User login
Eccrine Porocarcinoma Presenting as a Recurrent Wart
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
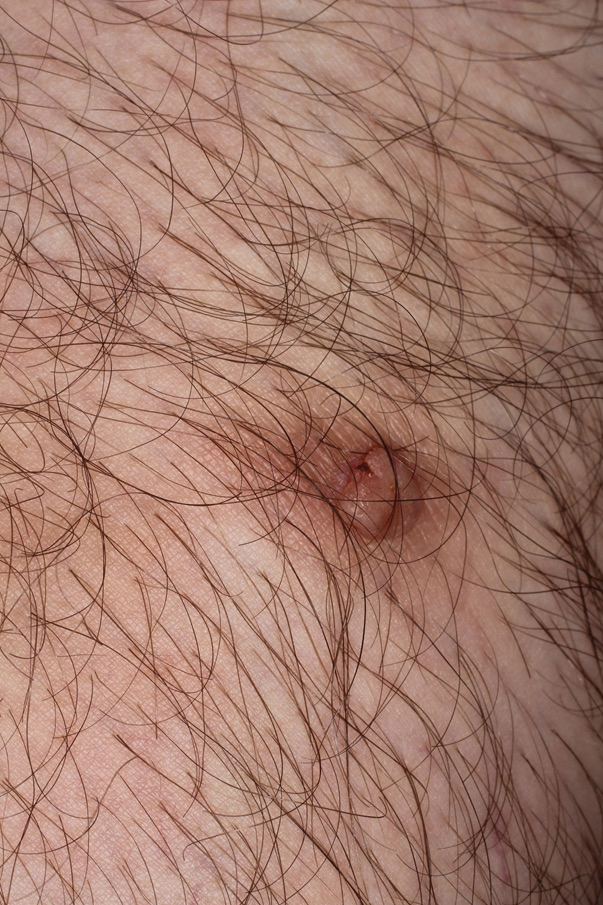
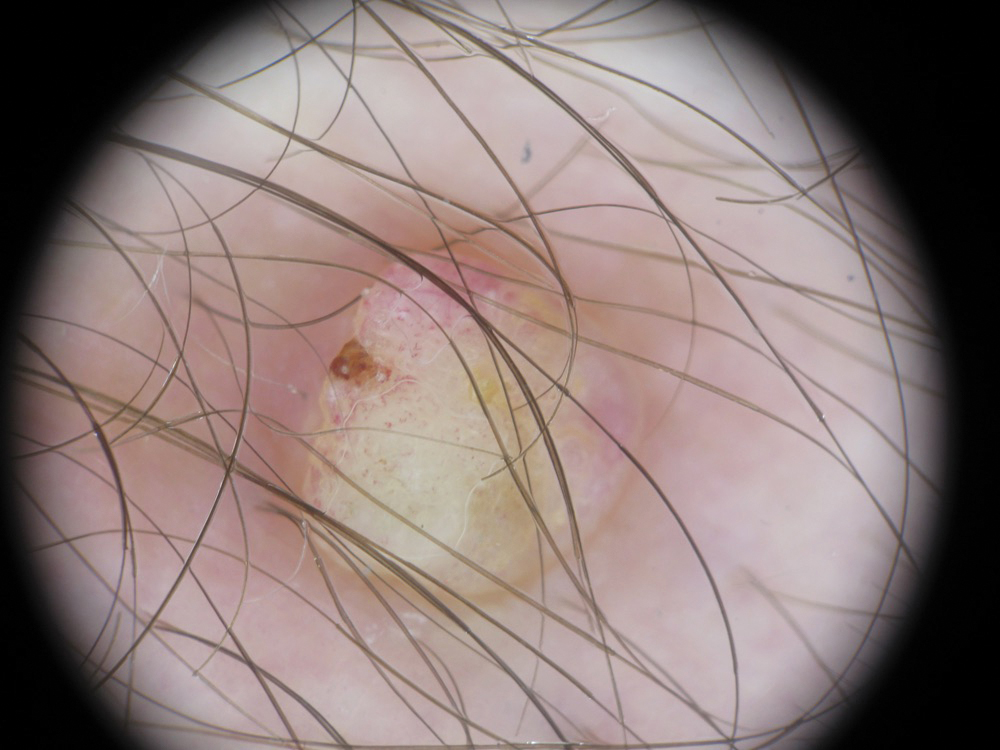
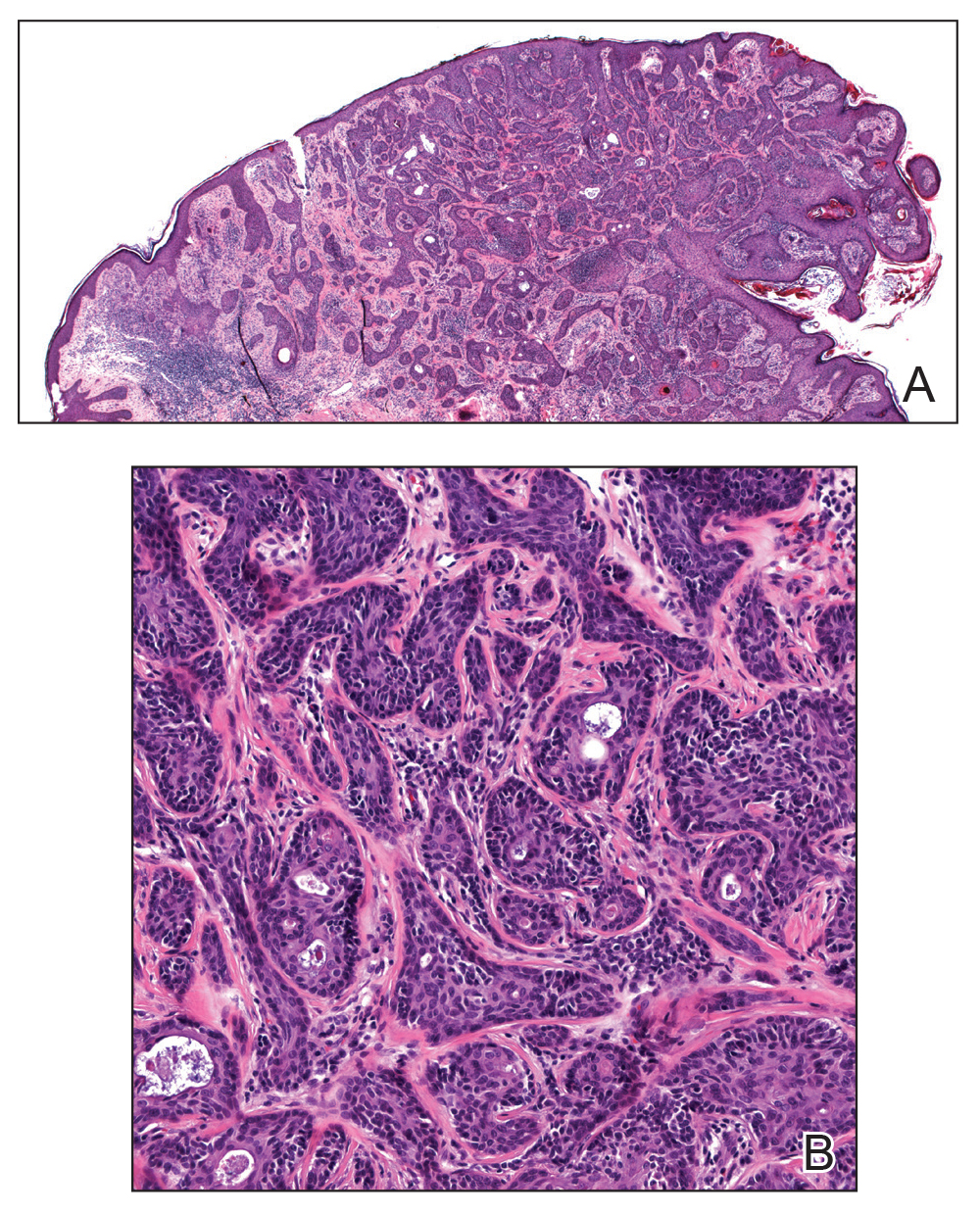
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
Practice Points
- Eccrine porocarcinoma is more common in older patients (age range, 71–75 years).
- Local recurrence and nodal metastasis are reported as high as 20% with wide local excision.
- Higher cure rates recently have been reported with Mohs micrographic surgery.
Hailey-Hailey Disease: A Diagnostic Challenge
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
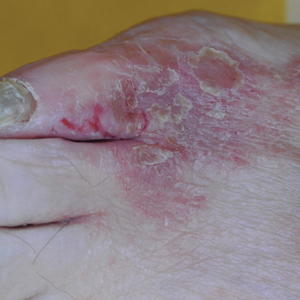
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
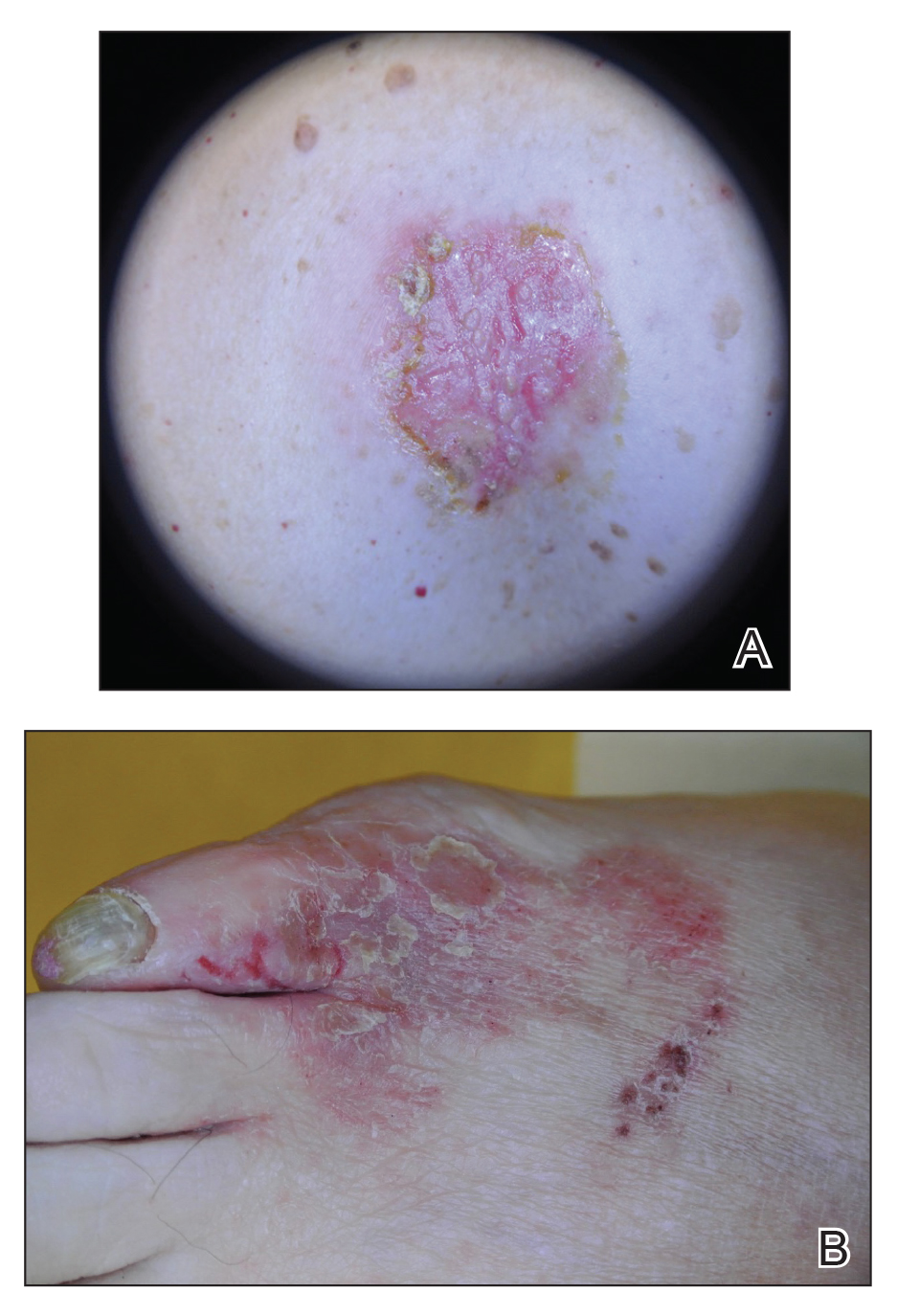
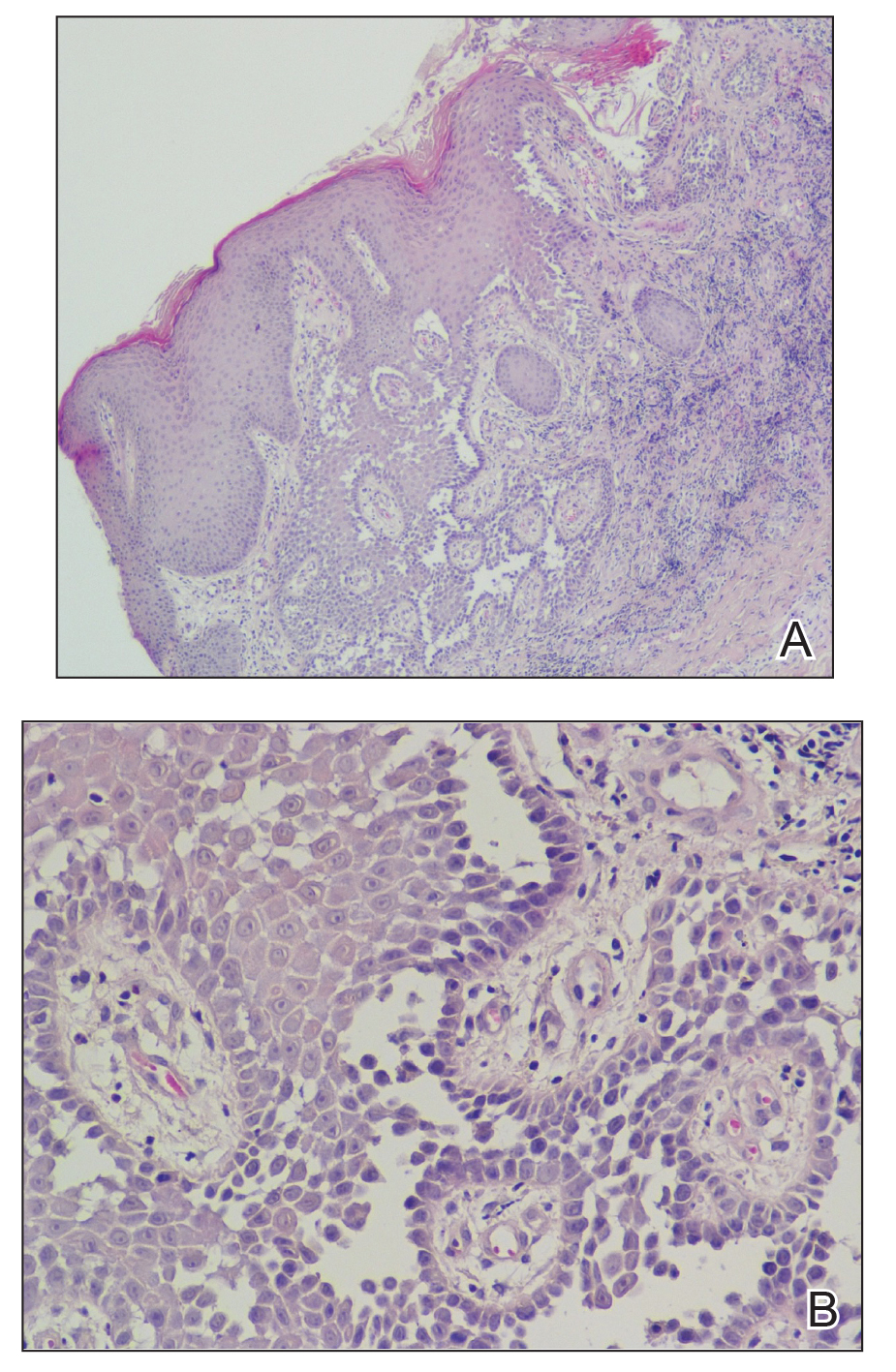
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
Hailey-Hailey disease (HHD), also known as benign familial chronic pemphigus, is an autosomal-dominant genodermatosis caused by mutations of the ATPase secretory pathway Ca2+ transporting 1 gene, ATP2C1.1 It is characterized by crusted macerated erosions and velvety, fissured, hypertrophic plaques classically involving the intertriginous areas. The diagnosis is suggested by characteristic clinical morphology, involvement of the intertriginous areas, and a positive family history. Histology often confirms the diagnosis and demonstrates a characteristic dilapidated brick wall appearance. If there is a need to distinguish HHD from pemphigus, direct immunofluorescence studies also should be performed, which would be negative.2,3 However, HHD often is misdiagnosed due to lack of knowledge of this uncommon disorder and its resemblance to other dermatoses of the intertriginous areas.4 We present an unusual presentation of HHD with late onset and involvement of the skin of the abdomen and foot.
Case Report
A 61-year-old woman presented with a 3×4-cm fissured plaque with erosions and a peripheral yellow crust on the left side of the anterior abdomen (Figure 1A). There was another fissured plaque with surrounding erythema and scaling on the fifth digit of the right foot (Figure 1B). For the last 11 years, she periodically experienced erosive and scabbing skin plaques under the breasts and on the axillae and groin. Her mother and maternal grandfather had a history of similar skin lesions. Due to a suspicion of HHD, a skin biopsy specimen of the abdominal plaque was performed, which demonstrated epidermal acanthosis and suprabasal acantholysis with lacunae formation (Figure 2). There was uneven thickening of the epidermal keratin layer with parakeratotic nests. The upper layer of the dermis demonstrated edema and focal fibrosis, enlarged capillaries, and pericapillary lymphohistiocytic infiltration with eosinophils and neutrophils. Accordingly, a diagnosis of HHD was established.
Comment
Hailey-Hailey disease occurs in 1 to 4 per 100,000 individuals without predilection for sex or ethnic group.5-9 Onset usually occurs after puberty, most commonly in the third decade of life.8,10-12 Mutations of the ATP2C1 gene on band 3q22.1 cause haploinsufficiency of Ca2+/Mn2+−ATPase protein 1 (hSPCA1) that alters the intracellular calcium gradient, leading to disruptions in assembly and trafficking of desmosomal proteins to the cell membrane. Consequently, altered intercellular connections and acantholysis of the epidermis occur.1,13-16
Hailey-Hailey disease initially manifests as grouped flaccid vesicles that rupture easily, leaving behind crusted erosions and dry, scaly, eczematous patches.17,18 Over time, velvety, fissured, and hypertrophic plaques develop. Up to 80% of patients experience secondary bacterial and fungal superinfections that may cause vegetative or malodorous plaques.9 Although HHD has no specific treatment, symptoms are managed with topical corticosteroids and antimicrobial agents. Patients should be advised to avoid irritants such as friction, sunlight, or sweat. For severe cases, botulinum toxin type A, laser therapy, dermabrasion, and surgery have been utilized with variable success.19-22 The responsiveness of HHD to corticosteroids and antimicrobial agents facilitates misdiagnosis as intertrigo, erythrasma, or dermatophytosis.
Our patient presented with late-onset HHD (age, 50 years) compared to the typical age of onset in the third decade of life.8 Furthermore, her presentation was atypical for HHD, which characteristically affects intertriginous areas due to sweat, heat, friction, and microorganisms. Hailey-Hailey disease involving the abdominal skin is unusual, as it typically occurs in regions of friction such as the belt area.23 Our patient lacked a history of friction or trauma at the site of the abdominal plaque. In addition, HHD involving the feet is exceedingly rare. It is plausible that friction and heat caused by footwear may have predisposed her to these skin changes.
Conclusion
This case highlights the difficulties of diagnosing HHD, especially if it appears in atypical locations.24 Obtaining a thorough family history and detailed dermatologic examination as well as maintaining a high level of suspicion can assist in diagnosing this uncommon disorder.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a alcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-65.
- Ohata C. Hailey-Hailey disease. Cutis. 2014;94:33-34.
- Abdullah L, Abbas O. Dermacase. can you identify this condition? benign familial chronic pemphigus. Can Fam Physician. 2011;57:1157-1158.
- Le Donne M, Lentini M, Moretti G, et al. Chronic vulvocrural dermatitis with burning and itching. CMAJ. 2008;179:555-556.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Saunders; 2012:887-897.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
- Godic A, Miljkovic J, Kansky A, et al. Epidemiology of Darier’s disease in Slovenia. Acta Dermatovenerol Alp Pannonica Adriat. 2005;14:43-48.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-282.
- Benmously-Mlika R, Bchetnia M, Deghais S, et al. Hailey-Hailey disease in Tunisia. Int J Dermatol. 2010;49:396-401.
- Bessa GR, Grazziotin TC, Manzoni AP, et al. Hailey-Hailey disease treatment with botulinum toxin type A. An Bras Dermatol. 2010;85:717-722.
- Gu H, Chang B, Chen W, et al. Clinical analysis of 69 patients with familial benign chronic pemphigus. Chin Med J (Engl). 1999;112:761-763.
- Dobson-Stone C, Fairclough R, Dunne E, et al. Hailey-Hailey disease: molecular and clinical characterization of novel mutations in the ATP2C1 gene. J Invest Dermatol. 2002;118:338-343.
- Fairclough RJ, Lonie L, Van Baelen K, et al. Hailey-Hailey disease: identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. J Invest Dermatol. 2004;123:6771.
- Shibata A, Sugiura K, Kimura U, et al. A novel ATP2C1 early truncation mutation suggests haploinsufficiency as a pathogenic mechanism in a patient with Hailey-Hailey disease. Acta Derm Venereol. 2013;93:719-720.
- Dhitavat J, Fairclough RJ, Hovnanian A, et al. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821-828.
- Raiko L, Siljamaki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21:586-591.
- Yadav N, Madke B, Kar S, et al. Hailey-Hailey disease. Indian Dermatol Online J. 2016;7:147-148.
- Vasudevan B, Verma R, Badwal S, et al. Hailey-Hailey disease with skin lesions at unusual sites and a good response to acitretin. Indian J Dermatol Venereol Leprol. 2015;81:88-91.
- Bagherani N, Smoller BR. The efficacy of botulinum toxin type A in the treatment of Hailey Hailey disease. Dermatol Ther. 2016;29:394-395.
- Hochwalt PC, Christensen KN, Cantwell SR, et al. Carbon dioxide laser treatment for Hailey-Hailey disease: a retrospective chart review with patient-reported outcomes. Int J Dermatol. 2015;54:1309-1314.
- Falto-Aizpurua LA, Griffith RD, Yazdani Abyaneh MA, et al. Laser therapy for the treatment of Hailey-Hailey disease: a systematic review with focus on carbon dioxide laser resurfacing. J Eur Acad Dermatol Venereol. 2015;29:1045-1052.
- Arora H, Bray FN, Cervantes J, et al. Management of familial benign chronic pemphigus. Clin Cosmet Investig Dermatol. 2016;9:281-290.
- Iijima S, Hamada T, Kanzaki M, et al. Sibling cases of Hailey-Hailey disease showing atypical clinical features and unique disease course. JAMA Dermatol. 2014;150:97-99.
- Saied NK, Schwartz RA, Hansen RC, et al. Atypical familial benign chronic pemphigus. Cutis. 1981;27:666-669.
Practice Points
- Hailey-Hailey disease may present atypically with a late age of onset, involvement of nonintertriginous areas, and lack of clear exacerbating factors such as friction.
- A detailed history and physical examination as well as a high degree of suspicion can aid in diagnosing this uncommon disorder.
Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
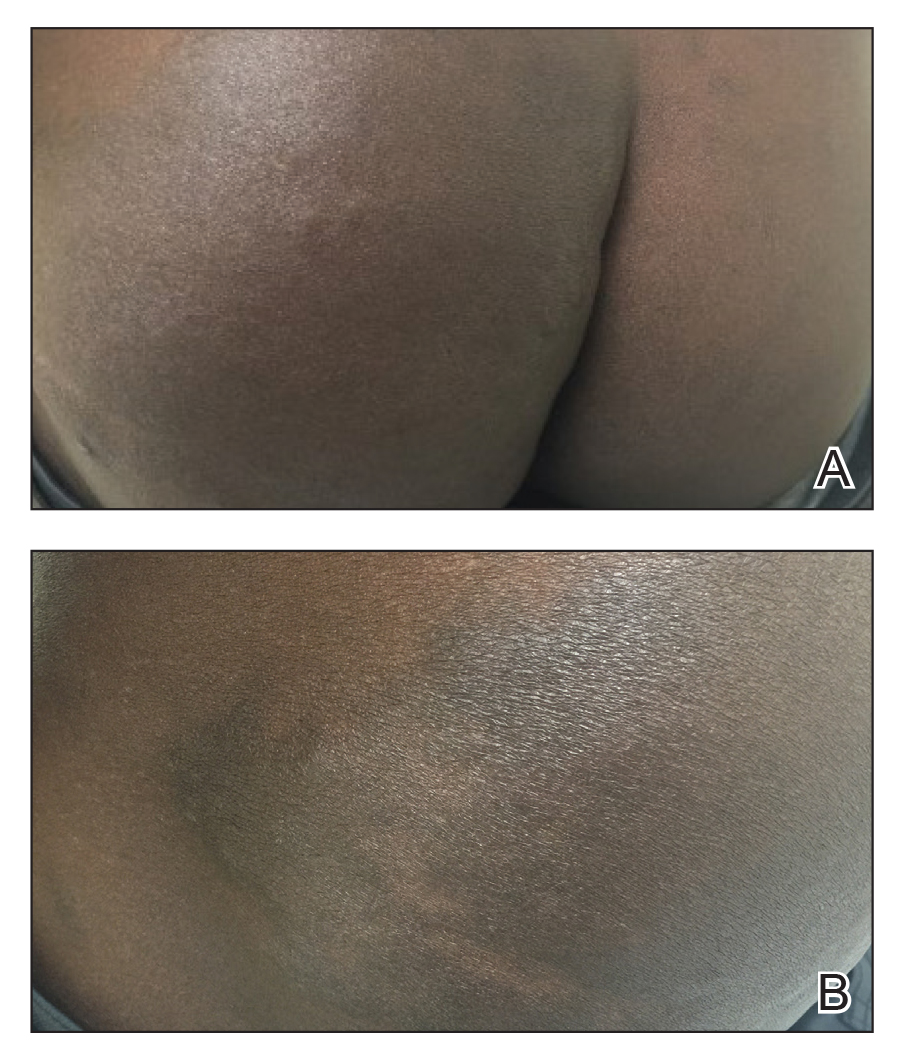
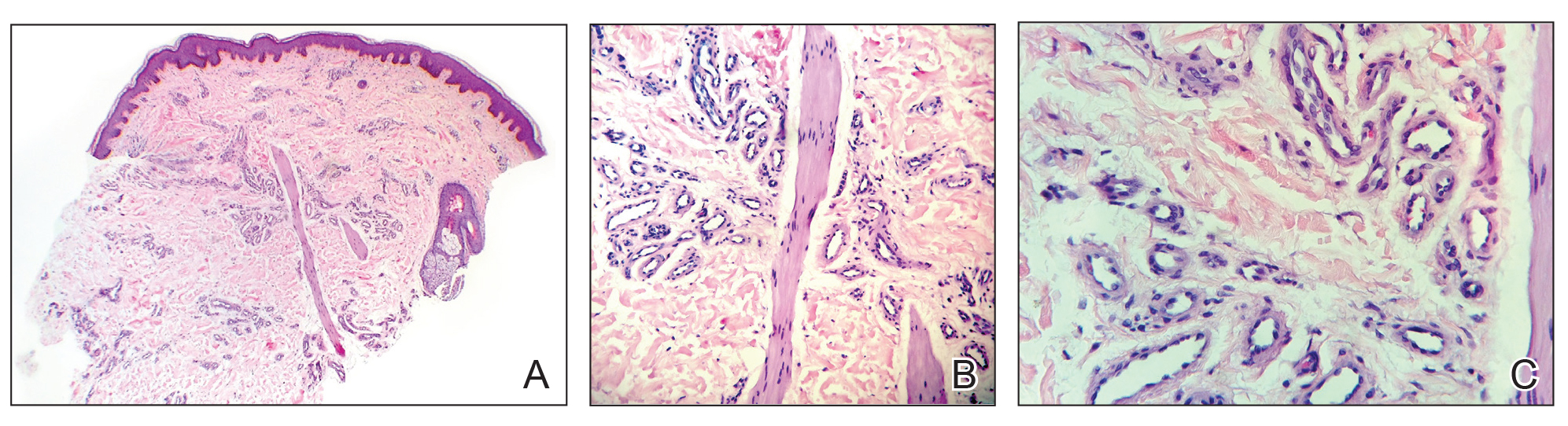
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
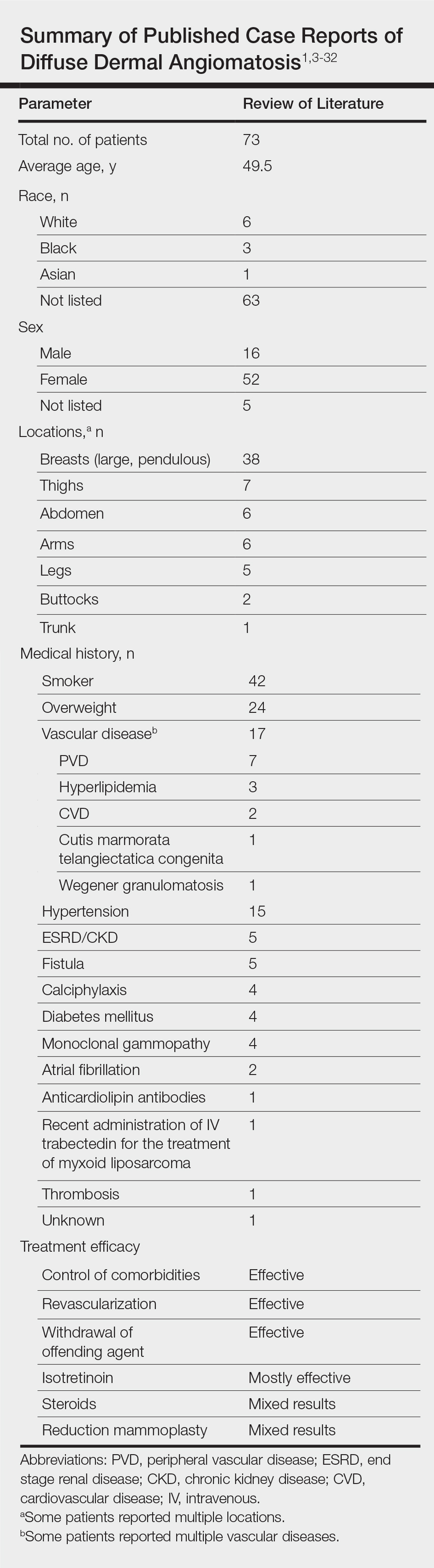
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
Practice Points
- Diffuse dermal angiomatosis is commonly reported in patients with hypoxic comorbidities such as smoking or vascular disease as well as in women with large pendulous breasts.
- Effective treatments include control of comorbidities, revascularization, withdrawal of the offending agent, steroids, and isotretinoin.
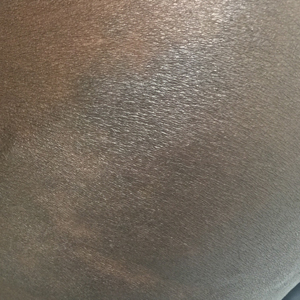
Breast Cancer Tumor Board
Case Presentation
This case represents a composite of many different patients and is not meant to represent an individual. Any resemblance to an actual patient is coincidental.
A 32-year-old African American woman presented with a self-palpated left breast mass (axillary tail at 9 o’clock position). The patient was a nonsmoker, was otherwise healthy, and had no family history of breast or any other cancer. She had never used oral contraceptives or hormones, was never pregnant, her menarche was at age 12 years, and she had regular menstrual periods. On physical examination she had a 1-cm left breast mass and a palpable left axillary lymph node. A complete diagnostic workup revealed a 2-cm left breast mass. An ultrasound-guided biopsy of the axillary lymph node was positive for invasive ductal carcinoma (IDC). The final diagnosis was left breast cancer, stage IIB IDC, T1N1M0, ER+, PR+, HER2 2+ by immunohistochemistry, fluorescence in situ hybridization (FISH) was 2.4, confirming a HER2+ tumor.
Anita Aggarwal, DO, PhD. What is the role of genetic counseling and testing in this young patient who does not have a family history of breast cancer?
Vickie L. Venne, MS. This patient absolutely would be a candidate for counseling and testing. From a genetic counseling perspective, one of the first points has to do with what “no family history of cancer” means. Typically, in a fast-paced clinic, a patient will be asked “Does anybody else in your family have cancer?” And it’s not uncommon to get the answer “no.” Genetic counselors collect specific information on at least the first- and second-degree relatives, so we end up with 3 generations. This includes both the maternal and the paternal histories. We find that people who initially report no family history of cancer are often just thinking of breast cancer, even if the provider’s question is broad. When we start digging, we often find other cancers because cancer is common.
The other issue is that she was diagnosed at a young age. Clearly, 32 years is much younger than we typically see in breast cancer, and we know that individuals with hereditary cancers often have an earlier age of onset. With no other information, her a priori risk of having a BRCA1/2 mutation would be < 2.5%.
Regardless, based on current National Comprehensive Cancer Network (NCCN) guidelines, she would be a testing candidate. We would also recommend testing for more than just BRCA1/2. In the last few decades, there have been many genes identified that are associated with an increased susceptibility to cancer. Many of these genes are part of syndromes, so if you had a mutation, that also would increase the risk for a cancer in another organ. If this woman’s mother and father lived into their 70s or 80s and she had a number of aunts on both sides who never developed breast cancer, it would be less likely to be BRCA1/2. However, P53 also can present in young women and as a de novo mutation. Therefore, we would offer her a panel of actionable genes. Genes that if, in fact, we identified a mutation in one of them, would mean we could do something different for this young lady.
JoAnn Manning, MD. Let’s say she does have testing, and she comes back BRCA+. Then what would be the recommendations or guidance?
Ms. Venne. Women (and men as well!) with mutations have an increased risk for a second primary breast cancer as well as cancer in other organs. Focusing first on the breast story and all the media around BRCA1/2 mutations and surgery, this is a woman who may consider a more aggressive surgery, including prophylactic contralateral mastectomy, if she is concerned. She is young, so we also would explore her fertility plans. While her next few months will be filled with breast cancer treatment choices, women with BRCA mutations also are at an increased risk to develop ovarian cancer, so that might be a decision she makes as well. Her health care team may also eventually discuss chemotherapeutic options available specifically to women with mutations.
However, we often see young women who are extremely nervous because there is a sense that if you’re younger, your cancer must be inherited. Part of the pretest counseling is to explore psychosocial issues and help these young ladies understand that, especially if she does not have a family history of cancer and the only indication is her age, then it’s highly likely that we’re not going to find an identifiable mutation. And in that circumstance, she probably could consider a more conservative surgical decision.
Dr. Aggarwal. How common is a BRCA1 or BRCA2 mutation in African American females?
Ms. Venne. I have not paid attention to the prevalence of mutations based on ethnicity, so I don’t know. While many of the initial mutations were discovered in women of European ancestry, there are large cohorts of women with African ancestry whose specimens are now available for identifying genetic markers that will improve breast cancer risk assessment in them.1 However, because those mutations are still being characterized, it is more common to find a variant of uncertain significance (VUS) in African American women. A VUS is an alteration—a change in the gene—that we simply don’t know what it means yet. Clinicians don’t have enough information to know if that alteration is pathogenic or benign. The problem is that people try to make sense out of everything in their lives, so they also will try to make a VUS mean something. We try hard to help people understand that a VUS is really no more significant than if we had not tested in the first place, and they should not act on that information. They should use their family history, their age, their other psychosocial concerns about their experiences with cancer as they make their treatment decisions. But they also should check back periodically with their genetic counselor because VUSs can be reclassified. And if that happens, the information might be more useful for not only them, but their family members.
Dr. Manning. Would you consider this patient for any neoadjuvant chemotherapy?
Dr. Aggarwal. The patient is a young female with a small tumor that is HER2+. The indication for neoadjuvant chemotherapy is typically a big tumor or inoperable disease. Neoadjuvant chemotherapy is considered the standard of care for patients with inflammatory breast cancer and may confer a survival benefit in these patients. Of all the breast cancer subtypes, triple negative and HER2+ are considered the most chemosensitive and may benefit from neoadjuvant therapy. This patient has a small tumor, and I don’t think she’s a candidate for neoadjuvant chemotherapy unless the patient wants to see if her tumor is chemosensitive or not.
Dr. Manning, What’s the role and benefit of lumpectomy vs mastectomy?
Dr. Manning. Historically, mastectomy would have been considered the standard of care, but luckily, in the 1970s and the 1980s, we had a significant number of randomized controlled trials that demonstrated that certain women with particular characteristics would get the same overall survival if they chose mastectomy vs lumpectomy, the removal of the tumor with negative margin and whole-breast radiation. The key thing to understand is that breast-conserving surgery is now very well established with more than 20 years of data to support it. And that breast irradiation after breast-conserving surgery is essential to maximizing the local control and the overall survival (OS).
There have been a lot of major studies, but the one with the greatest follow-up now is the National Surgical Adjuvant Breast and Bowel Project (NSABP) B06 protocol, which was the only trial to compare mastectomy to lumpectomy and radiation or lumpectomy alone. It required negative margins. With 20 years of follow-up, the data still support that mastectomy or lumpectomy with radiation offers equivalent OS and local control. It’s really about patient preference if they are candidates.
Who is a candidate? Clearly, there are contraindications. We tend to look primarily at the size of the tumor. However, removing an average-sized tumor (< 2 cm) with a margin may not have a good cosmetic result for a patient with very small breasts. That patient may opt to go forward with a mastectomy instead. Young patients who are candidates must have to have negative margins. If they have persistently positive resection margins after excision or reexcision, then they need to go forward with mastectomy.
A patient who has imaging evidence of multicentric disease with 2 or more primary tumors in separate quadrants would not be a candidate for breast-conserving therapy. Diffuse malignant-appearing microcalcifications on a mammogram also would suggest multicentric disease. And a patient with a prior history of radiation therapy to the breast or chest wall cannot go through breast-conserving therapy.
In the case we are discussing, we also should make sure this young lady is not pregnant. If the patient is adamant about breast-conserving surgery and pregnant, especially in the third trimester, radiation could be deferred until after delivery. Another relative contraindication is for patients who have connective tissue disorders. Sometimes if they are given whole-breast radiation, the cosmetic result is poor. So if you’re doing this procedure to save the breast, then having a good cosmetic result is an important consideration for many patients.
When you look at the size of the tumor for this patient, she seems to be a good candidate for breast-conserving surgery. I would recommend that she go forward with lumpectomy followed by whole-breast radiation.
Ms. Venne. Although the numbers aren’t nearly as large as they were in the original trials looking at the lumpectomy vs mastectomy, there are now survival data for women with BRCA1/2 mutations. With all of the caveats that Dr. Manning mentioned, even if you have an identifiable mutation, you may not necessarily need that more aggressive surgery.2 Clearly, individuals with identifiable mutations would have a higher chance of a contralateral breast cancer, a second primary, so some individuals consider a prophylactic bilateral mastectomy. But from a survival perspective, there are a fair amount of data now available that say that lumpectomy vs mastectomy should really be the conversation based on all of the information that Dr. Manning outlines rather than using primarily the mutation status
Dr. Manning. I agree.
Dr. Aggarwal. This patient had a lumpectomy and axillary lymph node dissection. Pathology reported 1.5-cm mass, grade 3 IDC; the margins were negative. There was no skin involvement, 27 lymph nodes removed were all negative. Dr. Manning, can you please discuss the role of radiation in early stage breast cancer in patients like this case?
Dr. Manning. One of the questions that is always controversial for radiation in these early stage breast cancer cases is what do you do with the nodal irradiation? Previously, radiation oncologists based treatment plans on retrospective data, but in 2015, there were 2 major studies, 1 from Canada, and 1 from the European Organisation for Research and Treatment of Cancer (EORTC).3,4 Both studies tried to determine whether there was an advantage to doing regional nodal irradiation in early breast cancer cases. That encompassed axillary, supraclavicular, and internal mammary nodes. The studies showed that there was no survival advantage, but there was a statistically significant improvement in disease free survival and in local regional recurrence and distant mets.
Unfortunately, there are still a lot of unanswered questions, like what group potentially would benefit the most? In the MA.20 Study, some observers questioned that maybe the ER-/PR- women had the most benefit, but then, in the other study the benefit wasn’t clear.4,5 One question is which lymph node group is having the most impact? Was the benefit from radiating the supraclavicular nodes or was it from radiating the internal mammary nodes? Determining the answer is important from a technical point of view because when you radiate the internal mammary nodes, you have the potential to expose more heart and lung to radiation. You have to put all these together and make a recommendation.
Clearly, for a patient with negative nodes there is no question: You would not treat the regional nodes. However, for a patient with positive nodes you really have to individualize the approach and consider age, anatomy, tumor location, and burden of axillary disease.
I would sit down and have a discussion with this young woman to weigh the risks and the benefits. There is a slight increased risk of lymphedema in these patients, and radiation pneumonitis increases, but not significantly. A key concern is to minimize the total dose of radiation to the heart. There have been great developments in radiation oncology technology and capabilities, so the cardiac dose is now less. But when you think about a 32-year-old patient and weigh the benefit of a 2% to 3% decrease in the incidence of distant metastases and no OS advantage, then you really need to have a conversation about how to safely treat her. At a minimum, I would treat the high axilla and the supraclavicular nodes because she had a pretty extensive lymph node dissection with more than 20 nodes, and then with her getting systemic therapy, that should be more than adequate.
Dr. Aggarwal. Is there any cutoff for age or size of the tumor where you would not do any radiation to the breast?
Dr. Manning. In this particular patient absolutely not because of the lymph node. She had breast-conserving therapy, and she’s only 32-years-old. The PRIME 2 study offered lumpectomy alone vs lumpectomy and radiation for women aged ≥ 65 years with tumors ≤ 3 cm, low grade.6 The study participants had to have negative lymph nodes, be ER+, and low grade. It was a very select group. The lumpectomy patients had a recurrence rate around 4%, and the other was closer to 1.3%.
You have to look at the whole picture. Is this a healthy 70-year-old woman? Is it an inconvenience for her to get treatment? Is she going to get hormone therapy and will she be adherent? There’s a very small group of women who underwent breast-conserving surgery that I would feel safe about not offering radiation.
Dr. Aggarwal. About 15% to 20% of all breast cancers are HER2 over expressors, which used to be a poor prognostic characteristic. However, the development of anti-HER2 therapies has changed the picture of HER2 prognosis. After the initial discovery of activity, the pivotal study by Slamon et al showed benefit in terms of progression-free survival (PFS) and OS with chemotherapy and trastuzumab. The NCCN guideline recommends anti-HER2 antibody trastuzumab in combination with chemotherapy.7
Patients with tumor < 0.5 cm who are HER2+ and ER+ may not benefit from trastuzumab, but those who are ER- and HER2+ will still benefit from trastuzumab. The combination is adriamycin/cyclophosphamide followed by a taxane with trastuzumab and to complete 1 year of trastuzumab or trastuzumab in combination with carboplatin and taxanes.
Pertuzumab, in combination with trastuzumab and docetaxel (PHT) has been FDA-approved in neoadjuvant and metastatic HER2+ disease, but is not FDA approved yet in the adjuvant setting. However, these are expensive drugs, and we don’t know how long these drugs should be given.
Mr. Crawford, What are the adverse effects (AEs) of an anti-HER2 or trastuzumab treatment, and what is the cost of trastuzumab?
Russell Crawford, BPharm. The anti-HER2 antibodies have certainly changed treatment plans and outcomes for patients with breast cancer who test HER2+. There are actually 3 of these anti-HER2 drugs on the U.S. market, and they can be used in a variety of settings. Trastuzumab and pertuzumab are indicated in women or patients who have HER2+ disease, and they work by binding to the extracellular domain of the HER2 proteins and mediate antibody-dependent cellular toxicity by inhibiting proliferation of the cells that overexpress HER2.
In this patient, we would be looking at using adjuvant trastuzumab to complete a 1-year course of therapy while she’s getting her dose-dense doxorubicin and cyclophosphamide (AC) on a weekly basis for the first 12 weeks. Trastuzumab is dosed with an initial loading dose of 4 mg/kg as the first dose, and then it’s 2 mg/kg/wk until adjuvant chemotherapy is completed. We usually extend the dosing out to 6 mg/kg every 3 weeks to complete the year of treatment.
These drugs are fairly well tolerated. They are monoclonal proteins, so a lot of the AEs that patients experience are the things that we’re used to seeing with other monoclonal proteins like the infusion-related reactions and some flulike symptoms. The biggest concern with these patients is that being on the drug for a year, there is a risk of decreasing the left ventricular ejection fraction (LVEF) of the heart. That risk is increased when these drugs are combined with anthracyclines that we know are cardiotoxic. As a single agent, the impact on left ventricular function is not significant, but when it is combined with chemotherapy, it does become a problem. Usually, we recommend routine and periodic monitoring of the LVEF with a multiple-gated acquisition or an echocardiogram to make sure that we’re not causing harm related to this treatment.
The cost of these drugs depends on the frequency, is it every week, every 2 weeks, or every 3 weeks? There are different ways to give trastuzumab, but for most patients, we prefer the every 3-week dose. And it’s estimated that for a 70-kg patient, a dose of trastuzumab at 6 mg/kg at the rate of every 3 weeks costs about $2,500 per dose. The VA pays about $6 a milligram, but it’s certainly money well spent because it has changed the playing field and the outcomes for these patients.
The cost of pertuzumab is dosed a little bit differently. It’s a flat dose not a weight-based dose. Patients get an initial loading dose of 840 mg and a continuation dose of 420 mg every 3 weeks. The cost of the 420-mg dose of pertuzumab is just under $3,000, so that first-time loading dose would be a $6,000 dose, and the continuing doses are about $3,000 per dose every 3 weeks. The AE profile is no different from what you would expect with trastuzumab. There is a similar toxicity profile for these 2 drugs. It does not appear that there is any additional cardiotoxicity if you are using the combination in the neoadjuvant setting.
The third targeted agent that goes after the HER2 is ado-trastuzumab, but it is only used in the metastatic setting, so we’ll reserve that for down the road for this patient should we ever need it.
Dr. Aggarwal. The patient received adriamycin/cyclophosphamide followed by paclitaxel weekly for 12 weeks with trastuzumab. After the 12 weekly doses, she went on trastuzumab every 3 weeks. Because she was ER+, she was a candidate for additional endocrine ablation therapy. She was started on tamoxifen and leuprolide acetate for complete hormonal ablation.
Tamoxifen was the first targeted therapy for breast cancer. In women with ER+ breast cancer, with tamoxifen given for 5 years as adjuvant treatment, the odds of recurrences decreased by 39%, and death decreased by 30% in both pre- and postmenopausal women.8 Then the ATLAS data came, which randomly allocated patients to continue another 5 years of tamoxifen vs placebo, for a total of 10 years of treatment with tamoxifen. With a mean of 7.6 years of further follow-up after entry at year 5 in this trial showed that recurrence and breast cancer mortality during the second decade after diagnosis are reduced more effectively by 10 years of adjuvant tamoxifen than by 5 years.9 The current recommendation for pre- and postmenopausal is 10 years of tamoxifen.
In addition we have 3 aromatase inhibitors (AIs), anastrozole, letrozole, and exemestane, which block the production of estrogen in postmenopausal females. Anastrozole and letrozole are nonsteroidal, and exemestane is steroidal. There are countless big randomized trials using all of these drug in different combinations. In most of these trials, AIs are shown to be equal to tamoxifen when they are compared with each other, but their AE profile is different.
The recommendation by the American Society of Clinical Oncology and NCCN guidelines is to use only AIs for 5 years. There are different combinations: You can give tamoxifen for 2 to 3 years, followed by 5 years of an AI, or 5 years of tamoxifen and 5 years of an AI. Some patients wants to stop because of AEs, but others want to continue. Patients can develop osteoporosis and arthritis from an AI and hot flashes from tamoxifen.
Mr. Crawford, How would you manage of these AEs from these treatments?
Mr. Crawford. Because this woman is young, age 32, and premenopausal, tamoxifen would be the recommended endocrine therapy for her being ER+/PR+. But the role of the leuprolide acetate is to induce a chemical oophorectomy. We are putting her into ovarian ablation by using the leuprolide acetate.
The tamoxifen is relatively well tolerated, but as an ER blocker, it has a different AE profile than does an estrogen production decreaser. With tamoxifen patients tend to complain about hot flashes, edema, fluid retention, altered menses, spotting vaginal discharge, vaginal bleeding, and dryness. These medications also increase the risk of venous thromboembolism (VTE), and there is some concern about increased risk of developing endometrial cancers with these medications. We can give it either once or twice daily. There’s nothing that really says 10 mg twice daily vs 20 mg once daily is any different. So we may play with dosing to see if patients tolerate it better one way or the other.
There are medications that we can offer to help manage the hot flashes. These medications don’t necessarily make the hot flashes go away, but they can decrease the hot flash intensity or and/or frequency. Many medications have been evaluated for hot flashes. The best data are for venlafaxine, which is usually given once a day at bedtime (dosage 37.5-75.0 mg). There has been success with gabapentin titrated up to a dose of about 300 mg 3 times daily. They are fairly similar for decreasing hot flash scores and intensities, but the patient preferences were more favorable toward the venlafaxine than for the gabapentin.
The AIs, on the other hand, have a different AE profile. With tamoxifen we see vaginal discharges, bleeding, endometrial cancer risk, and VTE risk, but these are not significant problems with any of the AIs. The AE profiles for AIs include hot flashes, but more often it is complaints of bone pain, arthralgias, and myalgias. Probably the top reason why most patients discontinue taking AIs is arthralgia and myalgia.
Because we have shut off estrogen production with the AIs, and estrogen is an important component of maintaining good bone health and bone homeostasis, patients are at an increased risked of losing or declining bone mineral density (BMD). It is recommended that these patients get placed on routine calcium and vitamin D supplementation with routine dual-energy X-ray absorptiometry scans, so we know whether we will need to initiate osteoporosis treatment, whether with oral bisphosphonates, intravenous bisphosphonates, or subcutaneous rank ligand inhibitors.
With bisphosphonates there may be a slight increase in fracture rates. But we have to balance that with the BMD concerns. If the patient progresses into the metastatic setting and we know that there’s a fair chance that there’s going to be some skeletal involvement, those people are also at an increased risk of fracture. While there is a slight concern about the increased risk of fractures with bisphosphonates, I tend to believe that the benefits outweigh the risks.
Go to www.fedprac.com/AVAHO for a discussion of the next steps in the treatment for the patient after she returned 2 years later with nausea, vomiting, acute onset headache, and 2 brain lesions that were about 2 cm.
Click here to read the digital edition.
1. Feng Y, Rhie SK, Huo D, et al. Characterizing genetic susceptibility to breast cancer in women of african ancestry. Cancer Epidemiol Biomarkers. 2017;26(7):1016-1026.
2. Copson ER, Maishman TC, Tapper WJ, et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 2018;19(2):169-180.
3. Poortmans PM, Collette S, Kirkove C, et al; EORTC Radiation Oncology and Breast Cancer Groups. Internal mammary and medial supraclavicular irradiation in breast cancer. N Engl J Med. 2015;373(4):317-327.
4. Whelan TJ, Olivotto IA, Parulekar WR, et al; MA.20 Study Investigators. Regional nodal irradiation in early-stage breast cancer. N Engl J Med. 2015;373(4):307-316.
5. EBCTCG (Early Breast Cancer Trialists’ Collaborative Group), McGale P, Taylor C, Correa C, et al. Effect of radiotherapy after mastectomy and axillary surgery on 10-year recurrence and 20-year breast cancer mortality: meta-analysis of individual patient data for 8135 women in 22 randomised trials. Lancet. 2014;383(9935):2127-2135.
6. Kunkler IH, Williams LJ, Jack WJ, Cameron DA, Dixon JM; PRIME II investigators. Breast-conserving surgery with or without irradiation in women aged 65 years or older with early breast cancer (PRIME II): a randomised controlled trial. Lancet Oncol. 2015;16(3):266-273.
7. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177-182.
8. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effect of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15 year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687-1717.
9. Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805-816.
Case Presentation
This case represents a composite of many different patients and is not meant to represent an individual. Any resemblance to an actual patient is coincidental.
A 32-year-old African American woman presented with a self-palpated left breast mass (axillary tail at 9 o’clock position). The patient was a nonsmoker, was otherwise healthy, and had no family history of breast or any other cancer. She had never used oral contraceptives or hormones, was never pregnant, her menarche was at age 12 years, and she had regular menstrual periods. On physical examination she had a 1-cm left breast mass and a palpable left axillary lymph node. A complete diagnostic workup revealed a 2-cm left breast mass. An ultrasound-guided biopsy of the axillary lymph node was positive for invasive ductal carcinoma (IDC). The final diagnosis was left breast cancer, stage IIB IDC, T1N1M0, ER+, PR+, HER2 2+ by immunohistochemistry, fluorescence in situ hybridization (FISH) was 2.4, confirming a HER2+ tumor.
Anita Aggarwal, DO, PhD. What is the role of genetic counseling and testing in this young patient who does not have a family history of breast cancer?
Vickie L. Venne, MS. This patient absolutely would be a candidate for counseling and testing. From a genetic counseling perspective, one of the first points has to do with what “no family history of cancer” means. Typically, in a fast-paced clinic, a patient will be asked “Does anybody else in your family have cancer?” And it’s not uncommon to get the answer “no.” Genetic counselors collect specific information on at least the first- and second-degree relatives, so we end up with 3 generations. This includes both the maternal and the paternal histories. We find that people who initially report no family history of cancer are often just thinking of breast cancer, even if the provider’s question is broad. When we start digging, we often find other cancers because cancer is common.
The other issue is that she was diagnosed at a young age. Clearly, 32 years is much younger than we typically see in breast cancer, and we know that individuals with hereditary cancers often have an earlier age of onset. With no other information, her a priori risk of having a BRCA1/2 mutation would be < 2.5%.
Regardless, based on current National Comprehensive Cancer Network (NCCN) guidelines, she would be a testing candidate. We would also recommend testing for more than just BRCA1/2. In the last few decades, there have been many genes identified that are associated with an increased susceptibility to cancer. Many of these genes are part of syndromes, so if you had a mutation, that also would increase the risk for a cancer in another organ. If this woman’s mother and father lived into their 70s or 80s and she had a number of aunts on both sides who never developed breast cancer, it would be less likely to be BRCA1/2. However, P53 also can present in young women and as a de novo mutation. Therefore, we would offer her a panel of actionable genes. Genes that if, in fact, we identified a mutation in one of them, would mean we could do something different for this young lady.
JoAnn Manning, MD. Let’s say she does have testing, and she comes back BRCA+. Then what would be the recommendations or guidance?
Ms. Venne. Women (and men as well!) with mutations have an increased risk for a second primary breast cancer as well as cancer in other organs. Focusing first on the breast story and all the media around BRCA1/2 mutations and surgery, this is a woman who may consider a more aggressive surgery, including prophylactic contralateral mastectomy, if she is concerned. She is young, so we also would explore her fertility plans. While her next few months will be filled with breast cancer treatment choices, women with BRCA mutations also are at an increased risk to develop ovarian cancer, so that might be a decision she makes as well. Her health care team may also eventually discuss chemotherapeutic options available specifically to women with mutations.
However, we often see young women who are extremely nervous because there is a sense that if you’re younger, your cancer must be inherited. Part of the pretest counseling is to explore psychosocial issues and help these young ladies understand that, especially if she does not have a family history of cancer and the only indication is her age, then it’s highly likely that we’re not going to find an identifiable mutation. And in that circumstance, she probably could consider a more conservative surgical decision.
Dr. Aggarwal. How common is a BRCA1 or BRCA2 mutation in African American females?
Ms. Venne. I have not paid attention to the prevalence of mutations based on ethnicity, so I don’t know. While many of the initial mutations were discovered in women of European ancestry, there are large cohorts of women with African ancestry whose specimens are now available for identifying genetic markers that will improve breast cancer risk assessment in them.1 However, because those mutations are still being characterized, it is more common to find a variant of uncertain significance (VUS) in African American women. A VUS is an alteration—a change in the gene—that we simply don’t know what it means yet. Clinicians don’t have enough information to know if that alteration is pathogenic or benign. The problem is that people try to make sense out of everything in their lives, so they also will try to make a VUS mean something. We try hard to help people understand that a VUS is really no more significant than if we had not tested in the first place, and they should not act on that information. They should use their family history, their age, their other psychosocial concerns about their experiences with cancer as they make their treatment decisions. But they also should check back periodically with their genetic counselor because VUSs can be reclassified. And if that happens, the information might be more useful for not only them, but their family members.
Dr. Manning. Would you consider this patient for any neoadjuvant chemotherapy?
Dr. Aggarwal. The patient is a young female with a small tumor that is HER2+. The indication for neoadjuvant chemotherapy is typically a big tumor or inoperable disease. Neoadjuvant chemotherapy is considered the standard of care for patients with inflammatory breast cancer and may confer a survival benefit in these patients. Of all the breast cancer subtypes, triple negative and HER2+ are considered the most chemosensitive and may benefit from neoadjuvant therapy. This patient has a small tumor, and I don’t think she’s a candidate for neoadjuvant chemotherapy unless the patient wants to see if her tumor is chemosensitive or not.
Dr. Manning, What’s the role and benefit of lumpectomy vs mastectomy?
Dr. Manning. Historically, mastectomy would have been considered the standard of care, but luckily, in the 1970s and the 1980s, we had a significant number of randomized controlled trials that demonstrated that certain women with particular characteristics would get the same overall survival if they chose mastectomy vs lumpectomy, the removal of the tumor with negative margin and whole-breast radiation. The key thing to understand is that breast-conserving surgery is now very well established with more than 20 years of data to support it. And that breast irradiation after breast-conserving surgery is essential to maximizing the local control and the overall survival (OS).
There have been a lot of major studies, but the one with the greatest follow-up now is the National Surgical Adjuvant Breast and Bowel Project (NSABP) B06 protocol, which was the only trial to compare mastectomy to lumpectomy and radiation or lumpectomy alone. It required negative margins. With 20 years of follow-up, the data still support that mastectomy or lumpectomy with radiation offers equivalent OS and local control. It’s really about patient preference if they are candidates.
Who is a candidate? Clearly, there are contraindications. We tend to look primarily at the size of the tumor. However, removing an average-sized tumor (< 2 cm) with a margin may not have a good cosmetic result for a patient with very small breasts. That patient may opt to go forward with a mastectomy instead. Young patients who are candidates must have to have negative margins. If they have persistently positive resection margins after excision or reexcision, then they need to go forward with mastectomy.
A patient who has imaging evidence of multicentric disease with 2 or more primary tumors in separate quadrants would not be a candidate for breast-conserving therapy. Diffuse malignant-appearing microcalcifications on a mammogram also would suggest multicentric disease. And a patient with a prior history of radiation therapy to the breast or chest wall cannot go through breast-conserving therapy.
In the case we are discussing, we also should make sure this young lady is not pregnant. If the patient is adamant about breast-conserving surgery and pregnant, especially in the third trimester, radiation could be deferred until after delivery. Another relative contraindication is for patients who have connective tissue disorders. Sometimes if they are given whole-breast radiation, the cosmetic result is poor. So if you’re doing this procedure to save the breast, then having a good cosmetic result is an important consideration for many patients.
When you look at the size of the tumor for this patient, she seems to be a good candidate for breast-conserving surgery. I would recommend that she go forward with lumpectomy followed by whole-breast radiation.
Ms. Venne. Although the numbers aren’t nearly as large as they were in the original trials looking at the lumpectomy vs mastectomy, there are now survival data for women with BRCA1/2 mutations. With all of the caveats that Dr. Manning mentioned, even if you have an identifiable mutation, you may not necessarily need that more aggressive surgery.2 Clearly, individuals with identifiable mutations would have a higher chance of a contralateral breast cancer, a second primary, so some individuals consider a prophylactic bilateral mastectomy. But from a survival perspective, there are a fair amount of data now available that say that lumpectomy vs mastectomy should really be the conversation based on all of the information that Dr. Manning outlines rather than using primarily the mutation status
Dr. Manning. I agree.
Dr. Aggarwal. This patient had a lumpectomy and axillary lymph node dissection. Pathology reported 1.5-cm mass, grade 3 IDC; the margins were negative. There was no skin involvement, 27 lymph nodes removed were all negative. Dr. Manning, can you please discuss the role of radiation in early stage breast cancer in patients like this case?
Dr. Manning. One of the questions that is always controversial for radiation in these early stage breast cancer cases is what do you do with the nodal irradiation? Previously, radiation oncologists based treatment plans on retrospective data, but in 2015, there were 2 major studies, 1 from Canada, and 1 from the European Organisation for Research and Treatment of Cancer (EORTC).3,4 Both studies tried to determine whether there was an advantage to doing regional nodal irradiation in early breast cancer cases. That encompassed axillary, supraclavicular, and internal mammary nodes. The studies showed that there was no survival advantage, but there was a statistically significant improvement in disease free survival and in local regional recurrence and distant mets.
Unfortunately, there are still a lot of unanswered questions, like what group potentially would benefit the most? In the MA.20 Study, some observers questioned that maybe the ER-/PR- women had the most benefit, but then, in the other study the benefit wasn’t clear.4,5 One question is which lymph node group is having the most impact? Was the benefit from radiating the supraclavicular nodes or was it from radiating the internal mammary nodes? Determining the answer is important from a technical point of view because when you radiate the internal mammary nodes, you have the potential to expose more heart and lung to radiation. You have to put all these together and make a recommendation.
Clearly, for a patient with negative nodes there is no question: You would not treat the regional nodes. However, for a patient with positive nodes you really have to individualize the approach and consider age, anatomy, tumor location, and burden of axillary disease.
I would sit down and have a discussion with this young woman to weigh the risks and the benefits. There is a slight increased risk of lymphedema in these patients, and radiation pneumonitis increases, but not significantly. A key concern is to minimize the total dose of radiation to the heart. There have been great developments in radiation oncology technology and capabilities, so the cardiac dose is now less. But when you think about a 32-year-old patient and weigh the benefit of a 2% to 3% decrease in the incidence of distant metastases and no OS advantage, then you really need to have a conversation about how to safely treat her. At a minimum, I would treat the high axilla and the supraclavicular nodes because she had a pretty extensive lymph node dissection with more than 20 nodes, and then with her getting systemic therapy, that should be more than adequate.
Dr. Aggarwal. Is there any cutoff for age or size of the tumor where you would not do any radiation to the breast?
Dr. Manning. In this particular patient absolutely not because of the lymph node. She had breast-conserving therapy, and she’s only 32-years-old. The PRIME 2 study offered lumpectomy alone vs lumpectomy and radiation for women aged ≥ 65 years with tumors ≤ 3 cm, low grade.6 The study participants had to have negative lymph nodes, be ER+, and low grade. It was a very select group. The lumpectomy patients had a recurrence rate around 4%, and the other was closer to 1.3%.
You have to look at the whole picture. Is this a healthy 70-year-old woman? Is it an inconvenience for her to get treatment? Is she going to get hormone therapy and will she be adherent? There’s a very small group of women who underwent breast-conserving surgery that I would feel safe about not offering radiation.
Dr. Aggarwal. About 15% to 20% of all breast cancers are HER2 over expressors, which used to be a poor prognostic characteristic. However, the development of anti-HER2 therapies has changed the picture of HER2 prognosis. After the initial discovery of activity, the pivotal study by Slamon et al showed benefit in terms of progression-free survival (PFS) and OS with chemotherapy and trastuzumab. The NCCN guideline recommends anti-HER2 antibody trastuzumab in combination with chemotherapy.7
Patients with tumor < 0.5 cm who are HER2+ and ER+ may not benefit from trastuzumab, but those who are ER- and HER2+ will still benefit from trastuzumab. The combination is adriamycin/cyclophosphamide followed by a taxane with trastuzumab and to complete 1 year of trastuzumab or trastuzumab in combination with carboplatin and taxanes.
Pertuzumab, in combination with trastuzumab and docetaxel (PHT) has been FDA-approved in neoadjuvant and metastatic HER2+ disease, but is not FDA approved yet in the adjuvant setting. However, these are expensive drugs, and we don’t know how long these drugs should be given.
Mr. Crawford, What are the adverse effects (AEs) of an anti-HER2 or trastuzumab treatment, and what is the cost of trastuzumab?
Russell Crawford, BPharm. The anti-HER2 antibodies have certainly changed treatment plans and outcomes for patients with breast cancer who test HER2+. There are actually 3 of these anti-HER2 drugs on the U.S. market, and they can be used in a variety of settings. Trastuzumab and pertuzumab are indicated in women or patients who have HER2+ disease, and they work by binding to the extracellular domain of the HER2 proteins and mediate antibody-dependent cellular toxicity by inhibiting proliferation of the cells that overexpress HER2.
In this patient, we would be looking at using adjuvant trastuzumab to complete a 1-year course of therapy while she’s getting her dose-dense doxorubicin and cyclophosphamide (AC) on a weekly basis for the first 12 weeks. Trastuzumab is dosed with an initial loading dose of 4 mg/kg as the first dose, and then it’s 2 mg/kg/wk until adjuvant chemotherapy is completed. We usually extend the dosing out to 6 mg/kg every 3 weeks to complete the year of treatment.
These drugs are fairly well tolerated. They are monoclonal proteins, so a lot of the AEs that patients experience are the things that we’re used to seeing with other monoclonal proteins like the infusion-related reactions and some flulike symptoms. The biggest concern with these patients is that being on the drug for a year, there is a risk of decreasing the left ventricular ejection fraction (LVEF) of the heart. That risk is increased when these drugs are combined with anthracyclines that we know are cardiotoxic. As a single agent, the impact on left ventricular function is not significant, but when it is combined with chemotherapy, it does become a problem. Usually, we recommend routine and periodic monitoring of the LVEF with a multiple-gated acquisition or an echocardiogram to make sure that we’re not causing harm related to this treatment.
The cost of these drugs depends on the frequency, is it every week, every 2 weeks, or every 3 weeks? There are different ways to give trastuzumab, but for most patients, we prefer the every 3-week dose. And it’s estimated that for a 70-kg patient, a dose of trastuzumab at 6 mg/kg at the rate of every 3 weeks costs about $2,500 per dose. The VA pays about $6 a milligram, but it’s certainly money well spent because it has changed the playing field and the outcomes for these patients.
The cost of pertuzumab is dosed a little bit differently. It’s a flat dose not a weight-based dose. Patients get an initial loading dose of 840 mg and a continuation dose of 420 mg every 3 weeks. The cost of the 420-mg dose of pertuzumab is just under $3,000, so that first-time loading dose would be a $6,000 dose, and the continuing doses are about $3,000 per dose every 3 weeks. The AE profile is no different from what you would expect with trastuzumab. There is a similar toxicity profile for these 2 drugs. It does not appear that there is any additional cardiotoxicity if you are using the combination in the neoadjuvant setting.
The third targeted agent that goes after the HER2 is ado-trastuzumab, but it is only used in the metastatic setting, so we’ll reserve that for down the road for this patient should we ever need it.
Dr. Aggarwal. The patient received adriamycin/cyclophosphamide followed by paclitaxel weekly for 12 weeks with trastuzumab. After the 12 weekly doses, she went on trastuzumab every 3 weeks. Because she was ER+, she was a candidate for additional endocrine ablation therapy. She was started on tamoxifen and leuprolide acetate for complete hormonal ablation.
Tamoxifen was the first targeted therapy for breast cancer. In women with ER+ breast cancer, with tamoxifen given for 5 years as adjuvant treatment, the odds of recurrences decreased by 39%, and death decreased by 30% in both pre- and postmenopausal women.8 Then the ATLAS data came, which randomly allocated patients to continue another 5 years of tamoxifen vs placebo, for a total of 10 years of treatment with tamoxifen. With a mean of 7.6 years of further follow-up after entry at year 5 in this trial showed that recurrence and breast cancer mortality during the second decade after diagnosis are reduced more effectively by 10 years of adjuvant tamoxifen than by 5 years.9 The current recommendation for pre- and postmenopausal is 10 years of tamoxifen.
In addition we have 3 aromatase inhibitors (AIs), anastrozole, letrozole, and exemestane, which block the production of estrogen in postmenopausal females. Anastrozole and letrozole are nonsteroidal, and exemestane is steroidal. There are countless big randomized trials using all of these drug in different combinations. In most of these trials, AIs are shown to be equal to tamoxifen when they are compared with each other, but their AE profile is different.
The recommendation by the American Society of Clinical Oncology and NCCN guidelines is to use only AIs for 5 years. There are different combinations: You can give tamoxifen for 2 to 3 years, followed by 5 years of an AI, or 5 years of tamoxifen and 5 years of an AI. Some patients wants to stop because of AEs, but others want to continue. Patients can develop osteoporosis and arthritis from an AI and hot flashes from tamoxifen.
Mr. Crawford, How would you manage of these AEs from these treatments?
Mr. Crawford. Because this woman is young, age 32, and premenopausal, tamoxifen would be the recommended endocrine therapy for her being ER+/PR+. But the role of the leuprolide acetate is to induce a chemical oophorectomy. We are putting her into ovarian ablation by using the leuprolide acetate.
The tamoxifen is relatively well tolerated, but as an ER blocker, it has a different AE profile than does an estrogen production decreaser. With tamoxifen patients tend to complain about hot flashes, edema, fluid retention, altered menses, spotting vaginal discharge, vaginal bleeding, and dryness. These medications also increase the risk of venous thromboembolism (VTE), and there is some concern about increased risk of developing endometrial cancers with these medications. We can give it either once or twice daily. There’s nothing that really says 10 mg twice daily vs 20 mg once daily is any different. So we may play with dosing to see if patients tolerate it better one way or the other.
There are medications that we can offer to help manage the hot flashes. These medications don’t necessarily make the hot flashes go away, but they can decrease the hot flash intensity or and/or frequency. Many medications have been evaluated for hot flashes. The best data are for venlafaxine, which is usually given once a day at bedtime (dosage 37.5-75.0 mg). There has been success with gabapentin titrated up to a dose of about 300 mg 3 times daily. They are fairly similar for decreasing hot flash scores and intensities, but the patient preferences were more favorable toward the venlafaxine than for the gabapentin.
The AIs, on the other hand, have a different AE profile. With tamoxifen we see vaginal discharges, bleeding, endometrial cancer risk, and VTE risk, but these are not significant problems with any of the AIs. The AE profiles for AIs include hot flashes, but more often it is complaints of bone pain, arthralgias, and myalgias. Probably the top reason why most patients discontinue taking AIs is arthralgia and myalgia.
Because we have shut off estrogen production with the AIs, and estrogen is an important component of maintaining good bone health and bone homeostasis, patients are at an increased risked of losing or declining bone mineral density (BMD). It is recommended that these patients get placed on routine calcium and vitamin D supplementation with routine dual-energy X-ray absorptiometry scans, so we know whether we will need to initiate osteoporosis treatment, whether with oral bisphosphonates, intravenous bisphosphonates, or subcutaneous rank ligand inhibitors.
With bisphosphonates there may be a slight increase in fracture rates. But we have to balance that with the BMD concerns. If the patient progresses into the metastatic setting and we know that there’s a fair chance that there’s going to be some skeletal involvement, those people are also at an increased risk of fracture. While there is a slight concern about the increased risk of fractures with bisphosphonates, I tend to believe that the benefits outweigh the risks.
Go to www.fedprac.com/AVAHO for a discussion of the next steps in the treatment for the patient after she returned 2 years later with nausea, vomiting, acute onset headache, and 2 brain lesions that were about 2 cm.
Click here to read the digital edition.
Case Presentation
This case represents a composite of many different patients and is not meant to represent an individual. Any resemblance to an actual patient is coincidental.
A 32-year-old African American woman presented with a self-palpated left breast mass (axillary tail at 9 o’clock position). The patient was a nonsmoker, was otherwise healthy, and had no family history of breast or any other cancer. She had never used oral contraceptives or hormones, was never pregnant, her menarche was at age 12 years, and she had regular menstrual periods. On physical examination she had a 1-cm left breast mass and a palpable left axillary lymph node. A complete diagnostic workup revealed a 2-cm left breast mass. An ultrasound-guided biopsy of the axillary lymph node was positive for invasive ductal carcinoma (IDC). The final diagnosis was left breast cancer, stage IIB IDC, T1N1M0, ER+, PR+, HER2 2+ by immunohistochemistry, fluorescence in situ hybridization (FISH) was 2.4, confirming a HER2+ tumor.
Anita Aggarwal, DO, PhD. What is the role of genetic counseling and testing in this young patient who does not have a family history of breast cancer?
Vickie L. Venne, MS. This patient absolutely would be a candidate for counseling and testing. From a genetic counseling perspective, one of the first points has to do with what “no family history of cancer” means. Typically, in a fast-paced clinic, a patient will be asked “Does anybody else in your family have cancer?” And it’s not uncommon to get the answer “no.” Genetic counselors collect specific information on at least the first- and second-degree relatives, so we end up with 3 generations. This includes both the maternal and the paternal histories. We find that people who initially report no family history of cancer are often just thinking of breast cancer, even if the provider’s question is broad. When we start digging, we often find other cancers because cancer is common.
The other issue is that she was diagnosed at a young age. Clearly, 32 years is much younger than we typically see in breast cancer, and we know that individuals with hereditary cancers often have an earlier age of onset. With no other information, her a priori risk of having a BRCA1/2 mutation would be < 2.5%.
Regardless, based on current National Comprehensive Cancer Network (NCCN) guidelines, she would be a testing candidate. We would also recommend testing for more than just BRCA1/2. In the last few decades, there have been many genes identified that are associated with an increased susceptibility to cancer. Many of these genes are part of syndromes, so if you had a mutation, that also would increase the risk for a cancer in another organ. If this woman’s mother and father lived into their 70s or 80s and she had a number of aunts on both sides who never developed breast cancer, it would be less likely to be BRCA1/2. However, P53 also can present in young women and as a de novo mutation. Therefore, we would offer her a panel of actionable genes. Genes that if, in fact, we identified a mutation in one of them, would mean we could do something different for this young lady.
JoAnn Manning, MD. Let’s say she does have testing, and she comes back BRCA+. Then what would be the recommendations or guidance?
Ms. Venne. Women (and men as well!) with mutations have an increased risk for a second primary breast cancer as well as cancer in other organs. Focusing first on the breast story and all the media around BRCA1/2 mutations and surgery, this is a woman who may consider a more aggressive surgery, including prophylactic contralateral mastectomy, if she is concerned. She is young, so we also would explore her fertility plans. While her next few months will be filled with breast cancer treatment choices, women with BRCA mutations also are at an increased risk to develop ovarian cancer, so that might be a decision she makes as well. Her health care team may also eventually discuss chemotherapeutic options available specifically to women with mutations.
However, we often see young women who are extremely nervous because there is a sense that if you’re younger, your cancer must be inherited. Part of the pretest counseling is to explore psychosocial issues and help these young ladies understand that, especially if she does not have a family history of cancer and the only indication is her age, then it’s highly likely that we’re not going to find an identifiable mutation. And in that circumstance, she probably could consider a more conservative surgical decision.
Dr. Aggarwal. How common is a BRCA1 or BRCA2 mutation in African American females?
Ms. Venne. I have not paid attention to the prevalence of mutations based on ethnicity, so I don’t know. While many of the initial mutations were discovered in women of European ancestry, there are large cohorts of women with African ancestry whose specimens are now available for identifying genetic markers that will improve breast cancer risk assessment in them.1 However, because those mutations are still being characterized, it is more common to find a variant of uncertain significance (VUS) in African American women. A VUS is an alteration—a change in the gene—that we simply don’t know what it means yet. Clinicians don’t have enough information to know if that alteration is pathogenic or benign. The problem is that people try to make sense out of everything in their lives, so they also will try to make a VUS mean something. We try hard to help people understand that a VUS is really no more significant than if we had not tested in the first place, and they should not act on that information. They should use their family history, their age, their other psychosocial concerns about their experiences with cancer as they make their treatment decisions. But they also should check back periodically with their genetic counselor because VUSs can be reclassified. And if that happens, the information might be more useful for not only them, but their family members.
Dr. Manning. Would you consider this patient for any neoadjuvant chemotherapy?
Dr. Aggarwal. The patient is a young female with a small tumor that is HER2+. The indication for neoadjuvant chemotherapy is typically a big tumor or inoperable disease. Neoadjuvant chemotherapy is considered the standard of care for patients with inflammatory breast cancer and may confer a survival benefit in these patients. Of all the breast cancer subtypes, triple negative and HER2+ are considered the most chemosensitive and may benefit from neoadjuvant therapy. This patient has a small tumor, and I don’t think she’s a candidate for neoadjuvant chemotherapy unless the patient wants to see if her tumor is chemosensitive or not.
Dr. Manning, What’s the role and benefit of lumpectomy vs mastectomy?
Dr. Manning. Historically, mastectomy would have been considered the standard of care, but luckily, in the 1970s and the 1980s, we had a significant number of randomized controlled trials that demonstrated that certain women with particular characteristics would get the same overall survival if they chose mastectomy vs lumpectomy, the removal of the tumor with negative margin and whole-breast radiation. The key thing to understand is that breast-conserving surgery is now very well established with more than 20 years of data to support it. And that breast irradiation after breast-conserving surgery is essential to maximizing the local control and the overall survival (OS).
There have been a lot of major studies, but the one with the greatest follow-up now is the National Surgical Adjuvant Breast and Bowel Project (NSABP) B06 protocol, which was the only trial to compare mastectomy to lumpectomy and radiation or lumpectomy alone. It required negative margins. With 20 years of follow-up, the data still support that mastectomy or lumpectomy with radiation offers equivalent OS and local control. It’s really about patient preference if they are candidates.
Who is a candidate? Clearly, there are contraindications. We tend to look primarily at the size of the tumor. However, removing an average-sized tumor (< 2 cm) with a margin may not have a good cosmetic result for a patient with very small breasts. That patient may opt to go forward with a mastectomy instead. Young patients who are candidates must have to have negative margins. If they have persistently positive resection margins after excision or reexcision, then they need to go forward with mastectomy.
A patient who has imaging evidence of multicentric disease with 2 or more primary tumors in separate quadrants would not be a candidate for breast-conserving therapy. Diffuse malignant-appearing microcalcifications on a mammogram also would suggest multicentric disease. And a patient with a prior history of radiation therapy to the breast or chest wall cannot go through breast-conserving therapy.
In the case we are discussing, we also should make sure this young lady is not pregnant. If the patient is adamant about breast-conserving surgery and pregnant, especially in the third trimester, radiation could be deferred until after delivery. Another relative contraindication is for patients who have connective tissue disorders. Sometimes if they are given whole-breast radiation, the cosmetic result is poor. So if you’re doing this procedure to save the breast, then having a good cosmetic result is an important consideration for many patients.
When you look at the size of the tumor for this patient, she seems to be a good candidate for breast-conserving surgery. I would recommend that she go forward with lumpectomy followed by whole-breast radiation.
Ms. Venne. Although the numbers aren’t nearly as large as they were in the original trials looking at the lumpectomy vs mastectomy, there are now survival data for women with BRCA1/2 mutations. With all of the caveats that Dr. Manning mentioned, even if you have an identifiable mutation, you may not necessarily need that more aggressive surgery.2 Clearly, individuals with identifiable mutations would have a higher chance of a contralateral breast cancer, a second primary, so some individuals consider a prophylactic bilateral mastectomy. But from a survival perspective, there are a fair amount of data now available that say that lumpectomy vs mastectomy should really be the conversation based on all of the information that Dr. Manning outlines rather than using primarily the mutation status
Dr. Manning. I agree.
Dr. Aggarwal. This patient had a lumpectomy and axillary lymph node dissection. Pathology reported 1.5-cm mass, grade 3 IDC; the margins were negative. There was no skin involvement, 27 lymph nodes removed were all negative. Dr. Manning, can you please discuss the role of radiation in early stage breast cancer in patients like this case?
Dr. Manning. One of the questions that is always controversial for radiation in these early stage breast cancer cases is what do you do with the nodal irradiation? Previously, radiation oncologists based treatment plans on retrospective data, but in 2015, there were 2 major studies, 1 from Canada, and 1 from the European Organisation for Research and Treatment of Cancer (EORTC).3,4 Both studies tried to determine whether there was an advantage to doing regional nodal irradiation in early breast cancer cases. That encompassed axillary, supraclavicular, and internal mammary nodes. The studies showed that there was no survival advantage, but there was a statistically significant improvement in disease free survival and in local regional recurrence and distant mets.
Unfortunately, there are still a lot of unanswered questions, like what group potentially would benefit the most? In the MA.20 Study, some observers questioned that maybe the ER-/PR- women had the most benefit, but then, in the other study the benefit wasn’t clear.4,5 One question is which lymph node group is having the most impact? Was the benefit from radiating the supraclavicular nodes or was it from radiating the internal mammary nodes? Determining the answer is important from a technical point of view because when you radiate the internal mammary nodes, you have the potential to expose more heart and lung to radiation. You have to put all these together and make a recommendation.
Clearly, for a patient with negative nodes there is no question: You would not treat the regional nodes. However, for a patient with positive nodes you really have to individualize the approach and consider age, anatomy, tumor location, and burden of axillary disease.
I would sit down and have a discussion with this young woman to weigh the risks and the benefits. There is a slight increased risk of lymphedema in these patients, and radiation pneumonitis increases, but not significantly. A key concern is to minimize the total dose of radiation to the heart. There have been great developments in radiation oncology technology and capabilities, so the cardiac dose is now less. But when you think about a 32-year-old patient and weigh the benefit of a 2% to 3% decrease in the incidence of distant metastases and no OS advantage, then you really need to have a conversation about how to safely treat her. At a minimum, I would treat the high axilla and the supraclavicular nodes because she had a pretty extensive lymph node dissection with more than 20 nodes, and then with her getting systemic therapy, that should be more than adequate.
Dr. Aggarwal. Is there any cutoff for age or size of the tumor where you would not do any radiation to the breast?
Dr. Manning. In this particular patient absolutely not because of the lymph node. She had breast-conserving therapy, and she’s only 32-years-old. The PRIME 2 study offered lumpectomy alone vs lumpectomy and radiation for women aged ≥ 65 years with tumors ≤ 3 cm, low grade.6 The study participants had to have negative lymph nodes, be ER+, and low grade. It was a very select group. The lumpectomy patients had a recurrence rate around 4%, and the other was closer to 1.3%.
You have to look at the whole picture. Is this a healthy 70-year-old woman? Is it an inconvenience for her to get treatment? Is she going to get hormone therapy and will she be adherent? There’s a very small group of women who underwent breast-conserving surgery that I would feel safe about not offering radiation.
Dr. Aggarwal. About 15% to 20% of all breast cancers are HER2 over expressors, which used to be a poor prognostic characteristic. However, the development of anti-HER2 therapies has changed the picture of HER2 prognosis. After the initial discovery of activity, the pivotal study by Slamon et al showed benefit in terms of progression-free survival (PFS) and OS with chemotherapy and trastuzumab. The NCCN guideline recommends anti-HER2 antibody trastuzumab in combination with chemotherapy.7
Patients with tumor < 0.5 cm who are HER2+ and ER+ may not benefit from trastuzumab, but those who are ER- and HER2+ will still benefit from trastuzumab. The combination is adriamycin/cyclophosphamide followed by a taxane with trastuzumab and to complete 1 year of trastuzumab or trastuzumab in combination with carboplatin and taxanes.
Pertuzumab, in combination with trastuzumab and docetaxel (PHT) has been FDA-approved in neoadjuvant and metastatic HER2+ disease, but is not FDA approved yet in the adjuvant setting. However, these are expensive drugs, and we don’t know how long these drugs should be given.
Mr. Crawford, What are the adverse effects (AEs) of an anti-HER2 or trastuzumab treatment, and what is the cost of trastuzumab?
Russell Crawford, BPharm. The anti-HER2 antibodies have certainly changed treatment plans and outcomes for patients with breast cancer who test HER2+. There are actually 3 of these anti-HER2 drugs on the U.S. market, and they can be used in a variety of settings. Trastuzumab and pertuzumab are indicated in women or patients who have HER2+ disease, and they work by binding to the extracellular domain of the HER2 proteins and mediate antibody-dependent cellular toxicity by inhibiting proliferation of the cells that overexpress HER2.
In this patient, we would be looking at using adjuvant trastuzumab to complete a 1-year course of therapy while she’s getting her dose-dense doxorubicin and cyclophosphamide (AC) on a weekly basis for the first 12 weeks. Trastuzumab is dosed with an initial loading dose of 4 mg/kg as the first dose, and then it’s 2 mg/kg/wk until adjuvant chemotherapy is completed. We usually extend the dosing out to 6 mg/kg every 3 weeks to complete the year of treatment.
These drugs are fairly well tolerated. They are monoclonal proteins, so a lot of the AEs that patients experience are the things that we’re used to seeing with other monoclonal proteins like the infusion-related reactions and some flulike symptoms. The biggest concern with these patients is that being on the drug for a year, there is a risk of decreasing the left ventricular ejection fraction (LVEF) of the heart. That risk is increased when these drugs are combined with anthracyclines that we know are cardiotoxic. As a single agent, the impact on left ventricular function is not significant, but when it is combined with chemotherapy, it does become a problem. Usually, we recommend routine and periodic monitoring of the LVEF with a multiple-gated acquisition or an echocardiogram to make sure that we’re not causing harm related to this treatment.
The cost of these drugs depends on the frequency, is it every week, every 2 weeks, or every 3 weeks? There are different ways to give trastuzumab, but for most patients, we prefer the every 3-week dose. And it’s estimated that for a 70-kg patient, a dose of trastuzumab at 6 mg/kg at the rate of every 3 weeks costs about $2,500 per dose. The VA pays about $6 a milligram, but it’s certainly money well spent because it has changed the playing field and the outcomes for these patients.
The cost of pertuzumab is dosed a little bit differently. It’s a flat dose not a weight-based dose. Patients get an initial loading dose of 840 mg and a continuation dose of 420 mg every 3 weeks. The cost of the 420-mg dose of pertuzumab is just under $3,000, so that first-time loading dose would be a $6,000 dose, and the continuing doses are about $3,000 per dose every 3 weeks. The AE profile is no different from what you would expect with trastuzumab. There is a similar toxicity profile for these 2 drugs. It does not appear that there is any additional cardiotoxicity if you are using the combination in the neoadjuvant setting.
The third targeted agent that goes after the HER2 is ado-trastuzumab, but it is only used in the metastatic setting, so we’ll reserve that for down the road for this patient should we ever need it.
Dr. Aggarwal. The patient received adriamycin/cyclophosphamide followed by paclitaxel weekly for 12 weeks with trastuzumab. After the 12 weekly doses, she went on trastuzumab every 3 weeks. Because she was ER+, she was a candidate for additional endocrine ablation therapy. She was started on tamoxifen and leuprolide acetate for complete hormonal ablation.
Tamoxifen was the first targeted therapy for breast cancer. In women with ER+ breast cancer, with tamoxifen given for 5 years as adjuvant treatment, the odds of recurrences decreased by 39%, and death decreased by 30% in both pre- and postmenopausal women.8 Then the ATLAS data came, which randomly allocated patients to continue another 5 years of tamoxifen vs placebo, for a total of 10 years of treatment with tamoxifen. With a mean of 7.6 years of further follow-up after entry at year 5 in this trial showed that recurrence and breast cancer mortality during the second decade after diagnosis are reduced more effectively by 10 years of adjuvant tamoxifen than by 5 years.9 The current recommendation for pre- and postmenopausal is 10 years of tamoxifen.
In addition we have 3 aromatase inhibitors (AIs), anastrozole, letrozole, and exemestane, which block the production of estrogen in postmenopausal females. Anastrozole and letrozole are nonsteroidal, and exemestane is steroidal. There are countless big randomized trials using all of these drug in different combinations. In most of these trials, AIs are shown to be equal to tamoxifen when they are compared with each other, but their AE profile is different.
The recommendation by the American Society of Clinical Oncology and NCCN guidelines is to use only AIs for 5 years. There are different combinations: You can give tamoxifen for 2 to 3 years, followed by 5 years of an AI, or 5 years of tamoxifen and 5 years of an AI. Some patients wants to stop because of AEs, but others want to continue. Patients can develop osteoporosis and arthritis from an AI and hot flashes from tamoxifen.
Mr. Crawford, How would you manage of these AEs from these treatments?
Mr. Crawford. Because this woman is young, age 32, and premenopausal, tamoxifen would be the recommended endocrine therapy for her being ER+/PR+. But the role of the leuprolide acetate is to induce a chemical oophorectomy. We are putting her into ovarian ablation by using the leuprolide acetate.
The tamoxifen is relatively well tolerated, but as an ER blocker, it has a different AE profile than does an estrogen production decreaser. With tamoxifen patients tend to complain about hot flashes, edema, fluid retention, altered menses, spotting vaginal discharge, vaginal bleeding, and dryness. These medications also increase the risk of venous thromboembolism (VTE), and there is some concern about increased risk of developing endometrial cancers with these medications. We can give it either once or twice daily. There’s nothing that really says 10 mg twice daily vs 20 mg once daily is any different. So we may play with dosing to see if patients tolerate it better one way or the other.
There are medications that we can offer to help manage the hot flashes. These medications don’t necessarily make the hot flashes go away, but they can decrease the hot flash intensity or and/or frequency. Many medications have been evaluated for hot flashes. The best data are for venlafaxine, which is usually given once a day at bedtime (dosage 37.5-75.0 mg). There has been success with gabapentin titrated up to a dose of about 300 mg 3 times daily. They are fairly similar for decreasing hot flash scores and intensities, but the patient preferences were more favorable toward the venlafaxine than for the gabapentin.
The AIs, on the other hand, have a different AE profile. With tamoxifen we see vaginal discharges, bleeding, endometrial cancer risk, and VTE risk, but these are not significant problems with any of the AIs. The AE profiles for AIs include hot flashes, but more often it is complaints of bone pain, arthralgias, and myalgias. Probably the top reason why most patients discontinue taking AIs is arthralgia and myalgia.
Because we have shut off estrogen production with the AIs, and estrogen is an important component of maintaining good bone health and bone homeostasis, patients are at an increased risked of losing or declining bone mineral density (BMD). It is recommended that these patients get placed on routine calcium and vitamin D supplementation with routine dual-energy X-ray absorptiometry scans, so we know whether we will need to initiate osteoporosis treatment, whether with oral bisphosphonates, intravenous bisphosphonates, or subcutaneous rank ligand inhibitors.
With bisphosphonates there may be a slight increase in fracture rates. But we have to balance that with the BMD concerns. If the patient progresses into the metastatic setting and we know that there’s a fair chance that there’s going to be some skeletal involvement, those people are also at an increased risk of fracture. While there is a slight concern about the increased risk of fractures with bisphosphonates, I tend to believe that the benefits outweigh the risks.
Go to www.fedprac.com/AVAHO for a discussion of the next steps in the treatment for the patient after she returned 2 years later with nausea, vomiting, acute onset headache, and 2 brain lesions that were about 2 cm.
Click here to read the digital edition.
1. Feng Y, Rhie SK, Huo D, et al. Characterizing genetic susceptibility to breast cancer in women of african ancestry. Cancer Epidemiol Biomarkers. 2017;26(7):1016-1026.
2. Copson ER, Maishman TC, Tapper WJ, et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 2018;19(2):169-180.
3. Poortmans PM, Collette S, Kirkove C, et al; EORTC Radiation Oncology and Breast Cancer Groups. Internal mammary and medial supraclavicular irradiation in breast cancer. N Engl J Med. 2015;373(4):317-327.
4. Whelan TJ, Olivotto IA, Parulekar WR, et al; MA.20 Study Investigators. Regional nodal irradiation in early-stage breast cancer. N Engl J Med. 2015;373(4):307-316.
5. EBCTCG (Early Breast Cancer Trialists’ Collaborative Group), McGale P, Taylor C, Correa C, et al. Effect of radiotherapy after mastectomy and axillary surgery on 10-year recurrence and 20-year breast cancer mortality: meta-analysis of individual patient data for 8135 women in 22 randomised trials. Lancet. 2014;383(9935):2127-2135.
6. Kunkler IH, Williams LJ, Jack WJ, Cameron DA, Dixon JM; PRIME II investigators. Breast-conserving surgery with or without irradiation in women aged 65 years or older with early breast cancer (PRIME II): a randomised controlled trial. Lancet Oncol. 2015;16(3):266-273.
7. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177-182.
8. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effect of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15 year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687-1717.
9. Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805-816.
1. Feng Y, Rhie SK, Huo D, et al. Characterizing genetic susceptibility to breast cancer in women of african ancestry. Cancer Epidemiol Biomarkers. 2017;26(7):1016-1026.
2. Copson ER, Maishman TC, Tapper WJ, et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 2018;19(2):169-180.
3. Poortmans PM, Collette S, Kirkove C, et al; EORTC Radiation Oncology and Breast Cancer Groups. Internal mammary and medial supraclavicular irradiation in breast cancer. N Engl J Med. 2015;373(4):317-327.
4. Whelan TJ, Olivotto IA, Parulekar WR, et al; MA.20 Study Investigators. Regional nodal irradiation in early-stage breast cancer. N Engl J Med. 2015;373(4):307-316.
5. EBCTCG (Early Breast Cancer Trialists’ Collaborative Group), McGale P, Taylor C, Correa C, et al. Effect of radiotherapy after mastectomy and axillary surgery on 10-year recurrence and 20-year breast cancer mortality: meta-analysis of individual patient data for 8135 women in 22 randomised trials. Lancet. 2014;383(9935):2127-2135.
6. Kunkler IH, Williams LJ, Jack WJ, Cameron DA, Dixon JM; PRIME II investigators. Breast-conserving surgery with or without irradiation in women aged 65 years or older with early breast cancer (PRIME II): a randomised controlled trial. Lancet Oncol. 2015;16(3):266-273.
7. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177-182.
8. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effect of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15 year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687-1717.
9. Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805-816.
Raising More Than Moods: Escitalopram-Associated Priapism
Selective serotonin reuptake inhibitors are a common first choice medication for anxiety and depression treatment, but health care providers should be aware of priapism as a possible adverse effect.
Case Presentation
An 80-year-old white male was evaluated in a primary care clinic following a recent hospitalization for a suicide attempt. His past medical history included type 2 diabetes mellitus, chronic atrial fibrillation, essential hypertension and hyperlipidemia, and no prior psychiatric illness. Six weeks after his wife died of cancer, the patient attempted suicide by slitting his wrists, which resulted in significant blood loss and tendon damage.
After medical stabilization he was treated at an inpatient psychiatric facility for 10 days. There was no evidence of impaired memory nor psychosis during his hospitalization. He was prescribed doxazosin 1 mg twice daily and finasteride 5 mg daily for obstructive urinary symptoms, along with escitalopram 5 mg daily for depression and continuation of prior medications, including glipizide 10 mg twice daily, simvastatin 20 mg daily, metformin 500 mg twice daily, and lisinopril 20 mg daily. The patient’s estimated glomerular filtration rate was 85 at the time of these events.
He was evaluated by the mental health staff at the time of his primary care outpatient visit and noted to have a Patient Health Questionnaire (PHQ-9) score of 5 (mild depression symptoms) and a Generalized Anxiety Disorder 7 Item Scale (GAD-7) score of 1 (minimum anxiety symptoms). Eleven days later during his counseling appointment, he mentioned to staff that he had experienced a painful erection the day before, which lasted 4 hours. The primary care pharmacist was consulted for review of potential medication triggers. It was noted that there was a low frequency of priapism with both doxazosin and escitalopram, a selective serotonin reuptake inhibitor (SSRI). The provider team felt that the α blocker (doxazosin) was more likely than was the SSRI to cause the reported priapism event. Doxazosin was discontinued, and escitalopram 5 mg daily was maintained. His mood remained stable with no further suicidal ideation.
Eighteen days after discontinuation of doxazosin, the patient experienced a second priapism episode. He reported 2 days later that he experienced a prolonged, painful erection that lasted 4 hours and resolved without intervention. The patient’s mood continued without further suicidal thoughts, his appetite was normal, he had good social support and played cards with friends regularly. At that time, the decision was made to discontinue the escitalopram. The SSRI was felt to be a possible cause of priapism due to the length of time off doxazosin in relation to the second event.
The patient continued to do well 15 months after discontinuation of these medications. Unfortunately, he did not seek medical care during either episode of priapism, but he was felt to be reliable in his report based on a normal mental status exam. He does not have any of the other known risk factors for priapism, suggesting a possible association with his α blocker and SSRI.
Discussion
Priapism is a prolonged, painful erection lasting more than 4 hours and is considered a urologic emergency. It is divided into ischemic and nonischemic types. Ischemic priapism occurs with blood dyscrasias, such as sickle cell disease, thalassemia, leukemia, neurologic conditions affecting the spinal cord, and malignancies of bladder/prostate. The lifetime probability of priapism in patients affected by sickle cell disease is estimated at 29% to 42%.1 Medications associated with priapism include cocaine, ondansetron, antipsychotics, excessive use of erectile dysfunction drugs, and increasingly, antidepressants.2-8
Nonischemic priapism is usually associated with pelvic trauma. Cavernous blood gas obtained at the time of the event can help distinguish between the 2 types. The color of the aspirated blood sample is black in patients with ischemic priapism. Corporal blood gas analysis shows hypoxemia and acidemia. The color of blood is red in patients with nonischemic priapism and shows normal oxygen and pH. Priapism is a urologic emergency requiring aspiration of blood from the cavernous sinus to prevent ischemic tissue damage. At times surgical decompression may be required if aspiration is not successful.
Adrenergic α-blocking agents were developed for treatment of hypertension. They have become popular for management of lower urinary tract symptoms (LUTS) secondary to prostate enlargement. Doxazosin, prazosin, and terazosin are nonuroselective and have a higher risk of cardiac adverse effects (AEs), including dizziness and orthostatic hypotension. Lexicomp lists < 1% incidence of priapism associated with doxazosin.9 The drug is metabolized by CYP3A4 with secondary pathways, including CYP2D6 and 2C9 with a drug half-life of 22 hours. Newer agents (eg, tamsulosin, alfuzosin) are considered more uroselective, targeting the α-1b receptors. The older agents have more effect on the α-1a receptors, which are also present at higher level in the cardiovascular system.10 By blocking sympathetic stimuli responsible for penile detumescence, the nonselective α blockers have a higher propensity to cause priapism. There seems to be a direct correlation between higher doses and increased risk of priapism.11 Our patient was at a relatively low dose (1 mg twice daily) of the nonselective agent doxazosin for treatment of his LUTS.
Primary care providers and psychiatrists treating depression are familiar with common sexual AEs of the SSRI class of medications. Decreased sexual desire and delayed orgasm and ejaculation are all issues that lead patients to discontinue treatment. Although SSRIs are considered first-line treatment for depression, reports indicate that up to 60% of patients with prior normal sexual function started on paroxetine may experience sexual AEs.12 The exact frequency is difficult to estimate due to underreporting of these issues by patients.
A review of the literature for cases of priapism associated with SSRIs shows that often there is more than 1 possible drug trigger, and medications used in combination may be a risk factor. It is hypothesized that SSRI action on 5-HT3 receptors may be responsible for priapism occurring in patients treated with SSRIs.13 One study cites a case of priapism in a veteran being treated with escitalopram, prazosin, and trazodone for posttraumatic stress disorder.14 Trazodone inhibits the neuronal uptake of serotonin and is used to treat depression in addition to off-label use in treatment of insomnia. Trazodone is implicated in cases of priapism via its α-blocking properties. In the aforementioned case, trazodone was initially thought to be the causative agent and was discontinued. The patient had recurrent symptoms at which time his prazosin was discontinued, and he had no further events.
Another case cites citalopram-induced priapism that occurred with an accidental overdose of citalopram 80 mg, when a patient confused his antidepressant with 81-mg aspirin tablets.15 He also had a prior history of priapism while taking trazodone. We found only 1 case listing escitalopram as the probable causative agent of priapism.16 Similar to our patient, that case had no risk factors prior to escitalopram administration. Lexicomp notes < 1% incidence of priapism reported in postmarketing studies.
Our patient had been off doxazosin for 18 days when his second event of priapism occurred. It is less likely given the half-life of doxazosin (t ½ = 22 hours) that the α blocker was the causative agent, though a combination of the 2 agents cannot be excluded as a significant factor. The Naranjo Score is an algorithm for determining the likelihood of whether an adverse drug reaction is due to the drug or other factors.17 Scoring ranks the event as probable, possible, or doubtful. This case scored +3 (+2 = appeared after suspected drug given, +1 = improved when drug discontinued), indicating possible association of escitalopram and priapism.
Conclusion
In view of the frequent use of SSRIs in treatment of depression, it may be prudent to advise patients of this uncommon but serious medication AE. Recent use of α blockers may be a risk factor in combination with SSRI therapy. Patients should be counseled to seek emergency care in the event of prolonged erection when discussing potential AEs of SSRI therapy.
1. Manjunath AS, Hofer MD. Urologic emergencies. Med Clin North Am. 2018;102(2):373-385.
2. Altman AL, Seftel AD, Brown SL, et al. Cocaine associated priapism. J Urol. 1999;161(6):1817-1818.
3. Pivot D, Javot L, Swiegot D, et al. Two Cases of recurrent priapism during antineoplastic chemotherapy: think about ondansetron. Therapie. 2013;68(6):409-410.
4. Fu E, Kovach JG, Dubin WR. Priapism associate with antipsychotic medication use: case report. J Clin Pshychopharmacol. 2017;37(4):477-478.
5. Saghafi O, Kao A, Druck J. Recurrent priapism from therapeutic quetiapine. West J Emerg Med, 2014;15(1):114-116.
6. King SH, Hallock M, Strote J, et al. Tadalafil-associated priapism. Urology. 2005;66(2):432.
7. Giuliano F, Jackson G, Montorsi F, et al. Safety of sildenafil citrate: review of 67 double-blind placebo-controlled trials and the postmarketing safety database. Int J Clin Pract. 2010;64(2):240-255.
8. Bhat IA, Shannon KD, Ara A, et al. Ninety-six hours ordeal of priapism induced by paroxetine: a case report and literature review. Int J Psychiatry Med. 2015;50(3):326-334.
9. Cardura [package insert]. New York, NY: Pfizer; 2009.
10. Spagnul SJ, Cabral PH, Verndl DO, Glina S. Adrenergic α-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23(3):95-98.
11. Avisrro MU, Fernandez IA, Sánchez AS, García-Pando AC, Arias LM, del Pozo JG. Doxazosin and priapism. J Urol. 2000;163(1): 238.
12. Higgins A, Nash M, Lynch AM. Antidepressant-associated sexual dysfunction: impact, effects, and treatments. Drug Healthc Patient Saf. 2010;2:141-150.
13. Bonnot O, Warot D, Cohen D. Priapism associated with sertraline. J Am Acad Child Adolesc Psychiatry. 2007;46(7):790-791.
14. Mann RA, George AK. Recurrent priapism in a military veteran receiving treatment for PTSD. Mil Med. 2017;182(11):e2014-e2017.
15. Dent LA, Brown WC, Murney JD. Citalopram-induced priapism. Pharmacotherapy. 2002;22(4):538-541.
16. Tulachan P, Chapagain M, Ojha SP, Dhungana S. Escitalopram induced priapism. J Inst Med. 2014;36(1):118-120.
17. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
Selective serotonin reuptake inhibitors are a common first choice medication for anxiety and depression treatment, but health care providers should be aware of priapism as a possible adverse effect.
Selective serotonin reuptake inhibitors are a common first choice medication for anxiety and depression treatment, but health care providers should be aware of priapism as a possible adverse effect.
Case Presentation
An 80-year-old white male was evaluated in a primary care clinic following a recent hospitalization for a suicide attempt. His past medical history included type 2 diabetes mellitus, chronic atrial fibrillation, essential hypertension and hyperlipidemia, and no prior psychiatric illness. Six weeks after his wife died of cancer, the patient attempted suicide by slitting his wrists, which resulted in significant blood loss and tendon damage.
After medical stabilization he was treated at an inpatient psychiatric facility for 10 days. There was no evidence of impaired memory nor psychosis during his hospitalization. He was prescribed doxazosin 1 mg twice daily and finasteride 5 mg daily for obstructive urinary symptoms, along with escitalopram 5 mg daily for depression and continuation of prior medications, including glipizide 10 mg twice daily, simvastatin 20 mg daily, metformin 500 mg twice daily, and lisinopril 20 mg daily. The patient’s estimated glomerular filtration rate was 85 at the time of these events.
He was evaluated by the mental health staff at the time of his primary care outpatient visit and noted to have a Patient Health Questionnaire (PHQ-9) score of 5 (mild depression symptoms) and a Generalized Anxiety Disorder 7 Item Scale (GAD-7) score of 1 (minimum anxiety symptoms). Eleven days later during his counseling appointment, he mentioned to staff that he had experienced a painful erection the day before, which lasted 4 hours. The primary care pharmacist was consulted for review of potential medication triggers. It was noted that there was a low frequency of priapism with both doxazosin and escitalopram, a selective serotonin reuptake inhibitor (SSRI). The provider team felt that the α blocker (doxazosin) was more likely than was the SSRI to cause the reported priapism event. Doxazosin was discontinued, and escitalopram 5 mg daily was maintained. His mood remained stable with no further suicidal ideation.
Eighteen days after discontinuation of doxazosin, the patient experienced a second priapism episode. He reported 2 days later that he experienced a prolonged, painful erection that lasted 4 hours and resolved without intervention. The patient’s mood continued without further suicidal thoughts, his appetite was normal, he had good social support and played cards with friends regularly. At that time, the decision was made to discontinue the escitalopram. The SSRI was felt to be a possible cause of priapism due to the length of time off doxazosin in relation to the second event.
The patient continued to do well 15 months after discontinuation of these medications. Unfortunately, he did not seek medical care during either episode of priapism, but he was felt to be reliable in his report based on a normal mental status exam. He does not have any of the other known risk factors for priapism, suggesting a possible association with his α blocker and SSRI.
Discussion
Priapism is a prolonged, painful erection lasting more than 4 hours and is considered a urologic emergency. It is divided into ischemic and nonischemic types. Ischemic priapism occurs with blood dyscrasias, such as sickle cell disease, thalassemia, leukemia, neurologic conditions affecting the spinal cord, and malignancies of bladder/prostate. The lifetime probability of priapism in patients affected by sickle cell disease is estimated at 29% to 42%.1 Medications associated with priapism include cocaine, ondansetron, antipsychotics, excessive use of erectile dysfunction drugs, and increasingly, antidepressants.2-8
Nonischemic priapism is usually associated with pelvic trauma. Cavernous blood gas obtained at the time of the event can help distinguish between the 2 types. The color of the aspirated blood sample is black in patients with ischemic priapism. Corporal blood gas analysis shows hypoxemia and acidemia. The color of blood is red in patients with nonischemic priapism and shows normal oxygen and pH. Priapism is a urologic emergency requiring aspiration of blood from the cavernous sinus to prevent ischemic tissue damage. At times surgical decompression may be required if aspiration is not successful.
Adrenergic α-blocking agents were developed for treatment of hypertension. They have become popular for management of lower urinary tract symptoms (LUTS) secondary to prostate enlargement. Doxazosin, prazosin, and terazosin are nonuroselective and have a higher risk of cardiac adverse effects (AEs), including dizziness and orthostatic hypotension. Lexicomp lists < 1% incidence of priapism associated with doxazosin.9 The drug is metabolized by CYP3A4 with secondary pathways, including CYP2D6 and 2C9 with a drug half-life of 22 hours. Newer agents (eg, tamsulosin, alfuzosin) are considered more uroselective, targeting the α-1b receptors. The older agents have more effect on the α-1a receptors, which are also present at higher level in the cardiovascular system.10 By blocking sympathetic stimuli responsible for penile detumescence, the nonselective α blockers have a higher propensity to cause priapism. There seems to be a direct correlation between higher doses and increased risk of priapism.11 Our patient was at a relatively low dose (1 mg twice daily) of the nonselective agent doxazosin for treatment of his LUTS.
Primary care providers and psychiatrists treating depression are familiar with common sexual AEs of the SSRI class of medications. Decreased sexual desire and delayed orgasm and ejaculation are all issues that lead patients to discontinue treatment. Although SSRIs are considered first-line treatment for depression, reports indicate that up to 60% of patients with prior normal sexual function started on paroxetine may experience sexual AEs.12 The exact frequency is difficult to estimate due to underreporting of these issues by patients.
A review of the literature for cases of priapism associated with SSRIs shows that often there is more than 1 possible drug trigger, and medications used in combination may be a risk factor. It is hypothesized that SSRI action on 5-HT3 receptors may be responsible for priapism occurring in patients treated with SSRIs.13 One study cites a case of priapism in a veteran being treated with escitalopram, prazosin, and trazodone for posttraumatic stress disorder.14 Trazodone inhibits the neuronal uptake of serotonin and is used to treat depression in addition to off-label use in treatment of insomnia. Trazodone is implicated in cases of priapism via its α-blocking properties. In the aforementioned case, trazodone was initially thought to be the causative agent and was discontinued. The patient had recurrent symptoms at which time his prazosin was discontinued, and he had no further events.
Another case cites citalopram-induced priapism that occurred with an accidental overdose of citalopram 80 mg, when a patient confused his antidepressant with 81-mg aspirin tablets.15 He also had a prior history of priapism while taking trazodone. We found only 1 case listing escitalopram as the probable causative agent of priapism.16 Similar to our patient, that case had no risk factors prior to escitalopram administration. Lexicomp notes < 1% incidence of priapism reported in postmarketing studies.
Our patient had been off doxazosin for 18 days when his second event of priapism occurred. It is less likely given the half-life of doxazosin (t ½ = 22 hours) that the α blocker was the causative agent, though a combination of the 2 agents cannot be excluded as a significant factor. The Naranjo Score is an algorithm for determining the likelihood of whether an adverse drug reaction is due to the drug or other factors.17 Scoring ranks the event as probable, possible, or doubtful. This case scored +3 (+2 = appeared after suspected drug given, +1 = improved when drug discontinued), indicating possible association of escitalopram and priapism.
Conclusion
In view of the frequent use of SSRIs in treatment of depression, it may be prudent to advise patients of this uncommon but serious medication AE. Recent use of α blockers may be a risk factor in combination with SSRI therapy. Patients should be counseled to seek emergency care in the event of prolonged erection when discussing potential AEs of SSRI therapy.
Case Presentation
An 80-year-old white male was evaluated in a primary care clinic following a recent hospitalization for a suicide attempt. His past medical history included type 2 diabetes mellitus, chronic atrial fibrillation, essential hypertension and hyperlipidemia, and no prior psychiatric illness. Six weeks after his wife died of cancer, the patient attempted suicide by slitting his wrists, which resulted in significant blood loss and tendon damage.
After medical stabilization he was treated at an inpatient psychiatric facility for 10 days. There was no evidence of impaired memory nor psychosis during his hospitalization. He was prescribed doxazosin 1 mg twice daily and finasteride 5 mg daily for obstructive urinary symptoms, along with escitalopram 5 mg daily for depression and continuation of prior medications, including glipizide 10 mg twice daily, simvastatin 20 mg daily, metformin 500 mg twice daily, and lisinopril 20 mg daily. The patient’s estimated glomerular filtration rate was 85 at the time of these events.
He was evaluated by the mental health staff at the time of his primary care outpatient visit and noted to have a Patient Health Questionnaire (PHQ-9) score of 5 (mild depression symptoms) and a Generalized Anxiety Disorder 7 Item Scale (GAD-7) score of 1 (minimum anxiety symptoms). Eleven days later during his counseling appointment, he mentioned to staff that he had experienced a painful erection the day before, which lasted 4 hours. The primary care pharmacist was consulted for review of potential medication triggers. It was noted that there was a low frequency of priapism with both doxazosin and escitalopram, a selective serotonin reuptake inhibitor (SSRI). The provider team felt that the α blocker (doxazosin) was more likely than was the SSRI to cause the reported priapism event. Doxazosin was discontinued, and escitalopram 5 mg daily was maintained. His mood remained stable with no further suicidal ideation.
Eighteen days after discontinuation of doxazosin, the patient experienced a second priapism episode. He reported 2 days later that he experienced a prolonged, painful erection that lasted 4 hours and resolved without intervention. The patient’s mood continued without further suicidal thoughts, his appetite was normal, he had good social support and played cards with friends regularly. At that time, the decision was made to discontinue the escitalopram. The SSRI was felt to be a possible cause of priapism due to the length of time off doxazosin in relation to the second event.
The patient continued to do well 15 months after discontinuation of these medications. Unfortunately, he did not seek medical care during either episode of priapism, but he was felt to be reliable in his report based on a normal mental status exam. He does not have any of the other known risk factors for priapism, suggesting a possible association with his α blocker and SSRI.
Discussion
Priapism is a prolonged, painful erection lasting more than 4 hours and is considered a urologic emergency. It is divided into ischemic and nonischemic types. Ischemic priapism occurs with blood dyscrasias, such as sickle cell disease, thalassemia, leukemia, neurologic conditions affecting the spinal cord, and malignancies of bladder/prostate. The lifetime probability of priapism in patients affected by sickle cell disease is estimated at 29% to 42%.1 Medications associated with priapism include cocaine, ondansetron, antipsychotics, excessive use of erectile dysfunction drugs, and increasingly, antidepressants.2-8
Nonischemic priapism is usually associated with pelvic trauma. Cavernous blood gas obtained at the time of the event can help distinguish between the 2 types. The color of the aspirated blood sample is black in patients with ischemic priapism. Corporal blood gas analysis shows hypoxemia and acidemia. The color of blood is red in patients with nonischemic priapism and shows normal oxygen and pH. Priapism is a urologic emergency requiring aspiration of blood from the cavernous sinus to prevent ischemic tissue damage. At times surgical decompression may be required if aspiration is not successful.
Adrenergic α-blocking agents were developed for treatment of hypertension. They have become popular for management of lower urinary tract symptoms (LUTS) secondary to prostate enlargement. Doxazosin, prazosin, and terazosin are nonuroselective and have a higher risk of cardiac adverse effects (AEs), including dizziness and orthostatic hypotension. Lexicomp lists < 1% incidence of priapism associated with doxazosin.9 The drug is metabolized by CYP3A4 with secondary pathways, including CYP2D6 and 2C9 with a drug half-life of 22 hours. Newer agents (eg, tamsulosin, alfuzosin) are considered more uroselective, targeting the α-1b receptors. The older agents have more effect on the α-1a receptors, which are also present at higher level in the cardiovascular system.10 By blocking sympathetic stimuli responsible for penile detumescence, the nonselective α blockers have a higher propensity to cause priapism. There seems to be a direct correlation between higher doses and increased risk of priapism.11 Our patient was at a relatively low dose (1 mg twice daily) of the nonselective agent doxazosin for treatment of his LUTS.
Primary care providers and psychiatrists treating depression are familiar with common sexual AEs of the SSRI class of medications. Decreased sexual desire and delayed orgasm and ejaculation are all issues that lead patients to discontinue treatment. Although SSRIs are considered first-line treatment for depression, reports indicate that up to 60% of patients with prior normal sexual function started on paroxetine may experience sexual AEs.12 The exact frequency is difficult to estimate due to underreporting of these issues by patients.
A review of the literature for cases of priapism associated with SSRIs shows that often there is more than 1 possible drug trigger, and medications used in combination may be a risk factor. It is hypothesized that SSRI action on 5-HT3 receptors may be responsible for priapism occurring in patients treated with SSRIs.13 One study cites a case of priapism in a veteran being treated with escitalopram, prazosin, and trazodone for posttraumatic stress disorder.14 Trazodone inhibits the neuronal uptake of serotonin and is used to treat depression in addition to off-label use in treatment of insomnia. Trazodone is implicated in cases of priapism via its α-blocking properties. In the aforementioned case, trazodone was initially thought to be the causative agent and was discontinued. The patient had recurrent symptoms at which time his prazosin was discontinued, and he had no further events.
Another case cites citalopram-induced priapism that occurred with an accidental overdose of citalopram 80 mg, when a patient confused his antidepressant with 81-mg aspirin tablets.15 He also had a prior history of priapism while taking trazodone. We found only 1 case listing escitalopram as the probable causative agent of priapism.16 Similar to our patient, that case had no risk factors prior to escitalopram administration. Lexicomp notes < 1% incidence of priapism reported in postmarketing studies.
Our patient had been off doxazosin for 18 days when his second event of priapism occurred. It is less likely given the half-life of doxazosin (t ½ = 22 hours) that the α blocker was the causative agent, though a combination of the 2 agents cannot be excluded as a significant factor. The Naranjo Score is an algorithm for determining the likelihood of whether an adverse drug reaction is due to the drug or other factors.17 Scoring ranks the event as probable, possible, or doubtful. This case scored +3 (+2 = appeared after suspected drug given, +1 = improved when drug discontinued), indicating possible association of escitalopram and priapism.
Conclusion
In view of the frequent use of SSRIs in treatment of depression, it may be prudent to advise patients of this uncommon but serious medication AE. Recent use of α blockers may be a risk factor in combination with SSRI therapy. Patients should be counseled to seek emergency care in the event of prolonged erection when discussing potential AEs of SSRI therapy.
1. Manjunath AS, Hofer MD. Urologic emergencies. Med Clin North Am. 2018;102(2):373-385.
2. Altman AL, Seftel AD, Brown SL, et al. Cocaine associated priapism. J Urol. 1999;161(6):1817-1818.
3. Pivot D, Javot L, Swiegot D, et al. Two Cases of recurrent priapism during antineoplastic chemotherapy: think about ondansetron. Therapie. 2013;68(6):409-410.
4. Fu E, Kovach JG, Dubin WR. Priapism associate with antipsychotic medication use: case report. J Clin Pshychopharmacol. 2017;37(4):477-478.
5. Saghafi O, Kao A, Druck J. Recurrent priapism from therapeutic quetiapine. West J Emerg Med, 2014;15(1):114-116.
6. King SH, Hallock M, Strote J, et al. Tadalafil-associated priapism. Urology. 2005;66(2):432.
7. Giuliano F, Jackson G, Montorsi F, et al. Safety of sildenafil citrate: review of 67 double-blind placebo-controlled trials and the postmarketing safety database. Int J Clin Pract. 2010;64(2):240-255.
8. Bhat IA, Shannon KD, Ara A, et al. Ninety-six hours ordeal of priapism induced by paroxetine: a case report and literature review. Int J Psychiatry Med. 2015;50(3):326-334.
9. Cardura [package insert]. New York, NY: Pfizer; 2009.
10. Spagnul SJ, Cabral PH, Verndl DO, Glina S. Adrenergic α-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23(3):95-98.
11. Avisrro MU, Fernandez IA, Sánchez AS, García-Pando AC, Arias LM, del Pozo JG. Doxazosin and priapism. J Urol. 2000;163(1): 238.
12. Higgins A, Nash M, Lynch AM. Antidepressant-associated sexual dysfunction: impact, effects, and treatments. Drug Healthc Patient Saf. 2010;2:141-150.
13. Bonnot O, Warot D, Cohen D. Priapism associated with sertraline. J Am Acad Child Adolesc Psychiatry. 2007;46(7):790-791.
14. Mann RA, George AK. Recurrent priapism in a military veteran receiving treatment for PTSD. Mil Med. 2017;182(11):e2014-e2017.
15. Dent LA, Brown WC, Murney JD. Citalopram-induced priapism. Pharmacotherapy. 2002;22(4):538-541.
16. Tulachan P, Chapagain M, Ojha SP, Dhungana S. Escitalopram induced priapism. J Inst Med. 2014;36(1):118-120.
17. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
1. Manjunath AS, Hofer MD. Urologic emergencies. Med Clin North Am. 2018;102(2):373-385.
2. Altman AL, Seftel AD, Brown SL, et al. Cocaine associated priapism. J Urol. 1999;161(6):1817-1818.
3. Pivot D, Javot L, Swiegot D, et al. Two Cases of recurrent priapism during antineoplastic chemotherapy: think about ondansetron. Therapie. 2013;68(6):409-410.
4. Fu E, Kovach JG, Dubin WR. Priapism associate with antipsychotic medication use: case report. J Clin Pshychopharmacol. 2017;37(4):477-478.
5. Saghafi O, Kao A, Druck J. Recurrent priapism from therapeutic quetiapine. West J Emerg Med, 2014;15(1):114-116.
6. King SH, Hallock M, Strote J, et al. Tadalafil-associated priapism. Urology. 2005;66(2):432.
7. Giuliano F, Jackson G, Montorsi F, et al. Safety of sildenafil citrate: review of 67 double-blind placebo-controlled trials and the postmarketing safety database. Int J Clin Pract. 2010;64(2):240-255.
8. Bhat IA, Shannon KD, Ara A, et al. Ninety-six hours ordeal of priapism induced by paroxetine: a case report and literature review. Int J Psychiatry Med. 2015;50(3):326-334.
9. Cardura [package insert]. New York, NY: Pfizer; 2009.
10. Spagnul SJ, Cabral PH, Verndl DO, Glina S. Adrenergic α-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23(3):95-98.
11. Avisrro MU, Fernandez IA, Sánchez AS, García-Pando AC, Arias LM, del Pozo JG. Doxazosin and priapism. J Urol. 2000;163(1): 238.
12. Higgins A, Nash M, Lynch AM. Antidepressant-associated sexual dysfunction: impact, effects, and treatments. Drug Healthc Patient Saf. 2010;2:141-150.
13. Bonnot O, Warot D, Cohen D. Priapism associated with sertraline. J Am Acad Child Adolesc Psychiatry. 2007;46(7):790-791.
14. Mann RA, George AK. Recurrent priapism in a military veteran receiving treatment for PTSD. Mil Med. 2017;182(11):e2014-e2017.
15. Dent LA, Brown WC, Murney JD. Citalopram-induced priapism. Pharmacotherapy. 2002;22(4):538-541.
16. Tulachan P, Chapagain M, Ojha SP, Dhungana S. Escitalopram induced priapism. J Inst Med. 2014;36(1):118-120.
17. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
Clearance of Psoriasis After Ischemic Stroke
The etiology of psoriasis is multifactorial, and it is attributed to both genetic and environmental components.1 One of the lesser-studied aspects of psoriasis pathogenesis is the involvement of the nervous system. It is thought that the pathogenesis involves inflammation of the cutaneous nerves,2 and cutaneous denervation has been shown to improve acanthosis and IL-23 expression in mice with psoriasiform skin.3 There also have been reports of psoriasis remission following peripheral and central nervous system injury from surgical nerve resection4 as well as cerebrovascular accident.5 We present a case of total psoriasis clearance following ischemic stroke.
Case Report
A 52-year-old man with psoriasis presented to the dermatology clinic for follow-up. The patient had been using topical clobetasol and apremilast with limited success but had not previously tried biologics. On physical examination he was noted to have erythematous, scaly, indurated papules and plaques on the chest, abdomen, back, arms, and legs, consistent with psoriasis. Affected body surface area was approximately 10%. Ustekinumab was prescribed, but the patient did not pick it up from the pharmacy.
Approximately 1 month later, the patient presented to the emergency department with left-sided weakness and numbness. He was hospitalized for treatment of stroke. During hospitalization, the patient was started on lisinopril, aspirin, and atorvastatin. He also was given subcutaneous enoxaparin with plans to initiate warfarin as an outpatient. His psoriasis was not treated with topical or systemic medications during the course of his admission. He was discharged to a skilled nursing facility after 3 days.
Three months following discharge, the patient returned to the dermatology clinic for follow-up. After his stroke, he reported that his psoriasis had cleared and had not returned. On physical examination his skin was clear of psoriatic lesions.
Comment
The nervous system is thought to play an important role in the pathophysiology of psoriasis. Evidence for this involvement includes the exacerbation of psoriasis with stress and the often symmetric distribution of psoriatic lesions.6
Moreover, numerous neuropeptides have been identified in the pathophysiology of psoriasis. Farber et al7 first proposed that release of substance P (SP) from cutaneous sensory nerve fibers causes a local neurogenic response that triggers psoriasis in predisposed individuals. The role of SP in psoriasis is unclear, as there have been reports of both higher8 and lower9 levels in involved and noninvolved skin of psoriatic patients compared to skin in healthy individuals. It has been suggested that numerous other neuropeptides, including nerve growth factor (NGF), calcitonin gene-related peptide, and vasoactive intestinal peptide, play a part in psoriasis.2,10 Specifically, NGF prevents apoptosis of keratinocytes11 and is found in higher levels in psoriatic skin compared to controls.12 Calcitonin gene-related peptide has been shown to stimulate keratinocyte proliferation13 and has been found at increased levels in psoriatic skin.14 Vasoactive intestinal peptide-positive nerve fibers in the epidermis and dermis are found in higher quantities in psoriatic plaques compared to nonlesional and normal skin.8
Neuropeptides also might play a role in the itching and Köbner phenomenon that accompany psoriasis. Increased levels of NGF in nonlesional skin of patients with psoriasis is thought to contribute to the development of psoriatic plaques following trauma by inducing an inflammatory response that upregulates other neuropeptides, such as SP and calcitonin gene-related peptide. These neuropeptides induce keratinocyte proliferation, which further increases NGF expression, thus creating a cycle of inflammation and formation of psoriatic lesions.6 Moreover, there is a notable correlation between pruritus severity and density of NGF-immunoreactive keratinocytes, high-affinity NGF receptors, protein gene product 9.5–immunoreactive intraepidermal fibers, and immunoreactive vessels for E-selectin.15
Spontaneous remission of psoriasis after cerebrovascular accident was first reported in 1998.5 Moreover, there have been cases of protective effects from psoriasis and psoriatic arthritis in limbs affected by poliomyelitis.16,17 In cases in which patients regained neurologic function, Zhu et al10 found that recurrence of skin lesions in areas corresponding to nervous system injury also occurred. However, in cases of permanent nerve damage, psoriasis did not return,10 confirming the role of peripheral nerves in the pathogenesis of psoriasis. It is thought that peripheral nerve damage results in decreased secretion of neuropeptides3 and that central nervous system injury also can cause similar downstream effects.10
Other reasons for the patient’s remission also were considered. Although it is possible that the sudden change in the patient’s usual environment could have induced remission of psoriasis, it seems more likely that the stress of the situation would have worsened his symptoms. Medications used during the patient’s hospitalization also were considered as reasons for symptom improvement. One study using a case-control and case-crossover design found psoriasis to be associated with nonsteroidal anti-inflammatory drugs and angiotensin-converting enzyme inhibitors (odds ratio, 4.0 and 2.1, respectively).18 Atorvastatin has been investigated as a potential treatment of psoriasis, though no therapeutic benefit has been proven.19,20 Heparin has been shown in case reports to improve psoriasis symptoms but was used in addition to standard psoriasis therapies and not as monotherapy.21
A more thorough understanding of which neuropeptides are directly implicated in the neurologic-mediated clearance of psoriasis might contribute to better targeted therapies. For example, infusion of peptide T, a vasoactive intestinal peptide analogue, was shown to have some effect in clearing the skin in 14 psoriasis patients.22 Although this finding has not been replicated, it demonstrates the potential utility of therapies targeted toward the neurologic aspects of psoriasis. More research is needed to evaluate the potential of targeting other neuropeptides for treatment of psoriatic plaques.
- Boehncke WH. Etiology and pathogenesis of psoriasis. Rheum Dis Clin North Am. 2015;41:665-675.
- Saraceno R, Kleyn CE, Terenghi G, et al. The role of neuropeptides in psoriasis. Br J Dermatol. 2006;155:876-882.
- Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
- Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
- Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
- Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
- Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
- Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
- Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
- Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
- Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
- Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
- He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
- Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
- Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
- Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomeylitis residual paralysis. Br J Dermatol. 2014;171:429-431.
- Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
- Cohen AD, Bonneh DY, Reuveni H, et al. Drug exposure and psoriasis vulgaris: case control and case-crossover studies. Acta Derm Venereol. 2005;85:299-303.
- Faghihi T, Radfar M, Mehrabian Z, et al. Atorvastatin for the treatment of plaque-type psoriasis. Pharmacotherapy. 2011;31:1045-1050.
- Chua SHH, Tioleco GMS, Dayrit CAF, et al. Atorvastatin as adjunctive therapy for chronic plaque type psoriasis versus betamethasone valerate alone: a randomized, double-blind, placebo-controlled trial. Indian J Dermatol Venereol Leprol. 2017;83:441-447.
- Jekel LG. Use of heparin in treatment of psoriasis. AMA Arch Derm Syphilol. 1953;68:80-82.
- Farber EM, Cohen EN, Trozak DJ, et al. Peptide T improves psoriasis when infused into lesions in nanogram amounts. J Am Acad Dermatol. 1991;25:658-664.
The etiology of psoriasis is multifactorial, and it is attributed to both genetic and environmental components.1 One of the lesser-studied aspects of psoriasis pathogenesis is the involvement of the nervous system. It is thought that the pathogenesis involves inflammation of the cutaneous nerves,2 and cutaneous denervation has been shown to improve acanthosis and IL-23 expression in mice with psoriasiform skin.3 There also have been reports of psoriasis remission following peripheral and central nervous system injury from surgical nerve resection4 as well as cerebrovascular accident.5 We present a case of total psoriasis clearance following ischemic stroke.
Case Report
A 52-year-old man with psoriasis presented to the dermatology clinic for follow-up. The patient had been using topical clobetasol and apremilast with limited success but had not previously tried biologics. On physical examination he was noted to have erythematous, scaly, indurated papules and plaques on the chest, abdomen, back, arms, and legs, consistent with psoriasis. Affected body surface area was approximately 10%. Ustekinumab was prescribed, but the patient did not pick it up from the pharmacy.
Approximately 1 month later, the patient presented to the emergency department with left-sided weakness and numbness. He was hospitalized for treatment of stroke. During hospitalization, the patient was started on lisinopril, aspirin, and atorvastatin. He also was given subcutaneous enoxaparin with plans to initiate warfarin as an outpatient. His psoriasis was not treated with topical or systemic medications during the course of his admission. He was discharged to a skilled nursing facility after 3 days.
Three months following discharge, the patient returned to the dermatology clinic for follow-up. After his stroke, he reported that his psoriasis had cleared and had not returned. On physical examination his skin was clear of psoriatic lesions.
Comment
The nervous system is thought to play an important role in the pathophysiology of psoriasis. Evidence for this involvement includes the exacerbation of psoriasis with stress and the often symmetric distribution of psoriatic lesions.6
Moreover, numerous neuropeptides have been identified in the pathophysiology of psoriasis. Farber et al7 first proposed that release of substance P (SP) from cutaneous sensory nerve fibers causes a local neurogenic response that triggers psoriasis in predisposed individuals. The role of SP in psoriasis is unclear, as there have been reports of both higher8 and lower9 levels in involved and noninvolved skin of psoriatic patients compared to skin in healthy individuals. It has been suggested that numerous other neuropeptides, including nerve growth factor (NGF), calcitonin gene-related peptide, and vasoactive intestinal peptide, play a part in psoriasis.2,10 Specifically, NGF prevents apoptosis of keratinocytes11 and is found in higher levels in psoriatic skin compared to controls.12 Calcitonin gene-related peptide has been shown to stimulate keratinocyte proliferation13 and has been found at increased levels in psoriatic skin.14 Vasoactive intestinal peptide-positive nerve fibers in the epidermis and dermis are found in higher quantities in psoriatic plaques compared to nonlesional and normal skin.8
Neuropeptides also might play a role in the itching and Köbner phenomenon that accompany psoriasis. Increased levels of NGF in nonlesional skin of patients with psoriasis is thought to contribute to the development of psoriatic plaques following trauma by inducing an inflammatory response that upregulates other neuropeptides, such as SP and calcitonin gene-related peptide. These neuropeptides induce keratinocyte proliferation, which further increases NGF expression, thus creating a cycle of inflammation and formation of psoriatic lesions.6 Moreover, there is a notable correlation between pruritus severity and density of NGF-immunoreactive keratinocytes, high-affinity NGF receptors, protein gene product 9.5–immunoreactive intraepidermal fibers, and immunoreactive vessels for E-selectin.15
Spontaneous remission of psoriasis after cerebrovascular accident was first reported in 1998.5 Moreover, there have been cases of protective effects from psoriasis and psoriatic arthritis in limbs affected by poliomyelitis.16,17 In cases in which patients regained neurologic function, Zhu et al10 found that recurrence of skin lesions in areas corresponding to nervous system injury also occurred. However, in cases of permanent nerve damage, psoriasis did not return,10 confirming the role of peripheral nerves in the pathogenesis of psoriasis. It is thought that peripheral nerve damage results in decreased secretion of neuropeptides3 and that central nervous system injury also can cause similar downstream effects.10
Other reasons for the patient’s remission also were considered. Although it is possible that the sudden change in the patient’s usual environment could have induced remission of psoriasis, it seems more likely that the stress of the situation would have worsened his symptoms. Medications used during the patient’s hospitalization also were considered as reasons for symptom improvement. One study using a case-control and case-crossover design found psoriasis to be associated with nonsteroidal anti-inflammatory drugs and angiotensin-converting enzyme inhibitors (odds ratio, 4.0 and 2.1, respectively).18 Atorvastatin has been investigated as a potential treatment of psoriasis, though no therapeutic benefit has been proven.19,20 Heparin has been shown in case reports to improve psoriasis symptoms but was used in addition to standard psoriasis therapies and not as monotherapy.21
A more thorough understanding of which neuropeptides are directly implicated in the neurologic-mediated clearance of psoriasis might contribute to better targeted therapies. For example, infusion of peptide T, a vasoactive intestinal peptide analogue, was shown to have some effect in clearing the skin in 14 psoriasis patients.22 Although this finding has not been replicated, it demonstrates the potential utility of therapies targeted toward the neurologic aspects of psoriasis. More research is needed to evaluate the potential of targeting other neuropeptides for treatment of psoriatic plaques.
The etiology of psoriasis is multifactorial, and it is attributed to both genetic and environmental components.1 One of the lesser-studied aspects of psoriasis pathogenesis is the involvement of the nervous system. It is thought that the pathogenesis involves inflammation of the cutaneous nerves,2 and cutaneous denervation has been shown to improve acanthosis and IL-23 expression in mice with psoriasiform skin.3 There also have been reports of psoriasis remission following peripheral and central nervous system injury from surgical nerve resection4 as well as cerebrovascular accident.5 We present a case of total psoriasis clearance following ischemic stroke.
Case Report
A 52-year-old man with psoriasis presented to the dermatology clinic for follow-up. The patient had been using topical clobetasol and apremilast with limited success but had not previously tried biologics. On physical examination he was noted to have erythematous, scaly, indurated papules and plaques on the chest, abdomen, back, arms, and legs, consistent with psoriasis. Affected body surface area was approximately 10%. Ustekinumab was prescribed, but the patient did not pick it up from the pharmacy.
Approximately 1 month later, the patient presented to the emergency department with left-sided weakness and numbness. He was hospitalized for treatment of stroke. During hospitalization, the patient was started on lisinopril, aspirin, and atorvastatin. He also was given subcutaneous enoxaparin with plans to initiate warfarin as an outpatient. His psoriasis was not treated with topical or systemic medications during the course of his admission. He was discharged to a skilled nursing facility after 3 days.
Three months following discharge, the patient returned to the dermatology clinic for follow-up. After his stroke, he reported that his psoriasis had cleared and had not returned. On physical examination his skin was clear of psoriatic lesions.
Comment
The nervous system is thought to play an important role in the pathophysiology of psoriasis. Evidence for this involvement includes the exacerbation of psoriasis with stress and the often symmetric distribution of psoriatic lesions.6
Moreover, numerous neuropeptides have been identified in the pathophysiology of psoriasis. Farber et al7 first proposed that release of substance P (SP) from cutaneous sensory nerve fibers causes a local neurogenic response that triggers psoriasis in predisposed individuals. The role of SP in psoriasis is unclear, as there have been reports of both higher8 and lower9 levels in involved and noninvolved skin of psoriatic patients compared to skin in healthy individuals. It has been suggested that numerous other neuropeptides, including nerve growth factor (NGF), calcitonin gene-related peptide, and vasoactive intestinal peptide, play a part in psoriasis.2,10 Specifically, NGF prevents apoptosis of keratinocytes11 and is found in higher levels in psoriatic skin compared to controls.12 Calcitonin gene-related peptide has been shown to stimulate keratinocyte proliferation13 and has been found at increased levels in psoriatic skin.14 Vasoactive intestinal peptide-positive nerve fibers in the epidermis and dermis are found in higher quantities in psoriatic plaques compared to nonlesional and normal skin.8
Neuropeptides also might play a role in the itching and Köbner phenomenon that accompany psoriasis. Increased levels of NGF in nonlesional skin of patients with psoriasis is thought to contribute to the development of psoriatic plaques following trauma by inducing an inflammatory response that upregulates other neuropeptides, such as SP and calcitonin gene-related peptide. These neuropeptides induce keratinocyte proliferation, which further increases NGF expression, thus creating a cycle of inflammation and formation of psoriatic lesions.6 Moreover, there is a notable correlation between pruritus severity and density of NGF-immunoreactive keratinocytes, high-affinity NGF receptors, protein gene product 9.5–immunoreactive intraepidermal fibers, and immunoreactive vessels for E-selectin.15
Spontaneous remission of psoriasis after cerebrovascular accident was first reported in 1998.5 Moreover, there have been cases of protective effects from psoriasis and psoriatic arthritis in limbs affected by poliomyelitis.16,17 In cases in which patients regained neurologic function, Zhu et al10 found that recurrence of skin lesions in areas corresponding to nervous system injury also occurred. However, in cases of permanent nerve damage, psoriasis did not return,10 confirming the role of peripheral nerves in the pathogenesis of psoriasis. It is thought that peripheral nerve damage results in decreased secretion of neuropeptides3 and that central nervous system injury also can cause similar downstream effects.10
Other reasons for the patient’s remission also were considered. Although it is possible that the sudden change in the patient’s usual environment could have induced remission of psoriasis, it seems more likely that the stress of the situation would have worsened his symptoms. Medications used during the patient’s hospitalization also were considered as reasons for symptom improvement. One study using a case-control and case-crossover design found psoriasis to be associated with nonsteroidal anti-inflammatory drugs and angiotensin-converting enzyme inhibitors (odds ratio, 4.0 and 2.1, respectively).18 Atorvastatin has been investigated as a potential treatment of psoriasis, though no therapeutic benefit has been proven.19,20 Heparin has been shown in case reports to improve psoriasis symptoms but was used in addition to standard psoriasis therapies and not as monotherapy.21
A more thorough understanding of which neuropeptides are directly implicated in the neurologic-mediated clearance of psoriasis might contribute to better targeted therapies. For example, infusion of peptide T, a vasoactive intestinal peptide analogue, was shown to have some effect in clearing the skin in 14 psoriasis patients.22 Although this finding has not been replicated, it demonstrates the potential utility of therapies targeted toward the neurologic aspects of psoriasis. More research is needed to evaluate the potential of targeting other neuropeptides for treatment of psoriatic plaques.
- Boehncke WH. Etiology and pathogenesis of psoriasis. Rheum Dis Clin North Am. 2015;41:665-675.
- Saraceno R, Kleyn CE, Terenghi G, et al. The role of neuropeptides in psoriasis. Br J Dermatol. 2006;155:876-882.
- Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
- Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
- Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
- Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
- Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
- Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
- Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
- Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
- Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
- Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
- He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
- Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
- Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
- Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomeylitis residual paralysis. Br J Dermatol. 2014;171:429-431.
- Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
- Cohen AD, Bonneh DY, Reuveni H, et al. Drug exposure and psoriasis vulgaris: case control and case-crossover studies. Acta Derm Venereol. 2005;85:299-303.
- Faghihi T, Radfar M, Mehrabian Z, et al. Atorvastatin for the treatment of plaque-type psoriasis. Pharmacotherapy. 2011;31:1045-1050.
- Chua SHH, Tioleco GMS, Dayrit CAF, et al. Atorvastatin as adjunctive therapy for chronic plaque type psoriasis versus betamethasone valerate alone: a randomized, double-blind, placebo-controlled trial. Indian J Dermatol Venereol Leprol. 2017;83:441-447.
- Jekel LG. Use of heparin in treatment of psoriasis. AMA Arch Derm Syphilol. 1953;68:80-82.
- Farber EM, Cohen EN, Trozak DJ, et al. Peptide T improves psoriasis when infused into lesions in nanogram amounts. J Am Acad Dermatol. 1991;25:658-664.
- Boehncke WH. Etiology and pathogenesis of psoriasis. Rheum Dis Clin North Am. 2015;41:665-675.
- Saraceno R, Kleyn CE, Terenghi G, et al. The role of neuropeptides in psoriasis. Br J Dermatol. 2006;155:876-882.
- Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
- Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
- Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
- Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
- Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
- Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
- Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
- Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
- Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
- Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
- He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
- Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
- Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
- Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomeylitis residual paralysis. Br J Dermatol. 2014;171:429-431.
- Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
- Cohen AD, Bonneh DY, Reuveni H, et al. Drug exposure and psoriasis vulgaris: case control and case-crossover studies. Acta Derm Venereol. 2005;85:299-303.
- Faghihi T, Radfar M, Mehrabian Z, et al. Atorvastatin for the treatment of plaque-type psoriasis. Pharmacotherapy. 2011;31:1045-1050.
- Chua SHH, Tioleco GMS, Dayrit CAF, et al. Atorvastatin as adjunctive therapy for chronic plaque type psoriasis versus betamethasone valerate alone: a randomized, double-blind, placebo-controlled trial. Indian J Dermatol Venereol Leprol. 2017;83:441-447.
- Jekel LG. Use of heparin in treatment of psoriasis. AMA Arch Derm Syphilol. 1953;68:80-82.
- Farber EM, Cohen EN, Trozak DJ, et al. Peptide T improves psoriasis when infused into lesions in nanogram amounts. J Am Acad Dermatol. 1991;25:658-664.
Practice Points
- Psoriasis is exacerbated in the presence of stress, and psoriatic lesions often have a symmetric distribution, which is evidence that the nervous system is involved in the pathophysiology of the condition.
- Various neuropeptides are involved in the pathophysiology of psoriasis, including substance P, nerve growth factor, calcitonin gene-related peptide, and vasoactive intestinal peptide.
- Peripheral nerve damage results in decreased secretion of neuropeptides, which can lead to remission of psoriasis.
Psoriasis Treatment in Patients With Sickle Cell Disease
Plaque psoriasis is a chronic inflammatory disease with a complex pathogenesis. Cutaneous dendritic cells drive the activation and proliferation of T cells with production of several immunomodulators, such as tumor necrosis factor (TNF) α, IL-17, IL-12, and IL-23. Because multiple systemic therapies are efficacious, treatment selection depends on side-effect profiles, availability, and patient preference. Activation of the TNF-α pathway is not unique to psoriasis. Tumor necrosis factor α plays a key role in multiple inflammatory conditions, including psoriatic arthritis, rheumatoid arthritis, and hidradenitis suppurativa. One study in mice demonstrated that TNF-α drives endothelial and vascular wall dysfunction in sickle cell anemia. In this study, use of the TNF-α blocker etanercept in mice with homozygous sickle cell anemia (HbSS) disease resulted in amelioration of TNF-mediated clinical features shared by sickle mice and humans.1
Sickle cell anemia is caused by a structural defect in hemoglobin that results in hemolysis and chronic anemia. The most common type of hemoglobin in adults without sickle cell anemia is HbAA. Homozygous sickle cell anemia patients carry 2 abnormal S alleles, whereas in sickle cell trait, patients carry both the S and normal A alleles (HbSA). Hemoglobin C is a structural variant of HbA that results in lower solubility in red blood cells. Patients with hemoglobin SC disease (HbSC) have S and C alleles.2 We present a case of a patient with moderate to severe plaque psoriasis and heterozygous sickle cell anemia treated with adalimumab.
Case Report
A 31-year-old woman presented with moderate to severe plaque psoriasis (70% body surface area) and HbSC. She reported chronic dull arthralgia in the ankles that was worse at night. Radiographs of the feet and ankles showed erosive changes of the distal tarsal row and metatarsal bases. The diffuse bone pain had gradually worsened over the years and was treated by hematology with ibuprofen and ketorolac. At presentation, her HbSC pain was 8/10 on a visual analog scale. She described her sickle cell pain crises as sharp 10/10 pain in the back, elbows, and ankles, associated with mild edema lasting 1 to 2 days. Radiographs of the spine, hands, and ankles were unremarkable.
Adalimumab was chosen as a systemic therapy for psoriasis based on the potential for improvement in HbSC. Within 17 weeks of starting adalimumab, the psoriasis body surface area decreased from 70% to 40%, and the HbSC pain decreased from 8/10 to 4/10 at 8-week follow-up and to 0/10 at 17-week follow-up. After initiation of adalimumab, she reported decreased use of pain medication with no sickle cell pain crises.
Comment
Tumor necrosis factor α blockers are commonly used for moderate to severe plaque psoriasis. To our knowledge, there have been no reported human studies showing TNF-α blockade as a potential treatment of sickle cell disease. Increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and severity of sickle cell disease by playing an integral role in the development of vascular wall dysfunction and ischemia.3 Inflammatory mediators in HbSS disease, such as heparan sulfate from the endothelial glycocalyx and heme from hemolysis, act on monocytes to release TNF-α.1 Through this effect on the endothelium, TNF-α impedes blood flow during sickle cell crisis, leading to worsening ischemia and resultant painful infarction.3 Analysis of cytokine levels in HbSS patients showed significantly (P<.05) elevated levels of TNF
Although these findings were observational and limited to a single patient, the 50% decrease in pain level and use of pain medications reported to her hematologist independent of her dermatology visits coincided with the initiation of adalimumab. Although radiographs showed possible psoriatic changes of the distal metatarsal row, her described sickle cell pain and pain crises were atypical for psoriatic arthralgia. Tumor necrosis factor α inhibitors could be the drug of choice to treat patients with psoriasis with concomitant HbSS or HbSC disease due to the blockade of a common inflammatory mediator. Further studies are indicated to analyze the in vivo role of TNF-α inhibition in sickle cell disease.
- Solovey A, Somani A, Belcher JD, et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am J Hematol. 2017;92:1119-1130.
- Mais DD. Diseases of red blood cells. In: Laposata M, ed. Laposata’s Laboratory Medicine: Diagnosis of Disease in the Clinical Laboratory. 3rd ed. New York, NY: McGraw-Hill; 2018:247-280.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
Plaque psoriasis is a chronic inflammatory disease with a complex pathogenesis. Cutaneous dendritic cells drive the activation and proliferation of T cells with production of several immunomodulators, such as tumor necrosis factor (TNF) α, IL-17, IL-12, and IL-23. Because multiple systemic therapies are efficacious, treatment selection depends on side-effect profiles, availability, and patient preference. Activation of the TNF-α pathway is not unique to psoriasis. Tumor necrosis factor α plays a key role in multiple inflammatory conditions, including psoriatic arthritis, rheumatoid arthritis, and hidradenitis suppurativa. One study in mice demonstrated that TNF-α drives endothelial and vascular wall dysfunction in sickle cell anemia. In this study, use of the TNF-α blocker etanercept in mice with homozygous sickle cell anemia (HbSS) disease resulted in amelioration of TNF-mediated clinical features shared by sickle mice and humans.1
Sickle cell anemia is caused by a structural defect in hemoglobin that results in hemolysis and chronic anemia. The most common type of hemoglobin in adults without sickle cell anemia is HbAA. Homozygous sickle cell anemia patients carry 2 abnormal S alleles, whereas in sickle cell trait, patients carry both the S and normal A alleles (HbSA). Hemoglobin C is a structural variant of HbA that results in lower solubility in red blood cells. Patients with hemoglobin SC disease (HbSC) have S and C alleles.2 We present a case of a patient with moderate to severe plaque psoriasis and heterozygous sickle cell anemia treated with adalimumab.
Case Report
A 31-year-old woman presented with moderate to severe plaque psoriasis (70% body surface area) and HbSC. She reported chronic dull arthralgia in the ankles that was worse at night. Radiographs of the feet and ankles showed erosive changes of the distal tarsal row and metatarsal bases. The diffuse bone pain had gradually worsened over the years and was treated by hematology with ibuprofen and ketorolac. At presentation, her HbSC pain was 8/10 on a visual analog scale. She described her sickle cell pain crises as sharp 10/10 pain in the back, elbows, and ankles, associated with mild edema lasting 1 to 2 days. Radiographs of the spine, hands, and ankles were unremarkable.
Adalimumab was chosen as a systemic therapy for psoriasis based on the potential for improvement in HbSC. Within 17 weeks of starting adalimumab, the psoriasis body surface area decreased from 70% to 40%, and the HbSC pain decreased from 8/10 to 4/10 at 8-week follow-up and to 0/10 at 17-week follow-up. After initiation of adalimumab, she reported decreased use of pain medication with no sickle cell pain crises.
Comment
Tumor necrosis factor α blockers are commonly used for moderate to severe plaque psoriasis. To our knowledge, there have been no reported human studies showing TNF-α blockade as a potential treatment of sickle cell disease. Increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and severity of sickle cell disease by playing an integral role in the development of vascular wall dysfunction and ischemia.3 Inflammatory mediators in HbSS disease, such as heparan sulfate from the endothelial glycocalyx and heme from hemolysis, act on monocytes to release TNF-α.1 Through this effect on the endothelium, TNF-α impedes blood flow during sickle cell crisis, leading to worsening ischemia and resultant painful infarction.3 Analysis of cytokine levels in HbSS patients showed significantly (P<.05) elevated levels of TNF
Although these findings were observational and limited to a single patient, the 50% decrease in pain level and use of pain medications reported to her hematologist independent of her dermatology visits coincided with the initiation of adalimumab. Although radiographs showed possible psoriatic changes of the distal metatarsal row, her described sickle cell pain and pain crises were atypical for psoriatic arthralgia. Tumor necrosis factor α inhibitors could be the drug of choice to treat patients with psoriasis with concomitant HbSS or HbSC disease due to the blockade of a common inflammatory mediator. Further studies are indicated to analyze the in vivo role of TNF-α inhibition in sickle cell disease.
Plaque psoriasis is a chronic inflammatory disease with a complex pathogenesis. Cutaneous dendritic cells drive the activation and proliferation of T cells with production of several immunomodulators, such as tumor necrosis factor (TNF) α, IL-17, IL-12, and IL-23. Because multiple systemic therapies are efficacious, treatment selection depends on side-effect profiles, availability, and patient preference. Activation of the TNF-α pathway is not unique to psoriasis. Tumor necrosis factor α plays a key role in multiple inflammatory conditions, including psoriatic arthritis, rheumatoid arthritis, and hidradenitis suppurativa. One study in mice demonstrated that TNF-α drives endothelial and vascular wall dysfunction in sickle cell anemia. In this study, use of the TNF-α blocker etanercept in mice with homozygous sickle cell anemia (HbSS) disease resulted in amelioration of TNF-mediated clinical features shared by sickle mice and humans.1
Sickle cell anemia is caused by a structural defect in hemoglobin that results in hemolysis and chronic anemia. The most common type of hemoglobin in adults without sickle cell anemia is HbAA. Homozygous sickle cell anemia patients carry 2 abnormal S alleles, whereas in sickle cell trait, patients carry both the S and normal A alleles (HbSA). Hemoglobin C is a structural variant of HbA that results in lower solubility in red blood cells. Patients with hemoglobin SC disease (HbSC) have S and C alleles.2 We present a case of a patient with moderate to severe plaque psoriasis and heterozygous sickle cell anemia treated with adalimumab.
Case Report
A 31-year-old woman presented with moderate to severe plaque psoriasis (70% body surface area) and HbSC. She reported chronic dull arthralgia in the ankles that was worse at night. Radiographs of the feet and ankles showed erosive changes of the distal tarsal row and metatarsal bases. The diffuse bone pain had gradually worsened over the years and was treated by hematology with ibuprofen and ketorolac. At presentation, her HbSC pain was 8/10 on a visual analog scale. She described her sickle cell pain crises as sharp 10/10 pain in the back, elbows, and ankles, associated with mild edema lasting 1 to 2 days. Radiographs of the spine, hands, and ankles were unremarkable.
Adalimumab was chosen as a systemic therapy for psoriasis based on the potential for improvement in HbSC. Within 17 weeks of starting adalimumab, the psoriasis body surface area decreased from 70% to 40%, and the HbSC pain decreased from 8/10 to 4/10 at 8-week follow-up and to 0/10 at 17-week follow-up. After initiation of adalimumab, she reported decreased use of pain medication with no sickle cell pain crises.
Comment
Tumor necrosis factor α blockers are commonly used for moderate to severe plaque psoriasis. To our knowledge, there have been no reported human studies showing TNF-α blockade as a potential treatment of sickle cell disease. Increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and severity of sickle cell disease by playing an integral role in the development of vascular wall dysfunction and ischemia.3 Inflammatory mediators in HbSS disease, such as heparan sulfate from the endothelial glycocalyx and heme from hemolysis, act on monocytes to release TNF-α.1 Through this effect on the endothelium, TNF-α impedes blood flow during sickle cell crisis, leading to worsening ischemia and resultant painful infarction.3 Analysis of cytokine levels in HbSS patients showed significantly (P<.05) elevated levels of TNF
Although these findings were observational and limited to a single patient, the 50% decrease in pain level and use of pain medications reported to her hematologist independent of her dermatology visits coincided with the initiation of adalimumab. Although radiographs showed possible psoriatic changes of the distal metatarsal row, her described sickle cell pain and pain crises were atypical for psoriatic arthralgia. Tumor necrosis factor α inhibitors could be the drug of choice to treat patients with psoriasis with concomitant HbSS or HbSC disease due to the blockade of a common inflammatory mediator. Further studies are indicated to analyze the in vivo role of TNF-α inhibition in sickle cell disease.
- Solovey A, Somani A, Belcher JD, et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am J Hematol. 2017;92:1119-1130.
- Mais DD. Diseases of red blood cells. In: Laposata M, ed. Laposata’s Laboratory Medicine: Diagnosis of Disease in the Clinical Laboratory. 3rd ed. New York, NY: McGraw-Hill; 2018:247-280.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
- Solovey A, Somani A, Belcher JD, et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am J Hematol. 2017;92:1119-1130.
- Mais DD. Diseases of red blood cells. In: Laposata M, ed. Laposata’s Laboratory Medicine: Diagnosis of Disease in the Clinical Laboratory. 3rd ed. New York, NY: McGraw-Hill; 2018:247-280.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
Practice Points
• Tumor necrosis factor α contributes both to the vascular inflammatory state seen in sickle cell disease as well as the cycle of inflammation seen in the development of psoriasis.
• Tumor necrosis factor α inhibitors may be the drug of choice for patients with both psoriasis and sickle cell disease.
History of melanoma in situ • dyspnea • rib pain • Dx?
THE CASE
A 56-year-old woman with a history of melanoma in situ presented with progressive dyspnea on exertion, cough productive of clear sputum, and right-sided rib pain radiating to the upper back of 5 weeks’ duration.
Twenty-four years earlier, the patient had undergone excision of a skin lesion from the right side of her back. Pathology revealed melanoma in situ with no evidence of invasion of the underlying dermis. Because of close margins, she underwent wider excision 2 weeks later and no residual tumor was found. The patient subsequently underwent routine biannual dermatologic follow-up and transitioned (within the past few years) to annual dermatologic follow-up. At a recent dermatologic visit (9 months earlier), there were no suspicious skin lesions.
At current presentation, she denied fever, chills, night sweats, or unintentional weight loss. On examination, her vital signs were normal. Her pulse oximetry on room air was 95% at rest and 94% with ambulation. She had decreased breath sounds at the right lung base and a fixed 2 × 2-cm nontender, indurated mass in the right upper anterior chest wall, superior to the right breast. A skin examination was not performed at this time.

A complete blood count revealed a white blood cell count of 8220/mcL (reference range, 4500–11,000/mcL), hemoglobin of 13.6 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 162 × 103/mcL (reference range, 150–350 × 103/mcL). The patient’s electrolytes were within normal limits, with a creatinine level of 0.67 mg/dL (reference range, 0.1–1.2 mg/dL) and a calcium level of 9.4 mg/dL (reference range, 8.2–10.2 mg/dL). Lactate dehydrogenase was elevated at 308 U/L (reference range, 100–200 U/L).

A chest radiograph revealed a right upper lobe mass, right lower lobe consolidation, and a large right-sided pleural effusion (FIGURE 1). Chest computed tomography (CT) with intravenous contrast revealed a 5.2 × 5.7–cm right pleural-based mass with extension to the anterior chest wall, 3 left-sided pulmonary nodules, numerous right-sided pleural-based nodules (FIGURE 2), and multiple low-density liver lesions. An abdominal and pelvic CT scan revealed a 5.6 × 2.5 × 4.8–cm hepatic lesion (FIGURE 3) with scattered hepatic cysts.

THE DIAGNOSIS
A diagnostic thoracentesis was performed, and pleural fluid cytology results revealed metastatic melanoma. Magnetic resonance imaging (MRI) of the brain showed no evidence of metastases.
After the patient’s initial presentation, her dermatologist performed a biopsy on a pre-existing skin lesion on the patient’s left abdomen, which initially was thought to be a cherry angioma. This left abdominal skin lesion was in a location different from her previous melanoma in situ, which was located on the right side of the back. Biopsy results of the presumed cherry angioma revealed a nodular malignant melanoma (which was partially removed), adjacent to a cherry angioma (which was completely excised).
Continue to: Two primary melanomas
Two primary melanomas. Our patient had 2 different primary melanomas: a melanoma in situ on the right back diagnosed 24 years prior to the current presentation and the more recently identified melanoma on the left abdomen with metastases to the lung and liver.
We referred the patient to Oncology and she was enrolled in a clinical study with ipilimumab and nivolumab, monoclonal antibodies directed against negative regulators of T-cell activation.
DISCUSSION
In the United States, melanoma is the fifth leading cancer in men and the sixth leading cancer in women.1 A prior history of melanoma or melanoma in situ increases the risk for a second melanoma,2-4 and the risk remains elevated for more than 20 years after the initial diagnosis.2 One- and 2-year survival rates for metastatic melanoma are 32% to 65% and 18% to 40%, respectively5; the 5-year survival rate of metastatic melanoma to the lung is approximately 16%.6
Recommendations regarding the appropriate follow-up of patients with a history of melanoma in situ and melanoma vary widely.7 For patients with a history of melanoma in situ, the American Academy of Dermatology and the National Comprehensive Cancer Network recommend annual skin examination indefinitely and self-examination of the skin and lymph nodes monthly.4,7,8
Novel therapies are powerful allies in fight against melanoma
Previous standard treatment of metastatic melanoma included surgery, radiation, and cytotoxic chemotherapy. Resection rarely is curative in distant metastatic melanoma, and cytotoxic chemotherapy has low response rates, has a response duration of 4 to 6 months, and does not improve overall survival in advanced melanoma.9-12
Continue to: Novel therapies...
Novel therapies, such as immunotherapy and molecular-targeted therapies, are dramatically increasing survival rates in metastatic melanoma. Melanoma frequently is associated with somatic mutations, and each patient may have a unique collection of mutations resulting in the expression of antigens that bind to certain T-cell receptors, which serve as targets for inhibitor immunotherapy.
Ipilimumab and nivolumab are monoclonal antibodies directed against negative regulators of T-cell activation. When ipilimumab and nivolumab bind to their receptors, feedback inhibition is prevented, which results in an immune response against the tumor. In a trial of 53 patients with advanced melanoma treated with both drugs, the overall survival rate at 1 and 2 years was 94% and 88%, respectively.13
Dabrafenib and trametinib. Mutations that activate the serine/threonine kinase gene, BRAF, are present in approximately 40% to 60% of advanced melanomas and lead to clonal expansion and tumor progression.14,15 Inhibition of BRAF produces rapid tumor regression—even in extensive disease. Treatment with dabrafenib, a BRAF inhibitor, and trametinib, a mitogen-activated protein kinase inhibitor, has been shown to be superior to a BRAF inhibitor alone and is associated with a survival rate of 72% at 1 year.16
Our patient. Seven months after enrolling in the clinical trial with ipilimumab and nivolumab, our patient developed brain metastases and was withdrawn from the trial. A resection of her brain metastases and radiation therapy followed. The patient was then started on molecular-targeted therapy with dabrafenib and trametinib. Twelve weeks later, a repeat CT scan of the chest, abdomen, and pelvis demonstrated an interval decrease in the size of the majority of the metastatic lesions, and a repeat brain MRI showed no additional metastases.
More than 4 years after her diagnosis, our patient remains on dabrafenib and trametinib therapy and her metastatic lesions to the lung and liver remain stable.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Patients with a prior melanoma in situ or invasive melanoma have a higher risk for a subsequent invasive melanoma, and this risk remains elevated for more than 20 years. While patients with a history of melanoma in situ do not require specific oncologic follow-up, they do require annual dermatologic follow-up indefinitely and should perform monthly self-examination of their skin and lymph nodes.
Heightened awareness of the risk for a second primary melanoma should prompt primary care physicians to conduct ongoing patient surveillance. Family physicians should also keep in mind that novel therapies for metastatic melanoma, such as molecular-targeted therapies and immunotherapy, are associated with a much higher survival rate than previous standard therapy.
CORRESPONDENCE
Iris Tong, MD, Associate Professor, Division of General Internal Medicine, Department of Medicine, Alpert Medical School of Brown University, 146 W River St, Providence, RI 02904; [email protected]
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018 [published online January 4, 2018]. Cancer J Clin. 2018;68:7-30.
2. Bradford PT, Freedman DM, Goldstein AM, et al. Increased risk of second primary cancers after diagnosis of melanoma. Arch Dermatol. 2010;146:265-272.
3. Balamurugan A, Rees JR, Kosary C, et al. Subsequent primary cancers among men and women with in situ and invasive melanoma of the skin. J Am Acad Dermatol. 2011;65(5) (suppl 1):S69-S77.
4. Pomerantz H, Huang D, Weinstock MA. Risk of subsequent melanoma after melanoma in situ and invasive melanoma: a population-based study from 1973 to 2011. J Am Acad Dermatol. 2015;72:794-800.
5. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
6. American Cancer Society. Cancer facts & figures 2015. www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/index. Accessed November 26, 2018.
7. Coit DG, Andtbacka R, Anker CJ, et al. Melanoma: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2012;3:366-400.
8. Bichakjian CK, Halpern AC, Johnson TM. Guidelines of care for the management of primary cutaneous melanoma. J Am Acad Dermatol. 2011;5:1032-1047.
9. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention & Therapy of Melanoma. New York, NY: Marcel Dekker; 1997:219.
10. Patel PM, Suciu S, Mortier L, et al; EORTC Melanoma Group. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032) [published online May 18, 2011]. Eur J Cancer. 2011;47:1476-1483.
11. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma [published online December 17, 2012]. J Clin Oncol. 2013;31:373-379.
12. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430 [published online March 31, 2011]. Cancer. 2011;117:4740-4746.
13. Sznol M, Kluger HM, Callahan MK, et al. Abstract LBA9003. Presented at: 2014 American Society of Clinical Oncology (ASCO) Annual Meeting; May 30–June 3, 2014; Chicago, IL.
14. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-6488.
15. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
16. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
THE CASE
A 56-year-old woman with a history of melanoma in situ presented with progressive dyspnea on exertion, cough productive of clear sputum, and right-sided rib pain radiating to the upper back of 5 weeks’ duration.
Twenty-four years earlier, the patient had undergone excision of a skin lesion from the right side of her back. Pathology revealed melanoma in situ with no evidence of invasion of the underlying dermis. Because of close margins, she underwent wider excision 2 weeks later and no residual tumor was found. The patient subsequently underwent routine biannual dermatologic follow-up and transitioned (within the past few years) to annual dermatologic follow-up. At a recent dermatologic visit (9 months earlier), there were no suspicious skin lesions.
At current presentation, she denied fever, chills, night sweats, or unintentional weight loss. On examination, her vital signs were normal. Her pulse oximetry on room air was 95% at rest and 94% with ambulation. She had decreased breath sounds at the right lung base and a fixed 2 × 2-cm nontender, indurated mass in the right upper anterior chest wall, superior to the right breast. A skin examination was not performed at this time.
A complete blood count revealed a white blood cell count of 8220/mcL (reference range, 4500–11,000/mcL), hemoglobin of 13.6 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 162 × 103/mcL (reference range, 150–350 × 103/mcL). The patient’s electrolytes were within normal limits, with a creatinine level of 0.67 mg/dL (reference range, 0.1–1.2 mg/dL) and a calcium level of 9.4 mg/dL (reference range, 8.2–10.2 mg/dL). Lactate dehydrogenase was elevated at 308 U/L (reference range, 100–200 U/L).
A chest radiograph revealed a right upper lobe mass, right lower lobe consolidation, and a large right-sided pleural effusion (FIGURE 1). Chest computed tomography (CT) with intravenous contrast revealed a 5.2 × 5.7–cm right pleural-based mass with extension to the anterior chest wall, 3 left-sided pulmonary nodules, numerous right-sided pleural-based nodules (FIGURE 2), and multiple low-density liver lesions. An abdominal and pelvic CT scan revealed a 5.6 × 2.5 × 4.8–cm hepatic lesion (FIGURE 3) with scattered hepatic cysts.
THE DIAGNOSIS
A diagnostic thoracentesis was performed, and pleural fluid cytology results revealed metastatic melanoma. Magnetic resonance imaging (MRI) of the brain showed no evidence of metastases.
After the patient’s initial presentation, her dermatologist performed a biopsy on a pre-existing skin lesion on the patient’s left abdomen, which initially was thought to be a cherry angioma. This left abdominal skin lesion was in a location different from her previous melanoma in situ, which was located on the right side of the back. Biopsy results of the presumed cherry angioma revealed a nodular malignant melanoma (which was partially removed), adjacent to a cherry angioma (which was completely excised).
Continue to: Two primary melanomas
Two primary melanomas. Our patient had 2 different primary melanomas: a melanoma in situ on the right back diagnosed 24 years prior to the current presentation and the more recently identified melanoma on the left abdomen with metastases to the lung and liver.
We referred the patient to Oncology and she was enrolled in a clinical study with ipilimumab and nivolumab, monoclonal antibodies directed against negative regulators of T-cell activation.
DISCUSSION
In the United States, melanoma is the fifth leading cancer in men and the sixth leading cancer in women.1 A prior history of melanoma or melanoma in situ increases the risk for a second melanoma,2-4 and the risk remains elevated for more than 20 years after the initial diagnosis.2 One- and 2-year survival rates for metastatic melanoma are 32% to 65% and 18% to 40%, respectively5; the 5-year survival rate of metastatic melanoma to the lung is approximately 16%.6
Recommendations regarding the appropriate follow-up of patients with a history of melanoma in situ and melanoma vary widely.7 For patients with a history of melanoma in situ, the American Academy of Dermatology and the National Comprehensive Cancer Network recommend annual skin examination indefinitely and self-examination of the skin and lymph nodes monthly.4,7,8
Novel therapies are powerful allies in fight against melanoma
Previous standard treatment of metastatic melanoma included surgery, radiation, and cytotoxic chemotherapy. Resection rarely is curative in distant metastatic melanoma, and cytotoxic chemotherapy has low response rates, has a response duration of 4 to 6 months, and does not improve overall survival in advanced melanoma.9-12
Continue to: Novel therapies...
Novel therapies, such as immunotherapy and molecular-targeted therapies, are dramatically increasing survival rates in metastatic melanoma. Melanoma frequently is associated with somatic mutations, and each patient may have a unique collection of mutations resulting in the expression of antigens that bind to certain T-cell receptors, which serve as targets for inhibitor immunotherapy.
Ipilimumab and nivolumab are monoclonal antibodies directed against negative regulators of T-cell activation. When ipilimumab and nivolumab bind to their receptors, feedback inhibition is prevented, which results in an immune response against the tumor. In a trial of 53 patients with advanced melanoma treated with both drugs, the overall survival rate at 1 and 2 years was 94% and 88%, respectively.13
Dabrafenib and trametinib. Mutations that activate the serine/threonine kinase gene, BRAF, are present in approximately 40% to 60% of advanced melanomas and lead to clonal expansion and tumor progression.14,15 Inhibition of BRAF produces rapid tumor regression—even in extensive disease. Treatment with dabrafenib, a BRAF inhibitor, and trametinib, a mitogen-activated protein kinase inhibitor, has been shown to be superior to a BRAF inhibitor alone and is associated with a survival rate of 72% at 1 year.16
Our patient. Seven months after enrolling in the clinical trial with ipilimumab and nivolumab, our patient developed brain metastases and was withdrawn from the trial. A resection of her brain metastases and radiation therapy followed. The patient was then started on molecular-targeted therapy with dabrafenib and trametinib. Twelve weeks later, a repeat CT scan of the chest, abdomen, and pelvis demonstrated an interval decrease in the size of the majority of the metastatic lesions, and a repeat brain MRI showed no additional metastases.
More than 4 years after her diagnosis, our patient remains on dabrafenib and trametinib therapy and her metastatic lesions to the lung and liver remain stable.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Patients with a prior melanoma in situ or invasive melanoma have a higher risk for a subsequent invasive melanoma, and this risk remains elevated for more than 20 years. While patients with a history of melanoma in situ do not require specific oncologic follow-up, they do require annual dermatologic follow-up indefinitely and should perform monthly self-examination of their skin and lymph nodes.
Heightened awareness of the risk for a second primary melanoma should prompt primary care physicians to conduct ongoing patient surveillance. Family physicians should also keep in mind that novel therapies for metastatic melanoma, such as molecular-targeted therapies and immunotherapy, are associated with a much higher survival rate than previous standard therapy.
CORRESPONDENCE
Iris Tong, MD, Associate Professor, Division of General Internal Medicine, Department of Medicine, Alpert Medical School of Brown University, 146 W River St, Providence, RI 02904; [email protected]
THE CASE
A 56-year-old woman with a history of melanoma in situ presented with progressive dyspnea on exertion, cough productive of clear sputum, and right-sided rib pain radiating to the upper back of 5 weeks’ duration.
Twenty-four years earlier, the patient had undergone excision of a skin lesion from the right side of her back. Pathology revealed melanoma in situ with no evidence of invasion of the underlying dermis. Because of close margins, she underwent wider excision 2 weeks later and no residual tumor was found. The patient subsequently underwent routine biannual dermatologic follow-up and transitioned (within the past few years) to annual dermatologic follow-up. At a recent dermatologic visit (9 months earlier), there were no suspicious skin lesions.
At current presentation, she denied fever, chills, night sweats, or unintentional weight loss. On examination, her vital signs were normal. Her pulse oximetry on room air was 95% at rest and 94% with ambulation. She had decreased breath sounds at the right lung base and a fixed 2 × 2-cm nontender, indurated mass in the right upper anterior chest wall, superior to the right breast. A skin examination was not performed at this time.
A complete blood count revealed a white blood cell count of 8220/mcL (reference range, 4500–11,000/mcL), hemoglobin of 13.6 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 162 × 103/mcL (reference range, 150–350 × 103/mcL). The patient’s electrolytes were within normal limits, with a creatinine level of 0.67 mg/dL (reference range, 0.1–1.2 mg/dL) and a calcium level of 9.4 mg/dL (reference range, 8.2–10.2 mg/dL). Lactate dehydrogenase was elevated at 308 U/L (reference range, 100–200 U/L).
A chest radiograph revealed a right upper lobe mass, right lower lobe consolidation, and a large right-sided pleural effusion (FIGURE 1). Chest computed tomography (CT) with intravenous contrast revealed a 5.2 × 5.7–cm right pleural-based mass with extension to the anterior chest wall, 3 left-sided pulmonary nodules, numerous right-sided pleural-based nodules (FIGURE 2), and multiple low-density liver lesions. An abdominal and pelvic CT scan revealed a 5.6 × 2.5 × 4.8–cm hepatic lesion (FIGURE 3) with scattered hepatic cysts.
THE DIAGNOSIS
A diagnostic thoracentesis was performed, and pleural fluid cytology results revealed metastatic melanoma. Magnetic resonance imaging (MRI) of the brain showed no evidence of metastases.
After the patient’s initial presentation, her dermatologist performed a biopsy on a pre-existing skin lesion on the patient’s left abdomen, which initially was thought to be a cherry angioma. This left abdominal skin lesion was in a location different from her previous melanoma in situ, which was located on the right side of the back. Biopsy results of the presumed cherry angioma revealed a nodular malignant melanoma (which was partially removed), adjacent to a cherry angioma (which was completely excised).
Continue to: Two primary melanomas
Two primary melanomas. Our patient had 2 different primary melanomas: a melanoma in situ on the right back diagnosed 24 years prior to the current presentation and the more recently identified melanoma on the left abdomen with metastases to the lung and liver.
We referred the patient to Oncology and she was enrolled in a clinical study with ipilimumab and nivolumab, monoclonal antibodies directed against negative regulators of T-cell activation.
DISCUSSION
In the United States, melanoma is the fifth leading cancer in men and the sixth leading cancer in women.1 A prior history of melanoma or melanoma in situ increases the risk for a second melanoma,2-4 and the risk remains elevated for more than 20 years after the initial diagnosis.2 One- and 2-year survival rates for metastatic melanoma are 32% to 65% and 18% to 40%, respectively5; the 5-year survival rate of metastatic melanoma to the lung is approximately 16%.6
Recommendations regarding the appropriate follow-up of patients with a history of melanoma in situ and melanoma vary widely.7 For patients with a history of melanoma in situ, the American Academy of Dermatology and the National Comprehensive Cancer Network recommend annual skin examination indefinitely and self-examination of the skin and lymph nodes monthly.4,7,8
Novel therapies are powerful allies in fight against melanoma
Previous standard treatment of metastatic melanoma included surgery, radiation, and cytotoxic chemotherapy. Resection rarely is curative in distant metastatic melanoma, and cytotoxic chemotherapy has low response rates, has a response duration of 4 to 6 months, and does not improve overall survival in advanced melanoma.9-12
Continue to: Novel therapies...
Novel therapies, such as immunotherapy and molecular-targeted therapies, are dramatically increasing survival rates in metastatic melanoma. Melanoma frequently is associated with somatic mutations, and each patient may have a unique collection of mutations resulting in the expression of antigens that bind to certain T-cell receptors, which serve as targets for inhibitor immunotherapy.
Ipilimumab and nivolumab are monoclonal antibodies directed against negative regulators of T-cell activation. When ipilimumab and nivolumab bind to their receptors, feedback inhibition is prevented, which results in an immune response against the tumor. In a trial of 53 patients with advanced melanoma treated with both drugs, the overall survival rate at 1 and 2 years was 94% and 88%, respectively.13
Dabrafenib and trametinib. Mutations that activate the serine/threonine kinase gene, BRAF, are present in approximately 40% to 60% of advanced melanomas and lead to clonal expansion and tumor progression.14,15 Inhibition of BRAF produces rapid tumor regression—even in extensive disease. Treatment with dabrafenib, a BRAF inhibitor, and trametinib, a mitogen-activated protein kinase inhibitor, has been shown to be superior to a BRAF inhibitor alone and is associated with a survival rate of 72% at 1 year.16
Our patient. Seven months after enrolling in the clinical trial with ipilimumab and nivolumab, our patient developed brain metastases and was withdrawn from the trial. A resection of her brain metastases and radiation therapy followed. The patient was then started on molecular-targeted therapy with dabrafenib and trametinib. Twelve weeks later, a repeat CT scan of the chest, abdomen, and pelvis demonstrated an interval decrease in the size of the majority of the metastatic lesions, and a repeat brain MRI showed no additional metastases.
More than 4 years after her diagnosis, our patient remains on dabrafenib and trametinib therapy and her metastatic lesions to the lung and liver remain stable.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Patients with a prior melanoma in situ or invasive melanoma have a higher risk for a subsequent invasive melanoma, and this risk remains elevated for more than 20 years. While patients with a history of melanoma in situ do not require specific oncologic follow-up, they do require annual dermatologic follow-up indefinitely and should perform monthly self-examination of their skin and lymph nodes.
Heightened awareness of the risk for a second primary melanoma should prompt primary care physicians to conduct ongoing patient surveillance. Family physicians should also keep in mind that novel therapies for metastatic melanoma, such as molecular-targeted therapies and immunotherapy, are associated with a much higher survival rate than previous standard therapy.
CORRESPONDENCE
Iris Tong, MD, Associate Professor, Division of General Internal Medicine, Department of Medicine, Alpert Medical School of Brown University, 146 W River St, Providence, RI 02904; [email protected]
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018 [published online January 4, 2018]. Cancer J Clin. 2018;68:7-30.
2. Bradford PT, Freedman DM, Goldstein AM, et al. Increased risk of second primary cancers after diagnosis of melanoma. Arch Dermatol. 2010;146:265-272.
3. Balamurugan A, Rees JR, Kosary C, et al. Subsequent primary cancers among men and women with in situ and invasive melanoma of the skin. J Am Acad Dermatol. 2011;65(5) (suppl 1):S69-S77.
4. Pomerantz H, Huang D, Weinstock MA. Risk of subsequent melanoma after melanoma in situ and invasive melanoma: a population-based study from 1973 to 2011. J Am Acad Dermatol. 2015;72:794-800.
5. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
6. American Cancer Society. Cancer facts & figures 2015. www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/index. Accessed November 26, 2018.
7. Coit DG, Andtbacka R, Anker CJ, et al. Melanoma: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2012;3:366-400.
8. Bichakjian CK, Halpern AC, Johnson TM. Guidelines of care for the management of primary cutaneous melanoma. J Am Acad Dermatol. 2011;5:1032-1047.
9. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention & Therapy of Melanoma. New York, NY: Marcel Dekker; 1997:219.
10. Patel PM, Suciu S, Mortier L, et al; EORTC Melanoma Group. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032) [published online May 18, 2011]. Eur J Cancer. 2011;47:1476-1483.
11. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma [published online December 17, 2012]. J Clin Oncol. 2013;31:373-379.
12. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430 [published online March 31, 2011]. Cancer. 2011;117:4740-4746.
13. Sznol M, Kluger HM, Callahan MK, et al. Abstract LBA9003. Presented at: 2014 American Society of Clinical Oncology (ASCO) Annual Meeting; May 30–June 3, 2014; Chicago, IL.
14. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-6488.
15. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
16. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018 [published online January 4, 2018]. Cancer J Clin. 2018;68:7-30.
2. Bradford PT, Freedman DM, Goldstein AM, et al. Increased risk of second primary cancers after diagnosis of melanoma. Arch Dermatol. 2010;146:265-272.
3. Balamurugan A, Rees JR, Kosary C, et al. Subsequent primary cancers among men and women with in situ and invasive melanoma of the skin. J Am Acad Dermatol. 2011;65(5) (suppl 1):S69-S77.
4. Pomerantz H, Huang D, Weinstock MA. Risk of subsequent melanoma after melanoma in situ and invasive melanoma: a population-based study from 1973 to 2011. J Am Acad Dermatol. 2015;72:794-800.
5. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
6. American Cancer Society. Cancer facts & figures 2015. www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/index. Accessed November 26, 2018.
7. Coit DG, Andtbacka R, Anker CJ, et al. Melanoma: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2012;3:366-400.
8. Bichakjian CK, Halpern AC, Johnson TM. Guidelines of care for the management of primary cutaneous melanoma. J Am Acad Dermatol. 2011;5:1032-1047.
9. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention & Therapy of Melanoma. New York, NY: Marcel Dekker; 1997:219.
10. Patel PM, Suciu S, Mortier L, et al; EORTC Melanoma Group. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032) [published online May 18, 2011]. Eur J Cancer. 2011;47:1476-1483.
11. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma [published online December 17, 2012]. J Clin Oncol. 2013;31:373-379.
12. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430 [published online March 31, 2011]. Cancer. 2011;117:4740-4746.
13. Sznol M, Kluger HM, Callahan MK, et al. Abstract LBA9003. Presented at: 2014 American Society of Clinical Oncology (ASCO) Annual Meeting; May 30–June 3, 2014; Chicago, IL.
14. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-6488.
15. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
16. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.

Leg-length discrepancy • asymmetric gluteal folds and popliteal fossae • positive Galeazzi test • Dx?
THE CASE
A healthy 6-month-old girl born via spontaneous vaginal delivery to a 33-year-old mother presented to her family physician (FP) for a routine well-child examination. The mother’s prenatal anatomy scan, delivery, and personal and family history were unremarkable. The patient was not firstborn or breech, and there was no family history of hip dysplasia. On prior infant well-child examinations, Ortolani and Barlow maneuvers were negative, and the patient demonstrated spontaneous movement of both legs. There was no evidence of hip dysplasia, lower extremity weakness, musculoskeletal abnormalities, or abnormal skin markings. The patient had normal growth and development (50th percentile for height and weight, average Ages & Stages Questionnaire scores) and no history of infection or trauma.
At the current presentation, the FP noted a leg-length discrepancy while palpating the bony (patellar and malleolar) landmarks of the lower extremities, but the right and left anterior superior iliac spine was symmetrical. The gluteal folds and popliteal fossae were asymmetric, a Galeazzi test was positive, and the right leg measured approximately 2 cm shorter than the left leg. There was no evidence of scoliosis or pelvic abnormalities. Physical examination revealed no ecchymosis or trauma. Orthopedic evaluation by the FP of the hips, knees, and ankles was normal, including negative repeat Ortolani and Barlow maneuvers and normal range of motion. We obtained x-rays of the lower extremities and ordered an orthopedic consultation.
THE DIAGNOSIS
The differential diagnosis included congenital, traumatic, infectious, inflammatory, idiopathic, and neurologic causes.1-3 The most common etiologies of leg-length discrepancies are summarized in TABLE 1.1-3 Radiographic imaging showed a femur length discrepancy, which was determined to be congenital without indication of trauma or disease; therefore, a diagnosis of congenital femoral bowing was made.

Initial orthopedic evaluation revealed a femur length discrepancy of approximately 2 cm. Plain films showed lateral femoral bowing (FIGURE 1A).

DISCUSSION
Congenital femoral bowing, which can present as a leg-length discrepancy in infants, is a relatively rare finding with an incidence of 1 per 52,000 births.4 Our patient presented with an isolated limb deformity, but congenital femoral bowing is recognized as a clinical feature of several skeletal dysplasias (TABLE 2).5

What’s recommended
The American Academy of Pediatrics recommends routine age-appropriate physical examination without specifying leg-length assessment.6 There is insufficient evidence, according to
Congenital femoral bowing requires plain film diagnosis
Following physical examination, diagnosis of congenital femoral bowing should be confirmed by plain films. Plain radiography remains the main imaging modality for proximal focal femoral deficiency and fibular hemimelia, and appropriate identification of the osseous abnormalities seen on radiographs allows for accurate classification of congenital femoral bowing, prognosis, and surgical planning.
Continue to: Early diagnosis can improve treatment outcome
Early diagnosis can improve treatment outcome
Both early diagnosis of congenital femoral bowing and prediction of leg-length discrepancy at skeletal maturity can influence potential treatment options, which range from conservative management (eg, watchful waiting, physical therapy, shoe lifts, orthotics, bracing) to surgical intervention. Several models have been used to predict skeletal growth, including the Moseley straight line graph, Green and Anderson growth curve, Amstutz method, and Paley’s multiplier method.4,9-14
Intervention for leg-length discrepancy generally is dictated by the magnitude of the inequality and the presence of functional deficits and/or pain.2 If the degree of femur angulation begins to affect structural development, surgical intervention should be considered to align development and/or correct the discrepancy. Physical therapy, shoe lifts, orthotics, and bracing are treatment options for managing smaller discrepancies.2,15
Our patient. The physician (CP) reviewed treatment options with the family that included watchful waiting, use of a shoe lift and/or orthotics, and bracing. The family chose watchful waiting due to the structural integrity of the patient’s other major joints and her relatively preserved function.
Surgery. Ultimately the patient underwent medial distal femoral hemiepiphysiodesis of the right lower extremity at 6 years of age due to increasing leg-length discrepancy and lateralization of the patella from the valgus deformity. The patient’s mother reported that she did well postoperatively, with increased range of motion, improved physical capabilities, and reduced discomfort in the right leg.
A second surgery. At approximately 8 years and 9 months of age, the orthopedist noted that the patient’s leg-length discrepancy had increased, and she had right extensor mechanism malalignment and severe patellar subluxation. The patient subsequently underwent surgery to remove the existing hardware, including right extensor mechanism realignment via a Roux-Goldthwait procedure (with reconstruction of the medial patellofemoral ligament and anterior cruciate ligament), as well as left distal femoral epiphysiodesis. She did very well postoperatively and continues to participate in physical therapy approximately once weekly. She has had an improvement in her gait and stability using shoe lifts.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A routine well-child examination can be an opportunity to identify congenital musculoskeletal problems. Congenital femoral bowing is a relatively rare finding4 that may present as a leg-length discrepancy. With proper evaluation, including visual inspection, palpation, range-of-motion testing, and special tests as needed (eg, Galeazzi test, Ortolani and Barlow maneuvers), early intervention is possible if a leg-length discrepancy is noted. Close monitoring of gait abnormalities at routine well-child visits is essential.
Physical therapy, shoe lift therapy, and surgical approaches are treatment options for leg-length discrepancy,2 and early intervention can improve treatment outcomes.14 Understanding how to manage congenital femoral bowing over time is important in providing options and counselling patients and their families.15
Treatment of leg-length discrepancy in pediatric patients requires long-term management with a team approach that includes patients and their families. The goal of intervention is to reduce physical and emotional trauma, while addressing complications and maintaining function of the affected limb, as well as the whole body.15
CORRESPONDENCE
Beth P. Davis, DPT, MBA, FNAP, Emory University School of Medicine, Department of Rehabilitation Medicine, Division of Physical Therapy, 1462 Clifton Road NE, Suite 312, Atlanta, GA 30342; [email protected].
1. Shailam R, Jaramillo D, Kan JH. Growth arrest and leg-length discrepancy. Pediatr Radiol. 2013:43(suppl 1):S155-S165.
2. Brady RJ, Dean JB, Skinner TM, et al. Limb length inequality: clinical implications for assessment and intervention. J Orthop Sports Phys Ther. 2003;33:221-234.
3. Stanitski DF. Limb-length inequality: assessment and treatment options. J Am Acad Orthop Surg. 1999;7:143-153.
4. Bedoya MA, Chauvin NA, Jaramillo D, et al. Common patterns of congenital lower extremity shortening: diagnosis, classification, and follow-up. Radiographics. 2015;35:1191-1207.
5. Alanay Y, Krakow D, Rimoin DL, et al. Angulated femurs and the skeletal dysplasias: experience of the International Skeletal Dysplasia Registry (1988-2006). Am J Med Genet. 2007;143A:1159-1168.
6. Hagan JF, Shaw JS, Duncan PM, eds. Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents. 4th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2017.
7. Shipman SA, Hefland M, Moyer VA, et al. Screening for developmental dysplasia of the hip: a systematic literature review for the US Preventive Services Task Force. Pediatrics. 2006;117:557-576.
8. US Preventive Services Task Force. Screening for developmental dysplasia of the hip: recommendation statement. Pediatrics. 2006;117:898-902.
9. Paley D, Bhave A, Herzenberg JE, et al. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82-A:1432-1446.
10. Castaneda P, Urquhart B, Sullivan E, et al. Hemiepiphysiodesis for the correcting of angular deformity about the knee. J Pediatr Orthop. 2008;28:188-191.
11. Mosley CF. A straight-line graph for leg-length discrepancies. J Bone Joint Surg Am. 1977; 59:174-179.
12. Anderson M, Messner MB, Green WT. Distribution of lengths of the normal femur and tibia in children from one to eighteen years of age. J Bone Joint Surg Am. 1964;46:1197-1202.
13. Amstutz HC. The morphology, natural history, and treatment of proximal femoral focal deficiency. In: GT Aitken, ed. Proximal Femoral Focal Deficiency: A Congenital Anomaly. Washington, DC: National Academy of Sciences; 1969: 50-76.
14. Amstutz HC. Natural history and treatment of congenital absence of the fibula. J Bone and Joint Surg Am. 1972;54(A):1349.
15. Guidera KJ, Helal AA, Zuern KA. Management of pediatric limb length inequality. Adv Pediatr. 1995;42:501-543.
THE CASE
A healthy 6-month-old girl born via spontaneous vaginal delivery to a 33-year-old mother presented to her family physician (FP) for a routine well-child examination. The mother’s prenatal anatomy scan, delivery, and personal and family history were unremarkable. The patient was not firstborn or breech, and there was no family history of hip dysplasia. On prior infant well-child examinations, Ortolani and Barlow maneuvers were negative, and the patient demonstrated spontaneous movement of both legs. There was no evidence of hip dysplasia, lower extremity weakness, musculoskeletal abnormalities, or abnormal skin markings. The patient had normal growth and development (50th percentile for height and weight, average Ages & Stages Questionnaire scores) and no history of infection or trauma.
At the current presentation, the FP noted a leg-length discrepancy while palpating the bony (patellar and malleolar) landmarks of the lower extremities, but the right and left anterior superior iliac spine was symmetrical. The gluteal folds and popliteal fossae were asymmetric, a Galeazzi test was positive, and the right leg measured approximately 2 cm shorter than the left leg. There was no evidence of scoliosis or pelvic abnormalities. Physical examination revealed no ecchymosis or trauma. Orthopedic evaluation by the FP of the hips, knees, and ankles was normal, including negative repeat Ortolani and Barlow maneuvers and normal range of motion. We obtained x-rays of the lower extremities and ordered an orthopedic consultation.
THE DIAGNOSIS
The differential diagnosis included congenital, traumatic, infectious, inflammatory, idiopathic, and neurologic causes.1-3 The most common etiologies of leg-length discrepancies are summarized in TABLE 1.1-3 Radiographic imaging showed a femur length discrepancy, which was determined to be congenital without indication of trauma or disease; therefore, a diagnosis of congenital femoral bowing was made.
Initial orthopedic evaluation revealed a femur length discrepancy of approximately 2 cm. Plain films showed lateral femoral bowing (FIGURE 1A).
DISCUSSION
Congenital femoral bowing, which can present as a leg-length discrepancy in infants, is a relatively rare finding with an incidence of 1 per 52,000 births.4 Our patient presented with an isolated limb deformity, but congenital femoral bowing is recognized as a clinical feature of several skeletal dysplasias (TABLE 2).5
What’s recommended
The American Academy of Pediatrics recommends routine age-appropriate physical examination without specifying leg-length assessment.6 There is insufficient evidence, according to
Congenital femoral bowing requires plain film diagnosis
Following physical examination, diagnosis of congenital femoral bowing should be confirmed by plain films. Plain radiography remains the main imaging modality for proximal focal femoral deficiency and fibular hemimelia, and appropriate identification of the osseous abnormalities seen on radiographs allows for accurate classification of congenital femoral bowing, prognosis, and surgical planning.
Continue to: Early diagnosis can improve treatment outcome
Early diagnosis can improve treatment outcome
Both early diagnosis of congenital femoral bowing and prediction of leg-length discrepancy at skeletal maturity can influence potential treatment options, which range from conservative management (eg, watchful waiting, physical therapy, shoe lifts, orthotics, bracing) to surgical intervention. Several models have been used to predict skeletal growth, including the Moseley straight line graph, Green and Anderson growth curve, Amstutz method, and Paley’s multiplier method.4,9-14
Intervention for leg-length discrepancy generally is dictated by the magnitude of the inequality and the presence of functional deficits and/or pain.2 If the degree of femur angulation begins to affect structural development, surgical intervention should be considered to align development and/or correct the discrepancy. Physical therapy, shoe lifts, orthotics, and bracing are treatment options for managing smaller discrepancies.2,15
Our patient. The physician (CP) reviewed treatment options with the family that included watchful waiting, use of a shoe lift and/or orthotics, and bracing. The family chose watchful waiting due to the structural integrity of the patient’s other major joints and her relatively preserved function.
Surgery. Ultimately the patient underwent medial distal femoral hemiepiphysiodesis of the right lower extremity at 6 years of age due to increasing leg-length discrepancy and lateralization of the patella from the valgus deformity. The patient’s mother reported that she did well postoperatively, with increased range of motion, improved physical capabilities, and reduced discomfort in the right leg.
A second surgery. At approximately 8 years and 9 months of age, the orthopedist noted that the patient’s leg-length discrepancy had increased, and she had right extensor mechanism malalignment and severe patellar subluxation. The patient subsequently underwent surgery to remove the existing hardware, including right extensor mechanism realignment via a Roux-Goldthwait procedure (with reconstruction of the medial patellofemoral ligament and anterior cruciate ligament), as well as left distal femoral epiphysiodesis. She did very well postoperatively and continues to participate in physical therapy approximately once weekly. She has had an improvement in her gait and stability using shoe lifts.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A routine well-child examination can be an opportunity to identify congenital musculoskeletal problems. Congenital femoral bowing is a relatively rare finding4 that may present as a leg-length discrepancy. With proper evaluation, including visual inspection, palpation, range-of-motion testing, and special tests as needed (eg, Galeazzi test, Ortolani and Barlow maneuvers), early intervention is possible if a leg-length discrepancy is noted. Close monitoring of gait abnormalities at routine well-child visits is essential.
Physical therapy, shoe lift therapy, and surgical approaches are treatment options for leg-length discrepancy,2 and early intervention can improve treatment outcomes.14 Understanding how to manage congenital femoral bowing over time is important in providing options and counselling patients and their families.15
Treatment of leg-length discrepancy in pediatric patients requires long-term management with a team approach that includes patients and their families. The goal of intervention is to reduce physical and emotional trauma, while addressing complications and maintaining function of the affected limb, as well as the whole body.15
CORRESPONDENCE
Beth P. Davis, DPT, MBA, FNAP, Emory University School of Medicine, Department of Rehabilitation Medicine, Division of Physical Therapy, 1462 Clifton Road NE, Suite 312, Atlanta, GA 30342; [email protected].
THE CASE
A healthy 6-month-old girl born via spontaneous vaginal delivery to a 33-year-old mother presented to her family physician (FP) for a routine well-child examination. The mother’s prenatal anatomy scan, delivery, and personal and family history were unremarkable. The patient was not firstborn or breech, and there was no family history of hip dysplasia. On prior infant well-child examinations, Ortolani and Barlow maneuvers were negative, and the patient demonstrated spontaneous movement of both legs. There was no evidence of hip dysplasia, lower extremity weakness, musculoskeletal abnormalities, or abnormal skin markings. The patient had normal growth and development (50th percentile for height and weight, average Ages & Stages Questionnaire scores) and no history of infection or trauma.
At the current presentation, the FP noted a leg-length discrepancy while palpating the bony (patellar and malleolar) landmarks of the lower extremities, but the right and left anterior superior iliac spine was symmetrical. The gluteal folds and popliteal fossae were asymmetric, a Galeazzi test was positive, and the right leg measured approximately 2 cm shorter than the left leg. There was no evidence of scoliosis or pelvic abnormalities. Physical examination revealed no ecchymosis or trauma. Orthopedic evaluation by the FP of the hips, knees, and ankles was normal, including negative repeat Ortolani and Barlow maneuvers and normal range of motion. We obtained x-rays of the lower extremities and ordered an orthopedic consultation.
THE DIAGNOSIS
The differential diagnosis included congenital, traumatic, infectious, inflammatory, idiopathic, and neurologic causes.1-3 The most common etiologies of leg-length discrepancies are summarized in TABLE 1.1-3 Radiographic imaging showed a femur length discrepancy, which was determined to be congenital without indication of trauma or disease; therefore, a diagnosis of congenital femoral bowing was made.
Initial orthopedic evaluation revealed a femur length discrepancy of approximately 2 cm. Plain films showed lateral femoral bowing (FIGURE 1A).
DISCUSSION
Congenital femoral bowing, which can present as a leg-length discrepancy in infants, is a relatively rare finding with an incidence of 1 per 52,000 births.4 Our patient presented with an isolated limb deformity, but congenital femoral bowing is recognized as a clinical feature of several skeletal dysplasias (TABLE 2).5
What’s recommended
The American Academy of Pediatrics recommends routine age-appropriate physical examination without specifying leg-length assessment.6 There is insufficient evidence, according to
Congenital femoral bowing requires plain film diagnosis
Following physical examination, diagnosis of congenital femoral bowing should be confirmed by plain films. Plain radiography remains the main imaging modality for proximal focal femoral deficiency and fibular hemimelia, and appropriate identification of the osseous abnormalities seen on radiographs allows for accurate classification of congenital femoral bowing, prognosis, and surgical planning.
Continue to: Early diagnosis can improve treatment outcome
Early diagnosis can improve treatment outcome
Both early diagnosis of congenital femoral bowing and prediction of leg-length discrepancy at skeletal maturity can influence potential treatment options, which range from conservative management (eg, watchful waiting, physical therapy, shoe lifts, orthotics, bracing) to surgical intervention. Several models have been used to predict skeletal growth, including the Moseley straight line graph, Green and Anderson growth curve, Amstutz method, and Paley’s multiplier method.4,9-14
Intervention for leg-length discrepancy generally is dictated by the magnitude of the inequality and the presence of functional deficits and/or pain.2 If the degree of femur angulation begins to affect structural development, surgical intervention should be considered to align development and/or correct the discrepancy. Physical therapy, shoe lifts, orthotics, and bracing are treatment options for managing smaller discrepancies.2,15
Our patient. The physician (CP) reviewed treatment options with the family that included watchful waiting, use of a shoe lift and/or orthotics, and bracing. The family chose watchful waiting due to the structural integrity of the patient’s other major joints and her relatively preserved function.
Surgery. Ultimately the patient underwent medial distal femoral hemiepiphysiodesis of the right lower extremity at 6 years of age due to increasing leg-length discrepancy and lateralization of the patella from the valgus deformity. The patient’s mother reported that she did well postoperatively, with increased range of motion, improved physical capabilities, and reduced discomfort in the right leg.
A second surgery. At approximately 8 years and 9 months of age, the orthopedist noted that the patient’s leg-length discrepancy had increased, and she had right extensor mechanism malalignment and severe patellar subluxation. The patient subsequently underwent surgery to remove the existing hardware, including right extensor mechanism realignment via a Roux-Goldthwait procedure (with reconstruction of the medial patellofemoral ligament and anterior cruciate ligament), as well as left distal femoral epiphysiodesis. She did very well postoperatively and continues to participate in physical therapy approximately once weekly. She has had an improvement in her gait and stability using shoe lifts.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A routine well-child examination can be an opportunity to identify congenital musculoskeletal problems. Congenital femoral bowing is a relatively rare finding4 that may present as a leg-length discrepancy. With proper evaluation, including visual inspection, palpation, range-of-motion testing, and special tests as needed (eg, Galeazzi test, Ortolani and Barlow maneuvers), early intervention is possible if a leg-length discrepancy is noted. Close monitoring of gait abnormalities at routine well-child visits is essential.
Physical therapy, shoe lift therapy, and surgical approaches are treatment options for leg-length discrepancy,2 and early intervention can improve treatment outcomes.14 Understanding how to manage congenital femoral bowing over time is important in providing options and counselling patients and their families.15
Treatment of leg-length discrepancy in pediatric patients requires long-term management with a team approach that includes patients and their families. The goal of intervention is to reduce physical and emotional trauma, while addressing complications and maintaining function of the affected limb, as well as the whole body.15
CORRESPONDENCE
Beth P. Davis, DPT, MBA, FNAP, Emory University School of Medicine, Department of Rehabilitation Medicine, Division of Physical Therapy, 1462 Clifton Road NE, Suite 312, Atlanta, GA 30342; [email protected].
1. Shailam R, Jaramillo D, Kan JH. Growth arrest and leg-length discrepancy. Pediatr Radiol. 2013:43(suppl 1):S155-S165.
2. Brady RJ, Dean JB, Skinner TM, et al. Limb length inequality: clinical implications for assessment and intervention. J Orthop Sports Phys Ther. 2003;33:221-234.
3. Stanitski DF. Limb-length inequality: assessment and treatment options. J Am Acad Orthop Surg. 1999;7:143-153.
4. Bedoya MA, Chauvin NA, Jaramillo D, et al. Common patterns of congenital lower extremity shortening: diagnosis, classification, and follow-up. Radiographics. 2015;35:1191-1207.
5. Alanay Y, Krakow D, Rimoin DL, et al. Angulated femurs and the skeletal dysplasias: experience of the International Skeletal Dysplasia Registry (1988-2006). Am J Med Genet. 2007;143A:1159-1168.
6. Hagan JF, Shaw JS, Duncan PM, eds. Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents. 4th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2017.
7. Shipman SA, Hefland M, Moyer VA, et al. Screening for developmental dysplasia of the hip: a systematic literature review for the US Preventive Services Task Force. Pediatrics. 2006;117:557-576.
8. US Preventive Services Task Force. Screening for developmental dysplasia of the hip: recommendation statement. Pediatrics. 2006;117:898-902.
9. Paley D, Bhave A, Herzenberg JE, et al. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82-A:1432-1446.
10. Castaneda P, Urquhart B, Sullivan E, et al. Hemiepiphysiodesis for the correcting of angular deformity about the knee. J Pediatr Orthop. 2008;28:188-191.
11. Mosley CF. A straight-line graph for leg-length discrepancies. J Bone Joint Surg Am. 1977; 59:174-179.
12. Anderson M, Messner MB, Green WT. Distribution of lengths of the normal femur and tibia in children from one to eighteen years of age. J Bone Joint Surg Am. 1964;46:1197-1202.
13. Amstutz HC. The morphology, natural history, and treatment of proximal femoral focal deficiency. In: GT Aitken, ed. Proximal Femoral Focal Deficiency: A Congenital Anomaly. Washington, DC: National Academy of Sciences; 1969: 50-76.
14. Amstutz HC. Natural history and treatment of congenital absence of the fibula. J Bone and Joint Surg Am. 1972;54(A):1349.
15. Guidera KJ, Helal AA, Zuern KA. Management of pediatric limb length inequality. Adv Pediatr. 1995;42:501-543.
1. Shailam R, Jaramillo D, Kan JH. Growth arrest and leg-length discrepancy. Pediatr Radiol. 2013:43(suppl 1):S155-S165.
2. Brady RJ, Dean JB, Skinner TM, et al. Limb length inequality: clinical implications for assessment and intervention. J Orthop Sports Phys Ther. 2003;33:221-234.
3. Stanitski DF. Limb-length inequality: assessment and treatment options. J Am Acad Orthop Surg. 1999;7:143-153.
4. Bedoya MA, Chauvin NA, Jaramillo D, et al. Common patterns of congenital lower extremity shortening: diagnosis, classification, and follow-up. Radiographics. 2015;35:1191-1207.
5. Alanay Y, Krakow D, Rimoin DL, et al. Angulated femurs and the skeletal dysplasias: experience of the International Skeletal Dysplasia Registry (1988-2006). Am J Med Genet. 2007;143A:1159-1168.
6. Hagan JF, Shaw JS, Duncan PM, eds. Bright Futures: Guidelines for Health Supervision of Infants, Children, and Adolescents. 4th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2017.
7. Shipman SA, Hefland M, Moyer VA, et al. Screening for developmental dysplasia of the hip: a systematic literature review for the US Preventive Services Task Force. Pediatrics. 2006;117:557-576.
8. US Preventive Services Task Force. Screening for developmental dysplasia of the hip: recommendation statement. Pediatrics. 2006;117:898-902.
9. Paley D, Bhave A, Herzenberg JE, et al. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82-A:1432-1446.
10. Castaneda P, Urquhart B, Sullivan E, et al. Hemiepiphysiodesis for the correcting of angular deformity about the knee. J Pediatr Orthop. 2008;28:188-191.
11. Mosley CF. A straight-line graph for leg-length discrepancies. J Bone Joint Surg Am. 1977; 59:174-179.
12. Anderson M, Messner MB, Green WT. Distribution of lengths of the normal femur and tibia in children from one to eighteen years of age. J Bone Joint Surg Am. 1964;46:1197-1202.
13. Amstutz HC. The morphology, natural history, and treatment of proximal femoral focal deficiency. In: GT Aitken, ed. Proximal Femoral Focal Deficiency: A Congenital Anomaly. Washington, DC: National Academy of Sciences; 1969: 50-76.
14. Amstutz HC. Natural history and treatment of congenital absence of the fibula. J Bone and Joint Surg Am. 1972;54(A):1349.
15. Guidera KJ, Helal AA, Zuern KA. Management of pediatric limb length inequality. Adv Pediatr. 1995;42:501-543.

Pain in right shoulder • recent influenza vaccination • history of hypertension and myocardial infarction • Dx?
THE CASE
A 61-year-old Caucasian woman presented with acute right shoulder pain that began after she received an influenza vaccination at a local pharmacy 2 weeks earlier. She pointed to the proximal-most aspect of her lateral right upper arm as the vaccination site. Her pain intensified with shoulder abduction, forward flexion, and reaching movements. She denied recent and past injury to her shoulder, fever, chills, rash, or skin changes at the injection site. She said her left shoulder did not bother her.
The patient had continued to participate in her aerobics class, despite the discomfort. Her medical history included hypertension and myocardial infarction, and the medications she was taking included lisinopril 20 mg/d, atenolol 50 mg/d, and aspirin 81 mg/d.
The physical exam revealed a thin female with no visible rashes or erythema on her right shoulder. While there was no deltoid atrophy in comparison to her unaffected shoulder, she generally had low muscle mass in both arms. A painful arc of abduction was present, as was pain with palpation of the supraspinatus insertion. No pain was appreciated over the short or long head of the biceps tendon or the sternoclavicular or acromioclavicular joints. Strength was 5/5 for all movements of the rotator cuff, but pain was reproduced with resisted shoulder abduction. A Hawkin’s test was positive, while Speed’s, Yergason’s, cross-arm abduction, and O’Brien’s tests were all negative.
THE DIAGNOSIS
Anteroposterior, Grashey, Y-view, and axillary view radiographs of the right shoulder were normal without any calcific tendinopathy, degenerative changes, or acute fractures. The patient’s history and physical exam were consistent with a rotator cuff tendinitis secondary to an immune response to an influenza vaccination that infiltrated the supraspinatus tendon.
DISCUSSION
Soreness, redness and swelling at the injection site, fever, body aches, and headache are common adverse effects of the influenza vaccine.1Although rare, acute brachial neuritis, infection, rotator cuff injuries, and contusions of the humeral head have also been reported. 2-5 Collectively, these conditions are referred to as shoulder injuries related to vaccination administration (SIRVA). There have been multiple SIRVA cases reported in the United States, and the US Court of Federal Claims has compensated >100 patients for SIRVA since 2011.6 There is currently no listing of SIRVA as a potential adverse reaction to the influenza vaccine on the package inserts or on the Centers for Disease Control and Prevention (CDC) Web site.
Shoulder soreness lasting <72 hours without functional impairment is likely due to soreness at the injection site. If symptoms do not resolve within 72 to 96 hours, consider a more thorough workup, with SIRVA being a possible diagnosis.1,7 The etiology of SIRVA remains uncertain, but an inflammatory reaction from a vaccine mistakenly administered into the subacromial/subdeltoid bursa has been suggested. Whether this reaction is dependent on the nonantigenic or antigenic components of the vaccine has yet to be determined.
Symptoms of SIRVA include pain with arm movement, pain that is worse at night or awakens the patient from sleep, restricted range of motion, or arm weakness. Examination will reveal pain when resisting rotator cuff movements, particularly shoulder abduction. Advanced imaging can be considered when the diagnosis is in question. In previous cases of vaccine-associated rotator cuff tendinopathy in the authors’ practice, T2 magnetic resonance imaging (MRI) has shown focal inflammatory signal within the supraspinatus tendon and subacromial bursa.
Continue to: With support from the CDC...
With support from the CDC, the Immunization Action Coalition (IAC), a source of immunization information for health care professionals, recommends that vaccines be administered into the deltoid or vastus lateralis for individuals between the ages of 3 and 18 years and recommends the deltoid as the preferred location in adults ≥19 years. The IAC suggests increasing the needle length for intramuscular (IM) immunizations (depending on the weight of the patient), although in the authors’ experience, the adjustment of needle length may often be overlooked (TABLE7).

The majority of reported SIRVA cases caused by overpenetration have occurred in individuals weighing <140 lb or those who had little deltoid muscle bulk. An MRI study to evaluate optimal intramuscular needle length in pediatric patients found that the IAC-recommended needle lengths still allowed penetration of the subdeltoid space in a substantial number of patients.8 Classic teaching of IM deltoid injection landmarks is 3 fingerbreadths distal to the acromion, and a more proximal administration of a vaccine would allow penetration of the rotator cuff structures below.
How to manage the patient
Patients who develop SIRVA should be managed similarly to patients with tendinopathy from other causes. Treatment options include: physical therapy, anti-inflammatory medications, and subacromial corticosteroid injections. Given the significant discomfort and nighttime pain associated with rotator cuff tendinopathy, corticosteroid injections can offer rapid relief.
Limited data exist on the effect of corticosteroids on the suppression of the immune response in immunocompetent patients. Vaccinations are generally thought to stimulate an adequate immune response 14 days following administration, so our suggestion would be to re-vaccinate patients if a corticosteroid injection to treat SIRVA is completed prior to this.9
Our patient’s outcome
We talked to the patient about treatment options, which included physical therapy and nonsteroidal anti-inflammatory drugs (NSAIDs), but the patient elected to go forward with a corticosteroid injection. We administered 2 cc of Depo-Medrol 40 mg/mL with 2 cc of 1% lidocaine without epinephrine and 2 cc of 0.5% ropivacaine into her right shoulder subacromial space using a posterior approach. The patient noticed a 70% improvement in her pain immediately following the injection.
Continue to: Considering her influenza vaccine...
Considering her influenza vaccine was administered more than 14 days prior to her corticosteroid injection, we felt that she had mounted enough of an immune response for the vaccination to have been adequate for protection.9 Therefore, we told her that she didn’t need to be revaccinated for influenza this season. The case was reported to the Vaccine Adverse Event Reporting System (VAERS).
At the patient’s 2-month follow up, she reported an overall 80% improvement in pain. She continued to have occasional discomfort with certain movements, although the pain was relieved with over-the-counter anti-inflammatory medication. On physical exam she had an intact arc of abduction of the right shoulder to 150° without pain. Forward flexion and external and internal rotation were normal and pain free. She had mild pain with resisted abduction and a positive Hawkin’s test. The patient agreed to go to physical therapy to work on rotator cuff strengthening. She denied any known influenza infection up to that time.
THE TAKEAWAY
It’s important to consider rotator cuff injuries or SIRVA as a potential adverse effect of influenza vaccination administration. Thin patients and those with low deltoid muscle mass are at risk of vaccine over-penetration, and proximally placed deltoid vaccines may reach the rotator cuff structures below. Staff should be trained on appropriate techniques for administering influenza vaccinations to avoid causing SIRVA. Specifically:
- Intramuscular vaccines injected into the deltoid muscle should be 3 fingerbreadths distal to the acromion. A more proximal approach could potentially contact the rotator cuff muscles.
- Vaccine administration should mirror the position of the patient (eg, if the patient is sitting, the administrator should be sitting; if the patient is standing, the administrator should be standing).
- Needle length for vaccine administration should be adjusted according to the patient’s weight (TABLE7).
Following vaccination, it is important to keep 2 other points in mind. First, if a subacromial corticosteroid injection is used for treatment of SIRVA within the first 2 weeks of vaccine administration, consider revaccination. Second, be sure to use the VAERS to report any clinically significant medical event that occurs after vaccination. VAERS is a national vaccine safety surveillance program that is supported by the CDC and the US Food and Drug Administration. The VAERS reporting system can be accessed through www.vaers.hhhs.gov.
CORRESPONDENCE
Dusty Marie Narducci, MD, 5290 Big Island Drive, Unit 1303, Jacksonville, FL 32246; [email protected]
1. Centers for Disease Control and Prevention. Flu vaccine safety information. https://www.cdc.gov/flu/protect/vaccine/general.htm. Updated October 23, 2018. Accessed January 2, 2019.
2. Barnes MG, Ledford C, Hogan K. A “needling” problem: shoulder injury related to vaccine administration. J Am Board Fam Med. 2012;25:919-922.
3. Shaikh MF, Baqai TJ, Tahir H. Acute brachial neuritis following influenza vaccine. BMJ Case Rep. 2012. doi:10.1136/bcr-2012-007673.
4. Miller JD, Pruitt S, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
5. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
6. Dugan IJ. Vaccine injury payouts rise. The Wall Street Journal. August 24, 2015. https://www.wsj.com/articles/vaccine-injury-payouts-rise-1440430702. Accessed December 3, 2018.
7. Immunization Action Coalition. Administering vaccines: dose, route, site, and needle size. www.immunize.org/catg.d/p3085.pdf. Accessed January 3, 2019.
8. Lippert WC, Wall EJ. Optimal intramuscular needle-penetration depth. Pediatrics. 2008;122:e556-e563.
9. Kroger AT, Sumaya CV, Pickering LK, et al. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). Centers of Disease Control and Prevention Web site. https://www.cdc.gov/mmwr/preview/mmwrhtml/rr6002a1.htm. Published January 28, 2011. Accessed December 3, 2018.
THE CASE
A 61-year-old Caucasian woman presented with acute right shoulder pain that began after she received an influenza vaccination at a local pharmacy 2 weeks earlier. She pointed to the proximal-most aspect of her lateral right upper arm as the vaccination site. Her pain intensified with shoulder abduction, forward flexion, and reaching movements. She denied recent and past injury to her shoulder, fever, chills, rash, or skin changes at the injection site. She said her left shoulder did not bother her.
The patient had continued to participate in her aerobics class, despite the discomfort. Her medical history included hypertension and myocardial infarction, and the medications she was taking included lisinopril 20 mg/d, atenolol 50 mg/d, and aspirin 81 mg/d.
The physical exam revealed a thin female with no visible rashes or erythema on her right shoulder. While there was no deltoid atrophy in comparison to her unaffected shoulder, she generally had low muscle mass in both arms. A painful arc of abduction was present, as was pain with palpation of the supraspinatus insertion. No pain was appreciated over the short or long head of the biceps tendon or the sternoclavicular or acromioclavicular joints. Strength was 5/5 for all movements of the rotator cuff, but pain was reproduced with resisted shoulder abduction. A Hawkin’s test was positive, while Speed’s, Yergason’s, cross-arm abduction, and O’Brien’s tests were all negative.
THE DIAGNOSIS
Anteroposterior, Grashey, Y-view, and axillary view radiographs of the right shoulder were normal without any calcific tendinopathy, degenerative changes, or acute fractures. The patient’s history and physical exam were consistent with a rotator cuff tendinitis secondary to an immune response to an influenza vaccination that infiltrated the supraspinatus tendon.
DISCUSSION
Soreness, redness and swelling at the injection site, fever, body aches, and headache are common adverse effects of the influenza vaccine.1Although rare, acute brachial neuritis, infection, rotator cuff injuries, and contusions of the humeral head have also been reported. 2-5 Collectively, these conditions are referred to as shoulder injuries related to vaccination administration (SIRVA). There have been multiple SIRVA cases reported in the United States, and the US Court of Federal Claims has compensated >100 patients for SIRVA since 2011.6 There is currently no listing of SIRVA as a potential adverse reaction to the influenza vaccine on the package inserts or on the Centers for Disease Control and Prevention (CDC) Web site.
Shoulder soreness lasting <72 hours without functional impairment is likely due to soreness at the injection site. If symptoms do not resolve within 72 to 96 hours, consider a more thorough workup, with SIRVA being a possible diagnosis.1,7 The etiology of SIRVA remains uncertain, but an inflammatory reaction from a vaccine mistakenly administered into the subacromial/subdeltoid bursa has been suggested. Whether this reaction is dependent on the nonantigenic or antigenic components of the vaccine has yet to be determined.
Symptoms of SIRVA include pain with arm movement, pain that is worse at night or awakens the patient from sleep, restricted range of motion, or arm weakness. Examination will reveal pain when resisting rotator cuff movements, particularly shoulder abduction. Advanced imaging can be considered when the diagnosis is in question. In previous cases of vaccine-associated rotator cuff tendinopathy in the authors’ practice, T2 magnetic resonance imaging (MRI) has shown focal inflammatory signal within the supraspinatus tendon and subacromial bursa.
Continue to: With support from the CDC...
With support from the CDC, the Immunization Action Coalition (IAC), a source of immunization information for health care professionals, recommends that vaccines be administered into the deltoid or vastus lateralis for individuals between the ages of 3 and 18 years and recommends the deltoid as the preferred location in adults ≥19 years. The IAC suggests increasing the needle length for intramuscular (IM) immunizations (depending on the weight of the patient), although in the authors’ experience, the adjustment of needle length may often be overlooked (TABLE7).
The majority of reported SIRVA cases caused by overpenetration have occurred in individuals weighing <140 lb or those who had little deltoid muscle bulk. An MRI study to evaluate optimal intramuscular needle length in pediatric patients found that the IAC-recommended needle lengths still allowed penetration of the subdeltoid space in a substantial number of patients.8 Classic teaching of IM deltoid injection landmarks is 3 fingerbreadths distal to the acromion, and a more proximal administration of a vaccine would allow penetration of the rotator cuff structures below.
How to manage the patient
Patients who develop SIRVA should be managed similarly to patients with tendinopathy from other causes. Treatment options include: physical therapy, anti-inflammatory medications, and subacromial corticosteroid injections. Given the significant discomfort and nighttime pain associated with rotator cuff tendinopathy, corticosteroid injections can offer rapid relief.
Limited data exist on the effect of corticosteroids on the suppression of the immune response in immunocompetent patients. Vaccinations are generally thought to stimulate an adequate immune response 14 days following administration, so our suggestion would be to re-vaccinate patients if a corticosteroid injection to treat SIRVA is completed prior to this.9
Our patient’s outcome
We talked to the patient about treatment options, which included physical therapy and nonsteroidal anti-inflammatory drugs (NSAIDs), but the patient elected to go forward with a corticosteroid injection. We administered 2 cc of Depo-Medrol 40 mg/mL with 2 cc of 1% lidocaine without epinephrine and 2 cc of 0.5% ropivacaine into her right shoulder subacromial space using a posterior approach. The patient noticed a 70% improvement in her pain immediately following the injection.
Continue to: Considering her influenza vaccine...
Considering her influenza vaccine was administered more than 14 days prior to her corticosteroid injection, we felt that she had mounted enough of an immune response for the vaccination to have been adequate for protection.9 Therefore, we told her that she didn’t need to be revaccinated for influenza this season. The case was reported to the Vaccine Adverse Event Reporting System (VAERS).
At the patient’s 2-month follow up, she reported an overall 80% improvement in pain. She continued to have occasional discomfort with certain movements, although the pain was relieved with over-the-counter anti-inflammatory medication. On physical exam she had an intact arc of abduction of the right shoulder to 150° without pain. Forward flexion and external and internal rotation were normal and pain free. She had mild pain with resisted abduction and a positive Hawkin’s test. The patient agreed to go to physical therapy to work on rotator cuff strengthening. She denied any known influenza infection up to that time.
THE TAKEAWAY
It’s important to consider rotator cuff injuries or SIRVA as a potential adverse effect of influenza vaccination administration. Thin patients and those with low deltoid muscle mass are at risk of vaccine over-penetration, and proximally placed deltoid vaccines may reach the rotator cuff structures below. Staff should be trained on appropriate techniques for administering influenza vaccinations to avoid causing SIRVA. Specifically:
- Intramuscular vaccines injected into the deltoid muscle should be 3 fingerbreadths distal to the acromion. A more proximal approach could potentially contact the rotator cuff muscles.
- Vaccine administration should mirror the position of the patient (eg, if the patient is sitting, the administrator should be sitting; if the patient is standing, the administrator should be standing).
- Needle length for vaccine administration should be adjusted according to the patient’s weight (TABLE7).
Following vaccination, it is important to keep 2 other points in mind. First, if a subacromial corticosteroid injection is used for treatment of SIRVA within the first 2 weeks of vaccine administration, consider revaccination. Second, be sure to use the VAERS to report any clinically significant medical event that occurs after vaccination. VAERS is a national vaccine safety surveillance program that is supported by the CDC and the US Food and Drug Administration. The VAERS reporting system can be accessed through www.vaers.hhhs.gov.
CORRESPONDENCE
Dusty Marie Narducci, MD, 5290 Big Island Drive, Unit 1303, Jacksonville, FL 32246; [email protected]
THE CASE
A 61-year-old Caucasian woman presented with acute right shoulder pain that began after she received an influenza vaccination at a local pharmacy 2 weeks earlier. She pointed to the proximal-most aspect of her lateral right upper arm as the vaccination site. Her pain intensified with shoulder abduction, forward flexion, and reaching movements. She denied recent and past injury to her shoulder, fever, chills, rash, or skin changes at the injection site. She said her left shoulder did not bother her.
The patient had continued to participate in her aerobics class, despite the discomfort. Her medical history included hypertension and myocardial infarction, and the medications she was taking included lisinopril 20 mg/d, atenolol 50 mg/d, and aspirin 81 mg/d.
The physical exam revealed a thin female with no visible rashes or erythema on her right shoulder. While there was no deltoid atrophy in comparison to her unaffected shoulder, she generally had low muscle mass in both arms. A painful arc of abduction was present, as was pain with palpation of the supraspinatus insertion. No pain was appreciated over the short or long head of the biceps tendon or the sternoclavicular or acromioclavicular joints. Strength was 5/5 for all movements of the rotator cuff, but pain was reproduced with resisted shoulder abduction. A Hawkin’s test was positive, while Speed’s, Yergason’s, cross-arm abduction, and O’Brien’s tests were all negative.
THE DIAGNOSIS
Anteroposterior, Grashey, Y-view, and axillary view radiographs of the right shoulder were normal without any calcific tendinopathy, degenerative changes, or acute fractures. The patient’s history and physical exam were consistent with a rotator cuff tendinitis secondary to an immune response to an influenza vaccination that infiltrated the supraspinatus tendon.
DISCUSSION
Soreness, redness and swelling at the injection site, fever, body aches, and headache are common adverse effects of the influenza vaccine.1Although rare, acute brachial neuritis, infection, rotator cuff injuries, and contusions of the humeral head have also been reported. 2-5 Collectively, these conditions are referred to as shoulder injuries related to vaccination administration (SIRVA). There have been multiple SIRVA cases reported in the United States, and the US Court of Federal Claims has compensated >100 patients for SIRVA since 2011.6 There is currently no listing of SIRVA as a potential adverse reaction to the influenza vaccine on the package inserts or on the Centers for Disease Control and Prevention (CDC) Web site.
Shoulder soreness lasting <72 hours without functional impairment is likely due to soreness at the injection site. If symptoms do not resolve within 72 to 96 hours, consider a more thorough workup, with SIRVA being a possible diagnosis.1,7 The etiology of SIRVA remains uncertain, but an inflammatory reaction from a vaccine mistakenly administered into the subacromial/subdeltoid bursa has been suggested. Whether this reaction is dependent on the nonantigenic or antigenic components of the vaccine has yet to be determined.
Symptoms of SIRVA include pain with arm movement, pain that is worse at night or awakens the patient from sleep, restricted range of motion, or arm weakness. Examination will reveal pain when resisting rotator cuff movements, particularly shoulder abduction. Advanced imaging can be considered when the diagnosis is in question. In previous cases of vaccine-associated rotator cuff tendinopathy in the authors’ practice, T2 magnetic resonance imaging (MRI) has shown focal inflammatory signal within the supraspinatus tendon and subacromial bursa.
Continue to: With support from the CDC...
With support from the CDC, the Immunization Action Coalition (IAC), a source of immunization information for health care professionals, recommends that vaccines be administered into the deltoid or vastus lateralis for individuals between the ages of 3 and 18 years and recommends the deltoid as the preferred location in adults ≥19 years. The IAC suggests increasing the needle length for intramuscular (IM) immunizations (depending on the weight of the patient), although in the authors’ experience, the adjustment of needle length may often be overlooked (TABLE7).
The majority of reported SIRVA cases caused by overpenetration have occurred in individuals weighing <140 lb or those who had little deltoid muscle bulk. An MRI study to evaluate optimal intramuscular needle length in pediatric patients found that the IAC-recommended needle lengths still allowed penetration of the subdeltoid space in a substantial number of patients.8 Classic teaching of IM deltoid injection landmarks is 3 fingerbreadths distal to the acromion, and a more proximal administration of a vaccine would allow penetration of the rotator cuff structures below.
How to manage the patient
Patients who develop SIRVA should be managed similarly to patients with tendinopathy from other causes. Treatment options include: physical therapy, anti-inflammatory medications, and subacromial corticosteroid injections. Given the significant discomfort and nighttime pain associated with rotator cuff tendinopathy, corticosteroid injections can offer rapid relief.
Limited data exist on the effect of corticosteroids on the suppression of the immune response in immunocompetent patients. Vaccinations are generally thought to stimulate an adequate immune response 14 days following administration, so our suggestion would be to re-vaccinate patients if a corticosteroid injection to treat SIRVA is completed prior to this.9
Our patient’s outcome
We talked to the patient about treatment options, which included physical therapy and nonsteroidal anti-inflammatory drugs (NSAIDs), but the patient elected to go forward with a corticosteroid injection. We administered 2 cc of Depo-Medrol 40 mg/mL with 2 cc of 1% lidocaine without epinephrine and 2 cc of 0.5% ropivacaine into her right shoulder subacromial space using a posterior approach. The patient noticed a 70% improvement in her pain immediately following the injection.
Continue to: Considering her influenza vaccine...
Considering her influenza vaccine was administered more than 14 days prior to her corticosteroid injection, we felt that she had mounted enough of an immune response for the vaccination to have been adequate for protection.9 Therefore, we told her that she didn’t need to be revaccinated for influenza this season. The case was reported to the Vaccine Adverse Event Reporting System (VAERS).
At the patient’s 2-month follow up, she reported an overall 80% improvement in pain. She continued to have occasional discomfort with certain movements, although the pain was relieved with over-the-counter anti-inflammatory medication. On physical exam she had an intact arc of abduction of the right shoulder to 150° without pain. Forward flexion and external and internal rotation were normal and pain free. She had mild pain with resisted abduction and a positive Hawkin’s test. The patient agreed to go to physical therapy to work on rotator cuff strengthening. She denied any known influenza infection up to that time.
THE TAKEAWAY
It’s important to consider rotator cuff injuries or SIRVA as a potential adverse effect of influenza vaccination administration. Thin patients and those with low deltoid muscle mass are at risk of vaccine over-penetration, and proximally placed deltoid vaccines may reach the rotator cuff structures below. Staff should be trained on appropriate techniques for administering influenza vaccinations to avoid causing SIRVA. Specifically:
- Intramuscular vaccines injected into the deltoid muscle should be 3 fingerbreadths distal to the acromion. A more proximal approach could potentially contact the rotator cuff muscles.
- Vaccine administration should mirror the position of the patient (eg, if the patient is sitting, the administrator should be sitting; if the patient is standing, the administrator should be standing).
- Needle length for vaccine administration should be adjusted according to the patient’s weight (TABLE7).
Following vaccination, it is important to keep 2 other points in mind. First, if a subacromial corticosteroid injection is used for treatment of SIRVA within the first 2 weeks of vaccine administration, consider revaccination. Second, be sure to use the VAERS to report any clinically significant medical event that occurs after vaccination. VAERS is a national vaccine safety surveillance program that is supported by the CDC and the US Food and Drug Administration. The VAERS reporting system can be accessed through www.vaers.hhhs.gov.
CORRESPONDENCE
Dusty Marie Narducci, MD, 5290 Big Island Drive, Unit 1303, Jacksonville, FL 32246; [email protected]
1. Centers for Disease Control and Prevention. Flu vaccine safety information. https://www.cdc.gov/flu/protect/vaccine/general.htm. Updated October 23, 2018. Accessed January 2, 2019.
2. Barnes MG, Ledford C, Hogan K. A “needling” problem: shoulder injury related to vaccine administration. J Am Board Fam Med. 2012;25:919-922.
3. Shaikh MF, Baqai TJ, Tahir H. Acute brachial neuritis following influenza vaccine. BMJ Case Rep. 2012. doi:10.1136/bcr-2012-007673.
4. Miller JD, Pruitt S, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
5. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
6. Dugan IJ. Vaccine injury payouts rise. The Wall Street Journal. August 24, 2015. https://www.wsj.com/articles/vaccine-injury-payouts-rise-1440430702. Accessed December 3, 2018.
7. Immunization Action Coalition. Administering vaccines: dose, route, site, and needle size. www.immunize.org/catg.d/p3085.pdf. Accessed January 3, 2019.
8. Lippert WC, Wall EJ. Optimal intramuscular needle-penetration depth. Pediatrics. 2008;122:e556-e563.
9. Kroger AT, Sumaya CV, Pickering LK, et al. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). Centers of Disease Control and Prevention Web site. https://www.cdc.gov/mmwr/preview/mmwrhtml/rr6002a1.htm. Published January 28, 2011. Accessed December 3, 2018.
1. Centers for Disease Control and Prevention. Flu vaccine safety information. https://www.cdc.gov/flu/protect/vaccine/general.htm. Updated October 23, 2018. Accessed January 2, 2019.
2. Barnes MG, Ledford C, Hogan K. A “needling” problem: shoulder injury related to vaccine administration. J Am Board Fam Med. 2012;25:919-922.
3. Shaikh MF, Baqai TJ, Tahir H. Acute brachial neuritis following influenza vaccine. BMJ Case Rep. 2012. doi:10.1136/bcr-2012-007673.
4. Miller JD, Pruitt S, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
5. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
6. Dugan IJ. Vaccine injury payouts rise. The Wall Street Journal. August 24, 2015. https://www.wsj.com/articles/vaccine-injury-payouts-rise-1440430702. Accessed December 3, 2018.
7. Immunization Action Coalition. Administering vaccines: dose, route, site, and needle size. www.immunize.org/catg.d/p3085.pdf. Accessed January 3, 2019.
8. Lippert WC, Wall EJ. Optimal intramuscular needle-penetration depth. Pediatrics. 2008;122:e556-e563.
9. Kroger AT, Sumaya CV, Pickering LK, et al. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). Centers of Disease Control and Prevention Web site. https://www.cdc.gov/mmwr/preview/mmwrhtml/rr6002a1.htm. Published January 28, 2011. Accessed December 3, 2018.
