a macrolide (azithromycin, clarithromycin, or erythromycin) (strong recommendation, level I evidence) or doxycycline (weak recommendation; level III evidence).
- In the presence of comorbidities that increase the risk for DRSP, these antibiotics are appropriate: a respiratory fluoroquinolone (moxifloxacin, gemifloxacin, or levofloxacin [750 mg/day dose]) (strong recommendation, level I evidence); or β-lactam plus a macrolide: (high-dose amoxicillin [eg, 1 g 3x daily] [strong recommendation, level I evidence] or amoxicillin-clavulanate [2 g twice daily] is preferred; but alternatives include ceftriaxone, cefpodoxime, and cefuroxime [500 mg twice daily]. Doxycycline [level II evidence] is an alternative to the macrolide.)
Inpatient non-ICU treatment
- β-lactam plus a macrolide (strong recommendation, level I evidence) (cefotaxime, ceftriaxone, and ampicillin; ertapenem for selected patients; doxycycline [level III evidence] is an alternative to the macrolide.).
- A respiratory fluoroquinolone (strong recommendation, level I evidence) is the treatment of choice for penicillin-allergic patients.
5. Assess MRSA risk factors
Although similar to the prior ATS guidelines, the new guidelines have added specific risk factors for community-acquired methicillin-resistant S aureus (MRSA). This change reflects the increasing prevalence of community-acquired MRSA as an etiology for CAP.
The new guidelines state that the overwhelming majority of CAP pathogens will be adequately treated with the recommended empiric regimens. Exceptions are infections due to community-acquired methicillin-resistant S aureus and Pseudomonas aeruginosa.
ICU treatment
- A β-lactam (cefotaxime, ceftriaxone, or ampicillin-sulbactam) plus either azithromycin (strong recommendation, level II evidence) or a respiratory fluoroquinolone (strong recommendation, level I evidence).
- For penicillin-allergic patients, a respiratory fluoroquinolone and aztreonam are recommended.
- For Pseudomonas infection (see FIGURE for risk factors), use an anti-pneumococcal, antipseudomonal β-lactam (piperacillin-tazobactam, cefepime, imipenem, or meropenem) plus either ciprofloxacin or levofloxacin (750-mg dose) or the above β-lactam plus an aminoglycoside and azithromycin or the above β-lactam plus aminoglycoside and a respiratory fluoroquinolone (moderate recommendation, level II evidence). Fifteen days of therapy may be more effective in Pseudomonas CAP based on nosocomial infection data.
- For community-acquired methicillin-resistant S aureus infection (see FIGURE for risk factors), add vancomycin or linezolid (moderate recommendation, level III evidence).
Diagnostic testing is of high yield for patients with severe CAP requiring ICU admission, allowing for early de-escalation of empirical treatment if results are negative.
FIGURE
Treatment of community-acquired pneumonia*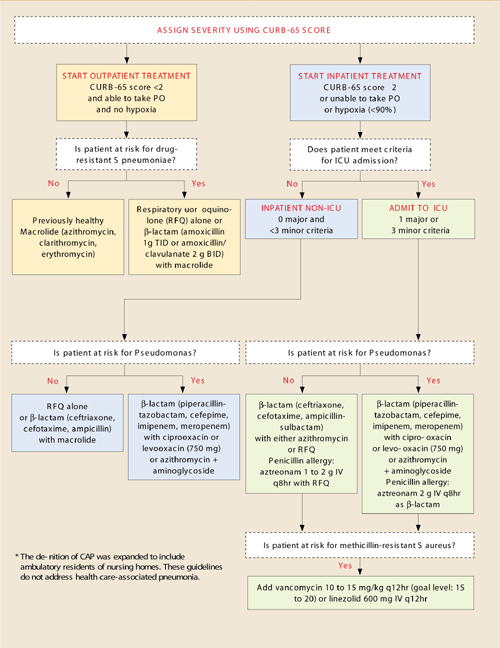
Adapted from: Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
CURB-65 score for assessing severity of illness
Confusion
Uremia BUN >20
Respiration ≥30
BP <90/≤60 mm Hg
≥65 years of age
| TOTAL POINTS | MORTALITY RATE |
|---|---|
| 0 | 0.7% |
| 1 | 2.1% |
| 2 | 9.2% |
| 3 | 14.5% |
| 4 | 40.0% |
| 5 | 57.0% |
Criteria for ICU admission
Major
Invasive mechanical ventilation
Septic shock with need for vasopressors
Minor
- Respiration ≥30
- PaO2/FiO2 ≤250
- Multilobar infiltrates
- Disorientation
- BUN ≥20
- Hypotension requiring aggressive fluids
- Temperature <36°C
- Platelets <100,000
- WBC<4000
Risk factors
Drug-resistant S pneumoniae
- Any antibiotics within 3 months
- Comorbidities: Cardiac disease, diabetes mellitus, alcoholism, pulmonary disease, renal disease, liver disease, asplenia, malignancy, immunosuppressed.
- age >65 years
Pseudomonal infection
- Structural lung disease (bronchiectasis)
- Severe COPD with frequent admissions, recent antibiotic and steroid use
Methicillin-resistant S aureus
Pulmonary abscess
End-stage renal disease
IV drug abuse
Recent influenza
Recent fluroquinolone use
Duration of antibiotic treatment
- At least 5 days
- After 5 days, antibiotics may be discontinued when patient is afebrile 72 hours and has no more than 1 criteria for instability: temperature ≥37.8°C, heart rate ≥100, respiration ≥24, systolic BP ≤90 mm Hg, O2 saturation ≤90%, inability to maintain oral intake, altered mental status
- 15 days for CAP due to Pseudomonas
Criteria for discharge
Temperature ≤37.8°C, heart rate ≤100, respiration ≤24, systolic BP ≥ 90 mm Hg,O2 saturation ≥90%, ability to maintain oral intake, normal mental status
Pathogen-directed therapy
6. Identify the pathogen
New guidelines recommend that, once the pathogen is identified by reliable microbiological methods, therapy should be directed towards that specific pathogen to prevent increased resistance in the community (moderate recommendation, level III evidence).
Influenza. Treatment within 48 hours of symptom onset with oseltamivir or zanamivir is recommended for influenza A (strong recommendation, level I evidence).
While these antimicrobials should not be used in uncomplicated influenza with symptoms for >48 hours (level I evidence), they may be used in hospitalized patients or influenza pneumonia to reduce viral shedding (moderate recommendation, level III evidence).
LEVEL I HIGH
Well-conducted, randomized controlled trials
LEVEL II MODERATE
Well-designed, controlled trials without randomization (including cohort, patient series, and case-controlled studies); large case series in which systematic analysis of disease patterns and/or microbial etiology was conducted; and reports of data on new therapies that were not collected in a randomized fashion.
LEVEL III LOW
Case studies and expert opinion. In some instances, therapy recommendations may come from antibiotic susceptibility data without clinical observations.
Adapted from: Mandell et al.1
Time to first dose
7. Start treatment in the ED
Rather than designating a time window for starting treatment, the IDSA/ATS committee recommended that patients receive the first antibiotic dose in the Emergency Department (moderate recommendation, level III evidence).
This newly added statement contrasts with some current quality measures that grade hospitals according to whether antibiotics are started within a specific time frame.
Duration of antibiotics
8. Base duration on specific criteria
Reflecting evidence that shorter courses appear to be as effective as longer courses, the newer guidelines recommend discontinuation when the patient meets specific clinical criteria. Before discontinuing antibiotics, all patients with CAP should:
- Be treated for at least 5 days (level I evidence),
- Be afebrile for 48 to 72 hours, and
- Have no more than 1 of these criteria for clinical instability (moderate recommendation, level II evidence): temperature ≥37.8°C; heart rate ≥100 beats/min; respiratory rate ≥24 breaths/min; systolic blood pressure ≤90 mm Hg; arterial oxygen saturation ≤90% or pO2 ≤60 mm Hg on room air; inability to maintain oral intake; altered mental status.
Switching from IV to oral
The guidelines, similar to the prior guidelines, recommend switching to oral therapy for hemodynamically stable patients who are clinically improving, able to ingest medications, and have a normally functioning gastrointestinal tract (strong recommendation, level II evidence).
Hospital discharge
The guidelines recommend that patients be discharged as soon as they are clinically stable and have a safe environment for continued care. Patients receiving oral therapy do not require inpatient observation (moderate recommendation, level II evidence). This is unchanged from prior recommendations.
1. Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell D, Dean NC, et al. IDSA/ATS consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007;44:(Suppl 2) S27-S72.
The 2007 guidelines from the Infectious Diseases Society of America (IDSA)/American Thoracic Society (ATS)1 are a blend of level-of-evidence strength and consensus opinion—a unified, evidence-based document. these new recommendations address prior discrepancies between the 2 specialties. We developed a CAP treatment algorithm based on the new advisory. (The following text includes levels of evidence.)
Site-of-care decisions
1. Let severity score be your guide
Based on evidence that physicians often hospitalize patients for CAP who could be managed as outpatients, the new guidelines recommend that we use an illness severity score (strong recommendation, level I evidence).
Previous guidelines advised only that we consider using a severity score.
- Use the validated Pneumonia Severity Index or the easier-to-use CURB-65. Patients with a CURB-65 score of 2 or more generally require hospitalization (moderate recommendation, level III evidence).
- Ability to reliably and safely take medications at home must also be taken into account (strong recommendation, level II evidence.)
2. Admit to ICU promptly if needed
The criteria for admission to the ICU is similar to the previous ATS guidelines, but the list of minor criteria is more extensive. This change reflects evidence demonstrating worse outcomes in patients whose transfer to the ICU was delayed. This new criteria has not been validated.
- Patients requiring vasopressors for blood pressure support or with hypoxemic respiratory failure should be admitted to the ICU—these are major criteria (strong recommendation, level II evidence).
- Patients with 3 or more minor criteria should also be directly admitted to the ICU (moderate recommendation, level II evidence).
3. Identify who needs more tests
In the wake of controversy about diagnostic testing recommendations, the new guidelines attempt to better identify patients who would benefit from further testing (TABLE).
- 12 indications. Prior ATS guidelines lacked specifics on required additional testing, but the new guidelines give 12 clinical indications for more extensive evaluation, and identify which tests are recommended for each indication (strong recommendation, level II evidence).
- Routine testing to identify the cause of CAP in outpatients is optional (moderate recommendation, level III evidence).
TABLE
Clinical indications for more extensive diagnostic testing
| CLINICAL INDICATION | RECOMMENDED DIAGNOSTIC TESTS | |||
|---|---|---|---|---|
| BLOOD CULTURE | SPUTUM CULTURE | LEGIONELLA URINARY ANTIGEN TEST | PNEUMOCOCCAL URINARY ANTIGEN TEST | |
| ICU admission* | ||||
| Failed outpatient therapy | ||||
| Cavitary infiltrates† | ||||
| Leukopenia | ||||
| Active alcohol abuse | ||||
| Chronic severe liver disease | ||||
| Severe lung disease | ||||
| Asplenia | ||||
| Anatomic or functional | ||||
| Recent travel‡ | ||||
| Within past 2 weeks | ||||
| Positive Legionella urinary antigen test | N/A | |||
| Positive Pneumococcal urinary antigen test | N/A | |||
| Pleural effusion** | ||||
| Additional tests: | ||||
| * Endotracheal aspirate if intubated, possibly bronchoscopy or nonbronchoscopic bronchoalveolar lavage. | ||||
| † Fungal and tuberculosis cultures. | ||||
| ‡ Region/type of travel related to Legionella, Coccidioides, Hantavirus, B pseudomallei, avian influenza, SARS. | ||||
| ** Thoracentesis and pleural fluid cultures. | ||||
| Adapted from Mandell et al.1 | ||||
Empiric antibiotics
The recommendations of IDSA/ATS are generally for a class of antibiotics rather than a specific drug, unless noted.
4. Assess DRSP risk factors
Growth of drug-resistant Streptococcus pneumoniae (DRSP) necessitated a more extensive list of risk factors for DRSP. Other recommendations did not change.
Outpatient treatment
- Adults who were previously healthy and who do not have risk factors for DRSP CAP should be treated with either a macrolide (azithromycin, clarithromycin, or erythromycin) (strong recommendation, level I evidence) or doxycycline (weak recommendation; level III evidence).
- In the presence of comorbidities that increase the risk for DRSP, these antibiotics are appropriate: a respiratory fluoroquinolone (moxifloxacin, gemifloxacin, or levofloxacin [750 mg/day dose]) (strong recommendation, level I evidence); or β-lactam plus a macrolide: (high-dose amoxicillin [eg, 1 g 3x daily] [strong recommendation, level I evidence] or amoxicillin-clavulanate [2 g twice daily] is preferred; but alternatives include ceftriaxone, cefpodoxime, and cefuroxime [500 mg twice daily]. Doxycycline [level II evidence] is an alternative to the macrolide.)
Inpatient non-ICU treatment
- β-lactam plus a macrolide (strong recommendation, level I evidence) (cefotaxime, ceftriaxone, and ampicillin; ertapenem for selected patients; doxycycline [level III evidence] is an alternative to the macrolide.).
- A respiratory fluoroquinolone (strong recommendation, level I evidence) is the treatment of choice for penicillin-allergic patients.
5. Assess MRSA risk factors
Although similar to the prior ATS guidelines, the new guidelines have added specific risk factors for community-acquired methicillin-resistant S aureus (MRSA). This change reflects the increasing prevalence of community-acquired MRSA as an etiology for CAP.
The new guidelines state that the overwhelming majority of CAP pathogens will be adequately treated with the recommended empiric regimens. Exceptions are infections due to community-acquired methicillin-resistant S aureus and Pseudomonas aeruginosa.
ICU treatment
- A β-lactam (cefotaxime, ceftriaxone, or ampicillin-sulbactam) plus either azithromycin (strong recommendation, level II evidence) or a respiratory fluoroquinolone (strong recommendation, level I evidence).
- For penicillin-allergic patients, a respiratory fluoroquinolone and aztreonam are recommended.
- For Pseudomonas infection (see FIGURE for risk factors), use an anti-pneumococcal, antipseudomonal β-lactam (piperacillin-tazobactam, cefepime, imipenem, or meropenem) plus either ciprofloxacin or levofloxacin (750-mg dose) or the above β-lactam plus an aminoglycoside and azithromycin or the above β-lactam plus aminoglycoside and a respiratory fluoroquinolone (moderate recommendation, level II evidence). Fifteen days of therapy may be more effective in Pseudomonas CAP based on nosocomial infection data.
- For community-acquired methicillin-resistant S aureus infection (see FIGURE for risk factors), add vancomycin or linezolid (moderate recommendation, level III evidence).
Diagnostic testing is of high yield for patients with severe CAP requiring ICU admission, allowing for early de-escalation of empirical treatment if results are negative.
FIGURE
Treatment of community-acquired pneumonia*
Adapted from: Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
CURB-65 score for assessing severity of illness
Confusion
Uremia BUN >20
Respiration ≥30
BP <90/≤60 mm Hg
≥65 years of age
| TOTAL POINTS | MORTALITY RATE |
|---|---|
| 0 | 0.7% |
| 1 | 2.1% |
| 2 | 9.2% |
| 3 | 14.5% |
| 4 | 40.0% |
| 5 | 57.0% |
Criteria for ICU admission
Major
Invasive mechanical ventilation
Septic shock with need for vasopressors
Minor
- Respiration ≥30
- PaO2/FiO2 ≤250
- Multilobar infiltrates
- Disorientation
- BUN ≥20
- Hypotension requiring aggressive fluids
- Temperature <36°C
- Platelets <100,000
- WBC<4000
Risk factors
Drug-resistant S pneumoniae
- Any antibiotics within 3 months
- Comorbidities: Cardiac disease, diabetes mellitus, alcoholism, pulmonary disease, renal disease, liver disease, asplenia, malignancy, immunosuppressed.
- age >65 years
Pseudomonal infection
- Structural lung disease (bronchiectasis)
- Severe COPD with frequent admissions, recent antibiotic and steroid use
Methicillin-resistant S aureus
Pulmonary abscess
End-stage renal disease
IV drug abuse
Recent influenza
Recent fluroquinolone use
Duration of antibiotic treatment
- At least 5 days
- After 5 days, antibiotics may be discontinued when patient is afebrile 72 hours and has no more than 1 criteria for instability: temperature ≥37.8°C, heart rate ≥100, respiration ≥24, systolic BP ≤90 mm Hg, O2 saturation ≤90%, inability to maintain oral intake, altered mental status
- 15 days for CAP due to Pseudomonas
Criteria for discharge
Temperature ≤37.8°C, heart rate ≤100, respiration ≤24, systolic BP ≥ 90 mm Hg,O2 saturation ≥90%, ability to maintain oral intake, normal mental status
Pathogen-directed therapy
6. Identify the pathogen
New guidelines recommend that, once the pathogen is identified by reliable microbiological methods, therapy should be directed towards that specific pathogen to prevent increased resistance in the community (moderate recommendation, level III evidence).
Influenza. Treatment within 48 hours of symptom onset with oseltamivir or zanamivir is recommended for influenza A (strong recommendation, level I evidence).
While these antimicrobials should not be used in uncomplicated influenza with symptoms for >48 hours (level I evidence), they may be used in hospitalized patients or influenza pneumonia to reduce viral shedding (moderate recommendation, level III evidence).
LEVEL I HIGH
Well-conducted, randomized controlled trials
LEVEL II MODERATE
Well-designed, controlled trials without randomization (including cohort, patient series, and case-controlled studies); large case series in which systematic analysis of disease patterns and/or microbial etiology was conducted; and reports of data on new therapies that were not collected in a randomized fashion.
LEVEL III LOW
Case studies and expert opinion. In some instances, therapy recommendations may come from antibiotic susceptibility data without clinical observations.
Adapted from: Mandell et al.1
Time to first dose
7. Start treatment in the ED
Rather than designating a time window for starting treatment, the IDSA/ATS committee recommended that patients receive the first antibiotic dose in the Emergency Department (moderate recommendation, level III evidence).
This newly added statement contrasts with some current quality measures that grade hospitals according to whether antibiotics are started within a specific time frame.
Duration of antibiotics
8. Base duration on specific criteria
Reflecting evidence that shorter courses appear to be as effective as longer courses, the newer guidelines recommend discontinuation when the patient meets specific clinical criteria. Before discontinuing antibiotics, all patients with CAP should:
- Be treated for at least 5 days (level I evidence),
- Be afebrile for 48 to 72 hours, and
- Have no more than 1 of these criteria for clinical instability (moderate recommendation, level II evidence): temperature ≥37.8°C; heart rate ≥100 beats/min; respiratory rate ≥24 breaths/min; systolic blood pressure ≤90 mm Hg; arterial oxygen saturation ≤90% or pO2 ≤60 mm Hg on room air; inability to maintain oral intake; altered mental status.
Switching from IV to oral
The guidelines, similar to the prior guidelines, recommend switching to oral therapy for hemodynamically stable patients who are clinically improving, able to ingest medications, and have a normally functioning gastrointestinal tract (strong recommendation, level II evidence).
Hospital discharge
The guidelines recommend that patients be discharged as soon as they are clinically stable and have a safe environment for continued care. Patients receiving oral therapy do not require inpatient observation (moderate recommendation, level II evidence). This is unchanged from prior recommendations.
The 2007 guidelines from the Infectious Diseases Society of America (IDSA)/American Thoracic Society (ATS)1 are a blend of level-of-evidence strength and consensus opinion—a unified, evidence-based document. these new recommendations address prior discrepancies between the 2 specialties. We developed a CAP treatment algorithm based on the new advisory. (The following text includes levels of evidence.)
Site-of-care decisions
1. Let severity score be your guide
Based on evidence that physicians often hospitalize patients for CAP who could be managed as outpatients, the new guidelines recommend that we use an illness severity score (strong recommendation, level I evidence).
Previous guidelines advised only that we consider using a severity score.
- Use the validated Pneumonia Severity Index or the easier-to-use CURB-65. Patients with a CURB-65 score of 2 or more generally require hospitalization (moderate recommendation, level III evidence).
- Ability to reliably and safely take medications at home must also be taken into account (strong recommendation, level II evidence.)
2. Admit to ICU promptly if needed
The criteria for admission to the ICU is similar to the previous ATS guidelines, but the list of minor criteria is more extensive. This change reflects evidence demonstrating worse outcomes in patients whose transfer to the ICU was delayed. This new criteria has not been validated.
- Patients requiring vasopressors for blood pressure support or with hypoxemic respiratory failure should be admitted to the ICU—these are major criteria (strong recommendation, level II evidence).
- Patients with 3 or more minor criteria should also be directly admitted to the ICU (moderate recommendation, level II evidence).
3. Identify who needs more tests
In the wake of controversy about diagnostic testing recommendations, the new guidelines attempt to better identify patients who would benefit from further testing (TABLE).
- 12 indications. Prior ATS guidelines lacked specifics on required additional testing, but the new guidelines give 12 clinical indications for more extensive evaluation, and identify which tests are recommended for each indication (strong recommendation, level II evidence).
- Routine testing to identify the cause of CAP in outpatients is optional (moderate recommendation, level III evidence).
TABLE
Clinical indications for more extensive diagnostic testing
| CLINICAL INDICATION | RECOMMENDED DIAGNOSTIC TESTS | |||
|---|---|---|---|---|
| BLOOD CULTURE | SPUTUM CULTURE | LEGIONELLA URINARY ANTIGEN TEST | PNEUMOCOCCAL URINARY ANTIGEN TEST | |
| ICU admission* | ||||
| Failed outpatient therapy | ||||
| Cavitary infiltrates† | ||||
| Leukopenia | ||||
| Active alcohol abuse | ||||
| Chronic severe liver disease | ||||
| Severe lung disease | ||||
| Asplenia | ||||
| Anatomic or functional | ||||
| Recent travel‡ | ||||
| Within past 2 weeks | ||||
| Positive Legionella urinary antigen test | N/A | |||
| Positive Pneumococcal urinary antigen test | N/A | |||
| Pleural effusion** | ||||
| Additional tests: | ||||
| * Endotracheal aspirate if intubated, possibly bronchoscopy or nonbronchoscopic bronchoalveolar lavage. | ||||
| † Fungal and tuberculosis cultures. | ||||
| ‡ Region/type of travel related to Legionella, Coccidioides, Hantavirus, B pseudomallei, avian influenza, SARS. | ||||
| ** Thoracentesis and pleural fluid cultures. | ||||
| Adapted from Mandell et al.1 | ||||
Empiric antibiotics
The recommendations of IDSA/ATS are generally for a class of antibiotics rather than a specific drug, unless noted.
4. Assess DRSP risk factors
Growth of drug-resistant Streptococcus pneumoniae (DRSP) necessitated a more extensive list of risk factors for DRSP. Other recommendations did not change.
Outpatient treatment
- Adults who were previously healthy and who do not have risk factors for DRSP CAP should be treated with either a macrolide (azithromycin, clarithromycin, or erythromycin) (strong recommendation, level I evidence) or doxycycline (weak recommendation; level III evidence).
- In the presence of comorbidities that increase the risk for DRSP, these antibiotics are appropriate: a respiratory fluoroquinolone (moxifloxacin, gemifloxacin, or levofloxacin [750 mg/day dose]) (strong recommendation, level I evidence); or β-lactam plus a macrolide: (high-dose amoxicillin [eg, 1 g 3x daily] [strong recommendation, level I evidence] or amoxicillin-clavulanate [2 g twice daily] is preferred; but alternatives include ceftriaxone, cefpodoxime, and cefuroxime [500 mg twice daily]. Doxycycline [level II evidence] is an alternative to the macrolide.)
Inpatient non-ICU treatment
- β-lactam plus a macrolide (strong recommendation, level I evidence) (cefotaxime, ceftriaxone, and ampicillin; ertapenem for selected patients; doxycycline [level III evidence] is an alternative to the macrolide.).
- A respiratory fluoroquinolone (strong recommendation, level I evidence) is the treatment of choice for penicillin-allergic patients.
5. Assess MRSA risk factors
Although similar to the prior ATS guidelines, the new guidelines have added specific risk factors for community-acquired methicillin-resistant S aureus (MRSA). This change reflects the increasing prevalence of community-acquired MRSA as an etiology for CAP.
The new guidelines state that the overwhelming majority of CAP pathogens will be adequately treated with the recommended empiric regimens. Exceptions are infections due to community-acquired methicillin-resistant S aureus and Pseudomonas aeruginosa.
ICU treatment
- A β-lactam (cefotaxime, ceftriaxone, or ampicillin-sulbactam) plus either azithromycin (strong recommendation, level II evidence) or a respiratory fluoroquinolone (strong recommendation, level I evidence).
- For penicillin-allergic patients, a respiratory fluoroquinolone and aztreonam are recommended.
- For Pseudomonas infection (see FIGURE for risk factors), use an anti-pneumococcal, antipseudomonal β-lactam (piperacillin-tazobactam, cefepime, imipenem, or meropenem) plus either ciprofloxacin or levofloxacin (750-mg dose) or the above β-lactam plus an aminoglycoside and azithromycin or the above β-lactam plus aminoglycoside and a respiratory fluoroquinolone (moderate recommendation, level II evidence). Fifteen days of therapy may be more effective in Pseudomonas CAP based on nosocomial infection data.
- For community-acquired methicillin-resistant S aureus infection (see FIGURE for risk factors), add vancomycin or linezolid (moderate recommendation, level III evidence).
Diagnostic testing is of high yield for patients with severe CAP requiring ICU admission, allowing for early de-escalation of empirical treatment if results are negative.
FIGURE
Treatment of community-acquired pneumonia*
Adapted from: Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
CURB-65 score for assessing severity of illness
Confusion
Uremia BUN >20
Respiration ≥30
BP <90/≤60 mm Hg
≥65 years of age
| TOTAL POINTS | MORTALITY RATE |
|---|---|
| 0 | 0.7% |
| 1 | 2.1% |
| 2 | 9.2% |
| 3 | 14.5% |
| 4 | 40.0% |
| 5 | 57.0% |
Criteria for ICU admission
Major
Invasive mechanical ventilation
Septic shock with need for vasopressors
Minor
- Respiration ≥30
- PaO2/FiO2 ≤250
- Multilobar infiltrates
- Disorientation
- BUN ≥20
- Hypotension requiring aggressive fluids
- Temperature <36°C
- Platelets <100,000
- WBC<4000
Risk factors
Drug-resistant S pneumoniae
- Any antibiotics within 3 months
- Comorbidities: Cardiac disease, diabetes mellitus, alcoholism, pulmonary disease, renal disease, liver disease, asplenia, malignancy, immunosuppressed.
- age >65 years
Pseudomonal infection
- Structural lung disease (bronchiectasis)
- Severe COPD with frequent admissions, recent antibiotic and steroid use
Methicillin-resistant S aureus
Pulmonary abscess
End-stage renal disease
IV drug abuse
Recent influenza
Recent fluroquinolone use
Duration of antibiotic treatment
- At least 5 days
- After 5 days, antibiotics may be discontinued when patient is afebrile 72 hours and has no more than 1 criteria for instability: temperature ≥37.8°C, heart rate ≥100, respiration ≥24, systolic BP ≤90 mm Hg, O2 saturation ≤90%, inability to maintain oral intake, altered mental status
- 15 days for CAP due to Pseudomonas
Criteria for discharge
Temperature ≤37.8°C, heart rate ≤100, respiration ≤24, systolic BP ≥ 90 mm Hg,O2 saturation ≥90%, ability to maintain oral intake, normal mental status
Pathogen-directed therapy
6. Identify the pathogen
New guidelines recommend that, once the pathogen is identified by reliable microbiological methods, therapy should be directed towards that specific pathogen to prevent increased resistance in the community (moderate recommendation, level III evidence).
Influenza. Treatment within 48 hours of symptom onset with oseltamivir or zanamivir is recommended for influenza A (strong recommendation, level I evidence).
While these antimicrobials should not be used in uncomplicated influenza with symptoms for >48 hours (level I evidence), they may be used in hospitalized patients or influenza pneumonia to reduce viral shedding (moderate recommendation, level III evidence).
LEVEL I HIGH
Well-conducted, randomized controlled trials
LEVEL II MODERATE
Well-designed, controlled trials without randomization (including cohort, patient series, and case-controlled studies); large case series in which systematic analysis of disease patterns and/or microbial etiology was conducted; and reports of data on new therapies that were not collected in a randomized fashion.
LEVEL III LOW
Case studies and expert opinion. In some instances, therapy recommendations may come from antibiotic susceptibility data without clinical observations.
Adapted from: Mandell et al.1
Time to first dose
7. Start treatment in the ED
Rather than designating a time window for starting treatment, the IDSA/ATS committee recommended that patients receive the first antibiotic dose in the Emergency Department (moderate recommendation, level III evidence).
This newly added statement contrasts with some current quality measures that grade hospitals according to whether antibiotics are started within a specific time frame.
Duration of antibiotics
8. Base duration on specific criteria
Reflecting evidence that shorter courses appear to be as effective as longer courses, the newer guidelines recommend discontinuation when the patient meets specific clinical criteria. Before discontinuing antibiotics, all patients with CAP should:
- Be treated for at least 5 days (level I evidence),
- Be afebrile for 48 to 72 hours, and
- Have no more than 1 of these criteria for clinical instability (moderate recommendation, level II evidence): temperature ≥37.8°C; heart rate ≥100 beats/min; respiratory rate ≥24 breaths/min; systolic blood pressure ≤90 mm Hg; arterial oxygen saturation ≤90% or pO2 ≤60 mm Hg on room air; inability to maintain oral intake; altered mental status.
Switching from IV to oral
The guidelines, similar to the prior guidelines, recommend switching to oral therapy for hemodynamically stable patients who are clinically improving, able to ingest medications, and have a normally functioning gastrointestinal tract (strong recommendation, level II evidence).
Hospital discharge
The guidelines recommend that patients be discharged as soon as they are clinically stable and have a safe environment for continued care. Patients receiving oral therapy do not require inpatient observation (moderate recommendation, level II evidence). This is unchanged from prior recommendations.
1. Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell D, Dean NC, et al. IDSA/ATS consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007;44:(Suppl 2) S27-S72.
1. Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell D, Dean NC, et al. IDSA/ATS consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007;44:(Suppl 2) S27-S72.
What we really need to do to reduce cardiovascular events in hypertensive patients
- In your efforts to reduce cardiovascular events in hypertensive patients, concentrate on getting patients to goal, rather than on which drugs to use to get them there (A).
- Beta-blockers—especially atenolol—should not be the drug of first choice when treating older patients with hypertension (A).
- Multiple drugs are required for adequate blood pressure control in most patients (A).
Strength of recommendation (SOR)
- Good quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Forget about a silver bullet.
Researchers have conducted numerous trials over the last decade to find an antihypertensive drug that best reduces cardiovascular events while reducing blood pressure. However, this objective review of 13 comparative antihypertensive drug trials over the past decade involving more than 168,000 patients reveals no great differences in the cardiovascular protective effects of diuretics, beta-blockers, calcium channel blockers, angiotensin receptor blockers (ARBs), and angiotensin-converting enzyme (ACE) inhibitors.
In fact, this review indicates that there were no significant differences in the primary cardiovascular endpoints in more than 90% of the patients studied. Where a difference in secondary clinical outcome was demonstrated, fewer events consistently occurred in the regimen that reached the lower blood pressure level.
This assessment will likely fly in the face of the way that many would view this body of research. That’s understandable. At first glance, it would appear that these 13 trials, with different methodology and endpoints, have produced conflicting conclusions with the confusion worsened by pharmaceutical companies seeking to interpret the results to best suit their marketing needs.1-3
It is not the quality of the data, however, that is in question; the controversy lies in the interpretation. Subjecting the studies to further statistical analysis would simply obscure the information.
By reviewing the data impartially and objectively as a whole, though, and interpreting individual studies in light of similar studies, it becomes evident that there is more consensus than conflict. The studies support the notion that we should concentrate on getting patients to goal, rather than focusing on which drugs we’ll use to get them there.
Methods
I performed a PubMed search of the last 10 years using the keywords hypertension, comparative, drug trials. I supplemented my search with references from the JNC 7, WHO, BHS/NICE, and European hypertension guidelines. For this review, I included only randomized controlled trials with clinical cardiovascular primary endpoints. The studies had to have enrolled at least 500 patients and followed them for at least 3 years. Thirteen trials satisfied these criteria.4-17 All 13 are summarized in the TABLE, but I will review 5 of the more recent trials here. They are:
- ASCOT-BPLA—Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm
- ALLHAT—Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial
- ANBP2—Second Australian National Blood Pressure Study
- LIFE—Losartan Intervention For Endpoint reduction
- VALUE—Valsartan Antihypertensive Long-term Use Evaluation.
Calcium channel blockers vs beta-blockers
ASCOT-BPLA studied 19,257 high-risk hypertensive patients on amlodipine (Norvasc), adding perindopril, or atenolol (Tenormin), adding bendroflumethiazide.17 After 5.5 years, the primary end-point of nonfatal myocardial infarction (MI) and cardiovascular death was similar (relative risk [RR]=0.90; 95% confidence interval [CI], 0.79–1.02; P=.1052).
Total coronary endpoint, stroke, and mortality were all lower on amlodipine. Blood pressure was significantly lower on amlodipine compared with atenolol, with an average difference of 2.7/1.9 mm Hg over the trial duration.18
At the end of the trial, patients on amlodipine also had a significantly higher HDL cholesterol, and lower body mass index, triglyceride, creatinine, and glucose levels. However, when researchers made a multivariate adjustment for all of these risk factors, cardiovascular event rate differences between the 2 groups disappeared, underscoring the importance of controlling for all risk factors in reducing clinical cardiovascular events.18,19
A careful reading of ASCOT-BPLA, then, makes it clear that this study does not support the notion that newer antihypertensives (calcium channel blockers and ACE inhibitors) are superior to older ones (beta-blockers and diuretics).20,21 This study actually demonstrates that while blood pressure reduction is vital, the differences between regimens are less important.
TABLE
More consensus than conflict among 13 comparative antihypertensive drug trials with cardiovascular primary endpoints
| YEAR | TRIAL | N | DRUGS COMPARED | PRIMARY ENDPOINT | RELATIVE RISK (95% CI) | P VALUE |
|---|---|---|---|---|---|---|
| 1998 | UKPDS4 | 758 | Captopril vs atenolol | Clinical diabetic event | 1.1 (0.86–1.41) | .43 |
| Diabetic death | 1.27 (0.82–1.97) | .28 | ||||
| Total mortality | 1.14 (0.81–1.61) | .44 | ||||
| 1999 | CAPPP5 | 10,985 | Captopril vs diuretic/beta-blocker | MI+stroke+CV death | 1.05 (0.90–1.22) | .52 |
| 1999 | STOP 26 | 6614 | New vs conventional drugs | CV death | 0.99 (0.84–1.16) | .89 |
| 4418 | ACE I vs conventional drugs | CV death | 1.01 (0.84–1.22) | .89 | ||
| 4409 | CCB vs conventional drugs | CV death | 0.97 (0.80–1.17) | .72 | ||
| 2000 | INSIGHT7 | 6321 | Nifedipine LA vs diuretic | CV death, MI, HF, stroke | 1.1 (0.91–1.34) | .35 |
| 2000 | NORDIL8 | 10,881 | Diltiazem vs beta-blocker/diuretic | Stroke, MI, CV death | 1.00 (0.87–1.15) | .97 |
| 2002 | LIFE9 | 9193 | Losartan vs atenolol | CV death, stroke, MI | 0.87 (0.77–0.98) | .021 |
| 2002–3 | ALLHAT10,11 | 24,303 | Amlodipine vs chlorthalidone | Fatal CHD, nonfatal MI | 0.98 (0.90–1.07) | .65 |
| 24,309 | Lisinopril vs chlorthalidone | Fatal CHD, nonfatal MI | 0.99 (0.91–1.08) | .81 | ||
| 24,314 | Doxazosin vs chlorthalidone | Fatal CHD, nonfatal MI | 1.03 (0.93–1.15) | .62 | ||
| 2003 | ANBP212 | 6083 | ACE I vs diuretic | CV event,* death | 0.89 (0.79–1.00) | .05 |
| 2003 | CONVINCE13 | 16,602 | Verapamil vs atenolol/thiazide | Stroke, MI, CV death | 1.02 (0.88–1.18) | .77 |
| 2003 | INVEST14 | 22,576 | Verapamil vs atenolol | Death, nonfatal MI, nonfatal stroke | 0.98 (0.90–1.06) | .57 |
| 2004 | VALUE15 | 15,245 | Valsartan vs amlodipine | CV event† | 1.04 (0.94–1.15) | .49 |
| 2004 | JMIC-B16 | 1650 | Nifedipine retard vs ACE I | Cardiac events‡ | 1.05 (0.81–1.37) | .86 |
| 2005 | ASCOT17 | 19,257 | Amlodipine (+ perindopril) vs atenolol (+ thiazide) | Nonfatal MI, fatal CHD | 0.90 (0.79–1.02) | .1052 |
| ACE I, angiotensin-converting enzyme inhibitor; CCB, calcium channel blocker; CHD, coronary heart disease; CI, confidence interval; CV, cardiovascular; HF, heart failure; MI, myocardial infarction. | ||||||
| *Defined as coronary events including MI, heart failure, acute occlusion of artery, dissecting or ruptured aortic aneurysm, and cerebrovascular events including stroke and transient ischemic attacks. | ||||||
| † Defined as cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. | ||||||
| ‡ Defined as cardiac death or sudden death, MI, angina pectoris requiring hospitalization, heart failure requiring hospitalization, serious arrhythmia, and coronary interventions. | ||||||
Largest hypertensive trial ever studied 4 drugs
ALLHAT, the largest hypertensive trial ever conducted, randomized 15,255 patients to chlorthalidone, 9061 to doxazosin (Cardura), 9048 to amlodipine, and 9054 to lisinopril (Prinivil/Zestril).10,11 (The arm involving doxazosin was terminated after 3.2 years.11,22)
Compared with the beta-blocker, more patients achieved target blood pressure control on chlorthalidone (63% vs 58%), and systolic blood pressure was about 2 mm Hg lower. Although the primary outcome of fatal coronary heart disease and nonfatal MI was equal in both groups (doxazosin=7.91%; chlorthalidone=7.76%; RR=1.03 [95% CI, 0.93–1.15]; P=.62), the doxazosin arm had more stroke, heart failure, and combined cardiovascular events.
Patients on amlodipine and lisinopril had a longer follow-up of 4.9 years. Systolic blood pressure was higher on amlodipine (0.8 mm Hg, P=.03) and lisinopril (2 mm Hg, P<.001) than on chlorthalidone. The primary endpoint (fatal coronary heart disease and nonfatal MI) was similar on the diuretic (11.5%), calcium channel blocker (11.3%; RR=0.98 [95% CI, 0.90–1.07]; P=.65), and ACE inhibitor (11.4%; RR=0.99 [95% CI, 0.91–1.08]; P=.81).
Compared with the diuretic arm, the calcium channel blocker arm had a higher incidence of heart failure, while the ACE inhibitor arm had a higher incidence of heart failure, stroke, and combined cardiovascular disease. The results were similar whatever the initial glycemic state, renal function status, and racial makeup of the patients studied.23-26 More than 60% of patients in ALLHAT required 2 or more drugs for good blood pressure control.27
“Diuretics first” for patients with or without diabetes?
In ALLHAT, although diabetes occurred more frequently and fasting glucose rose in patients on diuretics, these metabolic abnormalities did not result in more cardiovascular events. Even among patients with diabetes, heart failure was more common on doxazosin, amlodipine, and lisinopril compared with those on chlorthalidone.23,24
Given that the ultimate aim of hypertensive therapy is to reduce clinical disease—not just to improve laboratory profiles—ALLHAT should put to rest any apprehension physicians have about diuretic use. These findings have even led to suggestions that diuretics be the first line antihypertensive agent, in both diabetic and nondiabetic patients.28-30
ACE inhibitor vs diuretic
ANBP2 randomized hypertensive patients to initial treatment with an ACE inhibitor (n=3044) or a diuretic (n=3039).12 With similar blood pressure reduction in both arms (26/12 mm Hg), treatment with the ACE inhibitor resulted in a lower incidence of the composite primary end-point of cardiovascular events or total death that was of borderline significance (ACE inhibitor=22.8%; diuretic=24.2%; RR=0.89 [95% CI, 0.79–1.00]; P=.05).
Among women, there was no difference between the ACE inhibitor and diuretic groups. In the overall population, there was also no difference individually of total mortality or incidence of first cardiovascular event or death.
Thus ANBP2 actually confirms the results from ALLHAT by showing that ACE inhibitors and diuretics are equivalent in reducing cardiovascular events in hypertension.31
Losartan vs atenolol
In the LIFE study, 9193 hypertensive patients with left ventricular hypertrophy were randomized to either losartan (Cozaar) or atenolol.9 Losartan treatment resulted in a marked reduction in stroke incidence, which produced a significant reduction in the composite primary end-point of death, MI, or stroke (11% vs 13%; RR=0.87 [95% CI, 0.77–0.98]; P=.021).
When only the 1195 patients with diabetes were assessed, there was a significant reduction not only in the primary endpoint but also in cardiovascular and total mortality.32 Surprisingly, the reduction of stroke incidence did not reach statistical significance in this diabetic population (RR=0.79 [95% CI, 0.55–1.14]; P=.204).
A word of caution, though: The results of LIFE should be taken together with data from other trials. No other study has demonstrated a special benefit from the renin-angiotensin antagonists in preventing stroke. In fact, ACE inhibitors were weaker than the comparator drugs in preventing stroke in both CAPPP (TABLE) and ALLHAT.5,10 Various reviews have suggested that among antihypertensive drugs, it is the diuretics and calcium channel blockers that may be more useful in stroke reduction.33,34
Chalk the benefit up to the drop in blood pressure
In the LIFE study, the treated mean systolic blood pressure was lower with losartan in the overall (1.1 mm Hg; P=.017) and diabetic (2 mm Hg; P value not stated) populations, and thus the clinical benefit could possibly have been from the better blood pressure reduction on losartan. Furthermore, there is evidence that beta-blockers are less useful in the older hypertensive patient, and are especially weak in preventing stroke incidence.35,36
Rather than showing the superiority of the ARB, it is fair to say that LIFE actually confirms the importance of blood pressure reduction, and reveals the weaker cardiovascular protective effect of atenolol in older hypertensive patients.
Valsartan, amlodipine in high-risk patients
VALUE randomized 15,245 high-risk hypertensive patients to valsartan (Diovan) and amlodipine.15,37 Trial researchers sought to study the difference—for the same level of blood pressure reduction—between the 2 regimens in the incidence of cardiac events defined as sudden cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. That said, the attained blood pressure was lower on the calcium channel blocker: 4.0/2.1 mm Hg at 1 month and 2.1/1.7 mm Hg at the end of study.
After 4.2 years, there was no significant difference in the primary endpoint of first cardiac event (10.6% valsartan/10.4% amlodipine; RR=1.04 [95% CI, 0.94–1.15]; P=.49). Diabetes was lower, but the rate of MI was higher on valsartan. After correction for the blood pressure difference, the composite of cardiac events, stroke, death, or MI was similar in the 2 groups.38
VALUE patients reaching adequate blood pressure control by 6 months fared better, regardless of drug type used. Thus demonstrating that the benefit from good blood pressure control was more important than the subtle differences between antihypertensive drugs. The better metabolic profile in the angiotensin receptor blocker arm did not translate into a reduction in adverse clinical disease.
The VALUE trial suggests (as did ALLHAT) that drugs targeting the reninangiotensin system do not provide special cardiovascular protection.10,15
Choosing an antihypertensive drug according to the clinical disease and target organ most at risk of damage is logical and in keeping with numerous guidelines.42-45 Thus, you’ll want to treat hypertensive patients with these conditions as follows:
- Angina pectoris. Therapy should include a beta-blocker or calcium channel blocker, given their definite antianginal and possible anti-atherosclerotic effects.16,46,47
- Prior MI. Start the patient on a beta-blocker.47
- Poor left ventricular function. Start the patient on a diuretic, and then add an ACE inhibitor and beta-blocker, as needed.10,49,50
- Prior stroke (or a patient at special risk of stroke). Begin therapy with a calcium channel blocker or a diuretic.33,34
- Diabetic proteinuria. An ARB or an ACE inhibitor is best suited to prevent and delay nephropathy.51-54
Consensus emerges from studies spanning 10 years
This objective review of the comparative hypertension drug trials shows that there are no great differences in the cardiovascular protective efficacy of the diuretics, beta-blockers, calcium channel blockers, ARBs, and ACE inhibitors.
There was no significant difference in the cardiovascular primary endpoint in 11 of the 13 trials reviewed, involving 91% of the randomized 168,593 patients (TABLE).4-8,10,11,13-17 Of the remaining 2 trials, the difference in ANBP2 just reached a P value of .05, while the result in LIFE was driven by a lower stroke incidence on ARB treatment that is not noted in any of the other studies involving an ARB or ACE inhibitor.4-6,10,12-15
Focus on controlling blood pressure with combination of drugs
Given the very large number of patients studied in these well-conducted trials, if there were any especially useful, or detrimental, cardiovascular effect of a particular class of antihypertensive drug, it would have been obvious by now. Since most patients will require multiple drugs, the equivalent protective efficacy of different antihypertensive drugs is reassuring and suggests that physicians should not worry too much about which drug to start the patient on.28 Rather, the emphasis should be on how best to reach adequate blood pressure control by combining several antihypertensive drugs.
Small blood pressure differences, big impact
In LIFE (losartan vs atenolol), ALLHAT (doxazosin, amlodipine, lisinopril vs chlorthalidone), VALUE (amlodipine vs valsartan), and ASCOT (amlodipine vs atenolol), where a secondary cardiovascular endpoint was lower in one of the treatment arms, it was always the arm with the lower achieved blood pressure that had the better clinical outcome.9-11,15,17
Marya Ostrowski, JFP Editor
With only 50% of patients typically taking their medications as prescribed and the cost of poor adherence reaching an estimated $177 billion annually in direct and indirect health care costs, one medication safety group is saying enough is enough.
The National Council on Patient Information and Education (NCPIE), a nonprofit coalition that includes health professional associations, government agencies, and pharmaceutical companies, issued a report this summer detailing a 10-step action plan for reducing the adverse health and economic consequences of poor medication adherence.
The plan, developed by a panel of experts that NCPIE convened, calls on the government and health care community to, among other things:
- address the barriers to patient adherence for patients with low health literacy.
- develop a curriculum on medication adherence for use in medical schools.
- mount a unified national education campaign to make patient adherence a national health priority.
“Medication adherence is America’s new drug problem,” said Carolyn M. Clancy, MD, director of the Agency for Healthcare Research and Quality. AHRQ has been working with NCPIE, the FDA, and the National Consumers League to develop a public education campaign on medication adherence, according to Clancy. The NCPIE report helps to bolster those ongoing efforts, she said.
On the heels of the report, NCPIE is planning on releasing videos that will teach seniors about properly taking their medications, according to Ray Bullman, NCPIE’s executive vice president.
To learn more about NCPIE’s initiatives, or for a copy of the report, Enhancing Prescription Medicine Adherence: A National Action Plan, point your browser to: www.talkaboutrx.org.
These achieved blood pressure differences although small, were significant. Small overall mean blood pressure differences could mask much larger blood pressure differences in the individual patient. Consider, for instance, the HOPE (Heart Outcomes Prevention Evaluation) trial, where a reported overall blood pressure difference of only 3/1 mm Hg between the 2 treatment arms masked a difference of 10/4 mm Hg in 24-hour ambulatory blood pressure and a difference of 17/8 mm Hg in night-time blood pressure.39,40
Thus, instead of trying to work out why antihypertensive drugs could exert apparently different cardiovascular protective efficacy in different trials, the simple and consistent message is that the lower the achieved blood pressure, the lower the adverse clinical cardiovascular outcome.
What makes sense for your patient?
In selecting antihypertensive drugs, physicians should be guided by data supporting a particular drug in coexisting clinical conditions. (See “Where to begin when there are coexisting conditions,”.) In the hypertensive patient who is free of clinical disease, a case can be made for a diuretic as the first-line drug, although calcium channel blockers, ARBs, and ACE inhibitors can also claim evidence to support their use. In the older patient, beta-blockers—especially atenolol—should not be the drug of first choice.35,36,41
As this review of comparative hypertension drug trials shows, multiple drugs are required for adequate blood pressure control in most patients. Thus, physicians should not be too preoccupied about how to initiate treatment, but remember to add drugs until adequate control is achieved.
Correspondence
H T Ong, FRCP, FACC, FESC, H T Ong Heart Clinic, 251C Burma Road, Penang 10350, Malaysia; [email protected]
1. Psaty BM, Weiss NS, Furberg CD. Recent trials in hypertension. Compelling science or commercial speech? JAMA 2006;295:1704-1706.
2. Abramson J, Starfield B. The effect of conflict of interest on biomedical research and clinical practice guidelines: Can we trust the evidence in evidence-based medicine? J Am Board Fam Pract 2005;18:414-418.
3. Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167-1170.
4. UK Prospective Diabetes Study Group. Efficacy of Atenolol and Captopril in Reducing Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 39. BMJ 1998;317:713-720.
5. Hansson L, Lindholm LH, Niskanen L, et al. Effect of ACE inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP). Lancet 1999;353:611-616.
6. Hansson L, Lindholm LH, Ekbom T, et al. Randomized trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension 2 (STOP-Hypertension 2) study. Lancet 1999;354:1751-1756.
7. Brown MJ, Palmer CR, Castaigne A, et al. Morbidity and mortality in patients randomized to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 2000;356:366-372.
8. Hansson L, Hedner T, Lund-Johansen P, et al. For the NORDIL Study Group. Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) Study. Lancet 2000;356:359-365.
9. Dahlof B, Devereux RB, Kjeldsen SE, et al. For the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:995-1003.
10. The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to prevent Heart Attack trial (ALLHAT). JAMA 2002;288:2981-2997.
11. ALLHAT officers and Coordinators for the ALLHAT Collaborative research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and lipidlowering treatment to prevent Heart Attack trial (ALLHAT). Hypertension 2003;42:239-246.
12. Wing LMH, Reid CM, Ryan P, et al. For the Second Australian National blood pressure Study Group. A comparison of outcomes with angiotensin converting enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med 2003;348:583-592.
13. Black HR, Elliott WJ, Grandits G, et al. For the CONVINCE research Group. Principal results of the Controlled ONset Verapamil INvestigation of Cardiovascular End points (CONVINCE) trial. JAMA 2003;289:2073-2082.
14. Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al. For the INVEST Investigators. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): A randomized controlled trial. JAMA 2003;290:2805-2816.
15. Julius S, Kjeldsen SE, Weber M. for the VALUE trial group. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomized trial. Lancet 2004;363:2022-2031.
16. Yui Y, Sumiyoshi T, Kodama K, et al. Comparison of nifedipine retard with angiotensin-converting enzyme inhibitors in Japanese hypertensive patients with coronary artery disease: the Japan multicenter Investigation for Cardiovascular Diseases-B (JMIC-B) randomized trial. Hypertens Res 2004;27:181-191.
17. Dahlof B, Sever PS, poulter NR. for the ASCOT investigators. prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA): a multicentre randomized controlled trial. Lancet 2005;366:895-906.
18. Poulter NR, Wedel H, Dahlof B. For the ASCOT investigators. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA). Lancet 2005;366:907-913.
19. Daviglus ML, Liu K. today’s Agenda. We must focus on achieving favorable levels of all risk factors simultaneously. Arch Intern Med 2004;164:2086-2087.
20. Duerden M. ASCOT-BPLA. Lancet 2006;367:206.-
21. Cave JA. ASCOT: A tale of two treatment regimes. Is ASCOT all it’s cracked up to be? BMJ 2005;331:1023.-
22. The ALLHAT officers and Coordinators for the AllHAt Collaborative research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2000;283:1967-1975.
23. Barzilay JI, Davis BR, Bettencourt J, et al. For the ALLHAT Collaborative research Group. Cardiovascular outcomes using doxazosin vs chlorthalidone for the treatment of hypertension in older adults with and without glucose disorders: a report from the ALLHAT Study. J Clin Hypertens 2004;6:116-125.
24. Whelton PK, Barzilay J, Cushman WC, et al. For the ALLHAT Collaborative research group. Clinical outcomes in antihypertensive treatment of type 2 diabetes, impaired fasting glucose concentration, and normoglycemia. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med. 2005;165:1401-1409.
25. Rahman M, Pressel S, Davis BR, et al. For the ALLHAT Collaborative research Group. Renal outcomes in high-risk hypertensive patients treated with an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: a report from the ALLHAT Study. Arch Inten Med 2005;165:936-946.
26. Wright JT, Dunn JK, Cutler JA, et al. for the ALLHAT Collaborative research Group. outcomes in hyper-tensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA 2005;293:1595-1607.
27. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens 2002;4:393-504.
28. Williams B. Drug treatment for hypertension: most patients will need a treatment cocktail –including a thiazide diuretic. BMJ 2003;326:61-62.
29. Appel LJ. the verdict from ALLHAT—thiazide diuretics are the preferred initial therapy for hypertension. JAMA 2002;288:3039-3042.
30. Salvetti A, Ghiadoni L. Guidelines for antihypertensive treatment: An update after the ALLHAT study. J Am Soc Nephrol 2004;15:S51-S54.
31. Frohlich ED. Treating hypertension-what are we to believe? N Engl J Med 2003;348:639-641.
32. Lindholm LH, Ibsen H, Dahlof B, et al. For the LIFE study group. Cardiovascular morbidity and mortality in patients with diabetes in the losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:1004-1010.
33. Messerli FH, Grossman E, Lever AF. Do thiazide diuretics confer specific protection against strokes? Arch Intern Med 2003;163:2557-2560.
34. Opie LH, Schall R. Evidence-based evaluation of calcium channel blockers for hypertension. J Am Coll Cardiol 2002;39:315-322.
35. Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004;364:1684-1689.
36. Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006;174:1737-1742.
37. Kjeldsen SE, Julius S, Brunner H, et al. for the VALUE Trial Group. Characteristics of 15314 hypertensive patients at high coronary risk. the VALUE trial. Blood Press 2001;10:83-91.
38. Weber MA, Julius S, Kjeldsen SE, et al. Blood pressure dependent and independent effects of antihypertensive treatment on clinical events in the VALUE trial. Lancet 2004;363:2049-2051.
39. The Heart outcomes prevention evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000;342:145-153.
40. Svensson P, De Faire U, Sleight P, Yusuf S, Ostergren J. Comparative effects of ramipril on ambulatory and office blood pressures: a Hope substudy. Hypertension 2001;38:28-32.
41. Ong HT. Beta-blockers in hypertension and cardiovascular disease. BMJ 2007;334:946-949.
42. Chobanian AV, Bakris GL, Black HR, et al. The National High blood pressure education program Coordinating Committee.The Seventh report of the Joint National Committee on prevention, Detection, evaluation, and treatment of High blood pressure. the JNC 7 report. JAMA 2003;289:2560-2572.
43. Guidelines Committee. 2003 european Society of Hypertension-european Society of Cardiology guidelines for the management of arterial hypertension. J Hypertens 2003;21:1011-1053.
44. World Health Organization. International Society of Hypertension Writing Group. 2003 World Health Organisation (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens 2003;21:1983-1992.
45. Williams B, Poulter NR, Brown MJ, et al. Guidelines for management of hypertension: report of the fourth working party of the british Hypertension Society, 2004—BHS IV. J Hum Hypertens 2004;18:139-185.
46. Nissen SE, Tuzcu EM, Libby P. for the CAMELOT investigators. effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure. the CAMELOT Study: a randomized controlled trial. JAMA 2004;292:2217-2226.
47. Hedblad B, Wikstrand J, Janzon L, Wedel H, Berglund G. Low dose Metoprolol CR/XL and Fluvastatin slow progression of carotid intima-media thickness: main results from the beta-blocker Cholesterol-lowering Asymptomatic plaque Study (BCAPS). Circulation 2001;103:1721-1726.
48. Freemantle N, Cleland J, Young P, Mason J, Harrison J. Beta-blockade after myocardial infarction: systematic review and meta regression analysis. BMJ 1999;318:1730-1737.
49. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992;372:685-691.
50. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised Intervention trial in Congestive Heart Failure (MERIT-HF). Lancet 1999;353:2001-2006.
51. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-860.
52. Parving HH, Lehnert H, Mortensen JB, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-878.
53. Brenner BM, Cooper ME, Zeeuw D de, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-869.
54. Wright JT, Bakris G, Greene T, et al. For the African American Study of Kidney Disease and Hypertension Study Group. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 2002;288:2421-2431.
- In your efforts to reduce cardiovascular events in hypertensive patients, concentrate on getting patients to goal, rather than on which drugs to use to get them there (A).
- Beta-blockers—especially atenolol—should not be the drug of first choice when treating older patients with hypertension (A).
- Multiple drugs are required for adequate blood pressure control in most patients (A).
Strength of recommendation (SOR)
- Good quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Forget about a silver bullet.
Researchers have conducted numerous trials over the last decade to find an antihypertensive drug that best reduces cardiovascular events while reducing blood pressure. However, this objective review of 13 comparative antihypertensive drug trials over the past decade involving more than 168,000 patients reveals no great differences in the cardiovascular protective effects of diuretics, beta-blockers, calcium channel blockers, angiotensin receptor blockers (ARBs), and angiotensin-converting enzyme (ACE) inhibitors.
In fact, this review indicates that there were no significant differences in the primary cardiovascular endpoints in more than 90% of the patients studied. Where a difference in secondary clinical outcome was demonstrated, fewer events consistently occurred in the regimen that reached the lower blood pressure level.
This assessment will likely fly in the face of the way that many would view this body of research. That’s understandable. At first glance, it would appear that these 13 trials, with different methodology and endpoints, have produced conflicting conclusions with the confusion worsened by pharmaceutical companies seeking to interpret the results to best suit their marketing needs.1-3
It is not the quality of the data, however, that is in question; the controversy lies in the interpretation. Subjecting the studies to further statistical analysis would simply obscure the information.
By reviewing the data impartially and objectively as a whole, though, and interpreting individual studies in light of similar studies, it becomes evident that there is more consensus than conflict. The studies support the notion that we should concentrate on getting patients to goal, rather than focusing on which drugs we’ll use to get them there.
Methods
I performed a PubMed search of the last 10 years using the keywords hypertension, comparative, drug trials. I supplemented my search with references from the JNC 7, WHO, BHS/NICE, and European hypertension guidelines. For this review, I included only randomized controlled trials with clinical cardiovascular primary endpoints. The studies had to have enrolled at least 500 patients and followed them for at least 3 years. Thirteen trials satisfied these criteria.4-17 All 13 are summarized in the TABLE, but I will review 5 of the more recent trials here. They are:
- ASCOT-BPLA—Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm
- ALLHAT—Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial
- ANBP2—Second Australian National Blood Pressure Study
- LIFE—Losartan Intervention For Endpoint reduction
- VALUE—Valsartan Antihypertensive Long-term Use Evaluation.
Calcium channel blockers vs beta-blockers
ASCOT-BPLA studied 19,257 high-risk hypertensive patients on amlodipine (Norvasc), adding perindopril, or atenolol (Tenormin), adding bendroflumethiazide.17 After 5.5 years, the primary end-point of nonfatal myocardial infarction (MI) and cardiovascular death was similar (relative risk [RR]=0.90; 95% confidence interval [CI], 0.79–1.02; P=.1052).
Total coronary endpoint, stroke, and mortality were all lower on amlodipine. Blood pressure was significantly lower on amlodipine compared with atenolol, with an average difference of 2.7/1.9 mm Hg over the trial duration.18
At the end of the trial, patients on amlodipine also had a significantly higher HDL cholesterol, and lower body mass index, triglyceride, creatinine, and glucose levels. However, when researchers made a multivariate adjustment for all of these risk factors, cardiovascular event rate differences between the 2 groups disappeared, underscoring the importance of controlling for all risk factors in reducing clinical cardiovascular events.18,19
A careful reading of ASCOT-BPLA, then, makes it clear that this study does not support the notion that newer antihypertensives (calcium channel blockers and ACE inhibitors) are superior to older ones (beta-blockers and diuretics).20,21 This study actually demonstrates that while blood pressure reduction is vital, the differences between regimens are less important.
TABLE
More consensus than conflict among 13 comparative antihypertensive drug trials with cardiovascular primary endpoints
| YEAR | TRIAL | N | DRUGS COMPARED | PRIMARY ENDPOINT | RELATIVE RISK (95% CI) | P VALUE |
|---|---|---|---|---|---|---|
| 1998 | UKPDS4 | 758 | Captopril vs atenolol | Clinical diabetic event | 1.1 (0.86–1.41) | .43 |
| Diabetic death | 1.27 (0.82–1.97) | .28 | ||||
| Total mortality | 1.14 (0.81–1.61) | .44 | ||||
| 1999 | CAPPP5 | 10,985 | Captopril vs diuretic/beta-blocker | MI+stroke+CV death | 1.05 (0.90–1.22) | .52 |
| 1999 | STOP 26 | 6614 | New vs conventional drugs | CV death | 0.99 (0.84–1.16) | .89 |
| 4418 | ACE I vs conventional drugs | CV death | 1.01 (0.84–1.22) | .89 | ||
| 4409 | CCB vs conventional drugs | CV death | 0.97 (0.80–1.17) | .72 | ||
| 2000 | INSIGHT7 | 6321 | Nifedipine LA vs diuretic | CV death, MI, HF, stroke | 1.1 (0.91–1.34) | .35 |
| 2000 | NORDIL8 | 10,881 | Diltiazem vs beta-blocker/diuretic | Stroke, MI, CV death | 1.00 (0.87–1.15) | .97 |
| 2002 | LIFE9 | 9193 | Losartan vs atenolol | CV death, stroke, MI | 0.87 (0.77–0.98) | .021 |
| 2002–3 | ALLHAT10,11 | 24,303 | Amlodipine vs chlorthalidone | Fatal CHD, nonfatal MI | 0.98 (0.90–1.07) | .65 |
| 24,309 | Lisinopril vs chlorthalidone | Fatal CHD, nonfatal MI | 0.99 (0.91–1.08) | .81 | ||
| 24,314 | Doxazosin vs chlorthalidone | Fatal CHD, nonfatal MI | 1.03 (0.93–1.15) | .62 | ||
| 2003 | ANBP212 | 6083 | ACE I vs diuretic | CV event,* death | 0.89 (0.79–1.00) | .05 |
| 2003 | CONVINCE13 | 16,602 | Verapamil vs atenolol/thiazide | Stroke, MI, CV death | 1.02 (0.88–1.18) | .77 |
| 2003 | INVEST14 | 22,576 | Verapamil vs atenolol | Death, nonfatal MI, nonfatal stroke | 0.98 (0.90–1.06) | .57 |
| 2004 | VALUE15 | 15,245 | Valsartan vs amlodipine | CV event† | 1.04 (0.94–1.15) | .49 |
| 2004 | JMIC-B16 | 1650 | Nifedipine retard vs ACE I | Cardiac events‡ | 1.05 (0.81–1.37) | .86 |
| 2005 | ASCOT17 | 19,257 | Amlodipine (+ perindopril) vs atenolol (+ thiazide) | Nonfatal MI, fatal CHD | 0.90 (0.79–1.02) | .1052 |
| ACE I, angiotensin-converting enzyme inhibitor; CCB, calcium channel blocker; CHD, coronary heart disease; CI, confidence interval; CV, cardiovascular; HF, heart failure; MI, myocardial infarction. | ||||||
| *Defined as coronary events including MI, heart failure, acute occlusion of artery, dissecting or ruptured aortic aneurysm, and cerebrovascular events including stroke and transient ischemic attacks. | ||||||
| † Defined as cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. | ||||||
| ‡ Defined as cardiac death or sudden death, MI, angina pectoris requiring hospitalization, heart failure requiring hospitalization, serious arrhythmia, and coronary interventions. | ||||||
Largest hypertensive trial ever studied 4 drugs
ALLHAT, the largest hypertensive trial ever conducted, randomized 15,255 patients to chlorthalidone, 9061 to doxazosin (Cardura), 9048 to amlodipine, and 9054 to lisinopril (Prinivil/Zestril).10,11 (The arm involving doxazosin was terminated after 3.2 years.11,22)
Compared with the beta-blocker, more patients achieved target blood pressure control on chlorthalidone (63% vs 58%), and systolic blood pressure was about 2 mm Hg lower. Although the primary outcome of fatal coronary heart disease and nonfatal MI was equal in both groups (doxazosin=7.91%; chlorthalidone=7.76%; RR=1.03 [95% CI, 0.93–1.15]; P=.62), the doxazosin arm had more stroke, heart failure, and combined cardiovascular events.
Patients on amlodipine and lisinopril had a longer follow-up of 4.9 years. Systolic blood pressure was higher on amlodipine (0.8 mm Hg, P=.03) and lisinopril (2 mm Hg, P<.001) than on chlorthalidone. The primary endpoint (fatal coronary heart disease and nonfatal MI) was similar on the diuretic (11.5%), calcium channel blocker (11.3%; RR=0.98 [95% CI, 0.90–1.07]; P=.65), and ACE inhibitor (11.4%; RR=0.99 [95% CI, 0.91–1.08]; P=.81).
Compared with the diuretic arm, the calcium channel blocker arm had a higher incidence of heart failure, while the ACE inhibitor arm had a higher incidence of heart failure, stroke, and combined cardiovascular disease. The results were similar whatever the initial glycemic state, renal function status, and racial makeup of the patients studied.23-26 More than 60% of patients in ALLHAT required 2 or more drugs for good blood pressure control.27
“Diuretics first” for patients with or without diabetes?
In ALLHAT, although diabetes occurred more frequently and fasting glucose rose in patients on diuretics, these metabolic abnormalities did not result in more cardiovascular events. Even among patients with diabetes, heart failure was more common on doxazosin, amlodipine, and lisinopril compared with those on chlorthalidone.23,24
Given that the ultimate aim of hypertensive therapy is to reduce clinical disease—not just to improve laboratory profiles—ALLHAT should put to rest any apprehension physicians have about diuretic use. These findings have even led to suggestions that diuretics be the first line antihypertensive agent, in both diabetic and nondiabetic patients.28-30
ACE inhibitor vs diuretic
ANBP2 randomized hypertensive patients to initial treatment with an ACE inhibitor (n=3044) or a diuretic (n=3039).12 With similar blood pressure reduction in both arms (26/12 mm Hg), treatment with the ACE inhibitor resulted in a lower incidence of the composite primary end-point of cardiovascular events or total death that was of borderline significance (ACE inhibitor=22.8%; diuretic=24.2%; RR=0.89 [95% CI, 0.79–1.00]; P=.05).
Among women, there was no difference between the ACE inhibitor and diuretic groups. In the overall population, there was also no difference individually of total mortality or incidence of first cardiovascular event or death.
Thus ANBP2 actually confirms the results from ALLHAT by showing that ACE inhibitors and diuretics are equivalent in reducing cardiovascular events in hypertension.31
Losartan vs atenolol
In the LIFE study, 9193 hypertensive patients with left ventricular hypertrophy were randomized to either losartan (Cozaar) or atenolol.9 Losartan treatment resulted in a marked reduction in stroke incidence, which produced a significant reduction in the composite primary end-point of death, MI, or stroke (11% vs 13%; RR=0.87 [95% CI, 0.77–0.98]; P=.021).
When only the 1195 patients with diabetes were assessed, there was a significant reduction not only in the primary endpoint but also in cardiovascular and total mortality.32 Surprisingly, the reduction of stroke incidence did not reach statistical significance in this diabetic population (RR=0.79 [95% CI, 0.55–1.14]; P=.204).
A word of caution, though: The results of LIFE should be taken together with data from other trials. No other study has demonstrated a special benefit from the renin-angiotensin antagonists in preventing stroke. In fact, ACE inhibitors were weaker than the comparator drugs in preventing stroke in both CAPPP (TABLE) and ALLHAT.5,10 Various reviews have suggested that among antihypertensive drugs, it is the diuretics and calcium channel blockers that may be more useful in stroke reduction.33,34
Chalk the benefit up to the drop in blood pressure
In the LIFE study, the treated mean systolic blood pressure was lower with losartan in the overall (1.1 mm Hg; P=.017) and diabetic (2 mm Hg; P value not stated) populations, and thus the clinical benefit could possibly have been from the better blood pressure reduction on losartan. Furthermore, there is evidence that beta-blockers are less useful in the older hypertensive patient, and are especially weak in preventing stroke incidence.35,36
Rather than showing the superiority of the ARB, it is fair to say that LIFE actually confirms the importance of blood pressure reduction, and reveals the weaker cardiovascular protective effect of atenolol in older hypertensive patients.
Valsartan, amlodipine in high-risk patients
VALUE randomized 15,245 high-risk hypertensive patients to valsartan (Diovan) and amlodipine.15,37 Trial researchers sought to study the difference—for the same level of blood pressure reduction—between the 2 regimens in the incidence of cardiac events defined as sudden cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. That said, the attained blood pressure was lower on the calcium channel blocker: 4.0/2.1 mm Hg at 1 month and 2.1/1.7 mm Hg at the end of study.
After 4.2 years, there was no significant difference in the primary endpoint of first cardiac event (10.6% valsartan/10.4% amlodipine; RR=1.04 [95% CI, 0.94–1.15]; P=.49). Diabetes was lower, but the rate of MI was higher on valsartan. After correction for the blood pressure difference, the composite of cardiac events, stroke, death, or MI was similar in the 2 groups.38
VALUE patients reaching adequate blood pressure control by 6 months fared better, regardless of drug type used. Thus demonstrating that the benefit from good blood pressure control was more important than the subtle differences between antihypertensive drugs. The better metabolic profile in the angiotensin receptor blocker arm did not translate into a reduction in adverse clinical disease.
The VALUE trial suggests (as did ALLHAT) that drugs targeting the reninangiotensin system do not provide special cardiovascular protection.10,15
Choosing an antihypertensive drug according to the clinical disease and target organ most at risk of damage is logical and in keeping with numerous guidelines.42-45 Thus, you’ll want to treat hypertensive patients with these conditions as follows:
- Angina pectoris. Therapy should include a beta-blocker or calcium channel blocker, given their definite antianginal and possible anti-atherosclerotic effects.16,46,47
- Prior MI. Start the patient on a beta-blocker.47
- Poor left ventricular function. Start the patient on a diuretic, and then add an ACE inhibitor and beta-blocker, as needed.10,49,50
- Prior stroke (or a patient at special risk of stroke). Begin therapy with a calcium channel blocker or a diuretic.33,34
- Diabetic proteinuria. An ARB or an ACE inhibitor is best suited to prevent and delay nephropathy.51-54
Consensus emerges from studies spanning 10 years
This objective review of the comparative hypertension drug trials shows that there are no great differences in the cardiovascular protective efficacy of the diuretics, beta-blockers, calcium channel blockers, ARBs, and ACE inhibitors.
There was no significant difference in the cardiovascular primary endpoint in 11 of the 13 trials reviewed, involving 91% of the randomized 168,593 patients (TABLE).4-8,10,11,13-17 Of the remaining 2 trials, the difference in ANBP2 just reached a P value of .05, while the result in LIFE was driven by a lower stroke incidence on ARB treatment that is not noted in any of the other studies involving an ARB or ACE inhibitor.4-6,10,12-15
Focus on controlling blood pressure with combination of drugs
Given the very large number of patients studied in these well-conducted trials, if there were any especially useful, or detrimental, cardiovascular effect of a particular class of antihypertensive drug, it would have been obvious by now. Since most patients will require multiple drugs, the equivalent protective efficacy of different antihypertensive drugs is reassuring and suggests that physicians should not worry too much about which drug to start the patient on.28 Rather, the emphasis should be on how best to reach adequate blood pressure control by combining several antihypertensive drugs.
Small blood pressure differences, big impact
In LIFE (losartan vs atenolol), ALLHAT (doxazosin, amlodipine, lisinopril vs chlorthalidone), VALUE (amlodipine vs valsartan), and ASCOT (amlodipine vs atenolol), where a secondary cardiovascular endpoint was lower in one of the treatment arms, it was always the arm with the lower achieved blood pressure that had the better clinical outcome.9-11,15,17
Marya Ostrowski, JFP Editor
With only 50% of patients typically taking their medications as prescribed and the cost of poor adherence reaching an estimated $177 billion annually in direct and indirect health care costs, one medication safety group is saying enough is enough.
The National Council on Patient Information and Education (NCPIE), a nonprofit coalition that includes health professional associations, government agencies, and pharmaceutical companies, issued a report this summer detailing a 10-step action plan for reducing the adverse health and economic consequences of poor medication adherence.
The plan, developed by a panel of experts that NCPIE convened, calls on the government and health care community to, among other things:
- address the barriers to patient adherence for patients with low health literacy.
- develop a curriculum on medication adherence for use in medical schools.
- mount a unified national education campaign to make patient adherence a national health priority.
“Medication adherence is America’s new drug problem,” said Carolyn M. Clancy, MD, director of the Agency for Healthcare Research and Quality. AHRQ has been working with NCPIE, the FDA, and the National Consumers League to develop a public education campaign on medication adherence, according to Clancy. The NCPIE report helps to bolster those ongoing efforts, she said.
On the heels of the report, NCPIE is planning on releasing videos that will teach seniors about properly taking their medications, according to Ray Bullman, NCPIE’s executive vice president.
To learn more about NCPIE’s initiatives, or for a copy of the report, Enhancing Prescription Medicine Adherence: A National Action Plan, point your browser to: www.talkaboutrx.org.
These achieved blood pressure differences although small, were significant. Small overall mean blood pressure differences could mask much larger blood pressure differences in the individual patient. Consider, for instance, the HOPE (Heart Outcomes Prevention Evaluation) trial, where a reported overall blood pressure difference of only 3/1 mm Hg between the 2 treatment arms masked a difference of 10/4 mm Hg in 24-hour ambulatory blood pressure and a difference of 17/8 mm Hg in night-time blood pressure.39,40
Thus, instead of trying to work out why antihypertensive drugs could exert apparently different cardiovascular protective efficacy in different trials, the simple and consistent message is that the lower the achieved blood pressure, the lower the adverse clinical cardiovascular outcome.
What makes sense for your patient?
In selecting antihypertensive drugs, physicians should be guided by data supporting a particular drug in coexisting clinical conditions. (See “Where to begin when there are coexisting conditions,”.) In the hypertensive patient who is free of clinical disease, a case can be made for a diuretic as the first-line drug, although calcium channel blockers, ARBs, and ACE inhibitors can also claim evidence to support their use. In the older patient, beta-blockers—especially atenolol—should not be the drug of first choice.35,36,41
As this review of comparative hypertension drug trials shows, multiple drugs are required for adequate blood pressure control in most patients. Thus, physicians should not be too preoccupied about how to initiate treatment, but remember to add drugs until adequate control is achieved.
Correspondence
H T Ong, FRCP, FACC, FESC, H T Ong Heart Clinic, 251C Burma Road, Penang 10350, Malaysia; [email protected]
- In your efforts to reduce cardiovascular events in hypertensive patients, concentrate on getting patients to goal, rather than on which drugs to use to get them there (A).
- Beta-blockers—especially atenolol—should not be the drug of first choice when treating older patients with hypertension (A).
- Multiple drugs are required for adequate blood pressure control in most patients (A).
Strength of recommendation (SOR)
- Good quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Forget about a silver bullet.
Researchers have conducted numerous trials over the last decade to find an antihypertensive drug that best reduces cardiovascular events while reducing blood pressure. However, this objective review of 13 comparative antihypertensive drug trials over the past decade involving more than 168,000 patients reveals no great differences in the cardiovascular protective effects of diuretics, beta-blockers, calcium channel blockers, angiotensin receptor blockers (ARBs), and angiotensin-converting enzyme (ACE) inhibitors.
In fact, this review indicates that there were no significant differences in the primary cardiovascular endpoints in more than 90% of the patients studied. Where a difference in secondary clinical outcome was demonstrated, fewer events consistently occurred in the regimen that reached the lower blood pressure level.
This assessment will likely fly in the face of the way that many would view this body of research. That’s understandable. At first glance, it would appear that these 13 trials, with different methodology and endpoints, have produced conflicting conclusions with the confusion worsened by pharmaceutical companies seeking to interpret the results to best suit their marketing needs.1-3
It is not the quality of the data, however, that is in question; the controversy lies in the interpretation. Subjecting the studies to further statistical analysis would simply obscure the information.
By reviewing the data impartially and objectively as a whole, though, and interpreting individual studies in light of similar studies, it becomes evident that there is more consensus than conflict. The studies support the notion that we should concentrate on getting patients to goal, rather than focusing on which drugs we’ll use to get them there.
Methods
I performed a PubMed search of the last 10 years using the keywords hypertension, comparative, drug trials. I supplemented my search with references from the JNC 7, WHO, BHS/NICE, and European hypertension guidelines. For this review, I included only randomized controlled trials with clinical cardiovascular primary endpoints. The studies had to have enrolled at least 500 patients and followed them for at least 3 years. Thirteen trials satisfied these criteria.4-17 All 13 are summarized in the TABLE, but I will review 5 of the more recent trials here. They are:
- ASCOT-BPLA—Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm
- ALLHAT—Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial
- ANBP2—Second Australian National Blood Pressure Study
- LIFE—Losartan Intervention For Endpoint reduction
- VALUE—Valsartan Antihypertensive Long-term Use Evaluation.
Calcium channel blockers vs beta-blockers
ASCOT-BPLA studied 19,257 high-risk hypertensive patients on amlodipine (Norvasc), adding perindopril, or atenolol (Tenormin), adding bendroflumethiazide.17 After 5.5 years, the primary end-point of nonfatal myocardial infarction (MI) and cardiovascular death was similar (relative risk [RR]=0.90; 95% confidence interval [CI], 0.79–1.02; P=.1052).
Total coronary endpoint, stroke, and mortality were all lower on amlodipine. Blood pressure was significantly lower on amlodipine compared with atenolol, with an average difference of 2.7/1.9 mm Hg over the trial duration.18
At the end of the trial, patients on amlodipine also had a significantly higher HDL cholesterol, and lower body mass index, triglyceride, creatinine, and glucose levels. However, when researchers made a multivariate adjustment for all of these risk factors, cardiovascular event rate differences between the 2 groups disappeared, underscoring the importance of controlling for all risk factors in reducing clinical cardiovascular events.18,19
A careful reading of ASCOT-BPLA, then, makes it clear that this study does not support the notion that newer antihypertensives (calcium channel blockers and ACE inhibitors) are superior to older ones (beta-blockers and diuretics).20,21 This study actually demonstrates that while blood pressure reduction is vital, the differences between regimens are less important.
TABLE
More consensus than conflict among 13 comparative antihypertensive drug trials with cardiovascular primary endpoints
| YEAR | TRIAL | N | DRUGS COMPARED | PRIMARY ENDPOINT | RELATIVE RISK (95% CI) | P VALUE |
|---|---|---|---|---|---|---|
| 1998 | UKPDS4 | 758 | Captopril vs atenolol | Clinical diabetic event | 1.1 (0.86–1.41) | .43 |
| Diabetic death | 1.27 (0.82–1.97) | .28 | ||||
| Total mortality | 1.14 (0.81–1.61) | .44 | ||||
| 1999 | CAPPP5 | 10,985 | Captopril vs diuretic/beta-blocker | MI+stroke+CV death | 1.05 (0.90–1.22) | .52 |
| 1999 | STOP 26 | 6614 | New vs conventional drugs | CV death | 0.99 (0.84–1.16) | .89 |
| 4418 | ACE I vs conventional drugs | CV death | 1.01 (0.84–1.22) | .89 | ||
| 4409 | CCB vs conventional drugs | CV death | 0.97 (0.80–1.17) | .72 | ||
| 2000 | INSIGHT7 | 6321 | Nifedipine LA vs diuretic | CV death, MI, HF, stroke | 1.1 (0.91–1.34) | .35 |
| 2000 | NORDIL8 | 10,881 | Diltiazem vs beta-blocker/diuretic | Stroke, MI, CV death | 1.00 (0.87–1.15) | .97 |
| 2002 | LIFE9 | 9193 | Losartan vs atenolol | CV death, stroke, MI | 0.87 (0.77–0.98) | .021 |
| 2002–3 | ALLHAT10,11 | 24,303 | Amlodipine vs chlorthalidone | Fatal CHD, nonfatal MI | 0.98 (0.90–1.07) | .65 |
| 24,309 | Lisinopril vs chlorthalidone | Fatal CHD, nonfatal MI | 0.99 (0.91–1.08) | .81 | ||
| 24,314 | Doxazosin vs chlorthalidone | Fatal CHD, nonfatal MI | 1.03 (0.93–1.15) | .62 | ||
| 2003 | ANBP212 | 6083 | ACE I vs diuretic | CV event,* death | 0.89 (0.79–1.00) | .05 |
| 2003 | CONVINCE13 | 16,602 | Verapamil vs atenolol/thiazide | Stroke, MI, CV death | 1.02 (0.88–1.18) | .77 |
| 2003 | INVEST14 | 22,576 | Verapamil vs atenolol | Death, nonfatal MI, nonfatal stroke | 0.98 (0.90–1.06) | .57 |
| 2004 | VALUE15 | 15,245 | Valsartan vs amlodipine | CV event† | 1.04 (0.94–1.15) | .49 |
| 2004 | JMIC-B16 | 1650 | Nifedipine retard vs ACE I | Cardiac events‡ | 1.05 (0.81–1.37) | .86 |
| 2005 | ASCOT17 | 19,257 | Amlodipine (+ perindopril) vs atenolol (+ thiazide) | Nonfatal MI, fatal CHD | 0.90 (0.79–1.02) | .1052 |
| ACE I, angiotensin-converting enzyme inhibitor; CCB, calcium channel blocker; CHD, coronary heart disease; CI, confidence interval; CV, cardiovascular; HF, heart failure; MI, myocardial infarction. | ||||||
| *Defined as coronary events including MI, heart failure, acute occlusion of artery, dissecting or ruptured aortic aneurysm, and cerebrovascular events including stroke and transient ischemic attacks. | ||||||
| † Defined as cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. | ||||||
| ‡ Defined as cardiac death or sudden death, MI, angina pectoris requiring hospitalization, heart failure requiring hospitalization, serious arrhythmia, and coronary interventions. | ||||||
Largest hypertensive trial ever studied 4 drugs
ALLHAT, the largest hypertensive trial ever conducted, randomized 15,255 patients to chlorthalidone, 9061 to doxazosin (Cardura), 9048 to amlodipine, and 9054 to lisinopril (Prinivil/Zestril).10,11 (The arm involving doxazosin was terminated after 3.2 years.11,22)
Compared with the beta-blocker, more patients achieved target blood pressure control on chlorthalidone (63% vs 58%), and systolic blood pressure was about 2 mm Hg lower. Although the primary outcome of fatal coronary heart disease and nonfatal MI was equal in both groups (doxazosin=7.91%; chlorthalidone=7.76%; RR=1.03 [95% CI, 0.93–1.15]; P=.62), the doxazosin arm had more stroke, heart failure, and combined cardiovascular events.
Patients on amlodipine and lisinopril had a longer follow-up of 4.9 years. Systolic blood pressure was higher on amlodipine (0.8 mm Hg, P=.03) and lisinopril (2 mm Hg, P<.001) than on chlorthalidone. The primary endpoint (fatal coronary heart disease and nonfatal MI) was similar on the diuretic (11.5%), calcium channel blocker (11.3%; RR=0.98 [95% CI, 0.90–1.07]; P=.65), and ACE inhibitor (11.4%; RR=0.99 [95% CI, 0.91–1.08]; P=.81).
Compared with the diuretic arm, the calcium channel blocker arm had a higher incidence of heart failure, while the ACE inhibitor arm had a higher incidence of heart failure, stroke, and combined cardiovascular disease. The results were similar whatever the initial glycemic state, renal function status, and racial makeup of the patients studied.23-26 More than 60% of patients in ALLHAT required 2 or more drugs for good blood pressure control.27
“Diuretics first” for patients with or without diabetes?
In ALLHAT, although diabetes occurred more frequently and fasting glucose rose in patients on diuretics, these metabolic abnormalities did not result in more cardiovascular events. Even among patients with diabetes, heart failure was more common on doxazosin, amlodipine, and lisinopril compared with those on chlorthalidone.23,24
Given that the ultimate aim of hypertensive therapy is to reduce clinical disease—not just to improve laboratory profiles—ALLHAT should put to rest any apprehension physicians have about diuretic use. These findings have even led to suggestions that diuretics be the first line antihypertensive agent, in both diabetic and nondiabetic patients.28-30
ACE inhibitor vs diuretic
ANBP2 randomized hypertensive patients to initial treatment with an ACE inhibitor (n=3044) or a diuretic (n=3039).12 With similar blood pressure reduction in both arms (26/12 mm Hg), treatment with the ACE inhibitor resulted in a lower incidence of the composite primary end-point of cardiovascular events or total death that was of borderline significance (ACE inhibitor=22.8%; diuretic=24.2%; RR=0.89 [95% CI, 0.79–1.00]; P=.05).
Among women, there was no difference between the ACE inhibitor and diuretic groups. In the overall population, there was also no difference individually of total mortality or incidence of first cardiovascular event or death.
Thus ANBP2 actually confirms the results from ALLHAT by showing that ACE inhibitors and diuretics are equivalent in reducing cardiovascular events in hypertension.31
Losartan vs atenolol
In the LIFE study, 9193 hypertensive patients with left ventricular hypertrophy were randomized to either losartan (Cozaar) or atenolol.9 Losartan treatment resulted in a marked reduction in stroke incidence, which produced a significant reduction in the composite primary end-point of death, MI, or stroke (11% vs 13%; RR=0.87 [95% CI, 0.77–0.98]; P=.021).
When only the 1195 patients with diabetes were assessed, there was a significant reduction not only in the primary endpoint but also in cardiovascular and total mortality.32 Surprisingly, the reduction of stroke incidence did not reach statistical significance in this diabetic population (RR=0.79 [95% CI, 0.55–1.14]; P=.204).
A word of caution, though: The results of LIFE should be taken together with data from other trials. No other study has demonstrated a special benefit from the renin-angiotensin antagonists in preventing stroke. In fact, ACE inhibitors were weaker than the comparator drugs in preventing stroke in both CAPPP (TABLE) and ALLHAT.5,10 Various reviews have suggested that among antihypertensive drugs, it is the diuretics and calcium channel blockers that may be more useful in stroke reduction.33,34
Chalk the benefit up to the drop in blood pressure
In the LIFE study, the treated mean systolic blood pressure was lower with losartan in the overall (1.1 mm Hg; P=.017) and diabetic (2 mm Hg; P value not stated) populations, and thus the clinical benefit could possibly have been from the better blood pressure reduction on losartan. Furthermore, there is evidence that beta-blockers are less useful in the older hypertensive patient, and are especially weak in preventing stroke incidence.35,36
Rather than showing the superiority of the ARB, it is fair to say that LIFE actually confirms the importance of blood pressure reduction, and reveals the weaker cardiovascular protective effect of atenolol in older hypertensive patients.
Valsartan, amlodipine in high-risk patients
VALUE randomized 15,245 high-risk hypertensive patients to valsartan (Diovan) and amlodipine.15,37 Trial researchers sought to study the difference—for the same level of blood pressure reduction—between the 2 regimens in the incidence of cardiac events defined as sudden cardiac death, hospitalized heart failure, nonfatal MI, and emergency procedures to prevent MI. That said, the attained blood pressure was lower on the calcium channel blocker: 4.0/2.1 mm Hg at 1 month and 2.1/1.7 mm Hg at the end of study.
After 4.2 years, there was no significant difference in the primary endpoint of first cardiac event (10.6% valsartan/10.4% amlodipine; RR=1.04 [95% CI, 0.94–1.15]; P=.49). Diabetes was lower, but the rate of MI was higher on valsartan. After correction for the blood pressure difference, the composite of cardiac events, stroke, death, or MI was similar in the 2 groups.38
VALUE patients reaching adequate blood pressure control by 6 months fared better, regardless of drug type used. Thus demonstrating that the benefit from good blood pressure control was more important than the subtle differences between antihypertensive drugs. The better metabolic profile in the angiotensin receptor blocker arm did not translate into a reduction in adverse clinical disease.
The VALUE trial suggests (as did ALLHAT) that drugs targeting the reninangiotensin system do not provide special cardiovascular protection.10,15
Choosing an antihypertensive drug according to the clinical disease and target organ most at risk of damage is logical and in keeping with numerous guidelines.42-45 Thus, you’ll want to treat hypertensive patients with these conditions as follows:
- Angina pectoris. Therapy should include a beta-blocker or calcium channel blocker, given their definite antianginal and possible anti-atherosclerotic effects.16,46,47
- Prior MI. Start the patient on a beta-blocker.47
- Poor left ventricular function. Start the patient on a diuretic, and then add an ACE inhibitor and beta-blocker, as needed.10,49,50
- Prior stroke (or a patient at special risk of stroke). Begin therapy with a calcium channel blocker or a diuretic.33,34
- Diabetic proteinuria. An ARB or an ACE inhibitor is best suited to prevent and delay nephropathy.51-54
Consensus emerges from studies spanning 10 years
This objective review of the comparative hypertension drug trials shows that there are no great differences in the cardiovascular protective efficacy of the diuretics, beta-blockers, calcium channel blockers, ARBs, and ACE inhibitors.
There was no significant difference in the cardiovascular primary endpoint in 11 of the 13 trials reviewed, involving 91% of the randomized 168,593 patients (TABLE).4-8,10,11,13-17 Of the remaining 2 trials, the difference in ANBP2 just reached a P value of .05, while the result in LIFE was driven by a lower stroke incidence on ARB treatment that is not noted in any of the other studies involving an ARB or ACE inhibitor.4-6,10,12-15
Focus on controlling blood pressure with combination of drugs
Given the very large number of patients studied in these well-conducted trials, if there were any especially useful, or detrimental, cardiovascular effect of a particular class of antihypertensive drug, it would have been obvious by now. Since most patients will require multiple drugs, the equivalent protective efficacy of different antihypertensive drugs is reassuring and suggests that physicians should not worry too much about which drug to start the patient on.28 Rather, the emphasis should be on how best to reach adequate blood pressure control by combining several antihypertensive drugs.
Small blood pressure differences, big impact
In LIFE (losartan vs atenolol), ALLHAT (doxazosin, amlodipine, lisinopril vs chlorthalidone), VALUE (amlodipine vs valsartan), and ASCOT (amlodipine vs atenolol), where a secondary cardiovascular endpoint was lower in one of the treatment arms, it was always the arm with the lower achieved blood pressure that had the better clinical outcome.9-11,15,17
Marya Ostrowski, JFP Editor
With only 50% of patients typically taking their medications as prescribed and the cost of poor adherence reaching an estimated $177 billion annually in direct and indirect health care costs, one medication safety group is saying enough is enough.
The National Council on Patient Information and Education (NCPIE), a nonprofit coalition that includes health professional associations, government agencies, and pharmaceutical companies, issued a report this summer detailing a 10-step action plan for reducing the adverse health and economic consequences of poor medication adherence.
The plan, developed by a panel of experts that NCPIE convened, calls on the government and health care community to, among other things:
- address the barriers to patient adherence for patients with low health literacy.
- develop a curriculum on medication adherence for use in medical schools.
- mount a unified national education campaign to make patient adherence a national health priority.
“Medication adherence is America’s new drug problem,” said Carolyn M. Clancy, MD, director of the Agency for Healthcare Research and Quality. AHRQ has been working with NCPIE, the FDA, and the National Consumers League to develop a public education campaign on medication adherence, according to Clancy. The NCPIE report helps to bolster those ongoing efforts, she said.
On the heels of the report, NCPIE is planning on releasing videos that will teach seniors about properly taking their medications, according to Ray Bullman, NCPIE’s executive vice president.
To learn more about NCPIE’s initiatives, or for a copy of the report, Enhancing Prescription Medicine Adherence: A National Action Plan, point your browser to: www.talkaboutrx.org.
These achieved blood pressure differences although small, were significant. Small overall mean blood pressure differences could mask much larger blood pressure differences in the individual patient. Consider, for instance, the HOPE (Heart Outcomes Prevention Evaluation) trial, where a reported overall blood pressure difference of only 3/1 mm Hg between the 2 treatment arms masked a difference of 10/4 mm Hg in 24-hour ambulatory blood pressure and a difference of 17/8 mm Hg in night-time blood pressure.39,40
Thus, instead of trying to work out why antihypertensive drugs could exert apparently different cardiovascular protective efficacy in different trials, the simple and consistent message is that the lower the achieved blood pressure, the lower the adverse clinical cardiovascular outcome.
What makes sense for your patient?
In selecting antihypertensive drugs, physicians should be guided by data supporting a particular drug in coexisting clinical conditions. (See “Where to begin when there are coexisting conditions,”.) In the hypertensive patient who is free of clinical disease, a case can be made for a diuretic as the first-line drug, although calcium channel blockers, ARBs, and ACE inhibitors can also claim evidence to support their use. In the older patient, beta-blockers—especially atenolol—should not be the drug of first choice.35,36,41
As this review of comparative hypertension drug trials shows, multiple drugs are required for adequate blood pressure control in most patients. Thus, physicians should not be too preoccupied about how to initiate treatment, but remember to add drugs until adequate control is achieved.
Correspondence
H T Ong, FRCP, FACC, FESC, H T Ong Heart Clinic, 251C Burma Road, Penang 10350, Malaysia; [email protected]
1. Psaty BM, Weiss NS, Furberg CD. Recent trials in hypertension. Compelling science or commercial speech? JAMA 2006;295:1704-1706.
2. Abramson J, Starfield B. The effect of conflict of interest on biomedical research and clinical practice guidelines: Can we trust the evidence in evidence-based medicine? J Am Board Fam Pract 2005;18:414-418.
3. Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167-1170.
4. UK Prospective Diabetes Study Group. Efficacy of Atenolol and Captopril in Reducing Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 39. BMJ 1998;317:713-720.
5. Hansson L, Lindholm LH, Niskanen L, et al. Effect of ACE inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP). Lancet 1999;353:611-616.
6. Hansson L, Lindholm LH, Ekbom T, et al. Randomized trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension 2 (STOP-Hypertension 2) study. Lancet 1999;354:1751-1756.
7. Brown MJ, Palmer CR, Castaigne A, et al. Morbidity and mortality in patients randomized to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 2000;356:366-372.
8. Hansson L, Hedner T, Lund-Johansen P, et al. For the NORDIL Study Group. Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) Study. Lancet 2000;356:359-365.
9. Dahlof B, Devereux RB, Kjeldsen SE, et al. For the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:995-1003.
10. The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to prevent Heart Attack trial (ALLHAT). JAMA 2002;288:2981-2997.
11. ALLHAT officers and Coordinators for the ALLHAT Collaborative research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and lipidlowering treatment to prevent Heart Attack trial (ALLHAT). Hypertension 2003;42:239-246.
12. Wing LMH, Reid CM, Ryan P, et al. For the Second Australian National blood pressure Study Group. A comparison of outcomes with angiotensin converting enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med 2003;348:583-592.
13. Black HR, Elliott WJ, Grandits G, et al. For the CONVINCE research Group. Principal results of the Controlled ONset Verapamil INvestigation of Cardiovascular End points (CONVINCE) trial. JAMA 2003;289:2073-2082.
14. Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al. For the INVEST Investigators. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): A randomized controlled trial. JAMA 2003;290:2805-2816.
15. Julius S, Kjeldsen SE, Weber M. for the VALUE trial group. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomized trial. Lancet 2004;363:2022-2031.
16. Yui Y, Sumiyoshi T, Kodama K, et al. Comparison of nifedipine retard with angiotensin-converting enzyme inhibitors in Japanese hypertensive patients with coronary artery disease: the Japan multicenter Investigation for Cardiovascular Diseases-B (JMIC-B) randomized trial. Hypertens Res 2004;27:181-191.
17. Dahlof B, Sever PS, poulter NR. for the ASCOT investigators. prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA): a multicentre randomized controlled trial. Lancet 2005;366:895-906.
18. Poulter NR, Wedel H, Dahlof B. For the ASCOT investigators. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA). Lancet 2005;366:907-913.
19. Daviglus ML, Liu K. today’s Agenda. We must focus on achieving favorable levels of all risk factors simultaneously. Arch Intern Med 2004;164:2086-2087.
20. Duerden M. ASCOT-BPLA. Lancet 2006;367:206.-
21. Cave JA. ASCOT: A tale of two treatment regimes. Is ASCOT all it’s cracked up to be? BMJ 2005;331:1023.-
22. The ALLHAT officers and Coordinators for the AllHAt Collaborative research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2000;283:1967-1975.
23. Barzilay JI, Davis BR, Bettencourt J, et al. For the ALLHAT Collaborative research Group. Cardiovascular outcomes using doxazosin vs chlorthalidone for the treatment of hypertension in older adults with and without glucose disorders: a report from the ALLHAT Study. J Clin Hypertens 2004;6:116-125.
24. Whelton PK, Barzilay J, Cushman WC, et al. For the ALLHAT Collaborative research group. Clinical outcomes in antihypertensive treatment of type 2 diabetes, impaired fasting glucose concentration, and normoglycemia. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med. 2005;165:1401-1409.
25. Rahman M, Pressel S, Davis BR, et al. For the ALLHAT Collaborative research Group. Renal outcomes in high-risk hypertensive patients treated with an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: a report from the ALLHAT Study. Arch Inten Med 2005;165:936-946.
26. Wright JT, Dunn JK, Cutler JA, et al. for the ALLHAT Collaborative research Group. outcomes in hyper-tensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA 2005;293:1595-1607.
27. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens 2002;4:393-504.
28. Williams B. Drug treatment for hypertension: most patients will need a treatment cocktail –including a thiazide diuretic. BMJ 2003;326:61-62.
29. Appel LJ. the verdict from ALLHAT—thiazide diuretics are the preferred initial therapy for hypertension. JAMA 2002;288:3039-3042.
30. Salvetti A, Ghiadoni L. Guidelines for antihypertensive treatment: An update after the ALLHAT study. J Am Soc Nephrol 2004;15:S51-S54.
31. Frohlich ED. Treating hypertension-what are we to believe? N Engl J Med 2003;348:639-641.
32. Lindholm LH, Ibsen H, Dahlof B, et al. For the LIFE study group. Cardiovascular morbidity and mortality in patients with diabetes in the losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:1004-1010.
33. Messerli FH, Grossman E, Lever AF. Do thiazide diuretics confer specific protection against strokes? Arch Intern Med 2003;163:2557-2560.
34. Opie LH, Schall R. Evidence-based evaluation of calcium channel blockers for hypertension. J Am Coll Cardiol 2002;39:315-322.
35. Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004;364:1684-1689.
36. Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006;174:1737-1742.
37. Kjeldsen SE, Julius S, Brunner H, et al. for the VALUE Trial Group. Characteristics of 15314 hypertensive patients at high coronary risk. the VALUE trial. Blood Press 2001;10:83-91.
38. Weber MA, Julius S, Kjeldsen SE, et al. Blood pressure dependent and independent effects of antihypertensive treatment on clinical events in the VALUE trial. Lancet 2004;363:2049-2051.
39. The Heart outcomes prevention evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000;342:145-153.
40. Svensson P, De Faire U, Sleight P, Yusuf S, Ostergren J. Comparative effects of ramipril on ambulatory and office blood pressures: a Hope substudy. Hypertension 2001;38:28-32.
41. Ong HT. Beta-blockers in hypertension and cardiovascular disease. BMJ 2007;334:946-949.
42. Chobanian AV, Bakris GL, Black HR, et al. The National High blood pressure education program Coordinating Committee.The Seventh report of the Joint National Committee on prevention, Detection, evaluation, and treatment of High blood pressure. the JNC 7 report. JAMA 2003;289:2560-2572.
43. Guidelines Committee. 2003 european Society of Hypertension-european Society of Cardiology guidelines for the management of arterial hypertension. J Hypertens 2003;21:1011-1053.
44. World Health Organization. International Society of Hypertension Writing Group. 2003 World Health Organisation (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens 2003;21:1983-1992.
45. Williams B, Poulter NR, Brown MJ, et al. Guidelines for management of hypertension: report of the fourth working party of the british Hypertension Society, 2004—BHS IV. J Hum Hypertens 2004;18:139-185.
46. Nissen SE, Tuzcu EM, Libby P. for the CAMELOT investigators. effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure. the CAMELOT Study: a randomized controlled trial. JAMA 2004;292:2217-2226.
47. Hedblad B, Wikstrand J, Janzon L, Wedel H, Berglund G. Low dose Metoprolol CR/XL and Fluvastatin slow progression of carotid intima-media thickness: main results from the beta-blocker Cholesterol-lowering Asymptomatic plaque Study (BCAPS). Circulation 2001;103:1721-1726.
48. Freemantle N, Cleland J, Young P, Mason J, Harrison J. Beta-blockade after myocardial infarction: systematic review and meta regression analysis. BMJ 1999;318:1730-1737.
49. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992;372:685-691.
50. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised Intervention trial in Congestive Heart Failure (MERIT-HF). Lancet 1999;353:2001-2006.
51. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-860.
52. Parving HH, Lehnert H, Mortensen JB, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-878.
53. Brenner BM, Cooper ME, Zeeuw D de, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-869.
54. Wright JT, Bakris G, Greene T, et al. For the African American Study of Kidney Disease and Hypertension Study Group. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 2002;288:2421-2431.
1. Psaty BM, Weiss NS, Furberg CD. Recent trials in hypertension. Compelling science or commercial speech? JAMA 2006;295:1704-1706.
2. Abramson J, Starfield B. The effect of conflict of interest on biomedical research and clinical practice guidelines: Can we trust the evidence in evidence-based medicine? J Am Board Fam Pract 2005;18:414-418.
3. Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167-1170.
4. UK Prospective Diabetes Study Group. Efficacy of Atenolol and Captopril in Reducing Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 39. BMJ 1998;317:713-720.
5. Hansson L, Lindholm LH, Niskanen L, et al. Effect of ACE inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP). Lancet 1999;353:611-616.
6. Hansson L, Lindholm LH, Ekbom T, et al. Randomized trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension 2 (STOP-Hypertension 2) study. Lancet 1999;354:1751-1756.
7. Brown MJ, Palmer CR, Castaigne A, et al. Morbidity and mortality in patients randomized to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 2000;356:366-372.
8. Hansson L, Hedner T, Lund-Johansen P, et al. For the NORDIL Study Group. Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) Study. Lancet 2000;356:359-365.
9. Dahlof B, Devereux RB, Kjeldsen SE, et al. For the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:995-1003.
10. The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to prevent Heart Attack trial (ALLHAT). JAMA 2002;288:2981-2997.
11. ALLHAT officers and Coordinators for the ALLHAT Collaborative research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and lipidlowering treatment to prevent Heart Attack trial (ALLHAT). Hypertension 2003;42:239-246.
12. Wing LMH, Reid CM, Ryan P, et al. For the Second Australian National blood pressure Study Group. A comparison of outcomes with angiotensin converting enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med 2003;348:583-592.
13. Black HR, Elliott WJ, Grandits G, et al. For the CONVINCE research Group. Principal results of the Controlled ONset Verapamil INvestigation of Cardiovascular End points (CONVINCE) trial. JAMA 2003;289:2073-2082.
14. Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al. For the INVEST Investigators. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): A randomized controlled trial. JAMA 2003;290:2805-2816.
15. Julius S, Kjeldsen SE, Weber M. for the VALUE trial group. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomized trial. Lancet 2004;363:2022-2031.
16. Yui Y, Sumiyoshi T, Kodama K, et al. Comparison of nifedipine retard with angiotensin-converting enzyme inhibitors in Japanese hypertensive patients with coronary artery disease: the Japan multicenter Investigation for Cardiovascular Diseases-B (JMIC-B) randomized trial. Hypertens Res 2004;27:181-191.
17. Dahlof B, Sever PS, poulter NR. for the ASCOT investigators. prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA): a multicentre randomized controlled trial. Lancet 2005;366:895-906.
18. Poulter NR, Wedel H, Dahlof B. For the ASCOT investigators. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac outcomes trial-blood pressure lowering Arm (ASCOT-BPLA). Lancet 2005;366:907-913.
19. Daviglus ML, Liu K. today’s Agenda. We must focus on achieving favorable levels of all risk factors simultaneously. Arch Intern Med 2004;164:2086-2087.
20. Duerden M. ASCOT-BPLA. Lancet 2006;367:206.-
21. Cave JA. ASCOT: A tale of two treatment regimes. Is ASCOT all it’s cracked up to be? BMJ 2005;331:1023.-
22. The ALLHAT officers and Coordinators for the AllHAt Collaborative research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2000;283:1967-1975.
23. Barzilay JI, Davis BR, Bettencourt J, et al. For the ALLHAT Collaborative research Group. Cardiovascular outcomes using doxazosin vs chlorthalidone for the treatment of hypertension in older adults with and without glucose disorders: a report from the ALLHAT Study. J Clin Hypertens 2004;6:116-125.
24. Whelton PK, Barzilay J, Cushman WC, et al. For the ALLHAT Collaborative research group. Clinical outcomes in antihypertensive treatment of type 2 diabetes, impaired fasting glucose concentration, and normoglycemia. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med. 2005;165:1401-1409.
25. Rahman M, Pressel S, Davis BR, et al. For the ALLHAT Collaborative research Group. Renal outcomes in high-risk hypertensive patients treated with an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: a report from the ALLHAT Study. Arch Inten Med 2005;165:936-946.
26. Wright JT, Dunn JK, Cutler JA, et al. for the ALLHAT Collaborative research Group. outcomes in hyper-tensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA 2005;293:1595-1607.
27. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens 2002;4:393-504.
28. Williams B. Drug treatment for hypertension: most patients will need a treatment cocktail –including a thiazide diuretic. BMJ 2003;326:61-62.
29. Appel LJ. the verdict from ALLHAT—thiazide diuretics are the preferred initial therapy for hypertension. JAMA 2002;288:3039-3042.
30. Salvetti A, Ghiadoni L. Guidelines for antihypertensive treatment: An update after the ALLHAT study. J Am Soc Nephrol 2004;15:S51-S54.
31. Frohlich ED. Treating hypertension-what are we to believe? N Engl J Med 2003;348:639-641.
32. Lindholm LH, Ibsen H, Dahlof B, et al. For the LIFE study group. Cardiovascular morbidity and mortality in patients with diabetes in the losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:1004-1010.
33. Messerli FH, Grossman E, Lever AF. Do thiazide diuretics confer specific protection against strokes? Arch Intern Med 2003;163:2557-2560.
34. Opie LH, Schall R. Evidence-based evaluation of calcium channel blockers for hypertension. J Am Coll Cardiol 2002;39:315-322.
35. Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004;364:1684-1689.
36. Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006;174:1737-1742.
37. Kjeldsen SE, Julius S, Brunner H, et al. for the VALUE Trial Group. Characteristics of 15314 hypertensive patients at high coronary risk. the VALUE trial. Blood Press 2001;10:83-91.
38. Weber MA, Julius S, Kjeldsen SE, et al. Blood pressure dependent and independent effects of antihypertensive treatment on clinical events in the VALUE trial. Lancet 2004;363:2049-2051.
39. The Heart outcomes prevention evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000;342:145-153.
40. Svensson P, De Faire U, Sleight P, Yusuf S, Ostergren J. Comparative effects of ramipril on ambulatory and office blood pressures: a Hope substudy. Hypertension 2001;38:28-32.
41. Ong HT. Beta-blockers in hypertension and cardiovascular disease. BMJ 2007;334:946-949.
42. Chobanian AV, Bakris GL, Black HR, et al. The National High blood pressure education program Coordinating Committee.The Seventh report of the Joint National Committee on prevention, Detection, evaluation, and treatment of High blood pressure. the JNC 7 report. JAMA 2003;289:2560-2572.
43. Guidelines Committee. 2003 european Society of Hypertension-european Society of Cardiology guidelines for the management of arterial hypertension. J Hypertens 2003;21:1011-1053.
44. World Health Organization. International Society of Hypertension Writing Group. 2003 World Health Organisation (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens 2003;21:1983-1992.
45. Williams B, Poulter NR, Brown MJ, et al. Guidelines for management of hypertension: report of the fourth working party of the british Hypertension Society, 2004—BHS IV. J Hum Hypertens 2004;18:139-185.
46. Nissen SE, Tuzcu EM, Libby P. for the CAMELOT investigators. effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure. the CAMELOT Study: a randomized controlled trial. JAMA 2004;292:2217-2226.
47. Hedblad B, Wikstrand J, Janzon L, Wedel H, Berglund G. Low dose Metoprolol CR/XL and Fluvastatin slow progression of carotid intima-media thickness: main results from the beta-blocker Cholesterol-lowering Asymptomatic plaque Study (BCAPS). Circulation 2001;103:1721-1726.
48. Freemantle N, Cleland J, Young P, Mason J, Harrison J. Beta-blockade after myocardial infarction: systematic review and meta regression analysis. BMJ 1999;318:1730-1737.
49. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992;372:685-691.
50. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised Intervention trial in Congestive Heart Failure (MERIT-HF). Lancet 1999;353:2001-2006.
51. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-860.
52. Parving HH, Lehnert H, Mortensen JB, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-878.
53. Brenner BM, Cooper ME, Zeeuw D de, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-869.
54. Wright JT, Bakris G, Greene T, et al. For the African American Study of Kidney Disease and Hypertension Study Group. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 2002;288:2421-2431.
MANAGING CAP: Are you up-to-date?
For our purposes, the evidence ratings are based on literature quality, not expert opinion, and are updated to comply with the SORT taxonomy*
Grade A Recommendations
- Severity-of-illness scores can be used to identify patients with CAP who are candidates for outpatient treatment.
- Appropriate outpatient antibiotic treatment for a previously healthy person, with no risk factors for drug-resistant S. pneumonia (DRSP) is a macrolide (azithromycin, clarithromycin, or erythromycin).
- High risk patients: those with co-morbidities (chronic heart, lung, liver, or renal disease), diabetes mellitus, alcoholism, malignancies, asplenia, immunosuppression, or antibiotics within 3 months should be treated with a respiratory flouroquinolone—moxifloxacin, gemifloxacin, or levofloxacin (750 mg dose).
- A beta-lactam (high-dose amoxicillin, amoxicillin clavulanate, ceftriaxone, cefpodoxime, of cefuroxime), plus a macrolide is an option for high risk patients.
- Blood cultures and sputum cultures are optional prior to treatment of outpatients.
- In geographic areas where >25% of pneumococcal organisms are macrolide resistant, a beta-lactam, plus docxycycline should be considered.
- Treat with antibiotics at least 5 days.
- Health care workers in inpatient and outpatient settings and long-term facilities should receive annual influenza immunization.
Grade B Recommendations
- Severity of illness scores should be supplemented with physician subjective opinion about individual patients. the ability to safely and reliably take oral medications and the availability of outpatient resources should be considered.
- Patients with Cap should be investigated for specific pathogens that would significantly alter standard (empirical) management decisions, when suspected on the basis of clinical assessment.
- A beta-lactam, plus doxycycline is an alternative to the beta-lactam, plus macrolide combination for high risk patients.
- Pneumococcal polysaccharide vaccine is recommended for persons >65 years of age and for those with selected high-risk concurrent diseases.
Grade C Recommendations
- In addition to clinical features, an infiltrate by chest radiograph or other imaging technique is required for the diagnosis of pneumonia.
- An appropriate outpatient treatment for previously healthy individuals with no risk factors for DRSP infection is doxycycline.
- Use respiratory hygiene measures (hand hygiene, masks, tissues) for patients with cough in outpatient settings.
*Ebell M, Siwek J, Weiss BD, et al. Strength of recommendation taxonomy (SORT): A patient-centered approach to grading evidence in the medical literature. J Fam Pract 2004; 53:111–120.
When is outpatient treatment appropriate for community-acquired pneumonia (CAP)? Which antibiotics are recommended for outpatient therapy? What are the best prevention strategies? The answers are in the consensus guidelines published earlier this year by the Infectious Diseases Society of America and the American Thoracic Society (IDSA/ATS). The new guidelines update an IDSA guideline published in 2003.
Background. Management (and prevention) of CAP is inconsistent, and there is also emerging resistance of pneumococcus to macrolides.
These guidelines were developed to hasten consistency among caregivers and hospitals in the care of patients with pneumonia. Appropriateness of outpatient care, severity of illness assessment, hospital treatment decisions, ICU care, and choice of antibiotics for high-risk patients and for drug-resistant S pneumonia were reviewed. The joint committee recommended that hospitals standardize care and create policies to increase the vaccination rate.
CASE 1
Your patient is a 45-year-old man with cough, fever, and chills. He has a history of metabolic syndrome, and a 40 pack-year smoking history. He was well until 1 week ago when he went camping in the rain. Over the last 2 days he has had shaking chills, cough productive of green phlegm, and he finds that he gets a bit short-winded when walking stairs. He wonders if he has pneumonia. He is overweight and in no acute distress.
T 101 • P 88 • RR 18 • WT 220 • HT 5 7
Exam Normal other than localized coarse rales in the left posterior lung field; spot O2 saturation is 96%
What is your diagnosis and initial management?
Which of the following statements are true regarding the outpatient management of pneumonia?
- If 2 or more CURB-65 criteria are present, the patient should be hospitalized
- A macrolide is an appropriate choice of treatment for a previously healthy person with no risk of drug resistance
- A positive chest x-ray or other imaging is required for the diagnosis
- Blood cultures and sputum cultures must be obtained
- Antibiotic treatment should be a minimum of 10 days
ANSWERS: A, B, AND C
Diagnosis Community acquired pneumonia—left lobar.
Initial management This patient can be treated as an outpatient based on the severity-of-illness scores in this guideline. He should be treated with antibiotics a minimum of 5 days. With his comorbidities, antibiotic choices include 1) a fluoroquinolone, 2) a beta-lactam plus macrolide, or 3) (in areas with high prevalence of macrolide resistance) a beta-lactam plus doxycycline.
CASE 2
A 76-year-old man is brought into the office by his niece. “I just don’t feel well,” he says. The patient has been increasingly ill over the past week, and his niece is concerned that he seems to have trouble breathing. The patient minimizes his symptoms, but in relaying his history, he is obviously short of breath and cannot talk continuously. He needed a wheelchair to come in from the parking lot (and you know that he is usually spry and ambulatory). He has a history of congestive heart failure, hypertension, type 2 diabetes mellitus, depression, and osteoarthritis. He takes furosemide, potassium, enalapril, lantus insulin, sertraline, and PRN acetaminophen. He has never smoked. He denies PND and orthopnea. He is clearly short of breath and in some mild distress.
T 99 • P 102 • RR 36 • WT 260 • HT 5 9
Exam Remarkable for diffuse rhonchi and wheezing across all lung fields; spot O2 saturation is 89%
What is your diagnosis and initial management?
The differential diagnosis for this patient includes:
- Bacterial pneumonia
- Viral pneumonia
- Depression
- Congestive heart failure
- Pulmonary embolus
ANSWERS: A, B, D, E
Diagnosis This interstitial pattern on the chest x-ray is associated with multiple etiologies, both infectious and non-infectious. Examples include viral pneumonia, opportunistic infections in HIV patients, atypical infections such as mycoplasm, congestive heart failure, and pulmonary embolus.
Initial management Based on severity-of-illness scores, this patient should be admitted to the hospital. He should have further evaluation to identify the etiology.
By definition, CAP is acquired outside a hospital or long-term care facility. However, the new guidelines include ambulatory residents of nursing homes.
Adults with CAP are the focus of the guidelines, not immunocompromised patients, cancer patients receiving chemotherapy, patients on high-dose steroid therapy, or children under 18 years.
Epidemiology. There are about 5.6 million cases of CAP in the United States annually, and the cost is about $8.4 billion.1 Death rates increase with comorbidities and older age. There are no race or gender differences in morbidity.
Limitations of the guidelines. The decision whether to admit a patient with CAP is crucial, since the majority of the pneumonia care expenditures are the result of in-patient care.2 The guidelines do not state the outcomes that were considered or adverse events associated with therapy. It is weakened by lack of cost analysis and absence of clinical algorithms.
How the evidence was graded. Electronic databases were searched through June 2006. Experts considered reviews and meta-analyses and weighted the evidence according to a rating scheme. They graded each recommendation on the quality of the literature (levels I, II, or II) and by expert interpretation (strong, moderate, or weak). A strong recommendation required that more than 50% of the experts grade it as strong and the majority of the remainder grade it as moderate.
Most patients with CAP should receive a strongly rated intervention, and the rationale for variation should be apparent from the medical record. With a moderate or weak recommendation, the committee suggested, most physicians would follow the recommended management, but many would not.
1. Lutfiyya MN, Henley E, Chang LF, Reyburn SW. Diagnosis and treatment of community-acquired pneumonia. Am Fam Physician 2006;73:442-50.
2. Niederman MS, McCombs JS, Unger AN, Kumar A, Popovian R. The cost of treating community-acquired pneumonia. Clin Ther 1998;20:820-837.
Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
For our purposes, the evidence ratings are based on literature quality, not expert opinion, and are updated to comply with the SORT taxonomy*
Grade A Recommendations
- Severity-of-illness scores can be used to identify patients with CAP who are candidates for outpatient treatment.
- Appropriate outpatient antibiotic treatment for a previously healthy person, with no risk factors for drug-resistant S. pneumonia (DRSP) is a macrolide (azithromycin, clarithromycin, or erythromycin).
- High risk patients: those with co-morbidities (chronic heart, lung, liver, or renal disease), diabetes mellitus, alcoholism, malignancies, asplenia, immunosuppression, or antibiotics within 3 months should be treated with a respiratory flouroquinolone—moxifloxacin, gemifloxacin, or levofloxacin (750 mg dose).
- A beta-lactam (high-dose amoxicillin, amoxicillin clavulanate, ceftriaxone, cefpodoxime, of cefuroxime), plus a macrolide is an option for high risk patients.
- Blood cultures and sputum cultures are optional prior to treatment of outpatients.
- In geographic areas where >25% of pneumococcal organisms are macrolide resistant, a beta-lactam, plus docxycycline should be considered.
- Treat with antibiotics at least 5 days.
- Health care workers in inpatient and outpatient settings and long-term facilities should receive annual influenza immunization.
Grade B Recommendations
- Severity of illness scores should be supplemented with physician subjective opinion about individual patients. the ability to safely and reliably take oral medications and the availability of outpatient resources should be considered.
- Patients with Cap should be investigated for specific pathogens that would significantly alter standard (empirical) management decisions, when suspected on the basis of clinical assessment.
- A beta-lactam, plus doxycycline is an alternative to the beta-lactam, plus macrolide combination for high risk patients.
- Pneumococcal polysaccharide vaccine is recommended for persons >65 years of age and for those with selected high-risk concurrent diseases.
Grade C Recommendations
- In addition to clinical features, an infiltrate by chest radiograph or other imaging technique is required for the diagnosis of pneumonia.
- An appropriate outpatient treatment for previously healthy individuals with no risk factors for DRSP infection is doxycycline.
- Use respiratory hygiene measures (hand hygiene, masks, tissues) for patients with cough in outpatient settings.
*Ebell M, Siwek J, Weiss BD, et al. Strength of recommendation taxonomy (SORT): A patient-centered approach to grading evidence in the medical literature. J Fam Pract 2004; 53:111–120.
When is outpatient treatment appropriate for community-acquired pneumonia (CAP)? Which antibiotics are recommended for outpatient therapy? What are the best prevention strategies? The answers are in the consensus guidelines published earlier this year by the Infectious Diseases Society of America and the American Thoracic Society (IDSA/ATS). The new guidelines update an IDSA guideline published in 2003.
Background. Management (and prevention) of CAP is inconsistent, and there is also emerging resistance of pneumococcus to macrolides.
These guidelines were developed to hasten consistency among caregivers and hospitals in the care of patients with pneumonia. Appropriateness of outpatient care, severity of illness assessment, hospital treatment decisions, ICU care, and choice of antibiotics for high-risk patients and for drug-resistant S pneumonia were reviewed. The joint committee recommended that hospitals standardize care and create policies to increase the vaccination rate.
CASE 1
Your patient is a 45-year-old man with cough, fever, and chills. He has a history of metabolic syndrome, and a 40 pack-year smoking history. He was well until 1 week ago when he went camping in the rain. Over the last 2 days he has had shaking chills, cough productive of green phlegm, and he finds that he gets a bit short-winded when walking stairs. He wonders if he has pneumonia. He is overweight and in no acute distress.
T 101 • P 88 • RR 18 • WT 220 • HT 5 7
Exam Normal other than localized coarse rales in the left posterior lung field; spot O2 saturation is 96%
What is your diagnosis and initial management?
Which of the following statements are true regarding the outpatient management of pneumonia?
- If 2 or more CURB-65 criteria are present, the patient should be hospitalized
- A macrolide is an appropriate choice of treatment for a previously healthy person with no risk of drug resistance
- A positive chest x-ray or other imaging is required for the diagnosis
- Blood cultures and sputum cultures must be obtained
- Antibiotic treatment should be a minimum of 10 days
ANSWERS: A, B, AND C
Diagnosis Community acquired pneumonia—left lobar.
Initial management This patient can be treated as an outpatient based on the severity-of-illness scores in this guideline. He should be treated with antibiotics a minimum of 5 days. With his comorbidities, antibiotic choices include 1) a fluoroquinolone, 2) a beta-lactam plus macrolide, or 3) (in areas with high prevalence of macrolide resistance) a beta-lactam plus doxycycline.
CASE 2
A 76-year-old man is brought into the office by his niece. “I just don’t feel well,” he says. The patient has been increasingly ill over the past week, and his niece is concerned that he seems to have trouble breathing. The patient minimizes his symptoms, but in relaying his history, he is obviously short of breath and cannot talk continuously. He needed a wheelchair to come in from the parking lot (and you know that he is usually spry and ambulatory). He has a history of congestive heart failure, hypertension, type 2 diabetes mellitus, depression, and osteoarthritis. He takes furosemide, potassium, enalapril, lantus insulin, sertraline, and PRN acetaminophen. He has never smoked. He denies PND and orthopnea. He is clearly short of breath and in some mild distress.
T 99 • P 102 • RR 36 • WT 260 • HT 5 9
Exam Remarkable for diffuse rhonchi and wheezing across all lung fields; spot O2 saturation is 89%
What is your diagnosis and initial management?
The differential diagnosis for this patient includes:
- Bacterial pneumonia
- Viral pneumonia
- Depression
- Congestive heart failure
- Pulmonary embolus
ANSWERS: A, B, D, E
Diagnosis This interstitial pattern on the chest x-ray is associated with multiple etiologies, both infectious and non-infectious. Examples include viral pneumonia, opportunistic infections in HIV patients, atypical infections such as mycoplasm, congestive heart failure, and pulmonary embolus.
Initial management Based on severity-of-illness scores, this patient should be admitted to the hospital. He should have further evaluation to identify the etiology.
By definition, CAP is acquired outside a hospital or long-term care facility. However, the new guidelines include ambulatory residents of nursing homes.
Adults with CAP are the focus of the guidelines, not immunocompromised patients, cancer patients receiving chemotherapy, patients on high-dose steroid therapy, or children under 18 years.
Epidemiology. There are about 5.6 million cases of CAP in the United States annually, and the cost is about $8.4 billion.1 Death rates increase with comorbidities and older age. There are no race or gender differences in morbidity.
Limitations of the guidelines. The decision whether to admit a patient with CAP is crucial, since the majority of the pneumonia care expenditures are the result of in-patient care.2 The guidelines do not state the outcomes that were considered or adverse events associated with therapy. It is weakened by lack of cost analysis and absence of clinical algorithms.
How the evidence was graded. Electronic databases were searched through June 2006. Experts considered reviews and meta-analyses and weighted the evidence according to a rating scheme. They graded each recommendation on the quality of the literature (levels I, II, or II) and by expert interpretation (strong, moderate, or weak). A strong recommendation required that more than 50% of the experts grade it as strong and the majority of the remainder grade it as moderate.
Most patients with CAP should receive a strongly rated intervention, and the rationale for variation should be apparent from the medical record. With a moderate or weak recommendation, the committee suggested, most physicians would follow the recommended management, but many would not.
For our purposes, the evidence ratings are based on literature quality, not expert opinion, and are updated to comply with the SORT taxonomy*
Grade A Recommendations
- Severity-of-illness scores can be used to identify patients with CAP who are candidates for outpatient treatment.
- Appropriate outpatient antibiotic treatment for a previously healthy person, with no risk factors for drug-resistant S. pneumonia (DRSP) is a macrolide (azithromycin, clarithromycin, or erythromycin).
- High risk patients: those with co-morbidities (chronic heart, lung, liver, or renal disease), diabetes mellitus, alcoholism, malignancies, asplenia, immunosuppression, or antibiotics within 3 months should be treated with a respiratory flouroquinolone—moxifloxacin, gemifloxacin, or levofloxacin (750 mg dose).
- A beta-lactam (high-dose amoxicillin, amoxicillin clavulanate, ceftriaxone, cefpodoxime, of cefuroxime), plus a macrolide is an option for high risk patients.
- Blood cultures and sputum cultures are optional prior to treatment of outpatients.
- In geographic areas where >25% of pneumococcal organisms are macrolide resistant, a beta-lactam, plus docxycycline should be considered.
- Treat with antibiotics at least 5 days.
- Health care workers in inpatient and outpatient settings and long-term facilities should receive annual influenza immunization.
Grade B Recommendations
- Severity of illness scores should be supplemented with physician subjective opinion about individual patients. the ability to safely and reliably take oral medications and the availability of outpatient resources should be considered.
- Patients with Cap should be investigated for specific pathogens that would significantly alter standard (empirical) management decisions, when suspected on the basis of clinical assessment.
- A beta-lactam, plus doxycycline is an alternative to the beta-lactam, plus macrolide combination for high risk patients.
- Pneumococcal polysaccharide vaccine is recommended for persons >65 years of age and for those with selected high-risk concurrent diseases.
Grade C Recommendations
- In addition to clinical features, an infiltrate by chest radiograph or other imaging technique is required for the diagnosis of pneumonia.
- An appropriate outpatient treatment for previously healthy individuals with no risk factors for DRSP infection is doxycycline.
- Use respiratory hygiene measures (hand hygiene, masks, tissues) for patients with cough in outpatient settings.
*Ebell M, Siwek J, Weiss BD, et al. Strength of recommendation taxonomy (SORT): A patient-centered approach to grading evidence in the medical literature. J Fam Pract 2004; 53:111–120.
When is outpatient treatment appropriate for community-acquired pneumonia (CAP)? Which antibiotics are recommended for outpatient therapy? What are the best prevention strategies? The answers are in the consensus guidelines published earlier this year by the Infectious Diseases Society of America and the American Thoracic Society (IDSA/ATS). The new guidelines update an IDSA guideline published in 2003.
Background. Management (and prevention) of CAP is inconsistent, and there is also emerging resistance of pneumococcus to macrolides.
These guidelines were developed to hasten consistency among caregivers and hospitals in the care of patients with pneumonia. Appropriateness of outpatient care, severity of illness assessment, hospital treatment decisions, ICU care, and choice of antibiotics for high-risk patients and for drug-resistant S pneumonia were reviewed. The joint committee recommended that hospitals standardize care and create policies to increase the vaccination rate.
CASE 1
Your patient is a 45-year-old man with cough, fever, and chills. He has a history of metabolic syndrome, and a 40 pack-year smoking history. He was well until 1 week ago when he went camping in the rain. Over the last 2 days he has had shaking chills, cough productive of green phlegm, and he finds that he gets a bit short-winded when walking stairs. He wonders if he has pneumonia. He is overweight and in no acute distress.
T 101 • P 88 • RR 18 • WT 220 • HT 5 7
Exam Normal other than localized coarse rales in the left posterior lung field; spot O2 saturation is 96%
What is your diagnosis and initial management?
Which of the following statements are true regarding the outpatient management of pneumonia?
- If 2 or more CURB-65 criteria are present, the patient should be hospitalized
- A macrolide is an appropriate choice of treatment for a previously healthy person with no risk of drug resistance
- A positive chest x-ray or other imaging is required for the diagnosis
- Blood cultures and sputum cultures must be obtained
- Antibiotic treatment should be a minimum of 10 days
ANSWERS: A, B, AND C
Diagnosis Community acquired pneumonia—left lobar.
Initial management This patient can be treated as an outpatient based on the severity-of-illness scores in this guideline. He should be treated with antibiotics a minimum of 5 days. With his comorbidities, antibiotic choices include 1) a fluoroquinolone, 2) a beta-lactam plus macrolide, or 3) (in areas with high prevalence of macrolide resistance) a beta-lactam plus doxycycline.
CASE 2
A 76-year-old man is brought into the office by his niece. “I just don’t feel well,” he says. The patient has been increasingly ill over the past week, and his niece is concerned that he seems to have trouble breathing. The patient minimizes his symptoms, but in relaying his history, he is obviously short of breath and cannot talk continuously. He needed a wheelchair to come in from the parking lot (and you know that he is usually spry and ambulatory). He has a history of congestive heart failure, hypertension, type 2 diabetes mellitus, depression, and osteoarthritis. He takes furosemide, potassium, enalapril, lantus insulin, sertraline, and PRN acetaminophen. He has never smoked. He denies PND and orthopnea. He is clearly short of breath and in some mild distress.
T 99 • P 102 • RR 36 • WT 260 • HT 5 9
Exam Remarkable for diffuse rhonchi and wheezing across all lung fields; spot O2 saturation is 89%
What is your diagnosis and initial management?
The differential diagnosis for this patient includes:
- Bacterial pneumonia
- Viral pneumonia
- Depression
- Congestive heart failure
- Pulmonary embolus
ANSWERS: A, B, D, E
Diagnosis This interstitial pattern on the chest x-ray is associated with multiple etiologies, both infectious and non-infectious. Examples include viral pneumonia, opportunistic infections in HIV patients, atypical infections such as mycoplasm, congestive heart failure, and pulmonary embolus.
Initial management Based on severity-of-illness scores, this patient should be admitted to the hospital. He should have further evaluation to identify the etiology.
By definition, CAP is acquired outside a hospital or long-term care facility. However, the new guidelines include ambulatory residents of nursing homes.
Adults with CAP are the focus of the guidelines, not immunocompromised patients, cancer patients receiving chemotherapy, patients on high-dose steroid therapy, or children under 18 years.
Epidemiology. There are about 5.6 million cases of CAP in the United States annually, and the cost is about $8.4 billion.1 Death rates increase with comorbidities and older age. There are no race or gender differences in morbidity.
Limitations of the guidelines. The decision whether to admit a patient with CAP is crucial, since the majority of the pneumonia care expenditures are the result of in-patient care.2 The guidelines do not state the outcomes that were considered or adverse events associated with therapy. It is weakened by lack of cost analysis and absence of clinical algorithms.
How the evidence was graded. Electronic databases were searched through June 2006. Experts considered reviews and meta-analyses and weighted the evidence according to a rating scheme. They graded each recommendation on the quality of the literature (levels I, II, or II) and by expert interpretation (strong, moderate, or weak). A strong recommendation required that more than 50% of the experts grade it as strong and the majority of the remainder grade it as moderate.
Most patients with CAP should receive a strongly rated intervention, and the rationale for variation should be apparent from the medical record. With a moderate or weak recommendation, the committee suggested, most physicians would follow the recommended management, but many would not.
1. Lutfiyya MN, Henley E, Chang LF, Reyburn SW. Diagnosis and treatment of community-acquired pneumonia. Am Fam Physician 2006;73:442-50.
2. Niederman MS, McCombs JS, Unger AN, Kumar A, Popovian R. The cost of treating community-acquired pneumonia. Clin Ther 1998;20:820-837.
Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
1. Lutfiyya MN, Henley E, Chang LF, Reyburn SW. Diagnosis and treatment of community-acquired pneumonia. Am Fam Physician 2006;73:442-50.
2. Niederman MS, McCombs JS, Unger AN, Kumar A, Popovian R. The cost of treating community-acquired pneumonia. Clin Ther 1998;20:820-837.
Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(Suppl 2):S27–S72.
Personalized medicine: The promise, the reality
Genetic tests to guide warfarin dosing could avert 85,000 serious bleeding events and 17,000 strokes annually, according to a report from the AEI-Brookings Joint Center for Regulatory Studies, a Washington, DC, think tank. The report further suggests that by integrating genetic testing into warfarin therapy, American health care spending could be reduced by $1.1 billion annually.1 Unfortunately, the promise of using genetic testing to guide such pharmacological treatment has largely gone unfulfilled.2
Case in point: Genetic testing can tell us whether a patient is likely to be an ultra-rapid metabolizer of warfarin (and need larger doses) or a poor metabolizer (and need lower doses), but there are no guidelines to tell us how to dose accordingly. International normalized ratios (INRs) still need to be ordered and the patient will likely have to pick up the tab for the genetic test ($250), since Medicare and private insurers don’t cover the cost. (See “Warfarin: An ideal, but far from ready, candidate”)
Hints that change may be on the horizon. The government—specifically the Department of Health and Human Services—created the Secretary’s Advisory Committee on Genetics, Health, and Society (SACGHS) to assess how genetic and genomic technologies are being integrated into health care and to identify opportunities and gaps in research. To that end, SACGHS issued a draft report earlier this year that notes that genetic-based treatment has “the potential to yield significant gains in personal health, population health, and cost-effective resource allocation.” Among its many recommendations, SACGHS calls for greater collaboration between the public and private sectors to expand our knowledge of the clinical validity and utility of using genetics to guide treatment.3
A standard of care, potentially. Readying ourselves for the ways that genetics is likely to shape the way we prescribe such drugs as anticoagulants, antidepressants, and antiarrhythmics requires that we step back and assess the progress made so far, and the work that still needs to be done before genetic testing becomes a common occurrence, and perhaps even a standard of care.
The goal: Avert adverse events
The wide variation in the way different people respond to the same dose of medications is a major contributor to the problem of adverse drug reactions. Lazarou and colleagues estimated that 6.7% of hospitalized patients—over 2 million patients in the US—experienced an adverse drug reaction and 0.32% (106,000) had a fatal adverse drug reaction.4
It would appear that warfarin dosing would be a perfect candidate for the clinical use of a pharmacogenetic test. Studies have shown that about 7% of the Caucasian population are poor metabolizers and at increased risk of bleeding from over coagulation and 1% are ultra-rapid metabolizers.4 Despite what we know about the polymorphisms to the CYP2C9 enzyme, which is the primary route of metabolism for warfarin, the package insert on Coumadin still doesn’t contain a recommendation for determining a patient’s genetic profile before initiating treatment.30 Similarly, the chapter on anticoagulation in Applied Therapeutics, a commonly used medical textbook, says nothing about the use of CYP polymorphisms for dosing decisions.13
At issue: Genotyping to guide dosing has not been tested in comparison to the usual monitoring using the international normalized ratio (INR).31 Specifically, the outcomes of bleeding complications and adequate anticoagulation of the 2 methods have not been compared in a clinical trial.
Here’s what we do know: In one study, the presence of specific polymorphisms was associated with a lower maintenance warfarin dose, but not with over-anticoagulation.32 In a review of 4 studies on CYP2C9 polymorphisms and warfarin daily dose, Lee and colleagues found that between the slowest and fastest metabolizers, the difference in dose was, at most, 4 mg/day. These studies did not explore if dosing decisions could accurately be made on genetic classifications and it is unlikely they could because of the wide overlap in maintenance dosages in the different classes.7
To complicate things further, the future use of warfarin in some conditions is problematic because fractionated heparin has been proven in many situations to be as effective and less risky than warfarin, and does not require frequent monitoring with blood tests. All of these unknowns make it unclear how useful genetic tests will be, and whether insurers will pay for them.
Individual response to medications is determined by a host of factors including age, environment, other medications being taken, and genetic differences in drug absorption and metabolism. These genetic differences have spawned the fields of pharmacogenomics and pharmacogenetics.
Pharmacogenomics is the biotechnological science that combines the techniques of medicine, pharmacology, and genomics and is concerned with developing drug therapies to compensate for genetic differences in patients, which cause varied responses to a single therapeutic regimen.
- A good example of pharmacogenomics at work is the use of trastuzumab in addition to chemotherapy for breast cancer patients who are positive for the human epidermal growth factor receptor 2 (HER2) oncogene.5
Pharmacogenetics is the branch of pharmacology that examines the relation of genetic factors to variations in response to drugs.
The use of pharmacogenetics to predict individualized responses to medications and to prevent adverse drug reactions through individualized dosing regimens or avoidance of certain medications hinges on our knowledge of genetic polymorphisms, that is, gene-based differences in drug absorption, distribution, metabolism, or excretion.
Polymorphisms of the cytochrome P450 family of drug metabolizing enzymes have been the most extensively studied. The names of these enzymes are abbreviated by using CYP and then a series of letters and numbers to describe individual enzymes. The 4 most extensively studied CYP enzymes are CYP2A6, CYP2C9, CYP2C19, and CYP2D6. These 4 metabolize an estimated 40% of all drugs; the distributions of polymorphisms of each vary considerably by race/ethnicity.6-8
A sampling of medications and classes of medications where polymorphisms play a significant role in drug metabolism is listed in the TABLE. Three with significant potential for prevention of adverse drug reactions are the antipsychotics, because of the severity of a specific drug adverse reaction, tardive dyskinesia;6,9 warfarin, because of the risk of bleeding complications and its narrow therapeutic index;7,10 and chemotherapeutic agents because of the serious nature of the disease and the potential for tailoring individualized therapies to maximize tumor response to medication and to minimize adverse reactions of very toxic drugs.11
TABLE
Polymorphisms to these enzymes affect drug metabolism
| DRUG/CLASS | ENZYMES |
|---|---|
| Antiarrhythmmics7 | CYP2D6 |
| Antidepressants7 | CYP2D6 |
| Antipsychotics6,9 | CYP2D6, CYP1A2, CYP2C19, CYP3A4 |
| Beta-blockers7 | CYP2D6 |
| Cancer chemotherapy11 | Varies by the agent |
| HMG-CoA reductase inhibitors (statins)29 | CYP2D6, CYP2C9, CYP2C19 |
| Losartan7 | CYP2C9 |
| Neuroleptics7 | CYP2D6 |
| NSAIDs29 | CYP2C9 |
| Phenytoin7 | CYP2C9, CYP2C19 |
| Proton pump inhibitors29 | CYP2C19 |
| Tolbutamide7 | CYP2C9 |
| Warfarin7,10 | CYP2C9 |
Clinical resources reflect an information gap
In spite of the potential for improved patient care, there remains very little clinical application of pharmacogenetic information in primary care practice. Zineh and colleagues reviewed prescribing information in the electronic version of the Physicians’ Desk Reference (PDR) in 2004 and found that only 76 package inserts out of 3382 contained pharmacogenetic information.12 In only 25 was there enough information to affect treatment decisions. Just 5 inserts mentioned that the chance of successful response to treatment could be predicted by genetic testing, and only one insert mentioned that a specific genetic subgroup should not take a drug.
The authors concluded that, generally, the pharmacogenetic information was inadequate to guide drug therapy and the majority of information was available for drugs that are not commonly prescribed. The FDA is addressing this issue by requiring the inclusion of pharmacogenetic information in package inserts more frequently.
Consider, too, the 2005 edition of Applied Therapeutics, a commonly used medical textbook.13 In it there is no mention of the use of pharmacogenetics in managing pharmacological therapies, which tells us that very little teaching on this topic is going on in medical schools and residencies.
But why? Why are clinicians and the tools we rely on so out of sync with the recommendations and expectations of personalized medicine advocates?
Certain conditions will need to exist before testing for CYP polymorphisms will become the standard of care in dosing certain drugs, such as warfarin. (see “Warfarin: An ideal, but far from ready, candidate”.) The most important is that this clinical approach needs to be proven superior to existing methods, such as INR monitoring. Should this occur, guidelines that are evidence-based will include it as a recommendation, physician continuing education courses will cover it, and it will enter into the curricula of medical schools and residency programs. But the process of adopting new, evidence-based best practices has historically been slow.32-36
One variable that may play a part in facilitating more rapid acceptance of genetic testing before warfarin use is litigation. Marchant and colleagues describe potential legal pressures that may drive medicine to adopt more personalized medicine.37 If genetic testing before warfarin use results in better outcomes (fewer catastrophic events or very bad outcomes) and there is plausible evidence that a catastrophic outcome (massive bleeding) could have been avoided with genetic information, then litigation will surely be close behind.
As news of successful litigation spreads, one of two results will likely occur: Either the use of the CYP polymorphism testing will increase, or the movement to use alternative medications, such as fractionated heparin, will accelerate.
There are 5 likely culprits:
- A lack of clinically useful pharmacogenetic tests.
- A lack of test standardization and availability.
- A lack of coverage by third-party payers.
- A low level of physician knowledge about genetic testing.
- A lack of evidence of improved outcomes.
How useful are pharmacogenetic tests?
For a laboratory test to be clinically useful, it should provide information that will influence a therapeutic decision. Decisions that could be influenced by this information include the dose of a particular drug and the potential use of an alternative because of a contraindication or likelihood of a poor outcome based on a particular genetic polymorphism.
The use of pharmacogenetic laboratory information can place a patient into one of several groups:
- ultra-rapid metabolizers, who need a larger dose of medication
- normal metabolizers, often called extensive metabolizers, who do not need dose modifications
- poor metabolizers, who need lower doses.9
Most medications have a wide therapeutic margin of effectiveness and safety. This means that the medication works within a wide range of serum drug levels and is safe at these different levels, making refinement in dosing based on genetic information unnecessary. In medications with narrower therapeutic windows for effectiveness or adverse reactions, there are frequently alternative means of drug level monitoring.
In many instances, the genetic test predictability of a patient’s actual metabolic responses—and resulting drug levels—is poor, leading to the need to monitor drug levels anyway.14-16 This occurs because there is often a great deal of overlap in the response to a medication dose among the different metabolism classifications.9,14,16-18 All of these realities have limited the clinical usefulness of pharmacogenetics up to this point.
Test standardization: Poised to improve?
Genotyping, the determination of the actual genetic makeup of the patient is not always predictive of an individual patient’s metabolic response. In other words, genotype does not always equate to a phenotype. Further complicating matters is the fact that many of the pharmacogenetic studies have been performed at research laboratories and have used tests that are not standardized, or widely available.19-21
The recent commercial availability of pharmacogenetic tests by well-established and reputable laboratories will probably improve both standardization and availability.
An example is the AmpliChip CYP450 test by Roche Diagnostics.22 This microassay-based test identifies 29 CYP-2D6 polymorphisms and 2 CYP-2C19 polymorphisms. These genes affect the metabolism of 25% of currently prescribed drugs. Using this test, patients can be classified as poor, intermediate, extensive, or ultra rapid metabolizers of CYP-2D6 affected drugs, including antidepressants, antiarrhythmics, and antipsychotics. They can also be classified as poor or extensive metabolizers of CYP-2C19 affected drugs, including phenytoin and proton pump inhibitors.
How costly?
The cost of genetic testing will also affect availability. Tests will be widely available only if covered by third-party payers. The AmpliChip test costs between $300 and $500. Clearly, then, both cost and insurance coverage are issues that impede the adoption of pharmacogenetic testing, though there is little written about this in the medical literature.19,23
AAFP explores ways to teach genomics
Physicians who are currently in practice received little or no training in the clinical use of pharmacogenetics or other genetic tests, such as genetic testing for the prediction of cancer risk.24,25 The main resources of pharmacological information for practicing physicians do not contain much, if any, useful genomic information. A recent continuing education monograph for family physicians on clinical genetics mentioned pharmacogenetics only as a promising future technology.26
For its part, the American Academy of Family Physicians has formed a genomics work group and is exploring how to educate family physicians on clinically useful genomic topics.
Evidence-based outcomes are needed
To date there has not been a head-to-head comparison of the outcomes of using clinical pharmacogenetics with those obtained from standard drug level monitoring practices. The CDC has formed a committee modeled after the USPSTF, the Evaluations of Genomics Applications in Practice and Prevention (EGAPP), which will evaluate the effectiveness of genomic clinical tests and make recommendations to physicians on their use.27 The group’s first report, on the use of CYP450 testing in depression, concluded that there is a paucity of good quality data that addresses whether testing for CYP450 polymorphisms in adults entering SSRI treatment leads to improved outcomes.28
In addition, SACGHS, the Department of Health and Human Services’ Committee, recommends in its draft report that HHS “provide resources to identify and address evidentiary gaps in the analytic validity, clinical validity, clinical utility, and cost effectiveness of pharmacogenomics.”3
Outcomes data will undoubtedly be key. With it, pharmacogenetic testing has the potential to grow by leaps and bounds—perhaps even becoming a standard of care in guiding pharmacological therapy. Without it, such testing will remain a promising, but as yet unrealized advance in personalized medicine.
Correspondence
Doug Campos-Outcalt, MD, MPA, 550 E. Van Buren, Phoenix, AZ 85004; [email protected].
1. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalized medicine using genetic testing: The case of warfarin. [Working Paper 06-23]. November 2006. Available at: www.aei-brookings.org/admin/authorpdfs/page.php?id=1337&PHPsEssID=7b3a3ae4b30d77cb76223e29535e7590. Accessed on June 27, 2007.
2. Tucker G. Pharmacogenetics—expectations and reality. BMJ 2004;329:4-6.
3. Department of Health and Human Services. Realizing the promise of pharmacogenomics: Opportunities and Challenges [Draft report of the Secretary’s Advisory Committee on Genetics, Health, and Society]. Available for public comment March 23–June 1, 2007. Available at: www4.od.nih.gov/oba/sacghs/SACGHS_Pgx_PCdraft.pdf. Accessed on June 27, 2007.
4. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients. JAMA 1998;279:1200-1205.
5. Dent R, Clemons M. Adjuvant trastuzumab for breast cancer. Br Med J 2005;331:1035-1036.
6. Wolf CR, Smith G. Pharmacokinetics. Br Med Bull 1999;55:366-386.
7. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 2002;12:251-263.
8. Bachman K. Genotyping and phenotyping the cytochrome P450 enzymes. Am J Ther 2002;9:309-316.
9. Dahl ML. Cytochrome P450 phenotyping/genotyping in patients receiving antipsychotics. Clin Pharmacokinet 2002;41:453-470.
10. Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003;13:247-252.
11. Van Schaik RHN. Implications of cytochrome P450 genetic polymorphisms on the toxicity of antitumor agents. Ther Drug Monit 2004;26:236-240.
12. Zineh I, Gerhard T, Aquilante CL, Beitelshees AL, Beasley BN, Hartzema AG. Availability of pharmacogenomics-based prescribing information in drug package inserts for currently approved drugs. Pharmacogenomics J 2004;4:354-358.
13. Koda Kimball M, ed. Applied Therapeutics. 8th ed. Baltimore, Md: Lippincott, Williams and Wilkins; 2005.
14. Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics 1995;5:389-392.
15. Arthur H, Dahl ML, Siwers B. Polymorphic drug metabolism in schizophrenic patients with tardive dyskinesia. J Clin Psychopharmacology 1995;15:211-216.
16. Mulder AB, Van Lijf HJ, Bon MAM, et al. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin Pharmacol Ther 2001;70:546-551.
17. Takahashi H, Ehizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmakinetics 2001;40:587-603.
18. Caraco J. Genetic determinants of drug responsiveness and drug interactions. Ther Drug Monit 1998;20:517-524.
19. Logue LJ. Genetic testing coverage and reimbursement: a provider’s dilemma. Clin Leadersh Manag Rev 2003;17:346-350.
20. Schwartz MK. Genetic testing and the clinical laboratory improvement amendments of 1988: present and future. Clin Chem 1999;45:739-745.
21. US Food and Drug Administration. Guidance for industry: Pharmacogenomic data submissions 2003. Available at: www.fda.gov/cber/gdlns/pharmdtasub.pdf. Accessed on July 3, 2007.
22. Howard RH. Personalized drug therapy with pharmacogenetics. Part I; pharmacokinetics. J Psychosoc Nurs Ment Health Serv 2006;44:13-16.
23. Veenstra DL, Higashi MK, Phillips KA. Assessing the cost effectiveness of pharmacogenics. AAPS PharmSci 2000;2:E29.-
24. Gramling R, Trask P, Nash J, Culpepper L. Family physicians’ beliefs about genetic testing. Fam Med 2004;36:691-692.
25. Caulfield TA. The informed gatekeeper? A commentary on genetic tests, marketing pressure and the role of primary care physicians. Health Law Rev 2000;9:14-17.
26. Mueller C, Feero WG. Clinical genetics. AAFP Home study self assessment Program No 317. Leawood, Kan: AAFP; 2005.
27. CDC Activities page. Programs in Brief: Evaluations of Genomics Applications and Prevention (EGAPP). Available at: www.cdc.gov/genomics/activities/pib/egapp.htm. Accessed on June 27, 2007.
28. Agency for Healthcare Research and Quality. Testing for cytochrome P450 polymorphisms in adults with non-psychotic depression treated with selective serotonin reuptake inhibitors (SSRIs) [AHRQ Publication No. 07-E002]. Available at: www.ahrq.gov/downloads/pub/evidence/pdf/cyp450/cyp450.pdf. Accessed on July 3, 2007.
29. Vermes A, Vermes I. Genetic polymorphisms in cytochrome P450 enzymes. Effect on efficacy and tolerability of HMG-CoA reductase inhibitors. Am J Cardiovasc Drugs 2004;4:247-255.
30. Yasar U, Eliasson E, Dahl ML, Johansson I, Ingelman-Sundberg M, Sjöqvist F. Validation of methods for CYP2C9 genotyping of mutant alleles in a swedish population. Biochem Biophys Res Commun 1999;254:628-631.
31. Aithal GP, Day CP, Kesteven PJL, Daly AK. Association of polymorphisms in the cytochrome p450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999;353:717-719.
32. Taube J, Halsall D, Baglin T. Influence of cytochrome P450 CYP2C9 polymorphisms on warfarin sensitivity and risk of over anticoagulation in patients on long-term therapy. Blood 2000;96:1816-1819.
33. Reeves MJ, Bohm SR, Korzeniewski SJ, Brown MD. Asthma care and management before an emergency department visit in children in western Michigan: how well does care adhere to guidelines? Pediatrics 2006;117:118-126.
34. Rastogi D, Shetty A, Neugebauer R, Harijith A. National heart, lung, and blood institute guidelines and asthma management practices among innercity pediatric primary care providers. Chest 2006;129:619-623.
35. Bishop PB, Wing PC. Knowledge transfer in family physicians managing patients with acute low back pain: a prospective randomized control trial. Spine J 2006;6:282-288.
36. Bauchner H, Marchant CD, Bisbee A, et al. Effectiveness of centers for disease control and prevention recommendations for outcomes of acute otitis media. Pediatrics 2006;117:1009-1017.
37. Marchant GE, Milligan RJ, Wilhelmi B. Legal pressures and incentives for personalized medicine. Personalized Medicine 2007;3:391-399.
Genetic tests to guide warfarin dosing could avert 85,000 serious bleeding events and 17,000 strokes annually, according to a report from the AEI-Brookings Joint Center for Regulatory Studies, a Washington, DC, think tank. The report further suggests that by integrating genetic testing into warfarin therapy, American health care spending could be reduced by $1.1 billion annually.1 Unfortunately, the promise of using genetic testing to guide such pharmacological treatment has largely gone unfulfilled.2
Case in point: Genetic testing can tell us whether a patient is likely to be an ultra-rapid metabolizer of warfarin (and need larger doses) or a poor metabolizer (and need lower doses), but there are no guidelines to tell us how to dose accordingly. International normalized ratios (INRs) still need to be ordered and the patient will likely have to pick up the tab for the genetic test ($250), since Medicare and private insurers don’t cover the cost. (See “Warfarin: An ideal, but far from ready, candidate”)
Hints that change may be on the horizon. The government—specifically the Department of Health and Human Services—created the Secretary’s Advisory Committee on Genetics, Health, and Society (SACGHS) to assess how genetic and genomic technologies are being integrated into health care and to identify opportunities and gaps in research. To that end, SACGHS issued a draft report earlier this year that notes that genetic-based treatment has “the potential to yield significant gains in personal health, population health, and cost-effective resource allocation.” Among its many recommendations, SACGHS calls for greater collaboration between the public and private sectors to expand our knowledge of the clinical validity and utility of using genetics to guide treatment.3
A standard of care, potentially. Readying ourselves for the ways that genetics is likely to shape the way we prescribe such drugs as anticoagulants, antidepressants, and antiarrhythmics requires that we step back and assess the progress made so far, and the work that still needs to be done before genetic testing becomes a common occurrence, and perhaps even a standard of care.
The goal: Avert adverse events
The wide variation in the way different people respond to the same dose of medications is a major contributor to the problem of adverse drug reactions. Lazarou and colleagues estimated that 6.7% of hospitalized patients—over 2 million patients in the US—experienced an adverse drug reaction and 0.32% (106,000) had a fatal adverse drug reaction.4
It would appear that warfarin dosing would be a perfect candidate for the clinical use of a pharmacogenetic test. Studies have shown that about 7% of the Caucasian population are poor metabolizers and at increased risk of bleeding from over coagulation and 1% are ultra-rapid metabolizers.4 Despite what we know about the polymorphisms to the CYP2C9 enzyme, which is the primary route of metabolism for warfarin, the package insert on Coumadin still doesn’t contain a recommendation for determining a patient’s genetic profile before initiating treatment.30 Similarly, the chapter on anticoagulation in Applied Therapeutics, a commonly used medical textbook, says nothing about the use of CYP polymorphisms for dosing decisions.13
At issue: Genotyping to guide dosing has not been tested in comparison to the usual monitoring using the international normalized ratio (INR).31 Specifically, the outcomes of bleeding complications and adequate anticoagulation of the 2 methods have not been compared in a clinical trial.
Here’s what we do know: In one study, the presence of specific polymorphisms was associated with a lower maintenance warfarin dose, but not with over-anticoagulation.32 In a review of 4 studies on CYP2C9 polymorphisms and warfarin daily dose, Lee and colleagues found that between the slowest and fastest metabolizers, the difference in dose was, at most, 4 mg/day. These studies did not explore if dosing decisions could accurately be made on genetic classifications and it is unlikely they could because of the wide overlap in maintenance dosages in the different classes.7
To complicate things further, the future use of warfarin in some conditions is problematic because fractionated heparin has been proven in many situations to be as effective and less risky than warfarin, and does not require frequent monitoring with blood tests. All of these unknowns make it unclear how useful genetic tests will be, and whether insurers will pay for them.
Individual response to medications is determined by a host of factors including age, environment, other medications being taken, and genetic differences in drug absorption and metabolism. These genetic differences have spawned the fields of pharmacogenomics and pharmacogenetics.
Pharmacogenomics is the biotechnological science that combines the techniques of medicine, pharmacology, and genomics and is concerned with developing drug therapies to compensate for genetic differences in patients, which cause varied responses to a single therapeutic regimen.
- A good example of pharmacogenomics at work is the use of trastuzumab in addition to chemotherapy for breast cancer patients who are positive for the human epidermal growth factor receptor 2 (HER2) oncogene.5
Pharmacogenetics is the branch of pharmacology that examines the relation of genetic factors to variations in response to drugs.
The use of pharmacogenetics to predict individualized responses to medications and to prevent adverse drug reactions through individualized dosing regimens or avoidance of certain medications hinges on our knowledge of genetic polymorphisms, that is, gene-based differences in drug absorption, distribution, metabolism, or excretion.
Polymorphisms of the cytochrome P450 family of drug metabolizing enzymes have been the most extensively studied. The names of these enzymes are abbreviated by using CYP and then a series of letters and numbers to describe individual enzymes. The 4 most extensively studied CYP enzymes are CYP2A6, CYP2C9, CYP2C19, and CYP2D6. These 4 metabolize an estimated 40% of all drugs; the distributions of polymorphisms of each vary considerably by race/ethnicity.6-8
A sampling of medications and classes of medications where polymorphisms play a significant role in drug metabolism is listed in the TABLE. Three with significant potential for prevention of adverse drug reactions are the antipsychotics, because of the severity of a specific drug adverse reaction, tardive dyskinesia;6,9 warfarin, because of the risk of bleeding complications and its narrow therapeutic index;7,10 and chemotherapeutic agents because of the serious nature of the disease and the potential for tailoring individualized therapies to maximize tumor response to medication and to minimize adverse reactions of very toxic drugs.11
TABLE
Polymorphisms to these enzymes affect drug metabolism
| DRUG/CLASS | ENZYMES |
|---|---|
| Antiarrhythmmics7 | CYP2D6 |
| Antidepressants7 | CYP2D6 |
| Antipsychotics6,9 | CYP2D6, CYP1A2, CYP2C19, CYP3A4 |
| Beta-blockers7 | CYP2D6 |
| Cancer chemotherapy11 | Varies by the agent |
| HMG-CoA reductase inhibitors (statins)29 | CYP2D6, CYP2C9, CYP2C19 |
| Losartan7 | CYP2C9 |
| Neuroleptics7 | CYP2D6 |
| NSAIDs29 | CYP2C9 |
| Phenytoin7 | CYP2C9, CYP2C19 |
| Proton pump inhibitors29 | CYP2C19 |
| Tolbutamide7 | CYP2C9 |
| Warfarin7,10 | CYP2C9 |
Clinical resources reflect an information gap
In spite of the potential for improved patient care, there remains very little clinical application of pharmacogenetic information in primary care practice. Zineh and colleagues reviewed prescribing information in the electronic version of the Physicians’ Desk Reference (PDR) in 2004 and found that only 76 package inserts out of 3382 contained pharmacogenetic information.12 In only 25 was there enough information to affect treatment decisions. Just 5 inserts mentioned that the chance of successful response to treatment could be predicted by genetic testing, and only one insert mentioned that a specific genetic subgroup should not take a drug.
The authors concluded that, generally, the pharmacogenetic information was inadequate to guide drug therapy and the majority of information was available for drugs that are not commonly prescribed. The FDA is addressing this issue by requiring the inclusion of pharmacogenetic information in package inserts more frequently.
Consider, too, the 2005 edition of Applied Therapeutics, a commonly used medical textbook.13 In it there is no mention of the use of pharmacogenetics in managing pharmacological therapies, which tells us that very little teaching on this topic is going on in medical schools and residencies.
But why? Why are clinicians and the tools we rely on so out of sync with the recommendations and expectations of personalized medicine advocates?
Certain conditions will need to exist before testing for CYP polymorphisms will become the standard of care in dosing certain drugs, such as warfarin. (see “Warfarin: An ideal, but far from ready, candidate”.) The most important is that this clinical approach needs to be proven superior to existing methods, such as INR monitoring. Should this occur, guidelines that are evidence-based will include it as a recommendation, physician continuing education courses will cover it, and it will enter into the curricula of medical schools and residency programs. But the process of adopting new, evidence-based best practices has historically been slow.32-36
One variable that may play a part in facilitating more rapid acceptance of genetic testing before warfarin use is litigation. Marchant and colleagues describe potential legal pressures that may drive medicine to adopt more personalized medicine.37 If genetic testing before warfarin use results in better outcomes (fewer catastrophic events or very bad outcomes) and there is plausible evidence that a catastrophic outcome (massive bleeding) could have been avoided with genetic information, then litigation will surely be close behind.
As news of successful litigation spreads, one of two results will likely occur: Either the use of the CYP polymorphism testing will increase, or the movement to use alternative medications, such as fractionated heparin, will accelerate.
There are 5 likely culprits:
- A lack of clinically useful pharmacogenetic tests.
- A lack of test standardization and availability.
- A lack of coverage by third-party payers.
- A low level of physician knowledge about genetic testing.
- A lack of evidence of improved outcomes.
How useful are pharmacogenetic tests?
For a laboratory test to be clinically useful, it should provide information that will influence a therapeutic decision. Decisions that could be influenced by this information include the dose of a particular drug and the potential use of an alternative because of a contraindication or likelihood of a poor outcome based on a particular genetic polymorphism.
The use of pharmacogenetic laboratory information can place a patient into one of several groups:
- ultra-rapid metabolizers, who need a larger dose of medication
- normal metabolizers, often called extensive metabolizers, who do not need dose modifications
- poor metabolizers, who need lower doses.9
Most medications have a wide therapeutic margin of effectiveness and safety. This means that the medication works within a wide range of serum drug levels and is safe at these different levels, making refinement in dosing based on genetic information unnecessary. In medications with narrower therapeutic windows for effectiveness or adverse reactions, there are frequently alternative means of drug level monitoring.
In many instances, the genetic test predictability of a patient’s actual metabolic responses—and resulting drug levels—is poor, leading to the need to monitor drug levels anyway.14-16 This occurs because there is often a great deal of overlap in the response to a medication dose among the different metabolism classifications.9,14,16-18 All of these realities have limited the clinical usefulness of pharmacogenetics up to this point.
Test standardization: Poised to improve?
Genotyping, the determination of the actual genetic makeup of the patient is not always predictive of an individual patient’s metabolic response. In other words, genotype does not always equate to a phenotype. Further complicating matters is the fact that many of the pharmacogenetic studies have been performed at research laboratories and have used tests that are not standardized, or widely available.19-21
The recent commercial availability of pharmacogenetic tests by well-established and reputable laboratories will probably improve both standardization and availability.
An example is the AmpliChip CYP450 test by Roche Diagnostics.22 This microassay-based test identifies 29 CYP-2D6 polymorphisms and 2 CYP-2C19 polymorphisms. These genes affect the metabolism of 25% of currently prescribed drugs. Using this test, patients can be classified as poor, intermediate, extensive, or ultra rapid metabolizers of CYP-2D6 affected drugs, including antidepressants, antiarrhythmics, and antipsychotics. They can also be classified as poor or extensive metabolizers of CYP-2C19 affected drugs, including phenytoin and proton pump inhibitors.
How costly?
The cost of genetic testing will also affect availability. Tests will be widely available only if covered by third-party payers. The AmpliChip test costs between $300 and $500. Clearly, then, both cost and insurance coverage are issues that impede the adoption of pharmacogenetic testing, though there is little written about this in the medical literature.19,23
AAFP explores ways to teach genomics
Physicians who are currently in practice received little or no training in the clinical use of pharmacogenetics or other genetic tests, such as genetic testing for the prediction of cancer risk.24,25 The main resources of pharmacological information for practicing physicians do not contain much, if any, useful genomic information. A recent continuing education monograph for family physicians on clinical genetics mentioned pharmacogenetics only as a promising future technology.26
For its part, the American Academy of Family Physicians has formed a genomics work group and is exploring how to educate family physicians on clinically useful genomic topics.
Evidence-based outcomes are needed
To date there has not been a head-to-head comparison of the outcomes of using clinical pharmacogenetics with those obtained from standard drug level monitoring practices. The CDC has formed a committee modeled after the USPSTF, the Evaluations of Genomics Applications in Practice and Prevention (EGAPP), which will evaluate the effectiveness of genomic clinical tests and make recommendations to physicians on their use.27 The group’s first report, on the use of CYP450 testing in depression, concluded that there is a paucity of good quality data that addresses whether testing for CYP450 polymorphisms in adults entering SSRI treatment leads to improved outcomes.28
In addition, SACGHS, the Department of Health and Human Services’ Committee, recommends in its draft report that HHS “provide resources to identify and address evidentiary gaps in the analytic validity, clinical validity, clinical utility, and cost effectiveness of pharmacogenomics.”3
Outcomes data will undoubtedly be key. With it, pharmacogenetic testing has the potential to grow by leaps and bounds—perhaps even becoming a standard of care in guiding pharmacological therapy. Without it, such testing will remain a promising, but as yet unrealized advance in personalized medicine.
Correspondence
Doug Campos-Outcalt, MD, MPA, 550 E. Van Buren, Phoenix, AZ 85004; [email protected].
Genetic tests to guide warfarin dosing could avert 85,000 serious bleeding events and 17,000 strokes annually, according to a report from the AEI-Brookings Joint Center for Regulatory Studies, a Washington, DC, think tank. The report further suggests that by integrating genetic testing into warfarin therapy, American health care spending could be reduced by $1.1 billion annually.1 Unfortunately, the promise of using genetic testing to guide such pharmacological treatment has largely gone unfulfilled.2
Case in point: Genetic testing can tell us whether a patient is likely to be an ultra-rapid metabolizer of warfarin (and need larger doses) or a poor metabolizer (and need lower doses), but there are no guidelines to tell us how to dose accordingly. International normalized ratios (INRs) still need to be ordered and the patient will likely have to pick up the tab for the genetic test ($250), since Medicare and private insurers don’t cover the cost. (See “Warfarin: An ideal, but far from ready, candidate”)
Hints that change may be on the horizon. The government—specifically the Department of Health and Human Services—created the Secretary’s Advisory Committee on Genetics, Health, and Society (SACGHS) to assess how genetic and genomic technologies are being integrated into health care and to identify opportunities and gaps in research. To that end, SACGHS issued a draft report earlier this year that notes that genetic-based treatment has “the potential to yield significant gains in personal health, population health, and cost-effective resource allocation.” Among its many recommendations, SACGHS calls for greater collaboration between the public and private sectors to expand our knowledge of the clinical validity and utility of using genetics to guide treatment.3
A standard of care, potentially. Readying ourselves for the ways that genetics is likely to shape the way we prescribe such drugs as anticoagulants, antidepressants, and antiarrhythmics requires that we step back and assess the progress made so far, and the work that still needs to be done before genetic testing becomes a common occurrence, and perhaps even a standard of care.
The goal: Avert adverse events
The wide variation in the way different people respond to the same dose of medications is a major contributor to the problem of adverse drug reactions. Lazarou and colleagues estimated that 6.7% of hospitalized patients—over 2 million patients in the US—experienced an adverse drug reaction and 0.32% (106,000) had a fatal adverse drug reaction.4
It would appear that warfarin dosing would be a perfect candidate for the clinical use of a pharmacogenetic test. Studies have shown that about 7% of the Caucasian population are poor metabolizers and at increased risk of bleeding from over coagulation and 1% are ultra-rapid metabolizers.4 Despite what we know about the polymorphisms to the CYP2C9 enzyme, which is the primary route of metabolism for warfarin, the package insert on Coumadin still doesn’t contain a recommendation for determining a patient’s genetic profile before initiating treatment.30 Similarly, the chapter on anticoagulation in Applied Therapeutics, a commonly used medical textbook, says nothing about the use of CYP polymorphisms for dosing decisions.13
At issue: Genotyping to guide dosing has not been tested in comparison to the usual monitoring using the international normalized ratio (INR).31 Specifically, the outcomes of bleeding complications and adequate anticoagulation of the 2 methods have not been compared in a clinical trial.
Here’s what we do know: In one study, the presence of specific polymorphisms was associated with a lower maintenance warfarin dose, but not with over-anticoagulation.32 In a review of 4 studies on CYP2C9 polymorphisms and warfarin daily dose, Lee and colleagues found that between the slowest and fastest metabolizers, the difference in dose was, at most, 4 mg/day. These studies did not explore if dosing decisions could accurately be made on genetic classifications and it is unlikely they could because of the wide overlap in maintenance dosages in the different classes.7
To complicate things further, the future use of warfarin in some conditions is problematic because fractionated heparin has been proven in many situations to be as effective and less risky than warfarin, and does not require frequent monitoring with blood tests. All of these unknowns make it unclear how useful genetic tests will be, and whether insurers will pay for them.
Individual response to medications is determined by a host of factors including age, environment, other medications being taken, and genetic differences in drug absorption and metabolism. These genetic differences have spawned the fields of pharmacogenomics and pharmacogenetics.
Pharmacogenomics is the biotechnological science that combines the techniques of medicine, pharmacology, and genomics and is concerned with developing drug therapies to compensate for genetic differences in patients, which cause varied responses to a single therapeutic regimen.
- A good example of pharmacogenomics at work is the use of trastuzumab in addition to chemotherapy for breast cancer patients who are positive for the human epidermal growth factor receptor 2 (HER2) oncogene.5
Pharmacogenetics is the branch of pharmacology that examines the relation of genetic factors to variations in response to drugs.
The use of pharmacogenetics to predict individualized responses to medications and to prevent adverse drug reactions through individualized dosing regimens or avoidance of certain medications hinges on our knowledge of genetic polymorphisms, that is, gene-based differences in drug absorption, distribution, metabolism, or excretion.
Polymorphisms of the cytochrome P450 family of drug metabolizing enzymes have been the most extensively studied. The names of these enzymes are abbreviated by using CYP and then a series of letters and numbers to describe individual enzymes. The 4 most extensively studied CYP enzymes are CYP2A6, CYP2C9, CYP2C19, and CYP2D6. These 4 metabolize an estimated 40% of all drugs; the distributions of polymorphisms of each vary considerably by race/ethnicity.6-8
A sampling of medications and classes of medications where polymorphisms play a significant role in drug metabolism is listed in the TABLE. Three with significant potential for prevention of adverse drug reactions are the antipsychotics, because of the severity of a specific drug adverse reaction, tardive dyskinesia;6,9 warfarin, because of the risk of bleeding complications and its narrow therapeutic index;7,10 and chemotherapeutic agents because of the serious nature of the disease and the potential for tailoring individualized therapies to maximize tumor response to medication and to minimize adverse reactions of very toxic drugs.11
TABLE
Polymorphisms to these enzymes affect drug metabolism
| DRUG/CLASS | ENZYMES |
|---|---|
| Antiarrhythmmics7 | CYP2D6 |
| Antidepressants7 | CYP2D6 |
| Antipsychotics6,9 | CYP2D6, CYP1A2, CYP2C19, CYP3A4 |
| Beta-blockers7 | CYP2D6 |
| Cancer chemotherapy11 | Varies by the agent |
| HMG-CoA reductase inhibitors (statins)29 | CYP2D6, CYP2C9, CYP2C19 |
| Losartan7 | CYP2C9 |
| Neuroleptics7 | CYP2D6 |
| NSAIDs29 | CYP2C9 |
| Phenytoin7 | CYP2C9, CYP2C19 |
| Proton pump inhibitors29 | CYP2C19 |
| Tolbutamide7 | CYP2C9 |
| Warfarin7,10 | CYP2C9 |
Clinical resources reflect an information gap
In spite of the potential for improved patient care, there remains very little clinical application of pharmacogenetic information in primary care practice. Zineh and colleagues reviewed prescribing information in the electronic version of the Physicians’ Desk Reference (PDR) in 2004 and found that only 76 package inserts out of 3382 contained pharmacogenetic information.12 In only 25 was there enough information to affect treatment decisions. Just 5 inserts mentioned that the chance of successful response to treatment could be predicted by genetic testing, and only one insert mentioned that a specific genetic subgroup should not take a drug.
The authors concluded that, generally, the pharmacogenetic information was inadequate to guide drug therapy and the majority of information was available for drugs that are not commonly prescribed. The FDA is addressing this issue by requiring the inclusion of pharmacogenetic information in package inserts more frequently.
Consider, too, the 2005 edition of Applied Therapeutics, a commonly used medical textbook.13 In it there is no mention of the use of pharmacogenetics in managing pharmacological therapies, which tells us that very little teaching on this topic is going on in medical schools and residencies.
But why? Why are clinicians and the tools we rely on so out of sync with the recommendations and expectations of personalized medicine advocates?
Certain conditions will need to exist before testing for CYP polymorphisms will become the standard of care in dosing certain drugs, such as warfarin. (see “Warfarin: An ideal, but far from ready, candidate”.) The most important is that this clinical approach needs to be proven superior to existing methods, such as INR monitoring. Should this occur, guidelines that are evidence-based will include it as a recommendation, physician continuing education courses will cover it, and it will enter into the curricula of medical schools and residency programs. But the process of adopting new, evidence-based best practices has historically been slow.32-36
One variable that may play a part in facilitating more rapid acceptance of genetic testing before warfarin use is litigation. Marchant and colleagues describe potential legal pressures that may drive medicine to adopt more personalized medicine.37 If genetic testing before warfarin use results in better outcomes (fewer catastrophic events or very bad outcomes) and there is plausible evidence that a catastrophic outcome (massive bleeding) could have been avoided with genetic information, then litigation will surely be close behind.
As news of successful litigation spreads, one of two results will likely occur: Either the use of the CYP polymorphism testing will increase, or the movement to use alternative medications, such as fractionated heparin, will accelerate.
There are 5 likely culprits:
- A lack of clinically useful pharmacogenetic tests.
- A lack of test standardization and availability.
- A lack of coverage by third-party payers.
- A low level of physician knowledge about genetic testing.
- A lack of evidence of improved outcomes.
How useful are pharmacogenetic tests?
For a laboratory test to be clinically useful, it should provide information that will influence a therapeutic decision. Decisions that could be influenced by this information include the dose of a particular drug and the potential use of an alternative because of a contraindication or likelihood of a poor outcome based on a particular genetic polymorphism.
The use of pharmacogenetic laboratory information can place a patient into one of several groups:
- ultra-rapid metabolizers, who need a larger dose of medication
- normal metabolizers, often called extensive metabolizers, who do not need dose modifications
- poor metabolizers, who need lower doses.9
Most medications have a wide therapeutic margin of effectiveness and safety. This means that the medication works within a wide range of serum drug levels and is safe at these different levels, making refinement in dosing based on genetic information unnecessary. In medications with narrower therapeutic windows for effectiveness or adverse reactions, there are frequently alternative means of drug level monitoring.
In many instances, the genetic test predictability of a patient’s actual metabolic responses—and resulting drug levels—is poor, leading to the need to monitor drug levels anyway.14-16 This occurs because there is often a great deal of overlap in the response to a medication dose among the different metabolism classifications.9,14,16-18 All of these realities have limited the clinical usefulness of pharmacogenetics up to this point.
Test standardization: Poised to improve?
Genotyping, the determination of the actual genetic makeup of the patient is not always predictive of an individual patient’s metabolic response. In other words, genotype does not always equate to a phenotype. Further complicating matters is the fact that many of the pharmacogenetic studies have been performed at research laboratories and have used tests that are not standardized, or widely available.19-21
The recent commercial availability of pharmacogenetic tests by well-established and reputable laboratories will probably improve both standardization and availability.
An example is the AmpliChip CYP450 test by Roche Diagnostics.22 This microassay-based test identifies 29 CYP-2D6 polymorphisms and 2 CYP-2C19 polymorphisms. These genes affect the metabolism of 25% of currently prescribed drugs. Using this test, patients can be classified as poor, intermediate, extensive, or ultra rapid metabolizers of CYP-2D6 affected drugs, including antidepressants, antiarrhythmics, and antipsychotics. They can also be classified as poor or extensive metabolizers of CYP-2C19 affected drugs, including phenytoin and proton pump inhibitors.
How costly?
The cost of genetic testing will also affect availability. Tests will be widely available only if covered by third-party payers. The AmpliChip test costs between $300 and $500. Clearly, then, both cost and insurance coverage are issues that impede the adoption of pharmacogenetic testing, though there is little written about this in the medical literature.19,23
AAFP explores ways to teach genomics
Physicians who are currently in practice received little or no training in the clinical use of pharmacogenetics or other genetic tests, such as genetic testing for the prediction of cancer risk.24,25 The main resources of pharmacological information for practicing physicians do not contain much, if any, useful genomic information. A recent continuing education monograph for family physicians on clinical genetics mentioned pharmacogenetics only as a promising future technology.26
For its part, the American Academy of Family Physicians has formed a genomics work group and is exploring how to educate family physicians on clinically useful genomic topics.
Evidence-based outcomes are needed
To date there has not been a head-to-head comparison of the outcomes of using clinical pharmacogenetics with those obtained from standard drug level monitoring practices. The CDC has formed a committee modeled after the USPSTF, the Evaluations of Genomics Applications in Practice and Prevention (EGAPP), which will evaluate the effectiveness of genomic clinical tests and make recommendations to physicians on their use.27 The group’s first report, on the use of CYP450 testing in depression, concluded that there is a paucity of good quality data that addresses whether testing for CYP450 polymorphisms in adults entering SSRI treatment leads to improved outcomes.28
In addition, SACGHS, the Department of Health and Human Services’ Committee, recommends in its draft report that HHS “provide resources to identify and address evidentiary gaps in the analytic validity, clinical validity, clinical utility, and cost effectiveness of pharmacogenomics.”3
Outcomes data will undoubtedly be key. With it, pharmacogenetic testing has the potential to grow by leaps and bounds—perhaps even becoming a standard of care in guiding pharmacological therapy. Without it, such testing will remain a promising, but as yet unrealized advance in personalized medicine.
Correspondence
Doug Campos-Outcalt, MD, MPA, 550 E. Van Buren, Phoenix, AZ 85004; [email protected].
1. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalized medicine using genetic testing: The case of warfarin. [Working Paper 06-23]. November 2006. Available at: www.aei-brookings.org/admin/authorpdfs/page.php?id=1337&PHPsEssID=7b3a3ae4b30d77cb76223e29535e7590. Accessed on June 27, 2007.
2. Tucker G. Pharmacogenetics—expectations and reality. BMJ 2004;329:4-6.
3. Department of Health and Human Services. Realizing the promise of pharmacogenomics: Opportunities and Challenges [Draft report of the Secretary’s Advisory Committee on Genetics, Health, and Society]. Available for public comment March 23–June 1, 2007. Available at: www4.od.nih.gov/oba/sacghs/SACGHS_Pgx_PCdraft.pdf. Accessed on June 27, 2007.
4. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients. JAMA 1998;279:1200-1205.
5. Dent R, Clemons M. Adjuvant trastuzumab for breast cancer. Br Med J 2005;331:1035-1036.
6. Wolf CR, Smith G. Pharmacokinetics. Br Med Bull 1999;55:366-386.
7. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 2002;12:251-263.
8. Bachman K. Genotyping and phenotyping the cytochrome P450 enzymes. Am J Ther 2002;9:309-316.
9. Dahl ML. Cytochrome P450 phenotyping/genotyping in patients receiving antipsychotics. Clin Pharmacokinet 2002;41:453-470.
10. Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003;13:247-252.
11. Van Schaik RHN. Implications of cytochrome P450 genetic polymorphisms on the toxicity of antitumor agents. Ther Drug Monit 2004;26:236-240.
12. Zineh I, Gerhard T, Aquilante CL, Beitelshees AL, Beasley BN, Hartzema AG. Availability of pharmacogenomics-based prescribing information in drug package inserts for currently approved drugs. Pharmacogenomics J 2004;4:354-358.
13. Koda Kimball M, ed. Applied Therapeutics. 8th ed. Baltimore, Md: Lippincott, Williams and Wilkins; 2005.
14. Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics 1995;5:389-392.
15. Arthur H, Dahl ML, Siwers B. Polymorphic drug metabolism in schizophrenic patients with tardive dyskinesia. J Clin Psychopharmacology 1995;15:211-216.
16. Mulder AB, Van Lijf HJ, Bon MAM, et al. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin Pharmacol Ther 2001;70:546-551.
17. Takahashi H, Ehizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmakinetics 2001;40:587-603.
18. Caraco J. Genetic determinants of drug responsiveness and drug interactions. Ther Drug Monit 1998;20:517-524.
19. Logue LJ. Genetic testing coverage and reimbursement: a provider’s dilemma. Clin Leadersh Manag Rev 2003;17:346-350.
20. Schwartz MK. Genetic testing and the clinical laboratory improvement amendments of 1988: present and future. Clin Chem 1999;45:739-745.
21. US Food and Drug Administration. Guidance for industry: Pharmacogenomic data submissions 2003. Available at: www.fda.gov/cber/gdlns/pharmdtasub.pdf. Accessed on July 3, 2007.
22. Howard RH. Personalized drug therapy with pharmacogenetics. Part I; pharmacokinetics. J Psychosoc Nurs Ment Health Serv 2006;44:13-16.
23. Veenstra DL, Higashi MK, Phillips KA. Assessing the cost effectiveness of pharmacogenics. AAPS PharmSci 2000;2:E29.-
24. Gramling R, Trask P, Nash J, Culpepper L. Family physicians’ beliefs about genetic testing. Fam Med 2004;36:691-692.
25. Caulfield TA. The informed gatekeeper? A commentary on genetic tests, marketing pressure and the role of primary care physicians. Health Law Rev 2000;9:14-17.
26. Mueller C, Feero WG. Clinical genetics. AAFP Home study self assessment Program No 317. Leawood, Kan: AAFP; 2005.
27. CDC Activities page. Programs in Brief: Evaluations of Genomics Applications and Prevention (EGAPP). Available at: www.cdc.gov/genomics/activities/pib/egapp.htm. Accessed on June 27, 2007.
28. Agency for Healthcare Research and Quality. Testing for cytochrome P450 polymorphisms in adults with non-psychotic depression treated with selective serotonin reuptake inhibitors (SSRIs) [AHRQ Publication No. 07-E002]. Available at: www.ahrq.gov/downloads/pub/evidence/pdf/cyp450/cyp450.pdf. Accessed on July 3, 2007.
29. Vermes A, Vermes I. Genetic polymorphisms in cytochrome P450 enzymes. Effect on efficacy and tolerability of HMG-CoA reductase inhibitors. Am J Cardiovasc Drugs 2004;4:247-255.
30. Yasar U, Eliasson E, Dahl ML, Johansson I, Ingelman-Sundberg M, Sjöqvist F. Validation of methods for CYP2C9 genotyping of mutant alleles in a swedish population. Biochem Biophys Res Commun 1999;254:628-631.
31. Aithal GP, Day CP, Kesteven PJL, Daly AK. Association of polymorphisms in the cytochrome p450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999;353:717-719.
32. Taube J, Halsall D, Baglin T. Influence of cytochrome P450 CYP2C9 polymorphisms on warfarin sensitivity and risk of over anticoagulation in patients on long-term therapy. Blood 2000;96:1816-1819.
33. Reeves MJ, Bohm SR, Korzeniewski SJ, Brown MD. Asthma care and management before an emergency department visit in children in western Michigan: how well does care adhere to guidelines? Pediatrics 2006;117:118-126.
34. Rastogi D, Shetty A, Neugebauer R, Harijith A. National heart, lung, and blood institute guidelines and asthma management practices among innercity pediatric primary care providers. Chest 2006;129:619-623.
35. Bishop PB, Wing PC. Knowledge transfer in family physicians managing patients with acute low back pain: a prospective randomized control trial. Spine J 2006;6:282-288.
36. Bauchner H, Marchant CD, Bisbee A, et al. Effectiveness of centers for disease control and prevention recommendations for outcomes of acute otitis media. Pediatrics 2006;117:1009-1017.
37. Marchant GE, Milligan RJ, Wilhelmi B. Legal pressures and incentives for personalized medicine. Personalized Medicine 2007;3:391-399.
1. McWilliam A, Lutter R, Nardinelli C. Health care savings from personalized medicine using genetic testing: The case of warfarin. [Working Paper 06-23]. November 2006. Available at: www.aei-brookings.org/admin/authorpdfs/page.php?id=1337&PHPsEssID=7b3a3ae4b30d77cb76223e29535e7590. Accessed on June 27, 2007.
2. Tucker G. Pharmacogenetics—expectations and reality. BMJ 2004;329:4-6.
3. Department of Health and Human Services. Realizing the promise of pharmacogenomics: Opportunities and Challenges [Draft report of the Secretary’s Advisory Committee on Genetics, Health, and Society]. Available for public comment March 23–June 1, 2007. Available at: www4.od.nih.gov/oba/sacghs/SACGHS_Pgx_PCdraft.pdf. Accessed on June 27, 2007.
4. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients. JAMA 1998;279:1200-1205.
5. Dent R, Clemons M. Adjuvant trastuzumab for breast cancer. Br Med J 2005;331:1035-1036.
6. Wolf CR, Smith G. Pharmacokinetics. Br Med Bull 1999;55:366-386.
7. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 2002;12:251-263.
8. Bachman K. Genotyping and phenotyping the cytochrome P450 enzymes. Am J Ther 2002;9:309-316.
9. Dahl ML. Cytochrome P450 phenotyping/genotyping in patients receiving antipsychotics. Clin Pharmacokinet 2002;41:453-470.
10. Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003;13:247-252.
11. Van Schaik RHN. Implications of cytochrome P450 genetic polymorphisms on the toxicity of antitumor agents. Ther Drug Monit 2004;26:236-240.
12. Zineh I, Gerhard T, Aquilante CL, Beitelshees AL, Beasley BN, Hartzema AG. Availability of pharmacogenomics-based prescribing information in drug package inserts for currently approved drugs. Pharmacogenomics J 2004;4:354-358.
13. Koda Kimball M, ed. Applied Therapeutics. 8th ed. Baltimore, Md: Lippincott, Williams and Wilkins; 2005.
14. Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics 1995;5:389-392.
15. Arthur H, Dahl ML, Siwers B. Polymorphic drug metabolism in schizophrenic patients with tardive dyskinesia. J Clin Psychopharmacology 1995;15:211-216.
16. Mulder AB, Van Lijf HJ, Bon MAM, et al. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin Pharmacol Ther 2001;70:546-551.
17. Takahashi H, Ehizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmakinetics 2001;40:587-603.
18. Caraco J. Genetic determinants of drug responsiveness and drug interactions. Ther Drug Monit 1998;20:517-524.
19. Logue LJ. Genetic testing coverage and reimbursement: a provider’s dilemma. Clin Leadersh Manag Rev 2003;17:346-350.
20. Schwartz MK. Genetic testing and the clinical laboratory improvement amendments of 1988: present and future. Clin Chem 1999;45:739-745.
21. US Food and Drug Administration. Guidance for industry: Pharmacogenomic data submissions 2003. Available at: www.fda.gov/cber/gdlns/pharmdtasub.pdf. Accessed on July 3, 2007.
22. Howard RH. Personalized drug therapy with pharmacogenetics. Part I; pharmacokinetics. J Psychosoc Nurs Ment Health Serv 2006;44:13-16.
23. Veenstra DL, Higashi MK, Phillips KA. Assessing the cost effectiveness of pharmacogenics. AAPS PharmSci 2000;2:E29.-
24. Gramling R, Trask P, Nash J, Culpepper L. Family physicians’ beliefs about genetic testing. Fam Med 2004;36:691-692.
25. Caulfield TA. The informed gatekeeper? A commentary on genetic tests, marketing pressure and the role of primary care physicians. Health Law Rev 2000;9:14-17.
26. Mueller C, Feero WG. Clinical genetics. AAFP Home study self assessment Program No 317. Leawood, Kan: AAFP; 2005.
27. CDC Activities page. Programs in Brief: Evaluations of Genomics Applications and Prevention (EGAPP). Available at: www.cdc.gov/genomics/activities/pib/egapp.htm. Accessed on June 27, 2007.
28. Agency for Healthcare Research and Quality. Testing for cytochrome P450 polymorphisms in adults with non-psychotic depression treated with selective serotonin reuptake inhibitors (SSRIs) [AHRQ Publication No. 07-E002]. Available at: www.ahrq.gov/downloads/pub/evidence/pdf/cyp450/cyp450.pdf. Accessed on July 3, 2007.
29. Vermes A, Vermes I. Genetic polymorphisms in cytochrome P450 enzymes. Effect on efficacy and tolerability of HMG-CoA reductase inhibitors. Am J Cardiovasc Drugs 2004;4:247-255.
30. Yasar U, Eliasson E, Dahl ML, Johansson I, Ingelman-Sundberg M, Sjöqvist F. Validation of methods for CYP2C9 genotyping of mutant alleles in a swedish population. Biochem Biophys Res Commun 1999;254:628-631.
31. Aithal GP, Day CP, Kesteven PJL, Daly AK. Association of polymorphisms in the cytochrome p450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999;353:717-719.
32. Taube J, Halsall D, Baglin T. Influence of cytochrome P450 CYP2C9 polymorphisms on warfarin sensitivity and risk of over anticoagulation in patients on long-term therapy. Blood 2000;96:1816-1819.
33. Reeves MJ, Bohm SR, Korzeniewski SJ, Brown MD. Asthma care and management before an emergency department visit in children in western Michigan: how well does care adhere to guidelines? Pediatrics 2006;117:118-126.
34. Rastogi D, Shetty A, Neugebauer R, Harijith A. National heart, lung, and blood institute guidelines and asthma management practices among innercity pediatric primary care providers. Chest 2006;129:619-623.
35. Bishop PB, Wing PC. Knowledge transfer in family physicians managing patients with acute low back pain: a prospective randomized control trial. Spine J 2006;6:282-288.
36. Bauchner H, Marchant CD, Bisbee A, et al. Effectiveness of centers for disease control and prevention recommendations for outcomes of acute otitis media. Pediatrics 2006;117:1009-1017.
37. Marchant GE, Milligan RJ, Wilhelmi B. Legal pressures and incentives for personalized medicine. Personalized Medicine 2007;3:391-399.
Syncope in athletes: A guide to getting them back on their feet
- Following inconclusive cardiovascular and neurological testing, an echocardiogram, stress test with Holter monitor, and possibly tilt table testing are appropriate ways to determine whether a vasovagal response is to blame for a young athlete’s exercise-induced fainting episodes (C).
- Advise patients with exercise-induced syncope to increase fluid and salt intake while exercising (B). Strength training can also be helpful (B). Most drug therapies, such as beta-blockers, vasoconstrictors, and anti-arrhythmics, have inconsistent results (A). use of permanent pacemakers are not effective (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
An 18-year-old woman was referred to us for evaluation after fainting on several occasions during a track workout. She’d been a competitive athlete for several years, and the episodes began 3 months earlier. She reported that they happen shortly after she started to run. She also told us that just before a spell, she had a sense of fatigue and severe leg pain.
Bystanders who witnessed one of the episodes reported that her eyes rolled upwards right before she fell; they saw no tonic-clonic activity that would indicate a seizure. We learned that the spells were short-lived and followed by immediate recovery with no mental deficits, such as confusion. On several occasions, she suffered superficial skin abrasions, but fortunately no serious injuries had occurred.
We suspected exercise-induced syncope
Prompt diagnosis of syncope in athletes is essential as it may be a marker for sudden cardiac death. The differential diagnosis, however, is broad (TABLE). Syncope in adolescents is often associated with fasting and eating disorders, excessive heat, alcohol, abdominal straining, exercise, hypoglycemia, growth spurts, lack of sleep, and low blood pressure.
TABLE
Differential diagnosis for exercise-induced syncope
| Anaphylaxis | Hypertrophic cardiomyopathy |
| Aortic stenosis | Hypoglycemia |
| Atrial fibrillation | Hypoxia |
| Brugada syndrome | Left ventricular hypertrophy |
| Cardiac dilatation | Pulmonary hypertension |
| Cardiac ischemia | Sick sinus syndrome |
| Catecholamine-dependant polymorphic ventricular tachycardia | Vasodepressor reflex |
| Commotio cordis | Ventricular fibrillation |
| Dehydration | Ventricular tachycardia |
| Eating disorders | Wolff-Parkinson-White abnormality |
Few clues from her history and exam
The clinical evaluation requires a careful and detailed history, including taking a family history to exclude familial causes of sudden death, such as long QT syndrome. In our patient’s case, there was no family history of sudden death or arrhythmias. She was a nonsmoker, nondrinker, and denied using any illicit drugs. She had been taking phenytoin for suspected seizure activity, but the spells continued.
On further investigation, we discovered that our patient would faint predictably 14 minutes into a competitive 5K run. She had recently adopted a more aggressive “early breakaway” strategy: she would run the initial part of the race at a pace of about 6 minutes per mile and slow down gradually towards the end.
We also learned that she had never fainted during weight training or when running at a less aggressive pace. Other than fatigue and extreme leg pain, she could not recall any other symptoms before falling, such as nausea, dizziness, weakness, or palpitations.
Her resting seated left arm blood pressure was 110/60 mm Hg and her heart rate was regular at 53 bpm. On cardiac auscultation, we heard no murmurs or abnormal cardiac sounds in the supine or standing positions. Cardiac palpation did not reveal a distal point of maximal impulse, which can suggest cardiac dilatation or cardiomyopathy.
On examination, she had normal results for muscle strength and reflexes. A sensory exam also had normal results. We ordered blood work; her CBC, electrolytes, thyroid-stimulating hormone level, creatinine phosphokinase, and liver function tests all had normal results.
The next step: Cardio, neuro tests
If the patient’s history and physical examination yield normal results, he or she will require further tests, including the ones listed below.
Exclude arrhythmias and myopathy
Electrocardiogram (ECG). All patients should have an ECG. Most young patients and athletes have a variation in the sinus rate due to sinus arrhythmia. This is a normal finding and does not suggest a cause of syncope.
Prolongation of the PR interval or QRS duration may suggest the presence of atrioventricular nodal or conduction disease, respectively. A prolonged PR interval or left bundle branch block is very unusual in young patients; in the presence of syncope, these require further evaluation. The presence of isolated right bundle branch block is a less significant finding.1
The presence of a short PR interval and slurring of the QRS upstroke suggests Wolff-Parkinson-White syndrome. Abnormalities in the QRS morphology may suggest left ventricular hypertrophy or hypertrophic cardiomyopathy. You should also exclude abnormalities in repolarization such as prolonged QT interval, Brugada syndrome, and catecholamine-dependent polymorphic ventricular tachycardia.
A careful measurement of the corrected QT interval (QTc) is essential. The normal value of the QTc interval is <0.43 seconds. Prolonged QT interval may be due to presence of genetic or acquired (ie, drug-related) long QT syndrome. Acquired long QT syndrome can occur when a patient is taking an anti-arrhythmics such as amiodarone, or, more rarely, an antipsychotic such as haloperidol and ziprasidone.
Echocardiogram. All patients should have an echocardiogram before exercise stress testing to look for abnormalities such as aortic stenosis, left ventricular hypertrophy, and hypertrophic cardiomyopathy. When assessing for left ventricular enlargement, keep in mind that the left ventricular mass may be larger than what is seen in nonathletes. This condition, known as “athlete’s heart,” is mainly due to an increase in the cardiac cavity size and some increases in the left ventricular thickness. These patients show a preserved left ventricular ejection fraction.2
Pulmonary hypertension can also be evaluated using echocardiography; however, this is a rare cause of exercise-induced syncope.
Stress testing. Stress testing can reveal several different abnormalities found among patients with exercise-induced syncope. It may reveal ischemic changes on the electrocardiogram that suggest atherosclerotic coronary artery disease or anomalous take-off of the coronary arteries. Stress testing may also provoke neurocardiogenic syncope.
EEG and CT. Electroencephalography can show changes that suggest epilepsy as a cause of syncope. Exercise-induced temporal lobe seizures are extremely rare, but are another possible cause of loss of consciousness.
Computerized tomography of the chest combined with coronary CT angiography can be very useful in evaluating patients for structural abnormalities such as aortic dissection, pulmonary embolism, and especially coronary anomalies.
The search for other mechanisms
After patients have undergone the standard round of testing as described above, any further investigation will need to be customized to the patient. If structural cardiac abnormalities and arrhythmias are excluded (as was the case with our patient), the potential diagnoses are narrowed to neurocardiogenic syncope and orthostatic hypotension—2 common causes of sudden-onset syncope with sudden recovery.
Neurocardiogenic syncope. A common final mechanism of any syncope is a drop in blood pressure and low cerebral perfusion. This may be triggered by stimulation of the carotid baroreceptor reflex via cardiac hypercontractility, inadequate cardiac filling, or volume depletion. Volume depletion is more likely in endurance athletes who are exposed to prolonged exertion, such as marathon runners. Some patients with vasovagal mechanism may experience an aura of nausea prior to the loss of consciousness.
Another important mechanism of exercise-induced syncope is sudden vasodilatation or lack of vasoconstriction.3 Syncope can occur at peak exercise or immediately after stopping exercise. Syncope occurring after stopping exercise may be due to overwhelming vagal input during sympathetic withdrawal.
Several of these physiologic changes are accentuated during exercise and contribute to syncope. A strong cardiac contraction associated with exercise can trigger an abnormal baroreceptor reflex by stimulating cardiac mechanoreceptors.
Orthostatic intolerance. Syncope in distance athletes is sometimes due to orthostatic intolerance. Orthostatic hypotension may occur due to volume depletion and the increased contractility of the athlete’s heart—this may subsequently induce a neurocardiogenic mechanism of syncope. This is evaluated by measuring upright blood pressure after prolonged exertion.
Echocardiograms performed in athletes during upright tilt testing have proven that strong cardiac contractions occur during orthostatic stress.4 It has also been shown that syncopal athletes have a stronger cardiac contraction than nonsyncopal athletes.5 Athletes have increased cardiac distensibility—ie, increased variations of internal cardiac diameter over a range of equivalent filling pressures. This causes the cardiac contraction to be on the steep part of the Starling pressure; volume curve, so that a sudden drop in filling pressure results in a sudden fall in cardiac output and low blood pressure.6 Other researchers have found evidence against this, however.7
Loss of blood pressure can be caused by either sudden sympathetic withdrawal or parasympathetic overload. In some patients, syncope cannot be blocked by the use of atropine.8 A short diastolic filling time resulting from a rapid rate during exercise may decrease the ventricular filling in athletes and also result in low cardiac output due to the mechanism described above.
Orthostatic intolerance has been reported in ultra-marathon runners due to volume depletion;9 however, in light of the relatively short time to onset in our patient, and the lack of orthostasis after recovery, we did not suspect that this mechanism was at work.
Tilt table testing can be helpful
Reports on exercise-induced syncope in athletes show that a significant percentage of patients have a positive tilt table test, suggesting either an associated or independent abnormality of autonomic regulation.10
Positive tilt tests vary between 26% and 76% in these patients. In a series of 24 athletes with recurrent exercise-induced syncope, 19/24 were initially positive on tilt testing; after therapy only 2/24 remained positive.11 However, some may have been patients with underlying vasodepressor syncope exacerbated by exercise and may not have truly had “exercise-induced” syncope. It is not possible to define the exact sensitivity and specificity of tilt tests for these patients because of small sample sizes. We feel that a tilt test should be ordered, as it may suggest a diagnosis in some cases.
Invasive tests are not usually needed
Invasive electrophysiology testing has little role in athletes with syncope. Most well-conditioned athletes have a relative bradycardia due to high vagal tone, but it is extremely rare that they require a permanent pacemaker.12 Although some of these patients may have abnormal sinus node function when assessed by electrophysiology testing, to adequately test sinus node function in these patients you should perform autonomic blockade using atropine and propranolol; then an intrinsic heart rate (without the effects of autonomic tone) can be measured.
In most cases, you need to see the rhythm during an episode for accurate diagnosis. This requires a 12-lead ECG, and possibly a Holter monitor or event recorder. Remember that abnormal findings during a tilt table or electrophysiology test may be unrelated to the cause of syncope.
Anomalous take-off of coronary arteries should be excluded. In the past this required cardiac catheterization, but can now be assessed noninvasively with magnetic resonance angiography or CT.
What testing revealed about our patient
Our patient’s ECG showed nonspecific ST-T wave abnormality with no evidence of Wolff-Parkinson-White abnormality or conduction system disease. An echocardiogram showed a left ventricular end diastolic dimension of 50 mm (normal range, 40-62 mm) and a left ventricular ejection fraction of 55% (normal is 55%-60%). We saw no valvular abnormality and no evidence of left ventricular hypertrophy to suggest hypertrophic cardiomyopathy.
A stress test was performed using the Bruce protocol, during which the patient exercised for 13 minutes and achieved a heart rate of 184 bpm. She did not have any symptoms of loss of consciousness, and we saw no abnormal ST-T segment changes.
A tilt test was performed with and without isoproterenol, during which time her heart rate reached 125 bpm. There was no syncope or loss of consciousness.
A cardiac CT test was performed, which excluded coronary anomalies. The exercise test was repeated using a modified protocol with a pace set to simulate her level of exercise. The treadmill was set at a speed of 9.6 mph (a 6.25-minute mile) with no incline. Sixteen minutes into the test she had a sudden loss of consciousness and fell. She was diaphoretic with a weak, thready, rapid pulse and cool extremities.
Shortly after her fall, we recorded her blood pressure using a cuff sphygmomanometer—it was 169/95 mm Hg with a heart rate of 140 bpm. However, the patient had recovered consciousness by the time the blood pressure was recorded. The duration of loss of consciousness was less than 15 seconds. No hypotension or arrhythmias were recorded.
The exercise test with new protocol was repeated, but with placement of an intra-arterial blood pressure monitor and continuous electroencephalographic monitoring. Sixteen minutes into the test the patient requested that we stop, due to fatigue and leg pain. Just prior to this, at peak exercise, her blood pressure was 220/30 mm Hg and heart rate was 185 bpm. Immediately after stopping the tread-mill, we measured a precipitous drop in intra-arterial blood pressure—down 45/4 mm Hg—and her heart rate slowed to 178 bpm. This was associated with reproduction of clinical symptoms. She lay down, and her blood pressure recovered to >120 mm systolic within seconds.
Electroencephalographic monitoring did not show any epileptiform activity. Serum blood glucose recorded during the spell was 86 mg/dL. Our interpretation was that she had a brief precipitous vasodepressor syncope triggered by exercise.
Management: Changing habits, medication
You can advise patients with exercise-induced syncope to avoid strenuous exercise, but this is not acceptable for many athletes. If the patient is on certain drugs, such as vasodilators for the treatment of hypertension, these must be discontinued and another category of drug substituted, such as angiotensin-converting enzyme inhibitors.
The athlete should also increase his or her fluid and salt intake. This can be achieved with increased use of sports drinks or salt tablets (strength of recommendation [SOR]: B). Of course this must be done in moderation and titrated to each individual athlete’s needs.
Many Rx options; we started with fludrocortisone
Pharmacotherapy for treatment of vasovagal syncope has been extensively tested, but no medication is reliably effective. Beta-blockers are sometimes prescribed, but randomized clinical trials have failed to show a predictable benefit (SOR: A).13 Pindolol may induces less bradycardia due to intrinsic sympathomimetic activity.14
Other possible medications include midodrine, fludrocortisone, theophylline, clonidine, and serotonin reuptake inhibitors. Vasoconstrictors (such as midodrine) and fludrocortisone are more likely to be effective for patients with orthostatic hypotension than vasovagal syncope (SOR: B).15 Fludrocortisone may cause weight gain and fatigue and affect performance in athletes.
Disopyramide is a class I anti-arrhythmic agent that has additional effects of negative inotropy and a vagolytic effect. This decreases the stimulation of the carotid baroreceptors and interrupts the vagal efferent pathway. We have found this to be very useful and well-tolerated. Theoretically, though, because of its negative inotropic effect, it may decrease cardiac output enough to affect athletic performance. Initiate the drug with telemetry monitoring to detect any pro-arrhythmic electrocardiographic changes, such as torsades de pointes or QT prolongation. Prevention of the hyperdynamic cardiac contraction can trigger an abnormal baroreceptor reflex and block the efferent limb of the vagal response to counter the bradycardia and vasodepressor response.16
If pharmacotherapy is required, we generally prescribe drugs in the following sequence—first fludrocortisone, then beta-blockers, and, if these do not help, disopyramide. These should be first given in the hospital with telemetry monitoring to check for QT prolongation. If these are ineffective, try paroxetine or midodrine. In refractory cases, we may use various combinations of these drugs.
Disopyramide and a changed running strategy
Several mechanisms related to vigorous exercise played a role in our patient’s case. During exercise, she developed a very high systolic pressure with a low diastolic pressure, resulting in a high pulse pressure that possibly led to stimulation of carotid baroreceptors. Cardiac hypercontractility related to exercise was probably a contributing factor as well. Severe leg pain prior to syncope suggested involvement of a pain-mediated mechanism.
We prescribed disopyramide 150 mg twice daily for our patient. We also recommended that she do specific exercises—to strengthen her leg muscles—and modify her running strategy.
After 2 months of therapy, she resumed competitive running successfully without any further episodes of syncope. She was able to achieve a new personal record for a cross country run by running a slower pace of 6 minutes 20 seconds per mile initially and running faster only at the end of the race. She had no recurrences of syncope during 6 months of follow-up.
CorrespondenceSumit Verma, MD, Pensacola Heart Institute, 5151 N, 9th Avenue, Cardiology Consultants, Pensacola, Fl 32504; [email protected].
1. Fahy GJ, Pinksi SL, Miller DP, et al. Natural history of isolated bundle branch block. Am J Cardiol 1996;77:1185-1190.
2. Maron BJ. Structural features of the athlete’s heart: As defined by echocardiography. J Am Coll Cardiol 1986;7:190-203.
3. Sneddon J, Scalia G, Ward D, McKenna W, Camm AJ, Frenneaux M. Exercise induced vasodepressor syncope. Br Heart J 1994;71:554-557.
4. Shalev Y, Gal R, Tchou P, et al. Echocardiographic demonstration of decreased left ventricular dimensions and vigorous myocardial contraction during syncope induced by head up tilt. J Am Coll Cardiol 1991;18:746-751.
5. Ferrario G, Nicoli J, Peci P, Ginni P. Cardiac adaptation to training. Is it the key to understand the high incidence of positive tilt test in athletes? (Abstract.) Pacing Electrophys 1996;19:578.-
6. Levine B. Regulation of central blood volume and cardiac filling in endurance athletes, the Frank-Starling mechanism as a determinant of orthostatic tolerance. Med Sci Sports Exer 1993;25:727-731.
7. Convertino VA, Sather TM, Goldwater D, Alford W, Hahn H. Aerobic fitness does not contribute to prediction of orthostatic intolerance. Med Sci Sports Exer 1989;18:551-556.
8. Sra J, Jazayeri M, Avitall B, Blanck Z, Akhtar M. Comparison of cardiac pacing with drug therapy in the treatment of neurocardiogenic syncope with bradycardia or hypotension. N Engl J Med 1993;328:1085-1090.
9. Holtzhausen LM, Noakes TD. The prevalence and significance of post-exercise (postural) hypotension in ultramarathon runners. Med Sci Sports Exerc 1995;27:1595-1601.
10. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69-74.
11. Grubb BP, Temesy-Armos P, Samoil D, Wolfe D, Hahn H, Elliot L. Tilt table testing in the evaluation and management of athletes with recurrent exercise induced syncope. Med Sci Sports Exerc 1993;25:24-28.
12. Rasmussen V, Hauns S, Skagen K. Cerebral attacks due to excessive vagal tone in heavily trained persons. Acta Med Scand 1978;204:401-405.
13. Muller G, Deal B, Strasburger JF, Benson DW, Jr. Usefulness of metoprolol for unexplained syncope and positive response to tilt table testing in young persons. Am J Cardiol 1993;71:592-595.
14. Lurie KG, Dutton J, Mangat R, Scheinman MM. Pindolol is effective in patients with vasovagal syncope. Pacing Clin Electrophysiol 1992;15:592.-Abstract.
15. Sra J, Maglio C, Biehl M, et al. Efficacy of midodrine hydrochloride in neurocardiogenic syncope refractory to standard therapy. J Cardiovasc Electrophysiol 1997;8:42-46.
16. Milstein S, Buetikofer J, Dunnigan A, et al. usefulness of disopyramide for prevention of upright tilt-induced hypotension-bradycardia. Am J Cardiol 1990;65:1339-1344.
- Following inconclusive cardiovascular and neurological testing, an echocardiogram, stress test with Holter monitor, and possibly tilt table testing are appropriate ways to determine whether a vasovagal response is to blame for a young athlete’s exercise-induced fainting episodes (C).
- Advise patients with exercise-induced syncope to increase fluid and salt intake while exercising (B). Strength training can also be helpful (B). Most drug therapies, such as beta-blockers, vasoconstrictors, and anti-arrhythmics, have inconsistent results (A). use of permanent pacemakers are not effective (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
An 18-year-old woman was referred to us for evaluation after fainting on several occasions during a track workout. She’d been a competitive athlete for several years, and the episodes began 3 months earlier. She reported that they happen shortly after she started to run. She also told us that just before a spell, she had a sense of fatigue and severe leg pain.
Bystanders who witnessed one of the episodes reported that her eyes rolled upwards right before she fell; they saw no tonic-clonic activity that would indicate a seizure. We learned that the spells were short-lived and followed by immediate recovery with no mental deficits, such as confusion. On several occasions, she suffered superficial skin abrasions, but fortunately no serious injuries had occurred.
We suspected exercise-induced syncope
Prompt diagnosis of syncope in athletes is essential as it may be a marker for sudden cardiac death. The differential diagnosis, however, is broad (TABLE). Syncope in adolescents is often associated with fasting and eating disorders, excessive heat, alcohol, abdominal straining, exercise, hypoglycemia, growth spurts, lack of sleep, and low blood pressure.
TABLE
Differential diagnosis for exercise-induced syncope
| Anaphylaxis | Hypertrophic cardiomyopathy |
| Aortic stenosis | Hypoglycemia |
| Atrial fibrillation | Hypoxia |
| Brugada syndrome | Left ventricular hypertrophy |
| Cardiac dilatation | Pulmonary hypertension |
| Cardiac ischemia | Sick sinus syndrome |
| Catecholamine-dependant polymorphic ventricular tachycardia | Vasodepressor reflex |
| Commotio cordis | Ventricular fibrillation |
| Dehydration | Ventricular tachycardia |
| Eating disorders | Wolff-Parkinson-White abnormality |
Few clues from her history and exam
The clinical evaluation requires a careful and detailed history, including taking a family history to exclude familial causes of sudden death, such as long QT syndrome. In our patient’s case, there was no family history of sudden death or arrhythmias. She was a nonsmoker, nondrinker, and denied using any illicit drugs. She had been taking phenytoin for suspected seizure activity, but the spells continued.
On further investigation, we discovered that our patient would faint predictably 14 minutes into a competitive 5K run. She had recently adopted a more aggressive “early breakaway” strategy: she would run the initial part of the race at a pace of about 6 minutes per mile and slow down gradually towards the end.
We also learned that she had never fainted during weight training or when running at a less aggressive pace. Other than fatigue and extreme leg pain, she could not recall any other symptoms before falling, such as nausea, dizziness, weakness, or palpitations.
Her resting seated left arm blood pressure was 110/60 mm Hg and her heart rate was regular at 53 bpm. On cardiac auscultation, we heard no murmurs or abnormal cardiac sounds in the supine or standing positions. Cardiac palpation did not reveal a distal point of maximal impulse, which can suggest cardiac dilatation or cardiomyopathy.
On examination, she had normal results for muscle strength and reflexes. A sensory exam also had normal results. We ordered blood work; her CBC, electrolytes, thyroid-stimulating hormone level, creatinine phosphokinase, and liver function tests all had normal results.
The next step: Cardio, neuro tests
If the patient’s history and physical examination yield normal results, he or she will require further tests, including the ones listed below.
Exclude arrhythmias and myopathy
Electrocardiogram (ECG). All patients should have an ECG. Most young patients and athletes have a variation in the sinus rate due to sinus arrhythmia. This is a normal finding and does not suggest a cause of syncope.
Prolongation of the PR interval or QRS duration may suggest the presence of atrioventricular nodal or conduction disease, respectively. A prolonged PR interval or left bundle branch block is very unusual in young patients; in the presence of syncope, these require further evaluation. The presence of isolated right bundle branch block is a less significant finding.1
The presence of a short PR interval and slurring of the QRS upstroke suggests Wolff-Parkinson-White syndrome. Abnormalities in the QRS morphology may suggest left ventricular hypertrophy or hypertrophic cardiomyopathy. You should also exclude abnormalities in repolarization such as prolonged QT interval, Brugada syndrome, and catecholamine-dependent polymorphic ventricular tachycardia.
A careful measurement of the corrected QT interval (QTc) is essential. The normal value of the QTc interval is <0.43 seconds. Prolonged QT interval may be due to presence of genetic or acquired (ie, drug-related) long QT syndrome. Acquired long QT syndrome can occur when a patient is taking an anti-arrhythmics such as amiodarone, or, more rarely, an antipsychotic such as haloperidol and ziprasidone.
Echocardiogram. All patients should have an echocardiogram before exercise stress testing to look for abnormalities such as aortic stenosis, left ventricular hypertrophy, and hypertrophic cardiomyopathy. When assessing for left ventricular enlargement, keep in mind that the left ventricular mass may be larger than what is seen in nonathletes. This condition, known as “athlete’s heart,” is mainly due to an increase in the cardiac cavity size and some increases in the left ventricular thickness. These patients show a preserved left ventricular ejection fraction.2
Pulmonary hypertension can also be evaluated using echocardiography; however, this is a rare cause of exercise-induced syncope.
Stress testing. Stress testing can reveal several different abnormalities found among patients with exercise-induced syncope. It may reveal ischemic changes on the electrocardiogram that suggest atherosclerotic coronary artery disease or anomalous take-off of the coronary arteries. Stress testing may also provoke neurocardiogenic syncope.
EEG and CT. Electroencephalography can show changes that suggest epilepsy as a cause of syncope. Exercise-induced temporal lobe seizures are extremely rare, but are another possible cause of loss of consciousness.
Computerized tomography of the chest combined with coronary CT angiography can be very useful in evaluating patients for structural abnormalities such as aortic dissection, pulmonary embolism, and especially coronary anomalies.
The search for other mechanisms
After patients have undergone the standard round of testing as described above, any further investigation will need to be customized to the patient. If structural cardiac abnormalities and arrhythmias are excluded (as was the case with our patient), the potential diagnoses are narrowed to neurocardiogenic syncope and orthostatic hypotension—2 common causes of sudden-onset syncope with sudden recovery.
Neurocardiogenic syncope. A common final mechanism of any syncope is a drop in blood pressure and low cerebral perfusion. This may be triggered by stimulation of the carotid baroreceptor reflex via cardiac hypercontractility, inadequate cardiac filling, or volume depletion. Volume depletion is more likely in endurance athletes who are exposed to prolonged exertion, such as marathon runners. Some patients with vasovagal mechanism may experience an aura of nausea prior to the loss of consciousness.
Another important mechanism of exercise-induced syncope is sudden vasodilatation or lack of vasoconstriction.3 Syncope can occur at peak exercise or immediately after stopping exercise. Syncope occurring after stopping exercise may be due to overwhelming vagal input during sympathetic withdrawal.
Several of these physiologic changes are accentuated during exercise and contribute to syncope. A strong cardiac contraction associated with exercise can trigger an abnormal baroreceptor reflex by stimulating cardiac mechanoreceptors.
Orthostatic intolerance. Syncope in distance athletes is sometimes due to orthostatic intolerance. Orthostatic hypotension may occur due to volume depletion and the increased contractility of the athlete’s heart—this may subsequently induce a neurocardiogenic mechanism of syncope. This is evaluated by measuring upright blood pressure after prolonged exertion.
Echocardiograms performed in athletes during upright tilt testing have proven that strong cardiac contractions occur during orthostatic stress.4 It has also been shown that syncopal athletes have a stronger cardiac contraction than nonsyncopal athletes.5 Athletes have increased cardiac distensibility—ie, increased variations of internal cardiac diameter over a range of equivalent filling pressures. This causes the cardiac contraction to be on the steep part of the Starling pressure; volume curve, so that a sudden drop in filling pressure results in a sudden fall in cardiac output and low blood pressure.6 Other researchers have found evidence against this, however.7
Loss of blood pressure can be caused by either sudden sympathetic withdrawal or parasympathetic overload. In some patients, syncope cannot be blocked by the use of atropine.8 A short diastolic filling time resulting from a rapid rate during exercise may decrease the ventricular filling in athletes and also result in low cardiac output due to the mechanism described above.
Orthostatic intolerance has been reported in ultra-marathon runners due to volume depletion;9 however, in light of the relatively short time to onset in our patient, and the lack of orthostasis after recovery, we did not suspect that this mechanism was at work.
Tilt table testing can be helpful
Reports on exercise-induced syncope in athletes show that a significant percentage of patients have a positive tilt table test, suggesting either an associated or independent abnormality of autonomic regulation.10
Positive tilt tests vary between 26% and 76% in these patients. In a series of 24 athletes with recurrent exercise-induced syncope, 19/24 were initially positive on tilt testing; after therapy only 2/24 remained positive.11 However, some may have been patients with underlying vasodepressor syncope exacerbated by exercise and may not have truly had “exercise-induced” syncope. It is not possible to define the exact sensitivity and specificity of tilt tests for these patients because of small sample sizes. We feel that a tilt test should be ordered, as it may suggest a diagnosis in some cases.
Invasive tests are not usually needed
Invasive electrophysiology testing has little role in athletes with syncope. Most well-conditioned athletes have a relative bradycardia due to high vagal tone, but it is extremely rare that they require a permanent pacemaker.12 Although some of these patients may have abnormal sinus node function when assessed by electrophysiology testing, to adequately test sinus node function in these patients you should perform autonomic blockade using atropine and propranolol; then an intrinsic heart rate (without the effects of autonomic tone) can be measured.
In most cases, you need to see the rhythm during an episode for accurate diagnosis. This requires a 12-lead ECG, and possibly a Holter monitor or event recorder. Remember that abnormal findings during a tilt table or electrophysiology test may be unrelated to the cause of syncope.
Anomalous take-off of coronary arteries should be excluded. In the past this required cardiac catheterization, but can now be assessed noninvasively with magnetic resonance angiography or CT.
What testing revealed about our patient
Our patient’s ECG showed nonspecific ST-T wave abnormality with no evidence of Wolff-Parkinson-White abnormality or conduction system disease. An echocardiogram showed a left ventricular end diastolic dimension of 50 mm (normal range, 40-62 mm) and a left ventricular ejection fraction of 55% (normal is 55%-60%). We saw no valvular abnormality and no evidence of left ventricular hypertrophy to suggest hypertrophic cardiomyopathy.
A stress test was performed using the Bruce protocol, during which the patient exercised for 13 minutes and achieved a heart rate of 184 bpm. She did not have any symptoms of loss of consciousness, and we saw no abnormal ST-T segment changes.
A tilt test was performed with and without isoproterenol, during which time her heart rate reached 125 bpm. There was no syncope or loss of consciousness.
A cardiac CT test was performed, which excluded coronary anomalies. The exercise test was repeated using a modified protocol with a pace set to simulate her level of exercise. The treadmill was set at a speed of 9.6 mph (a 6.25-minute mile) with no incline. Sixteen minutes into the test she had a sudden loss of consciousness and fell. She was diaphoretic with a weak, thready, rapid pulse and cool extremities.
Shortly after her fall, we recorded her blood pressure using a cuff sphygmomanometer—it was 169/95 mm Hg with a heart rate of 140 bpm. However, the patient had recovered consciousness by the time the blood pressure was recorded. The duration of loss of consciousness was less than 15 seconds. No hypotension or arrhythmias were recorded.
The exercise test with new protocol was repeated, but with placement of an intra-arterial blood pressure monitor and continuous electroencephalographic monitoring. Sixteen minutes into the test the patient requested that we stop, due to fatigue and leg pain. Just prior to this, at peak exercise, her blood pressure was 220/30 mm Hg and heart rate was 185 bpm. Immediately after stopping the tread-mill, we measured a precipitous drop in intra-arterial blood pressure—down 45/4 mm Hg—and her heart rate slowed to 178 bpm. This was associated with reproduction of clinical symptoms. She lay down, and her blood pressure recovered to >120 mm systolic within seconds.
Electroencephalographic monitoring did not show any epileptiform activity. Serum blood glucose recorded during the spell was 86 mg/dL. Our interpretation was that she had a brief precipitous vasodepressor syncope triggered by exercise.
Management: Changing habits, medication
You can advise patients with exercise-induced syncope to avoid strenuous exercise, but this is not acceptable for many athletes. If the patient is on certain drugs, such as vasodilators for the treatment of hypertension, these must be discontinued and another category of drug substituted, such as angiotensin-converting enzyme inhibitors.
The athlete should also increase his or her fluid and salt intake. This can be achieved with increased use of sports drinks or salt tablets (strength of recommendation [SOR]: B). Of course this must be done in moderation and titrated to each individual athlete’s needs.
Many Rx options; we started with fludrocortisone
Pharmacotherapy for treatment of vasovagal syncope has been extensively tested, but no medication is reliably effective. Beta-blockers are sometimes prescribed, but randomized clinical trials have failed to show a predictable benefit (SOR: A).13 Pindolol may induces less bradycardia due to intrinsic sympathomimetic activity.14
Other possible medications include midodrine, fludrocortisone, theophylline, clonidine, and serotonin reuptake inhibitors. Vasoconstrictors (such as midodrine) and fludrocortisone are more likely to be effective for patients with orthostatic hypotension than vasovagal syncope (SOR: B).15 Fludrocortisone may cause weight gain and fatigue and affect performance in athletes.
Disopyramide is a class I anti-arrhythmic agent that has additional effects of negative inotropy and a vagolytic effect. This decreases the stimulation of the carotid baroreceptors and interrupts the vagal efferent pathway. We have found this to be very useful and well-tolerated. Theoretically, though, because of its negative inotropic effect, it may decrease cardiac output enough to affect athletic performance. Initiate the drug with telemetry monitoring to detect any pro-arrhythmic electrocardiographic changes, such as torsades de pointes or QT prolongation. Prevention of the hyperdynamic cardiac contraction can trigger an abnormal baroreceptor reflex and block the efferent limb of the vagal response to counter the bradycardia and vasodepressor response.16
If pharmacotherapy is required, we generally prescribe drugs in the following sequence—first fludrocortisone, then beta-blockers, and, if these do not help, disopyramide. These should be first given in the hospital with telemetry monitoring to check for QT prolongation. If these are ineffective, try paroxetine or midodrine. In refractory cases, we may use various combinations of these drugs.
Disopyramide and a changed running strategy
Several mechanisms related to vigorous exercise played a role in our patient’s case. During exercise, she developed a very high systolic pressure with a low diastolic pressure, resulting in a high pulse pressure that possibly led to stimulation of carotid baroreceptors. Cardiac hypercontractility related to exercise was probably a contributing factor as well. Severe leg pain prior to syncope suggested involvement of a pain-mediated mechanism.
We prescribed disopyramide 150 mg twice daily for our patient. We also recommended that she do specific exercises—to strengthen her leg muscles—and modify her running strategy.
After 2 months of therapy, she resumed competitive running successfully without any further episodes of syncope. She was able to achieve a new personal record for a cross country run by running a slower pace of 6 minutes 20 seconds per mile initially and running faster only at the end of the race. She had no recurrences of syncope during 6 months of follow-up.
CorrespondenceSumit Verma, MD, Pensacola Heart Institute, 5151 N, 9th Avenue, Cardiology Consultants, Pensacola, Fl 32504; [email protected].
- Following inconclusive cardiovascular and neurological testing, an echocardiogram, stress test with Holter monitor, and possibly tilt table testing are appropriate ways to determine whether a vasovagal response is to blame for a young athlete’s exercise-induced fainting episodes (C).
- Advise patients with exercise-induced syncope to increase fluid and salt intake while exercising (B). Strength training can also be helpful (B). Most drug therapies, such as beta-blockers, vasoconstrictors, and anti-arrhythmics, have inconsistent results (A). use of permanent pacemakers are not effective (B).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
An 18-year-old woman was referred to us for evaluation after fainting on several occasions during a track workout. She’d been a competitive athlete for several years, and the episodes began 3 months earlier. She reported that they happen shortly after she started to run. She also told us that just before a spell, she had a sense of fatigue and severe leg pain.
Bystanders who witnessed one of the episodes reported that her eyes rolled upwards right before she fell; they saw no tonic-clonic activity that would indicate a seizure. We learned that the spells were short-lived and followed by immediate recovery with no mental deficits, such as confusion. On several occasions, she suffered superficial skin abrasions, but fortunately no serious injuries had occurred.
We suspected exercise-induced syncope
Prompt diagnosis of syncope in athletes is essential as it may be a marker for sudden cardiac death. The differential diagnosis, however, is broad (TABLE). Syncope in adolescents is often associated with fasting and eating disorders, excessive heat, alcohol, abdominal straining, exercise, hypoglycemia, growth spurts, lack of sleep, and low blood pressure.
TABLE
Differential diagnosis for exercise-induced syncope
| Anaphylaxis | Hypertrophic cardiomyopathy |
| Aortic stenosis | Hypoglycemia |
| Atrial fibrillation | Hypoxia |
| Brugada syndrome | Left ventricular hypertrophy |
| Cardiac dilatation | Pulmonary hypertension |
| Cardiac ischemia | Sick sinus syndrome |
| Catecholamine-dependant polymorphic ventricular tachycardia | Vasodepressor reflex |
| Commotio cordis | Ventricular fibrillation |
| Dehydration | Ventricular tachycardia |
| Eating disorders | Wolff-Parkinson-White abnormality |
Few clues from her history and exam
The clinical evaluation requires a careful and detailed history, including taking a family history to exclude familial causes of sudden death, such as long QT syndrome. In our patient’s case, there was no family history of sudden death or arrhythmias. She was a nonsmoker, nondrinker, and denied using any illicit drugs. She had been taking phenytoin for suspected seizure activity, but the spells continued.
On further investigation, we discovered that our patient would faint predictably 14 minutes into a competitive 5K run. She had recently adopted a more aggressive “early breakaway” strategy: she would run the initial part of the race at a pace of about 6 minutes per mile and slow down gradually towards the end.
We also learned that she had never fainted during weight training or when running at a less aggressive pace. Other than fatigue and extreme leg pain, she could not recall any other symptoms before falling, such as nausea, dizziness, weakness, or palpitations.
Her resting seated left arm blood pressure was 110/60 mm Hg and her heart rate was regular at 53 bpm. On cardiac auscultation, we heard no murmurs or abnormal cardiac sounds in the supine or standing positions. Cardiac palpation did not reveal a distal point of maximal impulse, which can suggest cardiac dilatation or cardiomyopathy.
On examination, she had normal results for muscle strength and reflexes. A sensory exam also had normal results. We ordered blood work; her CBC, electrolytes, thyroid-stimulating hormone level, creatinine phosphokinase, and liver function tests all had normal results.
The next step: Cardio, neuro tests
If the patient’s history and physical examination yield normal results, he or she will require further tests, including the ones listed below.
Exclude arrhythmias and myopathy
Electrocardiogram (ECG). All patients should have an ECG. Most young patients and athletes have a variation in the sinus rate due to sinus arrhythmia. This is a normal finding and does not suggest a cause of syncope.
Prolongation of the PR interval or QRS duration may suggest the presence of atrioventricular nodal or conduction disease, respectively. A prolonged PR interval or left bundle branch block is very unusual in young patients; in the presence of syncope, these require further evaluation. The presence of isolated right bundle branch block is a less significant finding.1
The presence of a short PR interval and slurring of the QRS upstroke suggests Wolff-Parkinson-White syndrome. Abnormalities in the QRS morphology may suggest left ventricular hypertrophy or hypertrophic cardiomyopathy. You should also exclude abnormalities in repolarization such as prolonged QT interval, Brugada syndrome, and catecholamine-dependent polymorphic ventricular tachycardia.
A careful measurement of the corrected QT interval (QTc) is essential. The normal value of the QTc interval is <0.43 seconds. Prolonged QT interval may be due to presence of genetic or acquired (ie, drug-related) long QT syndrome. Acquired long QT syndrome can occur when a patient is taking an anti-arrhythmics such as amiodarone, or, more rarely, an antipsychotic such as haloperidol and ziprasidone.
Echocardiogram. All patients should have an echocardiogram before exercise stress testing to look for abnormalities such as aortic stenosis, left ventricular hypertrophy, and hypertrophic cardiomyopathy. When assessing for left ventricular enlargement, keep in mind that the left ventricular mass may be larger than what is seen in nonathletes. This condition, known as “athlete’s heart,” is mainly due to an increase in the cardiac cavity size and some increases in the left ventricular thickness. These patients show a preserved left ventricular ejection fraction.2
Pulmonary hypertension can also be evaluated using echocardiography; however, this is a rare cause of exercise-induced syncope.
Stress testing. Stress testing can reveal several different abnormalities found among patients with exercise-induced syncope. It may reveal ischemic changes on the electrocardiogram that suggest atherosclerotic coronary artery disease or anomalous take-off of the coronary arteries. Stress testing may also provoke neurocardiogenic syncope.
EEG and CT. Electroencephalography can show changes that suggest epilepsy as a cause of syncope. Exercise-induced temporal lobe seizures are extremely rare, but are another possible cause of loss of consciousness.
Computerized tomography of the chest combined with coronary CT angiography can be very useful in evaluating patients for structural abnormalities such as aortic dissection, pulmonary embolism, and especially coronary anomalies.
The search for other mechanisms
After patients have undergone the standard round of testing as described above, any further investigation will need to be customized to the patient. If structural cardiac abnormalities and arrhythmias are excluded (as was the case with our patient), the potential diagnoses are narrowed to neurocardiogenic syncope and orthostatic hypotension—2 common causes of sudden-onset syncope with sudden recovery.
Neurocardiogenic syncope. A common final mechanism of any syncope is a drop in blood pressure and low cerebral perfusion. This may be triggered by stimulation of the carotid baroreceptor reflex via cardiac hypercontractility, inadequate cardiac filling, or volume depletion. Volume depletion is more likely in endurance athletes who are exposed to prolonged exertion, such as marathon runners. Some patients with vasovagal mechanism may experience an aura of nausea prior to the loss of consciousness.
Another important mechanism of exercise-induced syncope is sudden vasodilatation or lack of vasoconstriction.3 Syncope can occur at peak exercise or immediately after stopping exercise. Syncope occurring after stopping exercise may be due to overwhelming vagal input during sympathetic withdrawal.
Several of these physiologic changes are accentuated during exercise and contribute to syncope. A strong cardiac contraction associated with exercise can trigger an abnormal baroreceptor reflex by stimulating cardiac mechanoreceptors.
Orthostatic intolerance. Syncope in distance athletes is sometimes due to orthostatic intolerance. Orthostatic hypotension may occur due to volume depletion and the increased contractility of the athlete’s heart—this may subsequently induce a neurocardiogenic mechanism of syncope. This is evaluated by measuring upright blood pressure after prolonged exertion.
Echocardiograms performed in athletes during upright tilt testing have proven that strong cardiac contractions occur during orthostatic stress.4 It has also been shown that syncopal athletes have a stronger cardiac contraction than nonsyncopal athletes.5 Athletes have increased cardiac distensibility—ie, increased variations of internal cardiac diameter over a range of equivalent filling pressures. This causes the cardiac contraction to be on the steep part of the Starling pressure; volume curve, so that a sudden drop in filling pressure results in a sudden fall in cardiac output and low blood pressure.6 Other researchers have found evidence against this, however.7
Loss of blood pressure can be caused by either sudden sympathetic withdrawal or parasympathetic overload. In some patients, syncope cannot be blocked by the use of atropine.8 A short diastolic filling time resulting from a rapid rate during exercise may decrease the ventricular filling in athletes and also result in low cardiac output due to the mechanism described above.
Orthostatic intolerance has been reported in ultra-marathon runners due to volume depletion;9 however, in light of the relatively short time to onset in our patient, and the lack of orthostasis after recovery, we did not suspect that this mechanism was at work.
Tilt table testing can be helpful
Reports on exercise-induced syncope in athletes show that a significant percentage of patients have a positive tilt table test, suggesting either an associated or independent abnormality of autonomic regulation.10
Positive tilt tests vary between 26% and 76% in these patients. In a series of 24 athletes with recurrent exercise-induced syncope, 19/24 were initially positive on tilt testing; after therapy only 2/24 remained positive.11 However, some may have been patients with underlying vasodepressor syncope exacerbated by exercise and may not have truly had “exercise-induced” syncope. It is not possible to define the exact sensitivity and specificity of tilt tests for these patients because of small sample sizes. We feel that a tilt test should be ordered, as it may suggest a diagnosis in some cases.
Invasive tests are not usually needed
Invasive electrophysiology testing has little role in athletes with syncope. Most well-conditioned athletes have a relative bradycardia due to high vagal tone, but it is extremely rare that they require a permanent pacemaker.12 Although some of these patients may have abnormal sinus node function when assessed by electrophysiology testing, to adequately test sinus node function in these patients you should perform autonomic blockade using atropine and propranolol; then an intrinsic heart rate (without the effects of autonomic tone) can be measured.
In most cases, you need to see the rhythm during an episode for accurate diagnosis. This requires a 12-lead ECG, and possibly a Holter monitor or event recorder. Remember that abnormal findings during a tilt table or electrophysiology test may be unrelated to the cause of syncope.
Anomalous take-off of coronary arteries should be excluded. In the past this required cardiac catheterization, but can now be assessed noninvasively with magnetic resonance angiography or CT.
What testing revealed about our patient
Our patient’s ECG showed nonspecific ST-T wave abnormality with no evidence of Wolff-Parkinson-White abnormality or conduction system disease. An echocardiogram showed a left ventricular end diastolic dimension of 50 mm (normal range, 40-62 mm) and a left ventricular ejection fraction of 55% (normal is 55%-60%). We saw no valvular abnormality and no evidence of left ventricular hypertrophy to suggest hypertrophic cardiomyopathy.
A stress test was performed using the Bruce protocol, during which the patient exercised for 13 minutes and achieved a heart rate of 184 bpm. She did not have any symptoms of loss of consciousness, and we saw no abnormal ST-T segment changes.
A tilt test was performed with and without isoproterenol, during which time her heart rate reached 125 bpm. There was no syncope or loss of consciousness.
A cardiac CT test was performed, which excluded coronary anomalies. The exercise test was repeated using a modified protocol with a pace set to simulate her level of exercise. The treadmill was set at a speed of 9.6 mph (a 6.25-minute mile) with no incline. Sixteen minutes into the test she had a sudden loss of consciousness and fell. She was diaphoretic with a weak, thready, rapid pulse and cool extremities.
Shortly after her fall, we recorded her blood pressure using a cuff sphygmomanometer—it was 169/95 mm Hg with a heart rate of 140 bpm. However, the patient had recovered consciousness by the time the blood pressure was recorded. The duration of loss of consciousness was less than 15 seconds. No hypotension or arrhythmias were recorded.
The exercise test with new protocol was repeated, but with placement of an intra-arterial blood pressure monitor and continuous electroencephalographic monitoring. Sixteen minutes into the test the patient requested that we stop, due to fatigue and leg pain. Just prior to this, at peak exercise, her blood pressure was 220/30 mm Hg and heart rate was 185 bpm. Immediately after stopping the tread-mill, we measured a precipitous drop in intra-arterial blood pressure—down 45/4 mm Hg—and her heart rate slowed to 178 bpm. This was associated with reproduction of clinical symptoms. She lay down, and her blood pressure recovered to >120 mm systolic within seconds.
Electroencephalographic monitoring did not show any epileptiform activity. Serum blood glucose recorded during the spell was 86 mg/dL. Our interpretation was that she had a brief precipitous vasodepressor syncope triggered by exercise.
Management: Changing habits, medication
You can advise patients with exercise-induced syncope to avoid strenuous exercise, but this is not acceptable for many athletes. If the patient is on certain drugs, such as vasodilators for the treatment of hypertension, these must be discontinued and another category of drug substituted, such as angiotensin-converting enzyme inhibitors.
The athlete should also increase his or her fluid and salt intake. This can be achieved with increased use of sports drinks or salt tablets (strength of recommendation [SOR]: B). Of course this must be done in moderation and titrated to each individual athlete’s needs.
Many Rx options; we started with fludrocortisone
Pharmacotherapy for treatment of vasovagal syncope has been extensively tested, but no medication is reliably effective. Beta-blockers are sometimes prescribed, but randomized clinical trials have failed to show a predictable benefit (SOR: A).13 Pindolol may induces less bradycardia due to intrinsic sympathomimetic activity.14
Other possible medications include midodrine, fludrocortisone, theophylline, clonidine, and serotonin reuptake inhibitors. Vasoconstrictors (such as midodrine) and fludrocortisone are more likely to be effective for patients with orthostatic hypotension than vasovagal syncope (SOR: B).15 Fludrocortisone may cause weight gain and fatigue and affect performance in athletes.
Disopyramide is a class I anti-arrhythmic agent that has additional effects of negative inotropy and a vagolytic effect. This decreases the stimulation of the carotid baroreceptors and interrupts the vagal efferent pathway. We have found this to be very useful and well-tolerated. Theoretically, though, because of its negative inotropic effect, it may decrease cardiac output enough to affect athletic performance. Initiate the drug with telemetry monitoring to detect any pro-arrhythmic electrocardiographic changes, such as torsades de pointes or QT prolongation. Prevention of the hyperdynamic cardiac contraction can trigger an abnormal baroreceptor reflex and block the efferent limb of the vagal response to counter the bradycardia and vasodepressor response.16
If pharmacotherapy is required, we generally prescribe drugs in the following sequence—first fludrocortisone, then beta-blockers, and, if these do not help, disopyramide. These should be first given in the hospital with telemetry monitoring to check for QT prolongation. If these are ineffective, try paroxetine or midodrine. In refractory cases, we may use various combinations of these drugs.
Disopyramide and a changed running strategy
Several mechanisms related to vigorous exercise played a role in our patient’s case. During exercise, she developed a very high systolic pressure with a low diastolic pressure, resulting in a high pulse pressure that possibly led to stimulation of carotid baroreceptors. Cardiac hypercontractility related to exercise was probably a contributing factor as well. Severe leg pain prior to syncope suggested involvement of a pain-mediated mechanism.
We prescribed disopyramide 150 mg twice daily for our patient. We also recommended that she do specific exercises—to strengthen her leg muscles—and modify her running strategy.
After 2 months of therapy, she resumed competitive running successfully without any further episodes of syncope. She was able to achieve a new personal record for a cross country run by running a slower pace of 6 minutes 20 seconds per mile initially and running faster only at the end of the race. She had no recurrences of syncope during 6 months of follow-up.
CorrespondenceSumit Verma, MD, Pensacola Heart Institute, 5151 N, 9th Avenue, Cardiology Consultants, Pensacola, Fl 32504; [email protected].
1. Fahy GJ, Pinksi SL, Miller DP, et al. Natural history of isolated bundle branch block. Am J Cardiol 1996;77:1185-1190.
2. Maron BJ. Structural features of the athlete’s heart: As defined by echocardiography. J Am Coll Cardiol 1986;7:190-203.
3. Sneddon J, Scalia G, Ward D, McKenna W, Camm AJ, Frenneaux M. Exercise induced vasodepressor syncope. Br Heart J 1994;71:554-557.
4. Shalev Y, Gal R, Tchou P, et al. Echocardiographic demonstration of decreased left ventricular dimensions and vigorous myocardial contraction during syncope induced by head up tilt. J Am Coll Cardiol 1991;18:746-751.
5. Ferrario G, Nicoli J, Peci P, Ginni P. Cardiac adaptation to training. Is it the key to understand the high incidence of positive tilt test in athletes? (Abstract.) Pacing Electrophys 1996;19:578.-
6. Levine B. Regulation of central blood volume and cardiac filling in endurance athletes, the Frank-Starling mechanism as a determinant of orthostatic tolerance. Med Sci Sports Exer 1993;25:727-731.
7. Convertino VA, Sather TM, Goldwater D, Alford W, Hahn H. Aerobic fitness does not contribute to prediction of orthostatic intolerance. Med Sci Sports Exer 1989;18:551-556.
8. Sra J, Jazayeri M, Avitall B, Blanck Z, Akhtar M. Comparison of cardiac pacing with drug therapy in the treatment of neurocardiogenic syncope with bradycardia or hypotension. N Engl J Med 1993;328:1085-1090.
9. Holtzhausen LM, Noakes TD. The prevalence and significance of post-exercise (postural) hypotension in ultramarathon runners. Med Sci Sports Exerc 1995;27:1595-1601.
10. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69-74.
11. Grubb BP, Temesy-Armos P, Samoil D, Wolfe D, Hahn H, Elliot L. Tilt table testing in the evaluation and management of athletes with recurrent exercise induced syncope. Med Sci Sports Exerc 1993;25:24-28.
12. Rasmussen V, Hauns S, Skagen K. Cerebral attacks due to excessive vagal tone in heavily trained persons. Acta Med Scand 1978;204:401-405.
13. Muller G, Deal B, Strasburger JF, Benson DW, Jr. Usefulness of metoprolol for unexplained syncope and positive response to tilt table testing in young persons. Am J Cardiol 1993;71:592-595.
14. Lurie KG, Dutton J, Mangat R, Scheinman MM. Pindolol is effective in patients with vasovagal syncope. Pacing Clin Electrophysiol 1992;15:592.-Abstract.
15. Sra J, Maglio C, Biehl M, et al. Efficacy of midodrine hydrochloride in neurocardiogenic syncope refractory to standard therapy. J Cardiovasc Electrophysiol 1997;8:42-46.
16. Milstein S, Buetikofer J, Dunnigan A, et al. usefulness of disopyramide for prevention of upright tilt-induced hypotension-bradycardia. Am J Cardiol 1990;65:1339-1344.
1. Fahy GJ, Pinksi SL, Miller DP, et al. Natural history of isolated bundle branch block. Am J Cardiol 1996;77:1185-1190.
2. Maron BJ. Structural features of the athlete’s heart: As defined by echocardiography. J Am Coll Cardiol 1986;7:190-203.
3. Sneddon J, Scalia G, Ward D, McKenna W, Camm AJ, Frenneaux M. Exercise induced vasodepressor syncope. Br Heart J 1994;71:554-557.
4. Shalev Y, Gal R, Tchou P, et al. Echocardiographic demonstration of decreased left ventricular dimensions and vigorous myocardial contraction during syncope induced by head up tilt. J Am Coll Cardiol 1991;18:746-751.
5. Ferrario G, Nicoli J, Peci P, Ginni P. Cardiac adaptation to training. Is it the key to understand the high incidence of positive tilt test in athletes? (Abstract.) Pacing Electrophys 1996;19:578.-
6. Levine B. Regulation of central blood volume and cardiac filling in endurance athletes, the Frank-Starling mechanism as a determinant of orthostatic tolerance. Med Sci Sports Exer 1993;25:727-731.
7. Convertino VA, Sather TM, Goldwater D, Alford W, Hahn H. Aerobic fitness does not contribute to prediction of orthostatic intolerance. Med Sci Sports Exer 1989;18:551-556.
8. Sra J, Jazayeri M, Avitall B, Blanck Z, Akhtar M. Comparison of cardiac pacing with drug therapy in the treatment of neurocardiogenic syncope with bradycardia or hypotension. N Engl J Med 1993;328:1085-1090.
9. Holtzhausen LM, Noakes TD. The prevalence and significance of post-exercise (postural) hypotension in ultramarathon runners. Med Sci Sports Exerc 1995;27:1595-1601.
10. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69-74.
11. Grubb BP, Temesy-Armos P, Samoil D, Wolfe D, Hahn H, Elliot L. Tilt table testing in the evaluation and management of athletes with recurrent exercise induced syncope. Med Sci Sports Exerc 1993;25:24-28.
12. Rasmussen V, Hauns S, Skagen K. Cerebral attacks due to excessive vagal tone in heavily trained persons. Acta Med Scand 1978;204:401-405.
13. Muller G, Deal B, Strasburger JF, Benson DW, Jr. Usefulness of metoprolol for unexplained syncope and positive response to tilt table testing in young persons. Am J Cardiol 1993;71:592-595.
14. Lurie KG, Dutton J, Mangat R, Scheinman MM. Pindolol is effective in patients with vasovagal syncope. Pacing Clin Electrophysiol 1992;15:592.-Abstract.
15. Sra J, Maglio C, Biehl M, et al. Efficacy of midodrine hydrochloride in neurocardiogenic syncope refractory to standard therapy. J Cardiovasc Electrophysiol 1997;8:42-46.
16. Milstein S, Buetikofer J, Dunnigan A, et al. usefulness of disopyramide for prevention of upright tilt-induced hypotension-bradycardia. Am J Cardiol 1990;65:1339-1344.
B12 deficiency: A look beyond pernicious anemia
- Mild, preclinical B12 deficiency is associated with food-B12 malabsorption more often than with pernicious anemia. (C)
- The classic treatment for B12 deficiency—particularly when the cause is not a dietary deficiency—is 100 to 1000 mcg per month of cyanocobalamin, IM. (B)
- Oral crystalline cyanocobalamin is an effective treatment for food-B12 malabsorption, though it’s effectiveness in the long term has not been demonstrated. (B)
If an image of an elderly patient with pernicious anemia is the first thing that comes to mind when you think of B12 deficiency, take note: That image could obfuscate a more common case of B12 deficiency—one caused by food-B12 malabsorption.
Food-B12 malabsorption, characterized by the inability to release B12 from food or its binding proteins, is actually the leading cause of B12 malabsorption, especially in elderly patients.1-4 And unlike pernicious anemia, it’s more likely to be associated with mild, preclinical B12 deficiency.1,5
Spotting this form of B12 deficiency requires that you focus on its nuances, such as its link to Helicobacter pylori infection and long-term antacid and biguanide use. It also requires that you consider not only a patient’s serum B12 levels, but his homocysteine and methylmalonic acid levels, since they are considered more sensitive indicators of cobalamin deficiency.6 Keying in on these indicators early will ensure prompt treatment, which typically includes intramuscular injections of the vitamin, but which could revolve around a more convenient option: oral B12.
A common problem that comes in many shades
B12 deficiency is common in elderly patients7 and its incidence increases with age.7,8 The Framingham study revealed a prevalence of 12% among elderly people living in the community.8 Other studies focusing on those who are in institutions or who are sick and malnourished, have suggested a higher prevalence of 30% to 40%.3,9
The clinical manifestations of B12 deficiency are highly polymorphic and of varying severity ranging from milder conditions such as the common sensory neuropathy and isolated anomalies of macrocytosis and hypersegmentation of neutrophils, to severe disorders, including combined sclerosis of the spinal cord, hemolytic anemia and even pancytopenia (TABLE 1).1,5,6,10-13
B12 deficiency is often unrecognized or not investigated because the clinical manifestations can be very subtle. In fact, one of its manifestations—mild memory loss—can mimic the early stages of dementia.14
Further muddying the waters is the fact that B12 deficiency appears to be more common among patients who have a variety of chronic neurologic conditions such as stroke, Parkinson’s disease, dementia, Alzheimer’s disease, and depression—although it is unclear if these are causal relationships.1,15 In our own studies in which we administered B12 to patients with dementia, we did not observe any improvement.2,5 Other studies have had similar results.16,17
B12 deficiency is typically defined in terms of the serum concentration of B12, as well as the concentration of homocysteine and methyl malonic acid—2 components of the cobalamin metabolic pathway. A deficiency exists if the patient’s blood work reveals the following:2,18
- Serum B12 levels <150 pmol/L and either total serum homocysteine levels >13 μmol/L or methylmalonic acid levels >0.4 μmol/L (in the absence of renal failure and folate and vitamin B6 deficiencies).
- Low serum holotranscobalamin levels <35 pmol/L.
TABLE 1
Clinical features of B12 deficiency1,5,6,10-13
| HEMATOLOGIC |
| Frequent* |
| Macrocytosis |
| Hypersegmentation of the neutrophils |
| Aregenerative macrocytary anemia |
| Medullary megaloblastosis ("blue spinal cord") |
| Rare |
| Isolated thrombocytopenia and neutropenia |
| Pancytopenia |
| Hemolytic anemia |
| Thrombotic microangiopathy (presence of schistocytes) |
| DIGESTIVE |
| Classic |
| Hunter’s glossitis |
| Jaundice |
| LDH and bilirubin elevation |
| Rare |
| Resistant and recurring mucocutaneous ulcers |
| NEUROPSYCHIATRIC |
| Classic |
| Combined sclerosis of the spinal cord |
| Frequent* |
| Polyneurites (especially sensitive ones) |
| Ataxia |
| Babinski’s phenomenon |
| Rare |
| Cerebellar syndromes affecting the cranial nerves including optic neuritis, optic atrophy, urinary or fecal incontinence |
| Possible |
| Cognitive impairment |
| Stroke and atherosclerosis (hyperhomocysteinemia) |
| Parkinsonian syndromes |
| Multiple sclerosis |
| OTHER |
| Possible |
| Atrophy of the vaginal mucosa |
| Chronic vaginal and urinary infections (especially mycosis) |
| Hypofertility and repeated miscarriages |
| Venous thromboembolic disease |
| Angina (hyperhomocysteinemia) |
| * Reported in practice and recent literature. |
The “classic” cause is not the most common
The principal causes of B12 deficiency include pernicious anemia, dietary deficiency, postsurgical malabsorption, and food-B12 malabsorption. Of note is the fact that there is typically a 5- to 10-year delay between the onset of B12 deficiency and the development of clinical illness, in part because of hepatic stores of cobalamin (>1.5 mg).1,19
In elderly patients, B12 deficiency is classically caused by pernicious anemia,3,7 the principal characteristics of which have been reported in detail in several reviews.20-22 The one thing, of course, that bears repeating is that this form of anemia is associated with a lack of intrinsic factor, which facilitates the absorption of B12.
B12 deficiency caused by dietary deficiency is more rare. Dietary causes of deficiency are limited to elderly people who are already malnourished, such as those living in institutions (they may consume inadequate amounts of foods containing vitamin B12) and strict vegetarians.1,19 (A typical Western diet contributes 3–30 mcg of B12 per day towards the recommended dietary allowance set by the Food and Nutrition Board of the Institute of Medicine (US) of 2.4 mcg/day for adults and 2.6 to 2.8 mcg/day during pregnancy.23)
Over the past 20 years, postsurgical malabsorption of B12 has been on the decline, due in large part to the decreasing frequency of gastrectomy and surgical resection of the terminal small intestine.1,2,5 There are, however, several disorders commonly seen in gastroenterology practice that may be associated with cobalamin malabsorption. These include deficiency in the exocrine function of the pancreas after chronic pancreatitis (usually alcoholic), lymphomas or tuberculosis (of the intestine), Crohn’s disease, Whipple’s disease, and occasionally celiac disease.3,13
Rounding out the list of causes of B12 deficiency is food-B12 malabsorption, which is the leading cause of B12 malabsorption—especially in elderly patients.1-4 In our own studies in which we have followed more than 300 patients with a documented B12 deficiency, food-B12 malabsorption accounts for about 60% to 70% of the cases of B12 deficiency in elderly patients, whereas pernicious anemia accounts for only 15% to 25%.5,24 In our study of 172 hospitalized patients with B12 deficiency (median age, 70), 53% had food-B12 malabsorption.5
A form of malabsorption that’s tough to spot
Food-B12 malabsorption is a syndrome characterized by the inability to release B12 from food or intestinal transport proteins, particularly in the presence of hypochlorhydria, in which the absorption of “unbound” B12 is normal. As various studies have shown,4,5,24 this syndrome is defined by B12 deficiency in the presence of sufficient food-B12 intake and normal Schilling test results, which rules out pernicious anemia. In theory, indisputable evidence of food-B12 malabsorption comes from using a modified Schilling test, which uses radioactive B12 bound to animal proteins (eg, salmon, trout) and reveals malabsorption when the results of a standard Schilling test are normal.1,5,24
Some authors have speculated about the significance of B12 deficiency related to food-cobalamin malabsorption,1 because many patients have only mild clinical or hematological features. Several of our patients, however, have had significant features classically associated with pernicious anemia, including polyneuropathy, confusion, dementia, medullar-combined sclerosis, anemia, and pancytopenia.5 Nevertheless, the partial nature of this form of malabsorption might produce a more slowly progressive depletion of B12 than does the more complete malabsorption engendered by disruption of intrinsic factor–mediated absorption. The slower progression of depletion probably explains why mild, preclinical deficiency is associated with food-B12 malabsorption more often than with pernicious anemia.1,5
H pylori, antacid use should raise suspicions
Food-B12 malabsorption is caused primarily by atrophic gastritis.5 More than 40% of patients older than 80 years have gastric atrophy that might (or might not) be related to H pylori infection.3,25 Other factors that contribute to food-B12 malabsorption in elderly people include:
- Chronic carriage of H pylori and intestinal microbial proliferation (in which case B12 deficiency can be corrected by antibiotic treatment)25,26
- Long-term ingestion of antacids, including H2-receptor antagonists and proton-pump inhibitors,27,28 particularly among patients with Zollinger-Ellison syndrome29,30
- Long-term ingestion of biguanides (metformin)31-33
- Chronic alcoholism
- Surgery or gastric reconstruction (eg, bypass surgery for obesity)
- Partial pancreatic exocrine failure1,5
- Sjögren’s syndrome or systemic sclerosis34
In our research involving 92 elderly patients (mean age: 76 years) with food-B12 malabsorption,5 we found at least one of the associated conditions or agents listed at left in 60% of the patients. These conditions mainly included atrophic gastritis (H pylori infection) in 30% of the patients and long-term metformin or antacid intake in 20% of the elderly patients.
TABLE 2
French hospital findings support use of oral B12 treatment38-41,45
| STUDY CHARACTERISTICS (NUMBER OF PATIENTS) | THERAPEUTIC MODALITIES | RESULTS |
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 and malabsorption (n=10)39 | Oral crystalline cyanocobalamin: 650 mcg per day, for at least 3 months |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=20)40 | Oral crystalline cyanocobalamin: between 1000 mcg per day for at least 1 week |
|
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 malabsorption (n=30)38 | Oral crystalline cyanocobalamin: between 250 and 1000 mcg per day, for 1 month |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=30)41 | Oral crystalline cyanocobalamin: between 125 and 1000 mcg per day for at least 1 week |
|
| Open prospective study of low vitamin B12 levels related to pernicious anemia (n=10)45 | Oral crystalline cyanocobalamin: 1000 mcg per day, for at least 3 months |
|
IM injection is customary, though dosages vary
The classic treatment for B12 deficiency, particularly when the cause is not a dietary deficiency, is parenteral administration—usually by intramuscular injection—of cyanocobalamin (and in rare occasions, hydroxocobalamin).7,11,16,35 In the US and UK, dosages range from 100 to 1000 mcg per month (or every 2–3 months when hydroxocobalamin is given). The patient will receive this treatment for the rest of his life.1,35
In France, the recommended practice is to build up the tissue stores of the vitamin quickly and correct serum B12 hypovitaminosis, particularly in the case of pernicious anemia. The treatment involves administering 1000 mcg of cyanocobalamin per day for 1 week, followed by 1000 mcg per week for 1 month, followed by 1000 mcg per month, normally for the rest of the patient’s life.2,3,20
Oral therapy is a well-kept secret
In cases of B12 deficiency that don’t involve nutritional deficiency, alternative routes of cobalamin administration, including the oral16,35-42 and nasal43,44 routes have been used. These alternative routes offer patients a way to avoid the discomfort, inconvenience, and cost of an office visit for monthly injections.
Our research team has developed an effective oral treatment of food-B12 malabsorption38-41 and for pernicious anemia45 using crystalline cobalamin (cyanocobalamin). Our principal studies of oral B12 treatment (open, not randomized studies) are described in TABLE 2.38-41,45 Our data confirm the previously reported efficacy of oral crystalline cyanocobalamin, especially in food-B12 therapy.6,16,36 All of our patients who received oral therapy corrected their B12 levels and at least two thirds corrected their hematological abnormalities.38-41,45 Moreover, one third of patients experienced a clinical improvement on oral treatment. In most cases of food-B12 malabsorption, a “low” B12 dose (ie, 125–1000 mcg of oral crystalline cyanocobalamin per day) was used.
These data are in line with the results of the 2 prospective randomized controlled studies comparing oral B12 with intramuscular B12 therapy.35,37 An evidence-based analysis by the Vitamin B12 Cochrane Group supports the efficacy of oral B12 therapy, with doses between 1000 and 2000 mcg given daily in the beginning, and then weekly.46 In this analysis, serum B12 levels increased significantly in patients receiving oral vitamin B12 and both groups of patients (receiving oral and intramuscular treatment) had neurological improvement.
In a randomized, parallel-group, double-blind, dose-finding trial, Eussen et al showed that the lowest dose of oral cyanocobalamin required to normalize mild B12 deficiency is more than 200 times the recommended dietary allowance of approximately 3 mcg daily (ie, >500 mcg/day).47 The procedure for oral B12 treatment has, however, not been completely validated yet in “real life,” particularly as it relates to long-term efficacy.48 Nonetheless, several authors suggest that oral B12 therapy remains one of medicine’s “best-kept secrets.”49
Acknowledgements
We are indebted to Professor Marc Imler and Jean-Louis Schlienger who initiated this work and to Helen Fothergill who kindly edited the text for publication in this English-language journal.
Correspondence
Prof. E. Andrès, Service de Médecine Interne, Diabète et Maladies Métaboliques, Clinique Médicale B, Hôpital Civil–Hôpitaux Universitaires de Strasbourg, 1 porte de l’Hôpital, 67091 Strasbourg Cedex, France; emmanuel. [email protected]
1. Carmel R. Current concepts in cobalamin deficiency. Ann Rev Med 2000;51:357-375.
2. Andrès E, Perrin AE, Kraemer JP, et al. Anémies par carence en vitamine B12 chez le sujet âgé de plus de 75 ans: nouveaux concepts. A propos de 20 observations. Rev Med Interne 2000;21:946-955.
3. Pautas E, Chérin P, De Jaeger C, Godeau P. Carence en vitamine B12 chez le sujet âgé. Presse Med 1999;28:1767-1770.
4. Carmel R. Malabsorption of food-cobalamin. Bailliere’s Clin Haematol 1995;8:639-655.
5. Andrès E, Affenberger S, Vinzio S, et al. Food-cobalamin malabsorption in elderly patients: clinical manifestations and treatment. Am J Med 2005;118:1154-1159.
6. Carmel R, Sarrai M. Diagnosis and management of clinical and subclinical cobalamin deficiency: advances and controversies. Curr Hematol Rep 2006;5:23-33.
7. Matthews JH. Cobalamin and folate deficiency in the elderly. Baillère’s Clin Haematol 1995;54:245-253.
8. Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994;60:2-11.
9. van Asselt DZ, Blom HJ, Zuiderent R, et al. Clinical significance of low cobalamin levels in older hospital patients. Neth J Med 2000;57:41-49.
10. Stabler SP, Allen RH, Savage DG, Lindenbaum J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood 1990;76:871-881.
11. Dharmarajan TS, Adiga GU, Norkus EP. Vitamin B12 deficiency. Recognizing subtle symptoms in older adults. Geriatrics 2003;58:30-38.
12. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol 2006;28:50-56.
13. Andrès E, Loukili NH, Noel E, et al. Vitamin B12 (cobalamin) deficiency in elderly patients. CAMJ 2004;171:251-260.
14. Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol 2006;5:949-960.
15. Abyad A. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2002;6:254-260.
16. Lane LA, Rojas-Fernandez C. Treatment of vitamin B12 deficiency anemia: oral versus parenteral therapy. Ann Pharmacother 2002;36:1268-1272.
17. Andrès E, Kaltenbach G. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2003;7:309-310.
18. Klee GG. Cobalamin and folate evaluation: measurements of methylmalonic acid and homocystein vs vitamin B12 and folate. Clin Chem 2000;46:12e77-1283.
19. Nicolas JP, Guéant JL. Absorption, distribution et excrétion de la vitamine B12. Ann Gastroenterol Hepatol 1994;30:270-282.
20. Loukili NH, Noel E, Blaison G, et al. Données actuelles sur la maladie de Biermer. A propos d’une étude rétrospective de 49 patients. Rev Med Interne 2004;25:556-561.
21. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med 1997;337:1441-1448.
22. Pruthi RK, Tefferi A. Pernicious anemia revisited. Mayo Clin Proc 1994;69:144-150.
23. Institute of Medicine Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, panthothenic acid, biotin and choline. Food and Nutrition Board, Washington, DC. National Academies Press, 1998.
24. Andrès E, Noel E, Kaltenbach G, et al. Carences en vitamine B12 avec test de Schilling normal ou syndrome de non-dissociation de la vitamine B12 de ses protéines porteuses chez le sujet âgé. Etude de 60 patients. Rev Med Interne 2003;24:218-223.
25. Carmel R, Aurangzeb I, Ojan D. Associations of food-cobalamin malabsorption with ethnic origin, age, Helicobacter pylori infection, and serum markers of gastritis. Am J Gastroenterol 2001;96:63-70.
26. Kaptan K, Beyan C, Ural AU, et al. Helicobacter pylori—is it a novel causative agent in Vitamin B12 deficiency? Arch Intern Med 2000;160:1349-1353.
27. Howden CW. Vitamin B12 levels during prolonged treatment with proton pump inhibitors. J Clin Gastroenterol 2000;30:29-33.
28. Andrès E, Noel E, Ben Abdelghani M. Vitamin B12 deficiency associated with chronic acid suppression therapy. Ann Pharmacother 2003;37:1730.-
29. Termanini B, Gibril F, Sutliff VE, Yu F, Venzon DJ, Jensen RT. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med 1998;104:422-430.
30. Jensen RT. Consequences of long-term proton pump blockade: insights from studies of patients with gastrinomas. Basic Clin Pharmacol Toxicol 2006;98:4-19.
31. Bauman WA, Shaw S, Javatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000;23:1227-1231.
32. Andrès E, Noel E, Goichot B. Metformin-associated vitamin B12 deficiency. Arch Intern Med 2002;162:2251-2252.
33. Liu KW, Dai LK, Jean W. Metformin-related vitamin B12 deficiency. Age Aging 2006;35:200-201.
34. Andrès E, Goichot B, Perrin AE, Vinzio S, Demangeat C, Schlienger JL. Sjögren’s syndrome: a potential new cause of mild cobalamin deficiency. Rheumatology (Oxford) 2001;40:1196-1197.
35. Kuzminski AM, Del Giacco EI, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood 1998;92:1191-1198.
36. Elia M. Oral or parenteral therapy for B12 deficiency. Lancet 1998;352:1721-1722.
37. Bolaman Z, Kadikoylu G, Yukselen V, Yavasoglu I, Barutca S, Senturk T. Oral versus intramuscular cobalamin treatment in megaloblastic anemia: a singlecenter, prospective, randomized, open-label study. Clin Ther 2003;25:3124-3134.
38. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption. A study of 30 patients. Clin Lab Haematol 2003;25:161-166.
39. Andrès E, Kurtz JE, Perrin AE, et al. Oral cobalamin therapy for the treatment of patients with food-cobalamin malabsorption. Am J Med 2001;111:126-129.
40. Kaltenbach G, Noblet-Dick M, Andrès E, et al. Réponse précoce au traitement oral par vitamine B12 chez des sujets âgés hypovitaminiques. Ann Med Interne (Paris) 2003;154:91-95.
41. Andrès E, Kaltenbach G, Noblet-Dick M, et al. Hematological response to short-term oral cyanocobalamin therapy for the treatment of cobalamin deficiencies in elderly patients. J Nutr Health Aging 2006;10:3-6.
42. Butler CC, Vidal-Alaball J, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency: a systematic review of randomized controlled trials. Fam Pract 2006;23:279-285.
43. Slot WB, Merkus FW, Van Deventer SJ, Tytgat GN. Normalization of plasma vitamin B12 concentration by intranasal hydroxocobalamin in vitamin B12-deficient patients. Gastroenterology 1997;113:430-433.
44. van Asselt DZ, Merkus FW, Russel FG, Hoefnagels WH. Nasal absorption of hydroxocobalamin in healthy elderly adults. Br J Clin Pharmacol 1998;45:83-86.
45. Andrès E, Loukili NH, Noel E, et al. Oral cobalamin (daily dose of 1000 μg) therapy for the treatment of patients with pernicious anemia. An open label study of 10 patients. Curr Ther Res 2005;66:13-22.
46. Vidal-Alaball J, Butler CC, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev 2005;20:CD004655.-
47. Eussen SJ, de Groot LC, Clarke R, et al. Oral cyanocobalamin supplementation in older people with vitamin B12 deficiency: a dose-finding trial. Arch Intern Med 2005;165:1167-1172.
48. Roth M, Orija I. Oral vitamin B12 therapy in vitamin B12 deficiency. Am J Med 2004;116:358.-
49. Graham ID, Jette N, Tetroe J, Robinson N, Milne S, Mitchell SL. Oral cobalamin remains medicine’s best kept secret. Arch Gerontol Geriatr 2007;44:49-59.
- Mild, preclinical B12 deficiency is associated with food-B12 malabsorption more often than with pernicious anemia. (C)
- The classic treatment for B12 deficiency—particularly when the cause is not a dietary deficiency—is 100 to 1000 mcg per month of cyanocobalamin, IM. (B)
- Oral crystalline cyanocobalamin is an effective treatment for food-B12 malabsorption, though it’s effectiveness in the long term has not been demonstrated. (B)
If an image of an elderly patient with pernicious anemia is the first thing that comes to mind when you think of B12 deficiency, take note: That image could obfuscate a more common case of B12 deficiency—one caused by food-B12 malabsorption.
Food-B12 malabsorption, characterized by the inability to release B12 from food or its binding proteins, is actually the leading cause of B12 malabsorption, especially in elderly patients.1-4 And unlike pernicious anemia, it’s more likely to be associated with mild, preclinical B12 deficiency.1,5
Spotting this form of B12 deficiency requires that you focus on its nuances, such as its link to Helicobacter pylori infection and long-term antacid and biguanide use. It also requires that you consider not only a patient’s serum B12 levels, but his homocysteine and methylmalonic acid levels, since they are considered more sensitive indicators of cobalamin deficiency.6 Keying in on these indicators early will ensure prompt treatment, which typically includes intramuscular injections of the vitamin, but which could revolve around a more convenient option: oral B12.
A common problem that comes in many shades
B12 deficiency is common in elderly patients7 and its incidence increases with age.7,8 The Framingham study revealed a prevalence of 12% among elderly people living in the community.8 Other studies focusing on those who are in institutions or who are sick and malnourished, have suggested a higher prevalence of 30% to 40%.3,9
The clinical manifestations of B12 deficiency are highly polymorphic and of varying severity ranging from milder conditions such as the common sensory neuropathy and isolated anomalies of macrocytosis and hypersegmentation of neutrophils, to severe disorders, including combined sclerosis of the spinal cord, hemolytic anemia and even pancytopenia (TABLE 1).1,5,6,10-13
B12 deficiency is often unrecognized or not investigated because the clinical manifestations can be very subtle. In fact, one of its manifestations—mild memory loss—can mimic the early stages of dementia.14
Further muddying the waters is the fact that B12 deficiency appears to be more common among patients who have a variety of chronic neurologic conditions such as stroke, Parkinson’s disease, dementia, Alzheimer’s disease, and depression—although it is unclear if these are causal relationships.1,15 In our own studies in which we administered B12 to patients with dementia, we did not observe any improvement.2,5 Other studies have had similar results.16,17
B12 deficiency is typically defined in terms of the serum concentration of B12, as well as the concentration of homocysteine and methyl malonic acid—2 components of the cobalamin metabolic pathway. A deficiency exists if the patient’s blood work reveals the following:2,18
- Serum B12 levels <150 pmol/L and either total serum homocysteine levels >13 μmol/L or methylmalonic acid levels >0.4 μmol/L (in the absence of renal failure and folate and vitamin B6 deficiencies).
- Low serum holotranscobalamin levels <35 pmol/L.
TABLE 1
Clinical features of B12 deficiency1,5,6,10-13
| HEMATOLOGIC |
| Frequent* |
| Macrocytosis |
| Hypersegmentation of the neutrophils |
| Aregenerative macrocytary anemia |
| Medullary megaloblastosis ("blue spinal cord") |
| Rare |
| Isolated thrombocytopenia and neutropenia |
| Pancytopenia |
| Hemolytic anemia |
| Thrombotic microangiopathy (presence of schistocytes) |
| DIGESTIVE |
| Classic |
| Hunter’s glossitis |
| Jaundice |
| LDH and bilirubin elevation |
| Rare |
| Resistant and recurring mucocutaneous ulcers |
| NEUROPSYCHIATRIC |
| Classic |
| Combined sclerosis of the spinal cord |
| Frequent* |
| Polyneurites (especially sensitive ones) |
| Ataxia |
| Babinski’s phenomenon |
| Rare |
| Cerebellar syndromes affecting the cranial nerves including optic neuritis, optic atrophy, urinary or fecal incontinence |
| Possible |
| Cognitive impairment |
| Stroke and atherosclerosis (hyperhomocysteinemia) |
| Parkinsonian syndromes |
| Multiple sclerosis |
| OTHER |
| Possible |
| Atrophy of the vaginal mucosa |
| Chronic vaginal and urinary infections (especially mycosis) |
| Hypofertility and repeated miscarriages |
| Venous thromboembolic disease |
| Angina (hyperhomocysteinemia) |
| * Reported in practice and recent literature. |
The “classic” cause is not the most common
The principal causes of B12 deficiency include pernicious anemia, dietary deficiency, postsurgical malabsorption, and food-B12 malabsorption. Of note is the fact that there is typically a 5- to 10-year delay between the onset of B12 deficiency and the development of clinical illness, in part because of hepatic stores of cobalamin (>1.5 mg).1,19
In elderly patients, B12 deficiency is classically caused by pernicious anemia,3,7 the principal characteristics of which have been reported in detail in several reviews.20-22 The one thing, of course, that bears repeating is that this form of anemia is associated with a lack of intrinsic factor, which facilitates the absorption of B12.
B12 deficiency caused by dietary deficiency is more rare. Dietary causes of deficiency are limited to elderly people who are already malnourished, such as those living in institutions (they may consume inadequate amounts of foods containing vitamin B12) and strict vegetarians.1,19 (A typical Western diet contributes 3–30 mcg of B12 per day towards the recommended dietary allowance set by the Food and Nutrition Board of the Institute of Medicine (US) of 2.4 mcg/day for adults and 2.6 to 2.8 mcg/day during pregnancy.23)
Over the past 20 years, postsurgical malabsorption of B12 has been on the decline, due in large part to the decreasing frequency of gastrectomy and surgical resection of the terminal small intestine.1,2,5 There are, however, several disorders commonly seen in gastroenterology practice that may be associated with cobalamin malabsorption. These include deficiency in the exocrine function of the pancreas after chronic pancreatitis (usually alcoholic), lymphomas or tuberculosis (of the intestine), Crohn’s disease, Whipple’s disease, and occasionally celiac disease.3,13
Rounding out the list of causes of B12 deficiency is food-B12 malabsorption, which is the leading cause of B12 malabsorption—especially in elderly patients.1-4 In our own studies in which we have followed more than 300 patients with a documented B12 deficiency, food-B12 malabsorption accounts for about 60% to 70% of the cases of B12 deficiency in elderly patients, whereas pernicious anemia accounts for only 15% to 25%.5,24 In our study of 172 hospitalized patients with B12 deficiency (median age, 70), 53% had food-B12 malabsorption.5
A form of malabsorption that’s tough to spot
Food-B12 malabsorption is a syndrome characterized by the inability to release B12 from food or intestinal transport proteins, particularly in the presence of hypochlorhydria, in which the absorption of “unbound” B12 is normal. As various studies have shown,4,5,24 this syndrome is defined by B12 deficiency in the presence of sufficient food-B12 intake and normal Schilling test results, which rules out pernicious anemia. In theory, indisputable evidence of food-B12 malabsorption comes from using a modified Schilling test, which uses radioactive B12 bound to animal proteins (eg, salmon, trout) and reveals malabsorption when the results of a standard Schilling test are normal.1,5,24
Some authors have speculated about the significance of B12 deficiency related to food-cobalamin malabsorption,1 because many patients have only mild clinical or hematological features. Several of our patients, however, have had significant features classically associated with pernicious anemia, including polyneuropathy, confusion, dementia, medullar-combined sclerosis, anemia, and pancytopenia.5 Nevertheless, the partial nature of this form of malabsorption might produce a more slowly progressive depletion of B12 than does the more complete malabsorption engendered by disruption of intrinsic factor–mediated absorption. The slower progression of depletion probably explains why mild, preclinical deficiency is associated with food-B12 malabsorption more often than with pernicious anemia.1,5
H pylori, antacid use should raise suspicions
Food-B12 malabsorption is caused primarily by atrophic gastritis.5 More than 40% of patients older than 80 years have gastric atrophy that might (or might not) be related to H pylori infection.3,25 Other factors that contribute to food-B12 malabsorption in elderly people include:
- Chronic carriage of H pylori and intestinal microbial proliferation (in which case B12 deficiency can be corrected by antibiotic treatment)25,26
- Long-term ingestion of antacids, including H2-receptor antagonists and proton-pump inhibitors,27,28 particularly among patients with Zollinger-Ellison syndrome29,30
- Long-term ingestion of biguanides (metformin)31-33
- Chronic alcoholism
- Surgery or gastric reconstruction (eg, bypass surgery for obesity)
- Partial pancreatic exocrine failure1,5
- Sjögren’s syndrome or systemic sclerosis34
In our research involving 92 elderly patients (mean age: 76 years) with food-B12 malabsorption,5 we found at least one of the associated conditions or agents listed at left in 60% of the patients. These conditions mainly included atrophic gastritis (H pylori infection) in 30% of the patients and long-term metformin or antacid intake in 20% of the elderly patients.
TABLE 2
French hospital findings support use of oral B12 treatment38-41,45
| STUDY CHARACTERISTICS (NUMBER OF PATIENTS) | THERAPEUTIC MODALITIES | RESULTS |
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 and malabsorption (n=10)39 | Oral crystalline cyanocobalamin: 650 mcg per day, for at least 3 months |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=20)40 | Oral crystalline cyanocobalamin: between 1000 mcg per day for at least 1 week |
|
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 malabsorption (n=30)38 | Oral crystalline cyanocobalamin: between 250 and 1000 mcg per day, for 1 month |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=30)41 | Oral crystalline cyanocobalamin: between 125 and 1000 mcg per day for at least 1 week |
|
| Open prospective study of low vitamin B12 levels related to pernicious anemia (n=10)45 | Oral crystalline cyanocobalamin: 1000 mcg per day, for at least 3 months |
|
IM injection is customary, though dosages vary
The classic treatment for B12 deficiency, particularly when the cause is not a dietary deficiency, is parenteral administration—usually by intramuscular injection—of cyanocobalamin (and in rare occasions, hydroxocobalamin).7,11,16,35 In the US and UK, dosages range from 100 to 1000 mcg per month (or every 2–3 months when hydroxocobalamin is given). The patient will receive this treatment for the rest of his life.1,35
In France, the recommended practice is to build up the tissue stores of the vitamin quickly and correct serum B12 hypovitaminosis, particularly in the case of pernicious anemia. The treatment involves administering 1000 mcg of cyanocobalamin per day for 1 week, followed by 1000 mcg per week for 1 month, followed by 1000 mcg per month, normally for the rest of the patient’s life.2,3,20
Oral therapy is a well-kept secret
In cases of B12 deficiency that don’t involve nutritional deficiency, alternative routes of cobalamin administration, including the oral16,35-42 and nasal43,44 routes have been used. These alternative routes offer patients a way to avoid the discomfort, inconvenience, and cost of an office visit for monthly injections.
Our research team has developed an effective oral treatment of food-B12 malabsorption38-41 and for pernicious anemia45 using crystalline cobalamin (cyanocobalamin). Our principal studies of oral B12 treatment (open, not randomized studies) are described in TABLE 2.38-41,45 Our data confirm the previously reported efficacy of oral crystalline cyanocobalamin, especially in food-B12 therapy.6,16,36 All of our patients who received oral therapy corrected their B12 levels and at least two thirds corrected their hematological abnormalities.38-41,45 Moreover, one third of patients experienced a clinical improvement on oral treatment. In most cases of food-B12 malabsorption, a “low” B12 dose (ie, 125–1000 mcg of oral crystalline cyanocobalamin per day) was used.
These data are in line with the results of the 2 prospective randomized controlled studies comparing oral B12 with intramuscular B12 therapy.35,37 An evidence-based analysis by the Vitamin B12 Cochrane Group supports the efficacy of oral B12 therapy, with doses between 1000 and 2000 mcg given daily in the beginning, and then weekly.46 In this analysis, serum B12 levels increased significantly in patients receiving oral vitamin B12 and both groups of patients (receiving oral and intramuscular treatment) had neurological improvement.
In a randomized, parallel-group, double-blind, dose-finding trial, Eussen et al showed that the lowest dose of oral cyanocobalamin required to normalize mild B12 deficiency is more than 200 times the recommended dietary allowance of approximately 3 mcg daily (ie, >500 mcg/day).47 The procedure for oral B12 treatment has, however, not been completely validated yet in “real life,” particularly as it relates to long-term efficacy.48 Nonetheless, several authors suggest that oral B12 therapy remains one of medicine’s “best-kept secrets.”49
Acknowledgements
We are indebted to Professor Marc Imler and Jean-Louis Schlienger who initiated this work and to Helen Fothergill who kindly edited the text for publication in this English-language journal.
Correspondence
Prof. E. Andrès, Service de Médecine Interne, Diabète et Maladies Métaboliques, Clinique Médicale B, Hôpital Civil–Hôpitaux Universitaires de Strasbourg, 1 porte de l’Hôpital, 67091 Strasbourg Cedex, France; emmanuel. [email protected]
- Mild, preclinical B12 deficiency is associated with food-B12 malabsorption more often than with pernicious anemia. (C)
- The classic treatment for B12 deficiency—particularly when the cause is not a dietary deficiency—is 100 to 1000 mcg per month of cyanocobalamin, IM. (B)
- Oral crystalline cyanocobalamin is an effective treatment for food-B12 malabsorption, though it’s effectiveness in the long term has not been demonstrated. (B)
If an image of an elderly patient with pernicious anemia is the first thing that comes to mind when you think of B12 deficiency, take note: That image could obfuscate a more common case of B12 deficiency—one caused by food-B12 malabsorption.
Food-B12 malabsorption, characterized by the inability to release B12 from food or its binding proteins, is actually the leading cause of B12 malabsorption, especially in elderly patients.1-4 And unlike pernicious anemia, it’s more likely to be associated with mild, preclinical B12 deficiency.1,5
Spotting this form of B12 deficiency requires that you focus on its nuances, such as its link to Helicobacter pylori infection and long-term antacid and biguanide use. It also requires that you consider not only a patient’s serum B12 levels, but his homocysteine and methylmalonic acid levels, since they are considered more sensitive indicators of cobalamin deficiency.6 Keying in on these indicators early will ensure prompt treatment, which typically includes intramuscular injections of the vitamin, but which could revolve around a more convenient option: oral B12.
A common problem that comes in many shades
B12 deficiency is common in elderly patients7 and its incidence increases with age.7,8 The Framingham study revealed a prevalence of 12% among elderly people living in the community.8 Other studies focusing on those who are in institutions or who are sick and malnourished, have suggested a higher prevalence of 30% to 40%.3,9
The clinical manifestations of B12 deficiency are highly polymorphic and of varying severity ranging from milder conditions such as the common sensory neuropathy and isolated anomalies of macrocytosis and hypersegmentation of neutrophils, to severe disorders, including combined sclerosis of the spinal cord, hemolytic anemia and even pancytopenia (TABLE 1).1,5,6,10-13
B12 deficiency is often unrecognized or not investigated because the clinical manifestations can be very subtle. In fact, one of its manifestations—mild memory loss—can mimic the early stages of dementia.14
Further muddying the waters is the fact that B12 deficiency appears to be more common among patients who have a variety of chronic neurologic conditions such as stroke, Parkinson’s disease, dementia, Alzheimer’s disease, and depression—although it is unclear if these are causal relationships.1,15 In our own studies in which we administered B12 to patients with dementia, we did not observe any improvement.2,5 Other studies have had similar results.16,17
B12 deficiency is typically defined in terms of the serum concentration of B12, as well as the concentration of homocysteine and methyl malonic acid—2 components of the cobalamin metabolic pathway. A deficiency exists if the patient’s blood work reveals the following:2,18
- Serum B12 levels <150 pmol/L and either total serum homocysteine levels >13 μmol/L or methylmalonic acid levels >0.4 μmol/L (in the absence of renal failure and folate and vitamin B6 deficiencies).
- Low serum holotranscobalamin levels <35 pmol/L.
TABLE 1
Clinical features of B12 deficiency1,5,6,10-13
| HEMATOLOGIC |
| Frequent* |
| Macrocytosis |
| Hypersegmentation of the neutrophils |
| Aregenerative macrocytary anemia |
| Medullary megaloblastosis ("blue spinal cord") |
| Rare |
| Isolated thrombocytopenia and neutropenia |
| Pancytopenia |
| Hemolytic anemia |
| Thrombotic microangiopathy (presence of schistocytes) |
| DIGESTIVE |
| Classic |
| Hunter’s glossitis |
| Jaundice |
| LDH and bilirubin elevation |
| Rare |
| Resistant and recurring mucocutaneous ulcers |
| NEUROPSYCHIATRIC |
| Classic |
| Combined sclerosis of the spinal cord |
| Frequent* |
| Polyneurites (especially sensitive ones) |
| Ataxia |
| Babinski’s phenomenon |
| Rare |
| Cerebellar syndromes affecting the cranial nerves including optic neuritis, optic atrophy, urinary or fecal incontinence |
| Possible |
| Cognitive impairment |
| Stroke and atherosclerosis (hyperhomocysteinemia) |
| Parkinsonian syndromes |
| Multiple sclerosis |
| OTHER |
| Possible |
| Atrophy of the vaginal mucosa |
| Chronic vaginal and urinary infections (especially mycosis) |
| Hypofertility and repeated miscarriages |
| Venous thromboembolic disease |
| Angina (hyperhomocysteinemia) |
| * Reported in practice and recent literature. |
The “classic” cause is not the most common
The principal causes of B12 deficiency include pernicious anemia, dietary deficiency, postsurgical malabsorption, and food-B12 malabsorption. Of note is the fact that there is typically a 5- to 10-year delay between the onset of B12 deficiency and the development of clinical illness, in part because of hepatic stores of cobalamin (>1.5 mg).1,19
In elderly patients, B12 deficiency is classically caused by pernicious anemia,3,7 the principal characteristics of which have been reported in detail in several reviews.20-22 The one thing, of course, that bears repeating is that this form of anemia is associated with a lack of intrinsic factor, which facilitates the absorption of B12.
B12 deficiency caused by dietary deficiency is more rare. Dietary causes of deficiency are limited to elderly people who are already malnourished, such as those living in institutions (they may consume inadequate amounts of foods containing vitamin B12) and strict vegetarians.1,19 (A typical Western diet contributes 3–30 mcg of B12 per day towards the recommended dietary allowance set by the Food and Nutrition Board of the Institute of Medicine (US) of 2.4 mcg/day for adults and 2.6 to 2.8 mcg/day during pregnancy.23)
Over the past 20 years, postsurgical malabsorption of B12 has been on the decline, due in large part to the decreasing frequency of gastrectomy and surgical resection of the terminal small intestine.1,2,5 There are, however, several disorders commonly seen in gastroenterology practice that may be associated with cobalamin malabsorption. These include deficiency in the exocrine function of the pancreas after chronic pancreatitis (usually alcoholic), lymphomas or tuberculosis (of the intestine), Crohn’s disease, Whipple’s disease, and occasionally celiac disease.3,13
Rounding out the list of causes of B12 deficiency is food-B12 malabsorption, which is the leading cause of B12 malabsorption—especially in elderly patients.1-4 In our own studies in which we have followed more than 300 patients with a documented B12 deficiency, food-B12 malabsorption accounts for about 60% to 70% of the cases of B12 deficiency in elderly patients, whereas pernicious anemia accounts for only 15% to 25%.5,24 In our study of 172 hospitalized patients with B12 deficiency (median age, 70), 53% had food-B12 malabsorption.5
A form of malabsorption that’s tough to spot
Food-B12 malabsorption is a syndrome characterized by the inability to release B12 from food or intestinal transport proteins, particularly in the presence of hypochlorhydria, in which the absorption of “unbound” B12 is normal. As various studies have shown,4,5,24 this syndrome is defined by B12 deficiency in the presence of sufficient food-B12 intake and normal Schilling test results, which rules out pernicious anemia. In theory, indisputable evidence of food-B12 malabsorption comes from using a modified Schilling test, which uses radioactive B12 bound to animal proteins (eg, salmon, trout) and reveals malabsorption when the results of a standard Schilling test are normal.1,5,24
Some authors have speculated about the significance of B12 deficiency related to food-cobalamin malabsorption,1 because many patients have only mild clinical or hematological features. Several of our patients, however, have had significant features classically associated with pernicious anemia, including polyneuropathy, confusion, dementia, medullar-combined sclerosis, anemia, and pancytopenia.5 Nevertheless, the partial nature of this form of malabsorption might produce a more slowly progressive depletion of B12 than does the more complete malabsorption engendered by disruption of intrinsic factor–mediated absorption. The slower progression of depletion probably explains why mild, preclinical deficiency is associated with food-B12 malabsorption more often than with pernicious anemia.1,5
H pylori, antacid use should raise suspicions
Food-B12 malabsorption is caused primarily by atrophic gastritis.5 More than 40% of patients older than 80 years have gastric atrophy that might (or might not) be related to H pylori infection.3,25 Other factors that contribute to food-B12 malabsorption in elderly people include:
- Chronic carriage of H pylori and intestinal microbial proliferation (in which case B12 deficiency can be corrected by antibiotic treatment)25,26
- Long-term ingestion of antacids, including H2-receptor antagonists and proton-pump inhibitors,27,28 particularly among patients with Zollinger-Ellison syndrome29,30
- Long-term ingestion of biguanides (metformin)31-33
- Chronic alcoholism
- Surgery or gastric reconstruction (eg, bypass surgery for obesity)
- Partial pancreatic exocrine failure1,5
- Sjögren’s syndrome or systemic sclerosis34
In our research involving 92 elderly patients (mean age: 76 years) with food-B12 malabsorption,5 we found at least one of the associated conditions or agents listed at left in 60% of the patients. These conditions mainly included atrophic gastritis (H pylori infection) in 30% of the patients and long-term metformin or antacid intake in 20% of the elderly patients.
TABLE 2
French hospital findings support use of oral B12 treatment38-41,45
| STUDY CHARACTERISTICS (NUMBER OF PATIENTS) | THERAPEUTIC MODALITIES | RESULTS |
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 and malabsorption (n=10)39 | Oral crystalline cyanocobalamin: 650 mcg per day, for at least 3 months |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=20)40 | Oral crystalline cyanocobalamin: between 1000 mcg per day for at least 1 week |
|
| Open prospective study of well-documented vitamin B12 deficiency related to food-B12 malabsorption (n=30)38 | Oral crystalline cyanocobalamin: between 250 and 1000 mcg per day, for 1 month |
|
| Open prospective study of low vitamin B12 levels not related to pernicious anemia (n=30)41 | Oral crystalline cyanocobalamin: between 125 and 1000 mcg per day for at least 1 week |
|
| Open prospective study of low vitamin B12 levels related to pernicious anemia (n=10)45 | Oral crystalline cyanocobalamin: 1000 mcg per day, for at least 3 months |
|
IM injection is customary, though dosages vary
The classic treatment for B12 deficiency, particularly when the cause is not a dietary deficiency, is parenteral administration—usually by intramuscular injection—of cyanocobalamin (and in rare occasions, hydroxocobalamin).7,11,16,35 In the US and UK, dosages range from 100 to 1000 mcg per month (or every 2–3 months when hydroxocobalamin is given). The patient will receive this treatment for the rest of his life.1,35
In France, the recommended practice is to build up the tissue stores of the vitamin quickly and correct serum B12 hypovitaminosis, particularly in the case of pernicious anemia. The treatment involves administering 1000 mcg of cyanocobalamin per day for 1 week, followed by 1000 mcg per week for 1 month, followed by 1000 mcg per month, normally for the rest of the patient’s life.2,3,20
Oral therapy is a well-kept secret
In cases of B12 deficiency that don’t involve nutritional deficiency, alternative routes of cobalamin administration, including the oral16,35-42 and nasal43,44 routes have been used. These alternative routes offer patients a way to avoid the discomfort, inconvenience, and cost of an office visit for monthly injections.
Our research team has developed an effective oral treatment of food-B12 malabsorption38-41 and for pernicious anemia45 using crystalline cobalamin (cyanocobalamin). Our principal studies of oral B12 treatment (open, not randomized studies) are described in TABLE 2.38-41,45 Our data confirm the previously reported efficacy of oral crystalline cyanocobalamin, especially in food-B12 therapy.6,16,36 All of our patients who received oral therapy corrected their B12 levels and at least two thirds corrected their hematological abnormalities.38-41,45 Moreover, one third of patients experienced a clinical improvement on oral treatment. In most cases of food-B12 malabsorption, a “low” B12 dose (ie, 125–1000 mcg of oral crystalline cyanocobalamin per day) was used.
These data are in line with the results of the 2 prospective randomized controlled studies comparing oral B12 with intramuscular B12 therapy.35,37 An evidence-based analysis by the Vitamin B12 Cochrane Group supports the efficacy of oral B12 therapy, with doses between 1000 and 2000 mcg given daily in the beginning, and then weekly.46 In this analysis, serum B12 levels increased significantly in patients receiving oral vitamin B12 and both groups of patients (receiving oral and intramuscular treatment) had neurological improvement.
In a randomized, parallel-group, double-blind, dose-finding trial, Eussen et al showed that the lowest dose of oral cyanocobalamin required to normalize mild B12 deficiency is more than 200 times the recommended dietary allowance of approximately 3 mcg daily (ie, >500 mcg/day).47 The procedure for oral B12 treatment has, however, not been completely validated yet in “real life,” particularly as it relates to long-term efficacy.48 Nonetheless, several authors suggest that oral B12 therapy remains one of medicine’s “best-kept secrets.”49
Acknowledgements
We are indebted to Professor Marc Imler and Jean-Louis Schlienger who initiated this work and to Helen Fothergill who kindly edited the text for publication in this English-language journal.
Correspondence
Prof. E. Andrès, Service de Médecine Interne, Diabète et Maladies Métaboliques, Clinique Médicale B, Hôpital Civil–Hôpitaux Universitaires de Strasbourg, 1 porte de l’Hôpital, 67091 Strasbourg Cedex, France; emmanuel. [email protected]
1. Carmel R. Current concepts in cobalamin deficiency. Ann Rev Med 2000;51:357-375.
2. Andrès E, Perrin AE, Kraemer JP, et al. Anémies par carence en vitamine B12 chez le sujet âgé de plus de 75 ans: nouveaux concepts. A propos de 20 observations. Rev Med Interne 2000;21:946-955.
3. Pautas E, Chérin P, De Jaeger C, Godeau P. Carence en vitamine B12 chez le sujet âgé. Presse Med 1999;28:1767-1770.
4. Carmel R. Malabsorption of food-cobalamin. Bailliere’s Clin Haematol 1995;8:639-655.
5. Andrès E, Affenberger S, Vinzio S, et al. Food-cobalamin malabsorption in elderly patients: clinical manifestations and treatment. Am J Med 2005;118:1154-1159.
6. Carmel R, Sarrai M. Diagnosis and management of clinical and subclinical cobalamin deficiency: advances and controversies. Curr Hematol Rep 2006;5:23-33.
7. Matthews JH. Cobalamin and folate deficiency in the elderly. Baillère’s Clin Haematol 1995;54:245-253.
8. Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994;60:2-11.
9. van Asselt DZ, Blom HJ, Zuiderent R, et al. Clinical significance of low cobalamin levels in older hospital patients. Neth J Med 2000;57:41-49.
10. Stabler SP, Allen RH, Savage DG, Lindenbaum J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood 1990;76:871-881.
11. Dharmarajan TS, Adiga GU, Norkus EP. Vitamin B12 deficiency. Recognizing subtle symptoms in older adults. Geriatrics 2003;58:30-38.
12. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol 2006;28:50-56.
13. Andrès E, Loukili NH, Noel E, et al. Vitamin B12 (cobalamin) deficiency in elderly patients. CAMJ 2004;171:251-260.
14. Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol 2006;5:949-960.
15. Abyad A. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2002;6:254-260.
16. Lane LA, Rojas-Fernandez C. Treatment of vitamin B12 deficiency anemia: oral versus parenteral therapy. Ann Pharmacother 2002;36:1268-1272.
17. Andrès E, Kaltenbach G. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2003;7:309-310.
18. Klee GG. Cobalamin and folate evaluation: measurements of methylmalonic acid and homocystein vs vitamin B12 and folate. Clin Chem 2000;46:12e77-1283.
19. Nicolas JP, Guéant JL. Absorption, distribution et excrétion de la vitamine B12. Ann Gastroenterol Hepatol 1994;30:270-282.
20. Loukili NH, Noel E, Blaison G, et al. Données actuelles sur la maladie de Biermer. A propos d’une étude rétrospective de 49 patients. Rev Med Interne 2004;25:556-561.
21. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med 1997;337:1441-1448.
22. Pruthi RK, Tefferi A. Pernicious anemia revisited. Mayo Clin Proc 1994;69:144-150.
23. Institute of Medicine Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, panthothenic acid, biotin and choline. Food and Nutrition Board, Washington, DC. National Academies Press, 1998.
24. Andrès E, Noel E, Kaltenbach G, et al. Carences en vitamine B12 avec test de Schilling normal ou syndrome de non-dissociation de la vitamine B12 de ses protéines porteuses chez le sujet âgé. Etude de 60 patients. Rev Med Interne 2003;24:218-223.
25. Carmel R, Aurangzeb I, Ojan D. Associations of food-cobalamin malabsorption with ethnic origin, age, Helicobacter pylori infection, and serum markers of gastritis. Am J Gastroenterol 2001;96:63-70.
26. Kaptan K, Beyan C, Ural AU, et al. Helicobacter pylori—is it a novel causative agent in Vitamin B12 deficiency? Arch Intern Med 2000;160:1349-1353.
27. Howden CW. Vitamin B12 levels during prolonged treatment with proton pump inhibitors. J Clin Gastroenterol 2000;30:29-33.
28. Andrès E, Noel E, Ben Abdelghani M. Vitamin B12 deficiency associated with chronic acid suppression therapy. Ann Pharmacother 2003;37:1730.-
29. Termanini B, Gibril F, Sutliff VE, Yu F, Venzon DJ, Jensen RT. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med 1998;104:422-430.
30. Jensen RT. Consequences of long-term proton pump blockade: insights from studies of patients with gastrinomas. Basic Clin Pharmacol Toxicol 2006;98:4-19.
31. Bauman WA, Shaw S, Javatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000;23:1227-1231.
32. Andrès E, Noel E, Goichot B. Metformin-associated vitamin B12 deficiency. Arch Intern Med 2002;162:2251-2252.
33. Liu KW, Dai LK, Jean W. Metformin-related vitamin B12 deficiency. Age Aging 2006;35:200-201.
34. Andrès E, Goichot B, Perrin AE, Vinzio S, Demangeat C, Schlienger JL. Sjögren’s syndrome: a potential new cause of mild cobalamin deficiency. Rheumatology (Oxford) 2001;40:1196-1197.
35. Kuzminski AM, Del Giacco EI, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood 1998;92:1191-1198.
36. Elia M. Oral or parenteral therapy for B12 deficiency. Lancet 1998;352:1721-1722.
37. Bolaman Z, Kadikoylu G, Yukselen V, Yavasoglu I, Barutca S, Senturk T. Oral versus intramuscular cobalamin treatment in megaloblastic anemia: a singlecenter, prospective, randomized, open-label study. Clin Ther 2003;25:3124-3134.
38. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption. A study of 30 patients. Clin Lab Haematol 2003;25:161-166.
39. Andrès E, Kurtz JE, Perrin AE, et al. Oral cobalamin therapy for the treatment of patients with food-cobalamin malabsorption. Am J Med 2001;111:126-129.
40. Kaltenbach G, Noblet-Dick M, Andrès E, et al. Réponse précoce au traitement oral par vitamine B12 chez des sujets âgés hypovitaminiques. Ann Med Interne (Paris) 2003;154:91-95.
41. Andrès E, Kaltenbach G, Noblet-Dick M, et al. Hematological response to short-term oral cyanocobalamin therapy for the treatment of cobalamin deficiencies in elderly patients. J Nutr Health Aging 2006;10:3-6.
42. Butler CC, Vidal-Alaball J, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency: a systematic review of randomized controlled trials. Fam Pract 2006;23:279-285.
43. Slot WB, Merkus FW, Van Deventer SJ, Tytgat GN. Normalization of plasma vitamin B12 concentration by intranasal hydroxocobalamin in vitamin B12-deficient patients. Gastroenterology 1997;113:430-433.
44. van Asselt DZ, Merkus FW, Russel FG, Hoefnagels WH. Nasal absorption of hydroxocobalamin in healthy elderly adults. Br J Clin Pharmacol 1998;45:83-86.
45. Andrès E, Loukili NH, Noel E, et al. Oral cobalamin (daily dose of 1000 μg) therapy for the treatment of patients with pernicious anemia. An open label study of 10 patients. Curr Ther Res 2005;66:13-22.
46. Vidal-Alaball J, Butler CC, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev 2005;20:CD004655.-
47. Eussen SJ, de Groot LC, Clarke R, et al. Oral cyanocobalamin supplementation in older people with vitamin B12 deficiency: a dose-finding trial. Arch Intern Med 2005;165:1167-1172.
48. Roth M, Orija I. Oral vitamin B12 therapy in vitamin B12 deficiency. Am J Med 2004;116:358.-
49. Graham ID, Jette N, Tetroe J, Robinson N, Milne S, Mitchell SL. Oral cobalamin remains medicine’s best kept secret. Arch Gerontol Geriatr 2007;44:49-59.
1. Carmel R. Current concepts in cobalamin deficiency. Ann Rev Med 2000;51:357-375.
2. Andrès E, Perrin AE, Kraemer JP, et al. Anémies par carence en vitamine B12 chez le sujet âgé de plus de 75 ans: nouveaux concepts. A propos de 20 observations. Rev Med Interne 2000;21:946-955.
3. Pautas E, Chérin P, De Jaeger C, Godeau P. Carence en vitamine B12 chez le sujet âgé. Presse Med 1999;28:1767-1770.
4. Carmel R. Malabsorption of food-cobalamin. Bailliere’s Clin Haematol 1995;8:639-655.
5. Andrès E, Affenberger S, Vinzio S, et al. Food-cobalamin malabsorption in elderly patients: clinical manifestations and treatment. Am J Med 2005;118:1154-1159.
6. Carmel R, Sarrai M. Diagnosis and management of clinical and subclinical cobalamin deficiency: advances and controversies. Curr Hematol Rep 2006;5:23-33.
7. Matthews JH. Cobalamin and folate deficiency in the elderly. Baillère’s Clin Haematol 1995;54:245-253.
8. Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994;60:2-11.
9. van Asselt DZ, Blom HJ, Zuiderent R, et al. Clinical significance of low cobalamin levels in older hospital patients. Neth J Med 2000;57:41-49.
10. Stabler SP, Allen RH, Savage DG, Lindenbaum J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood 1990;76:871-881.
11. Dharmarajan TS, Adiga GU, Norkus EP. Vitamin B12 deficiency. Recognizing subtle symptoms in older adults. Geriatrics 2003;58:30-38.
12. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol 2006;28:50-56.
13. Andrès E, Loukili NH, Noel E, et al. Vitamin B12 (cobalamin) deficiency in elderly patients. CAMJ 2004;171:251-260.
14. Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol 2006;5:949-960.
15. Abyad A. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2002;6:254-260.
16. Lane LA, Rojas-Fernandez C. Treatment of vitamin B12 deficiency anemia: oral versus parenteral therapy. Ann Pharmacother 2002;36:1268-1272.
17. Andrès E, Kaltenbach G. Prevalence of vitamin B12 deficiency among demented patients and cognitive recovery with cobalamin replacement. J Nutr Health Aging 2003;7:309-310.
18. Klee GG. Cobalamin and folate evaluation: measurements of methylmalonic acid and homocystein vs vitamin B12 and folate. Clin Chem 2000;46:12e77-1283.
19. Nicolas JP, Guéant JL. Absorption, distribution et excrétion de la vitamine B12. Ann Gastroenterol Hepatol 1994;30:270-282.
20. Loukili NH, Noel E, Blaison G, et al. Données actuelles sur la maladie de Biermer. A propos d’une étude rétrospective de 49 patients. Rev Med Interne 2004;25:556-561.
21. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med 1997;337:1441-1448.
22. Pruthi RK, Tefferi A. Pernicious anemia revisited. Mayo Clin Proc 1994;69:144-150.
23. Institute of Medicine Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, panthothenic acid, biotin and choline. Food and Nutrition Board, Washington, DC. National Academies Press, 1998.
24. Andrès E, Noel E, Kaltenbach G, et al. Carences en vitamine B12 avec test de Schilling normal ou syndrome de non-dissociation de la vitamine B12 de ses protéines porteuses chez le sujet âgé. Etude de 60 patients. Rev Med Interne 2003;24:218-223.
25. Carmel R, Aurangzeb I, Ojan D. Associations of food-cobalamin malabsorption with ethnic origin, age, Helicobacter pylori infection, and serum markers of gastritis. Am J Gastroenterol 2001;96:63-70.
26. Kaptan K, Beyan C, Ural AU, et al. Helicobacter pylori—is it a novel causative agent in Vitamin B12 deficiency? Arch Intern Med 2000;160:1349-1353.
27. Howden CW. Vitamin B12 levels during prolonged treatment with proton pump inhibitors. J Clin Gastroenterol 2000;30:29-33.
28. Andrès E, Noel E, Ben Abdelghani M. Vitamin B12 deficiency associated with chronic acid suppression therapy. Ann Pharmacother 2003;37:1730.-
29. Termanini B, Gibril F, Sutliff VE, Yu F, Venzon DJ, Jensen RT. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med 1998;104:422-430.
30. Jensen RT. Consequences of long-term proton pump blockade: insights from studies of patients with gastrinomas. Basic Clin Pharmacol Toxicol 2006;98:4-19.
31. Bauman WA, Shaw S, Javatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000;23:1227-1231.
32. Andrès E, Noel E, Goichot B. Metformin-associated vitamin B12 deficiency. Arch Intern Med 2002;162:2251-2252.
33. Liu KW, Dai LK, Jean W. Metformin-related vitamin B12 deficiency. Age Aging 2006;35:200-201.
34. Andrès E, Goichot B, Perrin AE, Vinzio S, Demangeat C, Schlienger JL. Sjögren’s syndrome: a potential new cause of mild cobalamin deficiency. Rheumatology (Oxford) 2001;40:1196-1197.
35. Kuzminski AM, Del Giacco EI, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood 1998;92:1191-1198.
36. Elia M. Oral or parenteral therapy for B12 deficiency. Lancet 1998;352:1721-1722.
37. Bolaman Z, Kadikoylu G, Yukselen V, Yavasoglu I, Barutca S, Senturk T. Oral versus intramuscular cobalamin treatment in megaloblastic anemia: a singlecenter, prospective, randomized, open-label study. Clin Ther 2003;25:3124-3134.
38. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption. A study of 30 patients. Clin Lab Haematol 2003;25:161-166.
39. Andrès E, Kurtz JE, Perrin AE, et al. Oral cobalamin therapy for the treatment of patients with food-cobalamin malabsorption. Am J Med 2001;111:126-129.
40. Kaltenbach G, Noblet-Dick M, Andrès E, et al. Réponse précoce au traitement oral par vitamine B12 chez des sujets âgés hypovitaminiques. Ann Med Interne (Paris) 2003;154:91-95.
41. Andrès E, Kaltenbach G, Noblet-Dick M, et al. Hematological response to short-term oral cyanocobalamin therapy for the treatment of cobalamin deficiencies in elderly patients. J Nutr Health Aging 2006;10:3-6.
42. Butler CC, Vidal-Alaball J, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency: a systematic review of randomized controlled trials. Fam Pract 2006;23:279-285.
43. Slot WB, Merkus FW, Van Deventer SJ, Tytgat GN. Normalization of plasma vitamin B12 concentration by intranasal hydroxocobalamin in vitamin B12-deficient patients. Gastroenterology 1997;113:430-433.
44. van Asselt DZ, Merkus FW, Russel FG, Hoefnagels WH. Nasal absorption of hydroxocobalamin in healthy elderly adults. Br J Clin Pharmacol 1998;45:83-86.
45. Andrès E, Loukili NH, Noel E, et al. Oral cobalamin (daily dose of 1000 μg) therapy for the treatment of patients with pernicious anemia. An open label study of 10 patients. Curr Ther Res 2005;66:13-22.
46. Vidal-Alaball J, Butler CC, Cannings-John R, et al. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev 2005;20:CD004655.-
47. Eussen SJ, de Groot LC, Clarke R, et al. Oral cyanocobalamin supplementation in older people with vitamin B12 deficiency: a dose-finding trial. Arch Intern Med 2005;165:1167-1172.
48. Roth M, Orija I. Oral vitamin B12 therapy in vitamin B12 deficiency. Am J Med 2004;116:358.-
49. Graham ID, Jette N, Tetroe J, Robinson N, Milne S, Mitchell SL. Oral cobalamin remains medicine’s best kept secret. Arch Gerontol Geriatr 2007;44:49-59.