User login
Metastatic Melanoma and Prostatic Adenocarcinoma in the Same Sentinel Lymph Node
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
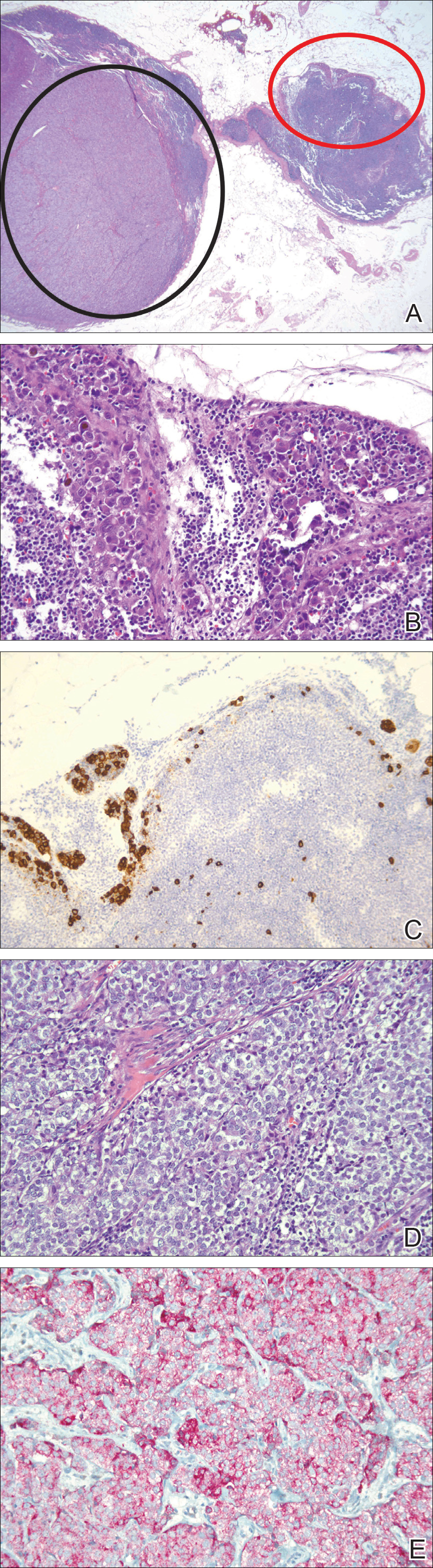
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
Practice Points
- Immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic sources of metastases.
- Thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis, enhancing the possibility of more effective treatment of earlier disease.
Periorbital Lupuslike Presentation of Graft-versus-host Disease
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
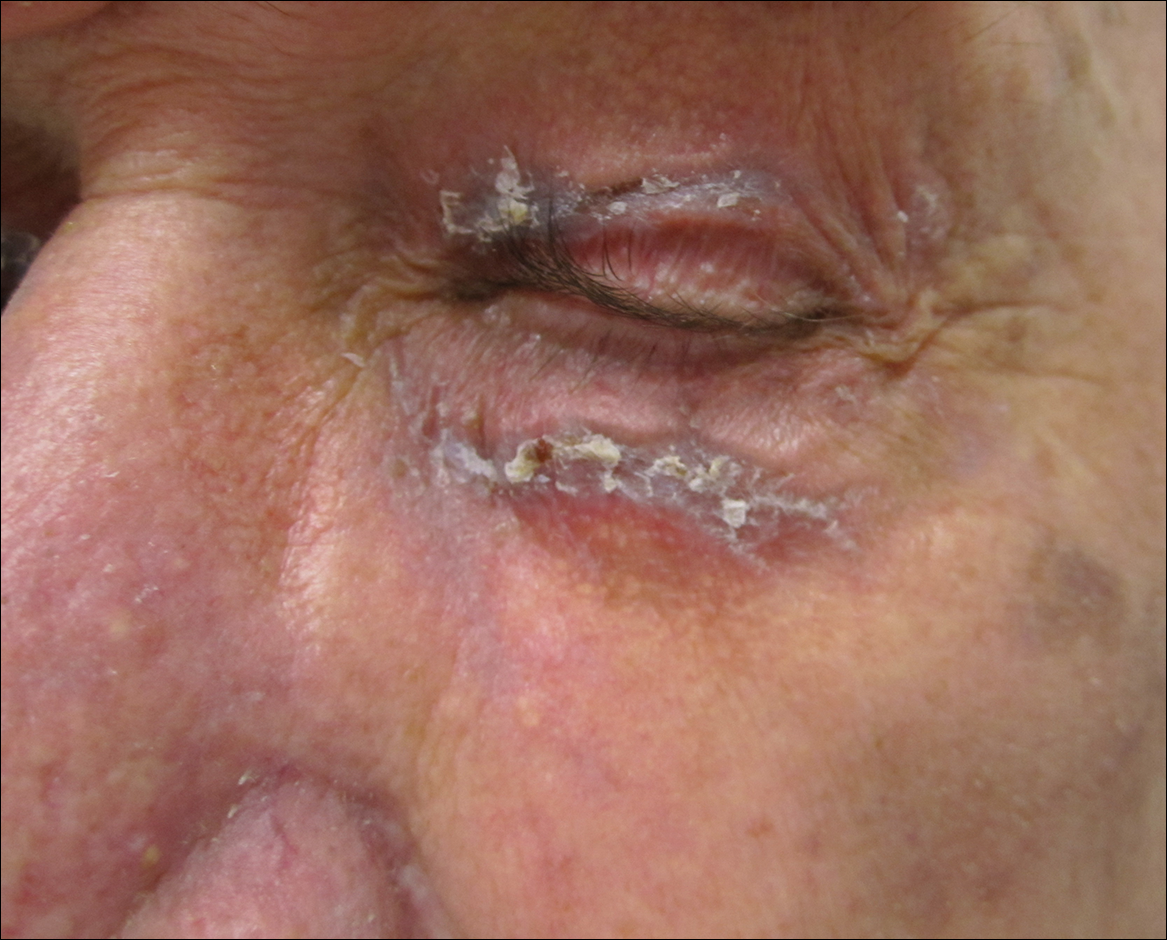
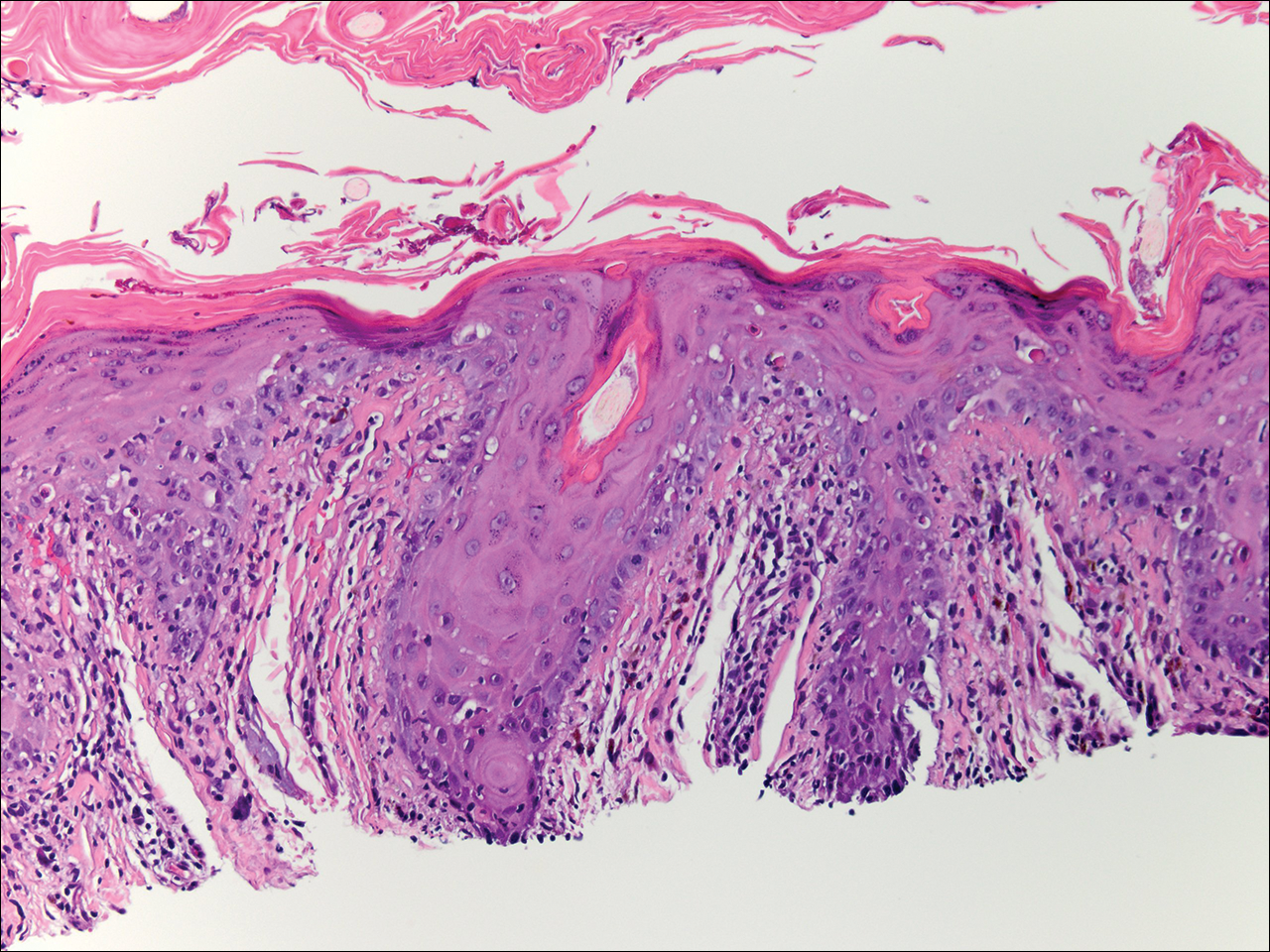
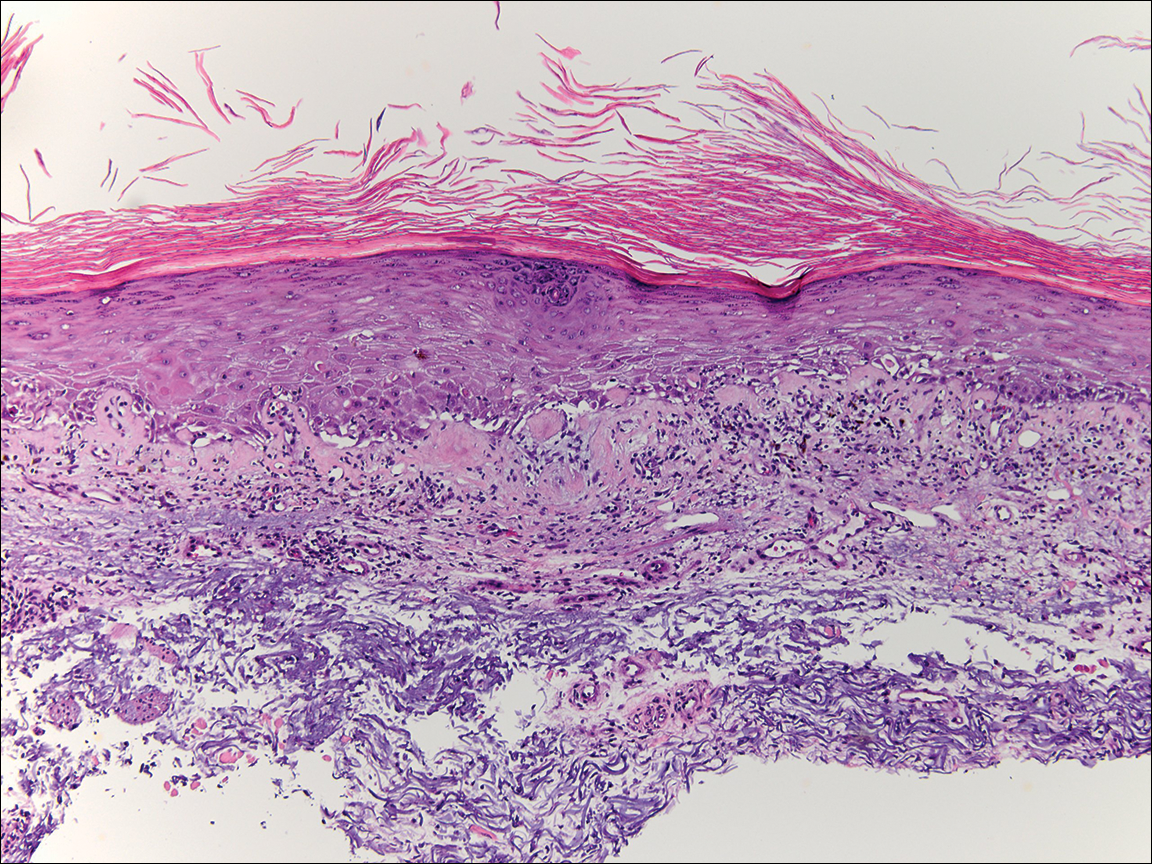
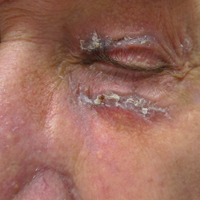
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
Complete Remission of Metastatic Merkel Cell Carcinoma in a Patient With Severe Psoriasis
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
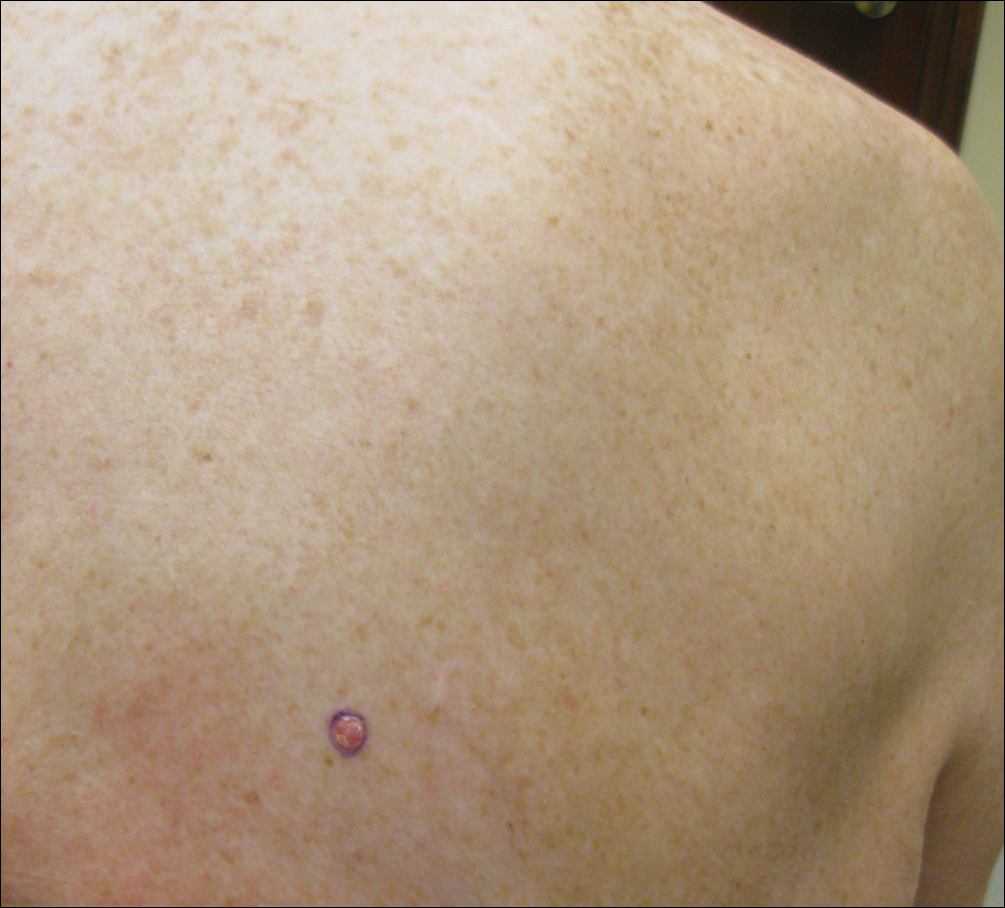
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
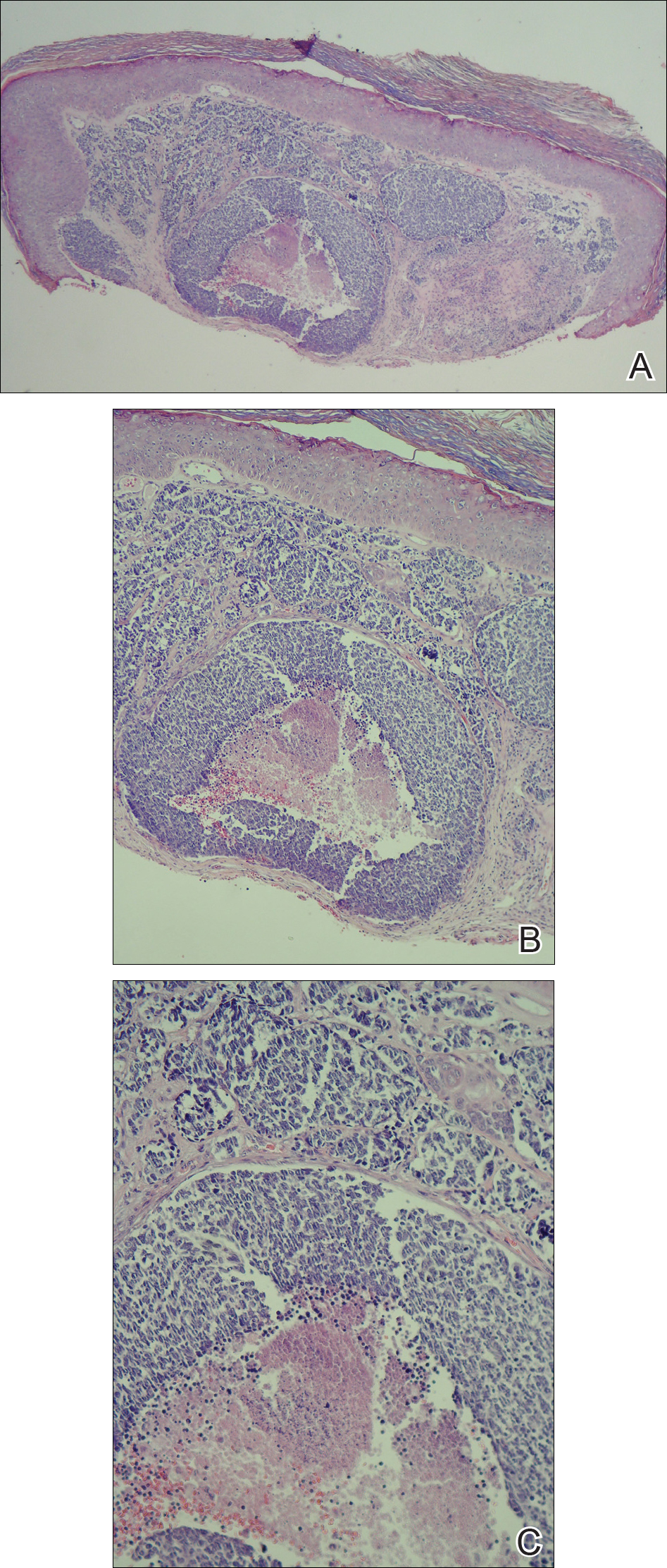
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
Practice Points
- Merkel cell carcinoma (MCC) has remarkable metastatic potential.
- Initial evaluation of patients with MCC should include clinical examination to detect cutaneous, lymph node, and distant metastasis.
- Risk factors associated with the development of MCC include immunosuppression, older age, and UV-exposed fair skin.
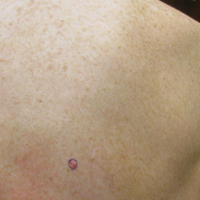
Differentiating Trigeminal Motor Neuropathy and Progressive Hemifacial Atrophy
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.

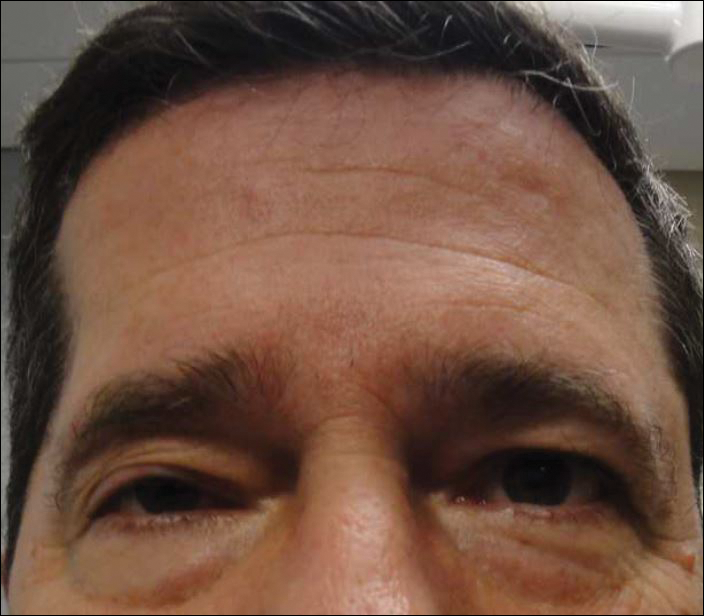
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
Practice Points
- The differential diagnosis of progressive hemifacial atrophy includes disorders of the trigeminal nerve.
- Trigeminal motor neuropathy presents with muscle weakness and atrophy without involvement of the skin, subcutis, or bone.
Nonmalignant Cutaneous Findings Associated With Vemurafenib
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).


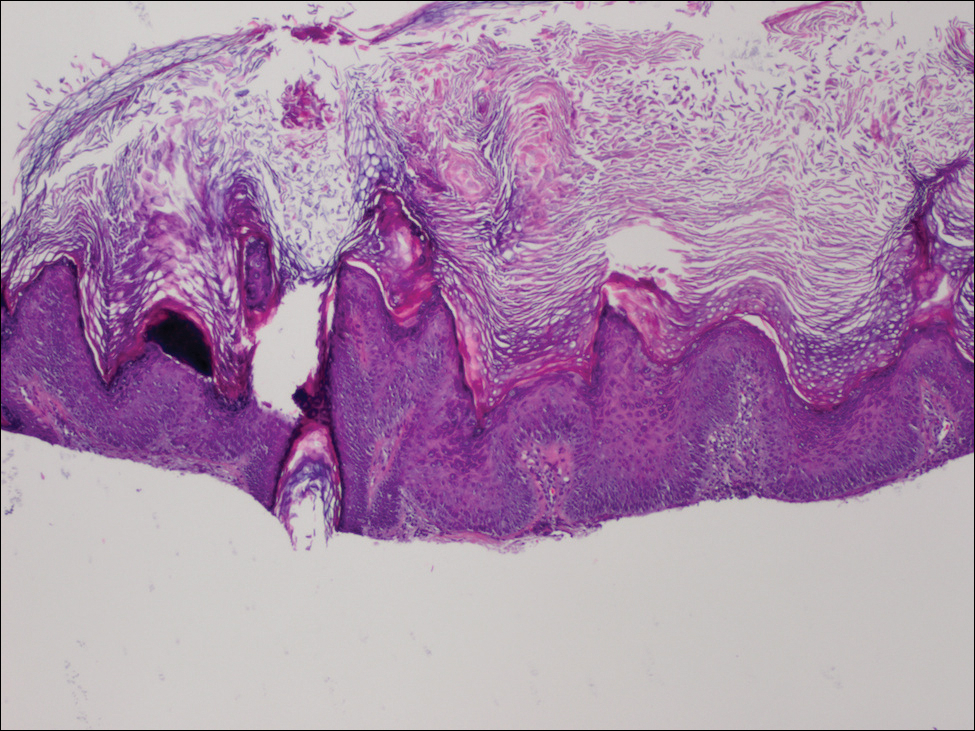
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
Practice Points
- Prior to starting a BRAF inhibitor, clinicians should perform a baseline total-body skin examination and follow-up every 2 months.
- Take photographs of the patient's entire body on initial total-body skin examination.
- Encourage sun protection for exposed areas on the body in all seasons.
Graft-versus-host Disease Presenting Along Blaschko Lines: Cutaneous Mosaicism
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
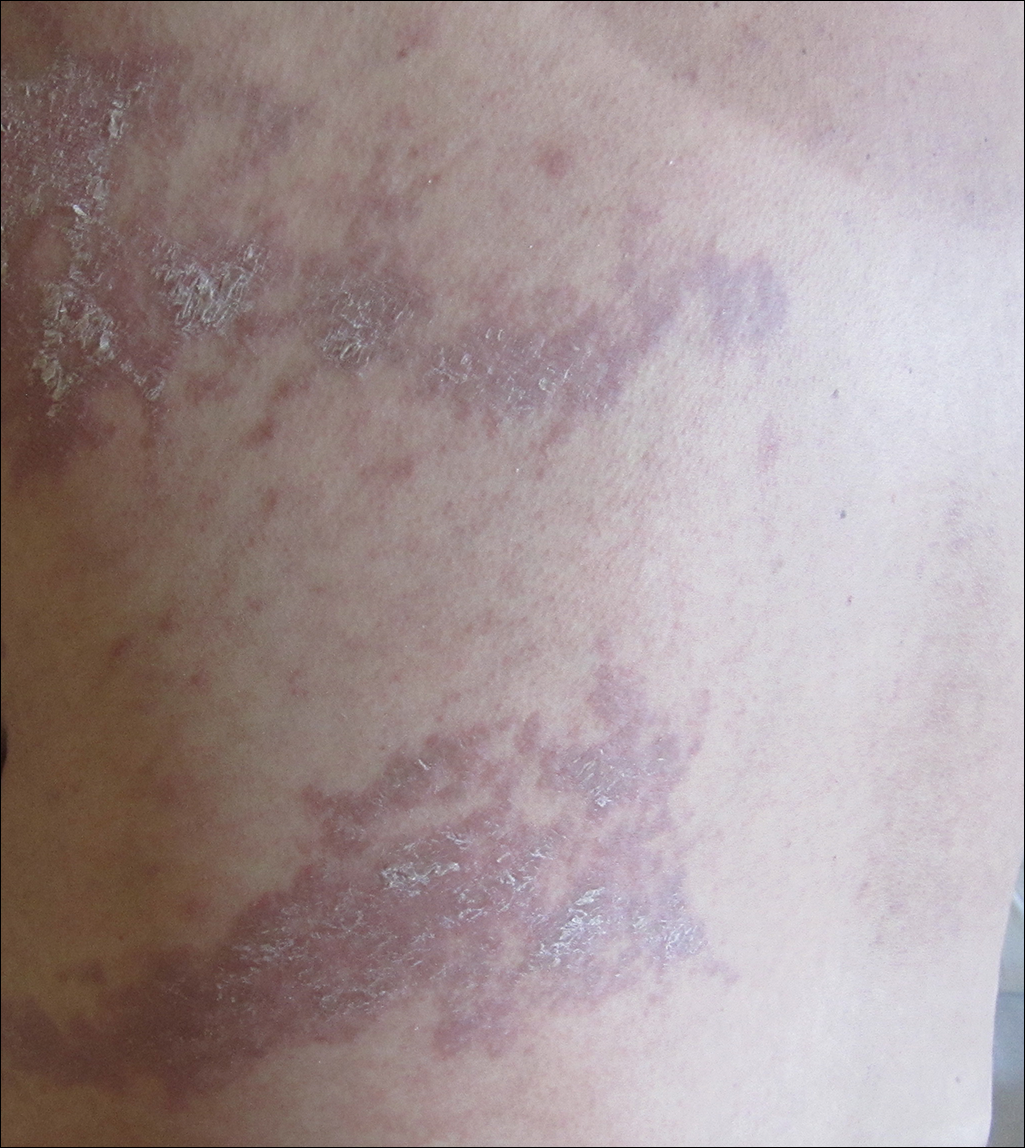
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
Practice Points
- Recognizing the characteristic manners in which different linear dermatoses present can aid in correctly identifying disorders that most commonly present in either a dermatomal or Blaschko-linear-type distribution.
- A blaschkoid-type distribution is the result of cutaneous mosaicism that occurs during embryological development and therefore subsequently produces a unique phenotypical presentation for various genetically influenced skin disorders, including graft-versus-host disease.
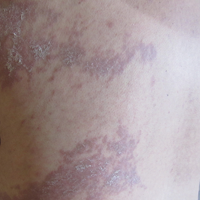
Richner-Hanhart Syndrome (Tyrosinemia Type II)
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
Practice Points
- Richner-Hanhart syndrome (tyrosinemia type II) should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Early diagnosis and initiation of a tyrosinephenylalanine–restricted diet in infancy is the most effective therapy to prevent mental retardation.
Over-the-counter Topical Musculoskeletal Pain Relievers Used With a Heat Source: A Dangerous Combination
To the Editor:
The combination of menthol and methyl salicylate found in a variety of over-the-counter (OTC) creams in conjunction with a heat source such as a heating pad used for musculoskeletal symptoms can be a dire combination due to increased systemic absorption with associated toxicity and localized effects ranging from contact dermatitis or irritation to burn or necrosis.1-6 We present a case of localized burn due a combination of topical methyl salicylate and heating pad use. We also discuss 2 commonly encountered side effects in the literature—localized burns and systemic toxicity associated with percutaneous absorption—and provide specific considerations related to the geriatric and pediatric populations.
A 62-year-old woman with a history of eczematous dermatitis and osteoarthritis with pain of the left shoulder presented to the dermatology clinic with painful skin-related changes on the left arm of 1 week’s duration. She was prescribed acetaminophen and ibuprofen. However, she self-medicated the left shoulder pain with 2 OTC products containing topical menthol and/or methyl salicylate in combination with a heating pad and likely fell asleep with this combination therapy applied. She noticed the burn the next morning. On examination, the left arm exhibited a geometric, irregularly shaped, erythematous, scaly plaque with a sharp transverse linear demarcation proximally and numerous erythematous linear scaly plaques oriented in an axial orientation with less-defined borders distally (Figure). The patient was diagnosed with burn secondary to combination of topical methyl salicylate and heating pad use. The patient was advised to discontinue the topical medication and to use caution with the heating pad in the future. She was prescribed pramoxine-hydrocortisone lotion to be applied to the affected area twice daily up to 5 days weekly until resolution. Subsequent evaluations revealed progressive improvement with only mild postinflammatory hyperpigmentation noted at 6 months after the burn.
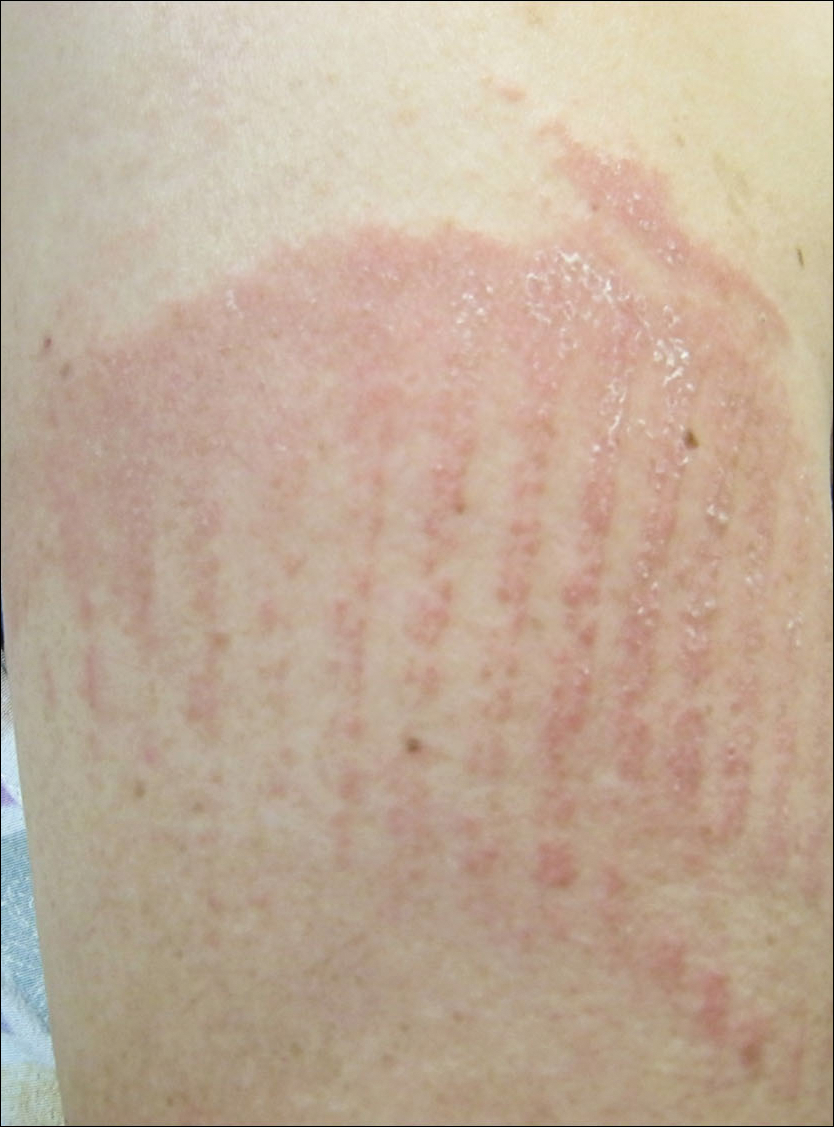
The US Food and Drug Administration (FDA) released statements in 2012 regarding concern for burns related to use of OTC musculoskeletal pain relievers, with 43 cases of burns reported due to methyl salicylate and menthol from 2004 to 2010. Most of the second- and third-degree burns occurred following topical applications of products containing either menthol monotherapy or a combination of methyl salicylate and menthol.1,2 In 2006, the FDA had already ordered 5 firms to stop compounding topical pain relief formulations containing these ingredients, with concerns that it puts patients at increased risk because the compounded formulations had not received FDA approval.3 Despite package warnings, patients may not be aware of the concerning side effects and risks associated with use of OTC creams, especially in combination with occlusion or heating pad use. Our case highlights the importance of ongoing patient education and physician counseling when encountering patients with arthritis or musculoskeletal pain who may often try various OTC self-treatments for pain relief.7
In 2012, the FDA reports stated that the cases of mild to serious burns were associated with methyl salicylate and menthol usage, in some cases 24 hours after first usage. Typically, these effects occur when concentrations are more than either 3% menthol alone or a combination of more than 3% menthol and more than 10% methyl salicylate.1,2 In our case, the patient had been using 2 different OTC products that may have contained as much as 11% menthol and/or 30% methyl salicylate. Electronic resources are available that disclose safety instructions including not to occlude the site, not to use on wounds, and not to be used in conjunction with a heating pad.8,9 Skin breakdown and vasodilation are more likely to occur in a setting of heat and occlusion, which allows for more absorption and localized side effects.4,10 Localized reactions may range from contact dermatitis4 to muscle necrosis.5
The most noteworthy case of localized destruction described a 62-year-old man who had applied topical methyl salicylate and menthol to the forearms, calves, and thighs, then intermittently used a heating pad for 15 to 20 minutes (total duration).5 He subsequently developed erythema and numerous 7.62- to 10.16-cm bullae, which was thought to be consistent with contact dermatitis. Three days later, he was found to have full-thickness cutaneous, fascial, and muscle necrosis in a linear pattern. He was hospitalized for approximately 1 year and treated with extensive debridement and a skin graft. His serum creatinine level increased from 0.7 mg per 100 mL to 2.7 mg per 100 mL (reference range, 0.6–1.2 mg/dL) with evidence of toxic nephrosis and persistent interstitial nephritis, demonstrating the severity of localized destruction that may result when combining these products with direct heat and potential subsequent systemic consequences of this combination.5
The systemic absorption of OTC formulations also has been studied. Morra et al10 studied 12 volunteers (6 women, 6 men) who applied either 5 g of methyl salicylate ointment 12.5% twice daily for 4 days to an area on the thigh (approximately equal to 567 mg salicylate) or trolamine cream 10% twice for 1 day. The participants underwent a break for 7 days and then switched to the alternate treatment. They found that 0.31 to 0.91 mg/L methyl salicylate was detected in the serum 1 hour after applying the ointment consisting of methyl salicylate, and 2 to 6 mg/L methyl salicylate was detected on day 4. Therapeutic serum salicylate levels are 150 to 300 mg/L. They found that approximately 22% of the methyl salicylate also was found in urine samples on day 4. Although these figures may appear small, this study was prompted when a 62-year-old man presented to the emergency department with symptoms of salicylate toxicity and a serum concentration of 518 mg/L from twice-daily use of an OTC formulation containing methyl salicylate over the course of multiple weeks.10 Additionally, those who have aspirin hypersensitivity should be cautious when using such products due to the risk for reported angioedema.4
Providers must exercise extreme caution while caring for geriatric patients, especially if patients are taking warfarin. The combined effects of warfarin and methyl salicylate have previously caused cutaneous purpura, gastrointestinal bleeding, and elevated international normalized ratio values.4,10 Older individuals also have increased skin fragility, allowing microtraumatic insult to easily develop. This fragility, along with an overall decreased intactness of the skin barrier, may lead to increased skin absorption. Furthermore, the addition of applying any heat source places the geriatric patient at greater risk for adverse events.10
In considering the limits of age, the pediatric population also has been studied regarding salicylate toxicity. Most commonly, oral ingestion has caused fatalities, as oil of wintergreen has been cited as extremely dangerous for children if swallowed; doses as small as a teaspoon (5 mL: 7000 mg salicylate) have resulted in fatalities.4,6 Although the consumption of a large amount of a cream- or ointment-based product is unlikely due to the consistency of the medication,6 the thought does merit consideration in the inquisitive toddler age group. For a 15-kg toddler, 150 mg/kg of aspirin or 2250 mg of aspirin, is considered the toxic level, which upon conversion to methyl salicylate levels using a 1.4 factor equates to 1607 mg of methyl salicylate to reach toxicity.6 If using a product with methyl salicylate 30% composition, 1 g of the product contains 300 mg of methyl salicylate; therefore if the toddler consumed approximately 5.3 g of the product (1607 mg methyl salicylate [toxic level] divided by 300 mg methyl salicylate per 1 g of product), he/she would reach toxic levels.6,11 To put this into perspective, a 2-oz tube contains 57 g (approximately 10 times the toxic dose) of the product.8 Thus, although there is less concern overall for consumption of cream- or ointment-based methyl salicylate, there still is potential for harm if a small child were to ingest such a product containing higher percentages of methyl salicylate.6
There also have been reports of pediatric toxicity related to percutaneous absorption, even leading to pediatric fatality.4,6 In particular, there was a case of a young boy hospitalized with ichthyosis who received escalating doses of percutaneous salicylate, which resulted in toxicity; when therapy was discontinued, he experienced full recovery.12 In 2007, a 17-year-old adolescent girl died from methyl salicylate toxicity after numerous applications of salicylate-containing products in conjunction with medicated pads.7
Although the FDA has drawn attention and encouraged caution with use of OTC topical musculoskeletal pain relievers, the importance of ensuring patients are fully aware of potential burns, permanent skin or muscle damage, and even death if used inappropriately cannot be overstated. The FDA consumer health information website has 2 patient-directed handouts2,3 that may be useful to post in patient waiting areas to increase overall understanding of the risks associated with OTC products containing methyl salicylate and menthol ingredients. Fortunately, our patient suffered only mild postinflammatory hyperpigmentation without substantial sustained consequences.
- US Food and Drug Administration. FDA Drug Safety Communication: rare cases of serious burns with the use of over-the-counter topical muscle and joint pain relievers. http://www.fda.gov/Drugs/DrugSafety/ucm318858.htm. Published September 13, 2012. Updated February 11, 2016. Accessed October 31, 2017.
- US Food and Drug Administration. Topical pain relievers may cause burns. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm318674.htm. Published September 13, 2012. Updated November 5, 2015. Accessed October 31, 2017.
- US Food and Drug Administration. Use caution with over-the-counter creams, ointments. http://www.fda.gov/forconsumers/consumerupdates/ucm049367.htm. Updated October 17, 2017. Accessed October 31, 2017.
- Chan TY. Potential dangers from topical preparations containing methyl salicylate. Hum Exp Toxicol. 1996;15:747-750.
- Heng MC. Local necrosis and interstitial nephritis due to topical methyl salicylate and menthol. Cutis. 1987;39:442-444.
- Davis JE. Are one or two dangerous? methyl salicylate exposure in toddlers. J Emerg Med. 2007;32:63-69.
- Associated Press. Sports cream warnings urged after teen’s death: track star’s overdose points to risks of popular muscle salve. NBC News. http://www.nbcnews.com/id/19208195. Updated June 13, 2007. Accessed October 31, 2017.
- Ultra Strength Bengay Cream. Bengay website. http://www.bengay.com/bengay-ultra-strength-cream. Accessed November 1, 2017.
- Tiger Balm Arthritis Rub. Tiger Balm website. http://www.tigerbalm.com/us/pages/tb_product?product_id=6. Accessed November 1, 2017.
- Morra P, Bartle WR, Walker SE, et al. Serum concentrations of salicylic acid following topically applied salicylate derivatives. Ann Pharmacother. 1996;9:935-940.
- US National Library of Medicine. Bengay Ultra Strength non greasy pain relieving- camphor (synthetic), menthol, and methyl salicylate cream. Daily Med website. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=5aa265f8-ab45-47b2-b5ab-d4df54daed01. Updated November 3, 2016. Accessed November 1, 2017.
- Aspinall JB, Goel KM. Salicylate poisoning in dermatological therapy. Br Med J. 1978;2:1373.
To the Editor:
The combination of menthol and methyl salicylate found in a variety of over-the-counter (OTC) creams in conjunction with a heat source such as a heating pad used for musculoskeletal symptoms can be a dire combination due to increased systemic absorption with associated toxicity and localized effects ranging from contact dermatitis or irritation to burn or necrosis.1-6 We present a case of localized burn due a combination of topical methyl salicylate and heating pad use. We also discuss 2 commonly encountered side effects in the literature—localized burns and systemic toxicity associated with percutaneous absorption—and provide specific considerations related to the geriatric and pediatric populations.
A 62-year-old woman with a history of eczematous dermatitis and osteoarthritis with pain of the left shoulder presented to the dermatology clinic with painful skin-related changes on the left arm of 1 week’s duration. She was prescribed acetaminophen and ibuprofen. However, she self-medicated the left shoulder pain with 2 OTC products containing topical menthol and/or methyl salicylate in combination with a heating pad and likely fell asleep with this combination therapy applied. She noticed the burn the next morning. On examination, the left arm exhibited a geometric, irregularly shaped, erythematous, scaly plaque with a sharp transverse linear demarcation proximally and numerous erythematous linear scaly plaques oriented in an axial orientation with less-defined borders distally (Figure). The patient was diagnosed with burn secondary to combination of topical methyl salicylate and heating pad use. The patient was advised to discontinue the topical medication and to use caution with the heating pad in the future. She was prescribed pramoxine-hydrocortisone lotion to be applied to the affected area twice daily up to 5 days weekly until resolution. Subsequent evaluations revealed progressive improvement with only mild postinflammatory hyperpigmentation noted at 6 months after the burn.
The US Food and Drug Administration (FDA) released statements in 2012 regarding concern for burns related to use of OTC musculoskeletal pain relievers, with 43 cases of burns reported due to methyl salicylate and menthol from 2004 to 2010. Most of the second- and third-degree burns occurred following topical applications of products containing either menthol monotherapy or a combination of methyl salicylate and menthol.1,2 In 2006, the FDA had already ordered 5 firms to stop compounding topical pain relief formulations containing these ingredients, with concerns that it puts patients at increased risk because the compounded formulations had not received FDA approval.3 Despite package warnings, patients may not be aware of the concerning side effects and risks associated with use of OTC creams, especially in combination with occlusion or heating pad use. Our case highlights the importance of ongoing patient education and physician counseling when encountering patients with arthritis or musculoskeletal pain who may often try various OTC self-treatments for pain relief.7
In 2012, the FDA reports stated that the cases of mild to serious burns were associated with methyl salicylate and menthol usage, in some cases 24 hours after first usage. Typically, these effects occur when concentrations are more than either 3% menthol alone or a combination of more than 3% menthol and more than 10% methyl salicylate.1,2 In our case, the patient had been using 2 different OTC products that may have contained as much as 11% menthol and/or 30% methyl salicylate. Electronic resources are available that disclose safety instructions including not to occlude the site, not to use on wounds, and not to be used in conjunction with a heating pad.8,9 Skin breakdown and vasodilation are more likely to occur in a setting of heat and occlusion, which allows for more absorption and localized side effects.4,10 Localized reactions may range from contact dermatitis4 to muscle necrosis.5
The most noteworthy case of localized destruction described a 62-year-old man who had applied topical methyl salicylate and menthol to the forearms, calves, and thighs, then intermittently used a heating pad for 15 to 20 minutes (total duration).5 He subsequently developed erythema and numerous 7.62- to 10.16-cm bullae, which was thought to be consistent with contact dermatitis. Three days later, he was found to have full-thickness cutaneous, fascial, and muscle necrosis in a linear pattern. He was hospitalized for approximately 1 year and treated with extensive debridement and a skin graft. His serum creatinine level increased from 0.7 mg per 100 mL to 2.7 mg per 100 mL (reference range, 0.6–1.2 mg/dL) with evidence of toxic nephrosis and persistent interstitial nephritis, demonstrating the severity of localized destruction that may result when combining these products with direct heat and potential subsequent systemic consequences of this combination.5
The systemic absorption of OTC formulations also has been studied. Morra et al10 studied 12 volunteers (6 women, 6 men) who applied either 5 g of methyl salicylate ointment 12.5% twice daily for 4 days to an area on the thigh (approximately equal to 567 mg salicylate) or trolamine cream 10% twice for 1 day. The participants underwent a break for 7 days and then switched to the alternate treatment. They found that 0.31 to 0.91 mg/L methyl salicylate was detected in the serum 1 hour after applying the ointment consisting of methyl salicylate, and 2 to 6 mg/L methyl salicylate was detected on day 4. Therapeutic serum salicylate levels are 150 to 300 mg/L. They found that approximately 22% of the methyl salicylate also was found in urine samples on day 4. Although these figures may appear small, this study was prompted when a 62-year-old man presented to the emergency department with symptoms of salicylate toxicity and a serum concentration of 518 mg/L from twice-daily use of an OTC formulation containing methyl salicylate over the course of multiple weeks.10 Additionally, those who have aspirin hypersensitivity should be cautious when using such products due to the risk for reported angioedema.4
Providers must exercise extreme caution while caring for geriatric patients, especially if patients are taking warfarin. The combined effects of warfarin and methyl salicylate have previously caused cutaneous purpura, gastrointestinal bleeding, and elevated international normalized ratio values.4,10 Older individuals also have increased skin fragility, allowing microtraumatic insult to easily develop. This fragility, along with an overall decreased intactness of the skin barrier, may lead to increased skin absorption. Furthermore, the addition of applying any heat source places the geriatric patient at greater risk for adverse events.10
In considering the limits of age, the pediatric population also has been studied regarding salicylate toxicity. Most commonly, oral ingestion has caused fatalities, as oil of wintergreen has been cited as extremely dangerous for children if swallowed; doses as small as a teaspoon (5 mL: 7000 mg salicylate) have resulted in fatalities.4,6 Although the consumption of a large amount of a cream- or ointment-based product is unlikely due to the consistency of the medication,6 the thought does merit consideration in the inquisitive toddler age group. For a 15-kg toddler, 150 mg/kg of aspirin or 2250 mg of aspirin, is considered the toxic level, which upon conversion to methyl salicylate levels using a 1.4 factor equates to 1607 mg of methyl salicylate to reach toxicity.6 If using a product with methyl salicylate 30% composition, 1 g of the product contains 300 mg of methyl salicylate; therefore if the toddler consumed approximately 5.3 g of the product (1607 mg methyl salicylate [toxic level] divided by 300 mg methyl salicylate per 1 g of product), he/she would reach toxic levels.6,11 To put this into perspective, a 2-oz tube contains 57 g (approximately 10 times the toxic dose) of the product.8 Thus, although there is less concern overall for consumption of cream- or ointment-based methyl salicylate, there still is potential for harm if a small child were to ingest such a product containing higher percentages of methyl salicylate.6
There also have been reports of pediatric toxicity related to percutaneous absorption, even leading to pediatric fatality.4,6 In particular, there was a case of a young boy hospitalized with ichthyosis who received escalating doses of percutaneous salicylate, which resulted in toxicity; when therapy was discontinued, he experienced full recovery.12 In 2007, a 17-year-old adolescent girl died from methyl salicylate toxicity after numerous applications of salicylate-containing products in conjunction with medicated pads.7
Although the FDA has drawn attention and encouraged caution with use of OTC topical musculoskeletal pain relievers, the importance of ensuring patients are fully aware of potential burns, permanent skin or muscle damage, and even death if used inappropriately cannot be overstated. The FDA consumer health information website has 2 patient-directed handouts2,3 that may be useful to post in patient waiting areas to increase overall understanding of the risks associated with OTC products containing methyl salicylate and menthol ingredients. Fortunately, our patient suffered only mild postinflammatory hyperpigmentation without substantial sustained consequences.
To the Editor:
The combination of menthol and methyl salicylate found in a variety of over-the-counter (OTC) creams in conjunction with a heat source such as a heating pad used for musculoskeletal symptoms can be a dire combination due to increased systemic absorption with associated toxicity and localized effects ranging from contact dermatitis or irritation to burn or necrosis.1-6 We present a case of localized burn due a combination of topical methyl salicylate and heating pad use. We also discuss 2 commonly encountered side effects in the literature—localized burns and systemic toxicity associated with percutaneous absorption—and provide specific considerations related to the geriatric and pediatric populations.
A 62-year-old woman with a history of eczematous dermatitis and osteoarthritis with pain of the left shoulder presented to the dermatology clinic with painful skin-related changes on the left arm of 1 week’s duration. She was prescribed acetaminophen and ibuprofen. However, she self-medicated the left shoulder pain with 2 OTC products containing topical menthol and/or methyl salicylate in combination with a heating pad and likely fell asleep with this combination therapy applied. She noticed the burn the next morning. On examination, the left arm exhibited a geometric, irregularly shaped, erythematous, scaly plaque with a sharp transverse linear demarcation proximally and numerous erythematous linear scaly plaques oriented in an axial orientation with less-defined borders distally (Figure). The patient was diagnosed with burn secondary to combination of topical methyl salicylate and heating pad use. The patient was advised to discontinue the topical medication and to use caution with the heating pad in the future. She was prescribed pramoxine-hydrocortisone lotion to be applied to the affected area twice daily up to 5 days weekly until resolution. Subsequent evaluations revealed progressive improvement with only mild postinflammatory hyperpigmentation noted at 6 months after the burn.
The US Food and Drug Administration (FDA) released statements in 2012 regarding concern for burns related to use of OTC musculoskeletal pain relievers, with 43 cases of burns reported due to methyl salicylate and menthol from 2004 to 2010. Most of the second- and third-degree burns occurred following topical applications of products containing either menthol monotherapy or a combination of methyl salicylate and menthol.1,2 In 2006, the FDA had already ordered 5 firms to stop compounding topical pain relief formulations containing these ingredients, with concerns that it puts patients at increased risk because the compounded formulations had not received FDA approval.3 Despite package warnings, patients may not be aware of the concerning side effects and risks associated with use of OTC creams, especially in combination with occlusion or heating pad use. Our case highlights the importance of ongoing patient education and physician counseling when encountering patients with arthritis or musculoskeletal pain who may often try various OTC self-treatments for pain relief.7
In 2012, the FDA reports stated that the cases of mild to serious burns were associated with methyl salicylate and menthol usage, in some cases 24 hours after first usage. Typically, these effects occur when concentrations are more than either 3% menthol alone or a combination of more than 3% menthol and more than 10% methyl salicylate.1,2 In our case, the patient had been using 2 different OTC products that may have contained as much as 11% menthol and/or 30% methyl salicylate. Electronic resources are available that disclose safety instructions including not to occlude the site, not to use on wounds, and not to be used in conjunction with a heating pad.8,9 Skin breakdown and vasodilation are more likely to occur in a setting of heat and occlusion, which allows for more absorption and localized side effects.4,10 Localized reactions may range from contact dermatitis4 to muscle necrosis.5
The most noteworthy case of localized destruction described a 62-year-old man who had applied topical methyl salicylate and menthol to the forearms, calves, and thighs, then intermittently used a heating pad for 15 to 20 minutes (total duration).5 He subsequently developed erythema and numerous 7.62- to 10.16-cm bullae, which was thought to be consistent with contact dermatitis. Three days later, he was found to have full-thickness cutaneous, fascial, and muscle necrosis in a linear pattern. He was hospitalized for approximately 1 year and treated with extensive debridement and a skin graft. His serum creatinine level increased from 0.7 mg per 100 mL to 2.7 mg per 100 mL (reference range, 0.6–1.2 mg/dL) with evidence of toxic nephrosis and persistent interstitial nephritis, demonstrating the severity of localized destruction that may result when combining these products with direct heat and potential subsequent systemic consequences of this combination.5
The systemic absorption of OTC formulations also has been studied. Morra et al10 studied 12 volunteers (6 women, 6 men) who applied either 5 g of methyl salicylate ointment 12.5% twice daily for 4 days to an area on the thigh (approximately equal to 567 mg salicylate) or trolamine cream 10% twice for 1 day. The participants underwent a break for 7 days and then switched to the alternate treatment. They found that 0.31 to 0.91 mg/L methyl salicylate was detected in the serum 1 hour after applying the ointment consisting of methyl salicylate, and 2 to 6 mg/L methyl salicylate was detected on day 4. Therapeutic serum salicylate levels are 150 to 300 mg/L. They found that approximately 22% of the methyl salicylate also was found in urine samples on day 4. Although these figures may appear small, this study was prompted when a 62-year-old man presented to the emergency department with symptoms of salicylate toxicity and a serum concentration of 518 mg/L from twice-daily use of an OTC formulation containing methyl salicylate over the course of multiple weeks.10 Additionally, those who have aspirin hypersensitivity should be cautious when using such products due to the risk for reported angioedema.4
Providers must exercise extreme caution while caring for geriatric patients, especially if patients are taking warfarin. The combined effects of warfarin and methyl salicylate have previously caused cutaneous purpura, gastrointestinal bleeding, and elevated international normalized ratio values.4,10 Older individuals also have increased skin fragility, allowing microtraumatic insult to easily develop. This fragility, along with an overall decreased intactness of the skin barrier, may lead to increased skin absorption. Furthermore, the addition of applying any heat source places the geriatric patient at greater risk for adverse events.10
In considering the limits of age, the pediatric population also has been studied regarding salicylate toxicity. Most commonly, oral ingestion has caused fatalities, as oil of wintergreen has been cited as extremely dangerous for children if swallowed; doses as small as a teaspoon (5 mL: 7000 mg salicylate) have resulted in fatalities.4,6 Although the consumption of a large amount of a cream- or ointment-based product is unlikely due to the consistency of the medication,6 the thought does merit consideration in the inquisitive toddler age group. For a 15-kg toddler, 150 mg/kg of aspirin or 2250 mg of aspirin, is considered the toxic level, which upon conversion to methyl salicylate levels using a 1.4 factor equates to 1607 mg of methyl salicylate to reach toxicity.6 If using a product with methyl salicylate 30% composition, 1 g of the product contains 300 mg of methyl salicylate; therefore if the toddler consumed approximately 5.3 g of the product (1607 mg methyl salicylate [toxic level] divided by 300 mg methyl salicylate per 1 g of product), he/she would reach toxic levels.6,11 To put this into perspective, a 2-oz tube contains 57 g (approximately 10 times the toxic dose) of the product.8 Thus, although there is less concern overall for consumption of cream- or ointment-based methyl salicylate, there still is potential for harm if a small child were to ingest such a product containing higher percentages of methyl salicylate.6
There also have been reports of pediatric toxicity related to percutaneous absorption, even leading to pediatric fatality.4,6 In particular, there was a case of a young boy hospitalized with ichthyosis who received escalating doses of percutaneous salicylate, which resulted in toxicity; when therapy was discontinued, he experienced full recovery.12 In 2007, a 17-year-old adolescent girl died from methyl salicylate toxicity after numerous applications of salicylate-containing products in conjunction with medicated pads.7
Although the FDA has drawn attention and encouraged caution with use of OTC topical musculoskeletal pain relievers, the importance of ensuring patients are fully aware of potential burns, permanent skin or muscle damage, and even death if used inappropriately cannot be overstated. The FDA consumer health information website has 2 patient-directed handouts2,3 that may be useful to post in patient waiting areas to increase overall understanding of the risks associated with OTC products containing methyl salicylate and menthol ingredients. Fortunately, our patient suffered only mild postinflammatory hyperpigmentation without substantial sustained consequences.
- US Food and Drug Administration. FDA Drug Safety Communication: rare cases of serious burns with the use of over-the-counter topical muscle and joint pain relievers. http://www.fda.gov/Drugs/DrugSafety/ucm318858.htm. Published September 13, 2012. Updated February 11, 2016. Accessed October 31, 2017.
- US Food and Drug Administration. Topical pain relievers may cause burns. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm318674.htm. Published September 13, 2012. Updated November 5, 2015. Accessed October 31, 2017.
- US Food and Drug Administration. Use caution with over-the-counter creams, ointments. http://www.fda.gov/forconsumers/consumerupdates/ucm049367.htm. Updated October 17, 2017. Accessed October 31, 2017.
- Chan TY. Potential dangers from topical preparations containing methyl salicylate. Hum Exp Toxicol. 1996;15:747-750.
- Heng MC. Local necrosis and interstitial nephritis due to topical methyl salicylate and menthol. Cutis. 1987;39:442-444.
- Davis JE. Are one or two dangerous? methyl salicylate exposure in toddlers. J Emerg Med. 2007;32:63-69.
- Associated Press. Sports cream warnings urged after teen’s death: track star’s overdose points to risks of popular muscle salve. NBC News. http://www.nbcnews.com/id/19208195. Updated June 13, 2007. Accessed October 31, 2017.
- Ultra Strength Bengay Cream. Bengay website. http://www.bengay.com/bengay-ultra-strength-cream. Accessed November 1, 2017.
- Tiger Balm Arthritis Rub. Tiger Balm website. http://www.tigerbalm.com/us/pages/tb_product?product_id=6. Accessed November 1, 2017.
- Morra P, Bartle WR, Walker SE, et al. Serum concentrations of salicylic acid following topically applied salicylate derivatives. Ann Pharmacother. 1996;9:935-940.
- US National Library of Medicine. Bengay Ultra Strength non greasy pain relieving- camphor (synthetic), menthol, and methyl salicylate cream. Daily Med website. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=5aa265f8-ab45-47b2-b5ab-d4df54daed01. Updated November 3, 2016. Accessed November 1, 2017.
- Aspinall JB, Goel KM. Salicylate poisoning in dermatological therapy. Br Med J. 1978;2:1373.
- US Food and Drug Administration. FDA Drug Safety Communication: rare cases of serious burns with the use of over-the-counter topical muscle and joint pain relievers. http://www.fda.gov/Drugs/DrugSafety/ucm318858.htm. Published September 13, 2012. Updated February 11, 2016. Accessed October 31, 2017.
- US Food and Drug Administration. Topical pain relievers may cause burns. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm318674.htm. Published September 13, 2012. Updated November 5, 2015. Accessed October 31, 2017.
- US Food and Drug Administration. Use caution with over-the-counter creams, ointments. http://www.fda.gov/forconsumers/consumerupdates/ucm049367.htm. Updated October 17, 2017. Accessed October 31, 2017.
- Chan TY. Potential dangers from topical preparations containing methyl salicylate. Hum Exp Toxicol. 1996;15:747-750.
- Heng MC. Local necrosis and interstitial nephritis due to topical methyl salicylate and menthol. Cutis. 1987;39:442-444.
- Davis JE. Are one or two dangerous? methyl salicylate exposure in toddlers. J Emerg Med. 2007;32:63-69.
- Associated Press. Sports cream warnings urged after teen’s death: track star’s overdose points to risks of popular muscle salve. NBC News. http://www.nbcnews.com/id/19208195. Updated June 13, 2007. Accessed October 31, 2017.
- Ultra Strength Bengay Cream. Bengay website. http://www.bengay.com/bengay-ultra-strength-cream. Accessed November 1, 2017.
- Tiger Balm Arthritis Rub. Tiger Balm website. http://www.tigerbalm.com/us/pages/tb_product?product_id=6. Accessed November 1, 2017.
- Morra P, Bartle WR, Walker SE, et al. Serum concentrations of salicylic acid following topically applied salicylate derivatives. Ann Pharmacother. 1996;9:935-940.
- US National Library of Medicine. Bengay Ultra Strength non greasy pain relieving- camphor (synthetic), menthol, and methyl salicylate cream. Daily Med website. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=5aa265f8-ab45-47b2-b5ab-d4df54daed01. Updated November 3, 2016. Accessed November 1, 2017.
- Aspinall JB, Goel KM. Salicylate poisoning in dermatological therapy. Br Med J. 1978;2:1373.
Practice Points
- Recognize the potential complication of burn from use of over-the-counter (OTC) musculoskeletal relievers in combination with a heat source.
- Screen for OTC product use as well as device application when evaluating an atypically patterned cutaneous eruption.
- Recognize potential toxicity associated with both topical application and accidental ingestion in the pediatric population.
- Physicians should become familiar with resources available, including patient handouts that describe risks associated with use of OTC musculoskeletal relievers containing methyl salicylate and menthol ingredients.
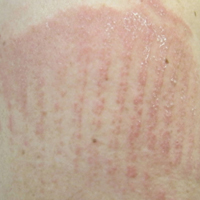
Primary Mucinous Carcinoma of the Eyelid Treated With Mohs Micrographic Surgery
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
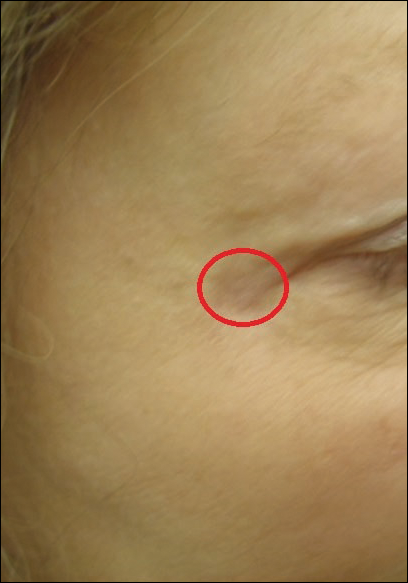
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
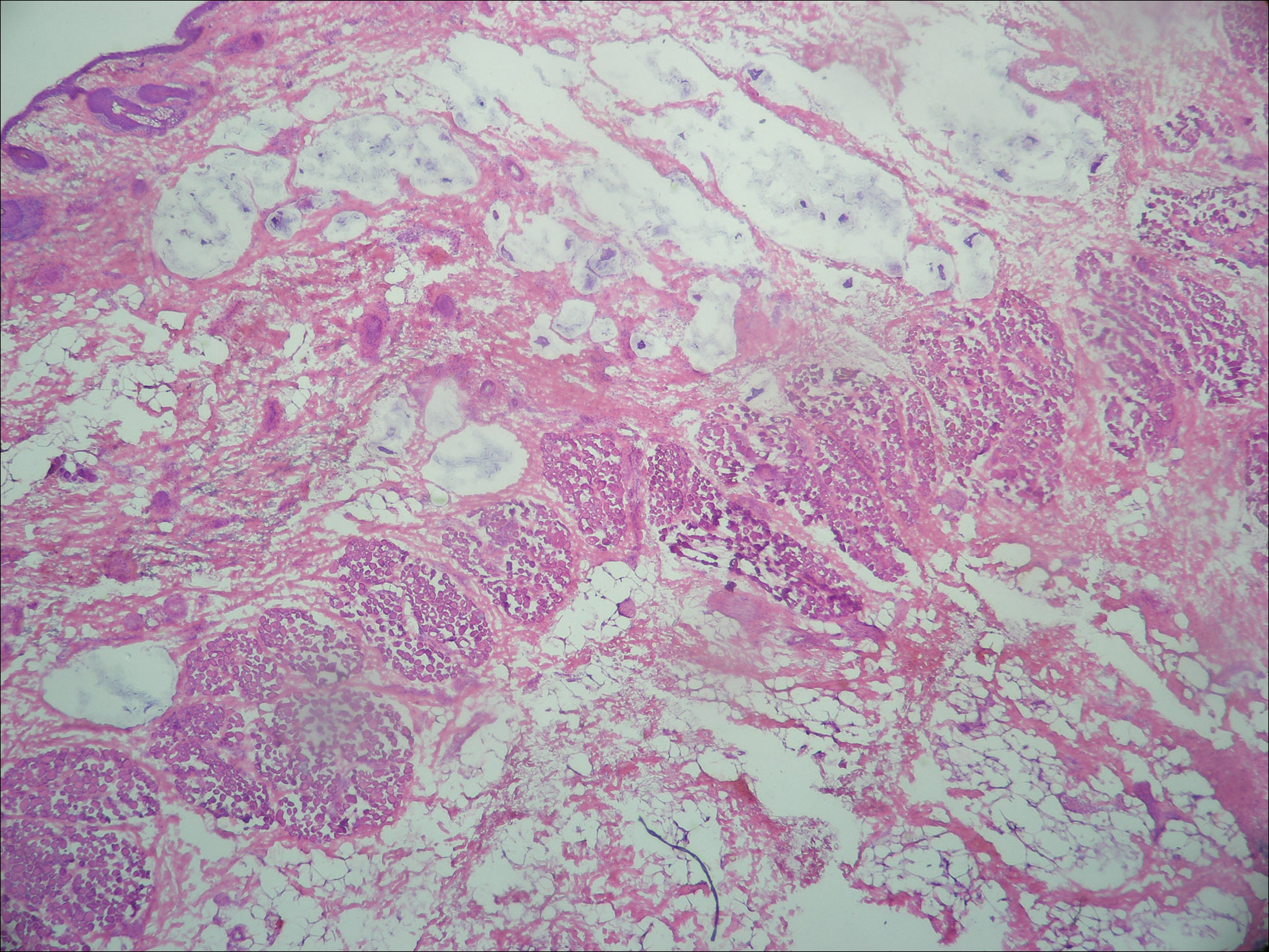
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
Practice Points
- Primary mucinous carcinoma (PMC) of the skin is a rare adnexal tumor.
- Prior to treatment, the diagnostic importance lies in distinguishing PMC from metastatic mucinous malignancies, which portend a poorer prognosis.
- Treatment primarily is surgical, with Mohs micrographic surgery offering improved tissue conservation and reduced recurrence rates.
Acrodermatitis Enteropathica in a Patient With Short Bowel Syndrome
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism

A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
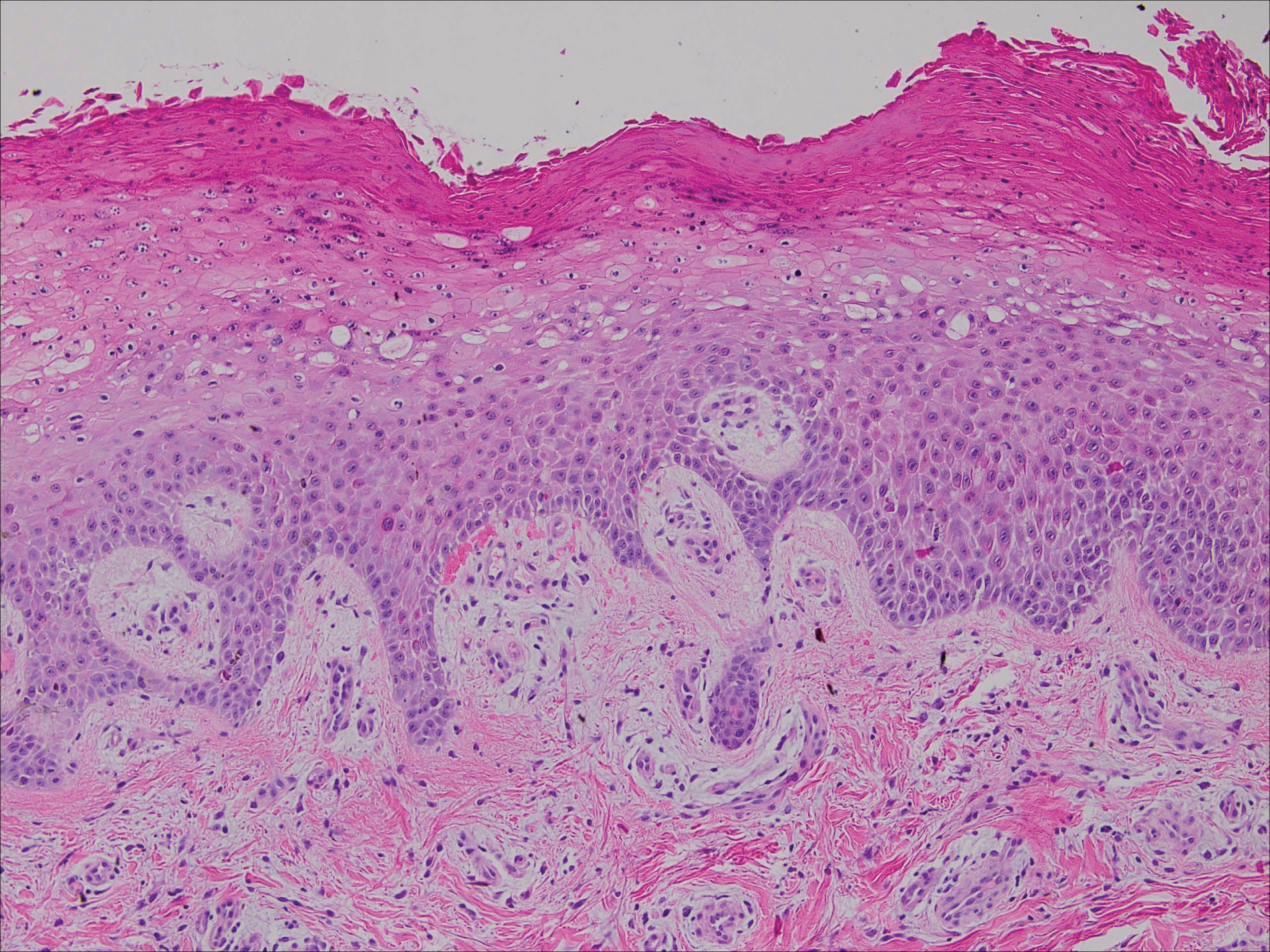
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
Practice Points
- Acrodermatitis enteropathica can be a manifestation of zinc deficiency.
- Acrodermatitis enteropathica should be considered in patients with poor intestinal absorption of nutrients.