User login
Nivolumab-Induced Lichen Planus Pemphigoides
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
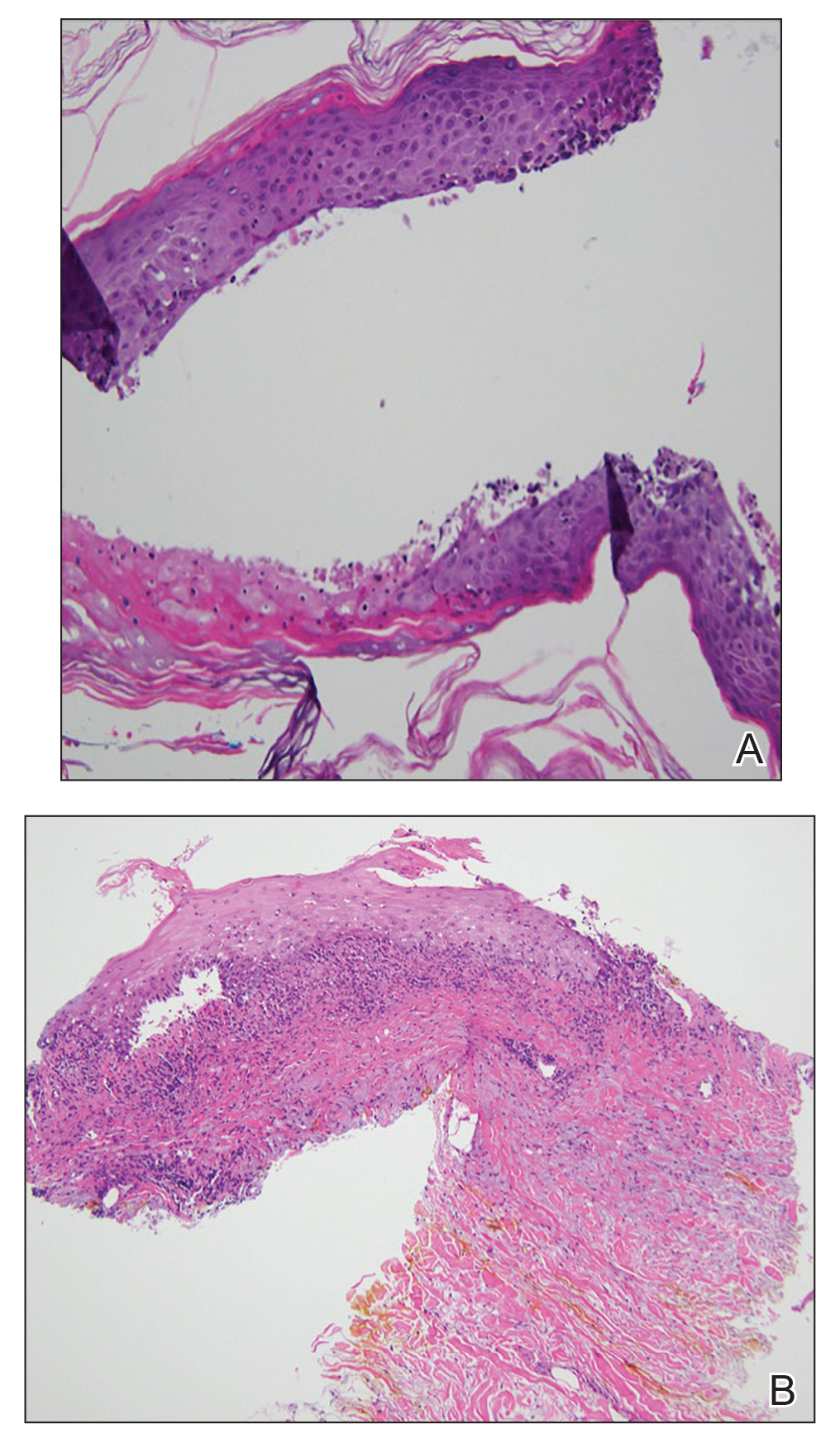
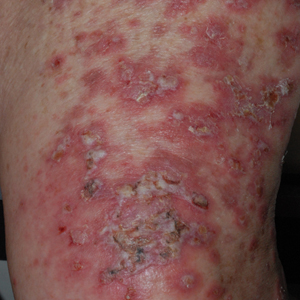
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
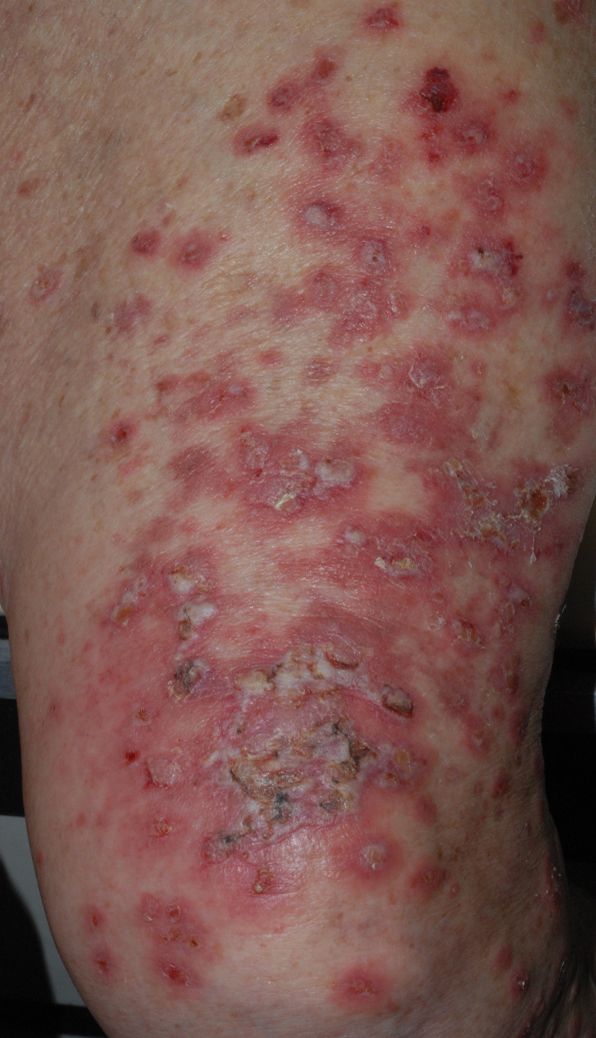
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
Practice Points
- Dermatologists should be aware that lichen planus pemphigoides is within the spectrum of toxicity for patients treated with nivolumab.
- Bullous eruptions related to anti–programmed cell death 1 agents tend to appear 4 months after initiation of therapy.
- A severe cutaneous toxicity of a checkpoint inhibitor should be managed using oral corticosteroids with consideration of withdrawing the offending agent.
Relapsing Polychondritis in Human Immunodeficiency Virus
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
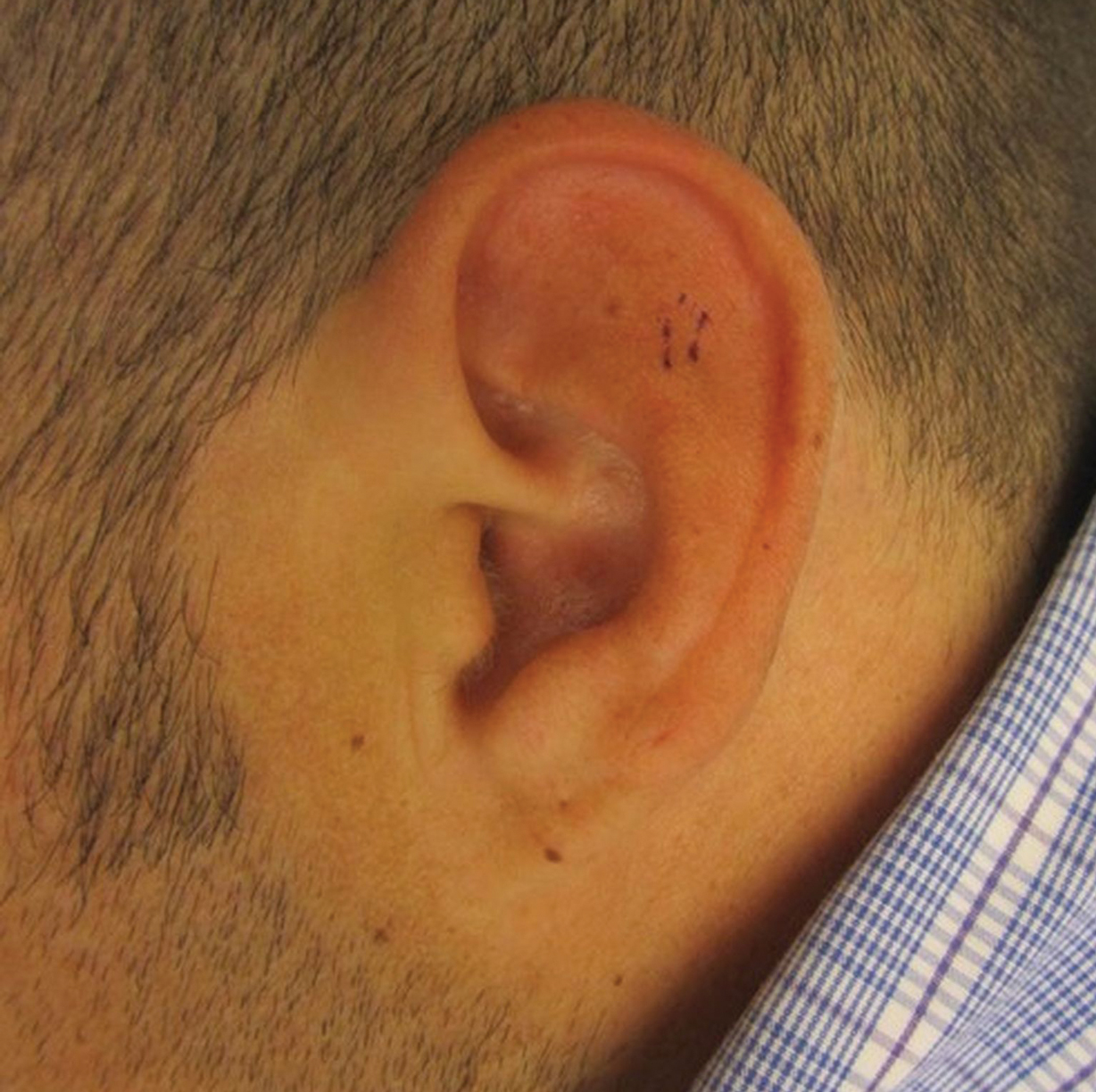
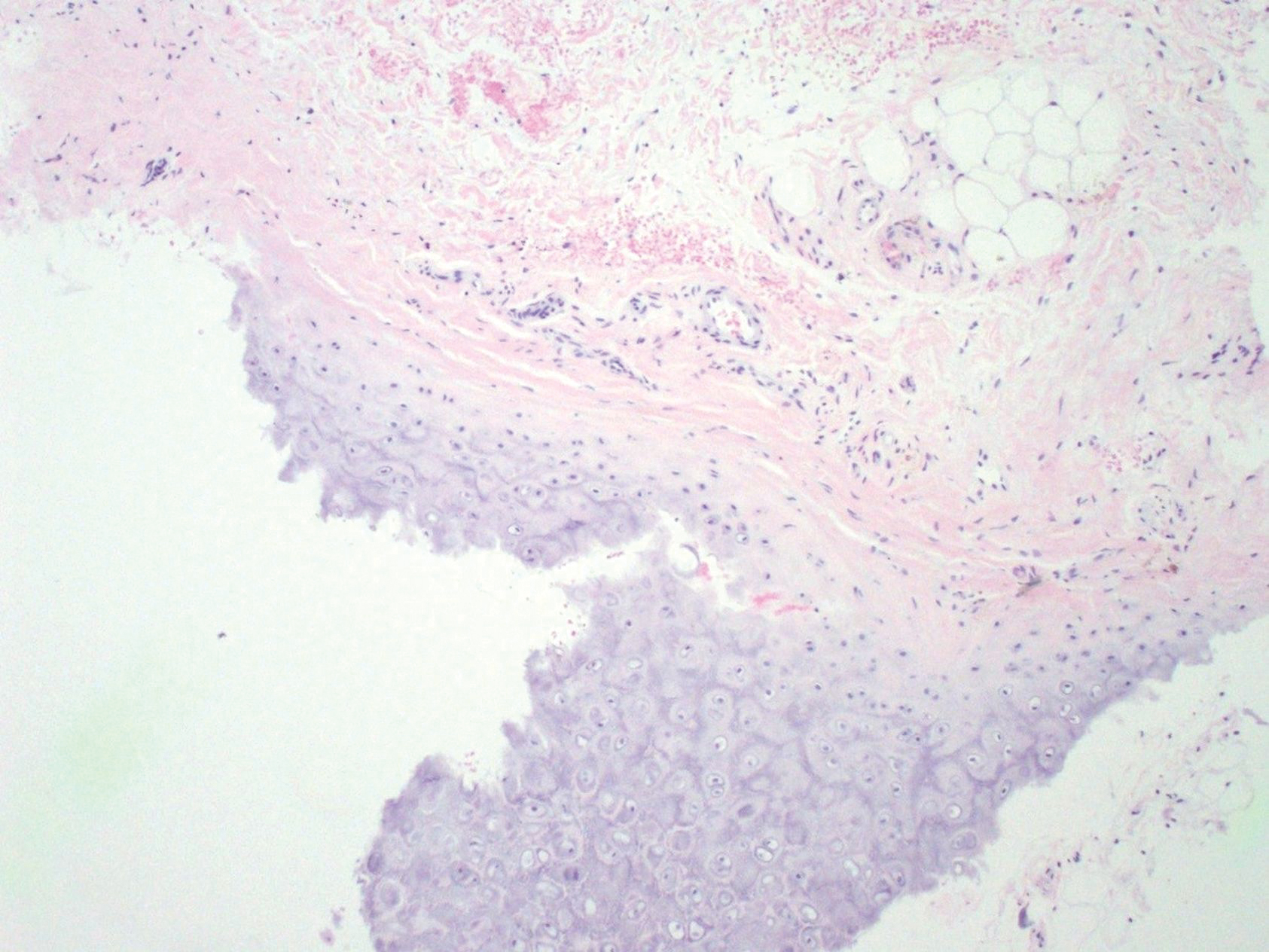
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Practice Points
- Relapsing polychondritis (RP) is characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures, most often manifesting as ear inflammation that involves the auricle but spares the lobe, nasal chondritis, and arthralgia.
- Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge.
- One-third of RP patients have coexisting autoimmune disease.
- Treatment of RP depends on severity of disease.
- Dermatologists must be aware of the potential for development of RP in the setting of human immunodeficiency virus infection; a missed diagnosis of this progressive disease has the potential to be life-threatening.
Cutaneous Metastasis of Endometrial Carcinoma: An Unusual and Dramatic Presentation
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
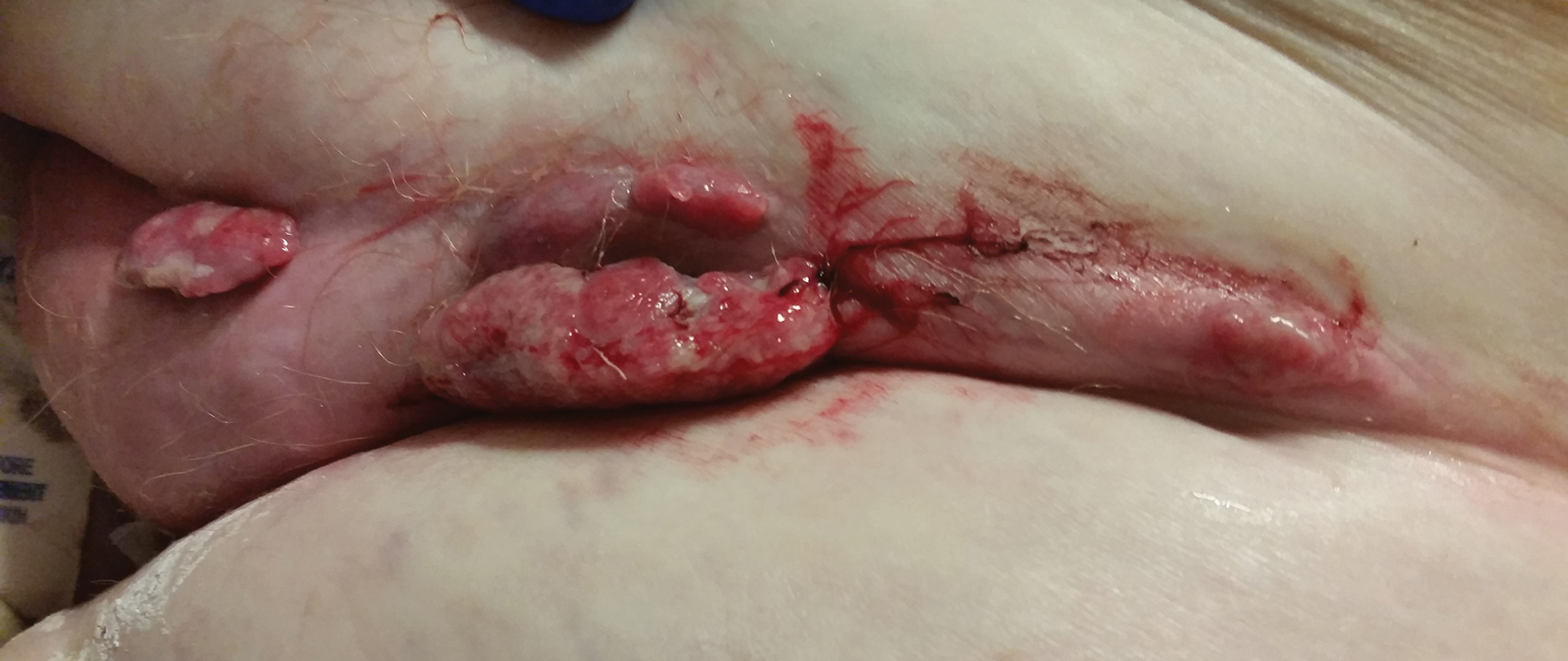
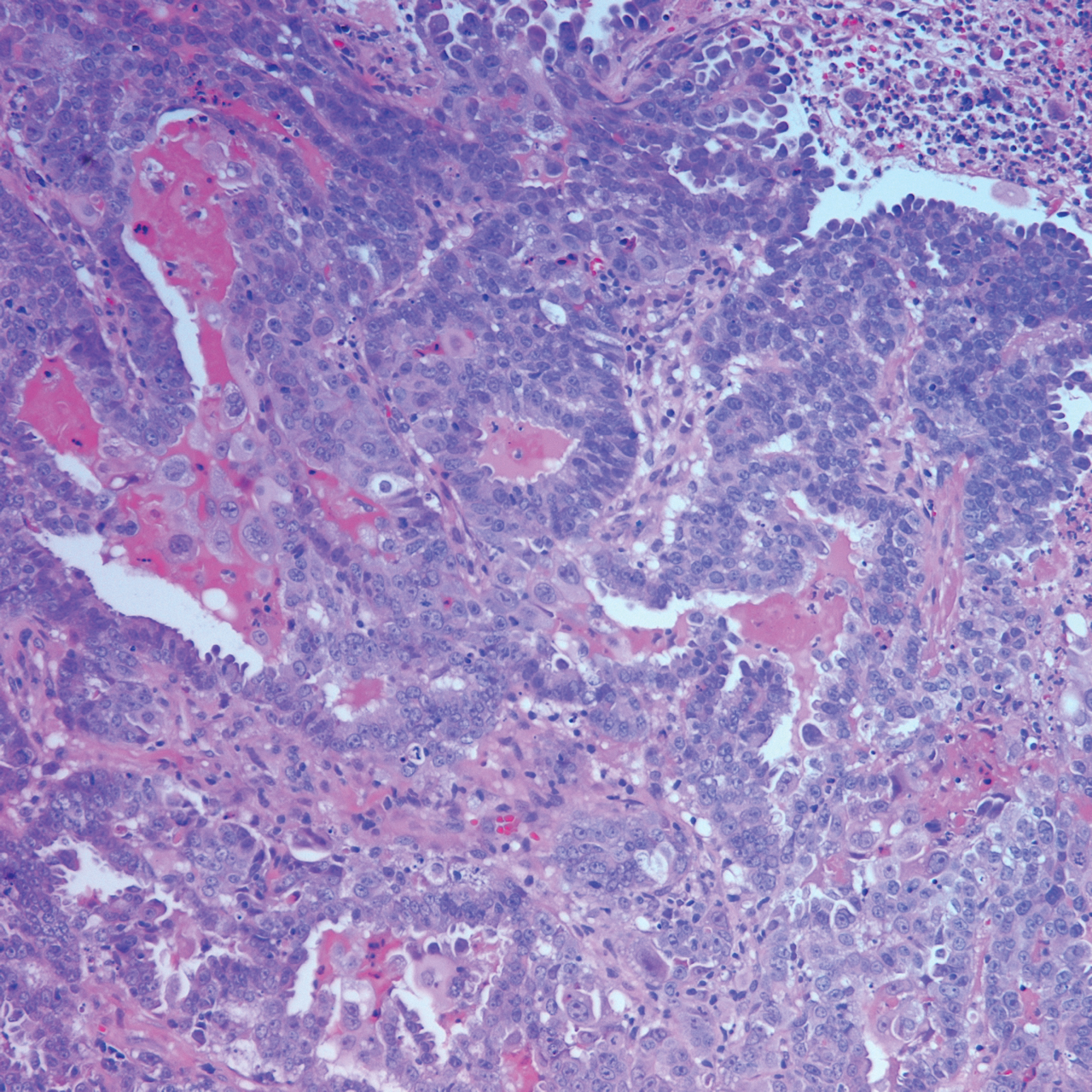
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Practice Points
- Cutaneous metastases of endometrial carcinoma are extremely rare and typically present in areas of direct local spread.
- As with other cutaneous metastases, lesions often are nonspecific, making history and histopathology essential for diagnosis.
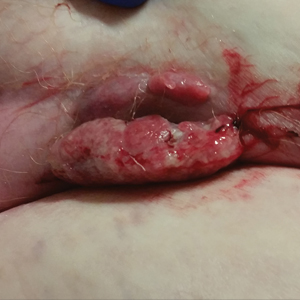
Recurrence of a small gastric gastrointestinal stromal tumor with high mitotic index
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A). 
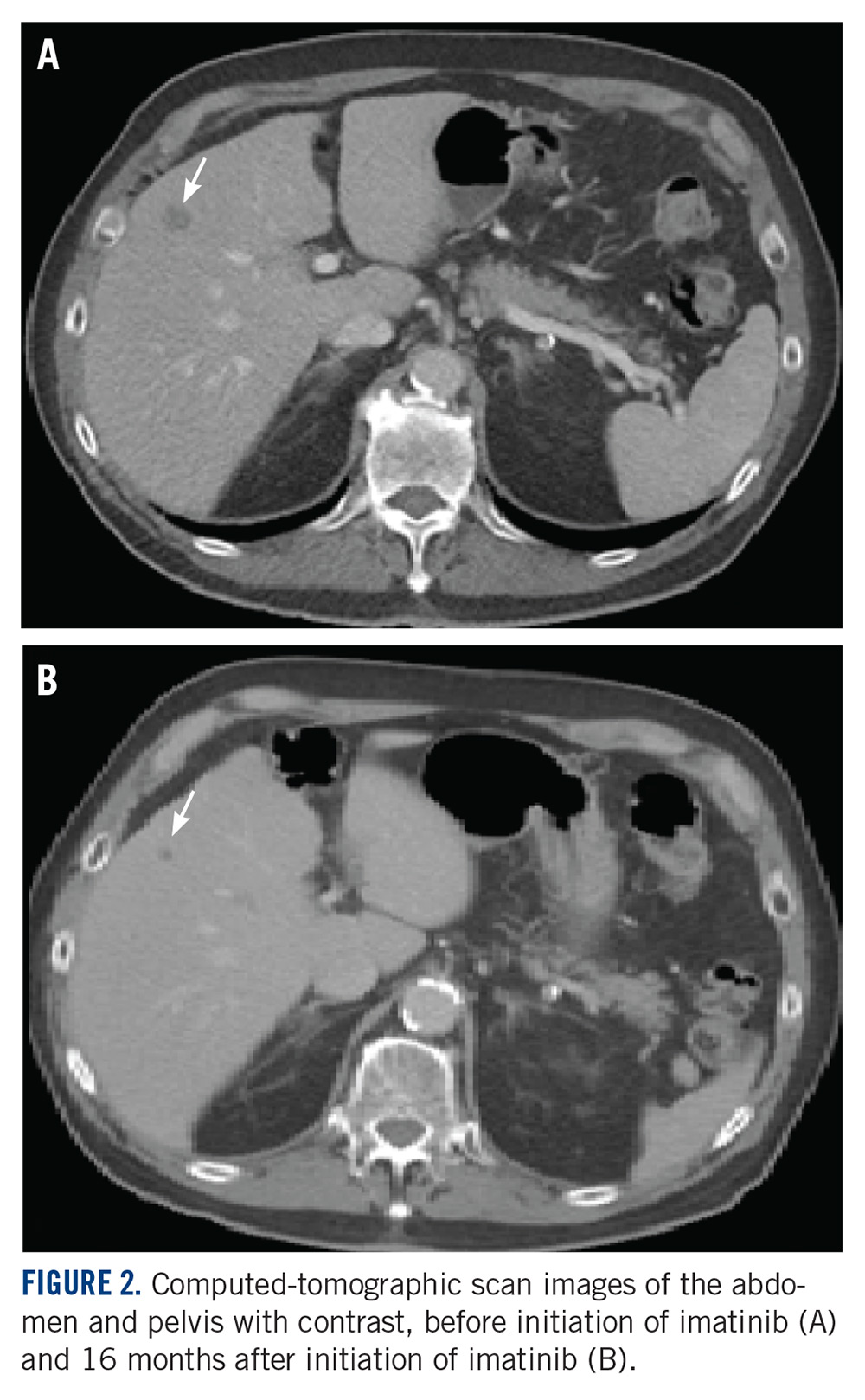
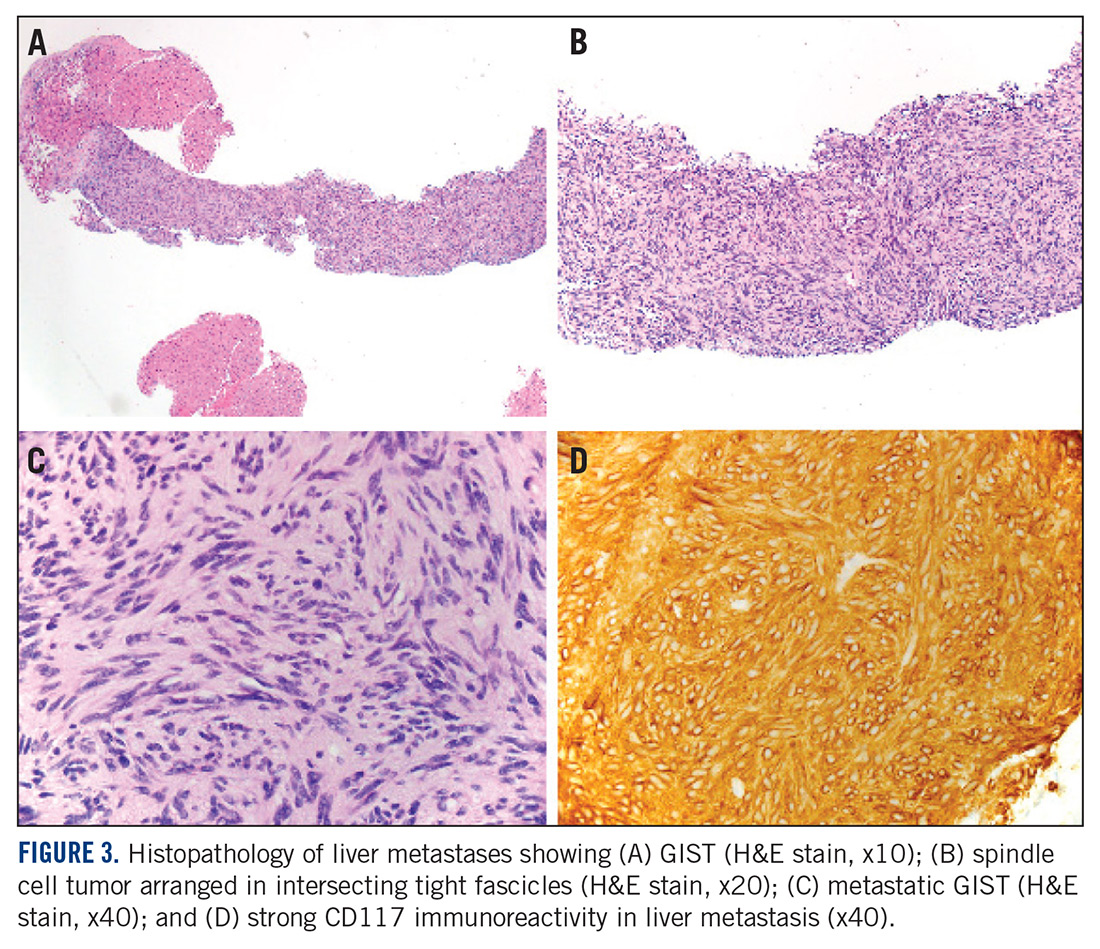
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
Abdominal Wall Schwannoma
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess. 
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1). 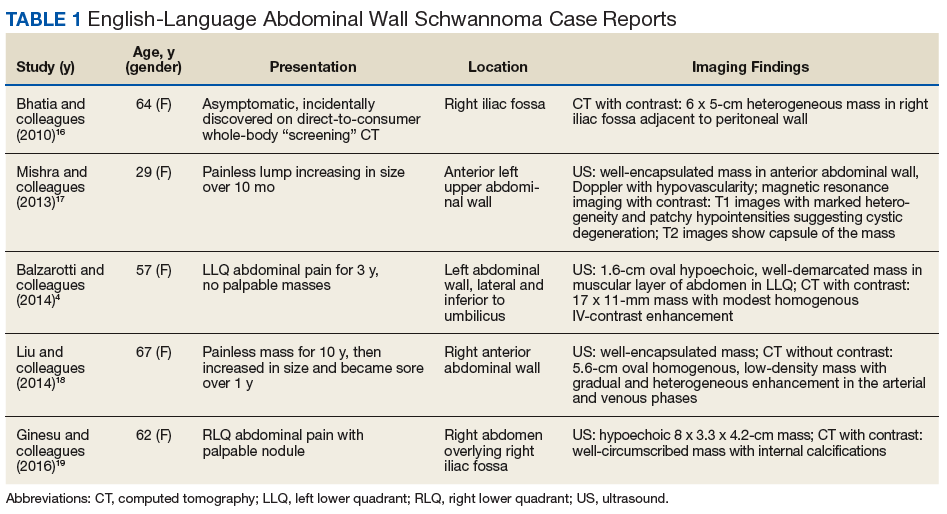
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve. 
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12 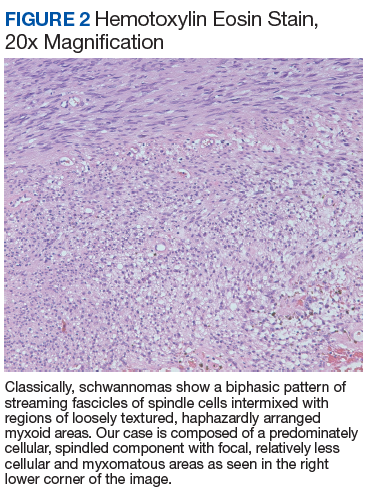
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess.
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1).
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve.
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess.
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1).
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve.
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
Sudden-onset rash on the trunk and limbs • morbid obesity • family history of diabetes mellitus • Dx?
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.

THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION

Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; [email protected]
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.
THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION
Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; [email protected]
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.
THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION
Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; [email protected]
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.

Cutaneous Gummatous Tuberculosis in a Kidney Transplant Patient
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
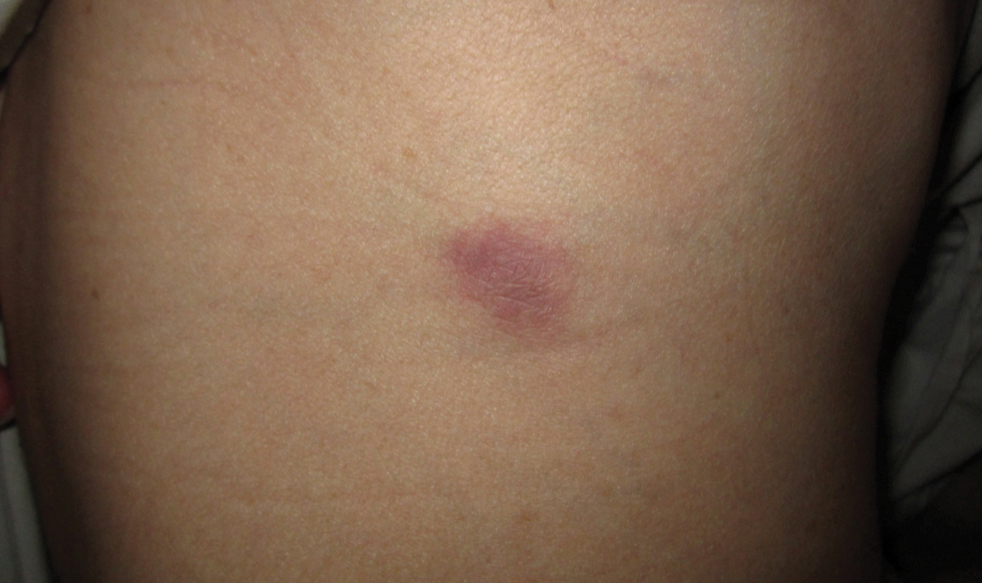
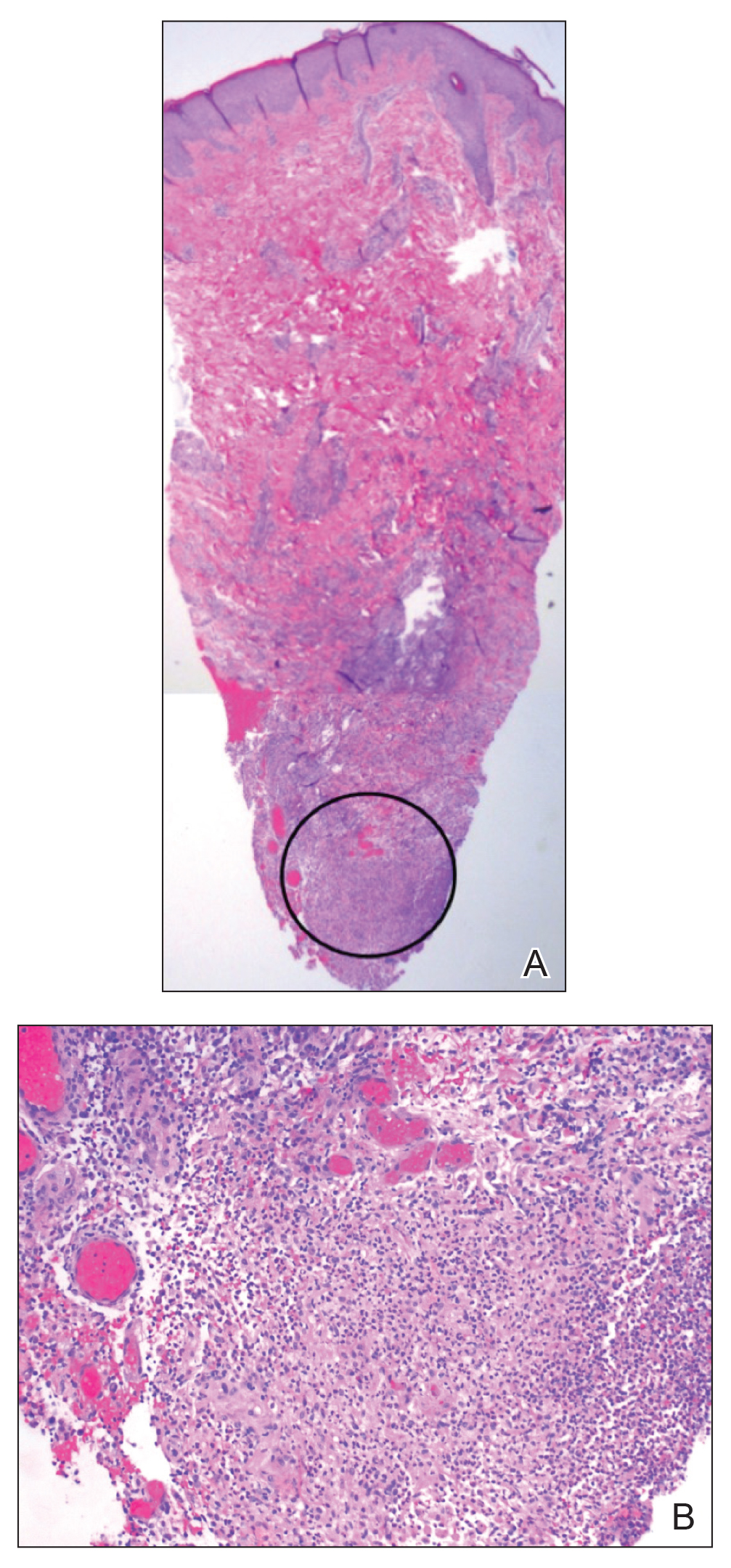
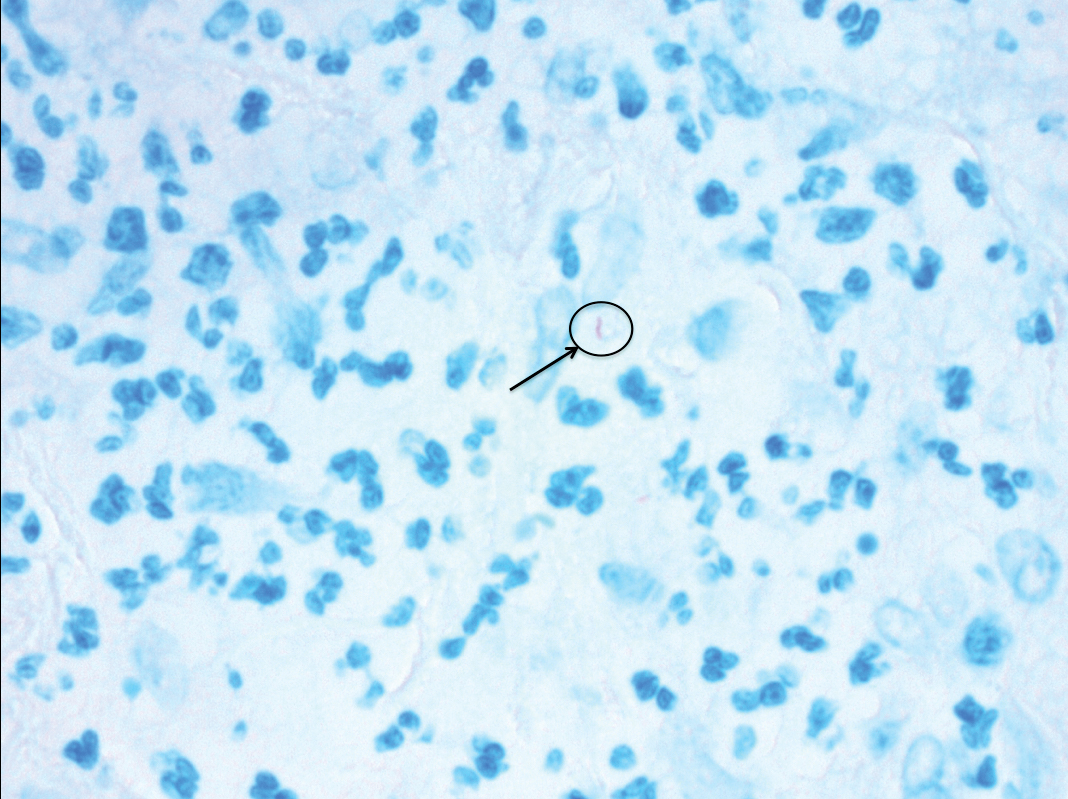
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12

Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12
Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
Case Report
A 60-year-old Cambodian woman presented with recurrent fever (temperature, up to 38.8°C) 7 months after receiving a kidney transplant secondary to polycystic kidney disease. Fever was attributed to recurrent pyelonephritis of the native kidneys while on mycophenolate mofetil, tacrolimus, and prednisone. As a result, she underwent a bilateral native nephrectomy and was found to have peritoneal nodules. Pathology of both native kidneys and peritoneal tissue revealed caseating granulomas and acid-fast bacilli (AFB) diagnostic for kidney and peritoneal tuberculosis (TB). She had no history of TB, and a TB skin test (purified protein derivative [PPD]) upon entering the United States from Cambodia a decade earlier was negative. Additionally, her pretransplantation PPD was negative.
Treatment with isoniazid, ethambutol, pyrazinamide, and levofloxacin was initiated immediately upon diagnosis, and all of her immunosuppressive medications—mycophenolate mofetil, tacrolimus, and prednisone—were discontinued. Her symptoms subsided within 1 week, and she was discharged from the hospital. Over the next 2 months, her immunosuppressive medications were restarted, and her TB medications were periodically discontinued by the Tuberculosis Control Program at the Department of Health (Philadelphia, Pennsylvania) due to severe thrombocytopenia. During this time, she was closely monitored twice weekly in the clinic with blood draws performed weekly.
Approximately 10 weeks after initiation of treatment, she noted recurrent subjective fever (temperature, up to 38.8°C) and painful lesions on the right side of the flank, left breast, and left arm of 3 days’ duration. Physical examination revealed a warm, dull red, tender nodule on the right side of the flank (Figure 1) and subcutaneous nodules with no overlying skin changes on the left breast and left arm. A biopsy of the lesion on the right side of the flank was performed, which resulted in substantial purulent drainage. Histologic analysis showed an inflammatory infiltrate within the deep dermis composed of neutrophils, macrophages, and giant cells, indicative of suppurative granulomatous dermatitis (Figure 2). Ziehl-Neelsen stain demonstrated rare AFB within the cytoplasm of macrophages, suggestive of Mycobacterium tuberculosis infection (Figure 3). A repeat chest radiograph was normal.
Based on the patient’s history and clinical presentation, she was continued on isoniazid, ethambutol, and levofloxacin, with complete resolution of symptoms and cutaneous lesions. Over the subsequent 2 months, the therapy was modified to rifabutin, pyrazinamide, and levofloxacin, and subsequently pyrazinamide was stopped. A subsequent biopsy of the left breast and histologic analysis indicated that the specimen was benign; stains for AFB were negative. Currently, both the fever and skin lesions have completely resolved, and she remains on anti-TB therapy.
Comment
Clinical Presentation
Cutaneous TB is an uncommon manifestation of TB that can occur either exogenously or endogenously.1 It tends to occur primarily in previously infected TB patients through hematogenous, lymphatic, or contiguous spread.2 Due to their immunocompromised state, solid organ transplant recipients have an increased incidence of primary and reactivated latent TB reported to be 20 to 74 times greater than the general population.3,4 One report stated the total incidence of posttransplant TB as 0.48% in the West and 11.8% in endemic regions such as India.5 The occurrence of cutaneous TB is rare among solid organ transplant recipients.1 On average, a diagnosis of latent TB is made 9 months after transplantation because of the opportunistic nature of M tuberculosis in an immunosuppressed environment.6
TB Subtypes
Cutaneous TB can be in the form of localized disease (eg, primary tuberculous chancre, TB verrucosa cutis, lupus vulgaris, smear-negative scrofuloderma), disseminated disease (eg, disseminated TB, TB gumma, orificial TB, miliary cutaneous TB), or tuberculids (eg, papulonecrotic tuberculid, lichen scrofulosorum, erythema induratum).7 Due to the pustular epithelioid cell granulomas and AFB positivity of the involved cutaneous lesions, our patient’s TB can be classified as a metastatic TB abscess or gummatous TB.7
Metastatic TB abscess, an uncommon subtype of cutaneous TB, generally is only seen in malnourished children and notably immunocompromised individuals.2,8,9 In these individuals, systemic failure of cell-mediated immunity enables M tuberculosis to hematogenously infect various organs of the body, resulting in alternative forms of TB, such as gummatous-type TB.10 One study reported that of the 0.1% of dermatology patients presenting with cutaneous TB, only 5.4% of these individuals had the rarer gummatous form.7 These metastatic TB abscesses begin as a single or multiple nontender subcutaneous nodule(s), which breaks down and softens to form a draining sinus abscess.2,8,9 Abscesses are most commonly seen on the trunk and extremities; however, they can be found nearly anywhere on the body.8 The pathology of cutaneous TB lesions demonstrates caseating necrosis with epithelioid and giant cells forming a surrounding rim.9
Diagnosis
Diagnosis may be difficult because of the vast number of dermatologic conditions that resemble cutaneous TB, including mycoses, sarcoidosis, leishmaniasis, leprosy, syphilis, other non-TB mycobacteria, and Wegener granulomatosis.9 Thus, confirmatory diagnosis is made via clinical presentation, detailed history and physical examination, and laboratory tests.11 These tests include the Mantoux tuberculin skin test (PPD or TST) or IFN-γ release assays (QuantiFERON-TB Gold test), identification of AFB on skin biopsy, and isolation of M tuberculosis from tissue culture or polymerase chain reaction.11
At-Risk Populations
The recommendation for the identification of at-risk populations for latent TB testing and treatment have been clearly defined by the World Health Organization (Table).12 Our patient met 2 of these criteria: she had been preparing for organ transplantation and was from a country with high TB burden. Such at-risk patients should be tested for a latent TB infection with either IFN-γ release assays or PPD.12
Treatment
The recommended treatment of active TB in transplant recipients is based on randomized trials in immunocompetent hosts, and thus the same as that used by the general population.16 This anti-TB regimen includes the use of 4 drugs—typically rifampicin, isoniazid, ethambutol, and pyrazinamide—for a 6-month duration.11 Unfortunately, the management of TB in an immunocompromised patient is more challenging due to the potential side effects and drug interactions.
Finally, thrombocytopenia is an infrequent, life-threatening complication that can be acquired by immunocompromised patients on anti-TB therapy.17 Drug-induced thrombocytopenia can be caused by a variety of medications, including rifampicin, isoniazid, ethambutol, and pyrazinamide. Diagnosis of drug-induced thrombocytopenia can be confirmed only after discontinuation of the suspected drug and subsequent resolution of the thrombocytopenia.17 Our patient initially became thrombocytopenic while taking isoniazid, ethambutol, pyrazinamide, and levofloxacin. However, her platelet levels improved once the pyrazinamide was discontinued, thereby suggesting pyrazinamide-induced thrombocytopenia.
Conclusion
The risk for infectious disease reactivation in an immunocompromised patient undergoing transplant surgery is notable. Our findings emphasize the value of a comprehensive pretransplant evaluation, vigilance even when test results appear negative, and treatment of latent TB within this population.16,18,19 Furthermore, this case illustrates a noteworthy example of a rare form of cutaneous TB, which should be considered and included in the differential for cutaneous lesions in an immunosuppressed patient.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
- Sakhuja V, Jha V, Varma PP, et al. The high incidence of tuberculosis among renal transplant recipients in India. Transplantation. 1996;61:211-215.
- Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
- Schultz V, Marroni CA, Amorim CS, et al. Risk factors for hepatotoxicity in solid organ transplants recipients being treated for tuberculosis. Transplant Proc. 2014;46:3606-3610.
- Tabarsi P, Farshidpour M, Marjani M, et al. Mycobacterial infection and the impact of rifabutin treatment in organ transplant recipients: a single-center study. Saudi J Kidney Dis Transpl. 2015;26:6-11.
- Rathi M, Gundlapalli S, Ramachandran R, et al. A rare case of cytomegalovirus, scedosporium apiospermum and mycobacterium tuberculosis in a renal transplant recipient. BMC Infect Dis. 2014;14:259.
- Hickey MD, Quan DJ, Chin-Hong PV, et al. Use of rifabutin for the treatment of a latent tuberculosis infection in a patient after solid organ transplantation. Liver Transpl. 2013;19:457-461.
- Kumar B, Muralidhar S. Cutaneous tuberculosis: a twenty-year prospective study. Int J Tuberc Lung Dis. 1999;3:494-500.
- Dekeyzer S, Moerman F, Callens S, et al. Cutaneous metastatic tuberculous abscess in patient with cervico-mediastinal lymphatic tuberculosis. Acta Clin Belg. 2013;68:34-36.
- Ko M, Wu C, Chiu H. Tuberculous gumma (cutaneous metastatic tuberculous abscess). Dermatol Sinica. 2005;23:27-31.
- Steger JW, Barrett TL. Cutaneous tuberculosis. In: James WD, ed. Textbook of Military Medicine: Military Dermatology. Washington, DC: Borden Institute; 1994:355-389.
- Santos JB, Figueiredo AR, Ferraz CE, et al. Cutaneous tuberculosis: diagnosis, histopathology and treatment - part II. An Bras Dermatol. 2014;89:545-555.
- Guidelines on the Management of Latent Tuberculosis Infection. Geneva, Switzerland: World Health Organization; 2015.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 pt 2):S221-S247.
- Mycobacterium tuberculosis. Am J Transplant. 2004;4(suppl 10):37-41.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis. 2009;48:1276-1284.
- Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
- Kant S, Verma SK, Gupta V, et al. Pyrazinamide induced thrombocytopenia. Indian J Pharmacol. 2010;42:108-109.
- Screening for tuberculosis and tuberculosis infection in high-risk populations. recommendations of the Advisory Council for the Elimination of Tuberculosis. MMWR Recomm Rep. 1995;44:19-34.
- Fischer SA, Avery RK; AST Infectious Disease Community of Practice. Screening of donor and recipient prior to solid organ transplantation. Am J Transplant. 2009;9(suppl 4):S7-S18.
Practice Points
- Transplant patients are at increased risk for infection given their immunosuppressed state.
- Although rare, cutaneous tuberculosis should be considered in the differential for cutaneous lesions in an immunosuppressed patient.
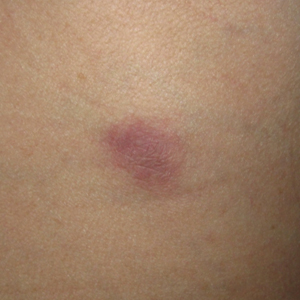
Cutaneous Rosai-Dorfman Disease
Case Report
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
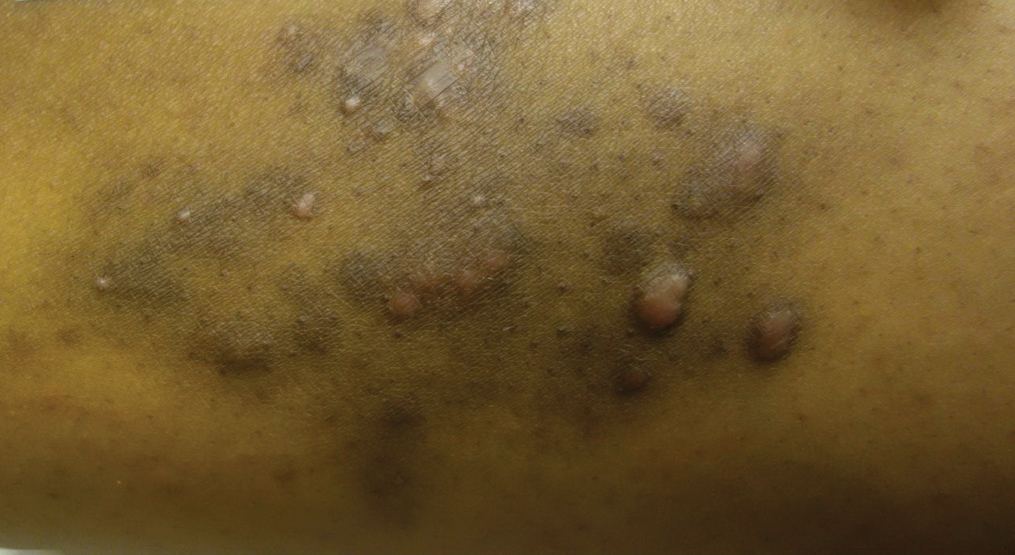
Histopathology
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
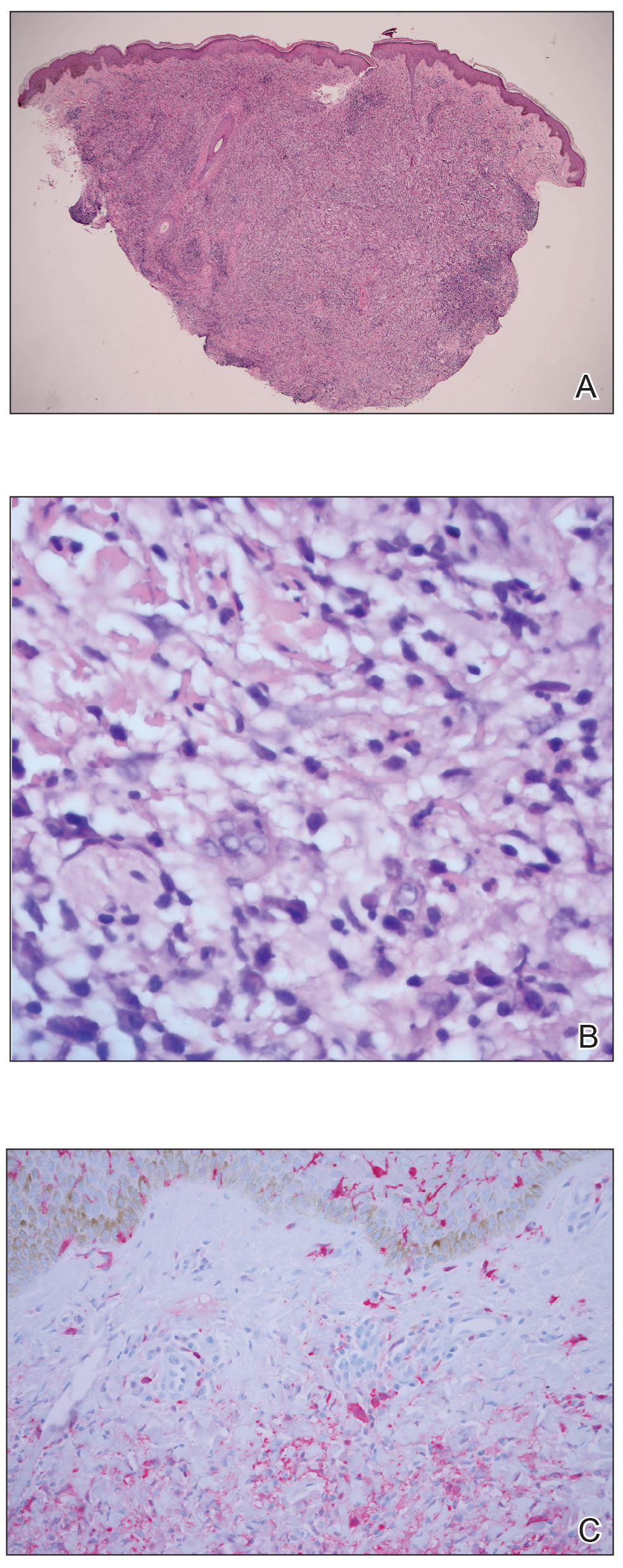
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
Case Report
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
Histopathology
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
Case Report
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
Histopathology
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
Practice Points
- Rosai-Dorfman disease generally is characterized by painless cervical lymphadenopathy and systemic involvement.
- Cutaneous Rosai-Dorfman disease can clinically present with great variety, mimicking many other dermatologic conditions.
- Patients presenting with cutaneous lesions and lymphadenopathy warrant workup with systemic imaging.