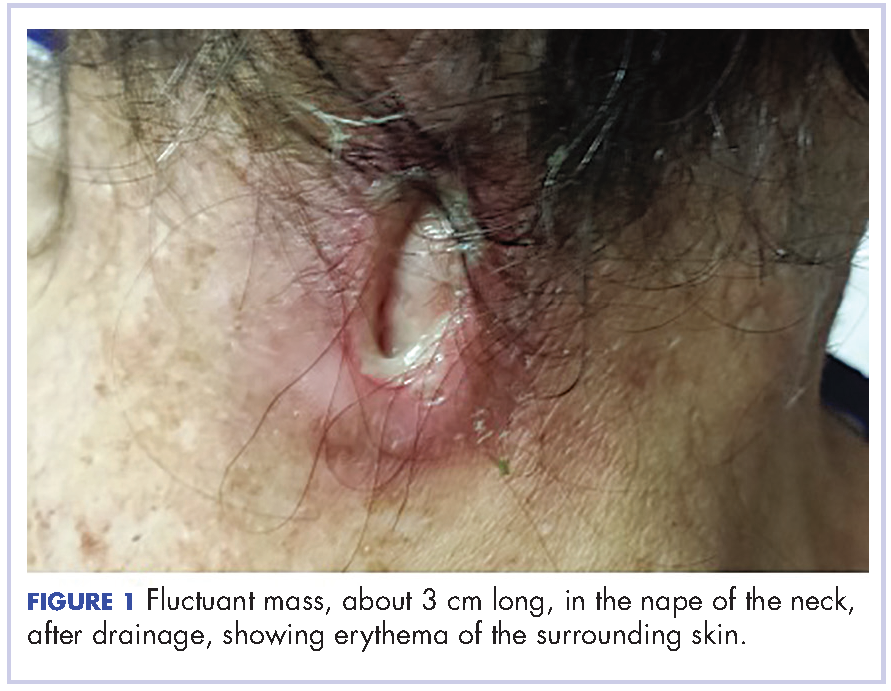
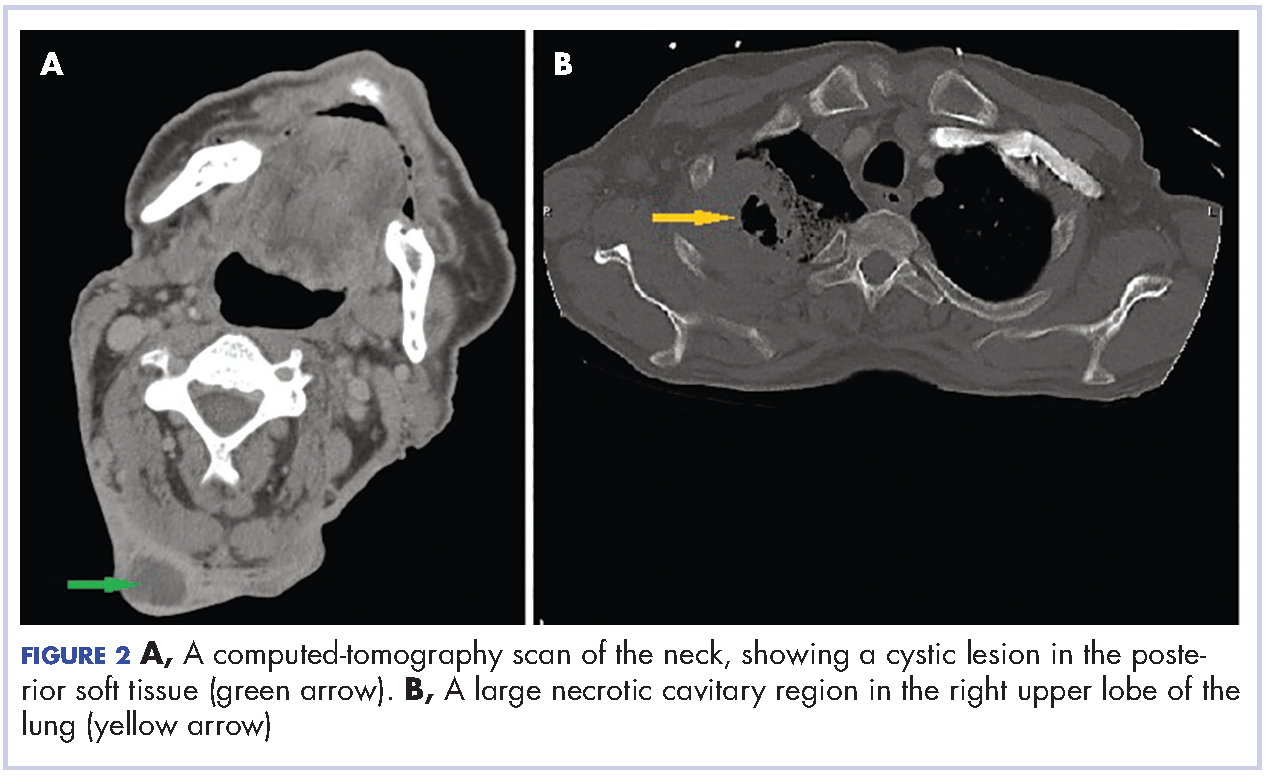
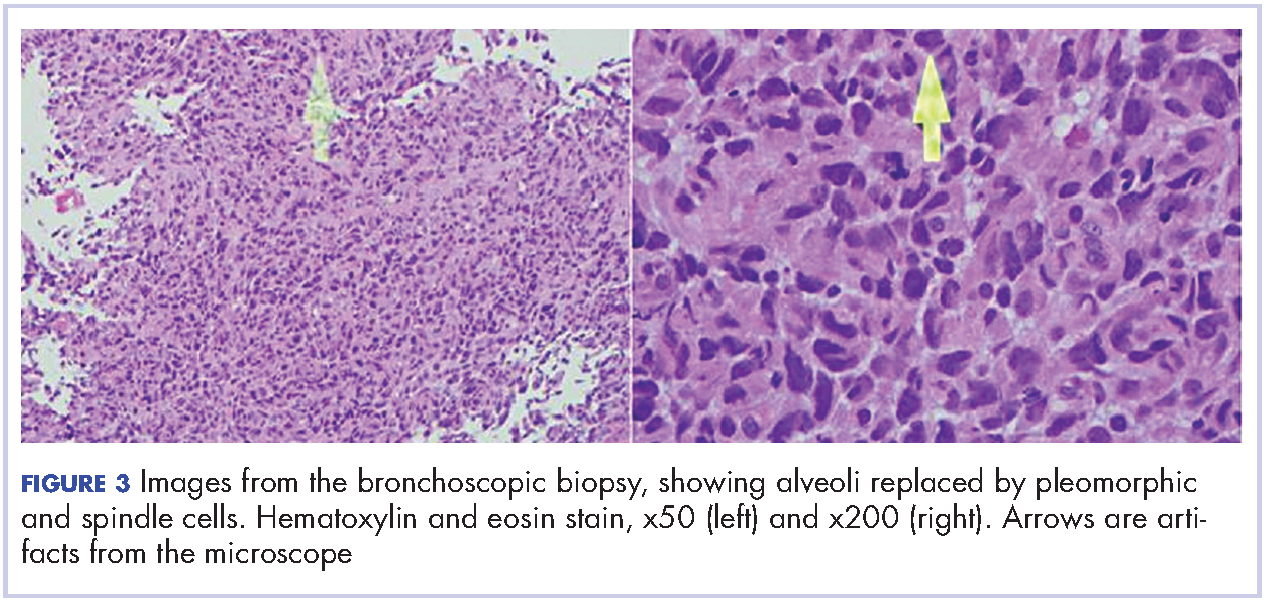
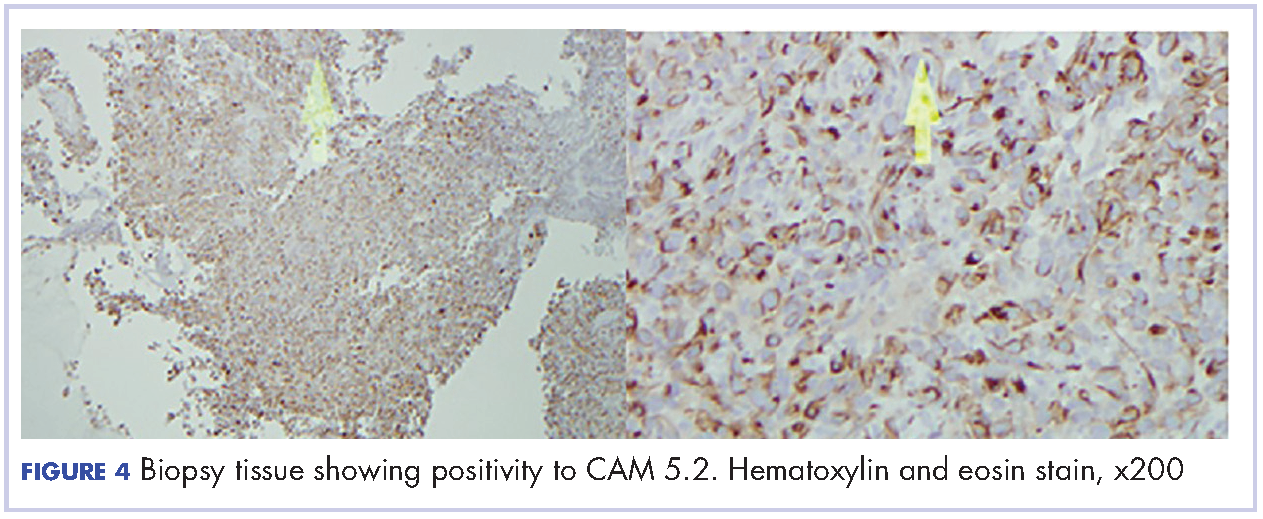
A 29-year-old man presented to the ED with a 3-day history of constant left-sided low back pain that radiated to his left buttock and groin. The patient stated the pain worsened with movement, making it difficult for him to walk. He reported lifting heavy boxes at work, but denied any trauma. The patient also denied recent fevers, chills, chest pain, dyspnea, abdominal pain, urinary or fecal incontinence, weakness, numbness, or saddle anesthesia. Regarding his medical history, he had an appendectomy as a child, but reported no other surgeries or medical issues. His social history was significant for narcotic and inhalant use and daily tobacco use. The patient also reported taking heroin intravenously (IV) 6 months prior.
Vital signs at presentation were: heart rate (HR), 92 beats/min; respiratory rate, 15 breaths/min; blood pressure, 118/80 mm Hg; and temperature, 98.2°F. Oxygen saturation was 98% on room air.
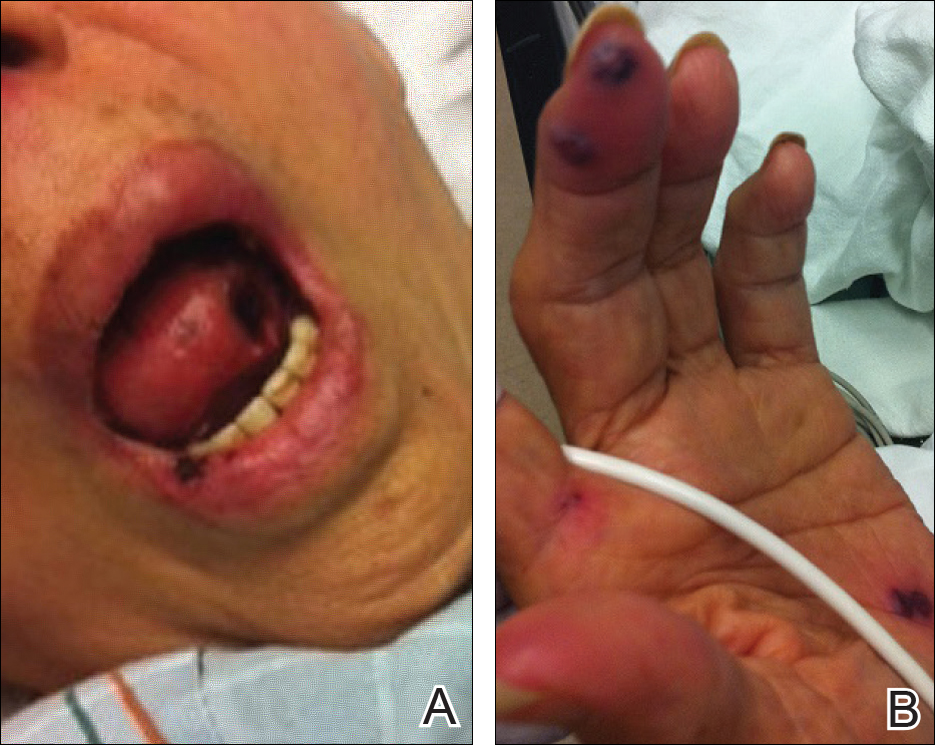
The patient was a well-developed young man in no apparent distress. Dermatological examination showed bilateral track marks in the antecubital fossa. The musculoskeletal (MSK) examination demonstrated left gluteal tenderness to palpation and decreased active and passive range of motion of the left hip, especially with internal rotation and flexion. He had no midline tenderness, and the lower extremities had normal pulses and no motor or sensory deficits.
The patient’s pain improved with IV fluids, diazepam, and ketorolac, and he was able to ambulate with assistance. He was clinically diagnosed with sciatica, and discharged home with prescriptions for diazepam and ibuprofen. He was also instructed to follow-up with an orthopedist within 7 days from discharge.
The patient returned to the ED the following day with similar complaints of unabating left-sided pain and difficulty ambulating. His vital signs were notable for an elevated HR of 106 beats/min. Physical examination findings were unchanged from his presentation the previous day, and an X-ray of the lumbar spine showed no abnormalities.
After receiving IV analgesics, the patient’s pain improved and his tachycardia resolved. He was discharged home with instructions to continue taking diazepam, and was also given prescriptions for prednisone and oxycodone/acetaminophen. He was instructed to follow-up with an orthopedist within 24 hours.
Over the next 9 days, the patient was seen twice by an orthopedist, who ordered imaging of the lumbar spine, including a repeat X-ray and contrast-enhanced magnetic resonance imaging (MRI), both of which were unremarkable. The patient completed the prescribed course of diclofenac, oxycodone/acetaminophen, and prednisone, but experienced only minimal pain relief.The orthopedist prescribed the diclofenac to supplement the medication regimen that he was already on.
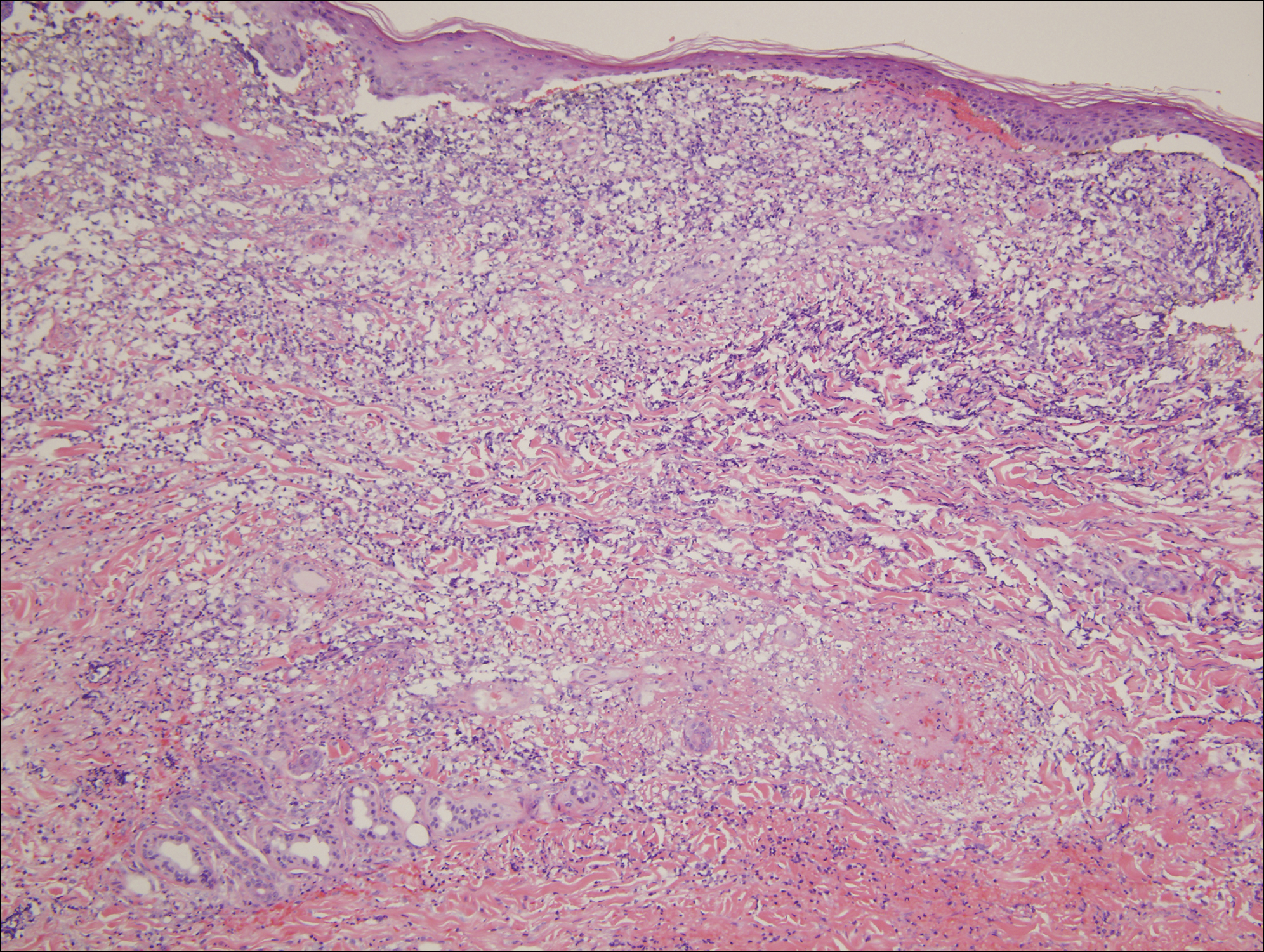
At the second follow-up visit, the orthopedist ordered an MRI of the patient’s left hip, which demonstrated inflammation of the left sacroiliac joint (SIJ) with effusion, and a 1-cm by 1-cm collection adjacent to the left psoas muscle; these findings were concerning for septic arthritis (Figure). Based on the MRI study, a computed tomography (CT)-guided arthrocentesis of the left SIJ was performed by an interventional radiologist.
Figure Following the arthrocentesis, the orthopedist referred the patient to the ED. At this presentation,the emergency physician (EP) ordered blood cultures, blood work, urinalysis, and a urinary toxicology screen, and started the patient on IV ceftriaxone and vancomycin. The laboratory studies were significant for the following elevated inflammatory markers: erythrocyte sedimentation rate (ESR), 19 mm/h; C-reactive protein (CRP), 2.45 mg/L; white blood cell count (WBC), 13.6 K/uL with normal differential; and lactate level, 2.6 mg/dL. The toxicology screen was positive for opioids. The basic metabolic panel, chest X-ray, and urinalysis were all unremarkable. An electrocardiogram showed sinus tachycardia.
The patient was admitted to the hospital, and infectious disease services was contacted. While awaiting transport to the inpatient floor, the patient admitted to IV drug use 4 weeks prior to his initial presentation—not the 6 months he initially reported at the first ED visit.
The blood cultures grew Candida parapsilosis, and culture from the SIJ arthrocentesis grew Pseudomonas aeruginosa. The infectious disease physician switched the patient’s antibiotic therapy to IV cefepime and fluconazole. The patient also was seen by an orthopedist, who determined that no surgical intervention was required.
Follow-up laboratory studies showed inflammatory markers peaking at the following levels: ESR, 36 mm/h; CRP, 4.84 mg/L; and WBC, 32.1 K/uL with 90% neutrophils. These markers normalized throughout his hospital stay. The patient was also tested for hepatitis and human immunodeficiency virus, both of which were negative. A transesophageal echocardiogram showed no obvious masses or vegetations.
The patient had an uncomplicated hospital course, and was discharged home on hospital day 6 with a 4-week prescription of oral fluconazole and levofloxacin, and instructed to follow-up with both infectious disease and the orthopedist. To address his history of IV drug use, he also was given follow-up with pain management.
One month later, the patient returned a fourth time to the ED for evaluation of bilateral lower extremity pain and swelling. He stated that he had been mostly bed-bound at home since his discharge from the hospital due to continued pain with weight-bearing.
The patient’s vital signs were normal. The EP ordered a duplex ultrasound study, which showed extensive bilateral lower extremity deep vein thrombosis. He was started on subcutaneous therapeutic enoxaparin and admitted to the inpatient hospital. During admission,a left lower lobe pulmonary artery embolism was found on chest CT angiography, though he had no cardiac or respiratory symptoms. He was discharged home with a 3-month prescription for oral rivaroxaban.
At a 4-month follow-up visit, the patient reported minimal residual disability after completing the course of treatment. During the follow-up, the patient denied using IV heroin; he was referred to a pain management specialist, who placed the patient on methadone.
Discussion
Infectious sacroiliitis (ISI) is a rare form of infectious arthritis affecting the SIJ, with an incidence of 1 to 2 reported cases per year.1 The literature on ISI currently consists only of case reports and case series. This infection is often diagnosed after the disease has progressed, with a mean time to diagnosis of 43.3 days.2
Infectious arthritis of any joint has a prevalence of 2 to 10 per 100,000 people. In 50% of cases, the knee is the joint most commonly affected, followed by the hip, shoulder, and elbow.3 Regardless of location, infectious arthritis is associated with significant morbidity and mortality due to sepsis and irreversible loss of joint function.4
Risk factors for ISI include IV drug use, pregnancy, trauma, endocarditis, and immunosuppression.1 The decision to initiate the workup for ISI can be difficult to make because the condition may present without signs of an infectious etiology, such as toxic appearance, inflammatory changes surrounding the joint, or even fever—only 41% of affected patients in one case series were febrile.2 The workup is often time-consuming, invasive, and expensive.
Although delayed diagnosis and treatment of septic arthritis is associated with significant adverse effects, there is unfortunately no consensus to guide the workup for ISI. As opposed to Kocher’s criteria for the differentiation of septic hip arthritis from transient synovitis in pediatric patients or well-known red-flags for further evaluation of low back pain, physicians are left without much guidance when considering laboratory workup or imaging decisions to evaluate for ISI.
Sacroiliac Joint
As previously noted, the SIJ is not commonly affected by infection. It is a diarthrodial, L-shaped joint comprised of the posterior ilium and sacrum, and is a near-rigid structure with very limited movement that provides stability to the axial skeleton.5 The SIJ is often overlooked as a secondary cause of low back pain in younger patients with rheumatologic conditions (eg, ankylosing spondylitis, Reiter syndrome), pregnancy-associated ligamentous laxity, and osteoarthritis in elderly patients. In one study, 88.2% of sacroiliitis cases were inflammatory, 8.8% infectious, and 2.9% degenerative.6
Signs and Symptoms
As our case illustrates, ISI often presents with nonspecific symptoms and physical findings.7 Patients typically present with fever, painful manipulation of the SIJ, and unilateral lumbo-gluteal pain.2 The components of the history and physical examination suspicious for an infectious etiology include the subacute presentation; unresolved pain despite treatment; tenderness to palpation; decreased range of motion; and recent IV drug use, which increases the risk of infectious disease due to unsterile practices and direct inoculation of pathogens into the bloodstream8 and a further predilection into the axial skeleton. 9 It is important to obtain an accurate social history; however, patients may not be forthright about disclosing sensitive information such as sexual history and illicit drug use.
Physical Assessment
The SIJ is best appreciated in the seated patient by palpating one fingerbreadth medial to the posterior superior iliac spine as he or she slowly bends forward.10 Tenderness elicited while in this position is suggestive of SIJ inflammation. The area of tenderness may be lower than anticipated and lateral to the gluteal cleft, as synovial fluid is typically relegated to the lower half of the joint.
Several adjunctive physical examination maneuvers, such as the Gaenslen test and Flexion Abduction External Rotation test (FABER test or Patrick’s test) can isolate SIJ pathology or dysfunction. The Gaenslen test is performed by asking the patient to lie supine and flex the affected hip and knee, with the lumbar spine flat against the examination table. Hyperextending the contralateral thigh downward will reproduce pain in the affected SIJ.
The FABER test is a simple but less specific examination technique to assess joint pain in the hip, lumbar, and sacroiliac joints.11 In this assessment, the clinician flexes the patient’s affected knee to 90°, externally rotates the hip, and applies downward pressure on the knee. Pain reproduced in the affected SI region is sensitive for joint inflammation.
Laboratory and Imaging Studies
Laboratory studies typically show inconsistent and nonspecific findings, such as the elevated ESR and CRP levels seen in our patient.2,12 Imaging studies to assess the SIJ for signs of infection are therefore essential for confirming infection.
Magnetic resonance imaging is the preferred imaging modality to assess for ISI, since it has the highest sensitivity in visualizing joint effusion and bone marrow edema compared to other modalities. Computed tomography, however, can be helpful in visualizing associated abscesses and guiding arthrocentesis.12 Plain X-ray may not demonstrate early changes in bone.13 The confirmatory study for ISI is synovial fluid analysis and culture.7
Treatment
Infectious sacroiliitis secondary to P aeruginosa, a gram-negative bacillus, is difficult to treat because of the glycocalyx and slime production that protects the pathogen from antibiotics, the development of multiple-antimicrobial resistance, and poor drug penetration into bones and abscesses.14 Antibiotic treatment should cover Staphylococcus aureus and may be broadened to cover gram-negative bacilli. The recommended duration of treatment is at least a 2-week course of IV antibiotics, followed by a 6-week course of oral antibiotics.2 Therapy also includes pain control and surgical intervention for abscesses, osteomyelitis, and refractory cases.7
Complications
Complications and long-term sequelae are common in ISI, often due to late diagnosis of the condition.Our case illustrates the delayed diagnosis of Pseudomonas ISI with candidemia in a young man with a history of IV drug use presenting with atraumatic low back pain. His clinical course was complicated by a thromboembolic event, likely secondary to immobility and a hypercoagulable state from infection and inflammation.15 Infectious sacroiliitis secondary to P aeruginosa is most commonly seen in patients with immunosuppression, hospitalization, and IV drug use.2
Summary
Infectious sacroiliitis remains a diagnostic challenge for physicians due to its rare incidence and nonspecific clinical manifestations. Our case illustrates the importance of maintaining a high level of clinical suspicion for infectious arthritis in young patients presenting with common MSK complaints in the presence of infectious risk factors. Emergency physicians should consider red flags, abnormal vital signs, and patient recidivism when deciding on the most appropriate workup.
References
1. Mancarella L, De Santis M, Magarelli N, Ierardi AM, Bonomo L, Ferraccioli G. Septic sacroiliitis: an uncommon septic arthritis. Clin Exp Rheumatol. 2009;27(6):1004-1008. 2. Hermet M, Minichiello E, Flipo RM, et al. Infectious sacroiliitis: a retrospective, multicentre study of 39 adults. BMC Infect Dis. 2012;12:305.doi:10.1186/1471-2334-12-305. 3. Abelson A. Septic Arthritis. Cleveland Clinic. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/septic-arthritis. Published August 2010. Accessed October 28, 2016. 4. Goldenberg DL. Septic arthritis. Lancet. 1998;351(9097):197-202. doi:10.1016/S0140-6736(97)09522-6. 5. Vleeming A, Schuenke MD, Masi AT, Carreiro JE, Danneels L, Willard FH. The sacroiliac joint: an overview of its anatomy, function and potential clinical implications. J Anat. 2012;221(6):537-567. doi:10.1111/j.1469-7580.2012.01564.x. 6. Owlia MB, Danesh-Ardakani M. Frequency of sacroiliitis among patients with low back pain. Electron Physician. 2016;8(3):2094-2100. doi:10.19082/2094. 7. Zimmermann B 3rd, Mikolich DJ, Lally EV. Septic sacroiliitis. Semin Arthritis Rheum. 1996;26(3):592-604. 8. Brtalik D, Pariyadath M. A case report of infectious sacroiliitis in an adult presenting to the emergency department with inability to walk. J Emerg Med. 2017:52(3)e65-e68. doi:10.1016/j.jemermed.2016.10.022. 9. Ferraro K, Cohen MA. Acute septic sacroiliitis in an injection drug user. Am J Emerg Med. 2004;22(1):60-61. 10. Safran M, Botser IB. Hip anatomy and biomechanics. In: Miller MD, Thompson SR, eds. DeLee & Drez’s Orthopaedic Sports Medicine. Vol 2. 4th ed. Philadelphia, PA: Elsevier Saunders; 2015:917-932.e1. 11. LeBlond RF, Brown DD, Suneja M, Szot JF. The spine, pelvic, and extremities. In: LeBlond RF, Brown DD, Suneja M, Szot JF. eds. DeGowin’s Diagnostic Examination. 10th ed. New York, NY: McGraw-Hill; 2015:508-576. 12. Scott KR, Rising KL, Conlon LW. Infectious sacroiliitis. J Emerg Med. 2014;47(3):83-84. doi:10.1016/j.jemermed.2014.05.001. 13. Cinar M, Sanal HT, Yilmaz S, et al. Radiological followup of the evolution of inflammatory process in sacroiliac joint with magnetic resonance imaging: a case with pyogenic sacroiliitis. Case Rep Rheumatol. 2012;2012:509136. doi:10.1155/2012/509136. 14. Calza L, Manfredi R, Marinacci G, Fortunato L, Chiodo F. Community-acquired Pseudomonas aeruginosa sacro-iliitis in a previously healthy patient. J Med Microbiol. 2002;51(7):620-622. 15. Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60(1):26-39.
A 29-year-old man presented for evaluation of unabating left-sided low back pain that radiated to his left buttock and groin.
A 29-year-old man presented for evaluation of unabating left-sided low back pain that radiated to his left buttock and groin.
Case
A 29-year-old man presented to the ED with a 3-day history of constant left-sided low back pain that radiated to his left buttock and groin. The patient stated the pain worsened with movement, making it difficult for him to walk. He reported lifting heavy boxes at work, but denied any trauma. The patient also denied recent fevers, chills, chest pain, dyspnea, abdominal pain, urinary or fecal incontinence, weakness, numbness, or saddle anesthesia. Regarding his medical history, he had an appendectomy as a child, but reported no other surgeries or medical issues. His social history was significant for narcotic and inhalant use and daily tobacco use. The patient also reported taking heroin intravenously (IV) 6 months prior.
Vital signs at presentation were: heart rate (HR), 92 beats/min; respiratory rate, 15 breaths/min; blood pressure, 118/80 mm Hg; and temperature, 98.2°F. Oxygen saturation was 98% on room air.
The patient was a well-developed young man in no apparent distress. Dermatological examination showed bilateral track marks in the antecubital fossa. The musculoskeletal (MSK) examination demonstrated left gluteal tenderness to palpation and decreased active and passive range of motion of the left hip, especially with internal rotation and flexion. He had no midline tenderness, and the lower extremities had normal pulses and no motor or sensory deficits.
The patient’s pain improved with IV fluids, diazepam, and ketorolac, and he was able to ambulate with assistance. He was clinically diagnosed with sciatica, and discharged home with prescriptions for diazepam and ibuprofen. He was also instructed to follow-up with an orthopedist within 7 days from discharge.
The patient returned to the ED the following day with similar complaints of unabating left-sided pain and difficulty ambulating. His vital signs were notable for an elevated HR of 106 beats/min. Physical examination findings were unchanged from his presentation the previous day, and an X-ray of the lumbar spine showed no abnormalities.
After receiving IV analgesics, the patient’s pain improved and his tachycardia resolved. He was discharged home with instructions to continue taking diazepam, and was also given prescriptions for prednisone and oxycodone/acetaminophen. He was instructed to follow-up with an orthopedist within 24 hours.
Over the next 9 days, the patient was seen twice by an orthopedist, who ordered imaging of the lumbar spine, including a repeat X-ray and contrast-enhanced magnetic resonance imaging (MRI), both of which were unremarkable. The patient completed the prescribed course of diclofenac, oxycodone/acetaminophen, and prednisone, but experienced only minimal pain relief.The orthopedist prescribed the diclofenac to supplement the medication regimen that he was already on.
At the second follow-up visit, the orthopedist ordered an MRI of the patient’s left hip, which demonstrated inflammation of the left sacroiliac joint (SIJ) with effusion, and a 1-cm by 1-cm collection adjacent to the left psoas muscle; these findings were concerning for septic arthritis (Figure). Based on the MRI study, a computed tomography (CT)-guided arthrocentesis of the left SIJ was performed by an interventional radiologist.
Figure Following the arthrocentesis, the orthopedist referred the patient to the ED. At this presentation,the emergency physician (EP) ordered blood cultures, blood work, urinalysis, and a urinary toxicology screen, and started the patient on IV ceftriaxone and vancomycin. The laboratory studies were significant for the following elevated inflammatory markers: erythrocyte sedimentation rate (ESR), 19 mm/h; C-reactive protein (CRP), 2.45 mg/L; white blood cell count (WBC), 13.6 K/uL with normal differential; and lactate level, 2.6 mg/dL. The toxicology screen was positive for opioids. The basic metabolic panel, chest X-ray, and urinalysis were all unremarkable. An electrocardiogram showed sinus tachycardia.
The patient was admitted to the hospital, and infectious disease services was contacted. While awaiting transport to the inpatient floor, the patient admitted to IV drug use 4 weeks prior to his initial presentation—not the 6 months he initially reported at the first ED visit.
The blood cultures grew Candida parapsilosis, and culture from the SIJ arthrocentesis grew Pseudomonas aeruginosa. The infectious disease physician switched the patient’s antibiotic therapy to IV cefepime and fluconazole. The patient also was seen by an orthopedist, who determined that no surgical intervention was required.
Follow-up laboratory studies showed inflammatory markers peaking at the following levels: ESR, 36 mm/h; CRP, 4.84 mg/L; and WBC, 32.1 K/uL with 90% neutrophils. These markers normalized throughout his hospital stay. The patient was also tested for hepatitis and human immunodeficiency virus, both of which were negative. A transesophageal echocardiogram showed no obvious masses or vegetations.
The patient had an uncomplicated hospital course, and was discharged home on hospital day 6 with a 4-week prescription of oral fluconazole and levofloxacin, and instructed to follow-up with both infectious disease and the orthopedist. To address his history of IV drug use, he also was given follow-up with pain management.
One month later, the patient returned a fourth time to the ED for evaluation of bilateral lower extremity pain and swelling. He stated that he had been mostly bed-bound at home since his discharge from the hospital due to continued pain with weight-bearing.
The patient’s vital signs were normal. The EP ordered a duplex ultrasound study, which showed extensive bilateral lower extremity deep vein thrombosis. He was started on subcutaneous therapeutic enoxaparin and admitted to the inpatient hospital. During admission,a left lower lobe pulmonary artery embolism was found on chest CT angiography, though he had no cardiac or respiratory symptoms. He was discharged home with a 3-month prescription for oral rivaroxaban.
At a 4-month follow-up visit, the patient reported minimal residual disability after completing the course of treatment. During the follow-up, the patient denied using IV heroin; he was referred to a pain management specialist, who placed the patient on methadone.
Discussion
Infectious sacroiliitis (ISI) is a rare form of infectious arthritis affecting the SIJ, with an incidence of 1 to 2 reported cases per year.1 The literature on ISI currently consists only of case reports and case series. This infection is often diagnosed after the disease has progressed, with a mean time to diagnosis of 43.3 days.2
Infectious arthritis of any joint has a prevalence of 2 to 10 per 100,000 people. In 50% of cases, the knee is the joint most commonly affected, followed by the hip, shoulder, and elbow.3 Regardless of location, infectious arthritis is associated with significant morbidity and mortality due to sepsis and irreversible loss of joint function.4
Risk factors for ISI include IV drug use, pregnancy, trauma, endocarditis, and immunosuppression.1 The decision to initiate the workup for ISI can be difficult to make because the condition may present without signs of an infectious etiology, such as toxic appearance, inflammatory changes surrounding the joint, or even fever—only 41% of affected patients in one case series were febrile.2 The workup is often time-consuming, invasive, and expensive.
Although delayed diagnosis and treatment of septic arthritis is associated with significant adverse effects, there is unfortunately no consensus to guide the workup for ISI. As opposed to Kocher’s criteria for the differentiation of septic hip arthritis from transient synovitis in pediatric patients or well-known red-flags for further evaluation of low back pain, physicians are left without much guidance when considering laboratory workup or imaging decisions to evaluate for ISI.
Sacroiliac Joint
As previously noted, the SIJ is not commonly affected by infection. It is a diarthrodial, L-shaped joint comprised of the posterior ilium and sacrum, and is a near-rigid structure with very limited movement that provides stability to the axial skeleton.5 The SIJ is often overlooked as a secondary cause of low back pain in younger patients with rheumatologic conditions (eg, ankylosing spondylitis, Reiter syndrome), pregnancy-associated ligamentous laxity, and osteoarthritis in elderly patients. In one study, 88.2% of sacroiliitis cases were inflammatory, 8.8% infectious, and 2.9% degenerative.6
Signs and Symptoms
As our case illustrates, ISI often presents with nonspecific symptoms and physical findings.7 Patients typically present with fever, painful manipulation of the SIJ, and unilateral lumbo-gluteal pain.2 The components of the history and physical examination suspicious for an infectious etiology include the subacute presentation; unresolved pain despite treatment; tenderness to palpation; decreased range of motion; and recent IV drug use, which increases the risk of infectious disease due to unsterile practices and direct inoculation of pathogens into the bloodstream8 and a further predilection into the axial skeleton. 9 It is important to obtain an accurate social history; however, patients may not be forthright about disclosing sensitive information such as sexual history and illicit drug use.
Physical Assessment
The SIJ is best appreciated in the seated patient by palpating one fingerbreadth medial to the posterior superior iliac spine as he or she slowly bends forward.10 Tenderness elicited while in this position is suggestive of SIJ inflammation. The area of tenderness may be lower than anticipated and lateral to the gluteal cleft, as synovial fluid is typically relegated to the lower half of the joint.
Several adjunctive physical examination maneuvers, such as the Gaenslen test and Flexion Abduction External Rotation test (FABER test or Patrick’s test) can isolate SIJ pathology or dysfunction. The Gaenslen test is performed by asking the patient to lie supine and flex the affected hip and knee, with the lumbar spine flat against the examination table. Hyperextending the contralateral thigh downward will reproduce pain in the affected SIJ.
The FABER test is a simple but less specific examination technique to assess joint pain in the hip, lumbar, and sacroiliac joints.11 In this assessment, the clinician flexes the patient’s affected knee to 90°, externally rotates the hip, and applies downward pressure on the knee. Pain reproduced in the affected SI region is sensitive for joint inflammation.
Laboratory and Imaging Studies
Laboratory studies typically show inconsistent and nonspecific findings, such as the elevated ESR and CRP levels seen in our patient.2,12 Imaging studies to assess the SIJ for signs of infection are therefore essential for confirming infection.
Magnetic resonance imaging is the preferred imaging modality to assess for ISI, since it has the highest sensitivity in visualizing joint effusion and bone marrow edema compared to other modalities. Computed tomography, however, can be helpful in visualizing associated abscesses and guiding arthrocentesis.12 Plain X-ray may not demonstrate early changes in bone.13 The confirmatory study for ISI is synovial fluid analysis and culture.7
Treatment
Infectious sacroiliitis secondary to P aeruginosa, a gram-negative bacillus, is difficult to treat because of the glycocalyx and slime production that protects the pathogen from antibiotics, the development of multiple-antimicrobial resistance, and poor drug penetration into bones and abscesses.14 Antibiotic treatment should cover Staphylococcus aureus and may be broadened to cover gram-negative bacilli. The recommended duration of treatment is at least a 2-week course of IV antibiotics, followed by a 6-week course of oral antibiotics.2 Therapy also includes pain control and surgical intervention for abscesses, osteomyelitis, and refractory cases.7
Complications
Complications and long-term sequelae are common in ISI, often due to late diagnosis of the condition.Our case illustrates the delayed diagnosis of Pseudomonas ISI with candidemia in a young man with a history of IV drug use presenting with atraumatic low back pain. His clinical course was complicated by a thromboembolic event, likely secondary to immobility and a hypercoagulable state from infection and inflammation.15 Infectious sacroiliitis secondary to P aeruginosa is most commonly seen in patients with immunosuppression, hospitalization, and IV drug use.2
Summary
Infectious sacroiliitis remains a diagnostic challenge for physicians due to its rare incidence and nonspecific clinical manifestations. Our case illustrates the importance of maintaining a high level of clinical suspicion for infectious arthritis in young patients presenting with common MSK complaints in the presence of infectious risk factors. Emergency physicians should consider red flags, abnormal vital signs, and patient recidivism when deciding on the most appropriate workup.
Case
A 29-year-old man presented to the ED with a 3-day history of constant left-sided low back pain that radiated to his left buttock and groin. The patient stated the pain worsened with movement, making it difficult for him to walk. He reported lifting heavy boxes at work, but denied any trauma. The patient also denied recent fevers, chills, chest pain, dyspnea, abdominal pain, urinary or fecal incontinence, weakness, numbness, or saddle anesthesia. Regarding his medical history, he had an appendectomy as a child, but reported no other surgeries or medical issues. His social history was significant for narcotic and inhalant use and daily tobacco use. The patient also reported taking heroin intravenously (IV) 6 months prior.
Vital signs at presentation were: heart rate (HR), 92 beats/min; respiratory rate, 15 breaths/min; blood pressure, 118/80 mm Hg; and temperature, 98.2°F. Oxygen saturation was 98% on room air.
The patient was a well-developed young man in no apparent distress. Dermatological examination showed bilateral track marks in the antecubital fossa. The musculoskeletal (MSK) examination demonstrated left gluteal tenderness to palpation and decreased active and passive range of motion of the left hip, especially with internal rotation and flexion. He had no midline tenderness, and the lower extremities had normal pulses and no motor or sensory deficits.
The patient’s pain improved with IV fluids, diazepam, and ketorolac, and he was able to ambulate with assistance. He was clinically diagnosed with sciatica, and discharged home with prescriptions for diazepam and ibuprofen. He was also instructed to follow-up with an orthopedist within 7 days from discharge.
The patient returned to the ED the following day with similar complaints of unabating left-sided pain and difficulty ambulating. His vital signs were notable for an elevated HR of 106 beats/min. Physical examination findings were unchanged from his presentation the previous day, and an X-ray of the lumbar spine showed no abnormalities.
After receiving IV analgesics, the patient’s pain improved and his tachycardia resolved. He was discharged home with instructions to continue taking diazepam, and was also given prescriptions for prednisone and oxycodone/acetaminophen. He was instructed to follow-up with an orthopedist within 24 hours.
Over the next 9 days, the patient was seen twice by an orthopedist, who ordered imaging of the lumbar spine, including a repeat X-ray and contrast-enhanced magnetic resonance imaging (MRI), both of which were unremarkable. The patient completed the prescribed course of diclofenac, oxycodone/acetaminophen, and prednisone, but experienced only minimal pain relief.The orthopedist prescribed the diclofenac to supplement the medication regimen that he was already on.
At the second follow-up visit, the orthopedist ordered an MRI of the patient’s left hip, which demonstrated inflammation of the left sacroiliac joint (SIJ) with effusion, and a 1-cm by 1-cm collection adjacent to the left psoas muscle; these findings were concerning for septic arthritis (Figure). Based on the MRI study, a computed tomography (CT)-guided arthrocentesis of the left SIJ was performed by an interventional radiologist.
Figure Following the arthrocentesis, the orthopedist referred the patient to the ED. At this presentation,the emergency physician (EP) ordered blood cultures, blood work, urinalysis, and a urinary toxicology screen, and started the patient on IV ceftriaxone and vancomycin. The laboratory studies were significant for the following elevated inflammatory markers: erythrocyte sedimentation rate (ESR), 19 mm/h; C-reactive protein (CRP), 2.45 mg/L; white blood cell count (WBC), 13.6 K/uL with normal differential; and lactate level, 2.6 mg/dL. The toxicology screen was positive for opioids. The basic metabolic panel, chest X-ray, and urinalysis were all unremarkable. An electrocardiogram showed sinus tachycardia.
The patient was admitted to the hospital, and infectious disease services was contacted. While awaiting transport to the inpatient floor, the patient admitted to IV drug use 4 weeks prior to his initial presentation—not the 6 months he initially reported at the first ED visit.
The blood cultures grew Candida parapsilosis, and culture from the SIJ arthrocentesis grew Pseudomonas aeruginosa. The infectious disease physician switched the patient’s antibiotic therapy to IV cefepime and fluconazole. The patient also was seen by an orthopedist, who determined that no surgical intervention was required.
Follow-up laboratory studies showed inflammatory markers peaking at the following levels: ESR, 36 mm/h; CRP, 4.84 mg/L; and WBC, 32.1 K/uL with 90% neutrophils. These markers normalized throughout his hospital stay. The patient was also tested for hepatitis and human immunodeficiency virus, both of which were negative. A transesophageal echocardiogram showed no obvious masses or vegetations.
The patient had an uncomplicated hospital course, and was discharged home on hospital day 6 with a 4-week prescription of oral fluconazole and levofloxacin, and instructed to follow-up with both infectious disease and the orthopedist. To address his history of IV drug use, he also was given follow-up with pain management.
One month later, the patient returned a fourth time to the ED for evaluation of bilateral lower extremity pain and swelling. He stated that he had been mostly bed-bound at home since his discharge from the hospital due to continued pain with weight-bearing.
The patient’s vital signs were normal. The EP ordered a duplex ultrasound study, which showed extensive bilateral lower extremity deep vein thrombosis. He was started on subcutaneous therapeutic enoxaparin and admitted to the inpatient hospital. During admission,a left lower lobe pulmonary artery embolism was found on chest CT angiography, though he had no cardiac or respiratory symptoms. He was discharged home with a 3-month prescription for oral rivaroxaban.
At a 4-month follow-up visit, the patient reported minimal residual disability after completing the course of treatment. During the follow-up, the patient denied using IV heroin; he was referred to a pain management specialist, who placed the patient on methadone.
Discussion
Infectious sacroiliitis (ISI) is a rare form of infectious arthritis affecting the SIJ, with an incidence of 1 to 2 reported cases per year.1 The literature on ISI currently consists only of case reports and case series. This infection is often diagnosed after the disease has progressed, with a mean time to diagnosis of 43.3 days.2
Infectious arthritis of any joint has a prevalence of 2 to 10 per 100,000 people. In 50% of cases, the knee is the joint most commonly affected, followed by the hip, shoulder, and elbow.3 Regardless of location, infectious arthritis is associated with significant morbidity and mortality due to sepsis and irreversible loss of joint function.4
Risk factors for ISI include IV drug use, pregnancy, trauma, endocarditis, and immunosuppression.1 The decision to initiate the workup for ISI can be difficult to make because the condition may present without signs of an infectious etiology, such as toxic appearance, inflammatory changes surrounding the joint, or even fever—only 41% of affected patients in one case series were febrile.2 The workup is often time-consuming, invasive, and expensive.
Although delayed diagnosis and treatment of septic arthritis is associated with significant adverse effects, there is unfortunately no consensus to guide the workup for ISI. As opposed to Kocher’s criteria for the differentiation of septic hip arthritis from transient synovitis in pediatric patients or well-known red-flags for further evaluation of low back pain, physicians are left without much guidance when considering laboratory workup or imaging decisions to evaluate for ISI.
Sacroiliac Joint
As previously noted, the SIJ is not commonly affected by infection. It is a diarthrodial, L-shaped joint comprised of the posterior ilium and sacrum, and is a near-rigid structure with very limited movement that provides stability to the axial skeleton.5 The SIJ is often overlooked as a secondary cause of low back pain in younger patients with rheumatologic conditions (eg, ankylosing spondylitis, Reiter syndrome), pregnancy-associated ligamentous laxity, and osteoarthritis in elderly patients. In one study, 88.2% of sacroiliitis cases were inflammatory, 8.8% infectious, and 2.9% degenerative.6
Signs and Symptoms
As our case illustrates, ISI often presents with nonspecific symptoms and physical findings.7 Patients typically present with fever, painful manipulation of the SIJ, and unilateral lumbo-gluteal pain.2 The components of the history and physical examination suspicious for an infectious etiology include the subacute presentation; unresolved pain despite treatment; tenderness to palpation; decreased range of motion; and recent IV drug use, which increases the risk of infectious disease due to unsterile practices and direct inoculation of pathogens into the bloodstream8 and a further predilection into the axial skeleton. 9 It is important to obtain an accurate social history; however, patients may not be forthright about disclosing sensitive information such as sexual history and illicit drug use.
Physical Assessment
The SIJ is best appreciated in the seated patient by palpating one fingerbreadth medial to the posterior superior iliac spine as he or she slowly bends forward.10 Tenderness elicited while in this position is suggestive of SIJ inflammation. The area of tenderness may be lower than anticipated and lateral to the gluteal cleft, as synovial fluid is typically relegated to the lower half of the joint.
Several adjunctive physical examination maneuvers, such as the Gaenslen test and Flexion Abduction External Rotation test (FABER test or Patrick’s test) can isolate SIJ pathology or dysfunction. The Gaenslen test is performed by asking the patient to lie supine and flex the affected hip and knee, with the lumbar spine flat against the examination table. Hyperextending the contralateral thigh downward will reproduce pain in the affected SIJ.
The FABER test is a simple but less specific examination technique to assess joint pain in the hip, lumbar, and sacroiliac joints.11 In this assessment, the clinician flexes the patient’s affected knee to 90°, externally rotates the hip, and applies downward pressure on the knee. Pain reproduced in the affected SI region is sensitive for joint inflammation.
Laboratory and Imaging Studies
Laboratory studies typically show inconsistent and nonspecific findings, such as the elevated ESR and CRP levels seen in our patient.2,12 Imaging studies to assess the SIJ for signs of infection are therefore essential for confirming infection.
Magnetic resonance imaging is the preferred imaging modality to assess for ISI, since it has the highest sensitivity in visualizing joint effusion and bone marrow edema compared to other modalities. Computed tomography, however, can be helpful in visualizing associated abscesses and guiding arthrocentesis.12 Plain X-ray may not demonstrate early changes in bone.13 The confirmatory study for ISI is synovial fluid analysis and culture.7
Treatment
Infectious sacroiliitis secondary to P aeruginosa, a gram-negative bacillus, is difficult to treat because of the glycocalyx and slime production that protects the pathogen from antibiotics, the development of multiple-antimicrobial resistance, and poor drug penetration into bones and abscesses.14 Antibiotic treatment should cover Staphylococcus aureus and may be broadened to cover gram-negative bacilli. The recommended duration of treatment is at least a 2-week course of IV antibiotics, followed by a 6-week course of oral antibiotics.2 Therapy also includes pain control and surgical intervention for abscesses, osteomyelitis, and refractory cases.7
Complications
Complications and long-term sequelae are common in ISI, often due to late diagnosis of the condition.Our case illustrates the delayed diagnosis of Pseudomonas ISI with candidemia in a young man with a history of IV drug use presenting with atraumatic low back pain. His clinical course was complicated by a thromboembolic event, likely secondary to immobility and a hypercoagulable state from infection and inflammation.15 Infectious sacroiliitis secondary to P aeruginosa is most commonly seen in patients with immunosuppression, hospitalization, and IV drug use.2
Summary
Infectious sacroiliitis remains a diagnostic challenge for physicians due to its rare incidence and nonspecific clinical manifestations. Our case illustrates the importance of maintaining a high level of clinical suspicion for infectious arthritis in young patients presenting with common MSK complaints in the presence of infectious risk factors. Emergency physicians should consider red flags, abnormal vital signs, and patient recidivism when deciding on the most appropriate workup.
References
1. Mancarella L, De Santis M, Magarelli N, Ierardi AM, Bonomo L, Ferraccioli G. Septic sacroiliitis: an uncommon septic arthritis. Clin Exp Rheumatol. 2009;27(6):1004-1008. 2. Hermet M, Minichiello E, Flipo RM, et al. Infectious sacroiliitis: a retrospective, multicentre study of 39 adults. BMC Infect Dis. 2012;12:305.doi:10.1186/1471-2334-12-305. 3. Abelson A. Septic Arthritis. Cleveland Clinic. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/septic-arthritis. Published August 2010. Accessed October 28, 2016. 4. Goldenberg DL. Septic arthritis. Lancet. 1998;351(9097):197-202. doi:10.1016/S0140-6736(97)09522-6. 5. Vleeming A, Schuenke MD, Masi AT, Carreiro JE, Danneels L, Willard FH. The sacroiliac joint: an overview of its anatomy, function and potential clinical implications. J Anat. 2012;221(6):537-567. doi:10.1111/j.1469-7580.2012.01564.x. 6. Owlia MB, Danesh-Ardakani M. Frequency of sacroiliitis among patients with low back pain. Electron Physician. 2016;8(3):2094-2100. doi:10.19082/2094. 7. Zimmermann B 3rd, Mikolich DJ, Lally EV. Septic sacroiliitis. Semin Arthritis Rheum. 1996;26(3):592-604. 8. Brtalik D, Pariyadath M. A case report of infectious sacroiliitis in an adult presenting to the emergency department with inability to walk. J Emerg Med. 2017:52(3)e65-e68. doi:10.1016/j.jemermed.2016.10.022. 9. Ferraro K, Cohen MA. Acute septic sacroiliitis in an injection drug user. Am J Emerg Med. 2004;22(1):60-61. 10. Safran M, Botser IB. Hip anatomy and biomechanics. In: Miller MD, Thompson SR, eds. DeLee & Drez’s Orthopaedic Sports Medicine. Vol 2. 4th ed. Philadelphia, PA: Elsevier Saunders; 2015:917-932.e1. 11. LeBlond RF, Brown DD, Suneja M, Szot JF. The spine, pelvic, and extremities. In: LeBlond RF, Brown DD, Suneja M, Szot JF. eds. DeGowin’s Diagnostic Examination. 10th ed. New York, NY: McGraw-Hill; 2015:508-576. 12. Scott KR, Rising KL, Conlon LW. Infectious sacroiliitis. J Emerg Med. 2014;47(3):83-84. doi:10.1016/j.jemermed.2014.05.001. 13. Cinar M, Sanal HT, Yilmaz S, et al. Radiological followup of the evolution of inflammatory process in sacroiliac joint with magnetic resonance imaging: a case with pyogenic sacroiliitis. Case Rep Rheumatol. 2012;2012:509136. doi:10.1155/2012/509136. 14. Calza L, Manfredi R, Marinacci G, Fortunato L, Chiodo F. Community-acquired Pseudomonas aeruginosa sacro-iliitis in a previously healthy patient. J Med Microbiol. 2002;51(7):620-622. 15. Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60(1):26-39.
References
1. Mancarella L, De Santis M, Magarelli N, Ierardi AM, Bonomo L, Ferraccioli G. Septic sacroiliitis: an uncommon septic arthritis. Clin Exp Rheumatol. 2009;27(6):1004-1008. 2. Hermet M, Minichiello E, Flipo RM, et al. Infectious sacroiliitis: a retrospective, multicentre study of 39 adults. BMC Infect Dis. 2012;12:305.doi:10.1186/1471-2334-12-305. 3. Abelson A. Septic Arthritis. Cleveland Clinic. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/rheumatology/septic-arthritis. Published August 2010. Accessed October 28, 2016. 4. Goldenberg DL. Septic arthritis. Lancet. 1998;351(9097):197-202. doi:10.1016/S0140-6736(97)09522-6. 5. Vleeming A, Schuenke MD, Masi AT, Carreiro JE, Danneels L, Willard FH. The sacroiliac joint: an overview of its anatomy, function and potential clinical implications. J Anat. 2012;221(6):537-567. doi:10.1111/j.1469-7580.2012.01564.x. 6. Owlia MB, Danesh-Ardakani M. Frequency of sacroiliitis among patients with low back pain. Electron Physician. 2016;8(3):2094-2100. doi:10.19082/2094. 7. Zimmermann B 3rd, Mikolich DJ, Lally EV. Septic sacroiliitis. Semin Arthritis Rheum. 1996;26(3):592-604. 8. Brtalik D, Pariyadath M. A case report of infectious sacroiliitis in an adult presenting to the emergency department with inability to walk. J Emerg Med. 2017:52(3)e65-e68. doi:10.1016/j.jemermed.2016.10.022. 9. Ferraro K, Cohen MA. Acute septic sacroiliitis in an injection drug user. Am J Emerg Med. 2004;22(1):60-61. 10. Safran M, Botser IB. Hip anatomy and biomechanics. In: Miller MD, Thompson SR, eds. DeLee & Drez’s Orthopaedic Sports Medicine. Vol 2. 4th ed. Philadelphia, PA: Elsevier Saunders; 2015:917-932.e1. 11. LeBlond RF, Brown DD, Suneja M, Szot JF. The spine, pelvic, and extremities. In: LeBlond RF, Brown DD, Suneja M, Szot JF. eds. DeGowin’s Diagnostic Examination. 10th ed. New York, NY: McGraw-Hill; 2015:508-576. 12. Scott KR, Rising KL, Conlon LW. Infectious sacroiliitis. J Emerg Med. 2014;47(3):83-84. doi:10.1016/j.jemermed.2014.05.001. 13. Cinar M, Sanal HT, Yilmaz S, et al. Radiological followup of the evolution of inflammatory process in sacroiliac joint with magnetic resonance imaging: a case with pyogenic sacroiliitis. Case Rep Rheumatol. 2012;2012:509136. doi:10.1155/2012/509136. 14. Calza L, Manfredi R, Marinacci G, Fortunato L, Chiodo F. Community-acquired Pseudomonas aeruginosa sacro-iliitis in a previously healthy patient. J Med Microbiol. 2002;51(7):620-622. 15. Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60(1):26-39.
A 20-year-old woman with no significant medical history presented to the ED with a several-month history of worsening abdominal pain. She reported that although she previously had been evaluated at multiple EDs, no cause of her abdominal pain had been identified. The patient further noted that the pain had significantly increased the day of this presentation.
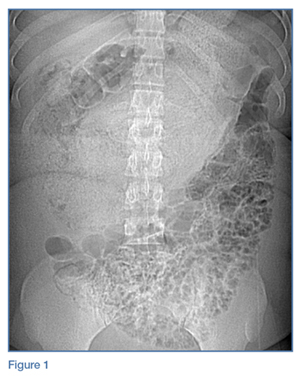
Figure 1
Physical examination revealed guarding and rebound tenderness in the midabdomen. Computed tomography (CT) studies of the abdomen and pelvis were performed; representative scout and axial images of the upper abdomen are shown above (Figures 1 and 2).
Figure 2
What is the suspected diagnosis?
Answer
The scout image of the abdomen revealed a distended stomach (white arrows, Figure 3), which displaced multiple loops of small bowel into the lower abdomen. The axial image through the upper abdomen showed air and solid material within the distended stomach (white arrows, Figure 4). Multiple foci of extraluminal (free) air were seen anteriorly (white asterisks, Figure 4). A coronal reformat of the CT better demonstrated the distended stomach filled with debris (white arrows, Figure 5), extraluminal air (white asterisk, Figure 5), and pneumatosis (air within the walls of multiple small bowel loops; red arrows, Figure 5).
These findings indicated a bowel obstruction and perforation due to the presence of a gastric bezoar. Upon further questioning, the patient admitted to a stress-related habit of eating her own hair (trichophagia) over the past 3 to 4 months.
Figure 3
Bezoars
Gastric bezoars are aggregates of nondigestible material that collect within the gastrointestinal system, usually fruit/vegetable matter (phytobezoars) or hair (trichobezoars). Phytobezoars are most common in patients with a history of reduced gastric motility and/or prior gastric surgery. Trichobezoars, similar to the one seen in this case, typically occur in young women and/or patients with psychiatric illness.1
Gastric bezoars are typically located in the gastric body but may extend into the small bowel and cause bowel obstruction. Trichobezoars that extend into the small bowel are referred to as “Rapunzel syndrome” (based on the fairy tale of the princess with long hair).
Figure 4
Clinical Presentation
Patients with gastric bezoars often present to the ED with nonspecific complaints of abdominal pain, including early satiety, weight loss, signs of anemia, abdominal pain, bloating, and symptoms of small bowel obstruction (SBO).2 Obtaining a thorough history is important to identify trichophagia, as only a small percentage of patients have evidence of alopecia on examination.
Figure 5
Workup
The workup for patients with gastric bezoars typically involves multiple imaging modalities. While abdominal radiography may demonstrate distention of the stomach, these findings are often nonspecific, and the characteristic feature of a mass with a diffusely mottled appearance is visualized in less than 20% of cases.
Computed tomography is the test of choice for detecting a bezoar, with a reported sensitivity of 97%.3 This modality is also useful for assessing the size of a bezoar and evaluating for complications such as SBO, perforation (free-air), or pneumatosis—all of which were revealed on this patient’s CT studies.
Treatment
The treatment for patients with large or obstructing gastric bezoars is surgical resection; both open and laparoscopic techniques have been described in the literature.2,4 The patient in this case was admitted to the hospital, where she underwent surgical removal of the bezoar. She was discharged home on hospital day 6 with outpatient psychiatric follow-up.
References
1. Guniganti P, Bradenham CH, Raptis C, Menias CO, Mellnick VM. Radiographics. 2015;35(7):1909-1921. doi:10.1148/rg.2015150062. 2. Fallon SC, Slater BJ, Larimer EL, Brandt ML, Lopez ME. The surgical management of Rapunzel syndrome: a case series and literature review. J Pediatr Surg. 2013;48(4):830-834. doi:10.1016/j.jpedsurg.2012.07.046. 3. Ripollés T, García-Aguayo J, Martínez MJ, Gil P. Gastrointestinal Bezoars: Sonographic and CT Characteristics. AJR Am J Roentgenol. 2001;177(1):65-69. doi:10.2214/ajr.177.1.1770065. 4. Flaherty DC, Aguilar F, Pradhan B, Grewal H. Rapunzel syndrome due to ingested hair extensions: Surgical and psychiatric considerations. Int J Surg Case Rep. 2015;17:155-157. doi:10.1016/j.ijscr.2015.11.009.
An otherwise healthy 20-year-old woman presented for evaluation of severe chronic abdominal pain.
An otherwise healthy 20-year-old woman presented for evaluation of severe chronic abdominal pain.
A 20-year-old woman with no significant medical history presented to the ED with a several-month history of worsening abdominal pain. She reported that although she previously had been evaluated at multiple EDs, no cause of her abdominal pain had been identified. The patient further noted that the pain had significantly increased the day of this presentation.
Figure 1
Physical examination revealed guarding and rebound tenderness in the midabdomen. Computed tomography (CT) studies of the abdomen and pelvis were performed; representative scout and axial images of the upper abdomen are shown above (Figures 1 and 2).
Figure 2
What is the suspected diagnosis?
Answer
The scout image of the abdomen revealed a distended stomach (white arrows, Figure 3), which displaced multiple loops of small bowel into the lower abdomen. The axial image through the upper abdomen showed air and solid material within the distended stomach (white arrows, Figure 4). Multiple foci of extraluminal (free) air were seen anteriorly (white asterisks, Figure 4). A coronal reformat of the CT better demonstrated the distended stomach filled with debris (white arrows, Figure 5), extraluminal air (white asterisk, Figure 5), and pneumatosis (air within the walls of multiple small bowel loops; red arrows, Figure 5).
These findings indicated a bowel obstruction and perforation due to the presence of a gastric bezoar. Upon further questioning, the patient admitted to a stress-related habit of eating her own hair (trichophagia) over the past 3 to 4 months.
Figure 3
Bezoars
Gastric bezoars are aggregates of nondigestible material that collect within the gastrointestinal system, usually fruit/vegetable matter (phytobezoars) or hair (trichobezoars). Phytobezoars are most common in patients with a history of reduced gastric motility and/or prior gastric surgery. Trichobezoars, similar to the one seen in this case, typically occur in young women and/or patients with psychiatric illness.1
Gastric bezoars are typically located in the gastric body but may extend into the small bowel and cause bowel obstruction. Trichobezoars that extend into the small bowel are referred to as “Rapunzel syndrome” (based on the fairy tale of the princess with long hair).
Figure 4
Clinical Presentation
Patients with gastric bezoars often present to the ED with nonspecific complaints of abdominal pain, including early satiety, weight loss, signs of anemia, abdominal pain, bloating, and symptoms of small bowel obstruction (SBO).2 Obtaining a thorough history is important to identify trichophagia, as only a small percentage of patients have evidence of alopecia on examination.
Figure 5
Workup
The workup for patients with gastric bezoars typically involves multiple imaging modalities. While abdominal radiography may demonstrate distention of the stomach, these findings are often nonspecific, and the characteristic feature of a mass with a diffusely mottled appearance is visualized in less than 20% of cases.
Computed tomography is the test of choice for detecting a bezoar, with a reported sensitivity of 97%.3 This modality is also useful for assessing the size of a bezoar and evaluating for complications such as SBO, perforation (free-air), or pneumatosis—all of which were revealed on this patient’s CT studies.
Treatment
The treatment for patients with large or obstructing gastric bezoars is surgical resection; both open and laparoscopic techniques have been described in the literature.2,4 The patient in this case was admitted to the hospital, where she underwent surgical removal of the bezoar. She was discharged home on hospital day 6 with outpatient psychiatric follow-up.
A 20-year-old woman with no significant medical history presented to the ED with a several-month history of worsening abdominal pain. She reported that although she previously had been evaluated at multiple EDs, no cause of her abdominal pain had been identified. The patient further noted that the pain had significantly increased the day of this presentation.
Figure 1
Physical examination revealed guarding and rebound tenderness in the midabdomen. Computed tomography (CT) studies of the abdomen and pelvis were performed; representative scout and axial images of the upper abdomen are shown above (Figures 1 and 2).
Figure 2
What is the suspected diagnosis?
Answer
The scout image of the abdomen revealed a distended stomach (white arrows, Figure 3), which displaced multiple loops of small bowel into the lower abdomen. The axial image through the upper abdomen showed air and solid material within the distended stomach (white arrows, Figure 4). Multiple foci of extraluminal (free) air were seen anteriorly (white asterisks, Figure 4). A coronal reformat of the CT better demonstrated the distended stomach filled with debris (white arrows, Figure 5), extraluminal air (white asterisk, Figure 5), and pneumatosis (air within the walls of multiple small bowel loops; red arrows, Figure 5).
These findings indicated a bowel obstruction and perforation due to the presence of a gastric bezoar. Upon further questioning, the patient admitted to a stress-related habit of eating her own hair (trichophagia) over the past 3 to 4 months.
Figure 3
Bezoars
Gastric bezoars are aggregates of nondigestible material that collect within the gastrointestinal system, usually fruit/vegetable matter (phytobezoars) or hair (trichobezoars). Phytobezoars are most common in patients with a history of reduced gastric motility and/or prior gastric surgery. Trichobezoars, similar to the one seen in this case, typically occur in young women and/or patients with psychiatric illness.1
Gastric bezoars are typically located in the gastric body but may extend into the small bowel and cause bowel obstruction. Trichobezoars that extend into the small bowel are referred to as “Rapunzel syndrome” (based on the fairy tale of the princess with long hair).
Figure 4
Clinical Presentation
Patients with gastric bezoars often present to the ED with nonspecific complaints of abdominal pain, including early satiety, weight loss, signs of anemia, abdominal pain, bloating, and symptoms of small bowel obstruction (SBO).2 Obtaining a thorough history is important to identify trichophagia, as only a small percentage of patients have evidence of alopecia on examination.
Figure 5
Workup
The workup for patients with gastric bezoars typically involves multiple imaging modalities. While abdominal radiography may demonstrate distention of the stomach, these findings are often nonspecific, and the characteristic feature of a mass with a diffusely mottled appearance is visualized in less than 20% of cases.
Computed tomography is the test of choice for detecting a bezoar, with a reported sensitivity of 97%.3 This modality is also useful for assessing the size of a bezoar and evaluating for complications such as SBO, perforation (free-air), or pneumatosis—all of which were revealed on this patient’s CT studies.
Treatment
The treatment for patients with large or obstructing gastric bezoars is surgical resection; both open and laparoscopic techniques have been described in the literature.2,4 The patient in this case was admitted to the hospital, where she underwent surgical removal of the bezoar. She was discharged home on hospital day 6 with outpatient psychiatric follow-up.
References
1. Guniganti P, Bradenham CH, Raptis C, Menias CO, Mellnick VM. Radiographics. 2015;35(7):1909-1921. doi:10.1148/rg.2015150062. 2. Fallon SC, Slater BJ, Larimer EL, Brandt ML, Lopez ME. The surgical management of Rapunzel syndrome: a case series and literature review. J Pediatr Surg. 2013;48(4):830-834. doi:10.1016/j.jpedsurg.2012.07.046. 3. Ripollés T, García-Aguayo J, Martínez MJ, Gil P. Gastrointestinal Bezoars: Sonographic and CT Characteristics. AJR Am J Roentgenol. 2001;177(1):65-69. doi:10.2214/ajr.177.1.1770065. 4. Flaherty DC, Aguilar F, Pradhan B, Grewal H. Rapunzel syndrome due to ingested hair extensions: Surgical and psychiatric considerations. Int J Surg Case Rep. 2015;17:155-157. doi:10.1016/j.ijscr.2015.11.009.
References
1. Guniganti P, Bradenham CH, Raptis C, Menias CO, Mellnick VM. Radiographics. 2015;35(7):1909-1921. doi:10.1148/rg.2015150062. 2. Fallon SC, Slater BJ, Larimer EL, Brandt ML, Lopez ME. The surgical management of Rapunzel syndrome: a case series and literature review. J Pediatr Surg. 2013;48(4):830-834. doi:10.1016/j.jpedsurg.2012.07.046. 3. Ripollés T, García-Aguayo J, Martínez MJ, Gil P. Gastrointestinal Bezoars: Sonographic and CT Characteristics. AJR Am J Roentgenol. 2001;177(1):65-69. doi:10.2214/ajr.177.1.1770065. 4. Flaherty DC, Aguilar F, Pradhan B, Grewal H. Rapunzel syndrome due to ingested hair extensions: Surgical and psychiatric considerations. Int J Surg Case Rep. 2015;17:155-157. doi:10.1016/j.ijscr.2015.11.009.
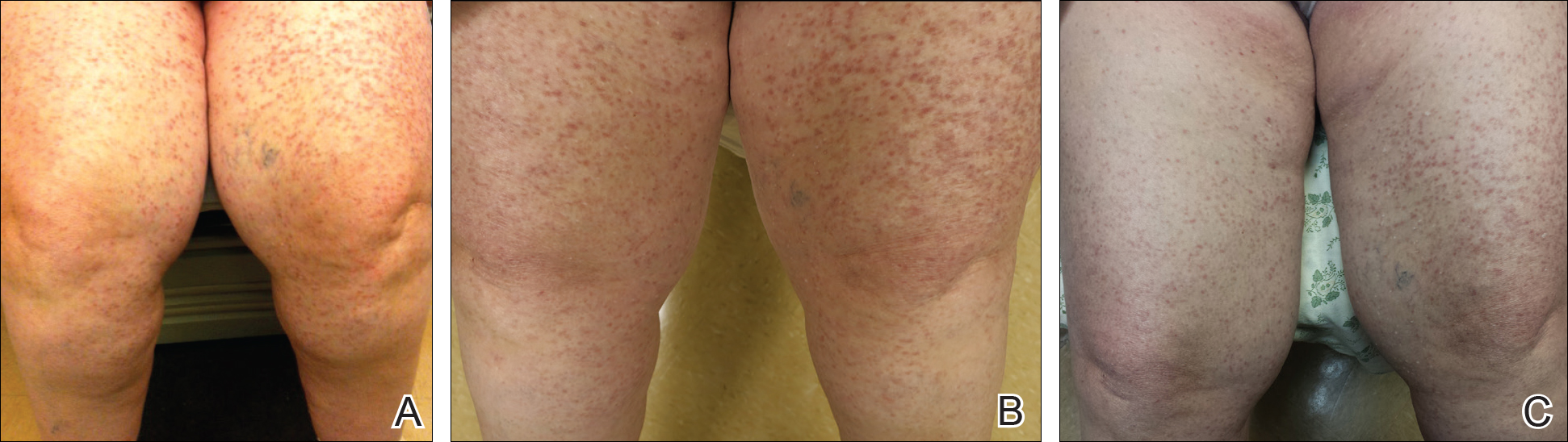
A 57-year-old African American woman was being treated at our clinicfor neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine.Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae,whichwe successfully treated with ciprofloxacin.A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UIother than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Duloxetine has been shown to be effective for the treatment of stress and mixed urinary incontinence. This case suggests it may be useful for neurogenic urinary incontinence, as well.
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categoriesof treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine.Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment,is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrineincrease in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic urinary incontinence would potentially address coexisting conditions, such as pain or depression.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
References
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.
Ark City Clinic, Arkansas City, Kans. (Dr. Keesling); University of Saint Francis, Fort Wayne, Ind. (Dr. Wilson); Fort Wayne Medical Education Program, Ind. (Dr. Wilkins) [email protected]
The authors reported no potential conflict of interest relevant to this article.
Adapted from a poster presentation, Indiana Academy of Family Physicians 2015 Research Day, May 7, 2015, Indianapolis, Ind
Ark City Clinic, Arkansas City, Kans. (Dr. Keesling); University of Saint Francis, Fort Wayne, Ind. (Dr. Wilson); Fort Wayne Medical Education Program, Ind. (Dr. Wilkins) [email protected]
The authors reported no potential conflict of interest relevant to this article.
Adapted from a poster presentation, Indiana Academy of Family Physicians 2015 Research Day, May 7, 2015, Indianapolis, Ind
Author and Disclosure Information
Ark City Clinic, Arkansas City, Kans. (Dr. Keesling); University of Saint Francis, Fort Wayne, Ind. (Dr. Wilson); Fort Wayne Medical Education Program, Ind. (Dr. Wilkins) [email protected]
The authors reported no potential conflict of interest relevant to this article.
Adapted from a poster presentation, Indiana Academy of Family Physicians 2015 Research Day, May 7, 2015, Indianapolis, Ind
A 57-year-old African American woman was being treated at our clinicfor neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine.Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae,whichwe successfully treated with ciprofloxacin.A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UIother than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Duloxetine has been shown to be effective for the treatment of stress and mixed urinary incontinence. This case suggests it may be useful for neurogenic urinary incontinence, as well.
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categoriesof treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine.Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment,is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrineincrease in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic urinary incontinence would potentially address coexisting conditions, such as pain or depression.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
THE CASE
A 57-year-old African American woman was being treated at our clinicfor neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine.Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae,whichwe successfully treated with ciprofloxacin.A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UIother than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Duloxetine has been shown to be effective for the treatment of stress and mixed urinary incontinence. This case suggests it may be useful for neurogenic urinary incontinence, as well.
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categoriesof treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine.Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment,is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrineincrease in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic urinary incontinence would potentially address coexisting conditions, such as pain or depression.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
References
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.
References
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Figure 1. Indolent systemic mastocytosis with red-brown macular and papular lesions on the thighs before (A) and after 20 cycles (B) and 40 cycles (C) of narrowband UVB phototherapy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.
Figure 2. Indolent systemic mastocytosis skin biopsy demonstrating acanthosis and dermal mononuclear cell proliferation (A)(H&E, original magnification ×20) as well as increased mast cell density in the upper dermis (B)(Giemsa, original magnification ×20).
The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
Drs. Husain, Waterman, and DeSimone are from Georgetown University Hospital/Washington Hospital Center, Washington, DC. Dr. Ellison is from the James H. Quillen College of Medicine, East Tennessee State University, Mountain Home.
The authors report no conflict of interest.
Correspondence: Zain Husain, MD, 8803 Old Courthouse Rd, Vienna, VA 22182 ([email protected]).
Drs. Husain, Waterman, and DeSimone are from Georgetown University Hospital/Washington Hospital Center, Washington, DC. Dr. Ellison is from the James H. Quillen College of Medicine, East Tennessee State University, Mountain Home.
The authors report no conflict of interest.
Correspondence: Zain Husain, MD, 8803 Old Courthouse Rd, Vienna, VA 22182 ([email protected]).
Author and Disclosure Information
Drs. Husain, Waterman, and DeSimone are from Georgetown University Hospital/Washington Hospital Center, Washington, DC. Dr. Ellison is from the James H. Quillen College of Medicine, East Tennessee State University, Mountain Home.
The authors report no conflict of interest.
Correspondence: Zain Husain, MD, 8803 Old Courthouse Rd, Vienna, VA 22182 ([email protected]).
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Figure 1. Indolent systemic mastocytosis with red-brown macular and papular lesions on the thighs before (A) and after 20 cycles (B) and 40 cycles (C) of narrowband UVB phototherapy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.
Figure 2. Indolent systemic mastocytosis skin biopsy demonstrating acanthosis and dermal mononuclear cell proliferation (A)(H&E, original magnification ×20) as well as increased mast cell density in the upper dermis (B)(Giemsa, original magnification ×20).
The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Figure 1. Indolent systemic mastocytosis with red-brown macular and papular lesions on the thighs before (A) and after 20 cycles (B) and 40 cycles (C) of narrowband UVB phototherapy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.
Figure 2. Indolent systemic mastocytosis skin biopsy demonstrating acanthosis and dermal mononuclear cell proliferation (A)(H&E, original magnification ×20) as well as increased mast cell density in the upper dermis (B)(Giemsa, original magnification ×20).
The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
Despite standardization of diagnostic criteria by the MSIS for the diagnosis of PJI, some low-grade inflections create a diagnostic challenge for clinicians.
P acnes infection following TJA can be present despite patients having normal serum inflammatory marker levels and synovial fluid aspirations.
Patients with a PJI with low virulence organisms can present with painful, arthrofibrotic joints that do not appear to be clinically infected.
Biopsy for pathology and culture can aid in the diagnosis of suspected PJI in patients who fail to meet MSIS criteria.
If detected and accurately diagnosed, PJI with P acnes can be successfully eradicated with IV antibiotics and 2-stage revision arthroplasty with a good functional outcome.
Total joint arthroplasty (TJA) is a routinely performed, highly efficacious procedure for patients with degenerative osteoarthritis.1,2 In the United States in 2003, more than 450,000 total knee arthroplasties (TKAs) were performed, and this number is projected to increase by more than 673% by 2030, as America’s population continues to age.3 With the increase in primary TJAs has come an increase in revision TJAs. The most common cause of revision TJA is infection (25.2%), which has a rate of 1% to 4% after primary TJA.1,4 Despite advancements in implant technology, preoperative preventive strategies, perioperative techniques, and postoperative management, a recent meta-analysis of patient follow-up data revealed that 15% to 20% of patients remained dissatisfied after TJA, despite having technically well-placed implants.5,6
Recent studies have suggested that prosthetic joint infection (PJI) may be underreported because of the difficulty in diagnosis, which may be one of the reasons why patients remain dissatisfied after TJA.7 As a result, new efforts have been made to develop uniform criteria for PJI diagnosis.8 In 2011, the Musculoskeletal Infection Society (MSIS) developed a new definition for the PJI diagnosis, based on clinical and laboratory criteria, in order to increase diagnostic accuracy. However, MSIS acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-grade infections, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. The biofilm-forming bacteria Propionibacterium acnes and Staphylococcus epidermidis are 2 such low-virulence organisms—once commonly considered contaminants but now recognized as potential pathogens for postoperative joint infections.9 In a review performed at a major orthopedic hospital, Bjerke-Kroll and colleagues10 found that the rate of PJI with P acnes has been increasing linearly over the past 14 years. According to reports in the literature,11-13P acnes has been isolated in 2% to 4% of all cases of PJI, and Zappe and colleagues13 found a P acnes PJI rate of 6% in a retrospective analysis performed at their institution. Given the high rate of P acnes colonization of the axilla, this organism is now increasingly recognized as a cause of infection after shoulder surgery, as found in a case series of 10 patients with P acnes PJI after total shoulder arthroplasty (TSA).14 However, there is still limited data on the role of P acnes in lower extremity PJI.
Although patients with P acnes PJI can present with overt signs of infection, more often they lack systemic or local signs of infection, making the diagnosis difficult.15 Surgeons may not consider PJI as a cause of TJA failure in patients who do not meet diagnostic criteria.7 In a case series of patients with P acnes PJI after TSA, Millett and colleagues14 concluded that erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are not always reliable indicators of infection with low-virulence organisms. Eighty percent of patients in their study had normal ESR and CRP level before surgery. Zappe and colleagues13 reported on P acnes PJI diagnoses in 4 total hip arthroplasties (THAs), 3 TKAs, and 1 TSA. Of the 8 patients, 6 (75%) had borderline elevated CRP levels, and 4 (50%) had normal synovial fluid analysis and cultures from joint aspirations. In a study using electron microscopy and fluorescence in situ hybridization (FISH) labeling, Stoodley and colleagues16 found, in 8 polyethylene liners removed from culture-negative THA patients for aseptic loosening, extensive biofilm colonization with S epidermidis.
Reports of PJI cases misdiagnosed as aseptic loosening also suggest that screening and diagnostic tools are not sensitive enough to detect all infections and that PJI likely is underdiagnosed. In a prospective cohort study, Portillo and colleagues17 categorized patients who were undergoing revision surgery after TJA by cause of failure: aseptic loosening, mechanical failure, or PJI based on current MSIS guidelines. Intraoperative cultures were taken during the revisions. P acnes was isolated in 2 (3%) of the 63 cases classified as PJI and in 12 (19%) of the 63 classified as aseptic loosening. Tsukayama and colleagues18 reported an 11% rate of positive intraoperative cultures for P acnes during revision surgery in cases that the operating surgeon considered aseptic, based on white blood cell (WBC) count, ESR, and CRP level. Rasouli and colleagues19 used an Ibis biosensor to perform polymerase chain reaction (PCR) on synovial fluid from 44 patients who underwent aseptic revision of TKA failures. The authors detected a pathogen in 17 (38%) of the 44 presumed aseptic patients and concluded some aseptic loosening cases are actually chronic low-grade organism PJIs not diagnosed according to current PJI criteria.
In this article, we present the case of a patient with a stiff, painful knee after TKA and with ESR, CRP level, and synovial fluid analysis within normal limits. Open biopsy for cultures showed P acnes PJI, which was successfully treated with 2-stage revision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 69-year-old man with a past medical history of hypertension underwent left primary TKA in 2012. In 2014, he presented to our office complaining of chronic left knee pain and stiffness that had developed insidiously over the first 3 months after surgery and never improved, despite rigorous physical therapy (Table).
With use of an assistive device, he could ambulate for a maximum of 1 city block, and he was on disability from his job as an electrician.
On presentation in 2014, radiographs of the left knee showed a well-seated, well-aligned TKA without any radiographic changes relative to the immediate postoperative radiographs (Figures 1A-1B, 2A-2B). Physical examination revealed no erythema or swelling of the joint. Skin was intact and incision well-healed. Left knee passive range of motion (ROM) was 10° to 30° of flexion and painful. A full infectious work-up was performed. Inflammatory markers were within normal limits: serum WBC count, 5.2 × 103/μL (normal, 4.0-10.5 × 103/uL); ESR, 9 mm/h (normal, <20 mm/h); and CRP, 0.29 mg/dL (normal, <0.8 mg/dL). Synovial fluid aspiration was performed for fluid analysis and cultures. Analysis revealed 422 WBCs/μL with 42% polymorphonuclear neutrophils (PMNs). MSIS criteria for using synovial fluid to diagnose PJI are >3000 WBC cells/uL with >65% PMNs. Cultures from synovial fluid were negative at 8 days of incubation.
Despite not meeting MSIS diagnostic criteria, the patient elected to undergo open biopsy for synovial culture as a last resort. During surgery, there was no purulence in the joint, and frozen section showed <5 neutrophils per high-power field. All cultures from 5 separate synovial tissue samples grew P acnes,confirming the PJI diagnosis. Cultures turned positive after being incubated an average of 12.2 days (range, 10-14 days). Sensitivities showed the organism was responsive to oxacillin. The risks and benefits of 2-stage revision surgery were discussed with the patient at the next office visit, and he decided on 2-stage revision. On November 4, 2014, he underwent open synovectomy, irrigation and débridement with iodine and Dakin solution, hardware removal, and cement antibiotic spacer placement without complication (Figures 3A, 3B).
Intravenous (IV) oxacillin was administered for 6 weeks, as directed by an infectious disease specialist, and the patient was monitored, both clinically and by ESR and CRP level, for signs of infection.
Just before stage 2 revision on January 6, 2015, preoperative inflammatory markers were within normal limits. During surgery, additional cultures were taken from synovial tissue. At 15 days, these cultures showed no growth, confirming eradication of the infection. The patient underwent reimplantation without complication and had an uneventful postoperative course with no wound-healing issues (Figures 4A, 4B).
At 1-month, 3-month, 6-month, and 1-year follow-up, he endorsed significantly improved pain and symptoms. ROM at 1-year follow-up was improved to 5° to 90° of flexion. The patient was ambulating pain-free, without an assistive device, and he had returned to work. He reported being satisfied with having undergone the 2-stage revision.
Discussion
Because PJIs with low-virulence organisms can present with normal levels of inflammatory markers and negative fluid analysis and culture from joint aspirations, they pose a diagnostic challenge for arthroplasty surgeons. In this case report, there was a low index of suspicion for PJI based on radiographic, physical examination, and laboratory findings. Our patient did not meet MSIS diagnostic criteria for PJI before undergoing open biopsy. Initial cultures from joint aspiration of synovial fluid were negative, and inflammatory markers were within normal limits. However, all 5 synovial tissue biopsy specimens that were cultured confirmed a low-grade periprosthetic infection with P acnes—likely the reason for the poor outcome. This case supports Zappe and colleagues13 and Millett and colleagues,14 who found that a subset of patients with a low-grade organism PJI had normal to mildly elevated inflammatory markers and negative fluid analysis and cultures from joint aspirations.
Hardware-involved orthopedic infections are often caused by bacteria that form a biofilm, which can be difficult to culture. Biofilm matrix binds cells into aggregates, which grow only a single colony on culture media, decreasing positive yield. Therefore, synovial fluid cultures are often negative, because of the low number of planktonic cells removed by aspirate. Using FISH and PCR, Stoodley and colleagues16 found biofilm on hardware removed for “culture-negative aseptic loosening.” This is especially important for low-grade organism infections that lack a strong inflammatory response in the joint and that may be missed with traditional screening. This may be one reason our patient’s synovial fluid cultures and inflammatory markers were negative.
Another reason these low-grade infections can be missed is that P acnes is notoriously difficult to culture—it may take up to 15 days to grow in a special medium.20 Intraoperative cultures may be read as false-negative if not incubated the right amount of time. In many hospitals, aerobic and anaerobic cultures are discarded if there is no growth after 3 to 5 days. In our patient’s case, the earliest that cultures turned positive was on day 10—which is consistent with other reports, including one by Butler-Wu and colleagues,15 who suggested a minimum incubation of 13 days for optimal recovery of organisms. Our case highlights the importance of lengthening incubation to allow for growth of low-virulent organisms. Given the different types of management used for PJI and aseptic loosening, it is imperative that surgeons take cultures during revision TJA and that cultures are held up to 14 days to allow enough time for low-virulence organisms to grow.
Fortunately, PJI with low-virulence organisms can be treated successfully. Treating P acnes PJI with exchange arthroplasty and IV antibiotics has documented success rates as high as 92%.21 Again, we emphasize the importance of obtaining intraoperative cultures to determine antibiotic sensitivities, which can guide treatment. Our patient’s infection was eradicated with 2-stage revision and IV antibiotics, and his symptoms, ROM, and function improved significantly.
Diagnosing PJI after TJA can be challenging, as there is no definitive test that is sensitive, specific, rapid, and minimally invasive. Researchers have looked for novel serum or synovial fluid biomarkers that may be elevated in PJI. Synovial interleukin 6 (IL-6) and synovial α-defensin show great promise. In 2 separate studies, elevated IL-6 levels strongly correlated with infection.22,23 Jacovides and colleagues23 found that a synovial IL-6 level higher than 4270 pg/mL had a 100% positive predictive value and a 91% negative predictive value for diagnosing PJI. In some trials, synovial α-defensin has shown up to 100% sensitivity and specificity for PJI diagnosis. Most notably, in a trial by Frangiamore and colleagues,24 α-defensin levels were elevated to statistically significant levels in P acnes PJI, indicating this test may help in diagnosing PJI with low-virulence organisms. Finally, PCR has also shown promise in detecting low-grade joint infections. PCR uses 16 primers that allow not only for the identification of pan-genomic bacterial markers, specific bacterial organisms, and Candida, but also for the presence of antibiotic resistance markers. Use of pan-genomic PCR also allows for detection of a wider variety of pathogens, including organisms commonly missed by conventional culture methods.25Early intervention can significantly improve outcomes in PJI. Therefore, we recommend maintaining a high index of suspicion for low-virulence PJI in patients with chronic pain and decreased functionality after TJA with well-placed implants, despite their not meeting current MSIS diagnostic criteria for PJI. As new microbiological tools for detecting PJI with low-grade organisms are developed, use of these technologies can be incorporated into the diagnosis algorithm. Screening tools more sensitive in detecting low-grade organisms can help avoid the morbidity associated with interoperative synovial biopsies for culture and can allow for more efficient surgical planning. These tools, along with increased clinical awareness of potential PJIs, ultimately will lead to earlier detection, accurate diagnosis, and optimal treatment.
Am J Orthop. 2017;46(3):E148-E153. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
References
1. Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total knee arthroplasty in the United States. Clin Orthop Relat Res. 2010;468(1):45-51.
2. Kamath AF, Ong KL, Lau E, et al. Quantifying the burden of revision total joint arthroplasty for periprosthetic infection. J Arthroplasty. 2015;30(9):1492-1497.
3. Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(suppl 3):144-151.
4. Zmistowski B, Restrepo C, Huang R, Hozack WJ, Parvizi J. Periprosthetic joint infection diagnosis: a complete understanding of white blood cell count and differential. J Arthroplasty. 2012;27(9):1589-1593.
5. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
6. Djahani O, Rainer S, Pietsch M, Hofmann S. Systematic analysis of painful total knee prosthesis, a diagnostic algorithm. Arch Bone Jt Surg. 2013;1(2):48-52.
7. Parvizi J, Suh DH, Jafari SM, Mullan A, Purtill JJ. Aseptic loosening of total hip arthroplasty: infection always should be ruled out. Clin Orthop Relat Res. 2011;469(5):1401-1405.
8. Della Valle C, Parvizi J, Bauer TW, et al. Diagnosis of periprosthetic joint infections of the hip and knee. J Am Acad Orthop Surg. 2010;18(12):760-770.
9. Dramis A, Aldlyami E, Grimer RJ, Dunlop DJ, O’Connell N, Elliott T. What is the significance of a positive Propionibacterium acnes culture around a joint replacement? Int Orthop. 2009;33(3):829-833.
10. Bjerke-Kroll BT, Christ AB, Mclawhorn AS, Sculco PK, Jules-Elysée KM, Sculco TP. Periprosthetic joint infections treated with two-stage revision over 14 years: an evolving microbiology profile. J Arthroplasty. 2014;29(5):877-882.
11. Pandey R, Berendt AR, Athanasou NA. Histological and microbiological findings in non-infected and infected revision arthroplasty tissues. The OSIRIS Collaborative Study Group. Oxford Skeletal Infection Research and Intervention Service. Arch Orthop Trauma Surg. 2000;120(10):570-574.
12. Segawa H, Tsukayama DT, Kyle RF, Becker DA, Gustilo RB. Infection after total knee arthroplasty. A retrospective study of the treatment of eighty-one infections. J Bone Joint Surg Am. 1999;81(10):1434-1445.
13. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
14. Millett PJ, Yen YM, Price CS, Horan MP, van der Meijden OA, Elser F. Propionibacterium acnes infection as an occult cause of postoperative shoulder pain: a case series. Clin Orthop Relat Res. 2011;469(10):2824-2830.
15. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
17. Portillo ME, Salvadó M, Alier A, et al. Prosthesis failure within 2 years of implantation is highly predictive of infection. Clin Orthop Relat Res. 2013;471(11):3672-3678.
18. Tsukayama DT, Strada R, Gustilo RB. Infection after total hip arthroplasty. A study of the treatment of one hundred and six infections. J Bone Joint Surg Am. 1996;78(4):512-523.
19. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
20. Schäfer P, Fink B, Sandow D, Margull A, Berger I, Frommelt L. Prolonged bacterial culture to identify late periprosthetic joint infection: a promising strategy. Clin Infect Dis. 2008;47(11):1403-1409.
21. Zeller V, Ghorbani A, Strady C, Leonard P, Mamoudy P, Desplaces N. Propionibacterium acnes: an agent of prosthetic joint infection and colonization. J Infect. 2007;55(2):119-124.
22. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
23. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
24. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
25. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
Despite standardization of diagnostic criteria by the MSIS for the diagnosis of PJI, some low-grade inflections create a diagnostic challenge for clinicians.
P acnes infection following TJA can be present despite patients having normal serum inflammatory marker levels and synovial fluid aspirations.
Patients with a PJI with low virulence organisms can present with painful, arthrofibrotic joints that do not appear to be clinically infected.
Biopsy for pathology and culture can aid in the diagnosis of suspected PJI in patients who fail to meet MSIS criteria.
If detected and accurately diagnosed, PJI with P acnes can be successfully eradicated with IV antibiotics and 2-stage revision arthroplasty with a good functional outcome.
Total joint arthroplasty (TJA) is a routinely performed, highly efficacious procedure for patients with degenerative osteoarthritis.1,2 In the United States in 2003, more than 450,000 total knee arthroplasties (TKAs) were performed, and this number is projected to increase by more than 673% by 2030, as America’s population continues to age.3 With the increase in primary TJAs has come an increase in revision TJAs. The most common cause of revision TJA is infection (25.2%), which has a rate of 1% to 4% after primary TJA.1,4 Despite advancements in implant technology, preoperative preventive strategies, perioperative techniques, and postoperative management, a recent meta-analysis of patient follow-up data revealed that 15% to 20% of patients remained dissatisfied after TJA, despite having technically well-placed implants.5,6
Recent studies have suggested that prosthetic joint infection (PJI) may be underreported because of the difficulty in diagnosis, which may be one of the reasons why patients remain dissatisfied after TJA.7 As a result, new efforts have been made to develop uniform criteria for PJI diagnosis.8 In 2011, the Musculoskeletal Infection Society (MSIS) developed a new definition for the PJI diagnosis, based on clinical and laboratory criteria, in order to increase diagnostic accuracy. However, MSIS acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-grade infections, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. The biofilm-forming bacteria Propionibacterium acnes and Staphylococcus epidermidis are 2 such low-virulence organisms—once commonly considered contaminants but now recognized as potential pathogens for postoperative joint infections.9 In a review performed at a major orthopedic hospital, Bjerke-Kroll and colleagues10 found that the rate of PJI with P acnes has been increasing linearly over the past 14 years. According to reports in the literature,11-13P acnes has been isolated in 2% to 4% of all cases of PJI, and Zappe and colleagues13 found a P acnes PJI rate of 6% in a retrospective analysis performed at their institution. Given the high rate of P acnes colonization of the axilla, this organism is now increasingly recognized as a cause of infection after shoulder surgery, as found in a case series of 10 patients with P acnes PJI after total shoulder arthroplasty (TSA).14 However, there is still limited data on the role of P acnes in lower extremity PJI.
Although patients with P acnes PJI can present with overt signs of infection, more often they lack systemic or local signs of infection, making the diagnosis difficult.15 Surgeons may not consider PJI as a cause of TJA failure in patients who do not meet diagnostic criteria.7 In a case series of patients with P acnes PJI after TSA, Millett and colleagues14 concluded that erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are not always reliable indicators of infection with low-virulence organisms. Eighty percent of patients in their study had normal ESR and CRP level before surgery. Zappe and colleagues13 reported on P acnes PJI diagnoses in 4 total hip arthroplasties (THAs), 3 TKAs, and 1 TSA. Of the 8 patients, 6 (75%) had borderline elevated CRP levels, and 4 (50%) had normal synovial fluid analysis and cultures from joint aspirations. In a study using electron microscopy and fluorescence in situ hybridization (FISH) labeling, Stoodley and colleagues16 found, in 8 polyethylene liners removed from culture-negative THA patients for aseptic loosening, extensive biofilm colonization with S epidermidis.
Reports of PJI cases misdiagnosed as aseptic loosening also suggest that screening and diagnostic tools are not sensitive enough to detect all infections and that PJI likely is underdiagnosed. In a prospective cohort study, Portillo and colleagues17 categorized patients who were undergoing revision surgery after TJA by cause of failure: aseptic loosening, mechanical failure, or PJI based on current MSIS guidelines. Intraoperative cultures were taken during the revisions. P acnes was isolated in 2 (3%) of the 63 cases classified as PJI and in 12 (19%) of the 63 classified as aseptic loosening. Tsukayama and colleagues18 reported an 11% rate of positive intraoperative cultures for P acnes during revision surgery in cases that the operating surgeon considered aseptic, based on white blood cell (WBC) count, ESR, and CRP level. Rasouli and colleagues19 used an Ibis biosensor to perform polymerase chain reaction (PCR) on synovial fluid from 44 patients who underwent aseptic revision of TKA failures. The authors detected a pathogen in 17 (38%) of the 44 presumed aseptic patients and concluded some aseptic loosening cases are actually chronic low-grade organism PJIs not diagnosed according to current PJI criteria.
In this article, we present the case of a patient with a stiff, painful knee after TKA and with ESR, CRP level, and synovial fluid analysis within normal limits. Open biopsy for cultures showed P acnes PJI, which was successfully treated with 2-stage revision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 69-year-old man with a past medical history of hypertension underwent left primary TKA in 2012. In 2014, he presented to our office complaining of chronic left knee pain and stiffness that had developed insidiously over the first 3 months after surgery and never improved, despite rigorous physical therapy (Table).
With use of an assistive device, he could ambulate for a maximum of 1 city block, and he was on disability from his job as an electrician.
On presentation in 2014, radiographs of the left knee showed a well-seated, well-aligned TKA without any radiographic changes relative to the immediate postoperative radiographs (Figures 1A-1B, 2A-2B). Physical examination revealed no erythema or swelling of the joint. Skin was intact and incision well-healed. Left knee passive range of motion (ROM) was 10° to 30° of flexion and painful. A full infectious work-up was performed. Inflammatory markers were within normal limits: serum WBC count, 5.2 × 103/μL (normal, 4.0-10.5 × 103/uL); ESR, 9 mm/h (normal, <20 mm/h); and CRP, 0.29 mg/dL (normal, <0.8 mg/dL). Synovial fluid aspiration was performed for fluid analysis and cultures. Analysis revealed 422 WBCs/μL with 42% polymorphonuclear neutrophils (PMNs). MSIS criteria for using synovial fluid to diagnose PJI are >3000 WBC cells/uL with >65% PMNs. Cultures from synovial fluid were negative at 8 days of incubation.
Despite not meeting MSIS diagnostic criteria, the patient elected to undergo open biopsy for synovial culture as a last resort. During surgery, there was no purulence in the joint, and frozen section showed <5 neutrophils per high-power field. All cultures from 5 separate synovial tissue samples grew P acnes,confirming the PJI diagnosis. Cultures turned positive after being incubated an average of 12.2 days (range, 10-14 days). Sensitivities showed the organism was responsive to oxacillin. The risks and benefits of 2-stage revision surgery were discussed with the patient at the next office visit, and he decided on 2-stage revision. On November 4, 2014, he underwent open synovectomy, irrigation and débridement with iodine and Dakin solution, hardware removal, and cement antibiotic spacer placement without complication (Figures 3A, 3B).
Intravenous (IV) oxacillin was administered for 6 weeks, as directed by an infectious disease specialist, and the patient was monitored, both clinically and by ESR and CRP level, for signs of infection.
Just before stage 2 revision on January 6, 2015, preoperative inflammatory markers were within normal limits. During surgery, additional cultures were taken from synovial tissue. At 15 days, these cultures showed no growth, confirming eradication of the infection. The patient underwent reimplantation without complication and had an uneventful postoperative course with no wound-healing issues (Figures 4A, 4B).
At 1-month, 3-month, 6-month, and 1-year follow-up, he endorsed significantly improved pain and symptoms. ROM at 1-year follow-up was improved to 5° to 90° of flexion. The patient was ambulating pain-free, without an assistive device, and he had returned to work. He reported being satisfied with having undergone the 2-stage revision.
Discussion
Because PJIs with low-virulence organisms can present with normal levels of inflammatory markers and negative fluid analysis and culture from joint aspirations, they pose a diagnostic challenge for arthroplasty surgeons. In this case report, there was a low index of suspicion for PJI based on radiographic, physical examination, and laboratory findings. Our patient did not meet MSIS diagnostic criteria for PJI before undergoing open biopsy. Initial cultures from joint aspiration of synovial fluid were negative, and inflammatory markers were within normal limits. However, all 5 synovial tissue biopsy specimens that were cultured confirmed a low-grade periprosthetic infection with P acnes—likely the reason for the poor outcome. This case supports Zappe and colleagues13 and Millett and colleagues,14 who found that a subset of patients with a low-grade organism PJI had normal to mildly elevated inflammatory markers and negative fluid analysis and cultures from joint aspirations.
Hardware-involved orthopedic infections are often caused by bacteria that form a biofilm, which can be difficult to culture. Biofilm matrix binds cells into aggregates, which grow only a single colony on culture media, decreasing positive yield. Therefore, synovial fluid cultures are often negative, because of the low number of planktonic cells removed by aspirate. Using FISH and PCR, Stoodley and colleagues16 found biofilm on hardware removed for “culture-negative aseptic loosening.” This is especially important for low-grade organism infections that lack a strong inflammatory response in the joint and that may be missed with traditional screening. This may be one reason our patient’s synovial fluid cultures and inflammatory markers were negative.
Another reason these low-grade infections can be missed is that P acnes is notoriously difficult to culture—it may take up to 15 days to grow in a special medium.20 Intraoperative cultures may be read as false-negative if not incubated the right amount of time. In many hospitals, aerobic and anaerobic cultures are discarded if there is no growth after 3 to 5 days. In our patient’s case, the earliest that cultures turned positive was on day 10—which is consistent with other reports, including one by Butler-Wu and colleagues,15 who suggested a minimum incubation of 13 days for optimal recovery of organisms. Our case highlights the importance of lengthening incubation to allow for growth of low-virulent organisms. Given the different types of management used for PJI and aseptic loosening, it is imperative that surgeons take cultures during revision TJA and that cultures are held up to 14 days to allow enough time for low-virulence organisms to grow.
Fortunately, PJI with low-virulence organisms can be treated successfully. Treating P acnes PJI with exchange arthroplasty and IV antibiotics has documented success rates as high as 92%.21 Again, we emphasize the importance of obtaining intraoperative cultures to determine antibiotic sensitivities, which can guide treatment. Our patient’s infection was eradicated with 2-stage revision and IV antibiotics, and his symptoms, ROM, and function improved significantly.
Diagnosing PJI after TJA can be challenging, as there is no definitive test that is sensitive, specific, rapid, and minimally invasive. Researchers have looked for novel serum or synovial fluid biomarkers that may be elevated in PJI. Synovial interleukin 6 (IL-6) and synovial α-defensin show great promise. In 2 separate studies, elevated IL-6 levels strongly correlated with infection.22,23 Jacovides and colleagues23 found that a synovial IL-6 level higher than 4270 pg/mL had a 100% positive predictive value and a 91% negative predictive value for diagnosing PJI. In some trials, synovial α-defensin has shown up to 100% sensitivity and specificity for PJI diagnosis. Most notably, in a trial by Frangiamore and colleagues,24 α-defensin levels were elevated to statistically significant levels in P acnes PJI, indicating this test may help in diagnosing PJI with low-virulence organisms. Finally, PCR has also shown promise in detecting low-grade joint infections. PCR uses 16 primers that allow not only for the identification of pan-genomic bacterial markers, specific bacterial organisms, and Candida, but also for the presence of antibiotic resistance markers. Use of pan-genomic PCR also allows for detection of a wider variety of pathogens, including organisms commonly missed by conventional culture methods.25Early intervention can significantly improve outcomes in PJI. Therefore, we recommend maintaining a high index of suspicion for low-virulence PJI in patients with chronic pain and decreased functionality after TJA with well-placed implants, despite their not meeting current MSIS diagnostic criteria for PJI. As new microbiological tools for detecting PJI with low-grade organisms are developed, use of these technologies can be incorporated into the diagnosis algorithm. Screening tools more sensitive in detecting low-grade organisms can help avoid the morbidity associated with interoperative synovial biopsies for culture and can allow for more efficient surgical planning. These tools, along with increased clinical awareness of potential PJIs, ultimately will lead to earlier detection, accurate diagnosis, and optimal treatment.
Am J Orthop. 2017;46(3):E148-E153. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
Despite standardization of diagnostic criteria by the MSIS for the diagnosis of PJI, some low-grade inflections create a diagnostic challenge for clinicians.
P acnes infection following TJA can be present despite patients having normal serum inflammatory marker levels and synovial fluid aspirations.
Patients with a PJI with low virulence organisms can present with painful, arthrofibrotic joints that do not appear to be clinically infected.
Biopsy for pathology and culture can aid in the diagnosis of suspected PJI in patients who fail to meet MSIS criteria.
If detected and accurately diagnosed, PJI with P acnes can be successfully eradicated with IV antibiotics and 2-stage revision arthroplasty with a good functional outcome.
Total joint arthroplasty (TJA) is a routinely performed, highly efficacious procedure for patients with degenerative osteoarthritis.1,2 In the United States in 2003, more than 450,000 total knee arthroplasties (TKAs) were performed, and this number is projected to increase by more than 673% by 2030, as America’s population continues to age.3 With the increase in primary TJAs has come an increase in revision TJAs. The most common cause of revision TJA is infection (25.2%), which has a rate of 1% to 4% after primary TJA.1,4 Despite advancements in implant technology, preoperative preventive strategies, perioperative techniques, and postoperative management, a recent meta-analysis of patient follow-up data revealed that 15% to 20% of patients remained dissatisfied after TJA, despite having technically well-placed implants.5,6
Recent studies have suggested that prosthetic joint infection (PJI) may be underreported because of the difficulty in diagnosis, which may be one of the reasons why patients remain dissatisfied after TJA.7 As a result, new efforts have been made to develop uniform criteria for PJI diagnosis.8 In 2011, the Musculoskeletal Infection Society (MSIS) developed a new definition for the PJI diagnosis, based on clinical and laboratory criteria, in order to increase diagnostic accuracy. However, MSIS acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-grade infections, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. The biofilm-forming bacteria Propionibacterium acnes and Staphylococcus epidermidis are 2 such low-virulence organisms—once commonly considered contaminants but now recognized as potential pathogens for postoperative joint infections.9 In a review performed at a major orthopedic hospital, Bjerke-Kroll and colleagues10 found that the rate of PJI with P acnes has been increasing linearly over the past 14 years. According to reports in the literature,11-13P acnes has been isolated in 2% to 4% of all cases of PJI, and Zappe and colleagues13 found a P acnes PJI rate of 6% in a retrospective analysis performed at their institution. Given the high rate of P acnes colonization of the axilla, this organism is now increasingly recognized as a cause of infection after shoulder surgery, as found in a case series of 10 patients with P acnes PJI after total shoulder arthroplasty (TSA).14 However, there is still limited data on the role of P acnes in lower extremity PJI.
Although patients with P acnes PJI can present with overt signs of infection, more often they lack systemic or local signs of infection, making the diagnosis difficult.15 Surgeons may not consider PJI as a cause of TJA failure in patients who do not meet diagnostic criteria.7 In a case series of patients with P acnes PJI after TSA, Millett and colleagues14 concluded that erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are not always reliable indicators of infection with low-virulence organisms. Eighty percent of patients in their study had normal ESR and CRP level before surgery. Zappe and colleagues13 reported on P acnes PJI diagnoses in 4 total hip arthroplasties (THAs), 3 TKAs, and 1 TSA. Of the 8 patients, 6 (75%) had borderline elevated CRP levels, and 4 (50%) had normal synovial fluid analysis and cultures from joint aspirations. In a study using electron microscopy and fluorescence in situ hybridization (FISH) labeling, Stoodley and colleagues16 found, in 8 polyethylene liners removed from culture-negative THA patients for aseptic loosening, extensive biofilm colonization with S epidermidis.
Reports of PJI cases misdiagnosed as aseptic loosening also suggest that screening and diagnostic tools are not sensitive enough to detect all infections and that PJI likely is underdiagnosed. In a prospective cohort study, Portillo and colleagues17 categorized patients who were undergoing revision surgery after TJA by cause of failure: aseptic loosening, mechanical failure, or PJI based on current MSIS guidelines. Intraoperative cultures were taken during the revisions. P acnes was isolated in 2 (3%) of the 63 cases classified as PJI and in 12 (19%) of the 63 classified as aseptic loosening. Tsukayama and colleagues18 reported an 11% rate of positive intraoperative cultures for P acnes during revision surgery in cases that the operating surgeon considered aseptic, based on white blood cell (WBC) count, ESR, and CRP level. Rasouli and colleagues19 used an Ibis biosensor to perform polymerase chain reaction (PCR) on synovial fluid from 44 patients who underwent aseptic revision of TKA failures. The authors detected a pathogen in 17 (38%) of the 44 presumed aseptic patients and concluded some aseptic loosening cases are actually chronic low-grade organism PJIs not diagnosed according to current PJI criteria.
In this article, we present the case of a patient with a stiff, painful knee after TKA and with ESR, CRP level, and synovial fluid analysis within normal limits. Open biopsy for cultures showed P acnes PJI, which was successfully treated with 2-stage revision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 69-year-old man with a past medical history of hypertension underwent left primary TKA in 2012. In 2014, he presented to our office complaining of chronic left knee pain and stiffness that had developed insidiously over the first 3 months after surgery and never improved, despite rigorous physical therapy (Table).
With use of an assistive device, he could ambulate for a maximum of 1 city block, and he was on disability from his job as an electrician.
On presentation in 2014, radiographs of the left knee showed a well-seated, well-aligned TKA without any radiographic changes relative to the immediate postoperative radiographs (Figures 1A-1B, 2A-2B). Physical examination revealed no erythema or swelling of the joint. Skin was intact and incision well-healed. Left knee passive range of motion (ROM) was 10° to 30° of flexion and painful. A full infectious work-up was performed. Inflammatory markers were within normal limits: serum WBC count, 5.2 × 103/μL (normal, 4.0-10.5 × 103/uL); ESR, 9 mm/h (normal, <20 mm/h); and CRP, 0.29 mg/dL (normal, <0.8 mg/dL). Synovial fluid aspiration was performed for fluid analysis and cultures. Analysis revealed 422 WBCs/μL with 42% polymorphonuclear neutrophils (PMNs). MSIS criteria for using synovial fluid to diagnose PJI are >3000 WBC cells/uL with >65% PMNs. Cultures from synovial fluid were negative at 8 days of incubation.
Despite not meeting MSIS diagnostic criteria, the patient elected to undergo open biopsy for synovial culture as a last resort. During surgery, there was no purulence in the joint, and frozen section showed <5 neutrophils per high-power field. All cultures from 5 separate synovial tissue samples grew P acnes,confirming the PJI diagnosis. Cultures turned positive after being incubated an average of 12.2 days (range, 10-14 days). Sensitivities showed the organism was responsive to oxacillin. The risks and benefits of 2-stage revision surgery were discussed with the patient at the next office visit, and he decided on 2-stage revision. On November 4, 2014, he underwent open synovectomy, irrigation and débridement with iodine and Dakin solution, hardware removal, and cement antibiotic spacer placement without complication (Figures 3A, 3B).
Intravenous (IV) oxacillin was administered for 6 weeks, as directed by an infectious disease specialist, and the patient was monitored, both clinically and by ESR and CRP level, for signs of infection.
Just before stage 2 revision on January 6, 2015, preoperative inflammatory markers were within normal limits. During surgery, additional cultures were taken from synovial tissue. At 15 days, these cultures showed no growth, confirming eradication of the infection. The patient underwent reimplantation without complication and had an uneventful postoperative course with no wound-healing issues (Figures 4A, 4B).
At 1-month, 3-month, 6-month, and 1-year follow-up, he endorsed significantly improved pain and symptoms. ROM at 1-year follow-up was improved to 5° to 90° of flexion. The patient was ambulating pain-free, without an assistive device, and he had returned to work. He reported being satisfied with having undergone the 2-stage revision.
Discussion
Because PJIs with low-virulence organisms can present with normal levels of inflammatory markers and negative fluid analysis and culture from joint aspirations, they pose a diagnostic challenge for arthroplasty surgeons. In this case report, there was a low index of suspicion for PJI based on radiographic, physical examination, and laboratory findings. Our patient did not meet MSIS diagnostic criteria for PJI before undergoing open biopsy. Initial cultures from joint aspiration of synovial fluid were negative, and inflammatory markers were within normal limits. However, all 5 synovial tissue biopsy specimens that were cultured confirmed a low-grade periprosthetic infection with P acnes—likely the reason for the poor outcome. This case supports Zappe and colleagues13 and Millett and colleagues,14 who found that a subset of patients with a low-grade organism PJI had normal to mildly elevated inflammatory markers and negative fluid analysis and cultures from joint aspirations.
Hardware-involved orthopedic infections are often caused by bacteria that form a biofilm, which can be difficult to culture. Biofilm matrix binds cells into aggregates, which grow only a single colony on culture media, decreasing positive yield. Therefore, synovial fluid cultures are often negative, because of the low number of planktonic cells removed by aspirate. Using FISH and PCR, Stoodley and colleagues16 found biofilm on hardware removed for “culture-negative aseptic loosening.” This is especially important for low-grade organism infections that lack a strong inflammatory response in the joint and that may be missed with traditional screening. This may be one reason our patient’s synovial fluid cultures and inflammatory markers were negative.
Another reason these low-grade infections can be missed is that P acnes is notoriously difficult to culture—it may take up to 15 days to grow in a special medium.20 Intraoperative cultures may be read as false-negative if not incubated the right amount of time. In many hospitals, aerobic and anaerobic cultures are discarded if there is no growth after 3 to 5 days. In our patient’s case, the earliest that cultures turned positive was on day 10—which is consistent with other reports, including one by Butler-Wu and colleagues,15 who suggested a minimum incubation of 13 days for optimal recovery of organisms. Our case highlights the importance of lengthening incubation to allow for growth of low-virulent organisms. Given the different types of management used for PJI and aseptic loosening, it is imperative that surgeons take cultures during revision TJA and that cultures are held up to 14 days to allow enough time for low-virulence organisms to grow.
Fortunately, PJI with low-virulence organisms can be treated successfully. Treating P acnes PJI with exchange arthroplasty and IV antibiotics has documented success rates as high as 92%.21 Again, we emphasize the importance of obtaining intraoperative cultures to determine antibiotic sensitivities, which can guide treatment. Our patient’s infection was eradicated with 2-stage revision and IV antibiotics, and his symptoms, ROM, and function improved significantly.
Diagnosing PJI after TJA can be challenging, as there is no definitive test that is sensitive, specific, rapid, and minimally invasive. Researchers have looked for novel serum or synovial fluid biomarkers that may be elevated in PJI. Synovial interleukin 6 (IL-6) and synovial α-defensin show great promise. In 2 separate studies, elevated IL-6 levels strongly correlated with infection.22,23 Jacovides and colleagues23 found that a synovial IL-6 level higher than 4270 pg/mL had a 100% positive predictive value and a 91% negative predictive value for diagnosing PJI. In some trials, synovial α-defensin has shown up to 100% sensitivity and specificity for PJI diagnosis. Most notably, in a trial by Frangiamore and colleagues,24 α-defensin levels were elevated to statistically significant levels in P acnes PJI, indicating this test may help in diagnosing PJI with low-virulence organisms. Finally, PCR has also shown promise in detecting low-grade joint infections. PCR uses 16 primers that allow not only for the identification of pan-genomic bacterial markers, specific bacterial organisms, and Candida, but also for the presence of antibiotic resistance markers. Use of pan-genomic PCR also allows for detection of a wider variety of pathogens, including organisms commonly missed by conventional culture methods.25Early intervention can significantly improve outcomes in PJI. Therefore, we recommend maintaining a high index of suspicion for low-virulence PJI in patients with chronic pain and decreased functionality after TJA with well-placed implants, despite their not meeting current MSIS diagnostic criteria for PJI. As new microbiological tools for detecting PJI with low-grade organisms are developed, use of these technologies can be incorporated into the diagnosis algorithm. Screening tools more sensitive in detecting low-grade organisms can help avoid the morbidity associated with interoperative synovial biopsies for culture and can allow for more efficient surgical planning. These tools, along with increased clinical awareness of potential PJIs, ultimately will lead to earlier detection, accurate diagnosis, and optimal treatment.
Am J Orthop. 2017;46(3):E148-E153. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
References
1. Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total knee arthroplasty in the United States. Clin Orthop Relat Res. 2010;468(1):45-51.
2. Kamath AF, Ong KL, Lau E, et al. Quantifying the burden of revision total joint arthroplasty for periprosthetic infection. J Arthroplasty. 2015;30(9):1492-1497.
3. Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(suppl 3):144-151.
4. Zmistowski B, Restrepo C, Huang R, Hozack WJ, Parvizi J. Periprosthetic joint infection diagnosis: a complete understanding of white blood cell count and differential. J Arthroplasty. 2012;27(9):1589-1593.
5. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
6. Djahani O, Rainer S, Pietsch M, Hofmann S. Systematic analysis of painful total knee prosthesis, a diagnostic algorithm. Arch Bone Jt Surg. 2013;1(2):48-52.
7. Parvizi J, Suh DH, Jafari SM, Mullan A, Purtill JJ. Aseptic loosening of total hip arthroplasty: infection always should be ruled out. Clin Orthop Relat Res. 2011;469(5):1401-1405.
8. Della Valle C, Parvizi J, Bauer TW, et al. Diagnosis of periprosthetic joint infections of the hip and knee. J Am Acad Orthop Surg. 2010;18(12):760-770.
9. Dramis A, Aldlyami E, Grimer RJ, Dunlop DJ, O’Connell N, Elliott T. What is the significance of a positive Propionibacterium acnes culture around a joint replacement? Int Orthop. 2009;33(3):829-833.
10. Bjerke-Kroll BT, Christ AB, Mclawhorn AS, Sculco PK, Jules-Elysée KM, Sculco TP. Periprosthetic joint infections treated with two-stage revision over 14 years: an evolving microbiology profile. J Arthroplasty. 2014;29(5):877-882.
11. Pandey R, Berendt AR, Athanasou NA. Histological and microbiological findings in non-infected and infected revision arthroplasty tissues. The OSIRIS Collaborative Study Group. Oxford Skeletal Infection Research and Intervention Service. Arch Orthop Trauma Surg. 2000;120(10):570-574.
12. Segawa H, Tsukayama DT, Kyle RF, Becker DA, Gustilo RB. Infection after total knee arthroplasty. A retrospective study of the treatment of eighty-one infections. J Bone Joint Surg Am. 1999;81(10):1434-1445.
13. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
14. Millett PJ, Yen YM, Price CS, Horan MP, van der Meijden OA, Elser F. Propionibacterium acnes infection as an occult cause of postoperative shoulder pain: a case series. Clin Orthop Relat Res. 2011;469(10):2824-2830.
15. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
17. Portillo ME, Salvadó M, Alier A, et al. Prosthesis failure within 2 years of implantation is highly predictive of infection. Clin Orthop Relat Res. 2013;471(11):3672-3678.
18. Tsukayama DT, Strada R, Gustilo RB. Infection after total hip arthroplasty. A study of the treatment of one hundred and six infections. J Bone Joint Surg Am. 1996;78(4):512-523.
19. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
20. Schäfer P, Fink B, Sandow D, Margull A, Berger I, Frommelt L. Prolonged bacterial culture to identify late periprosthetic joint infection: a promising strategy. Clin Infect Dis. 2008;47(11):1403-1409.
21. Zeller V, Ghorbani A, Strady C, Leonard P, Mamoudy P, Desplaces N. Propionibacterium acnes: an agent of prosthetic joint infection and colonization. J Infect. 2007;55(2):119-124.
22. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
23. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
24. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
25. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
References
1. Bozic KJ, Kurtz SM, Lau E, et al. The epidemiology of revision total knee arthroplasty in the United States. Clin Orthop Relat Res. 2010;468(1):45-51.
2. Kamath AF, Ong KL, Lau E, et al. Quantifying the burden of revision total joint arthroplasty for periprosthetic infection. J Arthroplasty. 2015;30(9):1492-1497.
3. Kurtz SM, Ong KL, Schmier J, et al. Future clinical and economic impact of revision total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(suppl 3):144-151.
4. Zmistowski B, Restrepo C, Huang R, Hozack WJ, Parvizi J. Periprosthetic joint infection diagnosis: a complete understanding of white blood cell count and differential. J Arthroplasty. 2012;27(9):1589-1593.
5. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
6. Djahani O, Rainer S, Pietsch M, Hofmann S. Systematic analysis of painful total knee prosthesis, a diagnostic algorithm. Arch Bone Jt Surg. 2013;1(2):48-52.
7. Parvizi J, Suh DH, Jafari SM, Mullan A, Purtill JJ. Aseptic loosening of total hip arthroplasty: infection always should be ruled out. Clin Orthop Relat Res. 2011;469(5):1401-1405.
8. Della Valle C, Parvizi J, Bauer TW, et al. Diagnosis of periprosthetic joint infections of the hip and knee. J Am Acad Orthop Surg. 2010;18(12):760-770.
9. Dramis A, Aldlyami E, Grimer RJ, Dunlop DJ, O’Connell N, Elliott T. What is the significance of a positive Propionibacterium acnes culture around a joint replacement? Int Orthop. 2009;33(3):829-833.
10. Bjerke-Kroll BT, Christ AB, Mclawhorn AS, Sculco PK, Jules-Elysée KM, Sculco TP. Periprosthetic joint infections treated with two-stage revision over 14 years: an evolving microbiology profile. J Arthroplasty. 2014;29(5):877-882.
11. Pandey R, Berendt AR, Athanasou NA. Histological and microbiological findings in non-infected and infected revision arthroplasty tissues. The OSIRIS Collaborative Study Group. Oxford Skeletal Infection Research and Intervention Service. Arch Orthop Trauma Surg. 2000;120(10):570-574.
12. Segawa H, Tsukayama DT, Kyle RF, Becker DA, Gustilo RB. Infection after total knee arthroplasty. A retrospective study of the treatment of eighty-one infections. J Bone Joint Surg Am. 1999;81(10):1434-1445.
13. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
14. Millett PJ, Yen YM, Price CS, Horan MP, van der Meijden OA, Elser F. Propionibacterium acnes infection as an occult cause of postoperative shoulder pain: a case series. Clin Orthop Relat Res. 2011;469(10):2824-2830.
15. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
17. Portillo ME, Salvadó M, Alier A, et al. Prosthesis failure within 2 years of implantation is highly predictive of infection. Clin Orthop Relat Res. 2013;471(11):3672-3678.
18. Tsukayama DT, Strada R, Gustilo RB. Infection after total hip arthroplasty. A study of the treatment of one hundred and six infections. J Bone Joint Surg Am. 1996;78(4):512-523.
19. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
20. Schäfer P, Fink B, Sandow D, Margull A, Berger I, Frommelt L. Prolonged bacterial culture to identify late periprosthetic joint infection: a promising strategy. Clin Infect Dis. 2008;47(11):1403-1409.
21. Zeller V, Ghorbani A, Strady C, Leonard P, Mamoudy P, Desplaces N. Propionibacterium acnes: an agent of prosthetic joint infection and colonization. J Infect. 2007;55(2):119-124.
22. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
23. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
24. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
25. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
Hydralazine-induced antineutrophilcytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
Figure 1. Erosions of the lower lip with ulceration and eschar of the distal aspect of the tongue (A) and multiple hemorrhagic and clear tense vesicles on the palm and fingers (B) in a patient with hydralazine-associated cutaneous vasculitis.
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virusscreening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.
Figure 2. Hydralazine-associated cutaneous vasculitis. A skin biopsy showed a striking necrotizing vascular reaction characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (H&E, original magnification ×200). Emanating from the zones of necrotizing leukocytoclastic vasculitis were marked extravascular neutrophilic infiltrates assuming a sheetlike pattern within the dermis in a fashion reminiscent of Sweet syndrome.
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
References
Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J(Clin Res Ed). 1981;282:1121-1122.
Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
From Weill Cornell Medical College, New York, New York. Drs. Levin and Harp are from the Department of Dermatology, Dr. Magro is from the Department of Pathology, and Dr. Horowitz is from the Department of Medicine.
The authors report no conflict of interest.
Correspondence: Laura Englander Levin, MD, Weill Cornell Medical College, Department of Dermatology, 1305 York Ave, 9th Floor, New York, NY 10021 ([email protected]).
From Weill Cornell Medical College, New York, New York. Drs. Levin and Harp are from the Department of Dermatology, Dr. Magro is from the Department of Pathology, and Dr. Horowitz is from the Department of Medicine.
The authors report no conflict of interest.
Correspondence: Laura Englander Levin, MD, Weill Cornell Medical College, Department of Dermatology, 1305 York Ave, 9th Floor, New York, NY 10021 ([email protected]).
Author and Disclosure Information
From Weill Cornell Medical College, New York, New York. Drs. Levin and Harp are from the Department of Dermatology, Dr. Magro is from the Department of Pathology, and Dr. Horowitz is from the Department of Medicine.
The authors report no conflict of interest.
Correspondence: Laura Englander Levin, MD, Weill Cornell Medical College, Department of Dermatology, 1305 York Ave, 9th Floor, New York, NY 10021 ([email protected]).
Hydralazine-induced antineutrophilcytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
Figure 1. Erosions of the lower lip with ulceration and eschar of the distal aspect of the tongue (A) and multiple hemorrhagic and clear tense vesicles on the palm and fingers (B) in a patient with hydralazine-associated cutaneous vasculitis.
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virusscreening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.
Figure 2. Hydralazine-associated cutaneous vasculitis. A skin biopsy showed a striking necrotizing vascular reaction characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (H&E, original magnification ×200). Emanating from the zones of necrotizing leukocytoclastic vasculitis were marked extravascular neutrophilic infiltrates assuming a sheetlike pattern within the dermis in a fashion reminiscent of Sweet syndrome.
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
Hydralazine-induced antineutrophilcytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
Figure 1. Erosions of the lower lip with ulceration and eschar of the distal aspect of the tongue (A) and multiple hemorrhagic and clear tense vesicles on the palm and fingers (B) in a patient with hydralazine-associated cutaneous vasculitis.
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virusscreening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.
Figure 2. Hydralazine-associated cutaneous vasculitis. A skin biopsy showed a striking necrotizing vascular reaction characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (H&E, original magnification ×200). Emanating from the zones of necrotizing leukocytoclastic vasculitis were marked extravascular neutrophilic infiltrates assuming a sheetlike pattern within the dermis in a fashion reminiscent of Sweet syndrome.
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
References
Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J(Clin Res Ed). 1981;282:1121-1122.
Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
References
Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J(Clin Res Ed). 1981;282:1121-1122.
Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
Hydralazine-induced small vessel vasculitis has a characteristic pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration, along with involvement of the aerodigestive tract.
Unlike systemic lupus erythematosus (SLE), hydralazine-induced vasculitis is associated with positive antineutrophil cytoplasmic antibodies, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.
Increased recognition of the clinical and serological features of hydralazine-induced small vessel vasculitis may lead to earlier recognition of this disease and decreased time to discontinuation of hydralazine when appropriate.
Preoperative orthogonal radiographs need to be carefully scrutinized in irreducible fractures.
Open reduction is often necessary when soft-tissue or bone is interposed in a fracture site.
The fibula’s size and interosseous connection to the tibia can lead to entrapment.
High-energy mechanisms can lead to significant deformity at time of injury, with spontaneous partial reduction prior to initial assessment.
Consider intramedullary entrapment of adjacent long bones.
The tibia is the most commonly fractured long bone; each year, almost 500,000 tibia fractures occur in the United States alone.1 Low-energy mechanisms of injury usually result from torsional forces and produce less comminuted fractures. Very high-energy injuries apply direct forces to the shin and are often highly comminuted or open, owing to the limited soft-tissue envelope. The extent of soft-tissue injury occurring with these fractures is the best predictor of the development of a complication, particularly nonunion or infection.1 Low-energy closed fractures with limited comminution and sufficient cortical apposition may be treated with closed reduction and casting, but the most common treatment for tibial shaft fractures is intramedullary nailing.2
In the acute setting, closed reduction allows for temporization of soft tissues and prevention of further damage to neurovascular structures. Whether eventual treatment consists of casting or intramedullary fixation, closed reduction must first be achieved.
In this article, we report a unique case of tibial shaft fracture irreducibility caused by telescoping of the distal fibula within the proximal tibial diaphysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 23-year-old unrestrained driver of a tow truck rear-ended another vehicle at high speed (~45 mph) and became trapped in the vehicle. Extrication time was prolonged. The driver was brought to the emergency department at a level I trauma center, where he was found to have an obvious closed deformity of the right lower leg but remained neurovascularly intact. Radiographs showed a highly comminuted fracture of the right tibial and fibular midshaft with more than 100% medial displacement of the distal fragment (Figure 1).
Immediate closed reduction was performed in the emergency department, with the patient under sedation. The procedure improved gross realignment and reduced tension from the overlying soft tissue. Postreduction radiographs showed improved alignment that, with some displacement and angulation remaining, was deemed sufficient to temporize the injury until surgery the next morning.
The next morning, the patient was taken to the operating room for planned closed intramedullary nailing through a suprapatellar approach. After entry to the tibial medullary canal was obtained, a ball-tipped guide wire was advanced proximal to the fracture site, until significant resistance was met. With the aid of intraoperative fluoroscopic imaging, it was determined that the distal fibula fracture segment was entrapped in the medullary canal of the proximal tibia (Figure 2).
Closed reduction and removal of the fibula fragment were attempted multiple times, to no avail. A limited open approach was deemed necessary, and an incision was made anterior to the fracture site. The soft tissue over the fracture segment was bluntly dissected until the point of intussusception was palpated and visualized. A bone hook was used to dislodge the fibula from the tibial diaphysis (Figure 3).
After the fibula was liberated, additional closed manipulative reduction techniques were used on the tibia to restore length, alignment, and rotation, and an appropriately sized nail was advanced distally past the fracture segment and fixed with interlocking screws proximally and distally (Figure 4).
The postoperative course was uncomplicated. At most recent follow-up, radiographs showed near anatomical alignment, and the patient was back to normal activities without use of any pain medication or assistive device.
Discussion
Although other irreducible tibia fracture patterns have been described, midshaft tibia fracture irreducibility caused by entrapment of the fibula within the intramedullary space was a previously unreported difficulty of this very common fracture. More commonly, irreducible tibia fractures are caused by entrapment of soft-tissue structures or fracture fragments.3,4
There are no previous documented cases of fracture patterns in which one long bone telescopes into another. Although not previously reported as occurring traumatically, the fibula was previously used as a vascularized graft in the intramedullary canal of the tibia and elsewhere.
The most well described irreducible fracture-dislocation of the lower leg is the Bosworth type, in which the proximal fragment of the fibula becomes displaced behind the tibia at the ankle joint. In addition, because of the torsional nature of most ankle fractures and the multitude of accompanying soft-tissue structures crossing at the joint, entrapment leading to irreducibility has had several different causes. These soft-tissue entrapment injures are often difficult to distinguish on plain radiographs and require further definition by computed tomography.
The high-energy mechanism of injury we have described is thought to result from lateral translation of the upper portion of the leg with the foot and lower leg fixed in place. We can posit that momentary hypervarus angulation of the tibia and the fibula with subsequent spontaneous partial reduction caused by tissue elasticity could lead to this unique injury pattern.
Although this is the first reported case of entrapment of the fibula within the intramedullary canal of the tibia, the injury should be considered when difficult closed reductions are encountered. It was only after attempted reduction for intramedullary nailing in the operating room that the telescoping fibula and the irreducibility were identified, and open reduction performed. There were no soft-tissue or neurovascular complications, but, had there been, they could have become of urgent concern and altered treatment.
In this case report, we have described a unique fracture pattern that could cause significant morbidity if not appropriately identified and treated in a timely manner.
Am J Orthop. 2017;46(3):E160-E162. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
References
1. Karladani AH, Granhed H, Kärrholm J, Styf J. The influence of fracture etiology and type on fracture healing: a review of 104 consecutive tibial shaft fractures. Arch Orthop Trauma Surg. 2001;121(6):325-328.
2. McGanity P. Tibial shaft fractures. In: Heckman JD, Schenck RC Jr, Agarwal A, eds. Current Orthopedic Diagnosis & Treatment. New York, NY: Springer; 2000:184-185.
3. Ermis MN, Yagmurlu MF, Kilinc AS, Karakas ES. Irreducible fracture dislocation of the ankle caused by tibialis posterior tendon interposition. J Foot Ankle Surg. 2010;49(2):166-171.
4. Green RN, Pullagura MK, Holland JP. Irreducible fracture-dislocation of the knee. Acta Orthop Traumatol Turc. 2014;48(3):363-366.
Preoperative orthogonal radiographs need to be carefully scrutinized in irreducible fractures.
Open reduction is often necessary when soft-tissue or bone is interposed in a fracture site.
The fibula’s size and interosseous connection to the tibia can lead to entrapment.
High-energy mechanisms can lead to significant deformity at time of injury, with spontaneous partial reduction prior to initial assessment.
Consider intramedullary entrapment of adjacent long bones.
The tibia is the most commonly fractured long bone; each year, almost 500,000 tibia fractures occur in the United States alone.1 Low-energy mechanisms of injury usually result from torsional forces and produce less comminuted fractures. Very high-energy injuries apply direct forces to the shin and are often highly comminuted or open, owing to the limited soft-tissue envelope. The extent of soft-tissue injury occurring with these fractures is the best predictor of the development of a complication, particularly nonunion or infection.1 Low-energy closed fractures with limited comminution and sufficient cortical apposition may be treated with closed reduction and casting, but the most common treatment for tibial shaft fractures is intramedullary nailing.2
In the acute setting, closed reduction allows for temporization of soft tissues and prevention of further damage to neurovascular structures. Whether eventual treatment consists of casting or intramedullary fixation, closed reduction must first be achieved.
In this article, we report a unique case of tibial shaft fracture irreducibility caused by telescoping of the distal fibula within the proximal tibial diaphysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 23-year-old unrestrained driver of a tow truck rear-ended another vehicle at high speed (~45 mph) and became trapped in the vehicle. Extrication time was prolonged. The driver was brought to the emergency department at a level I trauma center, where he was found to have an obvious closed deformity of the right lower leg but remained neurovascularly intact. Radiographs showed a highly comminuted fracture of the right tibial and fibular midshaft with more than 100% medial displacement of the distal fragment (Figure 1).
Immediate closed reduction was performed in the emergency department, with the patient under sedation. The procedure improved gross realignment and reduced tension from the overlying soft tissue. Postreduction radiographs showed improved alignment that, with some displacement and angulation remaining, was deemed sufficient to temporize the injury until surgery the next morning.
The next morning, the patient was taken to the operating room for planned closed intramedullary nailing through a suprapatellar approach. After entry to the tibial medullary canal was obtained, a ball-tipped guide wire was advanced proximal to the fracture site, until significant resistance was met. With the aid of intraoperative fluoroscopic imaging, it was determined that the distal fibula fracture segment was entrapped in the medullary canal of the proximal tibia (Figure 2).
Closed reduction and removal of the fibula fragment were attempted multiple times, to no avail. A limited open approach was deemed necessary, and an incision was made anterior to the fracture site. The soft tissue over the fracture segment was bluntly dissected until the point of intussusception was palpated and visualized. A bone hook was used to dislodge the fibula from the tibial diaphysis (Figure 3).
After the fibula was liberated, additional closed manipulative reduction techniques were used on the tibia to restore length, alignment, and rotation, and an appropriately sized nail was advanced distally past the fracture segment and fixed with interlocking screws proximally and distally (Figure 4).
The postoperative course was uncomplicated. At most recent follow-up, radiographs showed near anatomical alignment, and the patient was back to normal activities without use of any pain medication or assistive device.
Discussion
Although other irreducible tibia fracture patterns have been described, midshaft tibia fracture irreducibility caused by entrapment of the fibula within the intramedullary space was a previously unreported difficulty of this very common fracture. More commonly, irreducible tibia fractures are caused by entrapment of soft-tissue structures or fracture fragments.3,4
There are no previous documented cases of fracture patterns in which one long bone telescopes into another. Although not previously reported as occurring traumatically, the fibula was previously used as a vascularized graft in the intramedullary canal of the tibia and elsewhere.
The most well described irreducible fracture-dislocation of the lower leg is the Bosworth type, in which the proximal fragment of the fibula becomes displaced behind the tibia at the ankle joint. In addition, because of the torsional nature of most ankle fractures and the multitude of accompanying soft-tissue structures crossing at the joint, entrapment leading to irreducibility has had several different causes. These soft-tissue entrapment injures are often difficult to distinguish on plain radiographs and require further definition by computed tomography.
The high-energy mechanism of injury we have described is thought to result from lateral translation of the upper portion of the leg with the foot and lower leg fixed in place. We can posit that momentary hypervarus angulation of the tibia and the fibula with subsequent spontaneous partial reduction caused by tissue elasticity could lead to this unique injury pattern.
Although this is the first reported case of entrapment of the fibula within the intramedullary canal of the tibia, the injury should be considered when difficult closed reductions are encountered. It was only after attempted reduction for intramedullary nailing in the operating room that the telescoping fibula and the irreducibility were identified, and open reduction performed. There were no soft-tissue or neurovascular complications, but, had there been, they could have become of urgent concern and altered treatment.
In this case report, we have described a unique fracture pattern that could cause significant morbidity if not appropriately identified and treated in a timely manner.
Am J Orthop. 2017;46(3):E160-E162. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
Preoperative orthogonal radiographs need to be carefully scrutinized in irreducible fractures.
Open reduction is often necessary when soft-tissue or bone is interposed in a fracture site.
The fibula’s size and interosseous connection to the tibia can lead to entrapment.
High-energy mechanisms can lead to significant deformity at time of injury, with spontaneous partial reduction prior to initial assessment.
Consider intramedullary entrapment of adjacent long bones.
The tibia is the most commonly fractured long bone; each year, almost 500,000 tibia fractures occur in the United States alone.1 Low-energy mechanisms of injury usually result from torsional forces and produce less comminuted fractures. Very high-energy injuries apply direct forces to the shin and are often highly comminuted or open, owing to the limited soft-tissue envelope. The extent of soft-tissue injury occurring with these fractures is the best predictor of the development of a complication, particularly nonunion or infection.1 Low-energy closed fractures with limited comminution and sufficient cortical apposition may be treated with closed reduction and casting, but the most common treatment for tibial shaft fractures is intramedullary nailing.2
In the acute setting, closed reduction allows for temporization of soft tissues and prevention of further damage to neurovascular structures. Whether eventual treatment consists of casting or intramedullary fixation, closed reduction must first be achieved.
In this article, we report a unique case of tibial shaft fracture irreducibility caused by telescoping of the distal fibula within the proximal tibial diaphysis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 23-year-old unrestrained driver of a tow truck rear-ended another vehicle at high speed (~45 mph) and became trapped in the vehicle. Extrication time was prolonged. The driver was brought to the emergency department at a level I trauma center, where he was found to have an obvious closed deformity of the right lower leg but remained neurovascularly intact. Radiographs showed a highly comminuted fracture of the right tibial and fibular midshaft with more than 100% medial displacement of the distal fragment (Figure 1).
Immediate closed reduction was performed in the emergency department, with the patient under sedation. The procedure improved gross realignment and reduced tension from the overlying soft tissue. Postreduction radiographs showed improved alignment that, with some displacement and angulation remaining, was deemed sufficient to temporize the injury until surgery the next morning.
The next morning, the patient was taken to the operating room for planned closed intramedullary nailing through a suprapatellar approach. After entry to the tibial medullary canal was obtained, a ball-tipped guide wire was advanced proximal to the fracture site, until significant resistance was met. With the aid of intraoperative fluoroscopic imaging, it was determined that the distal fibula fracture segment was entrapped in the medullary canal of the proximal tibia (Figure 2).
Closed reduction and removal of the fibula fragment were attempted multiple times, to no avail. A limited open approach was deemed necessary, and an incision was made anterior to the fracture site. The soft tissue over the fracture segment was bluntly dissected until the point of intussusception was palpated and visualized. A bone hook was used to dislodge the fibula from the tibial diaphysis (Figure 3).
After the fibula was liberated, additional closed manipulative reduction techniques were used on the tibia to restore length, alignment, and rotation, and an appropriately sized nail was advanced distally past the fracture segment and fixed with interlocking screws proximally and distally (Figure 4).
The postoperative course was uncomplicated. At most recent follow-up, radiographs showed near anatomical alignment, and the patient was back to normal activities without use of any pain medication or assistive device.
Discussion
Although other irreducible tibia fracture patterns have been described, midshaft tibia fracture irreducibility caused by entrapment of the fibula within the intramedullary space was a previously unreported difficulty of this very common fracture. More commonly, irreducible tibia fractures are caused by entrapment of soft-tissue structures or fracture fragments.3,4
There are no previous documented cases of fracture patterns in which one long bone telescopes into another. Although not previously reported as occurring traumatically, the fibula was previously used as a vascularized graft in the intramedullary canal of the tibia and elsewhere.
The most well described irreducible fracture-dislocation of the lower leg is the Bosworth type, in which the proximal fragment of the fibula becomes displaced behind the tibia at the ankle joint. In addition, because of the torsional nature of most ankle fractures and the multitude of accompanying soft-tissue structures crossing at the joint, entrapment leading to irreducibility has had several different causes. These soft-tissue entrapment injures are often difficult to distinguish on plain radiographs and require further definition by computed tomography.
The high-energy mechanism of injury we have described is thought to result from lateral translation of the upper portion of the leg with the foot and lower leg fixed in place. We can posit that momentary hypervarus angulation of the tibia and the fibula with subsequent spontaneous partial reduction caused by tissue elasticity could lead to this unique injury pattern.
Although this is the first reported case of entrapment of the fibula within the intramedullary canal of the tibia, the injury should be considered when difficult closed reductions are encountered. It was only after attempted reduction for intramedullary nailing in the operating room that the telescoping fibula and the irreducibility were identified, and open reduction performed. There were no soft-tissue or neurovascular complications, but, had there been, they could have become of urgent concern and altered treatment.
In this case report, we have described a unique fracture pattern that could cause significant morbidity if not appropriately identified and treated in a timely manner.
Am J Orthop. 2017;46(3):E160-E162. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
References
1. Karladani AH, Granhed H, Kärrholm J, Styf J. The influence of fracture etiology and type on fracture healing: a review of 104 consecutive tibial shaft fractures. Arch Orthop Trauma Surg. 2001;121(6):325-328.
2. McGanity P. Tibial shaft fractures. In: Heckman JD, Schenck RC Jr, Agarwal A, eds. Current Orthopedic Diagnosis & Treatment. New York, NY: Springer; 2000:184-185.
3. Ermis MN, Yagmurlu MF, Kilinc AS, Karakas ES. Irreducible fracture dislocation of the ankle caused by tibialis posterior tendon interposition. J Foot Ankle Surg. 2010;49(2):166-171.
4. Green RN, Pullagura MK, Holland JP. Irreducible fracture-dislocation of the knee. Acta Orthop Traumatol Turc. 2014;48(3):363-366.
References
1. Karladani AH, Granhed H, Kärrholm J, Styf J. The influence of fracture etiology and type on fracture healing: a review of 104 consecutive tibial shaft fractures. Arch Orthop Trauma Surg. 2001;121(6):325-328.
2. McGanity P. Tibial shaft fractures. In: Heckman JD, Schenck RC Jr, Agarwal A, eds. Current Orthopedic Diagnosis & Treatment. New York, NY: Springer; 2000:184-185.
3. Ermis MN, Yagmurlu MF, Kilinc AS, Karakas ES. Irreducible fracture dislocation of the ankle caused by tibialis posterior tendon interposition. J Foot Ankle Surg. 2010;49(2):166-171.
4. Green RN, Pullagura MK, Holland JP. Irreducible fracture-dislocation of the knee. Acta Orthop Traumatol Turc. 2014;48(3):363-366.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear.The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsyof the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung,likely due to the underlying cavitary lesion (Figure 2).
Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic masswithin the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3).
Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms. The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine.
Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade and with equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment.
Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis andtimely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high platelet-derived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies.
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso.biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear.The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsyof the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung,likely due to the underlying cavitary lesion (Figure 2).
Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic masswithin the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3).
Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms. The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine.
Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade and with equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment.
Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis andtimely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high platelet-derived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear.The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsyof the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung,likely due to the underlying cavitary lesion (Figure 2).
Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic masswithin the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3).
Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms. The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine.
Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade and with equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment.
Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis andtimely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high platelet-derived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies.
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso.biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso.biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Issue
The Journal of Community and Supportive Oncology - 15(2)
Issue
The Journal of Community and Supportive Oncology - 15(2)