User login
Sunburn Purpura
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
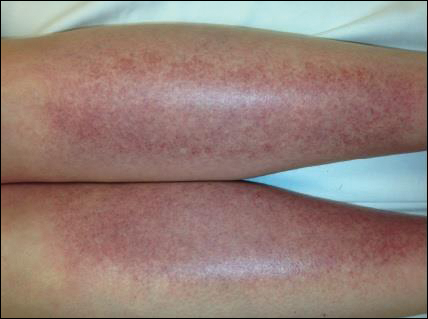
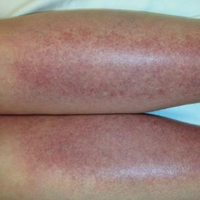
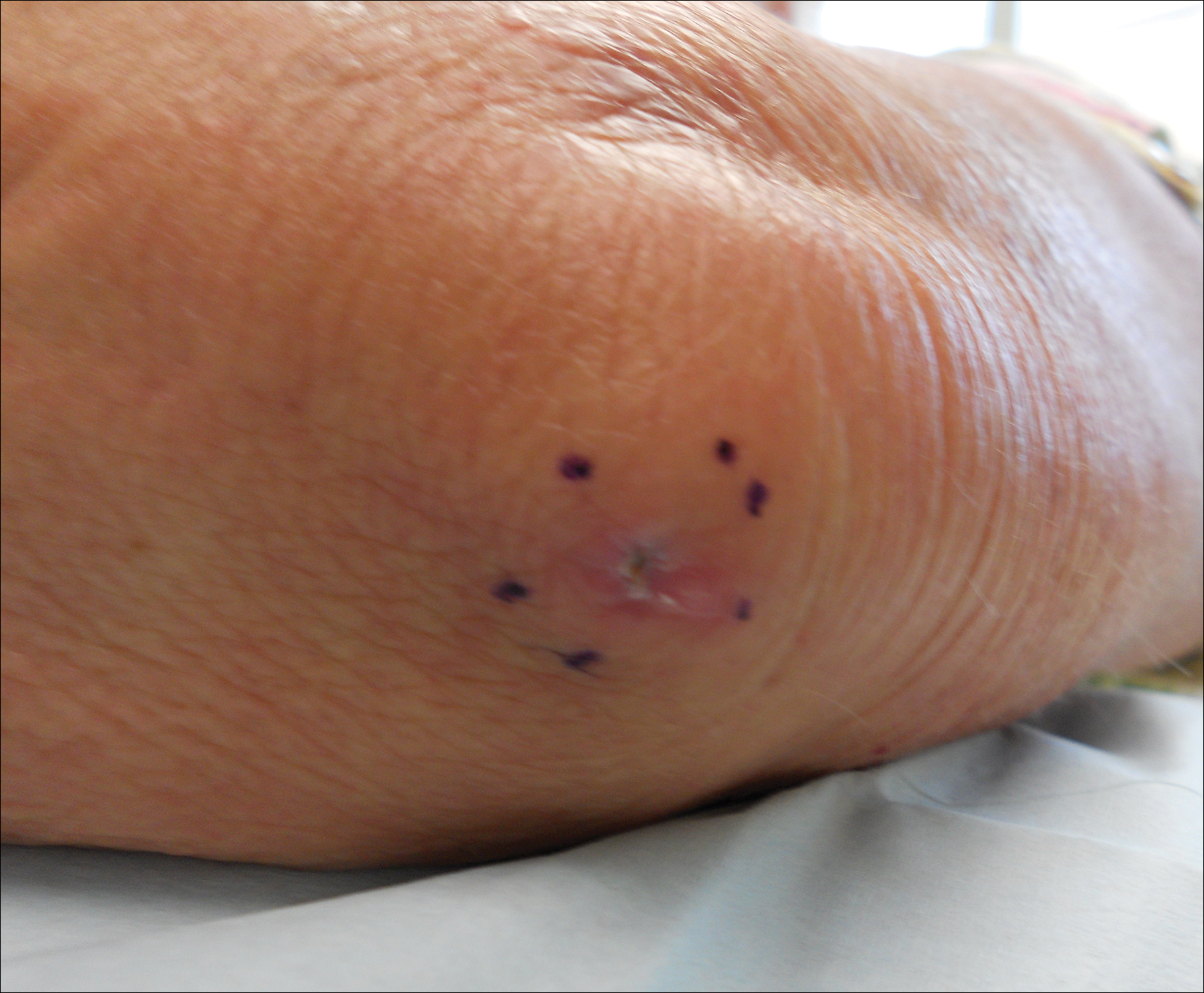
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
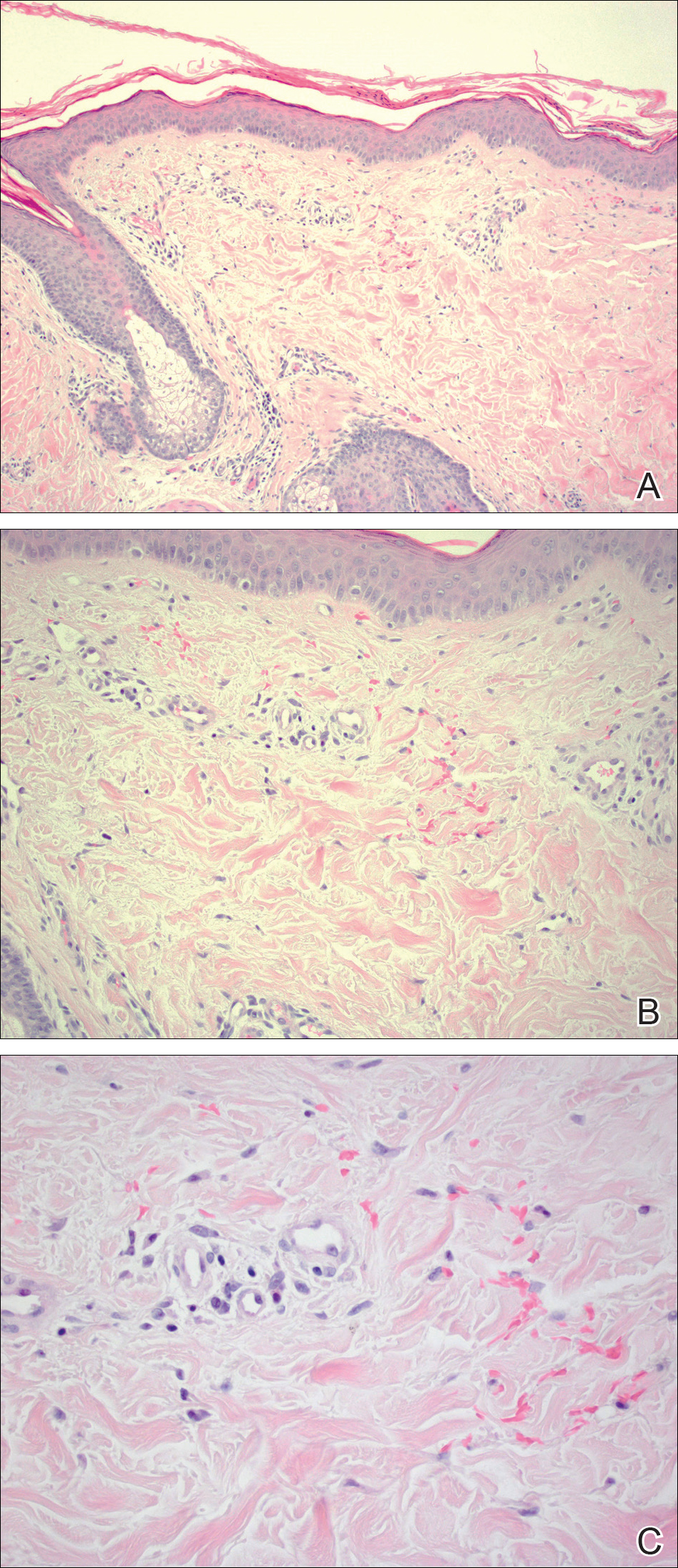
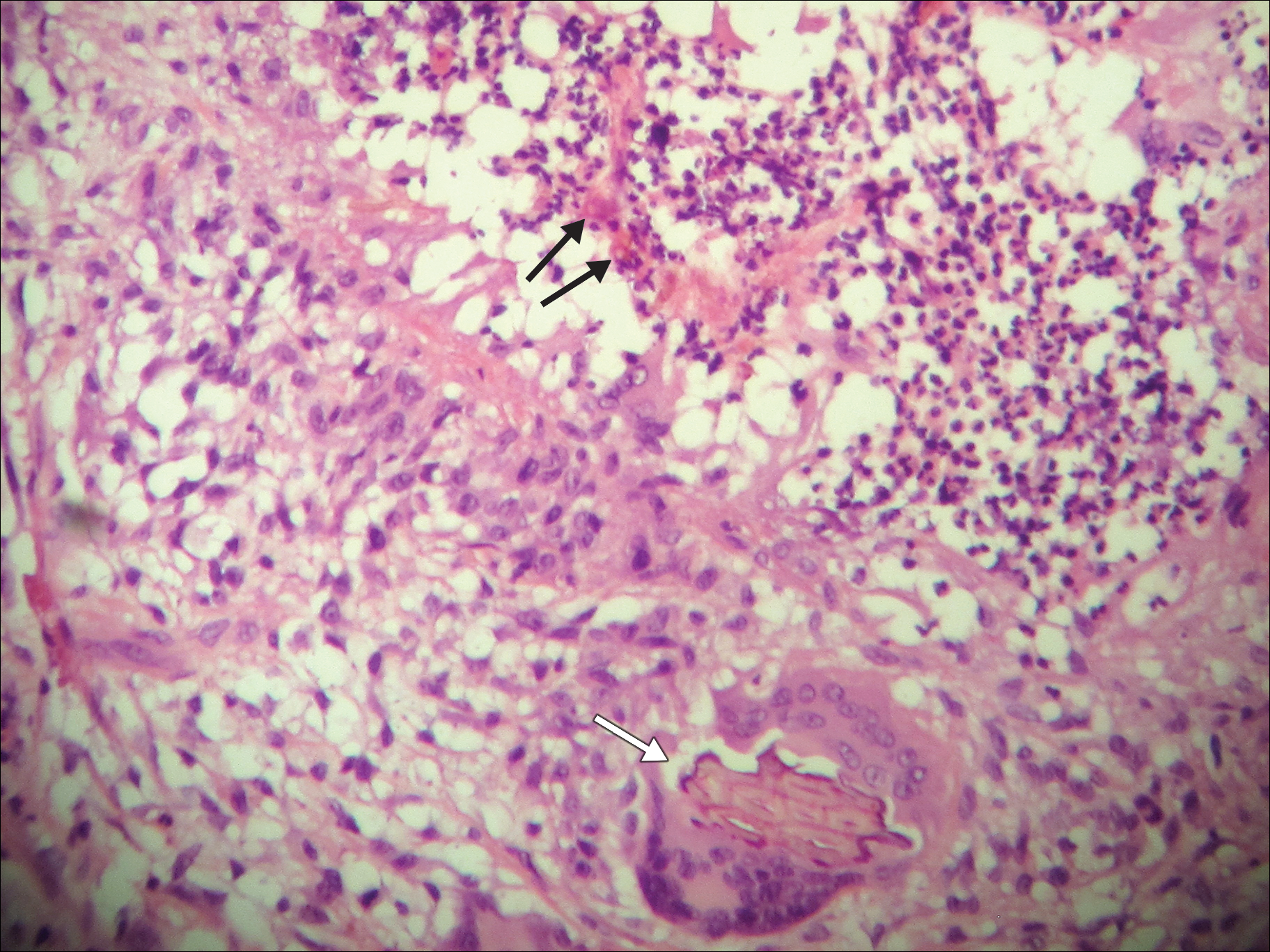
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
Practice Points
- Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
- Idiopathic sunburn purpura should only be diagnosed after vasculitis and/or thrombocytopenia is ruled out, which is best achieved through skin biopsy and a platelet count, respectively.
- The rash typically resolves without treatment in 1 to 2 weeks; however, a variety of UV protection modalities and education should be offered to the patient.
Levofloxacin-Induced Purpura Annularis Telangiectodes of Majocchi
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
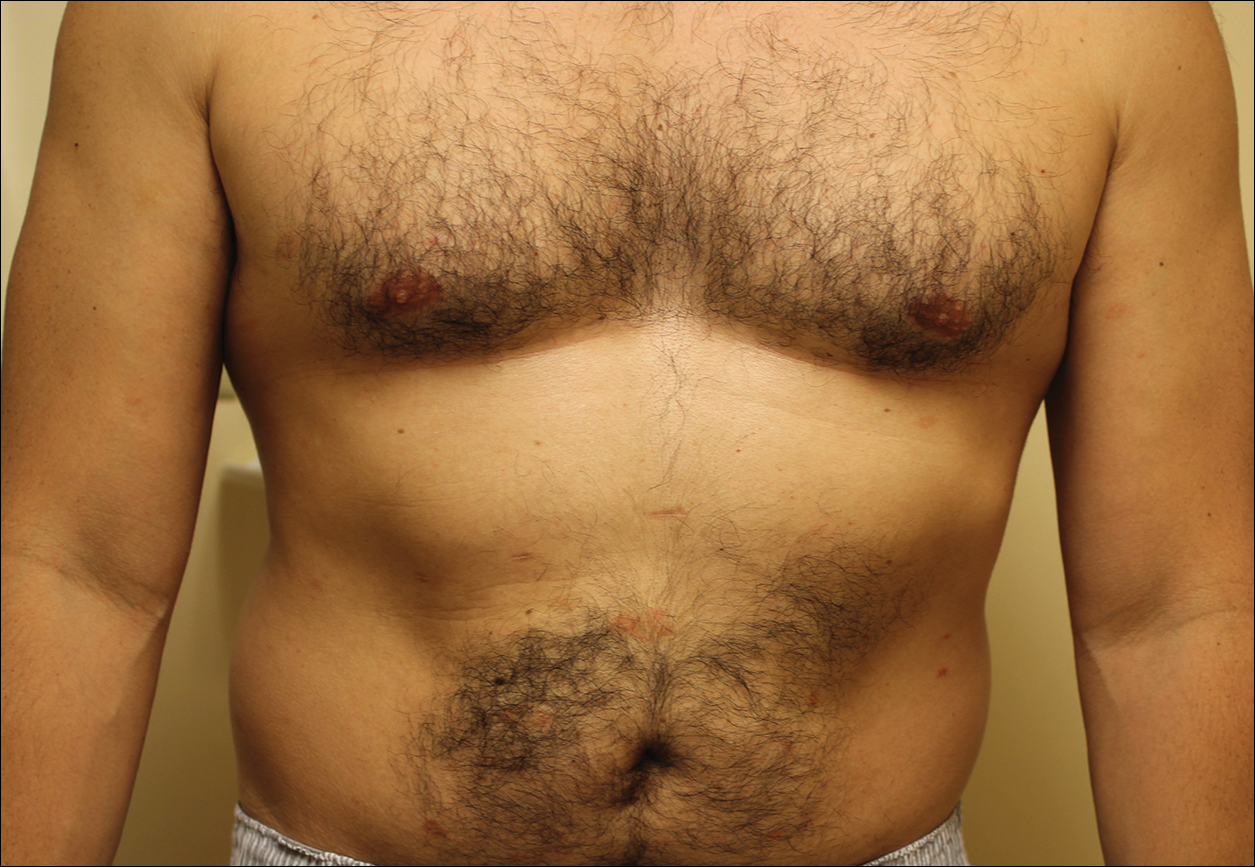
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
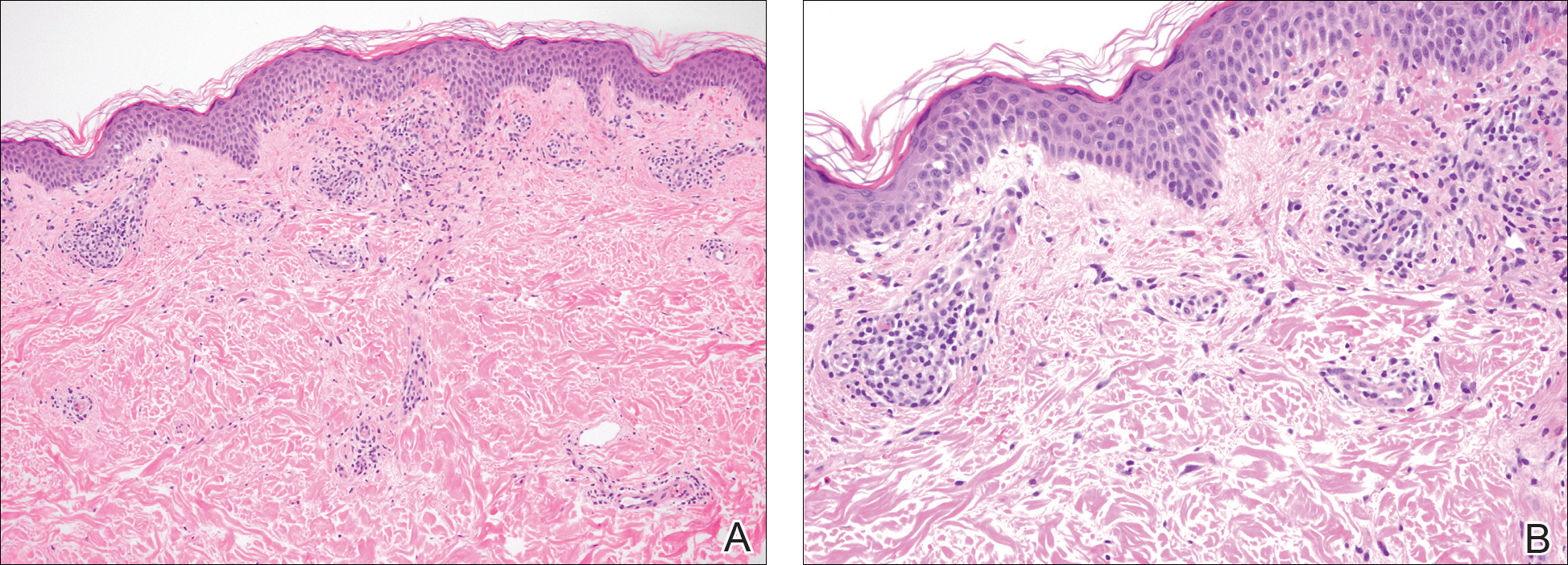
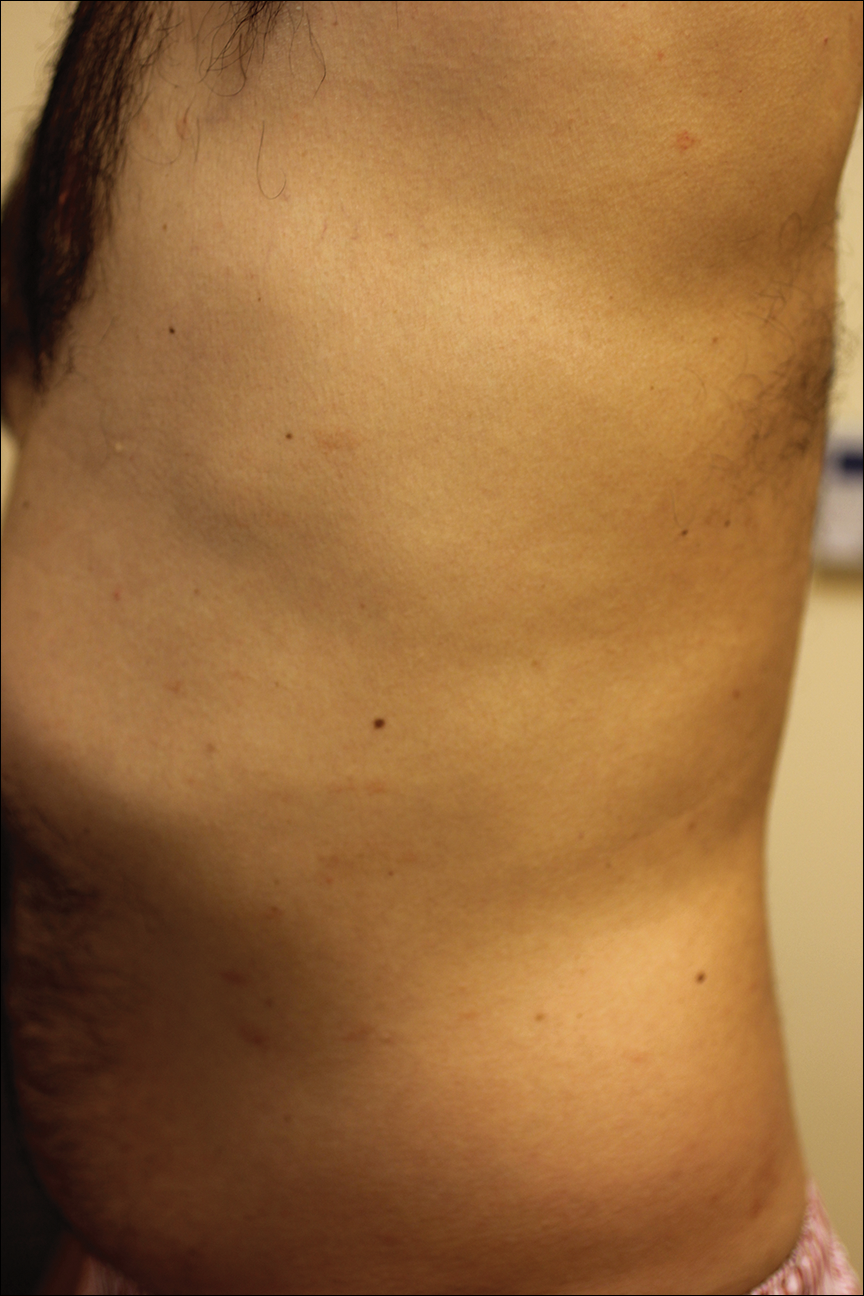
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
Practice Point
- Purpura annularis telangiectodes of Majocchi, a type of pigmented purpuric dermatosis, may on occasion be triggered by a medication; therefore, a careful medication history may prove to be an important part of the workup for this eruption.
Chromoblastomycosis Infection From a House Plant
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
Practice Points
- Chromoblastomycosis is an uncommon fungal infection that should be considered in cases of traumatic injuries to the skin.
- Biopsies of growing or nonhealing nodules will demonstrate characteristic golden brown spherules (medlar bodies).
- In localized cases, surgical excision may be curative.
Laugier-Hunziker Syndrome
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).

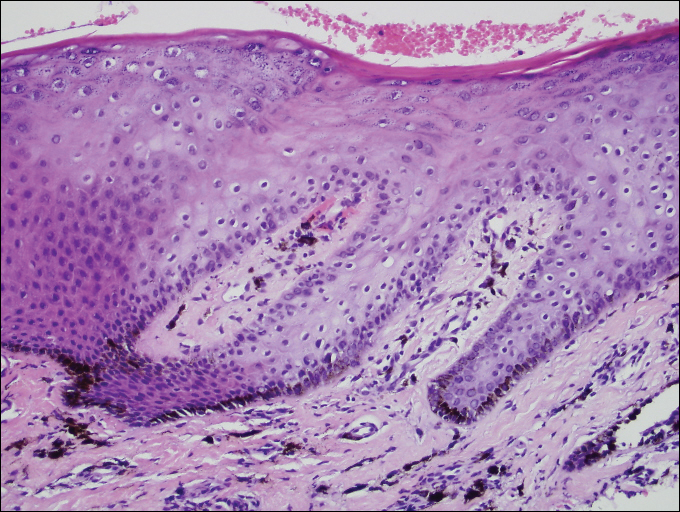
A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).
A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).
A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
Practice Points
- Laugier-Hunziker syndrome (LHS) comprises benign mucosal pigmentation in the absence of gastrointestinal pathology.
- Differentiating LHS from Peutz-Jeghers syndrome can prevent unnecessary aggressive cancer screening protocols.
- The average age of onset of LHS is 52 years and typically occurs in white adults.
- Pigmentation in LHS is most commonly distributed on the lower lips and oral mucosa.
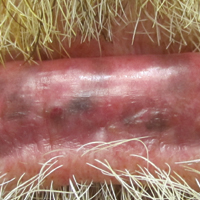
Pityriasis Rubra Pilaris and Severe Hypereosinophilia
To the Editor:
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
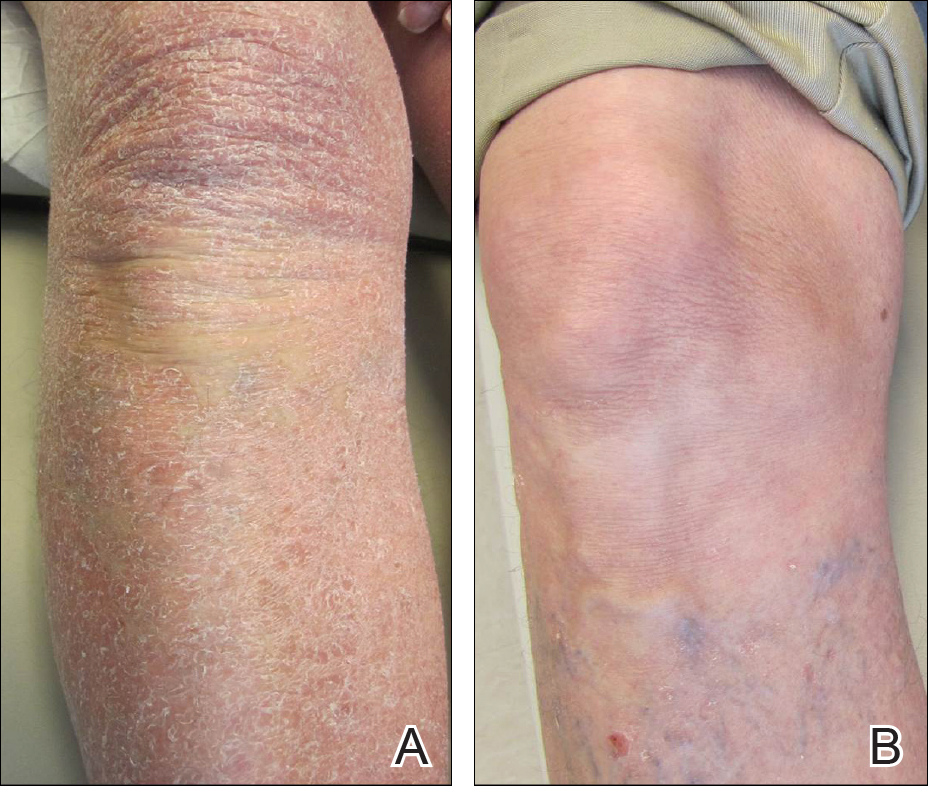
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.

The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
To the Editor:
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.
The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
To the Editor:
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.
The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
Practice Points
- Pityriasis rubra pilaris (PRP) can clinically mimic psoriasis. Look for islands of sparing and palmar and plantar hyperkeratosis to help diagnose PRP. A biopsy may be useful to help with this differentiation.
- Pityriasis rubra pilaris may be associated with eosinophilia, but one should rule out other causes of eosinophilia first.

Sporotrichoid Pattern of Mycobacterium chelonae-abscessus Infection
To the Editor:
We present a case of Mycobacterium chelonae-abscessus cutaneous infection in a sporotrichoid pattern, a rare presentation most often found in immunocompromised patients. A 34-year-old man with lupus nephritis who was taking oral prednisone, mycophenolate mofetil, and hydroxychloroquine presented with multiple erythematous fluctuant nodules and plaques on the left volar forearm in a sporotrichoid pattern of 3 months’ duration (Figure, A). He denied recent travel, exposure to fish or fish tanks, and penetrating wounds. Punch biopsy showed granulomatous inflammation and scarring with negative tissue cultures. Repeat biopsies and cultures were obtained when the lesions increased in number over 2 months.
Final biopsy showed upper dermal granulomatous inflammation with karyorrhectic debris, suggesting infection, and acid-fast bacilli. Culture grew M chelonae-abscessus on Löwenstein-Jensen agar at 37°C and blood culture media from which the complex was identified using high-performance liquid chromatography. Empiric therapy with renal dosing based on the Infectious Diseases Society of America statement of susceptibilities1 was initiated with clarithromycin, doxycycline, and ciprofloxacin for 4 months. Furthermore, the prednisone dose was tapered to 7.5 mg daily. Two months later, the lesions regressed and ciprofloxacin was discontinued (Figure, B).
The sporotrichoid spread of nodules suggests infection with mycobacteria, Sporothrix schenckii, Leishmania, Francisella tularensis, or Nocardia. Most cultures for nontuberculous mycobacteria will grow on Löwenstein-Jensen agar between 28°C and 37°C. Runyon rapidly growing (group IV) mycobacteria are defined by their ubiquitous presence in the environment and ability to develop colonies in 7 days.2 Cutaneous infections are increasing in prevalence, as reported in a retrospective study spanning nearly 30 years.3 The presentation is variable but often includes the distal extremities and usually is a nodule, ulcer, or abscess at a single site; a sporotrichoid pattern is more rare. Preceding skin trauma is the major risk factor for immunocompetent hosts, and the infection can spontaneously resolve in 8 to 12 months.1 In contrast, immunosuppressed patients may have no known source of infection and often have a progressive course with an increasing number of lesions and increased time until clearance.4
It is difficult to differentiate M chelonae and M abscessus based on growth characteristics, and they share the same 16S ribosomal RNA sequence commonly used to differentiate other mycobacterial species.2 Mycobacterium abscessus can be more difficult to treat, thus distinction via polymerase chain reaction of the heat-shock protein 65 gene, hsp65, can be valuable in cases recalcitrant to initial therapy.1
The likelihood of M chelonae and M abscessus isolates to be initially sensitive to clarithromycin is 100%,1 and this antibiotic remains the cornerstone of therapy. A clinical trial of treatments for M chelonae-abscessus found that clarithromycin monotherapy can be successful or complicated by resistance5; therefore, multidrug therapy is recommended. The antibiotic regimen for our patient was chosen to limit renal toxicity.
In summary, we report a case of M chelonae-abscessus cutaneous infection in a sporotrichoid pattern in a patient with lupus nephritis on immunosuppressive drugs. As the incidence of rapidly growing mycobacterial cutaneous infections rises, dermatologists must be aware of this pattern of infection.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- De Groote MA, Huitt G. Infections due to rapidly-growing Mycobacteria. Clin Infect Dis. 2006;42:1756-1763.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010:37:965-972.
- Wallace RJ, Tanner D, Brennan PJ, et al. Clinical trial of clarithromycin for cutaneous (disseminated) infection due to Mycobacterium chelonae. Ann Intern Med. 1993;119:482-486.
To the Editor:
We present a case of Mycobacterium chelonae-abscessus cutaneous infection in a sporotrichoid pattern, a rare presentation most often found in immunocompromised patients. A 34-year-old man with lupus nephritis who was taking oral prednisone, mycophenolate mofetil, and hydroxychloroquine presented with multiple erythematous fluctuant nodules and plaques on the left volar forearm in a sporotrichoid pattern of 3 months’ duration (Figure, A). He denied recent travel, exposure to fish or fish tanks, and penetrating wounds. Punch biopsy showed granulomatous inflammation and scarring with negative tissue cultures. Repeat biopsies and cultures were obtained when the lesions increased in number over 2 months.
Final biopsy showed upper dermal granulomatous inflammation with karyorrhectic debris, suggesting infection, and acid-fast bacilli. Culture grew M chelonae-abscessus on Löwenstein-Jensen agar at 37°C and blood culture media from which the complex was identified using high-performance liquid chromatography. Empiric therapy with renal dosing based on the Infectious Diseases Society of America statement of susceptibilities1 was initiated with clarithromycin, doxycycline, and ciprofloxacin for 4 months. Furthermore, the prednisone dose was tapered to 7.5 mg daily. Two months later, the lesions regressed and ciprofloxacin was discontinued (Figure, B).
The sporotrichoid spread of nodules suggests infection with mycobacteria, Sporothrix schenckii, Leishmania, Francisella tularensis, or Nocardia. Most cultures for nontuberculous mycobacteria will grow on Löwenstein-Jensen agar between 28°C and 37°C. Runyon rapidly growing (group IV) mycobacteria are defined by their ubiquitous presence in the environment and ability to develop colonies in 7 days.2 Cutaneous infections are increasing in prevalence, as reported in a retrospective study spanning nearly 30 years.3 The presentation is variable but often includes the distal extremities and usually is a nodule, ulcer, or abscess at a single site; a sporotrichoid pattern is more rare. Preceding skin trauma is the major risk factor for immunocompetent hosts, and the infection can spontaneously resolve in 8 to 12 months.1 In contrast, immunosuppressed patients may have no known source of infection and often have a progressive course with an increasing number of lesions and increased time until clearance.4
It is difficult to differentiate M chelonae and M abscessus based on growth characteristics, and they share the same 16S ribosomal RNA sequence commonly used to differentiate other mycobacterial species.2 Mycobacterium abscessus can be more difficult to treat, thus distinction via polymerase chain reaction of the heat-shock protein 65 gene, hsp65, can be valuable in cases recalcitrant to initial therapy.1
The likelihood of M chelonae and M abscessus isolates to be initially sensitive to clarithromycin is 100%,1 and this antibiotic remains the cornerstone of therapy. A clinical trial of treatments for M chelonae-abscessus found that clarithromycin monotherapy can be successful or complicated by resistance5; therefore, multidrug therapy is recommended. The antibiotic regimen for our patient was chosen to limit renal toxicity.
In summary, we report a case of M chelonae-abscessus cutaneous infection in a sporotrichoid pattern in a patient with lupus nephritis on immunosuppressive drugs. As the incidence of rapidly growing mycobacterial cutaneous infections rises, dermatologists must be aware of this pattern of infection.
To the Editor:
We present a case of Mycobacterium chelonae-abscessus cutaneous infection in a sporotrichoid pattern, a rare presentation most often found in immunocompromised patients. A 34-year-old man with lupus nephritis who was taking oral prednisone, mycophenolate mofetil, and hydroxychloroquine presented with multiple erythematous fluctuant nodules and plaques on the left volar forearm in a sporotrichoid pattern of 3 months’ duration (Figure, A). He denied recent travel, exposure to fish or fish tanks, and penetrating wounds. Punch biopsy showed granulomatous inflammation and scarring with negative tissue cultures. Repeat biopsies and cultures were obtained when the lesions increased in number over 2 months.
Final biopsy showed upper dermal granulomatous inflammation with karyorrhectic debris, suggesting infection, and acid-fast bacilli. Culture grew M chelonae-abscessus on Löwenstein-Jensen agar at 37°C and blood culture media from which the complex was identified using high-performance liquid chromatography. Empiric therapy with renal dosing based on the Infectious Diseases Society of America statement of susceptibilities1 was initiated with clarithromycin, doxycycline, and ciprofloxacin for 4 months. Furthermore, the prednisone dose was tapered to 7.5 mg daily. Two months later, the lesions regressed and ciprofloxacin was discontinued (Figure, B).
The sporotrichoid spread of nodules suggests infection with mycobacteria, Sporothrix schenckii, Leishmania, Francisella tularensis, or Nocardia. Most cultures for nontuberculous mycobacteria will grow on Löwenstein-Jensen agar between 28°C and 37°C. Runyon rapidly growing (group IV) mycobacteria are defined by their ubiquitous presence in the environment and ability to develop colonies in 7 days.2 Cutaneous infections are increasing in prevalence, as reported in a retrospective study spanning nearly 30 years.3 The presentation is variable but often includes the distal extremities and usually is a nodule, ulcer, or abscess at a single site; a sporotrichoid pattern is more rare. Preceding skin trauma is the major risk factor for immunocompetent hosts, and the infection can spontaneously resolve in 8 to 12 months.1 In contrast, immunosuppressed patients may have no known source of infection and often have a progressive course with an increasing number of lesions and increased time until clearance.4
It is difficult to differentiate M chelonae and M abscessus based on growth characteristics, and they share the same 16S ribosomal RNA sequence commonly used to differentiate other mycobacterial species.2 Mycobacterium abscessus can be more difficult to treat, thus distinction via polymerase chain reaction of the heat-shock protein 65 gene, hsp65, can be valuable in cases recalcitrant to initial therapy.1
The likelihood of M chelonae and M abscessus isolates to be initially sensitive to clarithromycin is 100%,1 and this antibiotic remains the cornerstone of therapy. A clinical trial of treatments for M chelonae-abscessus found that clarithromycin monotherapy can be successful or complicated by resistance5; therefore, multidrug therapy is recommended. The antibiotic regimen for our patient was chosen to limit renal toxicity.
In summary, we report a case of M chelonae-abscessus cutaneous infection in a sporotrichoid pattern in a patient with lupus nephritis on immunosuppressive drugs. As the incidence of rapidly growing mycobacterial cutaneous infections rises, dermatologists must be aware of this pattern of infection.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- De Groote MA, Huitt G. Infections due to rapidly-growing Mycobacteria. Clin Infect Dis. 2006;42:1756-1763.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010:37:965-972.
- Wallace RJ, Tanner D, Brennan PJ, et al. Clinical trial of clarithromycin for cutaneous (disseminated) infection due to Mycobacterium chelonae. Ann Intern Med. 1993;119:482-486.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- De Groote MA, Huitt G. Infections due to rapidly-growing Mycobacteria. Clin Infect Dis. 2006;42:1756-1763.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010:37:965-972.
- Wallace RJ, Tanner D, Brennan PJ, et al. Clinical trial of clarithromycin for cutaneous (disseminated) infection due to Mycobacterium chelonae. Ann Intern Med. 1993;119:482-486.
Practice Points
- Dermatologists should consider atypical mycobacterial infections, including rapidly growing mycobacteria, in the differential diagnosis for lesions with sporotrichoid-pattern spread.
- Multidrug therapy often is required for treatment of infection caused by Mycobacteria chelonae-abscessus complex.
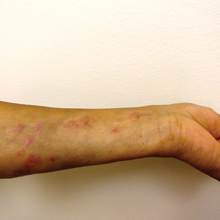
Stevens-Johnson Syndrome Secondary to Isolated Albuterol Use
To the Editor:
A 22-year-old obese man with untreated mild asthma diagnosed in childhood presented to the emergency department with cheilitis (Figure 1); conjunctivitis; and painful desquamation of the oral mucosa, penis (Figure 2), and perirectal area (Figure 3). Physical examination was notable for palpebral conjunctiva; mucosal involvement with stomatitis (Figure 1B); and isolated 0.5- to 2-cm erosions and ulcerations with positive Nikolsky sign of the scrotum (Figure 2), trunk, back, and arms and legs. Some areas had evidence of hemorrhagic crust, flaccid bullae, and denudation. Few scant targetoid lesions and dusky red macules on the trunk, face, palms, and soles also were present.
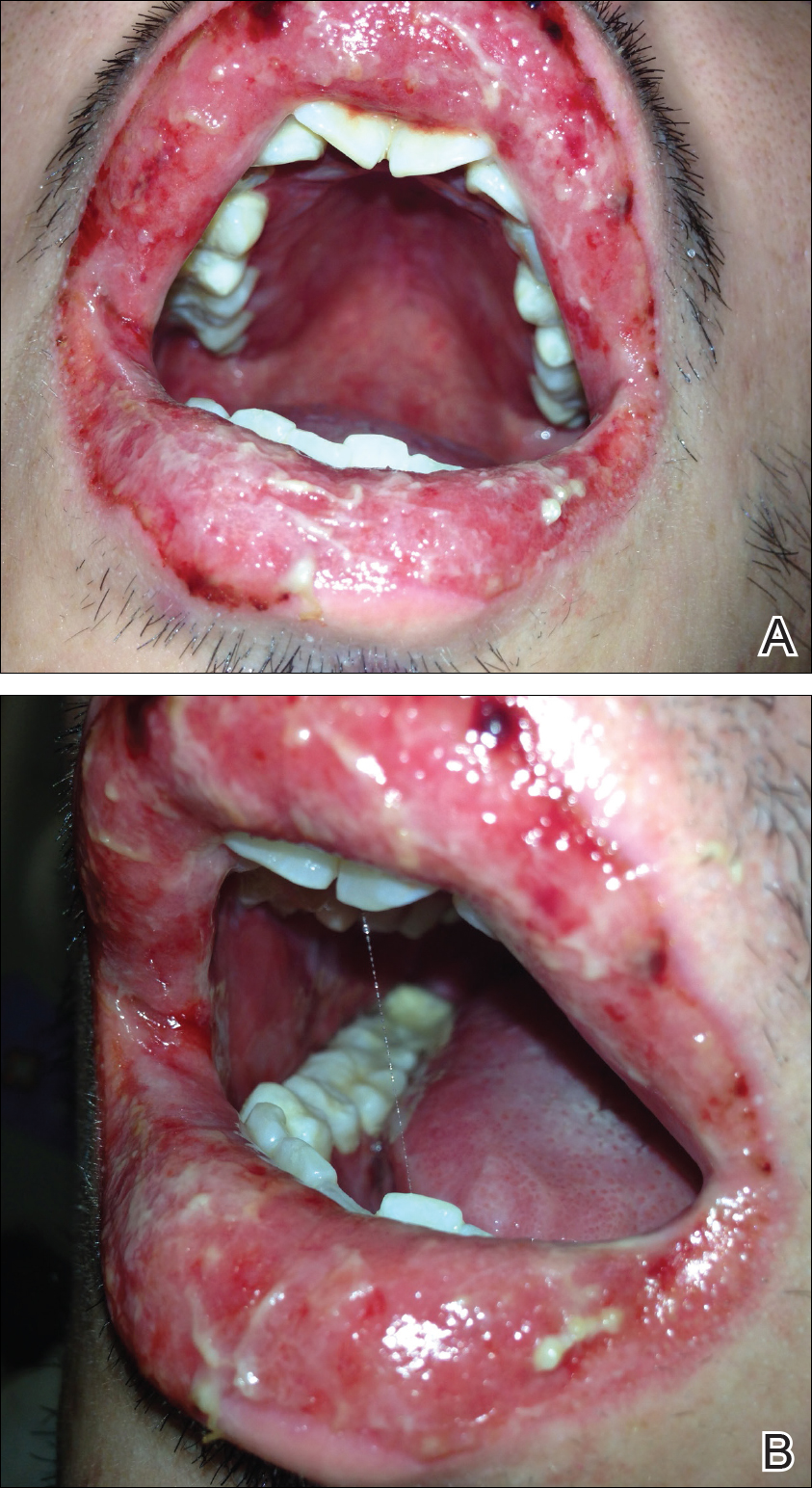

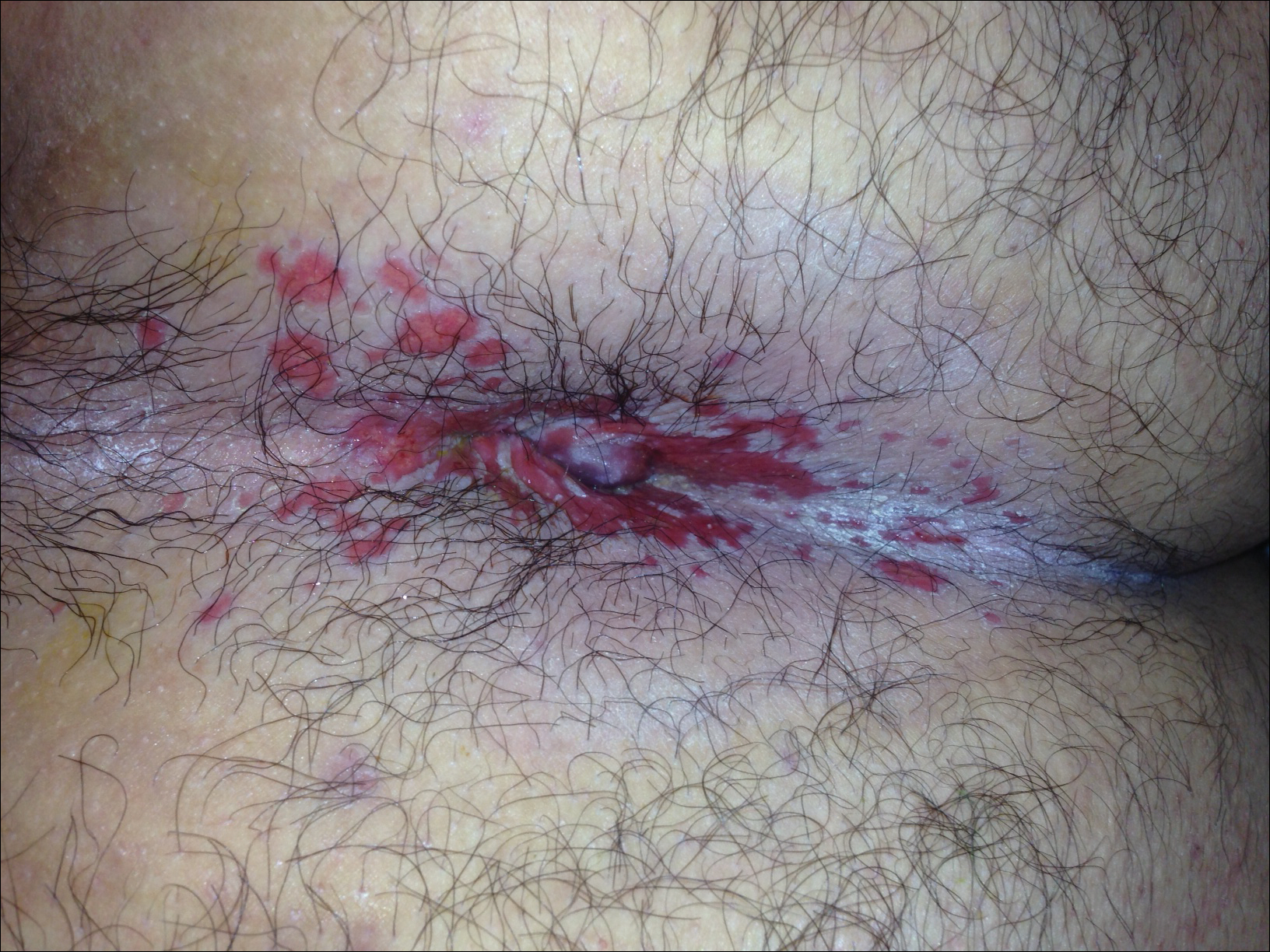
One week prior to presentation he had an episode of diarrhea and dyspnea with symptoms of mild heat stroke after working outdoors, and he self-treated with ibuprofen, which he had taken intermittently for years. He was subsequently seen at an outpatient clinic and was prescribed an albuterol inhaler for previously untreated childhood asthma. The patient stated that he inhaled 2 puffs every 6 hours for a total of 3 treatments. Shortly after the last dose, he noticed a tingling sensation of the oral mucosa that developed into a painful 2-cm bullous ulcer. Over the next 3 days, he developed several more oral ulcers and erosions. Three days before admission he developed dysuria and tense bullae at the glans penis. After admission, he developed cheilitis, conjunctivitis, dysuria, odynophagia, and dysphagia to solids. One day after admission, the patient had the onset of systemic symptoms, including cough with worsening dyspnea, fever, chills, hemoptysis, epistaxis, nausea, diarrhea, loss of appetite, joint pain, and myalgia. Review of systems was otherwise negative. A radiograph was performed at admission and was notable for mild atelectasis but was otherwise normal. The chest radiograph was negative for signs of perihilar lymphadenopathy, pleural effusion, pneumothorax, or lobar infiltrates suggestive of bacterial pneumonia. It also did not show signs of patchy opacities or air bronchograms suggestive of an interstitial pneumonia. On admission, he was started on acyclovir, fluconazole, methylprednisolone, nystatin, pantoprazole, acetaminophen, topical bacitracin, oxycodone, and topical silver nitrate.
At the time of admission our patient was afebrile with a normal heart rate, blood pressure, and respiratory rate. However, he was hypoxic, with a pulse oximetry of 86% on room air and 94% on 40% fraction of inspired oxygen. Complete blood cell count, electrolytes, and liver function tests were all within reference range. Urinalysis revealed evidence of scant red blood cells without pyuria, and the erythrocyte sedimentation rate and creatine kinase level were both elevated. Two blood cultures; sputum cultures; and polymerase chain reaction for Mycoplasma pneumoniae, herpes simplex virus, varicella-zoster virus, cytomegalovirus, and Epstein-Barr virus were negative. Human immunodeficiency virus panel, antinuclear antibody screen, and hepatitis B and C panels were all negative. Four punch biopsies were obtained showing full-thickness epidermal necrosis with neutrophils, few dyskeratotic cells, and sparse inflammatory infiltrate compatible with Stevens-Johnson syndrome (SJS).
After hospital admission, the patient’s mucosal desquamation progressively improved. By day 3, he required minimal supplemental oxygen with resolution of bowel symptoms and improved mucosal and skin findings. He was discharged on day 4 with supplemental oxygen and a 7-day course of prednisone, fluconazole, liquid oxycodone, pantoprazole, and acetaminophen. He showed continued improvement at a follow-up outpatient visit 2 days following discharge.
Stevens-Johnson syndrome is a rare severe drug reaction characterized by high fevers, mucosal erosions, tenderness, and skin detachment approximately 1 to 3 weeks after an inciting event.1,2 Although SJS has been linked to infections and less commonly to immunizations, in more than 80% of cases, SJS is strongly associated with a recent medication change.3 The classes of drugs that have been implicated in SJS most commonly include antibiotics, anticonvulsants, and nonsteroidal anti-inflammatory drugs.4 Stevens-Johnson syndrome from drug reactions is not uncommon; however, SJS secondary to isolated albuterol use is rare.
Although it is presumed that albuterol was the key inciting factor in our patient’s case of SJS, it also is recognized that mucosal SJS can be associated with M pneumoniae infection. For this reason, we performed polymerase chain reaction for M pneumoniae as well as a chest radiograph to rule out this possibility. In addition, our patient had denied prolonged respiratory symptoms suggestive of a mycoplasma pneumonia infection, such as a prodrome of cough, myalgia, headache, sore throat, or fever. A report of 8 patients with documented SJS and M pneumoniae as well as a review of the literature also demonstrated a mean of 10 days of prodromal symptoms prior to the onset of mucosal lesions and/or a rash.5 However, mucosal SJS associated with mycoplasma pneumonia is an important clinical entity that should not be forgotten during the workup of a young patient with mucosal lesions or rash suggestive of SJS.
The exact etiology and mechanism of drug-induced SJS is not well understood at this time; however, evidence suggests that SJS is strongly linked to the host’s inability to detoxify drug metabolites.6,7 It has been postulated that SJS occurs secondary to a cell-mediated immune response, which activates cytotoxic T lymphocytes and subsequently results in keratinocyte apoptosis. Keratinocyte apoptosis occurs via the CD95-CD95 death receptor and soluble or membrane-bound ligand interaction.3,8,9 Stevens-Johnson syndrome is thought to occur from an interaction involving an HLA antigen–restricted presentation of drug metabolites to cytotoxic T cells, which can be further supported by evidence of strong genetic associations with HLA antigen alleles B15.02 and B58.01 in the cases of carbamazepine- and allopurinol-induced SJS, respectively.6,7 However, the genetic associations of specific HLA antigen alleles and polymorphisms with SJS and other cutaneous reactions is thought to be drug specific and HLA antigen subtype specific.7 Therefore, it is difficult to determine or correlate the clinical outcomes and manifestations of drug reactions in individualized patients. The precise mechanism of antigenicity of albuterol in initiating this cascade has not yet been determined. However, these investigations provide strong evidence for a correlation between specific HLA antigen haplotypes and occurrence of drug antigenicity resulting in SJS and other cutaneous reactions in susceptible patient populations.
Although the specific molecular pathway and etiology of SJS is not well delineated, pathology in combination with clinical correlation allows for diagnosis. Early-stage biopsies in SJS typically show apoptotic keratinocytes throughout the epidermis. Late-stage biopsies exhibit subepidermal blisters and full-thickness epidermal necrosis.1 Histopathology was performed on 4-mm punch biopsies of the chest and back and demonstrated full-thickness epidermal necrosis with neutrophils and a few dyskeratotic cells, likely representing a late stage of epidermal involvement. Given the predominance of neutrophils, other diagnostic considerations based solely on the biopsy results included contact dermatitis or phototoxic dermatitis. The remaining inflammatory infiltrate was sparse. Immunofluorescence was pan-negative.
This report illustrates a rare case of SJS from isolated albuterol use. This adverse drug reaction has not been well reported in the literature and may be an important consideration in the management of a patient with asthma.
- Stern RS. Clinical practice. exanthematous drug eruptions. N Engl J Med. 2012;366:2492-2501.
- Tartarone A, Lerose R. Stevens-Johnson syndrome and toxic epidermal necrolysis: what do we know? Ther Drug Monit. 2010;32:669-672.
- Ferrandiz-Pulido C, Garcia-Patos V. A review of causes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Arch Dis Child. 2013;98:998-1003.
- Mockenhaupt M, Viboud C, Dunant A, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. the EuroSCAR-study. J Invest Dermatol. 2008;128:35-44.
- Levy M, Shear NH. Mycoplasma pneumoniae infections and Stevens-Johnson syndrome. report of eight cases and review of the literature. Clin Pediatr (Phila). 1991;30:42-49.
- Chung WH, Hung SI. Genetic markers and danger signals in Stevens-Johnson syndrome and toxic epidermal necrolysis [published online October 25, 2010]. Allergol Int. 2010;59:325-332.
- Chung WH, Hung SI. Recent advances in the genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci. 2012;66:190-196.
- Bharadwaj M, Illing P, Theodossis A, et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol. 2012;52:401-431.
- Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822-832.
To the Editor:
A 22-year-old obese man with untreated mild asthma diagnosed in childhood presented to the emergency department with cheilitis (Figure 1); conjunctivitis; and painful desquamation of the oral mucosa, penis (Figure 2), and perirectal area (Figure 3). Physical examination was notable for palpebral conjunctiva; mucosal involvement with stomatitis (Figure 1B); and isolated 0.5- to 2-cm erosions and ulcerations with positive Nikolsky sign of the scrotum (Figure 2), trunk, back, and arms and legs. Some areas had evidence of hemorrhagic crust, flaccid bullae, and denudation. Few scant targetoid lesions and dusky red macules on the trunk, face, palms, and soles also were present.
One week prior to presentation he had an episode of diarrhea and dyspnea with symptoms of mild heat stroke after working outdoors, and he self-treated with ibuprofen, which he had taken intermittently for years. He was subsequently seen at an outpatient clinic and was prescribed an albuterol inhaler for previously untreated childhood asthma. The patient stated that he inhaled 2 puffs every 6 hours for a total of 3 treatments. Shortly after the last dose, he noticed a tingling sensation of the oral mucosa that developed into a painful 2-cm bullous ulcer. Over the next 3 days, he developed several more oral ulcers and erosions. Three days before admission he developed dysuria and tense bullae at the glans penis. After admission, he developed cheilitis, conjunctivitis, dysuria, odynophagia, and dysphagia to solids. One day after admission, the patient had the onset of systemic symptoms, including cough with worsening dyspnea, fever, chills, hemoptysis, epistaxis, nausea, diarrhea, loss of appetite, joint pain, and myalgia. Review of systems was otherwise negative. A radiograph was performed at admission and was notable for mild atelectasis but was otherwise normal. The chest radiograph was negative for signs of perihilar lymphadenopathy, pleural effusion, pneumothorax, or lobar infiltrates suggestive of bacterial pneumonia. It also did not show signs of patchy opacities or air bronchograms suggestive of an interstitial pneumonia. On admission, he was started on acyclovir, fluconazole, methylprednisolone, nystatin, pantoprazole, acetaminophen, topical bacitracin, oxycodone, and topical silver nitrate.
At the time of admission our patient was afebrile with a normal heart rate, blood pressure, and respiratory rate. However, he was hypoxic, with a pulse oximetry of 86% on room air and 94% on 40% fraction of inspired oxygen. Complete blood cell count, electrolytes, and liver function tests were all within reference range. Urinalysis revealed evidence of scant red blood cells without pyuria, and the erythrocyte sedimentation rate and creatine kinase level were both elevated. Two blood cultures; sputum cultures; and polymerase chain reaction for Mycoplasma pneumoniae, herpes simplex virus, varicella-zoster virus, cytomegalovirus, and Epstein-Barr virus were negative. Human immunodeficiency virus panel, antinuclear antibody screen, and hepatitis B and C panels were all negative. Four punch biopsies were obtained showing full-thickness epidermal necrosis with neutrophils, few dyskeratotic cells, and sparse inflammatory infiltrate compatible with Stevens-Johnson syndrome (SJS).
After hospital admission, the patient’s mucosal desquamation progressively improved. By day 3, he required minimal supplemental oxygen with resolution of bowel symptoms and improved mucosal and skin findings. He was discharged on day 4 with supplemental oxygen and a 7-day course of prednisone, fluconazole, liquid oxycodone, pantoprazole, and acetaminophen. He showed continued improvement at a follow-up outpatient visit 2 days following discharge.
Stevens-Johnson syndrome is a rare severe drug reaction characterized by high fevers, mucosal erosions, tenderness, and skin detachment approximately 1 to 3 weeks after an inciting event.1,2 Although SJS has been linked to infections and less commonly to immunizations, in more than 80% of cases, SJS is strongly associated with a recent medication change.3 The classes of drugs that have been implicated in SJS most commonly include antibiotics, anticonvulsants, and nonsteroidal anti-inflammatory drugs.4 Stevens-Johnson syndrome from drug reactions is not uncommon; however, SJS secondary to isolated albuterol use is rare.
Although it is presumed that albuterol was the key inciting factor in our patient’s case of SJS, it also is recognized that mucosal SJS can be associated with M pneumoniae infection. For this reason, we performed polymerase chain reaction for M pneumoniae as well as a chest radiograph to rule out this possibility. In addition, our patient had denied prolonged respiratory symptoms suggestive of a mycoplasma pneumonia infection, such as a prodrome of cough, myalgia, headache, sore throat, or fever. A report of 8 patients with documented SJS and M pneumoniae as well as a review of the literature also demonstrated a mean of 10 days of prodromal symptoms prior to the onset of mucosal lesions and/or a rash.5 However, mucosal SJS associated with mycoplasma pneumonia is an important clinical entity that should not be forgotten during the workup of a young patient with mucosal lesions or rash suggestive of SJS.
The exact etiology and mechanism of drug-induced SJS is not well understood at this time; however, evidence suggests that SJS is strongly linked to the host’s inability to detoxify drug metabolites.6,7 It has been postulated that SJS occurs secondary to a cell-mediated immune response, which activates cytotoxic T lymphocytes and subsequently results in keratinocyte apoptosis. Keratinocyte apoptosis occurs via the CD95-CD95 death receptor and soluble or membrane-bound ligand interaction.3,8,9 Stevens-Johnson syndrome is thought to occur from an interaction involving an HLA antigen–restricted presentation of drug metabolites to cytotoxic T cells, which can be further supported by evidence of strong genetic associations with HLA antigen alleles B15.02 and B58.01 in the cases of carbamazepine- and allopurinol-induced SJS, respectively.6,7 However, the genetic associations of specific HLA antigen alleles and polymorphisms with SJS and other cutaneous reactions is thought to be drug specific and HLA antigen subtype specific.7 Therefore, it is difficult to determine or correlate the clinical outcomes and manifestations of drug reactions in individualized patients. The precise mechanism of antigenicity of albuterol in initiating this cascade has not yet been determined. However, these investigations provide strong evidence for a correlation between specific HLA antigen haplotypes and occurrence of drug antigenicity resulting in SJS and other cutaneous reactions in susceptible patient populations.
Although the specific molecular pathway and etiology of SJS is not well delineated, pathology in combination with clinical correlation allows for diagnosis. Early-stage biopsies in SJS typically show apoptotic keratinocytes throughout the epidermis. Late-stage biopsies exhibit subepidermal blisters and full-thickness epidermal necrosis.1 Histopathology was performed on 4-mm punch biopsies of the chest and back and demonstrated full-thickness epidermal necrosis with neutrophils and a few dyskeratotic cells, likely representing a late stage of epidermal involvement. Given the predominance of neutrophils, other diagnostic considerations based solely on the biopsy results included contact dermatitis or phototoxic dermatitis. The remaining inflammatory infiltrate was sparse. Immunofluorescence was pan-negative.
This report illustrates a rare case of SJS from isolated albuterol use. This adverse drug reaction has not been well reported in the literature and may be an important consideration in the management of a patient with asthma.
To the Editor:
A 22-year-old obese man with untreated mild asthma diagnosed in childhood presented to the emergency department with cheilitis (Figure 1); conjunctivitis; and painful desquamation of the oral mucosa, penis (Figure 2), and perirectal area (Figure 3). Physical examination was notable for palpebral conjunctiva; mucosal involvement with stomatitis (Figure 1B); and isolated 0.5- to 2-cm erosions and ulcerations with positive Nikolsky sign of the scrotum (Figure 2), trunk, back, and arms and legs. Some areas had evidence of hemorrhagic crust, flaccid bullae, and denudation. Few scant targetoid lesions and dusky red macules on the trunk, face, palms, and soles also were present.
One week prior to presentation he had an episode of diarrhea and dyspnea with symptoms of mild heat stroke after working outdoors, and he self-treated with ibuprofen, which he had taken intermittently for years. He was subsequently seen at an outpatient clinic and was prescribed an albuterol inhaler for previously untreated childhood asthma. The patient stated that he inhaled 2 puffs every 6 hours for a total of 3 treatments. Shortly after the last dose, he noticed a tingling sensation of the oral mucosa that developed into a painful 2-cm bullous ulcer. Over the next 3 days, he developed several more oral ulcers and erosions. Three days before admission he developed dysuria and tense bullae at the glans penis. After admission, he developed cheilitis, conjunctivitis, dysuria, odynophagia, and dysphagia to solids. One day after admission, the patient had the onset of systemic symptoms, including cough with worsening dyspnea, fever, chills, hemoptysis, epistaxis, nausea, diarrhea, loss of appetite, joint pain, and myalgia. Review of systems was otherwise negative. A radiograph was performed at admission and was notable for mild atelectasis but was otherwise normal. The chest radiograph was negative for signs of perihilar lymphadenopathy, pleural effusion, pneumothorax, or lobar infiltrates suggestive of bacterial pneumonia. It also did not show signs of patchy opacities or air bronchograms suggestive of an interstitial pneumonia. On admission, he was started on acyclovir, fluconazole, methylprednisolone, nystatin, pantoprazole, acetaminophen, topical bacitracin, oxycodone, and topical silver nitrate.
At the time of admission our patient was afebrile with a normal heart rate, blood pressure, and respiratory rate. However, he was hypoxic, with a pulse oximetry of 86% on room air and 94% on 40% fraction of inspired oxygen. Complete blood cell count, electrolytes, and liver function tests were all within reference range. Urinalysis revealed evidence of scant red blood cells without pyuria, and the erythrocyte sedimentation rate and creatine kinase level were both elevated. Two blood cultures; sputum cultures; and polymerase chain reaction for Mycoplasma pneumoniae, herpes simplex virus, varicella-zoster virus, cytomegalovirus, and Epstein-Barr virus were negative. Human immunodeficiency virus panel, antinuclear antibody screen, and hepatitis B and C panels were all negative. Four punch biopsies were obtained showing full-thickness epidermal necrosis with neutrophils, few dyskeratotic cells, and sparse inflammatory infiltrate compatible with Stevens-Johnson syndrome (SJS).
After hospital admission, the patient’s mucosal desquamation progressively improved. By day 3, he required minimal supplemental oxygen with resolution of bowel symptoms and improved mucosal and skin findings. He was discharged on day 4 with supplemental oxygen and a 7-day course of prednisone, fluconazole, liquid oxycodone, pantoprazole, and acetaminophen. He showed continued improvement at a follow-up outpatient visit 2 days following discharge.
Stevens-Johnson syndrome is a rare severe drug reaction characterized by high fevers, mucosal erosions, tenderness, and skin detachment approximately 1 to 3 weeks after an inciting event.1,2 Although SJS has been linked to infections and less commonly to immunizations, in more than 80% of cases, SJS is strongly associated with a recent medication change.3 The classes of drugs that have been implicated in SJS most commonly include antibiotics, anticonvulsants, and nonsteroidal anti-inflammatory drugs.4 Stevens-Johnson syndrome from drug reactions is not uncommon; however, SJS secondary to isolated albuterol use is rare.
Although it is presumed that albuterol was the key inciting factor in our patient’s case of SJS, it also is recognized that mucosal SJS can be associated with M pneumoniae infection. For this reason, we performed polymerase chain reaction for M pneumoniae as well as a chest radiograph to rule out this possibility. In addition, our patient had denied prolonged respiratory symptoms suggestive of a mycoplasma pneumonia infection, such as a prodrome of cough, myalgia, headache, sore throat, or fever. A report of 8 patients with documented SJS and M pneumoniae as well as a review of the literature also demonstrated a mean of 10 days of prodromal symptoms prior to the onset of mucosal lesions and/or a rash.5 However, mucosal SJS associated with mycoplasma pneumonia is an important clinical entity that should not be forgotten during the workup of a young patient with mucosal lesions or rash suggestive of SJS.
The exact etiology and mechanism of drug-induced SJS is not well understood at this time; however, evidence suggests that SJS is strongly linked to the host’s inability to detoxify drug metabolites.6,7 It has been postulated that SJS occurs secondary to a cell-mediated immune response, which activates cytotoxic T lymphocytes and subsequently results in keratinocyte apoptosis. Keratinocyte apoptosis occurs via the CD95-CD95 death receptor and soluble or membrane-bound ligand interaction.3,8,9 Stevens-Johnson syndrome is thought to occur from an interaction involving an HLA antigen–restricted presentation of drug metabolites to cytotoxic T cells, which can be further supported by evidence of strong genetic associations with HLA antigen alleles B15.02 and B58.01 in the cases of carbamazepine- and allopurinol-induced SJS, respectively.6,7 However, the genetic associations of specific HLA antigen alleles and polymorphisms with SJS and other cutaneous reactions is thought to be drug specific and HLA antigen subtype specific.7 Therefore, it is difficult to determine or correlate the clinical outcomes and manifestations of drug reactions in individualized patients. The precise mechanism of antigenicity of albuterol in initiating this cascade has not yet been determined. However, these investigations provide strong evidence for a correlation between specific HLA antigen haplotypes and occurrence of drug antigenicity resulting in SJS and other cutaneous reactions in susceptible patient populations.
Although the specific molecular pathway and etiology of SJS is not well delineated, pathology in combination with clinical correlation allows for diagnosis. Early-stage biopsies in SJS typically show apoptotic keratinocytes throughout the epidermis. Late-stage biopsies exhibit subepidermal blisters and full-thickness epidermal necrosis.1 Histopathology was performed on 4-mm punch biopsies of the chest and back and demonstrated full-thickness epidermal necrosis with neutrophils and a few dyskeratotic cells, likely representing a late stage of epidermal involvement. Given the predominance of neutrophils, other diagnostic considerations based solely on the biopsy results included contact dermatitis or phototoxic dermatitis. The remaining inflammatory infiltrate was sparse. Immunofluorescence was pan-negative.
This report illustrates a rare case of SJS from isolated albuterol use. This adverse drug reaction has not been well reported in the literature and may be an important consideration in the management of a patient with asthma.
- Stern RS. Clinical practice. exanthematous drug eruptions. N Engl J Med. 2012;366:2492-2501.
- Tartarone A, Lerose R. Stevens-Johnson syndrome and toxic epidermal necrolysis: what do we know? Ther Drug Monit. 2010;32:669-672.
- Ferrandiz-Pulido C, Garcia-Patos V. A review of causes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Arch Dis Child. 2013;98:998-1003.
- Mockenhaupt M, Viboud C, Dunant A, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. the EuroSCAR-study. J Invest Dermatol. 2008;128:35-44.
- Levy M, Shear NH. Mycoplasma pneumoniae infections and Stevens-Johnson syndrome. report of eight cases and review of the literature. Clin Pediatr (Phila). 1991;30:42-49.
- Chung WH, Hung SI. Genetic markers and danger signals in Stevens-Johnson syndrome and toxic epidermal necrolysis [published online October 25, 2010]. Allergol Int. 2010;59:325-332.
- Chung WH, Hung SI. Recent advances in the genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci. 2012;66:190-196.
- Bharadwaj M, Illing P, Theodossis A, et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol. 2012;52:401-431.
- Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822-832.
- Stern RS. Clinical practice. exanthematous drug eruptions. N Engl J Med. 2012;366:2492-2501.
- Tartarone A, Lerose R. Stevens-Johnson syndrome and toxic epidermal necrolysis: what do we know? Ther Drug Monit. 2010;32:669-672.
- Ferrandiz-Pulido C, Garcia-Patos V. A review of causes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Arch Dis Child. 2013;98:998-1003.
- Mockenhaupt M, Viboud C, Dunant A, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. the EuroSCAR-study. J Invest Dermatol. 2008;128:35-44.
- Levy M, Shear NH. Mycoplasma pneumoniae infections and Stevens-Johnson syndrome. report of eight cases and review of the literature. Clin Pediatr (Phila). 1991;30:42-49.
- Chung WH, Hung SI. Genetic markers and danger signals in Stevens-Johnson syndrome and toxic epidermal necrolysis [published online October 25, 2010]. Allergol Int. 2010;59:325-332.
- Chung WH, Hung SI. Recent advances in the genetics and immunology of Stevens-Johnson syndrome and toxic epidermal necrosis. J Dermatol Sci. 2012;66:190-196.
- Bharadwaj M, Illing P, Theodossis A, et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol. 2012;52:401-431.
- Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822-832.
Practice Points
- Think of Stevens-Johnson syndrome when new skin lesions are seen after any new medication is started.
- Perform a full-body examination to assess the extent of skin eruptions.
- When a medication is atypical for skin eruption, it becomes necessary to assess for other systemic causes and confirm pathologic results on skin biopsy.

Space Heater–Induced Bullous Erythema Ab Igne
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
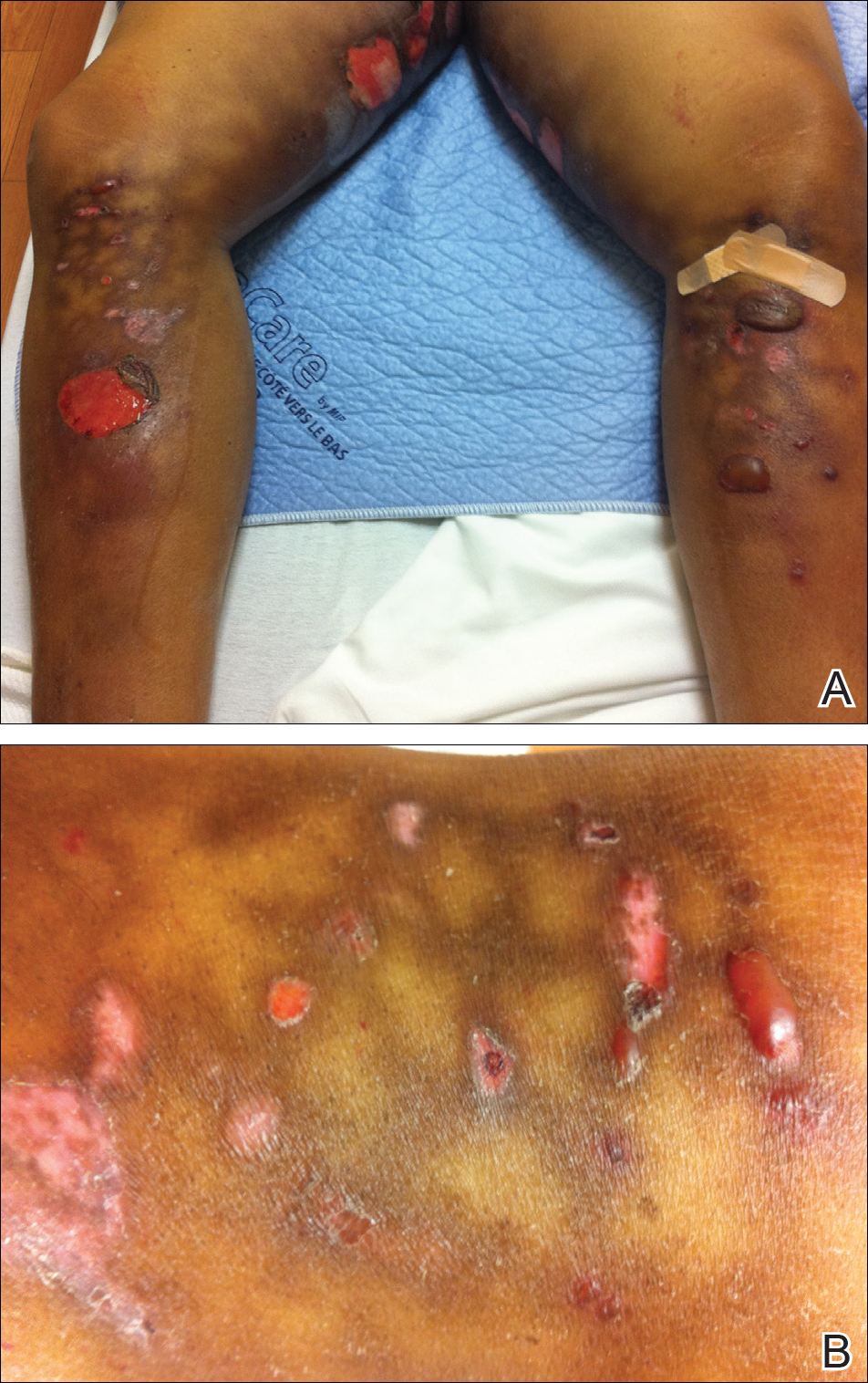
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
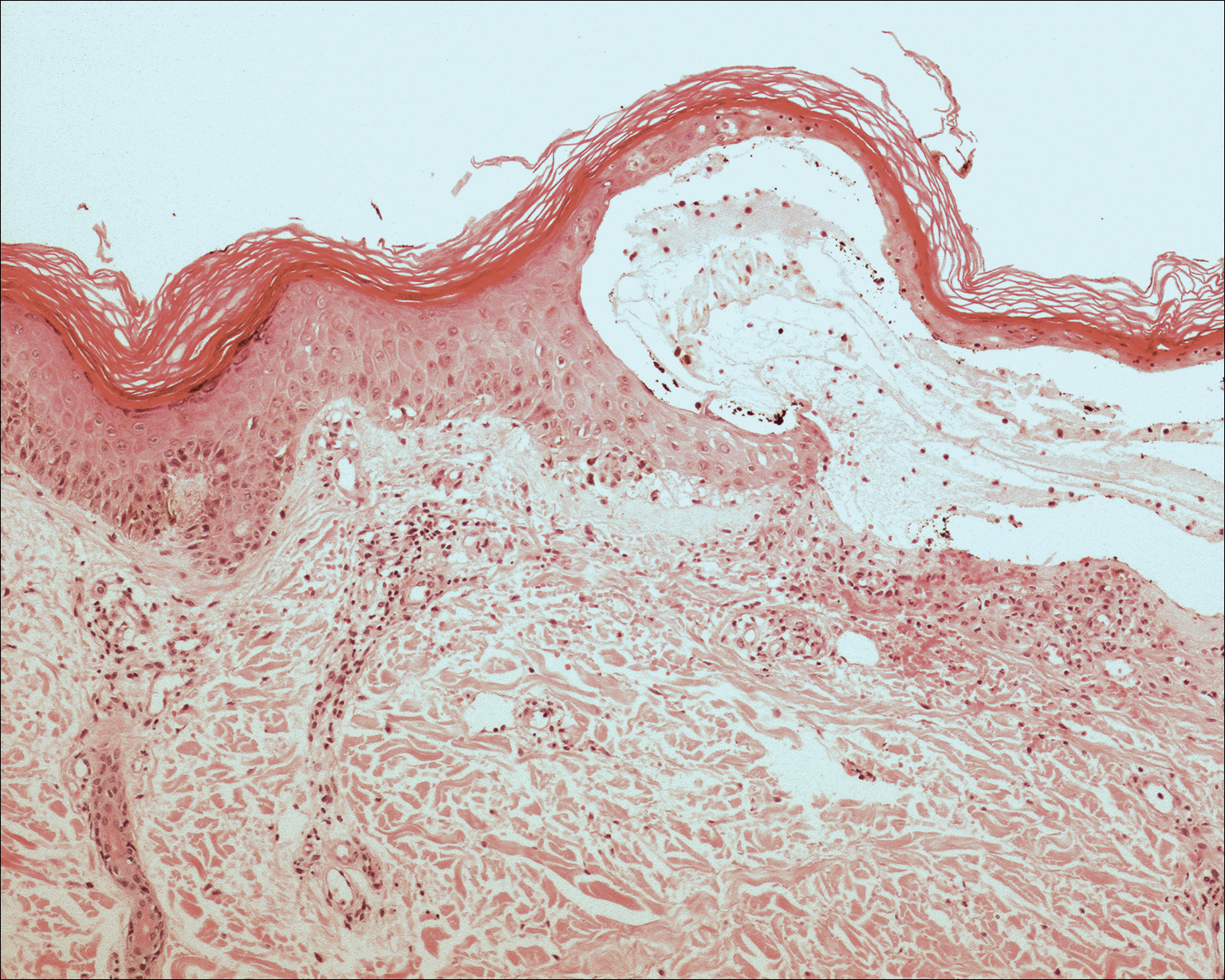
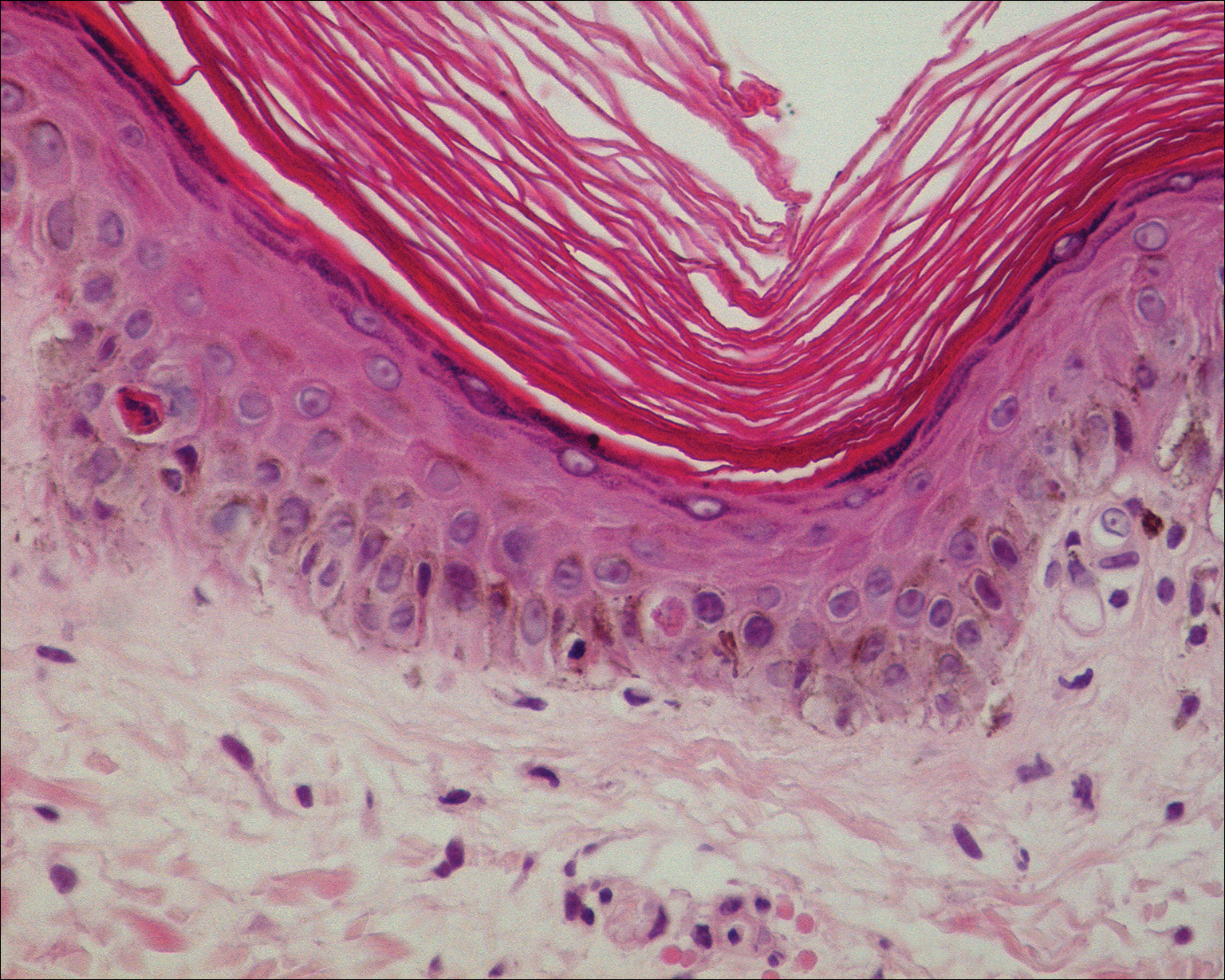
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
Practice Points
- Consider erythema ab igne (EAI) as a potential differential diagnosis in bullous eruptions.
- Space heaters, heating pads, and even laptop computers should be considered as potential causes of EAI.
Paraneoplastic Acrokeratosis Bazex Syndrome: Unusual Association With In Situ Follicular Lymphoma and Response to Acitretin
To the Editor:
Paraneoplastic acrokeratosis (PA), also known as Bazex syndrome, is a rare paraneoplastic dermatosis first described in 1965 by Bazex et al.1 This entity is clinically characterized by dusky erythematous to violaceous keratoderma of the acral sites and commonly affects men older than 40 years. In most reported cases, there has been an underlying primary malignant neoplasm of the upper aerodigestive tract2; however, some other associated malignancies also have been reported. Skin changes tend to occur before the diagnosis of the associated tumor in 67% of cases. The cutaneous lesions usually resolve after successful treatment of the tumor and relapse in case of recurrence of the malignancy.3
A 53-year-old woman who was a smoker with no relevant medical background was referred to the dermatology department with an itching psoriasiform dermatitis on the palms and soles of 2 months' duration. There were no signs of systemic disease. Physical examination revealed well-demarcated, dusky red, thick, scaly plaques on the soles with sparing of the insteps (Figure, A). Scattered symmetric hyperkeratotic plaques were present on the palms (Figure, B). We also detected onychodystrophy on the hands. Other dermatologic findings were normal. Histologic examination of a biopsy specimen of the left sole showed hyperkeratosis, focal parakeratosis, acanthosis, hypergranulosis, and a predominantly perivascular dermal lymphocytic infiltrate.

With the diagnostic suspicion of PA, blood tests, chest radiograph, and colonoscopy were performed without revealing abnormalities. Positron emission tomography and computed tomography also was performed, showing cervical, mesenteric, retroperitoneal, and inguinal adenopathies. Histologic examination of both inguinal adenectomy and cervical lymph node biopsy revealed Bcl-2-positive in situ follicular lymphoma (ISFL). Examination of an iliac crest marrow aspirate showed minimal involvement of lymphoma (10%). Follow-up imaging performed 4 months after diagnosis showed no changes. The patient was diagnosed with a low-grade chronic lymphoproliferative disorder with histologic findings consistent with ISFL presenting with small disperse adenopathies and minimal bone marrow involvement. The hematology department opted for a wait-and-see approach with 6-month follow-up imaging.
The skin lesions were first treated with salicylic acid cream 10%, psoralen plus UVA therapy, and methotrexate 20 mg weekly for 2 months without remission. Replacing the other therapies, we initiated acitretin 25 mg daily, achieving sustained remission after 6 months of treatment, and then continued with a scaled dose reduction. The patient remained lesion free 1 year after starting the treatment, with a daily dose of 10 mg of acitretin.
Paraneoplastic acrokeratosis has been traditionally described as a paraneoplastic entity mainly associated with primary squamous cell carcinoma (SCC) of the upper aerodigestive tract or a metastatic SCC of the cervical lymph nodes with an unknown origin.4,5 However, uncommon associations such as adenocarcinoma of the prostate, lung, esophagus, stomach, and colon; transitional cell carcinoma of the bladder; small cell carcinoma of the lung; cutaneous SCC; breast cancer; metastatic thymic carcinoma; metastatic neuroendocrine tumor; bronchial carcinoid tumor; SCC of the vulvar region; simultaneous multiple genitourinary tumors; and liposarcoma also have been described.6 Regarding the association with lymphoma, PA has been reported with peripheral T-cell lymphoma7 and Hodgkin disease8; however, ISFL underlying PA is rare.
Follicular lymphoma is the second most common non-Hodgkin lymphoma in Western countries and comprises approximately 20% of all lymphomas.9 It is slightly more prevalent in females, and the majority of patients present with advanced-stage disease. Generally considered to be an incurable disease, a watchful-waiting approach of conservative management has been advocated in most cases, deferring treatment until symptoms appear.9
Histology of PA is nonspecific, as in our case. However, it facilitates a differential diagnosis of major dermatoses including psoriasis vulgaris, pityriasis rubra pilaris, and lupus erythematosus.
Paraneoplastic palmoplantar keratoderma also is characteristic of Howel-Evans syndrome, which is a rare inherited condition associated with esophageal cancer. In contrast to our case, palmoplantar keratoderma in these patients usually begins around 10 years of age, is caused by a mutation in the RHBDF2 gene, and is inherited in an autosomal pattern.10
The diagnosis in our case was supported by a typical clinical picture, nonspecific histology, and the concurrent finding of the underlying lymphoma. Treatment of PA must focus on the removal of the underlying malignancy, which implies the remission of the cutaneous lesions. Taking into account that a recurrence of the primary tumor leads to a relapse of skin manifestations while distant metastases do not cause a reappearance of PA, it could be suggested that pathogenetically relevant factors are produced by the primary tumor and by lymph node metastases but not by metastases elsewhere.
In this case, due to the wait-and-see approach, a specific treatment for the skin lesions was established. Although management of the skin itself generally is ineffective, there are isolated reports of response after corticosteroids, antibiotics, antimycotics, keratolytic measures, or psoralen plus UVA therapy.6 Wishart11 used etretinate to achieve an improvement of PA. We also achieved good response with acitretin. Retinoids are known to have antineoplastic activity, which may have been helpful in both the patient we presented and the one reported by Wishart.11 In summary, we propose adding ISFL to the expanding list of malignant neoplasms associated with PA, noting the response of skin lesions after acitretin.
- Bazex A, Salvador R, Dupré A, et al. Syndrome paranéoplasique à type d'hyperkératose des extremités. Guérison après le traitement de l'épithelioma laryngé. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
- Bazex A, Griffiths A. Acrokeratosis paraneoplasticae--a new cutaneous marker of malignancy. Br J Dermatol. 1980;103:301-306.
- Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
- Witkowski JA, Parish LC. Bazex's syndrome. Paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
- Bolognia JL. Bazex's syndrome. Clin Dermatol. 1993;11:37-42.
- Sator PG, Breier F, Gschnait F. Acrokeratosis paraneoplastica (Bazex's syndrome): association with liposarcoma [published online August 28, 2006]. J Am Acad Dermatol. 2006;55:1103-1105.
- Lin YC, Chu CY, Chiu HC. Acrokeratosis paraneoplastica Bazex's syndrome: unusual association with a peripheral T-cell lymphoma. Acta Derm Venereol. 2001;81:440-441.
- Lucker GP, Steijlen PM. Acrokeratosis paraneoplastica (Bazex syndrome) occurring with acquired ichthyosis in Hodgkin's disease. Br J Dermatol. 1995;133:322-325.
- Jegalian AG, Eberle FC, Pack SD, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118:2976-2984.
- Sroa N, Witman P. Howel-Evans syndrome: a variant of ectodermal dysplasia. Cutis. 2010;85:183-185.
- Wishart JM. Bazex paraneoplastic acrokeratosis: a case report and response to Tigason. Br J Dermatol. 1986;115:595-599.
To the Editor:
Paraneoplastic acrokeratosis (PA), also known as Bazex syndrome, is a rare paraneoplastic dermatosis first described in 1965 by Bazex et al.1 This entity is clinically characterized by dusky erythematous to violaceous keratoderma of the acral sites and commonly affects men older than 40 years. In most reported cases, there has been an underlying primary malignant neoplasm of the upper aerodigestive tract2; however, some other associated malignancies also have been reported. Skin changes tend to occur before the diagnosis of the associated tumor in 67% of cases. The cutaneous lesions usually resolve after successful treatment of the tumor and relapse in case of recurrence of the malignancy.3
A 53-year-old woman who was a smoker with no relevant medical background was referred to the dermatology department with an itching psoriasiform dermatitis on the palms and soles of 2 months' duration. There were no signs of systemic disease. Physical examination revealed well-demarcated, dusky red, thick, scaly plaques on the soles with sparing of the insteps (Figure, A). Scattered symmetric hyperkeratotic plaques were present on the palms (Figure, B). We also detected onychodystrophy on the hands. Other dermatologic findings were normal. Histologic examination of a biopsy specimen of the left sole showed hyperkeratosis, focal parakeratosis, acanthosis, hypergranulosis, and a predominantly perivascular dermal lymphocytic infiltrate.
With the diagnostic suspicion of PA, blood tests, chest radiograph, and colonoscopy were performed without revealing abnormalities. Positron emission tomography and computed tomography also was performed, showing cervical, mesenteric, retroperitoneal, and inguinal adenopathies. Histologic examination of both inguinal adenectomy and cervical lymph node biopsy revealed Bcl-2-positive in situ follicular lymphoma (ISFL). Examination of an iliac crest marrow aspirate showed minimal involvement of lymphoma (10%). Follow-up imaging performed 4 months after diagnosis showed no changes. The patient was diagnosed with a low-grade chronic lymphoproliferative disorder with histologic findings consistent with ISFL presenting with small disperse adenopathies and minimal bone marrow involvement. The hematology department opted for a wait-and-see approach with 6-month follow-up imaging.
The skin lesions were first treated with salicylic acid cream 10%, psoralen plus UVA therapy, and methotrexate 20 mg weekly for 2 months without remission. Replacing the other therapies, we initiated acitretin 25 mg daily, achieving sustained remission after 6 months of treatment, and then continued with a scaled dose reduction. The patient remained lesion free 1 year after starting the treatment, with a daily dose of 10 mg of acitretin.
Paraneoplastic acrokeratosis has been traditionally described as a paraneoplastic entity mainly associated with primary squamous cell carcinoma (SCC) of the upper aerodigestive tract or a metastatic SCC of the cervical lymph nodes with an unknown origin.4,5 However, uncommon associations such as adenocarcinoma of the prostate, lung, esophagus, stomach, and colon; transitional cell carcinoma of the bladder; small cell carcinoma of the lung; cutaneous SCC; breast cancer; metastatic thymic carcinoma; metastatic neuroendocrine tumor; bronchial carcinoid tumor; SCC of the vulvar region; simultaneous multiple genitourinary tumors; and liposarcoma also have been described.6 Regarding the association with lymphoma, PA has been reported with peripheral T-cell lymphoma7 and Hodgkin disease8; however, ISFL underlying PA is rare.
Follicular lymphoma is the second most common non-Hodgkin lymphoma in Western countries and comprises approximately 20% of all lymphomas.9 It is slightly more prevalent in females, and the majority of patients present with advanced-stage disease. Generally considered to be an incurable disease, a watchful-waiting approach of conservative management has been advocated in most cases, deferring treatment until symptoms appear.9
Histology of PA is nonspecific, as in our case. However, it facilitates a differential diagnosis of major dermatoses including psoriasis vulgaris, pityriasis rubra pilaris, and lupus erythematosus.
Paraneoplastic palmoplantar keratoderma also is characteristic of Howel-Evans syndrome, which is a rare inherited condition associated with esophageal cancer. In contrast to our case, palmoplantar keratoderma in these patients usually begins around 10 years of age, is caused by a mutation in the RHBDF2 gene, and is inherited in an autosomal pattern.10
The diagnosis in our case was supported by a typical clinical picture, nonspecific histology, and the concurrent finding of the underlying lymphoma. Treatment of PA must focus on the removal of the underlying malignancy, which implies the remission of the cutaneous lesions. Taking into account that a recurrence of the primary tumor leads to a relapse of skin manifestations while distant metastases do not cause a reappearance of PA, it could be suggested that pathogenetically relevant factors are produced by the primary tumor and by lymph node metastases but not by metastases elsewhere.
In this case, due to the wait-and-see approach, a specific treatment for the skin lesions was established. Although management of the skin itself generally is ineffective, there are isolated reports of response after corticosteroids, antibiotics, antimycotics, keratolytic measures, or psoralen plus UVA therapy.6 Wishart11 used etretinate to achieve an improvement of PA. We also achieved good response with acitretin. Retinoids are known to have antineoplastic activity, which may have been helpful in both the patient we presented and the one reported by Wishart.11 In summary, we propose adding ISFL to the expanding list of malignant neoplasms associated with PA, noting the response of skin lesions after acitretin.
To the Editor:
Paraneoplastic acrokeratosis (PA), also known as Bazex syndrome, is a rare paraneoplastic dermatosis first described in 1965 by Bazex et al.1 This entity is clinically characterized by dusky erythematous to violaceous keratoderma of the acral sites and commonly affects men older than 40 years. In most reported cases, there has been an underlying primary malignant neoplasm of the upper aerodigestive tract2; however, some other associated malignancies also have been reported. Skin changes tend to occur before the diagnosis of the associated tumor in 67% of cases. The cutaneous lesions usually resolve after successful treatment of the tumor and relapse in case of recurrence of the malignancy.3
A 53-year-old woman who was a smoker with no relevant medical background was referred to the dermatology department with an itching psoriasiform dermatitis on the palms and soles of 2 months' duration. There were no signs of systemic disease. Physical examination revealed well-demarcated, dusky red, thick, scaly plaques on the soles with sparing of the insteps (Figure, A). Scattered symmetric hyperkeratotic plaques were present on the palms (Figure, B). We also detected onychodystrophy on the hands. Other dermatologic findings were normal. Histologic examination of a biopsy specimen of the left sole showed hyperkeratosis, focal parakeratosis, acanthosis, hypergranulosis, and a predominantly perivascular dermal lymphocytic infiltrate.
With the diagnostic suspicion of PA, blood tests, chest radiograph, and colonoscopy were performed without revealing abnormalities. Positron emission tomography and computed tomography also was performed, showing cervical, mesenteric, retroperitoneal, and inguinal adenopathies. Histologic examination of both inguinal adenectomy and cervical lymph node biopsy revealed Bcl-2-positive in situ follicular lymphoma (ISFL). Examination of an iliac crest marrow aspirate showed minimal involvement of lymphoma (10%). Follow-up imaging performed 4 months after diagnosis showed no changes. The patient was diagnosed with a low-grade chronic lymphoproliferative disorder with histologic findings consistent with ISFL presenting with small disperse adenopathies and minimal bone marrow involvement. The hematology department opted for a wait-and-see approach with 6-month follow-up imaging.
The skin lesions were first treated with salicylic acid cream 10%, psoralen plus UVA therapy, and methotrexate 20 mg weekly for 2 months without remission. Replacing the other therapies, we initiated acitretin 25 mg daily, achieving sustained remission after 6 months of treatment, and then continued with a scaled dose reduction. The patient remained lesion free 1 year after starting the treatment, with a daily dose of 10 mg of acitretin.
Paraneoplastic acrokeratosis has been traditionally described as a paraneoplastic entity mainly associated with primary squamous cell carcinoma (SCC) of the upper aerodigestive tract or a metastatic SCC of the cervical lymph nodes with an unknown origin.4,5 However, uncommon associations such as adenocarcinoma of the prostate, lung, esophagus, stomach, and colon; transitional cell carcinoma of the bladder; small cell carcinoma of the lung; cutaneous SCC; breast cancer; metastatic thymic carcinoma; metastatic neuroendocrine tumor; bronchial carcinoid tumor; SCC of the vulvar region; simultaneous multiple genitourinary tumors; and liposarcoma also have been described.6 Regarding the association with lymphoma, PA has been reported with peripheral T-cell lymphoma7 and Hodgkin disease8; however, ISFL underlying PA is rare.
Follicular lymphoma is the second most common non-Hodgkin lymphoma in Western countries and comprises approximately 20% of all lymphomas.9 It is slightly more prevalent in females, and the majority of patients present with advanced-stage disease. Generally considered to be an incurable disease, a watchful-waiting approach of conservative management has been advocated in most cases, deferring treatment until symptoms appear.9
Histology of PA is nonspecific, as in our case. However, it facilitates a differential diagnosis of major dermatoses including psoriasis vulgaris, pityriasis rubra pilaris, and lupus erythematosus.
Paraneoplastic palmoplantar keratoderma also is characteristic of Howel-Evans syndrome, which is a rare inherited condition associated with esophageal cancer. In contrast to our case, palmoplantar keratoderma in these patients usually begins around 10 years of age, is caused by a mutation in the RHBDF2 gene, and is inherited in an autosomal pattern.10
The diagnosis in our case was supported by a typical clinical picture, nonspecific histology, and the concurrent finding of the underlying lymphoma. Treatment of PA must focus on the removal of the underlying malignancy, which implies the remission of the cutaneous lesions. Taking into account that a recurrence of the primary tumor leads to a relapse of skin manifestations while distant metastases do not cause a reappearance of PA, it could be suggested that pathogenetically relevant factors are produced by the primary tumor and by lymph node metastases but not by metastases elsewhere.
In this case, due to the wait-and-see approach, a specific treatment for the skin lesions was established. Although management of the skin itself generally is ineffective, there are isolated reports of response after corticosteroids, antibiotics, antimycotics, keratolytic measures, or psoralen plus UVA therapy.6 Wishart11 used etretinate to achieve an improvement of PA. We also achieved good response with acitretin. Retinoids are known to have antineoplastic activity, which may have been helpful in both the patient we presented and the one reported by Wishart.11 In summary, we propose adding ISFL to the expanding list of malignant neoplasms associated with PA, noting the response of skin lesions after acitretin.
- Bazex A, Salvador R, Dupré A, et al. Syndrome paranéoplasique à type d'hyperkératose des extremités. Guérison après le traitement de l'épithelioma laryngé. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
- Bazex A, Griffiths A. Acrokeratosis paraneoplasticae--a new cutaneous marker of malignancy. Br J Dermatol. 1980;103:301-306.
- Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
- Witkowski JA, Parish LC. Bazex's syndrome. Paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
- Bolognia JL. Bazex's syndrome. Clin Dermatol. 1993;11:37-42.
- Sator PG, Breier F, Gschnait F. Acrokeratosis paraneoplastica (Bazex's syndrome): association with liposarcoma [published online August 28, 2006]. J Am Acad Dermatol. 2006;55:1103-1105.
- Lin YC, Chu CY, Chiu HC. Acrokeratosis paraneoplastica Bazex's syndrome: unusual association with a peripheral T-cell lymphoma. Acta Derm Venereol. 2001;81:440-441.
- Lucker GP, Steijlen PM. Acrokeratosis paraneoplastica (Bazex syndrome) occurring with acquired ichthyosis in Hodgkin's disease. Br J Dermatol. 1995;133:322-325.
- Jegalian AG, Eberle FC, Pack SD, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118:2976-2984.
- Sroa N, Witman P. Howel-Evans syndrome: a variant of ectodermal dysplasia. Cutis. 2010;85:183-185.
- Wishart JM. Bazex paraneoplastic acrokeratosis: a case report and response to Tigason. Br J Dermatol. 1986;115:595-599.
- Bazex A, Salvador R, Dupré A, et al. Syndrome paranéoplasique à type d'hyperkératose des extremités. Guérison après le traitement de l'épithelioma laryngé. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
- Bazex A, Griffiths A. Acrokeratosis paraneoplasticae--a new cutaneous marker of malignancy. Br J Dermatol. 1980;103:301-306.
- Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
- Witkowski JA, Parish LC. Bazex's syndrome. Paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
- Bolognia JL. Bazex's syndrome. Clin Dermatol. 1993;11:37-42.
- Sator PG, Breier F, Gschnait F. Acrokeratosis paraneoplastica (Bazex's syndrome): association with liposarcoma [published online August 28, 2006]. J Am Acad Dermatol. 2006;55:1103-1105.
- Lin YC, Chu CY, Chiu HC. Acrokeratosis paraneoplastica Bazex's syndrome: unusual association with a peripheral T-cell lymphoma. Acta Derm Venereol. 2001;81:440-441.
- Lucker GP, Steijlen PM. Acrokeratosis paraneoplastica (Bazex syndrome) occurring with acquired ichthyosis in Hodgkin's disease. Br J Dermatol. 1995;133:322-325.
- Jegalian AG, Eberle FC, Pack SD, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118:2976-2984.
- Sroa N, Witman P. Howel-Evans syndrome: a variant of ectodermal dysplasia. Cutis. 2010;85:183-185.
- Wishart JM. Bazex paraneoplastic acrokeratosis: a case report and response to Tigason. Br J Dermatol. 1986;115:595-599.
Practice Points
- Paraneoplastic acrokeratosis may mimic palmo-plantar acrokeratosis in both clinical presentation and treatment.
- Uncommon associations of paraneoplastic acrokeratosis with different types of lymphoma have been described.

Hereditary Hypotrichosis Simplex of the Scalp
To the Editor:
Hereditary hypotrichosis simplex (HHS)(Online Mendelian Inheritance in Man [OMIM] 146520) is a rare form of hypotrichosis that typically presents in school-aged children as worsening hair loss localized to the scalp.1 Most patients are unaffected at birth and otherwise healthy without abnormalities of the nails, teeth, or perspiration. Examination of the scalp reveals normal follicular ostia and absence of scale and erythema; however, decreased follicular density may be noted.1 The histopathologic findings of HHS reveal velluslike hair follicles without associated fibrosis or inflammation.2 Examination of hair follicles with light microscopy is unremarkable.3,4 Historically, this condition has been largely regarded as autosomal dominant, with variable severity also described within families. Herein, we report a case of this rare disease in a child, with 2 family members displaying a less severe phenotype.
A 7-year-old girl presented with gradual thinning of the scalp hair of 3 to 4 years’ duration. Her mother reported the patient had normal hair density at birth. Over the next several years, she was noted to have an inability to grow lengthy hair. At approximately 3 years of age, thinning of scalp hair was identified. There was no prior history of increased shedding, hypohidrosis, or tooth or nail abnormalities. Family history revealed fine hair in her older sister and fine thin hair at the frontal scalp in her mother. Her mother reported similar inability to grow lengthy hair. Physical examination of the patient demonstrated short blonde hair with diffuse thinning of the crown (Figure 1). The longest hair was approximately 10 cm in length. Follicular ostia were without erythema or scale but notably fewer in number on the crown. Eyebrows, eyelashes, teeth, and fingernails were without abnormalities. A hair pull test was negative and hair mount revealed normal bulb and shaft. Microscopy of hair shafts under polarized light was unremarkable.
Two punch biopsies were obtained and submitted for vertical and horizontal sectioning. Sections demonstrated an intact epidermis, decreased follicle number, and small follicles with hypoplastic velluslike appearance (Figure 2). Fibrosis and inflammation were not seen; there was no increase in catagen or telogen hairs. Clinical and histopathological findings were consistent with HHS.
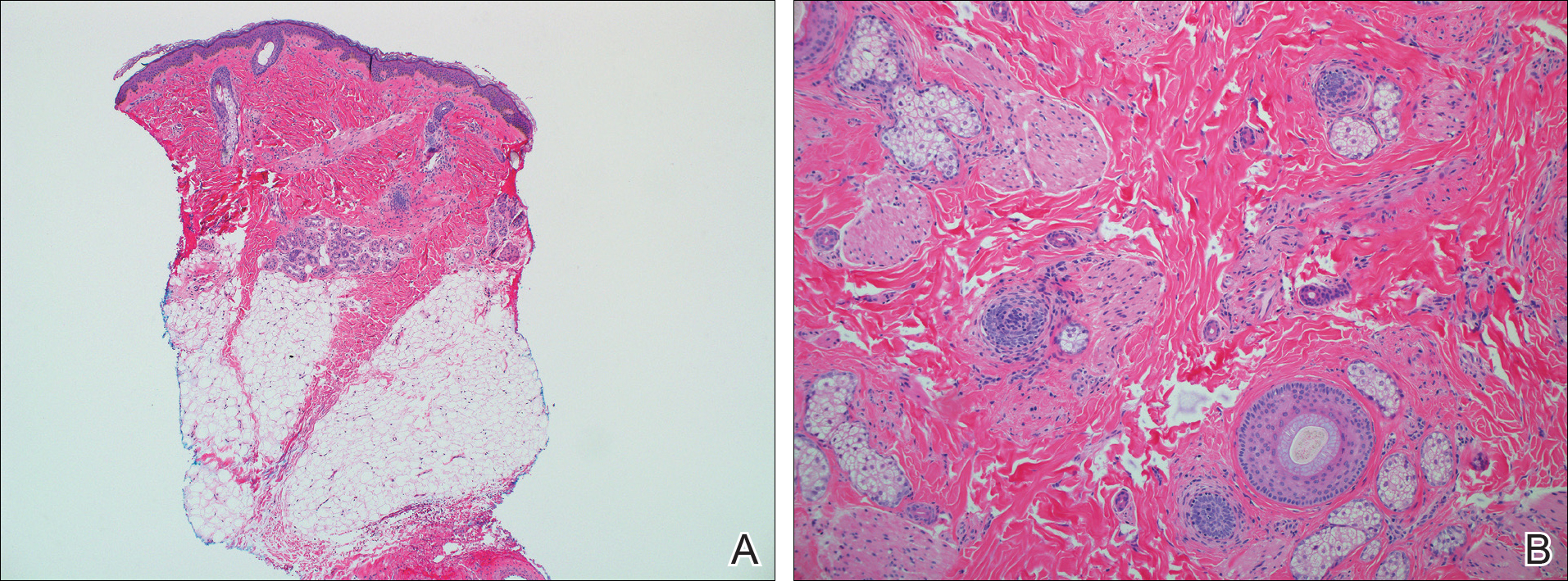
Hereditary hypotrichosis localized to the scalp was first described by Toribio and Quinones5 in 1974 in a large Spanish family presenting with normal scalp hair at birth followed by gradual diffuse hair loss. Hair loss that usually began in school-aged children with subsequent few fine hairs remaining on the scalp by the third decade of life was identified in these individuals.Eyelashes, eyebrows, pubic, axillary, and other truncal hairs were normal.5 Several similar cases of HHS localized to the scalp have since been reported.2,6 Hereditary hypotrichosis simplex is inherited in an autosomal-dominant fashion, with the exception of 1 reported sporadic case.3
Research on HHS has primarily focused on genetic analyses of several affected families. Betz et al7 mapped the gene for HHS to band 6p21.3 in 2 families of Danish origin and in the Spanish family initially described by Toribio and Quinones.5 Three years later, a nonsense mutation in the CDSN gene encoding corneodesmosin was described.8 Despite these genetic advances, the pathogenesis of HHS and the role that corneodesmosin may play remain unclear.
Generalized forms of hypotrichosis (OMIM #605389) have long been reported and described as loss of scalp hair with involvement of eyebrows, eyelashes, and other body hair.9 Genetic studies have allowed for genome-wide linkage analysis, linking 3 families with this more generalized HHS phenotype to chromosome 18; specifically, an Italian family with sparse scalp and body hair but normal eyelashes and eyebrows,4 and 2 Pakistani families with thinning scalp hair and sparse truncal hair.10 A mutation in the APC downregulated 1 gene, APCDD1, also has been identified in these families.10 These genetic findings indicate that the generalized form of HHS is a distinct syndrome.
The differential diagnosis of HHS includes Marie-Unna hereditary hypotrichosis, loose anagen hair syndrome, trichothiodystrophy, and androgenetic alopecia. Marie-Unna hereditary hypotrichosis usually presents as near-complete absence of scalp hair at birth, development of wiry twisted hair in childhood, and progressive alopecia.3 Loose anagen hair syndrome usually demonstrates a ruffled cuticle on hair pull test and remits in late childhood. Polarization of the hair shaft can identify patients with trichothiodystrophy. Follicular miniaturization may lead one to consider early-onset androgenetic alopecia in some patients.
There is no effective treatment of HHS. Due to potential phenotypic variation, patients should be counseled that they may experience progressive or possible total loss of scalp hair by the third decade of life.2,3,5 As with other hair loss disorders, wigs or additional over-the-counter cosmetic options may be considered.3 Currently, there are no known patient resources specific for HHS. Therefore, our patient’s family was referred to the National Alopecia Areata Foundation website (https://naaf.org/) for resources on discussing alopecia with school-aged children. The psychological impact of alopecia should not be overlooked and psychiatric referral should be provided, if needed. Examination of family members along with clinical monitoring are recommended. Genetic counseling also may be offered.3
- Rodríguez Díaz E, Fernández Blasco G, Martín Pascual A, et al. Heredity hypotrichosis simplex of the scalp. Dermatology. 1995;191:139-141.
- Ibsen HH, Clemmensen OJ, Brandrup F. Familial hypotrichosis of the scalp. autosomal dominant inheritance in four generations. Acta Derm Venereol. 1991;71:349-351.
- Cambiaghi S, Barbareschi M. A sporadic case of congenital hypotrichosis simplex of the scalp: difficulties in diagnosis and classification. Pediatr Dermatol. 1999;16:301-304.
- Baumer A, Belli S, Trueb RM, et al. An autosomal dominant form of hereditary hypotrichosis simple maps to 18p11.32-p11.23 in an Italian family. Eur J Hum Genet. 2000;8:443-448.
- Toribio J, Quinones PA. Heredity hypotrichosis simplex of the scalp. evidence for autosomal dominant inheritance. Br J Dermatol. 1974;91:687-696.
- Kohn G, Metzker A. Hereditary hypotrichosis simplex of the scalp. Clin Genet. 1987;32:120-124.
- Betz RC, Lee YA, Bygum A, et al. A gene for hypotrichosis simplex of the scalp maps to chromosome 6p21.3. Am J Hum Genet. 2000;66:1979-1983.
- Levy-Nissenbaum E, Betz R, Frydman M, et al. Hypotrichosis of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet. 2003;34:151-153.
- Just M, Ribera M, Fuente MJ, et al. Hereditary hypotrichosis simplex. Dermatology. 1998;196:339-342.
- Shimomura Y, Agalliu D, Vonica A, et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature. 2011;44:1043-1047.
To the Editor:
Hereditary hypotrichosis simplex (HHS)(Online Mendelian Inheritance in Man [OMIM] 146520) is a rare form of hypotrichosis that typically presents in school-aged children as worsening hair loss localized to the scalp.1 Most patients are unaffected at birth and otherwise healthy without abnormalities of the nails, teeth, or perspiration. Examination of the scalp reveals normal follicular ostia and absence of scale and erythema; however, decreased follicular density may be noted.1 The histopathologic findings of HHS reveal velluslike hair follicles without associated fibrosis or inflammation.2 Examination of hair follicles with light microscopy is unremarkable.3,4 Historically, this condition has been largely regarded as autosomal dominant, with variable severity also described within families. Herein, we report a case of this rare disease in a child, with 2 family members displaying a less severe phenotype.
A 7-year-old girl presented with gradual thinning of the scalp hair of 3 to 4 years’ duration. Her mother reported the patient had normal hair density at birth. Over the next several years, she was noted to have an inability to grow lengthy hair. At approximately 3 years of age, thinning of scalp hair was identified. There was no prior history of increased shedding, hypohidrosis, or tooth or nail abnormalities. Family history revealed fine hair in her older sister and fine thin hair at the frontal scalp in her mother. Her mother reported similar inability to grow lengthy hair. Physical examination of the patient demonstrated short blonde hair with diffuse thinning of the crown (Figure 1). The longest hair was approximately 10 cm in length. Follicular ostia were without erythema or scale but notably fewer in number on the crown. Eyebrows, eyelashes, teeth, and fingernails were without abnormalities. A hair pull test was negative and hair mount revealed normal bulb and shaft. Microscopy of hair shafts under polarized light was unremarkable.
Two punch biopsies were obtained and submitted for vertical and horizontal sectioning. Sections demonstrated an intact epidermis, decreased follicle number, and small follicles with hypoplastic velluslike appearance (Figure 2). Fibrosis and inflammation were not seen; there was no increase in catagen or telogen hairs. Clinical and histopathological findings were consistent with HHS.
Hereditary hypotrichosis localized to the scalp was first described by Toribio and Quinones5 in 1974 in a large Spanish family presenting with normal scalp hair at birth followed by gradual diffuse hair loss. Hair loss that usually began in school-aged children with subsequent few fine hairs remaining on the scalp by the third decade of life was identified in these individuals.Eyelashes, eyebrows, pubic, axillary, and other truncal hairs were normal.5 Several similar cases of HHS localized to the scalp have since been reported.2,6 Hereditary hypotrichosis simplex is inherited in an autosomal-dominant fashion, with the exception of 1 reported sporadic case.3
Research on HHS has primarily focused on genetic analyses of several affected families. Betz et al7 mapped the gene for HHS to band 6p21.3 in 2 families of Danish origin and in the Spanish family initially described by Toribio and Quinones.5 Three years later, a nonsense mutation in the CDSN gene encoding corneodesmosin was described.8 Despite these genetic advances, the pathogenesis of HHS and the role that corneodesmosin may play remain unclear.
Generalized forms of hypotrichosis (OMIM #605389) have long been reported and described as loss of scalp hair with involvement of eyebrows, eyelashes, and other body hair.9 Genetic studies have allowed for genome-wide linkage analysis, linking 3 families with this more generalized HHS phenotype to chromosome 18; specifically, an Italian family with sparse scalp and body hair but normal eyelashes and eyebrows,4 and 2 Pakistani families with thinning scalp hair and sparse truncal hair.10 A mutation in the APC downregulated 1 gene, APCDD1, also has been identified in these families.10 These genetic findings indicate that the generalized form of HHS is a distinct syndrome.
The differential diagnosis of HHS includes Marie-Unna hereditary hypotrichosis, loose anagen hair syndrome, trichothiodystrophy, and androgenetic alopecia. Marie-Unna hereditary hypotrichosis usually presents as near-complete absence of scalp hair at birth, development of wiry twisted hair in childhood, and progressive alopecia.3 Loose anagen hair syndrome usually demonstrates a ruffled cuticle on hair pull test and remits in late childhood. Polarization of the hair shaft can identify patients with trichothiodystrophy. Follicular miniaturization may lead one to consider early-onset androgenetic alopecia in some patients.
There is no effective treatment of HHS. Due to potential phenotypic variation, patients should be counseled that they may experience progressive or possible total loss of scalp hair by the third decade of life.2,3,5 As with other hair loss disorders, wigs or additional over-the-counter cosmetic options may be considered.3 Currently, there are no known patient resources specific for HHS. Therefore, our patient’s family was referred to the National Alopecia Areata Foundation website (https://naaf.org/) for resources on discussing alopecia with school-aged children. The psychological impact of alopecia should not be overlooked and psychiatric referral should be provided, if needed. Examination of family members along with clinical monitoring are recommended. Genetic counseling also may be offered.3
To the Editor:
Hereditary hypotrichosis simplex (HHS)(Online Mendelian Inheritance in Man [OMIM] 146520) is a rare form of hypotrichosis that typically presents in school-aged children as worsening hair loss localized to the scalp.1 Most patients are unaffected at birth and otherwise healthy without abnormalities of the nails, teeth, or perspiration. Examination of the scalp reveals normal follicular ostia and absence of scale and erythema; however, decreased follicular density may be noted.1 The histopathologic findings of HHS reveal velluslike hair follicles without associated fibrosis or inflammation.2 Examination of hair follicles with light microscopy is unremarkable.3,4 Historically, this condition has been largely regarded as autosomal dominant, with variable severity also described within families. Herein, we report a case of this rare disease in a child, with 2 family members displaying a less severe phenotype.
A 7-year-old girl presented with gradual thinning of the scalp hair of 3 to 4 years’ duration. Her mother reported the patient had normal hair density at birth. Over the next several years, she was noted to have an inability to grow lengthy hair. At approximately 3 years of age, thinning of scalp hair was identified. There was no prior history of increased shedding, hypohidrosis, or tooth or nail abnormalities. Family history revealed fine hair in her older sister and fine thin hair at the frontal scalp in her mother. Her mother reported similar inability to grow lengthy hair. Physical examination of the patient demonstrated short blonde hair with diffuse thinning of the crown (Figure 1). The longest hair was approximately 10 cm in length. Follicular ostia were without erythema or scale but notably fewer in number on the crown. Eyebrows, eyelashes, teeth, and fingernails were without abnormalities. A hair pull test was negative and hair mount revealed normal bulb and shaft. Microscopy of hair shafts under polarized light was unremarkable.
Two punch biopsies were obtained and submitted for vertical and horizontal sectioning. Sections demonstrated an intact epidermis, decreased follicle number, and small follicles with hypoplastic velluslike appearance (Figure 2). Fibrosis and inflammation were not seen; there was no increase in catagen or telogen hairs. Clinical and histopathological findings were consistent with HHS.
Hereditary hypotrichosis localized to the scalp was first described by Toribio and Quinones5 in 1974 in a large Spanish family presenting with normal scalp hair at birth followed by gradual diffuse hair loss. Hair loss that usually began in school-aged children with subsequent few fine hairs remaining on the scalp by the third decade of life was identified in these individuals.Eyelashes, eyebrows, pubic, axillary, and other truncal hairs were normal.5 Several similar cases of HHS localized to the scalp have since been reported.2,6 Hereditary hypotrichosis simplex is inherited in an autosomal-dominant fashion, with the exception of 1 reported sporadic case.3
Research on HHS has primarily focused on genetic analyses of several affected families. Betz et al7 mapped the gene for HHS to band 6p21.3 in 2 families of Danish origin and in the Spanish family initially described by Toribio and Quinones.5 Three years later, a nonsense mutation in the CDSN gene encoding corneodesmosin was described.8 Despite these genetic advances, the pathogenesis of HHS and the role that corneodesmosin may play remain unclear.
Generalized forms of hypotrichosis (OMIM #605389) have long been reported and described as loss of scalp hair with involvement of eyebrows, eyelashes, and other body hair.9 Genetic studies have allowed for genome-wide linkage analysis, linking 3 families with this more generalized HHS phenotype to chromosome 18; specifically, an Italian family with sparse scalp and body hair but normal eyelashes and eyebrows,4 and 2 Pakistani families with thinning scalp hair and sparse truncal hair.10 A mutation in the APC downregulated 1 gene, APCDD1, also has been identified in these families.10 These genetic findings indicate that the generalized form of HHS is a distinct syndrome.
The differential diagnosis of HHS includes Marie-Unna hereditary hypotrichosis, loose anagen hair syndrome, trichothiodystrophy, and androgenetic alopecia. Marie-Unna hereditary hypotrichosis usually presents as near-complete absence of scalp hair at birth, development of wiry twisted hair in childhood, and progressive alopecia.3 Loose anagen hair syndrome usually demonstrates a ruffled cuticle on hair pull test and remits in late childhood. Polarization of the hair shaft can identify patients with trichothiodystrophy. Follicular miniaturization may lead one to consider early-onset androgenetic alopecia in some patients.
There is no effective treatment of HHS. Due to potential phenotypic variation, patients should be counseled that they may experience progressive or possible total loss of scalp hair by the third decade of life.2,3,5 As with other hair loss disorders, wigs or additional over-the-counter cosmetic options may be considered.3 Currently, there are no known patient resources specific for HHS. Therefore, our patient’s family was referred to the National Alopecia Areata Foundation website (https://naaf.org/) for resources on discussing alopecia with school-aged children. The psychological impact of alopecia should not be overlooked and psychiatric referral should be provided, if needed. Examination of family members along with clinical monitoring are recommended. Genetic counseling also may be offered.3
- Rodríguez Díaz E, Fernández Blasco G, Martín Pascual A, et al. Heredity hypotrichosis simplex of the scalp. Dermatology. 1995;191:139-141.
- Ibsen HH, Clemmensen OJ, Brandrup F. Familial hypotrichosis of the scalp. autosomal dominant inheritance in four generations. Acta Derm Venereol. 1991;71:349-351.
- Cambiaghi S, Barbareschi M. A sporadic case of congenital hypotrichosis simplex of the scalp: difficulties in diagnosis and classification. Pediatr Dermatol. 1999;16:301-304.
- Baumer A, Belli S, Trueb RM, et al. An autosomal dominant form of hereditary hypotrichosis simple maps to 18p11.32-p11.23 in an Italian family. Eur J Hum Genet. 2000;8:443-448.
- Toribio J, Quinones PA. Heredity hypotrichosis simplex of the scalp. evidence for autosomal dominant inheritance. Br J Dermatol. 1974;91:687-696.
- Kohn G, Metzker A. Hereditary hypotrichosis simplex of the scalp. Clin Genet. 1987;32:120-124.
- Betz RC, Lee YA, Bygum A, et al. A gene for hypotrichosis simplex of the scalp maps to chromosome 6p21.3. Am J Hum Genet. 2000;66:1979-1983.
- Levy-Nissenbaum E, Betz R, Frydman M, et al. Hypotrichosis of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet. 2003;34:151-153.
- Just M, Ribera M, Fuente MJ, et al. Hereditary hypotrichosis simplex. Dermatology. 1998;196:339-342.
- Shimomura Y, Agalliu D, Vonica A, et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature. 2011;44:1043-1047.
- Rodríguez Díaz E, Fernández Blasco G, Martín Pascual A, et al. Heredity hypotrichosis simplex of the scalp. Dermatology. 1995;191:139-141.
- Ibsen HH, Clemmensen OJ, Brandrup F. Familial hypotrichosis of the scalp. autosomal dominant inheritance in four generations. Acta Derm Venereol. 1991;71:349-351.
- Cambiaghi S, Barbareschi M. A sporadic case of congenital hypotrichosis simplex of the scalp: difficulties in diagnosis and classification. Pediatr Dermatol. 1999;16:301-304.
- Baumer A, Belli S, Trueb RM, et al. An autosomal dominant form of hereditary hypotrichosis simple maps to 18p11.32-p11.23 in an Italian family. Eur J Hum Genet. 2000;8:443-448.
- Toribio J, Quinones PA. Heredity hypotrichosis simplex of the scalp. evidence for autosomal dominant inheritance. Br J Dermatol. 1974;91:687-696.
- Kohn G, Metzker A. Hereditary hypotrichosis simplex of the scalp. Clin Genet. 1987;32:120-124.
- Betz RC, Lee YA, Bygum A, et al. A gene for hypotrichosis simplex of the scalp maps to chromosome 6p21.3. Am J Hum Genet. 2000;66:1979-1983.
- Levy-Nissenbaum E, Betz R, Frydman M, et al. Hypotrichosis of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet. 2003;34:151-153.
- Just M, Ribera M, Fuente MJ, et al. Hereditary hypotrichosis simplex. Dermatology. 1998;196:339-342.
- Shimomura Y, Agalliu D, Vonica A, et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature. 2011;44:1043-1047.
Practice Points
- Hereditary hypotrichosis simplex (HHS) is a rare form of hypotrichosis that typically presents in school-aged children as worsening hair loss localized to the scalp.
- Historically, HHS has been largely regarded as autosomal dominant, with variable severity also described within families.
- There is no effective treatment of HHS. Due to potential phenotypic variation, patients should be counseled that they may experience progressive or possible total loss of scalp hair by the third decade of life.